
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 202)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Raw Material Variation into QbD Risk Assessment
DRUG REGULATORY AFFAIRS INTERNATIONAL
Areas of discussion included how expectations for raw material control are evolving within changing regulatory and business paradigms including quality by design (QbD), counterfeiting, complex supply chains, and sourcing changes. discussed risk assessment and mitigation strategies along with supplier risk management plans.

Regulatory Considerations
the lack of a consistent definition of raw materials in regulations pertaining to the pharmaceutical industry. In its Q7 guideline, the International Conference on Harmonisation of Technical Requirements for the Registration of Pharmaceuticals for Human Use (ICH) defines raw materials as “starting materials, reagents, and solvents intended for use in the production of intermediates or APIs.” However, the term as defined by different speakers could cover a wide range of materials including the following:
• starting or source materials (cell lines, viral or bacterial stocks, media components, chemicals, tissues, serum, water)
• in-process materials (resins, buffers, filters, column housings, tubing, reagents)
• excipients
• packaging components, both…
View original post 3,002 more words
FDA Warning Letter on Data Integrity
DRUG REGULATORY AFFAIRS INTERNATIONAL

The integrity of data is currently in the focus of international authorities In particular the US FDA issued serious violations in Warning Letters to the companies concerned. Read more about the current complaints in a Warning Letter issued to the API manufacturer VUAB Pharma.
For authorities the integrity of data is an essential quality attribute in the manufacture of pharmaceutical products. After some serious deviations international authorities have moved the topic into the centre of their interest. In particular the US FDA issued serious violations in Warning Letters to the companies concerned.

In a current letter to the API manufacturer VUAB Pharma in the Czech Republic the inspector and the authority criticised multiple aspects with regard to “failure to prevent unauthorized access or change to data and to provide controls preventing data omissions”:
- ‘The firm did not retain complete raw data from testing performed to assure the quality…
View original post 408 more words
Development of monographs for Indian pharmacopoeia
DRUG REGULATORY AFFAIRS INTERNATIONAL

Development of monographs for Indian pharmacopoeia
http://www.ipapharma.org/events/speakerpresentation/8-%20Dr%20S%20C%20Mathur.pdf

ALOGLIPTIN
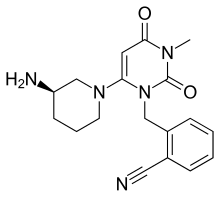
ALOGLIPTIN
Alogliptin is a potent, selective inhibitor of DPP-4 with IC50 of <10 nM, exhibits greater than 10,000-fold selectivity over DPP-8 and DPP-9.
Alogliptin (trade name Nesina in the US[1] and Vipidia in Europe[2]) is an orally administered anti-diabetic drug in the DPP-4 inhibitor class,[3] developed by Syrrx, a company which was acquired by Takeda Pharmaceutical Company in 2005. Like other medications for the treatment of Type 2 diabetes, alogliptin does not decrease the risk of heart attack and stroke. Like other members of the gliptin class, it causes little or no weight gain, exhibits relatively little risk of causing hypoglycemia, and exhibits relatively modest glucose-lowering activity. Alogliptin and other gliptins are commonly used in combination with metformin in patients whose diabetes cannot adequately be controlled with metformin alone.[4]
Clinical study
Alogliptin is a dipeptidyl peptidase-4 inhibitor (DPP-4i) that is designed to slow the inactivation of incretin hormones GLP-1 (glucagon-like peptide-1) and GIP (glucose-dependent insulinotropic peptide). [5]
A randomized clinical trial reporting in 2011 aimed to determine the efficacy and safety of alogliptin versus placebo and vogliboseamong newly diagnosed Type 2 diabetes patients in Japan. The main outcome indicated that alogliptin was statistically superior to both comparitors.[6]
A randomized clinical trial reporting in 2012 aimed to demonstrate that alogliptin was “non-inferior” to a “very low fat/calorie traditional Japanese diet” among newly diagnosed Type 2 diabetes patients in Japan. The outcome indicated that both the drug and dietary treatments comparably impacted indicators of the diabetic condition, such as HbA1c levels and glycemic efficacy. The drug treatment had its impact without changing body mass index (BMI), but the dietary treatment was accompanied by a significant reduction in the BMI.[7]
A randomized clinical trial reporting in 2011 aimed to demonstrate the efficacy of alogliptin as an add-on agent in combination withmetformin and pioglitazone versus simply increasing the dosage of pioglitazone in combination with metformin; in other words, this was a study to look at a three-agent therapy versus a two-agent therapy. The outcome of this study suggested that the addition of alogliptin to metformin and pioglitazone provided superior impact on diabetes biomarkers (e.g. HbA1c) than increasing the dose of pioglitazone in a two agent therapy with metformin.[8]
Reported adverse events
Adverse events appear to be restricted to mild hypoglycemia based on clinical studies.[6][7][8]
Alogliptin is not associated with increased weight, increased risk of cardiovasular events, or heart failure.[9][10]
Market access
In December 2007, Takeda submitted a New Drug Application (NDA) for alogliptin to the United States Food and Drug Adminiistration (USFDA),[11] after positive results from Phase III clinical trials.[1] In September of 2008, the company also filed for approval in Japan,[12] winning approval in April 2010.[11] The company also filed a Marketing Authorization Application (MAA) elsewhere outside the United States, which was withdrawn in June 2009 needing more data.[12] The first USFDA NDA failed to gain approval and was followed by a pair of NDAs (one for alogliptin and a second for a combination of alogliptin and pioglitazone) in July 2011.[11] In 2012, Takeda received a negative response from the USFDA on both of these NDAs, citing a need for additional data.[11]
In 2013 the FDA approved the drug in three formulations: As a stand-alone with the brand-name Nesina. Combined with metforminusing the name Kazano, and when combined with pioglitazone as Oseni.
Diabetes affects millions of people worldwide and is considered one of the main threats to human health in the 21st century. In 2006, the World Health Organization (WHO) estimated that over 180 million people worldwide had diabetes, and the number is projected to double by 2030. Over time, uncontrolled diabetes can damage body systems, including the heart, blood vessels, eyes, kidneys and nerves. According to the WHO, approximately 1.1 million people died from diabetes in 2005, and it is estimated that diabetes-related deaths will increase by more than 50% in the next decade. Globally, the socioeconomic burden of diabetes is substantial.
There are two main types of diabetes, designated type 1 and type 2, with type 2 diabetes accounting for over 90% of all diabetes cases globally. Type 1 diabetes is characterized by insulin deficiency, primarily caused by autoimmune-mediated destruction of pancreatic islet β-cells, and type 2 diabetes is characterized by abnormal insulin secretion and concomitant insulin resistance. To prevent the development of ketoacidosis, people with type 1 diabetes must take exogenous insulin for survival. Although those with type 2 diabetes are not dependent on exogenous insulin as much as subjects with type 1 diabetes, they may require exogenous insulin to control blood glucose levels.
As diabetes has become a global health concern, research interest in the condition has rapidly increased. In addition to studies on prevention, many studies with the aim of developing new interventions for the treatment of diabetes, especially type 2 diabetes, have been conducted. Currently available medications for the treatment and management of type 2 diabetes include metformin, sulfonylureas, thiazolidinediones and insulin. However, these therapies are commonly associated with secondary failure and may cause hypoglycemia. Insulin resistance and progressively worsening hyperglycemia caused by reduced β-cell function are major challenges in managing type 2 diabetes. Evidence suggests that patients with insulin resistance do not develop hyperglycemia until their β-cells are unable to produce enough insulin. New agents that can enhance insulin secretion from islet β-cells in a sustained glucose-dependent manner could therefore hold promise for the treatment of type 2 diabetes.
One promising approach is based on inhibition of the serine protease dipeptidyl- peptidase IV (DPP IV), a postproline dipeptidyl aminopeptidase that belongs to the S9b peptidase family of proteolytic enzymes. It is known that DPP IV plays a key role in maintaining glucose homeostasis by controlling the incretin activity of glucagon-like peptide 1 (GLP-I) and glucose-dependent insulinotropic polypeptide (GIP, also known as gastric inhibitory polypeptide). Inhibition of DPP IV is therefore recognized as a novel therapeutic approach for the treatment of type 2 diabetes.
Recently, a series of DPP IV inhibitors were developed. Among these highly potent compounds, alogliptin benzoate (SYR-322) and its analogs demonstrated encouraging antidiabetic efficacy (EP 1586571 (WO 2005/095381); WO 2008/067465; WO 2007/035379, and US 2004/097510).
Alogliptin benzoate can be prepared as described in EP 1586571 (WO 2005/095381) according to the process set forth in Scheme 1 :

Scheme 1
In accordance with this process, 6-Chlorouracil (1) is alkylated with 2- (bromomethyl)benzonitrile in the presence of NaH and LiBr in a mixture of DMF- DMSO to produce the TV-benzyluracil derivative (2) in 54% yield. Compound (2) is further alkylated with iodomethane and NaH in DMF/THF to give the 1 ,3 disubstituted uracil (3) in 72% yield. Subsequent displacement of chlorouracil (IV) with 3(R)- aminopiperidine dihydrochloride in the presence of either NaHCO3 in hot methanol or K2CO3 in aqueous isopropanol provides alogliptin (4), which is isolated as the corresponding benzoate salt by treatment with benzoic acid in ethanol. The overall yield of this three-stage process is -20-25%. One of the disadvantages of above described process is the difficulty to separate and purify mixtures of solvents with high boiling point (for example, DMF/DMSO) for recycling. Another disadvantage is the usage of hazardous materials such as sodium hydride, which requires anhydrous solvents as a reaction media.
Intermediate 2-((6-chloro-3-methyl-2,4-dioxo-3 ,4-dihydropyrimidin- 1 (2H)-yl)methyl) benzonitrile (3) is alternatively obtained by alkylation of 6-chloro-3 methyluracil with 2-(bromomethyl)benzonitrile by means of diisopropylethylamine in hot NMP (WO 2007/035629). Although this process is more technological than the previously described process (EP 1586571), the overall yield is still moderate (50-55%). The problem of mixed solvents (toluene, NMP, diisopropylethylamine) separation persists for this process as well.

………….
http://www.google.com/patents/EP2410855A1?cl=en
EXAMPLE 1
Preparation of (R)-2-((6-(3 -aminopiperidin-l-yl)-3 -methyl-2,4-dioxo-3 ,4- dihydropyrimidin-1 (2H)-yl) methyl)benzonitrile (alogliptin) via 6-chloro-l-(2- isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)-dione (Scheme 3):

Scheme 3
Preparation of l-(2-isocyanobenzyl)-3-methylurea
2-cyanobenzylamine hydrochloride (90 g) and Dichloromethane (800 ml) were taken into a round bottomed (RB) flask. Methyl isocyanate (45.6 g) was added at 5°C. Triethylamine (81 g) in Dichloromethane (300 ml) was added at the same temperature and stirred at room temperature for 16h. Water (1 L) was added and stirred for 30 min. The obtained solid was collected by filtration and dried in oven at 50°C for 12h. The yield is 85% and the purity 99.8%.
Preparation of l-(2-isocyanobenzyl)-3-methyIpyrimidine-2,4,6(lH,3H,5H)-trione
a). To a stirred solution of 0.11 mol of sodium ethanolate in 80 ml of ethanol abs. was added 0.1 mol of l-(2-isocyanobenzyl)-3-methylurea and 0.1 mol diethyl malonate. The mixture was refluxed for 3-5 h. The cooled residue was acidified with 0.1 M hydrochloric acid (60 ml). The solid which separated was filtered off and recrystallized from ethanol or any suitable solvent. The yield is 78-85% and purity >95%.
b). In an alternate embodiment, l-(2-isocyanobenzyl)-3-methylurea (30 g), acetic acid (105 ml) and malonic acid (18 g) were mixed and heated to 60°C. Acetic anhydride (60 ml) was added at 60°C and heating was continued for two hours at 80°C. The reaction mixture was poured over ice water (300 ml) and the obtained solid was filtered, washed with water (1×500 ml) and methyl-tert-butylether (100 ml). The yield is 60% with 93.4% purity.
The compound thus prepared can be used for the next step without purification or purified by crystallization or column chromatography.
Preparation of 6-chloro-l-(2-isocyanobenzyl)-3-methylpyriinidine-2,4(lH,3H)- dione
a). l-(2-isocyanobenzyl)-3-methylpyrimidine-2,4,6(lH,3H,5H)-trione (30 g) was mixed with phosphorus oxychloride (300 ml) and cooled to 0°C. Water (9 ml) was added slowly, stirred for 10 min. and heated to reflux at 110°C for 5h. Progress of the reaction was monitored by TLC (50% Ethyl acetate/Hexane). On completion of the reaction, phosphorus oxychloride was distilled off. The crude compound was dissolved in dichloromethane (500 ml) and poured into ice water (500 ml) by small portions. The layers were separated and the aqueous layer was extracted with dichloromethane (200 ml). The combined organic extracts were washed with water and brine, dried over sodium sulphate and concentrated under reduced pressure. The mixture of two isomers (4-chloro and 6-chloro derivatives = 1:1) was isolated and separated by column chromatography using neutral alumina and eluent – 25-50% of ethylacetate and hexane). The off-white solid was obtained, yield – 37%, purity – 99.8%. 1H NMR corresponds to literature data (J. Med. Chem. 2007, 50, 2297-2300).
b). In an alternate embodiment, a solution of l-(2-isocyanobenzyl)-3-methylpyrimidine- 2,4,6(1 H,3H,5H)-trione (18 mmol), phosphorus oxychloride (85 ml), benzyltriethylammonium chloride (16.5 g, 72 mmol) and phosphorus pentachloride (3.8 g, 18 mol) in acetonitrile (80 ml) was refluxed for 4-5 h with stirring. After evaporation under reduced pressure, the resulting oily residue was mixed with methylene chloride (or chloroform) and the mixture was poured into water and ice (50 ml). The layers were separated and the aqueous layer was extracted with dichloromethane (200 ml). The combined organic extracts were washed with water and brine, dried over sodium sulphate and concentrated under reduced pressure. Crude product was crystallized from THF-hexanes to give desired compound in 70.5% yield.
c). In an alternate embodiment, a solution of l-(2-isocyanobenzyl)-3-methylpyrimidine- 2,4,6(1 H,3H,5H)-trione (13.1 mmol) in POCl3 (30 ml) was refluxed for 1-3 h. The solvent was concentrated and then partitioned with CH2Cl2 (100 ml) and water (100 ml). The organic layer was washed with brine, dried over Na2SO4, and concentrated to give 6-chloro compound as a solid (-95%). Compound can be also precipitated from concentrated methylene chloride solution by hexanes and used as a crude for the next step or purified by reslurring in isopropanol, filtered off, washed with isopropanol, and dried under vacuum at 55-60° C.
Preparation of (R)-tert-butyl l-(3-(2-isocyanobenzyI)-l-methyl-2,6-dioxo-l,2,3,6- tetrahydropyrimidin-4-yl)piperidin-3-yl carbamate
a). 6-chloro- l-(2-isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)-dione (13 g), Dimethylformamide (130 ml), Potassium carbonate (13 g) and tert-butyl (R)-piperidin- 3-ylcarbamate (10.4 g) were heated to 80°C for 7 hrs. The mixture was then allowed to come to room temperature and poured over ice water (500 ml). The obtained solid was filtered and washed with cold water (500 ml). The solid thus obtained was taken in Methyl-tert-butylether (50 ml) stirred for 10 min. filtered and washed with Hexane (50 ml), to give the N-tert-butyloxycarbonyl protected compound in -75% yield. b). In an alternate embodiment, a flask charged with a stir bar, 6-chloro-l-(2- isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)-dione (4.10 mmol), (Λ)-3- terrtnityloxycarbonylaminopiperidine (4.64 mmol), K2CO3 (1.15 g, 8.32 mmol) and DMF (12 mL) was stirred at 75 °C for 6 h. Then, water was added and the mixture was extracted with methylene chloride. The organic layer was washed with brine, dried over Na2SO4, and concentrated to give the N-ter/butyloxycarbonyl protected compound in -93-96% yield.
Preparation of (R)-2-((6-(3-aminopiperidin-l-yl)-3-methyl-2,4-dioxo-3,4- dihydropyrimidin-1 (2H)-yl) methyl)benzonitrile salts
a). Preparation of (R)-2-((6-(3-aminopiperidin-l-yl)-3-methyl-2,4-dioxo-3,4- dihydropyrimidin-1 (2H)-yl) methyl)benzonitrile hydrochloride
The crude (R)-tert-butyl l-(3-(2-isocyanobenzyl)-l-methyl-2,6-dioxo-l,2,3,6- tetrahydropyrimidin-4-yl)piperidin-3-yl carbamate from previous procedure was dissolved in THF and acidified with 6M hydrochloric acid while maintaining the temperature below 15° C. The resultant slurry was cooled to 0-5° C, stirred at this temperature for 3-5 h and then filtered. The filter cake was washed twice with isopropanol and dried in vacuum at 45-5O0C to provide hydrochloride as a white crystalline solid.
b). Preparation of (R)-2-((6-(3-aminopiperidin-l-yl)-3-methyl-2,4-dioxo-3,4- dihydropyrimidin-1 (2H)-yl) methyl)benzonitrile trifluoroacetate
TFA (ImL) was added into the methylene chloride solution of (R)-tert-butyl l-(3-(2- isocyanobenzyl)- 1 -methyl-2,6-dioxo- 1 ,2,3,6-tetrahydropyrimidin-4-yl)piperidin-3-yl carbamate from the above-mentioned procedure. The solution was stirred at room temperature for 1 h and then the mixture was concentrated in vacuo. The residue was dissolved in a small amount of MeOH or isopropanol and the desired salt was precipitated by addition of diisopropyl ether. The solids were filtered off, washed with diisopropyl ether and dried in vacuum at 45-5O0C to provide trifluoroacetate as an off- white powder. c). Preparation of (R)-2-((6-(3-aminopiperidin-l-yl)-3-methyl-2,4-dioxo-3,4- dihydropyrimidin-1 (2H)-yl) methyl)benzonitrile benzoate (Alogliptin)
The crude (R)-tert-butyl l-(3-(2-isocyanobenzyl)-l-methyl-2,6-dioxo-l,2,3,6- tetrahydropyrimidin-4-yl)piperidin-3-yl carbamate was dissolved in ethanol. A solution of benzoic acid in ethanol was added and the mixture was slowly heated to 65-70°C. The solution was stirred at this temperature for Ih and was then crystallized by cooling to 0-5° C and stirring for 12 hrs. The solution was filtered, washed with alcohol. The wet cake was then conditioned under nitrogen for 2 hours. The cake was dried for 8 hrs at 40-50° C to provide the benzoic acid salt of alogliptin as a white crystalline solid.
EXAMPLE 2:
Preparation of (R)-2-((6-(3-aminopiperidin-l-yl)-3-methyl-2,4-dioxo-3,4- dihydropyrimidin-1 (2H)-yl) methyl)benzonitrile (alogliptin) via 6-amino-l-(2- isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)-dione (Scheme 4)


Scheme 4 Preparation of 6-amino-l-(2-isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)- dione
a). l-(2-isocyanobenzyl)-3-methylurea (0.2 mol) and cyanoacetic acid (0.22 mol) were dissolved in acetic anhydride (400 ml), and the mixture was heated at 80°C for 2 hours. Acetic anhydride was distilled off under reduced pressure and water (200 ml) was added. The mixture was cooled to 0-5 0C and 2N NaOH solution (220 ml) was added and stirring was continued for 2 hours. The obtained solids were filtered off, washed with cold methanol and dried under vacuum. The yield of 6-amino-l-(2- isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)-dione was 72 %.
b). Under nitrogen atmosphere, l-(2-isocyanobenzyl)-3-methylurea (98.4 g) and cyanoacetic acid (80.0 g) was added to N,N-dimethylformamide (836 ml). The mixture was stirred at room temperature and methanesulfonyl chloride (72.8 ml) was added dropwise with stirring at this temperature. The mixture was stirred at room temperature for 4 hrs, cooled with water, and water-isopropanol [2:1 (volume ratio), 1670 ml] was added drop wise. The mixture was stirred under water-cooling for 1 hr, and the precipitated crystals were collected by filtration and dried to give 3-(2-cyano-acetyl)-3- methyl-l-(2-isocyanobenzyl)-urea with 68% yield.
To 3-(2-cyano-acetyl)-3-methyl-l-(2-isocyanobenzyl)-urea (120 g) were added water (962 ml) and 2N aqueous sodium hydroxide solution (24.9 ml), and the mixture was stirred with heating at 80° C for 1 hr. After allowing to cool to room temperature, the crystals were collected by filtration and dried to give 6-amino-l-(2-isocyanobenzyl)-3- methylpyrimidine-2,4(lH,3H)-dione in 76% yield.
c). 6-amino-l-(2-isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)-dione (0.1 mol) was mixed with (R)-piperidin-3-yl-carbamic acid tert.-butyl ester hydrochloride (0.1 mol) of the appropriate amine hydrochloride and (R)-piperidin-3-yl-carbamic acid tert.-butyl ester (0.1 mol). The mixture was heated at 100°C and bubbling continued for 3 hr. Water was added to the cooled mixture and the mixture was extracted with methylene chloride. The organic layer was washed with brine, dried over Na2SO4, and concentrated to give N-tert-butyloxycarbonyl protected compound in ~93-96% yield.
d). Benzoate salt of alogliptin was prepared as described above. While certain embodiments of the invention have been illustrated and described, it will be clear that the invention is not limited to the embodiments described herein. Numerous modifications, changes, variations, substitutions and equivalents will be apparent to those skilled in the art without departing from the spirit and scope of the present invention as described by the claims, which follow.
………………
Patent EP2410855A1
http://www.google.com/patents/EP2410855A1?cl=en

…………..

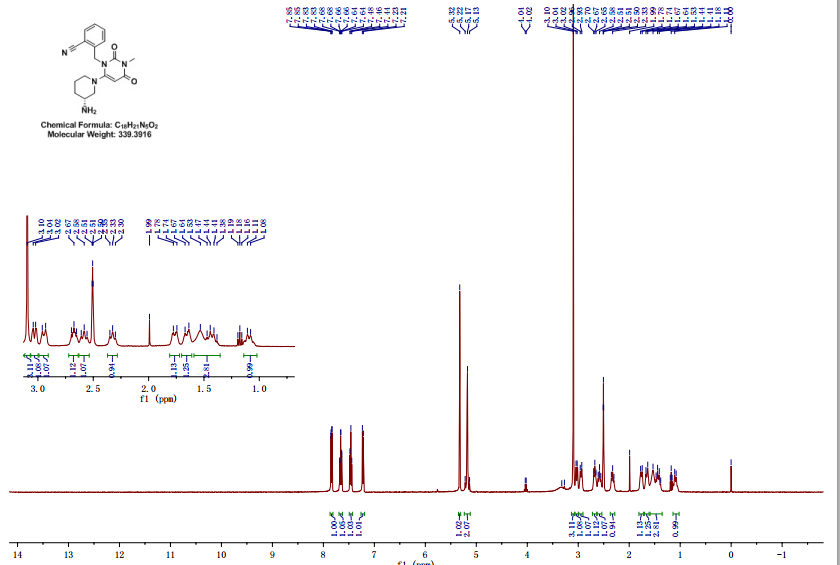
NMR
SOURCE APEXBT

References
- “Takeda Submits New Drug Application for Alogliptin (SYR-322) in the U.S.” (Press release). Takeda Pharmaceutical Company. January 4, 2008. Retrieved January 9, 2008.
- Vipidia: EPAR summary for the public (European Medicines Agency)
- Feng, Jun; Zhang, Zhiyuan; Wallace, Michael B.; Stafford, Jeffrey A.; Kaldor, Stephen W.; Kassell, Daniel B.; Navre, Marc; Shi, Lihong; Skene, Robert J.; Asakawa, Tomoko; Takeuchi, Koji; Xu, Rongda; Webb, David R.; Gwaltney II, Stephen L. (2007). “Discovery of alogliptin: a potent, selective, bioavailable, and efficacious inhibitor of dipeptidyl peptidase IV”. J. Med. Chem.50 (10): 2297–2300.doi:10.1021/jm070104l.PMID 17441705.
- “www.aace.com” (PDF).
- http://www.takeda.com/news/2013/20130618_5841.html
- Seino, Yutaka; Fujita, Tetsuya; Hiroi, Shinzo; Hirayama, Masashi; Kaku, Kohei (September 2011), “Efficacy and safety of alogliptin in Japanese patients with type 2 diabetes mellitus: a randomized, double-blind, dose-ranging comparison with placebo, followed by a long-term extension study (abstract only)”, Current Medical Research and Opinion 27 (9): 1781–1792,doi:10.1185/03007995.2011.599371,PMID 21806314, retrieved April 26,2012
- Kutoh, Eiji; Ukai, Yasuhiro (2012),“Alogliptin as an initial therapy in patients with newly diagnosed, drug naïve type 2 diabetes: a randomized, control trial (abstract only)”, Endocrine(January 17, 2012), doi:10.1007/s12020-012-9596-0, PMID 22249941, retrieved April 26, 2012
- Bosi, Emanuele; Ellis, G.C.; Wilson, C.A.; Fleck, P.R. (October 2011), “Alogliptin as a third oral antidiabetic drug in patients with type 2 diabetes and inadequate glycaemic control on metformin and pioglitazone: a 52-week, randomized, double-blind, active-controlled, parallel-group study”, Diabetes, Obesity and Metabolism (October 27, 2011) 13 (12): 1088–1096, doi:10.1111/j.1463-1326.2011.01463.x, retrieved April 26,2012
- White WB, Cannon CP, Heller SR et al. (October 2013). “Alogliptin after acute coronary syndrome in patients with type 2 diabetes”. N. Engl. J. Med. 369(14): 1327–35.doi:10.1056/NEJMoa1305889.PMID 23992602.
- White WB, Zannad F (January 2014). “Saxagliptin, alogliptin, and cardiovascular outcomes”. N. Engl. J. Med. 370 (5): 484.doi:10.1056/NEJMc1313880.PMID 24482824.
- Grogan, Kevin (April 26, 2012),“FDA wants yet more data on Takeda diabetes drug alogliptin”,PharmaTimes (PharmaTimes), PharmaTimes online, retrieved April 26,2012
- “GEN News Highlights: Takeda Pulls MAA for Type 2 Diabetes Therapy”. Genetic Engineering & Biotechnology News. June 4, 2009.
| EP1083172A1 * | May 26, 1998 | Mar 14, 2001 | Rimma Iliinichna Ashkinazi | N-substituted derivatives of 5-oxyiminobarbituric acid |
| US2598936 * | Apr 13, 1950 | Jun 3, 1952 | Searle & Co | Disubstituted cyanoalkanoylureas and thioureas and methods for their production |
| US6066641 * | Dec 12, 1995 | May 23, 2000 | Euro-Celtique S.A. | Aryl thioxanthines |
| US6248746 * | Jan 7, 1999 | Jun 19, 2001 | Euro-Celtique S.A. | 3-(arylalkyl) xanthines |
| US20080194593 * | Jan 11, 2008 | Aug 14, 2008 | Rao Kalla | A2b adenosine receptor antagonists |
| WO1994003456A1 * | Aug 5, 1993 | Feb 17, 1994 | Boehringer Ingelheim Kg | Asymmetrically substituted xanthine with adenosine-antagonistic properties |
| WO2001029010A1 * | Oct 18, 2000 | Apr 26, 2001 | Amjad Ali | Gram-positive selective antibacterial compounds, compositions containing such compounds and methods of treatment |
| WO2007035629A2 * | Sep 15, 2006 | Mar 29, 2007 | Takeda Pharmaceutical | Process for the preparation of pyrimidinedione derivatives |
| WO2007150011A2 * | Jun 22, 2007 | Dec 27, 2007 | Smithkline Beecham Corp | Prolyl hydroxylase inhibitors |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
2-({6-[(3R)-3-aminopiperidin-1-yl]-3-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl}methyl)benzonitrile
|
|
| Clinical data | |
| Trade names | Nesina, Vipidia Kazano, Vipidomet (withmetformin) Oseni, Incresync (withpioglitazone) |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 100% |
| Protein binding | 20% |
| Metabolism | Limited, hepatic (CYP2D6– and3A4-mediated) |
| Biological half-life | 12–21 hours |
| Excretion | Renal (major) and fecal (minor) |
| Identifiers | |
| CAS Registry Number | 850649-62-6 |
| ATC code | A10BH04 |
| PubChem | CID: 11450633 |
| IUPHAR/BPS | 6319 |
| ChemSpider | 9625485 |
| UNII | JHC049LO86 |
| KEGG | D06553 |
| ChEBI | CHEBI:72323 |
| ChEMBL | CHEMBL376359 |
| Synonyms | SYR-322 |
| Chemical data | |
| Formula | C18H21N5O2 |
| Molecular mass | 339.39 g/mol |
 Alogliptin benzoate
Alogliptin benzoate
| MF: | C18H21N5O2.C7H6O2 |
| MW: | 461.519 |
| Melting Point: | 185-188°C |
| Optical Rotation: | -56.3° (c=1, MeOH) |
Solubility:Soluble in MeOH; Insoluble in ACN
850649-62-6 CAS
Alogliptin
-
- Synonyms:SYR-322
- ATC:A10BH04
- Use:antidiabetic, DPP-4 inhibitor
- Chemical name:2-[[5-[(3R)-3-amino-1-piperidinyl]-3,4-dihydro-3-methyl-2,4-dioxo-2H-pyrimidin-1(2H)-yl]methyl]benzonitrile
- Formula:C18H21N5O2
- MW:339.40 g/mol
- CAS-RN:850649-61-5
Derivatives
benzoate
- Formula:C19H19NO2
- MW:293.37 g/mol
- CAS-RN:850649-62-6
Substance Classes
Synthesis Path
Substances Referenced in Synthesis Path
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 22115-41-9 | C8H6BrN | 2-(bromomethyl)benzonitril | |
| C12H8ClN3O2 | 2-[[6-chloro-3,4-dihydro-2,4-dioxo-1(2H)-pyrimidinyl]methyl]benzonitrile | ||
| C13H10ClN3O2 | 2-[[6-chloro-3,4-dihydro-3-methyl-2,4-dioxo-1(2H)-pyrimidinyl]methyl]benzonitrile | ||
| 4270-27-3 | C4H3ClN2O2 | 6-chloro-2,4(1H,3H)-pyrimidinedione | |
| 74-88-4 | CH3I | methyl iodide | Methane, iodo- |
| 127294-73-9 | C5H12N2 | (3R)-3-piperidinamine |
Trade Names
| Country | Trade Name | Vendor | Annotation |
|---|---|---|---|
| J | Nesina | Takeda ,2010 |
Formulations
- tabl. 12.5 and 25 mg
References
-
- Feng, J. et al.: J. Med. Chem. (JMCMAR) 50, 2297-2300 (2007).
- WO 2 005 095 381 (SYRRX; 13.10.2005; appl. 15.12.2004; USA-prior. 15.3.2004).
- WO 2 010 109 468 (MAPI Pharma; 30.9.2010; appl. 25.3.2010; USA-prior. 26.3.2009).
-
solid preparation comprising Alogliptin and Pioglitazone:
- US 20 100 092 551 (Takeda Pharm.; 15.4.2010; appl. 30.1.2008; J-prior. 1.2.2007).
-
solid preparation comprising Alogliptin and Metformin:
- US 20 200 136 127 (Takeda Pharm.; 3.6.2010; appl. 16.7.2008; J-prior. 19.7.2007).
 LIONEL MY SON
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

GEMIGLIPTIN

GEMIGLIPTIN
1-[2(S)-Amino-4-[2,4-bis(trifluoromethyl)-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-7-yl]-4-oxobutyl]-5,5-difluoropiperidin-2-one
PHASE 3, DPP-IV inhibitor, Lg Life Sciences Ltd.
CAS 911637-19-9
Mol. Formula: C18H19F8N5O2
Gemigliptin (rINN), previously identified as LC15-0444, is an oral anti-hyperglycemic agent (anti-diabetic drug) of the new dipeptidyl peptidase-4 (DPP-4) inhibitor class of drugs.[1] It is well known that glucose lowering effects of DPP-4 inhibitors are mainly mediated by GLP-1 and gastric inhibitory polypeptide (GIP) incretin hormones which are inactivated by DPP-4.
Gemigliptin was initially developed solely by LG Life Sciences. In 2010, Double-Crane Pharmaceutical Co. (DCPC) joined with LGLS to co-develop the final compound and collaborate on the marketing of the drug in China. LGLS also announced on Nov., 2010 that NOBEL Ilac has been granted rights to develop and commercialize gemigliptin in Turkey.
Gemigliptin, a dipeptidyl peptidase IV (CD26; DPP-IV; DP-IV) inhibitor, is currently undergoing phase III clinical trials at LG Life Sciences as an oral treatment for type II diabetes. The company is also testing the compound in phase II/III clinical studies for the treatment of patients with cisplatin-induced acute kidney injury.
DPP IV inhibitors have glucose-lowering effects mediated by GLP-1 incretin hormone which is inactivated by DPP IV. In 2010, gemigliptin was licensed to Beijing Double-Crane Pharmaceutical by LG Life Sciences for distribution and supply in China for the treatment of type 2 diabetes.
A New Drug Application (NDA) for gemigliptin in the treatment of type 2 diabetes was submitted to the Korea Food & Drug Administration (KFDA) in July 2011. Then on June 27, 2012, the KFDA has approved the manufacture and distribution of LG Life Sciences’ diabetes treatment, Zemiglo, the main substance of which is gemigliptin. Clinical trials for evaluating the safety and efficacy of gemigliptin in combination with metformin have been completed.
…………
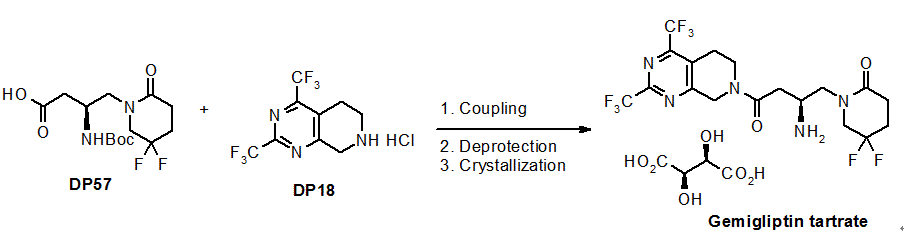
Efficient synthesis of gemigliptin, a potent and selective DPP-4 inhibitor for the treatment of type 2 diabetes mellitus, has been developed. Gemigliptin were prepared from two key API starting materials, DP18 and DP57, in 75~80% yield and >99% purity over three steps under the GMP control: coupling, deprotection of N-Boc group, and final crystallization with L-tartaric acid. All steps were conducted in the same solvent system and the intermediates were isolated by simple filtration without distillation of solvent. The established process was validated obviously through the three consecutive batches for a commercial production.
………..IN CASE IMAGES NOT VISIBLE …….SEE THIS AT ………http://www.allfordrugs.com/2015/07/06/gemigliptin/
GO TO MY OTHER SITE FOR SYNTHESIS
|
(3S)-3-amino-4-(5,5-difluoro-2-oxopiperidino)-1-[2,4-di(trifluoromethyl)-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-7-yl]butan-1-one
|
|
| Clinical data | |
|---|---|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 94% (rat), 73% (dog), 26% (monkey) |
| Biological half-life | 3.6 h (rat), 5.2 h (dog), 5.4 h (monkey) |
| Identifiers | |
| CAS Registry Number | 911637-19-9 |
| ATC code | A10BH06 |
| PubChem | CID: 11953153 |
| ChemSpider | 10127461 |
| UNII | 5DHU18M5D6 |
| Synonyms | LC15-0444 |
| Chemical data | |
| Formula | C18H19F8N5O2 |
| Molecular mass | 489.36 g/mol |
……………….
History
The NDA for gemigliptin was submitted to KFDA in July, 2011 and it was approved on June 27, 2012. By the end of 2012, gemigliptin will be marketed in Korea as Zemiglo which is the fifth new DPP-4 inhibitor diabetes treatment in the world.
Mechanism of action
DPP-4 is a serine protease located on the cell surfaces throughout the body. In plasma, DPP-4 enzyme rapidly inactivates incretins including GLP-1 and GIP which are produced in the intestine depending on the blood glucose level and contribute to the physiological regulation of glucose homeostatis. Active GLP-1 and GIP increase the production and release of insulin by pancreatinc beta cells. GLP-1 also reduces the scretion of glucacon by pancreatic alpha cells, thereby resulting in a decreased hepatic glucose production. However these incretins are rapidly cleaved by DPP-4 and their effects last only for a few minutes. DPP-4 inhibitors block the cleavage of the gliptins and thus lead to an increasee insulin level and a reduced glucagon level in a glucose-dependent way. This results in a decrease of fasting and postprandial glycemia, as well as HbA1c levels.[2]
Preclinical studies
Gemigliptin is a competitive, reversible DPP-4 inhibitor (IC50 = 16 nM) with excellent selectivity over other critical human proteases such as DPP-2, DPP-8, DPP-9, elastase,trypsin, urokinase and cathepsin G. Gemigliptin was rapidly absorbed after single oral dosing and the compound was eliminated with a half-life of 3.6 h, 5.2 h, and 5.4 h in the rat, dog, and monkey, respectively.
The bioavailability of gemigliptin in the rat, dog, and monkey was species-dependent with the values of 94%, 73%, and 26%, respectively. Following the oral administration of gemigliptin in the rat, dog and monkey, about 80% inhibition of plasma DPP-4 activity were observed at the plasma levels of 18 nM, 14 nM and 4 nM, respectively.
In the diet-induced obese (DIO) mice, gemigliptin reduced glucose excursion during OGTT in a dose dependent manner with the minimum effective dose of 0.3 mg/kg and enhanced glucose-stimulated plasma GLP-1 increase in a dose dependent manner reaching the maximum effect at the dose of 1 mg/kg.
Following 4 week oral repeat dosing in the DIO mice, gemigliptin reduced significantly HbA1c with the minimum effective dose of 3 mg/kg. In the beagle dog, gemigliptin significantly enhanced active GLP-1, decreased glucagon, and reduced glucose excursion during OGTT following a single dosing.
Studies on animals suggest its positive effect on hepatic and renal fibrosis .[3][4] Data on human patients are still inconclusive .[5]
Clinical studies
The dose-range finding phase 2 study was performed and 145 patients (91men and 54 women) with type 2 diabetes mellitus were enrolled. All three doses (50,100 and 200 mg groups) of gemigliptin significantly reduced the HbA1c from baseline compared to the placebo group without a significant difference between the doses.
Subjects with a higher baseline HbA1c (≥8.5%) had a greater reduction in HbA1c. Insulin secretory function, as assessed using homeostasis model assessment-beta cell, C-peptide and the insulinogenic index, improved significantly with gemigliptin treatment. Insulin sensitivity, as assessed using homeostasis model assessment-insulin resistance, also improved significantly after 12 weeks of treatment.
The 50 and 200 mg groups had significantly reduced total cholesterol and low-density lipoprotein cholesterol levels at 12 weeks compared to the placebo group.
The incidences of adverse events were similar in all study subjects. Gemigliptin monotherapy (50 mg for 12 weeks) improved the HbA1c, FPG level, oral glucose tolerance testresults, β-cell function and insulin sensitivity measures, and was well tolerated in subjects with type 2 diabetes.
Results of Phase 3 clinical trials which have been finished recently will be updated near future.
…………..
WO 2006104356
http://www.google.co.in/patents/WO2006104356A1?cl=en
EXAMPLE 83: Synthesis of l-(f2SV2-amino-4-r2.4-bisftrifluoromethylV5.8-dihvdropyridor3.4-d]pyrimidin-7f6H‘)
-yl1-4-oxobutyll-5.5-difluoropiperidin-2-one [1960]

[1961] 21 mg of the title compound was obtained in a yield of 56% at the same manner as in EXAMPLE 1, except that 42 mg (0.071 mmol) of t-butyl
{(lS)-3-[2,4-bis(trifluoromethyl)-5,8-dihydropyrido[3,4-d]pyrimidin-7(6H)-yl]-l-[(5,5
-difluoro-2-oxpiperidin-l-yl)methyl]-3-oxpropyl}carbamate obtained in
PREPARATION 143 was used. [1962] 1K NMR (CD3OD) δ 5.05-4.92 (2H, m), 3.98-3.91 (2H, m), 3.85-3.79 (2H, m),
3.70-3.59 (2H, m), 3.54-3.48 (IH, m), 3.36-3.33 (2H, m), 3.24 (IH, bra), 3.14 (IH, bra), 2.83-2.76 (IH, m), 2.72-2.53 (3H, m), 2.43-2.34 (2H, m) [1963] Mass (m/e) 490 (M+l)
[1964]
[1965] PREPARATION 144: Synthesis of t-butyl
(riSV3-r2.4-bisrtrifluoromethylV5.8-dihvdropyridor3.4-d]pyrimidin-7r6HVyl]-l-(rr2 S)-2-methyl-5-oxomorpholin-4-yl1methyl 1 -3-oxpropyl 1 carbamate
[1966] 14 mg of the title compound was obtained in a yield of 17% at the same manner as in PREPARATION 45, except that 43.7 mg (0.138 mmol) of (3S)-3-[(t-butoxycarbonyl)amino]-4-[2(S)-2-methyl-5-oxomoφholin-4-yl]-butanoic acid obtained in PREPARATION 55 and 42.5 mg (0.138 mmol) of 2,4-bis(trifluoromethyl)-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidine hydrochloric acid salt (product of PREPARATION 127) were used.
[1967] 1K NMR (CDCl3) δ 5.85-5.83 (IH, m), 5.09-4.92 (IH, m), 4.95-4.78 (IH, m),
4.23-4.08 (3H, m), 4.04-3.76 (3H, m), 3.73-3.66 (IH, m), 3.46-3.38 (IH, m), 3.36-3.21 (2H, m), 3.18-3.10 (2H, m), 2.96-2.81 (IH, m), 2.61-2.50 (IH, m), 1.43-1.41 (9H, m), 1.28-1.24 (3H, m)
[1968] Mass (m/e) 470 (M+l-Boc)
…………..
WO 2012030106
https://www.google.com/patents/WO2012030106A2?cl=en
Reaction Scheme 1

PREPARATION 1: Synthesis of diethyl 2,2-difluoropentanedioate

To a solution of ethyl bromodifluoroacetate (33.2 g) in tetrahydrofuran (94.0 g) was added ethyl acrylate (8.2 g) and copper powder (10.9 g). After heating to 50℃, TMEDA (9.5 g) was added dropwise and the reaction mixture was then stirred for 3 hours at the same temperature. Upon disappearance of ethyl acrylate as the starting material, to the reaction solution was added methyl t-butyl ether (MTBE, 73.7 g) followed by addition of 10% aqueous ammonium chloride solution (49.8 g) dropwise, and the mixture was then stirred for 30 minutes. The remaining copper residue was removed by filtration through a celite, and methyl t-butyl ether (MTBE, 66.3 g) was added to separate the layers. The separated organic layer was washed successively with 10% aqueous NH4Cl solution (66.3 g) and 3 N aqueous hydrochloric acid solution (99.6 g) in order and then distilled under reduced pressure to obtain 55.0 g of the desired title compound.
1H NMR (400 MHz, CDCl3) δ 1.26 (t, J=7.2 Hz, 3H), 1.37 (t, J=7.2 Hz, 3H), 2.37-2.49 (m, 2H), 2.55 (t, J=7.2 Hz, 2H), 4.16 (q, J=7.2 Hz, 2H), 4.29 (q, J=7.2 Hz, 2H).
PREPARATION 2: Synthesis of ethyl 4,4-difluoro-5-hydroxypentanoate

14.8 g of the compound obtained from the above Preparation 1 was diluted with ethanol (20.4 g) and tetrahydrofuran (69.1 g) and then cooled to 0℃. To this solution was slowly added sodium borohydride (NaBH4, 3.5 g) stepwise while keeping the internal temperature below 30℃. After confirming completion of the reaction by 1H NMR, the reaction solution was cooled to the temperature of 10℃ and 10% aqueous ammonium chloride solution (77.7 g) was slowly added. The remaining boron compound was filtered through celite, and the filtrate was distilled under reduced pressure to remove tetrahydrofuran. Then, ethyl acetate (105.2 g) was added to separate the layers, and the organic layer was distilled under reduced pressure to obtain 10.8 g of the title compound.
1H NMR (400 MHz, CDCl3) δ 1.23 (t, J=7.2 Hz, 3H), 2.15-2.29 (m, 2H), 2.49 (t, J=7.2 Hz, 2H), 3.69 (t, J=12.0 Hz, 2H), 4.12 (q, J=4.0 Hz, 2H).
EXAMPLE 1: Synthesis of ethyl 4,4-difluoro-5-{[(trifluoromethyl)sulfonyl]oxy}- pentanoate

To the solution of 10.8 g of the compound, as obtained from the above Preparation 2, dissolved in dichloromethane (100.2 g) was added pyridine (7.0 g), and then the mixture was cooled to -5.0℃. After completion of cooling, trifluoromethane sulfonic acid anhydride (20.1 g) was slowly added dropwise while keeping the reaction temperature below 6.3℃. After stirring the reaction solution for 30 minutes, 1.5 N hydrochloric acid solution was added dropwise at 0℃ to separate the layers. The aqueous layer as separated was back-extracted twice with dichloromethane (33.4 g), and the extracts were combined with the organic layer separated from the above and then distilled under reduced pressure to obtain 19.7 g of the title compound as a yellow oil.
1H NMR (500 MHz, CDCl3) δ 1.27 (t, J=7.2 Hz, 3H), 2.29-2.39 (m, 2H), 2.59 (t, J=7.6 Hz, 2H), 4.18 (q, J=7.2 Hz, 2H), 4.55 (t, J=11.6 Hz, 2H).
EXAMPLE 2-1: Synthesis of ethyl 4,4-difluoro-5-{[(nonafluorobutyl)sulfonyl]- oxy}pentanoate

To the solution of 100.0 g of the compound, as obtained from the above Preparation 2, dissolved in dichloromethane (300.0 ml) was added pyridine (65.7 g), and the mixture was then cooled to -10.0℃. After completion of cooling, nonafluorobutanesulfonic anhydride (477.4 g) was slowly added dropwise. After stirring the reaction solution for 3 hours, 1.0 N hydrochloric acid solution (300.0 ml) was added dropwise to separate the layers. The aqueous layer as separated was back extracted once with dichloromethane (500.0 ml), and the extracts were combined with the organic layer separated from the above and then distilled under reduced pressure to obtain 177.5 g of the title compound.
1H NMR (500 MHz, CDCl3) δ 1.26 (t, 3H, J=7.3 Hz), 2.30-2.36 (m, 2H), 2.58 (t, 2H, J=7.4 Hz), 4.16 (q, 2H, J=7.3 Hz), 4.57 (t, 2H, J=11 Hz).
EXAMPLE 2-2: Synthesis of ethyl 4,4-difluoro-5-{[(nonafluorobutyl)sulfonyl]- oxy}pentanoate
To the solution of 500.0 g of the compound, as obtained from the above Preparation 2, dissolved in dichloromethane (1000.0 ml) was added triethylamine (389.0 g), and the mixture was then cooled to 0℃. After completion of cooling, perfluorobutanesulfonyl chloride (948.80 g) was slowly added dropwise. The reaction solution was stirred for 3 hours at room temperature, distilled under reduced pressure, dissolved in methyl t-butyl ether (MTBE, 3000.0 ml) and then washed three times with water. The organic layer thus obtained was dehydrated with magnesium sulfate, filtered through a celite and then distilled under reduced pressure to obtain 960.0 g of the title compound.
EXAMPLE 3: Synthesis of methyl (2S)-2-[(tert-butoxycarbonyl)amino]-4-oxo- pentanoate

To 25.0 g of the starting material, (3S)-3-[(t-butoxycarbonyl)amino]-4-oxo- pentanoic acid, was added t-butanol (96.9 g) followed by the addition of Boc2O (25.4 g) and dimethylaminopyridine (DMAP, 62.0 g, 0.5 mol%) at room temperature, and the reaction mixture was then stirred for 23 hours at 40℃. Upon completion of the reaction, ethylene dichloride (62.3 g) in t-butanol was added, and the mixture was then distilled under reduced pressure to obtain 30.7 g of the title compound.
1H NMR (400 MHz, CDCl3) δ 1.45 (s, 9H), 1.47 (s, 9H), 2.71 (dd, J=4.8, 16.4 Hz, 1H), 2.88 (dd, J=4.4, 16.4 Hz, 1H), 3.75 (s, 3H), 4.53 (m, 1H), 5.44 (br d, J=8.0 Hz, 1H).
EXAMPLE 4: Synthesis of tert-butyl (3S)-3-[(tert-butoxycarbonyl)amino]-4-hydroxy- butanoate

30.7 g of the compound obtained from the above Example 3 was dissolved in ethanol (112.3 g) and, after lowering the internal temperature to 10.5℃ sodium borohydride (NaBH4, 5.7 g) was slowly added dropwise. This reaction solution was stirred while maintaining the temperature below 22℃. After confirming completion of the reaction by 1H NMR and TLC, to the reaction solution was slowly added 3.0 N hydrochloric acid solution (30.7 g) dropwise at the internal temperature of 10℃ followed by addition of diluted 0.2% hydrochloric acid solution (100.0 g). The reaction solution was adjusted to pH 3~4 with addition of 9.0% aqueous hydrochloric acid solution, and then back-extracted twice with ethyl acetate (100.0 g) and toluene (44.0 g). The organic layer thus obtained was distilled under reduced pressure to obtain 25.1 g of the title compound.
1H NMR (500 MHz, CDCl3) δ 1.44 (s, 9H), 1.45 (s, 9H), 2.48-2.57 (m, 2H), 3.69 (d, J=4.9 Hz, 1H), 3.97 (m, 1H), 5.22 (bs, 1H).
EXAMPLE 5: tert-butyl (3S)-[(tert-butoxycarbonyl)amino]-4-[(methylsulfonyl)oxy]- butanoate

To 25.1 g of the compound obtained from the above Example 4 was added dichloromethane (133.0 g) and triethylamine (148.0 g), and the mixture was then cooled to 0℃. To this reaction solution was slowly added methanesulfonyl chloride (11.8 g) diluted with dichloromethane (39.9 g) dropwise for 50 minutes while maintaining the internal temperature below 12℃. After completion of the reaction, the reaction solution was washed with 0.5 N aqueous hydrochloric acid solution (120.0 g) and water (100.4 g), and then distilled under reduced pressure to obtain 31.5 g of the title compound.
1H NMR (500 MHz, CDCl3) δ 1.44 (s, 9H), 1.46 (s, 9H), 2.62 (d, J=6.0 Hz, 2H), 3.04 (s, 3H), 4.21 (m, 1H), 4.30 (d, J=5.2 Hz, 2H), 5.16 (br d, J=7.2 Hz, 1H).
EXAMPLE 6: Synthesis of tert-butyl (3S)-4-azido-3-[(tert-butoxycarbonyl)amino]- butanoate

Sodium azide (NaN3, 11.6 g) was diluted with dimethylacetamide (DMAc, 260.0 g). After elevating the internal temperature to 80℃, a solution of 31.5 g of the compound, as obtained from the above Example 5, diluted with dimethylacetamide (DMAc, 45.0 g) was added thereto. The reaction proceeded at 80℃ for 2 hours. To the reaction solution were added toluene (251.0 g) and water (320.0 g) to separate the layers. The organic layer thus obtained was distilled under reduced pressure to obtain 24.0 g of the title compound.
1H NMR (500 MHz, CDCl3) δ 1.47 (s, 9H), 1.49 (s, 9H), 2.49 (d, J=6.0 Hz, 2H), 3.44-3.55 (m, 2H), 4.09 (br s, 1H), 5.14 (br s, 1H).
EXAMPLE 7: Synthesis of tert-butyl (3S)-4-amino-3-[(tert-butoxycarbonyl)amino]- butanoate

To 21.0 g of the compound obtained from the above Example 6 was added tetrahydrofuran (93.3 g) followed by the addition of triphenylphosphine (PPh3, 21.0 g) at 40℃, the mixture was stirred for 2 hours at the same temperature, and water (3.8 g) was then added thereto. The reaction solution was distilled under reduced pressure, and the resulting triphenylphosphine oxide solid was diluted with toluene (26.0 g) and n-hexane (41.0 g), and then filtered off. The filtrate was adjusted to pH 2~3 with 1.0 N aqueous hydrochloric acid solution (110.0 g) and then subjected to separation of the layers. To remove any residual triphenylphosphine oxide solid, the aqueous layer obtained above was washed with dichloromethane (100.0 g) and then adjusted to pH 8~9 with 28% aqueous ammonia solution (7.6 g). The aqueous solution thus obtained was extracted with dichloromethane (100.0 g) and distilled under reduced pressure to obtain 8.5 g of the title compound as a white solid.
1H NMR (500 MHz, CDCl3) δ 1.44 (s, 9H), 1.45 (s, 9H), 2.45 (d, J=6.1 Hz, 2H), 2.77 (d, J=5.5 Hz, 2H), 3.87 (br s, 1H), 5.22 (br s, 1H).
EXAMPLE 8: Synthesis of N,N-dibenzyl-L-N(Boc)-aspartamide 4-tert-butyl ester

N-Boc-L-aspartic acid 4-t-butyl ester (29.0 g, 0.10 mol) was added to THF (200 ml). After cooling to temperature below -5℃, to the reaction solution was added isobutylchloroformate (13.0 ml, 0.10 mol) followed by addition of N-methyl morpholine (12.0 ml, 0.10 mol) dropwise, and the reaction mixture was stirred for over 30 minutes. To the reaction mixture was added dropwise dibenzylamine (21.1 ml, 0.11 mol), and the mixture was then stirred for over 3 hours and monitored for the reaction progress by TLC (EtOAc: Hexane=1:4). Upon completion of the reaction, the reaction solution was stirred with addition of ethyl acetate (300.0 mL) and 1 N hydrochloric acid to separate the layers, and distilled under reduced pressure to precipitate a solid. The solid was filtered and washed with ethyl acetate (100 ml), and then the washings were concentrated by distillation again under reduced pressure. The residue was then subjected to silica gel column to obtain the purified desired product (41.7 g, 0.89 mol).
1H NMR (400 MHz, CDCl3) δ: 7.32 (m, 5H), 7.20 (m, 5H), 5.39 (d, J=7.2 Hz, 1H), 5.30 (m, 1H), 4.87-4.77 (m, 2H), 4.48-4.39 (m, 2H), 2.72 (dd, J=15.8 Hz, J=8.0 Hz, 1H), 2.56 (dd, J=15.8 Hz, J=6.4 Hz, 1H), 1.43 (s, 9H), 1.37 (s, 9H).
Mass (ESI, m/z): 491 (M+Na), 469 (M+H), 413 (M-55).
EXAMPLE 9: Synthesis of N, N-diallyl-L-N(Boc)-aspartamide 4-tert-butyl ester

L-N(Boc)-aspartic acid 4-t-butyl ester (5.00 g, 17.3 mol) was added to THF (50 ml). After cooling to temperature below -5℃, to the reaction solution was added isobutylchloroformate (2.26 ml, 17.3 mol) followed by addition of N-methyl morpholine (1.90 ml, 17.3 mol) dropwise, and the reaction mixture was stirred for over 30 minutes. To the reaction mixture was added dropwise diallylamine (2.35 ml, 19.0 mol), and the mixture was then stirred for over 3 hours and monitored for the reaction progress by TLC (EtOAc: Hexane=1:4). Upon completion of the reaction, the reaction solution was stirred with addition of ethyl acetate (60 ml) and 1 N hydrochloric acid and, after separating the layers, concentrated by distillation under reduced pressure. The residue was then subjected to silica gel column to obtain the purified desired product (6.0 g, 16.3 mol).
1H NMR (400 MHz, CDCl3) δ: 5.78 (m, 2H), 5.30 (m, 1H), 5.23-5.11 (m, 1H), 5.30 (m, 1H), 4.93 (m, 1H), 4.11-3.84 (m, 4H), 2.68 (dd, J=15.8 Hz, J=8.0 Hz, 1H), 2.51 (dd, J=15.8 Hz, J=8.0 Hz, 1H), 1.44 (s, 9H), 1.42 (s, 9H).
Mass (ESI, m/z): 391 (M+Na), 369 (M+H), 313 (M-55).
EXAMPLE 10: Synthesis of N,N-dibenzyl-4-amino-3(S)-N(Boc)-aminobutanoic acid 4-tert-butyl ester

10.0 g of the compound obtained from the above Example 8, Ru3(CO)12 (136 mg, 1mol%), and diphenylsilane (19.7 ml, 106.7 mmol) were added to tetrahydrofuran (50 ml), and the reaction solution was stirred under reflux for over 40 hours. The reaction solution was extracted with ethyl acetate (200 ml) and concentrated by distillation under reduced pressure. The residue was then subjected to silica gel column to obtain the purified desired product (4.7 g, 10.5 mmol).
1H NMR (400 MHz, CDCl3) δ: 7.31-7.20 (m, 10H), 5.12 (bs, 1H), 3.90 (bs, 1H), 3.63 (d, J=12.0 Hz, 2H), 3.48 (d, J=12.0 Hz, 2H), 3.24 (m, 1H), 3.16 (bs, 1H), 2.42 (m, 2H), 1.81 (m, 1H), 1.59 (m, 9H), 1.46 (s, 9H), 1.06 (s, 9H).
Mass (ESI, m/z): 455 (M+H), 441 (M-13).
EXAMPLE 11: Synthesis of tert-butyl (3S)-4-amino-3-[(tert-butoxycarbonyl)amino]- 4-oxobutanoate

360.0 g of the starting material, N-Boc-Asp(O-t-Bu)OH, together with Boc2O (353.0 g) and ammonium bicarbonate (NH4HCO3, 123.9 g) was added to dimethylformamide (1174.6 g), and pyridine (61.0 g) was added dropwise thereto at room temperature, and the reaction mixture was then stirred for about 3 hours. Upon completion of the reaction, water (1440 ml) and toluene (1800 ml) were added to the reaction solution and stirred for 30 minutes to separate the layers. The organic layer thus obtained was distilled under reduced pressure to remove t-butanol and toluene to obtain the title compound, which was directly used in the next reaction.
EXAMPLE 12: Synthesis of (S)-tert-butyl 3-(tert-butoxycarbonylamino)-3-cyanopropanoate

To the compound obtained from Example 11 was added dimethylformamide (1019.5 g) followed by addition of cyanuric chloride (112.0 g) dropwise for 1.5 hours at temperature below 25℃. The reaction solution was stirred for one hour at room temperature, and then 0.1 N aqueous sodium hydroxide solution (1850.0 g) and toluene (1860 ml) were added thereto to separate the layers. The organic layer thus obtained was washed once again with water (700 ml) and then distilled under reduced pressure to obtain 318.3 g of the title compound.
1H NMR (500 MHz, CDCl3) δ: 1.44 (s, 9H), 1.45 (s, 9H), 2.45 (d, J=6.1 Hz, 2H), 2.77 (d, J=5.5 Hz, 2H), 3.87 (br s, 1H), 5.22 (br s, 1H).
EXAMPLE 13: Synthesis of tert-butyl (3S)-4-amino-3-[(tert-butoxycarbonyl)amino]- butanoate

To 212.1 g of the compound obtained from the above Example 12 was added acetic acid (4000 ml) followed by addition of 20 wt% Pd(OH)2 (1.1 g) at 40℃. The mixture was stirred for 8 hours while keeping the internal temperature below 45℃ and 3 atmospheric pressure of hydrogen. Upon completion of the reaction, the reaction solution was distilled under reduced pressure to remove acetic acid, diluted with toluene (640 L) and then filtered through a celite. To the filtrate was added 0.25 N aqueous hydrochloric acid solution (1060 ml) to separate the layers. The aqueous layer thus obtained was basified with aqueous ammonia solution (543.1 g) and then extracted with methyl t-butyl ether (MTBE, 1000 ml). The organic layer thus obtained was distilled under reduced pressure to obtain 185.0 g of the title compound.
EXAMPLE 14: Synthesis of 3-t-butoxycarbonylamino-4-(5,5-difluoro-2-oxo- piperidin-1-yl)-butyric acid t-butyl ester

Triethylamine (13.2 g) was added to 16.0 g of the compound obtained from the above Example 1 or 2-1 or 2-2, and 14.1 g of the compound obtained from the above Example 7 or 13, and the mixture was then stirred for 21 hours at 40℃. Then, dichloromethane (154.8 g) and acetic acid (18.3 g) were added, and the mixture was stirred for 5 hours at room temperature. To the resulting reaction solution was added 0.5 N aqueous hydrochloric acid solution (116.8 g) and then, the mixture was stirred for 30 minutes to separate the layers. The organic layer thus obtained was distilled under reduced pressure to obtain 23.6 g of the title compound.
1H NMR (500 MHz, CDCl3) δ: 1.42 (s, 9H), 1.46 (s, 9H), 2.27 (m, 2H), 2.40-2.64 (m, 4H), 3.20 (dd, J=4.3, 13.5 Hz, 1H), 3.56-3.70 (m, 2H), 3.76-3.91 (m, 2H), 4.16 (m, 1H), 5.20 (d, J=8.6 Hz, 1H).
EXAMPLE 15: Synthesis of 3-t-butoxycarbonylamino-4-(5,5-difluoro-2-oxo- piperidin-1-yl)-butyric acid

23.6 g of the compound obtained from the above Example 14 was added to dichloromethane (20.0 g) followed by addition of H3PO4 (30.0 g), and the mixture was stirred for 16 hours at room temperature. After confirming the detachment of all of t-butyl group and t-butyloxycarbonyl group, the reaction solution was adjusted to pH 7.0~8.0 with 10 N aqueous hydrogen peroxide, and Boc2O (16.0 g) was added thereto. After completion of the addition, 10 N aqueous hydrogen peroxide was used to maintain the pH of the reaction solution at 8.0~9.0. After stirring for 3 hours, the resulting sodium phosphate was filtered off, and the filtrate was then adjusted to pH 2.0~3.0 with 3.0 N aqueous hydrochloric acid solution. The resulting solid was filtered and dried under nitrogen to obtain 14.5 g of the title compound.
1H NMR (500 MHz, CDCl3) δ: 1.32 (s, 9H), 2.20-2.43 (m, 6H), 3.26-3.31 (m, 2H), 3.61 (m, 1H), 3.81 (m, 1H), 4.02 (m, 1H), 6.73 (d, J=8.6 Hz, 1H), 12.16 (s, 1H).
For the title compound resulting from the above, its enantiomeric isomers―i.e. S-form and R-form―were measured by HPLC (high-performance liquid chromatography), and an excess of the enantiomeric isomers (S vs. R form) (enantiomeric excess; ee) was then calculated as being ee > 99%. On the other hand, in case of the Comparative Example prepared according to the prior method based on WO 06/104356, as described below, the excess (ee) of enantiomeric isomers (S vs. R form) was 80%. From this, it can be identified that the compound of formula (2) having an optically high purity could be obtained according to the method of the present invention.
COMPARATIVE EXAMPLE 1: Synthesis of 3-t-butoxycarbonylamino-4-(5,5- difluoro-2-oxo-piperidin-1-yl)-butyric acid t-butyl ester
COMPARATIVE EXAMPLE 1-1: Synthesis of methyl 5-amino-4,4-difluoro- pentanoate HCl

To 10.0 g of the compound obtained from Example 1 was added 40 ml of anhydrous ammonia solution (7 M solution in methanol), and the mixture was stirred for 3 hours. The reaction solution was distilled and 30 ml of hydrochloric acid solution saturated with methanol was added dropwise thereto. The reaction mixture was stirred at room temperature and then distilled to obtain 7.2 g of the title compound as a white solid.
1H NMR (500 MHz, CD3OD) δ: 2.35 (m, 2H), 2.59 (t, J=7.6 Hz, 2H), 3.49 (t, J=15.3 Hz, 2H), 3.68 (s, 3H).
COMPARATIVE EXAMPLE 1-2: Synthesis of 3-t-butoxycarbonylamino-4-(5,5- difluoro-2-oxo-piperidin-1-yl)-butyric acid t-butyl ester
To the solution of the compound (1.93 g), as obtained from the above Example 4, dissolved in dichloromethane (20.0 g) and H2O (4.0 g) were added NaBr (0.8 g) and TEMPO (11 mg, 1 mol%). To this reaction solution was slowly added a solution of 5% NaOCl (11.5 g) and NaHCO3 (1.7 g) dissolved in H2O (12.0 g) dropwise for about 2 hours while maintaining the temperature below 5℃. Upon completion of dropwise addition, the reaction solution was stirred for 30 minutes to separate the layers. To the organic layer thus obtained was added the compound (1.6 g) obtained from the above Comparative Example 1-1. After stirring for 15 minutes at room temperature, NaBH(OAc)3 (2.23 g) was added to the reaction solution. After stirring for about 19 hours, 10% aqueous NaHCO3 solution (20.0 g) and 0.5 N aqueous hydrochloric acid solution (20.0 g) were added dropwise to the reaction solution to separate the layers. The organic layer thus obtained was dehydrated under anhydrous MgSO4 to obtain 2.0 g (yield 73%) of the same title compound as Example 14, as a yellow solid. For the title compound resulting from the above, its enantiomeric isomers―i.e., S-form and R-form―were measured by HPLC (high-performance liquid chromatography), and an excess (ee) of the enantiomeric isomers (S vs. R form) was then calculated as being ee = 80%.
| WO2006104356A1 | Mar 30, 2006 | Oct 5, 2006 | Seong Cheol Bu | Dipeptidyl peptidase-iv inhibiting compounds, methods of preparing the same, and pharmaceutical compositions containing the same as an active agent |
| EP0279435A2 * | Feb 18, 1988 | Aug 24, 1988 | BASF Aktiengesellschaft | Process for the reduction of mono- and dicarboxylic acids |
| US5556982 * | Jul 12, 1993 | Sep 17, 1996 | Neorx Corporation | Metal radionuclide labeled proteins for diagnosis and therapy |
| US20080039517 * | Aug 7, 2007 | Feb 14, 2008 | Washburn David G | Pyrrolidinone anilines as progesterone receptor modulators |
Footnotes
- Lim KS, Kim JR, Choi YJ, Shin KH, Kim KP, Hong JH, Cho JY, Shin HS, Yu KS, Shin SG, Kwon OH, Hwang DM, Kim JA, Jang IJ (October 2008). “Pharmacokinetics, pharmacodynamics, and tolerability of the dipeptidyl peptidase IV inhibitor LC15-0444 in healthy Korean men: a dose-block-randomized, double-blind, placebo-controlled, ascending single-dose, Phase I study”. Clin Ther 30 (10): 1817–30. doi:10.1016/j.clinthera.2008.10.013. PMID 19014837.
- Ábel T. “A New Therapy of Type 2 Diabetes: DPP-4 Inhibitors”. In Rigobelo EC. Hypoglycemia – Causes and Occurrences. Croatia: InTech. pp. 3–52. doi:10.5772/23604. ISBN 978-953-307-657-7.
- Kaji K (Mar 2014). “Dipeptidyl peptidase-4 inhibitor attenuates hepatic fibrosis via suppression of activated hepatic stellate cell in rats.”. J Gastroenterol.. 49 (3): 481–91.doi:10.1007/s00535-013-0783-4. PMID 23475323.
- Min HS (Jun 2014). “Dipeptidyl peptidase IV inhibitor protects against renal interstitial fibrosis in a mouse model of ureteral obstruction.”. Lab Invest. 94 (5): 598–607.doi:10.1038/labinvest.2014.50. PMID 24687121.
- Gouni-Berthold I (2014). “The role of oral antidiabetic agents and incretin mimetics in type 2 diabetic patients with non-alcoholic Fatty liver disease.”. Curr Pharm Des. 20 (5): 3705–15.PMID 24040873.
Further reading
| Kim SE, Yi S, Shin KH, Kim TE, Kim MJ, Kim YH, Yoon SH, Cho JY, Shin SG, Jang IJ, Yu KS (January 2012). “Evaluation of the pharmacokinetic interaction between the dipeptidyl peptidase IV inhibitor LC15-0444and pioglitazone in healthy volunteers”. Int J Clin Pharmacol Ther. 50 (1): 17–23. doi:10.5414/cp201568. PMID 22192641. |
- Rhee EJ, Lee WY, Yoon KH, Yoo SJ, Lee IK, Baik SH, Kim YK, Lee MK, Park KS, Park JY, Cha BS, Lee HW, Min KW, Bae HY, Kim MJ, Kim JA, Kim DK, Kim SW (December 2010). “A multicenter, randomized, placebo-controlled, double-blind phase II trial evaluating the optimal dose, efficacy and safety of LC 15-0444 in patients with type 2 diabetes”. Diabetes Obes Metab.12 (12): 1113–1119. doi:10.1111/j.1463-1326.2010.01303.x. PMID 20977584.
- Lim KS, Cho JY, Kim BH, Kim JR, Kim HS, Kim DK, Kim SH, Yim HJ, Lee SH, Shin SG, Jang IJ, Yu KS (December 2009). “Pharmacokinetics and pharmacodynamics of LC15-0444, a novel dipeptidyl peptidase IV inhibitor, after multiple dosing in healthy volunteers”. Br J Clin Pharmacol. 68 (6): 883–890. doi:10.1111/j.1365-2125.2009.03376.x. PMC 2810799.PMID 20002082.
- Dal-Mi Hwang, Sung-Ho Kim, Min-Kyung Yoon, O Hwan Kwon, Ki Dong Koo, Changhee Min, Sung-Hack Lee, Jaeick Lee, Chang-Seok Lee, Hyeon Joo Yim (June 2008). “LC15-0444 is a novel, potent, selective, and orally active dipeptidyl peptidase IV inhibitor” (PDF). American Diabetes Association 68th Scientific Sessions.
External links
| DAVID G. WASHBURN ET AL.: ‘Discovery or orally active, pyrrolidinone-based progesterone receptor partial agonist‘ BIOORGANIC & MEDICINAL CHEMISTRY LETTERS vol. 19, no. 16, 2009, pages 4664 – 4667, XP026419052 | ||
| 2 | * | MONICA LOPEZ-GARCIA ET AL.: ‘Synthesis of (R)-3,4- diaminobutanoic acid by desymmetrization of dimethyl 3-(benzylamino)-glutarate through enzymatic ammonolysis‘ JOURNAL OF ORGANIC CHEMISTRY vol. 68, no. 2, 2003, pages 648 – 651, XP055105976 |
Google+
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy
TRELAGLIPTIN
1 TRELAGLIPTIN

Trelagliptin succinate (SYR-472)
2-[[6-[(3R)-3-aminopiperidin-1-yl]-3-methyl-2, 4-dioxopyrimidin-1-yl]methyl]-4-fluorobenzonitrile; butanedioic acid
2-[6-[3(R)-Aminopiperidin-1-yl]-3-methyl-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-ylmethyl]-4-fluorobenzonitrile
2- [ [6- [ (3R) -3-amino-l-piperidinyl] -3, 4-dihydro-3- methyl-2, 4-dioxo-l (2H) -pyrimidinyl]methyl] -4-fluorobenzonitrile
succinic acid salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile
Sponsor/Developer: Takeda Pharmaceuticals and Furiex Pharmaceuticals
Mechanism of action: DPP-4 inhibitor
865759-25-7 cas FREE BASE
1029877-94-8 succinate
- SYR 111472 succinate
- SYR 472
- Syr-472
- Syr111472 succinate
- Trelagliptin succinate
- UNII-4118932Z90
- clinical trials….http://clinicaltrials.gov/search/intervention=SYR+472
Trelagliptin-succinate M. Wt: 475.47
Trelagliptin-succinate Formula: C22H26FN5O6
SYR-472 is an oral dipeptidyl peptidase IV inhibitor originated by Takeda. It is in phase III clinical trials for the treatment of type 2 diabetes.
- Diabetes affects 25.8 million people of all ages, or roughly 8.3 percent of the U.S. population.
- The World Health Organization predicts that there will be 366 million people worldwide affected by diabetes by the year 2030.
- The advent of trelagliptin succinate, a unique once weekly medication for patients with type 2 Diabetes is now the focus of clinical trials and exciting research and development.
- Phase III clinical trials of trelagliptin succinate commenced in September 2011, and are estimated to be complete by the second half of 2013.
TRELAGLIPTIN (SYR-472)
Trelagliptin is a novel DPP-4 inhibitor that is being developed by Takeda. In contrast to alogliplitin, which is once a day, trelagliptin is a once-weekly oral agent which should provide patients with a convenient therapeutic alternative and has the potential to improve compliance. Takeda has commenced Phase III trials of trelagliptin in Japan for the treatment of Type 2 diabetes.

Indication (Phase): Japan—Once-weekly oral treatment for type 2 diabetes (Phase III; study expected to be completed in second half of 2013)


Compound I, A, TRELAGLIPTIN which has the formula:

is a DPP-IV inhibitor that is described in U.S. patent application Ser. No. 11/080,992 filed Mar. 15, 2005 (see Compound 34). Its dosing, administration and biological activities are described in U.S. patent application Ser. No. 11/531,671 filed Sep. 13, 2006. U.S. patent application Ser. No. 11/080,992 and Ser. No. 11/531,671 are incorporated herein by reference in their entirety.
Dipeptidyl peptidase IV (IUBMB Enzyme Nomenclature EC.3.4.14.5) (referred herein as “DPP-IV”) is a type II membrane protein and a non-classical serine aminodipeptidase that removes Xaa-Pro dipeptides from the amino terminus (N-terminus) of polypeptides and proteins. DPP-IV is constitutively expressed on epithelial and endothelial cells of a variety of different tissues (e.g., intestine, liver, lung, kidney and placenta), and is also found in body fluids. DPP-IV is also expressed on circulating T-lymphocytes and has been shown to be synonymous with the cell-surface antigen, CD-26. DPP-IV has been implicated in a number of human disease states, including, but are not limit to, diabetes, particularly type II diabetes mellitus, diabetic dislipidemia, conditions of impaired glucose tolerance (IGT), conditions of impaired fasting plasma glucose (IFG), metabolic acidosis, ketosis, appetite regulation and obesity; autoimmune diseases such as inflammatory bowel disease, multiple sclerosis and rheumatoid arthritis; AIDS; and cancers.
DPP-IV inhibitors are believed to be useful agents for the prevention, delay of progression, and/or treatment of conditions mediated by DPP-IV.
Compound (A) or a salt thereof has been reported as an inhibitor of dipeptidyl peptidase (DPP-IV) , which is an enzyme that decomposes glucagon-like peptide-1 (GLP-1) , a hormone increasing insulin secretion (patent document 1) .
In addition, a method including administering 1 – 250 mg of compound (A) or a salt thereof to a patient once per week (patent documents 2, 3), crystal polymorphs of compound (A) (patent documents 4, 5) , and a preparation of compound (A)
(patent documents 6, 7) have also been reported. Compound (A) and a salt thereof are recommended for oral administration in view of the easiness of self-administration, and a tablet, particularly a tablet in the dosage form for administration once per week, is desired. [0006]
The dosage form of once per week is expected to improve drug compliance of patients, whereas it requires supply of compound (A) or a salt thereof to patients in a high dose as compared to, for example, the dosage form of once per day. Since a solid preparation containing compound (A) or a salt thereof in a high dose increases its size, it may conversely degrade the drug compliance for patients, particularly infants and elderly patients having difficulty in swallowing
……………………..
SYNTHESIS
Compound 34 IS TRELAGLIPTIN

4-Fluoro-2-methylbenzonitrile (31).
A mixture of 2-bromo-5-fluorotoluene (3.5 g, 18.5 mmol) and CuCN (2 g, 22 mmol) in DMF (100 mL) was refluxed for 24 hours. The reaction was diluted with water and extracted with hexane. The organics were dried over MgSO4 and the solvent removed to give product 31 (yield 60%). 1H-NMR (400 MHz, CDCl3): δ 7.60 (dd, J=5.6, 8.8 Hz, 1H), 6.93-7.06 (m, 2H), 2.55 (s, 3H).
2-Bromomethyl-4-fluorobenzonitrile (32).
A mixture of 4-fluoro-2-methylbenzonitrile (2 g, 14.8 mmol), NBS (2.64 g, 15 mmol) and AIBN (100 mg) in CCl4 was refluxed under nitrogen for 2 hours. The reaction was cooled to room temperature. The solid was removed by filtration. The organic solution was concentrated to give crude product as an oil, which was used in the next step without further purification. 1H-NMR (400 MHz, CDCl3): δ 7.68 (dd, J=5.2, 8.4 Hz, 1H), 7.28 (dd, J=2.4, 8.8 Hz, 1H), 7.12 (m, 1H), 4.6 (s, 2H).
Alternatively, 32 was made as follows.
4-Fluoro-2-methylbenzonitrile (1 kg) in DCE (2 L) was treated with AIBN (122 g) and heated to 75° C. A suspension of DBH (353 g) in DCE (500 mL) was added at 75° C. portionwise over 20 minutes. This operation was repeated 5 more times over 2.5 hours. The mixture was then stirred for one additional hour and optionally monitored for completion by, for example, measuring the amount of residual benzonitrile using HPLC. Additional AIBN (e.g., 12.5 g) was optionally added to move the reaction toward completion. Heating was stopped and the mixture was allowed to cool overnight. N,N-diisopropylethylamine (1.3 L) was added (at <10° C. over 1.5 hours) and then diethyl phosphite (1.9 L) was added (at <20° C. over 30 min). The mixture was then stirred for 30 minutes or until completion. The mixture was then washed with 1% sodium metabisulfite solution (5 L) and purified with water (5 L). The organic phase was concentrated under vacuum to afford 32 as a dark brown oil (3328 g), which was used without further purification (purity was 97% (AUC)).
2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (33).
A mixture of crude 3-methyl-6-chlorouracil (0.6 g, 3.8 mmol), 2-bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol) and K2CO3 (0.5 g, 4 mmol) in DMSO (10 mL) was stirred at 60° C. for 2 hours. The reaction was diluted with water and extracted with EtOAc. The organics were dried over MgSO4 and the solvent removed. The residue was purified by column chromatography. 0.66 g of the product was obtained (yield: 60%). 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, J=7.2, 8.4 Hz, 1H), 7.26 (d, J=4.0 Hz, 1H), 7.11-7.17 (m, 1H), 6.94 (dd, J=2.0, 9.0 Hz, 1H), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [m+H] calc’d for C13H9ClFN3O2, 293.68; found 293.68.
Alternatively, 33 was made as follows.
To a solution of 6-chloro-3-methyluracil (750 g) and N,N-diisopropylethylamine (998 mL) in NMP (3 L) was added (at <30° C. over 25 min) a solution of 32 (2963 g crude material containing 1300 g of 32 in 3 L of toluene). The mixture was then heated at 60° C. for 2 hours or until completion (as determined, for example, by HPLC). Heating was then stopped and the mixture was allowed to cool overnight. Purified water (3.8 L) was added, and the resultant slurry was stirred at ambient temperature for 1 hour and at <5° C. for one hour. The mixture was then filtered under vacuum and the wet cake was washed with IPA (2×2.25 L). The material was then dried in a vacuum oven at 40±5° C. for 16 or more hours to afford 33 as a tan solid (>85% yield; purity was >99% (AUC)).
TFAsalt OF TRELAGLIPTIN
2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile (34).
2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (300 mg, 1.0 mmol), (R)-3-amino-piperidine dihydrochloride (266 mg, 1.5 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) were stirred in a sealed tube in EtOH (3 mL) at 100° C. for 2 hrs. The final compound was obtained as TFA salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.16-7.27 (m, 2H), 5.46 (s, 1H), 5.17-5.34 (ABq, 2H, J 35.2, 15.6 Hz), 3.33-3.47 (m, 2H), 3.22 (s, 3H), 2.98-3.08 (m, 1H), 2.67-2.92 (m, 2H), 2.07-2.17 (m, 1H), 1.82-1.92 (m, 1H), 1.51-1.79 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
FREE BASE OF TRELAGLIPTIN
Alternatively, the free base of 34 was prepared as follows. A mixture of 33 (1212 g), IPA (10.8 L), (R)-3-amino-piperidine dihydrochloride (785 g), purified water (78 mL) and potassium carbonate (2.5 kg, powder, 325 mesh) was heated at 60° C. until completion (e.g., for >20 hours) as determined, for example, by HPLC. Acetonitrile (3.6 L) was then added at 60° C. and the mixture was allowed to cool to <25° C. The resultant slurry was filtered under vacuum and the filter cake was washed with acetonitrile (2×3.6 L). The filtrate was concentrated at 45° C. under vacuum (for >3 hours) to afford 2.6 kg of the free base of 34.
HCL salt OF TRELAGLIPTIN
The HCl salt of 34 was prepared from the TFA salt as follows. The TFA salt (34) was suspended in DCM, and then washed with saturated Na2CO3. The organic layer was dried and removed in vacuo. The residue was dissolved in acetonitrile and HCl in dioxane (1.5 eq.) was added at 0° C. The HCl salt was obtained after removing the solvent. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.12-7.26 (m, 2H), 5.47 (s, 1H), 5.21-5.32 (ABq, 2H, J=32.0, 16.0 Hz), 3.35-3.5 (m, 2H), 3.22 (s, 3H), 3.01-3.1 (m, 1H), 2.69-2.93 (m, 2H), 2.07-2.17 (m, 1H), 1.83-1.93 (m, 1H), 1.55-1.80 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
Alternatively, the HCl salt was prepared from the free base as follows. To a solution of free base in CH2Cl2 (12 L) was added (at <35° C. over 18 minutes) 2 M hydrochloric acid (3.1 L). The slurry was stirred for 1 hour and then filtered. The wet cake was washed with CH2Cl2 (3.6 L) and then THF (4.8 L). The wet cake was then slurried in THF (4.8 L) for one hour and then filtered. The filter cake was again washed with THF (4.8 L). The material was then dried in a vacuum oven at 50° C. (with a nitrogen bleed) until a constant weight (e.g., >26 hours) to afford 34 as the HCl salt as a white solid (1423 g, >85% yield).
Succinate salt OF TRELAGLIPTIN

The succinate salt of 34 was prepared from the HCl salt as follows. To a mixture of the HCl salt of 34 (1414 g), CH2Cl2 (7 L) and purified water (14 L) was added 50% NaOH solution (212 mL) until the pH of the mixture was >12. The biphasic mixture was stirred for 30 min and the organic layer was separated. The aqueous layer was extracted with CH2Cl2 (5.7 L) and the combined organic layers were washed with purified water (6 L). The organic layer was then passed through an in-line filter and concentrated under vacuum at 30° C. over three hours to afford the free base as an off-white solid. The free base was slurried in prefiltered THF (15 L) and prefiltered IPA (5.5 L). The mixture was then heated at 60° C. until complete dissolution of the free base was observed. A prefiltered solution of succinic acid (446 g) in THF (7 L) was added (over 23 min) while maintaining the mixture temperature at >57° C. After stirring at 60° C. for 15 min, the heat was turned off, the material was allowed to cool, and the slurry was stirred for 12 hours at 25±5° C. The material was filtered under vacuum and the wet cake was washed with prefiltered IPA (2×4.2 L). The material was then dried in a vacuum oven at 70±5° C. (with a nitrogen bleed) for >80 hours to afford the succinate salt of 34 as a white solid (1546 g, >90% yield).
The product was also converted to a variety of corresponding acid addition salts. Specifically, the benzonitrile product (approximately 10 mg) in a solution of MeOH (1 mL) was treated with various acids (1.05 equivalents). The solutions were allowed to stand for three days open to the air. If a precipitate formed, the mixture was filtered and the salt dried. If no solid formed, the mixture was concentrated in vacuo and the residue isolated. In this way, salts of 34 were prepared from the following acids: benzoic, p-toluenesulfonic, succinic, R-(−)-Mandelic and benzenesulfonic. The succinate was found to be crystalline as determined by x-ray powder diffraction analysis.
Methanesulfonate salt
In addition, the methanesulfonate salt was prepared as follows. A 10.5 g aliquot of the benzonitrile product was mixed with 400 mL of isopropylacetate. The slurry was heated to 75° C. and filtered through #3 Whatman filter paper. The solution was heated back to 75° C. and a 1M solution of methanesulfonic acid (30.84 mL) was added slowly over 10 minutes while stirring. The suspension was cooled to room temperature at a rate of about 20° C./hr. After 1 hr at room temperature, the solid was filtered and dried in an oven overnight to obtain the methanesulfonate salt.
…………………………
FORMULATION
COMPD A IS TRELAGLIPTIN
Examples (Comparative Example IA)
Succinate of compound (A) (26.6 mg) was weighed in a glass bottle and used as Comparative Example IA. (Comparative Example 2A)
The succinate of compound (A) and microcrystalline cellulose were uniformly mixed in a mortar at a ratio of 1:10, and the mixture (226.6 mg) was weighed in a glass bottle and used as Comparative Example 2A. (Comparative Example 3A)
The succinate of compound (A) and corn starch were uniformly mixed in a mortar at a ratio of 1:5, and the mixture (126.6 mg) was weighed in a glass bottle and used as Comparative Example 3A. (Example IA) Succinate of compound (A) , mannitol and corn starch according to the formulation of Table IA were uniformly mixed in a fluid bed granulator (LAB-I, POWREX CORPORATION) , and the mixture was granulated by spraying an aqueous solution of dissolved hypromellose 2910, and dried therein. The obtained granules were passed through a sieve -(16M) to give milled granules. To the milled granules were added croscarmellose sodium, microcrystalline cellulose and magnesium stearate, and they were mixed in a bag to give granules for tableting. The granules were punched by a rotary tableting machine (Correct 19K, Kikusui Seisakusho, Ltd.) with a 6.5 mmφ punch to give a plain tablet weighting 121 mg. On the other hand, titanium oxide, yellow ferric oxide and talc were dispersed in a hypromellose 2910 aqueous solution to prepare a film coating liquid. The aforementioned coating liquid was sprayed onto the above-mentioned plain tablet in a film coating machine (Hicoater HCP-75, Freund Corporation), to give 2500 film- coated tablets containing 3.125 mg of compound (A) (free form) per tablet. Table IA

………………………..
POLYMORPHS AND SYNTHESIS
FORM A
Form A may be prepared by crystallization from the various solvents and under the various crystallization conditions used during the polymorph screen (e.g., fast and slow evaporation, cooling of saturated solutions, slurries, and solvent/antisolvent additions). Tables B and C of Example 3 summarize the procedures by which Form A was prepared. For example, Form A was obtained by room temperature slurry of an excess amount of Compound I in acetone, acetonitrile, dichloromethane, 1,4-dioxane, diethyl ether, hexane, methanol, isopropanol, water, ethylacetate, tetrahydrofuran, toluene, or other like solvents on a rotating wheel for approximately 5 or 7 days. The solids were collected by vacuum filtration, and air dried in the hood. Also, Form A was precipitated from a methanol solution of Compound I by slow evaporation (SE).
[0091] Form A was characterized by XRPD, TGA, hot stage microscopy, IR, Raman spectroscopy, solution 1H-NMR, and solid state 13C-NMR.
[0092] Figure 1 shows a characteristic XRPD spectrum (CuKa, λ=1.5418A) of Form A. The XRPD pattern confirmed that Form A was crystalline. Major X-Ray diffraction lines expressed in °2Θ and their relative intensities are summarized in Table 1.
Table 1. Characteristic XRPD Peaks (CuKa) of Form A


Characterization Data of Form A of Compound I

8. Amorphous Form
[0137] The Amorphous Form of Compound I was prepared by lyophilization of an aqueous solution of Compound I (Example 10). The residue material was characterized by XRPD and the resulting XRPD spectrum displayed in Figure 26. The XRPD spectrum shows a broad halo with no specific peaks present, which confirms that the material is amorphous. The material was further characterized by TGA, DSC, hot stage microscopy, and moisture sorption analysis.
Table A. Approximate Solubilities of Compound I
Compound I having the formula



POLYMORPH SCREEN
Crystallization Experiments of Compound I from Solvents




a) FE = fast evaporation; SE = slow evaporation; RT = room temperature; SC = slow cool;CC = crash cool, MB = moisture sorption/desorption analysis b) qty = quantity; PO = preferred orientation
…………………………
SYNTHESIS
EXAMPLES
1. Preparation of 2-[6-(3-Amino-piperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydro- 2H-pyrimidin-l-ylmethyl]-4-fluoro-benzonitrile and pharmaceutically acceptable salts


4-Fluoro-2-methylbenzonitrile (3)
[0166] A mixture of 2-bromo-5fluorotoluene ( 2) (3.5 g, 18.5 mmol) and CuCN (2 g, 22 mmol) in DMF (100 mL) was re fluxed for 24 hours. The reaction was diluted with water and extracted with hexane. The organics were dried over MgSO4 and the solvent removed to give product 3 (yield 60%). 1H-NMR (400 MHz, CDCl3): δ 7.60 (dd, J=5.6, 8.8 Hz, IH), 6.93-7.06 (m, 2H), 2.55 (s, 3H). 2-Bromomethyl-4-fluorobenzonitrile (4)
[0167] A mixture of 4-fluoro-2-methylbenzonitrile (3) (2 g, 14.8 mmol), NBS (2.64 g, 15 mmol) and AIBN (100 mg) in CCl4 was refluxed under nitrogen for 2 hours. The reaction was cooled to room temperature. The solid was removed by filtration. The organic solution was concentrated to give crude product as an oil, which was used in the next step without further purification.1H-NMR (400 MHz, CDCl3): δ 7.68 (dd, J= 5.2, 8.4 Hz, IH), 7.28 (dd, J= 2.4, 8.8 Hz, IH), 7.12 (m, IH), 4.6 (s, 2H).
2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l-ylmethyl)-4-fluoro- benzonitrile (6)
[0168] A mixture of crude 3-methyl-6-chlorouracil (5) (0.6 g, 3.8 mmol), 2- Bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol) and K2CO3 (0.5 g, 4 mmol) in DMSO
(10 mL) was stirred at 60 C for 2 hours. The reaction was diluted with water and extracted with EtOAc. The organics were dried over MgSO4 and the solvent removed. The residue was purified by column chromatography. 0.66 g of the product was obtained (yield: 60%). 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, 1=12, 8.4Hz, IH), 7.26 (d, J- 4.0Hz, IH), 7.11-7.17 (m, IH), 6.94 (dd, J=2.0, 9.0 Hz, IH), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [m+H] calc’d for Ci3H9ClFN3O2, 293.68; found 293.68.
2-[6-(3-Amino-piperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l- ylmethyl]-4-fluoro-benzonitrile, TFA salt (1) (TFA salt of Compound I)

[0169] 2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l-ylmethyl)-4- fluoro-benzonitrile (5) (300 mg, 1.0 mmol), (i?)-3-amino-piperidine dihydrochloride (266 mg, 1.5 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) were stirred in a sealed tube in EtOH (3 mL) at 100 0C for 2 hrs. The final compound was obtained as a TFA salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, IH), 7.16-7.27 (m, 2H), 5.46 (s, IH), 5.17-5.34 (ABq, 2H, J = 35.2, 15.6 Hz), 3.33-3.47 (m, 2H), 3.22 (s, 3H), 2.98-3.08 (m, IH), 2.67-2.92 (m, 2H), 2.07-2.17 (m, IH), 1.82-1.92 (m, IH), 1.51-1.79 (m, 2H). MS (ES) [m+H] calc’d for Ci8H20FN5O2, 357.38; found, 357.38.
2-[6-(3-Amino-piperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l- ylmethyl]-4-fluoro-benzonitrile, HCl salt

[0170] The TFA salt of Compound I was suspended in DCM, and then washed with saturated Na2CO3. The organic layer was dried and removed in vacuo. The residue was dissolved in acetonitrile and HCl in dioxane (1.5 eq.) was added at 0 C. The HCl salt was obtained after removing the solvent. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, IH), 7.12-7.26 (m, 2H), 5.47 (s, IH), 5.21-5.32 (ABq, 2H, J = 32.0, 16.0 Hz), 3.35-3.5 (m, 2H), 3.22 (s, 3H), 3.01-3.1 (m, IH), 2.69-2.93 (m, 2H), 2.07-2.17 (m, IH), 1.83-1.93 (m, IH), 1.55-1.80 (m, 2H). MS (ES) [m+H] calc’d for Ci8H20FN5O2, 357.38; found, 357.38.
General procedure for the preparation of salts of Compound I.
[0171] The benzonitrile product may be isolated as the free base if desired, but preferably, the product may be further converted to a corresponding acid addition salt. Specifically, the benzonitrile product (approximately 10 mg) in a solution of MeOH (1 mL) was treated with various acids (1.05 equivalents). The solutions were allowed to stand for three days open to the air. If a precipitate formed, the mixture was filtered and the salt dried. If no solid formed, the mixture was concentrated in vacuo and the residue isolated. In this way, salts of Compound I were prepared from the following acids: benzoic, p-toluenesulfonic, succinic, R-(-)-Mandelic and benzenesulfonic. [0172] The isolation and/or purification steps of the intermediate compounds in the above described process may optionally be avoided if the intermediates from the reaction mixture are obtained as relatively pure compounds and the by-products or impurities of the reaction mixture do not interfere with the subsequent reaction steps. Where feasible, one or more isolation steps may be eliminated to provide shorter processing times, and the elimination of further processing may also afford higher overall reaction yields.
…………………..
TABLET
2. Exemplary formulations comprising succinate salt of 2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile
Provided are examples of tablet formulations that may be used to administer succinate salt of 2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile (Succinate salt of Compound I) according to the present invention. It is noted that the formulations provided herein may be varied as is known in the art.
The exemplary tablet formulations are as follows:
| 12.5 mg of Compound I (weight of free base form) per tablet | ||||
| Core Tablet Formulation | ||||
| (1) | 2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4- | 17.0 | mg | |
| dioxo-3,4-dihydro-2H-pyrimidin-1- | ||||
| ylmethyl]-4-fluoro-benzonitrile (succinate salt) | ||||
| (2) | Lactose Monohydrate, NF, Ph, Eur | 224.6 | mg | |
| (FOREMOST 316 FAST FLO) | ||||
| (3) | Microcrystalline Cellulose, NF, Ph, Eur | 120.1 | mg | |
| (AVICEL PH 102) | ||||
| (4) | Croscarmellose Sodium, NF, Ph, Eur | 32.0 | mg | |
| (AC-DO-SOL) | ||||
| (5) | Colloidal Silicon Dioxide, NF, Ph, Eur | 3.2 | mg | |
| (CAB-O-SIL M-5P) | ||||
| (6) | Magnesium Stearate, NF, Ph, Eur | 3.2 | mg | |
| (MALLINCKRODT, Non-bovine Hyqual) | ||||
| TOTAL | 400.0 | mg | ||
| (per tablet) | ||||
…………..
POLYMORPHS AND SYNTHESIS
EXAMPLES Example 1 Preparation of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile succinate (Compound I)

Compound I may be prepared by the follow synthetic route (Scheme 1)

A. Preparation of 4-fluoro-2-methylbenzonitrile (Compound B)

Compound B was prepared by refluxing a mixture of 2-bromo-5-fluoro-toluene (Compound A) (3.5 g, 18.5 mmol) and CuCN (2 g, 22 mmol) in DMF (100 mL) for 24 hours. The reaction was diluted with water and extracted with hexane. The organics were dried over MgSO4 and the solvent removed to give product B (yield 60%). 1H-NMR (400 MHz, CDCl3): δ 7.60 (dd, J=5.6, 8.8 Hz, 1H), 6.93-7.06 (m, 2H), 2.55 (s, 3H).
B. Preparation of 2-bromomethyl-4-fluorobenzonitrile (Compound C)

Compound C was prepared by refluxing a mixture of 4-fluoro-2-methylbenzonitrile (Compound B) (2 g, 14.8 mmol), N-bromosuccinimide (NBS) (2.64 g, 15 mmol) and azo-bis-isobutyronitrile (AIBN) (100 mg) in CCl4 under nitrogen for 2 hours. The reaction was cooled to room temperature. The solid was removed by filtration. The organic solution was concentrated to give the crude product the form of an oil, which was used in the next step without further purification. 1H-NMR (400 MHz, CDCl3): δ 7.68 (dd, J=5.2, 8.4 Hz, 1H), 7.28 (dd, J=2.4, 8.8 Hz, 1H), 7.12 (m, 1H), 4.6 (s, 2H).
C. Preparation of 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound D)

Compound E was prepared by stirring a mixture of crude 3-methyl-6-chlorouracil D (0.6 g, 3.8 mmol), 2-bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol) and K2CO3 (0.5 g, 4 mmol) in DMSO (10 mL) at 60° C. for 2 hours. The reaction was diluted with water and extracted with EtOAc. The organics were dried over MgSO4 and the solvent removed. The residue was purified by column chromatography. 0.66 g of the product was obtained (yield: 60%). 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, J=7.2, 8.4 Hz, 1H), 7.26 (d, J=4.0 Hz, 1H), 7.11-7.17 (m, 1H), 6.94 (dd, J=2.0, 9.0 Hz, 1H), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [m+H] calc’d for C13H9ClFN3O2, 293.68; found 293.68.
D. Preparation of 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound F)

Compound F was prepared by mixing and stirring 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound E) (300 mg, 1.0 mmol), (R)-3-amino-piperidine dihydrochloride (266 mg, 1.5 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) in a sealed tube in EtOH (3 mL) at 100° C. for 2 hrs. The final compound was obtained as trifluoroacetate (TFA) salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.16-7.27 (m, 2H), 5.46 (s, 1H), 5.17-5.34 (ABq, 2H, J=35.2, 15.6 Hz), 3.33-3.47 (m, 2H), 3.22 (s, 3H), 2.98-3.08 (m, 1H), 2.67-2.92 (m, 2H), 2.07-2.17 (m, 1H), 1.82-1.92 (m, 1H), 1.51-1.79 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
E. Preparation of Compound I: the succinic acid salt of 2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile

The TFA salt prepared in the above step (Example 1, Step D) was suspended in DCM, and then washed with saturated Na2CO3. The organic layer was dried and removed in vacuo. The benzonitrile product (approximately 10 mg) was dissolved in MeOH (1 mL) and to which succinic acid in THF (1.05 equivalents) was added. The solutions were allowed to stand for three days open to the air. If a precipitate formed, the solid was collected by filtration. If no solid formed, the mixture was concentrated in vacuo, and the succinate salt was obtained after removing the solvent.
SUCCINATE SALT OF TRELAGLIPTIN
1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.12-7.26 (m, 2H), 5.47 (s, 1H), 5.21-5.32 (ABq, 2H, J=32.0, 16.0 Hz), 3.35-3.5 (m, 2H), 3.22 (s, 3H), 3.01-3.1 (m, 1H), 2.69-2.93 (m, 2H), 2.07-2.17 (m, 1H), 1.83-1.93 (m, 1H), 1.55-1.80 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
Compound I such prepared was found to be crystalline as determined by x-ray powder diffraction analysis (FIG. 1). The crystal material was designated Form A.
……………
patents
1. US 2013172377
2. WO 2011013639
3. WO 2009099172
4.WO 2009099171
5. WO 2008114807
6.WO 2008114800
7. WO 2008033851
8. WO 2007074884
9WO 2007035629
patent document 1: US2005/0261271
patent document 2: US2007/0060530
patent document 3: US2008/0287476
patent document 4: US2008/0227798
patent document 5: US2008/0280931
patent document 6: WO2008/114800
patent document 7: WO2011/013639
| US7906523 * | Oct 30, 2007 | Mar 15, 2011 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8084605 * | Nov 29, 2007 | Dec 27, 2011 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| US8188275 * | Oct 30, 2007 | May 29, 2012 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8222411 * | Sep 15, 2006 | Jul 17, 2012 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US20090275750 * | Sep 15, 2006 | Nov 5, 2009 | Jun Feng | Dipeptidyl peptidase inhibitors |
| WO2013183784A1 | Jun 4, 2013 | Dec 12, 2013 | Takeda Pharmaceutical Company Limited | Solid preparation |
| US20080227798 * | Nov 29, 2007 | Sep 18, 2008 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2h-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| US20120197018 * | Feb 15, 2012 | Aug 2, 2012 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2h-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| WO2007033265A1 * | Sep 13, 2006 | Mar 22, 2007 | Takeda Pharmaceutical | Dipeptidyl peptidase inhibitors for treating diabetis |
| WO2007033266A2 * | Sep 13, 2006 | Mar 22, 2007 | Takeda Pharmaceutical | Dipeptidyl peptidase inhibitors for treating diabetis |
| WO2007033350A1 * | Sep 13, 2006 | Mar 22, 2007 | Takeda Pharmaceutical | Dipeptidyl peptidase inhibitors for treating diabetes |
| EP1586571A1 * | Dec 21, 2004 | Oct 19, 2005 | Takeda San Diego, Inc. | Dipeptidyl peptidase inhibitors |
EVOGLIPTIN
EVOGLIPTIN
CAS: 1222102-29-5 FREE
HCL……1246960-27-9
tartare.. 1222102 -51-3
Dong-A Pharmaceutical. Co., Ltd, 동아제약 주식회사
2-Piperazinone, 4-((3R)-3-amino-1-oxo-4-(2,4,5-trifluorophenyl)butyl)-3-((1,1-dimethylethoxy)methyl)-, (3R)-
R)-4-((R)-3-Amino-4-(2,4,5-trifluorophenyl)-butanoyl)-3-(t-butoxymethyl)-piperazin-2-one
4-[3(R)-Amino-4-(2,4,5-trifluorophenyl)butyryl]-3(R)-(tert-butoxymethyl)piperazin-2-one hydrochloride
DA-1229
see…http://www.allfordrugs.com/2015/07/03/evogliptin/
DA-1229 is a dipeptidyl peptidase IV (CD26) inhibitor currently being developed in phase III clinical studies at Dong-A for the treatment of type 2 diabetes.
In 2014, Eurofarma aquired rights for product development and commercialization in Brazil.


If above image is not clear then see at…….http://www.allfordrugs.com/2015/07/03/evogliptin/
86…………H. J. Kim, W. Y. Kwak, J. P. Min, J. Y. Lee, T. H. Yoon, H. D. Kim, C. Y. Shin, M. K.
Kim, S. H. Choi, H. S. Kim, E. K. Yang, Y. H. Cheong, Y. N. Chae, K. J. Park, J. M.
Jang, S. J. Choi, M. H. Son, S. H. Kim, M. Yoo and B. J. Lee, Bioorg. Med. Chem. Lett.,
2011, 21 (12), 3809-3812.
[87] …………K. S. Lim, J. Y. Cho, B. H. Kim, J. R. Kim, H. S. Kim, D. K. Kim, S. H. Kim, H. J. Yim,
S. H. Lee, S. G. Shin, I. J. Jang and K. S. Yu, Br. J. Clin. Pharmacol., 2009, 68 (6), 883-
890.
- Originator Dong-A Pharmaceutical
- Developer Dong-A ST
- Class Amides; Antihyperglycaemics; Fluorobenzenes; Piperazines; Small molecules
- Mechanism of Action CD26 antigen inhibitors
- Orphan Drug Status No
- On Fast track No
- New Molecular Entity Yes
- Available For Licensing Yes – Type 2 diabetes mellitus
Highest Development Phases
- Phase III Type 2 diabetes mellitus
Most Recent Events
- 01 Sep 2014 Phase-I clinical trials in Type-2 diabetes mellitus (In volunteers) in United Kingdom (PO)
- 31 Jul 2014 Phase-III clinical trials in Type-2 diabetes mellitus in South Korea (PO)
- 31 Jul 2014 Dong-A ST initiates enrolment in a phase I trial in patients with renal impairment in South Korea (NCT02214693)
…………………………………..
WO 2010114291
http://www.google.co.in/patents/WO2010114291A2?cl=en
Formula 1

Korea Patent Publication No. 2008-0094604 the call to the scheme, as indicated by A Ⅰ) of formula (II) beta-compound of formula 3 is already substituted heterocyclic compound having 1-hydroxy-benzotriazole group (HOBT) 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide (EDC) and reacting with a tertiary amine to prepare a compound of formula (4) connected by peptide bonds; Ⅱ) beta comprises the step of reacting under acidic conditions a compound of the formula (4) – a method of manufacturing the heterocyclic compounds of the formula I having an amino group is disclosed.
– Scheme A]

(Wherein, PG is a protecting group.)
In this case, the beta of the formula (2) of Scheme A – a compound having an amino group is prepared in addition to the DPP-IV inhibitor International Publication represented by Formula 1 WO03 / 000181, WO03 / 004498, WO03 / 082817, WO04 / 007468, WO04 / 032836, WO05 / 011581, WO06 / 097175, WO07 / 077508, WO07 / 063928, WO08 / 028662 WO08 / it may be used for the production of different DPP-IV inhibitors according 087,560 and can be prepared in a number of ways.
To, the compound of Formula 2 is an example as shown in Scheme J. Med.Chem. 2005; 141, and Synthesis 1997; it can be produced by the known method described in 873.

Specifically, (2S) – (+) – 2,5- dihydro-3,6-dimethoxy-2-isopropyl-pyrazine 2,4,5-trifluoro-react with benzyl bromide and acid treatment, and then the amine an ester compound obtained by the protection reaction. Ester compounds are hydrolyzed to re-3- (2,4,5-trifluoro-phenyl) -2-amino-propionic acid tert such as isobutyl chloroformate, triethylamine or diisopropylethylamine to give the amine, and then using diazomethane to form a diazo ketone, and then may be prepared by reaction with silver benzoate. However, the reaction can be performed at low temperature (-78 ℃) or high alpha-amino acid to purchase and use, and may have a risk of problems such as the need to use large diazomethane.
To a different process for preparing a compound of Formula 2 as shown in scheme Tetrahedron: Asymmetry 2006; It is known in 2622; 205 or similarly Bioorganic & Medicinal Chemistry Letters 2007.

That is, a 1,1′-carbonyl-2,4,5 which the phenyl trifluoroacetic acid activated using the following imidazole mono-methyl words potassium carbonate is reacted with the beta-keto ester compound is prepared. This produced an enamine ester using ammonium acetate and ammonium solution, the ester compound chloro (1,5-cyclooctadiene) rhodium (I) dimer using a chiral ferrocenyl ligands I the reaction of the high-pressure hydrogen with a chiral primary amine with a beta-amino ester compound after production and can lead to hydrolysis to prepare a compound of formula (2). However, use of expensive metal catalyst has a problem that must be performed in high pressure hydrogenation.
The method for preparing a compound of Formula 2 is disclosed in International Publication No. WO 04/87650.

Specifically, 2,4,5-fluorophenyl reagent is oxalyl chloride, the acid activated acid with 2,2-dimethyl-1,3-dioxane-4,6-dione, and after the reaction of methanol and the resulting material at reflux to prepare a corresponding compound. With a selective reducing reagents which enantiomers (S) -BINAP-RuCl 2 and hydrogen through a reaction (S) – producing a compound having coordinated to each other, it again after the decomposition, and the singer O- benzyl hydroxyl amine and the coupling reaction and the intermediate is prepared. To do this, the resulting intermediate tree azodicarboxylate and diisopropyl azodicarboxylate presence ring condensation reaction, treated with an aqueous solution of lithium hydroxide to (R) – while having the formula (II) coordinated to the amine group protected with a benzyl-O- the compound can be produced. However, the method has a problem as a whole to be prepared by the reaction yield to be low and a long processing time to perform the reaction.
Thus, the conventional known method for producing a compound of the general formula (2) has the disadvantage of using expensive reagents, or not suitable for commercial mass-production method by a long synthesis time yield is also low.
In addition, the compound represented by General Formula (3), as described in Korea Patent Publication No. 2008-0094604 call, can be prepared by way of reaction schemes.

Specifically, the starting material D- serine methyl ester is substituted by a hydroxy group when reflux again substituted by trityl chloride as methoxy groups converted to the aziridine compound.
[Scheme 3]

<Example 3> (R)-4-[(R)-3-아미노-4-(2,4,5-트리플루오로페닐)부타노일]-3-(t-부톡시메틸)피페라진-2-온(화학식 1) Preparation of the hydrochloride
Step 1: t- butyl (R)-4-[(R)-2-(t-부톡시메틸)-3-옥소피페라진-1-일]-4-옥소 – 1-(2,4,5-트리플루오로페닐)부탄-2-일카르바메이트(화학식 Preparation of 4)
2 L flask, prepared in Example 1 (R) -3-t- butoxycarbonyl-4- (2,4,5-trifluoro-phenyl) butanoate acid (Formula 2) 10.0 g of toluene was dissolved in 450 mL of bis (2,2′-benzothiazolyl) disulfide 13.0 g, was cooled and then 10.2 g triphenylphosphine was added to the reaction solution at 0 ℃. While stirring the reaction mixture was added to a solution of 0.8 mL of triethylamine in 20 mL of toluene was stirred at room temperature for 5 hours. The reaction mixture was cooled to 0 ℃ and prepared in Example 2 (R) -3- (t- butoxymethyl) piperazin-2-one (Formula 3) was dissolved in 5.6 g of toluene and 40 mL pyridine a 2.4 mL was added slowly. After 30 minutes the reaction mixture was heated to room temperature and stirred for 1 hour. Saturated sheet to be the aqueous acid solution to a pH of 2.5 and then diluted with ethyl acetate 400 mL. Washed twice with brine and the organic layer was dehydrated with magnesium sulfate and concentrated. The residue was purified by column chromatography to give the title compound 838 mg.
1 H NMR (400 MHz, CDCl 3) δ 7.03 (m, 1H), 6.88 (m, 1H), 5.97 (m, 1H), 5.48 (m, 1H), 4.16 ~ 4.07 (m, 1H), 4.02 ~ 3.91 (m, 1H), 3.74 (m, 2H) 3.37 (m, 2H), 3.24 (m, 1H), 2.92 (m, 2H), 2.80 (m, 1H), 2.59 (m, 2H), 1.34 ( d, 9H), 1.13 (s, 9H)
Step 2: (R) -4 – [(R) -3- amino-4- (2,4,5-trifluoro-phenyl) butane five days] -3- (t- butoxymethyl) piperazin-2- on the production of (I) hydrochloride
Prepared in Step 1 t- butyl (R)-4-[(R)-2-(t-부톡시메틸)-3-옥소피페라진-1-일]-4-옥소-1-(2,4,5-트리플루오로페닐)부탄-2-일카르바메이트 97 mg was dissolved in methanol was added 3 mL 2N- hydrochloric acid / diethyl ether 2 mL was stirred at room temperature for 3 hours. The reaction mixture was concentrated and dried under reduced pressure to give 64 mg of the title compound as a foaming solid.
1 H NMR (400 MHz, CD 3 OD) δ 7.37 (m, 1H), 7.23 (m, 1H), 4.80 (m, 1H), 4.59 ~ 4.40 (m, 1H), 3.93 (m, 1H), 3.90 ~ 3.83 (m, 2H), 3.70 (m, 1H), 3.38 (m, 2H), 3.27 (m, 1H), 3.07 (m, 2H), 2.89 ~ 2.66 (m, 2H), 1.18 (s, 3H ), 1.11 (s, 6H)
Mass (M + 1): 402
<Example 4> (R)-4-[(R)-3-아미노-4-(2,4,5-트리플루오로페닐)부타노일]-3-(t-부톡시메틸)피페라진-2-온(화학식 1) tartaric acid salts
Step 1: (R) -4 – [(R) -3- amino-4- (2,4,5-trifluoro-phenyl) butane five days] -3- (t- butoxymethyl) piperazin-2- Preparation of one (I)
Example 3 to give a compound of formula I in hydrochloride 60 mg 5% sodium hydrogen carbonate in dichloromethane was added to 10 mL of an aqueous solution / 2-propanol (4/1 (v / v)) was added to the mixed solution and extracted two times 10 mL The organic layer was dried under reduced pressure to give 55 mg of the title compound as a solid.
1 H NMR (400 MHz, CD 3 OD) δ 7.27 (m, 1H), 7.14 (m, 1H), 4.56 ~ 4.39 (m, 1H), 3.96 ~ 3.81 (m, 3H), 3.70 (m, 1H) , 3.46 (m, 1H), 3.43 ~ 3.32 (m, 1H), 2.83 ~ 2.65 (m, 3H), 2.58 ~ 2.40 (m, 2H), 1.16 (s, 3H), 1.11 (s, 6H)
Mass (M + 1): 402
Step 2: (R) -4 – [(R) -3- amino-4- (2,4,5-trifluorophenyl) butanoyl] -3- (t- butoxymethyl) piperazin-2- one (I) tartaric acid salt [
Was dissolved 55 mg of the compound of step 1 in 0.56 mL of acetone, L- tartrate 26 mg ethanol / water (9/1 (v / v)) was added slowly to a solution of 0.35 mL was stirred for 30 minutes. Here was added 0.56 mL of 2-propanol was stirred for 10 minutes and re-filtered to give 77 mg of the title compound as a solid.
1 H NMR (400 MHz, CD 3 OD) δ 7.38 (m, 1H), 7.22 (m, 1H), 4.80 (m, 1H), 4.59 ~ 4.40 (m, 1H), 4.40 (s, 2H), 3.93 (m, 1H), 3.90 ~ 3.83 (m, 2H), 3.70 (m, 1H), 3.38 (m, 2H), 3.27 (m, 1H), 3.07 (m, 2H), 2.89 ~ 2.66 (m, 2H ), 1.15 (s, 3H), 1.11 (s, 6H)
Mass (M + 1): 402
………………………………
WO 2010114292
http://www.google.com/patents/WO2010114292A2?cl=en
…………………………………
Discovery of DA-1229: a potent, long acting dipeptidyl peptidase-4 inhibitor for the treatment of type 2 diabetes
Bioorg Med Chem Lett 2011, 21(12): 3809
http://www.sciencedirect.com/science/article/pii/S0960894X11004859

A series of β-amino amide containing substituted piperazine-2-one derivatives was synthesized and evaluated as inhibitors of dipeptidyl pepdidase-4 (DPP-4) for the treatment of type 2 diabetes. As results of intensive SAR study of the series, (R)-4-[(R)-3-amino-4-(2,4,5-trifluorophenyl)-butanoyl]-3-(t-butoxymethyl)-piperazin-2-one (DA-1229) displayed potent DPP-4 inhibition pattern in several animal models, was selected for clinical development.
http://www.luye.cn/en/uploads//2014-07/21/_1405936452_zr21xh.pdf
Dong-A ST has licensed its new diabetes drug Evogliptin to 17 Latin American countries including Mexico, Venezuela, Argentina, Chile, Colombia, Ecuador, Peru, the Dominican Republic, and Uruguay, Jung Jae-wook, Dong-A ST’s PR manager, told Business Korea.
Dong-A ST and Eurofarma, a Brazilian pharmaceutical company, concluded the licensing contract at Dong-A ST’s headquarters on April 13 in Seoul.
Eurofarma will be responsible for Evogliptin’s product development and sales in the 17 Latin American countries, Dong-A ST said. Dong-A ST will receive royalties from Eurofarma, and export the raw material of the medicine.
Dong-A ST has been developing Evogliptin with the support of the Ministry of Health & Welfare of South Korea as an innovative new medicine research project since May 2008. Evogliptin is a DPP-4 remedy based on the inhibition mechanism which is “excellent” at reducing blood sugar, whilst “less likely” to cause weight increases and hypoglycemia, the company said.
Park Chan-il, president of Dong-A ST, said that Dong-A ST will pursue further out-licensing “over the globe,” through continuous investment in research and development.
Maurizio Billi, Eurofarma’s president, wished to expand both companies’ partnership in the innovative new remedy development sector, according to Dong-A ST.
Last July, Dong-A ST and Eurofarma concluded a contract out-licensing Evogliptin to Brazil itself, the company said.
see gliptins at…..http://drugsynthesisint.blogspot.in/p/gliptin-series.html
Dong-A Pharm. Co., Ltd, Yongin-si, Gyeonggi-do, Republic of Korea.




GOSOGLIPTIN
GOSOGLIPTIN
CAS 869490-23-3 FREE BASE
DIHYDROCHLORIDE..869490-47-1
GOSOGLIPTIN; UNII-GI718UO477; PF-00734200; PF-734200;
(3,3-difluoropyrrolidin-1-yl)-[(2S,4S)-4-(4-pyrimidin-2-ylpiperazin-1-yl)pyrrolidin-2-yl]methanone
| Molecular Formula: | C17H24F2N6O |
|---|---|
| Molecular Weight: | 366.408866 g/mol |
| Company | Pfizer Inc. |
| Description | Dipeptidyl peptidase-4 (DPP-4) inhibitor |
| Molecular Target | Dipeptidyl peptidase-4 (DPP-4) (CD26) |
| Mechanism of Action | Dipeptidyl peptidase-4 (DPP-4) inhibitor |
| Latest Stage of Development | Phase II |
| Standard Indication | Diabetes |
| Indication Details | Treat Type II diabetes |
Type 2 diabetes mellitus is a chronic disorder characterized by hyperglycemia coupled with a gradual decline in insulin sensitivity and insulin secretion. The incretin hormone glucagon-like peptide-1 (GLP-1), which is released post-prandially from the L-cells of the intestine, stimulates the release of insulin from pancreatic β-cells. However, GLP-1 is rapidly degraded in vivo by peptidases, including dipeptidyl peptidase IV (DPP-4), which is a widely distributed serine protease that specifically cleaves N-terminal dipeptides from polypeptides with proline or alanine at the penultimate position.
In vivo administration of DPP-4 inhibitors to human subjects results in higher circulating concentrations of endogenous GLP-1 and subsequent decrease in plasma glucose. Long term treatment with a DPP-4 inhibitor leads to a reduction in circulating HbA1c (glycosylated hemoglobin). DPP-4 inhibition also offers the potential to improve the insulin producing function of the pancreas through either β-cell preservation or regeneration. Therefore, DPP-4 inhibition has emerged as a promising new treatment of Type 2 diabetes
PF-734200 is a potent, selective, orally active dipeptidyl peptidase IV inhibitor. It had been in phase II clinical development at Pfizer for the treatment of type 2 diabetes; however, in 2010 the company discontinued these trials. In 2012, the product was licensed to SatRx, a spin-off of the ChemRar High Tech Center, by Pfizer on an exclusive worldwide basis (with the exception of China) for the development and commercialization as monotherapy or in combination with other therapies for the treatment of type 2 diabetes. SatRx is conducting phase II clinical trials for the treatment of type 2 diabetes.
……………………….
PAPER
New synthetic route to a dipeptidyl peptidase-4 inhibitor
Org Process Res Dev 2012, 16(3): 409
http://pubs.acs.org/doi/abs/10.1021/op200309z

A new synthetic route to a dipeptidyl peptidase-4 (DPP4) inhibitor was developed and demonstrated on a multigram scale. This approach takes advantage of the cheap and readily available Boc-trans-4-hydroxy-l-proline methyl ester as starting material which was derivatized through an SN2 reaction. Several leaving groups were studied, and the nosylate group showed superiority over other derivatives. Formation of an amide using the most costly starting material, 3,3-difluoropyrrolidine, was performed late in the synthesis to minimize its economical impact on the overall cost of the API.
(3,3-Difluoropyrrolidin-1-yl)-(2S,4S)-4-(4-(pyrimidin-2-yl)piperazin-1-yl)pyrrolidin-2-yl)methanone.FREE BASE
Mp 149 °C (decomp).
[α]d = −31.1 (T = 24 °C, c = 1, CHCl3). Specific rotation of product 4 prepared using the initial route: [α]d = −31.5 (T = 24 °C, c = 1, CHCl3).
1H NMR (400 MHz; CDCl3) δ 8.30 (d, J = 4 Hz, 2H), 6.48 (t, J = 4 Hz, 1H), 3.95–3.6 (m, 9H), 3.25–2.85 (m, 4H), 2.6–2.25 (m, 7H), 1.75–1.6 (m, 1H).
13C NMR (100 MHz; CDCl3) δ 172.28; 161.55; 157.70; 127.22 (t, 1J C–F = 248 Hz), 126.22 (t, 1J C–F = 246 Hz), 109.95; 66.54; 58.87; 57.99; 52.71 (t, 2 J C–F = 32 Hz); 52.00; 50.41; 43.03; 34.46, 34.37, 34.25; 19F NMR (377 MHz, CDCl3) δ −102.1 (m, 2F).
IR (neat): 2951w, 2864w, 2799w, 2759w, 1630s, 1585vs, 1547m, 1449m, 1172m, 1254m, 1129m, 982w, 923m, 796m, 638w.
HRMS (ES, N2) Calcd for C17H24F2N6O: 367.20524, found: 367.20592.
……………………….
PAPER
(3,3-difluoro-pyrrolidin-1-yl)-((2S,4S)-(4-(4-pyrimidin-2-yl-piperazin-1-yl)-pyrrolidin-2-yl)-methanone: A potent, selective, orally active dipeptidyl peptidase IV inhibitor
Bioorg Med Chem Lett 2009, 19(7): 1991
http://www.sciencedirect.com/science/article/pii/S0960894X09001966?np=y
- Pfizer Global Research & Development, Groton/New London Laboratories, Pfizer Inc, Groton, CT 06340, United States
A series of 4-substituted proline amides was evaluated as inhibitors of dipeptidyl pepdidase IV for the treatment of type 2 diabetes. (3,3-Difluoro-pyrrolidin-1-yl)-[(2S,4S)-(4-(4-pyrimidin-2-yl-piperazin-1-yl)-pyrrolidin-2-yl]-methanone (5) emerged as a potent (IC50 = 13 nM) and selective compound, with high oral bioavailability in preclinical species.

………………….
PATENT
WO 2005116014
http://www.google.co.in/patents/WO2005116014A1?cl=en
Example 113 (3.3-Difluoropyrrolidin-1-yl)-((2S,4S)-4-(4-(pyrimidin-2-yl)piperazin-1-yl)pyrrolidin-2-yl)-methanone

Step 1 – (S)-2-(3.3-Difluoro-pyrrolidine-1-carbonyl)-4-oxo-pyrrolidine-1 -carboxylic acid tert-butyl ester
(S)-4-Oxo-pyrrolidine-1 ,2-dicarboxylic acid 1-tert-butyl ester (6.6 kg, 1.0 equivalent) was charged to a reactor, followed by addition of dichloromethane (15 volumes). The reaction mixture was cooled to 0°C. Triethylamine (4.82 liters, 1.2 equiv) was added over 30 minutes. The mixture turned from suspension to a clear solution at the end of triethylamine addition. The mixture was held at 0°C to 5°C for 10 minutes. Pivaloyl chloride (3.65 kg, 1.05 equivalents) was added slowly while keeping the reaction temperature at 0°C to 5°C. The reaction mixture turned back to aslurry. The reaction mixture was sampled for completion by HPLC (using diethylamine to derivatize) after held for 1 hour at 0°C to 5°C.
3,3-Difluoro- pyrrolidine hydrochloride (4.13 kg, 1.0 equivalent) was charged to the above mixture over 10 minutes at – 10°C to 0°C. Triethylamine (4.0 liters, 1.0 equiv) was introduced slowly over 70 minutes at -10°C to 0°C. Upon completion of triethylamine addition, the mixture was stirred for 1h at 0 to 5°C. The reaction was complete by HPLC assay (-1% starting material). The reaction was quenched with water (10 volumes) at 0°C to 5 °C. The mixture was heated to 20°C to 25 °C. The layers were separated, and the organic layer was washed with 0.5 M HCI (5 volumes). The organic layer was again washed with combined 5% NaHC03 (2 volumes) and half saturated brine solution (1.64 M, 3 volumes). The organic solution was concentrated atmospherically to a low stirrable volume (approximately 20 liters). Ethyl acetate (12.6 volumes, 82.8 liters) was added, the solution was concentrated atmospherically to -6 volumes. The mixture was held at 60°C to 65 °C for 2 hours and cooled to room temperature over 3 hours. The mixture was held at 20°C to 25 °C for 8 hours. Heptane (8 volumes) was added, and the mixture was granulated for a minimum of 2 hours. The solid was filtered, rinsed with 2:1 heptane/ethyl acetate (1 volume), and dried in a tray dryer at 25°C to 35°C for a minimum of 12 h. Yield: 7.26 kg, 79%. HPLC purity: 99.7%. The mother liquor (86 liters) was concentrated to 12 liters under partial vacuum at 65°C to 70°C. The mixture was cooled to 60°C to 65 °C. Ethyl acetate (4.0 liters) was added slowly over 15 minutes. The mixture was cooled to 20°C to 25 °C over 2 hours and was held at that temperature for at least 2 hours. The solid was filtered and rinsed with heptane/ethyl acetate (3:1 v/v, 1.7 liters). Drying in a tray dryer for 12 hours at 35°C to 45 °C yielded 435 grams of product. HPLC purity: 96.4%.
Step 2 – (2S.4S)-2-(3.3-Dif luoro-pyrrolidine-1 -carbonyl)-4-(4-pyrimidin-2-yl-piperazin-1 -yl)-pyrrolidine-1 – carboxylic acid tert-butyl ester A reactor was charged with THF (20 volumes), 2-piperazin-1-yl-pyrimidine (2.17 kg, 1.05 equivalents) and the product from Step 1 (4.00 kg, 1.0 equivalent). The mixture was held at 20°C to 25°C until all material was dissolved over 30 minutes. Acetic acid (0.792 kg, 1.05 equivalents) as added. The mixture was stirred for 1 hour during which the reaction mixture turned to cloudy. The reaction mixture was refluxed for 30 minutes and then concentrated at 60°C to 70°C until a steady temperature of 66.9°C was observed in the overheads indicating complete removal of water from the system. More THF was added as necessary. At the end, THF was added to bring the total volume in the reactor to 15 volumes of the limit reagent. The reaction mixture was cooled to -3°C to 7°C and sampled for complete formation of imine by HPLC (using sodium triacetoxyborohydride to reduce imine). Sodium triacetoxyborohydride (5.33 kg, 2.0 equivalents) was added portion-wise to the suspension at -5°C to 15°C. The reaction mixture was heated to 20°C to 25°C and held for 12 hours. HPLC results confirmed the reaction was complete by 99.8%. Sodium bicarbonate aqueous solution (10% w/w, 10 volumes) was added. The slurry was concentrated to remove 10 volumes of THF under partial vacuum at 30°C to 60°C. Ethyl acetate (10 volumes) was added to the suspension after it cooled to 20°C to 25CC. The organic phase was separated and the aqueous phase was checked by HPLC. It contained less than 2% of the product. The organic phase was washed with water (5 volumes), saturated brine solution (5 volumes) and concentrated to a small volume (2 volumes) under partial vacuum at 45°C to 50°C. To the slurry was added heptane (10 volumes) at 45°C to 50°C over 30 minutes. The mixture was cooled to 20°C to 25°C and granulated for 2 hours. Solid was collected by filtration, rinsed with heptane (2 volumes). Drying in a tray dryer for 12 hours at 35°C to 45°C yield 5.35 kg (91.3%) of the product. Step 3 – (3.3-Dif luoro-pyrrolidin-1 -yl)-f(2S.4S)-4-(4-pyrimidin-2-yl-piperazin-1 -yl)-pyrrolidin-2-yll- methanone Water (19 liters, 2 volumes) was charged to a reactor followed by the product from Step 2 (9.57 kg,
1.0 equivalent). To the slurry was added concentrated HCI (37 wt% in water, 19.1 liters, 2 volumes) slowly at 20°C to 30°C over 4 hours. The slurry went into solution after 12 liters of HCI was added. After the addition completion, the reaction was complete by HPLC assay. The reaction mixture was cooled to 5°C to 15°C. To the mixture was added 50% NaOH aqueous solution slowly with agitation to pH 10 to pH 11. The pH was monitored with a pH meter closely during the neutralization. The total volume of 50% NaOH added was 12.45 liters. The mixture was warmed to 20°C to 25°C and extracted with ethyl acetate twice (115 liters, 12 volumes and 57 liters, 6 volumes, respectively). The sample from aqueous layer after second extraction was analyzed by HPLC and showed only 1% of the product in that aqueous solution.
The organic layers were combined and treated with magnesium sulfate (5 kg) for 1 hour. The mixture was filtered. The filter cake was rinsed with ethyl acetate (10 liters). The filtrate was charged back to the reactor via a 0.2 micron in-line filter for speck free operation. (The following operations were performed under speck free conditions.) The solution was concentrated to 20 liters (2 volumes) under partial vacuum at 50°C to 60°C. The mixture was cooled to 20°C to 25°C over 30 minutes. Upon cooling to room temperature, crystallization occurred. The mixture was held for 30 minutes. Hexanes (20 liters, 2 volumes) was added slowly over 1 hour. The mixture was granulated for 2 hours. The solid product was collected by filtration and rinsed with hexanes/ethyl acetate (10 liters, 1 :1 v/v). The filter was blown dry with nitrogen for a minimum of 2 hours. The product was dried in a tray dryer at 44°C for 12 hours.
Yield: 5.7 kg, 75.9%.
m.p. 156°C. MS m/z 367 (MH+).
FREE BASE
1H NMR (400 MHz, D20): δ 8.15 (d, 2H, J = 5.0 Hz, CH of pyrimidine), 6.55 (t, 1 H, J = 4.8 Hz, CH of pyrimidine), 3.87-3.81 (dd, 1 H, H2b of proline, rotomeric), 3.78-3.50 (m, 4H, N-CH2 of pyrrolidide), 3.55-3.40 (m, 4H, N-CH2 of piperazine), 2.97 (dd, 1 H, J = 10.2, 6.6 Hz, H5a of proline), 2.85-2.75 (m, 1 H, H4b of proline), 2.69 (dd, 1 H, J = 10.0, 9.1 Hz, H5b of proline), 2.55-2.20 (m, 7H, overlapping N-CH2 of piperazine, CH2 of pyrrolidide and H3b of proline), 1.47-1.38 (m, 1 H, H3a of proline).
Alternatively, the dihydrochloride salt of the titled compound was prepared according to the method of Example 1.
………………
US 2005/0256310
http://www.google.com/patents/US20050256310

This approach begins with N–t-Boc-4-oxo-l-proline (1) that undergoes a mixed anhydride activation with pivaloyl chloride at 0 °C, followed by amidation with 3,3-difluoropyrrolidine to yield the intermediate 2. Reductive amination with 1-(2-pyrimidyl)piperazine using sodium triacetoxyborohydride in THF/AcOH provided the desired stereoisomer 3 in high yield and selectivity, the undesired diastereomer being completely removed by crystallization. Deprotection of 3 with 6 N HCl, followed by neutralization with 50% NaOH and extraction provided PF-734200 (4) in good yield.
EXAMPLE 113 (3,3-Difluoropyrrolidin-1-yl)-((2S,4S)-4-(4-(pyrimidin-2-yl)piperazin-1-yl)pyrrolidin-2-yl)-methanone

Step 1—(S)-2-(3,3-Difluoro-pyrrolidine-1-carbonyl)-4-oxo-pyrrolidine-1-carboxylic acid tert-butyl
(S)-4-Oxo-pyrrolidine-1,2-dicarboxylic acid 1-tert-butyl ester (6.6 kg, 1.0 equivalent) was charged to a reactor, followed by addition of dichloromethane (15 volumes). The reaction mixture was cooled to 0° C. Triethylamine (4.82 liters, 1.2 equiv) was added over 30 minutes. The mixture turned from suspension to a clear solution at the end of triethylamine addition. The mixture was held at 0° C. to 5° C. for 10 minutes. Pivaloyl chloride (3.65 kg, 1.05 equivalents) was added slowly while keeping the reaction temperature at 0° C. to 5° C. The reaction mixture turned back to a slurry. The reaction mixture was sampled for completion by HPLC (using diethylamine to derivatize) after held for 1 hour at 0° C. to 5° C. 3,3-Difluoro-pyrrolidine hydrochloride (4.13 kg, 1.0 equivalent) was charged to the above mixture over 10 minutes at −10° C. to 0° C. Triethylamine (4.0 liters, 1.0 equiv) was introduced slowly over 70 minutes at −10° C. to 0° C. Upon completion of triethylamine addition, the mixture was stirred for 1 h at 0 to 5° C. The reaction was complete by HPLC assay (˜1% starting material). The reaction was quenched with water (10 volumes) at 0° C. to 5 ° C. The mixture was heated to 20° C. to 25 ° C. The layers were separated, organic layer was washed with 0.5 M HCl (5 volumes). The organic layer was again washed with combined 5% NaHCO3 (2 volumes) and half saturated brine solution (1.64 M, 3 volumes). The organic solution was concentrated atmospherically to a low stirrable volume (approximately 20 liters). Ethyl acetate (12.6 volumes, 82.8 liters) was added, the solution was concentrated atmospherically to ˜6 volumes. The mixture was held at 60° C. to 65° C. for 2 hours and cooled to room temperature over 3 hours. The mixture was held at 20° C. to 25 ° C. for 8 hours. Heptane (8 volumes) was added, and the mixture was granulated for a minimum of 2 hours. The solid was filtered, rinsed with 2:1 heptane/ethyl acetate (1 volume), and dried in a tray dryer at 25° C. to 35° C. for a minimum of 12 h. Yield: 7.26 kg, 79%. HPLC purity: 99.7%. The mother liquor (86 liters) was concentrated to 12 liters under partial vacuum at 65° C. to 70° C. The mixture was cooled to 60° C. to 65° C. Ethyl acetate (4.0 liters) was added slowly over 15 minutes. The mixture was cooled to 20° C. to 25° C. over 2 hours and was held at that temperature for at least 2 hours. The solid was filtered and rinsed with heptane/ethyl acetate (3:1 v/v, 1.7 liters). Drying in a tray dryer for 12 hours at 35° C. to 45° C. yielded 435 grams of product. HPLC purity: 96.4%.
Step 2—(2S,4S)-2-(3,3-Difluoro-pyrrolidine-1-carbonyl)-4-(4-pyrimidin-2-yl-piperazin-1-yl)-pyrrolidine-1-carboxylic acid tert-butyl ester
A reactor was charged with THF (20 volumes), 2-piperazin-1-yl-pyrimidine (2.17 kg, 1.05 equivalents) and the product from Step 1 (4.00 kg, 1.0 equivalent). The mixture was held at 20° C. to 25° C. until all material was dissolved over 30 minutes. Acetic acid (0.792 kg, 1.05 equivalents) as added. The mixture was stirred for 1 hour during which the reaction mixture turned to cloudy. The reaction mixture was refluxed for 30 minutes and then concentrated at 60° C. to 70° C. until a steady temperature of 66.9° C. was observed in the overheads indicating complete removal of water from the system. More THF was added as necessary. At the end, THF was added to bring the total volume in the reactor to 15 volumes of the limit reagent. The reaction mixture was cooled to −3° C. to 7° C. and sampled for complete formation of imine by HPLC (using sodium triacetoxyborohydride to reduce imine). Sodium triacetoxyborohydride (5.33 kg, 2.0 equivalents) was added portion-wise to the suspension at −5° C. to 15° C. The reaction mixture was heated to 20° C. to 25° C. and held for 12 hours. HPLC results confirmed the reaction was complete by 99.8%. Sodium bicarbonate aqueous solution (10% w/w, 10 volumes) was added. The slurry was concentrated to remove 10 volumes of THF under partial vacuum at 30° C. to 60° C. Ethyl acetate (10 volumes) was added to the suspension after it cooled to 20° C. to 25° C. The organic phase was separated and the aqueous phase was checked by HPLC. It contained less than 2% of the product. The organic phase was washed with water (5 volumes), saturated brine solution (5 volumes) and concentrated to a small volume (2 volumes) under partial vacuum at 45° C. to 50° C. To the slurry was added heptane (10 volumes) at 45° C. to 50° C. over 30 minutes. The mixture was cooled to 20° C. to 25° C. and granulated for 2 hours. Solid was collected by filtration, rinsed with heptane (2 volumes). Drying in a tray dryer for 12 hours at 35° C. to 45° C. yield 5.35 kg (91.3%) of the product.
Step 3—(3,3-Difluoro-pyrrolidin-1-yl)-[(2S,4S)-4-(4-pyrimidin-2-yl-piperazin-1-yl)-pyrrolidin-2-yl]-methanone
Water (19 liters, 2 volumes) was charged to a reactor followed by the product from Step 2 (9.57 kg, 1.0 equivalent). To the slurry was added concentrated HCl (37 wt % in water, 19.1 liters, 2 volumes) slowly at 20° C. to 30° C. over 4 hours. The slurry went into solution after 12 liters of HCl was added. After the addition completion, the reaction was complete by HPLC assay. The reaction mixture was cooled to 5° C. to 15° C. To the mixture was added 50% NaOH aqueous solution slowly with agitation to pH 10 to pH 11. The pH was monitored with a pH meter closely during the neutralization. The total volume of 50% NaOH added was 12.45 liters. The mixture was warmed to 20° C. to 25° C. and extracted with ethyl acetate twice (115 liters, 12 volumes and 57 liters, 6 volumes, respectively). The sample from aqueous layer after second extraction was analyzed by HPLC and showed only 1% of the product in that aqueous solution. The organic layers were combined and treated with magnesium sulfate (5 kg) for 1 hour. The mixture was filtered. The filter cake was rinsed with ethyl acetate (10 liters). The filtrate was charged back to the reactor via a 0.2 micron in-line filter for speck free operation. (The following operations were performed under speck free conditions.) The solution was concentrated to 20 liters (2 volumes) under partial vacuum at 50° C. to 60° C. The mixture was cooled to 20° C. to 25° C. over 30 minutes. Upon cooling to room temperature, crystallization occurred. The mixture was held for 30 minutes. Hexanes (20 liters, 2 volumes) was added slowly over 1 hour. The mixture was granulated for 2 hours. The solid product was collected by filtration and rinsed with hexanes/ethyl acetate (10 liters, 1:1 v/v). The filter was blown dry with nitrogen for a minimum of 2 hours. The product was dried in a tray dryer at 44° C. for 12 hours.
Yield: 5.7 kg, 75.9%. m.p. 156° C. MS m/z 367 (MH+).
1H NMR (400 MHz, D2O): δ 8.15 (d, 2H, J=5.0 Hz, CH of pyrimidine), 6.55 (t, 1H, J=4.8 Hz, CH of pyrimidine), 3.87-3.81 (dd, 1H, H2b of proline, rotomeric), 3.78-3.50 (m, 4H, N—CH2 of pyrrolidide), 3.55-3.40 (m, 4H, N—CH2 of piperazine), 2.97 (dd, 1H, J=10.2, 6.6 Hz, H5a of proline), 2.85-2.75 (m, 1H, H4b of proline), 2.69 (dd, 1H, J=10.0, 9.1 Hz, H5b of proline), 2.55-2.20 (m, 7H, overlapping N—CH2 of piperazine, CH2 of pyrrolidide and H3b of proline), 1.47-1.38 (m, 1H, H3a of proline).
Alternatively, the dihydrochloride salt of the titled compound was prepared according to the method of Example 1.
……………..
PAPER

Scheme 1.
Reagents and conditions: (a) 3,3-difluoropyrrolidine hydrochloride, EDC, HOBt, TEA, DCM, rt; (b) NaBH4, MeOH, (c) (1) trifluoromethane-sulphonyl chloride, DIPEA, DCM; (2) 2-(1-piperazinyl)pyrimidine, DCM, −10 °C; (d) 4 N HCl in dioxane, rt; (e) 2-(1-piperazinyl)pyrimidine, NaBH(OAc)3, AcOH, DCE; (f) R1R2NH hydrochloride, EDC, HOBt TEA, DCM, 0–rt; (g) N-heterocyclic piperazine, NaBH(OAc)3, AcOH, DCE.
……………………….
if image is not clear see at………..http://www.allfordrugs.com/2015/07/03/gosogliptin/
| Patent | Submitted | Granted |
|---|---|---|
| Therapeutic compounds [US7291618] | 2005-11-17 | 2007-11-06 |
| (2S,4S)-4-(piperazin-1-yl)pyrrolidine-2-methanone derivatives [US7465732] | 2007-05-03 | 2008-12-16 |
| THERAPEUTIC COMPOUNDS [US2007161664] | 2007-07-12 | |
| Therapeutic compounds [US2006079498] | 2006-04-13 |
//////////
see gliptins at…………http://drugsynthesisint.blogspot.in/p/gliptin-series.html
TENELIGLIPTIN

TENELIGLIPTIN
| Teneligliptin; 760937-92-6; UNII-28ZHI4CF9C; Teneligliptin (INN); 28ZHI4CF9C | |
| MF | C22H30N6OS |
|---|---|
| MW | 426.5782 g/mol |
Teneligliptin (INN; trade name Tenelia) is a pharmaceutical drug for the treatment of type 2 diabetes mellitus. It is approved for use in Japan.[1] It belongs to the class of anti-diabetic drugs known as dipeptidyl peptidase-4 inhibitors or “gliptins”.[2] {(2S,4S)-4-[4-(3-Methyl-1-phenyl-1H-pyrazol-5-yl)-1-piperazinyl]-2-pyrrolidinyl}(1,3-thiazolidin-3-yl)methanone
Teneligliptin was launched in Japan in 2012 by Mitsubishi Pharma and Daiichi Sankyo for the treatment of type 2 diabetes mellitus. In 2013, the indication was partially changed to include it as a combination therapy with existing oral hypoglycemic agents, such as biganides, alpha-glucosidaseinhibitors, rapid-acting insulin secretagogues, and insulin preparations, as well as sulfonylureas and thiazolidines that had been approved for the combination.
In 2014, the product was registered in KR for the treatment of type 2 diabetes mellitus.
In 2013, Mitsubishi Tanabe Pharma filed for approval in Japan for use of the compound as combination therapy for the treatment of diabetes type 2.
| CAS | 760937-92-6 |
|---|
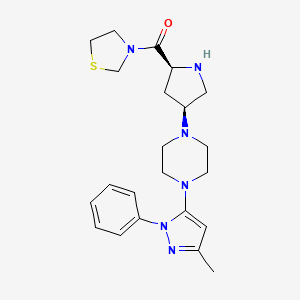
3-{(2S,4S)-4-[4-(3-methyl-l -phenyl- 1 H- pyrazol-5-yl)- l-piperazinyl]-2-pyrrolidinylcarbonyl}-l , 3-thiazolidine is represented structurally by a compound of formula (I):

Teneligliptin (CAS 760937-92-6) is a novel, potent and long-lasting dipeptidyl peptidase-4 inhibitor in treatment of type 2 diabetes. Dipeptidyl-peptidase-4 (DPP- 4) inhibitor has been demonstrated to improve glycemic control, in particular postparandial hyperglycemic control.
Despite of their common mechanism of action, DPP-4 inhibitors show marked structural heterogeneity. DPP-4 inhibitors may be classified into peptidomimetic (i.e. sitagliptin, vildagliptin, saxagliptin, and anagliptin) and non-peptidomimetic (i.e. alogliptin and linagliptin) subtypes.
Teneligliptin, is chemically known as a 3- {((2S,4S)-4-(4-(3-methyl-1-phenyl-1H-pyrazol-5-yl)piperazin-1-yl)pyrrolidin-2-yl 25 carbonyl}thiazolidine hemipentahydrobromide hydrate and is peptidomimetic with the molecular formula of C22H30N6OS.2½HBr.xH2O and molecular weight of 642.88 g/mol for hemipentahydrobromide. The hydrate can be from mono to dihydrate.
U.S. Patent No. 7,074,794 B2 (the US ‘794) discloses teneligliptin as L-proline derivative and its pharmaceutically acceptable salts which exhibits a Dipeptidyl 5 peptidase IV (DPP-IV) inhibitory activity, which is useful for the treatment or prophylaxis of diabetes, obesity, HIV infection, cancer metastasis, dermopathy, prostatic hyperplasia, periodontitis, autoimmune diseases and the like.
The example-222 of the US ‘794 discloses the process for the preparation of teneligliptin as trihydrochloride salt U.S. Patent No. 8,003,790 B2 (the US ‘790) discloses salts of proline derivative, solvate thereof and production method thereof. In particular, the US ‘790 discloses 2.0 hydrochloride or 2.5 hydrochloride; 2.0 hydrobromide or 2.5 hydrobromide, and hydrates thereof teneligliptin.
The US ‘790 B2 further discloses different salts 15 of teneligliptin which are incorporated herein as reference in their entirety U.S. PG-Pub. No. 2011/0282058 A1 discloses salts of 3-{((2S,4S)-4-(4-(3-methyl- 1-phenyl-1H-pyrazol-5-yl)piperazin-1-yl)pyrrolidin-2-ylcarbonyl}thiazolidine with mono-, di- and tri-basic acids or a solvate thereof. 20 International (PCT) publication No. WO 2012/165547 A1 discloses a process for preparation of teneligliptin and pharmaceutically acceptable salts thereof.
International (PCT) publication No. WO 2007/127635 A2 (the WO ‘635 A2) discloses a process for the preparation of diketo-piperazine and piperidine 25 derivatives. In particular, the WO ‘635 A2 discloses the process for preparation of 4-oxo-2-(thiazolidine-3-carbonyl)-pyrrolidine-1-carboxylic acid tert-butyl ester [herein compound (III)] by reacting piperazine with aryl halide.
International (PCT) publication No. WO 2012/099915 A1 (the WO ‘915 A1) 5 discloses the process for the preparation of deuterated thiazolidine derivatives. The WO ‘915 A1 also discloses the process for the preparation of 1-(3-methyl-1- phenyl-1H-pyrazol-5-yl)piperazine herein compound (V) by condensation of 5- chloro-3-methyl-1-phenyl-1H-pyrazole with piperazine.
Bioorganic & Medicinal Chemistry, 20(19), 5705-5719 (2012) discloses the process for the preparation of 1-(3-methyl-1-phenyl-1H-pyrazol-5-yl)piperazine herein compound (V) by deprotection of Boc-protected 1-(3-methyl-1-phenyl-1Hpyrazol-5-yl)piperazine with triflouroacetic acid.
U.S. Patent Nos. 7,807,676 B2 and 7,807,671 B2 discloses a process for the preparation of 1-(3-methyl-1-phenyl-1H-pyrazol-5-yl)piperazine by condensation of 5-chloro-3-methyl-1-phenyl-1H-pyrazole with piperazine in presence of n-BuLi in tetrahydrofuran. Bioorganic & Medicinal Chemistry, 14(11), 3662-3671 (2006),
Bioorganic & Medicinal Chemistry, 20(16), 5033-5041 (2012) and U.S. Patent Nos. 7,807,676 B2 and 7,807,671 B2 discloses a process for the preparation of (2S,4R)-tert-butyl 4-hydroxy-2-(thiazolidine-3-carbonyl)pyrrolidine-1-carboxylate by reacting (2S,4R)-1-(tert-butoxycarbonyl)-4-hydroxypyrrolidine-2-carboxylic acid with 25 thiazolidine in presence of HOBT and EDC.HCl in dimethylformamide solvent.
Bioorganic & Medicinal Chemistry, 15(2), 641-655 (2007) discloses a process for the preparation of (2S,4R)-tert-butyl 4-hydroxy-2-(thiazolidine-3- carbonyl)pyrrolidine-1-carboxylate by treating (2S,4S)-tert-butyl 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-(3-thiazolidinylcarbonyl)pyrrolidine-1- carboxylate with tetrabutylammonium fluoride in tetrahydrofuran.
Bioorganic & Medicinal Chemistry, 20(19), 5705-5719 (2012) discloses the 5 process for the preparation of herein compound (II) after by reacting 1-(3-methyl- 1-phenyl-1H-pyrazol-5-yl)piperazine herein compound (V) with (2S,4R)-tert-butyl 4-hydroxy-2-(thiazolidine-3-carbonyl)pyrrolidine-1-carboxylate in presence of sodium triacetoxyborohydride. There is provided different alternative processes for the preparation of teneligliptin and intermediates thereof.
Bioorganic & Medicinal Chemistry, 20(19), 5705-5719 (2012) also discloses the process for the preparation of 4-[4-(5-methyl-2-phenyl-2H-pyrazol-3-yl)-piperazin- 1-yl]-2-(thiazolidine-3-carbonyl)pyrrolidine-1-carboxylic acid tert-butyl ester [herein compound (II)] after by reacting 1-(3-methyl-1-phenyl-1H-pyrazol-5- 15 yl)piperazine [herein compound (V)] with (2S,4S)-tert-butyl 4-[[(1,1- dimethylethyl)dimethylsilyl]oxy]-2-(3-thiazolidinylcarbonyl)pyrrolidine-1- carboxylate in presence of trifluoromethylsulfonic anhydride and diisopropylethylamine. 3 – [[(2S, 4S) -4- [4- (3- methyl-1-phenyl–1H- pyrazol-5-yl) -1-piperazinyl ] -2-pyrrolidinyl] carbamoyl] thiazolidine, having the formula below, is a very novel DPP-4 inhibitor potential.

World Patent Application No. W02012099915 for Ge Lieting discloses a process for the preparation route is as follows:

Journal B10rganic & Medicinal Chemistry, 2012, 20, 5705-5719 also discloses a preparation method for Ge Lieting, the route is as follows:

[0009] 1- (3-methyl-1-phenyl-5-pyrazolyl) piperazine, was prepared for the Ge Lieting key intermediate. Journals B10rganic & Medicinal Chemistry, 2012,20,5705-5719 reported the preparation of the intermediates prepared route is as follows:

[0011] The preparative route after the N-Boc-N- acetoacetyl piperazine phenylhydrazine and methanesulfonic acid in an ethanol solution of the reaction at room temperature 14h, concentrated under reduced pressure after addition of pyridine.Was added phosphorus oxychloride in pyridine, 20h post treatment reaction at room temperature the reaction system. The compound obtained above was then added trifluoroacetic acid was dissolved in methylene chloride after, after treatment at room temperature for 1.5h to give 1- (3-methyl-1-phenyl-5-pyrazolyl) piperazine.
The reaction process requires mesylate mesylate flammable, easy-absorbent deliquescence, and has a strong corrosive and irritating, easy to cause the body burns; phosphorus oxychloride, a highly toxic substance, water violent hair in the air smoke, hydrolyzed into phosphoric acid and hydrogen chloride, is very unstable, to operate a lot of trouble; trifluoroacetic acid is highly corrosive and irritant, can cause the body burns; low yield of the reaction (10%). Seeking a simple operation, high reaction yield, low cost and suitable for industrial production production process 1- (3-methyl-1-phenyl-5-pyrazolyl) piperazine has a very important role in the field of medicine.
…………………………………….


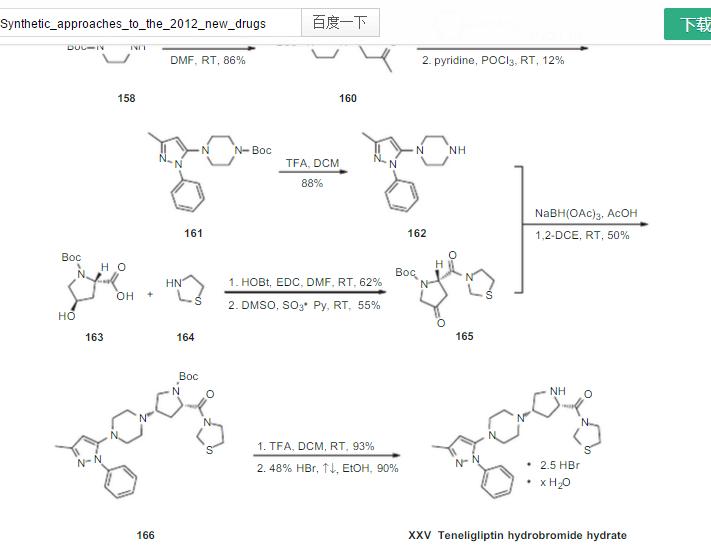
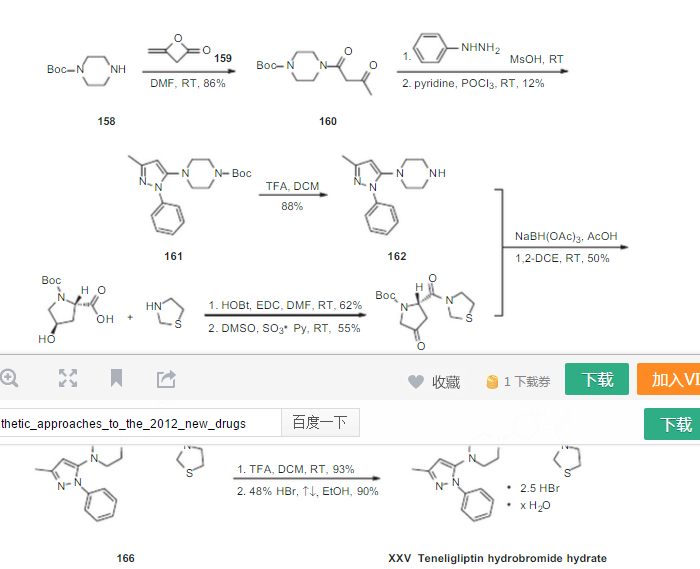
since the capture is staggered, compd 165 is not clear in above pic see below
…………
if above section iis not clear see at ……..http://www.allfordrugs.com/2015/07/03/teneligliptin/
…………………….
reaction scheme in http://www.google.com/patents/CN104177295A?cl=en

Description: LR as Lawesson reagent (Lawesson Reagent), is a sulfur oxygen exchange reagent. The present invention provides a method for preparing key intermediates Ge Lieting method, comprising the steps of: (I) N-Boc-N- acetoacetyl piperazine Lawesson’s reagent in the presence of an organic solvent, with a phenylhydrazine of the formula occurs ⑴ reaction shown:

(2) the step (1) The product was dissolved in an organic solvent, the following formula (II) in concentrated hydrochloric acid to deprotected shown:

Volume 20, Issue 19, 1 October 2012, Pages 5705–5719


………………………..
http://www.google.co.in/patents/WO2015019238A1?cl=en
Example 5: Preparation of {(2^,.4^)-4-r4-(3-methyl-l-phenyl-lH-pyrazol-5-yl)piperazin- 1 -vHpyrrolidin-2-yl } ( 1.3 -thiazolidin-3 -vDmethanone hemipentahydrobromide hydrate (Formula II)
Activated carbon (10 g) was added to a solution of the residue (obtained in Example 4) in isopropyl alcohol (1000 mL) at 30°C to 35°C. The reaction mixture was filtered through a Hyflo® bed. The filtrate was heated to a temperature of 70°C to 75°C. Hydrobromic acid (48%; 168 g) was slowly added to the filtrate at 70°C to 75°C over a period of 10 minutes to 15 minutes. The reaction mixture was stirred for 2.5 hours at 70°C to 77°C. The progress of the reaction was monitored by HPLC. After completion of the reaction, the reaction mixture was cooled to a temperature of 20°C to 25 °C, and stirred at the same temperature for 60 minutes. The reaction mixture was filtered to obtain a solid. The solid obtained was washed with isopropyl alcohol (2 x 200 mL), and dried at 50°C under reduced pressure for 15 hours to obtain crude {(25*,45)-4-[4-(3-methyl-l-phenyl-lH- pyrazol-5 -yl)piperazin- 1 -yl]pyrrolidin-2-yl} ( 1 ,3 -thiazolidin-3 -yl)methanone
hemipentahydrobromide hydrate.
Yield: 90%
Example 6: Purification of {(2^’.4^)-4-r4-(3-methyl-l-phenyl-lH-pyrazol-5-yl)piperazin- 1 -yllpyrrolidin-2-yl } ( 1.3 -thiazolidin-3 -vDmethanone hemipentahydrobromide hydrate (Formula II)
A reaction mixture containing {(2S,4S)-4-[4-(3-methyl-l-phenyl-lH-pyrazol-5- yl)piperazin- 1 -yl]pyrrolidin-2-yl } ( 1 ,3 -thiazolidin-3 -yl)methanone
hemipentahydrobromide hydrate (100 g; prepared according to the process of Example 5) in ethanol (700 mL) was heated at 70°C to 75°C to obtain a solution. The solution was filtered at the same temperature. The filtrate was allowed to cool to a temperature of 65 °C to 68°C, and deionized water (10 mL) was added at the same temperature. The solution was cooled to a temperature of 55°C to 60°C, and stirred at the same temperature for 2 hours. The solution was further cooled to a temperature of 20°C to 25 °C, and stirred at the same temperature for 60 minutes to obtain a solid. The solid was filtered, washed with ethanol (100 mL), and dried at 45°C to 50°C under reduced pressure for 18 hours to 20 hours to obtain pure {(2S,4S)-4-[4-(3-methyl-l-phenyl-lH-pyrazol-5-yl)piperazin-l- yl]pyrrolidin-2-yl } ( 1 ,3 -thiazolidin-3 -yl)methanone hemipentahydrobromide hydrate .
Yield: 90%
HPLC Purity: 99.93%
| WO2012099915A1 * | 18 Jan 2012 | 26 Jul 2012 | Hongwen Zhu | Thiazolidine derivatives and their therapeutic use |
| WO2012165547A1 * | 31 May 2012 | 6 Dec 2012 | Mitsubishi Tanabe Pharma Corporation | Method for manufacturing pyrazole derivative |
| WO2014041560A2 * | 28 Aug 2013 | 20 Mar 2014 | Glenmark Pharmaceuticals Limited; Glenmark Generics Limited | Process for the preparation of teneligliptin |
| US7074794 | 10 Aug 2001 | 11 Jul 2006 | Mitsubishi Pharma Corporation | Proline derivatives and the use thereof as drugs |
| US8003790 | 17 Feb 2006 | 23 Aug 2011 | Mitsubishi Tanabe Pharma Corporation | Salt of proline derivative, solvate thereof, and production method thereof |
| US20050256310 * | 12 May 2005 | 17 Nov 2005 | Pfizer Inc | Therapeutic compounds |
| EP1854795A1 * | 17 Feb 2006 | 14 Nov 2007 | Mitsubishi Pharma Corporation | Salt of proline derivative, solvate thereof, and production method thereof |
| EP1894567A1 * | 2 Jun 2006 | 5 Mar 2008 | Mitsubishi Tanabe Pharma Corporation | Concomitant pharmaceutical agents and use thereof |
| US20040106655 * | 10 Aug 2001 | 3 Jun 2004 | Hiroshi Kitajima | Proline derivatives and the use thereof as drugs |
| Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2015019238A1 * | 28 Jul 2014 | 12 Feb 2015 | Ranbaxy Laboratories Limited | Process for the preparation of n-protected (5s)-5-(1,3-thiazolidin-3-ylcarbonyl)pyrrolidin-3-one |
| Patent | Submitted | Granted |
|---|---|---|
| Proline derivatives and use thereof as drugs [US7060722] | 2005-11-03 | 2006-06-13 |
| Proline derivatives and the use thereof as drugs [US7074794] | 2004-06-03 | 2006-07-11 |
| Proline derivatives and use thereof as drugs [US2006173056] | 2006-08-03 | |
| SALT OF PROLINE DERIVATIVE, SOLVATE THEREOF, AND PRODUCTION METHOD THEREOF [US8003790] | 2009-08-27 | 2011-08-23 |
| METHOD OF TREATING ABNORMAL LIPID METABOLISM [US2010305139] | 2010-12-02 | |
| COMBINED USE OF DIPEPTIDYL PEPTIDASE 4 INHIBITOR AND SWEETENER [US2010113382] | 2010-05-06 | |
| CONCOMITANT PHARMACEUTICAL AGENTS AND USE THEREOF [US2009082256] | 2009-03-26 | |
| PROPHYLACTIC/THERAPEUTIC AGENT FOR ABNORMALITIES OF SUGAR/LIPID METABOLISM [US2009088442] | 2009-04-02 | |
| SALT OF PROLINE DERIVATIVE, SOLVATE THEREOF, AND PRODUCTION METHOD THEREOF [US2011282058] | 2011-11-17 |
- Joanne Bronson, Amelia Black, T. G. Murali Dhar, Bruce A. Ellsworth, and J. Robert Merritt. “Teneligliptin (Antidiabetic)”. Annual Reports in Medicinal Chemistry 48: 523–524. doi:10.1016/b978-0-12-417150-3.00028-4.
- Kishimoto, M (2013). “Teneligliptin: A DPP-4 inhibitor for the treatment of type 2 diabetes”. Diabetes, metabolic syndrome and obesity : targets and therapy 6: 187–95. doi:10.2147/DMSO.S35682. PMC 3650886. PMID 23671395.
see gliptins at…………http://drugsynthesisint.blogspot.in/p/gliptin-series.html
VILAZODONE SPECTRAL DATA


VILAZODONE
Vilazodone hydrochloride; 163521-08-2; Vilazodone HCl; Viibryd; UNII-U8HTX2GK8J; EMD-68843
NO SYNTHESIS IS THIS POST, ONLY SPECTRAL DATA DISCUSSED
SEE MORE SPECTROSCOPY DATA AT………..http://orgspectroscopyint.blogspot.in/2015/06/vilazodone.html
ENJOY THE INTERPRETATIONS
Vilazodone (United States trade name Viibryd veye-brid) is a serotonergic antidepressant developed by Clinical Data for the treatment of major depressive disorder. The chemical compound was originally developed by Merck KGaA (Germany).[2] Vilazodone was approved by the FDA for use in the United States to treat major depressive disorder in 2011.[3][4][5]
Medical uses
According to two eight-week, randomized, double-blind, placebo-controlled trials in adults, vilazodone elicits an antidepressant response after one week of treatment. After eight weeks, subjects assigned to vilazodone 40 mg daily dose (titrated over 2 weeks) experienced a significantly higher response rate than the group given placebo (44% vs 30%, P = .002). Remission rates for vilazodone were not significantly different versus placebo.[6]
According to an article on the United States approval of vilazodone written by FDA staff, “it is unknown whether [vilazodone] has any advantages compared to other drugs in the antidepressant class.”[7]
PAPER FROM OPRD
Scale-Up Synthesis of Antidepressant Drug Vilazodone

A scale-up synthesis of antidepressant drug vilazodone was accomplished in five steps. Friedel–Crafts acylation of 1-tosyl-1H-indole-5-carbonitrile with 4-chlorobutyryl chloride, selective deoxygenation in NaBH4/CF3COOH system coupled with ethyl 5-(piperazin-1-yl)-benzofuran-2-carboxylate hydrochloride, one-step deprotection and esterolysis, and the final ammonolysis led to the target molecule vilazodone in 52.4% overall yield and 99.7% purity. This convenient and economical procedure is remarkably applicable for scale-up production.
5-(4-(3-(5-Cyano-1H-indol-3-yl)butyl)piperazin-1-yl)benzofuran-2-carboxamide (1)


…………………………
SEE MORE SPECTROSCOPY DATA AT………..http://orgspectroscopyint.blogspot.in/2015/06/vilazodone.html
VIIBRYD Tablets for oral administration contain polymorph Form IV vilazodone hydrochloride (HCl), a selective serotonin reuptake inhibitor and a 5HT1A receptor partial agonist.
Vilazodone HCl is 2-benzofurancarboxamide, 5-[4-[4-(5cyano-1H-indol-3-yl)butyl]-1-piperazinyl]-, hydrochloride (1:1). Its molecular weight is 477.99. The structural formula is:
 |
In addition to the active ingredient, VIIBRYD Tablets contain lactose monohydrate, microcrystalline cellulose, magnesium stearate, colloidal silicon dioxide, polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, FD&C Blue #1 (40 mg only), FD&C Yellow #6 (20 mg only) and FD&C Red #40 (10 mg only).


- “VIIBRYD (vilazodone hydrochloride) tablet VIIBRYD (vilazodone hydrochloride) kit [Forest Laboratories, Inc.]”. DailyMed. Forest Laboratories, Inc. December 2012. Retrieved28 October 2013.
- “Clinical Data’s Vilazodone Patient Enrollment Over One Third Complete”. Business Wire. Berkshire Hathaway. 17 August 2006. Retrieved 12 April 2014.
- “FDA approves Clinical Data Inc’s antidepressant”. Reuters. January 22, 2011.
- “FDA approves Clinical Data Inc’s antidepressant”. Reuters. January 22, 2011. Retrieved 12 April 2014.
- “Clinical Data, Inc. – Clinical Data, Inc. Submits New Drug Application for Vilazodone for the Treatment of Major Depressive Disorder”. Retrieved 12 April 2014.
- Wang, SM; Han, C; Lee, SJ; Patkar, AA; Masand, PS; Pae, CU (August 2013). “A review of current evidence for vilazodone in major depressive disorder.”. International Journal of Psychiatry in Clinical Practice 17 (3): 160–9. doi:10.3109/13651501.2013.794245. PMID 23578403.
- Laughren TP, Gobburu J, Temple RJ, Unger EF, Bhattaram A, Dinh PV, Fossom L, Hung HM, Klimek V, Lee JE, Levin RL, Lindberg CY, Mathis M, Rosloff BN, Wang SJ, Wang Y, Yang P, Yu B, Zhang H, Zhang L, Zineh I (September 2011). “Vilazodone: clinical basis for the US Food and Drug Administration’s approval of a new antidepressant”. The Journal of Clinical Psychiatry 72 (9): 1166–73. doi:10.4088/JCP.11r06984. PMID 21951984.
