
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 235)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FDA Approves Vitekta (elvitegravir) for HIV-1 Infection
FDA Approves Vitekta (elvitegravir) for HIV-1 Infection
September 24, 2014 — The U.S. Food and Drug Administration (FDA) has approved Vitekta (elvitegravir), an integrase strand transfer inhibitor for the combination treatment of human immunodeficiency virus type 1 (HIV-1) infection in treatment-experienced adults.

Elvitegravir
697761-98-1 CAS

Elvitegravir (EVG, formerly GS-9137) is a drug used for the treatment of HIV infection. It acts as an integrase inhibitor. It was developed[1] by the pharmaceutical company Gilead Sciences, which licensed EVG from Japan Tobacco in March 2008.[2][3][4] The drug gained approval by U.S. Food and Drug Administration on August 27, 2012 for use in adult patients starting HIV treatment for the first time as part of the fixed dose combination known as Stribild.[5]
According to the results of the phase II clinical trial, patients taking once-daily elvitegravir boosted by ritonavir had greater reductions in viral load after 24 weeks compared to individuals randomized to receive a ritonavir-boosted protease inhibitor.[6]
。

Human immunodeficiency virus type 1 (HIV-1) is the causative agent of acquired immunodeficiency disease syndrome (AIDS). After over 26 years of efforts, there is still not a therapeutic cure or an effective vaccine against HIV/AIDS. The clinical management of HIV-1 infected people largely relies on antiretroviral therapy (ART). Although highly active antiretroviral therapy (HAART) has provided an effective way to treat AIDS patients, the huge burden of ART in developing countries, together with the increasing incidence of drug resistant viruses among treated people, calls for continuous efforts for the development of anti-HIV-1 drugs. Currently, four classes of over 30 licensed antiretrovirals (ARVs) and combination regimens of these ARVs are in use clinically including: reverse transcriptase inhibitors (RTIs) (e.g. nucleoside reverse transcriptase inhibitors, NRTIs; and non-nucleoside reverse transcriptase inhibitors, NNRTIs), protease inhibitors (PIs), integrase inhibitors and entry inhibitors (e.g. fusion inhibitors and CCR5 antagonists).

- Gilead Press Release Phase III Clinical Trial of Elvitegravir July 22, 2008
- Gilead Press Release Gilead and Japan Tobacco Sign Licensing Agreement for Novel HIV Integrase Inhibitor March 22, 2008
- Shimura K, Kodama E, Sakagami Y, et al. (2007). “Broad Anti-Retroviral Activity and Resistance Profile of a Novel Human Immunodeficiency Virus Integrase Inhibitor, Elvitegravir (JTK-303/GS-9137)”. J Virol 82 (2): 764. doi:10.1128/JVI.01534-07. PMC 2224569. PMID 17977962.
- Stellbrink HJ (2007). “Antiviral drugs in the treatment of AIDS: what is in the pipeline ?”. Eur. J. Med. Res. 12 (9): 483–95. PMID 17933730.
- Sax, P. E.; Dejesus, E.; Mills, A.; Zolopa, A.; Cohen, C.; Wohl, D.; Gallant, J. E.; Liu, H. C.; Zhong, L.; Yale, K.; White, K.; Kearney, B. P.; Szwarcberg, J.; Quirk, E.; Cheng, A. K.; Gs-Us-236-0102 Study, T. (2012). “Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir versus co-formulated efavirenz, emtricitabine, and tenofovir for initial treatment of HIV-1 infection: A randomised, double-blind, phase 3 trial, analysis of results after 48 weeks”.The Lancet 379 (9835): 2439–2448. doi:10.1016/S0140-6736(12)60917-9. PMID 22748591. edit
- Thaczuk, Derek and Carter, Michael. ICAAC: Best response to elvitegravir seen when used with T-20 and other active agents Aidsmap.com. 19 Sept. 2007.

The life cycle of HIV-1. 1. HIV-1 gp120 binds to CD4 and co-receptor CCR5/CXCR4 on target cell; 2. HIV-1 gp41 mediates fusion with target cell; 3. Nucleocapsid containing viral genome and enzymes enters cells; 4. Viral genome and enzymes are released; 5. Viral reverse transcriptase catalyzes reverse transcription of ssRNA, forming RNA-DNA hybrids; 6. RNA template is degraded by ribonuclease H followed by the synthesis of HIV dsDNA; 7. Viral dsDNA is transported into the nucleus and integrated into the host chromosomal DNA by the viral integrase enzyme; 8. Transcription of proviral DNA into genomic ssRNA and mRNAs formation after processing; 9. Viral RNA is exported to cytoplasm; 10. Synthesis of viral precursor proteins under the catalysis of host-cell ribosomes; 11. Viral protease cleaves the precursors into viral proteins; 12. HIV ssRNA and proteins assemble under host cell membrane, into which gp120 and gp41 are inserted; 13. Membrane of host-cell buds out, forming the viral envelope; 14. Matured viral particle is released
Elvitegravir, also known as GS 9137 or JTK 303, is an investigational new drug and a novel oral integrase inhibitor that is being evaluated for the treatment of HIV-1 infection. After HIVs genetic material is deposited inside a cell, its RNA must be converted (reverse transcribed) into DNA. A viral enzyme called integrase then helps to hide HIVs DNA inside the cell’s DNA. Once this happens, the cell can begin producing genetic material for new viruses. Integrase inhibitors, such as elvitegravir, are designed to block the activity of the integrase enzyme and to prevent HIV DNA from entering healthy cell DNA. Elvitegravir has the chemical name: 6-(3-chloro-2-fluorobenzyl)-1-[(S)-1 -hydroxy -methyl-2- methylpropyl]-7-methoxy-4-oxo-1, 4-dihydroquinoline-3-carboxylic acid and has the following structural formula:

WO 2000040561 , WO 2000040563 and WO 2001098275 disclose 4-oxo-1 , 4-dihydro-3- quinoline which is useful as antiviral agents. WO2004046115 provides certain 4- oxoquinoline compounds that are useful as HIV Integrase inhibitors.
US 7176220 patent discloses elvitegravir, solvate, stereoisomer, tautomer, pharmaceutically acceptable salt thereof or pharmaceutical composition containing them and their method of treatment. The chemistry involved in the above said patent is depicted below in the Scheme A. Scheme-A
Toluene, DIPEA
SOCl2 ,COCl (S)-(+)-Valinol
Toluene
,4-Difluoro-5-iodo- benzoic acid


THF
dichlorobis(triphenylphosphine)
palladium argon stream,

Elvitegravir Form ] Elvitegravir (residue) US 7635704 patent discloses certain specific crystalline forms of elvitegravir. The specific crystalline forms are reported to have superior physical and chemical stability compared to other physical forms of the compound. Further, process for the preparation of elvitegravir also disclosed and is depicted below in the Scheme B. The given processes involve the isolation of the intermediates at almost all the stages.
Scheme B
2,
–

Zn THF,
CK Br THF CU “ZnBr dιchlorobis(trιphenylphos
phine)palladium

Elvitegravir WO 2007102499 discloses a compound which is useful as an intermediate for the synthesis of an anti-HIV agent having an integrase-inhibiting activity; a process for production of the compound; and a process for production of an anti-HIV agent using the intermediate.
WO 2009036161 also discloses synthetic processes and synthetic intermediates that can be used to prepare 4-oxoquinolone compounds having useful integrase inhibiting properties.
The said processes are tedious in making and the purity of the final compound is affected because of the number of steps, their isolation, purification etc., thus, there is a need for new synthetic methods for producing elvitegravir which process is cost effective, easy to practice, increase the yield and purity of the final compound, or that eliminate the use of toxic or costly reagents.
US Patent No 7176220 discloses Elvitegravir, solvate, stereoisomer, tautomer, pharmaceutically acceptable salt thereof or pharmaceutical composition containing them and ■ their method of treatment. US Patent No 7635704 discloses Elvitegravir Form II, Form III and processes for their preparation. The process for the preparation of Form Il disclosed in the said patent is mainly by three methods – a) dissolution of Elvitegravir followed by seeding with Form II, b) recrystallisation of Elvitegravir, and c) anti-solvent method.
The process for the preparation of Form III in the said patent is mainly by three methods – a) dissolution of Form Il in isobutyl acetate by heating followed by cooling the reaction mass, b) dissolution of Form Il in isobutyl acetate by heating followed by seeding with Form III, and c) dissolving Form Il in 2-propanol followed by seeding with Form III.
Amorphous materials are becoming more prevalent in the pharmaceutical industry. In order to overcome the solubility and potential bioavailability issues, amorphous solid forms are becoming front-runners. Of special importance is the distinction between amorphous and crystalline forms, as they have differing implications on drug substance stability, as well as drug product stability and efficacy.
An estimated 50% of all drug molecules used in medicinal therapy are administered as salts. A drug substance often has certain suboptimal physicochemical or biopharmaceutical properties that can be overcome by pairing a basic or acidic drug molecule with a counter- ion to create a salt version of the drug. The process is a simple way to modify the properties of a drug with ionizable functional groups to overcome undesirable features of the parent drug. Salt forms of drugs have a large effect on the drugs’ quality, safety, and performance. The properties of salt-forming species significantly affect the pharmaceutical properties of a drug and can greatly benefit chemists and formulators in various facets of drug discovery and development.



chemical synthesis from a carboxylic acid 1 starts after conversion to the acid chloride iodide NIS 2 , and with three condensation 4 . 4 and the amino alcohol 5 addition-elimination reaction occurs 6 , 6 off under alkaline conditions with TBS protected hydroxy get the ring 7 , 7 and zinc reagent 8 Negishi coupling occurs to get 9 , the last 9 hydrolysis and methoxylated
Elvitegravir dimer impurity, WO2011004389A2
Isolation of 1-[(2S)-1-({3-carboxy-6-(3-chloro-2-fluorobenzyl)-1 -[(2S)-I- hydroxy-3-methylbutan-2-yl]-4-oxo-1 , 4-dihydroquinolin-7-yl}oxy)-3- methylbutan-2-yl 6-(3-chloro-2-fluorobenzyl)-7-methoxy-4-oxo-1 , 4-dihydroquinoline-3-carboxylic acid (elvitegravir dimer impurity, 13)
After isolation of the elvitegravir from the mixture of ethyl acetate-hexane, solvent from the filtrate was removed under reduced pressure. The resultant residue purified by column chromatography using a mixture of ethyl acetate-hexane (gradient, 20-80% EtOAc in hexane) as an eluent. Upon concentration of the required fractions, a thick solid was obtained which was further purified on slurry washing with ethyl acetate to get pure elvitegravir dimer impurity (13). The 1H-NMR, 13C-NMR and mass spectral data complies with proposed structure.

1H-NMR (DMSO-Cf6, 300 MHz, ppm) – δ 0.79 (m, d=6.3 Hz, 6H, 20 & 2O’)\ 1.18 & 1.20 (d, J=6.3 Hz & J=6.2 Hz, 6H, 21 & 21′)1, 2.42-2.49 (m, 2H, 19 & 19′), 3.81-3.89 (m, 3H, T & 17’Ha), 3.94-4.01 (m, 1 H, 17’Hb), 4.01 (s, 3H, 23), 4.11 (s, 2H, 7), 4.83-4.85 (m, 3H, 17 & 18′), 5.22 (t, J=4.7 Hz, 1H, OH), 5.41-5.44 (m, 1 H, 18), 6.73-6.78 (t, J=7.1 Hz, 1 H, 11)1‘ 2, 6.92-6.98 (t, J=8.0 Hz, 1H, 3′) 1‘2, 7.12-7.22 (m, 2H, 1 & 3), 7.34-7.39 (m, 1H, 2′),
7.45-7.48 (m, 1 H, 2), 7.49, 7.56 (s, 2H, 15 & 15′), 7.99, 8.02 (s, 2H, 9 & 9′), 8.89, 9.01 (s, 2H, 13 & 13′), 15.30, 15.33 (s, 2H, COOH’ & COOH”).
13C-NMR (DMSO-Cf6, 75 MHz, ppm)- δ 18.87, 19.03 (2OC, 20’C), 19.11 , 19.24 (21 C, 21 ‘C), 27.94 (7’C), 28.40 (7C), 28.91 , 30.08 (19C, 19’C), 56.80(23C), 60.11 (171C), 63.59 (18C), 66.52 (18’C), 68.53 (17C), 97.86, 98.97 (15, 15′), 107.43, 108.16 (12C, 12’C),
118.77, 119.38 (1OC, 10’C), 119.57 (d, J=17.6 Hz, 41C), 119.61 (d, J=17.9 Hz, 4C),
124.88 (d, J=4.3 Hz, 31C), 125.18 (d, J=4.2 Hz, 3C), 126.59, 126.96 (9C1 9’C), 127.14 (8’C), 127.62 (d, J=15.9 Hz, 61C), 127.73 (8C), 127.99 (d, J=15.2 Hz, 6C), 128.66 (2’C),
128.84 (11C), 128.84 (2C), 130.03 (d, J=3.4 Hz, 1C), 142.14, 142.44 (14C, 14’C), 144.37, 145.56 (13C, 131C), 155.24 (d, J=245.1 Hz, 5’C)1 155.61 (d, J=245.1 Hz, 5C),
160.17 (16’C), 162.04 (16C), 166.00, 166.14 (22C, 22’C), 176.17, 176.22 (11C, 111C).
DIP MS: m/z (%)- 863 [M+H]+, 885 [M+Na]+.

MAKE IN INDIA
http://makeinindia.com/sector/pharmaceuticals/

FDA Approves Spiriva Respimat (tiotropium) for the Maintenance Treatment of COPD

Ridgefield, Conn., September 25, 2014 – Boehringer Ingelheim Pharmaceuticals, Inc. announced today that the U.S. Food and Drug Administration (FDA) approved Spiriva Respimat (tiotropium bromide) inhalation spray for the long-term, once-daily maintenance treatment of bronchospasm associated with chronic obstructive pulmonary disease (COPD), including chronic bronchitis and emphysema and to reduce exacerbations in COPD patients. Boehringer Ingelheim anticipates Spiriva Respimat to be available in January 2015.

Spiriva Respimat provides a pre-measured amount of medicine in a slow-moving mist that helps patients inhale the medicine. Spiriva Respimat was developed to actively deliver medication in a way that does not depend of how fast air is breathed in from the inhaler.
READ AT



MAKE IN INDIA
FDA Approves Tybost (cobicistat) for use in the treatment of HIV-1 Infection

Cobicistat, GS-9350
1004316-88-4
| C 40 H 53 N 7 O 5 S 2 |
N-[1(R)-Benzyl-4(R)-[2(S)-[3-(2-isopropylthiazol-4-ylmethyl)-3-methyl]ureido]-4-(4-morpholinyl)butyramido]-5-phenylpentyl]carbamic acid thiazol-5-ylmethyl ester
(1,3-thiazol-5-yl) methyl (5S, 8R, 11R) -8,11-dibenzyl-2-methyl-5-[2 – (morpholin-4-yl) ethyl] -1 – [2 – (propan-2-yl) -1,3-thiazol-4-yl] -3,6-dioxo-2 ,4,7,12-tetraazatridecan-13-oate
cytochrome P450 3A4 (CYP3A4) inhibitor
FDA Approves Tybost (cobicistat) for use in the treatment of HIV-1 Infection
September 24, 2014 — The U.S. Food and Drug Administration (FDA) has approved Tybost (cobicistat), a CYP3A inhibitor used in combination with atazanavir or darunavir for the treatment of human immunodeficiency virus type 1 (HIV-1) infection
Cobicistat is a pharmacokinetic enhancer that works by inhibiting the enzyme (CYP3A) that metabolizes atazanavir and darunavir. It increases the systemic exposure of these drugs and prolongs their effect. Cobicistat is also one of the ingredients in the combination HIV drug Stribild, which was approved by the FDA in August, 2012.
Tybost comes in 150 mg tablets and is administered once daily in combination with the protease inhibitors atazanavir (Reyataz), or darunavir (Prezista).
Because Tybost inhibits CYP3A, other medications metabolized by CYP3A may result in increased plasma concentrations and potentially severe side effects, which may be life-threatening or even fatal. Extra care should be exercised by healthcare professionals to ensure than other medications are reviewed and their concentrations monitored, especially when initiating new medicines or changing doses.
The approval of Tybost was based on the following clinical trials:
•The data to support the use of atazanavir and Tybost were from a phase 2 and 3 trial in treatment-naïve adults comparing atazanavir/cobicistat 300/150 mg and atazanavir/ritonavir 300/100 mg once daily each in combination with Truvada. The atazanavir/cobicistat based regimen was non-inferior to the atazanavir/ritonavir based regimen.
•The data to support the use of cobicistat with darunavir is from a multiple dose trial in healthy subjects comparing the relative bioavailability of darunavir/cobicistat 800/150 mg to darunavir/ritonavir 800/100 mg.
The most common adverse drug reactions observed with Tybost (in combination with atazanavir) in clinical trials were jaundice, ocular icterus, and nausea.
Tybost is a product of Gilead Sciences, Foster City, CA.

Cobicistat (formerly GS-9350) is a licensed drug for use in the treatment of infection with the human immunodeficiency virus (HIV).
Like ritonavir (Norvir), cobicistat is of interest not for its anti-HIV properties, but rather its ability to inhibit liver enzymes that metabolize other medications used to treat HIV, notablyelvitegravir, an HIV integrase inhibitor currently under investigation itself. By combining cobicistat with elvitegravir, higher concentrations of elvitgravir are achieved in the body with lower dosing, theoretically enhancing elvitgravir’s viral suppression while diminishing its adverse side-effects. In contrast with ritonavir, the only currently approved booster, cobicistat has no anti-HIV activity of its own.[1]
Cobicistat, a cytochrome P450 CYP3A4 inhibitor, was approved in the E.U. in 2013 as a pharmacokinetic enhancer of the HIV-1 protease inhibitors atazanavir and darunavir in adults. First launch took place in 2014 in United Kingdom. In 2012, Gilead filed a New Drug Application in the U.S. for the same indication. In April 2013, the FDA issued a Complete Response Letter from the FDA. In 2014 the FDA accepted Gilead’s resubmission.
Cobicistat is a component of the four-drug, fixed-dose combination HIV treatmentelvitegravir/cobicistat/emtricitabine/tenofovir (known as the “Quad Pill” or Stribild).[1][2] The Quad Pill/Stribild was approved by the FDA in August 2012 for use in the United States and is owned by Gilead Sciences.
Cobicistat is a potent inhibitor of cytochrome P450 3A enzymes, including the importantCYP3A4 subtype. It also inhibits intestinal transport proteins, increasing the overall absorption of several HIV medications, including atazanavir, darunavir and tenofovir alafenamide fumarate.[3]
The drug candidate acts as a pharmaco-enhancer to boost exposure of HIV protease inhibitors. In 2011, cobicistat was licensed to Japan Tobacco by Gilead for development and commercialization in Japan as a stand-alone product for the treatment of HIV infection. In 2012, orphan drug designation was assigned in Japan for the pharmacokinetic enhancement of anti-HIV agent.
Oxidative metabolism by cytochrome P450 enzymes is one of the primary mechanisms of drug metabolism.. It can be difficult to maintain therapeutically effective blood plasma levels of drugs which are rapidly metabolized by cytochrome P450 enzymes. Accordingly, the blood plasma levels of drugs which are susceptible to cytochrome P450 enzyme degradation can be maintained or enhanced by co-administration of cytochrome P450 inhibitors, thereby improving the pharmacokinetics of the drug.
While certain drugs are known to inhibit cytochrome P450 enzymes, more and/or improved inhibitors for cytochrome P450 monooxygenase are desirable. Particularly, it would be desirable to have cytochrome P450 monooxygenase inhibitors which do not have appreciable biological activity other than cytochrome P450 inhibition. Such inhibitors can be useful for minimizing undesirable biological activity, e.g., side effects. In addition, it would be desirable to have P450 monooxygenase inhibitors that lack significant or have a reduced level of protease inhibitor activity. Such inhibitors could be useful for enhancing the effectiveness of antiretroviral drugs, while minimizing the possibility of eliciting viral resistance, especially against protease inhibitors.
…………………………….
Cobicistat (GS-9350): A potent and selective inhibitor of human CYP3A as a novel pharmacoenhancer
ACS Med Chem Lett 2010, 1(5): 209
http://pubs.acs.org/doi/abs/10.1021/ml1000257
http://pubs.acs.org/doi/suppl/10.1021/ml1000257/suppl_file/ml1000257_si_001.pdf

Cobicistat (3, GS-9350) is a newly discovered, potent, and selective inhibitor of human cytochrome P450 3A (CYP3A) enzymes. In contrast to ritonavir, 3 is devoid of anti-HIV activity and is thus more suitable for use in boosting anti-HIV drugs without risking selection of potential drug-resistant HIV variants. Compound 3 shows reduced liability for drug interactions and may have potential improvements in tolerability over ritonavir. In addition, 3 has high aqueous solubility and can be readily coformulated with other agents.
…………………………………
http://www.google.com/patents/CN103694196A?cl=en
CN 103694196
oxidative metabolism by cytochrome P450 enzymes is one of the main mechanisms of drug metabolism, generally by administration of cytochrome P450 inhibitors to maintain or increase the degradation of cytochrome P450 enzymes are sensitive to the drug plasma levels, in order to improve the pharmacokinetics of drugs dynamics, can be used to enhance the effectiveness of anti-retroviral drugs. For example W02008010921 discloses compounds of formula I as a cytochrome P450 monooxygenase specific compounds (Cobicistat):

W02008010921 discloses the synthesis of compounds of formula I with a variety of, as one of the methods of the following routes
Shows:

The reagents used in the method is expensive, and more difficult to remove by-products, long reaction time, high cost, is not conducive to industrial
Production.
W02010115000 on these routes has been improved:

The first step in the route used for the ring-opening reaction reagent trimethylsilyl iodide, trimethylsilyl iodide expensive. W02010115000 reports this step and the subsequent ring-opening reaction of morpholine substitution reaction yield of two steps is not high, only 71%, so that only iodotrimethylsilane a high cost of raw material is not suitable for industrial production.



Preparation of compounds of formula I
Example [0126] Implementation
[0127] I1-a (20g) was dissolved in dichloromethane, was added 50% K0H (5.5g) solution, control the internal temperature does not exceed 25 ° C, TLC analysis ΙΙ-a disappears. Was cooled to O ~ 10 ° C, was added (2R, 5R) -5 – amino-1 ,6 – diphenyl-2 – hexyl-carbamic acid 5 – methyl-thiazole ester hydrochloride (14.8g), stirred for I ~ 2 h, 1 – hydroxybenzotriazole triazole (5.5g), stirred for I h, 1 – ethyl – (3 – dimethylaminopropyl) carbodiimide hydrochloride (15g), and incubated for 5 ~ 10 hours, TLC analysis of the starting material disappeared, the reaction was completed. The reaction was quenched with aqueous acetic acid, methylene chloride layer was separated, washed with saturated aqueous NaHCO3, washed with water, dried and concentrated. By HPLC purity of 99.1%. Adding ethanol, the ethanol was evaporated to give the product compound of part I of a solution in ethanol. Molar yield 88%, LC-MS: M +1 = 777.1 [0128] All publications mentioned in the present invention are incorporated by reference as if each reference was individually incorporated by reference, as cited in the present application. It should also be understood that, after reading the foregoing teachings of the present invention, those skilled in the art that various modifications of the present invention or modifications, and these equivalents falling as defined by the appended claims scope of claims of the present application.
…………………………
US 2014088304
http://www.google.com/patents/US20140088304
International Patent Application Publication Number WO 2008/010921 and International Patent Application Publication Number WO 2008/103949 disclose certain compounds that are reported to be useful to modify the pharmacokinetics of a co-administered drug, e.g. by inhibiting cytochrome P450 monooxygenase. One specific compound identified therein is a compound of the following formula I:

There is currently a need for improved synthetic methods and intermediates that can be used to prepare the compound of formula I and its salts
Schemes 1-4 below.




Preparation of a Compound of Formula IV

Scheme V.
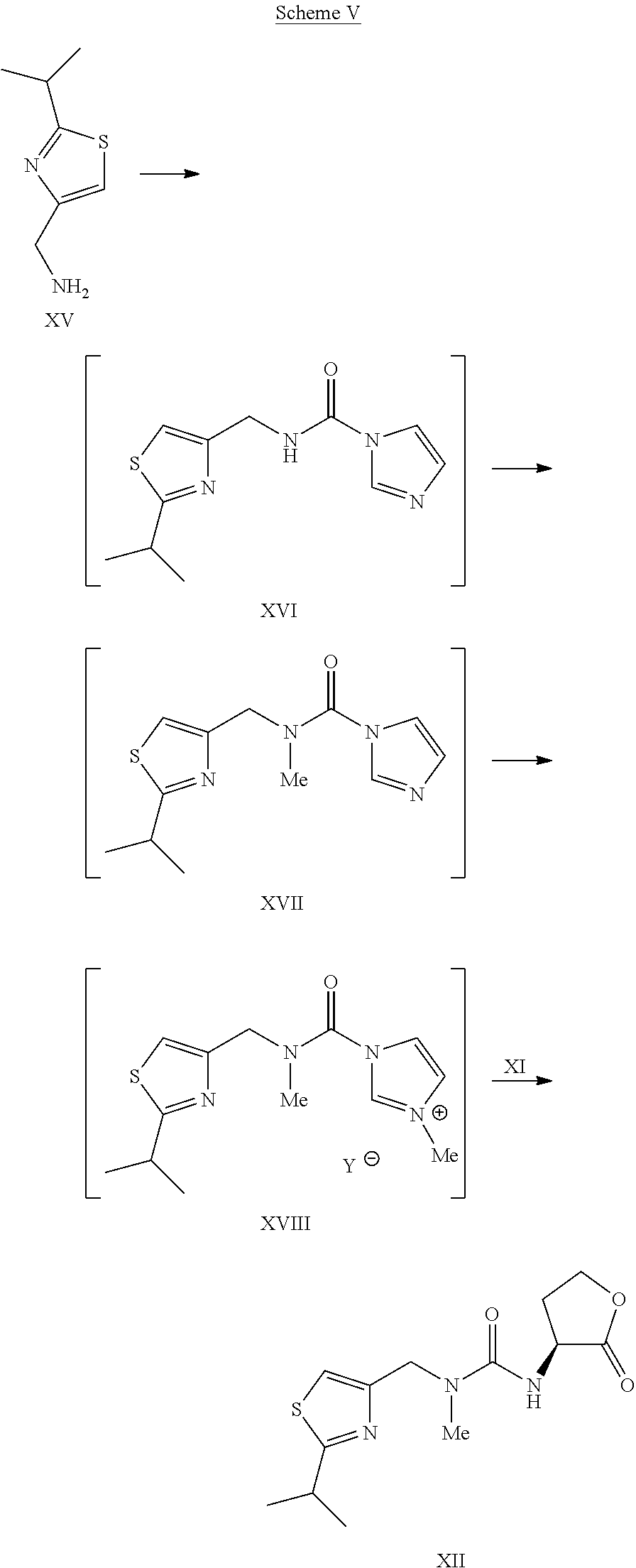
Example 14Preparation of Compound I

To the solution of L-thiazole morpholine ethyl ester oxalate salt XIVa (35.6 kg) in water (66.0 kg) was charged dichloromethane (264 kg), followed by a slow addition of 15 wt % KHCO3 solution (184.8 kg). The resulting mixture was agitated for about 1 hour. The layers were separated and the organic layer was washed with water (132 kg). The organic layer was concentrated under vacuum to dryness. Water (26.5 kg) was charged and the content temperature was adjusted to about 10° C., followed by slow addition of 45% KOH solution (9.8 kg) while maintaining the content temperature at less than or equal to 20° C. The mixture was agitated at less than or equal to 20° C. until the reaction was judged complete by HPLC. The reaction mixture was concentrated under vacuum to dryness and co-evaporated five times with dichloromethane (132 kg each time) under reduced pressure to dryness. Co-evaporation with dichloromethane (132 kg) was continued until the water content was <4% by Karl Fischer titration. Additional dichloromethane (264 kg) was charged and the content temperature was adjusted to −18° C. to −20° C., followed by addition of monocarbamate.HCl salt IXa (26.4 kg). The resulting mixture was agitated at −18° C. to −20° C. for about 1 hour. HOBt (11.4 kg) was charged and the reaction mixture was again agitated at −18° C. to −20° C. for about 1 hour. A pre-cooled solution (−20° C.) of EDC.HCl (21.4 kg) in dichloromethane (396 kg) was added to the reaction mixture while the content temperature was maintained at less than or equal to −20° C. The reaction mixture was agitated at −18° C. to −20° C. until the reaction was judged complete. The content temperature was adjusted to about 3° C. and the reaction mixture quenched with a 10 wt % aqueous citric acid solution (290 kg). The layers were separated and the organic layer was washed once with 15 wt % potassium bicarbonate solution (467 kg) and water (132 kg). The organic layer was concentrated under reduced pressure and then co-evaporated with absolute ethanol.
The product I was isolated as the stock solution in ethanol (35.0 kg product, 76.1% yield).
1H NMR (dDMSO) δ□ 9.05 (s, 1H), 7.85 (s, 1H), 7.52 (d, 1H), 7.25-7.02 (m, 12H), 6.60 (d, 1H), 5.16 (s, 2H), 4.45 (s, 2H), 4.12-4.05 (m, 1H), 3.97-3.85 (m, 1H), 3.68-3.59 (m, 1H), 3.57-3.45 (m, 4H), 3.22 (septets, 1H), 2.88 (s, 3H), 2.70-2.55 (m, 4H), 2.35-2.10 (m, 6H), 1.75 (m, 1H), 1.62 (m, 1H), 1.50-1.30 (m, 4H), 1.32 (d, 6H).
13C NMR (CD3OD) δ 180.54, 174., 160.1, 157.7, 156.9, 153.8, 143.8, 140.1, 140.0, 136.0, 130.53, 130.49, 129.4, 127.4, 127.3, 115.5, 67.7, 58.8, 56.9, 55.9, 54.9, 53.9, 51.6, 49.8, 42.7, 42.0, 35.4, 34.5, 32.4, 32.1, 29.1, 23.7.
Example 13Preparation of L-Thiazole Morpholine Ethyl Ester Oxalate Salt XIVa

To a solution of (L)-thiazole amino lactone XII (33.4 kg) in dichloromethane (89.5 kg) was charged dichloromethane (150 kg) and absolute ethanol (33.4 kg). The content temperature was then adjusted to about 10° C., followed by slow addition of TMSI (78.8 kg) while the content temperature was maintained at less than or equal to 22° C. and agitated until the reaction was judged complete. The content temperature was adjusted to about 10° C., followed by a slow addition of morpholine (49.1 kg) while the content temperature was maintained at less than or equal to 22° C. Once complete, the reaction mixture was filtered to remove morpholine.HI salt and the filter cake was rinsed with two portions of dichloromethane (33.4 kg). The filtrate was washed twice with water (100 kg). The organic layer was concentrated under vacuum to dryness. Acetone (100 kg) was then charged to the concentrate and the solution was concentrated under reduced pressure to dryness. Acetone (233.8 kg) was charged to the concentrate, followed by a slow addition of the solution of oxalic acid (10 kg) in acetone (100 kg). The resulting slurry was refluxed for about 1 hour before cooling down to about 3° C. for isolation. The product XIVa was filtered and rinsed with acetone (66.8 kg) and dried under vacuum at 40° C. to afford a white to off-white solid (40 kg, 71% yield). 1H NMR (CDCl3) δ □7.00 (s, 1H), 6.35 (broad s, 1H), 4.60-4.40 (m, 3H), 4.19 (quartets, 2H), 4.00-3.90 (m, 4H), 3.35-3.10 (m, 7H), 3.00 (s, 3H), 2.40-2.30 (m, 1H), 2.15-2.05 (m, 1H), 1.38 (d, 6H), 1.25 (triplets, 3H).
……………………………………..
W02008010921
http://www.google.co.in/patents/WO2008010921A2?cl=en
Preparation of Example A
Scheme 1

Example A Compound 2
To a solution of Compound 1 (ritonavir) (1.8 g, 2.5 mmol) in 1,2- dichloroethane (15 mL) was added l,l’-thiocarbonyldiimidazole (890 mg, 5.0 mmol). The mixture was heated at 75 SC for 6 hours and cooled to 25 SC. Evaporation under reduced pressure gave a white solid. Purification by flash column chromatography (stationary phase: silica gel; eluent: EtOAc) gave Compound 2 (1.6 g). m/z: 831.1 (M+H)+. Example A
To the refluxing solution of tributyltin hydride (0.78 mL, 2.9 mmol) in toluene (130 mL) was added a solution of Compound 2 (1.6 g, 1.9 mmol) and 2,2′- azobisisobutyronitrile (31 mg, 0.19 mmol) in toluene (30 mL) over 30 minutes. The mixture was heated at 1152C for 6 hours and cooled to 25 BC. Toluene was removed under reduced pressure. Purification by flash column chromatography (stationary phase: silica gel; eluent: hexane/EtOAc = 1/10) gave Example A (560 mg). m/z: 705.2 (M+H)+. 1H-NMR (CDCl3) δ 8.79 (1 H, s), 7.82 (1 H, s), 7.26-7.05 (10 H, m), 6.98 (1 H, s), 6.28 (1 H, m), 6.03 (1 H, m), 5.27 (1 H7 m), 5.23 (2 H, s), 4.45-4.22 (2 H, m), 4.17 (1 H, m), 3.98 (1 H, m), 3.75 (1 H, m), 3.25 (1 H7 m), 2.91 (3 H, s), 2.67 (4 H, m), 2.36 (1 H, m), 1.6-1.2 (10 H, m), 0.85 (6 H, m).
| EP1183026A2 * | 25 May 2000 | 6 Mar 2002 | Abbott Laboratories | Improved pharmaceutical formulations |
| US20060199851 * | 2 Mar 2006 | 7 Sep 2006 | Kempf Dale J | Novel compounds that are useful for improving pharmacokinetics |
| Thiazol-5-ylmethyl N-[1-benzyl-4-[[2-[[(2-isopropylthiazol-4-yl)methyl-methyl-carbamoyl]amino]-4-morpholino-butanoyl]amino]-5-phenyl-pentyl]carbamate | |
| Clinical data | |
|---|---|
| Legal status |
fda approved sept 2014
|
| Identifiers | |
| CAS number | 1004316-88-4 |
| ATC code | V03AX03 |
| PubChem | CID 25151504 |
| ChemSpider | 25084912 |
| UNII | LW2E03M5PG |
| Chemical data | |
| Formula | C40H53N7O5S2 |
| Mol. mass | 776.023 g/mol |
| US7939553 * | Jul 6, 2007 | May 10, 2011 | Gilead Sciences, Inc. | co-administered drug (as HIV protease inhibiting compound, an HIV (non)nucleoside/nucleotide inhibitor of reverse transcriptase, capsid polymerization inhibitor, interferon, ribavirin analog) by inhibiting cytochrome P450 monooxygenase; ureido- or amido-amine derivatives; side effect reduction |
- Highleyman, L.
Elvitegravir “Quad” Single-tablet Regimen Shows Continued HIV Suppression at 48 Weeks
- R Elion, J Gathe, B Rashbaum, and others. The Single-Tablet Regimen of Elvitegravir/Cobicistat/Emtricitabine/Tenofovir Disoproxil Fumarate (EVG/COBI/FTC/TDF; Quad) Maintains a High Rate of Virologic Suppression, and Cobicistat (COBI) is an Effective Pharmacoenhancer Through 48 Weeks. 50th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC 2010). Boston, September 12–15, 2010.
- Lepist, E. -I.; Phan, T. K.; Roy, A.; Tong, L.; MacLennan, K.; Murray, B.; Ray, A. S. (2012). “Cobicistat Boosts the Intestinal Absorption of Transport Substrates, Including HIV Protease Inhibitors and GS-7340, in Vitro”. Antimicrobial Agents and Chemotherapy 56 (10): 5409–5413. doi:10.1128/AAC.01089-12. PMC 3457391. PMID 22850510.
-
Patent No all US
Expiry 5814639 Sep 29, 2015 5814639*PED Mar 29, 2016 5914331 Jul 2, 2017 5914331*PED Jan 2, 2018 5922695 Jul 25, 2017 5922695*PED Jan 25, 2018 5935946 Jul 25, 2017 5935946*PED Jan 25, 2018 5977089 Jul 25, 2017 5977089*PED Jan 25, 2018 6043230 Jul 25, 2017 6043230*PED Jan 25, 2018 6642245 Nov 4, 2020 6642245*PED May 4, 2021 6703396 Mar 9, 2021 6703396*PED Sep 9, 2021 7176220 Nov 20, 2023 7635704 Oct 26, 2026 8148374 Sep 3, 2029
WHO publishes New Version of the Draft on “Hold-Time” Studies
DRUG REGULATORY AFFAIRS INTERNATIONAL
WHO publishes New Version of the Draft on “Hold-Time” Studies
The 2nd revision for comment was published already in February this year (we reported). Now, a 3rd version is available – also for comment. The document describes the design of hold-time studies for the determination of time limits which have to be determined according to the generally applicable intermediate and bulk products. This should avoid that the storage of intermediate or bulk products from having any negative influence on their quality or the quality of a finished before processing to the next stage.
Chapter 2 which defines what intermediate and bulk products are has been added. It is now explicitly pointed out that hold-time investigations are part of the process validation. In turn, the reference to retrospective observation has been removed from the current version as well as – fortunately – the incomprehensible paragraph on the ‚most probable /…
View original post 113 more words
How to identify Out-of-Trend Results in Stability Studies?
DRUG REGULATORY AFFAIRS INTERNATIONAL

How to identify Out-of-Trend Results in Stability Studies?
http://www.gmp-compliance.org/enews_4522_How-to-identify-Out-of-Trend-Results-in-Stability-Studies_8360,8348,8430,Z-QCM_n.html
An article in PharmTech from June 2013 (by Trajkovic-Jolevska et. al) deals with the methods to identify Out-of-Trend (OOT) results in ongoing stability studies.
With regard to stability studies, it is important to make the difference between Out-of-Specification (OOS) and Out-of-Trend (OOT). Both the pharmaceutical industry and authorities often misuse these two terms.
The article defines OOT results as those results which don’t follow the expected trend, either in comparison with other stability batches or compared to previous results collected during a stability study. OOT results aren’t necessarily OOS, but they don’t look like a typical data point.
Although OOT results are a serious problem, neither the scientific literature nor regulatory guidelines fully address them.
The aim of the study described in this Pharmtech article by Trajkovic-Jolevska et. al was to perform a statistical evaluation of the statistical methods used in…
View original post 112 more words
Pirenperone, R 47465

ON THE LEFT OR ABOVE
IS
3-(2- {4-[(4-fluorophenyl)carbonyl]piperidin- 1 -yl} ethyl)-2-methyl-4H-pyrido[ 1 ,2- α]pyrimidin-4-one
3-(2-{4-[(4-fluorophenyl)carbonyl]piperidin-1-yl}ethyl)-2-methyl-4H-pyrido[1,2-α]pyrimidin-4-one
3-[2-[4-(4-fluorobenzoyl)-1-piperidinyl]ethyl]-2-methyl-4H-pyrido[1,2-a]pyri- midin-4-one
pirenperone CAS : 75444-65-4
- C23 H24 F N3 O2
- 4H-Pyrido[1,2-a]pyrimidin-4-one, 3-[2-[4-(4-fluorobenzoyl)-1-piperidinyl]ethyl]-2-methyl-
- R 47465
Cardiovascular disease; Inflammatory disease; Neoplasm; Pain
Calcium channel modulator T-type
……………………………..
http://www.google.co.in/patents/US4342870
http://www.google.co.in/patents/EP0037265A1
Example XXIV
-
A solution of 2 parts of 3-[2-[4-(4-fluorobenzoyl)-1 -piperidinyl]ethyl]-2 -methyl-4H -pyricio[1, 2 -a]pyrimidin-4-one in 64 parts of 2-propanol is warm acidified with 2-propanol saturated with hydrogen chloride. The formed hydrochloride salt is allowed to crystallize. It is filtered off and dried, yielding 2 parts (85. 5%) of 3-[2-[4-(4-fluorobenzoyl)-1-piperidinyl]ethyl]-2-methyl-4H-pyrido[1,2-a]pyri- midin-4-one dihydrochloride; mp. + 300°C.
-
In a similar manner there are also prepared:
- 3-[2 -[4-(4-fluorobenzoyl)-1-piperidiny]yethyl7-2 -methyl-4H-pyrido-[1, 2 -a]pyrimidin-4-one sulfate (1 : 2); mp. 254. 7°C; and
- 3-[2-[4-(4-fluorobenzoyl)-1 -piperidinyl]ethyl]-2-methyl-4H-pyrido-[1, 2-a]pyrimidin-4-one phosphate (1 : 2) ; mp. 243.8°C.
WO-2014143915
http://www.google.com/patents/WO2014143915A1?cl=en
Novelcrystalline polymorphic forms of pirenperone, useful for treating disorders associated with T-type calcium ion channels such as pain syndrome, neoplasm, cardiovascular disease or inflammation. VM Discovery, from which VM Therapeutics was spun out, was investigating the VMD-C300 series of compounds which act as T-type calcium channel modulators, including VMD-3816 and VMD-3222, for treating cancer, pain, neurological diseases and cardiovascular diseases; but as of September 2014, this program was assumed to be discontinued. See WO2009108798, (by the inventor, assigned to VM Discovery) claiming use of the same compound for treating same indications.
It was first disclosed in the now-expired US Patent No. 4,342,870 (Claim 5), and intended to be used as potential anti-anxiety drug. However, the early human clinical studies has shown that the compound did not show any dose-related anti-anxiety effects as hoped, but otherwise the compound was safe in human (ref. Ansseau M, Doumo t A, Thlry D, Gelders Y. “Pilot study of a specific serotonergic antagonist, pirenperone, in the treatment of anxiety disorders”, Acta Psychiatr Belg, 1983 Sep-Oct;83(5):517-24). in the US Patent No. 4,342,870, there is no crystalline polymorphic form disclosed, nor disclosure of potential uses for management of pain and treatment of other related diseases or disorders.
It was further disclosed in the PCT patent application WO/2009/108798 as “Compound 10 (pirenperone)‘” to be used for novel T-type calcium ion channel antagonist for management of pain and treatment of other diseases or disorders associated to the T-type calcium ion channels.
Surprisingly, we have found that there are many crystalline polymorphic forms of this compound which may affect the compound’s pharmaceutical safety and pharmacology properties.
MAKE IN INDIA
http://makeinindia.com/sector/pharmaceuticals/
Amgen files ‘breakthrough’ leukaemia drug Blinatumomab (AMG103) in the US

Blinatumomab
Biotechnology giant Amgen has filed its investigational cancer immunotherapy blinatumomab in the US for the treatment of certain forms of acute lymphoblastic leukaemia (ALL).
Specifically, the Biologic License Application seeks approval to market the drug for patients with Philadelphia-negative (Ph-) relapsed/refractory B-precursor forms of the aggressive blood/bone marrow cancer.
![]()
Blinatumomab (AMG103) is a drug that has anti-cancer properties. It belongs to a new class of constructed monoclonal antibodies, bi-specific T-cell engagers (BiTEs), that exert action selectively and direct the human immune system to act against tumor cells. Blinatumomab specifically targets the CD19 antigen present on B cells.[1]
The drug was developed by a German-American company Micromet, Inc. in cooperation with Lonza; Micromet was later purchases byAmgen, which has furthered the drug’s clinical trials.
Structure and mechanism of action

Blinatumomab linking a T cell to a malignant B cell.
Blinatumomab enables a patient’s T cells to recognize malignant B cells. A molecule of blinatumomab combines two binding sites: a CD3 site for T cells and a CD19 site for the target B cells. CD3 is part of the T cell receptor. The drug works by linking these two cell types and activating the T cell to exert cytotoxic activity on the target cell.[2]
Therapeutic use
Clinical trials
In a phase 1 clinical study with blinatumomab, patients with non-Hodgkin’s lymphoma showed tumor regression, and in some cases completeremission.[3] There are ongoing phase 1 and phase 2 clinical trials of blinatumomab in patients with acute lymphoblastic leukemia (ALL),[4]lung or gastrointestinal cancers.[citation needed] One phase II trial for ALL reported good results in 2010 and another is starting.[5]
| Monoclonal antibody | |
|---|---|
| Type | Bi-specific T-cell engager |
| Source | Mouse |
| Target | CD19, CD3 |
| Clinical data | |
| Legal status |
?
|
| Identifiers | |
| CAS number | 853426-35-4 |
| ATC code | None |
| UNII | 4FR53SIF3A |
| Chemical data | |
| Formula | C2367H3577N649O772S19 |
| Mol. mass | 54.1 kDa |
References
- Statement on a Nonproprietary Name adopted by the USAN Council: Blinatumomab
- Mølhøj, M; Crommer, S; Brischwein, K; Rau, D; Sriskandarajah, M; Hoffmann, P; Kufer, P; Hofmeister, R; Baeuerle, PA (March 2007). “CD19-/CD3-bispecific antibody of the BiTE class is far superior to tandem diabody with respect to redirected tumor cell lysis”. Mol Immunol 44 (8): 1935–43. doi:10.1016/j.molimm.2006.09.032. PMID 17083975.
- Bargou, R; et al. (2008). “Tumor regression in cancer patients by very low doses of a T cell-engaging antibody”. Science 321 (5891): 974–977. doi:10.1126/science.1158545.PMID 18703743.
- ClinicalTrials.gov NCT00560794 Phase II Study of the BiTE Blinatumomab (MT103) in Patients With Minimal Residual Disease of B-precursor Acute ALL
- “Micromet initiates MT103 phase 2 trial in adult ALL patients”. 20 Sep 2010.
External links
 MAKE IN INDIA
MAKE IN INDIA
http://makeinindia.com/
http://makeinindia.com/sector/pharmaceuticals/
Pharmaceuticals; Make in India
 PM, MODI
PM, MODI
Brand India Pharma aims to make the most of a booming domestic pharma industry
India’s pharma exports stood at 90,000 crore rupees ($15 billion) for the year 2013-2014, and are set to cross the 1 lakh crore rupees ($16.4 billion) mark in the current financial year. The Brand India Pharma campaign aims to tap into this value proposition, under the guidance of the Indian Ministry of Commerce and Industry, aiming to showcase the strengths of the Indian pharma industry.
With more than 10,500 manufacturing units and more than 3,000 pharma companies, India is ranked among the top six producers of pharmaceuticals worldwide, and is well-positioned to take advantage of its place in a global landscape.
READ AT
List of WHO Approved Pharma Plant in India

India’s spacecraft cost $74 million, a fraction of the $671 million spent by NASA’s MAVEN ……….SEPT 24 2014
READ AT
http://www.brandindiapharma.in/infographic-business/

India’s spacecraft reaches Mars orbit … and history
India’s spacecraft cost $74 million, a fraction of the $671 million spent by NASA’s MAVEN
24 sept 2014
India’s Mars Orbiter Mission successfully entered Mars’ orbit Wednesday morning, becoming the first nation to arrive on its first attempt and the first Asian country to reach the Red Planet.
“We have gone beyond the boundaries of human enterprise and human imagination,” declared India’s Prime Minister Narendra Modi, who watched from the space agency’s nerve center in Bangalore. “We have accurately navigated our spacecraft through a route known to a very few.”
The staff at the Indian Space Research Organization erupted into applause and cheers after learning that the Mars Orbiter Mission, also known as Mangalyaan, reached the planet’s orbit and made history.
Before Wednesday, only the United States, Europe and the Soviets have successfully sent spacecraft to Mars.

 Photos: India’s first Mars orbiter
Photos: India’s first Mars orbiter“The odds were stacked against us,” Modi said. “Of the 51 missions attempted so far, a mere 21 had succeeded. But we have prevailed.”
And India reached Mars with significantly less money.
With a price tag of $74 million, the Mars Orbiter Mission cost a mere fraction of the $671 million NASA spent on its MAVEN spacecraft, which arrived to Mars earlier this week. Some space observers noted that India’s Mars orbiter cost less than the $100 million budget for the space thriller film “Gravity.”
Interactive: Exploring Mars from Viking to MAVEN
“It shows how optimal is the design, that way we’re able to cut cost and we’re not compromising quality,” said S. Satish, a space expert based in Bangalore.
The groundbreaking Mars mission wasn’t without controversy — with some critics who said India should spend the money on other issues.
The spacecraft launched on November 5, and has traveled over 650 million kilometers to enter Mars orbit. Its mission is to orbit the Red Planet, mapping its surface and studying the atmosphere. The Mars Orbiter kicked off its interplanetary debut with its own Twitter account.
The mission has been freighted with patriotic significance for India since its inception and is seen as a symbolic coup over its rival, China, which is also ramping up its space ambitions.
 India launches mission to Mars
India launches mission to Mars
China’s joint mission with Russia in 2011, which contained the Chinese Mars satellite Yinhuo-1, stalled and eventually fell back to Earth. Japan’s 1998 attempt with the spacecraft Nozomi was also unsuccessful due to fuel problems.
Once nears Mars’ orbit, India’s spacecraft had to execute a series of complicated and critical maneuvers. About half of all spacecraft sent on missions to the planet have veered off course, malfunctioned or crashed.
India’s Mars Orbiter Mission is in the company of NASA’s two Mars rovers on the ground, a European orbiter and NASA orbiters including the MAVEN, which has been there since Sunday.
The United States has expressed interest in cooperating with India as their spacecraft gather data about the planet.

MAKE IN INDIA
http://makeinindia.com/sector/pharmaceuticals/
A CASE OF ICHCHTHYOSIS ; A TYPICAL SKIN DISORDER ; E.T.G AYURVEDASCAN TEST EVALUATION ; “इक्थियासिस” जैसे लाइलाज चर्म रोग का ई०टी०जी० आयुर्वेदास्कैन आधारित आन्कलन
चर्म रोग ICHCHTHYOSIS या इख्तोयासिस एक तरह की ऐसी तकलीफ है जो त्वचा के टिश्यूज से जुड़ी हुयी बीमारी है / इस बीमारी मे त्वचा का रन्ग काला पड़ जाता है और त्वचा मोटी हो जाती है / इसके अलावा त्वचा का रन्ग काला और वर्ण cracks यानी फटी हुयी और आकार मछली की खाल जैसा हो जाता है /
जिस मरीज का नीचे दिया गया चित्र है उसे लगभग चार साल से यह तकलीफ रही है / अन्ग्रेजी और देशी और होम्योपैथी का इलाज कराने के बाद इसे आराम नही मिला / हमारे यहा से इलाज करा चुके एक मरीज द्वारा हमारे सन्स्थान मे इलाज कराने के लिये प्रोत्साहित किये जाने के बाद यह मरीज इलाज के लिये हमारे यहां आया है /
मरीज के दोनो पैरो और शरीर के लगभग सभी हिस्सो मे इसी तरह के scabs मौजूद है /.
नीचे दिये गये चित्र मे यह चर्म रोग…
View original post 339 more words
Ibritumomab tiuxetan

Ibritumomab tiuxetan, sold under the trade name Zevalin, is a monoclonal antibody radioimmunotherapy treatment for relapsed or refractory, low grade or transformed B cell non-Hodgkin’s lymphoma, a lymphoproliferative disorder. The drug uses the monoclonal mouse IgG1 antibody ibritumomab (pronounced as <ih bri TYOO mo mab>)[1] in conjunction with the chelator tiuxetan, to which a radioactive isotope (either yttrium-90 or indium-111) is added. Tiuxetan is a modified version of DTPA whose carbon backbone contains an isothiocyanatobenzyl and a methyl group.[2][3]

Mechanism of action
The antibody binds to the CD20 antigen found on the surface of normal and malignant B cells (but not B cell precursors), allowing radiation from the attached isotope (mostly beta emission) to kill it and some nearby cells. In addition, the antibody itself may trigger cell death via antibody-dependent cell-mediated cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), and apoptosis. Together, these actions eliminate B cells from the body, allowing a new population of healthy B cells to develop from lymphoid stem cells.

Zevalin (Ibritumomab tiuxetan) is a radio-labeled antibody. The antibody seeks and binds to cells that have a receptor called CD20 — present on both normal and malignant mature b-cells.
Once bound to the target cells, Zevalin delivers radiation, which enhances the killing effect of the antibody.
Because immature b-cells do not have the CD20 receptor, normal b-cells will recover in about nine months after treatment.
Rituxan (the naked antibody) is administered prior to Zevalin with the goal of clearing the majority of normal b-cells so that the therapeutic dose (the radio-labeled antibody) is more focused on tumor cells.

Preparation
Zevalin is supplied as a single dosage kit supplied by IDEC Pharmaceuticals Corp. It consists of Ibritumomab covalently conjugated to the metal chelator tiuxetan, which forms a stable complex with indium-111 for imaging and yttrium-90 for therapy.
The kit is supplied with four vials – a vial containing 3.2 mg of conjugated antibody in 2 ml saline, a vial containing 2 ml 50mM sodium acetate, a vial containing phosphate buffer, and a fourth empty reaction vial. Prior to labeling, a volume of sodium acetate buffer equivalent to 1.2 times the volume of the tracer solution is transferred to the reaction vial. Then 5.5 mCi (203.5 MBq) indium-111 or 40mCi (1.48 GBq) yttrium-90 is added to the reaction vial and mixed thoroughly without shaking. Next, 1.3 ml of conjugated antibody is added. The mixture is incubated for exactly 30min for indium-111 and for 5 min with yttrium-90 labeling, followed by the addition of enough phosphate buffer to make the final volume 10 ml. The labeling yield is determined by ITLC-SG with 0.9% saline as the mobile phase. Labeling efficiency should be greater than 95%.[4]

http://pubs.rsc.org/en/content/articlelanding/2006/cs/b514859f/unauth#!divAbstract
A cartoon depiction of the radiolabelled monoclonal antibody 90Y-ibritumomab tiuxetan 18.
Administration
In order to qualify for ibritumomab, a patient needs to have bone marrow involvement of < 25% and > 15% bone marrow cellularity. Since ibritumomab is known to cause cytopenia, platelet and neutrophil counts are also taken pretreatment. Refractory/relapsed patients should have platelet counts of 100,000 per cubic millimetre (100,000/cmm) or greater; consolidation patients should have counts of 150,000/cmm or greater. Since a murine antibody is used, the patient might also be tested for human anti mouse antibodies (HAMA). Having bulky disease does not disqualify a patient.
The ibritumomab regimen takes 7–9 days. An imaging dose of the drug is no longer required in the U.S. Rituxan 250 mg/sq.m is given day 1, then on day 7-9 the Rituxan dose is repeated and Zevalin given within four hours. The dose of Zevalin 0.4 mCi/kg (= 14.8MBq/kg) if platelet counts are above 150,000/cmm; 0.3 mCi/kg (= 11.1MBq/kg) if 100,000-150,000/cmm. The Zevalin dose never exceeds 32 mCi (= 1184MBq).[5]
Ibritumomab tiuxetan is administered by intravenous infusion which usually lasts around 10 minutes. Only acrylic shielding is needed, not lead. A trained nuclear medicine technologist performs the infusion and safely disposes of waste.
Efficacy
Treatment with ibritumomab showed higher response rates in clinical trials compared to treatment with only rituximab (similar to ibritumomab, but without the attached radioisotope), and showed very promising results for patients who no longer respond to rituximab.
In patients with relapsed or refractory low-grade, follicular, or transformed B-cell NHL, where no prior anti-CD20 therapy was allowed, the ORR was 83% / 55% and CR was 38% / 18%, comparing ibritumomab to rituximab. [6]
Recently, extended follow-up data for the ZEVALIN ([90Y]-ibritumomab tiuxetan) First-line Indolent (FIT) study presented at the American Society of Hematology (ASH) Annual Meeting demonstrated the continued improvement in progression-free survival (PFS) following ibritumomab consolidation therapy for patients with follicular B-cell non-Hodgkin’s lymphoma who achieved a response to first-line therapy over chemotherapy alone. Additionally, ibritumomab consolidation did not adversely affect the use of various effective second-line treatments including stem cell transplants in patients who relapsed.[7]
In a Phase II study on patients with relapsed and refractory mantle cell lymphoma, the OR was 42% and CR was 26%.[8]
A study demonstrated that rituximab followed by single agent ibritumomab in a front-line setting for patients with MALT lymphoma and low-grade follicular lymphoma that primarily involved the conjunctiva or orbit, produced a complete response rate of 83 percent.[9]

http://rd.springer.com/article/10.2165%2F00024669-200201050-00004#page-1
History
Developed by the IDEC Pharmaceuticals, which is now part of Biogen Idec, ibritumomab tiuxetan was the first radioimmunotherapy drug approved by the Food and Drug Administration (FDA) in 2002 to treat cancer. It was approved for the treatment of patients with relapsed or refractory, low‑grade or follicular B‑cell non‑Hodgkin’s lymphoma (NHL), including patients with rituximab refractory follicular NHL.
In December 2007, Cell Therapeutics Inc acquired the U.S. rights to sell, market, and distribute this radioimmunotherapy antibody from Biogen for approximately US$30 million, or the equivalent of about two years’ net sales revenue in the U.S. for the drug.[10] Outside of the U.S., Bayer Schering Pharma continues to have the rights to the drug.
In March 2009, Spectrum Pharmaceuticals acquired 100% control of RIT Oncology, LLC, to commercialize Zevalin in the US. Now Spectrum Pharmaceuticals is responsible for all activities relating to Zevalin in the US.
In September 2009, ibritumomab received approval from the FDA for an expanded label for the treatment of patients with previously untreated follicular non-Hodgkin’s Lymphoma (NHL), who achieve a partial or complete response to first-line chemotherapy.

Costs
Ibritumomab which is not available in a generic form because it is still under patent protection, is currently the most expensive drug available given in a single dose, costing over US$ 37,000 (€ 30,000) for the average dose. However, ibritumomab is essentially an entire course of lymphoma therapy which is delivered in 7–9 days, with one visit for pre-dosing Rituxan, and one visit a week later for the actual Zevalin therapeutic dose preceded by Rituxan. Compared to other monoclonal antibody treatments (many of which are well over US$ 40,000 for a course of therapy), this drug is priced in the middle for many of these therapies.
 |
|
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Mouse |
| Target | CD20 |
| Clinical data | |
| Trade names | Zevalin |
| AHFS/Drugs.com | monograph |
| Licence data | US FDA:link |
| Legal status | |
| Routes | intravenous |
| Identifiers | |
| CAS number | 174722-31-7 |
| ATC code | V10XX02 (90Y) |
| DrugBank | DB00078 |
External links
- http://www.zevalin.com/ – Official Zevalin web site
- http://www.spectrumpharm.com/ – Spectrum Pharmaceuticals, Inc. web site
- http://www.accessdata.fda.gov/drugsatfda_docs/label/2002/ibriide021902LB.pdf – Package Insert

http://www.fda.gov/ohrms/dockets/ac/01/slides/3782s2_02_idec/sld015.htm
References
- Ibritumomab: Pronunciation
- Milenic, Diane E.; Brady, Erik D.; Brechbiel, Martin W. (June 2004). “Antibody-targeted radiation cancer therapy”. Nat Rev Drug Discov 3 (6): 488–499. doi:10.1038/nrd1413. ISSN 1474-1776. PMID 15173838.
- WHO Drug Information
- http://www.accessdata.fda.gov/drugsatfda_docs/label/2002/ibriide021902LB.pdf
- Ibritumomab: Indications
- Ibritumomab: Efficacy
- ZEVALIN Consolidation in First-Line Therapy in Patients with Non-Hodgkin’s Lymphoma Resulted in a Progression-Free Survival of Greater Than 67 Months
- Zevalin and mantle cell
- ZEVALIN(R) Produced 83 Percent Complete Response Rate in Mucosa-Associated Lymphoid Tissue (MALT) Orbital Lymphoma Study
- [1]
// // // // //
September 23, 2014
// CASI Signs China Licensing Deal With Spectrum For 3 Cancer Drugs…http://www.outsourcedpharma.com/doc/casi-signs-china-licensing-deal-with-spectrum-for-cancer-drugs-0001
// CASI Signs China Licensing Deal With Spectrum For 3 Cancer Drugs// // // // //
CASI Pharmaceuticals and Spectrum Pharmaceuticals (SPPI) announced the signing of a license agreement that gives CASI exclusive rights to develop three cancer drugs from Spectrum and market them in China, including Macau, Hong Kong, and Taiwan.
The agreement concerns the two approved cancer drugs Zevalin (ibritumomab tiuxetan) Injection non-Hodgkin’s lymphoma (NHL) and Marqibo (vinCRIStine sulfate LIPOSOME injection) for acute lymphoblastic leukemia (ALL) as well as the investigational Phase 3 drug Captisol-Enabled Melphalan (CE melphalan) being studied as a conditioning treatment before autologous stem cell transplant in patients with multiple myeloma. Spectrum recently reported that Melphalan met its primary endpoint in its pivotal safety and efficacy trial. In view of the results, Spectrum said it intends to file a New Drug Application (NDA) with the U.S. Food and Drug Administration (FDA) for the drug in the second half of 2014.
// // // // //

{kind=link}