
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 226)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Time to back in the flow of things ;)
OK it was an extended time away from posting — I totally blame the Turkey, Ham, Beer, Stuffing, Pie — at least I have tapered off over the years.
So what’s sitting on my desk — after several pontifications, I have gotten back to thinking about how chemists think about their chemistry and where it can go in flow processes — so, OK, retrosynthesis — but I often think in classes of fragments and what they can do (think of it as a review on enaminone transformations so to speak). In this case, Ian Baxendale got me thinking about ynones or alpha, beta-acteylenic ketones — used quite a bit right? furans, flavones, pyrazoles, pyrimidines and heck back at Bayer I used them in a number of dipolarcycloadditions and intramolecular cyclizations to isoxazoles and pyrroles……you get the point……if interested in a nice article on using a flow approach to ynones and…
View original post 204 more words
APD 334 to treat to autoimmune diseases

APD 334
Arena Pharmaceuticals, Inc. innovator
2-[7-[4-Cyclopentyl-3-(trifluoromethyl)benzyloxy]-1,2,3,4-tetrahydrocyclopenta[b]indol-3(R)-yl]acetic acid
| Company | Arena Pharmaceuticals Inc. |
| Description | Sphingosine 1-phosphate receptor 1 (S1PR1; S1P1; EDG1) agonist |
| Molecular Target | Sphingosine 1-phosphate receptor 1 (S1PR1) (S1P1) (EDG1) |
| Mechanism of Action | Sphingosine 1-phosphate (S1P) receptor agonist |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Phase I |
| Standard Indication | Autoimmune (unspecified) |
| Indication Details | Treat autoimmune diseases |
APD334, an orally available agonist of the S1P1 receptor, is an internally discovered investigational drug candidate intended for the potential treatment of a number of conditions related to autoimmune diseases, including multiple sclerosis, psoriasis and rheumatoid arthritis. S1P1 receptors have been demonstrated to be involved in the modulation of several biological responses, including lymphocyte trafficking from lymph nodes to the peripheral blood. By isolating lymphocytes in lymph nodes, fewer immune cells are available in the circulating blood to effect tissue damage. We have optimized APD334 as a potent and selective small molecule S1P1 receptor agonist that reduces the severity of disease in preclinical autoimmune disease models.
Autoimmune diseases are characterized by an inappropriate immune response against substances and tissues that are normally present in the body. In an autoimmune reaction, a person’s antibodies and immune cells target healthy tissues, triggering an inflammatory response. Reducing the immune and/or inflammatory response is an important goal in the treatment of autoimmune disease.



The present invention relates to processes and intermediates useful in the preparation of of (R)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-l,2,3,4-tetrahydrocyclopenta[b]indol- 3-yl)acetic acid of Formula (la) or salts thereof, an SlPl receptor modulator that is useful in the treatment of SlPl receptor-associated disorders, for example, diseases and disorders mediated by lymphocytes, transplant rejection, autoimmune diseases and disorders, inflammatory diseases and disorders (e.g. , acute and chronic inflammatory conditions), cancer, and conditions characterized by an underlying defect in vascular integrity or that are associated with angiogenesis such as may be pathologic (e.g. , as may occur in inflammation, tumor development and atherosclerosis).
BACKGROUND OF THE INVENTION
SlPl receptor agonists have been shown to possess at least immunosuppressive, antiinflammatory, and/or hemostatic activities, e.g. by virtue of modulating leukocyte trafficking, sequestering lymphocytes in secondary lymphoid tissues, and/or enhancing vascular integrity. Accordingly, SlPl receptor agonists can be useful as immunosuppressive agents for at least autoimmune diseases and disorders, inflammatory diseases and disorders (e.g. , acute and chronic inflammatory conditions), transplant rejection, cancer, and/or conditions that have an underlying defect in vascular integrity or that are associated with angiogenesis such as may be pathologic (e.g., as may occur in inflammation, tumor development, and atherosclerosis) with fewer side effects such as the impairment of immune responses to systemic infection.
The sphingosine-1 -phosphate (SIP) receptors 1-5 constitute a family of G protein- coupled receptors containing a seven-transmembrane domain. These receptors, referred to as SlPl to S1P5 (formerly termed endothelial differentiation gene (EDG) receptor-1, -5, -3, -6, and -8, respectively; Chun et al., Pharmacological Reviews, 54:265-269, 2002), are activated via binding by sphingosine-1 -phosphate, which is produced by the sphingosine kmase-catalyzed phosphorylation of sphingosine. SlPl, S1P4, and S1P5 receptors activate Gi but not Gq, whereas S1P2 and S1P3 receptors activate both Gi and Gq. The S1P3 receptor, but not the SlPl receptor, responds to an agonist with an increase in intracellular calcium.
In view of the growing demand for S 1P1 agonists useful in the treatment of S 1P1 receptor-associated disorders, the compound (R)-2-(7-(4-cyclopentyl-3- (trifluoromethyl)benzyloxy)-l ,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid of Formula
(la):

has emerged as an important new compound, see PCT patent application, Serial No.
PCTVUS2009/004265 hereby incorporated by reference in its entirety. Accordingly, new and efficient routes leading to (R)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-l, 2,3,4- tetrahydrocyclopenta[b]indol-3-yl)acetic acid of Formula (la), salts, and intermediates related thereto are needed. The processes and compounds described herein help meet these and other needs.
Example 7: Preparation of (i?)-2-(7-(4-Cyclopentyl-3-(trifluoromethyl)benzyloxy)-l,2,3,4- tetrahydrocyclopenta[b]indol-3-yl)acetic acid (Compound of Formula (la)) and L-Arginine Salt of (JR)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-l,2,3,4- tetrahydrocyclopenta[b]indol-3-yl)acetic acid (Compound of Formula (la)).
Method 1
Preparation of (/?)-2-(7-(4-Cyclopentyl-3-(trifluoromethyl)benzyloxy)-l ,2,3,4- tetrahydrocyclopenta[b]indol-3-yl)acetic acid (Compound of Formula (la)) and L-Arginine Salt Thereof.
Step A: Preparation of (i?)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-l,2,3,4- tetrahydrocyclopenta [b] indol-3-yl)acetic acid.
To a solution of rac-ethyl 2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-l,2,3,4- tetrahydrocyclopenta[b]indol-3-yl)acetate (20.00 g, 41.19 mmol) in acetonitrile (185 ml) in a 500 mL three-neck RBF equipped with magnetic stir bar, N2 inlet, thermocouple, and condenser was added potassium phosphate buffer (15 ml, 1.0 M, pH = 7.80) and followed by addition of lipase B, Candida antarctica, immobilized recombinant from yeast (1.0 g, 5865 U/g, 5865 U). The resultant yellow suspension was stirred at about 40 °C under N2 for 16 hours. To the mixture, 1 M citric acid was added to adjust the pH to 3.96 which was then filtered on a Whatman filter cup. The solids were washed with ACN (3 x 15 mL). The combined filtrate and washings were concentrated at about 30 °C under vacuum to give an orange residue, which was partitioned between EtOAc (60 mL) and brine (60 mL). The layers were separated and the aqueous layer was extracted with EtOAc (2 x 40 mL). The combined organic layers were washed with H20 (2 x 80 mL), brine (2 x 80 mL), dried over Na2S04, decanted, and concentrated at 30 °C under vacuum to give an orange oil, which was dried under vacuum at room temperature overnight to give a light orange oil (22.203 g) containing (R)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-l ,2,3,4-tetrahydrocyclopenta|¾]indol-3- yl)acetic acid. The crude was assayed to be 41.41 wt % (9.194 g) with 99.42% ee.
Step B: Preparation of L-Arginine Salt of (i?)-2-(7-(4-Cyclopentyl-3- (trifluoromethyl)benzyloxy)-l,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid (Compound of Formula (la)).
To the crude (21.837 g) (R)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-l,2,3,4- tetrahydrocyclopenta[b]indol-3-yl)acetic acid (41.41 %w/w; 9.043 g, 19.77 mmol) containing the (5)-isomer as the ester impurity in a 200 mL round bottom flask was added IPA (150.72 mL). The mixture was heated at 60 °C under N2 till the oily residue dissolved completely. The resultant orange solution was heated at about 60 °C for 5 min. Seeds of L-arginine salt of (R)-2- (7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-l,2,3,4-tetrahydrocyclopenta[b]indol-3- yl)acetate (362 mg) were added. The seeds were suspended in the orange solution. A 2.27 M aqueous solution of L-arginine (8.709 mL, 3.44 g, 19.77 mmol) pre-warmed to about 60 °C was added into the mixture dropwise over 30 min. A light yellow precipitate formed gradually during the addition. The suspension was stirred for about an additional 30 min. The temperature of the suspension was allowed to drop at about 0.4 °C per minute to room temperature. The mixture was agitated occasionally at room temperature overnight. The suspension was filtered and the cake was washed with IP A (3 6 mL) and EtOAc (3 x 15 mL). The filter cake was dried at room temperature under vacuum overnight to give L-arginine salt of (R)-2-(7-(4- cyclopentyl-3-(trifluoromethyl)benzyloxy)-l,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetate as a white solid (11.631 g, 44.7%): HPLC 99.38 Area %, 99.6 % ee. TGA, PXRD, PLM, SEM and DSC indicated the solid as a non-solvated, crystalline compound with an average aggregates size of 18.05 microns and a melting point of 202.69 °C.
Ή NMR (400 MHz, DMSO-d6) δ ppm 1.53-1.80 (m, 8H), 1.81-1.92 (m, 2H), 1.93-2.13 (m, 3H), 2.19 (dd, J= 15.12, 8.18 Hz, 1H), 2.46 (dd, J= 15.12, 6.61 Hz, 1H), 2.57-2.77 (m, 3H), 3.03-3.19 (m, 2H), 3.21-3.35 (m, 2H), 3.39-3.51 (m, 1H), 5.13 (s, 2H), 6.70 (dd, J= 8.75, 2.40 Hz, 1H), 6.93 (d, J= 2.40 Hz, 1H), 7.23 (d, 7= 8.75 Hz, 1H), 7.64 (d, J= 8.08 Hz, 1H), 7.72 (d, 7= 8.08 Hz, 1H), 7.74 (s, 1 H), 7.10-8.70 (br. s, 6H), 10.49 (s, 1H). LCMS m/z calcd for C32H40F3N5O5: 631.69, found: 632.1 (Msalt+H)+, 458.3 (100, (Macid+H)+).
Method 2
Preparation of (l?)-2-(7-(4-Cyclopentyl-3-(trifluoromethyl)benzyloxy)-l ,2,3,4- tetrahydrocyclopenta[b]indol-3-yl)acetic acid (Compound of Formula (la)).
Additional procedures to prepare (R)-2-(7-(4-Cyclopentyl-3- (1xiiluoromethyl)benzyloxy)-l,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid (Compound of Formula (la)) using other lipases were utilized, for example, the following were shown to hydrolyze rac-ethyl 2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-l ,2,3,4- tetrahydrocyclopenta[b]indol-3-yl)acetate to (R)-2-(7-(4-Cyclopentyl-3- (trifluoromethyl)benzyloxy)-l ,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid (Compound of Formula (la)). General hydrolysis conditions and % enantiomeric excess (% ee) are shown below for the following enzymes, lipase B Candida Antarctica, lipase Mucor miehei (MML), and P. fluorescens.

5% DMF inP. fluorescens 7.5 30 C 19-20 phosphate Buffer
Free enzyme (i.e., non-immoblized)
Each of the above enzymes provided the desired (R)-2-(7-(4-Cyclopentyl-3- (trifluoromethyl)benzyloxy)-l ,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid (Compound of Formula (la)) with varying degrees of % ee.
Example 8: Preparation of L-Arginine Salt of (l?)-2-(7-(4-Cyclopentyl-3- (trifluoromethyl)benzyloxy)-l,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid.
Method 1
(R)-2-(7-(4-Cyclopentyl-3-(trifluoromethyl)benzyloxy)-l,2,3,4- tetrahydrocyclopenta[b]indol-3-yl)acetic acid (174.7 mg, 0.381 mmol) was dissolved in EPA (1.57 mL) and L-arginine (66.4 mg, 0.381 mmol) was added as a solution in water (263 μΕ,). The homogeneous solution was warmed to 40 °C. After 15 min at this temperature, a precipitate had formed. The reaction mixture was warmed to 70 °C causing the precipitate to dissolve. The heat bath was turned off. A precipitate began to form at 40 °C and the reaction mixture was allowed to cool to about 28 °C before collecting the solids by filtration. The solids were washed with 14% water in EPA to give the L-arginine salt of (R)-2-(7-(4-cyclopentyl-3- (1riiluoromethyl)benzyloxy)-l,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid (130 mg).
Method 2
Example 8: Preparation of L-Arginine Salt of (i?)-2-(7-(4-Cyclopentyl-3- (trifluoromethyl)benzyloxy)-l,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid.
Step A: Preparation of l-Cyclopentyl-2-(trifluoromethyl)benzene (Compound of Formula (lib)).

To a 50 L three-neck round-bottom flask equipped with a mechanical stirrer, thermocouple, and nitrogen inlet, was added dry THF (35 L) and cooled to 0-5 °C. To the flask was added Iron (III) chloride (2.7 kg, 0.15 eq) portion wise over 30-60 min. and stirred for 15- 30 min. resulting in a clear greenish solution. Under a nitrogen atmosphere in a dry 100 gallon glass lined reactor was added THF (87.5 L) and magnesium turnings (4.05 kg, 1.5 eq), and cooled to 0-5 °C. To the THF and magnesium mixture was added the solution of FeCl3 in THF at a rate to maintain the internal temperature below 10 °C. To the resulting yellow/green mixture was added TMEDA (15.5 kg, 1.2 eq) at a rate to maintain the internal temperature below 20 °C. The resulting reaction mixture was heated to 40-45 °C for 1 hour and a mixture of 1 bromo-2-
(trifluoromethyl) benzene (25 kg, 1.0 eq) and bromocyclopentane (19.9 kg, 1.2 eq) was added to the reaction mixture at a rate to maintain an internal temperature below 25 °C. The resulting reaction mixture was allowed to stir at room temperature overnight and subsequently cooled to an internal temperature of 0-5 °C. To the resulting mixture was added 6 N HC1 (100 L, 1.5 h) at such a rate as to maintain the internal temperature below 15 °C (caution, very exothermic). After the quench, MTBE (200 L) was added and the reactor contents was stirred for 30 min. The phases were separated and the aqueous layer back extracted with MTBE (75 L). The combined organic layers were washed with H20 (50 L), brine (50 L) and dried (MgS04). The mixture was filtered through an in-line (1 micron) filter cartridge followed by an additional in-line (0.45 micron) filter cartridge into a clean dry reactor. The solvent was evaporated under vacuum (jacket < 30 °C) and co-evaporated with heptanes (2 x 25 L) to provide a viscous liquid. The viscous liquid was dissolved in heptanes (100 L) and passed through a silica plug (25 kg). The silica plug was eluted with heptanes (TLC, Rf ~ 0.8, silica gel, heptanes) and the fractions containing the product were evaporated to provide the title compound as a yellow liquid, 11.7 kg (49.2%), purity as determined by HPLC was 94.1%. Ή NMR conforms to reference standard.
Step B: Preparation of 4-(Chloromethyl)-l-cyclopentyl-2-(trifluoromethyl)benzene (Compound of Formula (He)).

To a 100 gallon glass lined reactor equipped with a stirrer was added concentrated sulphuric acid (48.6 L) and cooled to an internal temperature between about -5 to -10 °C under an atmosphere of N2. To the sulfuric acid was added thionyl chloride (26.99 kg, 2 eq) at a rate to maintain the internal temperature below -5 °C. To the resulting mixture 1,3,5-trioxane (15.3 kg, 1.5 eq) was added portion wise at a rate to maintain the internal temperature below -5 °C. After the addition of 1,3,5-trioxane, l-cyclopentyl-2-(trifluoromethyl) benzene (24.0 kg) was added drop wise over a period of approximately 2-3 hours. The reaction mixture was stirred at 0 °C for approximately 3-4 hours, allowed to warm to room temperature overnight and subsequently cooled to an internal temperature of 0-5 °C. To the resulting mixture was added water (316 L) drop wise over a period of approximately 5-6 hours (Note: Very exothermic). After the quench with water, the resulting aqueous mixture was extracted with MTBE (243 L and 123 L). The combined organics were washed with saturated NaHC03 (100 L), brine (100 L), water (100 L), brine (100 L), and dried (MgS04). The mixture was filtered through an in-line (1 micron) filter cartridge followed by an additional in-line (0.45 micron) filter cartridge into a clean dry reactor. The solvent was evaporated under vacuum (jacket < 30 °C) and further evaporated under vacuum at 35-40 °C. The resulting oil was distilled under high vacuum to provide the title compound as a yellow liquid, 24.8 kg (83%), purity as determined by HPLC was 99.47%. Ή
NMR conforms to reference standard.
Step C: Preparation of Ethyl 2-(2-Morpholinocyclopent-2-enylidene)acetate (Compound of Formula (Kg), Whe

Cyclopentanone (22.00 kg), morpholine (22.88 kg) and cyclohexane (43.78 kg) were charged to a 400 L glass-lined reactor equipped with overhead agitation, jacket temperature control, a nitrogen inlet, and a Dean-Stark trap. The reactor contents were heated to about 85 °C to 95 °C for approximately 26 h while removing water using the Dean-Stark trap. The reaction to form the enamine (i.e., 4-cyclopentenylmorpholine, Compound of Formula (lie) wherein R1 and R2 together with the nitrogen atom form a morpholine ring) is deemed complete when the morpholine amount is verified to be < 3% by GC peak area.
The reactor contents were cooled to about 60 °C and ethyl glyoxalate (Compound of Formula (ΠΤ) wherein R3 is ethyl; 58.74 kg, 50% solution in toluene) was added to the mixture slowly so as to maintain an internal temperature of < 80 °C. The reactor contents were heated to about 85 °C to 95 °C for at least 25 hours while removing water using the Dean-Stark trap. The reaction was deemed complete when the eneamine (i.e., 4-cyclopentenylmorpholine) amount by GC was verified to be less than 0.5% by GC peak area. The cyclohexane/toluene mixture was distilled under vacuum, ethanol (261.80kg) was charged to the reactor, and the resulting solution was again distilled under vacuum. Ethanol (34.76 kg) and water 44.00 kg) were charged to the reactor and the reactor contents stirred at 25 °C. The mixture was stirred further for 6 h at about 0-5 °C.
The resulting product slurry was collected by filtration, washed with aqueous ethanol (34.76 kg ethanol dissolved in 176.00 kg water). The filter-cake was further washed with water (110.00 kg), dried initially at approximately 36 °C for 1 hour under vacuum and subsequently at approximately 50 °C under vacuum for 17 h. The title compound was obtained as a tan solid (23.48 kg, 37.8% yield).
Step D: Preparation of Ζί/ZEthyl 2-(7-(Benzyloxy)-l,2-dihydrocyclopenta[b]indol- 3(4H)-ylidene)acetate

To a 400 L glass-lined reactor equipped with overhead agitation, jacket temperature control, and a nitrogen inlet was added (4-(benzyloxy)phenyl)hydrazine hydrochloride (21.08 kg, 1.000 mole equiv.), ethyl 2-(2-mo holinocyclopent-2-en lidene)acetate (22.02 kg, 1.104 mole equiv.), ethanol (51.2 kg, 2.429 mass equiv.), and acetic acid (36.8 kg, 1.746 mass eq.). After the reactor contents are allowed to stand for 10 minutes, agitation and then heating to 60°C to 65°C (60°C target) was started. While stirring at that temperature, samples of the reaction mixture were taken over intervals of approximately 30 minutes and analyzed by HPLC for (4-
(benzyloxy)phenyl)hydrazine, ethyl 2-(2-morpholinocyclopent-2-enylidene)acetate, and hydrazone content. When (4-(benzyloxy)phenyl)hydrazine HPLC % area was < 1, TFA (11.6 kg, 101.7 mol, 1.200 mole equiv., 0.550 mass equiv.) was charged over approximately 1 hour while the stirred reaction mixture was maintained at 60°C ± 5°C with reactor jacket cooling. As stirring at 60°C to 65°C was continued, the hydrazone and imine content of the reaction mixture was monitored by HPLC. After stirring at 60°C to 65°C for at least 12 hours the imine content of the reaction mixture was < 5% area by HPLC, and the stirred reaction mixture was cooled to 20°C to 25°C over approximately 3 hours. Stirring was maintained at that temperature to allow isomerization of the Z isomer to the desired E isomer. The E isomer crystallizes from the reaction mixture. The Z isomer and E isomer % area content of the reaction mixture was monitored by HPLC during this period of stirring at 20°C to 25°C, which was continued until the Z-isomer content of the reaction mixture was < 15% area by HPLC.
The stirred reaction mixture was cooled (0°C to 5°C) over at least 2 hours and then filtered. The reactor was charged with ethanol (27.4 kg, 1.300 mass equiv.), which was stirred and chilled to 0°C to 5°C and then used in two approximately equal portions to slurry-wash the product filter cake twice. The reactor was charged with ethanol (13.8 kg, 0.655 mass equiv.), which was stirred and chilled to 0°C to 5°C and then used to wash the product filter cake by displacement. The reactor was charged with USP purified water (100 kg, 4.744 mass equiv.), and the temperature was adjusted to 20°C to 25°C. The USP purified water was then used in three approximately equal portions to wash the product filter cake three times, the first two by reslurrying and the third by displacement. The reactor was charged with ethanol (16.4 kg, 0.778 mass equiv.), stirred and chilled to 0°C to 5°C, and then used to wash the product filter cake by displacement. The washed product filter cake was dried under full vacuum first with a jacket temperature of 35°C for 1 hour and then with a jacket temperature of 50°C. While drying continues with a jacket temperature of 50°C, the product solids are turned over every 1 hour to 3 hours, and product samples are analyzed for loss on drying (LOD) every >4 hours. When LOD was < 1%, the product was cooled to < 30°C. The yield of the title compound was 13.06 kg (37.59 mol, 44.7%). Step E: Preparation of Ethyl 2-(7-Hydroxy-l,2,3,4-tetrahydrocyclopenta[b]indol-3- yl)acetate.

To a 200 liter Hastelloy reactor was added ethyl 2-(7-(benzyloxy)-l ,2- dihydrocyclopenta[b]indol-3(4H)-ylidene)acetate (E/Z mixture, 12 kg), 10% Pd/C (50% wet with H20; 1.80 kg) and ethyl acetate (108 kg). The suspension was degassed 3x with N2 and triethylamine (1.76 kg) was added. To the resulting mixture was added formic acid (3.34 kg) while maintaining the internal temperature at below 35 °C. The reaction progression was followed by HPLC to monitor the complete consumption of starting material (i.e., E/Z mixture of ethyl 2-(7-(benzyloxy)-l ,2-dihydrocyclopenta[b]indol-3(4H)-ylidene)acetate) and the debenzylated intermediate. After approximately 30 minutes an additional amount of formic acid (0.50 kg) was added and the combined peak area of ethyl 2-(7-(benzyloxy)-l ,2- dihydrocyclopenta[b]indol-3(4H)-ylidene)acetate and the related debenzylated intermediate was determined to be < 1 % area by HPLC. The reactor contents were filtered through a 1.2 μιη cartridge filter followed by an in-line 0.2 μπι inline polishing filter. To the filtrate was added water (60 kg) and the biphasic mixture was partitioned. The organics were separated and concentrated under vacuum at approximately 60°C ± 5°C to a minimum stir volume, ethyl acetate (21.6 kg) was added and the mixture was further concentrated under vacuum to a minimum stir volume. Once again ethyl acetate (16.8 kg) was charged to the crude mixture and the resulting solution was heated to approximately 60 °C. Heptanes (37.2 kg) were charged maintaining the internal temperature at 60 °C. The solution was slowly cooled to approximately 0 to 5 °C and approximately 2-3 hr to facilitate crystallization. The slurry was filtered, the filter cake was reslurried in heptanes (27.12 kg) and ethyl acetate (7.08 kg). The resulting suspension was filtered and the solids dried under vacuum at approximately 40 ± 5 °C (until the loss on drying (LOD) is < 1%) to afford the title compound (6.23 kg, 70.3 % yield) as a solid.
Step F: Preparation of ( ft^-Ethyl 2-(7-(4-Cyclopentyl-3- (trifluoromethyl)benzyloxy)-l,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetate (Compound of Formula (Ilk), Wher

To a 50 liter glass reactor containing ethyl 2-(7 -hydroxy- 1 ,2,3, 4- tetrahydrocyclopenta[b]indol-3-yl)acetate (2.000 kg, 1.000 equiv.) was added cesium carbonate
(3.266 kg, 1.300 equiv.) and acetonitrile (15.720 kg) under nitrogen. To the resulting mixture was added 4-(chloromethyl)-l-cyclopentyl-2-(trifluoromethyl)benzene (2.228 kg, 1.100 equiv.) over approximately one hour while maintaining the stirred reactor contents at 40°C ± 5°C. After the addition of 4-(chloromethyl)-l-cyclopentyl-2-(trifluoromethyl)benzene the reactor contents were heated to 65°C ± 5°C with stirring until the concentration of ethyl 2-(7-hydroxy-l , 2,3,4- tetrahydrocyclopenta[b]indol-3-yl)acetate in the reaction mixture was less than 2.0 % area by
HPLC. The reaction mixture was cooled to 50°C ± 5°C and filtered under nitrogen through a fine filter cloth with suction to remove cesium salts (Note: ethyl 2-(7-(4-cyclopentyl-3-
(trifluoromethyl)benzyloxy)-l ,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetate may precipitate below 30 °C). The filter cake was washed with fresh hot (50°C ± 5 °C) acetonitrile (5.658 kg divided in approximately three equal portions). The filtrates were returned to the reactor. The combined filtrates were concentrated by vacuum distillation with a jacket temperature of 60°C ± 10°C. To the reactor was added ethyl alcohol (3.156 kg) and once again concentrated with stirring by vacuum distillation with a jacket temperature of 60°C ± 10 °C. Once again, ethyl alcohol (3.156 kg) was added to the reactor and the contents were concentrated by vacuum distillation with a jacket temperature of 60 °C ± 10 °C to a reactor volume of approximately 14 L. The stirred reactor contents were cooled to 0 °C ± 5°C and the temperature maintained for 4 hours to facilitate the crystallization of the product. The resulting slurry was filtered. The filter cake was washed with cold 0 °C ± 5 °C ethyl alcohol (2 x 3.156 kg). The filter cake was dried under vacuum at 35 °C ± 5 °C until the weight loss over >1 hour was <2% to provide 3.0943 kg (81.0% yield) of the title compound as a solid.
Step G: Preparation of (!?)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)- l,2,3,4-tetrahydrocyclo

A 1.0 M buffer solution was prepared containing potassium phosphate monobasic (29.1 g, 0.0335 equiv.) in USP purified water (213 g) and potassium phosphate dibasic (368.2 g, 0.331 equiv.) in USP purified water (2.107 g). To a 50 liter glass reactor was added ethyl 2-(7-(4- cyclopentyl-3-(trifluoromethyl)benzyloxy)-l,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetate
(3.094 kg, 1.000 equiv.), Lipase B, Candida antarctica, immobilized (88.18 g, 293250 units/kg of ethyl ester starting material) and acetonitrile (22.32 kg). To the stirred contents of the reactor was added the previously prepared 1.0 M potassium phosphate buffer. The resulting mixture was stirred under nitrogen at a temperature of 40°C ± 5°C until the (R)-2-(7-(4-cyclopentyl-3-
(rrifluoromethyl)benzyloxy)-l ,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid concentration was >35% area as determined by HPLC (Note: although the reaction usually is complete after about 10 hours, the reaction mixture may be held at 40°C ± 5°C overnight). The stirred reactor contents were cooled to 25 °C ± 5°C and the pH was adjusted to between 4 and 5 by addition of a solution of citric acid (278.5 g, 0.228 equiv.) dissolved in USP purified water (1.454 kg). The reactor contents were filtered to remove immobilized lipase and phosphate and citrate salts. The reactor and solids were washed with acetonitrile (4.827 kg) and the combined filtrates were added backed into the reactor. The stirred reactor contents were concentrated to a volume of 1.0 L to 2.0 L by vacuum distillation at a jacket temperature of 55 °C ± 5°C. To the reactor was added ethyl acetate (5.582 kg) and USP purified water (6.188 kg). The contents were stirred at 20°C ± 5°C for at least 10 minutes and a solution of sodium chloride (1 kg) in USP purified water (1 kg) was added to facilitate phase separation. After phase separation was complete, the lower aqueous layer was drained. A solution of sodium chloride (5.569 kg) in USP purified water (12.38 kg) was divided in two approximately equal portions and the ethyl acetate phase was washed (2x). The ethyl acetate phase was transferred into a carboy and the reactor was rinsed with ethyl acetate (838.5 g) and added to the carboy containing the ethyl acetate phase. The reactor was washed sequentially with USP purified water (12.38 kg), acetone (4.907 kg), and ethyl acetate (838.5 g) and the ethyl acetate mixture from the carboy was transferred back to the reactor and concentrated with stirring to a volume of 1 L to 2 L by vacuum distillation at a jacket temperature of 55°C ± 5°C. To the reactor was added 2-propanol (14.67 kg) and after stirring the resulting mixture was concentrated to a volume of 1 L to 2 L by vacuum distillation at a jacket temperature of 55°C ± 5°C. To the reactor was added 2-propanol (7.333 kg) and heated with stirring at 60°C ± 5°C until the contents dissolved. The stirred reactor contents were cooled to 20°C ± 5°C and filtered through a medium-porosity fritted-glass filter to remove any inorganic solids to provide a 2-propanol solution containing 1.3188 kg of the title compound.
Step H: Preparation of L-Arginine Salt of (i?)-2-(7-(4-Cyclopentyl-3- (trifluoromethyl)benzyloxy)-l ,2,3?4-tetrahydrocyclopenta [b] indol-3-yl)acetic acid
(Compound of For

To a 50 liter glass reactor containing the 2-propanol solution prepared in Step G of (R)- 2-(7-(4-cyclopen1yl-3-(trifluoromethyl)ben2yloxy)-l,2,3,4-tetrahydrocyclopenta[b]indol-3- yl)acetic acid (1.3188 kg, 1.000 equiv.) was added an additional amount of 2-propanol (6.3389 kg) to adjust the total volume to approximately 16.7 L/kg of (R)-2-(7-(4-cyclopentyl-3- (trifluoromethyl)benzyloxy)-l,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid. The reactor contents were stirred and heated to 60 °C ± 5 °C. To the reactor was added seed material (L- arginine salt of (R)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-l , 2,3,4- tetrahydrocyclopenta[b]indol-3-yl)acetic acid, 26.4 g, 0.0145 equiv.). The reactor contents were stirred for approximately 5 minutes at 60 °C ± 5 °C and a solution of L-arginine (502.5 g, 1.000 equiv.) in USP purified water (1.27 kg) preheated to 60°C ± 5°C was added over approximately
1 hour while maintaining the stirred reactor contents at 60°C ± 5°C. The stirring of the reactor contents at 60°C ± 5°C was maintained for approximately 1 hour and then allowed to cool at an approximate rate of 0.2°C/min to 1.0°C/min. to a temperature of 25°C ± 5°C. Once at approximately 25°C the contents of the reactor were stirred for approximately 1 hour maintaining the temperature of 25°C ± 5°C. The resulting slurry was filtered and the filter cake was washed with 2- propanol (6.2511 kg divided in three approximately equal portions) and with ethyl acetate (13.560 kg divided in six approximately equal portions. The filter cake was dried under vacuum at 40°C ± 5°C (until the weight loss over >1 hour is <2%) to provide 1.657 kg of the title compound (32.9% yield) as a crystalline solid.
HPLC purity: 99.64 Area %; Enantiomeric purity: 99.3%; DSC melting onset temperature 203.46 °C; TGA Weight Loss out to ~1 10 °C was 0.05%. NMR confirms the structure of the L-salt.
Five additional lots of the L-arg salt have been prepared using substantially this same synthetic method as described above, the DSC melting onset temperatures for a sample from each of the lots is as follows: 203.96 °C, 203.00 °C, 203.11 °C, 203.79 °C and 203.97 °C; the TGA Weight Loss out to ~1 10 °C for a sample from each of the lots is as follows: 0.04%, 0.04%, 0.03%, 0.10%, and 0.12%.
| WO2009078983A1 * | Dec 15, 2008 | Jun 25, 2009 | Arena Pharm Inc | Tetrahydrocyclopenta[b]indol-3-yl carboxylic acid derivatives useful in the treatment of autoimmune and inflammatory disorders |
| WO2010011316A1 * | Jul 22, 2009 | Jan 28, 2010 | Arena Pharmaceuticals, Inc. | SUBSTITUTED 1,2,3,4- TETRAHYDROCYCLOPENTA[b]INDOL-3-YL) ACETIC ACID DERIVATIVES USEFUL IN THE TREATMENT OF AUTOIMMUNE AND INFLAMMATORY DISORDERS |
| US20090004265 | Jan 19, 2006 | Jan 1, 2009 | Bayer Healthcare Ag | Prevention and Treatment of Thromboembolic Disorders |
FDA Approves Blincyto (blinatumomab) for Precursor B-Cell Acute Lymphoblastic Leukemia
 Blinatumomab linking a T cell to a malignant B cell.
Blinatumomab linking a T cell to a malignant B cell.
FDA Approves Blincyto (blinatumomab) for Precursor B-Cell Acute Lymphoblastic Leukemia
December 3, 2014 — The U.S. Food and Drug Administration today
approved Blincyto (blinatumomab) to treat patients with Philadelphia
chromosome-negative precursor B-cell acute lymphoblastic leukemia
(B-cell ALL), an uncommon form of ALL.

Blinatumomab (AMG103) is a drug that has anti-cancer properties. It belongs to a new class of constructed monoclonal antibodies,bi-specific T-cell engagers (BiTEs), that exert action selectively and direct the human immune system to act against tumor cells. Blinatumomab specifically targets the CD19 antigen present on B cells.[1]
The drug was developed by a German-American company Micromet, Inc. in cooperation with Lonza; Micromet was later purchases by Amgen, which has furthered the drug’s clinical trials. In July 2014, the FDA granted breakthrough therapy status to blinatumomab for the treatment of acute lymphoblastic leukemia (ALL).[2] In October 2014, Amgen’s Biologics License Application for blinatumomab was granted priority review designation by the FDA, thus establishing a deadline of May 19, 2015 for completion of the FDA review process.[3]
Structure and mechanism of action
Blinatumomab linking a T cell to a malignant B cell.
Blinatumomab enables a patient’s T cells to recognize malignant B cells. A molecule of blinatumomab combines two binding sites: a CD3site for T cells and a CD19 site for the target B cells. CD3 is part of the T cell receptor. The drug works by linking these two cell types andactivating the T cell to exert cytotoxic activity on the target cell.[4] CD3 and CD19 are expressed in both pediatric and adult patients, making blinatumomab a potential therapeutic option for both pediatric and adult populations.[5]
Therapeutic use
Clinical trials
In a phase 1 clinical study with blinatumomab, patients with non-Hodgkin’s lymphoma showed tumor regression, and in some cases complete remission.[6] There are ongoing phase 1 and phase 2 clinical trials of blinatumomab in patients with acute lymphoblastic leukemia (ALL).[7] One phase II trial for ALL reported good results in 2010 and another is starting.[8]
Adverse effects
Common side effects observed in Phase 2 trials are listed below; they were temporary and typically occurred during the first treatment cycle:[5]
- Flu-like symptoms (i.e. fever, headache, and fatigue)
- Tremor
- Weight increase
- Hypokalemia
- Decrease of blood immunoglobulin
CNS effects were also observed during clinical trials and were treated via a lower dose of blinatumomab, administration of dexamethasone, or treatment discontinuation. Because the side effects were reversible, early monitoring for the CNS symptoms listed below is important:[5]
- Seizure
- Encephalopathy
- Tremor
- Apraxia
- Speech disorders
- Disorientation
Less common side effects include cytokine release syndrome and immunogenicity.[5]
References
- Statement on a Nonproprietary Name adopted by the USAN Council: Blinatumomab
- Amgen Receives FDA Breakthrough Therapy Designation For Investigational BiTE® Antibody Blinatumomab In Acute Lymphoblastic Leukemia
- Amgen’s BiTE® Immunotherapy Blinatumomab Receives FDA Priority Review Designation In Acute Lymphoblastic Leukemia
- Mølhøj, M; Crommer, S; Brischwein, K; Rau, D; Sriskandarajah, M; Hoffmann, P; Kufer, P; Hofmeister, R; Baeuerle, PA (March 2007). “CD19-/CD3-bispecific antibody of the BiTE class is far superior to tandem diabody with respect to redirected tumor cell lysis”. Mol Immunol 44 (8): 1935–43. doi:10.1016/j.molimm.2006.09.032. PMID 17083975.
- Background Information for the Pediatric Subcommittee of the Oncologic Drugs Advisory Committee Meeting 04 December 2012
- Bargou, R; et al. (2008). “Tumor regression in cancer patients by very low doses of a T cell-engaging antibody”. Science 321 (5891): 974–977. doi:10.1126/science.1158545.PMID 18703743.
- ClinicalTrials.gov NCT00560794 Phase II Study of the BiTE Blinatumomab (MT103) in Patients With Minimal Residual Disease of B-precursor Acute ALL
- “Micromet initiates MT103 phase 2 trial in adult ALL patients”. 20 Sep 2010.
External links
| Monoclonal antibody | |
|---|---|
| Type | Bi-specific T-cell engager |
| Source | Mouse |
| Target | CD19, CD3 |
| Clinical data | |
| Legal status |
?
|
| Identifiers | |
| CAS number | 853426-35-4 |
| ATC code | None |
| UNII | 4FR53SIF3A |
| Chemical data | |
| Formula | C2367H3577N649O772S19 |
| Mol. mass | 54.1 kDa |
US FDA issued a Warning Letter to the company Hikma Pharmaceuticals
DRUG REGULATORY AFFAIRS INTERNATIONAL

| Warning Letter: Deficiencies in Visual Inspection |
| In October 2014, the US FDA issued a Warning Letter to the company Hikma Pharmaceuticals justified by deficiencies in the visual inspection of vials. Read more here. |
In October 2014, the US FDA issued a Warning Letter to the company Hikma Pharmaceuticals because of deficiencies in the visual inspection of vials and environmental monitoring.

Already in a Warning Letter issued in 2011, a deficiency in the visual inspection was noted as the detection and evaluation of particulate matter failed to be sufficient. Now, the current complaint in the area of visual control explicitly refers to the qualification of staff for the performance of the manual visual inspection. Here, the FDA inspectors noticed that visible markings were present on the qualifcation test sets which enabled operators for visual inspection to recognize – thanks to these markings – vials with particles. The qualification of staff…
View original post 52 more words
Pi-Process Intensification Experts LLP at CPhI Mumbai India 3rd Dec 2014…My visit

Process Intensification
Creating competitive advantage through Improved and consistent quality, high efficiencies and maximum flexibility.
Safer, Cleaner, Smaller, Cheaper and Smarter processes , The basic principle of Process Intensification is to fit the equipment to the process and not process to the equipment, as is the case now.
Process Intensification can achieve drastic improvement in the time cycle and yields as well as converting batch processes to continuous process using specialized set of equipment. The design philosophy in process intensification is to design a process which has Chemical Kinetics as its only limitation. See the illustration below
“Process Intensification by Kinetics alone controlling the reaction, using specialized equipments; modification / telescoping of process steps achieves drastic reduction in time cycles and converts batch processes to continuous ; Reduced energy consumption, Reduced by-product formation; sustainability , hazard-containment, compliance to QbD and PAT and importantly a much faster time-to-market”

Illustrative examples are as follows:
- Watt’s aldol reaction: Time needed to reach 100 % conversion 20 minutes against 24 hours in batch process
- Fisher Esterification:
 gives 83% yield against 15% in batch process
gives 83% yield against 15% in batch process - Grignard Reaction: gives 78% yield against 49% in batch process
- Nitration Reaction: Product purity increase from 56% to 78% and yield of mononitrate increases 55% to 75%.
- Other Reactions: Acetylation, Amine Protection, Carbonylation, Claisen Schmidt Reaction, Esterification, Hydrogenation, Hydrolysis, Methylation, Oxidation, Phosgenation, Sulphonation, Suzuki Coupling Ring Expansion

Benefits of Process Intensification (PI) Techniques
Sponsored Projects
Scale-up for Retrofitting in existing plant as well as greenfield projects based on flow chemistry data generated in our laboratory. A well-equipped Laboratory and Pilot Plant set-up is available at our “Pi-Lab” for carrying out “FLOW Chemistry” based Reactions and utilizing numerous Process Intensification techniquesfor Unit-Processes & Unit-Operations for the industry to reap the benefits of Process Intensification.
The laboratory and pilot plant data will be utilized to provide the plant scale design using specialized equipments like micro-reactors, micro-plate-reactors in SiC, monolithic loop reactors, spinning disk reactors-cum-heat exchangers, FUMI reactors, dynamic mixing reactors, oscillatory baffled reactors (OBR), Bio-catalytic impregnated membrane Reactors, and other modern state-of-the-art equipments enabling conversion of batch to continuous flow processes.
We handle hazardous chemistries with very high exotherms (upto 1300 J/gm) safely in the range of -70oC to + 250oC with pressures upto 200 bar, and with reaction times from 0.03 sec to 1 hour and reactor volumes from 0.2 ml to 100 ml (Lab) and 1 L (Pilot) — yielding from 20 gms to 8 Kgs/hour (Lab) and 500 gms to 200 Kgs/hour (Pilot).
Scale Up – Flexibility & Adaptability
![]() …… will provide all the services for scale up to the sizes desired by clients by utilizing data from Laboratory trials.
…… will provide all the services for scale up to the sizes desired by clients by utilizing data from Laboratory trials.


Rental
A range of Flow Chemistry and Process Intensification equipments can be offered on rent. This enables the users to get the hands-on experience so as to select the apt equipments for their needs.
CEO
Pi-Process Intensification Experts LLP
Plot-W-33, M.I.D.C. Industrial Area
TALOJA – 410208, Navi Mumbai, INDIA
email : vk@pi-inc.co
www.pi-inc.co
Tel: +91-9321342022 // +91-9821342022

some pics from hall 5 -H-47 at cphi mumbai india dec 3 2014





Sanofi Gets US FDA Approval For Priftin, Rifapentine 利福喷汀 Tablets To Treat Latent TB Infection

French drug maker Sanofi Tuesday said it has received approval from the U.S. Food and Drug Administration for its Priftin (rifapentine) tablets to treat latent tuberculosis infection, or LTBI.

Following a priority review, FDA has approved Priftin in combination with isoniazid, or INH, for a new indication for treatment of LTBI in patients two years of age and older at high risk of progression to tuberculosis or TB disease.

Rifapentine
Antibiotic DL 473IT;Cyclopentylrifampicin;DL 473;KTC 1;MDL 473;Prifitin;Priftin;R 77-3;Rifamycin AF/ACPP;
Rifapentine is an antibiotic drug used in the treatment of tuberculosis. It inhibits DNA-dependent RNA polymerase activity in susceptible cells. Specifically, it interacts with bacterial RNA polymerase but does not inhibit the mammalian enzyme.
For the treatment of pulmonary tuberculosis
3-(((4-Cyclopentyl-1-piperazinyl)imino)methyl)rifamycin
| C47H64N4O12 |
| 61379-65-5 |
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (7S,9E,11S,12R,13S,14R,15R,16R,17S,18S,19E,21Z,26E)-26-{[(4-cyclopentylpiperazin-1-yl)amino]methylidene}-2,15,17,29-tetrahydroxy-11-methoxy-3,7,12,14,16,18,22-heptamethyl-6,23,27-trioxo-8,30-dioxa-24-azatetracyclo[23.3.1.14,7.05,28]triaconta-1(28),2,4,9,19,21,25(29)-heptaen-13-yl acetate | |
| Clinical data | |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a602026 |
| Legal status |
?
|
| Pharmacokinetic data | |
| Bioavailability | increases when administered with food |
| Identifiers | |
| CAS number | 61379-65-5 |
| ATC code | J04AB05 |
| PubChem | CID 5462354 |
| DrugBank | DB01201 |
| ChemSpider | 10482075 |
| UNII | XJM390A33U |
| KEGG | D00879 |
| ChEBI | CHEBI:45304 |
| ChEMBL | CHEMBL1660 |
| NIAID ChemDB | 007686 |
| Synonyms | 3{[(4-cyclopentyl-1-piperazinyl)imino]methyl}rifamycin |
| Chemical data | |
| Formula | C47H64N4O12 |
| Mol. mass | 877.031 g/mol |
Rifapentine (INN, marketed under the brand name Priftin by Sanofi-Aventis) is an antibiotic drug used in the treatment of tuberculosis.
Rifapentine was first synthesized in 1965 by the same company that produced rifampin. The drug was approved by the Food and Drug Administration (FDA) in June 1998.

Medical uses
A review of alternative regimens for prevention of active tuberculosis in HIV-negative individuals with latent TB found that a weekly, directly observed regimen of rifapentine with isoniazid for three months was as effective as a daily, self -administered regimen of isoniazid for nine months. But the rifapentine-isoniazid regimen had higher rates of treatment completion and lower rates of hepatotoxicity. However, the rate of treatment-limiting adverse events was higher in the rifapentine-isoniazid regimen. [1]
PRIFTIN (rifapentine) for oral administration contains 150 mg of the active ingredient rifapentine per tablet.
The 150 mg tablets also contain, as inactive ingredients: calcium stearate, disodium EDTA, FD&C Blue No. 2 aluminum lake, hydroxypropyl cellulose, hypromellose USP, microcrystalline cellulose, polyethylene glycol, pregelatinized starch, propylene glycol, sodium ascorbate, sodium lauryl sulfate, sodium starch glycolate, synthetic red iron oxide, and titanium dioxide.
Rifapentine is a rifamycin derivative antibiotic and has a similar profile of microbiological activity to rifampin (rifampicin). The molecular weight is 877.04.
The molecular formula is C47H64N4O12.
The chemical name for rifapentine is rifamycin, 3-[[(4-cyclopentyl-1-piperazinyl)imino]methyl]-or 3-[N-(4-Cyclopentyl – 1-piperazinyl)formimidoyl] rifamycin or 5,6,9,17,19,21-hexahydroxy-23-methoxy-2,4,12,16,18,20,22-heptamethyl-8-[N-(4-cyclopentyl-l-piperazinyl)-formimidoyl]-2,7-(epoxypentadeca[1,11,13]trienimino)naphtho[2,1-b]furan-1,11(2H)-dione 21-acetate. It has the following structure:
 |
Use in special populations
Pregnancy
Rifapentine has been assigned a Pregnancy Category C by the FDA. Rifapentine in pregnant women has not been studied, but animal reproduction studies have resulted in fetal harm and were teratogenic. If rifapentine and rifampin are used together in pregnancy, coagulation should be monitored due to a possible increased risk of maternal postpartum hemorrhage and infant bleeding. [2]
Adverse effects
Common side effects are hyperuricemia, pyuria, hematuria, urinary tract infection, proteinuria, neutropenia, anemia, and hypoglycemia. [2]
Contraindications
Rifapentine should be avoided in patients with an allergy to the rifamycin class of drugs. [2] This drug class includes rifampin and rifabutin. [3]
Interactions
Rifapentine induces metabolism by CYP3A4, CYP2C8 and CYP2C9 enzymes. It may be necessary to adjust the dosage of drugs metabolized by these enzymes if they are taken with rifapentine. Examples of drugs that may be affected by rifapentine include warfarin, propranolol, digoxin, protease inhibitors and oral contraceptives.[2]
History
Rifapentine was first synthesized in 1965 by the same company that produced rifampin. The drug was approved by the Food and Drug Administration (FDA) in June 1998. It is synthesized in one step from rifampicine.


Rifapentine was first synthesized in 1965 by the same company that produced rifampin. The drug was approved by the Food and Drug Administration (FDA) in June 1998.
(7S,11S,12S,13S,14R,15S,16R,17R,18R,26E)-26-{[(4-Cyclopentyl-1-piperazinyl)amino]methylene}-2,15,17,29-tetrahydroxy-11-methoxy-3,7,12,14,16,18,22-heptamethyl-6,23,27-trioxo-8,30-dioxa-24-azatetracyclo [23.3.1.14,7.05,28]triaconta-1(28),2,4,9,19,21,25(29)-heptaen-13-yl acetate. Rifapentine is an antibiotic drug used in the treatment of tuberculosis.
Preparation of Rifapentine: this chemical can be prepared by 3-aldehyde rifamycin SV with 1-Amino-4-cyclopentylpiperazine. This reaction needs reagent tetrahydrofuran. The yield is 55 %
References
- Sharma SK et al . (2013). “Rifamycins (rifampicin, rifabutin and rifapentine) compared to isoniazid for preventing tuberculosis in HIV-negative people at risk of active TB.”. Cochrane Database of Systematic Reviews 7: CD007545. doi:10.1002/14651858.CD007545.pub2. PMID 23828580.
- Sanofi-Aventis. (2010) Priftin (rifapentine): Highlights of Prescribing Information. Retrieved from http://products.sanofi.us/priftin/Priftin.pdf.
- CDC. (2013) Core Curriculum on Tuberculosis: What the Clinician Should Know. Retrieved from http://www.cdc.gov/TB/education/corecurr/default.htm
- http://www.mdpi.com/1424-8247/5/7/690/htm
IMPROVED CONTINUOUS FLOW PROCESSING: BENZIMIDAZOLE RING FORMATION VIA CATALYTIC HYDROGENATION OF AN AROMATIC NITRO COMPOUND

Improved Continuous Flow Processing: Benzimidazole Ring Formation via Catalytic Hydrogenation of an Aromatic Nitro Compound
http://pubs.acs.org/doi/full/10.1021/op400179f
In the development of a new route to bendamustine hydrochloride, the API in Treanda, the key benzimidazole intermediate 5 was generated via catalytic heterogeneous hydrogenation of an aromatic nitro compound using a batch reactor. Because of safety concerns and a site limitation on hydrogenation at scale, a continuous flow hydrogenation for the reaction was investigated at lab scale using the commercially available H-Cube. The process was then scaled successfully, generating kilogram quantities on the H-Cube Midi. This flow process eliminated the safety concerns about the use of hydrogen gas and pyrophoric catalysts and also showed 1200-fold increase in…
View original post 6 more words
BENDAMUSTINE


TREANDA contains bendamustine hydrochloride, an alkylating drug, as the active ingredient. The chemical name of bendamustine hydrochloride is 1H-benzimidazole-2-butanoic acid, 5-[bis(2-chloroethyl)amino]-1 methyl-, monohydrochloride. Its empirical molecular formula is C16H21Cl2N3O2 • HCl, and the molecular weight is 394.7. Bendamustine hydrochloride contains a mechlorethamine group and a benzimidazole heterocyclic ring with a butyric acid substituent, and has the following structural formula:
|
TREANDA (bendamustine hydrochloride) for Injection is intended for intravenous infusion only after reconstitution with Sterile Water for Injection, USP, and after further dilution with either 0.9% Sodium Chloride Injection, USP, or 2.5% Dextrose/0.45% Sodium Chloride Injection, USP. It is supplied as a sterile non-pyrogenic white to off-white lyophilized powder in a single-use vial. Each 25-mg vial contains 25 mg of bendamustine hydrochloride and 42.5 mg of mannitol, USP. Each 100-mg vial contains 100 mg of bendamustine hydrochloride and 170 mg of mannitol, USP. The pH of the reconstituted solution is 2.5 -3.5.
Bendamustine hydrochloride, 4-{5-[Bis(2-chloroethyl) amino]- l-methyl-2- benzimidazolyl} butyric acid hydrochloride, of the formula (VI) :

was initially synthesized in 1963 in the German Democratic Republic (GDR) and was available from 1971 to 1992 there, as the hydrochloride salt, under the trade name Cytostasan®. Since that time, it has been marketed in Germany under the trade name Ribomustin®. Bendamustine Hydrochloride as injection is available in the United States under the tradename Treanda®. Bendamustine hydrochloride is an alkylating agent that is approved for the treatment of non-Hodgkin’s lymphoma, multiple myeloma and chronic lymphocytic leukemia.
Bendamustine hydrochloride is a benzimidazole analog. While bendamustine has been demonstrated as efficacious, it is known to be unstable, especially in aqueous solutions, leading to formation of non-bendamustine products (i.e. “degradation impurities”) which leads to technical difficulties in its preparation and administration. In light of its instability in aqueous solution, bendamustine is supplied as a lyophilized cake of bendamustine hydrochloride salt. US2006/159713, US 2006/128777 and WO2010/036702 disclose various impurities of Bendamustine hydrochloride which are as follows:

PC-1 PC-2
Jena et al. were the first to disclose the synthesis of Bendamustine hydrochloride in German (GDR) Patent No. 34727. Krueger et al. in German (GDR) Patent No. 159877 recite a method as summarized in scheme-1, for the synthesis of bendamustine hydrochloride comprising the reaction of the 4-[l-methyl-5-bis-(2- hydroxyethyl)-benzimidazolyl-2]butyric acid ethyl ester (4) (or the corresponding methyl, propyl or butyl ester) with thionyl chloride in chloroform at 0-5°C to form 4-[l- methyl-5-bis-(2-chloroethyl)-benzimidazolyl-2]butyric acid ethyl ester (5). Excess of thionyl chloride is destroyed by stirring the reaction mixture in aqueous HCl. Finally chloroform is distilled off and stirred at 95°C for 3 hours. The reaction mixture is partially concentrated and the residue is diluted with water and stirred upto crystallization. Further purification is done by recrystallization from water.
Scheme-1: Method disclosed by Krueger et al. in DD159877 for the synthesis of Bendamustine hydrochloride

Bendamustine hydrochloride (6)
Ozegowski et al in Zentralblatt fuer Pharmazie, Pharmakotherapie und Laboratoriumsdiagnostik 1 10 (10), 1013-1019 (1971) discloses a process for the preparation of bendamustine hydrochloride monohydrate. The Chinese journal “Chinese journal of New Drugs “, 2007, No. 23, Vol. 16, 1960-61 and J. Prakt. Chem. 20, 178-186 (1963) disclose another method for the synthesis of Bendamustine hydrochloride monohydrate starting from 2,4-dinitrochlorobenzene as summarized in scheme-2.

The crucial conversions are reaction of l-methyl-2-(4′-ethyl butyrate)-5- amino]-lH-benzimidazole 6 with ethylene oxide in the presence of water, sodium acetate and acetic acid, by maintaining at 5°C for 5 hours and overnight at 20°C to give 4-{5-[bis-(2-hydroxy-ethyl)-amino]-l-methyl-lH-benzimidazol-2-yl}-butyric acid ethyl ester (dihydroxy ester) 7 as a jelly mass, which on chlorination using thionyl chloride in chloroform and subsequent in situ hydrolysis with concentrated HCI gave bendamustine hydrochloride. It also discloses a process for the recrystallization of bendamustine hydrochloride from water and the product obtained is a monohydrate with a melting point of 148-151°C.
IP.com Journal 2009, 9(7B), 21 discloses another process as shown below for the preparation of ethyl-4-[5-[bis(2-hydroxyethyl) amino]- l-methylbenzimidazol-2- yl]butanoate (III) wherein ethyl-4-(5 -amino- 1 -methyl- lH-benzo[d]imidazol-2-yl) butanoate (II) is reacted with 2-halo ethanol in the presence of an inorganic base selected from the group consisting potassium carbonate, potassium bicarbonate, sodium

The PCT application WO 2010/042568 assigned to Cephalon discloses the synthesis of Bendamustine hydrochloride as summarized in schem-3 starting from 2,4- dintroaniline in six steps. The crucial step is reductive alkylation of Il-a, using borane- tetrahydrofuran and chloroacetic acid at ambient temperature, producing compound of formula I-a. Acid mediated hydrolysis of I-a using concentrated hydrochloric acid at reflux produced bendamustine hydrochloride which has a purity of 99.1%. The above PCT Patent application also discloses a method of purification of Bendamustine hydrochloride by agitating the Bendamustine hydrochloride in a mixture of DMF and THF at 75°C for about 30 minutes followed by cooling to ambient temperature and isolating the solid by filtration.
Scheme-3:


iil-a


Bemdamuatine hydrochloride
The PCT application WO 2011/079193 assigned to Dr. Reddy’s Laboratories discloses the synthesis of Bendamustine hydrochloride as summarized in schem-4 starting from compound of formula (II). The crucial step is alkylation of compound of formula II with 2-haloethanol in the presence of an organic base to give a compound of formula (III) which on chlorination with a chlorinating agent affords a compound of formula (IV). Compound of formula (IV) on hydrolysis in acidic medium gives bendamustine hydrochloride. It further discloses purification of bendamustine hydrochloride using aqueous hydrochloric acid and acetonitrile.
Scheme-4:

Bendamustine hydrochloride (Pure)
The most of the prior art processes described above involve
• The use of ethylene oxide for the preparation of bendamustine hydrochloride, which is often not suitable for industrial scale processes due to difficulty in handling ethylene oxide, since it is shipped as a refrigerated liquid.
• Further, the known processes involve the use of strongly acidic conditions and high temperatures for the hydrolysis of ethyl ester of bendamustine and subsequent in-situ formation of bendamustine hydrochloride, thereby resulting in increased levels of various process-related impurities IMP. -A (RRT-0.46), IMP. -B (RRT-1.27) and IMP. -C (RRT-1.31) whose removal is quite difficult and make the process less economically viable.

IMP.-B
International Application Publication No. WO 2009/120386 describes various solid forms of bendamustine hydrochloride designated as bendamustine hydrochloride Form 1, bendamustine hydrochloride Form 2, bendamustine hydrochloride Form 3, bendamustine hydrochloride Form 4, amorphous bendamustine hydrochloride or a mixture thereof, processes for their preparation and lyophilized composition comprising the solid forms. According to the disclosure, monohydrate of bendamustine hydrochloride has been prepared previously. The monohydrate has a reported melting point of 152-156°C which is similar to that of the observed melting point of bendamustine hydrochloride Form 2.
It is known that synthetic compounds can contain extraneous compounds or impurities resulting from their synthesis or degradation. The impurities can be unreacted starting materials, by-products of the reaction, products of side reactions, or degradation products. Generally, impurities in an active pharmaceutical ingredient (API) may arise from degradation of the API itself, or during the preparation of the API. Impurities in Bendamustine hydrochloride or any active pharmaceutical ingredient (API) are undesirable and might be harmful.
Regulatory authorities worldwide require that drug manufacturers isolate, identify and characterize the impurities in their products. Furthermore, it is required to control the levels of these impurities in the final drug compound obtained by the manufacturing process and to ensure that the impurity is present in the lowest possible levels, even if structural determination is not possible. The product mixture of a chemical reaction is rarely a single compound with sufficient purity to comply with pharmaceutical standards. Side products and byproducts of the reaction and adjunct reagents used in the reaction will, in most cases, also be present in the product mixture. At certain stages during processing of the active pharmaceutical ingredient, the product is analyzed for purity, typically, by HPLC, TLC. or GC analysis, to determine if it is suitable for continued processing and, ultimately, for use in a pharmaceutical product. Purity standards are set with the intention of ensuring that an API is as free of impurities as possible, and, thus, are as safe as possible for clinical use. The United States Food and Drug Administration guidelines recommend that the amounts of some impurities are limited to less than 0.1 percent.
Generally, impurities are identified spectroscopically and by other physical methods, and then the impurities are associated with a peak position in a chromatogram (or a spot on a TLC plate). Thereafter, the impurity can be identified by its position in the chromatogram, which is conventionally measured in minutes between injection of the sample on the column and elution of the particular component through the detector, known as the “retention time” (“RT”). This time period varies daily based upon the condition of the instrumentation and many other factors. To mitigate the effect that such variations have upon accurate identification of an impurity, practitioners use “relative retention time” (“RRT”) to identify impurities. The RRT of an impurity is its retention time divided by the retention time of a reference marker.
It is known by those skilled in the art, the management of process impurities is greatly enhanced by understanding their chemical structures and synthetic pathways, and by identifying the parameters that influence the amount of impurities in the final product.
Therefore, there remains a need for improved process for the preparation of bendamustine hydrochloride, producing high yield and purity, and well-suited for use on an industrial scale. Despite the existence of various polymorphic forms of bendamustine hydrochloride, there exists a need for a simple process for the preparation of the stable form of bendamustine hydrochloride which is amenable to scale up and results in high yield and purity.
Bendamustine (INN, trade names Treakisym, Ribomustin, Levact and Treanda; also known as SDX-105) is a nitrogen mustardused in the treatment of chronic lymphocytic leukemia[1] and lymphomas. It belongs to the family of drugs called alkylating agents. It is also being studied for the treatment of sarcoma.[2] It is also being investigated in phase II trials for the non-cancer treatment of AL Amyloidosis.
Bendamustine hydrochloride, initially synthesized in 1963 in the German Democratic Republic, is an alkylating agent that has been shown to have therapeutic utility in treating diseases such as chronic lymphocytic leukemia, Hodgkin’s disease, non-Hodgkin’s lymphoma, multiple myeloma, and breast cancer.It was available from 1971 to 1992 under the trade name Cytostasanand, since that time, has been marketed in Germany as Ribomustin.In March 2008 the FDA approved bendamustine hydrochloride under the trade name Treanda for the treatment of chronic lymphocytic leukemia (CLL). Approval for use in indolent B-cell non-Hodgkin’s lymphoma (NHL) was received in 2009.
History
Bendamustine was first synthesized in 1963 by Ozegowski and Krebs in East Germany (the former German Democratic Republic). Until 1990 it was available only in East Germany. East German investigators found that it was useful for treating chronic lymphocytic leukemia, Hodgkin’s disease, non-Hodgkin’s lymphoma, multiple myeloma and lung cancer.
Bendamustine received its first marketing approval in Germany, where it is marketed under the tradename Ribomustin, by Astellas Pharma GmbH’s licensee, Mundipharma International Corporation Limited. It is indicated as a single-agent or in combination with other anti-cancer agents for indolent non-Hodgkin’s lymphoma, multiple myeloma, and chronic lymphocytic leukemia. SymBio Pharmaceuticals Ltd holds exclusive rights to develop and market bendamustine HCl in Japan and selected Asia Pacific Rim countries.
In March 2008, Cephalon received approval from the United States Food and Drug Administration to market bendamustine in the US, where it is sold under the tradename Treanda, for treatment of chronic lymphocytic leukemia.[3]
In October 2008, the FDA granted further approval to market Treanda for the treatment of indolent B-cell non-Hodgkin’s lymphoma that has progressed during or within six months of treatment with rituximab or a rituximab-containing regimen.[4]
Pharmacology
Bendamustine is a white, water soluble microcrystalline powder with amphoteric properties. It acts as an alkylating agent causing intra-strand and inter-strand cross-links between DNA bases.
After intravenous infusion it is extensively metabolised in the liver by cytochrome p450. More than 95% of the drug is bound to protein – primarily albumin. Only free bendamustine is active. Elimination is biphasic with a half-life of 6–10 minutes and a terminal half-life of approximately 30 minutes. It is eliminated primarily through the kidneys. This paragraph is inconsistent with sidebar for primary excretion pathway.
Chemotherapeutic uses
Bendamustine has been used both as sole therapy and in combination with other agents including etoposide, fludarabine,mitoxantrone, methotrexate, prednisone, rituximab, vincristine and 90Y-ibritumomab tiuxetan.
Lymphomas
One combination for stage III/IV relapsed or refractory indolent lymphomas and mantle cell lymphoma (MCL), with or without prior rituximab-containing chemoimmunotherapy treatment, is bendamustine with mitoxantrone and rituximab.[5] In Germany in 2012 it has become the first line treatment of choice for indolent lymphoma.[6] after Trial results released in June 2012 showed that it more than doubled disease progression-free survival when given along with rituximab. The combination also left patients with fewer side effects than the older R-CHOP treatment.[7]
Adverse effects
Common adverse reactions are typical for the class of nitrogen mustards, and include nausea, fatigue, vomiting, diarrhea, fever, constipation, loss of appetite, cough, headache, unintentional weight loss, difficulty breathing, rashes, and stomatitis, as well as immunosuppression, anemia, and low platelet counts. Notably, this drug has a low incidence of hair loss (alopecia) unlike most other chemotherapy drugs.[8]
……………………
http://www.google.com/patents/WO2013046223A1?cl=en
First aspect of the present invention provides an improved process for the preparation of Bendamustine hydrochloride of the formula (VI)

comprising the steps of:
a) reacting a compound of the formula (II), wherein R is Ci-C6 alkyl

with a 2-haloethanol in the presence of a base to give a compound of formula (III);

b) reacting the compound of formula (III) with a chlorinating agent to provide a compound of formula (IV);

c) hydrolyzing the compound of formula (IV) with Lithium source to give a compound of formula (V); and

d) converting the compound of formula (V) to bendamustine or bendamustine hydrochloride of Formula VI .

Reference Example- 1
Preparation of Bendamustine Hydrochloride as per Patent No. DD159877
Ethyl 4-[l-methyl-5-bis-(2-hydroxyethyl)-amino-benzimidazolyl- 2]butanoate (4, 4.305g) was added to chloroform (36mL) and agitated till clear solution is formed. The solution was cooled to 0°C. Thionyl chloride (2.175g) was added to the above solution within 40 minutes maintaining the temperature of the solution to 0-5°C by cooling. The reaction mixture was agitated at 0-5°C for 1 hour. The temperature was raised slowly to room temperature by removing cooling within 2.5 to 3 hrs and subsequently agitated at room temperature for 15 to 16 hrs. The solution was dispersed by agitating in 37.5mL concentrated hydrochloric acid whereby the excessive thionyl chloride was decomposed under increased hydrochloric acid and S02development. The chloroform was distilled away and further stirred for 3 hrs at around 95°C. Activated carbon (0.78g) was added to the solution and stirred for further 30 minutes at around 95 °C. The solution was concentrated to almost 8mL under vacuum and the residue was diluted with 24mL of water and stirred up to crystallization. The further purification was done by recrystallization from water.
Example-4
Preparation of Bendamustine hydrochloride (VI) through Lithium 4-[l-methyl-5- bis-(2-chloroethyl)-benzimidazoIyl-2] butanoate (V)
Activated charcoal (11. Og) was added to Cone. HC1 (165.0 mL) under stirring and cooled to 5-10°C. Lithium 4-[l-methyl-5-bis-(2-chloroethyl)- benzimidazolyl-2] butanoate (V, HO.Og, 0.302 mol) was added below 65°C under agitation and agitated for 30-45 minutes. The reaction mass was filtered on celite bed prewashed with cone. HC1 and the celite bed was washed with cone. HC1 (27.5mL). The filtrate and washings were combined. DM water (550.0mL) was added to combined filtrate and washings and agitated for 15 minutes. DM water (1.1L) was added and stirred at 20-30°C for 30 minutes. The resulting mass was cooled to 0-5°C and maintained at a temperature of 0 to 5°C for 30 minutes under agitation. The solid was filtered, washed with chilled (0-5°C) DM water twice (220.0 mL each X 2 = 440.0mL) followed by with chilled acetone (0-5°C) (55. OmL) and sucked dried for 1 hour. The solid cake was agitated with acetone (1 lOO.OmL) for 10 minutes and filtered. The solid material was dried at 20-25°C under 100-200 mbar vacuum for one hour till moisture content is between 4.4-6.0% w/w to give the title compound (VI, 80.0g; 67.10%), with a purity of 99.86%.
…………………………..

Gao, L.; Wang, Y.; Song, D. Chinese J. New Drugs 2007, 16, 1960
Ozegowski, V. W.; Krebs, D. J. Prakt. Chem. 1963, 20, 178
Werner, W.; Letsch, G.; Ihn, W.; Sohr, R.; Preiss, R. Pharmazie 1991, 46, 113
Ozegowski, W.; Krebs, D. J. Prakt. Chem. 1963, 20, 178
Werner, W.; Letsch, G.; Ihn, W. Pharmazie 1987,42, 272
………………………………..

-
(a) Chen, J., Przyuski, K., and Roemmele, R. U.S. Patent 8,420,829, April 16, 2013;
Chem. Abstr. 2010, 152, 454105.
(b) Chen, J.; Przyuski, K.; Roemmele, R.; Bakale, R. P.Org. Process Res. Dev. 2011, 15, 1063………………………………………Org. Process Res. Dev., 2011, 15 (5), pp 1063–1072DOI: 10.1021/op200176f Process Research and Development activities leading to a new and efficient route to bendamustine hydrochloride, 1, the active ingredient in Treanda, a treatment for blood cancers, are disclosed. Two key features of this new process include a one-pot hydrogenation/dehydration sequence to construct the benzimidazole moiety and a novel reductive alkylation using chloroacetic acid and borane to install the bischloroethyl side chain. The number of synthetic steps has been significantly reduced to five from the eight in the current commercial process. The overall yield has been improved from 12% to 45%. Additionally, this new route eliminates chloroform, ethylene oxide, and sodium sulfide. Scale-up of the new route has been successfully demonstrated to prepare kilogram quantities of bendamustine hydrochloride.…………………………Org. Process Res. Dev., 2011, 15 (5), pp 1063–1072DOI: 10.1021/op200176f
Process Research and Development activities leading to a new and efficient route to bendamustine hydrochloride, 1, the active ingredient in Treanda, a treatment for blood cancers, are disclosed. Two key features of this new process include a one-pot hydrogenation/dehydration sequence to construct the benzimidazole moiety and a novel reductive alkylation using chloroacetic acid and borane to install the bischloroethyl side chain. The number of synthetic steps has been significantly reduced to five from the eight in the current commercial process. The overall yield has been improved from 12% to 45%. Additionally, this new route eliminates chloroform, ethylene oxide, and sodium sulfide. Scale-up of the new route has been successfully demonstrated to prepare kilogram quantities of bendamustine hydrochloride.…………………………Org. Process Res. Dev., 2011, 15 (5), pp 1063–1072DOI: 10.1021/op200176fPreparation of Bendamustine Hydrochloride (1)
A……../…………..purity of 99.9 A%.1H NMR (400 MHz, DMSO-d6) δ 12.3 (br s, 1H), 7.72 (d, J = 9.3 Hz, 1H), 7.14 (d, J = 2.3 Hz, 1H), 6.89 (dd, J = 9.3, 2.3 Hz, 1H), 3.90 (s, 3H), 3.80 (m, 8H), 3.14 (t, J = 7.6 Hz, 2H), 2.42 (t, J = 7.2 Hz, 2H), 2.01 (quint, J = 7.6 Hz, 2H);LC/MS (ESI, m/z) 358.2 Da (M + 1).……………………..Bendamustine, 4-[5-[bis(2-chloroethyl)amino]-l-methyl-2-benzimidazolyl]butyric acid of formula (1)

, is a cytostatic agent currently approved, in a form of a hydrochloride salt, for treatment of various cancer diseases, e.g. chronic lymphocytic leukemia. It is marketed in the form of a lyophilized powder for intravenous injection, e.g., under the brand name Ribomustin.
Bendamustine, including bendamustine hydrochloride, was first disclosed in DD 34727. Bendamustine hydrochloride may exist, in solid state, in various polymorphic forms, which are disclosed, e.g. in WO 2009/120386. The hydrochloride product disclosed in DD 34727 is a monohydrate. The original process for making bendamustine in DD 34727 comprises the following synthetic pathway:

The group R in the above process is an ethyl group.
The last step of the above process was subsequently technologically improved in DD 159877.
Without providing any experimental detail, DD 34727 also teaches that the starting compound of formula (4) for the above process may be prepared from 2-methylamino-5-nitro- aniline of formula (2) and glutaric acid anhydride. The obtained anilide of formula (3) is cyclized in diluted hydrochloric acid.

Li-Mei et al, in Zhongguo Xinyao Zazhi, Chinese Journal of New Drugs (2007), 16(23), 1960-1961, disclose a process for the preparation of bendamustine hydrochloride in a total yield of 33.5%, which also involves reacting the compound of formula (10) with ethylene oxide to give compound (11). Starting from 2,4-dinitro-l-chlorobenzene, compound (11) is obtained in an overall yield of about 40%.IP.com Journal 2009, 9(7B), 21 discloses a process for the preparation of ethyl-4-[5-[bis(2- hydroxyethyl)amino]-l-methylbenzimidazol-2-yl]butanoate (11) [R=Et], wherein the corresponding compound of formula (10) reacts, instead of ethylene oxide, with 2-halo ethanol in the presence of an inorganic base.
A similar process has been disclosed in WO 2011/079193, wherein the base employed in the reaction of the compound of formula (10) with the 2-haloethanol is an organic base, which is advantageous over inorganic base. The preferred ester group R in the compounds (10) and (11) is the 2-propyl group.
WO 2010/042568 discloses a second basic process for making bendamustine, which is based on providing the compound of formula (5)

(5)
, wherein R is typically a methyl group, by a two step synthesis starting from 2,4- dinitroaniline of formula (6) via the dinitroanilide of formula (7)

This compound of formula (5) is subjected, at reductive conditions (preferably hydrogenation over a platinum catalyst), to a cyclization reaction forming a compound of formula (8)

(8)
, which subsequently may be dehydrated by a strong acid to yield the compound of formula
(10) above. The substituent R in both formulas is a methyl group.
The compound of formula (10) is advantageously subjected to a reductive alkylation with a chloroacetic acid or chloroacetylaldehyde. The reductive agent in the alkylation is suitably a borane or a borohydride. This way, the bendamustine ester of formula (la)

, wherein R is a methyl group, is made directly, without need of forming an intermediate bis-hydroxyethyl compound (11). In the last step of the overall process, the ester (la) is hydrolyzed by a strong acid.
In any process of making bendamustine, various impurities are formed due to various reactive groups in the molecule.
The subject of the present invention is a novel synthetic route to intermediates involved in the synthesis of bendamustine of formula (1) as well as of salts and esters thereof. The approach is based on a novel use of a compound of formula (13) below as the starting material in a synthetic transformation leading to bendamustine, or a pharmaceutically acceptable salt thereof.
In a first aspect, the invention provides a process for making a compound of formula (11), or a salt thereof,

wherein R is hydrogen or a C1-C4 alkyl group,
said process comprising the following steps:
a] providing the compound of formula (13), preferably by reaction of the compound of formula (12) with methylamine,

(12) (13)b] reduction of the compound of formula (13), preferably by hydrogen under catalysis by a transition metal, to an amino compound of formula (14),

c] condensation of the compound of formula (14) with glutaric acid anhydride, or a functional analogue thereof, providing a tertiary alcohol compound of formula (15)

and/or any tautomeric forms thereof according to formula (14A) or (14B)

(14A), (14B),
d] dehydratation and, optionally, esterification of the product of the step c) , preferably in the presence of a strong acid, to yield the compound of formula (11).
In a particular aspect, the above process sequence leading to a compound of formula (11) further comprises a subsequent step of converting the compound of formula (11) to
bendamustine, a salt thereof or an ester thereof, conventionally by reaction with thionyl chloride, followed by ester hydrolysis and salt formation using hydrochloric acid. In yet another aspect, the process sequence of the above steps a) to c) or, optionally, of the above steps a) to d), is performed without isolation or purification of intermediates.
The compounds of formula (14), (14A), (14B) and (15), the above processes of making them, and the use thereof as a starting material for making compounds of formula (11) and/or bendamustine of formula (1), or a pharmaceutically acceptable salt thereof, form next particular aspects of the present invention.

Example 3
A solution of [11, R = Me] (4.0 g, 12 mmol) in dichloromethane (40.0ml) was prepared.
A 100 ml, three -necked, round -bottomed flask equipped with a magnetic stirring bar and a reflux condenser was charged with thionyl chloride (3.60 g, 2.20 ml, 30 mmol) and dichloromethane (12.0 ml) to produce a clear solution. The latter was stirred at 500 rpm at 23 °C and the solution of [11 , R = Me] was added over 15 min via a syringe pump. The resulting mixture was stirred at 500 rpm at 23 °C for 15 min and then at 35 °C for 3.0 h. An aqueous solution of hydrochloric acid (19.4 %) was prepared by mixing of concentrated hydrochloric acid (4.7 g, 4.0 ml) with water (4.0 g, 4.0 ml) and the solution was charged to the reaction mixture. The mixture was further stirred at 500 rpm at 60 °C for 2.0 h under reduced pressure of 100 mbar and the escaping volatiles were condensed and collected outside the reaction vessel. An aqueous solution of hydrochloric acid (4.0 ml, 5 M) was charged in order to dilute the reaction mixture. The mixture was filtered through diatomaceous earth and the filter cake was washed with aqueous solution of hydrochloric acid (2x 1.0 ml, 5 M). The collected filtrate was treated with activated carbon, the used carbon was filtered off, washed with aqueous solution of hydrochloric acid (2x 1.0 ml, 5M), and the filtrate was collected to a 100 ml, round -bottomed flask equipped with a magnetic stirring bar. The filtrate was stirred and diluted with water (40.0 g, 40.0 ml). The slurry was filtered and the filter cake was washed with water (2x 1.0 ml). The filter cake (3.8 g) was charged to a 25 ml, round-bottomed flask equipped with a magnetic stirring bar containing an aqueous solution of hydrochloric acid (8.5 ml, 5M). The mixture was stirred at 60 °C until the solids were dissolved and activated carbon (0.38 g) was charged. The mixture was stirred at 40 °C for additional 5 min and the suspension was filtered through a diatomaceous earth pad. The filter cake was washed with aqueous solution of hydrochloric acid (2x 1.0 ml, 5M) and the filtrate was collected to a 100 ml, round -bottomed equipped with a magnetic stirring bar. The filtrate was stirred at 23 °C and water (42.0 g, 42.0 ml) was added and the mixture was stirred at 23 °C for additional 1 h. The slurry of crystals was filtered and the filter cake was washed with aqueous solution of hydrochloric acid (2x 3.0 ml, 5M). The filtrate was discarded and the filter cake was dried to produce Bendamustine hydrochloride monohydrate (3.3 g) with an overall isolated yield of 67 % and with a chemical purity of 99.9 % by HPLC peak area normalization.
Characteriz ation
lU NMR (400 MHz, DMSO-J<5): δ (ppm) = 2.05 (q, J = 7.51 Hz, 2H), 2.41 (t, J = 7.19 Hz, 2H), 3.18 (t, / = 7.63 Hz, 2H), 3.79 (m, 8H), 3.90 (s, 3H), 6.95 (d, J = 2.31 Hz, 1H), 7.11 (dd, Jx = 2.40 Hz, J2 = 9.20 Hz, 1H), 7.80 (d, J = 9.20 Hz, 1H).
Assay H20 (Karl-Fisher titration): 4.7 %
Assay HC1 (argentometric titration): 8.8 %
Bendamustine 
Systematic (IUPAC) name 4-[5-[Bis(2-chloroethyl)amino]-1-methylbenzimidazol-2-yl]butanoic acid Clinical data Trade names Treanda AHFS/Drugs.com Consumer Drug Information MedlinePlus a608034 Licence data US FDA:link Pregnancy cat. - US: D
Legal status Routes Intravenous infusion Pharmacokinetic data Bioavailability NA (intravenous only) Protein binding 94–96% Metabolism Hydrolyzed to inactive metabolites. Two minor metabolites (M3 and M4) formed by CYP1A2 Half-life 40 min (bendamustine), 3 h (M3), 30 min (M4) Excretion Mostly fecal Identifiers CAS number 16506-27-7 
ATC code L01AA09 PubChem CID 65628 ChemSpider 59069 UNII 9266D9P3PQ ChEMBL CHEMBL487253 Chemical data Formula C16H21Cl2N3O2 Mol. mass 358.262 g/mol References
- Kath R, Blumenstengel K, Fricke HJ, Höffken K (January 2001). “Bendamustine monotherapy in advanced and refractory chronic lymphocytic leukemia”. J. Cancer Res. Clin. Oncol. 127(1): 48–54. doi:10.1007/s004320000180. PMID 11206271.
- Bagchi S (August 2007). “Bendamustine for advanced sarcoma”. Lancet Oncol. 8 (8): 674. doi:10.1016/S1470-2045(07)70225-5. PMID 17726779.
- “Cephalon press release – Cephalon Receives FDA Approval for TREANDA, a Novel Chemotherapy for Chronic Lymphocytic Leukemia”. Retrieved 2008-03-23.
- “Cephalon press release -Cephalon Receives FDA Approval for TREANDA to Treat Patients with Relapsed Indolent Non-Hodgkin’s Lymphoma”. Retrieved 2008-11-03.
- Weide R, Hess G, Köppler H, et al. (2007). “High anti–lymphoma activity of bendamustine/mitoxantrone/rituximab in rituximab pretreated relapsed or refractory indolent lymphomas and mantle cell lymphomas. A muticenter phase II study of the German Low Grade Lymphoma Study Group (GLSG)”. Leuk. Lymphoma. 48 (7): 1299–1306. doi:10.1080/10428190701361828.PMID 17613757.
- New Combo Replaces CHOP for Lymphoma. Dec 2012
- “‘Rediscovered’ Lymphoma Drug Helps Double Survival: Study”. June 3, 2012.
- Tageja, Nishant; Nagi, Jasdeepa; “Bendamustine: something old, something new”; Cancer Chemotherapy and Pharmacology, 2010 Aug;66(3):413-23. doi: 10.1007/s00280-010-1317-x.
External links
- Manufacturer’s official website intended for US patients
References:Bifunctional alkylating agent. Prepn: W. Ozegowski, D. Krebs, J. Prakt. Chem. 20, 178 (1963); eidem,Zentralbl. Pharm. Pharmakother. Laboratoriumsdiagn. 110, 1013 (1971). Antitumor activity: W. Jungstand et al., ibid. 1021.Capillary GC determn in plasma: H. Weber et al., J. Chromatogr. 525, 459 (1990).Toxicity study: U. Horn et al., Arch. Toxicol.Suppl. 8, 504 (1985).Clinical evaluation in non-Hodgkin’s lymphomas: K. Bremer, J. Cancer Res. Clin. Oncol. 128, 603 (2002); in chronic lymphocytic leukemia: T. Lissitchkov et al., ibid. 132, 99 (2006); with prednisone in multiple myeloma: W. Pönisch et al.,ibid 205.Review of pharmacology and clinical development: K. Bremer, W. Roth, Tumordiagn. Ther. 17, 1-6 (1996); J. A. Barman Balfour, K. L. Goa, Drugs 61, 631-638 (2001).WO2009120386A2 Mar 26, 2009 Oct 1, 2009 Cephalon, Inc. Novel solid forms of bendamustine hydrochloride WO2010036702A1 Sep 23, 2009 Apr 1, 2010 Cephalon, Inc. Liquid formulations of bendamustine WO2010042568A1 Oct 7, 2009 Apr 15, 2010 Cephalon, Inc. Processes for the preparation of bendamustine WO2011079193A2 Dec 22, 2010 Jun 30, 2011 Dr. Reddy’s Laboratories Ltd. Preparation of bendamustine and its salts DD34727A Title not available DD159877A1 Title not available US20060128777 Nov 4, 2005 Jun 15, 2006 Bendall Heather H Cancer treatments US20060159713 Jan 12, 2006 Jul 20, 2006 Cephalon, Inc. Bendamustine pharmaceutical compositions













Ramatroban

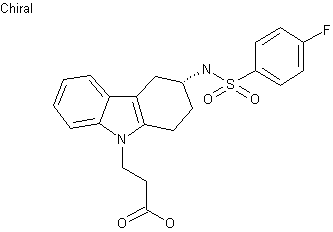

| Formula |
C21H21FN2O4S
|
|---|---|
| CAS |
116649-85-5
|
| Mol weight |
416.4658
|
- 3-(4-fluorophenylsulfonamido)-1,2,3,4-tetrahydro-9-carbazole propanoic acid
- BAY u 3405
- BAY u 3406
- BAY u-3405
- BAY u3405
- ramatroban
Ramatroban (INN) (also known as Bay-u3405)[1] is a thromboxane receptor antagonist.[2]
It is also a DP2 receptor antagonist.[3]
It is indicated for the treatment of coronary artery disease.[4] It has also been used for the treatment of asthma.[5]
It was developed by the German pharmaceutical company Bayer AG and is co-marketed in Japan by Bayer and Nippon Shinyaku Co. Ltd. under the trade name Baynas.
SYN
Science 1976,193163-5
Proc Natl Acad Sci USA 1975,72(8),2994-8
The synthesis of Bay u 3405 was carried out as follows: Reductive amination of 3-oxo-1,2,3,4-tetrahydrocarbazole (I) with S-phenethylamine (II) afforded a mixture of diastereomeric amines, of which the desired isomer (III) crystallized in high diastereomeric purity as the hydrogensulfate. Cleavage of the phenethyl group by transfer hydrogenolysis with amminium formate and palladium on charcoal yielded the enantiomerically pure (3R)-3-amino-1,2,3,4-tetrahydrocarbazole (IV). Sulfonylation of (IV) with 4-fluorobenzenesulfonyl chloride (V) to the sulfonamide (VI) followed by addition of acrylonitrile and subsequent hydrolysis gave Bay u 3405.
SYN
J Label Compd Radiopharm 1994,34(12),1207
The synthesis of [14C]-labeled Bay-u-3405 by two closely related ways has been described: 1) [14C]-Labeled aniline (I) is diazotized and reduced with sodium sulfite, yielding the labeled hydrazine (II), which is condensed with the monoketal of cyclohexane-1,4-dione (III) under Fisher’s indole synthesis (ZnCl2) to afford the tetrahydrocarbazole (IV). The hydrolysis of (IV) with HCl in THF/water yields 1,2,3,4-tetrahydrocarbazol-3-one (V), which is submitted to a reductive condensation with (S)-1-phenylethylamine (VI) by means of tetrabutylammonium borohydride, yielding preferentially the secondary amine (VII), which, after purification, is dealkylated with ammonium formate and Pd/C to afford 1,2,3,4-tetrahydrocarbazole-3(R)-amine (VIII). The acylation of (VIII) with 4-fluorophenylsulfonyl chloride (IX) gives the corresponding sulfonamide (X), which is condensed with acrylonitrile by means of NaH, yielding 3-[3(R)-(4-fluorophenylsulfonamido)-1,2,3,4-tetrahydrocarbazol-9-yl]pro pionitrile (XI). Finally, this compound is hydrolyzed in the usual way. 2) The condensation of the sulfonamide (X) with methyl acrylate by means of NaH as before gives 3-[3(R)-(4-fluorophenylsulfonamido)-1,2,3,4-tetrahydrocarbazol-9-yl]propionic acid methyl ester (XII), which is finally hydrolyzed in the usual way.
……………………

http://pubs.rsc.org/en/content/articlelanding/2012/oc/c2oc90018a#!divAbstract
………………….
http://onlinelibrary.wiley.com/doi/10.1002/adsc.201300993/abstract












135−137 °C; IR (KBr) ν 3276, 2926, 1712, 1591, 1494, 1467, 1153
cm−1
; 1
H NMR (CD3OD, 300.13 MHz) δ 1.87−2.09 (m, 2H14),
2.47−2.54 (m, 1H11), 2.67 (t, 3
JHH = 6.7 Hz, 2H2), 2.75−2.91 (m,
2H13+1H11), 3.61−3.71 (m, 1H12), 4.30 (t, 3
JHH = 6.7 Hz, 2H3), 6.96
(t, 3
JHH = 6.9 Hz, 1H7), 7.08 (t, 3
JHH = 7.1 Hz, 1H6), 7.21−7.31 (m,
2H18+H5+H7), 7.93−7.98 (m, 2H17); 13C NMR (CD3OD, 75.5 MHz)
δ 21.2 (C14), 29.7 (C11), 31.1 (C13), 35.8 (C2), 40.0 (C3), 51.5 (C12),
108.0 (C10), 110.2 (C5), 117.4 (d, 2
JCF= 23.3 Hz, 2C18), 118.7 (C8),
120.2 (C7), 122.4 (C6), 128.9 (C19), 131.0 (d, 3
JCF= 9.7 Hz, 2C17),
135.3 (C15), 138.0 (C4), 139.8 (d, 4
JCF= 3.5 Hz, C16), 166.6 (d, 1
JCF=
251.4 Hz, C19), 175.2 (C1); HRMS (ESI+, m/z) calcd for
(C21H22FN2O4S)+ (M + H)+ 417.1279, found 417.1273; [α]D
20=
+64.4 (c 1, MeOH) for 99% ee.
This invention relates to 2-amino- tetrahydrocarbazole-propanoic acid and a new process for its synthesis .
2-Amino-tetrahydrocarbazole-propanoic acid is a key intermediate for the synthesis of Ramatroban, a thromboxaneA2 receptor (TP) antagonist with clinical efficacy in asthma and allergic rhinitis.

Ramatroban l-Amino-tetrahydrocarbazole-proanoic acid
US Patent 4988820 discloses the synthesis of this compound stating from compound 1, which is condensed with phenylhydrazine and ring-closed to give indole 2. Deprotection of 2 using acid provides ketone 3. Reductive amination of ketone with s-phenylethylamine in the presence of tetrabutylammonium borohydride provides compound 4, which undergoes palladium catalyzed hydrogenation to give key intermediate 5.

Ramatroban

The process, however, has disadvantages: the starting material 1 is relatively expensive, and the yield of the amination step is only 40% and needs expensive tetrabutylammonium borohydride as the reducing agent. And also the subsequent hydrogenation provides only 70% of the desired compound 5. [0006] US Patent 4988820 also describes an alternative synthesis of compound 5 starting from compound 6, which is oxidized by chromium trioxide to afford ketone 7. Condensation of compound 7 with phenylhydrazine and ring closure give indole 8. The subsequent hydrolysis using HCl provides indole 9. The intermediate 5 is obtained by resolution of racemic 9 using ( + ) -mandelic acid as the resolving agent.

9 5
However, this process has crucial disadvantages: the first step oxidation reaction needs the heavy metal reagent chromium trioxide, which is toxic and expensive, and the resolution of indole 9 using (+) -mandelic acid affords only -10 % of compound 5.
US Patent 5684158 discloses the synthesis of 2- amino-tetrahydrocarbazole-propanoic acid ethyl ester 10 by the alkylation of compound 5 in the presence of about 1 mol of alkali metal hydroxides and phase-transfer catalysts such as potassium hydroxide and benzyltriethylammonium chloride.

The problem with this reaction is that the insoluble material in the reaction mixture becomes very sticky during the reaction. The reaction mixture must be filtered in hot solvent in order to remove insoluble material during work up and the sticky material tents to block the filtration. [0010] Therefore, there is a great need for a new process for the synthesis of 2-amino-tetrahydrocarbazole- propanoic acid.
-
9- (2-carboxyethyl) -4- (4-fluorphenylsulfonamidomethyl) -1,2,3,4-tetrahydrocarbazole

-
0.91 g 9-(2-Cyanoethyl)-4-[N-(4-fluorphenylsulfonyl)-N-(2-cyanoethyl)aminomethyl]-1,2,3,4-tetrahydrocarbazol be hydrolyzed analogously to Example 7. One obtains 0.77 g (89% of theory) of crystalline product as the sodium salt.
-
M.p .: 160 ° CR f = 0.57 CH 2 Cl 2: CH 3 0H = 9: 1

-
-
(+) – 3- (4-fluorophenylsulphonamido) -9- (2-carboxyethyl) -1,2,3,4-tetrahydrocarbazole

-
5.8 g (0.0128 mol) of Example 67 are dissolved in 60 ml isopropanol, treated with 130 ml of 10% potassium hydroxide solution, after 16 hours heating under reflux, is cooled, diluted with water and extracted with ethyl acetate. The aqueous phase is concentrated in vacuo and then treated dropwise with vigorous stirring with conc.Hydrochloric acid. The case precipitated acid is filtered off, washed with water and dried thoroughly in vacuo.Obtained 4.4 g (86.6% of theory) of the product. .: Mp 85-95 ° C rotation [α] 20 = 42.55 ° (CHCl 3) D
-

Example 70
-
(-) – 3- (4-fluorophenylsulphonamido) -9- (2-tarboxyethyl) -1,2,3,4-tetrahydrocarbazole

-
The preparation of Example 70 from Example 68 is carried out analogously to the preparation of Example 69 from Example 67. m.p .: 85-95 ° C optical rotation: [α] 20 = -37.83 ° (CHCl 3) D

Synthesis pathway
| Synthesis of a) |
|---|
     |
Trade names
| Country | Trade name | Manufacturer |
|---|---|---|
| Japan | Baynas | Bayer |
| Ukraine | no | no |
Formulations
-
50 mg tablet 75 mg
Reference
-
DE 3631824 (Bayer AG; appl. 19.9.1986; prior. 21.2.1986).
-
EP 728 743 (Bayer AG; appl. 14.2.1996; D-prior. 27.2.1995).
| Patent | Submitted | Granted |
|---|---|---|
| Phenylsulfonamid substituted pyridinealken- and aminooxyalkan-carboxylic-acid derivatives. [EP0471259] | 1992-02-19 | 1995-05-17 |
| Heterocyclic substituted cycloalkano(b)-indolesulfonamides. [EP0473024] | 1992-03-04 | |
| Cycloalkano[b]dihydroindoles and -indolesulphonamides substituted by heterocycles. [EP0451634] | 1991-10-16 | 1994-03-09 |
| Respiratory Drug Condensation Aerosols and Methods of Making and Using Them [US2009258075] | 2009-10-15 | |
| ANTITHROMBOTIC SUBSTITUTED CYCLOALKANO(B)DIHYDROINDOLE- AND -INDOLE-SULPHONAMIDES [US5096897] | 1992-03-17 | |
| Indolesulphonamide-substituted dihydropyridines [US5272161] | 1993-12-21 | |
| THERMODYNAMICALLY STABLE FORM OF (R)-3-[ [(4-FLUOROPHENYL) SULPHONYL]AMINO] -1,2,3,4- TETRAHYDRO -9H-CARBAZOLE -9-PROPANOIC ACID (RAMATROBAN) [WO9933803] | 1999-07-08 |
| DE1695703B2 * | Mar 15, 1967 | Nov 20, 1975 | Sumitomo Chemical Co., Ltd., Osaka (Japan) | Title not available |
| DE2125926A1 * | May 25, 1971 | Jan 27, 1972 | Title not available | |
| DE2226702A1 * | May 25, 1972 | Dec 13, 1973 | Schering Ag | Neue mittel zur behandlung des diabetes mellitus |
| FR1415322A * | Title not available | |||
| GB1487989A * | Title not available | |||
| US4235901 * | May 14, 1979 | Nov 25, 1980 | American Home Products Corporation | 1-Hydroxyalkanamine pyrano(3,4-b)indole compositions and use thereof |
-
References
- ^ “Ramatroban (compound)”. PubChem. National Center for Biotechnology Information. Retrieved 22 June 2019.
- ^ Sugimoto H, Shichijo M, Iino T, et al. (April 2003). “An orally bioavailable small molecule antagonist of CRTH2, ramatroban (BAY u3405), inhibits prostaglandin D2-induced eosinophil migration in vitro”. J. Pharmacol. Exp. Ther. 305 (1): 347–52. doi:10.1124/jpet.102.046748. PMID 12649388.
- ^ Royer JF, Schratl P, Carrillo JJ, et al. (September 2008). “A novel antagonist of prostaglandin D2 blocks the locomotion of eosinophils and basophils”. Eur. J. Clin. Invest. 38 (9): 663–71. doi:10.1111/j.1365-2362.2008.01989.x. PMID 18837743.
- ^ Fiedler VB, Seuter F, Perzborn E (December 1990). “Effects of the novel thromboxane antagonist Bay U 3405 on experimental coronary artery disease” (PDF). Stroke. 21 (12 Suppl): IV149–51. PMID 2260140.
- ^ Endo S, Akiyama K (November 1996). “[Thromboxane A2 receptor antagonist in asthma therapy]”. Nippon Rinsho (in Japanese). 54 (11): 3045–8. PMID 8950952.
External links
- (in Japanese) Baynas Tablets Prescribing Information
 |
|
| Systematic (IUPAC) name | |
|---|---|
| 3-((3R)-3-{[(4-fluorophenyl)sulfonyl]amino}-1,2,3,4-tetrahydro-9H-carbazol-9-yl)propanoic acid | |
| Clinical data | |
| Legal status |
|
| Routes | Oral |
| Identifiers | |
| CAS number | 116649-85-5 |
| ATC code | None |
| PubChem | CID 123879 |
| IUPHAR ligand | 1910 |
| ChemSpider | 110413 |
| UNII | P1ALI72U6C |
| ChEMBL | CHEMBL361812 |
| Chemical data | |
| Formula | C21H21FN2O4S |
| Mol. mass | 416.46 g/mol |
 |
|
| Clinical data | |
|---|---|
| Trade names | Baynas |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration |
Oral (tablets) |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.159.668 |
| Chemical and physical data | |
| Formula | C21H21FN2O4S |
| Molar mass | 416.47 g·mol−1 |
| 3D model (JSmol) | |

NEW DRUG APPROVALS
ONE TIME
$10.00
A Flow Reactor with Inline Analytics: Design and Implementation

A Flow Reactor with Inline Analytics: Design and Implementation
-
Wu, H., Dong, Z., Haitao, L., and Khan, M. Org. Process Res. Dev. 2014, 18, 10.1021/op500056a.
-
(a) Razzaq, T.; Kappe, C. O. Chem.—Asian J. 2010, 5, 1274– 1289
(b) Wiles, C.; Watts, P. Green Chem. 2014, 16, 55– 62
(c) Moseley, J. D.; Woodman, E. K. Org. Process Res. Dev. 2008, 12, 967– 981
-
Ullah, F.; Samarakoon, T.; Rolfe, A.; Kurtz, R. D.; Hanson, P. R.; Organ, M. G. Chem.—Eur. J. 2010, 16, 10959– 10962
and references cited therein
-
Guidance for Industry, Q8(R2) Pharmaceutical Development; U. S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research: Silver Springs, MD, 2009; p 18.
-
Calibrese, G. S.; Pissavini, S. AIChE J. 2011, 54, 828– 834
-
(a) Alsten, J. G.; Reeder, L. M.; Stanchina, C. L.; Knoechel, D. J. Org. Process Res. Dev. 2008, 12, 989– 994
(b) Roberge, D. M.; Zimmermann, B.; Rainonee, F.; Gottsponer, M.; Eyholzer, M.; Kockmann, N. Org. Process Res. Dev. 2008, 12, 905– 910

Vijay Kirpalani
vk@pi-inc.co
CEO
Pi-inc (Process Intensification Experts LLP)
READ AT….https://newdrugapprovals.org/2014/11/11/flow-chemistry-test-facility-in-india/
Toronto, canada


{kind=link}