
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 169)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
PF 06650833
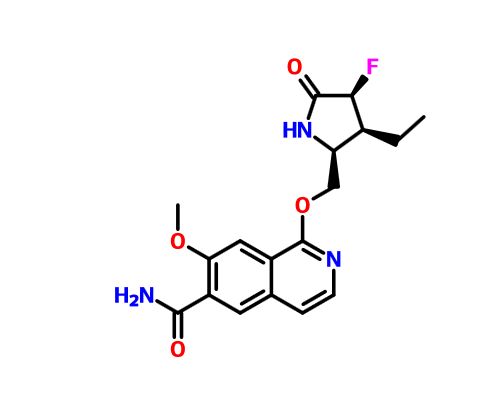
 .
.
Picture credit….Bethany Halford
PF 06650833
MFC18H20FN3O4, MW361.37
1-{[(2S,3S,4S)-3-ethyl-4-fluoro-5-oxopyrrolidin-2-yl]methoxy}-7-methoxyisoquinoline-6-carboxamide
6-Isoquinolinecarboxamide, 1-[[(2S,3S,4S)-3-ethyl-4-fluoro-5-oxo-2-pyrrolidinyl]methoxy]-7-methoxy-
CAS 1817626-54-2
WO 2015150995
1st disclosures is @pfizer‘s on inflammatory disease treatment targeting IRAK4
$PFE IRAK4 inhibitor
Phase I Lupus vulgaris
- 01 Feb 2016 Pfizer completes a phase I pharmacokinetics trial in Healthy volunteers in USA (PO) (NCT02609139)
- 01 Nov 2015 Pfizer initiates a phase I pharmacokinetics trial in Healthy volunteers in USA (PO) (NCT02609139)
- 01 Jun 2015 Pfizer completes a phase I trial for Lupus (In volunteers) in USA (PO) (NCT02224651)
Compounds useful for the treatment of autoimmune and inflammatory diseases associated with lnterleukin-1 Receptor Associated Kinase (IRAK) and more particularly compounds that modulate the function of IRAK4.
Protein kinases are families of enzymes that catalyze the phosphorylation of specific residues in proteins, broadly classified in tyrosine and serine/threonine kinases. Inappropriate activity arising from dysregulation of certain kinases by a variety of mechanisms is believed to underlie the causes of many diseases, including but not limited to, cancer, cardiovascular diseases, allergies, asthma, respiratory diseases, autoimmune diseases, inflammatory diseases, bone diseases, metabolic disorders, and neurological and neurodegenerative diseases. As such, potent and selective inhibitors of kinases are sought as potential treatments for a variety of human diseases.
There is considerable interest in targeting the innate immune system in the treatment of autoimmune diseases and sterile inflammation. Receptors of the innate immune system provide the first line of defense against bacterial and viral insults. These receptors recognize bacterial and viral products as well as pro-inflammatory cytokines and thereby initiate a signaling cascade that ultimately results in the up-regulation of inflammatory cytokines such as TNFa, IL6, and interferons. Recently it has become apparent that self-generated ligands such as nucleic acids and products of inflammation such as high-mobility group protein B1 (HMGB1) and Advanced Glycated End-products (AGE) are ligands for Toll-like receptors (TLRs) which are key receptors of the innate immune system (O’Neill 2003, Kanzler et al 2007, Wagner 2006). This demonstrates the role of TLRs in the initiation and perpetuation of inflammation due to autoimmunity.
lnterleukin-1 receptor associated kinase 4 (I RAK4) is a ubiquitously expressed serine/threonine kinase involved in the regulation of innate immunity (Suzuki & Saito 2006). IRAK4 is responsible for initiating signaling from TLRs and members of the I L- 1/18 receptor family. Kinase-inactive knock-ins and targeted deletions of IRAK4 in mice were reported to cause reductions in TLR and IL-1 induced pro-inflammatory cytokines (Kawagoe et al 2007; Fraczek et al. 2008; Kim et al. 2007). IRAK4 kinase-dead knock-in mice have also been shown to be resistant to induced joint inflammation in the antigen-induced-arthritis (AIA) and serum transfer-induced (K/BxN) arthritis models (Koziczak-Holbro 2009). Likewise, humans deficient in IRAK4 also appear to display the inability to respond to challenge by Toll ligands and IL-1 (Hernandez & Bastian 2006). However, the immunodeficient phenotype of IRAK4-null individuals is narrowly restricted to challenge by gram positive bacteria, but not gram negative bacteria, viruses or fungi. This gram positive sensitivity also lessens with age, implying redundant or compensating mechanisms for innate immunity in the absence of IRAK4 (Lavine et al 2007).
These data indicate that inhibitors of IRAK4 kinase activity should have therapeutic value in treating cytokine driven autoimmune diseases while having minimal immunosuppressive side effects. Additional recent studies suggest that targeting IRAK4 may be useful in other inflammatory pathologies such as atherosclerosis and diffuse large B-cell lymphoma (Rekhter et al 2008; Ngo et al 2011). Therefore, inhibitors of IRAK4 kinase activity are potential therapeutics for a wide variety of diseases including but not limited to autoimmunity, inflammation, cardiovascular diseases, cancer, and metabolic diseases. See the following references for additional information: N. Suzuki and T. Saito, Trends in Immunology, 2006, 27, 566. T. Kawagoe, S. Sato, A. Jung, M. Yamamoto, K. Matsui, H. Kato, S. Uematsu, O. Takeuchi and S. Akira, Journal of Experimental Medicine, 2007, 204, 1013. J. Fraczek, T. W. Kim, H. Xiao, J. Yao, Q. Wen, Y. Li, J.-L. Casanova, J. Pryjma and X. Li, Journal of Biological Chemistry, 2008, 283, 31697. T. W. Kim, K. Staschke, K. Bulek, J. Yao, K. Peters, K.-H. Oh, Y. Vandenburg, H. Xiao, W. Qian, T. Hamilton, B. Min, G. Sen, R. Gilmour and X. Li, Journal of Experimental Medicine, 2007, 204, 1025. M. Koziczak-Holbro, A. Littlewood- Evans,
B. Pollinger, J. Kovarik, J. Dawson, G. Zenke, C. Burkhart, M. Muller and H. Gram, Arthritis & Rheumatism, 2009, 60, 1661. M. Hernandez and J. F. Bastian, Current Allergy and Asthma Reports, 2006, 6, 468. E. Lavine, R. Somech, J. Y. Zhang, A. Puel, X. Bossuyt, C. Picard, J. L. Casanova and C. M. Roifman, Journal of Allergy and Clinical Immunology, 2007, 120, 948. M. Rekhter, K. Staschke, T. Estridge, P. Rutherford, N. Jackson, D. Gifford-Moore, P. Foxworthy,
C. Reidy, X.-d. Huang, M. Kalbfleisch, K. Hui, M.S. Kuo, R. Gilmour and C. J. Vlahos, Biochemical and Biophysical Research Communications, 2008, 367, 642. O’Neill, L. A. (2003). “Therapeutic targeting of Toll-like receptors for inflammatory and infectious diseases.” Curr Opin Pharmacol 3(4): 396. Kanzler, H et al. (2007) “Therapeutic targeting of innate immunity with toll-like receptor agonists and antagonists.” Nature Medicine 13:552. Wagner, H. (2006) “Endogenous TLR ligands and autoimmunity” /Advances in Immunol 91 : 159. Ngo, V. N. et al. (2011) “Oncogenically active MyD88 mutations in human lymphoma” Nature 470: 115.
PATENT
Preparation 1 : 1-chloro-7-methoxyisoquinoline-6-carbonitrile (P1) Step 1. Synthesis of methyl 4-iodo-3-methoxybenzoate (CAS 35387-92-9. CD.
To a solution of 3-hydroxy-4-iodobenzoic acid (CAS 58123-77-6, C12) (10800 g, 40.9 moles) in DMF (65 L) was added K2C03 (25398 g, 184 moles), followed by the slow addition of dimethyl sulfate (11352 g, 90 moles). This mixture was heated to about 50 °C for over night. The reaction mixture was cooled to about 25 °C, diluted with EtOAc (50 L) and filtered through a plug of Celite®. The solid was thoroughly washed with EtOAc (10 L X 3). The combined EtOAc filtrates were poured into water. After stirring for about 30 min, the EtOAc layer was separated and it was further washed sequentially with water, 1 M NaOH and brine. The EtOAc layer was separated, dried over Na2S04, filtered and concentrated to provide the title compound C1. Yield: 11750 g (98%).
Step 2. Synthesis of (4-iodo-3-methoxyphenyl)methanol (CAS 244257-61-2, C2).
To a solution of compound C1 (11750 g, 40.2 moles) in THF (35 L) was added NaBH4 (7645 g, 201.09 moles) and refluxed. While refluxing, MeOH (25 L) was slowly added into the reaction mixture at a rate of about 1 L per hour. After completion of the reaction, it was poured into a solution of cold dilute HCI. Once the excess of NaBH4was quenched, the solution was filtered and extracted with EtOAc (2.5 L X 3). The combined EtOAc extracts were washed sequentially with water, brine and dried over Na2S04. The solvent was evaporated under reduced pressure and the resulting crude material was treated with MTBE. The resulting solid was filtered and filtrate was washed with water, brine, dried over Na2S0 , and filtered. The solvent was evaporated under reduced pressure to provide the title compound C2. Yield: 9900 g (93%).
Step 3. Synthesis of 4-iodo-3-methoxybenzaldehyde (CAS 121404-83-9, C3).
To a solution of compound C2 (9900 g, 34.5 moles) in CHCI3 (186 L), was added manganese dioxide (18000 g, 207 moles) and the resulting mixture was refluxed for about 16 h. The mixture was cooled to about 25 °C and filtered through a Celite pad, which was then washed thoroughly with CHCI3. The CHCI3 was evaporated under reduced pressure to provide the title compound C3. Yield: 9330 g (95%). 1 H NMR (400 MHz, CDCI3): δ 9.95 (s, 1 H), 7.99 (d, 1 H), 7.14 (dd, 1 H), 3.95 (s, 3 H).
Step 3. Synthesis of 6-iodo-7-methoxyisoquinoline (CAS 244257-63-4. C4).
To a solution of compound C3 (9300 g, 35 moles) in toluene (60 L) was added amino acetaldehyde dimethyl acetal (5590 g, 53 moles) and the mixture was refluxed for about 4 h, while removing the liberated water by the use of a Dean – Stark water separator. The reaction mixture was cooled to about 0 °C, after which trifluoroacetic anhydride (22305 g, 106 moles) followed by BF3-Et20 (15080 g, 106 moles) were added, keeping internal temperature below 5 °C. The reaction mixture was stirred at about 25 °C for about 16 h and quenched by pouring into a mixture of ice and ammonium hydroxide. The product was extracted with EtOAc (10 L X 3), and the combined EtOAc extracts were washed sequentially with water and brine. The combined EtOAc extracts were dried over Na2S04, filtered, and concentrated to afford a dark tan colored residue. This was treated with a mixture of MTBE and hexane (1 :1 v/v, 30 L), followed by 6 M HCI (9 L), with stirring. The precipitated solid was filtered and washed with MTBE. The solid was suspended in EtOAc (5 L) and made alkaline with ammonium hydroxide. The EtOAc layer was separated, washed with brine, dried over Na2S04, filtered, and concentrated to afford crude compound C4 as a brown solid. HPLC (230 nm) showed it to be about 83% pure.
The crude material (1000 g) was taken in AcOH (2.5 L) and stirred for about 90 min at about 25 °C. The solid was filtered and washed with AcOH (500 ml_). The filtrate was neutralized with saturated aqueous Na2C03 solution. The resulting precipitated solid was filtered, washed with water (4 L), and oven dried at about 70 – 75 °C for about 5 h to afford about 780 g of pure C4. Similarly, the remaining crude C4 (4 kg) was purified to provide the title compound C4. Yield: 4300 g (42%). 1H NMR (400 MHz, CDCI3): δ 9.15 (s, 1 H), 8.45 (d, 1 H), 8.35 (s, 1 H), 7.45 (d, 1 H), 7.15 (s, 1 H) 4.00 (s, 3 H).
Step 4. Synthesis of 7-methoxyisoquinoline-6-carbonitrile (C5).
To a solution of compound C4 (4300 g , 15 moles) in DMSO (39 L) was added copper(l) cyanide (2954 g, 33 moles) and the mixture was heated to about 120 °C for about 3 h. The reaction mixture was quenched by pouring into a mixture of ice and ammonium hydroxide (40 L) and filtered. The filtrate was extracted with EtOAc (10 L X 2). While stirring, the solid residue was again treated with ammonium hydroxide solution (10 L) and EtOAc (10 L). After filtration, the precipitated material was repeatedly washed with a mixture of MeOH and CHCI3 (1 :9, v/v) several times and the combined extracts were washed with brine. The extracts were dried over Na2S04, filtered, and concentrated under reduced pressure. The resulting crude material was triturated with hexane to provide the title compound C5. Yield: 2250 g (87%). 1H NMR (400 MHz, CDCI3): δ 9.25 (br. s, 1 H), 8.55 (br. s, 1 H), 8.15 (s, 1 H), 7.60 (d, 1 H), 7.30 (s, 1 H), 4.05 (s, 3 H).

A solution of a reactant such as 1-(((2S,3S,4S)-3-ethyl-4-fluoro-5-oxopyrrolidin-2-yl)methoxy)-7-methoxyisoquinoline-6-carbonitrile (200 mg, 0.5 mmol) in concentrated H2SO4 (1.5 ml.) was warmed to about 55 °C for about two hours, then cooled to about 20 °C. The reaction mixture was added dropwise with vigorous stirring to 7.3 ml_ of ice cold concentrated ammonium hydroxide with cooling in ice. The precipitated solid was filtered and washed with water, heptane, ether, and dried under vacuum. The residue may be used directly for subsequent work, or it may be purified by chromatography or HPLC.
ABSTRACTS
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 261




//////////PF 06650833, IRAK4 inhibitor, inflammatory disease treatment , PFIZER, 1817626-54-2
N1C([C@H](C([C@H]1COc3c2cc(c(cc2ccn3)C(=O)N)OC)CC)F)=O
NC(=O)c2cc3ccnc(OC[C@H]1NC(=O)[C@@H](F)[C@H]1CC)c3cc2OC
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
LY 2922470

LY 2922470

LY 2922470
Picture credit….Bethany Halford
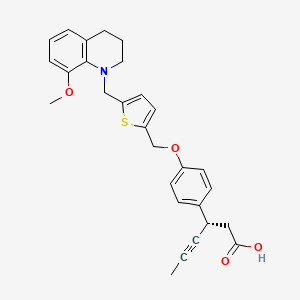
(3S)-3-[4-[[5-[(8-methoxy-3,4-dihydro-2H-quinolin-1-yl)methyl]thiophen-2-yl]methoxy]phenyl]hex-4-ynoic acid
Benzenepropanoic acid, 4-[[5-[(3,4-dihydro-8-methoxy-1(2H)-quinolinyl)methyl]-2-thienyl]methoxy]-β-1-propyn-1-yl-, (βS)-
Glucose Lowering Agents, Signal Transduction Modulators
| CAS | 1423018-12-5 |
|---|---|
| Molecular Formula: | C28H29NO4S |
| Molecular Weight: | 475.59916 g/mol |
|---|
https://clinicaltrials.gov/ct2/show/NCT01867216
- Phase I Type 2 diabetes mellitus
Eli Lilly
Antihyperglycaemics
- 28 Jan 2014 Eli Lilly completes a phase I trial in Type-2 diabetes mellitus in USA (NCT01867216)
- 30 Jun 2013 Phase-I clinical trials in Type-2 diabetes mellitus in USA (PO)
- 14 Jun 2013 Eli Lilly plans a phase I trial for Type-2 diabetes mellitus in USA (NCT01867216)
PATENT
WO 2013025424
https://www.google.com/patents/US20130045990?cl=de
| Also published as | CA2843474A1, CA2843474C, CN103687856A, CN103687856B, EP2744806A1, US8431706, WO2013025424A1, Less « |
| Inventors | Chafiq Hamdouchi |
| Original Assignee | Eli Lilly And Company |



Preparation 18-Methoxyquinoline
Add potassium hydroxide (435 g, 7.76 mol) to a solution of 8-hydroxy quinoline (250 g, 1.724 mol) in THF (10 L) at ambient temperature and stir. Add methyl iodide (435 g, 2.58 mol) dropwise and stir overnight. Filter the reaction mixture and wash the solid with THF (2 L). Concentrate the solution to dryness; add water; extract with dichloromethane (2×3 L); combine the organic layers; and wash with brine. Collect the organic layers and dry over sodium sulfate. Remove the solids by filtration. Collect the filtrate and concentrate under reduced pressure to give a red oil, which solidifies on standing, to give the title compound (281 g, 102%), which can be used without further purification. ESI (m/z) 160(M+H).
Preparation 2
8-Methoxy-1,2,3,4-tetrahydroquinoline
Add sodium cyanoborohydride (505 g, 8.11 mol) in EtOH (1 L) to a solution of 8-methoxy quinoline (425 g, 2.673 mol) in EtOH (9 L), and stir. Cool the reaction mixture to an internal temperature of 0° C. and add HCl (35%, 1.12 L, 10.962 mol) dropwise over 60 min so that the internal temperature did not rise above 20° C. Allow the reaction mixture to warm to ambient temperature and then heat to reflux for 2.5 hours. Cool to ambient temperature and stir overnight. Add ammonium hydroxide (25%, 1 L); dilute with water (15 L); and extract the mixture with dichloromethane (3×10 L). Combine the organic layers and dry over sodium sulfate. Remove the solids by filtration. Collect the filtrate and concentrate under reduced pressure to give a residue. Purify the residue by silica gel flash chromatography, eluting with ethyl acetate: hexane (1:10) to give the title compound (357 g, 82%). ESI (m/z) 164(M+H).
Preparation 3
Methyl-5-methylthiophene-2-carboxylate
Add thionyl chloride (153 ml, 2.1 mol) dropwise over 20 min to a solution of 5-methylthiophene-2-carboxylic acid (100 g, 0.703 mol) in MeOH (1 L) at 0° C. and stir. After the addition is complete, heat the reaction mixture to reflux for 3.5 hours. Cool and concentrate in vacuo to give a thick oil. Dilute the oil with EtOAc (500 ml) and sequentially wash with water (300 ml) then brine (300 ml). Dry the organic layer over sodium sulfate. Remove the solids by filtration. Collect the filtrate and concentrate under reduced pressure to give the title compound (106 g, 97%), which is used without further purification. ESI (m/z) 156(M+H).
Preparation 4
Methyl 5-(bromomethyl)thiophene-2-carboxylate
Add freshly recrystallised NBS (323.8 g, 1.81 mol) to a solution of methyl-5-methylthiophene-2-carboxylate (258 g, 1.65 mol) in chloroform (2.6 L) at room temperature, and stir. Add benzoyl peroxide (3.99 g, 0.016 mol) and heat the reaction mixture to reflux for 7 hours. Cool the reaction mixture to ambient temperature and filter through diatomaceous earth. Wash the filter cake with chloroform (250 ml). Collect the organic layers and remove the solvent to give the title compound (388 g, 100%), which is used without further purification. ESI (m/z) 236(M+H).
Preparation 5
Methyl-5-[8-methoxy-3,4-dihydro-2H-quinolin-1-yl)methyl]thiophene-2-carboxylate
Add methyl-5-(bromoethyl)thiophene-2-carboxylate (432.5 g, 1.84 mol) in EtOH (500 ml) to a solution of 8-methoxy-1,2,3,4-tetrahydroquinoline (300 g 1.84 mol) in EtOH (1 L) and stir. Add DIPEA (641 ml, 3.67 mol) dropwise and stir at room temperature overnight. After completion of the reaction, remove the EtOH in vacuo, and add water (5 L). Extract the aqueous with EtOAc (3×3 L); combine the organic layers; and dry over sodium sulfate. Filter the solution and concentrate under reduced pressure to give a residue. Purify the residue by silica gel flash chromatography eluting with ethyl acetate: hexane (6:94) to give the title compound (325 g, 56%). ESI (m/z) 318(M+H).
Preparation 6
[5-[(8-Methoxy-3,4-dihydro-2H-quinolin-1-yl)methyl]-2-thienyl]methanol
Add DIBAL-H (1 M in toluene 2.7 L, 2.66 mol) slowly via a cannula over a period of 1.5 h to a stirred solution of methyl-5-(8-methoxy-3,4-dihydroquinolin-1(2H)-yl)methyl)thiophene-2-carboxylate (281 g, 0.886 mol) in THF (4 L) at −70° C. Monitor the reaction via thin layer chromatography (TLC) for completion. After completion of the reaction, allow the reaction mixture to warm to 20° C. and add a saturated solution of ammonium chloride. Add a solution of sodium potassium tartrate (1.3 Kg in 5 L of water), and stir overnight. Separate the organic layer; extract the aqueous phase with EtOAc (2×5 L); then combine the organic layers; and dry the combined organic layers over sodium sulfate. Remove the solids by filtration. Remove the solvent from the filtrate under reduced pressure to give the title compound as a white solid (252 g, 98%). ESI (m/z) 290(M+H).
Preparation 7
Ethyl(3S)-3-[4-[[5-[(8-methoxy-3,4-dihydro-2H-quinolin-1-yl)methyl]-2-thienyl]methoxy]phenyl]hex-4-ynoate
Add tributylphosphine (50% solution in EtOAc, 543 ml, 1.34 mol) to a solution of ADDP (282.5 g, 1.5 eq) in THF (3 L) and cool the mixture to an internal temperature of 0° C., then stir for 15 minutes. Add (S)-ethyl 3-(4-hydroxyphenyl)hex-4-ynoate (173.5 g, 0.747 mol) in THF (3 L) dropwise over 15 min; then add 5-((8-methoxy-3,4-dihydroquinolin-1(2H)-yl)methyl)thiophene-2-yl)methanol (216 g, 0747 mol) in THF (5 L) dropwise. Allow the reaction mixture to warm to ambient temperature and stir overnight. Filter the reaction mixture through diatomaceous earth and wash the filter cake with ethyl acetate (2 L). Concentrate the organic filtrate to dryness. Add water (4 L); extract with ethyl acetate (3×5 L); combine the organic layers; and dry the combined organic layers over sodium sulfate. Remove the solids by filtration and concentrate under reduced pressure to give an oil. Purify the residue by silica gel flash chromatography by eluting with ethyl acetate: hexane (6:94) to give the title compound (167 g, 44%). ESI (m/z) 504(M+H).
Example 1
(3S)-3-[4-[[5-[(8-Methoxy-3,4-dihydro-2H-quinolin-1-yl)methyl]-2-thienyl]methoxy]phenyl]hex-4-ynoic acid

Add a solution of potassium hydroxide (49.76 g, 0.88 mol) in water (372 ml) to a solution of (S)-ethyl-3-(4-((5-8-methoxy-3,4-dihydroquinolin-1(2H)-yl)methyl)thiophen-2-yl)methoxy) phenyl)hex-4-ynoate (149 g, 0.296 mol) in EtOH (1.49 L) at room temperature and stir overnight. Concentrate the reaction mixture to dryness and add water (1.3 L). Extract the resulting solution with EtOAc (2×300 ml) and separate. Adjust the pH of the aqueous layer to pH=6 with 2 N HCl. Collect the resulting solids. Recrystallise the solids from hot MeOH (298 ml, 2 vol) to give the title compound (91 g, 65%). ESI (m/z) 476(M+H).
Abstract
GPR40 agonists for the treatment of type 2 diabetes: From the laboratory to the patient
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 260

Presenter

Chafiq Hamdouchi
Senior Research Advisor at Eli Lilly and Company
https://www.linkedin.com/in/chafiq-hamdouchi-4988126
Summary
Dr. Hamdouchi earned his bachelor’s degree and doctorate in organic chemistry from Louis Pasteur University, Strasbourg-France.
Following two postdoctoral fellowships, sponsored by the National Science Foundation-USA and Ministerio de Educación y Ciencia-Spain, he joined Eli Lilly and Company in 1995.
Throughout his 20 years of career at Lilly, he has contributed to a sustainable drug discovery portfolio from preclinical hypothesis to clinical proof-of-concept that spans the oncology, neuroscience and endocrinology therapeutic areas. He has led multidisciplinary (chemistry, pharmacology, ADMET, PK, medical) scientific teams in USA, Europe and Asia to deliver a number of compounds that achieved first human dose.
He is a co-inventor of six innovative molecules being pursued in clinical development for the treatment of Diabetes, Cancer and Neurodegenerative Diseases.
He has an extensive patent and publication record and deep experience in conducting drug discovery and development in Asia through effective partnership and mentorship.
SEE AT…………ONE ORGANIC CHEMIST ONE DAY BLOG
LINK……http://oneorganichemistoneday.blogspot.in/2016/03/chafiq-hamdouchi-senior-research.html
| Patent ID | Date | Patent Title |
|---|---|---|
| US8431706 | 2013-04-30 | 1,2,3,4-tetrahydroqinoline derivative useful for the treatment of diabetes |
References
GPR40 agonists for the treatment of type 2 diabetes: From the laboratory to the patient
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 260
//////Phase 1, LY2922470, LY 2922470, Eli Lilly, Type 2 diabetes mellitus, 1423018-12-5, Chafiq Hamdouchi
CC#CC(CC(=O)O)C1=CC=C(C=C1)OCC2=CC=C(S2)CN3CCCC4=C3C(=CC=C4)OC
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Googleplus amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
HELP, Need one time help to pay 10 year concessional subscription to this, your favorite blog to WordPress

Just One viewer please come forward
Dear Kind Viewer’s
WordPress is kind to me and negotiated a one time 10 year concessional subscription of 260 US dollars…….https://newdrugapprovals.org/
I need one time help to pay this one time 10 year concessional subscription to our favorite blog.
This is done to keep this blog running even after my death.
Currently I am paying 99 US Dollars per annum
email me
amcrasto@gmail.com
call +919323115463
Paypal will work for me via email request to you by me, Indian govt does not allow automatic transfer via paypal buttons on the blog.
email me at amcrasto@gmail.com and tell me amount, i will request you via paypal

DR ANTHONY CRASTO
LIONEL MY SON, MY MOTIVATION
.
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, He cried bitterly and we had never seen him so depressed
Now I keep Lionel as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son and family happy.

ps
The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent,
///////////
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Facebook 
Googleplus amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
DISCLAIMER
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
AVORALSTAT
Avoralstat, BCX4161,
CAS 918407-35-9
UNII: UX17773O15
513.5513, C28-H27-N5-O5
2-Pyridinecarboxylic acid, 3-(2-(((4-(aminoiminomethyl)phenyl)amino)carbonyl)-4-ethenyl-5-methoxyphenyl)-6-(((cyclopropylmethyl)amino)carbonyl)-
3-(2-((4-Carbamimidoylphenyl)carbamoyl)-4-ethenyl-5-methoxyphenyl)-6-((cyclopropylmethyl)carbamoyl)pyridine-2-carboxylic acid
Hereditary angioedema (HAE)
Kallikrein inhibitor
BioCryst Pharmaceuticals
![]()
BioCryst is also investigating second-generation plasma kallikrein inhibitors to avoralstat, for treating HAE (in February 2016, this program was listed as being in preclinical development).
Prevent acute attacks in patients with hereditary angioedema (HAE); Treat hereditary angioedema (HAE)
U.S. – Fast Track (Treat hereditary angioedema (HAE));
U.S. – Orphan Drug (Prevent acute attacks in patients with hereditary angioedema (HAE))
26 Feb 2016Clinical trials in Hereditary angioedema (Prevention) in USA (PO, Hard-gelatin capsule) before February 2016
24 Feb 2016Discontinued – Phase-III for Hereditary angioedema (Prevention) in France (PO, Soft-gelatin capsule)
24 Feb 2016Discontinued – Phase-III for Hereditary angioedema (Prevention) in Germany (PO, Soft-gelatin capsule)

| Conditions | Interventions | Phases | Recruitment | Sponsor/Collaborators |
|---|---|---|---|---|
| Hereditary Angioedema|HAE | Drug: BCX4161|Drug: Placebo | Phase 2|Phase 3 | Recruiting | BioCryst Pharmaceuticals |
| Hereditary Angioedema | Drug: BCX4161|Drug: Placebo | Phase 2 | Completed | BioCryst Pharmaceuticals |
| Hereditary Angioedema | Drug: BCX4161 | Phase 1 | Completed | BioCryst Pharmaceuticals |
| Hereditary Angioedema | Drug: BCX4161 | Phase 1 | Completed | BioCryst Pharmaceuticals |
Avoralstat, also known as BCX-4161, is a potent and orally active Kallikrein inhibitor and Bradykinin inhibitor. Avoralstat may be potentially useful for treatment for Hereditary angioedema. Avoralstat inhibits plasma kallikrein and suppresses bradykinin production. Bradykinin is the mediator of acute swelling attacks in HAE patients.

Selective inhibitor of plasma kallikrein that subsequently suppresses bradykinin production
Hereditary angioedema (HAE) is a serious and potentially life-threatening rare genetic illness, caused by mutations in the C1-esterase inhibitor (C1 INH) gene, located on chromosome 11q. HAE is inherited as an autosomal dominant condition, although one quarter of diagnosed cases arise from a new mutation. HAE has been classed as an orphan disease in Europe, with an estimated prevalence of 1 in 50,000. Individuals with HAE experience recurrent acute attacks of painful subcutaneous or submucosal edema of the face, larynx, gastrointestinal tract, limbs or genitalia which, if untreated, may last up to 5 days. Attacks vary in frequency, severity and location and can be life-threatening. Laryngeal attacks, with the potential for asphyxiation, pose the greatest risk. Abdominal attacks are especially painful, and often result in exploratory procedures or unnecessary surgery. Facial and peripheral attacks are disfiguring and debilitating.
HAE has a number of subtypes. HAE type I is defined by C1 INH gene mutations which produce low levels of C1 -inhibitor, whereas HAE type II is defined by mutations which produce normal levels of ineffective C1 protein. HAE type III has separate pathogenesis, being caused by mutations in the F12 gene which codes for the serine protease known as Factor XII. Diagnostic criteria for distinguishing the subtypes of HAE, and distinguishing HAE from other angioedemas, can be found in Ann Allergy Asthma Immunol 2008; 100(Suppl 2): S30-S40 and J Allergy Clin Immunol 2004; 114: 629-37, incorporated herin by reference.
Current treatments for HAE fall into two main types. Older non-specific treatments including androgens and antifibrinolytics are associated with significant side effects, particularly in females. Newer treatments are based on an understanding of the molecular pathology of the disease, namely that C1 INH is the most important inhibitor of kallikrein in human plasma and that C1 INH deficiency leads to unopposed activation of the kallikrein-bradykinin cascade, with bradykinin the most important mediator of the locally increased vascular permeability that is the hallmark of an attack.
Approved therapies include purified plasma-derived C1 INH (Cinryze®, Berinert), the recombinant peptide kallikrein inhibitor ecallantide (Kalbitor®), and the bradykinin receptor B2 inhibitor iticabant (Firazyr®). All of the currently available targeted therapies are administered by intravenous or subcutaneous injection. There is currently no specific targeted oral chronic therapy for HAE.
There are many delivery routes for active pharmaceutical ingredients (APIs). Generally, the oral route of administration is favored. Oral administration provides a number of advantages, such as, but not limited to, patient convenience, flexibility of timing of administration, location of administration and non-invasiveness. Oral administration also provides more prolonged drug exposure compared with intermittent intravenous infusion, which may be important for drugs with schedule-dependent efficacy. For example, a drug with a short half-life can achieve a greater exposure time by either continuous infusion or by continuous oral dosing. The use of oral therapy further has the potential to reduce the cost of healthcare resources for inpatient and ambulatory patient care services.
In the pharmaceutical arts, it is known that a number of APIs cannot be administered effectively by the oral route. The main reasons why these compounds cannot be administered by the oral route are: a) rapid enzymatic and metabolic degradation; b) chemical and/or biological instability; c) low solubility in aqueous medium; and/or d) limited permeability in the gastrointestinal tract. For such compounds, non-oral routes of delivery, such as parenteral administration, mainly via intramuscular or subcutaneous injections, may be developed. However, non-oral administration poses a disadvantage for the patient as well as healthcare providers, and for this reason, it is important to develop alternative routes of administration for such compounds, such as oral routes of administration.
While the oral route of administration is the most convenient for the patient and the most economical, designing formulations for administration by the oral route involves many complications. Several methods are available to predict the ease by which an API may be formulated into a formulation suitable for administration by the oral route. Such methods include, but are not limited to, and Lipinski rule (also referred to as the Rule of Five) and the Biopharmaceutical Drug Disposition Classification System (BDDCS).
The BDDCS divides APIs into four classifications, depending on their solubility and permeability. Class I APIs have high solubility and high permeability; Class II APIs have low solubility and high permeability; Class III APIs have high solubility and low permeability; and Class IV APIs have low solubility and low permeability. APIs in higher classes in the BDDCS face greater challenges in formulating into an effective, pharmaceutically acceptable product than those in lower classes. Of the four classes, APIs falling into Class IV are the most difficult to formulate into a formulation for administration by the oral route that is capable of delivering an effective amount of the API as problems of both solubility and permeability must be addressed (note the BDDCS does not inherently address chemical stability). The role of BDDCS in drug development is described generally in L.Z. Benet J Pharm Sci. 2013, 102(1), 34-42.
Lipinski’s rule (described in Lipinski et al. Adv. Drug Deliv. Rev. 46 (1-3): 3-26) states, in general, that in order to develop a successful formulation for administration by the oral route, an API can have no more than one violation of the following criteria:
i) not more than 5 hydrogen bond donors (nitrogen or oxygen atoms with one or more hydrogen atoms)
ii) not more than 10 hydrogen bond acceptors (nitrogen or oxygen atoms) iii) a molecular mass less than 500 daltons
iv) an octanol-water partition coefficient log P not greater than 5.
J. Zhang et al. Medicinal Chemistry, 2006, 2, 545-553, describes a number of small molecule amidine compounds which have activity as inhibitors of kallikrein. The molecules described in this document fall into Class IV of the BDDCS as described above. The compounds are poorly soluble in aqueous and physiological fluids, and are poorly permeable as demonstrated by oral dosing in rats and in vitro experiments with Caco-2 cells.
Furthermore, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid, one of the compounds described in Zhang et al., is a Class IV API and violates criteria iii) and iv) as set forth in the Lipinski Rule.
Furthermore, the compounds described in Zhang et al., including 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid, exhibit poor stability with respect to oxidation in air, to light
(photodegradation) and in aqueous and physiological fluids, as well as to elevated temperatures.
Therefore, the compounds described by Zhang et al. including, but not limited to, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid, not only exhibit poor solubility and permeability characteristics, but also poor stability characteristics. As a result, such compounds are predicted to be especially difficult to formulate into an effective, orally deliverable
pharmaceutical composition that is capable of delivering an effective amount of the compound to a subject.
Polymorphism, the occurrence of different crystal forms, is a property of some molecules. A single molecule may give rise to a variety of polymorphs having distinct crystal structures and physical properties, such as, but not limited to, melting point, thermal behaviors (e.g. measured by thermogravimetric analysis (TGA), or differential scanning calorimetry (DSC), x-ray diffraction pattern, infrared absorption fingerprint, and solid state NMR spectrum. One or more of these techniques may be used to distinguish different polymorphic forms of a compound.
Discovering new polymorphic forms and solvates of a pharmaceutical product can provide alternate forms of the compound that display a number of desirable and advantageous properties, such as, but not limited to, ease of handling, ease of processing, ease of formulation, storage stability, and/or ease of purification. Further, new polymorphic forms and solvates of a pharmaceutically useful compound or salts thereof may further provide for improved pharmaceutical products, by providing compounds that are more soluble in a set of pharmaceutical excipients. Still further, the provision of new polymorphic forms and solvates of a pharmaceutically useful compound or salts thereof enlarges the repertoire of compounds that a formulation scientist has available for formulation optimization, for example by providing a pharmaceutical product with different properties, such as, but not limited to, improved processing characteristics, improved handling characteristics, improved solubility profiles, improved dissolution profile and/or improved shelf-life. Therefore, there is a need for additional polymorphs of pharmaceutically useful compounds, such as, but not limited to, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6- (cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid and the compounds disclosed herein.
In one aspect, the present invention provides an oral formulation that is capable of delivering an effective amount of the amidine compounds described by Zhang et al. to a subject. In particular, the present invention provides an oral formulation that is capable of delivering an effective amount of 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid to a subject. In one specific aspect, the 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid is present in a particular crystal form designated Form A. In light of the art suggesting the difficulties in formulating such an oral formulation, this result was unexpected.
As described herein, the amidine compounds described in Zhang et al., including, but not limited to, 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6- (cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (specifically including particular crystal Form A), may now be conveniently used in oral administration and further used in oral administration for the treatment of a number of diseases and conditions in a subject, such as, but not limited to, HAE as described herein.
Avoralstat & next generation kallikrein inhibitors for HAE
Avoralstat
![]()
May 16 is HAE awareness day
See BioCryst’s video regarding HAE to learn more
Avoralstat is being developed as an oral prophylactic treatment for patients suffering from Hereditary Angioedema (HAE). Avoralstat inhibits plasma kallikrein and suppresses bradykinin production. Bradykinin is the mediator of acute swelling attacks in HAE patients.
In May 2014 BioCryst, announced that the OPuS-1 (OralProphylaxiS-1) Phase 2a proof of concept clinical trial met its primary efficacy endpoint, several secondary endpoints and all other objectives established for the trial. OpuS-1 enrolled 24 HAE patients with a history of HAE attack frequency of at least 1 per week. Treatment with avoralstat demonstrated a statistically significant mean attack rate reduction of 0.45 attacks per week versus placebo, p<0.001. The mean attack rate per week was 0.82 on BCX4161 treatment, compared to 1.27 on placebo.
In December 2014, BioCryst initiated enrollment in OPuS-2 (Oral ProphylaxiS-2). OPuS-2 is a blinded, randomized, 12-week, three-arm, parallel cohort design trial evaluating the efficacy and safety of two different dose regimens of avoralstat administered three-times daily, 300 mg and 500 mg, compared with placebo. The primary efficacy endpoint for the trial will be the mean angioedema attack rate, which will be reported for each avoralstat dose group compared to placebo. The trial is being conducted in the U.S., Canada and Europe. On October 8, 2015, announced that it has completed enrollment of approximately 100 HAE patients with a history of moderately frequent to very frequent attacks in OPuS-2. BioCryst expects to report the OPuS-2 trial results in early 2016.
PATENT
WO200234711
http://www.google.com/patents/WO2002034711A1?cl=en
PATENT
PATENT
Examples
Example 1 – Synthesis of 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl- phenyll-6-(cvclopropylmethyl-carbarnoyl)-pyridine-2-carboxylic acid
The synthesis of the above compound and intermediates is described below. In this section, the following abbreviations are used:

The synthesis of starting material, (4-(benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) is described in Scheme 1.
f 0HCY ° ΒΓΥΥ°

Preparation of 6-bromobenzofdl[1,3ldioxole-5-carbaldehvde (1b)

1a 1b
To a mixture of piperonal (1a) (498 g, 3.32 mol) in glacial acetic acid (1000 mL) was added a solution of bromine (200 mL, 3.89 mol) in glacial acetic acid (500 mL) over a period of 30 min and stirred at room temperature for 24h. The reaction mixture was poured into water (2000 mL) and the solid that separated was collected by filtration. The solid was dissolved in boiling ethanol (4000 mL) and cooled to room temperature. The solid obtained on cooling was collected by filtration to furnish 6-bromobenzo[d][1 ,3]dioxole-5-carbaldehyde (lb) (365 g, 48 %) as a white solid, MP 126 °C; HNMR (300 MHz, DMSO-d6): δ 10.06 (s, 1 H), 7.42 (s,1 H), 7.29 (s, 1 H), 6.20 (d, J=12.3, 2H); IR (KBr) 3434, 2866, 1673,1489, 1413, 259, 1112, 1031 , 925 cm“1; Analysis calculated for CeH5BrO3.O 25H C, 41.15; H, 2.37; Found: C, 41.07; H, 2.11.
Preparation of 2-bromo-5-hvdroxy-4-methoxybenzaldehyde (1c)

1c
A solution of potassium tert-butoxide (397 g, 3.36 mol) in DMSO (1.5 L) was heated at 50 °C for 30 min. Methanol (1.5 L) was added to it and continued heating at 50 °C for additional 30 min. To the hot reaction mixture was added 6-bromo-benzo[d][1,3]dioxole-5-carbaldehyde (1 b) (350g, 1.53 mol) and continued heating at 50 °C for 30 min. The reaction mixture was cooled to room temperature and quenched with water (2.3 L) and sodium hydroxide (61.2 g, 1.53 mol). The reaction mixture was washed with ether (2 x 1.5 L), acidified to pH 2 using cone. HCI and extracted with ethyl acetate ( 1 L). The ethyl acetate layers were combined and concentrated under vacuum to dryness. The residue obtained was treated with water (1.5 L) and ethyl acetate (1 L). The solid obtained was collected by filtration to furnish 2-bromo-5-hydroxy-4-methoxybenzaldehyde (1c) (97 g, 27.5% as a first crop). The layers from the filtrate were separated and aqueous layer was extracted with ethyl acetate (200 ml_). The ethyl acetate layers were combined dried over MgS04 and concentrated under vacuum to dryness to furnish 2-bromo-5-hydroxy-4-methoxybenzaldehyde (1c) (192 g, 54.4%, second crop) as an orange solid, MP 108 °C; ‘HNMR (300MHz, DMSO-cfe): S 10.00 (s, 1 H), 9.92 (s,1 H), 7.27 (s, 1 H), 7.26 (s, 1 H), 3.93 (s, 3H); IR (KBr) 3477, 2967, 2917,
2837, 2767, 2740, 1657, 1595, 1428, 1270, 1210, 1164, 1022 cm“‘; Analysis calculated for C8H7Br03.H20: C, 38.58; H, 3.64: Found: C, 38.60; H, 3.60.
Preparation of 5-(benzyloxy)-2-bromo-4-methoxybenzaldehvde ( d)

To a solution 2-bromo-5-hydroxy-4-methoxybenzaldehyde (1c) (120 g, 520 mmol) in DMF (1000 mL) was added potassium carbonate (79 g, 572 mmol) and benzyl bromide (68 mL, 572 mmol). The reaction mixture was stirred at room temperature overnight and quenched with water (3000 mL). The solid obtained was collected by filtration, washed with ether and dried under vacuum to furnish 5-(benzyloxy)-2-bromo-4-methoxybenzaldehyde (1d) (113.19 g, 67.9%) as a white solid, MP 144 °C;1HNMR (300 MHz, DMSO-c/6): δ 10.06 (s, 1H), 7.47-7.34 (m, 7H), 5.17 (s, 2H), 3.92 (s, 3H); IR (KBr) 2898, 2851 , 1673, 1592, 1502, 1437, 1402, 1264, 1210, 1158, 1017, 754 cm“1; Analysis calculated for C 5H13Br03: C, 56.10; H, 4.08; Found: C, 55.44; H, 4.08.
Preparation of 1-(benzyloxy)-4-bromo-5-(diethoxymethyl)-2-methoxybenzene (1e)
15 046578
146

1d 1e
To a solution of 5-(benzyloxy)-2-bromo-4-methoxybenzaldehyde (1d) (100 g, 311 mmol) in
ethanol (1500 mL) was added triethyl orthoformate (103 mL, 622 mmol), ammonium nitrate
(7.5 g, 93.3 mmol) and stirred at room temperature overnight. The reaction mixture was
treated with ether (1200 mL) and stirred for 15 min before filtration. The filtrate was
concentrated under vacuum to dryness to give 1-(benzyloxy)-4-bromo-5-(diethoxymethyl)-2-methoxybenzene (1e) (134 g) as a brown syrup; The product was used in the next step
without further purification; 1H N R (300 MHz, DMSO-cf6) δ 7.45 – 7.37 (m, 4H), 7.36 – 7.33
(m, 1 H), 7.17 – 7.14 (m, 1 H), 7.10 (s, 1 H), 5.10 (s, 2H), 3.80 (s, 3H), 3.58 – 3.33 (m, 5H),
1.13 – 1.07 (m, 6H); IR (KBr) 2974, 2879, 1601 , 1503, 1377, 1260, 1163, 1060 cm“1;
Analysis calculated for C19H23Br04: C, 57.73; H, 5.86; Found: C, 57.21 ; H, 5.94.
acid (1fi

To a solution of 1-(benzyloxy)-4-bromo-5-(diethoxymethyl)-2-methoxybenzene (1e) (120 g,
300 mmol) in dry ether (1000 mL) at -78 °C was added n-butyllithium (1.6 M solution in
hexanes, 244 mL, 390 mmol) over a period of 30 min and further stirred at -78 °C for 30 min.
A solution of tri-n-butylborate (110 mL, 405 mmol) in dry ether (300 mL) was added to this
solution at -78 °C over a period of 30 min. The reaction mixture was further stirred for 2 h at -78 °C and warmed to 0 °C. The reaction mixture was quenched with 3N HCI (300 mL) at 0
°C and heated at reflux for 1 h. After cooling to room temperature, the solid obtained was
collected by filtration washed with water (250 mL) dried in vaccum to afford (4-(benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) (30.85 gm, 37.6% as a white solid. The organic
layer from above filtrate was extracted with 1.5 N NaOH (3 x 200 mL). The combined basic
extracts were acidified with cone. HCI (pH about 4). The solid obtained was collected by
filtration, washed with water and dried under vacuum to furnish a second crop of (4-(benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) (22.3 g, 26%) as a light orange solid
MP 158 °C; 1H NMR (300 MHz, DMSO-cfe) δ 10.08 (s, 1 H), 7.52 (s, 1 H), 7.48 – 7.33 (m, 5H),
7.24 (s, 1H), 5.18 (s, 2H), 3.89 (s, 3H); 1H NMR (300 MHz, DMSO-d6/D20) δ 10.06 (s, 1H),
7.52 (s, 1H), 7.49 – 7.32 (m, 5H), 7.23 (s, 1 H), 5.18 (s, 2H), 3.89 (s, 3H); MS (ES+) 309.1 (M+Na); IR (KBr) 3335, 2937, 1647, 1545, 1388, 1348, 1268, 1146, 1095 cm-1; Analysis calculated for C15H15BO5.0.25H2O: C, 62.00; H, 5.38; Found: C, 61.77; H, 5.19.
Synthesis of methyl-6-(cvclopropylmethylcarbamoyl¾-3-ftrifluoromethylsulfonyloxyVpicolinate
The synthesis of the intermediate methyl 6-(cyclopropylmethylcarbamoyl)-3-(trifluoromethyl sulfonyloxy)picolinate (2h) is described in Scheme 2.

Preparation of 2-bromo-3-hvdroxy-6-methylpyridine (2b)

H3C N Br
2a 2b
To a solution of 3-hydroxy-6-methylpyridine (2a) (3000 g, 27.5 mol) in pyridine (24 L) cooled to 15 °C was added a solution of bromine (4.83 kg, 1.55 L, 30.2 mol) in pyridine (3 L) over a period of 50 min maintaining the internal temperature between 20 to 25 DC. After stirring for 19 h at room temperature the solvent was removed under vacuum and the residue was triturated with water. The solid separated was collected by filtration, washed with water and dried under vacuum to give 2-bromo-3-hydroxy-6-methylpyridine (2b) (3502 g, 67.7 %) as a light brown solid which was used as such without further purification; 1H NMR (300 MHz, DMSO-d6) δ 10.43 (s, 1H), 7.18 (d, J = 8.0 Hz, 1 H), 7.08 (d, J
MS (ES+) 188.35, 186.36 (M+1).
(2c)

2b 2c
A mixture of 2-bromo-3-hydroxy-6-methylpyridine (2b) (3000 g, 15.96 mol), anhydrous potassium carbonate (3308 g, 23.94 mol), and iodomethane (2.491 kg, 1.09 L, 17.556 mol) in 30 L of acetone was heated at 40 °C overnight. The reaction mixture was cooled to room temperature and filtered through Celite. Evaporation of the solvent followed by silica gel chromatography (Hexane: ethyl acetate = 7:3) afforded the desired compound, 2-bromo-3-methoxy-6-methylpyridine (2c) which was used as such for the next step; 1H NMR (300 MHz, DMSO-cfe) δ 7.42 (dd, J = 8.3, 1.5 Hz, 1H), 7.29 – 7.19 (m, 1H), 3.84 (d, J = 1.6 Hz, 3H), 2.37 (d, J = 1.7 Hz, 3H).

2c
2d
To a solution of 2-bromo-3-methoxy-6-methylpyridine (2c) (310 g, 1.53 mol) in 6000 mL of water at 60 °C was added KMnO, (725 g, 4.59 mol) in small portions over a 90 min period with vigorous mechanical stirring. A dark purple solution resulted. This solution was kept at 90 °C for a further 3 h and filtered through Celite while still hot to give a colourless filtrate.
After cooling, the aqueous solution was acidified to pH 1-2 by adding 6 N HCI. The white solid obtained was collected by filtration to give on drying 6-bromo-5-methoxy-2-pyridinecarboxylic acid (2d) (302g, 85%) of product, which was used as such in the next reaction without further purification. An analytical sample was obtained by recrystallization from methanol to give 6-bromo-5-methoxy-2-pyridinecarboxylic acid; 1H NMR (300 MHz, DMSO-tfe) δ 7.40 – 7.28 (m, 1H), 7.17 (d, J = 8.3 Hz, 1 H), 3.83 (d, J = 1.7 Hz, 3H).
Preparation of 6-bromo-N-(cvclopropylmethyl)-5-methoxypicolinamide (2e)

To a solution of 6-bromo-5-methoxy-2-pyridinecarboxylic acid (2d) (12 g, 52 mol) in pyridine (70 mL) was added EDCI (11.5 g, 59 mmol) and cyclopropylmethylamine (3.6 g, 52 mmol). The reaction mixture was stirred at room temperature overnight and then concentrated under vacuum. The reaction mixture was diluted with water (100 mL) and ethyl acetate (100 mL). The organic layer was separated and the water layer was extracted with ethyl acetate (2 x 100 mL). The organic layers were combined and washed with water (2 x 50 mL), brine (500 mL), dried over magnesium sulphate, filtered and concentrated under vacuum to furnish 10.43g of crude product. The crude product was converted into a slurry (silica gel 20 g) and purified by flash column chromatography (silica gel 230 g, eluting with 0-100% ethyl acetate in hexane) to yield compound 6-bromo-N-(cyclopropylmethyl)-5-methoxypicolinamide (2e) (8.02 g, 54%) as off white solid, mp 67-70 °C; 1HNMR (300 MHz, DMSO-d6) δ 8.51 (t, J = 5.8, 1 H), 8.02 (d, J = 8.4, 1 H), 7.65 (d, J = 8.5, 1 H), 3.96 (s, 3H), 3.14 (t, J = 6.5, 2H), 1.11 -0.99 (m, 1 H), 0.47 – 0.36 (m, 2H), 0.27 – 0.20 (m, 2H); MS (ES+) 307.0, 309.0 (100%
M+Na)
Preparation of methyl 6-(cvclopropylmethylcarbamoyl)-3-methoxypicolinate (2f)

To a solution of 6-bromo-N-(cyclopropylmethyl)-5-methoxypicolinamide (2e) (7.5 g, 27.6 mol) in methanol (300 mL) in a 2-L stainless steel bomb was added Pd(OAc)2(750 mg), 1 ,1-bis(diphenylphosphino)-ferrocene (750 mg), and triethylamine (3.9 mL, 27.6 mmol). The reaction mixture was vacuum flushed and charged with CO gas to 150 psi. The reaction mixture was and heated with stirring at 150°C overnight and cooled to room temperature. The catalyst was filtered through a pad of celite, and concentrated to dryness to furnish crude product. The crude was purified by flash column chromatography (silica gel 150 g,
eluting with, 0%, 5%, 10%, 20%, 30%, 50% ethyl acetate/hexanes (250 mL each) as eluents to give methyl 6-(cyclopropylmethyl-carbamoyl)-3-methoxypicolinate (2f) (6.29 g, 86.1 %) as a salmon coloured solid, MP 107 °C; 1HNMR (300 MHz, DMSO-cfe) δ 8.28 (t, J = 6.0, 1H), 7.91 (d, J = 8.8, 1H), 7.55 (d, J = 8.8, 1 H), 3.68 (s, 3H), 3.64 (s, 3H), 2.90 (t, J = 6.5, 2H), 0.89 – 0.68 (m, 1 H), 0.26 – 0.09 (m, 2H), 0.08 – 0.00 (m, 2H); MS (ES+) 287.1 (M+Na); IR (KBr) 3316, 2921 , 1730, 1659, 1534, 1472, 1432, 1315, 1272, 1228, 1189, 1099, 1003, 929, 846, 680 cm“1; Analysis calculated for C13H16 204: C, 59.08; H, 6.10; N, 10.60; Found: C, 58.70; H, 5.97; N, 10.23.
Preparation of 6-(cvclopropylmethylcarbamoyl 3-hvdroxypicolinic acid (2q)

2f 2g
Aluminium chloride method:
To a solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-methoxypicolinate (2f) (0.16 mmol) in dichloromethane (840 mL) was added AICI3 (193 g, 1.5 mol). The reaction mixture was heated at reflux for 12 h under nitrogen. After slowly adding ~2L of 1 N HCI, the organic layer was separated. The aqueous layer was re-extracted several times with ethyl acetate/DME. The combined organic layer was washed with brine, dried (MgSO.4), and evaporated in vacuo to furnish crude 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid. To a solution of 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid was added a solution of acetyl chloride (1 10 mL) in methanol (1.1 L). The reaction mixture was stirred for 12 h at room temperature and then concentrated to dryness in vacuo. After co-evaporating once with methanol, the compound was purified by flash-column chromatography (silica gel, 500 g, eluted with chloroform and 3% methanol in chloroform) to furnish 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid (2g).
Boron tribromide method:
To a stirring solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-ethoxypicolinate (2f) (58.0 g, 208 mmol) was added BBr3 (79 mL, 834 mmol) in CH2CI2 (1.3 L) at 0-5 °C. The reaction mixture was allowed to warm to room temperature and stirred for 18h. The reaction mixture was evaporated to dryness and anhydrous methanol (1 L) was added to the light yellowish solid residue. Insoluble solid was collected by filtration (36 g). Mother liquor was evaporated and co-evaporated with MeOH (2 x 200 mL). The insoluble solid (36 g) was treated with MeOH (500 mL) and acetyl chloride (50 mL) and stirred at room temperature for 18 h (at this point reaction mixture was clear). The mixture was evaporated to dryness and diluted with water and extracted with EtOAc. White solid that separated out from EtOAc layer was collected by filtration, washed with water (2 x 20 mL), dried in vacuo at 50 °C to afford 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid (2g) (5.36 g, 10 %) as a white solid, MP 92-95 °C. 1HNMR (DMSO-cfe) δ 11.04 (s, 1 H, exchangeable with D20), 8.37 (t, J = 6.0, 1 H, exchangeable with D20), 8.12 (d, J = 8.7 Hz, 1 H), 7.57 (d, J = 8.7 Hz, 1 H), 3.90 (m, 3 H), 3.15 (m, 2 H), 1.04 ( m, 1 H), 0.41 (m, 2 H), 0.24 (m, 2 H). IR (KBr): 3346, 3205, 1684 cm“1; MS (ES+): 251.1 (M+1); Analysis calculated for C12H14N2O4.0.1 H2O: C, 57.18; H, 5.67; N, 11.14; Found: C, 57.11 ; H, 5.61; N, 11.09.
Preparation of methyl-6-(cvclopropylmethylcarbamoyl)-3-(trifluoromethylsulfonyloxy) picolinate (2h

To a solution of 6-(cyclopropylmethylcarbamoyl)-3-hydroxypicolinic acid (2g) (28 mmol) in DMF (200 mL) were added triethylamine (12 mL, 84 mmol) and N-phenyl-bis(trifluoromethanesulfonimide) (12 g, 34 mmol). The reaction mixture was stirred for 1.5 h at room temperature and then poured into ice. After diluting with water and extracting with ethyl acetate, the aqueous phase was re-extracted, and then the combined organic layer was washed with water and concentrated under vacuum to give methyl-6-(cyclopropylmethylcarbamoyl)-3-(trifluoromethylsulfonyloxy)picolinate (2h), which was used in the next step without purification.
1H NMR (300 MHz, CDCI3) δ 8.50 (d, J = 8.6, 1 H), 8.07 (s, 1 H), 7.88 (d, J = 8.6, 1 H), 4.09 (d, J = 12.6, 3H), 3.48 – 3.24 (m, 2H), 1.18 – 1.01 (m, 1 H), 0.69 – 0.44 (m, 2H), 0.42 – 0.20 (m, 2H). MS (ES*): 405.17, 100%, M+Na.
Synthesis of 3-f2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyll-6-(cvclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid:
The synthesis of 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (3i) is described as shown in Scheme 3.

3-f4-Benzyloxy-2-formyl-5-methoxy-phenylV6-(cvcloDroDvlmethvl-carbarnovn-pyridine-2-carboxylic acid methyl ester (3a)
5 046578
153

3a
To a solution of methyl-6-(cyclopropylmethylcarbamoyl)-3-(trifluoromethylsulfonyloxy)
picolinate (2h) (24.3g, 63 mmol) in DME (225 mL) were added water (25 mL), (4- (benzyloxy)-2-formyl-5-methoxyphenyl)boronic acid (1f) (27.3 g, 95 mmol), NaHC03(15.9 g,
5 189 mmol), and bis(triphenylphosphine)palladium(ll) chloride (0.885 g). The reaction
mixture was stirred at 70°C overnight under nitrogen. After extracting with ethyl acetate, the organic layer was washed with water and brine and dried (MgSO^), and then concentrated
under vacuum. The compound was purified by flash-column chromatography (silica gel, 300 g, eluting with 10%, 20%, 30% and 40% ethyl acetate in hexane) to furnish 3-(4-benzyloxy- 10 2-formyl-5-methoxy-phenyl)-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid
methyl ester (3a) (25 g, 83%) as off white solid, MP 48-50°C: 1H NMR (300 MHz, DMSO-cfe) δ 9.61(s, 1 H), 8.40 (d, J= 7.9 Hz, 1H), 8.14 (t, J= 5.0 Hz, 1H), 7.87 (d, J= 8.1 Hz, 1 H), 7.58
(s, 1H), 7.54-7.30 (m, 5H), 6.71 (s, 1 H), 5.24 (s, 2H), 3.93 (s, 3H), 3.70 (s, 3H), 3.45-3.34 (m,
2H), 1.19-1.05 (m, 1 H), 0.64-0.54 (m, 2H), 0.37-0.30 (m, 2H); IR ( Br) 1735, 1678, 1594,
15 1513, 1437, 1283, 1217, 1141, 1092 cm“1; MS (ES+) 497.29 (M+Na); Analysis calculated for
C27H2eN206: C, 68.34; H, 5.52; N, 5.90; Found; C, 68.16; H, 5.62; N, 5.80.
2-(6-(Cvclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-vn-4-methoxy-5- vinylbenzoic acid (3b)

To a solution of 3-(4-benzyloxy-2-formyl-5-methoxy-phenyl)-6-(cyclopropylmethyl- carbamoyl)-pyridine-2-carboxylic acid methyl ester (3a) (24g, 50.6 mmol) in acetonitrile (50
mL), 2-methyl-2-propanol (350 mL), and water (125 mL) were added sodium dihydrogen
phosphate (12.5 g) and 2-methyl-2-butene (55 mL, 519 mmol). The reaction mixture was cooled in an ice bath and then sodium chlorite (28 g) was added. After stirring for 1 h, the reaction mixture was extracted with ethyl acetate and washed with water. The aqueous layer was re-extracted and then the combined organic layers were dried (MgS04). The solvent was evaporated in vacuo to furnish 5-(benzyloxy)-2-(6- ((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxybenzoic acid (3b) (29 g) which was used for the next step. MS (ES+): 513.24, (M+Na(; (ES ): 489.26, M-1.
Methyl 3-(4-(benzyloxy)-5-methoxy-2-(((2-methoxyethoxy)methoxytoarbonyltohenyl)-6-(cvclopropylmethylcarbamovnpicolinate (3c)

To a mixture of 5-(benzyloxy)-2-(6-(cyclopropylmethylcarbamoyl)-2-(methoxy-carbonyl)pyridin-3-yl)-4-methoxybenzoic acid (3b) (31 g, 63.2 mmol), and triethylamine (17.7 mL, 126.4 mmol) in dichloromethane (300 mL), was added MEM-chloride (9.03 mL, 79 mmol), and stirred at room temperature overnight. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was washed with water and dried over MgS04, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, 40 g) to furnish methyl 3-(4-(benzyloxy)-5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)phenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3c) (32.8 g, 89%) as a thick gum; H NMR (300 MHz, CDCI3) δ 8.35 (d, J = 8.0 Hz, 1 H), 8.15 (t, J = 5.7 Hz, 1 H), 7.78 (d, J = 8.0 Hz, 1H), 7.71 (s, 1H), 7.49 (d, J = 6.8 Hz, 2H), 7.36 (ddd, J = 7.5, 14.8, 22.4 Hz, 3H), 6.66 (s, 1 H), 5.37-5.13 (m, 4H), 3.90 (s, 3H), 3.69 (s, 3H), 3.60-3.49 (m, 2H), 3.49 (s, 2H), 3.39 (dd, J = 4.4, 8.4 Hz, 2H), 3.34 (s, 3H), 1.19-1.00 (m, 1H), 0.57 (q, J = 5.8 Hz, 2H), 0.38-0.25 (m, 2H). MS (ES+): 601.24 (M+Na); (ES“): 577.27 (M-1);1H NMR (300 MHz, DMSO-cfe) δ 8.69 (t, 7 = 6.1 Hz, 1H), 8.20 (d, J = 8.0 Hz, 1H), 7.97 (d, J = 8.0 Hz, 1 H), 7.63 (s, 1H), 7.41 (m, 5H), 6.92 (s, 1 H), 5.20 (m, 4H), 3.83 (s, 3H), 3.57 (s, 3H), 3.44 (m, 2H), 3:33 (m, 2H), 3.21 (m, 5H), 1.14 (m, 1H), 0.44 (m, 2H), 0.27 (m, 2H). IR (KBr):
1732, 1671 cm“1. MS (ES+): 601.1(M+Na); Analysis calculated for C31H 2Oe: C, 64.35; H, 5.92; N, 4.84; Found: C, 64.27; H, 6.04; N, 4.79.
Methyl 6-(cvclopropylmethylcarbamoyl)-3-(4-hvdroxy-5-methoxy-2-(((2-methoxyethoxy¾methoxy)carbonyl)phenyl)picolinate (3d)

3c 3d
To a solution of methyl 3-(4-(benzyloxy)-5-methoxy-2-(((2-methoxyethoxy)methoxy)-carbonyl)phenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3c) (32.8 g, 56.68 mmol) in ethanol (650 mL) was added 10% Pd/C (4 g) and hydrogenated at 45 psi for 5 h. The catalyst was removed by filtration through Celite and the filtrate was concentrated under vacuum to yield methyl 6-(cyclopropylmethylcarbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)phenyl)picolinate (3d) (31.87 g, 86%), which was pure enough to be used as such for the next step. An analytical sample of methyl 6-(cyclopropylmethylcarbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2-methoxyethoxy) methoxy)carbonyl)phenyl)picolinate (3d) was obtained by purification of 350 mg of above crude using flash column chromatography (silica gel, eluting with ethyl acetate in hexane) to afford methyl 6-(cyclopropylmethyl-carbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)-phenyl)picolinate (3d) as a clear gum; 1HNMR (300 MHz, DMSO-d6) δ 9.74 (s, 1 H), 8.68 (t, J = 6.1 Hz, 1H), 8.18 (d, J = 8.0 Hz, 1 H), 7.95 (d, J = 8.0 Hz, 1H), 7.47 (s, 1H), 6.83 (s, 1H), 5.19 (s, 2H), 3.77 (m, 3H), 3.58 (s, 3H), 3.44 (m, 2H), 3.34 (m, 2H), 3.21 (m, 5H), 1.04 (m, 1 H), 0.44 (m, 2H), 0.27 (m, 2H); IR (KBr): 1731 , 1664 cm‘1. MS (ES*): 489.0 (M+1); Analysis calculated for C^e^O,,: C, 59.01; H, 5.78; N, 5.73; Found: C, 58.92; H, 6.15; N, 5.29.
6-(Cvclopropylmethylcarbamovn-3-(5-methoxy-2-(((2-methoxyethoxy^methoxy)-carbonyl)-4- (trifluoromethylsulfonyloxy)phenyl)picolinate (3e)

To a solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-(4-hydroxy-5-methoxy-2-(((2- methoxyethoxy) methoxy)carbonyl)phenyl)picolinate (3d) (14.3 g, 29.3 mmol) in dichloromethane (150 mL) were added pyridine (12 mL, 146 mmol) and triflic anhydride (7.5 mL g, 44 mmol). After stirring overnight at room temperature under N2. the reaction mixture was poured into ice water and then extracted twice with dichloromethane. After washing the combined organic extracts with water and drying (MgS0 ), the solvent was evaporated in vacuo. The compound was purified by flash chromatography over silica gel column using ethyl acetate: hexane to afford methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2- methoxyethoxy)methoxy)-carbonyl)-4-(trifluoromethylsulfonyloxy)phenyl)picolinate (3e) (1 g, 93%); H NMR (300 MHz, CDCy a 8.41 (d, J = 8.0, 1H), 8.17 (s, 1H), 8.03 (s, 1H), 7.79 (d, J = 8.0, 1 H), 6.82 (s, 1H), 5.32 (q, J = 6.1, 2H), 3.97 (s, 3H), 3.74 (s, 3H), 3.67 – 3.57 (m, 2H), 3.55 – 3.45 (m, 2H), 3.41 (dd, J = 8.2, 14.5, 2H), 3.34 (s, 3H), 1.36 – 1.17 (m, 1H), 0.58 (d, J = 7.1 , 2H), 0.33 (d, J = 5.1 , 2H).
Methyl 6-(cvclopropylmethylcarbamoyl)-3-(5-methoxy-2-f((2-methoxyethoxy)- methoxy)carbonvn-4-vinylphenyl)picolinate (3f)

To a solution of methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2- methoxyethoxy)methoxy)carbonyl)-4-(trifluoromethylsulfonyloxy)phenyl)picolinate (3e) (37.4
g, 60.30 mmol) and potassium vinyltrifluoroborate (16.87 g, 120.6 mmol) in DMF (450 mL) and water (45 mL) was bubbled N2 for 5 min. To this mixture was added NaHC03 (20.26 g, 241.2 mmol) and dichloro-bis(triphenylphosphine)palladium (II) (6.34 g, 9.0 mmol). The reaction mixture was stirred at 70 °C for 20 h under N2(reaction progress was checked by 1H N R because product and starting material had same Rf in TLC). The reaction mixture was cooled down to room temperature and diluted with ethyl acetate. The organic layer was separated, washed with water, brine, dried ( gS04) and filtered. The filtrate was concentrated under vacuum to yield crude methyl 6-(cyclopropylmethyl-carbamoyl)-3-(5-methoxy-2-(((2-methoxyethoxy)methoxy)carbonyl)-4-vinylphenyl)-picolinate (3f). The crude product was purified by flash column chromatography (silica gel, 1 kg, eluting with 0-100% ethyl acetate in hexane) to afford methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2-methoxyethoxy)methoxy) carbonyl)-4-vinylphenyl)picolinate [31) (26.54 g, 88%) as an amber gum; H NMR (300 MHz, DMSO-c¾ δ 8.70 (t, J = 6.1 Hz, 1H), 8.23 (d, J = 8.0 Hz, 1 H), 8.12 (s, 1 H), 8.00 (d, J = 8.0 Hz, 1 H), 6.98 (m, 2H), 5.94 (dd, J = 1.2, 17.8 Hz, 1H), 5.43 (d, J = 12.5 Hz, 1 H), 5.21 (d, J = 6.5 Hz, 2H), 3.88 (s, 3H), 3.64 (s, 3H), 3.48 (d, J = 3.1 Hz, 2H), 3.35 (m, 5H), 3.22 (m, 2H), 1.11 (s, 1H), 0.44 (dt, J = 4.9, 5.5 Hz, 2H), 0.28 (q, J = 4.8 Hz, 2H). IR (KBr); 1732, 1670 cm“1. MS (ES+) 499.1 (M+1).
2-(6-(cvclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzolc acid (3g)

A mixture of methyl 6-(cyclopropylmethylcarbamoyl)-3-(5-methoxy-2-(((2-methoxyethoxy)methoxy) carbonyl)-4-vinylphenyl)picolinate (3f) (27.4 mmol) in DME (160 mL) and 6N HCI (40 mL) was stirred at room temperature for 6 h or till TLC showed complete conversion. The solvent was removed under vacuum. The residue obtained was suspended in water, the solid separated out was collected by filtration, washed with water and dried under vacuum to give 2-(6-(cyclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (3g) (7.0 g, 63%) as a white
solid MP 40 – 42 °C; H NMR (300 MHz, DMSO-de) δ 8.69 (t, J= 6.0 Hz, 1H, NH), 8.20 (d, J= 7.9 Hz, 1H), 8.09 (s, 1 H), 7.95 (d, J= 8.1 Hz, 1H), 6.97 (dd, J= 18.0, 11.3 Hz, 1H), 6.88 (s, 1H), 5.92 (d, J= 7.9 Hz, 1H), 5.38 (d, J= 11.1 Hz, 1H), 3.85 (s, 3H), 3.63 (s, 3H), 3.27-3.17 (m, 2H), 1.15-1.05 (m, 1 H), 0.48-0.40 (m, 2H), 0.31-0.24 (m, 2H); IR (KBr): 3084, 1728, 1650, 1533, 1212, 1143 cm-1; MS (ES+) 433.26 (M+Na); (ES-): 409.28 (M-1); Analysis calculated for θ22Η22Ν2Ο6.0.25Η2Ο; C, 63.68; H, 5.47; N, 6.75; Found C, 63.75; H, 5.56; N, 6.65
Methyl-3-(2-(4-carbamimidoylprienylcarbamoyl)-5-metrioxy-4-vinylphenyl)-6- (cvclopropylmethylcarbamoyl)picolinate (3h)

To a solution of 2-(6-(cyclopropylmethylcarbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (3g) (2.35 g, 5.7 mmol) and 4-aminobenzimidamide dihydrochloride (3j) (1.79 g, 8.6 mmol) in DMF (20 mL) and pyridine (30 mL) at 0 °C was added EDCI (1.65 g, 8.6 mmol) and allowed to warm to room temperature overnight. The reaction mixture was quenched with 6N HCI (60 mL) and extracted with chloroform (3 x 60 mL). The organic layer was dried over MgS04, filtered and purified by flash column chromatography (silica gel, 110 g, eluting with 0 to 100% chloroform in CMA 80 in CMA 50) yielding methyl-3-(2-(4-carbamimidoylphenyl-carbamoyl)-5-methoxy-4-vinylphenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3h) (2.2 g, 65%) as a white solid MP 266 °C; 1H NMR (300 MHz, DMSO-c/6) δ 10.78 (s, 1 H), 9.26 (s, 2H), 9.03 (s, 2H), 8.67 (t, J = 6.1 , 1 H), 8.22 (d, J = 8.0, 1 H), 8.06 (d, J = 8.0, 1 H), 7.96 (s, 1 H), 7.89 – 7.74 (m, 4H), 7.13 – 6.96 (m, 2H), 6.07 (d, J = 17.7, 1H), 5.45 (d, J = 12.4, 1 H), 3.91 (s, 3H), 3.61 (s, 3H), 3.20 (s, 2H), 1.09 (dd, J = 4.7, 8.2, 1H), 0.43 (dt, J = 4.9, 5.4, 2H), 0.34 – 0.21 (m, 2H); MS (ES+) 528.1 (M+1); Analysis calculated for 
C, 58.93; H, 5.63; N,11.85; Found: C, 58.75; H, 5.65; N, 11.92.
46578
159
3-r2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy -vinyl-phenyll-6-(cvclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (3i)

3h 3i
To a solution of methyl-3-(2-(4-carbamirriidoylphenylcarbarnoyl)-5-methoxy-4-vinylphenyl)-6-(cyclopropylmethylcarbamoyl)picolinate (3h) (1 g, 1.9 mmol) in methanol (10 mL) and THF
(10 mL) was added 2 N NaOH (10 mL). The reaction mixture was stirred at room
temperature for 3 h, and concentrated in vacuo to remove methanol and THF. The aqueous layer was acidified with 6N HCI to pH 6-7 and the solid obtained was collected by filtration
washed with water and ether to furnish on drying 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid
(3i)(0.775 g, 80%) as the hydrochloride salt as an off white solid.
1H NMR (300 MHz, DMSO-d6) δ 12.67 (s, 1 H), 9.11 (s, 2H), 8.97 (s, 2H), 8.74 (s, 1 H), 7.90
(d, J = 7.8, 1 H), 7.80 (s, 1 H), 7.72 – 7.58 (m, 4H), 6.99 (dd, J = 11.3, 17.7, 1 H), 6.78 (s, 1H),
5.95 (d, J = 17.2, 1H), 5.38 (d, J = 11.9, 1H), 3.82 (s, 3H), 3.18 (s, 2H), 1.06 (s, 1 H), 0.43 (d,
J = 7.9, 2H), 0.25 (d, J = 4.7, 2H); MS (ES+) 514.0 (M+1 ); Analysis calculated for
C2eH27N5O5.HCI.H2O: C, 59.21; H, 5.32; N, 12.33; Found: C, 59.43; H, 5.21; N, 12.06.
Example 1A- Preparation of 3-f2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyll-6-(cvclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride in Form
C

The jacket of a 10 L glass reactor was set to -5 °C. To the reactor was charged 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)-pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) prepared in Step (11) of Example 1 (500 g, 1.22 mol), 4-amino-benzamidine-2HCI (280 g, 1.34 mol), and 2-propanol (4.05 kg). The mixture was cooled to 0.3 °C, and pyridine (210 g, 2.62 mol) followed by EDCI HCI (310 g, 1.61 mol) was added. The mixture was stirred at -1.1 to -0.3 °C for 22 hrs followed by addition of the second portion of EDCI HCI (58 g, 0.30 mol). The temperature of jacket was set to 14.0 °C, and the mixture was stirred for 89 hrs. The precipitate was filtered, and washed with 1.32 kg of 2-propanol.
The wet product (8a) was recharged to the reactor followed by addition of acetonitrile (1.6 kg) and water (0.57 kg). The mixture was heated to 46 °C. Smopex-234 (21 g) and Acticarbone 2SW (10 g) were added and the mixture was stirred at this temperature for 1 hr. The solution was filtered, and filtrate was returned back to the reactor. The jacket of the reactor was set to -5 °C, and the mixture was cooled to -0.2 “C. NaOH solution (256 g 46% NaOH, 2.95 mol, in 960 g water) was added in 25 min keeping the temperature ❤ °C. The mixture was stirred at 0.2-2.0 °C for 1 hr 40 min and then quenched with cone, acetic acid (40 g, 0.66 mol). Diluted acetic acid (80 g, 1.33 mol AcOH in 1000 g water) was added during 1 hr 20 min (temperature 1.7-3.0 °C), followed by 1250 g water (30 min). The
suspension was stirred at 0-3.0 “for 1 hr, and filtered at 0-5 °C (ice mantle around the filter). The reactor and product (8d) was rinsed with 3.5 kg water.
The wet product (8d) was recharged to the reactor followed by 0.65 kg water and 1.69 kg acetonitrile. The mixture was heated to 57-60 °C, and stirred at this temperature for 14.5 hrs. The mixture was cooled to -2.2 °C (Tjackel= -5 °C), and a solution of NaOH (163 g 46%, 1.87 mol, in 580 g water) was added during 15 min. The temperature rose to -0.4 °C. Hydrochloric acid (407 g 37% HCI, 4 mol) was added in 10 min, the temperature rose to 7.5 °C. The suspension was agitated at -3 – 0 °C for 19 hrs. The product was filtered and the filter cake was rinsed with 2.87 kg water, compressed and pulled dry. The wet product (1.30 kg) was dried at 40-43 °C and 50 mbar for 11 hrs to furnish 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6- (cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) (484 g) as Form C.
Example-1 B: Preparation of 3-f2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyll-6-(cvclopropylmethylcarbartiovQpyridine-2-carboxylic acid hydrochloride in Form A
The procedure was carried out in an identical manner to Example 1 A, with the exception that after the final filtration the filter cake was rinsed with 2.87 kg methyl ierf-butyl ether instead of 2.87 kg water, and pulled dry. The product was dried at 40-43 °C and 50 mbar to furnish 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) as Form A.
PATENT
Methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (compound 6a) is (I) (pages 85 and 86). Avoralstat hydrochloride (compound of formula XVIII) is (II) (claim 40, page 109). A Markush structures is presented (claim 1, page 99).
The synthesis of (II) via intermediate (I) is described (example 1, pages 80-93).
A synthesis of the compound 3-[2-(4-carbamimidoyl-phenylcarbamoyl)-5-methoxy-4-vinyl-phenyl]-6-(cyclopropylmethyl-carbamoyl)-pyridine-2-carboxylic acid (Compound 3i) is described in Schemes A-C.
O y OHCk n Br^ ^OCH3
B Brr22,, AAccOOHH Y^ V”“ \ \ tt–BBuuOOKK
OHC^^^O ” Br^\^0 MeOH ” OHC
1a 1b 66%

1d 95% 1 e

1f
Scheme A


3h 31
Scheme C
Examples. In this section, the following abbreviations are used:



Example-1 : Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b)

7b
Step (1): Preparation of 6-Bromobenzo 1 ,3]dioxole-5-carbaldehyde (1 b):

1b
A solution of bromine (33.0 kg, 206.49 mol) in acetic acid (27.5 L) was added slowly to a solution of piperonal (1a) (29.9 kg, 199.16 mol) in acetic acid (105 L) at room
temperature over a period of 50 min and the reaction mixture was stirred at room temperature for 14.2 h. Additional solution of bromine (33 kg, 206.49 mol) in acetic acid (27.5 L) was added slowly to the reaction mixture over a period of 2 h and the reaction mixture was stirred for 22 h. The reaction mixture was quenched by addition of ice water (500 L) with stirring over a period of 6 h and continued stirring for additional 1.25 h. The mixture was allowed to settle and most of the supernatant liquid was decanted to a waste container using nitrogen pressure. Water (600 L) was added to the solid, stirred, mixture was allowed to settle and then most of the supernatant liquid was decanted to a waste container using nitrogen pressure. Water (100 L) was added to the decanted mixture, stirred for 15 min and the solid obtained was collected by filtration using a centrifuge. The solid was washed with water (2 x 100 L) and air-dried in a tray drier for 3.75 h to afford the crude product 1 b (52 kg). The crude product (51.2 kg) was stirred in n-hexane (178 L) for 3 h, collected by filtration, washed with n-hexane (25 L) and dried to afford 6-bromobenzo[1 ,3]dioxole-5-carbaldehyde (1b) (40.1 1 kg, 87.9%) as a light brown solid. MP: 109-112°C. 1H NMR (300 MHz, CDCI3) δ 10.21 (s, 1 H), 7.37 (s, 1 H), 7.07 (s, 1 H), 6.10 (s, 2H); HNMR (DMSO-cf6): δ 10.06 (s, 1 H), 7.42 (s, 1 H), 7.29 (s, 1 H), 6.20 (d, J =12.3 Hz, 2H)
The process is also illustrated in Fig. 1.
Average yield of isolated 1 b from step-1 is 78 – 88%.
Step (2): Preparation of 2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c)

A solution of potassium terf-butoxide (10.7 kg, 95.36 mol) in DMSO (49 L) was stirred at 50 °C for 30 min. Methanol (49 L) was added slowly over a period of 4.25 h and stirred at 50 °C for 30 min. 6-Bromobenzo[1 ,3]dioxole-5-carbaldehyde (1 b) (9.91 kg, 43.27 mol) was added to the reaction mixture in small portions over a period of 45 min and stirred at 50 °C for 1 h. The reaction mixture was cooled to room temperature and split into two equal portions. Each portion was quenched with water (50.9 L) and basified with 50% aqueous NaOH solution (2.4 L). Each portion was extracted with MTBE (4 x 36 L) to remove impurities. The aqueous layer was acidified with cone. HCI to pH ~ 3 to obtain
product as a yellow solid. The solid was collected by filtration using a centrifuge, washed with water (2 x 35 L) and air-dried to afford 2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c) (4.37 kg, 40.7%, contains 7 % water); Mp: 100-102°C; 1HNMR (300MHz, DMSO-d6): δ 10.00 (s, 1 H), 9.92 (s,1 H), 7.27 (s, 1 H), 7.26 (s, 1 H), 3.93 (s, 3H).
The process is also illustrated in Fig. 2.
Average yield of isolated product 2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c) from step-2 is 40-50%.
Step (3): 5-Hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxaborolan-2-y benzaldehyde (4a)

2-Bromo-5-hydroxy-4-methoxy-benzaldehyde (1c) [1.3 kg (93%, 7% water content), 5.25 mol] was dissolved in toluene (13 L) in a reaction flask equipped with a Dean Stark apparatus. The solution was heated at reflux with stirring to distil off about 25% of the toluene along with water (90 ml_). The solution was cooled to 90 °C then
bis(pinacolato)diboron (1.5 kg, 5.82 mol), KOAc (772.6 g, 7.87 mol) and Pd(PPh3) (24.3 g, 0.02 mol) were added and the reaction mixture was heated at reflux for 10h. After confirming the completion of reaction by TLC (mobile phase: 100% DCM), the reaction mixture was cooled to room temperature and was kept standing overnight. The reaction mixture was filtered through celite and the celite cake was washed with toluene (4 L). The filtrate of this batch was mixed with the filtrate of another batch (batch size 1.3 kg obtained from an identical reaction). The mixed filtrate was washed with water (17.5 L), brine (17.5 L), dried over Na2S04, filtered and the solution was passed through a pad of silica gel (2 kg, mesh size 230-400). The silica gel pad was washed with toluene. The combined filtrate and washing was concentrated under reduced pressure and the residual crude product was stirred with n-hexane (23 L) for 1 h to obtain a solid product. The solid was collected by filtration, washed with n-hexane (5 L) and dried to afford 5-hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxaborolan-2-yl)benzaldehyde (4a) (2.47 kg, 84.6%). H NMR (300 MHz, CDCI3) δ 10.54 (s, 1 H), 7.57 (s, 1 H), 7.33 (s, 1 H), 5.89 (s, 1 H), 4.01 (s, 3H), 1.37 (s, 12H); 1H NMR (300 MHz, DMSO-d6) δ 10.35 (s, 1 H), 9.95 (s, 1 H), 7.33 (s, 1 H), 7.23 (s, 1 H), 3.87 (s, 3H), 1.33 (s, 12H); MS (ES+) 301.1 (M+Na); 579.1 (2M+Na); Analysis calculated for C14H19B05: C, 60.46; H, 6.89; Found: C, 60.60; H, 6.87
The average yield of 5-hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxa-borolan-2-yl)benzaldehyde (4a) from step (3) is 78 – 90%.
The process is also illustrated in Fig. 3.
Step (4): Preparation of 3-Bromo-2,6-dimethylpyridine (5b)

2,6-lutidine (5a) (115 kg, 1073.3 mol) was added into pre-chilled oleum (20-23%, 1015 kg, 2276.7 mol) at 0 °C over a period of 4.5 h (temperature r6ached 14 °C during the addition). Bromine (88.18 kg, 1103.6 mol) was then added at 5-10 °C over a period of 1 h. The reaction mixture was slowly heated to 150 °C over a period of 12h. TLC analysis indicated about 40-50% conversion to product and the formation of a dimer by-product (5%). The reaction mixture was cooled to room temperature and then additional bromine (88.18 kg, 1103.6 mol) was added slowly. The reaction mixture was slowly heated to maintain a temperature of 65-75 °C over a period of 15h. TLC analysis indicated a 65-70 % conversion to product and the formation of 5% dimer by product. The reaction mixture was quenched by addition of water (500L) while maintaining the reaction temperature below 20 °C. The mixture was basified with 6.6 M NaOH (3800 L) while maintain the temperature at < 40 °C. EtOAc (220 L) was added and the mixture was stirred for 1 h then allowed to settle over a period of 2 h. The layers were separated and the aqueous layer was treated with NaOH (10 kg) in water (10 L) and extracted with EtOAc (160 L). The organic extracts were combined washed with brine (100 L), dried over Na2S04 (50.0 kg), filtered and the solvent was evaporated under atmospheric pressure. The residue was vacuum distilled and the desired product 3-bromo-2,6-dimethylpyridine (5b) was collected at 58-60 °C, 2 mmHg (98.45 kg, 49.2 %) as a colorless liquid.
The process is also illustrated in Fig. 4.
Step (5): Preparation of 3-Bromopyridine-2,6-dicarboxylic acid (5c)

5b 5c
To a stirred solution of 3-bromo-2,6-dimethylpyridine (5b) (98 kg, 5326 mol) in water (1310 L) was added KMn0 (225 kg, 1423.6 mol) in 5 equal portions in 1 h intervals at 70 °C. After stirring for 1 h at 70 °C, additional KMn04 (225 Kg, 1423.6 mol) was added in 5 equal portion in 1 h intervals at 90 °C. The reaction mixture was stirred for 12 h at 90 °C. The suspension was filtered hot through celite to obtain a clear solution. The solvent was distilled off to remove about 30% of the total volume. The remaining concentrated solution was chilled to 0 °C and made acidic (to pH 3-4) by the addition of cone. HCI (120 L). The white precipitate obtained was collected by filtration and dried at 70 °C to afford 3-bromopyridine-2,6-dicarboxylic acid (5c) as a white solid (109 kg, 84%).
The process is also illustrated in Fig. 5.
Step (6): Preparation of Dimethyl 3-Bromopyridine-2,6-dicarboxylate (5d)

To a stirred solution of 3-bromopyridine-2,6-dicarboxylic acid (5c) (20.0 kg, 81.29 mol) in methanol (100 L) was added cone. H2S04 (4.4 L) over a period of 30 min. The reaction mixture was heated to 65 °C and maintained at that temperature for 5 h (the reaction was monitored by TLC analysis to determine completion of reaction). The reaction mixture was cooled to room temperature basified by careful addition of aqueous NaHC03 solution (prepared from 10 kg NaHC03 in 120 L of water) and further diluted with water (120 L). The white solid obtained was collected by filtration, washed with plenty of water and then oven-dried at 40 °C to obtain dimethyl 3-bromopyridine-2,6-dicarboxylate (5d) (9.2 kg, 41.3%) as a white solid; 1HNMR (300 MHz, DMSO-cf6) δ 8.47 (d, J = 8.4, 1 H), 8.08 (dd, J = 4.5, 8.4, 1 H), 3.95 (s, 3H), 3.91 (s, 3H); MS (ES+) 570.6 (2M+Na); Analysis calculated for C9H8BrN04: C, 39.44; H, 2.94; Br, 29.15 N, 5. 1 ;
Found: C, 39.52; H, 2.92; Br, 29.28; N, 5.03.
The process is also illustrated in Fig. 6.
6582
Step (7): Preparation of Methyl 3-bromo-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate (

To a stirred solution of dimethyl 3-bromopyridine-2,6-dicarboxylate (5d) (27 kg, 98.52 mol) in ierf-butanol (135 L) was added at room temperature cyclopropylmethanamine (7.83 kg, 110.1 mol). The reaction mixture was heated at 65 °C for 17 h. The progress of reaction was monitored by TLC and HPLC (HPLC analysis showed the formation of 74% of the product 5e after 17 h. The reaction mixture was cooled to room temperature and then cone. HCI (2.7 L) was added slowly and the mixture was stirred for 15 min. The reaction mixture was concentrated under reduced pressure to obtain the crude product. The crude product was dissolved in hot /-PrOH (54 L) filtered through a celite pad. The filtrate was cooled with stirring to 10 °C to obtain a white precipitate. The solid obtained was collected by filtration, washed with cold
i-PrOH (13 kg), n-hexane (15 L) and dried to provide pure methyl 3-bromo-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate (5e) (15.7 kg, 50.9%). The filtrate was concentrated under reduced pressure and the crude product can be purified by silica gel column chromatography eluting with tert-butanol in hexanes to furnish additional 10% methyl 3-bromo-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate (5e). HNMR (300 MHz, DMSO-cf6) δ 8.83 (t, J = 5.9, 1 H), 8.47 – 8.41 (m, 1 H), 8.06 (d, J = 8.4, 1 H), 3.96 (s, 3H), 3.16 (t, J = 6.5, 2H), 1.14 – 0.99 (m, 1 H), 0.42 (m, 2H), 0.30 -0.19 (m, 2H); MS (ES+) 337.0 (M+23), 650.8 (2M+23); Analysis calculated for
C12H13BrN203: C, 46.03; H, 4.18; N, 8.95; Br, 25.52; Found: C, 46.15; H, 4.17; N, 8.72; Br, 25.26.
The average isolated yield for step (7) is 50% to 60%.
The process is also illustrated in Fig. 7.
Step (8): Preparation of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (6a)
2

6a
THF (37.5 L) was charged to a 100 L reactor followed by ethyl 3-bromo-6- (cyclopropylmethyl-carbamoyl)pyridine-2-carboxylate (5e) (2.5 kg, 7.98 mol) under a nitrogen atmosphere. The reaction mixture was degassed twice by applying alternate vacuum and nitrogen. 5-Hydroxy-4-methoxy-2-(4,4,5,5-tetramethyl-[1 ,3,2]dioxa-borolan-2-yl)benzaldehyde (4a) (2.88 kg, 10.36 mol) was added, followed by the addition of PPh3 (53.13 g, 0.20 mol), PdCI2(PPh3)2 (120.4 g, 0.17 mol) and a solution of Na2C03(2.12 kg, 20.00 mol) in demineralized water (10.0 L) under nitrogen atmosphere. The reaction mixture was degassed again two times by applying alternate vacuum and nitrogen. The reaction mixture was heated at reflux for 6.5 h, cooled to room temperature and filtered through a Celite bed. Water (75 L) was added to the filtrate and the product was extracted with ethyl acetate (75 L). The aqueous layer was back extracted with ethyl acetate (2 χ 60 L). The combined ethyl acetate extract was divided into two equal portions and each portion was washed with brine (37 L), dried over Na2S04, filtered and concentrated under reduced pressure to give crude methyl 6- ((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (6a) as a reddish viscous material (-4.5 Kg) which was used as such for the next step without further purification. An analytical sample was prepared by purification of a small sample by flash column chromatography (silica gel, eluting with 0-100% ethyl acetate in hexane) to furnish methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)-picolinate (6a) as an off-white solid; HNMR (300 MHz, DMSO-d6) δ 9.89 (s, 1 H), 9.52 (s, 1 H), 8.79 (t, J = 6.1 Hz, 1 H), 8.23 (d, J = 8.0 Hz, 1 H), 8.09 (d, J = 8.0 Hz, 1 H), 7.34 (s, 1 H), 6.90 (s, 1 H), 3.85 (s, 3H), 3.62 (s, 3H), 3.22 (m, 2H), 1.16 -1.02 (m, 1 H), 0.49 – 0.38 (m, 2H), 0.32 – 0.22 (m, 2H); MS (ES+) 791.0 (2M+Na), (ES-) 382.7 (M-1), 767.3 (2M-1); Analysis calculated for C20H20N2O6.0.25 H20: C, 61.77; H, 5.31 ; N, 7.20; Found: C, 61.54; H, 5.13; N, 7.05.
The process is also illustrated in Fig. 8.
46582
Step (9): Preparation of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-(((trifluoromethyl)sulfonyl)oxy)phenyl)picolinate (6b)

6a 6b
A solution of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-4-hydroxy-5-methoxyphenyl)picolinate (6a) (2.11 kg, estimated about 3.83 mol from step-8) in dichloromethane (16.0 L) and pyridine (1.4 L, 17.4 mol) cooled to -10°C and maintained at that temperature for 1 h was added a solution of triflic anhydride (980.0 ml_, 5.8 mol) in dichloromethane (6.0 L) drop wise over a period of 3 h at -10 °C. The reaction mixture was stirred at -5°C for 1.3 h, quenched with saturated aqueous NaHCO3(10.4 L) and stirred for 30 mins. The organic layer was separated, washed successively with saturated aqueous NaHC03 (10.4 L), 1 HCI (2 x 16.6 L), water (13.2 L), brine (13.2 L), dried over MgS04, filtered and concentrated under reduced pressure to give the crude product. The crude product was stirred with 15% ethyl acetate in n-hexane (7.0 L) for 1 h. The solid obtained was collected by filtration washed with 15% ethyl acetate in n-hexane (3.0 L). The solid was stirred again with 15% ethyl acetate in n-hexane (7.0 L) for 1 h, was collected by filtration and washed with 15% ethyl acetate in n-hexane (3.0 L). The solid was stirred again with 15% ethyl acetate in n-hexane (8.0 L) for 1 h, collected by filtration washed with 15% ethyl acetate in n-hexane (3.0 L). The solid was dried to afford methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-(((trifluoromethyl)sulfonyl)-oxy)phenyl)picolinate (6b) as a light brown solid (1.7 kg, 86% yield, for combined steps 8 & 9). Average isolated yield for combined steps 8 and 9 was 70% to 86%; Ή NMR (300 MHz, DMSO-cf6): δ 9.64 (s, 1 H), 8.78 (t, J = 6.1 , 1 H), 8.29 (d, J = 8.0, 1 H), 8.16 (d, J = 8.0, 1 H), 8.03 (s, 1H), 7.39 (s, 1 H), 4.00 (s, 3H), 3.63 (s, 3H), 3.22 (m, 2H), 1.11 (m, 1 H), 0.52 – 0.39 (m, 2H), 0.28 (m, 2H); MS (ES+) 538.9 (M+Na). The process is also illustrated in Fig. 9.
Step (10): Preparation of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-vinylphenyl)picolinate (6c)

A solution of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4- (((trifluoromethyl)sulfonyl)oxy)phenyl)picolinate (6b) (12 kg, 23.24 mol) in DME (106 L) was charged into reactor under nitrogen. The reaction mixture was degassed twice by applying alternate vacuum and nitrogen. Potassium trifluoro(vinyl)borate (3.9 kg, 29.1 1 mol), PdCI2(PPh3)2 (815 g, 1.13 mol), KHC03 (4.65 g, 46.44 mol) and demineralized water (12 L) was then added under a N2 atmosphere. The reaction mixture was degassed by applying alternate vacuum and nitrogen. The reaction mixture was heated at reflux for 5 h. The reaction mixture was cooled to room temperature and then filtered through a Celite bed. Demineralized water (118 L) was added to the filtrate followed by ethyl acetate (124 L). The mixture was stirred for 20 min and then the organic layer was separated. The aqueous layer was back-extracted with ethyl acetate (2 x 95 L). The combined organic extract was washed with brine (95 L), dried over Na2S04, and filtered. The solvent was evaporated under reduced pressure to give the crude product. The crude product was purified by column chromatography (silica gel, 120 kg, 230-400 mesh size, eluting with ethyl acetate in n-hexane) to obtain methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-vinylphenyl)picolinate (6c) (6 kg, 72%). 1H NMR (300 MHz, CDCI3): δ (ppm) 9.64 (s, 1 H), 8.35 (d, J = 7.8 Hz, 1 H), 8.06-8.03 (m, 2H), 7.78(d, J = 7.8 Hz, 1 H), 7.02-6.92 (m, 1 H), 6.61 (s, 1 H), 5.86 (d, J = 17.7 Hz, 1 H), 5.38 (d, J = 1 1.4 Hz, 1 H), 3.84 (s, 3H), 3.67 (s, 3H), 3.35-3.29 (m, 2H),1.08-1.03 (m, 1H), 0.55-0.49 (m, 2H), 0.29-0.2 4(m, 2H). 1HNMR (300 MHz, DMSO-d6) 6 9.68 (s, 1 H), 8.77 (t, J = 6.1 , 1 H), 8.35 – 8.21 (m, 1 H), 8.16 – 8.01 (m, 2H), 7.14 -6.87 (m, 2H), 6.01 (dd, J = 1.2, 17.8, 1 H), 5.45 (dd, J = 1.1 , 1 1.3, 1 H), 3.91 (s, 3H), 3.64 (s, 3H), 3.23 (m, 2H), 1.21 – 1.01 (m, 1H), 0.51 – 0.40 (m, 2H), 0.34 – 0.20 (m, 2H). MS
(ES+) 417.0 (M+Na); Analysis calculated for C22H22N205: C, 66.99; H, 5.62; N, 7.10;
Found: C, 66.75; H, 5.52; N, 7.06.
The process is also illustrated in Fig. 10.
Step (1 1): Preparation of 2-(6-((cyclopropylmethyl)carbamoyl)-2- (methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d)

To a stirred solution of methyl 6-((cyclopropylmethyl)carbamoyl)-3-(2-formyl-5-methoxy-4-vinylphenyl)picolinate (6c) (1.57 kg, 3.80 mol) in acetonitrile (15.4 L) was added ferf-butyl alcohol (22.2 L), demineralized water (3.2 L) and sodium dihydrogen phosphate monohydrate (323.74 g, 2.346 mol). The reaction mixture was cooled to 0 °C and added 2-methyl-2-butene (5.3 L, 50.0 mol) and stirred at 0 °C for 30 min. A solution of 80% sodium chlorite (1.36 kg, 12.0 mol) in demineralized water (5.2 L) was added to the reaction mixture over a period of 2.5 h at 0 °C [temperature rises to 7 °C during the addition]. The reaction mixture was stirred at 0 °C for 2 h, diluted with water (40 L) and ethyl acetate (24 L). After stirring the mixture, it was allowed to settle and the organic layer was separated. The aqueous layer was back-extracted with ethyl acetate (2 x 20 L) then acidified with 5.9 % aqueous acetic acid (2 L) and extracted once with ethyl acetate (10 L). The organic extracts were combined washed with water (2 x 20 L), a solution of acetic acid (125 mL) in water (20.0 L), brine (2 χ 20 L), dried over Na2S04, filtered and concentrated under reduced pressure (vapor temperature below 40 °C). The residue obtained was dissolved in acetone (7 L) (residue didn’t dissolve completely). The solution was poured slowly into a reactor containing stirred n-hexane (70.0 L) to precipitate the solid product and the mixture was stirred for 2 h. The solid obtained was collected by filtration, washed with 10% acetone in n-hexane (6.3 L), AJ-hexane (6.3 L), dried to afford 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4- methoxy-5-vinylbenzoic acid (6d) as an off-white solid (1.29 Kg, yield: 79.0%). Average isolated yield for step 1 1 is 74% to 84%. 1H NMR (300 MHz, DMSO-d6): δ (ppm) 12.50 (brs, 1 H), 8.69(t, J= 6.0 Hz, 1 H, NH), 8.20 (d, J= 7.9 Hz, 1 H), 8.09 (s, 1 H), 7.95 (d, J= 8.1 Hz, 1 H), 6.97 (dd, J= 18.0, 1 1.3 Hz, 1 H), 6.88 (s, 1 H), 5.92 (d, J= 7.9 Hz, 1 H), 5.38 (d, J= 1 1.1 Hz, 1 H), 3.85 (s, 3H), 3.63 (s, 3H), 3.27-3.17 (m, 2H), 1.15-1.05 (m, 1 H), 0.48-0.40 (m, 2H), 0.31-0.24 (m, 2H); MS (ES+) 433.26, (M+Na); (ES-) 409.28 (M-1). The process is also illustrated in Fig. 1 1.
Step (12): Preparation of Methyl 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylate methanesulfonate (7a

Pyridine (3.8 L, 47.17 mol) and EDCI (5.31 kg, 27.66 mol) were sequentially added to a cooled solution (0 °C) of 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)-pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) (9 kg, 21.92 mol) and 4-aminobenzamidine dihydrochloride (5.13 kg, 24.65 mol) in /-PrOH (90 L). The reaction mixture was allowed to warm to room temperature and stirred for 2 h. TLC analysis indicated incomplete reaction. Additional EDCI (1.08 kg, 5.6 mol) was added and the reaction mixture was stirred for 8 h. The reaction was still incomplete as indicated by TLC analysis, additional EDCI (0.54 kg, 2.8 mol) was added and the reaction mixture was stirred for 5 h. TLC analysis indicated there was trace amount of unreacted starting material remaining. The reaction mixture was cooled to 0 °C and a solution of
methanesulfonic acid (MSA) (9.13 kg, 95 mol) in MeOH (38.7 L) was added to the cooled mixture over a period of 4 h. The reaction mixture was allowed to warm to room temperature and stirred for 15 h. The product was collected by filtration, washed with a mixture of /‘-PrOH and MeOH (4:1 , 45 L). The wet cake was slurried in a mixture of /-PrOH and MeOH (2:1 , 135 L) stirred for 1 h and the product was collected by filtration and washed with a mixture of /‘-PrOH and MeOH (4:1 , 46.8 L). The product was dried in
2015/046582
a vacuum oven at 45 °C to afford methyl 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethyl-carbamoyl)pyridine-2-carboxylate methanesulfonate (7a) as a pink-colored solid (12.71 kg, 93%). Average isolated yield for this step: >90%.
1H NMR (300 MHz, DMSO-c/6) δ 10.71 (s, 1 H), 9.16 (s, 2H), 8.80 (s, 2H), 8.68 (t, J = 6.1 Hz, 1 H), 8.22 (d, J = 8.0 Hz, 1H), 8.06 (d, J = 8.1 Hz, 1 H), 7.93 (s, 1H), 7.84 – 7.72 (m, 4H), 7.12 – 6.97 (m, 2H), 6.04 (dd, J = 17.8, 1.3 Hz, 1 H), 5.45 (d, J = 12.6 Hz, 1H), 3.91 (s, 3H), 3.60 (s, 3H), 3.25 – 3.16 (m, 2H), 2.32 (s, 3H), 1.10 – 1.01 (m, 1 H), 0.48 – 0.37 (m, 2H), 0.30 – 0.22 (m, 2H); MS (ES+) 528.0 (M+1); Analysis calculated for
C29H29N5O5.CH3SO3H.2H2O. C, 54.62; H, 5.65; N, 10.62; S, 4.86; Found: C, 54.95; H, 5.55; N, 10.61 ; S, 4.87.
The process is also illustrated in Fig. 12.
Step (13): Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-rnethoxy-4- vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrate

(3i) ,a 3i
A pre-cooled (0-5 °C) aq. NaOH solution [prepared from solid NaOH (4 kg, 100 mol) in water (86 L)] was added to a suspension of methyl 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethyl-carbamoyl)pyridine-2-carboxylate methanesulfonate (7a) (28.7 kg, 46 mol) in acetonitrile (86 L) cooled to 0 to 5 °C over a period of 25 mins. The reaction mixture was stirred at 0 to 5 °C for 2.5 h (TLC analysis showed the reaction was complete). The reaction mixture was filtered through a sparkler filter, washed with a mixture of 1 :1 CH3CN / H20 ( 57.4 L). Acetic acid (3.2 L, 55.9 mol) in water (56 L) was added to the filtrate at room temperature over a period of 25 mins and the resulting mixture was stirred at room temperature for 2.5 h. The solid product obtained was collected by filtration, washed with a 1 :4 mixture of CH3CN / H20 (57.5 L). The solid was dried at 45°C in a vacuum oven to afford 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6- (cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrate (3i) as an off-white solid (12,77 kg, 54.1%). Average yield for this step is 50% to 75%. Mp: >200°C; H NMR (300 MHz, DMSO-d6): δ 13.49 (s, 1 H), 8.94 (bs, 4H), 8.56 (t, 1 H), 7.82 – 7.71 (m, 2H), 7.67 -7.56 (m, 4H), 7.51 (d, J = 7.8, 1 H), 6.98 (dd, J = 11.3, 17.8, 1 H), 6.68 (s, 1 H), 5.92 (d, J = 16.6, 1 H), 5.36 (d, J = 12.4, 1 H), 3.80 (s, 3H), 3.16 (m, 2H), 1.05 (m, 1 H), 0.43 (m, 2H), 0.24 (m, 2H); MS (ES+) 514.1 (M+1), 536.1 (M+Na), (ES-) 512.1 ; Analysis calculated for C28H27N5O5.3H2O: C, 59.25; H, 5.86; N, 12.34; Found C, 59.50; H,
5.75; N, 12.05. If needed this material can be crystallized from a mixture of acetone and water.
The process is also illustrated in Fig. 13.
Step 14: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b

A pre-cooled (5-8 °C) aqueous NaOH solution (prepared from solid NaOH (1.97 kg, 49.25 mol) in demineralized water (41 L) was added to a pre-cooled (0-5 °C) suspension of (3i) (13.8 kg, 26.9 mol) in acetonitrile (41 L). The reaction mixture was stirred at 0-5 °C for 30 min (until the reaction mixture becomes homogeneous). The reaction mixture was filtered through a sparkler filter washed with 50% acetonitrile in demineralized water (4.4 L). The filtrate was charged into a reactor and cooled to 0-5 °C. Aqueous HCI [prepared from cone. HCI (9.3 L) in demineralized water (36 L)] was added slowly with stirring to keep the reaction temperature at or below 15 °C, the resulting mixture was stirred at 10-15 °C for 13 h. The reaction mixture was cooled to 0-5 °C and stirred for 1 h. The solid obtained was collected by filtration and washed with demineralized water (36 L). The solid product was suspended in water (69 L) stirred for 30 mins and collected by filtration washed twice with water (20 L each). The solid product was dried in a vacuum oven at 45°C to afford 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-
(cyclopropylmethyl carbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) (1 1.21 Kg, 75.77%). Mp: >200°C; 1H NMR (300 MHz, DMSO-ci6): δ 12.98 (br s, 1 H), 10.86 (s, 1 H), 9.24 (s, 3H), 9.04 (s, 2H), 8.22 (d, J = 7.8 Hz, 1 H), 7.96 (d, J = 5.7 Hz, 2H), 7.78 (s, 4H), 7.09-6.99 (m, 2H), 6.07 (d, J = 17.7 Hz, 1 H), 5.45(d, J = 11.4 Hz, 1 H), 3.88 (s, 3H), 3.26-3.24 (m, 2H), 1.09 (m, 1 H), 0.47 (m, 2H), 0.28 (m, 2H).
Average isolated yield for this step varies from 63% to 80%.
The process is also illustrated in Fig. 14.
Example-2: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid sulfate salt (8b)

6d 8a
To a solution of 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) (2.35 g, 5.7 mmol) and 4-aminobenzamidine dihydrochloride (1.79 g, 8.6 mmol) in DMF (20 mL) and pyridine (30 ml_) at 0 °C was added EDCI (1.65 g, 8.6 mmol) and allowed to warm to room temperature overnight. The
reaction mixture was quenched with 6N HCI (60 mL) and extracted with chloroform (3 x 60 mL). The organic layer was dried over MgS04, filtered and concentrated in vacuum. The residue obtained was purified by flash column chromatography (silica gel, 110 g, eluting with 0 to 100% chloroform in CMA 80 and 0-100% chloroform in CMA 50) to furnish methyl 3-(2-((4-carbamimidoylphenyl)carbamoyl)-5-methoxy-4-vinylphenyl)-6-((cyclopropylmethyl)-carbamoyl)picolinate hydrochloride (8a) (2.2 g, 65%) as a white solid; MP 266 °C; 1HNMR (300 MHz, DMSO-d6) δ 10.78 (s, 1 H), 9.26 (s, 2H), 9.03 (s, 2H), 8.67 (t, J = 6.1 , 1 H), 8.22 (d, J = 8.0, 1 H), 8.06 (d, J = 8.0, 1 H), 7.96 (s, 1 H), 7.89 -7.74 (m, 4H), 7.13 – 6.96 (m, 2H), 6.07 (d, J = 17.7, 1 H), 5.45 (d, J = 12.4, 1 H), 3.91 (s, 3H), 3.61 (s, 3H), 3.20 (s, 2H), 1.09 (dd, J = 4.7, 8.2, 1 H), 0.43 (dt, J = 4.9, 5.4, 2H), 0.34 – 0.21 (m, 2H); MS (ES+) 528.1 (M+1); Analysis calculated for C29H29N505 (H20)1 5 (HCI): C, 58.93; H, 5.63; N, 1 1.85; Found: C, 58.75; H, 5.65; N, 1 1.92.
Step-2: preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid sulfate salt (8b)

8a 8b j0 a solution of methyl 3-(2-((4-carbamimidoylphenyl)carbamoyl)-5-methoxy-4-vinylphenyl)-6-((cyclopropylmethyl)carbamoyl)picolinate hydrochloride (8a) (1.128 g, 2 mmol) in acetonitrile (5 ml), was added 1 N aqueous sodium hydroxide (5.00 ml, 5.00 mmol) and stirred at room temperature for 2 h, TLC [CMA80/CMA50 (7/3)] shows reaction was complete. The reaction mixture was neutralized with a solution of sulfuric acid (0.483 ml, 9.00 mmol) in water (5 mL) and stirred for 10 min at room temperature. To this cold water (5 ml) was added and stirred at room temperature until product crystallized out. Cold water (5 mL) was added to the slurry and stir for additional 20 min, additional cold water (5 mL) was added prior to filtration of solid. The solid obtained was collected by filtration washed with water (5 mL and 2.5 mL), dried under vacuum overnight to afford 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-
(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid sulfate salt (8b) (1.103 g, 90 % yield) as a white solid; MP 221.7 °C; H NMR (300 MHz, DMSO-d6) δ 12.30 – 10.91 (bs, 1 H, D20 exchangeable), 10.69 (bs, 1 H, D20 exchangeable), 9.24 (t, J = 6.0 Hz, 1 H), 9.16 (s, 2H, D2O exchangeable), 8.78 (s, 2H, D2O exchangeable), 8.24 (d, J = 8.0 Hz, 1 H), 8.04 – 7.91 (m, 2H), 7.84 – 7.67 (m, 4H), 7.13 – 6.94 (m, 2H), 6.03 (dd, J = 17.8, 1 .4 Hz, 1 H), 5.51 – 5.37 (m, 1 H), 3.88 (s, 3H), 3.24 (t, J = 6.4 Hz, 2H), 1.16 – 1.01 (m, 1 H), 0.52 – 0.41 (m, 2H), 0.32 – 0.22 (m, 2H); MS (ES+) 514.0 (M+1); Analysis calculated for: C28H27N605 1.0H2SO4 1.5H20: C, 52.66; H, 5.05; N, 10.97; S, 5.02; Found: C, 52.81 ; H, 4.95; N, 10.94; S, 4.64.
Example-3: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid methane s

To a solution of methyl 3-(2-((4-carbamimidoylphenyl)carbamoyl)-5-methoxy-4-vinylphenyl)-6-((cyclopropylmethyl)carbamoyl)picolinate hydrochloride (8a) (1.128 g, 2 mmol) in acetonitrile (5 ml) was added 1 N aqueous sodium hydroxide (5.00 ml, 5.00 mmol) and stirred at room temperature for 2 h, TLC [CMA80/CMA50 (7/3)] shows reaction was complete. The reaction mixture was neutralized with methanesulfonic acid (0.584 ml, 9.00 mmol) and stirred for 1 h at room temperature. Cold water (5.00 ml) was added to the reaction mixture and stirred at room temperature until product crystallized out. To the slurry was added water (5 ml) of water stirred for additional 20 min, followed by the addition of water (5 ml) prior to filtration. The solid obtained was collected by filtration washed with water (5 ml and 2.5 ml), dried under vacuum to afford 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6- (cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid methane sulfonate salt (8c)
(1 .138 g, 1.867 mmol, 93 % yield) as a white solid; MP 221.2 °C; 1 H NMR (300 MHz,
DMSO-d6) δ 12.89 (s, 1 H, D2O exchangeable), 10.69 (s, 1 H, D2O exchangeable), 9.24
(t, J = 6.0 Hz, 1 H), 9.16 (s, 2H,), 8.85 (s, 2H), 8.24 (d, J = 8.0 Hz, 1 H), 8.06 – 7.91 (m, 2H), 7.86 – 7.70 (m, 4H), 7.15 – 6.96 (m, 2H), 6.03 (dd, J = 17.8, 1.4 Hz, 1 H), 5.52 – 5.35 (m, 1 H), 3.88 (s, 3H), 3.25 (t, J = 6.3 Hz, 2H), 2.34 (s, 3H), 1.17 – 1.01 (m, 1 H), 0.53 -0.43 (m, 2H), 0.32 – 0.23 (m, 2H); MS (ES+) 514.0 (M+1); Analysis calculated for:
CzeH^NsOsCHsSOsH 1.5H20: C, 54.71 ; H, 5.38; N, 11.00; S, 5.04; Found: C, 54.80; H, 5.14; N, 10.94; S, 4.90.
Example-4: Preparation of 3-[2-(4-Carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) in Form C (Compound XX)

The jacket of a 10 L glass reactor was set to -5 °C. To the reactor was charged 2-(6-((cyclopropylmethyl)carbamoyl)-2-(methoxycarbonyl)-pyridin-3-yl)-4-methoxy-5-vinylbenzoic acid (6d) prepared in Step (11) of Example 1 (500 g, 1.22 mol), 4-amino-benzamidine-2HCI (280 g, 1.34 mol), and 2-propanol (4.05 kg). The mixture was cooled
46582
to 0.3 °C, and pyridine (210 g, 2.62 mol) followed by EDCI HCI (310 g, 1.61 mol) was added. The mixture was stirred at -1.1 – -0.3 °C for 22 hrs followed by addition of the second portion of EDCI HCI (58 g, 0.30 mol). The temperature of jacket was set to 14.0 °C, and the mixture was stirred for 89 hrs. The precipitate was filtered, and washed with 1.32 kg of 2-propanol.
The wet product (8a) was recharged to the reactor followed by addition of acetonitrile (1 .6 kg) and 0.57 kg water. The mixture was heated to 46 °C. 21 g of Smopex-234 and 10 g Acticarbone 2SW were added and the mixture was stirred at this temperature for 1 hr. The solution was filtered, and filtrate was returned back to the reactor. The jacket of the reactor was set to -5 °C, and the mixture was cooled to -0.2 °C. NaOH solution (256 g 46% NaOH, 2.95 mol, in 960 g water) was added in 25 min keeping the temperature ❤ °C. The mixture was stirred at 0.2-2.0 °C for 1 hr 40 min and then quenched with cone, acetic acid (40 g, 0.66 mol). Diluted acetic acid (80 g, 1.33 mol AcOH in 1000 g water) was added during 1 hr 20 min (temperature 1.7-3.0 °C), followed by 1250 g water (30 min). The suspension was stirred at 0-3.0 °for 1 hr, and filtered at 0-5 °C (ice mantle around the filter). The reactor and product (8d) was rinsed with 3.5 kg water.
The wet product (8d) was recharged to the reactor followed by 0.65 kg water and 1.69 kg acetonitrile. The mixture was heated to 57-60 °C, and stirred at this temperature for 14.5 hrs. The mixture was cooled to -2.2 °C (Tjacke,= -5 °C), and a solution of NaOH (163 g 46%, 1.87 mol, in 580 g water) was added during 15 min. The temperature rose to -0.4 °C. Hydrochloric acid (407 g 37% HCI, 4 mol) was added in 10 min, the temperature rose to 7.5 °C. The suspension was agitated at -3 – 0 °C for 19 hrs. The product was filtered and the filter cake was rinsed with 2.87 kg water, compressed and pulled dry. The wet product (1.30 kg) was dried at 40-43 °C and 50 mbar for 1 17 hrs to furnish 3-[2-(4-carbamimidoylphenylcarbamoyl)-5-methoxy-4-vinylphenyl]-6-(cyclopropylmethylcarbamoyl)pyridine-2-carboxylic acid hydrochloride (7b) (484 g) as Form C (Compound XX).
/////avoralstat, BCX4161, Fast Track, Treat hereditary angioedema (HAE), Orphan Drug, PRECLINICAL
COc1cc(c(cc1C=C)C(=O)Nc2ccc(cc2)C(=N)N)c3cc(ncc3C(=O)O)C(=O)NCC4CC4
BRIVARACETAM
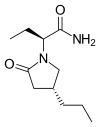
BRIVARACETAM, UCB-34714
(2S)-2-[(4R)-2-oxo-4-propylpyrrolidin-1-yl]butanamide
| Molecular Formula: | C11H20N2O2 |
|---|---|
| Molecular Weight: | 212.2887 g/mol |
UNII-U863JGG2IA
UCB; For the treatment of partial onset seizures related to epilepsy, Approved February 2016
Brivaracetam, the 4-n-propyl analog of levetiracetam, is a racetam derivative with anticonvulsant properties.[1][2] Brivaracetam is believed to act by binding to the ubiquitous synaptic vesicle glycoprotein 2A (SV2A).[3] Phase II clinical trials in adult patients with refractory partial seizures were promising. Positive preliminary results from stage III trials have been recorded,[4][5] along with evidence that it is around 10 times more potent[6] for the prevention of certain types of seizure in mouse models than levetiracetam, of which it is an analogue.
On 14 January 2016, the European Commission,[7] and on 18 February 2016, the USFDA[8] approved brivaracetam under the trade name Briviact (by UCB). The launch of this anti-epileptic is scheduled for the first quarter of that year. Currently, brivaracetam is still not approved in other countries like Australia, Canada and Switzerland.
Brivaracetam was approved by European Medicine Agency (EMA) on Jan 14, 2016 and approved by the U.S. Food and Drug Administration (FDA) on Feb 18, 2016. It was developed and marketed as Briviact® by UCB in EU/US.
Brivaracetam is a selective high-affinity synaptic vesicle protein 2A ligand, as an adjunctive therapy in the treatment of partial-onset seizures with or without secondary generalization in adult and adolescent patients from 16 years of age with epilepsy.
Briviact® is available in three formulations, including film-coated tablets, oral solution and solution for injection/infusion. And it will be available as 10 mg, 25 mg, 50 mg, 75 mg and 100 mg film-coated tablets, a 10 mg/ml oral solution, and a 10 mg/ml solution for injection/infusion. The recommended starting dose is either 25 mg twice a day or 50 mg twice a day, depending on the patient’s condition. The dose can then be adjusted according to the patient’s needs up to a maximum of 100 mg twice a day. Briviact can be given by injection or by infusion (drip) into a vein if it cannot be given by mouth.
European Patent No. 0 162 036 Bl discloses compound (S)-α-ethyl-2-oxo-l- pyrrolidine acetamide, which is known under the International Non-proprietary Name of Levetiracetam.

Levetiracetam
Levetiracetam is disclosed as a protective agent for the treatment and prevention of hypoxic and ischemic type aggressions of the central nervous system in European patent EP 0 162 036 Bl. This compound is also effective in the treatment of epilepsy.
The preparation of Levetiracetam has been disclosed in European Patent No. 0 162 036 and in British Patent No. 2 225 322.
International patent application having publication number WO 01/62726 discloses 2-oxo-l -pyrrolidine derivatives and methods for their preparation. It particularly discloses compound (2S)-2-[(4R)-2-oxo-4-propyl-pyrrolidin-l-yl] butanamide known under the international non propriety name of brivaracetam.

Brivaracetam
International patent application having publication number WO 2005/121082 describes a process of preparation of 2-oxo-l -pyrrolidine derivatives and particularly discloses a process of preparation of (2S)-2-[(4S)-4-(2,2-difluorovinyl)-2-oxo-pyrrolidin-l- yl]butanamide known under the international non propriety name of seletracetam.

Seletracetam
Kenda et al., in J. Med. Chem. 2004, 47, 530-549, describe processes of preparation of 2-oxo-l -pyrrolidine derivatives and particularly discloses compound 1-((1S)-I- carbamoyl-propyl)-2-oxo-pyrrolidone-3-carboxylic acid as a synthetic intermediate.
WO2005028435
CLIPS
Find better ways to make old and new epilepsy drugs. J. Surtees and co-inventors disclose alternative processes for making active pharmaceutical ingredients (APIs) that are used to treat epilepsy and seizures. One compound that can be prepared by their processes is the established drug levetiracetam (1, Figure 1), marketed under the trade name Keppra. Because 1 is now off-patent, there is obvious interest in new drugs.

The inventors also claim that seletracetam (2) and brivaracetam (3) (Figure 2) can be prepared by their processes. These drugs are apparently much more active than 1.
All of the drugs are used as single isomers, so a stereoselective synthesis is desirable. The inventors describe two routes for preparing the molecules; the first, shown in Figure 1, is the synthesis of 1 by the reaction between pyrrolidone (4) and chiral bromo amide 5 in the presence of a base. GC analysis showed that the conversion is 40.3% and that the product contains 51% of the (S)-enantiomer and 49% of the (R)-isomer. No details of their separation are given, although the use of chiral HPLC is discussed.
The same reaction is used to prepare derivative 6 of 1. Compound 7 is prepared from the corresponding hydroxy ester and then condensed with 4 to give 6. Chiral HPLC showed that the product is a mixture of 89.3% (S)-enantiomer 6 and 10.7% of its (R)-isomer.
The inventors do not describe the detailed preparation of 2, but they report that acid 8 is prepared in 41% yield from pyrrolidone 9 and acid 10 in the presence of NaH (Figure 2). Ammonolysis of 8 produces 2; no reaction details are provided.

In a reaction similar to the preparation of 8, acid 11 is prepared from 10 and pyrrolidone 12. The product is isolated in 77% yield and can be converted to 3 by ammonolysis. Again, no details are provided for this reaction.
The second route for preparing the substituted pyrrolidones does not start with simple pyrrolidones and is the subject of additional claims. The route involves a cyclization reaction, shown in Figure 3. The preparation of enantiomer 13 begins with the reaction of racemic salt 14 and optically pure bromo ester 15. This step produces intermediate 16, isolated as a yellow oil. The crude material is treated with 2-hydroxypyridine (2-HP) to cyclize it to 17. This ester is hydrolyzed to give acid 18. Conversion to 13 is carried out by adding ClCO2Et, followed by reaction with liquid NH3 in the presence of K2CO3. The overall yield of 13 is 32%.

This route is also used to prepare levetiracetam (1) by treating 5 with the HCl salt of amino ester 19 to give 20, recovered as its HCl salt in 49% yield. The salt is basified with Et3N and treated with 2-HP to cyclize it to 1, initially isolated as an oil. GC analysis showed 100% conversion, and chiral HPLC showed that the product contains 98.6% (S)-isomer and 1.4% (R)-isomer.
The inventors also prepared 1 and its (R)-enantiomer 21 by using a similar reaction scheme with alternative substrates to 5. Figure 4 outlines the route, which starts from protected hydroxy amide 22 and amino ester 23. When the reaction is carried out in the presence of Cs2CO3, the product is (R)-enantiomer24, which is used without purification to prepare 21 by treating it with 2-HP. Chiral HPLC showed that the product is 94% (R) and 6% (S).

When the reaction between 22 and 23 is run with K2CO3, the product is (S)-enantiomer 25. This is used to prepare 1, but the product contains only 79% (S)-isomer.
The inventors do not comment on the apparent stereoselectivity of the carbonate salts in the reaction of 22 with 23. This is an intriguing finding and worthy of investigation. (UCB S.A. [Brussels]. US Patent 8,338,621

SYNTHESIS
PATENT
WO2005028435
Example 1: Synthesis of (2S)-2-((4R)-2-oxo-4-n-propyl-l-pyrrolidinyl)butanamide 1.1 Synthesis of (2S)-2-aminobutyramide free base

1800 ml of isopropanol are introduced in a 5L reactor. 1800 g of (2S)-2- aminobutyramide tartrate are added under stirring at room temperature. 700 ml of a 25% aqueous solution of ammonium hydroxide are slowly added while maintaining the temperature below 25°C. The mixture is stirred for an additional 3 hours and then the reaction is allowed to complete at 18°C for 1 hour. The ammonium tartrate is filtered. Yield : 86%.
1.2 Synthesis of 5-hydroxy-4-n-propyl-furan-2-one

Heptane (394 ml) and morpholine (127.5 ml) are introduced in a reactor. The mixture is cooled to 0°C and glyoxylic acid (195 g, 150 ml, 50w% in water) is added. The mixture is heated at 20°C during 1 hour, and then valeraldehyde (148.8 ml) is added . The reaction mixture is heated at 43°C during 20 hours. After cooling down to 20CC, a 37 % aqueous solution of HCl (196.9 ml) is slowly added to the mixture, which is then stirred during 2 hours.
After removal of the heptane phase, the aqueous phase is washed three times with heptane. Diisopropyl ether is added to the aqueous phase. The organic phase is removed, and the aqueous phase further extracted with diisopropyl ether (2x). The diisopropyl ether phases are combined, washed with brine and then dried by azeotropic distillation. After filtration and evaporation of the solvent, 170g of 5- hydroxy-4-n-propyl-furan-2-one are obtained as a brown oil. Yield: 90.8 %
1.3 Synthesis of (2S)-2-((4R)-2-oxo-4-n-propyl-l-pyrrolidinyl)butanamide and (2S)-2-((4S)-2-oxo-4-n-propyl-l-pyrrolidinyl)butanamide

(S, R) (S, S) The (2S)-2-aιninobutyrarnide solution in isopropanol containing 250 g obtained as described here above is dried by azeotropic distillation under vacuum. To the dried (2S)-2-am obutyraιnide solution is added 5-hydroxy-4-n-propyl-furan-2-one (290 g) between 15°C and 25 °C; the mixture is heated to 30 °C and kept for at least 2 hours at that temperature. Acetic acid (1, 18 eq.), Pd/C catalyst (5 w/w%; Johnson Matthey 5% Pd on carbon – type 87L) are then added and hydrogen introduced into the system under pressure. The temperature is kept at 40 °C maximum and the H2 pressure maintained between 0,2 bar and 0,5 bar followed by stirring for at least 20 hours following the initial reaction. The solution is then cooled to between 15 °C and 25 °C and filtered to remove the catalyst. The solution of product in isopropanol is solvent switched to a solution of product in isopropyl acetate by azeotropic distillation with isopropyl acetate. The organic solution is washed with aqueous sodium bicarbonate followed by a brine wash and then filtered. After recristallisation, 349 g of (2S)-2-((4R)-2- oxo-4-n-propyl-l-pyrrolidinyl)butanamide and (2S)-2-((4S)-2-oxo-4-n-propyl-l- pyιτolidinyl)butanamide are obtained (Yield: 82.5%).
1.4 Preparation of (2S)-2-((4R)-2-oxo-4-n-propyl-l-pyrrolidinyl)butanamide The chromatographic separation of the two diastereoisomers obtained in 1.3 is performed using of (CHIRALPAK AD 20 um) chiral stationary phase and a 45/55 (volume /volume) mixture of n-heptane and ethanol as eluent at a temperature of 25 + 2°C. The crude (2S)-2-((4R)-2-oxo-4-n-propyl-l-pyrrolidinyl)butanamide thus obtained is recristallised in isopropylacetate, yielding pure (2S)-2-((4R)-2-oxo-4-n-propyl-l- pyrrolidinyl)butanamide (Overall yield: 80%) .
Example 2: Synthesis of (2S)-2-((4R)-2-oxo-4-n-propyl-l-pyrrolidinyl)butanamide

Example 1 is repeated except that in step 1.1 a solution of (2S)-2- aminoburyramide.HCl in isopropanol is used (27.72 g, 1.2 equivalent), which is neutralised with a NHs/isopropanol solution (3,4-3,7 mol/L). The resulting ainmonium chloride is removed from this solution by filtration and the solution is directly used for reaction -with 5-hydroxy-4-n-propyl-furan-2-one (23.62 g, 1.0 equivalent) without intermediate drying of the (2S)-2-aminobutyramide solution. Yield after separation of the two diastereoisomers and recristallisation: approximately 84%.

Ref ROUTE1
1. WO0162726A2.
2. WO2005028435A1 / US2007100150A1.
3. J. Med. Chem. 1988, 31, 893-897.
4. J. Org. Chem. 1981, 46, 4889-4894.
PATENT
https://www.google.com/patents/WO2007031263A1?cl=en
Example 3-Synthesis of brivaracetam (I)
3.a. Synthesis of (S) and (R) 2-((R)-2-oxo-4-propyl-pyrrolidin-l-yl)-butyric acid methyl ester fVIaa*) and (Wlab)

(VIaa) (VIab) A slurry of 60% sodium hydride suspension in mineral oil (0.94g, 23.4 mmol) in tetrahydrofuran (30 mL) is cooled at 0°C under a nitrogen atmosphere. A solution of substantially optically pure (R)-4-propyl-pyrrolidin-2-one (Ilia) (2g, 15.7 mmol) in tetrahydrofuran (2 mL) is added over a 15 minutes period. The reaction mixture is stirred 10 min at 0°C then a solution of methyl-2-bromo-butyric acid methyl ester (V) (3.69g, 20.4 mmol) in tetrahydrofuran (2mL) was added over a 20 minutes period. The reaction mixture is stirred at O0C until maximum conversion of starting material and the reaction mixture is then allowed to warm to room temperature and diluted with water (20 mL). Tetrahydrofuran is removed by evaporation and the residue is extracted with isopropyl acetate (20 ml + 10 mL). The combined organic layers are dried on anhydrous magnesium sulfate and evaporated to afford 3g (13.2 mmol, 86 %) of a mixture of epimers of compound (Via), as a mixture respectively of epimer (VIaa) and epimer (VIab). 1H NMR(400 MHz, CDCI3) of the mixture of epimers (VIaa) and (VIab) : δ = 4.68
(dd, J= 10.8, J= 5.1, 2×1 H) ; 3.71 (s, 2x3H); 3.60 (t app, J= 8.2, IH); 3.42 (t app, J= 8.7, IH); 313 (dd, J= 9.2, J = 6.8, IH); 2.95 (dd, J= 9.2, J= 6.8, IH); 2.56 (dd, J= 16.6, J = 8.7, 2xlH); 2.37 (dm, 2xlH); 2.10 (m, 2xlH); 2.00 (m, 2xlH); 1.68 (m, 2xlH); 1.46 (m, 2x2H); 1.36 (m, 2x2H); 0.92 (m, 2x6H).
13C NMR (400 MHz, CDCl3) of the mixture of epimers (VIaa) and (VIab) : δ =
175.9; 175.2; 171.9; 55.3; 52.4; 49.8; 49.5; 38.0; 37.8; 37.3; 36.9; 32.5; 32.2; 22.6; 22.4; 21.0; 14.4; 11.2; 11.1
HPLC (GRAD 90/10) of the mixture of epimers (VIaa) and (VIab): retention time= 9.84 minutes (100 %)
GC of the mixture of epimers (VIaa) and (VIab): retention time = 13.33 minutes (98.9 %)
MS of the mixture of epimers (VIaa) and (VIab) (ESI) : 228 MH+
3.b. Ammonolysis of compound of the mixture of (VIaa) and (VIab)

(VIaa) (VIab) (I) (VII)
A solution of (VIaa) and (VIab) obtained in previous reaction step (1.46g, 6.4 mmol) in aqueous ammonia 50 % w/w (18 mL) at 00C is stirred at room temperature for 5.5hours. A white precipitate that appears during the reaction, is filtered off, is washed with water and is dried to give 0.77g (3.6 mmol, yield = 56 %) of white solid which is a mixture of brivaracetam (I) and of compound (VII) in a 1 :1 ratio.
1H NMR of the mixture (I) and (VII) (400 MHz, CDCI3) : δ = 6.36 (s, broad, IH); 5.66 (s, broad, IH); 4.45 (m, IH); 3.53 (ddd, J= 28.8, J= 9.7, J= 8.1, IH); 3.02 (m, IH); 2.55 (m, IH); 2.35 (m, IH); 2.11 (m, IH); 1.96 (m, IH); 1.68 (m, IH); 1.38 (dm, 4H); 0.92 (m, 6H). 13c NMR of the mixture (I) and (VII) (400 MHz, CDCl3) : δ = 176.0; 175.9; 172.8;
172.5; 56.4; 56.3; 50.0; 49.9; 38.3; 38.1; 37.3; 37.0; 32.3; 32.2; 21.4; 21.3; 21.0; 20.9; 14.4; 10.9; 10.8
HPLC (GRAD 90/10) of the mixture of (I) and (VII) retention time= 7.67 minutes (100 %)
Melting point of the mixture of (I) and (VII) = 104.90C (heat from 400C to 1200C at 10°C/min)
Compounds (I) and (VII) are separated according to conventional techniques known to the skilled person in the art. A typical preparative separation is performed on a 11.7g scale of a 1 :1 mixture of compounds (I) and (VII) : DAICEL CHIRALPAK® AD 20 μm, 100*500 mm column at 300C with a 300 mL/minutes debit, 50 % EtOH – 50 % Heptane. The separation affords 5.28g (45 %) of compound (VII), retention time = 14 minutes and 5.2Og (44 %) of compounds (I), retention time = 23 minutes.
1H NMR of compound (I) (400 MHz, CDCl3): δ = 6.17 (s, broad, IH); 5.32 (s, broad, IH); 4.43 (dd, J= 8.6, J= 7.1, IH); 3.49 (dd, J= 9.8, J= 8.1, IH); 3.01 (dd, J= 9.8, J= 7.1, IH); 2.59 (dd, J= 16.8, J= 8.7, IH); 2.34 (m, IH); 2.08 (dd, J= 16.8, J= 7.9, IH); 1.95 (m, IH); 1.70 (m, IH); 1.47-1.28 (m, 4H); 0.91 (dt, J= 7.2, J= 2.1, 6H)
HPLC (GRAD 90/10) of compound (I) : retention time = 7.78 minutes
1H NMR of compound (VII) (400 MHz, CDCl3): δ = 6.14 (s, broad, IH); 5.27 (s, broad, IH); 4.43 (t app, J = 8.1, IH); 3.53 (t app, J = 9.1, IH); 3.01 (t app, J = 7.8, IH); 2.53 (dd, J = 16.5, J = 8.8, IH); 2.36 (m, IH); 2.14 (dd, J = 16.5, J = 8.1, IH); 1.97 (m, IH); 1.68 (m, IH); 1.43 (m, 2H); 1.34 (m, 2H); 0.92 (m, 6H)
3c. Epimerisation of compound of (2RV2-((R)-2-oxo-4-propyl-pyπOlidin-l-ylV butyramide (VID
Compound (VII) (200 mg, 0.94 mmol) is added to a solution of sodium tert- butoxide (20 mg, 10 % w/w) in isopropanol (2 mL) at room temperature. The reaction mixture is stirred at room temperature for 18h. The solvent is evaporated to afford 200 mg
(0.94 mmol, 100 %) of a white solid. Said white solid is a mixture of brivaracetam (I) and of (VII) in a ratio 49.3 / 50.7.
HPLC (ISO80): retention time= 7.45 min (49.3%) brivaracetam (I); retention time= 8.02 minutes (50.7%) compound (VII).

Reference:ROUTE 2
1. WO2007031263A1 / US2009318708A1.
PATENT
http://www.google.com/patents/WO2007065634A1?cl=en
(scheme 3).

Scheme 3
scheme 4.

5h. Synthesis of brivaracetam and (V) A suspension of (Id) and (Ie) (0.6 g, 2.3 mmol) in MIBK (10 mL) is heated at
120°C for 6 hours. The resulting solution is concentrated and separated on chromatography column (Silicagel 600.068-0.200 mm, cyclohexane/EtOAc : 10/90) to give 0.13 g of brivaracetam (0.6 mmol, 26 %, ee = 94 %) and (V).
1H NMR (400 MHz, CDCl3): δ = 6.17 (s, broad, IH); 5.32 (s, broad, IH); 4.43 (dd, J= 8.6, J= 7.1, IH); 3.49 (dd, J= 9.8, J= 8.1, IH); 3.01 (dd, J= 9.8, J= 7.1, IH); 2.59 (dd, J= 16.8, J= 8.7, IH); 2.34 (m, IH); 2.08 (dd, J= 16.8, J= 7.9, IH); 1.95 (m, IH); 1.70 (m, IH); 1.47-1.28 (m, 4H); 0.91 (dt, J= 7.2,J= 2.1, 6H).
HPLC (method 90/10) : Retention time = 7.78 minutes Chiral HPLC : Retention time = 9.66 minutes (97%) MS (ESI): 213 MH+

Route 3
Reference:1. WO2007065634A1 / US2009012313A1.
PAPER
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.6b00094
A Biocatalytic Route to the Novel Antiepileptic Drug Brivaracetam


AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- von Rosenstiel P (Jan 2007). “Brivaracetam (UCB 34714)”. Neurotherapeutics 4 (1): 84–7. doi:10.1016/j.nurt.2006.11.004.PMID 17199019.
- Malawska B, Kulig K (Jul 2005). “Brivaracetam UCB”. Current Opinion in Investigational Drugs 6 (7): 740–746. PMID 16044671.
- Rogawski MA, Bazil CW (Jul 2008). “New molecular targets for antiepileptic drugs: alpha(2)delta, SV2A, and K(v)7/KCNQ/M potassium channels”. Current Neurology and Neuroscience Reports 8 (4): 345–352. doi:10.1007/s11910-008-0053-7. PMC 2587091.PMID 18590620.
- Clinical trial number NCT00464269 for “Double-blind, Randomized Study Evaluating the Efficacy and Safety of Brivaracetam in Adults With Partial Onset Seizures” at ClinicalTrials.gov
- Rogawski MA (Aug 2008). “Brivaracetam: a rational drug discovery success story”. British Journal of Pharmacology 154 (8): 1555–7.doi:10.1038/bjp.2008.221. PMC 2518467. PMID 18552880.
- Matagne A, Margineanu DG, Kenda B, Michel P, Klitgaard H (Aug 2008). “Anti-convulsive and anti-epileptic properties of brivaracetam (ucb 34714), a high-affinity ligand for the synaptic vesicle protein, SV2A”. British Journal of Pharmacology 154 (8): 1662.doi:10.1038/bjp.2008.198. PMID 18500360.
- http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/003898/human_med_001945.jsp&mid=WC0b01ac058001d124
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm486827.htm
|
|||
| Names | |||
|---|---|---|---|
| IUPAC name
(2S)-2-[(4R)-2-oxo- 4-propylpyrrolidin-1-yl] butanamide
|
|||
| Identifiers | |||
| 357336-20-0 |
|||
| ChEMBL | ChEMBL607400 |
||
| ChemSpider | 8012964 |
||
| Jmol interactive 3D | Image | ||
| PubChem | 9837243 | ||
| UNII | U863JGG2IA |
||
| Properties | |||
| C11H20N2O2 | |||
| Molar mass | 212.15 g/mol | ||
| Pharmacology | |||
| ATC code | N03 | ||
| Legal status |
|
||
| Oral | |||
| Pharmacokinetics: | |||
| Nearly 100% | |||
| <20% | |||
| Hydrolysis, CYP2C8-mediated hydroxylation | |||
| 8 hrs | |||
| >75% renal | |||
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|||


//////BRIVARACETAM, UCB, 2016 FDA, UCB-34714
CCCC1CC(=O)N(C1)C(CC)C(=O)N
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021260721&_cid=P12-KXX1JU-33531-1
Brivaracetam is chemically known as (2S)-2-[(4R)-2-oxo-4-propyltetrahydro-1H-pyrrol-1-yl] butanamide, having the chemical structure of formula 1 as below:
Brivaracetam is basically a chemical analogue of Levetiracetam, marketed under the brand name of BRIVIACT for the treatment as adjunctive therapy in the treatment of partial-onset seizures in patients at 16 years of age and older with epilepsy. Brivaracetam has an advantage over Levetiracetam in that it gets into the brain “much more quickly,” which means that “it could be used for status epilepticus, or acute seizures than cluster, or prolonged seizures”. From the Phase III trials, the self-reported rate of irritability with Brivaracetam was 2% for both drug doses (100 mg and 200 mg) vs 1% for placebo, which compares to as much as 10% for Levetiracetam in some post-marketing studies.
With the improved safety profile and possibility to be used for wider range of epilepsy, Brivaracetam is considered as one of the most promising 3rd generation antiepileptic drugs.
Brivaracetam molecule is first disclosed in patent publication WO2001062726, which describes 2-oxo-1 -pyrrolidine derivatives and methods for their preparation. This patent publication further discloses compound (2S)-2-[(4R)-2-oxo-4-propyl-pyrrolidin-1-yl] butanamide which is known under the international non propriety name as Brivaracetam. As per Biopharmaceutics Classification System, Brivaracetam is a class I drug (high solubility and permeability).
Some prior arts US6784197 and US7629474 disclose a process for synthesizing a diastereomeric mixture of (2S)-2-[(4R)-2-oxo-4-propylpyrrolidin-1-yl]-butanamide and (2S)-2-[(4S)-2-oxo-4-propylpyrrolidin-1-yl]-butanamide (Brivaracetam) which is purified by chiral HPLC (Scheme-I & Scheme-II respectively, as provided below). This process used for chiral resolution makes it difficult for bulk manufacturing as well as it affects the overall yield making the process uneconomical.
Scheme-I
Synthesis of (2S)-2-(2-oxo-4-propyl-1-pyrrolidinyl)butanamide
[As disclosed in columns 37-38 of US 6784197 B2]
Scheme-II
1.1 Synthesis of (2S)-2-aminobutyramide-Free base
1.2 Synthesis of 5-hydroxy-4-n-propylfuran-2-one
1.3 Synthesis of (2S)-2-((4R)-2-oxo-4-n-propyl-1-pyrrolidinyl)butanamide and
(2S)-2-((4S)-2-oxo-4-n-propyl-1-pyrrolidinyl)butanamide
[As disclosed in columns 6-7 of US 7629474 B2]
Moreover, some prior arts such as US7122682B2, US8076493B2, US8338621B2 and US8957226B2 also describe processes for preparing Brivaracetam, wherein, the purifications are reportedly done by chiral HPLC methods resulting into similar shortcomings.
Kenda et al.: Journal of Medicinal Chemistry, 2004, 47, 530-549 further proposes selection of (2S)-2-[(4R)-2-oxo-4-propylpyrrolidin-1-yl]butanamide 83α (ucb 34714; Brivaracetam) as the most interesting candidate showing 10 times more potency than Levetiracetam as an antiseizure agent in audiogenic seizure-prone mice. This article further discloses methods for synthesizing the said compound Brivaracetam. However, here too these compounds are synthesized as mixtures of stereoisomers (racemic or diastereoisomeric mixtures), separated by preparative HPLC on silica gel and/or chiral phases.
All these processes for the preparation of Brivaracetam as described in the above- mentioned prior arts suffer from many disadvantages which includes difficulty in achieving desired chiral purity, tedious and cumbersome work up procedures, high temperature and longer time reaction, multiple crystallizations or isolation steps, use of excess reagents and solvents, column chromatographic separations & purifications etc. All these disadvantages affect the overall yield as well as the quality of the final product Brivaracetam and intermediates produced thereof; further, rendering such processes to be uneconomical and unsuitable for industrial scale-ups.
As a result, enantioselective synthesis of Brivaracetam was perceived to be a possible way of overcoming such problems in view of the large-scale synthesis. However, very few prior arts have been found to report successful reduction of such concept into practice.
WO2016191435A1 (also as IN201717005820A) relates to a process for a scalable synthesis of enantiomerically pure Brivaracetam from an intermediate (4R)-4- Propyldihydrofuran-2(3H)-one (compound IV):
, wherein, R is saturated or unsaturated C1-20 alkyl or C1 alkyl-unsubstituted or substituted aryl, comprising the steps of decarboxylation of
the compound of formula IV
to produce the compound of formula VI
ring-opening of the compound of formula VI to produce the compound of formula VII
, wherein Rl is saturated or unsaturated Cl-20 alkyl or Cl alkyl-unsubstituted or substituted aryl; and X is CI Br I OMs, OTs, ONs; or
the compound of formula X
reacting the compound of formula VI with (S)-2- aminobutanamide or its salt to produce the compound of formula XII in one step; or reacting the compound of formula VI with alkyl (S)-2- aminobutanoate to produce Xll-a
, wherein R in the compound of formula Xll-a is a saturated or unsaturated C1-C20 alkoxyl; and then converting Xll-a to XII that is Brivaracetam by aminolysis and amide formation reaction.
In above mentioned prior arts, the synthesis of chiral lactone which is the key starting material for making Brivaracetam involved Grignard addition, column chromatography and Krapcho decarboxylation techniques at high temperature, all of which are not at all recommendable in view of process perspective at industrial levels. Further the final step of the said reaction often involved cryogenic condition –30°C which is also difficult with respect to industrial scale up activities.
Furthermore, prior art IN201641030239A disclosed a process for the preparation of Brivaracetam of Formula (I) by means of converting enantiomerically pure compound of Formula VII to obtain enantiomerically pure compound of Formula XI:
, wherein X is each independently selected from halogen; alkyl or aryl sulfonyloxy; OR2; R2 is optionally substituted C1-C12 alkyl, aryl, alkyl aryl, aryl alkyl;
such that the said process further comprises steps of:
1) cyclizing compound of formula VII to give enantiomerically pure compound of formula IX:
, wherein R2 is optionally substituted C1-C12 alkyl, aryl, alkyl aryl or aryl alkyl;
2) converting the compound of formula IX to give a enantiomerically pure compound of formula X:
, wherein X is halogen;
3) converting compound of formula X to give a enantiomerically pure compound of formula XI:
wherein X is each independently selected from halogen; alkyl or aryl sulfonyloxy; OR2; R2 as defined above; followed by 4) treating the enantiomerically pure compound of formula XI with (S)- aminobutyramide of formula XII or its salt thereof to obtain Brivaracetam of Formula I.
However, this process suffered from drawbacks of handling acid chloride. Acid chlorides are unstable and storing a large amount of acid chloride is also not recommendable in view of safety and stability in industries. Moreover, this prior art process involves use of HBr in acetic acid, where HBr liberates Br that is hazardous and not recommendable for industrial scale-up activities in view of safety and handling.
Some other prior arts such as CN108503573A, CN105646319A, CN106588740A, CN106588831A, CN108689903B and CN108929289A report various processes of synthesizing Brivaracetam from its lactone intermediate by various ring opening techniques. Among these, in particular CN108929289A discloses a process of reacting a compound represented by the formula IV with (S) -2-aminobutyramide in order to obtain Brivaracetam. The synthetic route is as follows:
Also, CN108689903B relates to a new preparation method of Brivaracetam that comprises steps of: a) subjecting a compound of formula III and (S) -2-aminobutanamide or salt thereof to condensation reaction, in the presence of a condensing agent, in order to obtain a compound shown in a formula IV, wherein the compound has two chiral centres; b) removal of the hydroxy-protecting group R1 to obtain a compound of formula V; and c) carrying out chlorination reaction on the compound shown in the formula V using a chlorination reagent to obtain a compound shown in the formula VI; and d) carrying out substitution reaction on the compound shown in the formula VI in the presence of an
alkaline reagent, and closing a ring to obtain Brivaracetam of formula I having two chiral centres.
It has further been noted that although the above reaction goes through formation of intermediates V and/or VI; however, these intermediates are not essentially formed from the key lactone intermediate of Brivaracetam that is (4R)-4-propyldihydrofuran-2(3H)-one [or (R)-lactone].
Furthermore, CN111196771A relates to a preparation method of Brivaracetam which comprises the steps of: 1) carrying out ring-opening reaction on a compound R-4-propyldihydrofuran-2-ketone in a formula II and a compound S-2-aminobutanamide in a formula III to obtain an intermediate compound in a formula I; 2) condensing the said intermediate compound of formula I is followed by cyclization to produce Brivaracetam
However, the ring-opening reaction in step 1 of this process essentially occurs under acidic conditions, specifically in presence of Lewis acids like tetra-isopropyl titanate, anhydrous aluminium trichloride, anhydrous zinc chloride, boron trifluoride diethyl etherate etc.; and also in presence of organic solvents chosen from one or more of anhydrous tetrahydrofuran, 2-methyltetrahydrofuran, acetone, dimethyl sulfoxide and N, N-dimethylformamide; which makes this process both industrially non-scalable and environmentally unfriendly.
The prior art Org. Process Res. Dev.2016. v 20. no 9. p 1566-1575 in its scheme 4, on page 17 also discloses a scheme for synthesizing Brivaracetam from its lactone intermediate:
Nevertheless, it has been noted that the process reported in this prior art provides Brivaracetam (API) with a very poor yield of ~30% and also having an inferior chiral purity of 95.9% ee, which does not even meet the ICH-specification for the Finished Product (API).
Furthermore, a recently filed patent application WO2020148787A1 (also as IN201931002041) recites a new, improved and economical process for enantioselective synthesis and purification of a key intermediate of Brivaracetam that is the R-lactone, essentially utilizing a low chiral loading and without involving any chiral chromatographic resolution technique. Even though this prior art also discloses a process for the preparation of a chirally pure Brivaracetam of formula I utilizing the said intermediate; however, that process is mostly a conventional one.
Accordingly, there is still a need in the art for a more economical and improved process for the synthesis of Brivaracetam with better purity and yield which overcomes the drawbacks of above prior arts.
Therefore, the present inventors have developed a cost effective, novel and efficient process for the preparation of Brivaracetam which essentially avoids all the drawbacks involved in prior art as mentioned above. The currently developed process is advantageously capable of producing the key lactone intermediate with more than 80% ee applying transfer hydrogenation with a very simple operation in view of process perspective. Further, by means of using such chiral lactone with more than 80% ee, the currently developed process is also capable of delivering >99.9% chirally pure Brivaracetam with excellent yield.
EXAMPLES:
EXAMPLE 1: Synthesis of (3R)-N-[(1S)-1-carbamoylpropyl]-3-(hydroxymethyl) hexanamide [Intermediate 7 of scheme A of the present invention]
Example 1 illustrates one pot process for preparing purified (3R)-N-[(1S)-1- carbamoylpropyl]-3-(hydroxymethyl) hexanamide [Intermediate 7] from Intermediate 3 (80% ee) as developed in step 1 of scheme A of the present invention.
Procedure:
In the first step of scheme A of the present invention, a mixture of (R)/(S)-4-propyldihydrofuran-2(3H)-one (Intermediate 3, R: S isomer = 80: 20) (1 eq), (S)-2-aminobutanamide (1.1 eq), triethylamine (1.5 eq) is refluxed at a temperature of 95±5 °C for 24h. The mixture is then cooled to 60-65 °C, washed with a mixture of dichloromethane and diisopropyl ether (2.5 vol) in order to get Intermediate-7 [(3R)-N-[(1S)-1-carbamoyl-propyl]-3-(hydroxymethyl) hexanamide] (80% yield).
Results:
Formation of Intermediate 7 is confirmed further by following analytical studies: a) The 1H NMR analysis is conducted and the data as illustrated in accompanying figure 1 depicts: (400 MHz, DMSO-d6): δ 0.6 (t, J= Hz, 6H), 1.07-1.18 (m, 1H), 1.21-1.35 (m, 3H), 1.45-1.43 (m, 1H), 1.61-1.72 (m, 1H), 1.75-1.90 (m, 1H), 2.03 (dd, J=6.64 & 14.08 Hz, 1H), 2.18 (dd, J=7.0 & 14.08 Hz, 1H), 3.28 (t, J=5.36 Hz, 2H), 4.07-4.18 (m, 1H), 4.43 (t, J=5.2 Hz, 1H), 6.95 (s, 1H), 7.28 (s, 1H), 7.76 (d, J=8.08 Hz, 1H).; thus, confirming formation of Intermediate 7 of the present invention.
b) The LCMS analysis is further conducted and the data as graphically illustrated in accompanying figure 2 provides a (M+H+) value of 231.0; thus, confirming formation of Intermediate 7 of the present invention.
c) The HPLC study is also conducted and the data as graphically illustrated in accompanying figure 3 confirms formation of Intermediate 7 of the present invention with chiral purity of 97.38%
EXAMPLE 2: Preparation of (3R)-N-(1S)-1-amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide [Intermediate 8A of scheme A of the present invention]: Example 2 illustrates a process for preparing (3R)-N-(1S)-1-Amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide [Intermediate 8A] from Intermediate 7 of example 1 above as developed in the present invention.
Procedure:
In second step of scheme A of the present invention, the said Intermediate 7 of example 1 above that is (3R)-N-(1S)-1-Amino-1-oxobutan-2-yl)-3-(hydroxymethyl) hexanamide (~98% Chemical purity and ~97% Chiral purity) (1736.86 mmol) is dissolved in DCM (1.2 L) at RT into a RBF under N2 atm. Then the solution is cooled to 10-20 C and Oxaloyl chloride (2605.29 mmol) is added to this cooled solution at 10-20 °C. The mixture is stirred for 24 h at 25-40 °C under N2 atm. Completion of the reaction is monitored by TLC. After completion of reaction, the solvent is distilled off and the residual mass is diluted with water (6 L), stirred at 30-50 °C for 4 h. Slurry mass is then filtered and washed with water (2×400 mL) followed by MTBE (800 mL). The solid is dried under vacuum at 50-55 °C for 4-5 h to afford Intermediate 8A that is (3R)-N-(1S)-1-Amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide as a white solid (92% yield).
Results:
Formation of Intermediate 8A is confirmed further by following analytical studies: a) The 1H NMR analysis is conducted and the data as illustrated in accompanying figure 4 depicts: 1H NMR (400 MHz, DMSO-d6) : δ 0.83 (t, J=7.44 Hz, 3H), 0.85 (t, J=6.72 Hz, 3H), 1.20-1.40 (m, 4H), 1.43-1.56 (m, 1H), 2.08-2.18 (m, 1H), 2.20-2.28 (m, 2H), 3.61 (dd, J=4.6 & 10.8 Hz, 1 H), 3.67 (dd, J=4.6 & 10.8 Hz, 1H), 4.07-4.18 (m, 1H), 6.95 (s, 1H), 7.29 (s, 1H), 7.89 (d, J=8.12 Hz, 1H); thus confirming formation of Intermediate 8A of the present invention.
b) The LCMS analysis is further conducted and the data as graphically illustrated in accompanying figure 5 (a, b) provides a (M+H+) value of 249.20; thus, confirming formation of Intermediate 8A of the present invention.
EXAMPLE 3: Process for purification of Intermediate 8A forming Intermediate 8B Example 3 illustrates a process for purifying the said Intermediate 8A of example 2 above of the present invention.
Procedure:
The Intermediate 8A as obtained in example 2 above [that is (3R)-N-(1S)-1-Amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide] is first dissolved in a polar solvent like Acetonitrile raising the temperature to 50 to 60 °C; followed by stirring and then addition of another solvent methyl tert-butyl ether (MTBE) which is less polar in nature. The mixture is then cooled down to 0°C, the filtered mass thus obtained is dried in order to obtain a white solid of Intermediate 8B. The material thus obtained is further dissolved in THF (5 vol) at 60 °C, cooled to 20-30°C, followed by addition of heptane (5 vol), stirred at 10°C to 30 °C for 1 h. The mass obtained is filtered and washed with heptane (2×1 vol), dried under vacuum at 50-55°C in order to afford formation of purer form of Intermediate 8A that is Intermediate 8B that is (3R)-N-(1S)-1-amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide as a white solid having a chemical purity of 99.9% along with a chiral purity of 100% (yield: 390 g).
Results:
The purification of Intermediate 8A is further confirmed by the following analytical test results:
a) Chiral HPLC: A Chiral HPLC as illustrated in accompanying figure 6 confirmed formation of purest form of Intermediate 8B having 100% chiral purity [Peak 1; RT (min) = 6.244; %Area=100%].
b) GLP-HPLC: A GLP-HPLC as illustrated in accompanying figure 7 further confirmed formation of Intermediate 8B having 99.9% chemical purity [Peak 3; BRIV8; RT = 29.278; % Area=99.90%].
EXAMPLE 4: Synthesis of (3R)-N-[(1S)-1-carbamoylpropyl]-3-(hydroxymethyl) hexanamide [Intermediate 7’ of scheme B of the present invention]
Example 4 illustrates one pot process for preparing purified (3R)-N-[(1S)-1-carbamoylpropyl]-3-(hydroxymethyl) hexanamide [Intermediate 7’] from Intermediate 6 (99.99% ee) as developed in step-1 scheme B of the present invention.
Procedure:
In another method, in the first step of scheme B of the present invention, a mixture of (R)/(S)-4-propyldihydrofuran-2(3H)-one (Intermediate 6: S isomer = 99.99% : 0.1%) (1 eq), (S)-2-aminobutanamide (1.7 eq), triethylamine (5 eq) is refluxed at a temperature between 95±5 °C for 24 h. Then, the crude reaction mass is cooled and washed with dichloromethane and diisopropyl ether mixture (2.5 vol) in order to achieve Intermediate-7’ of scheme B of the present invention [(3R)-N-[(1S)-1-carbamoylpropyl]-3-(hydroxymethyl) hexanamide] (90% yield).
Results:
The chiral purity of the formed Intermediate 7’ is analyzed by HPLC method and the data as graphically illustrated in accompanying figure 8 confirms formation of Intermediate 7’ of the present invention with chiral purity of 99.11%
EXAMPLE 5: Preparation of (3R)-N-(1S)-1-amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide [Intermediate 8’ of scheme B of present invention]: Example 5 illustrates a process for preparing purest form of (3R)-N-(1S)-1-amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide [Intermediate 8’] from Intermediate 7’ of example 4 as developed in scheme B (step 2) of the present invention.
Procedure:
In the second step of scheme B of the present invention, the intermediate 7’ of the above example 4 that is (3R)-N-(1S)-1-amino-1-oxobutan-2-yl)-3-(hydroxymethyl) hexanamide (98% Chemical purity and >99%Chiral purity) (1736.86 mmol) is dissolved in DCM (1.2 L) at RT in a round bottomed flask under N2 atm. Then the solution is cooled to 10-30 °C and 1-Chloro-N,N,2-trimethyl-1-propenylamine (2605.29 mmol) is added to this cooled solution at 10-30 °C. The mixture is stirred for 24 h at 25-40 °C under N2 atm. Completion of the reaction is monitored by TLC. After completion of the reaction, the solvent is distilled off and the residual mass is diluted with water (6 L), stirring at 30-50 °C for 4 h. The slurry mass thus obtained is then filtered and washed with water (2×400 mL) followed by methyl tert-buty ether (MTBE) (800 mL). The solid thus obtained is dried under vacuum at 50-55 °C for 4-5 h in order to afford formation of Intermediate 8’ that is (3R)-N-(1S)-1-amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide as a white solid (92% yield).
Results:
Formation of Intermediate 8’ is confirmed further by following analytical studies: a) The 1H NMR analysis is conducted and the data as illustrated in accompanying figure 9 depicts: (400 MHz, DMSO-d6): 1H NMR (400 MHz, DMSO-d6) : δ 0.83 (t, J=7.44 Hz, 3H), 0.85 (t, J=6.72 Hz, 3H), 1.20-1.40 (m, 4H), 1.43-1.56 (m, 1H), 2.08-2.18 (m, 1H), 2.20-2.28 (m, 2H), 3.61 (dd, J=4.6 & 10.8 Hz, 1 H), 3.67 (dd, J=4.6 & 10.8 Hz, 1H), 4.07-4.18 (m, 1H), 6.95 (s, 1H), 7.29 (s, 1H), 7.89 (d, J=8.12 Hz, 1H); thus, confirming formation of Intermediate 8’ of the present invention.
b) The LCMS analysis is further conducted and the data as graphically illustrated in accompanying figure 10 provides a (M+H+) value of 249.1; thus, confirming formation of Intermediate 8’ of the present invention.
c) The HPLC data as illustrated in accompanying figure 11 confirms 100% chiral purity of Intermediate 8’.
EXAMPLE 6: Preparation of (2S)-2-[(4R)-2-oxo-4-propyl-pyrrolidin-1-yl] butanamide [Brivaracetam-API]:
Example 6 illustrates a process for preparing (2S)-2-[(4R)-2-oxo-4-propyl-pyrrolidin-1-yl] butanamide [Brivaracetam API] from Intermediate 8B of example 3 or from Intermediate 8’ of example 5 as developed in step 3 of scheme A or scheme B of the present invention respectively.
Procedure:
In the final step of scheme A or scheme B of the present invention, the intermediate 8B of example 3 or intermediate 8’ of example 5 above that is (3R)-N-((1S)-1-Amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide (1608.04 mmol) is dissolved in dimethyl acetamide (0.5 vol) and isopropylacetate (2 L) into a RBF at 25-30 °C under N2 atm. Then 18-Crown-6 (160.79 mmol) is added into the solution and stirred at RT for 30 min. Reaction mixture is then cooled to 0-10 °C and t-BuOK (1.5 eq) is added portion wise to the cooled solution over 1 h maintaining the temperature from – 0-10 °C to 25 °C under N2 atm. Stirring is then continued for 2 h at -10 °C to 0 °C and then for 12 h at 15-25 °C under N2 atm. Completion of reaction is monitored by TLC. After completion of reaction, the reaction mixture is quenched with addition of 1M HCl solution (pH~6.5-7.0). The resulting mixture is extracted with i-PrOAc (2 L) and MTBE (1 L). Water (0.5 L) is added to the combined organic extract and then filtered through celite bed, washed the bed with MTBE-i-PrOAc (1:1) (400 mL). The organic part is separated and the aqueous part is re-extracted with i-PrOAc-MTBE (1:1) (2 ×0.8 L). The combined organic phases are washed with brine solution (100 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuum under a rotary evaporator to afford crude API. Distillation of dimethylacetamide solvent from the crude is then done at high vacuum pressure (0.05 mm Hg) at 70 °C. Crude product is then dissolved in isopropyl acetate (1.6 L) and treated with activated charcoal (7% w/w) to afford a tech-grade crude of Brivaracetam API as a white solid (yield: 90%) with 97.82% chemical purity.
Results:
Formation of Brivaracetam API is confirmed further by following analytical studies: a) The 1H NMR analysis is conducted and the data as illustrated in accompanying figure 12 depicts: 1H NMR (400 MHz, DMSO-d6) : δ 0.77 (t, J=7.32 Hz, 3H), 0.87 (t, J=7.2 Hz, 3H), 1.21-1.31 (m, 2H), 1.33-1.43 (m, 2H), 1.50-1.62 (m, 1H), 1.73-1.84 (m, 1H), 1.97 (dd, J=8.0 & 16.12 Hz, 1H), 2.18-2.28 (m, 1H), 2.37 (dd, J=8.4 & 16.14 Hz, 1H), 3.11 (dd, J=7.16 & 9.44 Hz, 1H), 3.36 (dd, J=9.2 & 17.5 Hz, 1H), 4.30 dd, J=5.44 & 10.28 Hz, 1H), 6.98 (s, 1H), 7.32 (s, 1H); thus, confirming formation of Brivaracetam API of the present invention.
b) The LCMS analysis is further conducted and the data as graphically illustrated in accompanying figure 13 provides a (M+H+) value of 213.0; thus, confirming formation of Brivaracetam API of the present invention.
^ Purification of Brivaracetam API:
The Brivaracetam thus formed above is further purified by means of dissolving the said material (307 g) in 30% i-PrOAc -MTBE (1 vol) at 55-60 °C, cool to 20-30°C. A mixture of Heptane and MTBE and DIPE (2:2:1) is added, stirred at 10 °C to 30°C for 1 h. The obtained mass is filtered and washed with heptane, which is subsequently dried under vacuum at 40-45 °C to afford (3R)-N-((1S)-1-amino-1-oxobutan-2-yl)-3-(chloromethyl) hexanamide as a white solid (yield: 80%, chiral purity 99.93% and chemical purity 99.94%).
Results:
a) Chiral HPLC: A Chiral HPLC as illustrated in accompanying figure 14 confirmed formation of purest form of Brivaracetam API having 99.93% chiral purity [Peak 2; RT (min) = 9.45; %Area=99.93%].
b) GLP-HPLC: A GLP-HPLC as illustrated in accompanying figure 15 further confirmed formation of Brivaracetam API having 99.9% chemical purity [Peak 2; RT = 21.138; % Area=99.94%].
PF-06747775 (Pfizer) Third generation covalent EGFR inhibitors

 .
.

PF-06747775 (Pfizer)
PF06747775; PF06747775; PF 06747775; PF6747775; PF 6747775; PF6747775. PFE-X775
N-((3R,4R)-4-fluoro-1-(6-((3-methoxy-1-methyl-1H-pyrazol-4-yl)amino)-9-methyl-9H-purin-2-yl)pyrrolidin-3-yl)acrylamide
N-((3R,4R)-4-fluoro-1-(6-((3-methoxy-1-methyl-1H-pyrazol-4-yl)amino)-9-methyl-9H-purin-2-yl)pyrrolidin-3-yl)acrylamide
CAS 1776112-90-3
Chemical Formula: C18H22FN9O2
Exact Mass: 415.188
Recruiting, Phase I/II (NTC02349633)
Epidermal growth factor receptor antagonists
Antineoplastics
Non-small cell lung cancer
Dose escalation study to evaluate safety, PK, PD and efficacy in advanced EGFRm+ NSCLC
- 02 May 2015Phase-I clinical trials in Non-small cell lung cancer (Metastatic disease, Second-line therapy or greater) in USA (PO) (NCT02349633)
- 05 Feb 2015Pfizer plans a phase I trial for Non-small cell lung cancer (Second-line therapy or greater) in USA (NCT02349633)
- 05 Jan 2015Preclinical trials in Non-small cell lung cancer in USA (PO)
PF-06747775 is an orally available inhibitor of the epidermal growth factor receptor (EGFR) mutant form T790M, with potential antineoplastic activity. EGFR T790M inhibitor PF-06747775 specifically binds to and inhibits EGFR T790M, a secondarily acquired resistance mutation, which prevents EGFR-mediated signaling and leads to cell death in EGFR T790M-expressing tumor cells. Compared to some other EGFR inhibitors, PF-06747775 may have therapeutic benefits in tumors with T790M-mediated drug resistance.
for the oral treatment of patients with locally advanced or metastatic EGFR mutant (del19 or L858R) non-small cell lung cancer

Kinetic mechanism for two-step covalent inhibition of EGFR
PATENT
Example 7
(Scheme F): Preparation of N-((3R,4R)-4-fluoro-1-(6-((3-methoxy-1-methyl-1H-pyrazol-4-yl)amino)-9-methyl-9H-purin-2-yl)pyrrolidin-3-yl)acrylamide

Step 1: Preparation of 2-fluoro-N-(3-methoxy-1-methyl-1H-pyrazol-4-yl)-9H-purin -6-amine

Step 2: Preparation of 2-fluoro-N-(3-methoxy-1-methyl-1H-pyrazol-4-yl)-9-methyl -9H-purin-6-amine

Step 3: Preparation of N-((3R,4R)-4-fluoro-1-(6-((3-methoxy-1-methyl-1H-pyrazol -4-yl)amino)-9-methyl-9H-purin-2-yl)pyrrolidin-3-yl)acrylamide

Example 7A
(Scheme F): Preparation of N-((3R,4R)-4-fluoro-1-(6-((3-methoxy-1-methyl-1H-pyrazol-4-yl)amino)-9-methyl-9H-purin-2-yl)pyrrolidin-3-yl)acrylamide

Preparation Step 1A: Preparation of (3R,4R)-1-benzyl-3,4-dihydroxypyrrolidine-2,5-dione

Preparation Step 2A: Preparation of (3S,4S)-1-benzylpyrrolidine-3,4-diol

Preparation Step 3A: Preparation of (3aR,6aS)-5-benzyl-2,2-dioxo-tetrahydro-1-oxa-2λ6-thia-3-5-diaza-pentalene-3-carboxylic acid t-butyl ester

Preparation Step 4A: Preparation of (3R,4R)-1-benzyl-4-fluoropyrrolidin-3-amine bis-tosylate

Preparation Step 5A: N-((3R,4R)-1-benzyl-4-fluoropyrrolidin-3-yl)-3-(methylsulfonyl)propanamide

Preparation Step 6A: N-((3R,4R)-4-fluoropyrrolidin-3-yl)-3-(methylsulfonyl)propanamide

Step 1: Preparation of 2-fluoro-N-(3-methoxy-1-methyl-1H-pyrazol-4-yl)-9H-purin-6-amine

Step 2: Preparation of 2-fluoro-N-(3-methoxy-1-methyl-1H-pyrazol-4-yl)-9-methyl-9H-purin-6-amine

Step 3: Preparation of N-((3R,4R)-4-fluoro-1-(6-((3-methoxy-1-methyl-1H-pyrazol-4-yl)amino)-9-methyl-9H-purin-2-yl)pyrrolidin-3-yl)acrylamide


Summary of 1st generation and 2nd generation EGFR inhibitors

REFERENCES
Planken, S.; Murray, B. W.; Lafontaine, J.; Weinrich, S.; Hemkens, M.; Kath, J. C.; Nair, S. K.; Johnson, T. O.; Cheng, H.; Sutton, S. C.; Zientek, M.; Yin, M. -J.; Solowiej, J.; Nagata, A.; Gajiwala, K. Abstracts of Papers, 249th ACS National Meeting & Exposition, Denver, CO, United States, March 22–26, 2015; MEDI-248
//////Third generation, covalent EGFR inhibitors, PF-06747775, Pfizer, PFE-X775
Compound name AND SMILES string
Rociletinib COC(C=C(N1CCN(C(C)=O)CC1)C=C2)=C2NC3=NC=C(C(F)(F)F)C(NC4=CC=CC(NC(C=C)=O)=C4)=N3
Osimertinib CN(CCN(C)C)C(C(NC(C=C)=O)=C1)=CC(OC)=C1NC2=NC=CC(C3=CN(C)C4=C3C=CC=C4)=N2
EGF816 ClC1=C2C(N=C(NC(C3=CC(C)=NC=C3)=O)N2[C@H]4CN(C(/C=C/CN(C)C)=O)CCCC4)=CC=C1
PF-06747775 CN1C2=NC(N3C[C@@H](NC(C=C)=O)[C@H](F)C3)=NC(NC4=CN(C)N=C4OC)=C2N=C1
PF-06459988 CN(N=C1)C=C1NC2=NC3=C(C(Cl)=CN3)C(OC[C@H]4CN(C(C=C)=O)C[C@@H]4OC)=N2
WZ4002 ClC1=CN=C(NC2=C(OC)C=C(N3CCN(C)CC3)C=C2)N=C1OC4=CC=CC(NC(C=C)=O)=C4
罗西替尼 роцилетиниб روسيليتينيب Rociletinib, CO-1686. Third generation covalent EGFR inhibitors

Rociletinib (CO-1686)
AVL-301,CNX-419
Celgene (Originator) , Clovis Oncology
- Molecular FormulaC27H28F3N7O3
- Average mass555.552
- HYDROBROMIDE 1446700-26-0
Molecular Weight 636.46 Formula C27H28F3N7O3 ● HBr
Cellular proliferation IC507–32 nM against EGFRm+ NSCLC cells
547 nM against A431 cell with WT EGFR
Ongoing, not currently recruiting
Phase I/II (NCT01526928)
Recruiting
Phase III (NCT02322281, TIGER-3)
Evaluate safety, PK and efficacy of previously treated NSCLC patients, Compare the efficacy of oral single agent versus single agent cytotoxic chemotherapy in patients with EGFRm+ NSCLC after failure of at least 1 previous EGFR-directed TKI and at least 1 line of platinum-containing doublet therapy. Compare the safety and efficacy of CO-1686 versus erlotinib as first line treatment of patients with EGFRm+ NSCLC
Rociletinib (CO-1686): Rociletinib is an orally administered irreversible inhibitor currently in several clinical trials targeting both the activating EGFR mutations and the acquired T790M resistance mutation while sparing WT EGFR. It is a potent inhibitor of EGFR T790M/L858R double mutant with a kinact/Ki of 2.41 × 105 M−1 s−1. It has a 22-fold selectivity over WT EGFR (kinact/Ki of 1.12 × 104 M−1 s−1). In NSCLC cell lines containing EGFR mutations, rociletinib demonstrates the following cellular pEGFR IC50: 62 nM in NCI-1975 (L858R/T790M), 187 nM in HCC827 (exon 19 deletion), 211 nM in PC9 (exon 19 deletion). In cell lines expressing WT EGFR, cellular pEGFR IC50 are: >4331 nM in A431, >2000 nM in NCI-H1299, and >2000 nM in NCI-H358.
Rociletinib displayed good oral bioavailability (65%) and long half-life when dosed at 20 mg/kg in female Nu/Nu mice. In tumor bearing mice when rociletinib was dosed orally once daily as a single agent, the compound showed dose-dependent tumor growth inhibition in various EGFR-mutant models. In NCI-H1975 as well as in patient-derived LUM 1868 lines expressing the EGFR T790M/L858R double mutation that are erlotinib-resistant models, rociletinib caused tumor regressions at 100 mg/kg/d. In the HCC827 xenograft model that expresses the del-19 activating EGFR mutation, rociletinib showed antitumor activity that was comparable with erlotinib and the second-generation EGFR TKI, afatinib. The wild-type sparing feature of rociletinib was further demonstrated through its minimal inhibition (36%) of tumor growth in the A431 xenograft model that is dependent on WT EGFR for proliferation.
In a Phase I/II study (TIGER-X), rociletinib was administered to patients with EGFR mutated NSCLC who had disease progression during treatment with a previous line of EGFR TKI therapy.The Phase I trial was a dose escalation study to assess safety, side-effect profile and pharmacokinetic properties of rociletinib, and the Phase II trial was an expansion arm to evaluate efficacy. T790M positivity was confirmed before enrollment in the Phase II portion. At the dose of 500 mg BID, the objective response rate in 243 centrally confirmed tissues from T790M positive patients was 60% and the disease control rate was 90%. The estimated overall median PFS at the time of the publication (May 2015) was 8.0 months among all centrally confirmed T790M positive patients. Rociletinib also showed activity in centrally confirmed T790M negative patients with the overall response rate being 37%. The common dose-limiting adverse event was grade 3 hyperglycemia occurring in 17% of patients at a dose of 500 mg BID. Grade 3 QTc prolongation was observed in 2.5% of the patients at the same dose. Treatment-related adverse events leading to drug discontinuation was seen in only 2.5% of patients at 500 mg BID.
Patent
WO2012061299A1
http://www.google.co.in/patents/WO2012061299A1?cl=en
EXAMPLE 1
Intermediate 1
Scheme 1

Step 1 :
In a 25 mL 3-neck RBF previously equipped with a magnetic stirrer, Thermo pocket and CaCl2 guard tube, N-Boc-l,3-diaminobenzene (0.96 g) and n-butanol (9.00 mL) were charged. Reaction mixture was cooled to 0 °C. 2,4-Dichloro-5-trifluoromethylpyrimidine (1.0 g) was added dropwise to the above reaction mixture at 0 °C. The DIPEA (0.96 mL) was dropwise added to the above reaction mixture at 0 °C and the reaction mixture was stirred for 1 hr at 0 °C to 5 °C. Finally the reaction mixture was allowed to warm to room temperature. Reaction mixture was stirred for another 4 hrs at room temperature. Completion of reaction was monitored by TLC using hexane: ethyl acetate (7: 3). The solid precipitated out was filtered off and washed with 1-butanol (2 mL). Solid was dried under reduced pressure at 40 °C for 1 hr. ^-NMR (DMSO-d6, 400 MHz) δ 1.48 (S, 9 H), 7.02 (m, 1 H), 7.26 (m, 2 H), 7.58 (S, 1 H), 8.57 (S, 1 H), 9.48 (S, 1 H), 9.55 (S, 1 H).
Step 2:
To the above crude (3.1 g) in DCM (25 mL) was added TFA (12.4 mL) slowly at 0 °C. The reaction mixture was allowed to warm to room temperature. Reaction mixture was stirred for another 10 min at room temperature. The crude was concentrated under reduced pressure.
Step 3:
The concentrated crude was dissolved in DIPEA (2.0 mL) and DCM (25 mL), and then cooled to -30 °C. To the reaction mixture was slowly added acryloyl chloride (0.76 g) at -30 °C. The reaction mass was warmed to room temperature stirred at room temperature for 1.0 hr. The reaction was monitored on TLC using hexane: ethyl acetate (7:3) as mobile phase. Reaction got completed after 1 hr. 1H-NMR (DMSO-d6, 400 MHz) δ 5.76 (dd, J = 2.0, 10.0 Hz, 1 H), 6.24 (dd, J = 2.0, 17.2 Hz, 1 H), 6.48 (m, 1 H), 7.14 (d, J = 8.8 Hz, 1 H), 7.37 (t, J = 8.0 Hz, 1 H), 7.94 (S, 1 H), 8.59 (S, 1 H), 9.60 (S, 1 H), 10.26 (S, 1 H).
EXAMPLE 3
Compound 1-4 N- henylamino)-5-
(trifluor mide)

Using 2-methoxy-4-(4-acteylpiperazinyl)aniline and intermediate 1 in Example 1, the title compound 1-4 was prepared as described in Example 2. 1H-NMR (DMSO-d6, 400 MHz) δ 10.2 (S, 1 H), 8.2 (br, 1 H), 8.30 (S, 1 H), 7.73 (br, 1 H), 7.52 (d, J = 7.8 Hz, 1 H), 7.45 (d, J = 7.8 Hz, 1 H), 7.26 (J = 8.2 Hz, 1 H), 7.14 (be, 1 H), 6.60 (S, 1 H), 6.42 (dd, J = 11.4, 16.9 Hz, 1 H), 6.24 (d, J = 16.9 Hz, 1 H), 5.75 (d, J = 11.4 Hz, 1 H), 3.76 (S, 3 H), 3.04 (br, 4 H), 2.04 (S, 3 H); calculated mass for C27H28F3N7O3 : 555.2, found: 556.2 (M+H+).

| Patent ID | Date | Patent Title |
|---|---|---|
| US2015344441 | 2015-12-03 | SALTS OF AN EPIDERMAL GROWTH FACTOR RECEPTOR KINASE INHIBITOR |
| US2015246040 | 2015-09-03 | HETEROCYCLIC COMPOUNDS AND USES THEREOF |
| US2015225422 | 2015-08-13 | HETEROARYLS AND USES THEREOF |
| US8975249 | 2015-03-10 | Heterocyclic compounds and uses thereof |
| US2013267531 | 2013-10-10 | SALTS OF AN EPIDERMAL GROWTH FACTOR RECEPTOR KINASE INHIBITOR |
| US2013267530 | 2013-10-10 | SOLID FORMS OF AN EPIDERMAL GROWTH FACTOR RECEPTOR KINASE INHIBITOR |
References
////Rociletinib, CO-1686, Clovis, Third generation, covalent EGFR inhibitors, AVL-301, CNX-419
CC(=O)N1CCN(CC1)C2=CC(=C(C=C2)NC3=NC=C(C(=N3)NC4=CC(=CC=C4)NC(=O)C=C)C(F)(F)F)OC
//////
Compound name AND SMILES string
Rociletinib COC(C=C(N1CCN(C(C)=O)CC1)C=C2)=C2NC3=NC=C(C(F)(F)F)C(NC4=CC=CC(NC(C=C)=O)=C4)=N3
Osimertinib CN(CCN(C)C)C(C(NC(C=C)=O)=C1)=CC(OC)=C1NC2=NC=CC(C3=CN(C)C4=C3C=CC=C4)=N2
EGF816 ClC1=C2C(N=C(NC(C3=CC(C)=NC=C3)=O)N2[C@H]4CN(C(/C=C/CN(C)C)=O)CCCC4)=CC=C1
PF-06747775 CN1C2=NC(N3C[C@@H](NC(C=C)=O)[C@H](F)C3)=NC(NC4=CN(C)N=C4OC)=C2N=C1
PF-06459988 CN(N=C1)C=C1NC2=NC3=C(C(Cl)=CN3)C(OC[C@H]4CN(C(C=C)=O)C[C@@H]4OC)=N2
WZ4002 ClC1=CN=C(NC2=C(OC)C=C(N3CCN(C)CC3)C=C2)N=C1OC4=CC=CC(NC(C=C)=O)=C4
EGF 816 , Nazartinib

EGF 816, Nazartinib
EGF-816; EGFRmut-TKI EGF816
Novartis Ag innovator
(R,E)-N-(7-chloro-1-(1-(4-(dimethylamino)but-2-enoyl)azepan-3-yl)-1H-benzo[d]imidazol-2-yl)-2-methylisonicotinamide
(R,E)-N-(7-chloro-l-(l-(4-(dimethylamino)but-2-enoyl)azepan-3-yl)-lH-benzo[d]imidazol-2 -yl)-2-methylisonicotinamide
NCI-H1975 (L858R/T790M): 25 nM
H3255 (L858R): 9 nM
HCC827 (Del ex19): 11 nM
| M.Wt | 495.02 | ||
|---|---|---|---|
| Formula | C26H31ClN6O2 | ||
| CAS No | 1508250-71-2 |
EGF816 is a novel covalent inhibitor of mutant-selective EGFR; overcomes T790M-mediated resistance in NSCLC.
Epidermal growth factor receptor antagonists; Protein tyrosine kinase inhibitors
- Phase IINon-small cell lung cancer
- Phase I/IISolid tumours
-
- 01 Feb 2015Phase-II clinical trials in Non-small cell lung cancer (Late-stage disease, Combination therapy) in Singapore (PO) (NCT02323126)
- 24 Nov 2014Phase-I/II clinical trials in Non-small cell lung cancer (Combination therapy, Late-stage disease) in Spain (PO) after November 2014 (EudraCT2014-000726-37)
- 24 Nov 2014Phase-I/II clinical trials in Non-small cell lung cancer (Combination therapy, Late-stage disease) in Germany (PO)
| Determine MTD, or recommended phase II dose in patients with NSCLC harboring EGFR mutations, in combination with INC280 | Recruiting Phase I/II (NCT02335944) |
| Determine MTD, or recommended phase II dose in adult patients with EGFRm+ solid malignancies | Recruiting Phase I/II (NCT02108964) |
| Determine efficacy and safety in patients with previously treated NSCLC, in combination with nivolumab | Recruiting Phase II (NCT02323126) |
In November 2015, FDA approved osimertinib (Tagrisso™) for the treatment of patients with metastatic EGFR T790M mutation-positive NSCLC, who have progressed on or after EGFR TKI therapy. Based on the clinical performance of the third generation EGFR drugs, more regulatory approvals can be expected.
Nazartinib, also known as EGF816, is an orally available, irreversible, third-generation, mutant-selective epidermal growth factor receptor (EGFR) inhibitor, with potential antineoplastic activity. EGF816 covalently binds to and inhibits the activity of mutant forms of EGFR, including the T790M EGFR mutant, thereby preventing EGFR-mediated signaling. This may both induce cell death and inhibit tumor growth in EGFR-overexpressing tumor cells. EGF816 preferentially inhibits mutated forms of EGFR including T790M, a secondarily acquired resistance mutation, and may have therapeutic benefits in tumors with T790M-mediated resistance when compared to other EGFR tyrosine kinase inhibitors
PATENT
WO 2016016822
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016016822
PATENT
WO 2015081463
http://www.google.co.in/patents/WO2015081463A1?cl=en
PATENT
Intermediate 26
1055 (R)-tert-butyl 3-(2-amino-7-chloro- 1 H-benzo[dlimidazol- 1 -yOazepane- 1 -carboxylate

Step A: (R)-tert-butyl 3 -((2-chloro-6-nitrophenyl)amino)azepane-l -carboxylate (I-26a) was prepared following procedures analogous to 1-15, Step A, using the appropriate starting materials. JH-NMR (400MHz, CDC13): d 8.00-7.91 (m, 1H), 7.58-7.49 (m, 1H), 7.02-6.51
1060 (m, 2H), 4.31-4.03 (m, 1H), 3.84-2.98 (m, 4H), 1.98-1.60 (m, 5H), 1.46-1.39 (m, 10H); MS calculated for Ci7H25ClN304 (M+H+) 370.15, found 370.10.
Step B: A mixture of I-26a (7.5 g, 19.5 mmol) and Zn (12.8 mg, 195 mmol) in AcOH (22 mL) was stirred at room temperature for 2 h. The reaction was basified with saturated aqueous Na2C03 solution, filtered, and extracted with EtOAc (3 x 80 mL). The combined
1065 organic phase was washed with brine, dried with Na2S04 and concentrated in vacuo to afford (R)-tert-butyl 3-((2-amino-6-chlorophenyl)amino)azepane-l -carboxylate (I-26b). MS calculated for Ci7H27ClN302 (M+H+) 340.17, found 340.10. The crude was used in the next step without further purification.
Step C: The title compound (Intermediate 26) was prepared from I-26b following
1070 procedures analogous to 1-15, Step C. 1H-NMR (400MHz, CDC13): d Ί .34-126 (m, 1H),
7.04-6.97 (m, 2H), 6.05-5.85 (m, 1H), 5.84-5.72 (m, 1H), 5.50-5.37 (m, 0.5H), 5.10-4.80(m, 0.5H), 4.41-4.23(m, 1H), 4.09-3.96(m, 0.5H), 3.94-3.81 (m, 1H), 3.76-3.57 (m, 1H), 3.22-3.14 (m, 0.5H), 2.84-2.63 (m, 1H), 2.34-2.17 (m, 1H), 2.07-1.84 (m, 1H), 1.82-1.64 (m, 2H), 1.53 (s, 9H), 1.48-1.37 (m, 1H); MS calculated for C18H26CIN4O2 (M+H+) 365.17,
1075 found 365.10.
Intermediate 27
(R)-N-(l-(azepan-3-yl)-7-chloro-lH-benzo[dlimidazol-2-yl)-2-methylisonicotinamide hydrochloride

Intermediate 27
Step A
1080 Step A: A mixture of 2-methylisonicotinic acid (3.371 g, 24.6 mmol) and 2-(7-aza-lH- benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (9.345 g, 24.6 mmol) in CH2CI2 (120 ml) was treated at room temperature with NEt3 (4.1 mL, 29.4 mmol). The
reaction was stirred for 1 hour before it was slowly added into a CH2CI2 solution (45 ml) of 1-26 (5.98 g, 16.4 mmol). Ten minutes later, more NEt3 (4.1 mL, 29.4 mmol) was added and 1085 the mixture stirred for 2 h. The mixture was then diluted with CH2CI2 (240 mL), washed with H20 (2 x 80 mL), saturated aqueous NaHC03 solution (70 mL), and brine (70 mL). The organic phase was dried with Na2SC>4, and concentrated under reduced pressure. The crude material was purified by column chromatography (55% EtOAc/hexanes) to afford
(R)-tert-butyl
1090 3-(7-chloro-2-(2-methylisonicotinamido)-lH-benzo[d]imidazol-l-yl)azepane-l-carboxylate (I-27a) as a light yellow foam. 1H-NMR (400MHz, CDC13): d 12.81 (br s, 1H), 8.65-8.62 (m, 1H), 7.95-7.85 (m, 2H), 7.27-7.1 1 (m, 3H), 5.64 – 5.51 (m, 1H), 4.56-4.44 (m, 1H),
4.07-3.92 (m, 1H), 3.79-3.71 (m, 0.5H), 3.41-3.35 (m, 0.5H), 3.29-3.23 (m, 1H), 2.71-2.59 (m, 1H), 2.65 (s, 3H), 2.22-2.00 (m, 3H), 1.93-1.80 (m, 1H), 1.51-1.45 (m, 1H), 1.50 (s,
1095 3.5H), 1.41 (s, 5.5H); MS calculated for C25H3iClN503 (M+H+) 484.20, found 484.20.
Step B: A solution of I-27a (8.62 g, 16.4 mmol) in MeOH (67 mL) was treated with HC1 in dioxane (4M, 67 mL) and the mixture was stirred at room temperature for 7 h. The mixture was then concentrated under reduced pressure to afford the title compound (Intermediate 27). The product was used in the next step without further purification. A sample was treated
1 100 with 1M NaOH, extracted with EtOAc, dried with Na2SC>4 and concentrated under reduced pressure to afford 1-27 as a free base. 1H-NMR (400MHz, CD3CN): d 8.49 (d, J=5.0 Hz, 1H), 7.81 (s, 1H), 7.72 (d, J=4.8 Hz, 1H), 7.50 (br d, J=7.52 Hz, 1H), 7.16 – 7.09 (m, 2H), 5.66-5.59 (m, 1H), 3.77 (dd, J = 6.54, 14.3 Hz, 1H), 3.18 (dd, J = 5.3, 14.3 Hz, 1H), 3.05 – 2.98 (m, 1H), 2.76-2.69 (m, 1H), 2.63-2.53 (m, 1H), 2.47 (s, 3H), 2.10-2.03 (m, 1H),
1 105 1.96-1.93 (m, 2H), 1.86 – 1.75 (m, 2H), 1.61 – 1.54 (m, 2H); MS calculated for
C2oH23ClN50 (M+H+) 384.15, found 384.20.
(i?.E)-N-(7-chloro-l-(l-(4-(dimethylamino)but-2-enoyl)azepan-3-yl)-lH-benzo[dlimidazol-2
-yl)-2-methylisonicotinamide
1 1 10 
A mixture of (E)-4-(dimethylamino)but-2-enoic acid hydrochloride (58 mg, 0.35 mmol) and l-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (67 mg, 0.35 mmol) in DMF (2 mL) was treated with hydroxybenzotriazole (54 mg, 0.35 mmol) and stirred at room temperature for 1 h. The resulting mixture was added to a solution of 1-27 (100 mg, 0.22 1 1 15 mmol) in DMF (2 mL). Triethylamine (199 mg, 1.97 mmol) was then added and the mixture was stirred for 5 days. Water (2 mL) was added and the mixture was concentrated under
reduced pressure. The residue was diluted with IN NaOH (20 mL) and extracted with EtOAc (3 x 50 mL). The combined organic layers were washed with water (50 mL) and brine (2 x 50 mL), dried over Na2S04, and concentrated under reduced pressure. The crude was purified by
1 120 column chromatography (9: 1 :0.175N CH2Cl2/MeOH/NH3 in CH2C12, 0% to 100%) to afford the title compound. JH NM (400 MHz, DMSO-d6) δ 8.59 (d, J= 4.8 Hz, 1H), 7.89 (s, 1H), 7.79 (d, J = 4.8 Hz, 1H), 7.60 (d, J = 7.5 Hz, 1H), 7.30-7.22 (m, 2H), 6.71-6.65 (m, 1H), 6.57-6.54 (m, 1H), 5.54 (br. s, 1H), 4.54 (br. s, 1H), 4.20 (br s, 1H), 3.95 (br s, 1H), 3.48 (br s, 1H), 2.98 (br s, 2H), 2.72 (d, J = 12.0 Hz, 1H), 2.58 (s, 3H), 2.14 (br s, 6H), 2.05 (d, J =
1 125 6.7 Hz, 3H), 1.88 (br s, 1H), 1.46 (d, J=l 1.3 Hz, 1H); MS calculated for C26H32C1N602
(M+H+) 495.22, found 495.10. Melting point (1 14.6 °C).
WO 2015083059
https://www.google.com/patents/WO2015083059A1?cl=en
Intermediate 26
(RVtert-butyl 3-(2-amino-7-chloro-lH-benzo[dlimidazol-l-vf)azepane-l-carboxylate

Step A: (R)-tert- butyl 3-((2-chloro-6-nitrophenyl)amino)azepane-l-carboxylate (I-26a) was prepared following procedures analogous to 1-15, Step A, using the appropriate starting materials. 1H-NMR (400MHz, CDC13): d 8.00-7.91 (m, 1H), 7.58-7.49 (m, 1H), 7.02-6.51 (m, 2H), 4.31-4.03 (m, 1H), 3.84-2.98 (m, 4H), 1.98-1.60 (m, 5H), 1.46-1.39 (m, 10H); MS calculated for Ci7H25ClN304 (M+H+) 370.15, found 370.10.
Step B: A mixture of I-26a (7.5 g, 19.5 mmol) and Zn (12.8 mg, 195 mmol) in AcOH
(22 mL) was stirred at room temperature for 2 h. The reaction was basified with saturated aqueous Na2CC>3 solution, filtered, and extracted with EtOAc (3 x 80 mL). The combined organic phase was washed with brine, dried with Na2S04 and concentrated in vacuum to afford (R)-tert-butyl 3-((2-amino-6-chlorophenyl)amino)azepane-l-carboxylate (I-26b). MS calculated for C17H27CIN3O2 (M+H+) 340.17, found 340.10. The crude was used in the next step without further purification.
Step C: The title compound (Intermediate 26) was prepared from I-26b following procedures analogous to 1-15, Step C. ‘H-NMR (400MHZ, CDCI3): d 7.34-7.26 (m, 1H), 7.04-6.97 (m, 2H), 6.05-5.85 (m, 1H), 5.84-5.72 (m, 1H), 5.50-5.37 (m, 0.5H), 5.10-4.80(m, 0.5H), 4.41-4.23(m, 1H), 4.09-3.96(m, 0.5H), 3.94-3.81 (m, 1H), 3.76-3.57 (m, 1H), 3.22-3.14 (m, 0.5H), 2.84-2.63 (m, 1H), 2.34-2.17 (m, 1H), 2.07-1.84 (m, 1H), 1.82-1.64 (m, 2H), 1.53 (s, 9H), 1.48-1.37 (m, 1H); MS calculated for Ci8H26ClN402(M+H+) 365.17, found 365.10.
Intermediate 27
(R)-N-(l-(azepan-3-yl)-7-chloro-lH-benzo[dlimidazol-2-yl)-2-methylisonicotinamide hydrochloride

5-26 step A l~27a intermediate 27
Step A: A mixture of 2-methylisonicotinic acid (3.371 g, 24.6 mmol) and 2-(7-aza-lH-benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (9.345 g, 24.6 mmol) in CH2C12 (120 ml) was treated at room temperature with NEt3 (4.1 mL, 29.4 mmol). The reaction was stirred for 1 hour before it was slowly added into a CH2C12solution (45 ml) of 1-26 (5.98 g, 16.4 mmol). Ten minutes later, more NEt3 (4.1 mL, 29.4 mmol) was added and the mixture stirred for 2 h. The mixture was then diluted with CH2C12 (240 mL), washed with H20 (2 x 80 mL), saturated aqueous NaHCC solution (70 mL), and brine (70 mL). The organic phase was dried with Na2S04, and concentrated under reduced pressure. The crude material was purified by column chromatography (55% EtOAc/hexanes) to afford
(R)-tert-butyl
3-(7-chloro-2-(2-methylisonicotinamido)-lH-benzo[d]imidazol-l-yl)azepane-l-carboxylate (I-27a) as a light yellow foam. 1H-NMR (400MHz, CDCI3): d 12.81 (br s, 1H), 8.65-8.62 (m, 1H), 7.95-7.85 (m, 2H), 7.27-7.11 (m, 3H), 5.64 – 5.51 (m, 1H), 4.56-4.44 (m, 1H),
4.07-3.92 (m, 1H), 3.79-3.71 (m, 0.5H), 3.41-3.35 (m, 0.5H), 3.29-3.23 (m, 1H), 2.71-2.59 (m, 1H), 2.65 (s, 3H), 2.22-2.00 (m, 3H), 1.93-1.80 (m, 1H), 1.51-1.45 (m, 1H), 1.50 (s, 3.5H), 1.41 (s, 5.5H); MS calculated for C25H3iClN503 (M+H+) 484.20, found 484.20.
Step B: A solution of I-27a (8.62 g, 16.4 mmol) in MeOH (67 mL) was treated with HCI in dioxane (4M, 67 mL) and the mixture was stirred at room temperature for 7 h. The mixture was then concentrated under reduced pressure to afford the title compound (Intermediate 27). The product was used in the next step without further purification. A sample was treated with 1M NaOH, extracted with EtOAc, dried with Na2S04 and concentrated under reduced pressure to afford 1-27 as a free base. ‘H-NMR (400MHZ, CD3CN): d 8.49 (d, J=5.0 Hz, 1H), 7.81 (s, 1H), 7.72 (d, J=4.8 Hz, 1H), 7.50 (br d, J=7.52 Hz, 1H), 7.16 – 7.09 (m, 2H), 5.66-5.59 (m, 1H), 3.77 (dd, J = 6.54, 14.3 Hz, 1H), 3.18 (dd, J = 5.3, 14.3 Hz, 1H), 3.05 -2.98 (m, 1H), 2.76-2.69 (m, 1H), 2.63-2.53 (m, 1H), 2.47 (s, 3H), 2.10-2.03 (m, 1H), 1.96-1.93 (m, 2H), 1.86 – 1.75 (m, 2H), 1.61 – 1.54 (m, 2H); MS calculated for
C20H23CIN5O (M+H+) 384.15, found 384.20.
(i?,£,)-N-(7-chloro-l-(l-(4-(dimethylamino)but-2-enoyl)azepan-3-yl)-lH-benzo[dlimidazol-2
-νΠ-2-methylisonicotinamide

A mixture of (E)-4-(dimethylamino)but-2-enoic acid hydrochloride (58 mg, 0.35 mmol) and l -ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (67 mg, 0.35 mmol) in DMF (2 mL) was treated with hydroxybenzotriazole (54 mg, 0.35 mmol) and stirred at room temperature for 1 h. The resulting mixture was added to a solution of 1-27 (100 mg, 0.22 mmol) in DMF (2 mL). Triethylamine (199 mg, 1.97 mmol) was then added and the mixture was stirred for 5 days. Water (2 mL) was added and the mixture was concentrated under reduced pressure. The residue was diluted with IN NaOH (20 mL) and extracted with EtOAc (3 x 50 mL). The combined organic layers were washed with water (50 mL) and brine (2 x 50 mL), dried over Na2S04, and concentrated under reduced pressure. The crude was purified by column chromatography (9: 1 :0.175N CH2Cl2/MeOH/NH3 in CH2C12, 0% to 100%) to afford the title compound. 1H NMR (400 MHz, DMSO-d6) δ 8.59 (d, J = 4.8 Hz, 1H), 7.89 (s, 1H), 7.79 (d, J = 4.8 Hz, 1H), 7.60 (d, J = 7.5 Hz, 1H), 7.30-7.22 (m, 2H), 6.71-6.65 (m, 1H), 6.57-6.54 (m, 1H), 5.54 (br. s, 1H), 4.54 (br. s, 1H), 4.20 (br s, 1H), 3.95 (br s, 1H), 3.48 (br s, 1H), 2.98 (br s, 2H), 2.72 (d, J = 12.0 Hz, 1H), 2.58 (s, 3H), 2.14 (br s, 6H), 2.05 (d, J = 6.7 Hz, 3H), 1.88 (br s, 1H), 1.46 (d, J=11.3 Hz, 1H); MS calculated for C26H32C1N602 (M+H+) 495.22, found 495.10. Melting point (114.6 °C).
PATENT
WO 2015112705
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015112705
PATENT
WO 2013184757
https://www.google.com/patents/WO2013184757A1?cl=en
Intermediate 26
(R)-tert-butyl 3 -(2-amino-7-chloro- 1 H-benzo Tdlimidazol- 1 – vDazepane- 1 – carboxylate

Intermediate 26
Step A: (R)-tert-butyl 3-((2-chloro-6-nitrophenyl)amino)azepane-l-carboxylate (I- 26a) was prepared following procedures analogous to 1-15, Step A, using the appropriate starting materials. 1 H-NMR (400MHz, CDC13): d 8.00-7.91 (m, 1H), 7.58-7.49 (m, 1H), 7.02-6.51 (m, 2H), 4.31-4.03 (m, 1H), 3.84-2.98 (m, 4H), 1.98-1.60 (m, 5H), 1.46-1.39 (m, 10H); MS calculated for C17H25CIN3O4 (M+H+) 370.15, found 370.10. Step B: A mixture of I-26a (7.5 g, 19.5 mmol) and Zn (12.8 mg, 195 mmol) in AcOH (22 mL) was stirred at room temperature for 2 h. The reaction was basified with saturated aqueous Na2CC>3 solution, filtered, and extracted with EtOAc (3 x 80 mL). The combined organic phase was washed with brine, dried with Na2S04 and concentrated in vacuo to afford (R)-tert-butyl 3-((2-amino-6-chlorophenyl)amino)azepane-l-carboxylate (I-26b). MS calculated for Ci7H27ClN302 (M+H+) 340.17, found 340.10. The crude was used in the next step without further purification.
Step C: The title compound (Intermediate 26) was prepared from I-26b following procedures analogous to 1-15, Step C. ]H-NMR (400MHz, CDC13): d 7. ,34-7.26 (m, 1H), 7.04-6.97 (m, 2H), 6.05-5.85 (m, 1H), 5.84-5.72 (m, 1H), 5.50-5.37 (m, 0.5H), 5.10- 4.80(m, 0.5H), 4.41-4.23(m, 1H), 4.09-3.96(m, 0.5H), 3.94-3.81 (m, 1H), 3.76-3.57 (m, 1H), 3.22-3.14 (m, 0.5H), 2.84-2.63 (m, 1H), 2.34-2.17 (m, 1H), 2.07-1.84 (m, 1H), 1.82- 1.64 (m, 2H), 1.53 (s, 9H), 1.48-1.37 (m, 1H); MS calculated for Ci8H26ClN402 (M+H+) 365.17, found 365.10.
Intermediate 27
(R)-N-(l-(azepan-3-yl)-7-chloro-lH-benzordlimidazol-2-yl)-2-methylisonicotinamide hydrochloride

l-27a Intermediate 27
Step A: A mixture of 2-methylisonicotinic acid (3.371 g, 24.6 mmol) and 2-(7-aza- 1H- benzotriazole-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate (9.345 g, 24.6 mmol) in CH2C12 (120 ml) was treated at room temperature with NEt3 (4.1 mL, 29.4 mmol). The reaction was stirred for 1 hour before it was slowly added into a CH2C12 solution (45 ml) of 1-26 (5.98 g, 16.4 mmol). Ten minutes later, more NEt3 (4.1 mL, 29.4 mmol) was added and the mixture stirred for 2 h. The mixture was then diluted with CH2C12 (240 mL), washed with H20 (2 x 80 mL), saturated aqueous NaHC03 solution (70 mL), and brine (70 mL). The organic phase was dried with Na2S04, and concentrated under reduced pressure. The crude material was purified by column chromatography (55% EtOAc/hexanes) to afford (R)-tert-butyl 3-(7-chloro-2-(2-methylisonicotinamido)- lH-benzo[d]imidazol-l-yl)azepane-l-carboxylate (I-27a) as a light yellow foam. ]H- NMR (400MHz, CDC13): d 12.81 (br s, IH), 8.65-8.62 (m, IH), 7.95-7.85 (m, 2H), 7.27- 7.11 (m, 3H), 5.64 – 5.51 (m, IH), 4.56-4.44 (m, IH), 4.07-3.92 (m, IH), 3.79-3.71 (m, 0.5H), 3.41-3.35 (m, 0.5H), 3.29-3.23 (m, IH), 2.71-2.59 (m, IH), 2.65 (s, 3H), 2.22-2.00 (m, 3H), 1.93-1.80 (m, IH), 1.51-1.45 (m, IH), 1.50 (s, 3.5H), 1.41 (s, 5.5H); MS calculated for C25H31CIN5O3 (M+H+) 484.20, found 484.20.
Step B: A solution of I-27a (8.62 g, 16.4 mmol) in MeOH (67 mL) was treated with HCl in dioxane (4M, 67 mL) and the mixture was stirred at room temperature for 7 h. The mixture was then concentrated under reduced pressure to afford the title compound
(Intermediate 27). The product was used in the next step without further purification. A sample was treated with 1M NaOH, extracted with EtOAc, dried with Na2S04 and concentrated under reduced pressure to afford 1-27 as a free base. ]H-NMR (400MHz, CD3CN): d 8.49 (d, J=5.0 Hz, IH), 7.81 (s, IH), 7.72 (d, J=4.8 Hz, IH), 7.50 (br d, J=7.52 Hz, IH), 7.16 – 7.09 (m, 2H), 5.66-5.59 (m, IH), 3.77 (dd, J = 6.54, 14.3 Hz, IH), 3.18 (dd, J = 5.3, 14.3 Hz, IH), 3.05 – 2.98 (m, IH), 2.76-2.69 (m, IH), 2.63-2.53 (m, IH), 2.47 (s, 3H), 2.10-2.03 (m, IH), 1.96-1.93 (m, 2H), 1.86 – 1.75 (m, 2H), 1.61 – 1.54 (m, 2H); MS calculated for C20H23CIN5O (M+H+) 384.15, found 384.20.
Example 5
(/?,£,)-N-(7-chloro-l-(l-(4-(dimethylamino)but-2-enoyl)azepan-3-yl)- lH- benzordlimidazol-2-yl)-2-methylisonicotinamide

A mixture of (E)-4-(dimethylamino)but-2-enoic acid hydrochloride (58 mg, 0.35 mmol) and l-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (67 mg, 0.35 mmol) in DMF (2 mL) was treated with hydroxybenzotriazole (54 mg, 0.35 mmol) and stirred at room temperature for 1 h. The resulting mixture was added to a solution of 1-27 (100 mg, 0.22 mmol) in DMF (2 mL). Triethylamine (199 mg, 1.97 mmol) was then added and the mixture was stirred for 5 days. Water (2 mL) was added and the mixture was concentrated under reduced pressure. The residue was diluted with IN NaOH (20 mL) and extracted with EtOAc (3 x 50 mL). The combined organic layers were washed with water (50 mL) and brine (2 x 50 mL), dried over Na2SC>4, and concentrated under reduced pressure. The crude was purified by column chromatography (9: 1 :0.175N CH2Cl2/MeOH/NH3 in CH2C12, 0% to 100%) to afford the title compound (Example 5). ]H NMR (400 MHz, DMSO-d6) δ 8.59 (d, J = 4.8 Hz, IH), 7.89 (s, IH), 7.79 (d, J = 4.8 Hz, IH), 7.60 (d, / = 7.5 Hz, IH), 7.30-7.22 (m, 2H), 6.71-6.65 (m, IH), 6.57-6.54 (m, IH), 5.54 (br. s, IH), 4.54 (br. s, IH), 4.20 (br s, IH), 3.95 (br s, IH), 3.48 (br s, IH), 2.98 (br s, 2H), 2.72 (d, / = 12.0 Hz, IH), 2.58 (s, 3H), 2.14 (br s, 6H), 2.05 (d, / = 6.7 Hz, 3H), 1.88 (br s, IH), 1.46 (d, 7=11.3 Hz, IH); MS calculated for C26H32CIN6O2 (M+H+) 495.22, found 495.10. Melting point (114.6 °C).
(/?,E)-N-(7-chloro- l-(l-(4-(dimethylamino)but-2-enoyl)azepan-3-yl)-lH- benzo[d]imidazol-2-yl)-2-methylisonicotinamide (1.0 g) was dissolved in acetone (30 mL) by heating to 55°C to form a solution. Methanesulfonic acid (325 μί) was added to acetone (50 mL), and the methanesulfonic acid/acetone (22.2 mL) was added to the solution at 0.05ml/min. Following precipitation, the resulting suspension was cooled to room temperature at 0.5 °C/min, and crystals were collected by filtration, and dried for 4 hours at 40°C under vacuum. The collected crystals (300 mg) were suspended in acetone/H20 (6 mL; v/v=95/5) by heating to 50°C. The suspension was kept slurrying for 16 hours, and cooled to room temperature at 0.5 °C/min. The crystal was collected by filtration and dried for 4 hours at 40°C under vacuum.
The structure of (7?,£‘)-N-(7-chloro-l-(l-(4-(dimethylamino)but-2-enoyl)azepan-3-yl)- lH-benzo[d]imidazol-2-yl)-2-methylisonicotinamide mesylate was confirmed by Differential Scanning Calorimetry, X-Ray Powder Diffraction, and Elemental Analyses. Melting point (170.1 °C). Theoretical calculated: C (54.8); H (5.9); N (14.2); 0 (13.5); %S (5.4); and C1 (6.0); C:N ratio: 3.86. Found: C (52.0); H (5.8); N (13.3); C1 (5.9); C:N ratio: 3.91. Stoichiometry: 1.01.
References
AACR Annual Meeting 2014; April 5-9, 2014; San Diego, CA.
nmr http://www.medchemexpress.com/product_pdf/HY-12872/EGF816-NMR-HY-12872-17795-2015.pdf
/////EGF 816, EGF816, EGFR, Covalent inhibitor, T790M, Oncogenic mutation, Lung cancer, NSCLC, SBDD, Drug resistance, EGF-816, EGFRmut-TKI EGF816, Nazartinib
O=C(NC1=NC2=CC=CC(Cl)=C2N1[C@H]3CN(C(/C=C/CN(C)C)=O)CCCC3)C4=CC=NC(C)=C4
HELP, Need one time help to pay 10 year concessional subscription to this, your favorite blog to WordPress
Just One viewer please come forward
Dear Kind Viewer’s
WordPress is kind to me and negotiated a one time 10 year concessional subscription of 260 US dollars…….https://newdrugapprovals.org/
I need one time help to pay this one time 10 year concessional subscription to our favorite blog.
This is done to keep this blog running even after my death.
Currently I am paying 99 US Dollars per annum
email me
amcrasto@gmail.com
call +919323115463
Paypal will work for me via email request to you by me, Indian govt does not allow automatic transfer via paypal buttons on the blog
email me at amcrasto@gmail.com and tell me amount, i will request you via paypal
DR ANTHONY CRASTO
LIONEL MY SON, MY MOTIVATION
.
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, He cried bitterly and we had never seen him so depressed
Now I keep Lionel as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son and family happy.
ps
The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent,
///////////
LAMIVUDINE
Lamivudine (2′,3′-dideoxy-3′-thiacytidine, commonly called 3TC) is an antiretroviral medication used to prevent and treat HIV/AIDS and used to treat chronic hepatitis B.[1]
It is of the nucleoside analog reverse transcriptase inhibitor (NRTI) class. It is marketed in the United States under the tradenames Epivir and Epivir-HBV.
It is on the World Health Organization’s List of Essential Medicines, a list of the most important medication needed in a basic health system.[2] As of 2015 the cost for a typical month of medication in the United States is more than 200 USD.[3]
Medical uses
Lamivudine has been used for treatment of chronic hepatitis B at a lower dose than for treatment of HIV/AIDS. It improves the seroconversion of e-antigen positive hepatitis B and also improves histology staging of the liver. Long term use of lamivudine leads to emergence of a resistant hepatitis B virus (YMDD) mutant. Despite this, lamivudine is still used widely as it is well tolerated.
Resistance
In HIV, high level resistance is associated with the M184V/I mutation in the reverse transcriptase gene as reported by Raymond Schinazi’s group at Emory University. GlaxoSmithKline claimed that the M184V mutation reduces “viral fitness”, because of the finding that continued lamivudine treatment causes the HIV viral load to rebound but at a much lower level, and that withdrawal of lamivudine results in a higher viral load rebound with rapid loss of the M184V mutation; GSK therefore argued that there may be benefit in continuing lamivudine treatment even in the presence of high level resistance, because the resistant virus is “less fit”. The COLATE study has suggested that there is no benefit to continuing lamivudine treatment in patients with lamivudine resistance.[4] A better explanation of the data is that lamivudine continues to have a partial anti-viral effect even in the presence of the M184V mutation.
In hepatitis B, lamivudine resistance was first described in the YMDD (tyrosine–methionine–aspartate-aspartate) locus of the HBV reverse transcriptase gene. The HBV reverse transcriptase gene is 344 amino acids long and occupies codons 349 to 692 on the viral genome. The most commonly encountered resistance mutations are M204V/I/S.[5] The change in amino acid sequence from YMDD to YIDD results in a 3.2 fold reduction in the error rate of the reverse transcriptase, which correlates with a significant growth disadvantage of the virus. Other resistance mutations are L80V/I, V173L and L180M.[6]
Mechanism of action
Lamivudine is an analogue of cytidine. It can inhibit both types (1 and 2) of HIV reverse transcriptase and also the reverse transcriptase of hepatitis B virus. It is phosphorylated to active metabolites that compete for incorporation into viral DNA. They inhibit the HIV reverse transcriptase enzyme competitively and act as a chain terminator of DNA synthesis. The lack of a 3′-OH group in the incorporated nucleoside analogue prevents the formation of the 5′ to 3′ phosphodiester linkage essential for DNA chain elongation, and therefore, the viral DNA growth is terminated.
Lamivudine is administered orally, and it is rapidly absorbed with a bio-availability of over 80%. Some research suggests that lamivudine can cross the blood–brain barrier. Lamivudine is often given in combination with zidovudine, with which it is highly synergistic. Lamivudine treatment has been shown to restore zidovudine sensitivity of previously resistant HIV. Lamivudine showed no evidence of carcinogenicity or mutagenicity in in vivo studies in mice and rats at doses from 10 to 58 times those used in humans.[7]
History
Racemic BCH-189 (the minus form is known as Lamivudine) was invented by Dr. Bernard Belleau while at work at McGill University and Dr Paul Nguyen-Ba at the Montreal-based IAF BioChem International, Inc. laboratories in 1988 and the minus enantiomer isolated in 1989. Samples were first sent to Dr. Yung-Chi Cheng of Yale University for study of its toxicity. When used in combination with AZT, he discovered that Lamivudine’s negative form reduced side effects and increased the drug’s efficiency at inhibiting reverse transcriptase.[8] The combination of Lamivudine and AZT increased the efficiency at inhibiting an enzyme HIV uses to reproduce its genetic material. As a result, Lamivudine was identified as a less toxic agent to mitochondria DNA than other retroviral drugs.[9]
Lamivudine was approved by the Food and Drug Administration (FDA) on November 17, 1995 for use with zidovudine (AZT) and again in 2002 as a once-a-day dosed medication. The fifth antiretroviral drug on the market, it was the last NRTI for three years while the approval process switched to protease inhibitors. According to the manufacturer’s 2004 annual report, its patent will expire in the United States in 2010 and in Europe in 2011.
On September 2014, Dr. Gorbee Logan, a Liberian physician, reported positive results while treating Ebola virus disease with Lamivudine. Out of 15 patients treated with the antiviral, 13 (those treated within the third to fifth day of symptoms being manifested) survived the disease and were declared virus-free; the remaining cases (treated from the fifth day or later) died.[10][11]
Presentation
- Epivir 150 mg or 300 mg tablets (GlaxoSmithKline; US and UK) for the treatment of HIV;
- Epivir-HBV 100 mg tablets (GlaxoSmithKline; US only) for the treatment of hepatitis B;
- Zeffix 100 mg tablets (GlaxoSmithKline; UK only) for the treatment of hepatitis B.
- 3TC 150 mg tablets (GlaxoSmithKline; South Africa) for the treatment of HIV;
Lamivudine is also available in fixed combinations with other HIV drugs:
- Combivir (with zidovudine);
- Epzicom/Kivexa (with abacavir);
- Trizivir (with zidovudine and abacavir)
Lamivudine (I) (CAS No. 134678-17-4) is chemically known as (-)-[2R,5S]-4T amino- 1 – [2-(hydroxymethyl)- 1 ,3 -oxathiolan-5-yl] -2( 1 H)-pyrimidin-2-one.

Formula (I)
Lamivudine is a reverse transcriptase inhibitor used alone or in combination with other classes of Anti-HIV drugs in the treatment of HIV infection. It is available commercially as a pharmaceutical composition under the brand name EPIVIR®, marketed by GlaxoSmithKline, and is covered under US 5,047,407.
This molecule has two stereo-centres, thus giving rise to four stereoisomers: (±)- Cis Lamivudine and (±)-Trans Lamivudine. The pharmaceutically active isomer however is the (-)-Cis isomer which has the absolute configuration [2R,5S] as show in Formula (I).
US 5,047,407 discloses the 1,3-oxathiolane derivatives; their geometric (cis/trans) and optical isomers. This patent describes the preparation of Lamivudine as a mixture of cis and trans isomers (shown in scheme I). The diastereomers obtained are converted into N-acetyl derivatives before separation by column chromatography using ethylacetate and methanol (99:1); however, this patent remains silent about further resolution of the cis isomer to the desired (-)- [2R,5S]-Cis-Lamivudine. Secondly, as the ethoxy group is a poor leaving group, the condensation of cytosine with compound VI gives a poor yield, i.e. 30 – 40%, of compound VII. Thirdly, chromatographic separation that has been achieved only after acetylation requires a further step of de-acetylation of the cis-(±)- isomer. Also, separation of large volumes of a compound by column chromatography makes the process undesirable on a commercial scale.

(+/-) Cis (+/-) Cis Lamivudine (VIII)
Scheme – 1 Efforts have been made in the past to overcome the shortcomings of low yield and enantiomeric enrichment, hi general, there have been two approaches to synthesize (— )-[2R,5S]-Cis-Lamivudine. One approach involves stereoselective synthesis, some examples of which are discussed below.
US 5,248,776 describes an asymmetric process for the synthesis of enantiomerically pure β-L-(-)-l,3-oxathiolone-nucleosides starting from optically pure 1,6-thioanhydro-L-gulose, which in turn can be easily prepared from L- Gulose. The condensation of the 1,3-oxathiolane derivative with the heterocyclic base is carried out in the presence of a Lewis acid, most preferably SnCl4, to give the [2R,5R] and [2R,5S] diastereomers that are then separated chromatographically.
US 5,756,706 relates a process where compound A is esterified and reduced to compound B. The hydroxy group is then converted to a leaving group (like acetyl) and the cis- and trans-2R-tetrahydrofuran derivatives are treated with a pyrimidine base, like N-acetylcytosine, in the presence trimethylsilyl triflate to give compound C in the diastereomeric ratio 4: 1 of cis and trans isomers.

A B C
Z = S5 CH
Dissolving compound C in a mixture of 3:7 ethyl acetate-hexane separates the cis isomer. The product containing predominantly the cis-2R,5S isomer and some trans-2R,5R compound is reduced with NaBH4 and subjected to column chromatography (30% MeOH-EtOAc) to yield the below compound.

US 6,175,008 describes the preparation of Lamivudine by reacting mercaptoacetaldehyde dimer with glyoxalate and further with silylated pyrimidine base to give mainly the cis-isomer by using an appropriate Lewis acid, like TMS-
I5 TMS-Tf, TiCl4 et cetera. However the stereoselectivity is not absolute and although the cis isomer is obtained in excess, this process still requires its separation from the trans isomer. The separation of the diastereomers Js done by acetylation and chromatographic separation followed by deacetylation. Further separation of the enantiomers of the cis-isomer is not mentioned.
US 6,939,965 discloses the glycosylation of 5-fluoro-cytosine with compound F (configuration: 2R and 2S)

. F
The glycosylation is carried out in the presence of TiCl3(OiPr) which is stereoselective and the cis-2R,5S-isomer is obtained in excess over the trans- 2S,5S-isomer. These diastereomers are then separated by fractional crystallization.
US 6,600,044 relates a method for converting the undesired trans-l,3-oxathiolane nucleoside to the desired cis isomer by a method of anomerizatioή or transglycosylation and the separation of the hydroxy-protected form of cis-, trans- (-)-nucleosides by fractional crystallization of their hydrochloride, hydrobromide, methanesulfonate salts. However, these cis-trans isomers already bear the [R] configuration at C2 and only differ in their configuration at C5; i.e. the isomers are [2R,5R] and [2R,5S]. Hence diastereomeric separation directly yields the desired [2R, 5S] enantiomer of Lamivudine.
In the second approach to prepare enantiomerically pure Lamivudine the resolution of racemic mixtures of nucleosides is carried out. US 5,728,575 provides one such method by using enzyme-mediated enantioselective hydrolysis of esters of the formula

wherein, ‘R’ is an acyl group and ‘Rl ‘ represents the purine or pyrimidine base.
‘R’ may be alkyl carboxylic, substituted alkyl carboxylic and preferably an acyl group that is significantly electron-withdrawing, eg. α-haloesters. After selective hydrolysis, the process involves further separation of the unhydrolyzed ester from the‘ enantiomerically pure 1,3-oxathiolane-nucleoside. Three methods are suggested in this patent, which are:
1. Separation of the more lipophilic unhydrolyzed ester by solvent extraction with one of a wide variety of nonpolar organic solvents.
2. Lyophilization followed by extraction into MeOH or EtOH. 3. Using an HPLC column designed for chiral separations.
In another of its aspects, this patent also refers to the use of the enzyme cytidine- deoxycytidine deaminase, which is enantiomer-specific, Λo catalyze the deamination of the cytosine moiety and thereby converting it to uridine. Thus, the enantiomer that remains unreacted is still basic and can be extracted by using an acidic solution.
However, the above methods suffer from the following drawbacks, (a) Enzymatic hydrolysis sets down limitations on choice of solvents: alcohol solvents cannot be used as they denature enzymes. (b) Lyophilization on an industrial scale is tedious, (c) Chiral column chromatographic separations are expensive.
WO 2006/096954 describes the separation of protected or unprotected enantiomers of the cis nucleosides of below formula by using a chiral acid to form diastereomeric salts that are isolated by filtration. Some of the acids used are R-
(-)-Camphorsulfonic acid, L-(-)-Tartaric acid, L-(-)-Malic acid, et cetera.
However, the configuration of these CIS-nucleosides are [2R,4R] and [2S,4S] as the heterocyclic base is attached at the 4 position of the oxathiolane ring and the overall stereo-structure of the molecule changes from that of the 2,5-substituted oxathiolane ring.

Thus various methods are described for the preparation of Lamivudine. However there is no mention in the prior art about the separation of an enantiomeric pair, either cis-(±) or trans-(±), from a mixture containing cis-[2R,5S], [2S,5R] and trans-[2R,5R], [2S,5S] isomers. Further, there also is a need to provide resolution of the cis-(±) isomers to yield the desired enantiomer in high optical purity.
CN 1223262 (Deng et aϊ) teaches the resolution of a certain class of compounds called Prazoles by using chiral host compounds such as dinaphthalenephenols (BINOL), diphenanthrenols or tartaric acid derivatives. The method consists of the formation of a 1:1 complex between the chiral host (BINOL) and one of the enantiomers, the guest molecule. The other enantiomer remains in solution. (S)- Omeprazole, which is pharmaceutically active as a highly potent inhibitor of gastric acid secretion, has been isolated from its racemic mixture in this manner by using S-BINOL.
BINOL is a versatile chiral ligand that has found its uses in various reactions involving asymmetric synthesis (Noyori, R. Asymmetric Catalysis in Organic
Synthesis) and optical resolution (Cram, D. J. et al J. Org. Chem. 1977, 42, 4173-
4184). Some of these reactions include BINOL-mediated oxidation and reduction reactions, C-C bond formation reactions such as Aldol reaction, Michael addition,
Mannich reaction et cetera (Brunei Chem. Rev. 2005 105, 857-897) and kinetic resolution, resolution by inclusion complexation et cetera.
BINOL, or l,l’-bi-2-Naphthol, being an atropoisomer possesses the property of chiral recognition towards appropriate compounds. One of the uses of BINOL in resolution that is known in literature is in Host-Guest complexation. In one such example, 1,1-binaphthyl derivatives have been successfully incorporated into optically active crown ethers for the enantioselective complexation of amino acid esters and chiral primary ammonium ions (Cram, D. J. Ace. Chem. Res. 1978, 11, 8-14). The chiral ‘host’ is thus able to discriminate between enantiomeric compounds by the formation of hydrogen bonds between the ether oxygen and the enantiomers. The complex formed with one of the isomers, the ‘guest’, will be less stable on steric grounds and this forms the basis for its separation.
It is evident from the literature cited that there exists a need to (a) synthesize Lamivudine by a process requiring less expensive, less hazardous and easily available reagents, and (b) achieve good yields with superior quality of product without resorting to column chromatography as a means of separation, thereby making the process of Lamivudine manufacture more acceptable industrially.
CLIP
ideally, the chemical synthesis of APIs begins from simple, inexpensive building blocks or RMs that are used for multiple purposes and are available in the fine chemicals industry, though some require uncommon RMs that contribute significantly to API manufacturing cost. RMs are converted into APIs by multi-step processes of breaking old chemical bonds and making new ones. A synthesis of 3TC is shown in . In the seven-step sequence, six steps involve breaking existing chemical bonds and creating new ones to build the molecular architecture of the API. The final recrystallization of an API is a critical step; at this stage the crystalline form of the API is determined and related substances (impurities) are removed or reduced to acceptable levels. APIs are often milled in a final step so that their particle size distribution (PSD) falls within specified limits. The crystalline form and PSD of an API must be controlled, because these properties are often critical to the formulation, dissolution, absorption and bioavailability of a drug. Bioavailability is the fraction of a drug dose that reaches systemic circulation (that is, is present in blood plasma) after administration. By definition, a drug is 100% bioavailable when administered by injection; drugs for ART are taken every day and administration by injection is not possible.
The cost of ART is absolutely critical to ensuring access in LMICs. The cost of manufacturing an API is dependent upon the cost of RMs, the cost of overheads and labour (OHL) and volume demand for the product. OHL includes the capital investment to build a manufacturing facility and operating costs, including personnel and energy, waste disposal and the eventual cost of decommissioning of the facility. Increased volume demand generally decreases the cost contribution of RM and OHL. Substantial production volumes are required to obtain full economy of scale . Producing 1–5 metric tons per year is substantially more expensive per kilogram than producing 100 metric tons of an API. There is a practical limit of approximately 50–100 metric tons/year beyond which cost reductions are modest with increased volume, but this practical limit refers to the volumes of drug manufactured in any single manufacturing plant. Exceptions to these generalizations do occur, most often when demand exceeds either the existing manufacturing capacity for a specific API or the availability of critical RMs . Exceptions that have occurred include shortages of β-thymidine for producing AZT and a squeeze on the availability and price of adenine as a starting material for TDF. Another contributor to RM and OHL costs is the efficiency of a chemical synthesis. Since operating costs for a manufacturing facility may be USD2,000/h, the number of steps or processing time for a chemical synthesis affects manufacturing cost. The efficiency of a synthesis is often quoted as an E-factor representing the kilograms of waste produced per kilogram of product manufactured. Waste management is expensive in chemical manufacturing wherever environmental guidelines are both reasonable and followed. From a slightly different perspective, increasing the overall yield of an API synthesis reduces RM use and associated cost for manufacturing.
Jinliang L, Feng LV. inventors; Shanghai Desano Pharmaceutical, assignee. A process for stereoselective synthesis of lamivudine. European Patent Application EP 2161 267 A1. 2007 June 29.

Object of the invention
Thus, one object of the present invention is to provide a process for the synthesis of_Lamivudine which is cost effective, uses less hazardous and easily available reagents, yet achieves good yields with superior quality of product without resorting to column chromatography.
A further object of the present invention is to provide an improved process for the synthesis of Lamivudine, by separating the mixture of diastereomers: Cis-[2R,5S], [2S,5R] from Trans-[2R,5R], [2S,5S] and then resolving the Cis isomers using BINOL to obtain (-)-[2R,5S]^Cis-Lamivudine with at least 99% ee.
This 1,3-oxathiolane compound VIII is further condensed with silylated cytosine in the presence of a Lewis acid such as trimethylsilyliodide to get protected 6-amino-3 – {2-hydroxymethyl- 1 ,3 -oxathiolan-5-yl} -3 -hydropyrimidine- 2-one (compound IX). OH

Cis(±)and Trans (±) racemic mixtures

Lamivudine (-)-[2

Compound (IX) is mixture of following optical isomers

SCHEME 2 The separation of the four-component diastereomeric mixture of isomers bearing the following configuration: trans-[2R,5R], [2S.5S] and cis-[2R,5S], [2S,5R] forms the next step. The separation efficiency of the benzoyl-protected compound
Example 9
Preparation of Lamivudine: (-)-[2R,5S]-4-amino-l-[2-(hydroxymethyl)-l,3- oxathiolan-5 -yl] -2(1 H)-pyrimidin-2-one
Compound I 5mL of cone. HCl was slowly added to a solution of 2Og of Lamivudine-BINOL complex in 100ml of ethylacetate and 10OmL of DM water (pH 2-2.5). The layers. were separated and a 10OmL aliquot of ethylacetate was added to the aqueous layer. The layers were separated again and the aqueous layer was neutralized using 1OmL of 10% aqueous NaOH solution. The solvent was recovered under vacuum at 40-45 0C, the product obtained was dissolved in 160 mL of methanol, filtered, the filtrate was concentrated and 32 mL of water-ethanol mixture (3:1) was added to this product, heated to get a clear solution, cooled to 5 – 10 0C and then filtered. The residue was vacuum dried at 45-50 0C. Yield: 4-5g.
Enantiomeric excess = 99.74 % m.p. = 133-135 °C [<X]D at 25°C = 98.32° (c = 5 water)
1H NMR (DMSO d6): 2.99-3.07 (dd, IH), 3.35-3.38 (dd, IH), 3.72-3.74 (m, 2H), 5.14-5.18 (t, IH), 5.32-5.38 (t, IH), 5.71-5.75 (d, IH), 6.16-6.21 (t, IH), 7.22-
7.27 (d, 2H), 7.80-7.83 (d, IH)
Moisture content: 1.67%
IR (in KBr, cm“1): 3551, 3236, 2927, 1614, 1492, 1404, 1336, 1253, 1146, 1052,
967, 786. MS: M+l =230
XRD [2Θ] (Cu – Ka1=I.54060A, Ka2=1.54443A Kβ= 1.39225A; 4OmA, 45kV):
5.08, 9.89, 10.16, 11.40, 11.65, 12.96, 13.23, 15.26, 15.82, 17.74, 18.74, 18.88,
19.67, 20.69, 22.13, 22.88, 23.71, 25.47, 26.07.
PATENT
http://www.google.com/patents/WO2013021290A1?cl=en



| EP 0382526; EP 0711771; JP 1996119967; JP 2000143662; US 5047407 |

| J Org Chem 1992,57(8),2217-9 |

Lamivudine is a nucleoside reverse transcriptase inhibitor, and is a kind of deoxycytidine analogue, which can inhibit the reproduction of Human immunodeficiency virus (HIV) and hepatitis B virus (HBV), whose chemical name is (2R-cis)-4-amino-1-(2-hydroxymethyl-1,3-oxathiolan-5-yl)-1H-pyrimidin-2-one, and structural formula is as follows:
-

-
In 1990, Belleau et al firstly reported Lamivudine structure, and BioChem Pharma of Canada firstly developed Lamivudine to be used to treat AIDS ( WO91/17159 ) and hepatitis B ( EP0474119 ), and found that it had distinguished therapeutic effect on hepatitis B. Since Lamivudine has two chiral centers, it has 4 stereisomers, among which the 2R,5S (2R–cis)-isomer is the most potent in anti-HIV and anti-HBV activities, and its cytotoxicity on some cells is lower than its enatiomer or racemic body.
-
WO94/14802 mentioned two synthetic schemes (see Scheme 1 and Scheme 2):


-
In the above two schemes of this process, chirality was not controlled, and the final product was obtained by column chromatography, thus the yield was low and the requirement on the equipment was high, resulting in that the production cost was high and the operation in the production could not be controlled easily.


The specific reaction scheme is as follows:

synthetic route is preferably as follows:

. The specific reaction scheme is as follows:


The specific reaction scheme is as follows:

Example 8 The preparation of (2R,5S)-4-amino-1-(2-hydroxymethyl-1,3-oxathiolane-5-yl) -2(1H)-pyrimidone (Lamivudine)
-
The compound of Example 7 (41.0g, 0.1mol) and methanol (250ml) were added to a reaction flask, and then stirred to make the compound dissolved in methanol. The mixture was cooled to 0 °C, and then K2CO3 (41.2g, 0.3mol) was added. The mixture was further stirred at room temperature overnight and then was adjusted by 0.1N HCl to a pH of about 7. The mixture was filtered and the solvent was evaporated under reduced pressure from the filtrate, and then to the residue was added 150ml of water. The aqueous layer was extracted by 150ml of toluene (50ml X 3), and then p-nitrobenzoic acid (16.8g, 0.1mol) was added to the aqueous layer and refluxed for 30 minutes, after which, the reaction mixture was cooled and further stirred at 0-5 °C for 2 hours. Then the reaction mixture was filtered and dried to give 31.7g of a white solid.
-
The resulting salt and anhydrous ethanol (120ml) were added to a reaction flask, and warmed to 70-75 °C. Triethylamine (12ml) was added dropwise, and the reaction was conducted at that temperature for 2 hours. Then the mixture was cooled to 50 °C, at which point ethyl acetate (150ml) was added dropwsie. After the addition was complete, the mixture was cooled to 10 °C and further stirred for 4 hours. The mixture was filtered to give 15.6g of Lamivudine, and the yield was 68%. 1H-NMR (DMSO-d6) δ: 7.83(dd, 1H), 7.17∼7.23(dd, 2H), 6.21(t, 1H), 5.72 (dd, 1H), 5.29 (t, 2H), 5.16 (t, 1H), 3.70∼3.74 (m, 2H), 3.32∼3.43 (dd, 1H), 3.01∼3.05(dd, 1H); Elemental analysis: C8H11N3O3S found(%): C 41.85, H 4.88 N 18.25, S 13.94; calculated (%) C 41.91, H 4.84, N 18.33, S13.99.
PAPER

http://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-9-265
References
- 1″Lamivudine”. The American Society of Health-System Pharmacists. Retrieved 31 July 2015.
- 2
- “WHO Model List of EssentialMedicines” (PDF). World Health Organization. October 2013. Retrieved 22 April 2014.
- 3
- Hamilton, Richart (2015). Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. p. 65. ISBN 9781284057560.
- 4
- Fox Z, Dragsted UB, Gerstoft J, et al. (2006). “A randomized trial to evaluate continuation versus discontinuation of lamivudine in individuals failing a lamivudine-containing regimen: The COLATE trial”. Antiviral Therapy 11 (6): 761–70. PMID 17310820.
- 5
- http://hivdb.stanford.edu/index.html Stanford University Drug Resistance Database.
- 6
- Koziel MJ, Peters MG (2007). “Viral hepatitis in HIV infection”. N Engl J Med 356 (14): 1445–54. doi:10.1056/NEJMra065142. PMID 17409326.
- 7
- “Epivir package insert” (PDF). GlaxoSmithKline. Retrieved January 20, 2011.
- 8
- Curtis, John (June 20, 1998). “Hunting Down HIV”. Yale Medicine.
- 9
- Soderstrom, E John (July 10, 2003). “National Institutes of Health: Moving Research from the Bench to the Bedside”.
- 10
- Cohen, Elizabeth (September 29, 2014). “Doctor treats Ebola with HIV drug in Liberia — seemingly successfully”. CNN.
- 11 HIV drug may stop Ebola. Operonlabs.com, 27 September 2014
External links
- Epivir (manufacturer’s website)
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-amino-1-[(2R,5S)-2-(hydroxymethyl)-1,3-oxathiolan-5-yl]-1,2-dihydropyrimidin-2-one
|
|
| Clinical data | |
| Trade names | Epivir |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a696011 |
| Pregnancy category |
|
| Routes of administration |
Oral |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 86% |
| Protein binding | Less than 36% |
| Biological half-life | 5 to 7 hours |
| Excretion | Renal (circa 70%) |
| Identifiers | |
| CAS Number | 134678-17-4 |
| ATC code | J05AF05 (WHO) |
| PubChem | CID 73339 |
| DrugBank | DB00709 |
| ChemSpider | 66068 |
| UNII | 2T8Q726O95 |
| KEGG | D00353 |
| ChEMBL | CHEMBL141 |
| NIAID ChemDB | 000388 |
| Synonyms | L-2′,3′-dideoxy-3′-thiacytidine |
| PDB ligand ID | 3TC (PDBe, RCSB PDB) |
| Chemical data | |
| Formula | C8H11N3O3S |
| Molar mass | 229.26 g/mol |
