WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
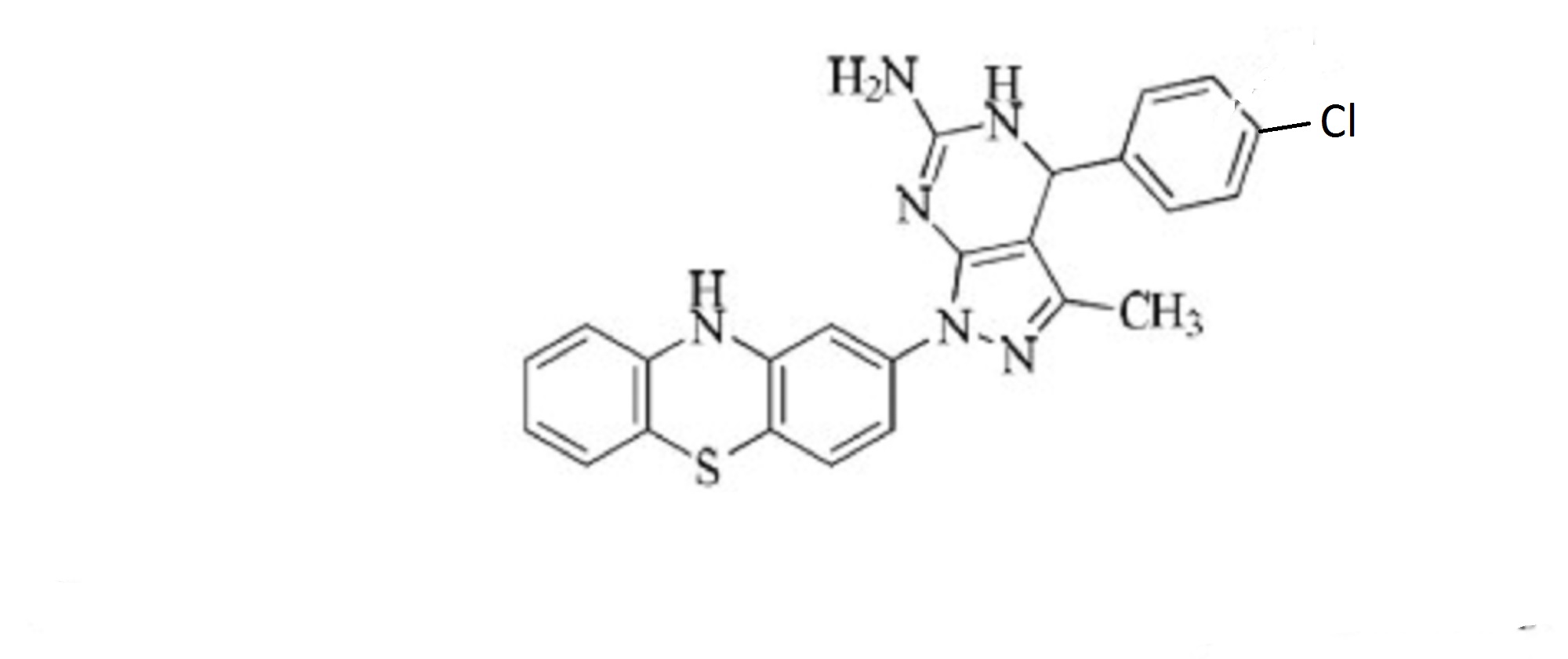
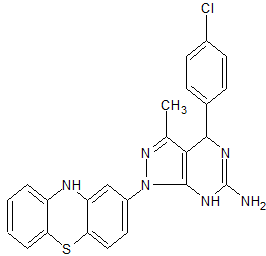
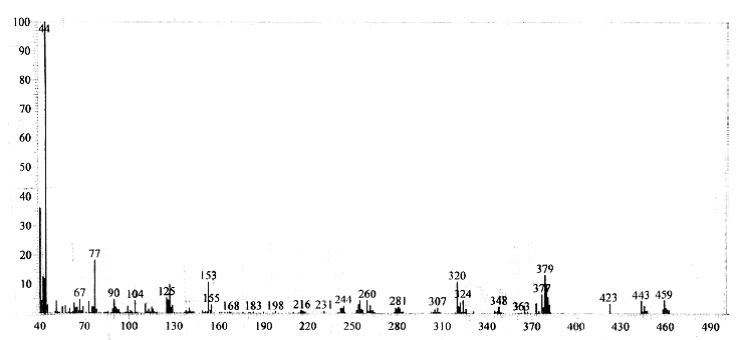
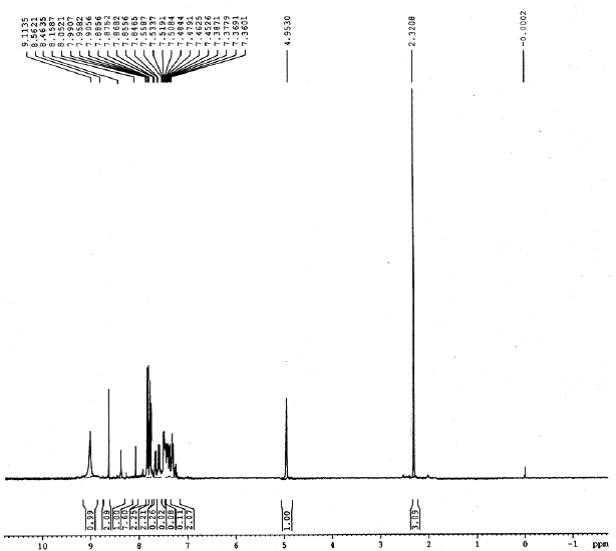
13C NMR (75 MHz, CDCl3): d 26.1, 41.2, 52.5, 59.8, 103.6, 104.2, 105.3, 114.2, 116.6, 122.7, 127.1, 127.9, 128.2, 128.6, 129.2, 132.5, 134.6, 142.4, 143.7, 155.3, 162.5. Mass (m/z): 459. Anal. (%) for C24H19ClN6S, Calcd. C, 62.81; H, 4.17; N, 18.31. Found: C, 62.75; H, 4.15; N, 18.26.
Mass spectrum of 4g
1H NMR spectrum of 4g
Tuberculosis (TB) is a highly infectious airborne disease caused by the pathogenic bacterium Mycobacterium tuberculosis (Mtb). 1According to the latest World Health Organization (WHO) report, an estimated 8.6 million people developed TB and 1.3 million died from the disease (including 320,000 deaths among HIV-positive people) in 2012. The majority of cases worldwide in 2012 were in the South-East Asia (29%), African (27%) and western Pacific (19%) regions. India and China alone accounted for 26% and 12% of total cases, respectively.2 The standard antitubercular treatment regimen, termed DOTS (Directly Observed Therapy, Short-course), is based on the co-administration of age-old drugs like isoniazid (INH), rifampin (RMP), ethambutol (EMB), and pyrazinamide (PZA) for the first two months, followed by a prolonged treatment with INH and RMP for additional 4–7 months with no guarantee of complete sterilization from the infection. 4 and 5 Furthermore, emergence of new virulent forms of TB such as multi drug resistant (MDR-TB) and extremely drug resistant (XDR-TB), and its synergy with human immunodeficiency virus (HIV) has fuelled its epidemic nature. These reasons make a compelling case for an urgent need for new and effective antitubercular agents with improved properties such as enhanced activity against MDR strains, reduced toxicity, rapid mycobactericidal mechanism of action and the ability to penetrate host cells and exert antimycobacterial effects in the intracellular environment.
Phenothiazines are important classes of compounds which have increasingly attracted attention, owing to their remarkable biological and pharmacological properties, such as antibacterial, antifungal, anticancer, antiviral, anti-inflammatory, antimalarial, antifilarial, trypanocidal, anticonvulsant, analgesic, immunosuppressive and multidrug resistance reversal. These activities are the results of the actions exerted by phenothiazines on biological systems via the interaction of the pharmacophoric substituent (in some cases of strict length), via the interaction multicyclic ring system (π–π interaction, intercalation in DNA) and via the lipophilic character permitting the penetration through the biological membranes to reach its site of action. Further, Phenothiazines have been shown to exhibit in vitro and in vivo activity against Mtb and multidrug-resistant Mtb. Some of the phenothiazine derived antipsychotic agents such as chlorpromazine, trifluoperazine (TPZ) and thioridazine are found to be effective inhibitors of Mtb.Phenothiazines are predicted to target the genetically validated respiratory chain component type II NADH:quinone oxidoreductase (Ndh)
aAmneal Pharmaceuticals India Pvt Ltd, 882/1-871, Village Rajoda, Tal.: Bavla Dist.: Ahmedabad 382220, Gujarat, India
bDivision of Medicinal Chemistry, Department of Chemistry (DST-FIST Sponsored), Mahatma Gandhi Campus, Maharaja Krishnakumarsinhji Bhavnagar University, Bhavnagar 364002, Gujarat, India
cDepartment of Chemistry, Saurashtra University, Kalawad Road, Rajkot 360005, Gujarat, India
A series of novel dihydropyrazolo[3,4-d]pyrimidine derivatives bearing a phenothiazine nucleus were synthesized in excellent yields via a modified Biginelli multicomponent reaction. The newly synthesized compounds were characterized by IR, 1H NMR, 13C NMR, Mass spectra and elemental analysis followed by antimycobacterial screening. Among all the screened compounds, compound 4g showed most pronounced activity against Mycobacterium tuberculosis (Mtb) with minimum inhibitory concentration (MIC) of 0.02 μg/mL, making it more potent than first line antitubercular drug isoniazid.
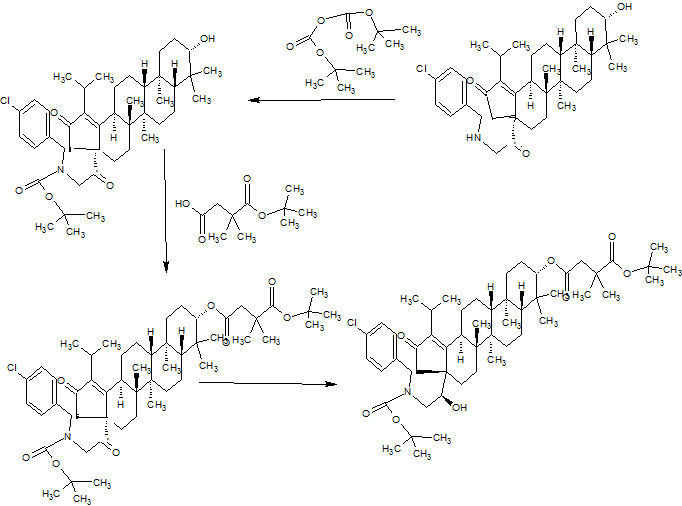
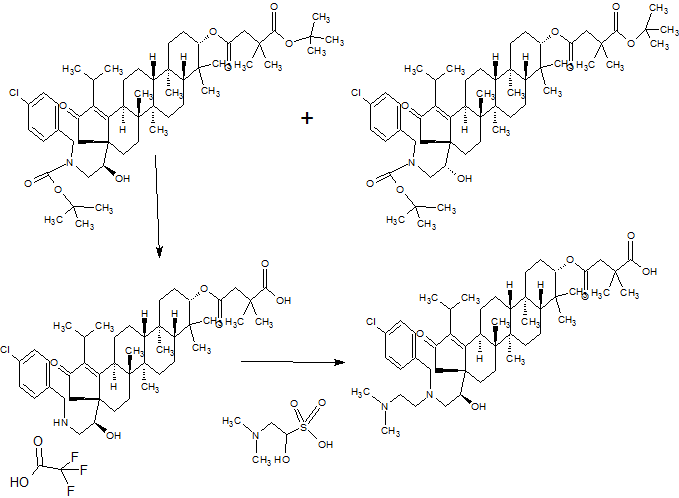
Scheme 1.
Synthetic protocol of title compounds 4a–k. Reagents and conditions: (a) NH2NH2, reflux; (b) ethyl acetoacetate, sodium ethoxide, reflux; (c) guanidine hydrochloride, aldehyde (R-CHO), P2O5, EtOH, reflux.
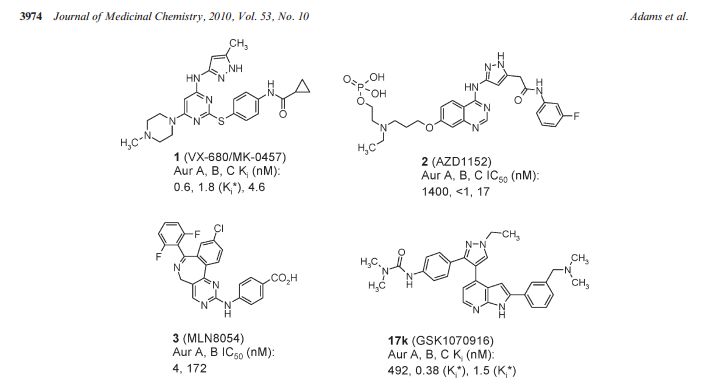
AMG 900 is a small-molecule inhibitor of Aurora kinases A, B and C with potential antineoplastic activity. Aurora kinase inhibitor AMG 900 selectively binds to and inhibits the activities of Aurora kinases A, B and C, which may result in inhibition of cellular division and proliferation in tumor cells that overexpress these kinases. Aurora kinases are serine-threonine kinases that play essential roles in mitotic checkpoint control during mitosis and are overexpressed by a wide variety of cancer cell types. Check for active clinical trials or closed clinical trials using this agent
AMG 900 is a potent and highly selective pan-Aurora kinases inhibitor for Aurora A/B/C with IC50 of 5 nM/4 nM /1 nM; >10-fold selective for Aurora kinases than p38α, Tyk2, JNK2, Met and Tie2. IC50 Value: 5 nM(Aurora A); 4 nM(Aurora B); 1 nM(Aurora C) Target: pan-Aurora in vitro: AMG 900 is a novel class of ATP-competitive phthalazinamine small molecule inhibitors of aurora kinases. In HeLa cells, AMG 900 inhibits autophosphorylation of aurora-A and -B as well as phosphorylation of histone H3 on Ser, a proximal substrate of aurora-B. The predominant cellular response of tumor cells to AMG 900 treatment is aborted cell division without a prolonged mitotic arrest, which ultimately results in cell death. AMG 900 inhibits the proliferation of 26 tumor cell lines, including cell lines resistant to the antimitotic drug paclitaxel and to other aurora kinase inhibitors (AZD1152, MK-0457, and PHA-739358), at low nanomolar concentrations (about 2- 3 nM). Furthermore, AMG 900 is active in an AZD1152-resistant HCT116 variant cell line that harbors an aurora-B mutation (W221L) [1]. in vivo: Oral administration of AMG 900 blocks the phosphorylation of histone H3 in a dose-dependent manner and significantly inhibited the growth of HCT116 tumor xenografts. AMG 900 is broadly active in multiple xenograft models, including 3 multidrugresistant xenograft models, representing 5 tumor types [1]. AMG 900 exhibits a low-to-moderate clearance and a small volume of distribution. Its terminal elimination half-life ranged from 0.6 to 2.4 hours. AMG 900 is well-absorbed in fasted animals with an oral bioavailability of 31% to 107%. Food intake has an effect on rate (rats) or extent (dogs) of AMG 900 oral absorption. The clearance and volume of distribution at steady state in humans are predicted to be 27.3 mL/h/kg and 93.9 mL/kg, respectively. AMG 900 exhibits acceptable PK properties in preclinical species and is predicted to have low clearance in humans [2].
In mammalian cells, the aurora kinases (aurora-A, -B, and -C) play essential roles in regulating cell division. The expression of aurora-A and -B is elevated in a variety of human cancers and is associated with high proliferation rates and poor prognosis, making them attractive targets for anticancer therapy. AMG 900 is an orally bioavailable, potent, and highly selective pan-aurora kinase inhibitor that is active in taxane-resistant tumor cell lines. In tumor cells, AMG 900 inhibited autophosphorylation of aurora-A and -B as well as phosphorylation of histone H3 on Ser(10), a proximal substrate of aurora-B. The predominant cellular response of tumor cells to AMG 900 treatment was aborted cell division without a prolonged mitotic arrest, which ultimately resulted in cell death. AMG 900 inhibited the proliferation of 26 tumor cell lines, including cell lines resistant to the antimitotic drug paclitaxel and to other aurora kinase inhibitors (AZD1152, MK-0457, and PHA-739358), at low nanomolar concentrations. Furthermore, AMG 900 was active in an AZD1152-resistant HCT116 variant cell line that harbors an aurora-B mutation (W221L). Oral administration of AMG 900 blocked the phosphorylation of histone H3 in a dose-dependent manner and significantly inhibited the growth of HCT116 tumor xenografts. Importantly, AMG 900 was broadly active in multiple xenograft models, including 3 multidrug-resistant xenograft models, representing 5 tumor types. AMG 900 has entered clinical evaluation in adult patients with advanced cancers and has the potential to treat tumors refractory to anticancer drugs such as the taxanes.
MG 900 is an orally bioavailable, potent, and highly selective pan-aurora kinase inhibitor that is active in taxane-resistant tumor cell lines. In tumor cells, AMG 900 inhibited autophosphorylation of aurora-A and -B as well as phosphorylation of histone H3 on Ser10, a proximal substrate of aurora-B. The predominant cellular response of tumor cells to AMG 900 treatment was aborted cell division without a prolonged mitotic arrest, which ultimately resulted in cell death. AMG 900 inhibited the proliferation of 26 tumor cell lines, including cell lines resistant to the antimitotic drug paclitaxel and to other aurora kinase inhibitors (AZD1152, MK-0457, and PHA-739358), at low nanomolar concentrations. Furthermore, AMG 900 was active in an AZD1152-resistant HCT116 variant cell line that harbors an aurora-B mutation (W221L). Oral administration of AMG 900 blocked the phosphorylation of histone H3 in a dose-dependent manner and significantly inhibited the growth of HCT116 tumor xenografts. Importantly, AMG 900 was broadly active in multiple xenograft models, including 3 multidrug-resistant xenograft models, representing 5 tumor types. AMG 900 has entered clinical evaluation in adult patients with advanced cancers and has the potential to treat tumors refractory to anticancer drugs such as the taxanes. (Source: Cancer Res; 70(23); 9846–54.)
The compound, N-(4-((3-(2-amino-4-pyrimidinyl)-2-pyridinyl)oxy)phenyl)- 4-(4-methyl-2-thienyl)-l-phthalazinamine, also chemically named as 4-((3-(2-amino- pyrimidin-4-yl)-pyridin-2-yl)oxy)phenyl-(4-(4-methyl-thiophen-2-yl)-phthalazin-l- yl)amine, and is referred to herein as “AMG 900” has a chemical structure of
AMG 900 is an ATP competitive small molecule Aurora kinase inhibitor that is highly potent and selective for Aurora kinases A, B and C. AMG 900 is disclosed in US patent publication no. 20070185111, which published on August 9, 2007 and issued as U.S. Patent No. 7,560,551. AMG 900 is further disclosed in US patent publication no.
20090163501, now US patent no 8,022,221. Various uses and applications of AMG 900 are described in patent publications US20120028917 and WO2013149026. AMG 900 is being clinically evaluated primarily for its safety, tolerability and pharmacokinetic (PK) profile in human phase I trials for (1) advanced solid tumors (US Clinical Trial Id No. NCT00858377), and (2) for acute leukemias (US Clinical Trial Id No. NCT1380756).
Different solid state forms of a given compound are typically investigated to determine whether or not a particular form possesses and/or exhibits desirable properties allowing that compound to be clinically and/or commercially developed. Such beneficial and advantageous properties, by way of example, include without limitation, crystallinity, improved thermodynamic stability, non-hygroscopicity, high purity, minimal to total absence of moisture and/or residual solvents, chemical stability, high yielding synthetic process and/or manufacturability and reproducibility, desirable biopharmaceutical properties including improved dissolution characteristics and increased bioavailability, absence or reduced toxicities due to reduced or limited exposure, rate of exposure or release, or related to counter ions, good bulk and formulation properties including good flow, bulk density, desirable particle size and the like, or a combination of the aforementioned characteristic attributes.
Generally when a compound, also referred to herein as drug substance (DS), has been identified as a developmental candidate, the DS is screened to identify potentially beneficial polymorphic, crystalline or solid state forms of the compound and/or a pharmaceutically acceptable salt thereof. X-ray diffraction, Raman, solid state NMR and a melting point temperature and/or a melting point temperature range have been typically used to monitor or screen and identify the different polymorphic form of the DS.
Different polymorphic forms of a given DS can have an impact on that compound’s solubility, stability and bioavailability. Also, it is important to monitor possible changes in polymorphic forms of the DS during stability studies.
AMG 900 was previously isolated and identified as a free base compound. This compound exhibited rather lack-luster pharmacokinetic (PK) and/or pharmacodynamic (PD) properties, including poor aqueous solubility, poor bioavailability, poor absorption, poor target exposure and overall, a not-so-attractive in-vivo efficacy profile. Thus, there is a need to address and solve the technical problem of identifying alternative forms of AMG 900 to achieve substantially the same effect or an improved effect, including improved PK and PD profiles, as that of AMG 900 known in the art.
Example 1
Synthesis of N-(4-((3-(2-amino-4-pyrimidinylN)-2-pyridinylN)oxyN)phenylN)-4-(4-methyl-2-thienvD-l-phthalazinamine (AMG 900)
In an argon purged 500 mL round bottom flask placed in an isopropanol bath, was added sodium metal (3.40g, 148mmol) slowly to methanol (180mL). The mixture was stirred at room temperature (RT) for about 30 minutes. To this was added guanidine hydrochloride (12.0 mL, 182 mmol) and the mixture was stirred at RT for 30 minutes, followed by addition of (E)-l-(2-chloropyridin-3-yl)-3-(dimethylamino)prop-2-en-l-one (12.0 g, 57.0 mmol), attached air condenser, moved reaction to an oil bath, where it was heated to about 50 °C for 24 hr. Approximately half of the methanol was evaporated under reduced pressure and the solids were filtered under vacuum, then washed with saturated sodium bicarbonate (NaHCO and H^O, air dried to yield 4-(2-chloropyridin-3-yl)pyrimidin-2-amine as off white solid. MS m/z = 207 [M+l]+. Calc’d for C9H7C1N4: 206.63.
To a resealable tube was added 4-aminophenol (1.3 g, 12 mmol), cesium carbonate (7.8 g, 24 mmol), and DMSO (16 ml, 0.75 M). The mixture was heated to 100 °C for 5 minutes, and then 4-(2-chloropyridin-3-yl)pyrimidin-2 -amine (2.5 g, 12 mmol) was added, and the reaction mixture was heated to 130 °C overnight. Upon completion, as judged by LCMS, the reaction mixture was allowed to cool to RT and diluted with water. The resulting precipitate was filtered, and the solid washed with water and diethyl ether. The solid was then taken up in 9: 1 CH2Cl2:MeOH and passed through a pad of silica gel with 9:1 CH2Cl2:MeOH as eluent. The solvent was concentrated in vacuo to provide the desired product, 4-(2-(4-aminophenoxy)pyridin-3-yl)pyrimidin-2-amine. MS m/z = 280
1 ,4-Dichlorophthalazine (1.40 g, 7.03 mmol), 4-methyltmophen-2-ylboronic acid (999 mg, 7.03 mmol), and PdCl2(DPPF) (721 mg, 985 μιηοΐ) were added into a sealed tube. The tube was purged with Argon. Then sodium carbonate (2.0 M in water) (7.74 ml, 15.5 mmol) and 1,4-dioxane (35.2 ml, 7.03 mmol) were added. The tube was sealed, stirred at RT for 5 min, and placed in a preheated oil bath at 110 °C. After 1 hr, LC-MS showed product and byproduct (double coupling), and starting material
dichlorophthalazme. The reaction was cooled to RT, filtered through a pad of celite with an aid of ethyl acetate (EtOAc), concentrated, and loaded onto column. The product was purified by column chromatography using Hex to remove the top spot, then 80:20 hexanes:EtOAc to collect the product. The product, 1 -chloro-4-(4-methylthiophen-2-yl)phthalazine was obtained as yellow solid. LC-MS showed that the product was contaminated with a small amount of dichlorophthalazme and biscoupling byproduct. MS m/z = 261 [M+l]+. Calcd for Ci3H9ClN2S: 260.12.
To 4-(2-(4-aminophenoxy)pyridin-3-yl)pyrimidin-2 -amine and l-chloro-4-(4-methyl-2-thienyl)phthalazine was added tBuOH. The resulting mixture was heated at 100 °C in a sealed tube for 16 hours. The rection was diluted with diethyl ether and saturated sodium carbonate and vigorously shaken. The resulting solids were filtered and washed with water, diethyl ether and air dried to yield N-(4-((3-(2-amino-4-pyrimidinyl)-2-pyridinyl)oxy)phenyl)-4-(4-methyl-2-thienyl)-l -phthalazinamine as an off-white solid. MS m/z = 504 [M+H]+. Calc’d for C28 H21 N7 O S: 503.58.
Example 2
Alternative Synthesis of N-(4-((3-(2-amino-4-pyrimidinylN)-2-pyridinylN)oxyN)phenylN)-4-(4-methyl-2-thienvD-l-phthalazinamine (AMG 900)
In an argon purged 500 mL round bottom flask placed in an isopropanol bath, was added sodium metal (3.40g, 148mmol) slowly to methanol (180mL). The mixture was stirred at room temperature (RT) for about 30 minutes. To this was added guanidine hydrochloride (12.0 mL, 182 mmol) and the mixture was stirred at RT for 30 minutes, followed by addition of (E)-l-(2-chloropyridin-3-yl)-3-(dimethylamino)prop-2-en-l-one (12.0 g, 57.0 mmol), attached air condenser, moved reaction to an oil bath, where it was heated to about 50 °C for 24 hr. Approximately half of the methanol was evaporated under reduced pressure and the solids were filtered under vacuum, then washed with saturated sodium bicarbonate (NaHCO and H^O, air dried to yield 4-(2-chloropyridin-3-yl)pyrimidin-2-amine as off white solid. MS m/z = 207 [M+l]+. Calc’d for C9H7C1N4: 206.63.
To a reaction vessel at ambient temperature was added 4-aminophenol (571 g, 5.25 mol, 1.05 equiv) followed by 4-(2-chloropyridin-3-yl)pyrimidin-2-amine (1064g, 97 wt%, 5.00 mol, 1.0 equiv) and DMSO (7110 ml, 7820 g, 7x the volume of 4-(2-chloropyridin-3-yl)pyrimidin-2 -amine). The reaction mixture was agitated and sparged with nitrogen gas for at least 10 minutes. Then a 50 weight % aqueous KOH solution (593 g, 5.25 mol, 1.05 equiv.) was added to the mixture while keeping the reaction
mixture temperature below about 40°C. The mixture was sparged with nitrogen gas for more than 5 minutes, the sparging tube was removed, and the reaction mixture was heated to 110 +/- 10 °C for at least 1.5 hrs. Upon completion, as judged by HPLC, the reaction mixture was allowed to cool to RT and diluted with 6N HC1 (42 mL, 0.25 mol, 0.05 equiv). The desired product, 4-(2-(4-aminophenoxy)pyridin-3-yl)pyrimidin-2 -amine was not isolated. Rather, it was formed in-situ and combined with the product of step 3 below, in step 4 to form the desired product.
A separate reaction vessel was fitted with a reflux condenser and an addition funnel, and 4-(4-methylthiophen-2-yl)phthalazin-l(2H)-one (1,537 mg, 6.34 mol, 1.0 equivalent) was added to the reaction vessel. Acetonitrile (7540 mL, 5859 g, 5 V), was added and the reaction vessel was agitated to allow the starting material to dissolve. The vessel was then charged with phosphorus oxychloride (709 ml, 1166 g, 7.44 mol, 1.2 equivalents) and the reaction was heated to about 75 +/- 5 °C for a least 4 hrs. The reaction was cooled to about about 25 +/- 5 °C and held there for more than 24 hrs. N,N-diisopropylethylamine (3046 g, 4100 mL, 3.8 equivalents) was added to the reaction vessel and the temperature was maintained at <30°C. Pyridine (97g, 1.24 mol, 0.2 equiv) was added in a single portion followed by water (4100 g, 2.7V) over more than 30 minutes. The reaction mixture was stirred at ambient temperature ofr about 24 hrs. the mixture was filtered through a <25uM polypropylene filter and the rsulting mother liquor was diluted with 1 : 1 ACN:water (9000 mL total) and stirred for a minimum of 2 minutes. Filter off product solids as they precipitate. Collect mother liquor and washes to obtain additional product. Dry the filter cake, and additional product crops, under a constant stream of nitrogen gas for at least 14 hrs. Unlike the previous method, the present method avoids contamination of impurities, such as dichlorophthalazine and biscoupling byproduct, as seen via LC-MS. Yield: 1537 g (97.2 weight %). MS m/z = 261 [M+l]+. Calcd for Ci3H9ClN2S: 260.12.
To the reaction mixture was added l-chloro-4(4-methylthiophen-2-yl)phthalazine
(1450g, 97.2 wt%, 5.40 mol, 1.08 equiv) rinding the addition port with DMSO (520 ml, 572 g, 0.5x the volume of 4-(2-chloropyridin-3-yl)pyrimidin-2-amine). The reaction mixture was again agitated and sparged with nitrogen gas for at least 10 minutes. The sparging tube was removed, and the reaction mixture was heated to 80 +/- 20 °C for at least 2 hrs. Upon completion, as judged by HPLC, the reaction mixture was allowed to cool to RT and N,N-diisopropylethylamine (776 g, 1045 mL, 6.0 mol, 1.2 equiv) was added and the mixture was kept at about 80 +/- 10°C. Filter the mixture at about 80oC into a separate reactor vessel rinsing with DMSO (1030 mL, 1133 g, 1 V). Then adjust the raction mixture temperature to about 70+/-5 °C and add 2-propanol (13200 mL, 10360 g, 12.75 V) over more than 15 minutes at about 70°C. As the reaction mistreu cools, the product begins to precipitate out of solution. Add more 2-propanol (8780 mL, 6900 g, 8.5V) to the solution slowly over more then 60 minutes at about 70°C. The reactor vessel was cooled to about 20°C over more than 60 minutes and let sit for over 2 hrs. The product was filtered through an Aurora filter with a >25uM polypropylene filter cloth. Additional crops were obtained from the mother liquors by diluting with additional 2-propanol. The filter cakes were dried under a constant stream of nitrogen gas for at least 14 hrs to provide the desired product, N-(4-((3-(2-amino-4-pyrimidinyl)-2-pyridinyl)oxy)phenyl)-4-(4-methyl-2-thienyl)-l-phthalazinamine as an off-white solid. Yield: 2831 g (88.8%); purity 99.7%. MS m/z = 504 [M+H]+. Calc’d for C28 H21 N7 O S: 503.58.
The starting material 1 used/shown in Example 2 was prepared as follows:
and starting material 3, thienyl substituted phthalazinone, shown in Example 2 was prepared as follows:
Starting material 3
Synthesis of 4-(5-methylthiophen-2-yl)phthalazin-l(2//)-one
Step 1 : 2-(Dimethylamino)isoindoline-1.3-dione
A solution of isobenzofuran-l,3-dione (5.00 g, 34 mmol) and N,N-dimethylhydrazine (2.9 ml, 37 mmol) in toluene (75 ml, 34 mmol) was added p-TsOH H20 (0.32 g, 1.7 mmol). The Dean-Stark apparatus and a condenser were attached. The mixture was refluxed. After 4 hr, LCMS showed mainly product. The reaction was cooled to rt. Toluene was removed under reduced pressure the crude was dissolved in DCM, washed with sat NaHC03, water, and brine. The organic was dried over MgS04, filtered, and concentrated. Light yellow solid was obtained. !H NMR showed mainly product, 2-(dimethylamino)isoindoline-l,3-dione. MS Calcd for C10H10N2O2: [M]+ = 190. Found: [M+H]+ = 191.
A solution of 2-bromo-5-methylthiophene (0.60 mL, 5.3 mmol) in THF (11 mL) was purged with nitrogen and cooled to -78 °C. «-Butyllithium (2.2 mL, 5.5 mmol; 2.5 M in THF) was added and the mixture was stirred under nitrogen for 30 min. This solution was cannulated into a flask containing a solution of 2-(dimethylamino)isoindoline-l,3-dione (1.5 g, 7.9 mmol) in THF (16 mL) at -78 °C under nitrogen. The reaction was allowed to warm to -30 °C over an hour, at which point LCMS showed complete conversion of 2-bromo-5-methylthiophene to product. The reaction was quenched by careful addition of saturated aqueous NH4C1. The reaction mixture was diluted with dichloromethane and water, and the layers were separated. The aqueous portion was extracted with additional dichloromethane, and the combined organic layers were dried with MgS04, filtered, concentrated, and purified by silica gel chromatography eluting with 0-2% MeOH in dichloromethane to provide intermediate A, as a light yellow solid, 2-(dimethylamino)-3-hydroxy-3-(5-methylthiophen-2-yl)isoindolin-l-one (1.2 g, 80% yield). !H NMR (400 MHz, DMSO-4) δ 7.68-7.65 (m, 1H). 7.63-7.59 (m, 1H), 7.57-7.51 (m, 1H), 7.37 (d, 1H, J=8), 7.09 (s, 1H), 6.69-6.66 (m, 1H), 6.65-6.62 (m, 1H), 2.81 (s, 6H), 2.40 (s, 3H). 13C NMR (400 MHz, DMSO-de) δ 165.0, 147.3, 141.6, 139.3, 132.7, 129.49, 129.46, 125.0, 124.7, 123.0, 122.1, 88.4, 44.7, 14.9. FT-IR (thin film, cm ) 3347, 3215, 1673. MS Calcd for Ci2H7ClN2S: [M]+ = 288. Found: [M+H]+= 289.
HRMS Calcd for Ci5H16N202S: [M+H]+= 288.1005, [M+Na]+ = 311.0825. Found:
[M+H]+ = 289.1022, [M+Na]+= 311.0838. mp = 138-140 °C.
2-(Dimethylamino)-3 -hydroxy-3 -(5 -methylthiophen-2-yl)isoindolin- 1 -one (1.1 g, 0.40 mmol), EtOH (4.0 mL), and hydrazine (0.19 mL, 59 mmol) were added into a RBF fitted with a reflux condenser. A nitrogen balloon was attached on top of the condenser. After refluxing overnight, the reaction was cooled to room temperature. An off-white solid precipitated. After cooling to 0 °C, water was added. The solid was filtered off with an aid of water and dried under vacuum to afford a white solid, 4-(5-methylthiophen-2-yl)phthalazin-l(2//)-one (0.82 g, 85% yield).
Alternatively, starting material 3 was prepared as follows:
The above scheme depicts the process by which intermediate-scale synthesis of thiophene-phthalazinone 5 (shown above) was prepared. Treatment of 50 grams of 3-methylthiophene with z‘-PrMgCl at 66 °C in the presence of catalytic TMP-H resulted in 98% conversion to the reactive species lb with a >40:1 regioisomeric ratio. After cooling to 20 °C, this mixture was added dropwise to a -20 °C slurry of phthalic anhydride in THF to provide keto acid 3 in 94% assay yield. While this intermediate could be crystallized from toluene/heptane, the crude reaction mixture was taken directly in a through -process conversion to the phthalazinone 5. To that end, removal of THF, MTBE, and residual 3-methylthiophene was accomplished through a distillative solvent switch into ethanol. The resulting solution of 3 was exposed to aqueous hydrazine at 80 °C. After 18 hours, the reaction was cooled and the precipitated product was filtered directly at 20 °C. This process provided 82.7 grams of 98.6 wt % thiophene-phthalazinone 5 in a weight-adjusted 85% yield over the two steps.
LCMS Method:
Samples were run on a Agilent model- 1100 LC-MSD system with an Agilent Technologies XDB-C8 (3.5 μ) reverse phase column (4.6 x 75 mm) at 30 °C. The flow rate was constant and ranged from about 0.75 mL/min to about 1.0 mL/min.
The mobile phase used a mixture of solvent A (H2O/0.1% HO Ac) and solvent B
(AcCN/O.1 HOAc) with a 9 min time period for a gradient from 10%> to 90%> solvent B. The gradient was followed by a 0.5 min period to return to 10% solvent B and a 2.5 min 10% solvent B re-equilibration (flush) of the column.
Other methods may also be used to synthesize AMG 900. Many synthetic chemistry transformations, as well as protecting group methodologies, useful in synthesizing AMG 900, are known in the art. Useful organic chemical transformation literature includes, for example, R. Larock, Comprehensive Organic Transformations, VCH Publishers (1989); T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 3rd edition, John Wiley and Sons (1999); L. Fieser and M. Fieser, Fieser and Fieser’s Reagents for Organic Synthesis, John Wiley and Sons (1994); A. Katritzky and
A. Pozharski, Handbook of Heterocyclic Chemistry, 2nd edition (2001); M. Bodanszky, A. Bodanszky, The Practice of Peptide Synthesis, Springer- Verlag, Berlin Heidelberg (1984); J. Seyden-Penne, Reductions by the Alumino- and Borohydrides in Organic Synthesis, 2nd edition, Wiley- VCH, (1997); and L. Paquette, editor, Encyclopedia of Reagents for Organic Synthesis, John Wiley and Sons (1995).
AMG 900 was tested for its ability to reduce or inhibit tumor progression in various cell lines (in-vitro) and multiple solid tumor types (in-vivo), some of which have previously been exposed to and developed resistance to standard-of-care antimitotic agents, including taxanes and vinca alkaloids, as well as to other chemotherapeutic agents. The following Examples and resulting data will illustrate the ability of AMG 900 to treat cancer, including cancer resistant to the presently standard-of-care therapies, including antimitotic agents, such as paclitaxel, and other drugs used in conjunction with chemotherapy, such as doxorubicin. Unless otherwise indicated, the free base form of AMG 900 was used in the Examples described hereinbelow.
The following Examples describe the efforts of identifying and characterizing various crystalline solid state forms of various salts of AMG 900. Some attempts at forming a solid state crystalline form of a given salt failed, as shown in table 1 hereinbelow. To this end, synthesizing and/or forming &isolating a crystalline solid state form of AMG 900 was not, in any way, straightforward or routine. Rather, the ability to prepare and identify a crystalline solid state form of AMG 900 depended upon the particular salt of AMG 900 and/or the crystallization conditions employed.
PAPER
Journal of Medicinal Chemistry (2015), 58(13), 5189-5207
Discovery of N-(4-(3-(2-Aminopyrimidin-4-yl)pyridin-2-yloxy)phenyl)-4-(4-methylthiophen-2-yl)phthalazin-1-amine (AMG 900), A Highly Selective, Orally Bioavailable Inhibitor of Aurora Kinases with Activity against Multidrug-Resistant Cancer Cell Lines
†Departments of Medicinal Chemistry, ‡Pharmaceutical Research and Development, §Pharmacokinetics and Drug Metabolism, ∥Molecular Structure, and ⊥Oncology Research, Amgen Inc., 360 Binney Street, Cambridge, Massachusetts 02142, United States, and Amgen Inc., One Amgen Center Drive, Thousand Oaks, California 91320, United States
ACS Editors’ Choice – This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes.
Abstract
Efforts to improve upon the physical properties and metabolic stability of Aurora kinase inhibitor14a revealed that potency against multidrug-resistant cell lines was compromised by increased polarity. Despite its high in vitro metabolic intrinsic clearance, 23r (AMG 900) showed acceptable pharmacokinetic properties and robust pharmacodynamic activity. Projecting from in vitro data to in vivo target coverage was not practical due to disjunctions between enzyme and cell data, complex and apparently contradictory indicators of binding kinetics, and unmeasurable free fraction in plasma. In contrast, it was straightforward to relate pharmacokinetics to pharmacodynamics and efficacy by following the time above a threshold concentration. On the basis of its oral route of administration, a selectivity profile that favors Aurora-driven pharmacology and its activity against multidrug-resistant cell lines, 23r was identified as a potential best-in-class Aurora kinase inhibitor. In phase 1 dose expansion studies with G-CSF support, 23r has shown promising single agent activity.
Applying similar SNAr conditions as for 23b, reaction of 22r and 20a in 2-butanol provided the title compound (2.08 g, 49%) as an off-white solid; mp (DSC) 216 °C.
USE OF DIANHYDROGALACTITOL AND ANALOGS OR DERIVATIVES THEREOF IN COMBINATION WITH PLATINUM-CONTAINING ANTINEOPLASTIC AGENTS TO TREAT NON-SMALL-CELL CARCINOMA OF THE LUNG AND BRAIN METASTASES
USE OF N-(4-((3-(2-AMINO-4-PYRIMIDINYL)-2-PYRIDINYL)OXY)PHENYL)-4-(4-METHYL-2-THIENYL)-1-PHTHALAZINAMINE IN COMBINATION WITH HISTONE DEACETYLASE INHIBITORS FOR TREATMENT OF CANCER
Use Of N-(4-((3-(2-Amino-4-Pyrimidinyl)-2-Pyridinyl)Oxy)Phenyl)-4-(4-Methyl-2-Thienyl)-1-Phthalazinamine In The Treatment Of Antimitotic Agent Resistant Cancer
N-4 ( – ( ( 3- ( 2 -amino-4 pyrimidinyl) -2 -pyridinyl) oxy) phenyl) -4- (4-methyl-2-thienyl) -1-phthalazinamine for use in the treatment of antimitotic agent resistant cancer
Crystalline forms of n-(4-((3-(2-amino-4-pyrimidinyl) – 2-pyridinyl)oxy)phenyl)-4-(4-methyl-2-thienyl)-1 -phthalazinamine pharmaceutically acceptable salts and uses thereof
N-(4-((3-(2-amino-4-pyrimidinyl)-2-pyridin yl)oxy)phenyl)-4-(4-methyl-2-thienyl)-1-phthalazinamine for use in the treatment of antimitotic agent resistant cancer
Benzene sulfonamide thiazole and oxazole compounds
///////////945595-80-2, AMG 900, aurora kinase (ARK) inhibitor, treatment of leukemia and solid tumours, AMGEN, 2014, orphan drug designation, U.S. for the treatment of ovarian cancer
MS m/z 508.4 [M + H]+. Anal. (C30H33N7O·1.0H2O) C, H, N.
GSK1070916 is a reversible and ATP-competitive inhibitor of Aurora B/C with IC50 of 3.5 nM/6.5 nM; displays >100-fold selectivity against the closely related Aurora A-TPX2 complex(IC50=490 nM).
NMI-900, an Aurora B/C kinase inhibitor, is under development at Cancer Research Technology in phase I/II clinical studies for the treatment of advanced and/or metastatic solid tumors. Other phase I clinical trials for the treatment of solid tumors had been previously completed, in a collaboration between GlaxoSmithKline and Cancer Research Technology, under the Cancer Research UK’s Clinical Development Partnerships (CDP) program.
The drug was originated by GlaxoSmithKline. The rights of the product were acquired by Cancer Research Technology from GlaxoSmithKline after the company elected not to take the program forward. In December 2015, the product was licensed by Cancer Research Technology to Nemucore Medical Innovations for the exclusive worldwide development and commercialization for the treatment of difficult-to-treat cancers.
GlaxoSmithKline, 1250 South Collegeville Road, Collegeville, Pennsylvania 19426
§ Tsukuba Research Laboratories, Japan
J. Med. Chem., 2010, 53 (10), pp 3973–4001
DOI: 10.1021/jm901870q
The Aurora kinases play critical roles in the regulation of mitosis and are frequently overexpressed or amplified in human tumors. Selective inhibitors may provide a new therapy for the treatment of tumors with Aurora kinase amplification. Herein we describe our lead optimization efforts within a 7-azaindole-based series culminating in the identification of GSK1070916 (17k). Key to the advancement of the series was the introduction of a 2-aryl group containing a basic amine onto the azaindole leading to significantly improved cellular activity. Compound 17k is a potent and selective ATP-competitive inhibitor of Aurora B and C with Ki* values of 0.38 ± 0.29 and 1.5 ± 0.4 nM, respectively, and is >250-fold selective over Aurora A. Biochemical characterization revealed that compound 17k has an extremely slow dissociation half-life from Aurora B (>480 min), distinguishing it from clinical compounds 1 and 2. In vitro treatment of A549 human lung cancer cells with compound 17k results in a potent antiproliferative effect (EC50 = 7 nM). Intraperitoneal administration of 17k in mice bearing human tumor xenografts leads to inhibition of histone H3 phosphorylation at serine 10 in human colon cancer (Colo205) and tumor regression in human leukemia (HL-60). Compound 17k is being progressed to human clinical trials.
GSK1070916 is a reversible and ATP-competitive inhibitor of Aurora B/C with IC50 of 3.5 nM/6.5 nM; displays >100-fold selectivity against the closely related Aurora A-TPX2 complex(IC50=490 nM). IC50 Value: 3.5 nM(Aurora B); 6.5 nM(Aurora C) Target: Aurora B/C in vitro: GSK1070916 selectively inhibits Aurora B and Aurora C with Ki of 0.38 nM and 1.5 nM over Aurora A with Ki of 490 nM. Inhibition of Aurora B and Aurora C is time-dependent, with an enzyme-inhibitor dissociation half-life of >480 min and 270 min respectively. In addition, GSK1070916 is also a competitive inhibitor with respect to ATP. Human tumor cells treated with GSK1070916 shows dose-dependent inhibition of phosphorylation on serine 10 of Histone H3, a substrate specific for Aurora B. Moreover, GSK1070916 inhibits the proliferation of tumor cells with EC50 values of <10 nM in over 100 cell lines spanning a broad range of tumor types, with a median EC50 of 8 nM. Although GSK1070916 has potent activity against proliferating cells, a dramatic shift in potency is observed in primary, nondividing, normal human vein endothelial cells. Furthermore, GSK1070916-treated cells do not arrest in mitosis but instead fails to divide and become polyploid, ultimately leading to apoptosis. In another study, it is also reported high chromosome number associated with resistance to the inhibition of Aurora B and C suggests cells with a mechanism to bypass the high ploidy checkpoint are resistant to GSK1070916. in vivo: GSK1070916 (25, 50, or 100 mg/kg) shows dose-dependent inhibition of phosphorylation of an Aurora B–specific substrate in mice and consistent with its broad cellular activity, has antitumor effects in 10 human tumor xenograft models including breast, colon, lung, and two leukemia models.
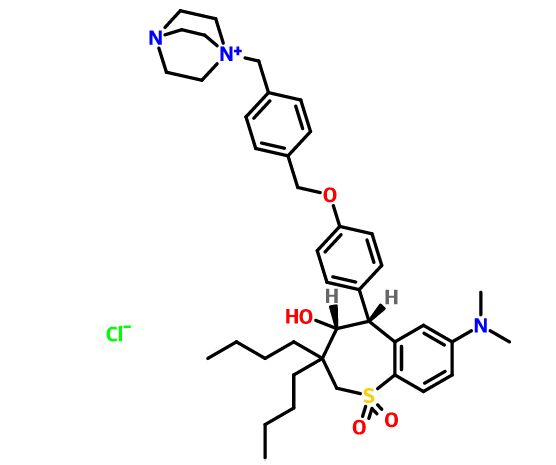
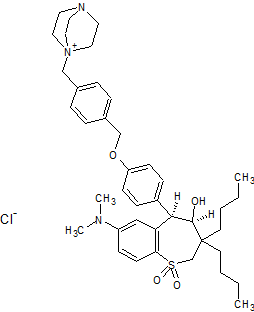
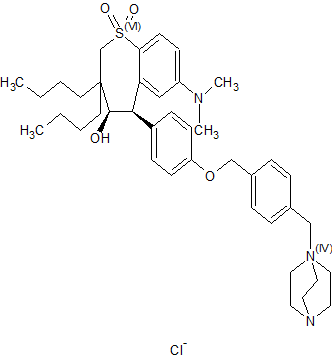
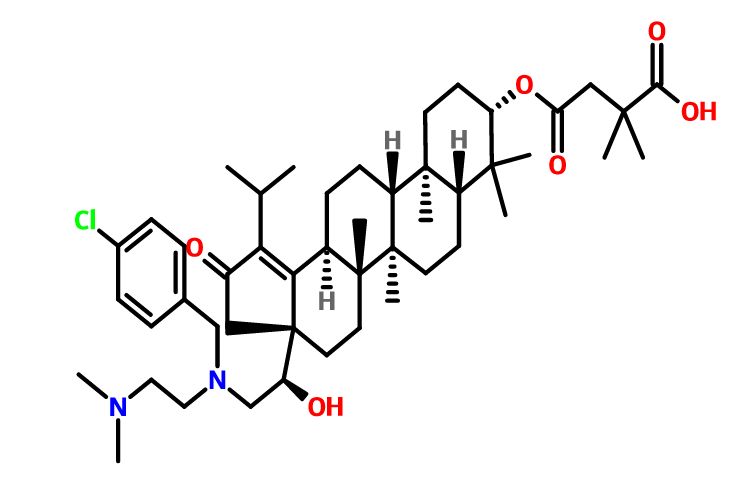
(4R–cis)-1-[[4-[[4-[3,3-Dibutyl-7-(dimethylamino)-2,3,4,5-tetrahydro-4-hydroxy-1,1-dioxido-1-benzothiepin-5-yl]phenoxy]methyl]phenyl]methyl]-4-aza-1-azoniabicyclo[2.2.2]octane Chloride Salt
It is well established that agents which inhibit the 20 transport of bile acids across the ileum can also cause a decrease in the level of cholesterol in blood serum. Stedronski, in “Interaction of bile acids and cholesterol with nonsystemic agents having hypocholesterolemic properties,” Biochimica et Biophysica Acta, 1210 (1994) 255- 25287, discusses biochemistry, physiology, and known active agents affecting bile acids and cholesterol.
A class of ileal bile acid transport-inhibiting compounds which was recently discovered to be useful for influencing the level of blood serum cholesterol is 30 tetrahydrobenzothiepine-l,l-dioxides (THBDO compounds). (U.S. Patent Application No. 08/816,065)
Some classes of compounds show enhanced potency as pharmaceutical therapeutics after they have been enantiomerically-enriched (see, for example, Richard B. Silverman, The Organic Chemistry of Drug Design and Drug Action, Academic Press, 1992, pp. 76-82) . Therefore, THBDO compounds that have been enantiomerically-enriched are of particular interest.
A class of chemistry useful as intermediates in the preparation of racemic THBDO compounds is tetrahydrobenzothiepine-1-oxides (THBO compounds) . THBDO compounds and THBO compounds possess chemical structures in which a phenyl ring is fused to a seven-member ring. A method of preparing enantiomerically-enriched samples of another phenyl/seven-member fused ring system, the benzothiazepines, is described by Higashikawa (JP 59144777) , where racemic benzothiazepine derivatives are optically resolved on a chromatographic column containing chiral crown ethers as a stationary phase. Although optical resolution is achieved, the Higashikawa method is limited to producing only small quantities of the enantiomerically-enriched benzothiazepine derivatives. Giordano (CA 2068231) reports the cyclization of (2S, 3S) -aminophenylthiopropionates in the presence of a phosphonic acid to produce (2S, 3S) -benzothiazepin-4-ones . However, that preparation is constrained by the need to use enantiomerically-enriched starting materials rather than racemic starting materials. In addition, the Giordano method controls the stereochemistry of the seven-member ring of the benzothiazepin-4-one only at the 2- and 3 -positions. The 4- and 5-positions of the seven-member ring of the benzothiazepin-4-one are not asymmetric centers, and the stereochemistry at these sites therefore cannot be controlled by the Giordano method. A method by which enantiomerically-enriched 1,5- benzothiazepin-3-hydroxy-4 (5H) -one compounds have been produced is through the asymmetric reduction of 1,5- benzothiazepin-3,4 (2H, 5H) -dione compounds, reported by Yamada, et al . (J“. Org. Chem. 1996, 61 (24), 8586-8590). The product is obtained by treating the racemic 1,5- benzothiazepin-3,4 (2H, 5H) -dione with the reaction product of an optically active alpha-amino acid and a reducing agent, for example sodium borohydride. Although a product with high optical purity was achieved, the method is limited by the use of a relatively expensive chemical reduction step.
The microbial reduction of racemic 1, 5-benzothiazepin- 3 , 4 (2H, 5H) -dione compounds to produce enantiomerically- enriched 1, 5-benzothiazepin-3-hydroxy-4 (5H) -one compounds is reported by Patel et al . , U.S. Patent 5,559,017. This method is limited by the inherent problems of maintaining a viable and pure bacterial culture of the appropriate species and variety. In addition, that method is limited in scale, producing only microgram quantities of the desired product. Until now, there have been no reported processes for preparing enantiomerically-enriched THBDO compounds or enantiomerically-enriched THBO compounds. Furthermore, there have been no reported processes for controlling the stereochemistry at the 4- and 5-positions of the seven- member rings of THBDO compounds or THBO compounds
FDA Grants Breakthrough Designation to Shire’s Rare GI Therapies
Tue, 06/14/2016
Shire announced that the U.S. Food and Drug Administration (FDA) has granted Breakthrough Therapy Designation for two investigational products for rare diseases: SHP621 (budesonide oral suspension, or BOS) for eosinophilic esophagitis (EoE), and SHP625 (maralixibat) for progressive familial intrahepatic cholestasis type 2 (PFIC2).
“Receiving Breakthrough Therapy Designation on two pipeline products this past week reflects the potential of our strong and innovative pipeline of more than 60 programs,” said Flemming Ornskov, M.D., MPH, and CEO, Shire. “Shire is committed to bringing innovation to the rare and specialty areas we focus on. We persevere to see compounds through the many stages of development through their challenges and successes, and always keep patients with unmet needs top of mind.”
EoE is a serious, chronic and rare disease that stems from an elevated number of eosinophils, a type of white blood cell, that infiltrate the walls of the esophagus. EoE is characterized by an inflammation of the esophagus that may lead to difficulty swallowing (dysphagia). The diagnosed prevalence of EoE ranges from approximately 15-55 cases per 100,000 persons, with high-end estimates reported by studies in Western regions.
PFIC refers to a group of autosomal-recessive liver disorders of childhood that disrupt bile formation and present with cholestasis. The symptoms of PFIC include severe itching of the skin (pruritus), and jaundice. PFIC is estimated to affect 1 in 50,000 to 1 in 100,000 births. PFIC2 is the most common type of PFIC, accounting for around half of cases.
According to the FDA, Breakthrough Therapy Designation is granted to a therapy that is intended to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement on one or more clinically significant endpoints over current standard of care. Under the designation, the FDA provides intensive guidance, organizational commitment involving senior managers, and eligibility for rolling and priority review of the application; this process helps ensure patients have access to therapies as soon as possible, pending approval. Breakthrough Therapy Designation does not guarantee that FDA will ultimately approve BOS for EoE or maralixibat for PFIC2, and the timing of any such approval is uncertain.
“On behalf of patients in the United States with EoE and PFIC2, we are so pleased that the FDA has granted Breakthrough Therapy Designation to BOS and maralixibat,” said Philip J. Vickers, Ph.D., Head of R&D, Shire. “We look forward to working with the agency to continue their development and, pending FDA approval, deliver these therapeutic options to the patients who need them most.”
It is well established that agents which inhibit the transport of bile acids across the tissue of the ileum can also cause a decrease in the levels of cholesterol in blood serum. Stedronski, in “Interaction of bile acids and cholesterol with nonsystemic agents having hypocholesterolemic properties,” Biochimica et Biophysica Acta, 1210 (1994) 255-287 discusses biochemistry, physiology, and known active agents surrounding bile acids and cholesterol. Bile acids are actively transported across the tissue of the ileum by an apical sodium co-dependent bile acid transporter (ASBT), alternatively known as an ileal bile acid transporter (IBAT). A class of ASBT-inhibiting compounds that was recently discovered to be useful for influencing the level of blood serum cholesterol comprises tetrahydrobenzothiepine oxides (THBO compounds, PCT Patent Application No. WO 96/08484). Further THBO compounds useful as ASBT inhibitors are described in PCT Patent Application No. WO 97/33882. Additional THBO compounds useful as ASBT inhibitors are described in U.S. Patent No. 5,994,391. Still further THBO compounds useful as ASBT inhibitors are described in PCT Patent Application No. WO 99/64409. Included in the THBO class are tetrahydrobenzo-thiepine-l -oxides and tetrahydrobenzothiepine- 1,1 -dioxides. THBO compounds possess chemical structures in which a phenyl ring is fused to a seven-member ring.
Published methods for the preparation of THBO compounds include the synthesis through an aromatic sulfone aldehyde intermediate. For example l-(2,2-dibutyl-3-oxopropylsulfonyl)-2-((4-methoxyphenyl)methyl)benzene (29) was cyclized with potassium t-butoxide to form tetrahydrobenzothiepine- 1,1 -dioxide (svn-24) as shown in Eq. 1.
Compound 29 was prepared by reacting 2-chloro-5-nitrobenzoic acid chloride with anisole in the presence of aluminum trichloride to produce a chlorobenzophenone compound; the chlorobenzophenone compound was reduced in the presence of trifluoromethanesulfonic acid and triethylsilane to produce a chlorodiphenylmethane compound; the chlorodiphenylmethane compound was treated with lithium sulfide and 2,2-dibutyl-3-(methanesulfonato)propanal to produce l-(2,2-dibutyl-3-oxopropylthio)-2-((4-methoxyphenyl)methyl)-4-dimethylaminobenzene (40); and 40 was oxidized with m-chloroperbenzoic acid to produce 29. The first step of that method of preparing compound 29 requires the use of a corrosive and reactive carboxylic acid chloride that was prepared by the reaction of the corresponding carboxylic acid with phosphorus pentachloride. Phosphorus pentachloride readily hydrolyzes to produce volatile and hazardous hydrogen chloride. The reaction of 2,2-dibutyl-3-(methanesulfonato)propanal with the lithium sulfide and the chlorodiphenylmethane compound required the intermediacy of a cyclic tin compound to make the of 2,2-dibutyl-3-(methanesulfonato)propanal. The tin compound is expensive and creates a toxic waste stream. In WO 97/33882 compound syn-24 was dealkylated using boron tribromide to produce the phenol compound 28. Boron tribromide is a corrosive and hazardous material that generates hydrogen bromide gas and requires special handling. Upon hydrolysis, boron tribromide also produces borate salts that are costly and time-consuming to separate and dispose of.
An alternative method of preparing THBO compounds was described in WO 97/33882, wherein a 1,3-propanediol was reacted with thionyl chloride to form a cyclic sulfite compound. The cyclic sulfite compound was oxidized to produce a cyclic sulfate compound. The cyclic sulfate was condensed with a 2-methylthiophenol that had been deprotonated with sodium hydride. The product of the condensation was a (2-methylphenyl) (3′-hydroxypropyl)thioether compound. The thioether compound was oxidized to form an thioether aldehyde compound. The thioether aldehyde compound was further oxidized to form an aldehyde sulfone compound which in turn was cyclized in the presence of potassium t-butoxide to form a 4-hydroxytetrahydrobenzothiepine 1,1 -dioxide compound. This cyclic sulfate route to THBO compounds requires an expensive catalyst. Additionally it requires the use of SOCI2, which in turn requires special equipment to handle. PCT Patent Application No. WO 97/33882 describes a method by which the phenol compound 28 was reacted at its phenol hydroxyl group to attach a variety of functional groups to the molecule, such as a quaternary ammonium group. For example, (4R,5R)-28 was reacted with l,4-bis(chloromethyl)benzene (?,??’-dichloro-p-xylene) to produce the chloromethyl benzyl- ether (4R,5R)-27. Compound (4R,5R)-27 was treated with diazabicyclo[2.2.2]octane (DABCO) to produce (4R,5R)-l-((4-(4-(3,3-dibutyl-7-(dimemylamino)-2,3,4,5-tetrahydro-4-hydroxy-l , 1 -dioxido-1 -benzothiepin-5-yl)phenoxy)methyl)phenyl)methyl-4-aza-l-azomabicyclo[2.2.2]octane chloride (41). This method suffers from low yields because of a propensity for two molecules of compound (4R,5R)-28 to react with one molecule of l,4-bis(chloromethyl)benzene to form a bis(benzothiepine) adduct. Once the bis-adduct forms, the reactive chloromethyl group of compound (4R,5R)-27 is not available to react with an amine to form the quaternary ammonium product.
A method of preparing enantiomerically enriched tetrahydrobenzothiepine oxides is described in PCT Patent Application No. WO 99/32478. In that method, an aryl-3- hydroxypropylsulfide compound was oxidized with an asymmetric oxidizing agent, for example (lR (->(8,9-dichloro-10-camphorsulfonyl)oxaziridine, to yield a chiral aryl-3-hydroxypropylsulfoxide. Reaction of the aryl-3-hydroxypropylsulfoxide with an oxidizing agent such as sulfur trioxide pyridine complex yielded an aryl-3-propanalsulfoxide. The aryl- 3-propanalsulfoxide was cyclized with a base such as potassium t-butoxide to enantioselectively produce a tetrahydrobenzothiepine- 1 -oxide. The tetrahydrobenzothiepine- 1 -oxide was further oxidized to produce a tetrahydrobenzothiepine- 1 , 1 -dioxide. Although this method could produce tetrahydrobenzothiepine- 1,1 -dioxide compounds of high enantiomeric purity, it requires the use of an expensive asymmetric oxidizing agent. Some 5-amidobenzothiepine compounds and methods to make them are described in
PCT Patent Application Number WO 92/18462. In Svnlett. 9, 943-944(1995) 2-bromophenyl 3-benzoyloxy-l-buten-4-yl sulfone was treated with tributyl tin hydride and AIBN to produce 3-benzoyloxytetrahydrobenzothiepine-1,1 -dioxide. In addition to forming the desired ASBT inhibitors, it is also desirable to form such
ASBT inhibitors of higher purity and having lower levels of residual solvent impurities. This is especially so with respect to ASBT inhibitors having a positively charged substituent, for example, the compounds designated as 41 (supra) and 60 (infra). It is further desirable to provide methods for making such high purity ASBT inhibitors.
( 4R, 5R) -26 A 1000 mL 4 neck jacketed Ace reactor flask was fitted with a mechanical stirrer, a nitrogen inlet, an addition funnel or condenser or distilling head with receiver, a thermocouple, four internal baffles and a 28 mm Teflon turbine agitator. The flask was purged with nitrogen gas and charged with 25.0 grams of (4R,5R)-28 and 125 mL of N,N-dimethylacetamide (DMAC). To this was added 4.2 grams of 50% sodium hydroxide. The mixture was heated to 50°C and stiπed for 15 minutes. To the flask was added 8.3 grams of 55 dissolved in 10 mL of DMAC, all at once. The temperature was held at 50°C for 24 hrs. To the flask was added 250 mL of toluene followed by 125 mL of dilution water. The mixture was stiπed for 15 minutes and the layers were then allowed to separate at 50°C. The flask was then charged with 125 mL of saturated sodium chloride solution and stiπed 15 minutes. Layers separated cleanly in 30 seconds at 50°C. Approximately half of the solvent was distilled off under vacuum at 50°C. The residual reaction mixture contained (4R,5R)-26.
Step 2. Preparation of (4R.5RV27.
( 4R, 5R) -27 Toluene was charged back to the reaction mixture of Step 1 and the mixture was cooled to 35°C. To the mixture was then added 7.0 grams of thionyl chloride over 5 minutes. The reaction was exothermic and reached 39°C. The reaction turned cloudy on first addition of thionyl chloride, partially cleared then finally remained cloudy. The mixture was stirred for 0.5 hr and was then washed with 0.25N NaOH. The mixture appeared to form a small amount of solids that diminished on stirring, and the layers cleanly separated. The solvent was distilled to a minimum stir volume under vacuum at 50°C. The residual reaction mixture contained (4R,5R)-27.
Step 3. Preparation of 41. To the reaction mixture of Step 2 was charged with 350 mL of methyl ethyl ketone (MEK) followed by 10.5 mL water and 6.4 grams of diazabicyclo[2.2.2]octane (DABCO) dissolved in 10 mL of MEK. The mixture was heated to reflux, and HPLC showed <0.5% of (4R,5R)-27. The reaction remained homogenous initially then crystallized at the completion of the reaction. An additional 5.3 mL of water was charged to the flask to redissolve product. Approximately 160 mL of solvent was then distilled off at atmospheric pressure. The mixture started to form crystals after 70 mL of solvent was distilled. Water separated out of distillate indicating a ternary azeotrope between toluene, water and methyl ethyl ketone (MEK). The mixture was then cooled to 25°C. The solids were filtered and washed with 150 mL MEK, and let dry under vacuum at 60°C. Isolated 29.8.0 g of off-white crystalline 4 Example 11a. Alternate Preparation of (4R,5R)-l-((4-(4-(3,3-dibutyl-7-(dimemylamino)-2,3,4,5-tetrahydro- 4-hydroxy- 1 , 1 -dioxido- 1 -benzithiepin-5-yl)phenoxy)methyl)phenyl)methyl-4-aza- 1 – azoniabicyclo[2.2.2]octane chloride, Form II of 41
A 1000 mL 4 neck jacketed Ace reactor flask is fitted with a mechanical stiπer, a nitrogen inlet, an addition funnel or condenser or distilling head with receiver, a thermocouple, four internal baffles and a 28 mm Teflon turbine agitator. The flask is purged with nitrogen gas and charged with 25.0 grams of (4R,5R)-28 and 100 mL of N,N-dimethylacetamide (DMAC). The mixture is heated to 50°C and to it is added 4.02 grams of 50% sodium hydroxide. The mixture is stiπed for 30 minutes. To the flask is added 8.7 grams of 55 dissolved in 12.5 mL of DMAC, all at once. The charge vessel is washed with 12.5 mL DMAC and the wash is added to the reactor. The reactor is stiπed for 3 hours. To the reactor is added 0.19 mL of 49.4% aq. NaOH and the mixture is stirred for 2 hours. To the mixture is added 0.9 g DABCO dissolved in 12.5 mL DMAC. The mixture is stiπed 30 to 60 minutes at 50°C. To the flask is added 225 mL of toluene followed by 125 mL of dilution water. The mixture is stiπed for 15 minutes and the layers are then allowed to separate at 50°C. The bottom aqueous layer is removed but any rag layer is retained. The flask is then charged with 175 mL of 5% hydrochloric acid solution and stiπed 15 minutes. Layers are separated at 50°C to remove the bottom aqueous layer, discarding any rag layer with the aqueous layer. Approximately half of the solvent is distilled off under vacuum at a maximum pot temperature of 80°C. The residual reaction mixture contains (4R,5R)-26.
Step 2. Preparation of (4R.5RV27.
Toluene (225 mL) is charged back to the reaction mixture of Step 1 and the mixture is cooled to 30°C. To the mixture is then added 6.7 grams of thionyl chloride over 30 to 45 minutes. The temperature is maintained below 35°C. The reaction turns cloudy on first addition of thionyl chloride, then at about 30 minutes the layers go back together and form a clear mixture. The mixture is stiπed for 0.5 hr and is then charged with 156.6 mL of 4% NaOH wash over a 30 minute period. The addition of the wash is stopped when the pH of the mixture reaches’ 8.0 to 10.0. The bottom aqueous layer is removed at 30°C and any rag layer is retained with the organic layer. To the mixture is charged 175 mL of saturated NaCl wash with agitation. The layers are separated at 30°C and the bottom aqueous layer is removed, discarding any rag layer with the aqueous layer. The solvent is distilled to a minimum stir volume under vacuum at 80°C. The residual reaction mixture contains (4R,5R)-27.
Step 3. Preparation of 41. To the reaction mixture of Step 2 is charged 325 mL of methyl ethyl ketone (MEK) and 13 mL water. Next, the reactor is charged 6.2 grams of diazabicyclo[2.2.2]octane (DABCO) dissolved in 25 mL of MEK. The mixture is heated to reflux and held for 30 minutes. Approximately 10% of solvent volume is then distilled off. The mixture starts to form crystals during distillation. The mixture is then cooled to 20°C for 1 hour. The off-white crystalline 41 (Form U) is filtered and washed with 50 mL MEK, and let dry under vacuum at 100°C.
Example lib. Alternate Preparation of (4R,5R)-1 -((4-(4-(3,3-dibutyl-7-(dimethylamino)-2,3,4,5-tetrahydro- 4-hydroxy- 1 , 1 -dioxido- 1 -benzithiepin-5-yl)phenoxy)methyl)phenyl)methyl-4-aza- 1 – azoniabicyclo[2.2.2]octane chloride, Form II of 41
A 1000 mL 4 neck jacketed Ace reactor flask is fitted with a mechanical stiπer, a nitrogen inlet, an addition funnel or condenser or distilling head with receiver, a thermocouple, four internal baffles and a Teflon turbine agitator. The flask is purged with nitrogen gas and charged with 25.0 grams of (4R,5R)-28 and 125 mL of N,N-dimethylacetamide (DMAC). The mixture is heated to 50°C and to it is added 7.11 grams of 30% sodium hydroxide over a period of 15 to 30 minutes with agitation. The mixture is stiπed for 30 minutes. To the flask is added 9.5 grams of solid 55. The reactor is stiπed for 3 hours. To the mixture is added 1.2 g of solid DABCO. The mixture is stiπed 30 to 60 minutes at 50°C. To the flask is added 225 mL of toluene followed by 125 mL of water. The mixture is stirred for 15 minutes and the layers are then allowed to separate at 50°C. The bottom aqueous layer is removed but any rag layer is retained with the organic layer. The flask is then charged with 175 mL of 5% hydrochloric acid solution and stirred 15 minutes. Layers are separated at 50°C to remove the bottom aqueous layer, discarding any rag layer with the aqueous layer. The flask is then charged with 225 mL of water and stirred 15 minutes. The layers are allowed to separate at 50°C. The bottom aqueous layer is removed, discarding any rag layer with the aqueous layer. Approximately half of the solvent is distilled off under vacuum at a maximum pot temperature of 80°C. The residual reaction mixture contains (4R,5R)-26.
Step 2. Preparation of (4R.5RV27.
Toluene (112.5 mL) is charged back to the reaction mixture of Step 1 and the mixture is cooled to 25°C. To the mixture is then added 7.3 grams of thionyl chloride over 15 to 45 minutes. The temperature of the mixture is maintained above 20°C and below 40°C. The reaction turns cloudy on first addition of thionyl chloride, then at about 30 minutes the layers go back together and form a clear mixture. The mixture is then charged with 179.5 mL of 4% NaOH wash over a 30 minute period. The mixture is maintained above 20°C and below 40°C during this time. The addition of the wash is stopped when the pH of the mixture reaches 8.0 to 10.0. The mixture is then allowed to separate at 40°C for at least one hour.
The bottom aqueous layer is removed and any rag layer is retained with the organic layer. To the mixture is charged 200 mL of dilution water. The mixture is stiπed for 15 minutes and then allowed to separate at 40°C for at least one hour. The bottom aqueous layer is removed, discarding any rag layer with the aqueous layer. The solvent is distilled to a minimum stir volume under vacuum at 80°C. The residual reaction mixture contains (4R,5R)-2 .
Step 3. Preparation of 41. To the reaction mixture of Step 2 is charged 350 mL of methyl ethyl ketone (MEK) and 7 mL water. The mixture is stiπed for 15 minutes and the temperature of the mixture is adjusted to 25°C. Next, the reactor is charged with 6.7 grams of solid diazabicyclo[2.2.2]octane (DABCO). The mixture is maintained at 25°C for three to four hours. It is then heated to 65°C and maintained at that temperature for 30 minutes. The mixture is then cooled to 25°C for 1 hour. The off-white crystalline 41 (Form II) is filtered and washed with 50 mL MEK, and let dry under vacuum at 100°C.
Example 12. Alternate preparation of (4R,5R)-1 -((4-(4-(3,3-dibutyl-7-(dimethylamino)-2,3,4,5-tetrahydro- 4-hydroxy- 1 , 1 -dioxido- 1 -benzithiepin-5-yl)phenoxy)methyl)phenyl)methyl-4-aza- 1 – azoniabicyclo[2.2.2]octane chloride, Form I of 41
(4R,5R)-27 (2.82 kg dry basis, 4.7 mol) was dissolved in MTBE (9.4 L). The solution of (4R,5R)-22 was passed through a 0.2 mm filter cartridge into the feeding vessel. The flask and was rinsed with MTBE (2 x 2.5 L). The obtained solution as passed through the cartridge filter and added to the solution of (4R,5R)-2 in the feeding vessel. DABCO (diazabicyclo[2.2.2]octane, 0.784 kg, 7.0 mol) was dissolved in MeOH (14.2 L). The DABCO solution was passed through the filter cartridge into the 100 L nitrogen-flushed reactor. The Pyrex bottle and the cartridge filter were rinsed with MeOH (7.5 L) and the solution was added to the reactor. The (4R,5R)-22 solution was added from the feeding vessel into the reactor at 37°C over a period of 10 min, while stirring. Methanol (6.5 L) was added to the Pyrex bottle and via the cartridge filter added to the feeding vessel to rinse the remaining (4R,5R)-2 into the reactor. The reaction mixture was brought to 50-60°C over 10-20 min and stiπed at that temperature for about 1 h. The mixture was cooled to 20-25°C over a period of 1 h. To the reaction mixture, methyl t-butyl ether (MTBE) (42 L) was added over a period of 1 h and stiπed for a minimum of 1 h at 20 – 25°C. The suspension was filtered through a Buchner funnel. The reactor and the filter cake were washed with MTBE (2 x 14 L). The solids were dried on a rotary evaporator in a 20 L flask at 400 – 12 mbar, 40°C, for 22 h. A white crystalline solid was obtained. The yield of 4 . (Form I) was 3.08 kg (2.97 kg dry, 93.8 %) and the purity 99.7 area % (HPLC; Kromasil C 4, 250 x 4.6 mm column; 0.05% TFA in H2O/0.05% TFA in ACN gradient, UV detection at 215 nm).
Example 12a. Conversion of Form I of Compound 41 into Form II of Compound 41.
To 10.0 grams of Form I of 4 . in a 400 mL jacketed reactor is added 140 mL of MEK. The reactor is stirred (358 φm) for 10 minutes at 23 °C for 10 minutes and the stirring rate is then changed to 178 φm. The suspension is heated to reflux over 1 hour using a programmed temperature ramp (0.95°C/minute) using batch temperature control (cascade mode). The delta Tmaχ is set to 5°C. The mixture is held at reflux for 1 hour. The mixture is cooled to
25°C. After 3 hours at 25°C, a sample of the mixture is collected by filtration. Filtration is rapid (seconds) and the filtrate is clear and colorless. The white solid is dried in a vacuum oven (80°C, 25 in. Hg) to give a white solid. The remainder of the suspension is stirred at 25°C for 18 hours. The mixture is filtered and the cake starts to shrink as the mother liquor reaches the top of the cake. The filtration is stopped and the reactor is rinsed with 14 mL of MEK. The reactor stirrer speed is increased from 100 to 300 φm to rinse the reactor. The rinse is added to the filter and the solid is dried with a rapid air flow for 5 minutes. The solid is dried in a vacuum oven at 25 in. Hg for 84 hours to give Form II of 4
Department of Discovery Chemistry and Department of Cardiovascular Disease, Pharmacia, 700 Chesterfield Parkway W, Chesterfield, Missouri 63017, Office of Science and Technology, Chemical Science Division, Pharmacia, 800 Lindbergh Boulevard, Creve Coeur, Missouri 63167, Department of Pharmaceutical Sciences, Pharmacia, Skokie, Illinois, and Department of Chemistry, University of Missouri, St. Louis, Missouri
In the preceding paper several compounds were reported as potent apical sodium-codependent bile acid transporter (ASBT) inhibitors. Since the primary site for active bile acid reabsorption is via ASBT, which is localized on the luminal surface of the distal ileum, we reasoned that a nonsystemic inhibitor would be desirable to minimize or eliminate potential systemic side effects of an absorbed drug. To ensure bioequivalency and product stability, it was also essential that we identify a nonhygroscopic inhibitor in its most stable crystalline form. A series of benzothiepines were prepared to refine the structure−activity relationship of the substituted phenyl ring at the 5-position of benzothiepine ring and to identify potent, crystalline, nonhygroscopic, and efficacious ASBT inhibitors with low systemic exposure.
compd
R
IC50 (nM)b
hygroscp I wt gain (%)c
hygroscp II % wt gain (%)d
crystallinitye
74
OCH2C6H4(p)CH2(N+)DB
0.28
1.59
2.1
yes
(4R–cis)-1-[[4-[[4-[3,3-Dibutyl-7-(dimethylamino)-2,3,4,5-tetrahydro-4-hydroxy-1,1-dioxido-1-benzothiepin-5-yl]phenoxy]methyl]phenyl]methyl]-4-aza-1-azoniabicyclo[2.2.2]octane Chloride Salt(74). Following a similar procedure as in General Method B, the title compound 74 was prepared from the corresponding chloromethyl benzyl ether and DABCO as a white solid, mp 223−230 °C (dec); 1H NMR (CDCl3) δ 0.89 (m, 6H), 1.27−1.52 (br m, 10H), 1.63 (m, 1H), 2.20 (m, 1H), 2.81 (s, 6H), 3.06 (ABq, JAB = 15.1 Hz, J = 43.3 Hz, 2H), 3.16 (s, 6H), 3.76 (s, 6H), 4.11 (d, J = 7.7 Hz, 1H), 5.09 (s, 2H), 5.14 (s, 2H), 5.48 (s, 1H), 5.96 (s, 1H), 6.49 (d, J = 8.9 Hz, 1H), 6.99 (d, J = 8.0 Hz, 2H), 7.26 (m, 1H), 7.44 (d, J = 8.0 Hz, 2H), 7.52 (d, J = 7.4 Hz, 2H), 7.68 (d, J = 7.4 Hz, 2H), 7.87 (d, J = 8.9 Hz, 1H). HRMS calcd for C40H56N3O4S: 674.3992; found, 674.4005. Anal. Calcd for C40H56N3O4S: ‘ C, 67.62; H, 7.95; N, 5.92; S, 4.51. Found: C, 67.48; H, 8.32; N, 5.85; S, 4.60.
a All compounds were prepared using method B in Scheme 3.b Taurocholate is transported across the baby hamster kidney cell membrane.c % weight gain in a 25 °C, 57% humidity chamber for 2 weeks.d % weight gain in a 40 °C, 80% humidity chamber for 2 weeks.e Crystallinity as determined by X-ray powder diffraction analysis.f (N+)DB is a DABCO terminal group with the quaternary ammonium attached to the linke
Example 10. Preparation of enantiomerically-enriched (4R.5R)-1- r.4- r _4- .3.3 -Dibutyl-7- (dimethylamino) -2.3 ,4.5- tetrahydro-4-hydroxy-1, l-dioxido-l-benzothiepin-5- yl] henoxy] ethyl] phenyl1methyl] -4-aza-l- azoniabicyclo [2.2.2] octane chloride ( (4R,5R) -XXVII) ♦
( (4R,5R) -XXVII) * = chiral center
Step 1. Preparation of 4-flUoro-2- ( (4- methoxyphenyl) methyl) -phenol To a stirred solution of 23.66 g of 95% sodium hydride (0.94 mol) in 600 mL of dry toluene was added 100.0 g of 4- fluorophenol (0.89 mol) at 0°C. The mixture was stirred at 90°C for 1 hour until gas evolution stopped. The mixture was cooled down to room temperature and a solution of 139.71 g of 3 -methoxybenzyl chloride (0.89 mol) in 400 mL of dry toluene was added. After refluxing for 24 hours, the mixture was cooled to room temperature and quenched with 500 mL of water. The organic layer was separated, dried over MgS04, and concentrated under high vacuum. The remaining starting materials were removed by distillation. The crude dark red oil was filtered through a layer of 1 L of silica gel with neat hexane to yield 53.00 g (25.6%) of the product as a pink solid: *H NMR (CDC13) d 3.79 (s, 3H) , 3.90 (s, 2H) , 4.58 (s, IH) , 6.70-6.74 (m, IH) , 6.79-6.88 (m, 4H) , 7.11-7.16 (m, 2H) .
Step 2. Preparation of 4-fluoro-2- ( (4- methoxyphenyl) methyl) -thiophenol
Step 2a. Preparation of thiocarbamate To a stirred solution of 50.00 g (215.30 mmol) of 4- fluoro-2- ( ( -methoxyphenyl) methyl) -phenol in 500 mL of dry DMF was added 11.20 g of 60% sodium hydride dispersion in mineral oil (279.90 mmol) at 2°C. The mixture was allowed to warm to room temperature and 26.61 g of dimethylthiocarbamoyl chloride (215.30 mmol) was added. The reaction mixture was stirred at room temperature overnight. The mixture was quenched with 100 mL of water in an ice bath. The solution was extracted with 500 mL of diethyl ether. The ether solution was washed with 500 mL of water and 500 mL of brine. The ether solution was dried over MgS04 and stripped to dryness. The crude product was filtered through a plug of 500 mL silica gel using 5% ethyl acetate/hexane to yield 48.00 g (69.8%) of the product as a pale white solid: XH NMR (CDC13) d 3.21 (s, 3H) , 3.46 (s, 3H) , 3.80 (s, 3H) , 3.82 (s, 2H) , 6.78-6.86 (m, 3H) , 6.90- 7.00 (m, 2H) , 7.09 (d, J = 8.7 Hz, 2H) .
Step 2b. Rearrangement and hydrolysis of thiocarbamate to 4-fluoro-2- ( (4 -methoxyphenyl) methyl) -thiophenol A stirred solution of 48.00 g (150.29 mmol) of thiocarbamate (obtained from Step 2a) in 200 mL of diphenyl ether was refluxed at 270°C overnight. The solution was cooled down to room temperature and filtered through 1 L of silica gel with 2 L of hexane to remove phenyl ether. The rearrangement product was washed with 5% ethyl acetate/hexane to give 46.00 g (95.8%) of the product as a pale yellow solid: XH NMR (CDC13) d 3.02 (s, 3H) , 3.10 (s, 3H) , 3.80 (s, 3H) , 4.07 (s, 2H) , 6.82-6.86 (m, 3H) , 6.93 (dt, J = 8.4 Hz, 2.7 Hz, IH) , 7.08 (d, J = 8.7 Hz, 2H) , 7.49 (dd, J = 6.0 Hz, 8.7 Hz, IH) . To a solution of 46.00 g (144.02 mmol) of the rearrangement product (above) in 200 mL of methanol and 200 mL of THF was added 17.28 g of NaOH (432.06 mmol) . The mixture was refluxed under nitrogen overnight . The solvents were evaporated off and 200 mL of water was added. The aqueous solution was washed with 200 mL of diethyl ether twice and placed in an ice bath. The aqueous mixture was acidified to pH 6 with concentrated HCl solution. The solution was extracted with 300 mL of diethyl ether twice. The ether layers were combined, dried over MgS04 and stripped to dryness to afford 27.00 g (75.5%) of the product as a brown oil: XH NMR (CDC13) d 3.24 (s, IH) , 3.80 (s, 3H) , 3.99 (s, 2H) , 6.81-6.87 (m, 4H) , 7.09 (d, J = 8.7 Hz, 2H) , 7.27- 7.33 (m, IH) .
Step 3. Preparation of dibutyl cyclic sulfate
Step 3a. Preparation of 2 , 2-dibutyl-l, 3-propanediol . To a stirred solution of di-butyl-diethylmalonate (Aldrich) (150g, 0.55 mol in dry THF (700ml) in an acetone/dry ice bath was added LAH (1 M THF) 662 ml (1.2 eq. , 0.66 mol) dropwise maintaining the temperature between -20 to 0°C. The reaction was stirred at RT overnight. The reaction was cooled to -20°C and 40 ml of water, and 80 mL of 10% NaOH and 80 ml of water were added dropwise. The resulting suspension was filtered. The filtrate was dried over sodium sulphate and concentrated in vacuo to give diol 598.4 g (yield 95%) as an oil. MS spectra and proton and carbon NMR spectra were consistent with the product.
Step 3b. Preparation of dibutyl cyclic sulfite
A solution of 2 , 2-dibutyl-l, 3-propanediol (103 g, 0.548 0 mol, obtained from Step 3a) and triethylamine (221 g, 2.19 mol) in anhydrous methylene chloride (500 ml) was stirred at 0°C under nitrogen. To the mixture, thionyl chloride (97.8* g, 0.‘82 mol) was added dropwise and within 5 min the solution turned yellow and then black when the addition was 5 completed within half an hour. The reaction mixture was stirred for 3 hrs. at 0°C. GC showed that there was no starting material left. The mixture was washed with ice water twice then with brine twice . The organic phase was dried over magnesium sulfate and concentrated under vacuum 0 to give 128 g (100%) of the dibutyl cyclic sulfite as a black oil. Mass spectrum (MS) was consistent with the product .
Step 3c. Oxidation of dibutyl cyclic sulfite to 5 dibutyl cyclic sulfate
To a solution of the dibutyl cyclic sulfite (127.5 g , 0.54 mol, obtained from Step 3b) in 600 ml acetonitrile and 500 ml of water cooled in an ice bath under nitrogen was added ruthenium (III) chloride (1 g) and sodium periodate 0 (233 g, 1.08 mol) . The reaction was stirred overnight and the color of the solution turned black. GC showed that there was no starting material left. The mixture was extracted with 300 ml of ether and the ether extract was washed three times with brine. The organic phase was dried over magnesium sulfate and passed through celite. The filtrate was 5 concentrated under vacuum and to give 133 g (97.8%) of the dibutyl cyclic sulfate as an oil. Proton and carbon NMR and MS were consistent with the product.
Step 4. Preparation of aryl-3-hydroxypropylsulfide
10 To a stirred solution of 27.00 g (108.73 mmol) of 4- fluoro-2- ( (4-methoxyphenyl) methyl) thiophenol (obtained from Step 2) in 270 mL of diglyme was added 4.35 g of 60% sodium-, hydride dispersion in mineral oil (108.73 mmol) at 0°C. After gas evolution ceased, 29.94 g (119.60 mmol) of the
15 dibutyl cyclic sulfate (obtained from Step 3c) was added at 0°C and stirred for 10 minutes. The mixture was allowed to warm up to room temperature and stirred overnight. The solvent was evaporated and 200 mL of water was added. The solution was washed with 200 mL of diethyl ether and added
2025 mL of concentrated sulfuric acid to make a 2.0 M solution that was refluxed overnight. The solution was extracted with ethyl acetate and the organic solution was dried over MgS04 and concentrated in vacuo. The crude aryl-3 – hydroxypropylsulfide was purified by silica gel
25 chromatography (Waters Prep 500) using 8% ethyl acetate/hexane to yield 33.00 g (72.5%) of the product as a light brown oil: E NMR (CDC13) d 0.90 (t, J = 7.1 Hz, 6H) , 1.14-1.34 (m, 12H) , 2.82 (s, 2H) , 3.48 (s, 2H) , 3.79 (s, 3H) , 4.10 (s, 2H) , 6.77-6.92 (m, 4H) , 7.09 (d, J = 8.7 Hz,
To a stirred solution of 20.00 g (47.78 mmol) of aryl- 53 -hydroxypropylsulfide (obtained from Step 4) in 1 L of methylene chloride was added 31.50 g of 96% (12?) – ( -) – (8 , 8- dichloro-10-camphor-sulfonyl) oxaziridine (100.34 mmol, Aldrich) at 2°C. After all the oxaziridine dissolved the mixture was placed into a -30 °C freezer for 72 hours. The
10 solvent was evaporated and the crude solid was washed with 1 L of hexane. The white solid was filtered off and the hexane solution was concentrated in vacuo. The crude oil was purified on a silica gel column (Waters Prep 500) using 15% ethyl acetate/hexane to afford 19.00 g (95%) of the
207.00 (d, J = 8.1 Hz, 2H) , 7.18-7.23 (m, IH) , 7.99-8.04 (m, IH) . Enantiomeric excess was determined by chiral HPLC on a (2?,2?) -Whelk-0 column using 5% ethanol/hexane as the eluent. It showed to be 78% e.e. with the first eluting peak as the major product.
25
Step 6. Preparation of enantiomerically-enriched aryl-3- propanalsulfoxide
To a stirred solution of 13.27 g of triethylamine (131.16 mmol, Aldrich) in 200 mL dimethyl sulfoxide were
30 added 19.00 g (43.72 mmol) of enantiomerically-enriched aryl-3 -hydroxypropylsulfoxide (obtained from Step 5) and 20.96 g of sulfur trioxide-pyridine (131.16 mmol, Aldrich) at room temperature. After the mixture was stirred at room temperature for 48 hours, 500 mL of water was added to the mixture and stirred vigorously. The mixture was then 5 extracted with 500 mL of ethyl acetate twice. The ethyl acetate layer was separated, dried over MgS04, and concentrated in vacuo. The crude oil was filtered through 500 mL of silica gel using 15% ethyl acetate/hexane to give 17.30 g (91%) of the enantiomerically-enriched aryl-3-
Step 7. Preparation of the enantiomerically-enriched tetrahydrobenzothiepine-1-oxide (4R, 5R)
20 To a stirred solution of 17.30 g (39.99 mmol) of enantiomerically-enriched aryl-3 -propanalsulfoxide (obtained from Step 6) in 300 mL of dry THF at -15°C was added 48 mL of 1.0 M potassium t-butoxide in THF (1.2 equivalents) under nitrogen. The solution was stirred at -15°C for 4 hours.
25 The solution was then quenched with 100 mL of water and neutralized with 4 mL of concentrated HCl solution at 0°C. The THF layer was separated, dried over MgS04, and concentrated in vacuo. The enantiomerically-enriched tetrahydrobenzothiepine-1-oxide (4R,5R) was purified by
To a stirred solution of 13.44 g (31.07 mmol) of enantiomerically-enriched tetrahydrobenzothiepine-1-oxide (obtained from Step 7) in 150 mL of methylene chloride was added 9.46 g of 68% m-chloroperoxybenzoic acid (37.28 mmol,
15 Sigma) at 0 °C. After stirring at 0 °C for 2 hours, the mixture was allowed to warm up to room temperature and stirred for 4 hours. 50 mL of saturated Na2S03 was added into the mixture and stirred for 30 minutes. The solution was then neutralized with 50 mL of saturated NaHC03 solution.
20 The methylene chloride layer was separated, dried over MgS04, and concentrated in vacuo to give 13.00 g (97.5%) of the enantiomerically-enriched tetrahydrobenzothiepine-1, 1- dioxide (4R,5R) as a light yellow solid: ‘H NMR (CDC13) d 0.89-0.95 (m, 6H) , 1.09-1.42 (m, 12H) , 2.16-2.26 (m, IH) ,
30 Step 9. Preparation of enantiomerically-enriched 7-
(dimethylamino) tetrahydrobenzothiepine-1 , 1-dioxide (4R.5R) – To a solution of 13.00 g (28.98 mmol) of enantiomerically-enriched tetrahydrobenzothiepine-1, 1- dioxide (obtained from Step 8) in 73 mL of dimethylamine (2.0 M in THF, 146 mmol) in a Parr Reactor was added ca . 20 5 mL of neat dimethylamine . The mixture was sealed and stirred at 110 °C overnight, and cooled to ambient temperature. The excess dimethylamine was evaporated. The crude oil was dissolved in 200 mL of ethyl acetate and washed with 100 mL of water, dried over MgS04 and
10 concentrated in vacuo. Purification on a silica gel column (Waters Prep 500) using 20% ethyl acetate/hexane gave 12.43 g (90.5%) of the enantiomerically- enriched 7- (dimethylamino) tetrahydrobenzothiepine-1, 1-dioxide (4R, 5R) as a colorless solid: *H NMR (CDC13) d 0.87-0.93 (m, 6H) ,
20 IH) . The product was determined to have 78% e.e. by chiral HPLC on a Chiralpak AD column using 5% ethanol/hexane as the eluent. Recrystallization of this solid from ethyl acetate/hexane gave 1.70 g of the racemic product. The remaining solution was concentrated and recrystallized to
25 give 9.8 g of colorless solid. Enantiomeric excess of this solid was determined by chiral HPLC on a Chiralpak AD column using 5% ethanol/hexane as the eluent. It showed to have 96% e.e with the first eluting peak as the major product.
30 Step 10: Demethylation of 5- (4 ‘ -methoxyphenyl) -7-
(dimethylamino) tetrahydrobenzothiepine-1.1-dioxide (4R, 5R) To a solution of 47 g (99 mmol) of enantiomeric- enriched (dimethylamino) tetrahydrobenzothiepine-1, 1-dioxide (obtained from Step 9) in 500 mL of methylene chloride at -10 °C was added dropwise a solution of boron tribromide (297 mL, 1M in methylene chloride, 297 mmol), and the resulting solution was stirred cold (-5 °C to 0 °C) for 1 hour or until the reaction was complete. The reaction was cooled in an acetone-dry ice bath at -10 °C, and slowly quenched with 300 mL of water. The mixture was warmed to 10 °C, and further diluted with 300 mL of saturated sodium bicarbonate solution to neutralize the mixture. The aqueous layer was separated and extracted with 300 mL of methylene chloride, and the combined extracts were washed with 200 mL of water, brine, dried over MgS04 and concentrated in vacuo. The residue was dissolved in 500 mL of ethyl acetate and stirred with 50 mL of glacial acetic acid for 30 minutes at ambient temperature. The mixture was washed twice with 200 mL of water, 200 mL of brine, dried over MgS04 and concentrated in vacuo to give the crude 4-hydroxyphenyl intermediate. The solid residue was recrystallized from methylene chloride to give 37.5 g (82%) of the desired (4R, 5R) -5- (4′ – hydoxyphenyl) -7- (dimethylamino) tetrahydrobenzothiepine-1, 1- dioxide as a white solid: *H NMR (CDC13) d 0.84-0.97 (m, 6H) , 1.1-1.5 (m, 10H) , 1.57-1.72 (m, IH) , 2.14-2.28 (m, IH) , 2.83 (s, 6H) , 3.00 (d, J = 15.3 Hz, IH) , 3.16 (d, J – 15.3 Hz, IH) , 4.11 (s, 2H) , 5.48 (s, IH) , 6.02 (d, J – 2.4 Hz, IH) , 6.55 (dd, J“ = 9, 2.4 Hz, IH) , 6.88 (d, 8 , 7 Hz , 2H) , 7.38 (d, J – 8.7 Hz, 2H) , 7.91 (d, J“ = 9 Hz, 2H) .
Step 11: Preparation of enantiomerically-enriched chlorobenzyl intermediate Treat a solution of enantiomerically-enriched (4R,5R)- 5- (4′ -hydoxypheny1) -7- (dimethylamino) tetrahydrobenzothiepine-1, 1-dioxide (5.0 g, 10.9 mmol, obtained from Step 10) in acetone (100 mL) at 25 °C under N2 with powdered 5 K2C03 (2.3 g, 16.3 mmol, 1.5 eq.) and a, a’ -dichloro-p-xylene (6.7 g, 38.1 mmol, 3.5 eq.) . Stir the resulting solution at 65 °C for about 48 hours. Cool the reaction mixture to 25 °C and concentrate to 1/5 of original volume. Dissolve the residue in EtOAc (150 mL) and wash with water (2 x 150 mL) .
10 Extract the aqueous layer with EtOAc (2 x 150 mL) and wash the combined organic extracts with saturated aqueous NaCI (2 x 150 mL. Dry the combined extracts with MgS04 and concentrate in vacuo to provide the crude product . Purification by flash chromatography (5.4 x 45 cm silica,
1525-40% EtOAc/hexane) will afford the enantiomerically- enriched chlorobenzyl intermediate .
Step 12: Preparation of enantiomerically-enriched (4R.5R)- 1- r [4- [ [4- [3 , 3-Dibutyl-7- (dimethylamino) -2,3 , 4 , 5-tetrahvdro-
Treat a solution of the enantiomerically-enriched chlorobenzyl intermediate (4.6 g, 7.7 mmol, obtained from
25 above in Step 11) in acetonitrile (100 mL) at 25 °C under N2 with diazabicyclo [2.2.2] -octane (DABCO, 0.95 g, 8.5 mmol, 1.1 eq.) and stir at 35 °C for 2 hours. Collect the precipitated solid and wash with CH3CN. Recrystallization from CH3OH/Et20 will give the desired title compound (XXVII) .
ANY ERROR, EMAIL amcrasto@gmail.com, +919323115463
Mechanism of Action Selective androgen receptor modulators
Phase I Cachexia
Most Recent Events
03 Sep 2015 GlaxoSmithKline initiates enrolment in a phase I trial for Cachexia (In volunteers) in USA (NCT02567773)
01 Mar 2015 GlaxoSmithKline completes a phase I trial in Cachexia (In volunteers) in USA (NCT02045940)
31 Jan 2014 Phase-I clinical trials in Cachexia (In volunteers) in USA (PO)
GSK2881078 is a selective androgen receptor modulator (SARM) that is being evaluated for effects on muscle growth and strength in subjects with muscle wasting to improve their physical function. Part A of this study will evaluate the safety, efficacy and pharmacokinetics of GSK2881078 in healthy, older men and post-menopausal women who will take daily dosing for 28 days and be followed for a total of 70 days. Part B of this study will characterize the effect of Cytochrome P450 3A4 (CYP3A4) inhibition on the GSK2881078 pharmacokinetics. Part B will only be conducted if safe and efficacious dose is identified in Part A. Part A will include healthy older males and post-menopausal females; and randomize approximately 60 subjects (about 15 per cohort [4 cohorts]) to complete approximately 48 (about 12 per cohort). Part B will enroll one cohort of approximately 15 healthy male subjects to complete approximately 12. The study duration will be approximately 115 days for Part A and 122 days for Part B.
Steroidal nuclear receptor (NR) ligands are known to play important roles in the health of both men and women. Testosterone (T) and dihydrotestosterone (DHT) are endogenous steroidal ligands for the androgen receptor (AR) that appear to play a role in every tissue type found in the mammalian body. During the development of the fetus, androgens play a role in sexual differentiation and development of male sexual organs. Further sexual development is mediated by androgens during puberty. Androgens play diverse roles in the adult, including stimulation and maintenance of male sexual accessory organs and maintenance of the musculoskeletal system. Cognitive function, sexuality, aggression, and mood are some of the behavioral aspects mediated by androgens. Androgens have a physiologic effect on the skin, bone, and skeletal muscle, as well as blood, lipids, and blood cells (Chang, C. and Whipple, G. Androgens and Androgen Receptors. Kluwer Academic Publishers: Boston, MA, 2002)
Many clinical studies with testosterone have demonstrated significant gains in muscle mass and function along with decreases in visceral fat. See, for example,
Bhasin (2003) S. J. Gerontol. A Biol. Sci. Med. Sci. 58:1002-8, and Ferrando, A. A. et al. (2002) Am. J. Phys. Endo. Met. 282: E601-E607. Androgen replacement therapy (ART) in men improves body composition parameters such as muscle mass, strength, and bone mineral density (see, for example, Asthana, S. et al. (2004) J. Ger, Series A: Biol. Sci. Med. Sci. 59: 461 -465). There is also evidence of improvement in less tangible parameters such as libido and mood. Andrologists and other specialists are increasingly using androgens for the treatment of the symptoms of androgen deficiency. ART, using T and its congeners, is available in transdermal, injectable, and oral dosage forms. All current treatment options have contraindications (e.g., prostate cancer) and side-effects, such as increased hematocrit, liver toxicity, and sleep apnoea. Side-effects from androgen therapy in women include: acne, hirsutism, and lowering of high-density lipoprotein (HDL) cholesterol levels, a notable side-effect also seen in men.
Agents that could selectively afford the benefits of androgens and greatly reduce the side-effect profile would be of great therapeutic value. Interestingly, certain NR ligands are known to exert their action in a tissue selective manner (see, for example, Smith et al. (2004) Endoc. Rev. 2545-71 ). This selectivity stems from the particular ability of these ligands to function as agonists in some tissues, while having no effect or even an antagonist effect in other tissues. The term “selective receptor modulator” (SRM) has been given to these molecules. A synthetic compound that binds to an intracellular receptor and mimics the effects of the native hormone is referred to as an agonist. A compound that inhibits the effect of the native hormone is called an antagonist. The term “modulators” refers to compounds that have a spectrum of activities ranging from full agonism to partial agonism to full antagonism.
SARMs (selective androgen receptor modulators) represent an emerging class of small molecule pharmacotherapeutics that have the potential to afford the important benefits of androgen therapy without the undesired side-effects. Many SARMs with demonstrated tissue-selective effects are currently in the early stages of development See, for example, Mohler, M. L. et al. (2009) J. Med. Chem. 52(12): 3597-617. One notable SARM molecule, Ostarine™, has recently completed phase I and II clinical studies. See, for example, Zilbermint, M. F. and Dobs, A. S. (2009) Future Oncology 5(8):121 1-20. Ostarine™ appears to increase total lean body mass and enhance functional performance. Because of their highly-selective anabolic properties and demonstrated androgenic-sparing activities, SARMs should be useful for the prevention and/or treatment of many diseases in both men and women, including, but not limited to sarcopenia, cachexias (including those associated with cancer, heart failure, chronic obstructive pulmonary disease (COPD), and end stage renal disease (ESRD), urinary incontinence, osteoporosis, frailty, dry eye and other conditions associated with aging or androgen deficiency. See, for example, Ho et al. (2004) Curr Opin Obstet Gynecol. 16:405-9; Albaaj et al. (2006) Postgrad Med J 82:693-6; Caminti et al. (2009) J Am Coll Cardiol. 54(10):919-27; lellamo et al. (2010) J Am Coll Cardiol. 56(16): 1310-6; Svartberg (2010) Curr Opin Endocrinol Diabetes Obes. 17(3):257-61 , and Mammadov et al. (201 1 ) Int Urol Nephrol 43:1003-8. SARMS also show promise for use in promoting muscle regeneration and repair (see, for example, Serra et al. (Epub 2012 Apr 12)
doi:10.1093/Gerona/gls083),in the areas of hormonal male contraception and benign prostatic hyperplasia (BPH), and in wound healing (see, for example, Demling (2009) ePIasty 9:e9).
Preclinical studies and emerging clinical data demonstrate the therapeutic potential of SARMs to address the unmet medical needs of many patients. The demonstrated advantages of this class of compounds in comparison with steroidal androgens (e.g. , tissue-selective activity, oral administration, AR selectivity, and lack of androgenic effect) position SARMs for a bright future of therapeutic applications.
Although amorphous forms of SARMs may be developed for some uses, compounds having high crystallinity are generally preferred for pharmaceutical use due to their improved solubility and stability. Accordingly, there remains a need in the art for crystalline form of SARMs for therapeutic use.
To a solution of commercially available (R)-2-aminopropan-1 -ol (5 g, 66.6 mmol) in MeCN (20 mL), in an ice bath, is added very slowly, dropwise, chlorosulfonic acid (4.46 mL, 66.6 mmol) (very exothermic). The reaction mixture is kept in the cold bath for ~10 min, and then at rt for ~ 30 min. After stirring for another ~ 10 minutes, the solids are collected by filtration, washed sequentially with MeCN (40 mL) and hexanes (100 mL), and dried by air suction for ~ 40 min. to produce the intermediate ((R)-2-aminopropyl hydrogen sulfate.
Step 2:
To a solution of sodium thiomethoxide (5.60 g, 80 mmol) in water (20 mL) is added solid NaOH (2.66 g, 66.6 mmol) in portions over ~ 10 min. Then the intermediate from step 1 is added as a solid over ~ 5 min. The mixture is then heated at 90 °C for ~10 h. The reaction mixture is biphasic. Upon cooling, MTBE (20 mL) is added, and the organic phase (brownish color) is separated. The aqueous phase is extracted with MTBE (2 x 20 mL). The original organic phase is washed with 1 N NaOH (15 mL). The basic aqueous phase is re-extracted with MTBE (2 x 20 mL). All the ether phases are combined, dried over Na2S04, filtered, and concentrated (carefully, since the product is volatile) to afford the crude product as a light yellow oil.
Method 2
(R)-1-(methylthio)propan-2 -amine hydrochloride
A. (R)-2-((tert-Butoxycarbonyl)amino)propyl methanesulfonate
Step 1
Commercially available (R)-2-aminopropan-1 -ol (135 g, 1797 mmol) is dissolved in MeOH 1350 mL). The solution is cooled to 5°C with an icebath, then Boc20 (392 g, 1797 mmol) is added as a solution in MeOH (1000 mL). The reaction temperature is kept below 10°C. After the addition, the cooling bath is removed, and the mixture is stirred for 3 h. The MeOH is removed under vacuum (rotavap bath: 50°C). This material is used as is for the next step.
Step 2
The residue is dissolved in CH2CI2 (1200 mL) and NEt3 (378 mL, 2717 mmol) is added, then the mixture is cooled on an ice bath. Next, MsCI (166.5 mL, 2152 mmol) is added over ~2 h, while keeping the reaction temperature below 15°C. The mixture is stirred in an icebath for 1 h then the bath was removed. The mixture is stirred for 3 d, then washed with a 10% NaOH solution (500 mL 3 x), then with water. The organic phase is dried with MgS04, filtered, then stripped off (rota, 50°C waterbath. The impure residue is dissolved in a mix of 500mL EtOAc (500 mL) and MTBE (500 mL) and then extracted with water to remove all water-soluble salts. The organic phase is dried with MgS04, filtered, then stripped off to afford a white solid residue.
B. (R)-tert-Butyl (1 -(methylthio)propan-2-yl)carbamate
NaSMe (30 g, 428 mmol) is stirred with DMF (200 mL) to afford a suspension. Next, (R)-2-((tertbutoxycarbonyl)amino)propyl methanesulfonate (97 g, 383 mmol) is added portionwise while the temperature is kept below 45°C (exothermic). After the addition, the mixture is stirred for 2 h, then toluene (100 mL) is added. The mixture is washed with water (500 mL, 4 x), then dried with MgS04, and filtered. The filtrate is stripped off (rotavap) to a pale yellow oil.
C. (R)-1 -(Methylthio)propan-2 -amine hydrochloride
Acetyl chloride (150 mL,) is added to a stirred solution of MeOH (600 mL) cooled with an icebath. The mixture is stirred for 30 min in an icebath, then added to (R)-tert-butyl (1 -(methylthio)propan-2-yl)carbamate (78 g, 380 mmol). The mixture is stirred at rt for 2 h, (C02, (CH3)2C=CI-l2 evolution) and then stripped off to a white solid.
D. 4-Fluoro-3-iodo-2-(trifluoromethyl)benzonitrile
To a freshly prepared solution of LDA (1 19 mmol) in anhyd THF (250 mL) at -45°C is added a solution of commercially available 4-fluoro-2-(trifluoromethyl)benzonitrile (21 .5 g, 1 14 mmol) in THF (30 mL), dropwise at a rate such that the internal temperature remained < -40°C (became dark brown during addition). The mixture is stirred 30 min at -45°C, cooled to -70°C and iodine (31 .7 g, 125 mmol) is added in one portion (-70°C→ -52°C). The mixture is stirred for 1 h, removed from the cooling bath and quenched by addition of 10% Na2S203 (ca. 250 mL) and 1 N HCI (ca. 125 mL). The mixture is extracted with EtOAc (x3). Combined organics are washed (water, brine), dried over Na2S04 and concentrated in vacuo. The residue is purified by low pressure liquid chromatography (silica gel, EtOAc / hexanes, gradient elution) followed by
recrystallization from heptane (30 mL), twice, affording 4-fluoro-3-iodo-2-(trifluoromethyl)benzonitrile (15.79 g, 50.1 mmol, 44.1 % yield) as a pale yellow solid.
E. 4-Fluoro-2-(trifluoromethyl)-3-((trimethylsilyl)ethynyl)benzonitrile
A 20 mL vial is charged with 4-fluoro-3-iodo-2-(trifluoromethyl)benzonitrile,(0.315 g, 1 .00 mmol), Pd(PPh3)2CI2 (0.014 g, 0.020 mmol) and Cul (0.0076 g, 0.040 mmol), and sealed with a rubber septum. Anhyd PhMe (5 mL) and DIPA (0.210 mL, 1 .500 mmol) are added via syringe and the mixture is degassed 10 min by sparging with N2while immersed in an ultrasonic bath. Ethynyltrimethylsilane (0.155 mL, 1 .100 mmol) is added dropwise via syringe and the septum is replaced by a PTFE-faced crimp top. The mixture is stirred in a heating block at 60°C. Upon cooling the mixture is diluted with EtOAc and filtered through Celite. The filtrate is washed (satd NH4CI, water, brine), dried over Na2S04 and concentrated in vacuo. The residue is purified by low pressure liquid chromatography (silica gel, EtOAc / hexanes, gradient elution) affording 4-fluoro-2-(trifluoromethyl)-3-((trimethylsilyl)ethynyl)benzonitrile .
F. (R)-1 -(1 -(methylthio)propan-2-yl)-4-(trifluoromethyl)-1 H-indole-5-carbonitrile
A mixture of 4-fluoro-2-(trifluoromethyl)-3-((trimethylsilyl)ethynyl)benzonitrile (1 .16 g, 4.07 mmol), (R)-1 -(methylthio)propan-2-amine (0.599 g, 5.69 mmol) and DIEA (1 .42 mL, 8.13 mmol) in DMSO (7 mL) is heated (sealed tube) at 100°C for 50 min. Upon cooling, the reaction mixture is diluted with EtOAc (50 mL) and washed with water (30 mL). The organic phase is washed with water and brine, dried over Na2S04, filtered and concentrated to give the intermediate aniline. This intermediate is dissolved in NMP (7 mL), treated with KOtBu (1 M in THF) (5.69 mL, 5.60 mmol) and heated at 50°C. The reaction is monitored by LCMS, and deemed complete after 40 min. Upon cooling, the reaction mixture is diluted with EtOAc (40 mL) and washed with water (30 mL). The organic phase is washed with more water and brine, dried over Na2S04, filtered and concentrated. The residue is chromatographed over silica gel using a 5-40% EtOAc-hexane gradient to give the thioether intermediate:
G. (R)-1 -(1-(methylsulfonyl)propan-2-yl)-4-(trifluoromethyl)-1 H-indole-5-carbonitrile
To an ice-cold solution of (R)-1 -(1 -(methylthio)propan-2-yl)-4-(trifluoromethyl)-1 H-indole-5-carbonitrile (0.560 g, 1.88 mmol) in MeOH (10 mL) is added a solution of Oxone (4.04 g, 6.57 mmol) in water (10 mL). After 50 min, the reaction mixture is diluted with water (30 mL) and extracted with EtOAc (50 mL). The organic phase is washed with brine, dried over Na2S04, filtered and concentrated. The residue is chromatographed over silica gel using 100% CH2CI2 to give (R)-1-(1 -(methylsulfonyl)propan-2-yl)-4-(trifluoromethyl)-l H-indole-5-carbonitrile as a white foam that is crystallized from
CH2CI2/hexanes to afford a white solid.
Example 2- Preparation of crystalline form 1 of (R)-1 -(1-(methylsulfonyl)propan-2-yl)-4-(trifluoromethyl)indoline-5-carbonitrile
(R)-1 -(1-(methylsulfonyl)propan-2-yl)-4-(trifluoromethyl)indoline-5-carbonitrile (1 .74kg, 1wt) was dissolved in ethyl acetate (12.0 Kg, 6.9 wt) at 20-30°C. The solution was transferred into a clean reaction vessel via an in-line cartridge filter. The solution was concentrated to ~3.0-5.0 volumes under reduced pressure, keeping the temperature below 50°C. The solution was cooled to 20-30°C, and n-heptane (23.0 Kg, 13.2 wt) was added slowly over ~1 hour. The solution was stirred 1 -2 hrs at 20-30°C, heated to 50-55°C for 2-3 hours, cooled back to 20-30°C and stirred for 1 -2 hours. The slurry was sampled and analyzed by XRPD. The solid was collected by filtration, washed with n-heptane (1 .4 Kg, 0.8 wt), and dried in vacuo at 40-50 °C to provide crystalline
(R)-1 -(1-(methylsulfonyl)propan-2-yl)-4-(trifluoromethyl)indoline-5-carbonitrile (1 .54 Kg, Form 1 ; 88.5 % yield, 99.5% purity) as a slightly colored solid.
Example 3- Preparation of crystalline form 2 of (R)-1 -(1-(methylsulfonyl)propan-2-yl)-4-(trifluoromethyl)indoline-5-carbonitrile
Crude (R)-1 -(1 -(methylsulfonyl)propan-2-yl)-4-(trifluoromethyl)indoline-5-carbonitrile (1 .54 g [theoretical], 1 wt) was dissolved in dichloromethane (5mL, 3.25 vol) and loaded onto a 12-g ISCO column (Si02). The column was eluted with DCM (-500 mL, 325 vol) and the product-containing fractions were combined and concentrated in vacuo. The resulting residue was triturated in n-heptane. The solid was collected by filtration, air-dried, and placed under high vacuum for 3 h to provide GSK2881078A (1 .009 g, Form 2; 65.1 % yield, 100% AUC HPLC-UV) as a white solid.
1-(1-(Methylsulfonyl)propan-2-yl)-4-(trifiuoromethyl)-1H-indole-5-carbonitrile Synthesized in a manner similar to Example 9 using 1-(1-(methylthio)propan-2-yl)-4-(trifluoromethyl)-1 H-indole-5-carbonitrile (Example 25): MS (ESI): m/z 331 (MH+).
To a solution of commercially available (R)-2-aminopropan-1-ol (5 g, 66.6 mmol) in MeCN (20 mL), in an ice bath, was added very slowly, dropwise, chlorosulfonic acid (4.46 mL, 66.6 mmol) (very exothermic). A gummy beige precipitate formed. The reaction mixture was kept in the cold bath for -10 min, and then at rt for ~ 30 min. The reaction mixture was scratched with a spatula to try to solidify the gummy precipitate. After a few minutes, a beige solid formed. After stirring for another ~ 10 minutes, the solids were collected by filtration, washed sequentially with MeCN (40 mL) and hexanes (100 mL), and dried by air suction for ~ 40 min. The intermediate ((R)-2-aminopropyl hydrogen sulfate, weighed 0.46 g (~ 96% yield).
Step 2:
To a solution of sodium thiomethoxide (5.60 g, 80 mmol) in water (20 mL) was added solid NaOH (2.66 g, 66.6 mmol) in portions over – 10 min. Then the intermediate from step 1 was added as a solid over ~ 5 min. The mixture was then heated at 90 °C for -10 h. The reaction mixture was biphasic. Upon cooling, MTBE (20 mL) was added, and the organic phase (brownish color) was separated. The aqueous phase was extracted with MTBE (2 x 20 mL). The original organic phase is washed with 1 NaOH (15 mL) (this removes most of the color). The basic aqueous phase was re-extracted with MTBE (2 x 20 mL). All the ether phases are combined, dried over Na2S04, filtered, and
concentrated (carefully, since the product is volatile) to afford the crude product as a light yellow oil: 1H NMR (400 MHz, DMSO-cf6) δ 2.91-2.87 (m, 1 H), 2.43-2.31 (m, 2 H), 2.04 (s, 3 H), 1.50 (bs, 2 H), 1.01 (d, J = 6.3 Hz, 3 H).
Alternative synthesis of example 27A:
(R)-1 -(Methylthio)propan-2 -amine hydrochloride
A. (R)-2-((tert-Butoxycarbonyl)amino)propyl methanesulfonate
Step 1
Commercially available (R)-2-aminopropan-1-ol (135 g, 1797 mmol) was dissolved in MeOH 1350 mL). The solution was cooled to 5°C with an icebath, then Boc20 (392 g, 1797 mmol) was added as a solution in MeOH (1000 mL). The reaction temperature was kept below 10°C. After the addition, the cooling bath was removed, and the mixture was stirred for 3 h. The MeOH was removed under vacuum (rotavap bath: 50°C). The resulting residue was a colorless oil that solidified overnight to a white solid. This material was used as is for the next step.
Step 2
The residue was dissolved in CH2CI2 (1200 mL) and NEt3 (378 mL, 2717 mmol) was added, then the mixture was cooled on an ice bath. Next, MsCI (166.5 mL, 2152 mmol) was added over ~2 h, while keeping the reaction temperature below 15°C. The mixture was stirred in an icebath for 1 h then the bath was removed. The mixture was stirred for 3 d, then washed with a 10% NaOH solution (500 mL 3 x), then with water. The organic phase was dried with MgS0 , filtered, then stripped off (rota, 50°C waterbath. The impure residue was dissolved in a mix of 500mL EtOAc (500 mL) and MTBE (500 mL) and then, extracted with water to remove all water-soluble salts.The organic phase was dried with MgS04, filtered, then stripped off to afford a white solid residue: 1H NMR (400 MHz, DMSO-ds) δ 6.94-6.92 (m, 1 H), 4.02 (d, J = 5.8 Hz, 2 H), 3.78-3.71 (m, 1 H), 3.16 (s, 3 H), 1.38 (s, 9 H), 1.06 (d, J = 6.8 Hz, 3 H).
B. (R)-tert-Butyl (1-(methylthio)propan-2-yl)carbamate
NaSMe (30 g, 428 mmol) was stirred with DMF (200 mL) to afford a suspension. Next, (R)-2-((tertbutoxycarbonyl)amino)propyl methanesulfonate (97 g, 383 mmol) was added
portionwise while the temperature was kept below 45°C (exothermic).. After the addition, the mixture was stirred for 2 h, then toluene (100 ml_) was added. The mixture was washed with water (500 ml_, 4 x), then dried with MgS04, and filtered. The filtrate was stripped off (rotavap) to a pale yellow oil: 1H NMR (400 MHz, DMSO-d6) δ 6.77-6.75 (m, 1 H), 3.60-3.54 (m, 1 H), 2.54-2.50 (m, 1 H), 2.43-2.38 (m, 1 H), 2.05 (s, 3 H), 1.38 (s, 9 H), 1.08 (d, J = 7.8 Hz, 3 H).
C. (R)-1-(Methylthio)propan-2-amine hydrochloride
Acetyl chloride (150 mL,) was added to a stirred solution of MeOH (600 mL) cooled with an icebath. The mixture was stirred for 30 min in an icebath, then added to (R)-tert-butyl (1-(methylthio)propan-2-yl)carbamate (78 g, 380 mmol). The mixture was stirred at rt for 2 h, (C02, (CH3)2C=CH2 evolution) and then stripped off to a white solid: 1H NMR (400 MHz, DMSO-d6) δ 8.22 (bs, 3 H), 3.36-3.29 (m, 1 H), 2.80-2.75 (m, 1 H), 2.64-2.59 (m, 1 H (d, J = 6.6 Hz, 3 H).
D. (R)-1 -(1 -(Methylthio)propan-2-yl)-4-(trif luoromethy l)-1 H-indole-5-carbonitrile
A mixture of 4-fluoro-2-(trifluoromethyl)-3-((trimethylsilyl)ethynyl)benzonitrile (Example 21 D,1.16 g, 4.07 mmol), (R)-1-(methylthio)propan-2-amine (0.599 g, 5.69 mmol) and DIEA (1.42 mL, 8.13 mmol) in DMSO (7 mL) was heated (sealed tube) at 100°C for 50 min. Upon cooling, the reaction mixture was diluted with EtOAc (50 mL) and washed with water (30 mL). The organic phase was washed with water and brine, dried over Na2S04, filtered and concentrated to give the intermediate aniline. This intermediate was dissolved in NMP (7 mL), treated with KOtBu (1 M in THF) (5.69 mL, 5.60 mmol) and heated at 50°C. The reaction was monitored by LCMS, and deemed complete after 40 min. Upon cooling, the reaction mixture was diluted with EtOAc (40 mL) and washed with water (30 mL). The organic phase was washed with more water and brine, dried over Na2S04, filtered and concentrated. The residue was chromatographed over silica
gel using a 5-40% EtOAc-hexane gradient to give the thioether intermediate: MS (ESI):
E. (R)-1-(1-(Methylsulfonyl)propan-2-yl)-4-(trifluoromethyl)-1H-indole-5-carbonitrile
To an ice-cold solution of (R)-1-(1-(methylthio)propan-2-yl)-4-(trifluoromethyl)-1 H-indole-5-carbonitrile (0.560 g, 1.88 mmol) in MeOH (10 mL) was added a solution of Oxone (4.04 g, 6.57 mmol) in water (10 mL). After 50 min, the reaction mixture was diluted with water (30 mL) and extracted with EtOAc (50 mL). The organic phase was washed with brine, dried over Na2S04, filtered and concentrated. The residue was chromatographed over silica gel using 100% CH2CI2 to give (R)-1-(1-(methylsulfonyl)propan-2-yl)-4-(trifluoromethyl)-l H-indole-5-carbonitrile as a white foam that was crystallized from CH2CI2/hexanes to afford a white solid (0.508 g, 79% yield): 1H NMR (400 MHz, DMSO-d6) δ 8.17 (d, J = 8.6 Hz, 1 H), 8.12 (d, J = 3.5 Hz, 1 H), 7.81 (d, J – 8.5 Hz, 1 H), 6.87-6.84 (m, 1 H), 5.43-5.35 (m, 1 H), 4.01 (dd, J = 14.8, 8.6 Hz, 1 H), 3.83 (dd, J = 14.8, 4.9 Hz, 1 H), 2.77 (s, 3 H), 1.59 (d, J = 6.8 Hz, 3 H); MS (ESI): m/z 331 (M+H).
A LSD1 inhibitor potentially for the treatment of small cell lung cancer and acute myeloid leukemia.
GSK2879552 is an orally available, irreversible, inhibitor of lysine specific demethylase 1 (LSD1), with potential antineoplastic activity. Upon administration, GSK2879552 binds to and inhibits LSD1, a demethylase that suppresses the expression of target genes by converting the dimethylated form of lysine at position 4 of histone H3 (H3K4) to mono- and unmethylated H3K4. LSD1 inhibition enhances H3K4 methylation and increases the expression of tumor-suppressor genes. This may lead to an inhibition of cell growth in LSD1-overexpressing tumor cells. LSD1, overexpressed in certain tumor cells, plays a key role in tumor cell growth and survival. Check for active clinical trials or closed clinical trials using this agent.
Formula: C23H29ClN2O2 M.Wt: 400.94
CAS 1902123-72-1
Molecular Weight:
437.41
Formula:
C23H28N2O2.2HCl
Chromatin modification plays an essential role in transcriptional regulation (T. Kouzarides, 2007, Cell 128: 693-705). These modifications, which include DNA methylation, histone acetylation and hsitone methylation, are disregulated in tumors. This epigenetic disregulation plays an important role in the silencing of tumor suppressors and overexpression of oncogenes in cancer (M. Esteller, 2008, N Engl J Med 358: 1148-59. P. Chi et al, 2010, Nat Rev Cane 10:457-469.). The enzymes that regulate histone methylation are the histone methyl transferases and the histone demethylases.
Lysine-specific demethylase 1 (LSDl; also known as BHC110) is a histone lysine demethylase reported to demethylate H3K4mel/2 (Y. Shi et al, 2004, Cell 119: 941-953) and H3K9mel/2 (R. Schule et al.,2005, Nature 437: 436-439). LSDl is overexpressed in multiple human cancers, including prostate where it is associated with more frequent relapse (P. Kahl et al, 2006, Cane. Res. 66: 11341-11347), breast (J. Kirfel et al, 2010, Carcinogenesis 31: 512-520) neuroblastoma (J. Kirfel et al, 2009, Cane. Res. 69: 2065-2071. G. Sun et al, 2010, Mol. Cell. Biol. 28: 1997-2000). LSDl is essential for transcriptional regulation mediated by a number of nuclear hormone receptors, including androgen receptor in prostate cancer (R. Schuele et al, 2005, Nature 437: 436-439. R. Schuele et al, 2007, Nat. Cell Biol. 9: 347-353. R. Schuele et al, 2010, Nature 464: 792-796), estrogen receptor in breast carcinomas (M.G. Rosenfeld et al, 2007, Cell 128: 505-518), and TLX receptor in neuorblastoma (S. Kato et al, 2008, Mol. Cell. Biol. 28: 3995-4003). These studies have shown that knockdown of LSDl expression results in decreased cancer cell proliferation. Additionally, LSDl is overexpressed in multiple cancer types that are nuclear hormone receptor-independent. Those tumors include ER-negative breast (J. Kirfel et al, 2010, Carcinogenesis 31: 512-520), small-cell lung, bladder, head & neck, colon, serous ovary, and kidney Wilm’s tumor. Therefore, potent selective small molecule inhibitors of LSDl may be useful for treatment of cancers that are nuclear hormone receptor-dependent and/or nuclear hormone receptor-independent.
The compositions and methods provided herein can potentially be useful for the treatment of cancer including tumors such as skin, breast, brain, cervical carcinomas, testicular carcinomas, etc. More particularly, cancers that may be treated by the compositions and methods of the invention include, but are not limited to tumor types such as astrocytic, breast, cervical, colorectal, endometrial, esophageal, gastric, head and neck, hepatocellular, laryngeal, lung, oral, ovarian, prostate and thyroid carcinomas and sarcomas. More specifically, these compounds can potentially be used to treat: Cardiac: sarcoma (angiosarcoma, fibrosarcoma, rhabdomyosarcoma, liposarcoma), myxoma, rhabdomyoma, fibroma, lipoma and teratoma; Lung: bronchogenic carcinoma (squamous cell, undifferentiated small cell, undifferentiated large cell, adenocarcinoma), alveolar (bronchiolar) carcinoma, bronchial adenoma, sarcoma, lymphoma, chondromatous hamartoma, mesothelioma; Gastrointestinal: esophagus (squamous cell carcinoma, adenocarcinoma, leiomyosarcoma, lymphoma), stomach (carcinoma, lymphoma, leiomyosarcoma), pancreas (ductal adenocarcinoma, insulinoma, glucagonoma, gastrinoma, carcinoid tumors, vipoma), small bowel (adenocarcinoma, lymphoma, carcinoid tumors, Kaposi’s sarcoma, leiomyoma, hemangioma, lipoma, neurofibroma, fibroma), large bowel (adenocarcinoma, tubular adenoma, villous adenoma, hamartoma, leiomyoma); Genitourinary tract: kidney (adenocarcinoma, Wilm’s tumor
(carcinoma); Hematologic: blood (myeloid leukemia (acute and chronic), acute lymphoblastic leukemia, chronic lymphocytic leukemia, myeloproliferative diseases, multiple myeloma, myelodysplasia syndrome), Hodgkin’s disease, non-Hodgkin’s lymphoma (malignant lymphoma); Skin: malignant melanoma, basal cell carcinoma, squamous cell carcinoma, Kaposi’s sarcoma, moles dysplastic nevi, lipoma, angioma, dermatofibroma, keloids, psoriasis; and Adrenal glands: neuroblastoma. Thus, the term “cancerous cell” as provided herein, includes a cell afflicted by any one of or related to the above identified conditions.
To the solution of 2,2,2-trifluoro-N-(trans-2-phenylcyclopropyl)-N-(piperidin-4-ylmethyl)acetamide (200 mg, 0.613 mmol, Example l ib) and 4-(bromomethyl)benzoic acid (198 mg, 0.919 mmol) in acetonitrile (6 mL) was added potasium carbonate (254 mg, 1.838 mmol). The reaction mixture was stirred for 3 hours at the 90 °C. The reaction mixture was then filtered and evaporated. The crude oil was mixed with 10 mL of 10 % acetic acid and 10 mL of ethyl acetate. Layers were separated, and the organic layer was discharged. Aqueous layer was neutralized with 1 M Na2C03, and the product was extracted into 10 mL of ethyl acetate. The organic layer was washed with brine, dried over MgS04, filtered and evaporated. The oil was dissolved in 6 ml of EtOH and 3 ml of 1 M NaOH. The reaction mixture was stirred for 20 min, and then it was concentrated. The solution was then partioned between 2 ml of water and 5 mL of ethyl acetate. The organic layer was separated and evaporated. The oil was purified on preparatory HPLC (2 to 10 % AcCN: H20 with 0.1 % formic acid modifier). The fractions were collected. To each
fraction was added 1 ml of 1 M HCl, and the fractions were evaporated to dryness. 4-((4-(((trans-2-phenylcyclopropyl)amino)methyl)piperidin-l-yl)methyl)benzoic acid (50 mg, 0.118 mmol, 19.33 % yield) was isolated as a white solid. 1H NMR (400 MHz,
tert-Butyl 4-(bromomethyl)benzoate (1 g, 3.13 mmol) and piperidin-4-ylmethanol (0.361 g, 3.13 mmol) were dissolved in acetonitrile (25 mL). K2CO3 (1.300 g, 9.40 mmol) was added and the reaction mixture was heated to reflux for 20 min. The reaction mixture was cooled down to room temperature, filtered and evaporated. The resulting solid was partitioned between ethyl acetate (50mL) and 1 M HC1 (50 mL). The layers were separated and the aqueous layer was washed with ethyl acetate and the organic layers were discarded. The aqueous layer was basified with 8 M NaOH to pH -10 and extracted 2 times with 50 mL of ethyl acetate. The organic layers were combined, washed with brine and dried over MgSC^, filtered and evaporated. tert-Butyl 4-((4- (hydroxymethyl)piperidin-l-yl)methyl)benzoate (0.95 g, 2.99 mmol, 95 % yield) was isolated as yellow oil. 1H NMR (400 MHz, CHLOROFORM-d) δ 7.95 (d, J= 8.34 Hz, 2H), 7.39 (d, J = 8.08 Hz, 2H), 3.56 (s, 2H), 3.51 (d, J = 6.57 Hz, 2H), 2.90 (d, J= 11.37 Hz, 2H), 1.94 – 2.04 (m, 2H), 1.73 (d, J= 14.15 Hz, 2H), 1.61 (s, 9H), 1.40 – 1.56 (m, 2H), 1.30 – 1.37 (m, 2H); LC-MS Rt = 0.67 min; MS (ESI): 306.2 [M+H]+.
To a solution of oxalyl chloride (0.408 mL, 4.67 mmol) in dichloromethane (5 mL) at -60 °C was added a solution of DMSO (0.508 mL, 7.15 mmol) in 15 mL of dichloromethane over 30 minutes. The reaction was stirred for 30 minutes at -60 °C A solution of tert-butyl 4-((4-(hydroxymethyl)piperidin-l-yl)methyl)benzoate (950 mg, 3.11 mmol) in 5 mL of dichloromethane was added over 10 minutes at -60 °C. The reaction mixture was stirred for 3 hours at – 60 °C, then triethylamine (2.168 mL, 15.55 mmol) was added and after 10 minutes 10 mL of water was added. The reaction mixture was allowed to warm up to the room temperature. The layers were separated. The pH of the water layer was adjusted to ~7 with 1 M HC1 and then extracted with 20 mL of dichloromethane. The combined organic layers were washed with water and brine, then dried over MgSO, filtered and evaporated. The resulting oil was purified on a silica column eluting with EtOAc to yield tert-butyl 4-((4-formylpiperidin-l-yl)methyl)benzoate (550 mg, 1.722 mmol, 55.4 % yield) as a yellow oil. 1H NMR (400 MHz, CHLOROFORM-d) δ 9.67 (d, J= 1.26 Hz, 1H), 7.96 (d, J= 8.34 Hz, 2H), 7.38 (d, J= 8.34 Hz, 2H), 3.56 (s, 2H), 2.75 – 2.92 (m, 2H), 2.21 – 2.35 (m, 1H), 2.14 (t, J= 10.48 Hz, 2H), 1.91 (dd, J= 2.78, 13.14 Hz, 2H), 1.65 – 1.81 (m, 2H), 1.58 – 1.64 (m, 9H); LC-MS Rt = 0.69 min; MS (ESI): 304.2
To a solution of tert-butyl 4-((4-formylpiperidin-l-yl)methyl)benzoate (6.7 g, 22.08 mmol) in methanol (50 mL) was added (lR,2S)-2-phenylcyclopropanamine (3.53 g, 26.5 mmol). The reaction mixture was refluxed for 5 minutes then cooled down to the room temperature. Sodium cyanotrihydroborate (2.082 g, 33.1 mmol) was added. The reaction mixture was stirred 1 hour at room temperature. Water (50 mL) was added. The reaction was concentrated and 50 mL of dichloromethane was added. The layers were separated. The organics were washed with 10 % acetic acid (50 mL). The layers were separated and 50 mL of brine was added slowly as a solid crashed out. The solid was filtered and suspended in isopropanol. The suspension was sonicated and filtered. tert-Butyl 4-((4-(((( 1 R,2S)-2-phenylcyclopropyl)amino)methyl)piperidin- 1 -yl)methyl)benzoate (5.8 g, 13.65 mmol, 61.8 % yield) was isolated as a white solid. 1H NMR (400 MHz,
An histone-lysine n-methyltransferase EZH2 inhibitor potentially for the treatment of B-cell lymphoma.
Research CodeGSK-2816126; GSK-126; 2816126
CAS No. 1346574-57-9
Originator GlaxoSmithKline
Class Antineoplastics
Mechanism of Action EZH2 enzyme inhibitors; Histone modulators
Phase I Diffuse large B cell lymphoma; Follicular lymphoma
Preclinical Acute myeloid leukaemia
Most Recent Events
31 Mar 2014Phase-I clinical trials in Follicular lymphoma (Second-line therapy or greater) in USA and United Kingdom (IV)
31 Mar 2014Phase-I clinical trials in Diffuse large B cell lymphoma (Second-line therapy or greater) in USA and United Kingdom (IV)
16 Jan 2014Preclinical trials in Diffuse large B cell lymphoma & Follicular lymphoma in United Kingdom (IV)
GSK-126 is an inhibitor of mutant EZH2, a histone methyltransferase (HMT) that exhibits point mutations at key residues Tyr641 and Ala677; this compound does not appreciably affect WT EZH2. EZH2 is responsible for modulating expression of a variety of genes. GSK-126 competes with cofactor S-adenylmethionine (SAM) for binding to EZH2. GSK-126 displays anticancer chemotherapeutic activity by inhibiting proliferation in in vitro and in vivo models of diffuse large B-cell lymphoma.
(S)-6-bromo-1 -(sec-butyl)-N-((4,6-dimethyl-2-oxo-1 ,2-dihydropyridin-3-yl)methyl)-3- methyl-1 H-indole-4-carboxamide (Ex 267) and (R)-6-Bromo-1 -(sec-butyl)-N-((4,6- dimethyl-2-oxo-1 ,2-dihydropyridin-3-yl)methyl)-3-methyl-1 H-indole-4-carboxamide (Ex 268)
6-Bromo-1-(sec-butyl)-N-((4,6-dimethyl-2-oxo-1 ,2-dihydropyridin-3-yl)methy methyl-1 H-indole-4-carboxamide (racemic mixture, 1.9 g) was resolved by chiral HPLC (column : Chiralpak AD-H, 5 microns, 50 mm x 250 mm, UV detection :240 nM, flow rate: 100 mL/min, T = 20 deg C, eluent: 60:40:0.1 n-heptane:ethanol:isopropylamine
(isocratic)). For each run, 100 mg of the racemic compound was dissolved in 30 volumes (3.0 ml.) of warm ethanol with a few drops of isopropylamine added. A total of 19 runs were performed. Baseline resolution was observed for each run. The isomer that eluted at 8.3-10.1 min was collected (following concentration) as a white solid, which was dried at 50 °C (< 5 mm Hg) to afford 901 mg, and was determined to be the S isomer* (Ex. 267; chiral HPLC: >99.5% ee (no R isomer detected). The isomer that eluted at 10.8-13.0 min was collected as a white solid, which was dried at 50 °C (< 5 mm Hg) to afford 865 mg, and was determined to be the R isomer* (Ex. 268; chiral HPLC: 99.2% ee; 0.4% S isomer detected). 1H NMR and LCMS were consistent with the parent racemate.
* The absolute configuration was determined by an independent synthesis of each enantiomer from the corresponding commercially available homochiral alcohols via Mitsunobu reaction. The sterochemical assignments were also consistent by vibrational circular dichroism (VCD) analysis.
Added sequentially to a reaction vial were 6-bromo-1 -(sec-butyl)-N-((4,6-dimethyl- 2-OXO-1 , 2-dihydropyridin-3-yl)methyl)-3-methyl-1 H-indole-4-carboxamide (0.15 g, 0.338 mmol), 1-(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridin-2-yl)piperazine (0.127 g, 0.439 mmol), and potassium phosphate (tribasic) (0.287 g, 1.350 mmol), followed by 1 ,4- Dioxane (3 mL) and water (0.75 mL). The suspension was stirred under N2 degassing for 10 min., and then added PdCI2(dppf)-CH2CI2adduct (0.028 g, 0.034 mmol). The reaction vial was sealed, placed into a heat block at 95 °C, and stirred for 1.5 h. The contents were removed from heating and allowed to cool to room temperature. The aq layer was removed from bottom of the reaction vial via pipette. The reaction mixture was diluted into EtOAc (20 mL) followed by addition of 0.2 g each of Thiol-3 silicycle resin and silica gel. The volatiles were removed in vacuo and the residue dried on hi-vac for 1 h. The contents were purified by silica gel chromatography (dry loaded, eluent : A:
To a 30 mL microwave vial were added (S)-6-bromo-1 -(sec-butyl)-N-((4,6- dimethyl-2-oxo-1 ,2-dihydropyridin-3-yl)methyl)-3-methyl-1 H-indole-4-carboxamide (100 mg, 0.225 mmol), 1 -(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridin-2-yl)piperazine (85 mg, 0.293 mmol), 1 ,2-Dimethoxyethane (DME) (3 mL), water (1.000 mL) and sodium carbonate (0.338 mL, 0.675 mmol), and the mixture was degassed for 5 min by bubbling nitrogen. PdCI2(dppf)-CH2CI2 adduct (14.70 mg, 0.018 mmol) was added and the tube was sealed. The mixture was irradiated (microwave) at 140 °C for 10 min. The mixture was concentrated and the residue was taken up into MeOH and filtered. The filtrate was purified using reverse-phase HPLC (eluent: 25%ACN/H20, 0.1 % NH4OH to 60%
To a 30 mL microwave vial were added (R)-6-bromo-1-(sec-butyl)-N-((4,6- dimethyl-2-oxo-1 ,2-dihydropyridin-3-yl)methyl)-3-methyl-1 H-indole-4-carboxamide (100 mg, 0.225 mmol), 1 -(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridin-2-yl)piperazine (85 mg, 0.293 mmol), 1 ,2-Dimethoxyethane (DME) (3 mL), water (1.000 mL) and sodium carbonate (0.338 mL, 0.675 mmol), and the mixture was degassed for 5 min by bubbling nitrogen. PdCI2(dppf)-CH2Cl2 adduct (14.70 mg, 0.018 mmol) was added and the tube was sealed. The mixture was irradiated (microwave) at 140 °C for 10 min. The mixture was concentrated and the residue was taken up into MeOH and filtered. The filtrate was purified using reverse-phase HPLC (eluent: 25%ACN/H20, 0.1 % NH4OH to 60%
A reverse transcriptase inhibitor potentially for the treatment of HIV infection.
GSK-2838232; GSK-8232; 2838232
CAS No. 1443460-91-0
C48H73ClN2O6,809.56
SYNTHESIS
PART 1
PART2
PART3
PART 4
AND UNWANTEDISOMER SHOWN BELOW
PART5
GSK2838232 is a novel human immune virus (HIV) maturation inhibitor being developed for the treatment of chronic HIV infection. GSK2838232 is a betulin derivative
Human immunodeficiency virus type 1 (HIV-1 ) leads to the contraction of acquired immune deficiency disease (AIDS). The number of cases of HIV continues to rise, and currently over twenty-five million individuals worldwide suffer from the virus. Presently, long-term suppression of viral replication with antiretroviral drugs is the only option for treating HIV-1 infection. Indeed, the U.S. Food and Drug Administration has approved twenty-five drugs over six different inhibitor classes, which have been shown to greatly increase patient survival and quality of life.
However, additional therapies are still required because of undesirable drug-drug interactions; drug-food interactions; non-adherence to therapy; and drug resistance due to mutation of the enzyme target.
Currently, almost all HIV positive patients are treated with therapeutic regimens of antiretroviral drug combinations termed, highly active antiretroviral therapy (“HAART”). However, HAART therapies are often complex because a combination of different drugs must be administered often daily to the patient to avoid the rapid emergence of drug-resistant HIV-1 variants. Despite the positive impact of HAART on patient survival, drug resistance can still occur. The emergence of multidrug-resistant HIV-1 isolates has serious clinical consequences and must be suppressed with a new drug regimen, known as salvage therapy.
Current guidelines recommend that salvage therapy includes at least two, and preferably three, fully active drugs. Typically, first-line therapies combine three to four drugs targeting the viral enzymes reverse transcriptase and protease. One option for salvage therapy is to administer different combinations of drugs from the same mechanistic class that remain active against the resistant isolates.
However, the options for this approach are often limited, as resistant mutations frequently confer broad cross-resistance to different drugs in the same class.
Alternative therapeutic strategies have recently become available with the development of fusion, entry, and integrase inhibitors. However, resistance to all three new drug classes has already been reported both in the lab and in patients. Sustained successful treatment of HIV-1 -infected patients with antiretroviral drugs will therefore require the continued development of new and improved drugs with new targets and mechanisms of action.
Presently, long-term suppression of viral replication with antiretroviral drugs is the only option for treating HIV-1 infection. To date, a number of approved drugs have been shown to greatly increase patient survival. However, therapeutic regimens known as highly active antiretroviral therapy (HAART) are often complex because a combination of different drugs must be administered to the patient to avoid the rapid emergence of drug-resistant HIV-1 variants. Despite the positive impact of HAART on patient survival, drug resistance can still occur.
The HIV Gag polyprotein precursor (Pr55Gag), which is composed of four protein domains – matrix (MA), capsid (CA), nucleocapsid (NC) and p6 – and two spacer peptides, SP1 and SP2, represents a new therapeutic target. Although the cleavage of the Gag polyprotein plays a central role in the progression of infectious virus particle production, to date, no antiretroviral drug has been approved for this mechanism.
In most cell types, assembly occurs at the plasma membrane, and the
MA domain of Gag mediates membrane binding. Assembly is completed by budding of the immature particle from the cell. Concomitant with particle release, the virally encoded PR cleaves Gag into the four mature protein domains, MA, CA, NC and p6, and the two spacer peptides, SP1 and SP2. Gag-Pol is also cleaved by PR, liberating the viral enzymes PR, RT and IN. Gag proteolytic processing induces a
morphological rearrangement within the particle, known as maturation. Maturation converts the immature, donut-shaped particle to the mature virion, which contains a condensed conical core composed of a CA shell surrounding the viral RNA genome in a complex with NC and the viral enzymes RT and IN. Maturation prepares the virus for infection of a new cell and is absolutely essential for particle infectivity.
Bevirimat (PA-457) is a maturation inhibitor that inhibits the final step in the processing of Gag, the conversion of capsid-SP1 (p25) to capsid, which is required for the formation of infectious viral particles. Bevirimat has activity against ART-resistant and wild-type HIV, and has shown synergy with antiretrovirals from all classes. Bevirimat reduced HIV viral load by a mean of 1.3 logi0/mL in patients who achieved trough levels of >= 20 μg/mL and who did not have any of the key baseline Gag polymorphisms at Q369, V370 or T371. However, Bevirimat users with Gag polymorphisms at Q369, V370 or T371 demonstrated significantly lower load reductions than patients without Gag polymorphisms at these sites.
Other examples of maturation inhibitors can be found in PCT Patent
Application No. WO201 1/100308, “Derivatives of Betulin”; PCT Patent Application No. PCT/US2012/024288, “Novel Anti-HIV Compounds and Methods of Use Thereof ; Chinese PCT Application No. PCT/CN201 1/001302, “Carbonyl Derivatives of Betulin”; Chinese PCT Application No. PCT/CN201 1/001303, “Methylene Derivatives of Betulin”; Chinese PCT Application Nos. PCT/CN201 1/002105 and PCT/CN201 1/002159, “Propenoate Derivatives of Betulin”. Maturation inhibitors in the prior art leave open gaps in the areas of polymorphism coverage whereby potency against a broad range of clinically relevant gag sequences is extremely important, along with overall potency including the clinically relevant protein adjusted antiviral activity that will be required for robust efficacy in long term durability trials. To date, no maturation inhibitor has achieved an optimal balance of these properties.
[00242] To a solution of 2-(dimethylamino)acetaldehyde, hydrochloride (6.75 g, 54.6 mmol) in methanol (20 ml_) was added 4-
(((3aR,5aR,5bR,7aR,9S, 1 1 aR, 1 1 bR, 13aS)-3a-((R)-2-((4-chlorobenzyl)amino)-1 – hydroxyethyl)-1 -isopropyl-5a,5b,8,8, 1 1 a-pentamethyl-2-oxo- 3,3a,4,5,5a,5b,6,7,7a,8,9,10,1 1 ,1 1 a,1 1 b,12,13,13a-octadecahydro-2H- cyclopenta[a]chrysen-9-yl)oxy)-2,2-dimethyl-4-oxobutanoic acid , Trifluoroacetic acid salt (46) (9.5 g, 10.92 mmol). The pH was adjusted to 7-8 with Et3N. The reaction mixture was stirred at rt for 2 h. Sodium cyanoborohydride (0.686 g, 10.92 mmol) was then added and the mixture was stirred at rt overnight. After the reaction was complete, water (15 ml_) and EtOAc (15 ml_) were added, and then the organic phase was removed and concentrated under reduced presure. The product was extracted with EtOAc (80 ml_x3), the combined organic phase was washed with brine, dried, and concentrated. The product was purified by flash chromatography (DCM:EtOAc=2: 1 to 1 : 1 , then DCM:MeOH=100: 1 to 20: 1 ) to give 4- (((3aR,5aR,5bR,7aR,9S, 1 1 aR, 1 1 bR, 13aS)-3a-((R)-2-((4-chlorobenzyl)(2- (dimethylamino)ethyl)amino)-1 -hydroxyethyl)-1 -isopropyl-5a,5b,8,8, 1 1 a-pentamethyl- 2-0X0-3, 3a,4, 5, 5a, 5b, 6, 7, 7a, 8, 9, 10, 1 1 , 1 1 a, 1 1 b, 12, 13, 13a-octadecahydro-2H- cyclopenta[a]chrysen-9-yl)oxy)-2,2-dimethyl-4-oxobutanoic acid (51 ) (6 g, 7.41 mmol, 67.9 % yield) as white solid. Multiple batches of this material (were combined 95 g), dissolved in 600 mL of dichloromethane and washed with NaHC03 (400 ml_*3) and the organic phase was dried over Na2S04, filtered and concentrated. The solids were washed with a mixture of EtOAc: petroleum ether (600 mL), and filtered followed by lyophilization to provide the final title compound 62 g as a white solid. 1H NMR (400MHz ,METHANOL-d4) δ = 7.47 – 7.29 (m, 4 H), 4.48 (dd, J = 5.8, 10.3 Hz, 1 H), 4.15 – 4.04 (m, 1 H), 3.80 (d, J = 13.8 Hz, 1 H), 3.57 (d, J = 14.1 Hz, 1 H), 3.21 – 2.82 (m, 5 H), 2.72 – 2.41 (m, 9 H), 2.37 – 2.05 (m, 4 H), 2.05 – 0.74 (m, 45 H);
LC/MS: m/z calculated 808.5, found 809.5 (M+1 )+.
REFERENCES
Hatcher, Mark Andrew; Johns, Brian Alvin; Martin, Michael Tolar; Tabet, Elie Amine; Tang, Jun. Preparation of betulin derivatives for the treatment of HIV, PCT Int. Appl. (2013), WO 2013090664 A1 20130620.
Mark Hatcher
Director, US R&D Policy and Scientific Affairs at GlaxoSmithKline
A glycopeptide-cephalosporin heterodimer potentially for the treatment of gram-positive bacterial infection.
CAS No. 827040-07-3
C95 H109 Cl3 N18 O31 S2,
Molecular Weight, 2169.47
Vancomycin, 29-[[[2-[[6-[[[1-[[(6R,7R)-7-[[(2Z)-2-(2-amino-5-chloro-4-thiazolyl)-2-(methoxyimino)acetyl]amino]-2-carboxy-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-3-yl]methyl]pyridinium-4-yl]methyl]amino]-1,6-dioxohexyl]amino]ethyl]amino]methyl]-, inner salt
Vancomycin, 29-[[[2-[[6-[[[1-[[(6R,7R)-7-[[(2Z)-(2-amino-5-chloro-4-thiazolyl)(methoxyimino)acetyl]amino]-2-carboxy-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-3-yl]methyl]pyridinium-4-yl]methyl]amino]-1,6-dioxohexyl]amino]ethyl]amino]methyl]-, inner salt
Originator Theravance
Developer Theravance Biopharma
Class Antibacterials; Cephalosporins; Glycopeptides
Mechanism of Action Cell wall inhibitors
Phase I Gram-positive infections
Most Recent Events
21 Apr 2016 Phase I development is ongoing in USA
01 Jul 2014 Theravance completes a phase I trial in Healthy volunteers in in USA (NCT01949103)
02 Jun 2014 Theravance Biopharma is formed as a spin-off of Theravance
The present invention provides novel cross-linked glycopeptide – cephalosporin compounds that are useful as antibiotics. The compounds of this invention have a unique chemical structure in which a glycopeptide group is covalently linked to a pyridinium moiety of a cephalosporin group. Among other properties, compounds of this invention have been found to possess surprising and unexpected potency against Gram-positive bacteria including methicillin-resistant Staphylococci aureus (MRSA). Accordingly, in one aspect, the invention provides a compound of formula I:
Mechanism of Action Serotonin 2A receptor inverse agonists
Phase I Arterial thrombosis
Most Recent Events
30 Mar 2016 Arena Pharmaceuticals has patents pending for Temanogrel in 12 regions, including Brazil (Arena Pharmaceuticals 10-K; march 2016)
30 Mar 2016 Arena Pharmaceuticals has patent protection for Temanogrel in 87 regions, including USA, Japan, China, Germany, France, Italy, the United Kingdom, Spain, Canada, Russia, India, Australia and South Korea
01 Mar 2015 Ildong Pharmaceutical initiates enrolment in a phase I trial for Arterial thrombosis in South Korea (NCT02419820)
A 5-HT2A inverse agonist potentially for the reduction of the risk of arterial thrombosis.
Temanogrel, also known as APD791, is a highly selective 5-hydroxytryptamine2A receptor inverse agonist under development for the treatment of arterial thrombosis. APD791 displayed high-affinity binding to membranes (K(i) = 4.9 nM) and functional inverse agonism of inositol phosphate accumulation (IC(50) = 5.2 nM) in human embryonic kidney cells stably expressing the human 5-HT(2A) receptor. APD791 was greater than 2000-fold selective for the 5-HT(2A) receptor versus 5-HT(2C) and 5-HT(2B) receptors. APD791 inhibited 5-HT-mediated amplification of ADP-stimulated human and dog platelet aggregation (IC(50) = 8.7 and 23.1 nM, respectively)
Arterial thrombosis is the formation of a blood clot or thrombus inside an artery or arteriole that restricts or blocks the flow of blood and, depending upon location, can result in acute coronary syndrome or stroke. The formation of a thrombus is usually initiated by blood vessel injury, which triggers platelet aggregation and adhesion of platelets to the vessel wall. Treatments aimed at inhibiting platelet aggregation have demonstrated clear clinical benefits in the setting of acute coronary syndrome and stroke. Current antiplatelet therapies include aspirin, which irreversibly inhibits cyclooxygenase (COXa
Abbreviations: COX, cyclooxygenase; ADP, adenosine diphosphate; SAR, structure−activity relationship; hERG, human ether-a-go-go-related gene; CNS, central nervous system; 5-HT, serotonin; AUC, area under the plasma concentration time curve, iv, intravenous; IP, inositol phosphate.
) and results in reduced thromboxane production, clopidogrel and prasugrel, which inhibit platelet adenosine diphosphate (ADP) P2Y12 receptors, and platelet glycoprotein IIb/IIIa receptor antagonists. Another class of antiplatelet drugs, protease-activated thrombin receptor (PAR-1) antagonists, are also being evaluated in the clinic for the treatment of acute coronary syndrome. The most advanced candidate in this class, N-[(1R,3aR,4aR,6R,8aR,9S,9aS)-9-{2-[5-(3-fluorophenyl)pyridin-2-yl]vinyl}-1-methyl-3-oxoperhydro-naphtho[2,3-c]furan-6-yl]-carbamic acid ethyl ester (SCH-530348), is currently in phase 3 trials for the prevention of arterial thrombosis.
The 5-HT2A receptor is one of 15 different serotonin receptor subtypes.
In the cardiovascular system, modulation of 5-HT2A receptors on vascular smooth muscle cells and platelets is thought to play an important role in the regulation of cardiovascular function. Platelets are activated by a variety of agonists such as ADP, thrombin, thromboxane, serotonin, epinephrine, and collagen. Upon platelet activation at the site of blood vessel injury, a number of factors including serotonin (5-HT) are released. Although by itself serotonin is a weak activator of platelet aggregation, in vitro it can amplify aggregation induced by other agonists as mentioned above. Therefore, serotonin released from activated platelets may induce further platelet aggregation and enhance thrombosis.
The 5-HT2A receptor antagonist ketanserin was shown in clinical studies to reduce early restenosis(7) and decrease myocardial ischemia during coronary balloon angioplasty.(8)However, in another study, ketanserin did not significantly improve clinical outcomes, and the rate of adverse events was higher than that observed in the control group.(9) Some of the adverse events reported in the latter study could be specific to ketanserin and resulted from its lack of 5-HT2A receptor selectivity. Other 5-HT2A antagonists with improved selectivity profiles have shown promise in clinical studies. For example, sarpogrelate was shown to inhibit restenosis following coronary stenting.
Figure 1. Serotonin and known 5-HT2A receptor antagonists.
Because the 5-HT2A receptor is expressed both in peripheral tissues and in the central nervous system (CNS), compounds with limited CNS partitioning would be preferred to maximize cardiovascular and blood platelet pharmacological activity while minimizing CNS effects. In addition, because 5-HT2A receptor inverse agonists are thought to reduce thrombus formation via inhibition of serotonin-mediated amplification of platelet aggregation without inhibiting agonist driven aggregation per se, it is possible that this class of inhibitors will have an improved bleeding risk side effect profile compared to what has been observed with other classes of antithrombotic drugs.
SYNTHESIS
PAPER
Journal of Medicinal Chemistry (2010), 53(11), 4412-4421.
Serotonin, which is stored in platelets and is released during thrombosis, activates platelets via the 5-HT2A receptor. 5-HT2A receptor inverse agonists thus represent a potential new class of antithrombotic agents. Our medicinal program began with known compounds that displayed binding affinity for the recombinant 5-HT2A receptor, but which had poor activity when tested in human plasma platelet inhibition assays. We herein describe a series of phenyl pyrazole inverse agonists optimized for selectivity, aqueous solubility, antiplatelet activity, low hERG activity, and good pharmacokinetic properties, resulting in the discovery of 10k (APD791). 10k inhibited serotonin-amplified human platelet aggregation with an IC50 = 8.7 nM and had negligible binding affinity for the closely related 5-HT2B and 5-HT2C receptors. 10k was orally bioavailable in rats, dogs, and monkeys and had an acceptable safety profile. As a result, 10k was selected further evaluation and advanced into clinical development as a potential treatment for arterial
Discovery and Structure−Activity Relationship of 3-Methoxy-N-(3-(1-methyl-1H-pyrazol-5-yl)-4-(2-morpholinoethoxy)phenyl)benzamide (APD791): A Highly Selective 5-Hydroxytryptamine2A Receptor Inverse Agonist for the Treatment of Arterial Thrombosis
10k was prepared in a manner similar to that described for 10c, using 9d (120 mg, 0.40 mmol) and 3-methoxybenzoyl chloride (81 mg, 0.48 mmol) to give the TFA salt of 10k as a white solid (88 mg, 51%); mp (HCl salt, recrystallized from iPrOH) 214−216 °C. 1H NMR (acetone-d6, 400 MHz) δ: 2.99−3.21 (m, 2H), 3.22−3.45 (m, 2H), 3.66 (t, J = 4.8 Hz, 2H), 3.75 (s, 3H), 3.85 (s, 3H), 3.79−3.89 (m, 4H), 4.58 (t, J = 4.8 Hz, 2H), 6.29 (d, J = 2.0 Hz, 1H), 7.13 (dd, J = 2.5, 8.3 Hz, 1H), 7.22 (d, J = 8.8 Hz, 1H), 7.42 (t, J = 7.8 Hz, 1H), 7.47 (d, J = 1.7 Hz, 1H), 7.52 (t, J = 1.7 Hz, 1H), 7.56 (d, J = 7.0 Hz, 1H), 7.80−7.83 (m, 1H), 7.91−7.96 (m, 1H), 9.54 (s, 1H). LCMSm/z = 437.5 [M + H]+.
Additional Information
Oral administration of APD791 to dogs resulted in acute (1-h) and subchronic (10-day) inhibition of 5-HT-mediated amplification of collagen-stimulated platelet aggregation in whole blood. Two active metabolites, APD791-M1 and APD791-M2, were generated upon incubation of APD791 with human liver microsomes and were also indentified in dogs after oral administration of APD791. The affinity and selectivity profiles of both metabolites were similar to APD791. These results demonstrate that APD791 is an orally available, high-affinity 5-HT(2A) receptor antagonist with potent activity on platelets and vascular smooth muscle.(http://www.ncbi.nlm.nih.gov/pubmed/19628629).
Example 1.88: Preparation of 3-methoxy-N-[3-(2-methyl-2H-pyrazol-3-yl)-4-(2-morpholin~
4-yl-ethoxy)-phenyl]-benzamide (Compound 733).
A mixture of 3-(2-methyl-2H-pyrazol-3-yl)-4-(2-morpholin-4-yl-ethoxy)-phenylamine (120 mg, 0.40 mmole), 3-methoxy-benzoyl chloride (81 mg, 0.48 mmole), and triethylamine (0.1 mL, 0.79 mmole) in 5 mL THF was stirred at room temperature for 10 minutes. The mixture was purified by HPLC to give the title compound as a white solid (TFA salt, 88 mg, 51 %). 1H NMR ( Acetone-^, 400 MHz) 2.99-3.21 (m, 2H), 3.22-3.45 (m, 2H), 3.66 (t, J= 4.80 Hz, 2H), 3.75 (s, 3H), 3.85 (s, 3H), 3.79-3.89 (m, 4H), 4.58 (t, J= 4.80 Hz, 2H), 6.29 (d, J= 2.02 Hz IH), 7.13 (dd, J= 8.34, 2.53 Hz, IH), 7.22 (d, J= 8.84 Hz, IH), 7.42 (t, J= 7.83 Hz, IH), 7.47 (d, J= 1.77 Hz, IH), 7.52 (t, J= 1.77 Hz, IH), 7.56 (d, J= 7.07 Hz, IH), 7.80-7.83 (m, IH), 7.91-7.96 (m, IH), 9.54 (s, NH). Exact mass calculated for C24H28N4O4 436.2, found 437.5 (MH+).
References
1: Xiong Y, Teegarden BR, Choi JS, Strah-Pleynet S, Decaire M, Jayakumar H, Dosa
PI, Casper MD, Pham L, Feichtinger K, Ullman B, Adams J, Yuskin D, Frazer J,
Morgan M, Sadeque A, Chen W, Webb RR, Connolly DT, Semple G, Al-Shamma H.
Discovery and structure-activity relationship of
3-methoxy-N-(3-(1-methyl-1H-pyrazol-5-yl)-4-(2-morpholinoethoxy)phenyl)benzamide
(APD791): a highly selective 5-hydroxytryptamine2A receptor inverse agonist for
the treatment of arterial thrombosis. J Med Chem. 2010 Jun 10;53(11):4412-21.
doi: 10.1021/jm100044a. PubMed PMID: 20455563.
2: Przyklenk K, Frelinger AL 3rd, Linden MD, Whittaker P, Li Y, Barnard MR, Adams
J, Morgan M, Al-Shamma H, Michelson AD. Targeted inhibition of the serotonin
5HT2A receptor improves coronary patency in an in vivo model of recurrent
thrombosis. J Thromb Haemost. 2010 Feb;8(2):331-40. doi:
10.1111/j.1538-7836.2009.03693.x. Epub 2009 Nov 17. PubMed PMID: 19922435; PubMed
Central PMCID: PMC2916638.
3: Adams JW, Ramirez J, Shi Y, Thomsen W, Frazer J, Morgan M, Edwards JE, Chen W,
Teegarden BR, Xiong Y, Al-Shamma H, Behan DP, Connolly DT. APD791,
3-methoxy-n-(3-(1-methyl-1h-pyrazol-5-yl)-4-(2-morpholinoethoxy)phenyl)benzamide,
a novel 5-hydroxytryptamine 2A receptor antagonist: pharmacological profile,
pharmacokinetics, platelet activity and vascular biology. J Pharmacol Exp Ther.
2009 Oct;331(1):96-103. doi: 10.1124/jpet.109.153189. Epub 2009 Jul 23. PubMed
PMID: 19628629.
CRYSTALLINE FORMS OF CERTAIN 3-PHENYL-PYRAZOLE DERIVATIVES AS MODULATORS OF THE 5-HT2A SEROTONIN RECEPTOR USEFUL FOR THE TREATMENT OF DISORDERS RELATED THERETO

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....