
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 146)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Varenicline (Chantix™) バレニクリン酒石酸塩

Varenicline (Chantix™)
Varenicline
- MF C13H13N3
- MW 211.26
(1R,12S)-5,8,14-Triazatétracyclo[10.3.1.02,11.04,9]hexadéca-2,4,6,8,10-pentaène [French] [ACD/IUPAC Name]
6,10-Methano-6H-azepino[4,5-g]quinoxaline, 7,8,9,10-tetrahydro-, (6R,10S)- [ACD/Index Name]
Champix
(1R,12S)-5,8,14-triazatetracyclo[10.3.1.02,11.04,9]hexadeca-2(11),3,5,7,9-pentaene
CP-526,555
MFCD08460603
MFCD10001497
UNII:W6HS99O8ZO
APPROVALS
FDA MAY 10, 2006
EMA SEPT 2006
PMDA JAPAN JAN 25 2008
Varenicline (trade name Chantix and Champix usually in the form of varenicline tartrate), is a prescription medication used to treatnicotine addiction. Varenicline is a nicotinic receptor partial agonist—it stimulates nicotine receptors more weakly than nicotine itself does. In this respect it is similar to cytisine and different from the nicotinic antagonist, bupropion, and nicotine replacement therapies(NRTs) like nicotine patches and nicotine gum. As a partial agonist it both reduces cravings for and decreases the pleasurable effects of cigarettes and other tobacco products. Through these mechanisms it can assist some patients to quit smoking.


Varenicline
CAS Registry Number: 249296-44-4
CAS Name: 7,8,9,10-Tetrahydro-6,10-methano-6H-pyrazino[2,3-h][3]benzazepine
Additional Names: 5,8,14-triazatetracyclo[10.3.1.02,11.04,9]hexadeca-2(11)-3,5,7,9-pentaene
Manufacturers’ Codes: CP-526555
Molecular Formula: C13H13N3
Molecular Weight: 211.26
Percent Composition: C 73.91%, H 6.20%, N 19.89%
Literature References: Nicotinic a4b2 acetylcholine receptor partial agonist. Prepn: P. R. P. Brooks, J. W. Coe, WO 0162736(2001 to Pfizer). Synthesis, receptor binding studies, and in vivo dopaminergic acitvity: J. W. Coe et al., J. Med. Chem. 48, 3474 (2005). Metabolism: R. S. Obach et al., Drug Metab. Dispos. 34, 121 (2006).

Derivative Type: Tartrate
CAS Registry Number: 375815-87-5
Trademarks: Champix (Pfizer)
Molecular Formula: C13H13N3.C4H6O6
Molecular Weight: 361.35
Percent Composition: C 56.51%, H 5.30%, N 11.63%, O 26.57%
Therap-Cat: Aid in smoking cessation.
バレニクリン酒石酸塩
Varenicline Tartrate

C13H13N3 C4H6O6 : 361.35
C4H6O6 : 361.35
[375815-87-5]
Varenicline Tartrate
C13H13N3
C4H6O6 : 361.35[375815-87-5]
Medical uses
Varenicline is used for smoking cessation. In a 2009 meta-analysis varenicline was found to be more effective than bupropion (odds ratio 1.40) and NRTs (odds ratio 1.56).[1]
A 2013 Cochrane overview and network meta-analysis concluded that varenicline is the most effective medication for tobacco cessation and that smokers were nearly three times more likely to quit on varenicline than with placebo treatment. Varenicline was more efficacious than bupropion or NRT and as effective as combination NRT for tobacco smoking cessation.[2][3]
The United States’ Food and Drug Administration (US FDA) has approved the use of varenicline for up to twelve weeks. If smoking cessation has been achieved it may be continued for another twelve weeks.[4]
Varenicline has not been tested in those under 18 years old or pregnant women and therefore is not recommended for use by these groups. Varenicline is considered a class C pregnancy drug, as animal studies have shown no increased risk of congenital anomalies, however, no data from human studies is available.[5] An observational study is currently being conducted assessing for malformations related to varenicline exposure, but has no results yet.[6] An alternate drug is preferred for smoking cessation during breastfeeding due to lack of information and based on the animal studies on nicotine.[7]

Varenicline L-tartrate (Compound I) is the international commonly accepted name for 7,8,9,10- tetrahydro-6, 10-methano-6i7-pyrazino [2, 3- h] [3 ] benzazepme, (2R, 3R) -2 , 3-dihydroxybutanedioate (1:1) (which is also known as 5,8,14- tπazatetracyclo [10.3.1. O2‘11. O4‘9] -hexadeca-2 (11) , 3, 5, 7, 9-pentaene, (2R, 3R)-2,3- dihydroxybutanedioate (1:1)) and has an empirical formula of C13H13N3 • C4H6O6 and a molecular weight of 361.35. Varenicline L-tartrate is a commercially marketed pharmaceutically active substance known to be useful for the treatment of smoking addiction.

(D
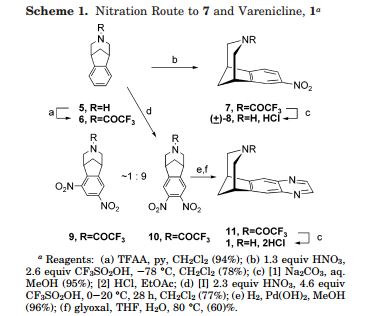
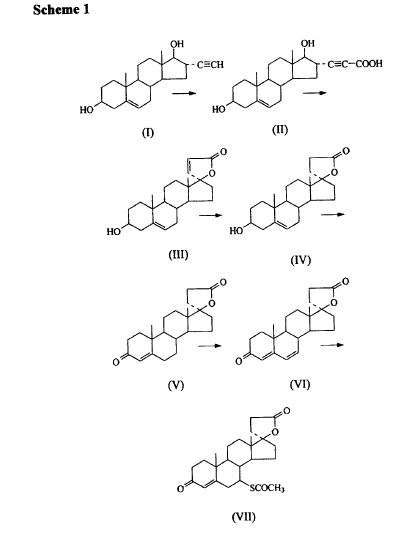
Varenicline L-tartrate is a partial agonist selective for (X4β2 nicotinic acetylcholine receptor subtypes. In the United States, varenicline L-tartrate is marketed under the name Chantix™ for the treatment of smoking cessation. Varenicline base and its pharmaceutically acceptable acid addition salts are described in U.S. Patent No. 6,410,550. In particular, Example 26 of U.S. Patent No. 6,410,550 describes the preparation of varenicline hydrochloride salt using 1- (4 , 5-dinitro-10- aza-tπcyclo [6.3.1.O2‘7] dodeca-2, 4, 6-trien-10-yl) -2,2,2- tπfluoroethanone (compound of formula (III)) as starting compound. On the other hand, Example HA) of U.S. Patent No. 6,410,550 illustrates the preparation of compound of formula (III) via nitration of compound of formula (II) using an excess of nitronium triflate (>4 equiv) as a nitrating agent. The process disclosed in U.S. Patent No. 6,410,550 is depicted in Scheme 1.

VareniclineΗCl
Scheme 1
However, Coe et al., J. Med. Chem., 48, 3474 (2005), describes the same process and examples as U.S. Patent No. 6,410,550, and it also reveals that this process affords intermediate ortho-4 , 5-dinitrocompound of formula (III) together with the meta-3, 5-dinitro- isomer (i.e. the meta-dinitrocompound) in a ratio 9:1. The presence of the meta-dinitrocompound may affect not only the purity of the intermediate compound of formula III but it may also have an effect on the purity of the final varenicline tartrate, given that it can be carried along the synthetic pathway and/or it can also give rise to other derivative impurities. Thereby, as well as in U.S. Patent No. 6,410,550, in order to isolate pure compound of formula (III) , the raw product is triturated with ethyl acetate/hexane to afford compound of formula (III) with 77% yield. Additionally, the mother liquor is purified by chromatography on silica gel to improve the yield to a total of 82.8%. However, this process is not desirable for industrial implementation since it requires extensive and complicated purification procedures, i.e. trituration of the solid product along with column chromatography purification of the mother liquor, which is not very efficient or suitable for industrial scale-up.
Several improved processes for the synthesis of varenicline or its salts have been reported in the literature (e.g. WO2006/090236) . However, none of these processes tackle the optimization of the purification step of compound of formula (III).
There is therefore the need for providing an improved process for the preparation of varenicline L- tartrate which involves simple experimental procedures well suited to industrial production, which avoids the use of column chromatography purifications, and which affords high pure varenicline L-tartrate which hence can be used directly as a starting product for the preparation of the marketed pharmaceutical speciality.
Additionally, it has been observed that varenicline L-tartrate is usually obtained as a yellow solid under – A –
standard synthetic conditions. In this regard, colour must be attributed to the presence of some specific impurities that may or may not be detectable by conventional methods such as HPLC. The presence of impurities may adversely affect the safety and shelf life of formulations. In this connection, International application No. WO2006/090236 describes the isolation of vareniclme L- tartrate as a white solid. However, in order to remove coloured impurities, the varenicline L-tartrate obtained in WO2006/090236 is treated with a particular activated carbon having a specific grade (i.e. Darco KB-B™) . In fact, Example 5 of WO2006/090236 describes a large reprocessing step which comprises: dissolving varenicline L-tartrate in water, adding toluene, basifying with NaOH aqueous solution, collecting the toluene phase containing varenicline free base, distilling, adding methanol, azeotropically distilling the mixture, and adding more methanol to obtain a methanolic solution containing varenicline free base, adding Darco KB-B™ (10% w/w) , stirring for one hour, filtering through a pad of celite, and treating with L-tartaric acid to give varenicline L- tartrate salt as a white solid. Further, WO2006/090236 provides the absorbance at 430 nm of a varenicline L- tartrate salt solution, either in dichloromethane or in toluene, with or without using Darco KB-B™ activated carbon. However, this measure cannot be used to corroborate the whiteness of the solid varenicline L- tartrate. In addition, Example 3 of International application No. WO2002/092089, also disclose the preparation of varenicline L-tartrate polymorphic form C (i.e. a hydrate polymorph) as a white precipitate. Therefore, there is also a need for a simple and efficient method for preparing varenicline L-tartrate with enhanced whiteness and having a high purity.
SYNTHESIS



Synthesis of Intermediate VIII
Paper
J. Med. Chem. 48, 3474 (2005).
http://pubs.acs.org/doi/pdf/10.1021/jm050069n
PATENT
https://www.google.com/patents/WO2001062736A1?cl=en
CLIP
Profiles of Drug Substances, Excipients and Related Methodology, Volume 37
edited by Harry G. Brittain



SYNTHESIS

|
DOI: 10.1021/jm00190a020
|
|
| DOI: 10.1021/jm050069n |
CLIP
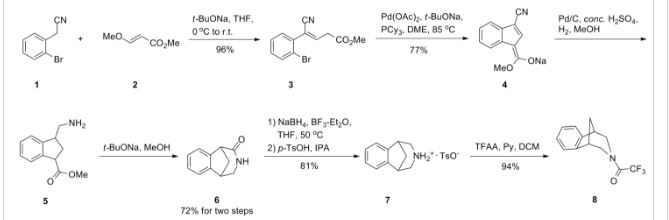
Scheme (I) compound patent US6410550B1 is provided adjacent difluorobromobenzene as raw materials by DA reaction, oxidation, cyclization, debenzylation get varenicline intermediate (II). The synthesis route is as follows:

CLIP
Patent CN101693712A mainly given varenicline intermediate (II) The preparation process is different from the compound patented. After the five-step method patents cited compounds. The entire route is longer, while using a large number of precious metal catalysts and reaction conditions need very strict control, inappropriate EVAL industry production.

CLIP
PATENT
A varenicline intermediate 2,3, 4, 5-tetrahydro-1,5-methylene bridge synthesis -1H-3- benzazepine hydrochloride, which comprises the following Step: (1) 2-indanone of formula 3 and the compound and paraformaldehyde under alkaline or acidic conditions Mannich reaction, as shown in general formula 2 intermediate; (2) the step (I) obtained through reaction of Formula 2 intermediate under basic or acidic conditions by reducing the role of the carbonyl group is reduced to a methylene group, and get varenicline intermediate (II) by debenzylation, the reaction is:

Wherein, R groups are selected from _H, _Me, _Et, _iPr> _t_Bu.
Figure 2;

Wherein, R group is -H, -Me, -Et, -iPr or -t_Bu.
(2) Step (I) obtained by the reaction intermediates of formula under basic or acidic conditions by reducing the role of the carbonyl group is reduced 2 methylene, and get by debenzylation cutting Lenk Lin intermediate (II);

CLIP
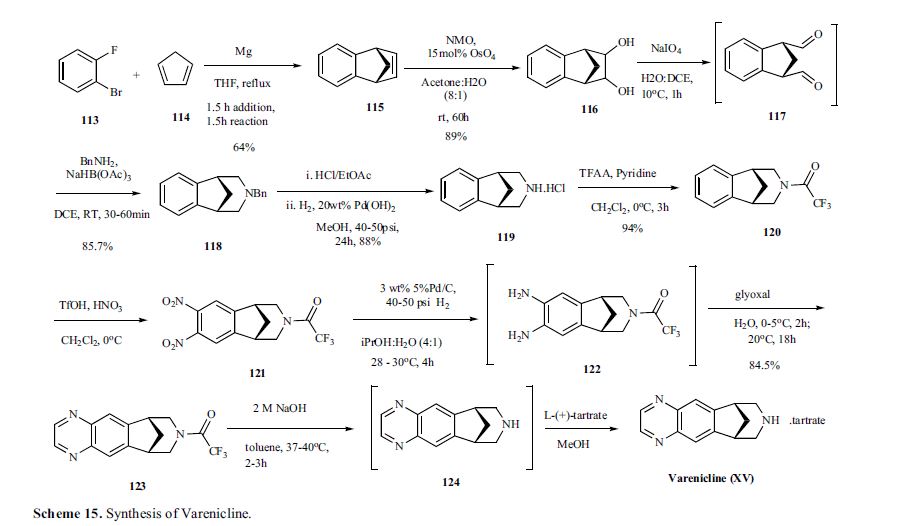
Varenicline, a nicotinic 42 partial agonist, was approved in the US for the treatment of smoking cessation in May of 2006. It was developed and marketed by Pfizer as a treatment for cigarette smokers who want to quit. Varenicline partially activates the nicotinic receptors and thus reduces the craving for cigarette that smokers feel when they try to quit smoking. By mitigating this craving and antagonizing nicotine activity without other symptoms, this novel drug helps quitting this dangerous addiction easier on the patients [6,52]. Several modifications [54,55] to the original synthesis [53,56] have been reported in the literature, including an improved process scale synthesis of the last few steps (Scheme 15) [57]. The Grignard reaction was initiated on a small scale by addition of 2-bromo fluorobenzene 113 to a slurry of Magnesium turnings and catalytic 1,2-dibromoethane in THF and heating the mixture until refluxing in maintained. To this refluxing mixture was added a mixture of the 2-bromo fluorobenzene 113 and cyclopentadiene 114 over a period of 1.5 h. After complete addition, the reaction was allowed to reflux for additional 1.5 h to give the Diels- Alder product 115 in 64% yield. Dihydroxylation of the olefin 115 by reacting with catalytic osmium tetraoxide in the presence of N-methylmorpholine N-oxide (NMO) in acetone: water mixture at room temperature provided the diol 116 in 89% yield. Oxidative cleavage of diol 116 with sodium periodate in biphasic mixture of water: DCE at 10ºC provided di-aldehyde 117 which was immediately reacted with benzyl amine in the presence of sodium acetoxyborohydride to give benzyl amine 118 in 85.7% yield. The removal of the benzyl group was effected by hydrogenation of the HCl salt in 40-50 psi hydrogen pressure with 20% Pd(OH)2 in methanol to give amine hydrochloride 119 in 88% yield. Treatment of amine 119 with trifluoroacetic anhydride and pyridine in dichloromethane at 0ºC gave trifluoroacetamide 120 in 94% yield. Dinitro compound 121 was prepared by addition of trifluoroacetamide 120 to a mixture of trifluoromethane sulfonic acid and nitric acid, which was premixed, in dichloromethane at 0ºC. Reduction of the dinitro compound 121 by hydrogenation at 40-50 psi hydrogen in the presence of catalytic 5%Pd/C in isopropanol:water mixture provided the diamine intermediate 122 which was quickly reacted with glyoxal in water at room temperature for 18h to give compound 123 in 85% overall yield. The trifluoroacetamide 123 was then hydrolyzed with 2 M sodium hydroxide in toluene at 37-40ºC for 2-3h followed by preparation of tartrate salt in methanol to furnish varenicline tartrate (XV).
[52]Keating, G.; Siddiqui, M. A. A. CNSdrugs, 2006, 11, 946.
[53] Coe, J. W.; Brooks, P. R.; Vetelino, M. G.; Wirtz, M. C.; Arnold,E. P. ; Huang, J.; Sands, S. B.; Davis, T. I.; Lebel, L. A.; Fox, C.
B.; Shrikhande, A.; Heym, J. H.; Schaeffer, E.; Rollema, H.; Lu,Y.; Mansbach, R. S.; Chambers, L. K.; Rovetti, C. C.; Schulz, D.
W.; Tingley, III, F. D.; O’Neill, B. T. J. Med. Chem., 2005, 48,3474.
[54] Brooks, P. R.; Caron, S.; Coe, J. W.; Ng, K. K.; Singer, R. A.;Vazquez, E.; Vetelino, M. G.; Watson, Jr. H. H.; Whritenour, D.
C.; Wirtz, M. C. Synthesis, 2004, 11, 1755.
[55] Singer, R. A.; McKinley, J. D.; Barbe, G.; Farlow, R. A. Org. Lett.,2004, 6, 2357.
[56] Coe, J. W.; Brooks, P. R. P. US-6410550 B1, 2002.
[57] Busch, F. R.; Hawkins, J. M.; Mustakis, L. G.; Sinay, T. G., Jr.;Watson, T. J. N.; Withbroe, G. J. WO-2006090236 A1, 2006.
PATENT
WO 2002085843
https://google.com/patents/WO2002085843A2?cl=en
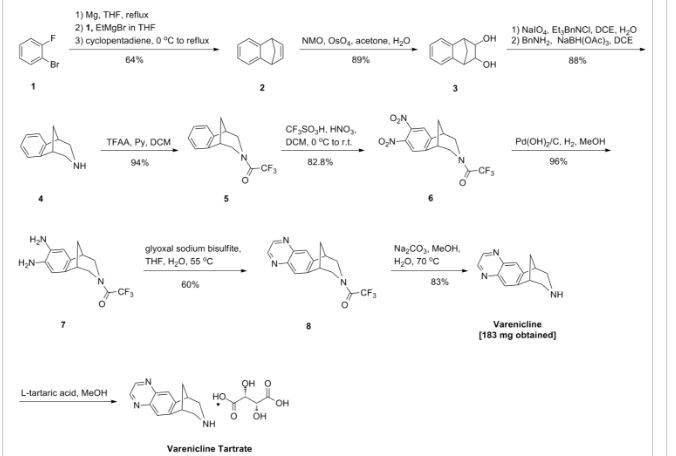
PATENT
https://www.google.com/patents/EP2204369A1?cl=en
Varenicline (a compound I of formula I) is the international commonly accepted non-proprietary name for 7,8,9,10-tetrahydro-6,10-methano-6H-pyrazino[2,3-h][3]benzazepine (which is also known as 5,8,14-triazatetracyclo[10.3.1.02,11.04,9]-hexadeca-2(11),3,5,7,9-pentaene), and has an empirical formula of C13H13N3 and a molecular weight of 211.26.

The L-tartrate salt of varenicline is known to be therapeutically useful and is commercially marketed for the treatment of smoking addiction. Varenicline L-tartrate is a partial agonist selective for α4β2 nicotinic acetylcholine receptor subtypes. In the United States, varenicline L-tartrate is marketed under the trade mark Chantix and is indicated as an aid to smoking cessation treatment.
Varenicline base and its pharmaceutically acceptable acid addition salts are described in U.S. Patent No. 6,410,550 . In particular, the preparation of varenicline provided in this reference makes use of 10-aza-tricyclo[6.3.1.02,7]-dodeca-2(7),3,5-triene (a compound of Formula VI), as a key intermediate compound (see Scheme 1 below). Specifically, Example 1 of U.S. Patent No. 6,410,550 describes the synthetic preparation of key intermediate compound of Formula VI as depicted in Scheme 1.

1,2,3,4-tetrahydro-1,4-methano-naphthalene-cis-2,3-diol (a compound of Formula III), and / or indane-1,3-dicarbaldehyde (a compound of Formula IV).
Example 1: Preparation of 1,2,3,4-tetrahydro-1,4-methano-naphthalene-cis-2,3-diol (a compound of Formula III)
A 10mL round bottom flask was charged with a compound of formula II (142mg, 1mmol), N-methylmorpholine-N-oxide (120mg, 1.03mmol), tert-butanol (3mL) and water (1mL). FibreCat™ 3003 (OsO4 anchored onto a polymeric support) (11.6mg, 0.0025mmol) was added to this solution and the mixture was heated to reflux. Complete conversion to a compound of formula III was detected by GC, method A, after 48h.
Example 2: Preparation of 1,2,3,4-tetrahydro-1,4-methano-naphthalene-cis-2,3-diol (a compound of Formula III)Step A) Preparation of hexadecyl-trimethylammoniumpermanganate (HTAP):
HTAP was prepared from ion exchange reaction between hexadecyltrimethylammoniumbromide and potassium permanganate.
Potassium permanganate (17.38g, 0.11mol, 1equiv.) was dissolved in 500mL water. A solution of hexadecyltrimethylammoniumbromide (40.10g, 0.11mol, 1equiv) in 500mL water was added drop-wise over 45 min at 20-22°C, and the mixture stirred for 30 minutes at this temperature. The precipitated solid was collected by filtration, washed with water (3 x 100mL) and dried under vacuum at 35°C for 24 hours to give 34.38g of HTAP as a light purple solid.
Step B) Preparation of a compound of formula III:
Compound II (3.52g, 24.8mmol, 1equiv.) was dissolved in anhydrous tetrahydrofuran (80mL) and a solution of HTAP (10g, 24.8mmol, 1.0equiv.) in anhydrous tetrahydrofuran (125mL) was added drop-wise at 23-30°C over 45min. The reaction was monitored by TLC (hexane-ethyl acetate = 1:1). After complete reaction the mixture was cooled to below 10°C, and methyl tert-butyl ether (50mL) and 5% aqueous NaOH solution (50mL) were added and the mixture stirred for 30min. The solid was removed by filtration, and washed with methyl tert-butyl ether (2 x 30mL). The combined layers of the filtrate were separated and the aqueous phase extracted with methyl tert-butyl ether (2 x 30mL). The organic layers were combined and washed with 5% aqueous NaOH solution (50mL), water (2 x 50mL), dried over MgSO4, filtered and concentrated to obtain a dark green solid. This residue was suspended in acetone (15mL) and collected by filtration, washing with additional acetone (3 x 5mL). The product was dried under vacuum at 40°C to give 2.215g (50.7% yield) as a white crystalline solid.
Analytical data: m.p. = 178.8-179.3°C; 1H-NMR: See Figure 1; 13C-NMR: See Figure 2.
Example 3: Preparation of indane-1,3-dicarbaldehyde (a compound of Formula IV)
A 25 mL round bottom flask was charged with a compound of formula I (142mg, 1mmol), Ruthenium (III) chloride hydrate (Aldrich, Reagent Plus™) (7.2mg, 0.035mmol), acetonitrile (8.5mL) and water (1.1mL). The solution was heated to 45°C and sodium periodate (449mg, 2.1mmol) was added portionwise over 25 minutes. After 1h, the reaction was cooled to ambient temperature and filtered. The solids were washed with ethyl acetate (3 x 2mL) and water (3mL). The filtrate was concentrated under vacuum and 5mL of water were added to the obtained residue. The mixture was extracted with ethyl acetate (2 x 5mL) and the combination of the organic layers was washed with water (3 x 5mL), dried with MgSO4 and concentrated under vacuum to obtain a compound of formula IV (118mg) in 68% yield, 70.9% purity (analyzed by GC, method A).
PATENT
WO 199935131, WO 2002092089, US 2013030179
PATENT
https://www.google.com/patents/WO2009065872A2?cl=en
Example 1: Preparation of 7,8,9,10- tetrahydro-6, 10-methano-6H-pyrazino [2, 3-h] [3] benzazepine L-tartrate (i.e. varenicline L-tartrate)
A) Preparation of compound of formula (III)
This example is based on U.S. Patent No. 6,410,550.
A 250 mL round bottom flask with thermometer, condenser, addition funnel and magnetic stirring was charged with 10-aza-tricyclo [ 6.3.1. O2‘7] dodeca-2, 4, 6- triene para-toluene sulfonic acid salt (12.4g, 37.5 mmol) and 44 mL of CH2Cl2. Triethylamine (8.3 g, 82.5 mmol) was added to the slurry and the resulting solution was cooled to 0-5 0C. The addition funnel was charged with a solution of (CF3CO)2O (8.1q, 41.25 mmol) in 19 mL of CH2Cl2. This solution was slowly added to the reaction mixture, maintaining the temperature < 15 0C. The resulting mixture was stirred for 1 hour, and the complete conversion was monitored by GC. The crude reaction mixture was washed with water (2 * 40 mL) and brine (40 mL) . The organic phase was used in the next step without further purification.
On the other hand, a 500 mL round bottom flask with thermometer, condenser, addition funnel and magnetic stirring was charged with CF3SO3H (25.9 g, 172.5 mmol), CH2Cl2 (110 mL) and cooled to 0-5 0C. At this temperature, fuming nitric acid (5.4 g, 86.25 mmol) was added slowly. To the resulting slurry at 0-5 0C, the solution obtained in the previous step was slowly added, maintaining the temperature < 15 0C. After the addition, the reaction mixture was stirred overnight. The complete dinitration was confirmed by GC. The crude reaction mixture was poured into water (60 mL) an ice (80 g) and stirred. The phases were separated and the aqueous phase was extracted with CH2Cl2 (3 x 50 mL) . The mixture of the organic phases was washed with aqueous saturated NaHCO3, dried over Na2SO4 and volatiles evaporated under vacuum to obtain 11.9 g of a solid that was suspended and stirred for 2 hours in AcOEt (12 mL) and hexanes (24 mL) . The solid was filtered and washed with hexanes to obtain the compound of formula (III), 9.1g with a purity of 88.9% by GC (9.8% of meta-dimtrocompound impurity) .
B) Preparation of compound of formula (IV)
This example is based on International Patent No. WO/2006/090236.
A 200 mL autoclave was charged with (III) (9.1 g, 26.3 mmol), damp 5% Pd/C 50% and 180 mL of a 2- propanol/water (80/20 wt/wt) . The reaction was stirred under 50 psi of hydrogen for 18 hours. The complete hydrogenation was confirmed by GC analysis. The reaction was filtered through Celite and washed with 2-propanol (40 mL) . To this solution, K2HPO4(458 mg, 2.63 mmol) was added. The mixture was cooled at 0-5 0C and a solution of 4.07 g of 40% aqueous glyoxal diluted with water (14.5 mL) was added slowly. The resulting solution was stirred 2 hours at this temperature and overnight at room temperature. The complete conversion was confirmed by GC analysis. The reaction was concentrated under vacuum to a volume of 68 mL and water (128 mL) was added drop- wise. The resulting suspension was stirred for 2 hours at room temperature, 1 hour in a ice/water bath, filtered, washed with water (20 mL) and dried m a oven at 50 0C to obtain the compound of formula (IV), 6.78 g.
C) Preparation of vareniclme L-tartrate (compound of formula (I) )
This example is based on International Patent No. WO/2006/090236.
A 250 mL round bottom flask with thermometer, condenser, and magnetic stirring was charged with compound of formula (IV) (6.78 g, 22 mmol) and toluene
(47 mL) . To this solution was added a solution of NaOH (2.7 g, 68.2 mmol) in water (34 mL) . The mixture was heated to 400C and stirred for 4 hours. The complete hydrolysis was confirmed by GC analysis. Toluene (68 mL) was added and the reaction was cooled. The phases were separated and the aqueous phase was extracted with toluene (30 mL) . The organic phases were evaporated under vacuum. The residue was dissolved in MeOH (90 mL) and evaporated again. The final residue was dissolved in 156 mL of MeOH. 1.3 g of activated carbon “Darco G-60 100 mesh” were added and the mixture was stirred for 30 min and filtered through Celite to obtain an intense yellow solution. The process with activated carbon was repeated without any improvement in the colour. This solution was added drop-wise over a solution of L- tartaric acid (3.63 g, 24.2 mmol) in MeOH (47 mL) . The slurry was stirred for 72 hours at room temperature, filtered, washed with MeOH and dried in an oven at 50 0C for 8 hours, to obtain 5.05 g of varenicline L-tartrate as a yellow solid with a 95.5% purity by HPLC (4.4% of unknown impurity A). Colour L: 92.75, a*: -7.19, b*:43.08.
Comparative Example 2: Preparation of 7,8,9,10- tetrahydro-6, 10-methano-6H-pyrazmo [2, 3-h] [3 ] benzazepine L-tartrate (i.e. varenicline L-tartrate) A) Preparation of compound of formula (IV)
This example is based on International Patent No. WO/2006/090236.
A 200 mL autoclave was charged with (III) prepared according to Comparative Example 1.A) (4.1 g) , 123 mg of damp 5% Pd/C 50% and 81 mL of a 2-propanol/water (80/20 wt/wt) . The reaction was stirred under 50 psi of hydrogen for 24 hours. The complete hydrogenation was confirmed by GC analysis. The reaction was filtered through Celite and washed with 2-propanol (16 mL) . To this solution, K2HPO4 (207 mg, 1.19 mmol) was added. The mixture was cooled at 0-5 0C and a solution of 1.84 g of 40% aqueous glyoxal diluted with water (6.6 mL) was added slowly. The resulting solution was stirred 2 hours at this temperature and overnight at room temperature. The complete conversion was confirmed by GC analysis. The reaction was concentrated under vacuum to a volume of 30 mL and water (56 mL) was added drop-wise. The resulting suspension was stirred for 2 hours at room temperature, 1 hour in a ice/water bath, filtered, washed with water and dried in a oven at 50 0C to obtain 3.15 g of compound of formula (IV) .
B) Preparation of vareniclme L-tartrate (compound of formula (I) )
This example is based on International application No. WO/2006/090236. A 100 mL round bottom flask with thermometer, condenser, and magnetic stirring was charged with
7, 8, 9, 10-tetrahydro-8- (tπfluoroacetyl) -6, 10-methano-6H- pyrazino [2 , 3-h] [3] benzazepine, i.e. compound of formula
(IV) (3.14 g, 10.2 mmol) and toluene (22 mL) . To this solution was added a solution of NaOH (1.3 g, 31.6 mmol) in water (16 mL) . The mixture was heated to 40 0C and stirred for 2.5 hours. The complete hydrolysis was confirmed by GC analysis. Toluene (30 mL) was added and the reaction was cooled. The phases were separated and the aqueous phase was extracted with toluene (15 mL) . The organic phases were evaporated under vacuum. The residue was dissolved in MeOH (45 mL) and evaporated again. The final residue was dissolved m 70 mL of MeOH. 314 mg of activated carbon “Darco G-60 100 mesh” were added and the mixture was stirred for 30 mm and filtered through Celite to obtain a yellow solution. This solution was added drop-wise over a solution of L- tartaπc acid (1.68 g, 11.22 mmol) m MeOH (22 mL) . The slurry was stirred for 1 hour at room temperature, filtered, washed with MeOH (2 x 5 mL) and dried under vacuum, to obtain vareniclme L-tartrate (2.48 g) as a yellow solid with a 95.6% purity by HPLC (4.4% of unknown impurity A). Colour L: 99.50, a*: -4.98, b*:43.02
Comparative Example 3: Preparation of 7,8,9,10- tetrahydro-6, 10-methano-6H-pyrazino [2, 3-h] [3 ] benzazepine L-tartrate (i.e. vareniclme L-tartrate)
This example is based on International application No. WO/2002/092089.
2 g of vareniclme L-tartrate as obtained from Comparative Example 1 were dissolved in 3 mL of water.
To this solution, 100 mL of CH3CN were added, and the resulting slurry was stirred for 10 mm and filtered.
After drying the product was analysed to be a 98.2% purity by HPLC (1.7% of unknown impurity A) . Colour L: 91.44, a*: -3.24, b* : 33.47
Example 1: Preparation of 7, 8, 9, lO-tetrahydro-6, 10- methano-6H-pyrazmo [2, 3-h] [3] benzazepine L-tartrate
(i.e. vareniclme L-tartrate)
A) Preparation of compound of formula (III) This example is based on U.S. Patent No. 6,410,550, except for the purification step, which is the object of the present invention (i.e. crystallization in toluene) .
A 500 mL round bottom flask with thermometer, condenser, addition funnel and magnetic stirring was charged with 10-aza-tricyclo [ 6.3.1. O2‘7] dodeca-2, 4, 6- tπene para-toluene sulfonic acid salt (32.5g, 98.2 mmol) and 115 mL of CH2Cl2. Triethylamine (21.8 g, 216 mmol) was added to the slurry and the resulting solution was cooled to 0-5 0C. The addition funnel was charged with a solution of (CF3CO)2O (22.7 g, 108 mmol) in 50 mL of CH2Cl2. This solution was slowly added to the reaction mixture, maintaining the temperature < 15 0C. The resulting mixture was stirred for 1 hour, and the complete conversion was monitored by GC. The crude reaction mixture was washed with water (2 x 100 mL) and brine (100 mL) . The organic phase was used in the next step without further purification.
A l L round bottom flask with thermometer, condenser, addition funnel and magnetic stirring was charged with CF3SO3H (67.8 g, 452 mmol), CH2Cl2 (280 mL) and cooled to 0-5 0C. At this temperature, fuming nitric acid (14.2 g, 226 mmol) was slowly added. To the resulting slurry at 0-5 0C, the solution obtained in the previous step was slowly added, maintaining the temperature < 15 0C. After the addition, the reaction mixture was stirred overnight. The complete dinitration was confirmed by GC. The crude reaction mixture was poured into water (150 mL) an ice (200 g) and stirred. The phases were separated and the aqueous phase was extracted with CH2Cl2 (100 mL) . The mixture of the organic phases was washed with aqueous saturated NaHCO3 (2×100 mL) , water (100 mL) , dried over Na2SO4 and volatiles evaporated under vacuum to obtain 30.5 g of a solid with a 83.6% purity by GC (12.5% of meta- dinitrocompound impurity) . 20 g of this solid were crystallized in toluene (100 mL) to obtain the compound of formula (III), 15 g of a pale brown solid with a 98.5 % purity by GC (meta-dinitrocompound impurity not detected) .
B) Preparation of compound of formula (IV) This example is based on International Patent No. WO/2006/090236.
A 200 mL autoclave was charged with (III) (9.1 g, 26.3 mmol, crystals from toluene), damp 5% Pd/C 50% and 180 mL of a 2-propanol/water (80/20 wt/wt) . The reaction was stirred under 50 psi of hydrogen for 18 hours. The complete hydrogenation was confirmed by GC analysis. The reaction was filtered over Celite and washed with 2- propanol (40 mL) . To this solution, K2HPO4 (458 mg, 2.63 mmol) was added. The mixture was cooled at 0-5 0C and a solution of 4.07 g of 40% aqueous glyoxal diluted with water (14.5 mL) was added slowly. The resulting solution was stirred 2 hours at this temperature and overnight at room temperature. The complete conversion was confirmed by GC analysis. The reaction was concentrated under vacuum to a volume of 68 mL and water (128 mL) was added drop-wise. The resulting suspension was stirred for 2 hours at room temperature, 1 hour in a ice/water bath, filtered, washed with water (20 mL) and dried m a oven at 50 0C to obtain the product, 7.16 g of compound of formula (IV) with a 99.9% purity by HPLC. C) Preparation of varenicline L-tartrate (compound of formula ( I) )
Thrs example rs based on International Patent No. WO/2006/090236. A 250 mL round bottom flask with thermometer, condenser, and magnetic stirring was charged with a solution of NaOH (2.89 g, 72.23 mmol) in water (36 mL) , compound of formula (IV) (7.15 g, 23.3 mmol) and toluene (50 mL) . The mixture was heated to 40 0C and stirred for 4 hours. The complete hydrolysis was confirmed by GC analysis. Toluene (71 mL) was added and the reaction was cooled. The phases were separated and the aqueous phase was extracted with toluene (36 mL) . The organic phases were evaporated under vacuum. The residue was dissolved in MeOH (110 mL) and evaporated again. The final residue was dissolved in 164 mL of MeOH. 750 mg of activated carbon “Darco G-60 100 mesh” were added and the mixture was stirred for 30 min and filtered through Celite to obtain a yellow solution. This solution was added drop- wise over a solution of L-tartaric acid (3.84 g, 25.6 mmol) in MeOH (50 mL) . The slurry was stirred for 14 hours at room temperature, filtered, washed with MeOH and dried under vacuum, to obtain varenicline L-tartrate
(7.04 g) as an off-white solid with a >99.9% purity by HPLC (unknown impurity A not detected) . Colour L: 94.39, a*: 2.27, b*:9.02.

Post-marketing surveillance
No evidence for increased risks of cardiovascular events, depression, or self-harm with varenicline versus nicotine replacement therapy has been found in one post-marketing surveillance study.[23]
Mechanism of action
Varenicline displays full agonism on α7 nicotinic acetylcholine receptors.[24][25] And it is a partial agonist on the α4β2, α3β4, and α6β2 subtypes.[26] In addition, it is a weak agonist on the α3β2 containing receptors.
Varenicline’s partial agonism on the α4β2 receptors rather than nicotine’s full agonism produces less effect of dopamine release than nicotine’s. This α4β2 competitive binding, reduces the ability of nicotine to bind and stimulate the mesolimbic dopamine system – similar to the method of action of buprenorphine in the treatment of opioid addiction.[3]
Pharmacokinetics
Most of the active compound is excreted by the kidneys (92–93%). A small proportion is glucuronidated, oxidised, N-formylated or conjugated to a hexose.[27] The elimination half-life is about 24 hours.
History
Use of Cytisus plant as a smoking substitute during World War II[28] led to use as a cessation aid in eastern Europe and extraction of cytisine.[29] Cytisine analogs led to varenicline at Pfizer.[30][31][32]
Varenicline received a “priority review” by the US FDA in February 2006, shortening the usual 10-month review period to 6 months because of its demonstrated effectiveness inclinical trials and perceived lack of safety issues.[33] The agency’s approval of the drug came on May 11, 2006.[4] On August 1, 2006, varenicline was made available for sale in the United States and on September 29, 2006, was approved for sale in the European Union.[34]
SEE
Busch FR, Concannon PE, Handfield RE, McKinley JD, McMahon ME, Singer RA, Watson TJ, Withbroe GJ, Stivanello M, Leoni L, Bezze C. Synthesis of (1 (Aminomethyl)-2,3-dihydro-1H-inden-3-yl)methanol: Structural Confirmation of the Main Band Impurity Found in Varenicline® Starting Material.Synth Commun. 2008;38:441–447. http://dx.doi.org/10.1080/00397910701771231.
Varenicline standards and impurity controls. www.freepatentsonline.com/US2007/0224690.html.
N-formyl and N-methyl degradation products. www.freepatentsonline.com/y2004/0235850.html.
Methods of reducing degradant formation in pharmaceutical compositions of Varenicline.www.freepatentsonline.com/y2008/0026059.html.
Varenicline standards and impurity controls. www.freepatentsonline.com/EP2004186.html.
Satheesh B, Kumarpulluru S, Raghavan V, Saravanan D. UHPLC Separation and Quantification of Related Substances of Varenicline Tartrate Tablet. Acta Chromatogr. 2010;22:207–218.http://dx.doi.org/10.1556/AChrom.22.2010.2.4.

| US6410550 | Nov 13, 1998 | Jun 25, 2002 | Pfizer Inc | Aryl fused azapolycyclic compounds |
| WO2009155403A2 * | Jun 18, 2009 | Dec 23, 2009 | Teva Pharmaceutical Industries Ltd. | Processes for the preparation of varenicline and intermediates thereof |
| Reference | ||
|---|---|---|
| 1 | * | BHUSHAN, VIDYA; RATHORE, RAJENDRA; CHANDRASEKARAN, S.: “A Simple and Mild Method for the cis-Hydroxylation of Alkenes with Cetyltrimethylammonium Permanganate” SYNTHESIS, no. 5, 1984, pages 431-433, XP002581198 |
| 2 | * | BROOKS P R ET AL: “Synthesis of 2,3,4,5-tetrahydro-1,5-methano-1H-3-benzaz epine via oxidative cleavage and reductive amination strategies” SYNTHESIS 20040803 DE, no. 11, 3 August 2004 (2004-08-03), pages 1755-1758, XP002581197 ISSN: 0039-7881 |
| 3 | * | SORBERA L A ET AL: “Varenicline tartrate: Aid to smoking cessation nicotinic [alpha]4[beta]2 partial agonist” DRUGS OF THE FUTURE 200602 ES LNKD- DOI:10.1358/DOF.2006.031.02.964028, vol. 31, no. 2, February 2006 (2006-02), pages 117-122, XP002581199 ISSN: 0377-8282 DOI: 10.1358/dof.2006.031.02.964028 |
| WO2001062736A1 * | Feb 8, 2001 | Aug 30, 2001 | Pfizer Products Inc. | Aryl fused azapolycyclic compounds |
| WO2002085843A2 * | Mar 4, 2002 | Oct 31, 2002 | Pfizer Products Inc. | Process for the preparation of 1,3-substituted indenes and aryl-fused azapolycyclic compounds |
| WO2006090236A1 * | Feb 21, 2006 | Aug 31, 2006 | Pfizer Products Inc. | Preparation of high purity substituted quinoxaline |
| WO2008060487A2 * | Nov 9, 2007 | May 22, 2008 | Pfizer Products Inc. | Polymorphs of nicotinic intermediates |
| Reference | ||
|---|---|---|
| 1 | * | COE J W ET AL: “Varenicline: an alpha4beta2 Nicotinic Receptor Partial Agonist for Smoking Cessation” JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY, WASHINGTON., US, vol. 48, no. 10, 1 January 2005 (2005-01-01), pages 3474-3477, XP002474642 ISSN: 0022-2623 cited in the application |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2010005643A1 * | May 28, 2009 | Jan 14, 2010 | Teva Pharmaceutical Industries Ltd. | Processes for purifying varenicline l-tartrate salt and preparing crystalline forms of varenicline l-tartrate salt |
| WO2011110954A1 * | Mar 8, 2011 | Sep 15, 2011 | Actavis Group Ptc Ehf | Highly pure varenicline or a pharmaceutically acceptable salt thereof substantially free of methylvarenicline impurity |
| WO2011154586A3 * | Jun 13, 2011 | Mar 22, 2012 | Medichem, S. A. | Improved methods for the preparation of quinoxaline derivatives |
| EP2581375A2 * | Jun 13, 2011 | Apr 17, 2013 | Medichem, S.A. | Improved methods for the preparation of quinoxaline derivatives |
| US8039620 | May 21, 2009 | Oct 18, 2011 | Teva Pharmaceutical Industries Ltd. | Varenicline tosylate, an intermediate in the preparation process of varenicline L-tartrate |
| US8178537 | Jun 22, 2010 | May 15, 2012 | Teva Pharmaceutical Industries Ltd. | Solid state forms of varenicline salts and processes for preparation thereof |
References
- Jump up^ Mills EJ, Wu P, Spurden D, Ebbert JO, Wilson K (2009). “Efficacy of pharmacotherapies for short-term smoking abstinance: a systematic review and meta-analysis” (PDF). Harm Reduct J 6: 25. doi:10.1186/1477-7517-6-25. PMC 2760513. PMID 19761618.
- ^ Jump up to:a b Cahill K, Stevens S, Perera R, Lancaster T (May 2013). “Pharmacological interventions for smoking cessation: an overview and network meta-analysis”. Cochrane Database Syst Rev (Systematic Review & Meta-Analysis) 5: CD009329.doi:10.1002/14651858.CD009329.pub2. PMID 23728690.
- ^ Jump up to:a b c d Elrashidi MY, Ebbert JO (June 2014). “Emerging drugs for the treatment of tobacco dependence: 2014 update”. Expert Opin Emerg Drug (Review) 19 (2): 243–60.doi:10.1517/14728214.2014.899580. PMID 24654737.
- ^ Jump up to:a b U.S. Food and Drug Administration.FDA Approves Novel Medication for Smoking Cessation. Press release, 11 May 2006.
- Jump up^ Cressman, AM; Pupco, A; Kim, E; Koren, G; Bozzo, P (May 2012). “Smoking cessation therapy during pregnancy.”. Canadian Family Physician 58 (5): 525–7. PMC 3352787.PMID 22586193.
- Jump up^ “Varenicline Pregnancy Cohort Study”. clinicaltrials.gov.
- Jump up^ “LactMed”. nih.gov.
- Jump up^ Leung, LK; Patafio, FM; Rosser, WW (September 28, 2011). “Gastrointestinal adverse effects of varenicline at maintenance dose: a meta-analysis”. BMC clinical pharmacology11 (1): 15. doi:10.1186/1472-6904-11-15. PMC 3192741. PMID 21955317.
- American Cancer Society. “Cancer Drug Guide: Varenicline”. Retrieved 2008-01-19.
- Jump up^ “DailyMed – CHANTIX- varenicline tartrate”. nih.gov.
- FDA. “Public Health Advisory: FDA Requires New Boxed Warnings for the Smoking Cessation Drugs Chantix and Zyban”. Retrieved 2009-07-01.
- ^ Jump up to:a b “www.accessdata.fda.gov” (PDF).
- Hughes, JR (8 January 2015). “Varenicline as a Cause of Suicidal Outcomes.”. Nicotine & tobacco research : official journal of the Society for Research on Nicotine and Tobacco.doi:10.1093/ntr/ntu275. PMID 25572451.
- “FDA Drug Safety Communication: Chantix (varenicline) may increase the risk of certain cardiovascular adverse events in patients with cardiovascular disease”. 2011-06-16.
- Jump up^ Singh, S; Loke, YK, Spangler, JG, Furberg, CD (Sep 6, 2011). “Risk of serious adverse cardiovascular events associated with varenicline: a systematic review and meta-analysis” (PDF). CMAJ : Canadian Medical Association 183 (12): 1359–66.doi:10.1503/cmaj.110218. PMC 3168618. PMID 21727225.
- Takagi, H; Umemoto, T (Sep 6, 2011). “Varenicline: quantifying the risk”. CMAJ : Canadian Medical Association 183 (12): 1404. doi:10.1503/cmaj.111-2063.PMC 3168634. PMID 21896705.
- Jump up^ Samuels, L (Sep 6, 2011). “Varenicline: cardiovascular safety”. CMAJ : Canadian Medical Association 183 (12): 1407–08. doi:10.1503/cmaj.111-2073. PMC 3168639.PMID 21896709.
- “European Medicine Agency confirms positive benefit-risk balance for Champix.”. 2011-07-21.
- ^ Jump up to:a b Prochaska JJ, Hilton JF (2012). “Risk of cardiovascular serious adverse events associated with varenicline use for tobacco cessation: systematic review and meta-analysis”. BMJ (Systematic Review & Meta-Analysis) 344: e2856.doi:10.1136/bmj.e2856. PMC 3344735. PMID 22563098.
- Mills EJ, Thorlund K, Eapen S, Wu P, Prochaska JJ (January 2014). “Cardiovascular events associated with smoking cessation pharmacotherapies: a network meta-analysis”.Circulation (Network Meta-Analysis) 129 (1): 28–41.doi:10.1161/CIRCULATIONAHA.113.003961. PMID 24323793.
- cessation in cardiovascular patients”. Evidence-Based Medicine (Review & Commentary) 19 (5): 193. doi:10.1136/eb-2014-110030.PMID 24917603.
- Rowland K (April 2014). “ACP Journal Club. Review: Nicotine replacement therapy increases CVD events; bupropion and varenicline do not”. Annals of Internal Medicine(Review & Commentary) 160 (8): JC2. doi:10.7326/0003-4819-160-8-201404150-02002.PMID 24733219.
- Jump up^ Kotz D, Viechtbauer W, Simpson C, van Schayck OC, West R, Sheikh A (2015).“Cardiovascular and neuropsychiatric risks of varenicline: a retrospective cohort study”.Lancet Respir Med (retrospective cohort) 3: 761–768. doi:10.1016/S2213-2600(15)00320-3. PMC 4593936. PMID 26355008.
- Jump up^ Mihalak KB, Carroll FI, Luetje CW; Carroll; Luetje (2006). “Varenicline is a partial agonist at alpha4beta2 and a full agonist at alpha7 neuronal nicotinic receptors”. Mol. Pharmacol.70 (3): 801–805. doi:10.1124/mol.106.025130. PMID 16766716.
- Jump up^ Mineur YS, Picciotto MR; Picciotto (December 2010). “Nicotine receptors and depression: revisiting and revising the cholinergic hypothesis”. Trends Pharmacol. Sci. 31 (12): 580–6. doi:10.1016/j.tips.2010.09.004. PMC 2991594. PMID 20965579.
- Tanuja Bordia. “Varenicline Is a Potent Partial Agonist at α6β2* Nicotinic Acetylcholine Receptors in Rat and Monkey Striatum”. aspetjournals.org.
- Obach, RS; Reed-Hagen, AE; Krueger, SS; Obach, BJ; O’Connell, TN; Zandi, KS; Miller, S; Coe, JW (2006). “Metabolism and disposition of varenicline, a selective alpha4beta2 acetylcholine receptor partial agonist, in vivo and in vitro”. Drug metabolism and disposition: the biological fate of chemicals 34 (1): 121–130.doi:10.1124/dmd.105.006767. PMID 16221753.
- “[Cytisine as an aid for smoking cessation].”. Med Monatsschr Pharm 15 (1): 20–1. Jan 1992. PMID 1542278.
- Prochaska, BMJ 347:f5198 2013 http://www.bmj.com/content/347/bmj.f5198
- Coe JW, Brooks PR, Vetelino MG, Wirtz MC, Arnold EP, Huang J, Sands SB, Davis TI, Lebel LA, Fox CB, Shrikhande A, Heym JH, Schaeffer E, Rollema H, Lu Y, Mansbach RS, Chambers LK, Rovetti CC, Schulz DW, Tingley FD 3rd, O’Neill BT (2005). “Varenicline: an alpha4beta2 nicotinic receptor partial agonist for smoking cessation”. J. Med. Chem. 48(10): 3474–3477. doi:10.1021/jm050069n. PMID 15887955.
- Schwartz JL (1979). “Review and evaluation of methods of smoking cessation, 1969–77. Summary of a monograph”. Public Health Rep 94 (6): 558–63. PMC 1431736.PMID 515342.
- Etter JF (2006). “Cytisine for smoking cessation: a literature review and a meta-analysis”. Arch. Intern. Med. 166 (15): 1553–1559. doi:10.1001/archinte.166.15.1553.PMID 16908787.
- Kuehn BM (2006). “FDA speeds smoking cessation drug review”. JAMA 295 (6): 614–614.doi:10.1001/jama.295.6.614. PMID 16467225.
- European Medicines Agency (2011-01-28). “EPAR summary for the public. Champix varenicline”. London. Retrieved 2011-02-14.
External links
Manufacturer’s website USA

|
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
7,8,9,10-Tetrahydro-6,10-methano-6H-pyrazino[2,3-h] [3]benzazepine
|
|
| Clinical data | |
| Trade names | Chantix |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a606024 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
Oral |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Protein binding | <20% |
| Metabolism | Limited (<10%) |
| Biological half-life | 24 hours |
| Excretion | Renal (81–92%) |
| Identifiers | |
| CAS Number | 249296-44-4 |
| ATC code | N07BA03 (WHO) |
| PubChem | CID 5310966 |
| IUPHAR/BPS | 5459 |
| DrugBank | DB01273 |
| ChemSpider | 4470510 |
| UNII | W6HS99O8ZO |
| KEGG | D08669 |
| ChEBI | CHEBI:84500 |
| ChEMBL | CHEMBL1076903 |
| Chemical data | |
| Formula | C13H13N3 |
| Molar mass | 211.267 g/mol |
////////////Varenicline, Chantix™, FDA 2006, 249296-44-4, 375815-87-5, Champix , Pfizer, バレニクリン酒石酸塩
n1c2cc3c(cc2ncc1)[C@@H]4CNC[C@H]3C4
Written Confirmation expired: Can an API still be imported when produced earlier?
DRUG REGULATORY AFFAIRS INTERNATIONAL

What needs to be considered if an API is produced in the time period of a valid written confirmation but imported after this confirmation has expired? This is answered in a revised Q&A Document of the EU Commission.
The EU Commission has updated its Question and Answers Document “Importation of active substances for medicinal products for human use” (now version 7). In this updated version, the question “Can an API batch manufactured during the period of validity of a written confirmation be imported into the EU once the written confirmation is expired?”
In the answer it is referred to Article 46(b)(2)(b) of Directive 2001/83/EC, where it is defined that APIs can only be imported if they are manufactured in accordance with EU GMP or equivalent, and accompanied by a written confirmation from the competent authority of the exporting third country certifying this.
But what if an…
View original post 167 more words
MHRA GxP Data Integrity Definitions and Guidance for Industry: New Draft Version for Consultation
DRUG REGULATORY AFFAIRS INTERNATIONAL

In January and March 2015, the U.K. Medicines and Healthcare Products Regulatory Agency (MHRA) published a “GMP Data Integrity Definitions and Guidance for Industry”. The agency has recently published a new version of the Guidance. Please find here a short overview of the new features in the “GxP Data Integrity Definitions and Guidance for Industry: Draft version for consultation”.
In recent years, regulatory authorities have been struggling with data integrity issues. In particular the U.S. American FDA has tightened the awareness regarding the topic in many Warning Letters. In the meantime, data integrity has also become a focus of European regulatory authorities’ inspections. One of the first regulatory authorities to publish a “GMP Data Integrity Definitions and Guidance for Industry” in January and March 2015 was the U.K. Medicines and Healthcare Products Regulatory Agency (MHRA). More information can be found in “MHRA revises its Guideline on Data Integrity in the short Term
View original post 172 more words
WHO Draft on Analytical Method Validation
DRUG REGULATORY AFFAIRS INTERNATIONAL

The World Health Organization (WHO) recently published a draft document on analytical method Validation for comment. Read more about the draft “Guidelines on Validation – Appendix 4 Analytical Method Validation“.
In June 2016 the World Health Organization (WHO) published a draft document “Guidelines on Validation – Appendix 4 Analytical Method Validation”. Comments on the text should be sent to WHO until July 30, 2016.
The appendix 4 of the published Supplementary guidelines on good manufacturing practices: validation (WHO Technical Report Series, No. 937, 2006, Annex 4) has been revised in view of current trends in validation. The appendix presents some information on the characteristics that should be considered during validation of analytical methods. Approaches other than those specified in the Appendix may be followed and may be acceptable.
The new Appendix 4 is structured as follows (New and revised):
1. Principle (revised):
- 1.5 The…
View original post 761 more words
SPIRONOLACTONE, спиронолактон , سبيرونولاكتون , 螺内酯 ,

Spironolactone
Spironolactone, Supra-puren, Suracton, спиронолактон, سبيرونولاكتون ,
螺内酯 , Abbolactone, Aldactide, SNL, Spiroctanie, Sprioderm, Verospirone, Opianin
7α-Acetylthio-17α-hydroxy-3-oxopregn-4-ene-21-carboxylic acid γ-lactone
(1’S,2R,2’R,9’R,10’R,11’S,15’S)-9′-(acetylsulfanyl)-2′,15′-dimethylspiro[oxolane-2,14′-tetracyclo[8.7.0.02,7.011,15]heptadecan]-6′-ene-5,5′-dione
(7a,17a)-7-(Acetylthio)-17-hydroxy-3-oxopregn-4-ene-21-carboxylic acid g-lactone
17-Hydroxy-7a-mercapto-3-oxo-17a-pregn-4-ene-21-carboxylic Acid g-Lactone Acetate
3-(3-Oxo-7a-acetylthio-17b-hydroxy-4-androsten-17a-yl)propionic Acid g-Lactone
| CAS 52-01-7 |
MF C24H32O4S, MW 416.573 Da
Spironolactone, marketed under the brand name Aldactone among others, is a medication primarily used to treatfluid build-up due to heart failure, liver scarring, or kidney disease.[1] Other uses include high blood pressure, low blood potassium that does not improve with supplementation, early puberty, excessive hair growth in women,[1] and as a component of hormone replacement therapy for transgender women.[6] It is taken by mouth.[1]
Common side effects include electrolyte abnormalities particularly high blood potassium, nausea, vomiting, headache, a rash, and a decreased desire for sex. In those with liver or kidney problems extra care should be taken.[1]Spironolactone has not been well studied in pregnancy and should not be used to treat high blood pressure of pregnancy.[7] It is a steroid that blocks mineralocorticoid receptors. It also blocks androgen, and blocks progesterone. It belongs to a class of medications known as potassium-sparing diuretics.[1]
Spironolactone was introduced in 1959.[8][9] It is on the World Health Organization’s List of Essential Medicines, the most important medications needed in a basic health system.[10] It is available as a generic medication.[1] The wholesale cost in the developing world as of 2014 is between 0.02 and 0.12 USD per day.[11] In the United States it costs about 0.50 USD per day.[1]
|
Title: Spironolactone
CAS Registry Number: 52-01-7
CAS Name: (7a,17a)-7-(Acetylthio)-17-hydroxy-3-oxopregn-4-ene-21-carboxylic acid g-lactone
Additional Names: 17-hydroxy-7a-mercapto-3-oxo-17a-pregn-4-ene-21-carboxylic acid g-lactone, acetate; 3-(3-oxo-7a-acetylthio-17b-hydroxy-4-androsten-17a-yl)propionic acid g-lactone
Manufacturers’ Codes: SC-9420
Trademarks: Aldactone (Pharmacia & Upjohn); Aquareduct (Azupharma); Practon (Pfizer); Osyrol (Aventis); Sincomen (Schering AG); Spirobeta (Betapharm); Spiroctan (Ferlux); Spirolone (APS); Spironone (Dexo); Verospiron (Richter Gedeon); Xenalon (Mepha)
Molecular Formula: C24H32O4S
Molecular Weight: 416.57
Percent Composition: C 69.20%, H 7.74%, O 15.36%, S 7.70%
Literature References: Aldosterone antagonist. Prepn: Cella, Tweit, J. Org. Chem. 24, 1109 (1959); US 3013012 (1961 to Searle); Tweit et al., J. Org. Chem. 27, 3325 (1962). Activity and metabolic studies: Gerhards, Engelhardt, Arzneim.-Forsch. 13, 972 (1963). Crystal and molecular structure: Dideberg, Dupont, Acta Crystallogr. B28, 3014 (1972). Comprehensive description: J. L. Sutter, E. P. K. Lau, Anal. Profiles Drug Subs. 4, 431-451 (1975). Review of carcinogenetic risk: IARC Monographs 24, 259-273 (1980). Review of antiandrogen effects and clinical use in hirsutism: R. R. Tremblay, Clin. Endocrinol. Metab. 15, 363-371 (1986); of clinical efficacy in hypertension: A. N. Brest, Clin. Ther. 8, 568-585 (1986). Review of pharmacology: H. A. Skluth, J. G. Gums,DICP Ann. Pharmacother. 24, 52-59 (1990). Clinical trial in congestive heart failure: B. Pitt et al., N. Engl. J. Med. 341, 709 (1999).
Properties: Crystals from methanol, mp 134-135° (resolidifies and dec 201-202°). [a]D20 -33.5° (chloroform). uv max: 238 nm (e20200). Practically insol in water. Sol in alcohol; freely sol in benzene, chloroform. LD50 in rats, mice, rabbits (mg/kg): 790, 360, 870 i.p. (IARC, 1980).
Melting point: mp 134-135° (resolidifies and dec 201-202°)
Optical Rotation: [a]D20 -33.5° (chloroform)
Absorption maximum: uv max: 238 nm (e 20200)
Toxicity data: LD50 in rats, mice, rabbits (mg/kg): 790, 360, 870 i.p. (IARC, 1980)
Therap-Cat: Diuretic.
Therap-Cat-Vet: Diuretic.
Keywords: Aldosterone Antagonist; Diuretic; Steroids
|
Medical uses
Spironolactone is used primarily to treat heart failure, edematous conditions such as nephrotic syndrome or ascites in people with liver disease, essential hypertension, hypokalemia, secondary hyperaldosteronism (such as occurs with hepatic cirrhosis), and Conn’s syndrome (primary hyperaldosteronism). On its own, spironolactone is only a weak diuretic because it primarily targets the distal nephron (collecting tubule), where only small amounts of sodium are reabsorbed, but it can be combined with other diuretics to increase efficacy.
Spironolactone is an antagonist of the androgen receptor (AR) as well as an inhibitor of androgen production. Due to the antiandrogenic effects that result from these actions, it is frequently used off-label to treat a variety of dermatological conditions in which androgens, such as testosterone and dihydrotestosterone (DHT), play a role. Some of these uses include androgenic alopecia in men (either at low doses or as a topical formulation) and women, and hirsutism, acne, and seborrhea in women.[12] Spironolactone is the most commonly used drug in the treatment of hirsutism in the United States.[13] Higher doses of spironolactone are not recommended in males due to the high risk of feminization and other side effects. Similarly, it is also commonly used to treat symptoms of hyperandrogenism in polycystic ovary syndrome.[14]
Spironolactone (SL) is known to be a potent aldosterone antagonist at mineralocorticoid steroid hormone receptors, and it is widely used in humans for the treatment of essential hypertension, congestive heat failure and refractory edema or hyperaldosteronism. However, the prolonged use of SL is associated with undesirable endocrine side effects such as gynecomastia and lose of libido in men and menstrual irregularities in women due to interaction of SL with gonadal steroid hormone biosynthesis and target cell gonadal steroid receptors.
The nature and prevalence of the undesirable side effects limit the usefulness of spironolactone as a therapeutic agent. Gynecomastia or tender breast enlargement has been found to occur in 10% of hypertensive patients using spironolactone for therapy as compared to 1% of men in the placebo group. Recent studies by Pitt, et al. with spironolactone have shown that in patients with congestive heart failure (CHF) taking digoxin and a loop diuretic—spironolactone therapy in conjunction with digitalis and ACE inhibitor—reduces mortality by 30%. See Pitt, B., et al., The Effect of Spironolactone on Morbidity and Mortality in Patients with Severe Heart Failure, Randomized Aldactone Evaluation Study Investigors; N. Engl. J. Med., 1999, 341:709-717. These authors stated that the 30% reduction in the risk of death among patients in the group receiving spironolactone could be attributed to a lower risk of both death from progressive heart failure and sudden death from cardiac arrhythmic causes. In addition, they found that the frequency of hospitalization for worsening heart failure is 35% lower in the spironolacotone treated group than in the placebo group. These authors concluded that patients who received spironolactone had a significant improvement in the symptoms of severe heart failure caused by systolic left ventricular dysfunction. Overall, 8% of the patients in the spironolactone group discontinued treatment because of adverse events. The purpose of the present invention is to make available the individual chiral isomers of spironolactone that would be effective in treating CHF and in reducing hypertension, and at the same time would be devoid of undesirable side effects such as gynecomastia, lose of libido in men, and menstrual irregularities in women.
Spironolactone is the name commonly used for a specific spirolactone that has the full chemical name 17-hydroxy-7-alpha-mercapto-3-oxo-17-alpha-pregn-4-ene-21-carboxylic acid gamma-lactone acetate. The term “spirolactone” denotes that a lactone 10 ring (i.e., a cyclic ester) is attached to another ring structure in a spiro configuration (i.e., the lactone ring shares a single carbon atom with the other ring). Spirolactones that are coupled to steroids are the most important class of spirolactones from a pharmaceutical perspective, so they are widely referred to in the pharmaceutical arts simply as spirolactones. As used herein, “spironolactone” refers to a molecule comprising a lactone structure coupled via a spiro configuration to a steroid structure or steroid derivative.
Spironolactone, its activities, and modes of synthesis and purification are described in a number of U.S. patents, notably U.S. Pat. Nos. 3,013,012, 4,529,811 and 4,603,128.
Intracellular receptors (IRs) form a class of structurally-related genetic regulators that act as ligand-dependent transcription factors. See Evans, R. M., “The Steroid and Thyroid Hormone Receptor Superfamily”, Science, May 13, 1988; 240(4854):889-95. Steroid receptors are a recognized subset of the IRs, including the progesterone receptor (PR), androgen receptor (AR), estrogen receptor (ER), which can be referred to collectively as the gonadal steroid receptors, glucocorticoid receptor (GR), and mineralocorticoid receptor (MR). Regulation of a gene by such factors requires both the IR itself and a corresponding ligand that has the ability to selectively bind to the IR in a way that affects gene transcription.
Ligands for the IRs can include low molecular weight native molecules, such as the hormones aldosterone, progesterone, estrogen and testosterone, as well as synthetic derivative compounds such as medroxyprogesterone acetate, diethylstilbesterol and 19-nortestosterone. These ligands, when present the fluid surrounding a cell, pass through the outer cell membrane by passive diffusion and bind to specific IR proteins to create a ligand/receptor complex. This complex then translocates to the cell’s nucleus, where it binds to a specific gene or genes present in the cell’s DNA. Once bound to DNA, the complex modulates the production of the protein encoded by that gene. In this regard, a compound that binds to an IR and mimics the effect of the native ligand is referred to as an “agonist”, while a compound that binds to an IR and inhibits the effect of the native ligand is called an “antagonist”.
The therapeutic mechanism of action of spironolactone involves binding to intracellular mineralocorticoid receptors (MRs) in kidney epithelial cells, thereby inhibiting the binding of aldosterone. Spironolactone has been found to counteract the sodium reabsorption and potassium excretion effects of aldosterone and other mineralocorticoids. Spironolactone has also been shown to interfere with testosterone biosynthesis, has anti-androgen action and inhibits adrenal aldosterone biosynthesis. Large doses of spironolactone in children appear to decrease the testosterone production rate.
Spironolactone is found to exhibit intra-individual variability of pharmacokinetic parameters and it presumably belongs to the group of drugs with high inter-subject variability. Spironolactone has poor water solubility and dissolution rate.
In order to prolong the half-life and decrease the side effects associated with spironolactone, syntheses of spironolactone derivatives have been developed (e.g. synthesis of mexrenone, prorenone, spirorenone). Slight modifications of the spironolactone steroid skeleton, e.g. such as formation of 11β-allenic and epoxy compounds, have been shown to effect important variations in the affinity and specificity for the mineralocorticoid receptor. These results suggest that it is possible to develop spironolactone analogues that do not interact with the androgen receptor or cytochrome P-450 and are therefore free of spironolactone undesirable side-effects.
METABOLISM

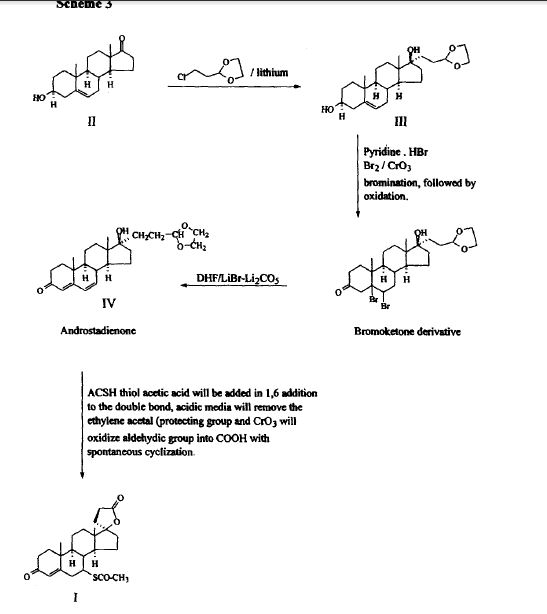
SYNTHESIS
METHOD 1 REF 150

REF 130, 150

METHOD 2 REF 140
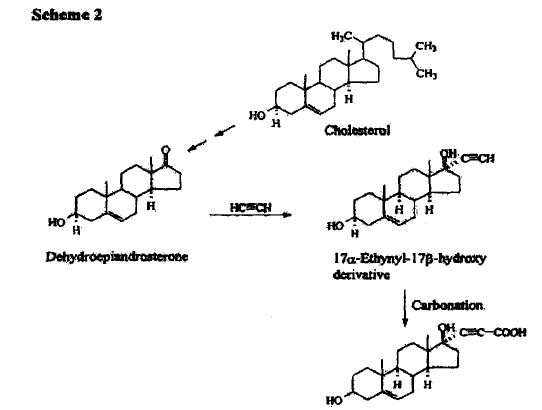
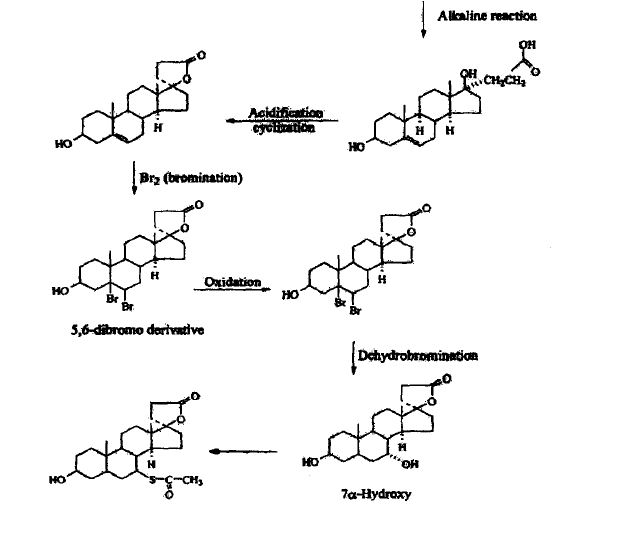

METHOD 3 REF 150
Synthesis
Cella, John A.; Tweit, Robert C. (1959). Journal of Organic Chemistry 24: 1109. doi:.
(See also part 1 and part 3)

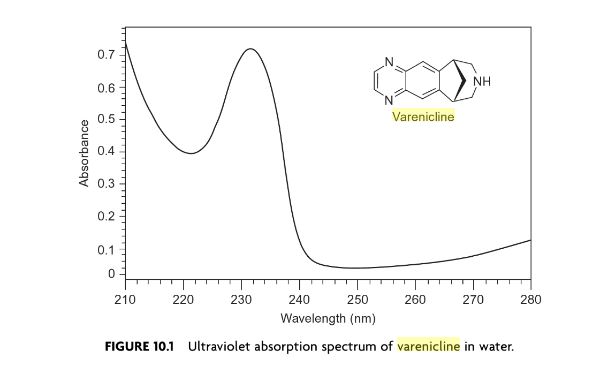
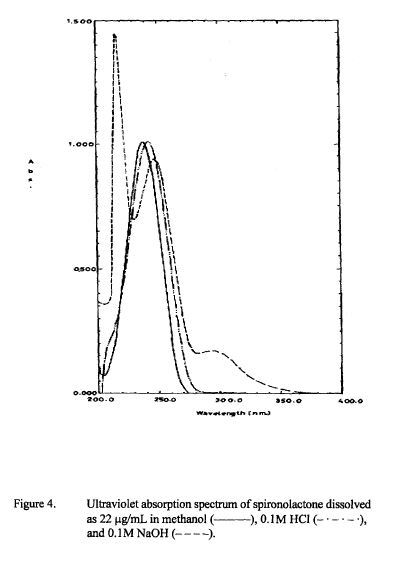
SPECTROSCOPY UV
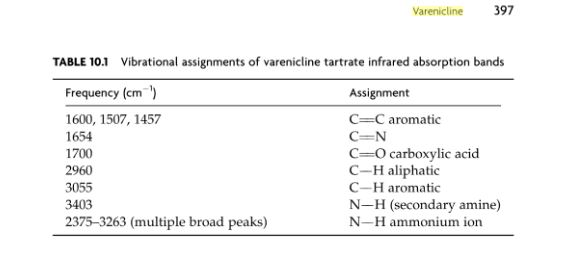
SPECTROSCOPY IR
KBR
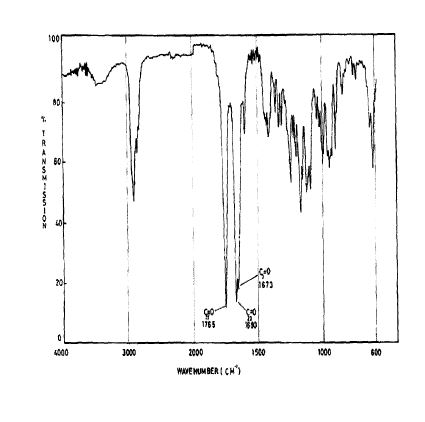
The principal absorption peaks of the spectrum shown in Figure 5 were noted at 1765,
1693, 1673, 1240, 1178, 1135, 1123 and 1193 cm -1.
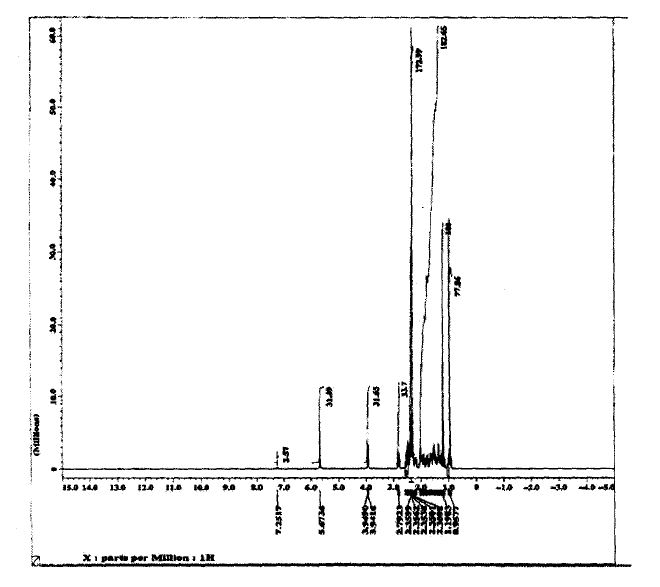
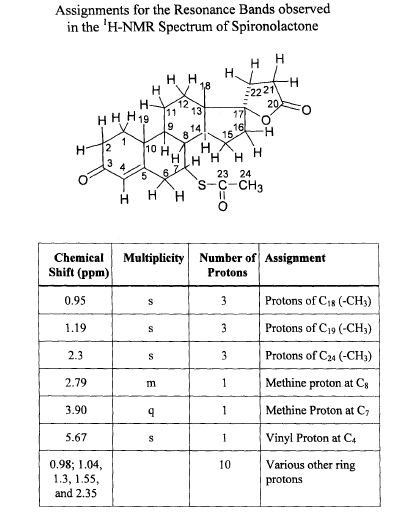
SPECTROSCOPY 1H NMR
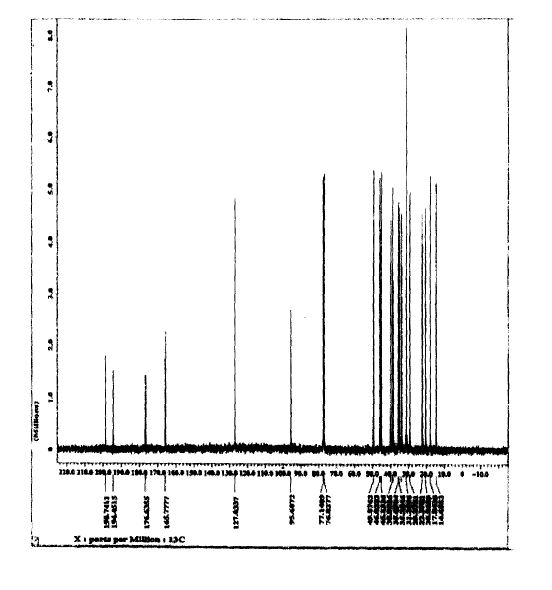
SPECTROSCOPY 13C NMR
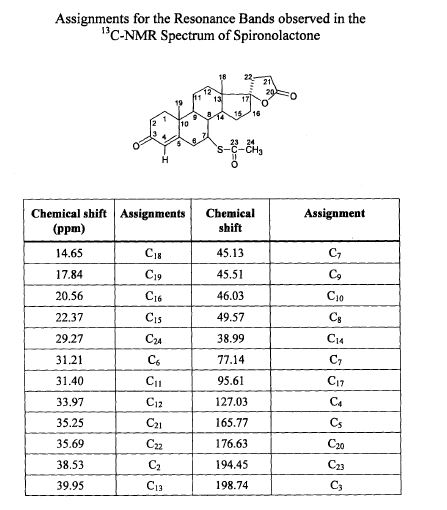
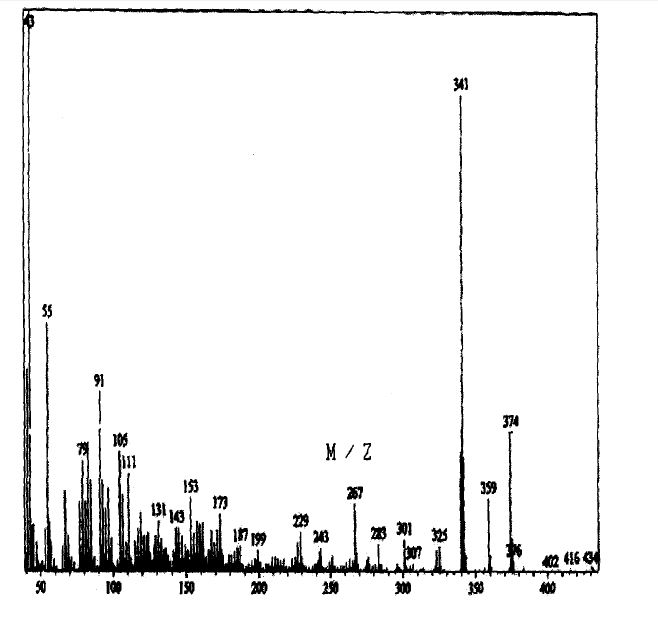
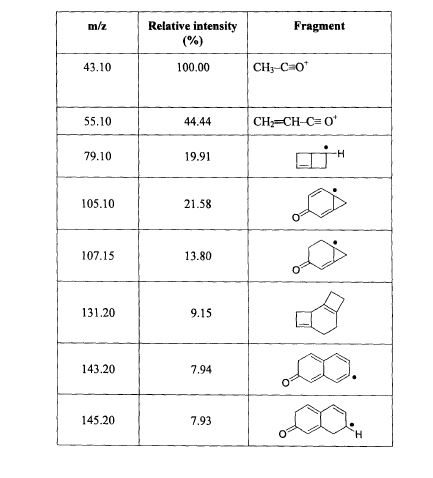
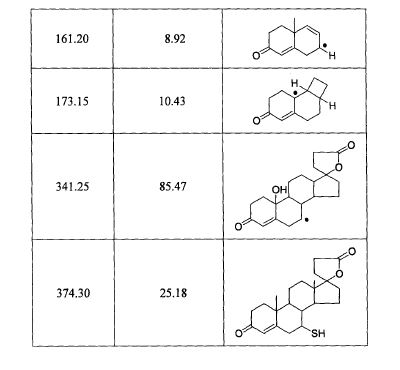
SPECTROSCOPY MASS SPECTRUM
130 J.A. Cola, E.A. Brown, and R.R. Burtner, 3. Org. Chem., 24, 1109(1959).
140 Remington’s: The Science and Practice of Pharmacy, 19 t~ edn.Volume II, K.G. Alfonso, ed.; Mack Publishing Co., Pennsylvania (1995) p.1048.
150. G. Anner and H. Wehrli (Ciba-Geigy, A.-G.), German Often 2,625,723 (cl.C07J21/00), Dec,1976; Swiss Appl. 75/7, 696, 13Jun. 1975; pp. 37.
ANALYTICAL
-
High-Performance Liquid Chromatographic Conditions Column LiChrosorb RP-8, 5 μm. 150 × 4.6 mm I.D. Eluent Acetonitrile-0.05 M phosphate buffer, pH 4 (45:55) Flow-rate 1 ml/min Temperature 25° C. Detector UV detector, wavelength 286 nm or 271 nm Recorder Chart speed 0.5 cm/min Sample loop 10 μl -
The concentration of canrenone is determined in plasma and urine samples by high-performance liquid chromatography (HPLC) with UV-detection. An aliquot of 300 ng of spironolactone derivative is added to the samples as internal standard, which are then extracted twice with 1 ml n-hexane-toluene (1:1, v/v). The organic phase is taken to dryness and re-dissolved in 250 μl HPLC eluent (methanol-water, 60:40, v/v). (25×4.6 mm; 5 μm). Detection is performed with the UV detector set at λ=285 nm.
Flurometric Method
- Five ml of water is a reagent blank and 5 ml of working standards containing 0.05 μg and 0.20 μg of SC-9376 are carried through the entire procedure. Lower sales are read vs. the 0.05 μg standard at full scale, and higher samples vs. the 0.20 μg standard. Fluorescence readings are proportional to the concentrations of the standards in this range.
- Pipette 0.2 ml of heparinized plasma into a 50-ml polyethylene-stoppered centrifuge tube, dilute to 5 ml with water and add 15 ml of methylene chloride (Du Pont refrigeration grade, redistilled). Shake for 30 seconds, centrifuge and discard the aqueous supernatant. Add 1 ml 0.1 N NaOH, shake 15 seconds, centrifuge and discard the supernatant. Transfer a 10-ml aliquot of the methylene chloride phase to another tube containing 2 ml of 65% aqueous sulfuric acid, shake 30 seconds, centrifuge and remove organic phase by aspiration. The material is allowed to stand at room temperature for about 1 hour and then about 1 ml of the sulfuric acid phase in transferred to a quartz cuvette. Fluorescence intensity is determined in an Aminco-Bowman spectrophotofluorometer (activation maximum, 465 nm).
- Gas Liquid Chromatography
- The GLC estimation is carried out on a Fractovap Model 251 series 2150 (Carlo Erba) instrument equipped with a Nickel-63 electron capture detector. A 6-foot, 0.4 mm internal diameter, U-shaped glass column, packed with OV-17 2% or XE-60 1% on gas chrom A, 100-120 mesh (Applied Science Lab) is conditioned for 3 days before use. Argon with 10% methane which passed through a molecular sieve before entering the column is used as the carrier gas. The conditions of analysis are: column 255° C., detector 275° C., carrier gas flow 30 ml/min. Samples are injected on the column with a 10 μl Hamilton syringe. The injector in not heated.
PATENT
https://www.google.com/patents/US20090325918
EXAMPLE 1Chiral Separation
The separation of 7 beta isomer of SL is schematically described below.
-
 Chromatographic Method for Isolation of SL IsomersThe basic method is described in Chan, Ky, et al., J. Chromatog, Nov. 15, 1991:571 (1-2) 291-297. The separation is performed using spectra-physics HPLC instrument and UV variable wavelength detector set at 254 nm. For chiral separation, the chromatographic column is either a pre-packed 25 mm×4.6 mm ID Cyclobond 1 (5 μm particle size), or a pre-packed 150 mm×4 mm ID Resolvosil BSA-7 column (5 μm) operated using the conditions described herein.Analysis of the isomers present in the peaks in the chromatograms and their chiral extract purity analysis can be determined in each case by high resolution NMR spectroscopy using a chiral shift reagent. Based on this information and the determination of molecular weight by mass spectrometry and/or optical activity, structural configuration is assigned to each isomer. Eluted samples of isomers may be re-chromatographed in order to obtain adequate quantities of isomers having desired optical purity for study. For future use, reference standards that are optically pure will be compared for confirmation of purity and identity to the isolated isomers that are obtained after their chromatographic separation.
Chromatographic Method for Isolation of SL IsomersThe basic method is described in Chan, Ky, et al., J. Chromatog, Nov. 15, 1991:571 (1-2) 291-297. The separation is performed using spectra-physics HPLC instrument and UV variable wavelength detector set at 254 nm. For chiral separation, the chromatographic column is either a pre-packed 25 mm×4.6 mm ID Cyclobond 1 (5 μm particle size), or a pre-packed 150 mm×4 mm ID Resolvosil BSA-7 column (5 μm) operated using the conditions described herein.Analysis of the isomers present in the peaks in the chromatograms and their chiral extract purity analysis can be determined in each case by high resolution NMR spectroscopy using a chiral shift reagent. Based on this information and the determination of molecular weight by mass spectrometry and/or optical activity, structural configuration is assigned to each isomer. Eluted samples of isomers may be re-chromatographed in order to obtain adequate quantities of isomers having desired optical purity for study. For future use, reference standards that are optically pure will be compared for confirmation of purity and identity to the isolated isomers that are obtained after their chromatographic separation.
EXAMPLE 2Chemical Synthesis of Optical Isomers
- As an example, the desire spironolactone 7-beta-isomer is synthesized following the scheme that is described below:
-
 Diene (i) is prepared from commercially available starting materials using methods well known in the art of chemical synthesis.Diene (i) is treated with acetic acid and the mixture is heated to reflux to yield 7-alpha-acetate ester (ii). The 7-alpha-ester (ii) is further subjected to nucleophilic substitution, followed by hydrolysis to obtain the 7-beta-isomer (iii). The 7-beta-isomer (iii) is then esterified with an acyl halide in the presence of a base to generate the desired spironolactone 7-beta-isomer (iv).
Diene (i) is prepared from commercially available starting materials using methods well known in the art of chemical synthesis.Diene (i) is treated with acetic acid and the mixture is heated to reflux to yield 7-alpha-acetate ester (ii). The 7-alpha-ester (ii) is further subjected to nucleophilic substitution, followed by hydrolysis to obtain the 7-beta-isomer (iii). The 7-beta-isomer (iii) is then esterified with an acyl halide in the presence of a base to generate the desired spironolactone 7-beta-isomer (iv).
EXAMPLE 3Preparation of Radiolabeled Probe Compounds of the Invention
- Using known methods, the compounds of the invention may be prepared as radiolabeled probes by carrying out their synthesis using precursors comprising at least one atom that is a radioisotope. The radioisotope is preferably selected from at least one of carbon (preferably
14
- C), hydrogen (preferably
3
- H), sulfur (preferably
35
- S), or iodine (preferably I). Such radiolabeled probes are conveniently synthesized by a radioisotope supplier specializing in customer synthesis of radiolabeled probe compounds. Such suppliers include Amersham Corporation, Arlington Heights, Ill.; Cambridge Isotope Laboratories, Inc., Andover, Mass.; SRI International, Menlo Park, Calif.; Wizard Laboratories, West Sacramento, Calif.; ChemSyn Laboratories, Lexena, Kans.; American Radiolabeled Chemicals, Inc., St. Louis, Mo.; and Moravek Biochemicals Inc., Brea, Calif.
- Tritium labeled probe compounds are also conveniently prepared catalytically via platinum-catalyzed exchange in tritiated acetic acid, acid-catalyzed exchange in tritiated trifluoroacetic acid, or heterogeneous-catalyzed exchange with tritium gas. Tritium labeled probe compounds can also be prepared, when appropriate, by sodium borotritide reduction. Such preparations are also conveniently carried out as a custom radiolabeling by any of the suppliers listed in the preceding paragraph using the compound of the invention as substrate.
- EXAMPLE 4Isolation and Purification Procedure
- The optical isomers of spironolactones may be isolated from fluid sample such as urine or blood as follows:
- Extraction from Urine
- The urine sample is extracted with dichloromethane and the extract washed with NaOH (0.1 N) and then with water to neutrality. The residue obtained after evaporation of the dichloromethane extract is purified on TLC in three different systems: benzene-acetone-water, (150:100:0.4); chloroform-ethanol, (90:10); ethyl acetate-cyclohexane-ethanol, (45:25:10), using aldosterone as reference standard.
- The extract is then purified by high performance liquid chromatography (HPLC) on a Waters 6000 A, 480 U.V. detector instrument with radial pressure. The extract is first run through a C
18
- 10μ column using methanol-water (70:30) as the eluent, followed by a silica 5μ column using dichloromethane-methanol (95:5). In both cases, the rate of the eluent is 1.5 ml/min. A small part of the extract is subjected to heptafluorobutyrylation for GLC investigation.
References
- “Spironolactone”. The American Society of Health-System Pharmacists. Retrieved Oct 24, 2015.
- “Spironolactone: MedlinePlus Drug Information”. Retrieved 2016-01-20.
- “Spironolactone”. Merriam-Webster Dictionary.
- “Spironolactone”. Dictionary.com Unabridged. Random House.
- Harry G. Brittain (26 November 2002). Analytical Profiles of Drug Substances and Excipients. Academic Press. p. 309. ISBN 978-0-12-260829-2. Retrieved 27 May 2012.
- Maizes, Victoria (2015). Integrative Women’s Health (2 ed.). p. 746.ISBN 9780190214807.
- “Spironolactone Pregnancy and Breastfeeding Warnings”. Retrieved 29 November2015.
- Camille Georges Wermuth (24 July 2008). The Practice of Medicinal Chemistry. Academic Press. p. 34. ISBN 978-0-12-374194-3. Retrieved 27 May 2012.
- Marshall Sittig (1988). Pharmaceutical Manufacturing Encyclopedia. William Andrew. p. 1385. ISBN 978-0-8155-1144-1. Retrieved 27 May 2012.
- “WHO Model List of EssentialMedicines” (PDF). World Health Organization. October 2013. Retrieved 22 April 2014.
- “Spironolactone”. International Drug Price Indicator Guide. Retrieved 29 November2015.
- Hughes BR, Cunliffe WJ (May 1988). “Tolerance of spironolactone”. The British Journal of Dermatology 118 (5): 687–91. doi:10.1111/j.1365-2133.1988.tb02571.x.PMID 2969259.
- Victor R. Preedy (1 January 2012). Handbook of Hair in Health and Disease. Springer Science & Business Media. pp. 132–. ISBN 978-90-8686-728-8.
- Loy R, Seibel MM (December 1988). “Evaluation and therapy of polycystic ovarian syndrome”. Endocrinology and Metabolism Clinics of North America 17 (4): 785–813.PMID 3143568.
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
7α-Acetylthio-17α-hydroxy-3-oxopregn-4-ene-21-carboxylic acid γ-lactone
|
|
| Clinical data | |
| Pronunciation | /spɪˌroʊnəˈlæktoʊn, spaɪ–, spə–, –ˈrɒ–, –noʊ–/or /ˌspaɪrənoʊˈlæktoʊn/[2][3][4] |
| Trade names | Aldactone |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682627 |
| Pregnancy category |
|
| Routes of administration |
Oral[1] |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Protein binding | 90%+[5] |
| Metabolism | Hepatic CYP450 |
| Biological half-life | 1.3-2 hours |
| Excretion | Urine, bile |
| Identifiers | |
| CAS Number | 52-01-7 |
| ATC code | C03DA01 (WHO) |
| PubChem | CID 5833 |
| IUPHAR/BPS | 2875 |
| DrugBank | DB00421 |
| ChemSpider | 5628 |
| UNII | 27O7W4T232 |
| KEGG | D00443 |
| ChEBI | CHEBI:9241 |
| ChEMBL | CHEMBL1393 |
| Chemical data | |
| Formula | C24H32O4S |
| Molar mass | 416.574 g/mol |
///////Spironolactone, Supra-puren, Suracton, спиронолактон, سبيرونولاكتون ,
螺内酯 , Abbolactone, Aldactide, SNL, Spiroctanie, Sprioderm, Verospirone, Opianin
O=C5O[C@@]4([C@@]3([C@H]([C@@H]2[C@H](SC(=O)C)C/C1=C/C(=O)CC[C@]1(C)[C@H]2CC3)CC4)C)CC5
Prucalopride succinate (Resolor)
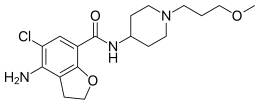
Prucalopride (Resolor)
CAS 179474-81-8 , R-093877; R-108512
4-Amino-5-chlor-N-[1-(3-methoxypropyl)-4-piperidinyl]-2,3-dihydro-1-benzofuran-7-carboxamid
R-093877|R-108512|Resolor®
Resolor;Resotran
Resotran
UNII:0A09IUW5TP
SHIRE 2010 LAUNCHED
JANNSEN PHASE 3 IRRITABLE BOWL SYNDROME
Janssen Pharmaceutica N.V., INNOVATOR
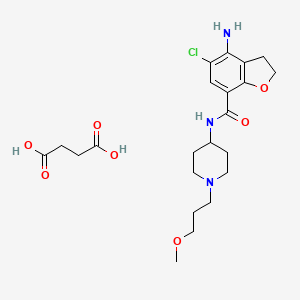
| Prucalopride succinate; 179474-85-2; Resolor; Prucalopride (succinate); UNII-4V2G75E1CK; R-108512; | |
| Molecular Formula: | C22H32ClN3O7 |
|---|---|
| Molecular Weight: | 485.95838 g/mol |
Drug Name:Prucalopride Succinate
Trade Name:Resolor®, MOA:Serotonin (5-HT4) receptor agonist, Indication:Chronic constipation
Company:Shire (Originator) , Johnson & Johnson
APPROVED EU 2009-10-15
CHINA 2014-01-21
Prucalopride (brand name Resolor, developed by Johnson & Johnson and licensed to Movetis) is a drug acting as a selective, high affinity 5-HT4 receptor agonist[1] which targets the impaired motility associated with chronic constipation, thus normalizing bowel movements.[2][3][4][5][6][7] Prucalopride was approved for use in Europe in 2009,[8] in Canada (named Resotran) on December 7, 2011[9] and in Israel in 2014[10] but it has not been approved by the Food and Drug Administration for use in the United States. The drug has also been tested for the treatment of chronic intestinal pseudo-obstruction.[11][12]
Mechanism of action
Prucalopride, a first in class dihydro-benzofuran-carboxamide, is a selective, high affinity serotonin (5-HT4) receptor agonist with enterokinetic activities.[13] Prucalopride alters colonic motility patterns via serotonin 5-HT4 receptor stimulation: it stimulates colonic mass movements, which provide the main propulsive force for defecation.

The observed effects are exerted via highly selective action on 5-HT4 receptors:[13] prucalopride has >150-fold higher affinity for 5-HT4 receptors than for other receptors.[1][14] Prucalopride differs from other 5-HT4 agonists such as tegaserod and cisapride, which at therapeutic concentrations also interact with other receptors (5-HT1B/D and the cardiac human ether-a-go-go K+ or hERG channelrespectively) and this may account for the adverse cardiovascular events that have resulted in the restricted availability of these drugs.[14] Clinical trials evaluating the effect of prucalopride on QT interval and related adverse events have not demonstrated significant differences compared with placebo.[13]
Pharmacokinetics
Prucalopride is rapidly absorbed (Cmax attained 2–3 hours after single 2 mg oral dose) and is extensively distributed. Metabolism is not the major route of elimination. In vitro, human liver metabolism is very slow and only minor amounts of metabolites are found. A large fraction of the active substance is excreted unchanged (about 60% of the administered dose in urine and at least 6% in feces).Renal excretion of unchanged prucalopride involves both passive filtration and active secretion. Plasma clearance averages 317 ml/min, terminal half-life is 24–30 hours,[15] and steady-state is reached within 3–4 days. On once daily treatment with 2 mg prucalopride, steady-state plasma concentrations fluctuate between trough and peak values of 2.5 and 7 ng/ml, respectively.[13]
In vitro data indicate that prucalopride has a low interaction potential, and therapeutic concentrations of prucalopride are not expected to affect the CYP-mediated metabolism of co-medicated medicinal products.[13]
Efficacy
The primary measure of efficacy in the clinical trials is three or more spontaneous complete bowel movements per week; a secondary measure is an increase of at least one complete spontaneous bowel movement per week.[7][16][17] Further measures are improvements in PAC-QOL[18] (a quality of life measure) and PAC-SYM[19] (a range of stool,abdominal, and rectal symptoms associated with chronic constipation). Infrequent bowel movements, bloating, straining, abdominal pain, and defecation urge with inability to evacuate can be severe symptoms, significantly affecting quality of life.[20][21][22][23][24]
In three large clinical trials, 12 weeks of treatment with prucalopride 2 and 4 mg/day resulted in a significantly higher proportion of patients reaching the primary efficacy endpoint of an average of ≥3 spontaneous complete bowel movements than with placebo.[7][16][17] There was also significantly improved bowel habit and associated symptoms, patient satisfaction with bowel habit and treatment, and HR-QOL in patients with severe chronic constipation, including those who did not experience adequate relief with prior therapies (>80% of the trial participants).[7][16][17] The improvement in patient satisfaction with bowel habit and treatment was maintained during treatment for up to 24 months; prucalopride therapy was generally well tolerated.[25][26]
Side effects
Prucalopride has been given orally to ~2700 patients with chronic constipation in controlled clinical trials. The most frequently reported side effects are headache andgastrointestinal symptoms (abdominal pain, nausea or diarrhea). Such reactions occur predominantly at the start of therapy and usually disappear within a few days with continued treatment.[13]
Approval
In the European Economic Area, prucalopride was originally approved for the symptomatic treatment of chronic constipation in women in whom laxatives fail to provide adequate relief.[13] Subsequently, it has been approved by the European Commission for use in adults – that is, including male patients – for the same indication.[27]
Contraindications
Prucalopride is contraindicated where there is hypersensitivity to the active substance or to any of the excipients, renal impairment requiring dialysis, intestinal perforation orobstruction due to structural or functional disorder of the gut wall, obstructive ileus, severe inflammatory conditions of the intestinal tract, such as Crohn’s disease, and ulcerative colitis and toxic megacolon/megarectum.[13]
CLIP
Prucalopride succinate, a first-in-class dihydrobenzofurancarboxamide, is a selective serotonin (5-HT4) receptor agonist.86–94 The drug, marketed under the brand name Resolor, possesses enterokinetic activity and was developed by the Belgian-based pharmaceutical firm Movetis. Prucalopride alters colonic motility patterns via serotonin 5-HT4 receptor stimulation, triggering the central propulsive force for defecation.95–97 The preparation of prucalopride succinate begins with the commercially available salicylic aniline 124 (Scheme 18). Acidic esterification, acetylation of the aniline nitrogen atom, and ambient-temperature chlorination via sulfuryl chloride (SO2Cl2) converted aminophenol 124 to acetamidoester 125 in 83% yield over the course of three steps.98–102 An unique set of conditions involving sodium tosylchloramide (chloramine T) trihydrate and sodium iodide were then employed to convert 125 to o-phenolic iodide 126, which then underwent sequential Sonogashira/cyclization reaction utilizing TMS-acetylene with tetramethylguanidine (TMG) in the presence of silica gel to furnish the benzofuran progenitor of 127.103 Hydrogenation of this intermediate benzofuranyl Sonagashira product saturated the 2,3-benzofuranyl bond while leaving the chlorine atom intact, ultimately delivering dihydrobenzofuran 127 in excellent yield for the two step sequence. Base-induced saponification and acetamide removal gave rise to acid 128. This acid was activated as the corresponding mixed anhydride and treated with commercial piperidine 129 to construct prucalopride which was stirred at room temperature for 24 h in ethanolic succinic acid to provide prucalopride succinate (XI). The yield for the formation of the salt was not provided.
86. Briejer, M. R.; Bosmans, J. P.; Van Daele, P.; Jurzak, M.; Heylen, L.; Leysen, J. E.;Prins, N. H.; Schuurkes, J. A. J. Eur. J. Pharmacol. 2001, 423, 71.
87. Briejer, M. R.; Prins, N. H.; Schuurkes, J. A. J. Neurogastroenterol. Motil. 2001, 13,465.
88. Coggrave, M.; Wiesel, P. H.; Norton, C. Cochrane Database Syst. Rev. 2006.CD002115.
89. Coremans, G.; Kerstens, R.; De Pauw, M.; Stevens, M. Digestion 2003, 67, 82.
90. De Winter, B. Y.; Boeckxstaens, G. E.; De Man, J. G.; Moreels, T. G.; Schuurkes, J.A. J.; Peeters, T. L.; Herman, A. G.; Pelckmans, P. A. Gut 1999, 45, 713.
91. Emmanuel, A. V.; Roy, A. J.; Nicholls, T. J.; Kamm, M. A. Aliment. Pharmacol.Ther. 2002, 16, 1347.
92. Frampton, J. E. Drugs 2009, 69, 2463.
93. Krogh, K.; Bach Jensen, M.; Gandrup, P.; Laurberg, S.; Nilsson, J.; Kerstens, R.;De Pauw, M. Scand. J. Gastroenterol. 2002, 37, 431.
94. Pau, D.; Workman, A. J.; Kane, K. A.; Rankin, A. C. J. Pharmacol. Exp. Ther. 2005,313, 146.
95. De Maeyer, J. H.; Schuurkes, J. A. J.; Lefebvre, R. A. Br. J. Pharmacol. 2009, 156,362.
96. Irving, H. R.; Tochon-Danguy, N.; Chinkwo, K. A.; Li, J. G.; Grabbe, C.; Shapiro,M.; Pouton, C. W.; Coupar, I. M. Pharmacology 2010, 85, 224.
97. Ray, A. M.; Kelsell, R. E.; Houp, J. A.; Kelly, F. M.; Medhurst, A. D.; Cox, H. M.;Calver, A. R. Eur. J. Pharmacol. 2009, 604, 1.
98. Baba, Y.; Usui, T.; Iwata, N. EP 640602 A1, 1995.
99. Fancelli, D.; Caccia, C.; Severino, D.; Vaghi, F.; Varasi, M. WO 9633186 A1,1996.
100. Hirokawa, Y.; Fujiwara, I.; Suzuki, K.; Harada, H.; Yoshikawa, T.; Yoshida, N.;Kato, S. J. Med. Chem. 2003, 46, 702.
101. Kakigami, T.; Usui, T.; Tsukamoto, K.; Kataoka, T. Chem. Pharm. Bull. 1998, 46,42.
102. Van Daele, G. H. P.; Bosmans, J.-P. R. M. A.; Schuurkes, J. A. J. WO 9616060 A1,1996.
103. Candiani, I.; DeBernadinis, S.; Cabri, W.; Marchi, M.; Bedeschi, A.; Penco, S.Synlett 1993, 269.
PAPER
Synlett 1993, 269
https://www.thieme-connect.com/products/ejournals/abstract/10.1055/s-1993-22663
PAPER
Chem. Pharm. Bull. 1998, 46,42.
https://www.jstage.jst.go.jp/article/cpb1958/46/1/46_1_42/_article
https://www.jstage.jst.go.jp/article/cpb1958/46/1/46_1_42/_pdf
PATENT
US5948794
http://www.google.co.in/patents/US5948794
EXAMPLE 1
In trichloromethane (135 ml) 4-amino-5-chloro-2,3-dihydro-7-benzofurancarboxylic acid (0.05 mol) (the preparation of which was described in EP-0,389,037-A) was suspended and cooled to ±5° C. N,N-diethylethanamine (0.05 mol) was added dropwise at a temperature below 10° C. Ethyl chloroformate (0.05 mol) was added dropwise and the reaction mixture was stirred for 40 min. while keeping the temperature below 10° C. The resulting mixture was added dropwise over a 20-min period to a solution of 1-(3-methoxypropyl)-4-piperidinamine (0.05 mol) in trichloromethane (35 ml). The cooling bath was removed and the reaction mixture was stirred for 150 min. Said mixture was washed with water (50 ml). The precipitate was filtered off over a glass filter and washed with water and CHCl3. The filtrate was separated in it’s layers. The separated organic layer was washed with water (50 ml)+a 50% NaOH solution (1 ml), dried, filtered and the solvent was evaporated. The residue was stirred in 2-propanol (100 ml). This mixture was acidified with HCl/2-propanol (7.2 ml; 5.29 N). The mixture was stirred for 16 hours at room temperature and the resulting precipitate was filtered off, washed with 2-propanol (15 ml) and dried (vacuum; 50° C.), yielding 12.6 g (62%) of 4-amino-5-chloro-2,3-dihydro-N- 1-(3-methoxypropyl)-4-piperidinyl!-7-benzofurancarboxamide monohydrochloride (comp. 1).
US5854260
http://www.google.co.in/patents/US5854260
EXPERIMENTAL PART EXAMPLE 1
In trichloromethane (135 ml) 4-amino-5-chloro-2,3-dihydro-7-benzofurancarboxylic acid (0.05 mol) (the preparation of which was described in EP-0,389,037-A) was suspended and cooled to ±5° C. N,N-diethylethanamine (0.05 mol) was added dropwise at a temperature below 10° C. Ethyl chloroformate (0.05 mol) was added dropwise and the reaction mixture was stirred for 40 min. while keeping the temperature below 10° C. The resulting mixture was added dropwise over a 20-min period to a solution of 1-(3-methoxypropyl)-4-piperidinamine (0.05 mol) in trichloromethane (35 ml). The cooling bath was removed and the reaction mixture was stirred for 150 min. Said mixture was washed with water (50 ml). The precipitate was filtered off over a glass filter and washed with water and CHCl3. The filtrate was separated in it’s layers. The separated organic layer was washed with water (50 ml)+ a 50% NaOH solution (1 ml), dried, filtered and the solvent was evaporated. The residue was stirred in 2-propanol (100 ml). This mixture was acidified with HCl/2-propanol (7.2 ml; 5.29 N). The mixture was stirred for 16 hours at room temperature and the resulting precipitate was filtered off, washed with 2-propanol (15 ml) and dried (vacuum; 50° C.), yielding 12.6 g (62%) of 4-amino-5-chloro-2,3-dihydro-N- 1-(3-methoxypropyl)-4-piperidinyl!-7-benzofurancarboxamide monohydrochloride (comp. 1).
PATENT
WO199616060A1
http://www.google.co.in/patents/WO1996016060A1?cl=en
EP-0,389,037-A, published on September 26, 1990, N-(3-hydroxy-4-piperidin- yl) (dihydrobenzofuran or dihydro-2H-benzopyran)carboxamide derivatives are disclosed as having gastrointestinal motility stimulating properties. In our EP-0,445,862-A, published on September 11, 1991, N-(4-piperidinyl) (dihydrobenzo¬ furan or dihydro-2H-benzopyran)carboxamide derivatives are disclosed also having gastrointestinal motility stimulating properties.
The compound subject to the present application differs therefrom by showing superior enterokinetic properties.
The present invention concerns a compound of formula

and the pharmaceutically acceptable acid addition salts thereof.
The chemical name of the compound of formula (I) is 4-amino-5-chloro-2,3-dihydro-N- [l-(3-methoxypropyl)-4-piperidinyl]-7-benzofurancarboxamide.

Example 1
In trichloromethane (135 ml) 4-amino-5-chloro-2,3-dihydro-7-benzofurancarboxylic acid (0.05 mol) (the preparation of which was described in EP-0,389,037-A) was suspended and cooled to ± 5 °C. H,N-diethylethanamine (0.05 mol) was added dropwise at a temperature below 10 °C. Ethyl chloroformate (0.05 mol) was added dropwise and the reaction mixture was stirred for 40 min. while keeping the temperature below 10°C. The resulting mixture was added dropwise over a 20-min period to a solution of l-(3-methoxypropyl)-4-piperidinamine (0.05 mol) in trichloromethane (35 ml). The cooling bath was removed and the reaction mixture was stirred for 150 min. Said mixture was washed with water (50 ml). The precipitate was filtered off over a glass filter and washed with water and CHCI3. The filtrate was separated in it’s layers. The separated organic layer was washed with water (50 ml) + a 50% NaOH solution (1 ml), dried, filtered and the solvent was evaporated. The residue was stirred in 2-propanol (100 ml). This mixture was acidified with HCl/2-propanol (7.2 ml; 5.29 N). The mixture was stirred for 16 hours at room temperature and the resulting precipitate was filtered off, washed with 2-propanol (15 ml) and dried (vacuum; 50 °C), yielding 12.6 g (62%) of 4-amino-5-chloro-2,3-dihydro-M-[ 1 -(3-methoxypropyl)-4-piperidinyl]-7- benzofurancarboxamide monohydrochloride (comp. 1).
Example 2
A mixture of 4-amino-5-chloro-2,3-dihydro-N-(4-piperidinyl)-7-benzofuran- carboxamide(O.Olmol), l-chloro-3-methoxypropane (0.012mol), M,M-diethyl- ethanamine (2Jml) and KI (catalytic amount) in N,M-dimethylformamide (75ml) was stirred overnight at 50°C. The reaction mixture was cooled. The solvent was evaporated. The residue was purified by column chromatography over silica gel (eluent: CHCl3/(CH3OH/NH3) 97/3). The pure fractions were collected and the solvent was evaporated. The residue was dissolved in 2-propanol and converted into the hydrochloric acid salt (1:1) with HCl/2-propanol. The precipitate was filtered off and dried (vacuum; 80°C), yielding 1.40g (35%) of 4-amino-5-chloro-2,3-dihydro-N-[l-(3-methoxypropyl)- 4-piperidinyl]-7-benzofurancarboxamide monohydrochloride (comp. 1).
PAPER
Chinese Journal of Pharmaceuticals 2012, 43, 5-8.
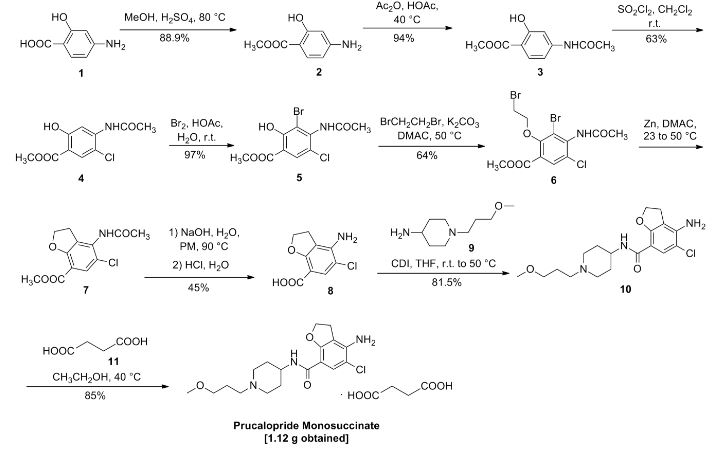
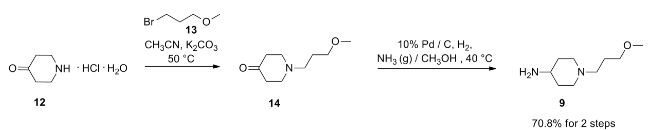
CLIP
Chinese Patent CN 103012337 A report is as follows:

PAPER
Pharmaceutical & Clinical Research 2011, 19, 306-307.
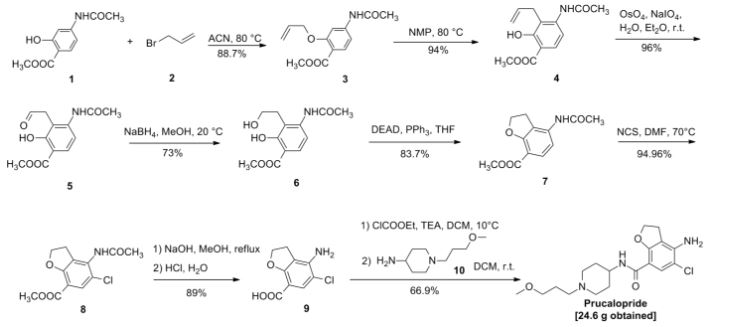
CLIP
US5374637 (CN1045781, EP389037) and J. Het Chem, 1980,17 (6): 1333-5 reported synthetic route, as follows:

CLIP
Chinese Patent CN 104016949 A synthetic route reported as follows:

PATENT
CN104529960A
https://www.google.com/patents/CN104529960A?cl=zh

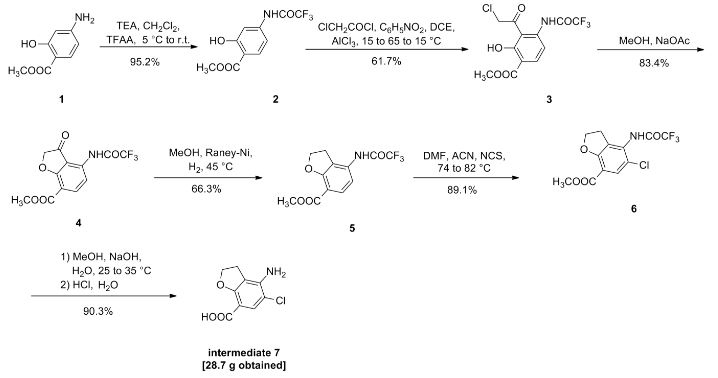 .
.

Example 1
1. Preparation of Compound II
Compound I (167. lg, Imol), triethylamine (111. lg, I. Imol) and methylene chloride (KMOg) added to the reaction flask, nitrogen cooled to 5 ° C, was slowly added dropwise trifluoroacetic anhydride (220. 5g, 1.05mol) / methylene chloride (150g) solution, maintaining the temperature throughout 5~15 ° C, dropping was completed, the reaction after 3 hours at room temperature, TLC (DCM = MeOH = 25: 1) The reaction was monitored to complete the reaction; the reaction mixture was slowly poured into ice water (560g) and stirred for 20 minutes, standing layer, the aqueous phase was separated, the organic phase was washed with saturated aqueous sodium bicarbonate (IOOg) wash sash; IM hydrochloric acid (IlOg) wash sash, then with saturated brine (200g) washed sash, magnesium sulfate (40g) dried, filtered and concentrated to give compound II (250. Ig), yield: 952%.
[0066] 2. Preparation of Compound III
[0067] Chloroacetyl chloride (101. 7g, 0. 9mol), nitrobenzene (20g) and dichloroethane (580 g) added to the reaction flask, nitrogen cooled to 5 ° C, was slowly added anhydrous trichloro aluminum powder (359. 2g, 2. 7mol), to keep the whole temperature 5~20 ° C, plus complete, insulation 15~25 ° C for 30 minutes to obtain a mixture A.
[0068] Compound II (. 236. 7g, 0 9mol) and dichloroethane (500g) added to the reaction flask, nitrogen cooled to 15 ° C; the mixture was added Compound II A quick solution, plus complete, rapid heating 65~75 ° C, 1 hours later once every 15 minutes in the control, monitoring TLC (DCM = MeOH = 50: 1) to complete the reaction; the reaction mixture was immediately poured into ice water (800g) and stirred for 30 minutes, controlling the temperature between 15~25 ° C, the organic phase was separated, the organic phase washed with water (180g) was washed with saturated brine (240g), dried over magnesium sulfate (45g) was dried, filtered and concentrated to give crude compound III (303 . 2g).
[0069] Take the crude compound III (291. 3g) / ethanol 1 dichloromethane: 1 solution (1500ml) was dissolved, and then adding activated carbon (14. 5g) was refluxed for one hour, cooled to room temperature filtered and the filtrate concentrated at room temperature to 600~ 650g, stop and concentrated down to 5~10 ° C, filtered to give a yellow solid (204. 7g); the resulting yellow solid (207. 6g) in tetrahydrofuran (510g) was purified, reduced to 10~15 ° C, filtered, The filter cake was washed with tetrahydrofuran (90g) dip, dried under vacuum to give compound III (181. 3g), yield: 61.7% billion
[0070] 3. Preparation of Compound IV
[0071] Compound 111 (! 169.68,0.5 11〇1), methanol (5,801,111) and sodium acetate (123.38,1.5111〇1) was added to the reaction flask. After 6 hours of reaction, began TLC (DCM: MeOH = 30: 1 ) the reaction was monitored to completion of the reaction; the reaction mixture was cooled to room temperature, concentrated, and the residue with ethyl acetate (500g) and water (200g) was dissolved, the organic phase was separated, the organic phase was washed with 2M sodium hydrogen carbonate (120g) was washed, then with saturated brine (IOOg), dried over magnesium sulfate (50g) was dried, filtered and concentrated to 250~280g, cooled to room temperature with stirring was added cyclohexane (200 g of), after stirring for 1 hour and then filtered and dried to obtain compound IV (126. 7g), yield: 83.4% billion
[0072] 4. Preparation of Compound V
[0073] Compound IV (12L 2g, 0. 4mol), methanol (380g) and Raney-Ni (12. 5g) added to the autoclave, purged with nitrogen, hydrogen is introduced (3. Ompa), the reaction was heated to 45 ° C after 8 hours, TLC (DCM = MeOH = 30: 1) to monitor the reaction, to complete the reaction, cooled to room temperature and pressure, and then purged with nitrogen, the reaction solution was filtered and concentrated to give crude compound V (103. 7g), taking compound V crude product (103g) was refluxed with ethyl acetate (420g) (1 hour) was purified, cooled to room temperature and stirred for 30 minutes and filtered to give a yellow solid was dried in vacuo to give compound V (76 8g.), yield: 663 %.
[0074] 5. Preparation of Compound VI
[0075] Compound ¥ (57.88,0.2111〇1), 1 ^ dimethylformamide (4.58) and acetonitrile (30 (^) was added to the reaction flask and heated 74~76 ° C; solution of N- chlorosuccinimide imide (. 26. 7g, 0 2mol) and acetonitrile (45g) was added dropwise over 30 minutes and maintaining the temperature finished 76~82 ° C, dropping was completed, the reaction was kept, after one hour the reaction started TLC (DCM: MeOH = 30: 1) to monitor the reaction, the reaction is complete the reaction solution cooled to 5~8 ° C, the filter cake was washed with water (210g) washed stirred, filtered, and dried in vacuo to give compound VI (57. 6g), yield. rate of 89.1%.
6. Preparation of Compound VII
Compound VI (48. 5g, 0. 15mol) and methanol (80g) added to the reaction flask, stirring at room temperature was added dropwise 4M aqueous sodium hydroxide (HOg), dropwise complete, for the reaction, 25 ° C~35 after 4 hours of reaction ° C, samples of about 7:00 adjust PH TLC (DCM = MeOH = 30: 1) to monitor the reaction, until the reaction was complete, down to 5~10 ° C, with 6M hydrochloric acid solution PH ~ 7. 5, half the solution was concentrated, then 2M hydrochloric acid solution PH ~ 7, reduced to 15~20 ° C was stirred for 30 minutes, filtered, the filter cake with methyl tert-butyl ether (70g) beating, filtration, and dried in vacuo to give compound VII (28. 7g), yield: 903%.
PAPER
Chem Pharm Bull 46 (1), 42-52 (1998) and Pharmaceutical and clinical study based on 2011 (4) 306-307 reported synthetic route is as follows:

Biological Activity
| Description | Prucalopride is a selective, high affinity 5-HT4 receptor agonist, inhibiting human 5-HT(4a) and 5-HT(4b) receptor with Ki value of 2.5 nM and 8 nM, respectively. | |||||
|---|---|---|---|---|---|---|
| Targets | 5-HT4A [1] | 5-HT4B [1] | ||||
| IC50 | 2.5 nM(Ki) | 8 nM(Ki) | ||||
| In vitro | Prucalopride induces contractions in a concentration-dependent manner with pEC50 of 7.5. Prucalopride (1 mM) significantly amplifies the rebound contraction of the guinea-pig proximal colon after electrical field stimulation. Prucalopride induces relaxation of the rat oesophagus preparation of rat oesophagus tunica muscularis mucosae with pEC50 of 7.8, yielding a monophasic concentration–response curve. [1] Prucalopride (0.1 μM) concentration-dependently increases the amplitude of submaximal cholinergic contractions and of acetylcholine release induced by electrical field stimulation in pig gastric circular muscle, and the effect is induced and enhanced IBMX (10 μM). [2] Prucalopride (1 μM) significantly enhances the electrically induced cholinergic contractions in pig descending colon, and the facilitating effect is significantly enhanced by Rolipram. [3] | |||||
| In vivo | Prucalopride alters colonic contractile motility patterns in a dose-dependent fashion by stimulating high-amplitude clustered contractions in the proximal colon and by inhibiting contractile activity in the distal colon of fasted dogs. Prucalopride also causes a dose-dependent decrease in the time to the first giant migrating contraction (GMC); at higher doses of prucalopride, the first GMC generally occurres within the first half-hour after treatment. [4] | |||||
| Features | ||||||
Conversion of different model animals based on BSA (Value based on data from FDA Draft Guidelines)
| Species | Mouse | Rat | Rabbit | Guinea pig | Hamster | Dog |
| Weight (kg) | 0.02 | 0.15 | 1.8 | 0.4 | 0.08 | 10 |
| Body Surface Area (m2) | 0.007 | 0.025 | 0.15 | 0.05 | 0.02 | 0.5 |
| Km factor | 3 | 6 | 12 | 8 | 5 | 20 |
| Animal A (mg/kg) = Animal B (mg/kg) multiplied by | Animal B Km |
| Animal A Km |
For example, to modify the dose of resveratrol used for a mouse (22.4 mg/kg) to a dose based on the BSA for a rat, multiply 22.4 mg/kg by the Km factor for a mouse and then divide by the Km factor for a rat. This calculation results in a rat equivalent dose for resveratrol of 11.2 mg/kg.
| Rat dose (mg/kg) = mouse dose (22.4 mg/kg) × | mouse Km(3) | = 11.2 mg/kg |
| rat Km(6) |
1
References
[1] Briejer MR, et al. Eur J Pharmacol, 2001, 423(1), 71-83.
[2] Priem E, et al. Neuropharmacology, 2012, 62(5-6), 2126-2135.
Clinical Trial Information( data from http://clinicaltrials.gov, updated on 2016-07-23)
| NCT Number | Recruitment | Conditions | Sponsor /Collaborators |
Start Date | Phases |
|---|---|---|---|---|---|
| NCT02806206 | Not yet recruiting | Gastrointestinal Hemorrhage|Crohn Disease|Celiac Disease|Intestinal Diseases|Inflammatory Bowel Diseases | University of British Columbia | July 2016 | Phase 4 |
| NCT02781493 | Not yet recruiting | Prucalopride Plus Polyethylene Glycol in Bowel Preparation for Colonoscopyp | Shandong University|Binzhou Peoples Hospital|Taian People …more | June 2016 | Phase 4 |
| NCT02538367 | Recruiting | Functional Constipation | Yuhan Corporation | August 2015 | Phase 1|Phase 2 |
| NCT02228616 | Recruiting | Constipation | Xian-Janssen Pharmaceutical Ltd. | October 2014 | Phase 4 |
| NCT02425774 | Recruiting | Postoperative Ileus | Katholieke Universiteit Leuven|Universitaire Ziekenhuizen …more | July 2014 | Phase 4 |
References
- Briejer, M. R.; Bosmans, J. P.; Van Daele, P.; Jurzak, M.; Heylen, L.; Leysen, J. E.; Prins, N. H.; Schuurkes, J. A. (2001). “The in vitro pharmacological profile of prucalopride, a novel enterokinetic compound”. European Journal of Pharmacology 423 (1): 71–83.doi:10.1016/S0014-2999(01)01087-1. PMID 11438309.
- Clinical trial number [1] for “NCT00793247” at ClinicalTrials.gov
- Emmanuel, A. V.; Kamm, M. A.; Roy, A. J.; Kerstens, R.; Vandeplassche, L. (2012).“Randomised clinical trial: The efficacy of prucalopride in patients with chronic intestinal pseudo-obstruction – a double-blind, placebo-controlled, cross-over, multiple n = 1 study”.Alimentary Pharmacology & Therapeutics 35 (1): 48–55. doi:10.1111/j.1365-2036.2011.04907.x. PMC 3298655. PMID 22061077.
- Smart, C. J.; Ramesh, A. N. (2011). “The successful treatment of acute refractory pseudo-obstruction with Prucalopride”. Colorectal Disease: no. doi:10.1111/j.1463-1318.2011.02929.x.
- Jump up^ Bouras, E. P.; Camilleri, M.; Burton, D. D.; McKinzie, S. (1999). “Selective stimulation of colonic transit by the benzofuran 5HT4 agonist, prucalopride, in healthy humans”. Gut44 (5): 682–686. doi:10.1136/gut.44.5.682. PMC 1727485. PMID 10205205.
- Jump up^ Bouras, E. P.; Camilleri, M.; Burton, D. D.; Thomforde, G.; McKinzie, S.; Zinsmeister, A. R. (2001). “Prucalopride accelerates gastrointestinal and colonic transit in patients with constipation without a rectal evacuation disorder”. Gastroenterology 120 (2): 354–360.doi:10.1053/gast.2001.21166. PMID 11159875.
- ^ Jump up to:a b c d Tack, J.; Van Outryve, M.; Beyens, G.; Kerstens, R.; Vandeplassche, L. (2008). “Prucalopride (Resolor) in the treatment of severe chronic constipation in patients dissatisfied with laxatives”. Gut 58 (3): 357–365. doi:10.1136/gut.2008.162404.PMID 18987031.
- European Medicines Agency -EPAR
- Health Canada, Notice of Decision for Resotran
- Digestive Remedies in Israel
- Briejer, M. R.; Prins, N. H.; Schuurkes, J. A. (2001). “Effects of the enterokinetic prucalopride (R093877) on colonic motility in fasted dogs”. Neurogastroenterology and motility : the official journal of the European Gastrointestinal Motility Society 13 (5): 465–472. doi:10.1046/j.1365-2982.2001.00280.x. PMID 11696108.
- Oustamanolakis, P.; Tack, J. (2012). “Prucalopride for chronic intestinal pseudo-obstruction”. Alimentary Pharmacology & Therapeutics 35 (3): 398–9. doi:10.1111/j.1365-2036.2011.04947.x. PMID 22221087.
- SmPC. Summary of product characteristics Resolor (prucalopride) October, 2009: 1-9.
- De Maeyer, JH; Lefebvre, RA; Schuurkes, JA (Feb 2008). “5-HT(4) receptor agonists: similar but not the same”. Neurogastroenterol Motil 20 (2): 99–112. doi:10.1111/j.1365-2982.2007.01059.x. PMID 18199093.
- Frampton, J. E. (2009). “Prucalopride”. Drugs 69 (17): 2463–2476.doi:10.2165/11204000-000000000-00000. PMID 19911858.
- Camilleri, M.; Kerstens, R.; Rykx, A.; Vandeplassche, L. (2008). “A Placebo-Controlled Trial of Prucalopride for Severe Chronic Constipation”. New England Journal of Medicine 358 (22): 2344–2354. doi:10.1056/NEJMoa0800670. PMID 18509121.
- ^ Jump up to:a b c Quigley, E. M. M.; Vandeplassche, L.; Kerstens, R.; Ausma, J. (2009). “Clinical trial: the efficacy, impact on quality of life, and safety and tolerability of prucalopride in severe chronic constipation – a 12-week, randomized, double-blind, placebo-controlled study”.Alimentary Pharmacology & Therapeutics 29 (3): 315–328. doi:10.1111/j.1365-2036.2008.03884.x. PMID 19035970.
- Marquis, P.; De La Loge, C.; Dubois, D.; McDermott, A.; Chassany, O. (2005). “Development and validation of the Patient Assessment of Constipation Quality of Life questionnaire”. Scandinavian Journal of Gastroenterology 40 (5): 540–551.doi:10.1080/00365520510012208. PMID 16036506.
- Frank, L.; Kleinman, L.; Farup, C.; Taylor, L.; Miner Jr, P. (1999). “Psychometric validation of a constipation symptom assessment questionnaire”. Scandinavian journal of gastroenterology 34 (9): 870–877. doi:10.1080/003655299750025327.PMID 10522604.
- Johanson, JF; Kralstein, J (2007). “Chronic constipation: a survey of the patient perspective.”. Alimentary pharmacology & therapeutics 25 (5): 599–608. doi:10.1111/j.1365-2036.2006.03238.x. PMID 17305761.
- Koch, A.; Voderholzer, W. A.; Klauser, A. G.; Müller-Lissner, S. (1997). “Symptoms in chronic constipation”. Diseases of the colon and rectum 40 (8): 902–906.doi:10.1007/BF02051196. PMID 9269805.
- McCrea, G. L.; Miaskowski, C.; Stotts, N. A.; MacEra, L.; Paul, S. M.; Varma, M. G. (2009). “Gender differences in self-reported constipation characteristics, symptoms, and bowel and dietary habits among patients attending a specialty clinic for constipation”.Gender Medicine 6 (1): 259–271. doi:10.1016/j.genm.2009.04.007. PMID 19467522.
- Pare, P.; Ferrazzi, S.; Thompson, W. G.; Irvine, E. J.; Rance, L. (2001). “An epidemiological survey of constipation in Canada: definitions, rates, demographics, and predictors of health care seeking”. The American Journal of Gastroenterology 96 (11): 3130–3137. doi:10.1111/j.1572-0241.2001.05259.x. PMID 11721760.
- Wald, A.; Scarpignato, C.; Kamm, M. A.; Mueller-Lissner, S.; Helfrich, I.; Schuijt, C.; Bubeck, J.; Limoni, C.; Petrini, O. (2007). “The burden of constipation on quality of life: results of a multinational survey”. Alimentary Pharmacology & Therapeutics 26 (2): 227–236. doi:10.1111/j.1365-2036.2007.03376.x. PMID 17593068.
- Camilleri, M; Beyens, G; Kerstens, R; Vandeplassche, L (2009). “Long-term follow-up of safety and satisfaction with bowel function in response to oral prucalopride in patients with chronic constipation [Abstract]”. Gastroenterology 136 (Suppl 1): 160. doi:10.1016/s0016-5085(09)60143-8.
- Van Outryve, MJ; Beyens, G; Kerstens, R; Vandeplassche, L (2008). “Long-term follow-up study of oral prucalopride (Resolor) administered to patients with chronic constipation [Abstract T1400]”. Gastroenterology 134 (4 (suppl 1)): A547. doi:10.1016/s0016-5085(08)62554-8.
- https://www.shire.com/newsroom/2015/june/resolor-eu-male-indication-press-release
External links
| EP0389037A1 * | 13 Mar 1990 | 26 Sep 1990 | Janssen Pharmaceutica N.V. | N-(3-hydroxy-4-piperidinyl)(dihydrobenzofuran, dihydro-2H-benzopyran or dihydrobenzodioxin)carboxamide derivatives |
| EP0445862A2 * | 22 Feb 1991 | 11 Sep 1991 | Janssen Pharmaceutica N.V. | N-(4-piperidinyl)(dihydrobenzofuran or dihydro-2H-benzopyran)carboxamide derivatives |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO1999058527A2 * | 13 May 1999 | 18 Nov 1999 | EGIS Gyógyszergyár Rt. | Benzofuran derivatives, pharmaceutical composition containing the same, and a process for the preparation of the active ingredient |
| WO1999058527A3 * | 13 May 1999 | 27 Jan 2000 | Bela Agai | Benzofuran derivatives, pharmaceutical composition containing the same, and a process for the preparation of the active ingredient |
| WO2000030640A1 * | 16 Nov 1999 | 2 Jun 2000 | Janssen Pharmaceutica N.V. | Use of prucalopride for the manufacture of a medicament for the treatment of dyspepsia |
| WO2000066170A1 * | 20 Apr 2000 | 9 Nov 2000 | Janssen Pharmaceutica N.V. | Prucalopride oral solution |
| WO2003059906A1 * | 13 Jan 2003 | 24 Jul 2003 | Janssen Pharmaceutica N.V. | Prucalopride-n-oxide |
| WO2012116976A1 | 28 Feb 2012 | 7 Sep 2012 | Shire – Movetis Nv | Prucalopride oral solution |
| WO2013024164A1 | 17 Aug 2012 | 21 Feb 2013 | Shire Ag | Combinations of a 5-ht4 receptor agonist and a pde4 inhibitor for use in therapy |
| US6413988 | 20 Apr 2000 | 2 Jul 2002 | Janssen Pharmaceutica N.V. | Prucalopride oral solution |
| US8063069 | 30 Oct 2007 | 22 Nov 2011 | Janssen Pharmaceutica N.V. | Prucalopride-N-oxide |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2016082123 | 2016-03-24 | Hydrogel-Linked Prodrugs Releasing Tagged Drugs |
| US2015202317 | 2015-07-23 | DIPEPTIDE-BASED PRODRUG LINKERS FOR ALIPHATIC AMINE-CONTAINING DRUGS |
| US2014323402 | 2014-10-30 | Protein Carrier-Linked Prodrugs |
| US2014296257 | 2014-10-02 | High-Loading Water-Soluable Carrier-Linked Prodrugs |
| US2014243254 | 2014-08-28 | Polymeric Hyperbranched Carrier-Linked Prodrugs |
| US2013053301 | 2013-02-28 | DIPEPTIDE-BASED PRODRUG LINKERS FOR ALIPHATIC AMINE-CONTAINING DRUGS |
| US2012220630 | 2012-08-30 | PRUCALOPRIDE ORAL SOLUTION |
| US2012156259 | 2012-06-21 | Biodegradable Polyethylene Glycol Based Water-Insoluble Hydrogels |
| US6413988 | 2002-07-02 | Prucalopride oral solution |
| US6310077 | 2001-10-30 | Enterokinetic benzamide |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-Amino-5-chloro-N-[1-(3-methoxypropyl)piperidin-4-yl]-2,3-dihydro-1-benzofuran-7-carboxamide
|
|
| Clinical data | |
| Trade names | Resolor, Resotran |
| AHFS/Drugs.com | International Drug Names |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Oral |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | 179474-81-8 |
| ATC code | A06AX05 (WHO) |
| PubChem | CID 3052762 |
| IUPHAR/BPS | 243 |
| ChemSpider | 2314539 |
| UNII | 0A09IUW5TP |
| Chemical data | |
| Formula | C18H26ClN3O3 |
| Molar mass | 367.870 g/mol |
//////////Prucalopride succinate, Resolor, R-093877, R-108512, Resolor®, Resolor, Resotran, UNII:0A09IUW5TP, 179474-81-8 , R-093877, R-108512, Shire , Johnson & Johnson, 179474-85-2, UNII-4V2G75E1CK, SHIRE, 2010, LAUNCHED, JANNSEN , PHASE 3, IRRITABLE BOWL SYNDROME
COCCCN1CCC(CC1)NC(=O)C2=CC(=C(C3=C2OCC3)N)Cl
Delamanid, (Deltyba) デラマニド

Delamanid
デラマニド
MKT as Deltyba® by Otsuka Pharmaceutical
http://www.ama-assn.org/resources/doc/usan/delamanid.pdf
(2R)-2-Methyl-6-nitro-2-[(4-{4-[4-(trifluoromethoxy)phenoxy]-1-piperidinyl}phenoxy)methyl]-2,3-dihydroimidazo[2,1-b][1,3]oxazole
2(R)-Methyl-6-nitro-2-[4-[4-[4-(trifluoromethoxy)phenoxy]piperidin-1-yl]phenoxymethyl]-2,3-dihydroimidazo[2,1-b]oxazole
(R) -2-methyl-6-nitro-2- { 4- [4- (4- trifluoromethoxyphenoxy) piperidin-l-yl] phenoxymethyl } -2 , 3- dihydroimidazo [2 , 1-b] oxazole
Imidazo[2,1-b]oxazole, 2,3-dihydro-2-methyl-6-nitro-2-[[4-[4-[4-(trifluoromethoxy)phenoxy]-1-piperidinyl]phenoxy]methyl]-, (2R)-
(R)-2-methyl-6-nitro-2-{4-[4-(4-trifluoromethoxyphenoxy)piperidin-1-yl]phenoxymethyl}-2,3-dihydroimidazo[2,1-b]oxazole
| (2R)-2-Methyl-6-nitro-2-[(4-{4-[4-(trifluoromethoxy)phenoxy]piperidin-1-yl}phenoxy)methyl]-2,3-dihydroimidazo[2,1-b]oxazole |
681492-22-8 CAS

Delamanid, 681492-22-8, Delamanid (JAN/USAN), Delamanid [USAN:INN],UNII-8OOT6M1PC7,
- OPC 67683
- OPC-67683
- UNII-8OOT6M1PC7
MW: C25H25F3N4O6
MW: 534.48441
Clinical trials……….http://clinicaltrials.gov/search/intervention=OPC+67683+OR+Delamanid
CLINICAL TRIALS
Primary Sponsor: Otsuka Pharmaceutical Development & Commercialization, Inc.
Trial ID / Reg # / URL: http://clinicaltrials.gov/ct2/show/NCT00685360

Delamanid

C25H25F3N4O6 : 534.48
[681492-22-8]
C25H25F3N4O6 : 534.48
[681492-22-8]
Delamanid (USAN, INN) is a drug for the treatment of multi-drug-resistant tuberculosis. It works by blocking the synthesis of mycolic acids in Mycobacterium tuberculosis, the organism which causes tuberculosis, thus destabilising its cell wall.[2][3][4] The drug is approved in the EU under the trade name Deltyba (made by Otsuka Pharmaceutical).
It is on the World Health Organization’s List of Essential Medicines, the most important medications needed in a basichealth system.[5]
Adverse effects
Delamanid prolongs QT interval.[6]
Interactions
Delamanid is metabolised by the liver enzyme CYP3A4, wherefore strong inducers of this enzyme can reduce its effectiveness.[6]
History
In phase II clinical trials, the drug was used in combination with standard treatments, such as four or five of the drugsethambutol, isoniazid, pyrazinamide, rifampicin, aminoglycoside antibiotics, and quinolones. Healing rates (measured as sputum culture conversion) were significantly better in patients who additionally took delamanid.[4][7]
The European Medicines Agency (EMA) recommended conditional marketing authorization for delamanid in adults with multidrug-resistant pulmonary tuberculosis without other treatment options because of resistance or tolerability. The EMA considered the data show that the benefits of delamanid outweigh the risks, but that additional studies were needed on the long-term effectiveness.[8]
Delamanid was first approved by European Medicine Agency (EMA) on Apr 28, 2014, then approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on July 4, 2014. It was developed and marketed as Deltyba® by Otsuka Pharmaceutical.
Delamanid is a novel bactericidal agent that interferes with the metabolism of the mycobacterium tuberculosis (MTB) cell walls. It is indicated for the treatment of pulmonary multi-drugresistant tuberculosis (MDR-TB) in adult patients.
Deltyba® is available as tablets for oral use, containing 50 mg of free Delamanid, and the recommended dose is 100 mg twice daily for 24 weeks.
Delamanid, an antibiotic active against Mycobacterium tuberculosis strains, has been filed for approval in the E.U. and by Otsuka for the treatment of multidrug-resistant tuberculosis. In 2013, a positive opinion was received in the E.U. for this indication. Phase III trials for treatment of multidrug-resistant tuberculosis are under way in the U.S. Phase II study for the pediatric use is undergone in the Europe.
The drug candidate’s antimycobacterial mechanism of action is via specific inhibition of the synthesis pathway of mycolic acid, which is a cell wall component unique to M. tuberculosis.
In 2008, orphan drug designation was received in Japan for the treatment of pulmonary tuberculosis.
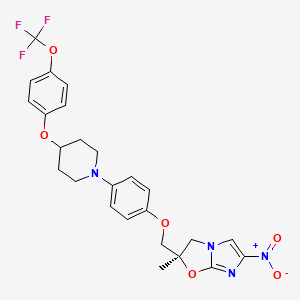
Tuberculosis (TB), an airborne lung infection, still remains a major public health problem worldwide. It is estimated that about 32% of the world population is infected with TB bacillus, and of those, approximately 8.9 million people develop active TB and 1.7 million die as a result annually according to 2004 figures. Human immunodeficiency virus (HIV) infection has been a major contributing factor in the current resurgence of TB. HIV-associated TB is widespread, especially in sub-Saharan Africa, and such an infectious process may further accelerate the resurgence of TB.
Moreover, the recent emergence of multidrug-resistant (MDR) strains ofMycobacterium tuberculosis that are resistant to two major effective drugs, isonicotinic acid hydrazide (INH) and rifampicin (RFP), has further complicated the world situation.
The World Health Organization (WHO) has estimated that if the present conditions remain unchanged, more than 30 million lives will be claimed by TB between 2000 and 2020. As for subsequent drug development, not a single new effective compound has been launched as an antituberculosis agent since the introduction of RFP in 1965, despite the great advances that have been made in drug development technologies.
Although many effective vaccine candidates have been developed, more potent vaccines will not become immediately available. The current therapy consists of an intensive phase with four drugs, INH, RFP, pyrazinamide (PZA), and streptomycin (SM) or ethambutol (EB), administered for 2 months followed by a continuous phase with INH and RFP for 4 months. Thus, there exists an urgent need for the development of potent new antituberculosis agents with low-toxicity profiles that are effective against both drug-susceptible and drug-resistant strains of M. tuberculosis and that are capable of shortening the current duration of therapy.
PATENT
(R)-2-bromo-4-nitro-1-(2-methyl-2-oxiranylmethyl)imidazole
4-[4-(4-Trifluoromethoxyphenoxy)piperidin-1-yl]phenol
ARE THE INTERMEDIATES
Example 1884
Production of (R)-2-methyl-6-nitro-2-{4-[4-(4-trifluoromethoxyphenoxy)piperidin-1-yl]phenoxymethyl}-2,3-dihydroimidazo[2,1-b]oxazole
4-[4-(4-Trifluoromethoxyphenoxy)piperidin-1-yl]phenol (693 mg, 1.96 mmol) was dissolved in N,N′-dimethylformamide (3 ml), and sodium hydride (86 mg, 2.16 mmol) was added while cooling on ice followed by stirring at 70-75° C. for 20 minutes. The mixture was cooled on ice. To the solution, a solution prepared by dissolving (R)-2-bromo-4-nitro-1-(2-methyl-2-oxiranylmethyl)imidazole (720 mg, 2.75 mmol) in N,N′-dimethylformamide (3 ml) was added followed by stirring at 70-75° C. for 20 minutes. The reaction mixture was allowed to return to room temperature, ice water (25 ml) was added, and the resultant solution was extracted with methylene chloride (50 ml) three times. The organic phases were combined, washed with water 3 times, and dried over magnesium sulfate. After filtration, the filtrate was concentrated, and the residue was purified by silica gel column chromatography (methylene chloride/ethyl acetate=3/1). Recrystallization from ethyl acetate/isopropyl ether gave (R)-2-methyl-6-nitro-2-{4-[4-(4-trifluoromethoxyphenoxy)piperidin-1-yl]phenoxymethyl}-2,3-dihydroimidazo[2,1-b]oxazole (343 mg, 33%) as a light yellow powder.
PATENT
WO 2010021409 AND http://worldwide.espacenet.com/publicationDetails/biblio?CC=IN&NR=203704A1&KC=A1&FT=D
FOR 2, 4 DINITROIMIDAZOLE
PATENT
These patent literatures disclose Reaction Schemes A and B below as the processes for producing the aforementioned 2, 3-dihydroimidazo [2, 1-b] oxazole compound.
Reaction Scheme A:

wherein R1 is a hydrogen atom or lower-alkyl group; R2 is a substituted pxperidyl group or a substituted piperazinyl group; and X1 is a halogen atom or a nitro group.
Reaction Scheme B:


wherein X2 is a halogen or a group causing a substitution reaction similar to that of a halogen; n is an integer from 1 to 6; and R1, R2 and X1 are the same as in Reaction Scheme A.
An oxazole com ound represented by Formula (la) :

, i.e., 2-methyl-6-nitro-2-{4- [4- (4- trifluoromethoxyphenoxy) piperidin-l-yl] phenoxymethyl }-2, 3- dihydroimidazo [2, 1-b] oxazole (hereunder, this compound may be simply referred to as “Compound la”) is produced, for example, by the method shown in the Reaction Scheme C below (Patent
Literature 3) . In this specification, the term “oxazole compound’ means an oxazole derivative that encompasses compounds that contain an oxazole ring or an oxazoline ring (dihydrooxazole ring) in the molecule.
Reaction Scheme C:


However, the aforementioned methods are unsatisfactory in terms of the yield of the objective compound. For example, the method of Reaction Scheme C allows the objective oxazole Compound (la) to be obtained from Compound (2a) at a yield as low as 35.9%. Therefore, alternative methods for producing the compound in an industrially advantageous manner are desired. Citation List
Patent Literature
PTL 1: WO2004/033463
PTL 2: WO2004/035547
PTL 3: WO2008/140090
Example 9
Production of (R) -2-methyl-6-nitro-2- { 4- [4- (4- trifluoromethoxyphenoxy) piperidin-l-yl] phenoxymethyl } -2 , 3- dihydroimidazo [2 , 1-b] oxazole
{R) -1- [ – {2 , 3-epoxy-2-methylpropoxy ) phenyl] -4- [4- ( trifluoromethoxy ) phenoxy ] piperidine (10.0 g, 23.6 mmol, optical purity of 94.3%ee), 2-chloro-4-nitroimidazole (4.0 g, 27.2 mmol), sodium acetate (0.4 g, 4.9 mmol), and t- butyl acetate (10 ml) were mixed and stirred at 100°C for 3.5 hours. Methanol (70 ml) was added to the reaction mixture, and then a 25% sodium hydroxide aqueous solution (6.3 g, 39.4 mmol) was added thereto dropwise while cooling with ice. The resulting mixture was stirred at 0°C for 1.5 hours, and further stirred at approximately room
temperature for 40 minutes. Water (15 ml) and ethyl acetate (5 ml) were added thereto, and the mixture was stirred at 45 to 55°C for 1 hour. The mixture was cooled to room temperature, and the precipitated crystals were collected by filtration. The precipitated crystals were subsequently washed with methanol (30 ml) and water (40 ml) . Methanol (100 ml) was added to the resulting
crystals, followed by stirring under reflux for 30 minutes. The mixture was cooled to room temperature. The crystals were then collected by filtration and washed with methanol (30 ml) . The resulting crystals were dried under reduced pressure, obtaining 9.3 g of the objective product (yield: 73%) .
Optical purity: 99.4%ee.
PATENT
Synthesis and antituberculosis activity of a novel series of optically active 6-nitro-2,3-dihydroimidazo[2,1-b]oxazoles
J Med Chem 2006, 49(26): 7854
http://pubs.acs.org/doi/abs/10.1021/jm060957y
(R)-2-Methyl-6-nitro-2-{4-[4-(4-trifluoromethoxyphenoxy)piperidin-1-yl]phenoxymethyl}-2,3-dihydroimidazo[2,1-b]oxazole (19, DELAMANID).
To a mixture of 27 (127.56 g, 586.56 mmol) and 4-[4-(4-trifluoromethoxyphenoxy)piperidin-1-yl]phenol (28g) (165.70 g, 468.95 mmol) in N,N-dimethylformamide (1600 mL) was added 60% sodium hydride (22.51 g, 562.74 mmol) at 0 °C portionwise. After the mixture was stirred at 50 °C for 2 h under a nitrogen atmosphere, the reaction mixture was cooled in an ice bath and carefully quenched with ethyl acetate (230 mL) and ice water (50 mL). The thus-obtained mixture was poured into water (3000 mL) and stirred for 30 min. The resulting precipitates were collected by filtration, washed with water, and dried at 60 °C overnight. This crude product was purified by silica gel column chromatography using a dichloromethane and ethyl acetate mixture (5/1) as solvent. The appropriate fractions were combined and evaporated under reduced pressure. The residue was recrystallized from ethyl acetate (1300 mL)−isopropyl alcohol (150 mL) to afford 19 (119.11 g, 48%) as a pale yellow crystalline powder.
Mp 195−196 °C.
1H NMR (CDCl3) δ 1.77 (3H, s), 1.87−2.16 (4H, m), 2.95−3.05 (2H, m), 3.32−3.41 (2H, m), 4.02 (1H, d, J = 10.2 Hz), 4.04 (1H, d, J = 10.2 Hz), 4.18 (1H, J = 10.2 Hz), 4.36−4.45 (1H, m), 4.49 (1H, d, J = 10.2 Hz), 6.76 (2H, d, J = 6.7 Hz), 6.87−6.94 (4H, m), 7.14 (2H, d, J = 8.6 Hz), 7.55 (1H, s).
[α  −9.9° (c 1.01, CHCl3).
−9.9° (c 1.01, CHCl3).
MS (DI) m/z 535 (M+ + 1). Anal. (C25H25F3N4O6) C, H, N.
http://pubs.acs.org/doi/suppl/10.1021/jm060957y/suppl_file/jm060957ysi20061113_095044.pdf
CLIPS
Delamanid (Deltyba)
Marketed by Otsuka, delamanid was approved in both the European Union and Japan in 2014 as part of combination therapies for
multi-drug resistant tuberculosis (TB). Because delamanid exhibited no adverse drug–drug interactions, it has found utility as a
combination therapy with standard antiretroviral drugs indicated for TB. Delamanid blocks mycolic acid biosynthesis in ycobacterium
tuberculosis, which allows its cell wall to be penetrated by small molecule antivirals.92
Although delamanid possesses a rather linear structure capable of a variety of retrosynthetic disconnections, the most likely scale
synthesis is a convergent approach involving two key synthons—diol 82 and piperidine 81, as is outlined in Scheme 13.93–95
Preparation of 82 proceeded through a Sharpless Asymmetric Epoxidation of commercial alcohol 86, followed by a diastereoselective
epoxide ring opening with 4-bromophenol to afford key diol 82 in 76% for the two step sequence (Scheme 14).93–96
Piperidine 81 was concurrently prepared by first generating biaryl ether 79, which arose from a substitution reaction between
pyridine N-oxide 77 and phenol 78 that proceeded in 86% yield. Next, removal of the N-oxide functionality by means of catalytic
hydrogenation under mild pressure and neutral conditions afforded diaryl ether 80 in excellent yield. Reduction of the pyridine
to the corresponding piperidine (81) was affected through the use of catalytic hydrogenation as well, this time under acidic
conditions and elevated pressures relative to the N-oxide reduction.95,97 At this juncture, subjection of piperidine 81 to Buchwald–
Hartwig conditions in the presence of diol subunit 82
(preparation described in Scheme 14) delivered diol 83. A two-step elimination to deliver enantiopure epoxide 84 set the stage for an
interesting cascade reaction to arrive at delamanid (XI) directly— the initial alkylation of the epoxide by imidazole 85 proceeded
under basic conditions with sodium acetate which then underwent an intramolecular nucleophilic substitution reaction by the liberated alcohol on the pendant imidazole chloride in the presence of sodium hydroxide. The reaction sequence proceeded in 73%
yield to provide delamanid (XI) as a free base.96
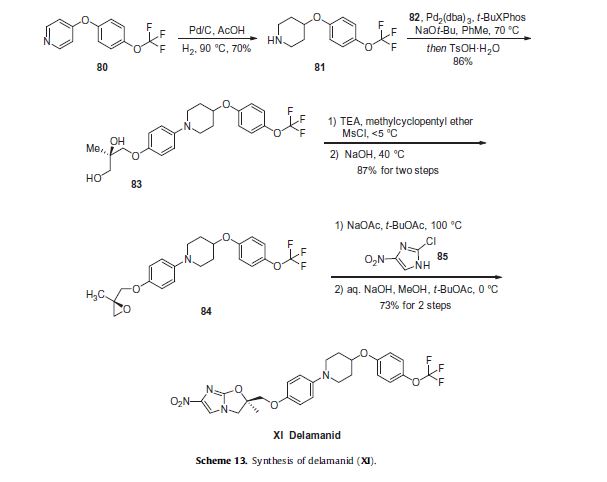
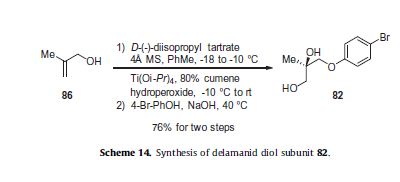
92. Blair, H. A.; Scott, L. J. Drugs 2015, 75, 91.
93. Tsubouchi, H.; Sasaki, H.; Kuroda, H.; Itotani, M.; Hasegawa, T.; Haraguchi, Y.;Kuroda, T.; Matsuzaki, T. US Patent 2006094767A1, 2006.
94. Sasaki, H.; Haraguchi, Y.; Itotani, M.; Kuroda, H.; Hashizume, H.; Tomishige,T.; Kawasaki, M.; Matsumoto, M.; Komatsu, M.; Tsubouchi, H. J. Med. Chem.2006, 49, 7854.
95. Goto, F.; Takemura, N.; Otani, T.; Hasegawa, T.; Tsubouchi, H.; Utsumi, N.; Fujita, S.; Kuroda, H.; Shitsuta, T.; Sasaki, H. US2012130082A1, 2012.
96. Yamamoto, A.; Shinhama, K.; Fujita, N.; Aki, S.; Ogasawara, S.; Utsumi, N. WOPatent 2011093529A1, 2011.
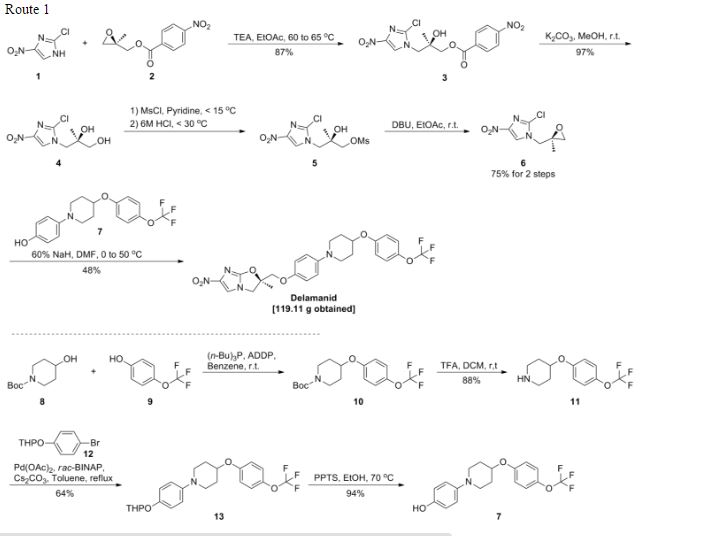

References
- “Deltyba (delamanid): Summary of Product Characteristics. 5.2. Pharmacokinetic Properties” (PDF). Otsuka Novel Products GmbH. p. 10. Retrieved 9 July 2016.
- Matsumoto, M.; Hashizume, H.; Tomishige, T.; Kawasaki, M.; Tsubouchi, H.; Sasaki, H.; Shimokawa, Y.; Komatsu, M. (2006). “OPC-67683, a Nitro-Dihydro-Imidazooxazole Derivative with Promising Action against Tuberculosis in Vitro and in Mice”. PLoS Medicine 3 (11): e466. doi:10.1371/journal.pmed.0030466. PMC 1664607. PMID 17132069.
- Skripconoka, V.; Danilovits, M.; Pehme, L.; Tomson, T.; Skenders, G.; Kummik, T.; Cirule, A.; Leimane, V.; Kurve, A.; Levina, K.; Geiter, L. J.; Manissero, D.; Wells, C. D. (2012). “Delamanid Improves Outcomes and Reduces Mortality for Multidrug-Resistant Tuberculosis”. European Respiratory Journal 41 (6): 1393–1400. doi:10.1183/09031936.00125812. PMC 3669462.PMID 23018916.
- H. Spreitzer (18 February 2013). “Neue Wirkstoffe – Bedaquilin und Delamanid”. Österreichische Apothekerzeitung (in German) (4/2013): 22.
- “WHO Model List of EssentialMedicines” (PDF). World Health Organization. October 2013. Retrieved 22 April 2014.
- Pharmazeutische Zeitung: Delamanid: Neuer Wirkstoff gegen multiresistente TB, 9 May 2014. (German)
- Gler, M. T.; Skripconoka, V.; Sanchez-Garavito, E.; Xiao, H.; Cabrera-Rivero, J. L.; Vargas-Vasquez, D. E.; Gao, M.; Awad, M.; Park, S. K.; Shim, T. S.; Suh, G. Y.; Danilovits, M.; Ogata, H.; Kurve, A.; Chang, J.; Suzuki, K.; Tupasi, T.; Koh, W. J.; Seaworth, B.; Geiter, L. J.; Wells, C. D. (2012). “Delamanid for Multidrug-Resistant Pulmonary Tuberculosis”. New England Journal of Medicine 366 (23): 2151–2160. doi:10.1056/NEJMoa1112433. PMID 22670901.
- Drug Discovery & Development. EMA Recommends Two New Tuberculosis Treatments. November 22, 2013.
- Japan PMDA.[7]. PLoS Med. 2006 Nov;3(11):e466.[8]. Drug@EMA, EMEA/H/C/002552 Deltyba: EPAR-Assessment Report.
|
12-28-2006
|
Synthesis and antituberculosis activity of a novel series of optically active 6-nitro-2,3-dihydroimidazo[2,1-b]oxazoles.
|
Journal of medicinal chemistry
|
|
|
11-1-2006
|
OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice.
|
PLoS medicine
|
|
1-1-2008
|
New anti-tuberculosis drugs with novel mechanisms of action.
|
Current medicinal chemistry
|
|
11-11-2010
|
Synthesis and Structure-Activity Relationships of Aza- and Diazabiphenyl Analogues of the Antitubercular Drug (6S)-2-Nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824).
|
Journal of medicinal chemistry
|
|
5-1-2012
|
Tuberculosis: the drug development pipeline at a glance.
|
European journal of medicinal chemistry
|
|
|
1-12-2012
|
Structure-activity relationships for amide-, carbamate-, and urea-linked analogues of the tuberculosis drug (6S)-2-nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824).
|
Journal of medicinal chemistry
|
|
9-11-2009
|
Pharmaceutical Composition Achieving Excellent Absorbency of Pharmacologically Active Substance
|
|
|
1-16-2009
|
Sulfonamide Derivatives for the Treatment of Bacterial Infections
|
| WO2004033463A1 | Oct 10, 2003 | Apr 22, 2004 | Otsuka Pharma Co Ltd | 2,3-DIHYDRO-6-NITROIMIDAZO[2,1-b]OXAZOLES |
| WO2004035547A1 | Oct 14, 2003 | Apr 29, 2004 | Otsuka Pharma Co Ltd | 1-substituted 4-nitroimidazole compound and process for producing the same |
| WO2008140090A1 | May 7, 2008 | Nov 20, 2008 | Otsuka Pharma Co Ltd | Epoxy compound and method for manufacturing the same |
| JP2009269859A * | Title not available |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(2R)-2-Methyl-6-nitro-2-[(4-{4-[4-(trifluoromethoxy)phenoxy]-1-piperidinyl}phenoxy)methyl]-2,3-dihydroimidazo[2,1-b][1,3]oxazole
|
|
| Clinical data | |
| Trade names | Deltyba |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration |
Oral (film-coated tablets) |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Protein binding | ≥99.5% |
| Metabolism | in plasma by albumin, in liver by CYP3A4 (to a lesser extent) |
| Biological half-life | 30–38 hours |
| Excretion | not excreted in urine[1] |
| Identifiers | |
| CAS Number | 681492-22-8 |
| ATC code | J04AK06 (WHO) |
| PubChem | CID 6480466 |
| ChemSpider | 4981055 |
| ChEMBL | CHEMBL218650 |
| Synonyms | OPC-67683 |
| Chemical data | |
| Formula | C25H25F3N4O6 |
| Molar mass | 534.48 g/mol |
//////////////////////////681492-22-8 , Delamanid, Deltyba, Otsuka Pharmaceutical
FC(F)(F)Oc5ccc(OC4CCN(c3ccc(OC[C@@]2(Oc1nc(cn1C2)[N+]([O-])=O)C)cc3)CC4)cc5
TB

It is estimated that a third of the world’s population is currently infected with tuberculosis, leading to 1.6 million deaths annually. The current drug regimen is 40 years old and takes 6-9 months to administer. In addition, the emergence of drug resistant strains and HIV co-infection mean that there is an urgent need for new anti-tuberculosis drugs. The twenty-first century has seen a revival in research and development activity in this area, with several new drug candidates entering clinical trials. This review considers new potential first-line anti-tuberculosis drug candidates, in particular those with novel mechanisms of action, as these are most likely to prove effective against resistant strains.
From among acid-fast bacteria, human Mycobacterium tuberculosis has been widely known. It is said that the one-third of the human population is infected with this bacterium. In addition to the human Mycobacterium tuberculosis, Mycobacterium africanum and Mycobacterium bovis have also been known to belong to the Mycobacterium tuberoculosis group. These bacteria are known as Mycobacteria having a strong pathogenicity to humans.
Against these tuberculoses, treatment is carried out using three agents, rifampicin, isoniazid, and ethambutol (or streptomycin) that are regarded as first-line agents, or using four agents such as the above three agents and pyrazinamide.
However, since the treatment of tuberculosis requires extremely long-term administration of agents, it might result in poor compliance, and the treatment often ends in failure.
Moreover, in respect of the above agents, it has been reported that: rifampicin causes hepatopathy, flu syndrome, drug allergy, and its concomitant administration with other drugs is contraindicated due to P450-associated enzyme induction; that isoniazid causes peripheral nervous system disorder and induces serious hepatopathy when used in combination with rifampicin; that ethambutol brings on failure of eyesight due to optic nerve disorder; that streptomycin brings on diminution of the hearing faculty due to the 8th cranial nerve disorder; and that pyrazinamide causes adverse reactions such a hepatopathy, gouty attack associated with increase of uric acid level, vomiting (A Clinician’s Guide To Tuberculosis, Michael D. Iseman 2000 by Lippincott Williams & Wilkins, printed in the USA, ISBN 0-7817-1749-3, Tuberculosis, 2nd edition, Fumiyuki Kuze and Takahide Izumi, Igaku-Shoin Ltd., 1992).
Actually, it has been reported that cases where the standard chemotherapy could not be carried out due to the adverse reactions to these agents made up 70% (approximately 23%, 52 cases) of the total cases where administration of the agents was discontinued (the total 228 hospitalized patients who were subject to the research) (Kekkaku, Vol. 74, 77-82, 1999).
In particular, hepatotoxicity, which is induced by rifampicin, isoniazid, and ethambutol out of the 5 agents used in combination for the aforementioned first-line treatment, is known as an adverse reaction that is developed most frequently. At the same time, Mycobacterium tuberculosis resistant to antitubercular agents, multi-drug-resistant Mycobacterium tuberculosis, and the like have been increasing, and the presence of these types of Mycobacterium tuberculosismakes the treatment more difficult.
According to the investigation made by WHO (1996 to 1999), the proportion ofMycobacterium tuberculosis that is resistant to any of the existing antitubercular agents to the total types of Mycobacterium tuberculosis that have been isolated over the world reaches 19%, and it has been published that the proportion of multi-drug-resistant Mycobacterium tuberculosis is 5.1%. The number of carriers infected with such multi-drug-resistant Mycobacterium tuberculosis is estimated to be 60,000,000, and concerns are still rising that multi-drug-resistantMycobacterium tuberculosis will increase in the future (April 2001 as a supplement to the journal Tuberculosis, the “Scientific Blueprint for TB Drug Development.”)
In addition, the major cause of death of AIDS patients is tuberculosis. It has been reported that the number of humans suffering from both tuberculosis and HIV reaches 10,700,000 at the time of year 1997 (Global Alliance for TB drug development). Moreover, it is considered that the mixed infection of tuberculosisand HIV has an at least 30 times higher risk of developing tuberculosis than the ordinary circumstances.
Taking into consideration the aforementioned current situation, the profiles of the desired antitubercular agent is as follows: (1) an agent, which is effective even for multi-drug-resistant Mycobacterium tuberculosis, (2) an agent enabling a short-term chemotherapy, (3) an agent with fewer adverse reactions, (4) an agent showing an efficacy to latent infecting Mycobacterium tuberculosis (i.e., latentMycobacterium tuberculosis), and (5) an orally administrable agent.
Examples of bacteria known to have a pathogenicity to humans include offending bacteria of recently increasing MAC infection (Mycobacterium avium—intracellulare complex infection) such as Mycobacterium avium andMycobacterium intracellulare, and atypical acid-fast bacteria such asMycobacterium kansasii, Mycobacterium marinum, Mycobacterium simiae, Mycobacterium scrofulaceum, Mycobacterium szulgai, Mycobacterium xenopi, Mycobacterium malmoense, Mycobacterium haemophilum, Mycobacterium ulcerans, Mycobacterium shimoidei, Mycobacterium fortuitum, Mycobacterium chelonae, Mycobacterium smegmatis, and Mycobacterium aurum.
Nowadays, there are few therapeutic agents effective for these atypical acid-fast bacterial infections. Under the presence circumstances, antitubercular agents such as rifampicin, isoniazid, ethambutol, streptomycin and kanamycin, a newquinolone agent that is a therapeutic agent for common bacterial infections, macrolide antibiotics, aminoglycoside antibiotics, and tetracycline antibiotics are used in combination.
However, when compared with the treatment of common bacterial infections, the treatment of atypical acid-fast bacterial infections requires a long-term administration-of agents, and there have been reported cases where the infection is changed to an intractable one, finally leading to death. To break the afore-mentioned current situation, the development of an agent having a stronger efficacy is desired.
For example, National Publication of International Patent Application No. 11-508270 (WO97/01562) discloses that a 6-nitro-1,2,3,4-tetrahydro[2,1-b]-imidazopyran compound has a bactericidal action in vitro to Mycobacterium tuberculosis (H37Rv strain) and multi-drug-resistant Mycobacterium tuberculosis, and that the above compound has a therapeutic effect to a tuberculosis-infected animal model when it is orally administered and thus useful as antitubercular agent.
Mifamurtide (Mepact) мифамуртид , ميفامورتيد , 米法莫肽 ,
Mifamurtide (Mepact)
- MF C59H109N6O19P
- MW 1237.499
CGP-19835, MFCD09954133, MTP-cephalin, Mtp-PE
Muramyl tripeptide phosphatidylethanolamine
N-(N-Acetylmuramoyl)-L-alanyl-D-α-glutaminyl-N-[(7R)-4-hydroxy-4-oxido-10-oxo-7-[(1-oxohexadecyl)oxy]-3,5,9-trioxa-4-phosphapentacos-1-yl]-L-alaninamide
N-Acetylmuramyl-L-alanyl-D-isoglutamine-L-alanine 2-(1′,2′-dipalmitoyl-sn-glycero-3′-hydroxyphosphoryloxy)ethylamide
(2R,5S,8R,13S,22R)-2-{[(3R,4R,5S,6R)-3-Acetamido-2,5-dihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-4-yl]oxy}-8-carbamoyl-19-hydroxy-5,13-dimethyl-19-oxido-3,6,11,14,25-pentaoxo-18,20,24-trioxa-4,7,12 ;,15-tetraaza-19λ5-phosphatetracontan-22-yl hexadecanoate
Mifamurtide (trade name Mepact, marketed by Takeda) is a drug against osteosarcoma, a kind of bone cancer mainly affecting children and young adults, which is lethal in about a third of cases. The drug was approved in Europe in March 2009.
History
The drug was invented by Ciba-Geigy (now Novartis) in the early 1980s and sold to Jenner Biotherapies in the 1990s. In 2003,IDM Pharma bought the rights and developed it further.[1] IDM Pharma was acquired by Takeda along with mifamurtide in June 2009.[2]
Mifamurtide had already been granted orphan drug status by the U.S. Food and Drug Administration (FDA) in 2001, and theEuropean Medicines Agency (EMA) followed in 2004. It was approved in the 27 European Union member states plus Iceland, Liechtenstein, and Norway by a centralized marketing authorization in March 2009. The drug was denied approval by the FDA in 2007.[3][4] Mifamurtide has been licensed by the EMA since March, 2009.[5]
Indications
Mifamurtide is indicated for the treatment of high-grade, nonmetastasizing, resectable osteosarcoma following complete surgical removal in children, adolescents, and young adults, aged two to 30 years.[1][6][7] Osteosarcoma is diagnosed in about 1,000 individuals in Europe and the USA per year, most under the age of 30.[8] The drug is used in combination with postoperative, multiagent chemotherapy to kill remaining cancer cells and improve a patient’s chance of overall survival.[6]
In a phase-III clinical trial in about 800 newly diagnosed osteosarcoma patients, mifamurtide was combined with the chemotherapeutic agents doxorubicin and methotrexate, with or without cisplatin and ifosfamide. The mortality could be lowered by 30% versus chemotherapy plus placebo. Six years after the treatment, 78% of patients were still alive. This equals an absolute risk reduction of 8% .[1]
Adverse effects
In a clinical study, mifamurtide was given to 332 subjects (half of whom were under age of 16) and most side effects were found to be mild to moderate in nature. Most patients experience fewer adverse events with subsequent administration.[9][10]Common side effects include fever (about 90%), vomiting, fatigue and tachycardia (about 50%), infections, anaemia, anorexia, headache, diarrhoea and constipation(>10%).[1][11]
Pharmacokinetics
After application of the liposomal infusion, the drug is cleared from the plasma within minutes and is concentrated in lung, liver, spleen, nasopharynx, and thyroid. The terminal half-life is 18 hours. In patients receiving a second treatment after 11–12 weeks, no accumulation effects were observed.[12]
Pharmacodynamics
Mifamurtide is a fully synthetic derivative of muramyl dipeptide (MDP), the smallest naturally occurring immune stimulatory component of cell walls from Mycobacterium species. It has similar immunostimulatory effects as natural MDP with the advantage of a longer half-life in plasma.
NOD2 is a pattern recognition receptor which is found in several kinds of white blood cells, mainly monocytes and macrophages. It recognises muramyl dipeptide, a component of the cell wall of bacteria. Mifamurtide simulates a bacterial infection by binding to NOD2, activating white cells. This results in an increased production of TNF-α, interleukin 1,interleukin 6, interleukin 8, interleukin 12, and other cytokines, as well as ICAM-1. The activated white cells attack cancer cells, but not, at least in vitro, other cells.[13]
Interactions
- Theoretical considerations suggest calcineurin inhibitors like ciclosporin and tacrolimus might interact with mifamurtide because of their effect on macrophages.
- High-dose NSAIDs block the mechanism of mifamurtide in vitro.
Consequently, the combination of mifamurtide with these types of drugs is contraindicated. However, mifamurtide can be coadministered with low doses of NSAIDs. No evidence suggests mifamurtide interacts with the studied chemotherapeutics, or with the cytochrome P450 system.[14]
Chemistry

Scheme of a liposome formed by phospholipids in an aqueous solution
Mifamurtide is muramyl tripeptide phosphatidylethanolamine (MTP-PE), a synthetic analogue of muramyl dipeptide. The side chains of the molecule give it a longer elimination half-life than the natural substance. The substance is applied encapsulated into liposomes (L-MTP-PE). Being a phospholipid, it accumulates in the lipid bilayer of the liposomes in the infusion.[15]
Synthesis
One method of synthesis (shown first) is based on N,N’-dicyclohexylcarbodiimide (DCC) assisted esterification of N-acetylmuramyl-L-alanyl-D–isoglutaminyl–L-alanine with N-hydroxysuccinimide, followed by a condensation with 2-aminoethyl-2,3-dipalmitoylglycerylphosphoric acid in triethylamine (Et3N).[16] A different approach (shown second) uses N-acetylmuramyl-L-alanyl-D-isoglutamine, hydroxysuccinimide and alanyl-2-aminoethyl-2,3-dipalmitoylglycerylphosphoric acid;[17] that is, the alanine is introduced in the second step instead of the first.
 |
 |
Synthesis
Mifamurtide is an anticancer agent for the treatment of osteosarcoma, the most common primary malignancy of bone tissue mainly affecting children and adolescents.10
The drug was invented by Ciba-Geigy (now Novartis) in the early 1980s and the agent was subsequently licensed to Jenner Biotherapies in the 1990s.
IDM Pharma bought the rights to the drug from Jenner in April 2003.78 In March 2009, mifamurtide was approved in the 27 European Union member states plus Iceland, Liechtenstein and Norway via a centralized marketing authorization.
After the approval, IDM Pharma was acquired by Takeda, which began launching mifamurtide, as Mepact, in February 2010.
Mifamurtide, a fully synthetic lipophilic derivative of muramyl dipeptide (MDP), is muramyl tripeptide phosphatidylethanolamine (MTP-PE), which is formulated as a liposomal infusion.79 Being a phospholipid, mifamurtide accumulates in the lipid bilayer of the liposomes upon infusion.
After application of the liposomal infusion, the drug is cleared from the plasma within minutes. However, it is concentrated in lung, liver, spleen, nasopharynx and thyroid, and the terminal half-life is 18 h, which is longer than the natural substance.
Two synthetic routes have been reported,80,81 and Scheme 16 describes the more processamenable route.
Commercially available 1,2-dipalmitoyl-sn-glycero- 3-phosphoethanolamine (110) was coupled with N-Boc-L-alanine (111) by means of N-hydroxysuccinimide (112), DCC in DMF to give amide 113, which was followed by hydrogenolysis of the CBZ group to give the corresponding L-alanyl-phosphoric acid 114.
Next, commercially available N-acetylmuramoyl-L-alanyl-Disoglutamine (115) was subjected to hydroxybenzotriazole (HOBT) and DIC in DMF to provide the corresponding succinimide ester 116 which was condensed with compound 114 to provide mifamurtide (IX).
No yields were provided for these transformations.
79. Prous, J. R.; Castaner, J. Drugs Future 1989, 14, 220.
80. Baschang, G.; Tarcsay, L.; Hartmann, A.; Stanek, J. EP 0027258 A1, 1980.
81. Brundish, D. E.; Wade, R. J. Labelled Compd. Radiopharm. 1985, 22, 29.
PATENT
https://www.google.com/patents/CN103408635A?cl=en
mifamurtide, the English called mifamurtide, formula C59Hltl9N6O19P, primarily for the treatment of non-metastatic
Resectable osteosarcoma (a rare but the main cause of death for children and young people osteoma), having the formula as follows:

mifamurtide by certain stimuli such as macrophages and other white blood cells to kill tumor cells. Currently, mifamurtide listed injections into spherical liposome vesicles are muramyl tripeptide (MTP). This lipid trigger macrophages to consume mifamurtide. Once consumed mifamurtide, MTP-stimulated macrophages, in particular we will look for tumors in the liver, spleen and lung macrophages and kill it.
mifamurtide injection approved for marketing based on the results of phase III clinical study. Taiwan’s National Cancer Institute Cooperative Group (NCI) established by the Children’s Oncology Group (COG) study, complete treatment of this product in patients with osteosarcoma largest research project in the book of about 800 cases. Evaluation of mifamurtide and 3-4 adjuvant chemotherapy (cis molybdenum, doxorubicin, methotrexate, cyclophosphamide with or the same as) the results of combination therapy. Studies have shown that mifamurtide used in combination with chemotherapy can reduce the mortality rate of about 30%, 78% of treated patients survived more than six years.
Shortcomings disclosed the full liquid phase synthesis technology route mifamurtide, but all-liquid phase synthesis: [0006] Currently, mifamurtide universal rely wholly liquid phase synthesis, relevant literature (220 Drugs Futl989, 14, (3)) that the synthesis requires intermediate purification steps cumbersome, time-consuming, and the total yield of the whole liquid phase synthesis is less than 30%, which has been the main factors affecting the productivity of mifamurtide
A method for logging meter synthetic peptide, characterized in that it comprises the following steps: Step 1, under the effect of coupling agent, an amino group, and Fmoc-D-Glu on the amino resin (OPG) -OH main chain carboxyl acylation, a compound of formula I; Step 2, Fmoc removal of the protecting group the compound of formula I, under the effect of coupling with Fmoc-L-Ala-OH acylation, a compound of formula 2; step 3, Fmoc removal of the protecting group the compound of formula 2, in the role of a coupling agent, with a compound of formula 3 for acylation, a compound of formula 4; step 4, PG protecting group removing compound of formula 4, the coupling the role of agent, and HL-Ala-OPG acylation, a compound of formula 5; Step 5, PG protecting group removal compound of formula 5, under the effect of coupling agent, and an amino acid performed on brain phospholipids reaction of a compound of formula 6, and then the resin was added Lysates deaminated compound of formula 7; Step 6, the compound of formula 7 to obtain the removal of benzyl mifamurtide;


Wherein Fmoc is the amino protecting group; wherein PG is a carboxy-protecting group for Allyl or Dmab; Resin as the amino resin.
Example: Synthesis of mifamurtide crude peptide
Example 11 to give the formula hydrogenolysis at atmospheric pressure to 16 hours Example 7 was added to 7.42 g compound 250ml single neck flask, dried 150ml of methanol was added to dissolve 0.4 g of 10% palladium on carbon.Completion of the reaction, palladium-carbon was filtered off, the filtrate was concentrated by rotary evaporation to 65ml, is mifamurtide crude peptide solution. Mifamurtide synthetic crude peptide: 15 [0173] Example
Example 12 to give the formula hydrogenolysis at atmospheric pressure to 16 hours Example 7 was added to 4.21 g compound 150ml single neck flask, dried 85ml of methanol was added to dissolve 0.2 g of 10% palladium on carbon.Completion of the reaction, palladium-carbon was filtered off, the filtrate was concentrated by rotary evaporation to 37ml, is mifamurtide crude peptide solution.
16 [0175] Example 2: Preparation of mifamurtide
The embodiment 14 of crude peptide solution obtained in Example 65ml, IOOOml round bottom flask was added, under magnetic stirring, 650ml of anhydrous diethyl ether was added dropwise. Upon completion, at room temperature for crystallization. After filtration and drying the filter cake, the filter cake was again dissolved in 65ml of methanol. This methanol solution was added IOOOml round bottom flask, under magnetic stirring, 650ml of anhydrous diethyl ether was added dropwise. Upon completion, at room temperature for crystallization. Filtered cake was dried in vacuo to give mifamurtide 5.62g, yield 86.5%, purity 99.4%, total yield 74.5%
Preparation of mifamurtide of: 17 Example
The embodiment of the crude peptide solution obtained in Example 15, 37ml, 500ml round bottom flask was added, under magnetic stirring, 370ml of anhydrous diethyl ether was added dropwise. Upon completion, at room temperature for crystallization. After filtration and drying the filter cake, the filter cake was again dissolved in 37ml of methanol. This solution was added to methanol 500ml round bottom flask, under magnetic stirring, 370ml of anhydrous diethyl ether was added dropwise. Upon completion, at room temperature for crystallization. Filtered, the filter cake was dried under vacuum to give · mifamurtide 3.16g, yield 85.8%, purity 99.5%, 72.2% overall yield.
References
- “Mifamurtide: CGP 19835, CGP 19835A, L-MTP-PE, liposomal MTP-PE, MLV 19835A, MTP-PE, muramyltripeptide phosphatidylethanolamine”. Drugs in R&D 9 (2): 131–5. 2008. doi:10.2165/00126839-200809020-00007. PMID 18298131.
- “First Treatment to Improve Survival in 20 Years Now Available for Patients With Osteosarcoma (Bone Cancer)”. Takeda. November 2009. Retrieved 23 March 2010.
- “IDM Pharma’s MEPACT (Mifamurtide, L-MTP-PE) Receives Approval in Europe for Treatment of Patients with Non-Metastatic, Resectable Osteosarcoma”. PR Newswire. 2009-03-09. Retrieved 2009-11-12.
- “IDM Pharma receives not approvable letter for Mifamurtide for treatment of osteosarcoma”. The Medical News. 2007-08-28. Retrieved 2009-11-12.
- Mepact for Healthcare Professionals, retrieved 2009-11-12
- ^ Jump up to:a b EMA (2009-03-06). “Mepact: Product Information. Annex I: Summary of Product Characteristics” (PDF). p. 2. Retrieved 2009-11-12.
- EMA (2009-05-06). “Mepact: European Public Assessment Report. Summary for the public” (PDF). p. 1. Retrieved 2009-11-12.
- Meyers, P. A. (2009). “Muramyl tripeptide (mifamurtide) for the treatment of osteosarcoma”. Expert Review of Anticancer Therapy 9 (8): 1035–1049.doi:10.1586/era.09.69. PMID 19671023.
- Meyers, P. A.; Schwartz, C. L.; Krailo, M. D.; Healey, J. H.; Bernstein, M. L.; Betcher, D.; Ferguson, W. S.; Gebhardt, M. C.; Goorin, A. M.; Harris, M.; Kleinerman, E.; Link, M. P.; Nadel, H.; Nieder, M.; Siegal, G. P.; Weiner, M. A.; Wells, R. J.; Womer, R. B.; Grier, H. E.; Children’s Oncology, G. (2008). “Osteosarcoma: the Addition of Muramyl Tripeptide to Chemotherapy Improves Overall Survival–A Report from the Children’s Oncology Group”.Journal of Clinical Oncology 26 (4): 633–638. doi:10.1200/JCO.2008.14.0095.PMID 18235123.
- Meyers, P. A.; Schwartz, C. L.; Krailo, M.; Kleinerman, E. S.; Betcher, D.; Bernstein, M. L.; Conrad, E.; Ferguson, W.; Gebhardt, M.; Goorin, A. M.; Harris, M. B.; Healey, J.; Huvos, A.; Link, M.; Montebello, J.; Nadel, H.; Nieder, M.; Sato, J.; Siegal, G.; Weiner, M.; Wells, R.; Wold, L.; Womer, R.; Grier, H. (2005). “Osteosarcoma: A Randomized, Prospective Trial of the Addition of Ifosfamide and/or Muramyl Tripeptide to Cisplatin, Doxorubicin, and High-Dose Methotrexate”. Journal of Clinical Oncology 23 (9): 2004–2011. doi:10.1200/JCO.2005.06.031. PMID 15774791.
- (EMA 2009, pp. 5–7)
- (EMA 2009, p. 8)
- (EMA 2009, pp. 7–8)
- (EMA 2009, p. 4)
- Fidler, I. J. (1982). “Efficacy of liposomes containing a lipophilic muramyl dipeptide derivative for activating the tumoricidal properties of alveolar macrophages in vivo”. Journal of Immunotherapy 1 (1): 43–55.
- Prous, J. R.; Castaner, J. (1989). “ENV 2-3/MTP-PE”. Drugs Fut. 14 (3): 220.
- Brundish, D. E.; Wade, R. (1985). “Synthesis of N-[2-3H]acetyl-D-muramyl-L-alanyl-D-iso-glutaminyl-L-alanyl-2-(1′,2′-dipalmitoyl-sn-glycero-3′-phosphoryl)ethylamide of high specific radioactivity”. J Label Compd Radiopharm 22 (1): 29–35. doi:10.1002/jlcr.2580220105.
| CN1055736A * | Jan 28, 1986 | Oct 30, 1991 | E·R·斯奎布父子公司 | Process for preparing 4,4-dialkyl-2-azetidinones |
| CN101709079A * | Dec 22, 2009 | May 19, 2010 | 江苏诺泰制药技术有限公司 | Synthesis method of romurtide |
| US4323560 * | Oct 6, 1980 | Apr 6, 1982 | Ciba-Geigy Corporation | Novel phosphorylmuramyl peptides and processes for the manufacture thereof |
| Reference | ||
|---|---|---|
| 1 | * | PROUS, J. ET AL: “ENV 2-3/MTP-PE“, 《DRUGS FUT》, vol. 14, no. 3, 31 March 1989 (1989-03-31), pages 220 |
| 2 | * | 黄胜炎: “抗肿瘤药新品与研发进展“, 《上海医药》, vol. 30, no. 9, 30 September 2009 (2009-09-30), pages 412 – 414 |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
2-[(N-{(2R)-[(2-acetamido-2,3-dideoxy-D-glucopyranos-3-yl)oxy]-propanoyl}-L-alanyl-D-isoglutaminyl-L-alanyl)amino]ethyl (2R)-2,3-bis(hexadecanoyloxy)propyl hydrogen phosphate
|
|
| Clinical data | |
| License data | |
| Pregnancy category |
|
| Routes of administration |
intravenous liposomal infusion over one hour |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | N/A |
| Biological half-life | minutes (in plasma) 18 hrs (terminal) |
| Identifiers | |
| CAS Number | 83461-56-7 838853-48-8 (mifamurtide sodium · xH2O) |
| ATC code | L03AX15 (WHO) |
| PubChem | CID 11672602 |
| ChemSpider | 9847332 |
| UNII | EQD2NNX741 |
| KEGG | D06619 |
| Chemical data | |
| Formula | C59H109N6O19P |
| Molar mass | 1237.499 g/mol |
//////////83461-56-7, 838853-48-8, CGP-19835, Mepact, MFCD09954133, Mifamurtide, mifamurtide sodium, MTP-cephalin, Mtp-PE, Muramyl tripeptide, phosphatidylethanolamine, PEPTIDE, мифамуртид, ميفامورتيد, 米法莫肽
CCCCCCCCCCCCCCCC(=O)OCC(COP(O)(=O)OCCNC(=O)[C@H](C)NC(=O)CC[C@@H](NC(=O)[C@H](C)NC(=O)[C@@H](C)O[C@H]1C(O)[C@@H](CO)O[C@@H](O)[C@@H]1NC(C)=O)C(N)=O)OC(=O)CCCCCCCCCCCCCCC
WO 2016113415, Sandoz, Riociguat, New Patent

WO 2016113415, Sandoz, Riociguat, New Patent
STEFINOVIC, Marijan; (AT).
RICHTER, Frank; (AT).
GRIESSER, Ulrich; (AT).
LANGES, Christoph; (AT)
SANDOZ AG [CH/CH]; Lichtstrasse 35 4056 Basel (CH)
Novel method for purifying riociguat, useful for treating chronic thromboembolic pulmonary hypertension, pulmonary arterial hypertension, systemic sclerosis and Raynaud’s phenomenon. Also claims novel crystalline solvates of riociguat (eg ethyl acetate or butan-2-one solvate), useful as intermediates in the purification of riociguat. Bayer and licensee Merck have developed and launched riociguat.
The present filing appears to be the first filing from Sandoz on riociguat; however see WO2015095515, assigned to Novartis, parent company of Sandoz, claiming an ophthalmic composition comprising a soluble guanylate cyclase activator (eg riociguat).
Riociguat (BAY 63-2521 ), having the chemical name N-[4,6-Diamino-2-[1-(2-fluorobenzyl)-1 H-pyrazolo[3,4-b]pyridin-3-yl]pyrimidin-5-yl]-N-methylcarbamic acid methyl ester, or sometimes also called or also sometimes called Methyl-(4,6-diamino-2-(1-(2-fluorobenzyl)-1 H-pyrazolo[3, 4-b]pyridin-3-yl)-5-pyrimidinyl)(methyl)carbamate is a stimulator of the soluble guanylate cyclase.
Riociguat has been approved for the treatment of inoperable, or persistent, recurrent chronic thromboembolic pulmonary hypertension (CTEPH) after surgery in adult patients and for the treatment of pulmonary arterial hypertension and is in development for the treatment of systemic sclerosis and Raynaud’s phenomenon.

(I)
The preparation of the compound of formula (I) and its purification are known. According to the experimental procedure of Example 8 of WO 03/095451 (comparable description in Chem. Med. Chem 2009, 4, 853-865), iodomethane is used as an alkylating agent in a late step and the purification of the crude riociguat either comprised preparatory HPLC steps or several steps of extracting, precipitating, suspending, washing, redissolving and reprecipitating riociguat, resulting in a long and tedious workup procedure with moderate yield.
In WO 201 1/064171 a potential genotoxic azo compound of formula III is used as a key intermediate, which under catalytic hydrogenation forms a compound of formula VIII.

The compound of formula VIII is further reacted with a methyl chloroformate or with a dimethyl carbonate derivative to form a compound of formula VI. The compound of formula VI is then methylated to form crude riociguat of formula (I).

Crude riociguat of formula (I) is then purified by a process comprising the intermediate isolation of a riociguat DMSO solvate of formula (II).
For the pharmaceutical use of riociguat, the solvent DMSO has to be removed. To that end, the compound of formula (II) is boiled in pharmaceutically acceptable solvents such as ketones, esters, ethers or alcohols. However, the riociguat obtained in this manner contains detectable amounts of DMSO.
These processes for the preparation of riociguat and their laborious purification protocols have a number of disadvantages which are unfavorable for industrial realization on a large scale.
On the one hand, the purification process according to WO 03/095451 require the repeated isolation of solid intermediates or preparatory HPLC, which ultimately results in a reduced yield of pure riociguat of formula (I) of pharmaceutical grade. Yet, traces of compound of formula (III) remain.
It is therefore one of the objects of the present invention to provide a process for the preparation of pure riociguat – compound of the formula (I) – which yields riociguat free from any genotoxic impurity and/or mutagenic impurity.
On the other hand, the process for the preparation of riociguat described in WO 201 1/064171 has a different serious drawback. It comprises the use of a DMSO solvate.
DMSO is an active pharmaceutical ingredient by itself. It is used as an active pharmaceutical ingredient in the treatment of interstitial cystitis. DMSO removal is difficult to achieve by the published processes. It is thus a further object of the invention to provide riociguat essentially free from DMSO and suitable for pharmaceutical use.
WO 2014/128109 discloses forms of riociguat, such as polymorphs and solvates, and describes a ¼ ethyl acetate solvate of riociguat in example 6. The X-ray powder
diffractogram in Tab.3 and figure 4 comprises reflexes at °2Theta positions of 9.1 and 25.6.
Thus, there is a need in the art for a process, which allows the preparation of pure riociguat free from any genotoxic impurity and/or mutagenic impurity which at the same time does not comprise residual DMSO.
Surprisingly, we have now identified a process for the purification of crude riociguat which yields riociguat which is essentially free from genotoxic impurities and DMSO. In particular, this novel process differs from the processes known to date in that the isolation of intermediates prior to the formation of riociguat is not required. This process allows to overcome the disadvantages of the processes known to date and to obtain riociguat in high yield and high purity and pharmaceutical acceptable quality essentially free of genotoxic impurities.
Examples
Preparative example
Preparation of crude riociguat
Riociguat was prepared as disclosed in example 7 of WO 201 1/064171 and had a chemical purity of 91.7% by the area of the riociguat peak in the HPLC-UV elution profile.
Comparative Example 1
Preparation of DMSO solvate
An amount of 4.505 g (0.0107 moles) of crude riociguat was dissolved in 8 ml DMSO at 100 °C. The obtained brownish, turbid solution was then cooled to 75 °C within 16 minutes. After that 55 ml of ethylacetate were added and the mixture was cooled to 25 °C (30 minutes). After 22 h the obtained precipiate was filtered off, washed with 14 ml EtOAc and dried for 4 hours at 50 °C at reduced pressure (50 mbar). The precipitate was analysed with XRPD, confirming that riociguat DMSO was obtained. The product was also analyzed by HPLC-UV-MS. Purity was calculated based on UV detection at 254nm. The so obtained riociguat DMSO solvate was 91 .92% pure.
Comparative Example 2
Preparation of riociguat form I from riociguat DMSO solvate
The entire product prepared in comparative example 1 (4.283 g = 0.009 moles) was reflux heated in 77 ml of ethylacetate at 78 °C for 1 h and then cooled to 25 °C. The white solid was filtered off with suction, washed with a total of 18 ml of ethyl acetate and dried at 50 °C under reduced pressure (50 mbar) for 5 hours. The dried product was then analyzed by XRPD, confirming identity of riociguat form I unequivocally.
Yield (dry): 3.224 g (0.0076 moles) = 75% for comparative example 2 and 72% overall (C.ex. 1 and 2). Total organic volatile impurity is higher than 1000 ppm and total DMSO content is higher than 100 ppm.
Example 1 ; Preparation of Riociguat ethylacetate solvate
Crude Riociguat (500 mg; Form I; 91 .7% percentage area purity) was dissolved in 2 ml DMF and heated to 100 °C to obtain a slightly turbid solution. After filtration through a 0.44 micron filter, 20 ml EtOAc were added to the hot solution (water bath 70°C) and allowed to stand. The temperature was slowly decreased to ambient temperature. Crystallization started after
10min. The yellowish, fine powder was filtered off and dried at ambient conditions. The PXRD indicated the formation of a new ethylacetate solvate. Yield 71 %, 97.8% purity.
Example 2; Preparation of the Methyl ethyl ketone (butan-2-one) solvate of Riociguat.
Crude Riociguat (500 mg; Form I; 91 .7% percentage area purity) was dissolved in 2 ml DMF at 100 °C to obtain a clear solution. After filtration through a 0.44 micron filter, 20 ml MEK were added. The hot solution (water bath 70 °C) was allowed stand. The temperature was then slowly decreased to ambient temperature. After 30 minutes yellowish, square-shaped crystals appeared, which were analyzed. Analysis confirmed that they were a new crystalline MEK-solvate. Yield 43%, 97.2% purity.
Example 3 ; Conversion of Solvated forms to Form I
Both the solvates from examples 1 and 2 can be converted to riociguat Form I by heating the material to 150°C under vacuum for an appropriate amount of time.
Example 4; Direct preparation of riociguat form I from crude riociguat using DMF-Acetone Crude Riociguat (200 mg; Form I; 91 .7% percentage area purity) was dissolved in 1.0 ml DMF at 100 °C to obtain a clear solution. After filtration through a 0.44 micron filter, 5 ml acetone was added. The hot solution (water bath 70 °C) was allowed to stand. Crystallisation occurred while the temperature was slowly decreased to ambient temperature. After 24 hours the precipitate was filtered off and dried at ambient conditions to obtain form I. Yield 78% ; 97.6% purity
///////////WO 2016113415, Sandoz, Riociguat, New Patent
Carbotegravir, Dolutegravir, New Patent, WO 2016113372, Lek Pharmaceutical and Chemical Co DD
![]()
Carbotegravir, New Patent, WO 2016113372, Lek Pharmaceutical and Chemical Co DD
LEK PHARMACEUTICALS D.D. [SI/SI]; Verovskova 57 1526 Ljubljana (SI)
MARAS, Nenad; (SI).
SELIC, Lovro; (SI).
CUSAK, Anja; (SI)
ViiV Healthcare is developing cabotegravir (first disclosed in WO2006088173), which in July 2016, was reported to be in phase 2 clinical development.
WO-2016113372
Process for preparing integrase inhibitors such as dolutegravir and cabotegravir and their analogs, useful for treating viral infections eg HIV infection. Also claims a process for preparing intermediates of dolutegravir and cabotegravir.
(4R, 12aS)-N-[(2,4-Difluorophenyl)methyl]-3 ,4,6,8, 12, 12a-hexahydro-7-hydroxy-4-methyl-6,8-dioxo-2H-pyrido[1 ‘,2’:4,5]pyrazino[2, 1-b][1 ,3]oxazine-9-carboxamide (Formula A):

Formula A
known by the INN name dolutegravir, is a new efficient antiviral agent from the group of HIV integrase inhibitors which is used in combination with some other antiviral agents for treatment of HIV infections, such as AIDS. The compound, which belongs to condensed polycyclic pyridines and was first disclosed in WO2006/1 16764, is marketed.
Another compound disclosed in WO2006/1 16764 is (3S, 1 1 aR)-N-[(2,4-difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7, 1 1 ,1 1 a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxamide (Formula

Formula C
known by the INN name cabotegravir.
The complex structures of dolutegravir and cabotegravir present a synthetic challenge. The first description of the synthesis in WO2006/1 16764 shows a 16-steps synthesis (see Scheme A), which is industrially impractical due to its length and low overall yield.

Scheme A
WO 2010/068253 and WO 2006/1 16764 describe an alternative synthesis. The 1 1 -step synthesis, shown in Scheme B1 and Scheme B2, is based on bromination of the 9-position for further introduction of the carboxylic group. The synthesis relies on the use of expensive palladium catalysts and toxic selenium compounds. Furthermore, some variations of these approaches involve pyrone intermediates in several steps. In some cases pyrones are liquids which can complicate purification, while further reactions form complex mixtures.
![]()

 doiutegravir
doiutegravir
Scheme B2
In further alternative syntheses, acetoacetates were used as starting materials. Such an approach is challenging in terms of introducing the hydroxy group in the 7-position. The variation in Scheme C1 , described in WO2012/018065, starts from 4-benzyloxyacetoacetate. The procedure requires 9 steps, but use expensive reagents like palladium catalysts. Moreover, there is described a possibility of formation a co-crystal between an intermediate and hydroquinone, wherein however the additional step may diminish yields and make the process longer and time consuming.

Scheme C1
The variation in Scheme C2, described in WO2012/018065, starts from 4-chloroacetoacetate. The process is not optimal because of problems in steps which include pyrones and because of problems with conversion of 7-chloro to 7-hydroxy group which includes a disadvantageous use of silanolates with low yield (25%).

Scheme C2
The variation in Scheme C3, described in WO201 1/1 19566, starts from unsubstituted acetoacetate. For the introduction of the 7-hydroxy group, bromination is used and substitution of bromo with hydroxy is performed by a use of silanolates. The substitution of the bromine is achieved in a 43% yield.

Scheme C3
The variation in Scheme C4, described in WO201 1/1 19566, starts from 4-methoxyacetoacetate aiming at preparing dolutegravir or cabotegravir. The process uses lithium bases to affect a difficult to control selective monohydrolysis of a diester.

The object of the present invention is to provide short, simple, cost-effective, environmentally friendly and industrially suitable processes for beneficially providing dolutegravir and analogues thereof and cabotegravir and analogues thereof, in particular dolutegravir.
Scheme 1

According to an embodiment of the process of the invention the building block 3-aminobutanol can suitably be substituted with other aminoalcohols to give dolutegravir analogues. For example, using (S)-alaninol gives cabotegravir as the final product. Similarly, using amines other than 2,4-difluorobenzylamine in the amidation step results in the synthesis of other dolutegravir analogues.
According to the another preferred embodiment cabotegravir or a pharmaceutically acceptable salt thereof is prepared by the analogue process, which comprises providing a compound of formula (5c)

5c
converting the compound of formula (5c) to a compound of formula (6c)

6c
by carrying out a chlorination reaction, and converting the compound of formula (6c) to cabotegravir and/or a pharmaceutically acceptable salt thereof.
The compound of formula (5c) can preferably be provided by converting a compound of formula (3) to a compound of formula (4c)

Scheme 2

1. ) EtOCOCI, Et3N / Me2CO
2. ) 2,4-difiuorobenzylamine

Scheme 3

Analogous compound of formula 7c is a useful intermediate in the synthesis of cabotegravir. Scheme 3a

Scheme 4

Examples
The following examples are merely illustrative of the present invention and they should not be considered as limiting the scope of the invention in any way. The examples and modifications or other equivalents thereof will become apparent to those versed in the art in the light of the present entire disclosure. Particularly, all Examples related to the preparation of dolutegravir and intermediates thereof can be used by the analogy for the preparation of cabotegravir and intermediates thereof.
Example 1 :

Methyl acetoacetate (1 , 25.22 g) and dimethylformamide dimethyl acetal (DMFDMA, 35 mL) was heated at 50-55°C for 2 h, then methanol (60 mL), aminoacetaldehyde dimethyl acetal (24 mL) and acetic acid (4 mL) was added an the mixture was heated under reflux for one hour, then concentrated. MTBE (100 mL) was added and the mixture was kept at 5 °C overnight to crystallize. Upon filtration 46 g (92%) of product 2 was recovered.
1H NMR (DMSO-d6): δ 2.31 (s, 3H), 3.30 (s, 6H), 3.49 (m, 2H), 3.61 (s, 3H), 4.43 (m, 1 H), 8.02 (d, 1 H), 10.8 (bs, 1 H). 13C NMR (DMSO-d6): δ 30.52, 35.48, 50.53, 54.23, 98.99, 102.47, 160.70, 166.92, 197.21 .
Example 2:

Compound 2 (5.00 g) was dissolved in 2-propanol, dimethyl oxalate (7.02 g) was added and heated to 40 °C. Sodium methylate (25% in methanol; 20 mL) was slowly (10 min) added, the mixture was then heated to 50-55 °C and stirred at that temperature for 2-2.5 h. The mixture was cooled to ambient temperature, then sodium hydroxide solution (1 M, 65 mL) was added to the mixture and stirred for another 2 h, followed by addition of concentrated hydrochloric acid (1 1 mL) and stirred for another 2 h. The precipitate was filtered and dried to give 8.08 g (NMR assay 47%; 65% yield) of compound 3.
1H NMR (DMSO-d6): δ 2.50 (m, 2H), 3.30 (s. 6H), 4.49 (m, 1 H), 7.06 (s, 1 H); 8.70 (s, 1 H). 13C NMR (DMSO-d6): δ 55.23, 55.37, 102.34, 1 15.47, 120.24, 145.17, 162.71 , 165.22, 178.55.
Example 3:

Compound 2 (158.37 g) was dissolved in methanol (548 mL), followed by the addition of dimethyl oxalate (202.2 g). While keeping the temperature below 30°C, potassium ferf-butoxide (192.1 g) was added and reaction mixture was heated at 50 °C overnight. The suspension was then filtered and the filter cake washed with methanol. The filtrate was concentrated (approximately to 680 mL), then water (680 mL) was added, followed by addition of lithium hydroxide hydrate (143.7 g) while keeping the temperature below 40 °C. The suspension was then stirred at ambient temperature overnight and filtered. To the obtained filtrate, concentrated hydrochloric acid (339 mL) was added while keeping the temperature below 30 °C. The suspension was aged for 2 h and filtered to give 4 as a white powder (95.6 g, NMR assay 100%; 52% yield).
Example 4:

Compound 2 (5.00 g) was dissolved in 2-propanol, dimethyl oxalate (7.02 g) was added and heated to 40 °C. Sodium methylate (25% in methanol; 15 mL) was slowly (10 min) added then the mixture was heated to 50-55 °C and stirred at that temperature for 72 h. The mixture was concentrated and components were separated by flash column chromatography (ethyl acetate/methanol 9:1 to 6:4). Early fractions gave compound 22 upon concentration, late fractions gave compound 23.
Compound 22: 1H NMR (DMSO-d6): δ 2.49 (m, 2H), 3.28 (s, 6H), 3.73 (s, 3H), 3.85 (s, 3H), 4.41 (m, 1 H), 4.50 (m, 1 H), 6.65 (s, 1 H), 8.36 (s, 1 H). 13C NMR (DMSO-d6): δ 51.63, 53.36, 54.25, 55.47, 102.71 , 1 18.24, 123.60, 140.81 , 150.21 , 162.44, 164.49, 173.43.
Compound 23: 1H NMR (DMSO-d6): δ 2.49 (m, 2H), 3.26 (s, 6H); 3.70 (s, 3H); 4.33 (d, 1 H); 4.60 (m, 1 H), 6.19 (s, 1 H), 8.12 (s, 1 H). 13C NMR (DMSO-d6): δ 50.03, 51.34, 54.59, 54.85, 102.91 , 1 16.04, 1 18.19, 148.32, 152.12, 163.46, 165.24, 174.99
Example 5:

Compound 3 (5.5 g; assay 53%) was suspended in acetonitrile, acetic acid (6 mL) and methanesulfonic acid (2.5 mL) were added followed by the heating of mixture to 70 °C for 4 h. The suspension was filtered and filtrate cooled to ambient temperature. Triethylamine (6.6 mL) and (R)-3-amino-butan-1 -ol (1.24 mL) was added followed by heating the mixture at reflux temperature for 20-24 h. The mixture was filtered, filtrate concentrated and 1 M HCI (100 mL) was added, followed by extraction with dichloromethane (3 x 50 mL). Combined organic fractions were concentrated, 2-propanol was added (10 mL) and suspension was stirred at 70-80 °C for 10 min, left to cool to ambient temperature then filtered to give 2.19 g of compound 4 (73%).
1H NMR (DMSO-de): δ 1.31 (d, 3H), 1.52 (m, 1 H), 1 .97 (m, 1 H), 3.89 (m, 1 H), 4.01 (m, 1 H), 4.46 (m, 1 H), 4.64 (m, 1 H), 4.78 (m, 1 H), 5.50 (m, 1 H), 7.29 (s, 1 H), 8.88 (s, 1 H), 15.83 (s, 1 H). 13C NMR (DMSO-d6): δ 15.22, 29.14, 45.26, 51.13, 62.09, 76.03, 1 16.31 , 1 18.79, 140.53, 146.79, 155.36, 165.24, 178.75.
Example 6:

Compound 3 (14.55 g; assay 49%) was suspended in acetonitrile (125 mL), acetic acid (15 mL) and methanesulfonic acid (6.25 mL) were added followed by the heating of mixture to 70 °C for 4 h. The suspension was filtered and filtrate cooled to ambient temperature. Triethylamine (16.5 mL) and (S)-2-aminopropanol (2.45 mL) was added followed by heating the mixture at reflux temperature for 24 h. The insoluble product was filtered, washed with 2-propanol (20 mL) and dried to give (3S, 1 1 aR)-3-methyl-5,7-dioxo-2,3,5,7, 1 1 ,1 1 a-hexahydrooxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxylic acid (5.2 g, 75%).
1H NMR (DMSO-d6): δ 1.31 (d, J = 6.3 Hz, 3H), 3.65 (dd, J = 8.6, 6.8 Hz, 1 H), 4.13 (dd, J = 1 1.7, 10.3 Hz, 1 H), 4.28 (m, 1 H), 4.39 (dd, J = 8.6, 6.8 Hz, 1 H), 4.92 (dd, J = 12.3, 4.2 Hz, 1 H), 5.45 (dd, J = 10.2, 4.1 Hz, 1 H), 7.16 (s, 1 H), 8.84 (s, 1 H), 15.74 (s, 1 H).
Example 7:

Compound 4 (0.63 g) was dissolved in dichloromethane (15 mL), cooled to 5°C, then triethylamine (0.31 mL) was added, followed by ethyl chloroformate (0.26 mL), followed by slow (30 min) addition of 2,4-difluorobenzylamine. The mixture was then stirred at ambient temperature for 24 h. Water (10 mL) was added, organic phase was separated and washed with 1 M HCI (15 mL) and water (15 mL), concentrated and treated with 2-propanol to give the product 5 in a quantitative yield.
1H NMR (CDCI3): δ 1.39 (d, 3H), 1.52 (s, 1 H), 2.19 (m, 1 H), 4.00 (m, 2H), 4.16 (m, 1 H), 4.31 (m, 1 H), 4.62 (d, 2H), 5.00 (m, 1 H), 5.27 (m, 1 H), 6.80 (m 2H), 7.33 (m, 2H), 8.49 (s, 1 H), 10.48 (s, 1 H). 13C NMR (CDCI3): 15.50, 29.22, 36.43, 45.19, 51.83, 62.79, 103.71 , 103.91 , 1 1 1 .0, 1 1 1 .18, 120.59, 123.04, 130.40, 137.41 , 144.58, 156.27, 163. 87, 177.83.
Example 8:

To a suspension of 4 (2.84 g, 10 mmol) in a mixture of triethylamine (2.24 mL, 16 mmol) and acetone (50 mL) stirring on an ice bath was added ethyl chloroformate (1 .20 mL, 12 mmol). After stirring for 10 min, 2,4-difluorobenzylamine (1.21 mL, 10 mmol) was added and the mixture left stirring at room temperature for 1 h. The product was isolated by slowly diluting the reaction mixture with water (50 mL), partial concentration, filtration, washing with water (2 50 mL) and drying. There was obtained 5 as a white powder (3.48 g, 86%): mp 181.0-184.7 °C. 1H NMR (DMSO-d6): δ 1.29 (d, J = 7.0 Hz, 3H), 1 .56 (dd, J = 13.9, 2.0 Hz, 1 H), 1 .93-2.06 (m, 1 H), 3.90 (ddd, J = 1 1.6, 5.0, 2.1 Hz, 1 H), 3.98 (td, J = 12.0, 2.2 Hz, 1 H), 4.45 (dd, J = 13.6, 6.6 Hz, 1 H), 4.72 (dd, J = 13.6, 3.8 Hz, 1 H), 4.74-4.81 (m, 1 H), 5.44 (dd, J = 6.6, 3.8 Hz, 1 H), 8.93 (s, 1 H), 15.14 (s, 1 H). 13C NMR (DMSO-d6): δ 15.78, 29.13, 44.89, 52.88, 61 .63, 75.61 , 1 13.54, 128.49, 136.42, 145.64, 154.62, 164.58, 174.58
Example 9:

To a suspension of 4 (1 1.36 g, 40 mmol) in acetonitrile (80 mL) stirring at room temperature was added TCCA (9.29 g, 38 mmol) and DABCO (0.23 g, 5 mol%). After stirring at room temperature for 1 h, the reaction was quenched with a mixture of DMSO (5.26 mL) and water (1.33 mL). The insoluble cyanuric acid was removed by filtration and the filtrate evaporated under reduced pressure to give viscous oil. This was triturated in methanol (20 mL) to induce crystallization. The product was filtered, washed with cold methanol (10 mL) and dried to give 7 as a yellowish powder (5.13 g, 41 %): mp 191 .3-198.7 °C.
Example 10:

Attempted chlorination of 23: Compound 23 (0.54g) was suspended in acetonitrile (10 mL) and trichlorocyanuric acid (0.44 g) was added and the solution was stirred at ambient temperature overnight. Precipitate was filtered. Only traces of a product corresponding to the compound 26 could be detected in the reaction mixture by LC-MS analysis. Conversion did not improve with time.
Example 11 :

Attempted chlorination of 3: Compound 3 (0.30 g) was suspended in acetonitrile (5 mL) and trichlorocyanuric acid (0.13 g) was added. The suspension was stirred at ambient temperature overnight. Only traces of a product corresponding to the compound 24 could be detected in the reaction mixture by LC-MS analysis.
Example 12:

9 10
Trichloroisocyanuric acid (0.23 g) was added in a single portion to a stirred solution of the diethyl 1 -(2,2-dimethoxyethyl)-4-oxo-1 ,4-dihydropyridine-2,5-dicarboxylate (9, 0.66 g) in dry acetonitrile (4 mL) at room temperature. The resulting suspension was stirred at room temperature for ca. 24 h. The reaction mixture was diluted with dichloromethane and filtrated. The filtrate was then concentrated in vacuo to afford crude oil (0.86 g). Purification by flash chromatography (eluting ethyl acetate/cyclohexane) furnished diethyl 3-chloro-1 -(2,2-dimethoxyethyl)-4-oxo-1 ,4-dihydropyridine-2,5-dicarboxylate, 10 as a yellow semi-solid (0.38 g). 1H NMR (CDCI3): δ 1.28 (t, J=7A Hz, 3H), 1 .37 (t, J=7.2 Hz, 3H), 3.35 (s, 6H), 3.89 (d, J=5.0 Hz, 2H), 4.27 (q, J=l A Hz, 2H), 4.43 (q, J=l A Hz, 2H), 4.48 (t, J=4.9 Hz, 1 H), 8.15 (s, 1 H). 13C NMR (CDCI3): δ 13.83, 14.13, 55.82, 57.09, 61.41 , 63.72, 102.52, 1 17.35, 126.90, 140.22, 146.92, 160.67, 164.13, 168.95.
Example 13:

Diethyl 1 -(2,2-dimethoxyethyl)-4-oxo-1 ,4-dihydropyridine-2,5-dicarboxylate (9, 0.64 g) was dissolved in anhydrous acetonitrile (6 mL) and treated sequentially with acetic acid (560 μί) and methanesulfonic acid (40 μί). The resulting mixture was heated to 62 °C and stirred for 4 h and more methanesulfonic acid (40 μΙ_) was added. After additional 2 h, more methanesulfonic acid (80 μΙ_) was added. This was repeated after additional 2 h, when more methanesulfonic acid (80 μΙ_) was added. The reaction mixture was stirred additional 17 h at 62 °C then was treated with a mixture of (R)-3-aminobutanol (0.22 g), triethylamine (0.5 mL) and acetonitrile (0.7 mL). The reaction mixture was stirred additional 22 h at 62 °C and then concentrated in vacuo. The crude material was partitioned between dichloromethane and 1 M HCI solution (15 mL). The combined organic phases were dried (Na2S04), filtered and concentrated in vacuo to afford the crude (4R, 12aS)-ethyl 4-methyl-6,8-dioxo-3,4,6,8, 12,12a-hexahydro-2H-pyrido[1 ‘,2’:4,5]pyrazino[2, 1 -b][1 ,3]oxazine-9-carboxylate (11 ) as a brownish oil (0.61 g).
1H NMR (CD3OD): δ 8.44 (s, 1 H), 7.16 (m, 1 H), 5.48 (t, J=4.8 Hz, 1 H), 4.86 (m, 1 H), 4.49 (dd, J=13.6, 4.0 Hz, 1 H), 4.30-4.25 (m, 3H), 4.09 (dt, J=12.1 , 2.3 Hz, 1 H), 3.96 (ddd, J=1 1.7, 5.0, 2.1 Hz, 1 H), 2.18-2.10 (m, 1 H), 1.60-1 .56 (m, 1 H) 1 .39 (d, J=7A Hz, 3H), 1.33 (t, J=7A Hz, 3H). 13C NMR (CDCI3): δ 8.45, 14.08, 15.39, 29.17, 45.04, 45.72, 51 .56, 60.86, 62.61 , 76.33, 1 19.54, 123.72, 136.96, 145.67, 156.26, 163.68, 175.43
Example 14:

10
Diethyl 3-chloro-1 -(2,2-dimethoxyethyl)-4-oxo-1 ,4-dihydropyridine-2,5-dicarboxylate (10, 1.23 g) was dissolved in 85% formic acid (25 mL) at room temperature. The mixture was warmed to 40 °C and stirred for 23 h. The reaction mixture was concentrated in vacuo, and then partitioned between dichloromethane and aqueous NaHC03 solution. The combined organic phases were dried (Na2S04), filtered and concentrated in vacuo to afford brownish oil (0.49 g). The crude oil was dissolved in anhydrous toluene (5 mL) and treated sequentially with (R)-3-aminobutanol (0.19 g), methanol (0.2 mL) and acetic acid (96 μί). The resulting mixture was heated to 90 °C and stirred for 20 h. The reaction mixture was cooled to room temperature and then partitioned between dichloromethane and aqueous NaHC03 solution. The combined organic phases were dried (Na2S04), filtered and concentrated in vacuo to afford the crude (4R,12aS)-Ethyl 7-chloro-4-methyl-6,8-dioxo-3,4,6,8,12, 12a-hexahydro-2H-pyrido[1 ‘,2’:4,5] pyrazino [2, 1-b][1 ,3]oxazine-9-carboxylate (12) as a brownish oil (0.24 g).
Example 15:

To a solution of 4 (5.68 g, 20 mmol) in dichloromethane (50 mL) stirring in an ice bath was added triethylamine (5.6 mL, 40 mmol), followed by ethyl chloroformate (2.61 mL, 26 mmol). After 20 min, ethanol (50 mL) was added. The mixture was then left stirring 24 h at room temperature and concentrated under reduced pressure. The residue was triturated in acetone (80 mL). The insoluble salt (triethylamine hydrochloride) was removed by filtration. The filtrate was evaporated under reduced pressure to give 11 as an amorphous solid in a quantitative yield (6.1 g).
Example 16:

To a stirring solution of 11 (0.94 g, 3.0 mmol) in acetonitrile (8 mL) heated at 40 °C was added TCCA in portions during 1 h (0.44 g, 1 .8 mmol). After an additional 1 h, the reaction mixture was diluted with a solution of NaHS03 (0.60 g) in water (60 mL), extracted with dichloromethane (50 mL) and the extract evaporated under reduced pressure to give a crude product which was purified by flash chromatography (CH2CI2 : MeOH, from 98 : 2 to 80 : 20) to give 12 (0.45 g, 44%).
1H NMR (CDCI3): δ 1.37 (t, J = 7.1 Hz, 3H), 1.38 (d, J = 7.0 Hz, 3H), 1 .56 (dq, J = 13.9, 2.2 Hz, 1 H), 2.21 (m, 1 H), 3.99 (d, J = 2.3 Hz, 1 H), 4.00 (t, J = 1.8 Hz, 1 H), 4.10 (dd, J = 13.2, 6.6 Hz, 1 H), 4.37-4.27 (m, 3H), 4.98 (m, 1 H), 5.35 (dd, J = 6.6, 3.8 Hz, 1 H), 8.07 (s, 1 H).
13C NMR (CDCI3): δ 14.20, 16.09, 29.34, 44.87, 53.73, 61.49, 62.29, 76.01 , 1 16.22, 133.1 1 , 134.18, 144.52, 155.48, 163.88, 169.98.
Example 17:

To a mixture of 7 (3.89 g, 12.2 mmol) in methanol (12 mL) was added sodium methylate (22.3 mL, 97.6 mmol). The reaction mixture was stirred for 24 h at 30 °C and then quenched with a slow addition of 3M hydrochloric acid (35 mL) while stirring in an ice bath. The mixture was concentrated under reduced pressure to remove most of the methanol, then extracted with dichloromethane (2 30 mL), the combined extracts washed with water (30 mL) and evaporated under reduced pressure. Methanol (20 mL) was added to the obtained amorphous residue and removed under reduced pressure to yield the solid 8 (3.69 g, 98%).
1H NMR (CDCI3): δ 15.04 (s, 1 H), 8.42 (s, 1 H), 5.29 (dd, J=5.6, 3.9 Hz, 1 H), 5.01 -4.96 (m, 1 H), 4.42 (dd, J=13.6, 3.6 Hz, 1 H), 4.25 (dd, J=13.6, 6.0 Hz, 1 H), 4.05 (s, 3H), 4.00-3.97 (m, 2H), 2.21 -2-14 (m, 1 H), 1.53 (dd, J=14.1 , 1.9 Hz, 1 H), 1.36 (d, J=7 Hz, 3H). 13C NMR (CDCI3): δ 176.35, 165.94, 155.03, 153.70, 143.08, 130.90, 1 15.94, 76.05, 62.65, 61.45, 53.86, 44.96, 29.43, 16.06.
Example 18:

To a suspension of 7 (2.55 g, 8.0 mmol) in a mixture of triethylamine (1 .46 mL, 10.4 mmol) and acetone (32 mL) stirring on an ice bath was added ethyl chloroformate (0.88 mL, 8.8 mmol). After stirring for 10 min, 2,4-difluorobenzylamine (1.07 mL, 8.8 mmol) was added and the mixture left stirring at room temperature for 1 h. The product was isolated by slowly diluting the reaction mixture with water (40 mL), filtration, washing with water (2 30 mL) and drying. There was obtained 2.91 g of 6 as a white powder (83%).
1H NMR (CDCI3): δ 1.30 (d, J = 7.0 Hz, 3H), 1 .49 (dd, J = 14.0, 2.2 Hz, 1 H), 2.14 (ddd, J = 14.6, 1 1.1 , 6.4 Hz, 1 H), 3.89-3.95 (m, 2H), 4.09-4.15 (m, 1 H), 4.26 (dd, J = 13.4, 3.8 Hz, 1 H), 4.55 (d, J = 5.8 Hz, 2H), 4.89-4.98 (m, 1 H), 5.18 (dd, J = 6.2, 3.8 Hz, 1 H), 6.68-6.79 (m, 2H), 7.23-7.31 (m, 1 H), 8.41 (s, 1 H), 10.24 (t, J = 5.8 Hz, 1 H). 13C NMR (CDCI3): δ 16.09, 26.95, 29.30, 36.79, 45.1 1 , 45.28, 53.86, 62.47, 75.93, 103.87 (t, J = 25.4 Hz), 1 1 1 .21 (dd, J = 21 .0, 3.4 Hz), 1 17.32, 130.58 (dd, J = 9.3, 5.8 Hz), 133.40, 143.54, 155.34, 163.16, 163.25, 163.35, 172.88.
Example 19:

To a suspension of 5 (1 .67 g, 4 mmol) in acetonitrile (20 mL) was added DABCO (23 mg, 5 mol%) and TCCA (0.62 g, 2.52 mmol). The mixture was stirred 18 h at 40 °C protected from light and then quenched with a mixture of DMSO (0.48 mL) and water (0.12 mL). The insoluble cyanuric acid was removed by filtration and washed with acetonitrile (5 mL). The filtrate was evaporated under reduced pressure to give viscous oil that was crystallized from a mixture of methanol (6 mL) and water (3 mL), by slowly cooling the solution from 60 °C to room
temperature. The product 6 was filtered, washed with cold methanol (5 mL) and dried to give an off-white powder (1.07 g, 61 %).
1H NMR (CDCI3): δ 1.30 (d, J = 7.0 Hz, 3H), 1 .49 (dd, J = 14.0, 2.2 Hz, 1 H), 2.14 (ddd, J = 14.6, 1 1.1 , 6.4 Hz, 1 H), 3.89-3.95 (m, 2H), 4.09-4.15 (m, 1 H), 4.26 (dd, J = 13.4, 3.8 Hz, 1 H), 4.55 (d, J = 5.8 Hz, 2H), 4.89-4.98 (m, 1 H), 5.18 (dd, J = 6.2, 3.8 Hz, 1 H), 6.68-6.79 (m, 2H), 7.23-7.31 (m, 1 H), 8.41 (s, 1 H), 10.24 (t, J = 5.8 Hz, 1 H). 13C NMR (CDCI3): δ 16.09, 26.95, 29.30, 36.79, 45.1 1 , 45.28, 53.86, 62.47, 75.93, 103.87 (t, J = 25.4 Hz), 1 1 1 .21 (dd, J = 21.0, 3.4 Hz), 1 17.32, 130.58 (dd, J = 9.3, 5.8 Hz), 133.40, 143.54, 155.34, 163.16, 163.25, 163.35, 172.88.
Example 20:

To a suspension of 6 (0.44 g) in anhydrous methanol (1 mL) was added a 25% methanolic solution of sodium methylate (1 .14 mL) and the mixture stirred for 4 h at 40 °C. The reaction was quenched with acetic acid (0.4 mL), diluted with water (8 mL), extracted with 2-methyltetrahydrofuran (12 mL), the extract washed with 1 M NaOH(aq) (8 mL), water (8 mL) and evaporated under reduced pressure. To the oily residue was added methanol (8 mL) and evaporated under reduced pressure to give 27 as a white solid (0.38 g, 88%).
Example 21 :

The suspension of (4R, 12aS)-7-chloro-N-(2,4-difluorobenzyl)-4-methyl-6,8-dioxo-3,4,6,8,12, 12a-hexahydro-2H-pyrido[1 ‘,2’:4,5]pyrazino[2, 1 -b][1 ,3]oxazine-9-carboxamide (6, 0.44 g) and solid sodium hydroxide (0.20 g) in absolute ethanol (2 mL) was stirred at room temperature for 24 h. The reaction was quenched with 2M H2S04 (1 .18 mL) and left stirring for 2 h at room temperature. The reaction mixture was filtered through fitted funnel rinsing with water (2 x 2 mL). The obtained white precipitate (0.38 g) was suspended in THF-water (1 :1 , 4.5 mL) and stirred at room temperature for ca. 2 h. The reaction mixture was filtered through fitted funnel rinsing with water (2 χ 1 mL) and dried in vacuo at 40°C to afford pure dolutegravir as a white solid (0.33 g, HPLC purity: 99.38%).
1H NMR (DMSO-d6): δ 12.51 (s, 1 H), 10.36 (t, J=5.9 Hz, 1 H), 8.50 (s, 1 H), 7.41-7.36 (m, 1 H), 7.26-7.21 (m, 1 H), 7.07-7.03 (m, 1 H), 5.45 (dd, J=5.4, 4.3 Hz, 1 H), 4.81 -4.76 (m, 1 H), 4.59-4.53 (m, 3H), 4.36 (dd, J=13.8, 5.8 Hz, 1 H), 4.05-4.00 (m, 1 H), 3.91-3.88 (m, 1 H), 2.05-1 .97 (m, 1 H), 1.55-1.52 (m, 1 H), 1 .33 (d, J=7.1 Hz, 3H). 13C NMR (DMSO-d6): δ 170.27, 163.68, 162.29, 161 .78 (dd), 159.82 (dd), 154.61 , 140.64, 130.74 (d), 130.67 (d), 122.37 (d), 1 16.73, 1 15.38, 1 1 1 .33 (d), 103.80 (t), 62.01 , 51 .16, 44.69, 35.74, 29.13, 15.21.
Example 22:

A suspension of dolutegravir (0.31 g) in methanol (4 mL) was cooled to 0 °C.25% Solution of sodium methoxide in methanol was added to the mixture and the resulting suspension was stirred at 0 °C for 2 h, then at room temperature for 23 h. The reaction mixture was then filtered through fitted funnel rinsing with methanol (3 x 10 mL). The white precipitate was dried overnight at room temperature to afford pure dolutegravir sodium as a white solid (0.26 g, HPLC purity: 99.84%).
1H NMR (DMSO-d6): δ 10.70 (t, J=5.8, 1H), 7.89 (s, 1H), 7.37-7.30 (m, 1H), 7.23-7.19 (m, 1H), 7.04-7.01 (m, 1H), 5.17 (m, 1H), 4.81 (t, J=6.4Hz, 1H), 4.51 (d, J=5.5Hz, 2H), 4.32-4.29 (m, 1H), 4.16 (dd, J=14.1, 4.8 Hz, 1H), 3.99-3.94 (m, 1H), 3.82-3.80 (m, 1H), 1.89-1.84 (m, 1H), 1.38 (d, J=12.9 Hz, 1H), 1.24 (d, J=7.0Hz, 3H).13C NMR (DMSO-d6): δ 177.93, 167.12, 166.08, 161.59 (dd), 161.13, 159.63 (dd), 134.26, 130.44 (d), 130.38 (d), 122.90 (d), 114.95, 111.23 (d), 108.78, 103.64 (t), 75.59, 61.95, 53.11, 43.01, 35.32, 29.22, 15.30.
Example 23:

The suspension of 6 (0.44 g) and solid sodium hydroxide (0.20 g) in absolute ethanol (2 mL) was stirred at room temperature for 24 h. The reaction was diluted with absolute ethanol (10 mL) and left stirring for ca. 30 min at room temperature. The reaction mixture was filtered through fitted funnel rinsing with absolute ethanol (3 x 10 mL) and dried in vacuo at room temperature to afford dolutegravir sodium as a pale yellow solid (0.43 g, HPLC purity: 98.80%). 1H NMR (DMSO-d6): δ 10.70 (t, J = 5.8 Hz, 1H), 7.89 (s, 1H), 7.37-7.30 (m, 1H), 7.23-7.19 (m, 1H), 7.04-7.01 (m, 1H), 5.17 (m, 1H), 4.81 (t, J = 6.4 Hz, 1H), 4.51 (d, J = 5.5 Hz, 2H), 4.32-4.29 (m, 1H), 4.16 (dd, J= 14.1, 4.8 Hz, 1H), 3.99-3.94 (m, 1H), 3.82-3.80 (m, 1H), 1.89-1.84 (m, 1H), 1.38 (d, J = 12.9 Hz, 1H), 1.24 (d, J = 7.0 Hz, 3H).13C NMR (DMSO-d6): δ 177.93, 167.12, 166.08, 161.59 (dd), 161.13, 159.63 (dd), 134.26, 130.44 (d), 130.38 (d), 122.90 (d), 114.95, 111.23 (d), 108.78, 103.64 (t), 75.59, 61.95, 53.11, 43.01, 35.32, 29.22, 15.30.
Example 24:

The suspension of (4R,12aS)-N-(2,4-difluorobenzyl)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12, 12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-6][1,3]oxazine-9-carboxamide (27, 0.43 g) and solid sodium hydroxide (0.20 g) in absolute ethanol (2.5 mL) was stirred at room temperature for ca.24 h. The reaction was diluted with mixture of water/ethanol (5 mL, 1:1) and left stirring for ca. 1.5 h at room temperature. The reaction mixture was filtered through fitted funnel rinsing with mixture of water/ethanol (3 x 5 mL, 1:1) and dried in vacuo at room temperature to afford 15 as a pale yellow solid (0.41 g, HPLC purity: 98.87%).
1H NMR (DMSO-de): δ 10.70 (t, J = 5.8 Hz, 1H), 7.89 (s, 1H), 7.37-7.30 (m, 1H), 7.23-7.19 (m, 1H), 7.04-7.01 (m, 1H), 5.17 (m, 1H), 4.81 (t, J = 6.4 Hz, 1H), 4.51 (d, J = 5.5 Hz, 2H), 4.32-4.29 (m, 1H), 4.16 (dd, J = 14.1, 4.8 Hz, 1H), 3.99-3.94 (m, 1H), 3.82-3.80 (m, 1H), 1.89-1.84 (m, 1H), 1.38 (d, J = 12.9 Hz, 1H), 1.24 (d, J = 7.0 Hz, 3H).13C NMR (DMSO-d6): δ 177.93, 167.12, 166.08, 161.59 (dd), 161.13, 159.63 (dd), 134.26, 130.44 (d), 130.38 (d), 122.90 (d), 114.95, 111.23 (d), 108.78, 103.64 (t), 75.59, 61.95, 53.11, 43.01, 35.32, 29.22, 15.30.
Example 25:

The suspension of {4R, 12aS)-7-chloro-4-methyl-6,8-dioxo-3,4, 6,8, 12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-6][1,3]oxazine-9-carboxylic acid (7, 0.31 g) and solid sodium hydroxide (0.20 g) in absolute ethanol (2.5 mL) was stirred at 50 °C for 3 days. The reaction was quenched with 2M H2S04 (1.2 mL) and left stirring for 7 h at room temperature. The reaction mixture was filtered through fitted funnel rinsing with water (3×5 mL) and ethanol (5 mL) dried in vacuo at 40°C to afford 28 as a pale yellow solid (0.17 g).
1H NMR (DMSO-d6): δ 15.37 (s, 1H), 12.76 (s, 1H), 8.66 (s, 1H), 5.51-5.49 (m, 1H), 4.80-4.78 (m, 1H), 4.65 (dd, J=13.8, 3.7 Hz, 1H), 4.43 (dd, J=13.8, 5.9 Hz, 1H), 4.05 (t, J^^.b Hz, 1H), 3.91 (dd, J=11.4, 3.1 Hz, 1H), 2.07-2.00 (m, 1H), 1.56 (d, J=13.8 Hz, 1H), 1.34 (d, J=7.0 Hz, 3H).13C NMR (DMSO-de): δ 172.21, 165.39, 161.73, 153.61, 141.11, 118.66, 112.99, 75.95, 62.03, 51.50, 44.90, 29.08, 15.18.
Example 26:

The suspension of (4R,12aS)-N-(2,4-difluorobenzyl)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8, 12, 12a-hexahydro-2H-pyrido[1 ‘,2’:4,5]pyrazino[2, 1 ,3]oxazine-9-carboxamide (27, 0.88 g) and solid sodium hydroxide (0.24 g) in absolute ethanol (20 mL) was stirred at 30 °C for 1.5 h. The reaction was quenched with 2M H2S04 (1 .5 mL) and left stirring for 3 hours at room temperature. The reaction mixture was filtered through fritted funnel and rinsed with water (3 x 2 mL) and ethanol (4 mL), and dried in vacuo at 40 °C to afford O-ethyl dolutegravir (29) as a pale yellow solid (0.25 g). The filtrate was extracted with ethyl acetate (3 x 5 mL). The combined organic layers were dried over MgS04, filtered and concentrated, then dried in vacuo at 40 °C to afford more 29 as a pale yellow solid (0.27 g).
1H NMR (CDCI3): δ 10.37 (t, J = 5.8 Hz, 1 H), 8.36 (s, 1 H), 7.37-7.32 (m, 1 H), 6.83-6.77 (m, 2H), 5.19 (dd, J = 5.9, 3.8 Hz, 1 H), 5.04-4.98 (m, 1 H), 4.61 (d, J = 6Hz, 2H), 4.26-4.22 (m, 3H), 4.1 1 (dd, J = 13.4, 5.9 Hz, 1 H), 3.97 (t, J = 2.4 Hz, 1 H), 3.96 (d, J = 2.4 Hz, 1 H), 2.21-2.14 (m, 1 H), 1.51 (dq, J = 14.0, 2.3 Hz, 1 H), 1 .47 (t, J = 7.0 Hz, 3H), 1 .35 (d, J = 7.1 Hz, 3H).
13C NMR (CDCI3): δ 174.78, 164.17, 162.49 (dd), 160.51 (dd), 155.72, 154.08, 142.32, 130.60 (dd), 129.33, 121 .51 (dd), 1 18.67, 1 1 1 .23 (dd), 103.78 (t), 76.15, 69.74, 62.58, 53.42, 44.58, 36.50 (d), 29.44, 16.04, 15.64.
Example 27:

The suspension of (4R, 12aS)-7-(benzyloxy)-4-methyl-3,4, 12,12a-tetrahydro-2H-pyrido[1 ‘,2’:4,5]pyrazino[2, 1-b][1 ,3]oxazine-6,8-dione (30, 0.68 g, prepared according to prior art) and solid sodium hydroxide (0.40 g) in absolute ethanol (5 mL) was stirred at 50 °C for 14 h. The reaction was quenched with formic acid (0.35 mL), water (2 mL) was added and mixture was left stirring for additional 1 h at room temperature. The reaction mixture was extracted with ethyl acetate (3 x 5 mL) and the combined organic layers concentrated to afford a crude oil. Purification by flash chromatography (eluting with CH2CI2/methanol) afforded 32 as an orange solid (0.26 g, 52 %).
The above procedure if done at room temperature in same time period, affords 31 as orange oil (0.24 g, 43 %).
Compound 32: 1H NMR (DMSO-d6): δ 7.64 (d, J = 7.4 Hz, 1 H), 6.20 (d, J = 7.3 Hz, 1 H), 5.40 (dd, J = 5.1 , 4.2 Hz, 1 H), 4.83-4.78 (m, 1 H), 4.35 (dd, J = 13.6, 3.9 Hz, 1 H), 4.13 (dd, J = 13.6, 5.4 Hz, 1 H), 4.05-4.00 (m, 1 H), 3.90-3.85 (m, 1 H), 2.03-1.95 (m, 1 H), 1.52 (dd, J = 13.9, 1 .9 Hz, 1 H), 1.33 (d, J = 7.1 Hz, 3H). 13C NMR (DMSO-d6): δ 170.96, 163.01 , 153.48, 137.96, 1 16.83, 1 13.52, 76.18, 62.05, 50.39, 44.53, 29.21 , 15.28.
Compound 31 : 1H NMR (DMSO-d6): δ 7.67 (d, J = 7.4 Hz, 1 H), 6.28 (d, J = 7.4 Hz, 1 H), 5.29 (dd, J = 5.4, 3.8 Hz, 1 H), 4.82-4.75 (m, 1 H), 4.32 (dd, J = 13.6, 3.6 Hz, 1 H), 4.10 (dd, J = 13.5, 5.6 Hz, 1 H), 4.03-3.93 (m, 3H), 3.85 (ddd, J = 1 1 .6, 5.0, 2.2 Hz, 1 H), 1.97-1 .89 (m, 1 H), 1 .48 (dd, J = 13.8, 2.1 Hz, 1 H), 1.27 (d, J = 7.1 Hz, 3H), 1.26 (d, J = 7.0 Hz, 3H). 13C NMR (DMSO-d6): δ 174.38, 156.1 1 , 150.82, 139.48, 1 16.39, 1 13.52, 75.92, 67.31 , 61 .80, 51 .36, 44.22, 29.29, 15.76, 15.36.
Exa

The transformation of 6 to dolutegravir with sodium hydroxide in ethanol was monitored for the interconversion of intermediates. The suspension of 6 (0.44 g) and solid sodium hydroxide (0.20 g) in ethanol (3.33 ml.) was stirred at 22 °C. Samples of the reaction mixture were taken after 3, 8 and 24 h for UPLC analysis. After 24 h, the reaction mixture was quenched with 2 M H2S04 (5 ml_), and left stirring at room temperature. The reaction mixture was filtered through fritted funnel, the product rinsed with water (30 ml.) and dried in vacuo at 50 °C overnight to afford dolutegravir as a white solid (0.27 g, 64 %).
The results of reaction monitoring:
Time UPLC analysis (area%)
Entry
(h) compound 6 compound 29 dolutegravir
1 3 h 37.50 20.63 39.99
2 8 h 0.78 15.46 80.32
3 24h 0.31 8.56 88.21
Example 29:

The effect of added water and reaction temperature was evaluated by monitoring 4 reactions in parallel. To the suspensions of 27 (0.86 g) in MeOH were added solid sodium hydroxide (0.40 g) or aqueous solution of NaOH (5 M, 2 ml.) (see Table below). The reactions were stirred in parallel at 50 °C or 22 °C. Samples were taken in timely intervals for UPLC analysis.
The results of reaction monitoring demethylation of 27 in MeOH:

Example 30:

The effect of added water and reaction temperature was evaluated by monitoring 4 reactions in parallel. To the suspensions of 6 (0.88 g) in EtOH were added solid sodium hydroxide (0.40 g) or aqueous solution of NaOH (5 M, 2 mL) (see Table below). The reactions were stirred in parallel at 50 °C or 22 °C. Samples were taken in timely intervals for UPLC analysis.
The results of reaction monitoring of the transformations of 6 in ethanol with NaOH:

dol. = dolutegravir
Exa

The effect of added water and reaction temperature was evaluated by monitoring 4 reactions in parallel. To the suspensions of 27 (0.88 g) in EtOH were added solid sodium hydroxide (0.40 g) or aqueous solution of NaOH (5 M, 2ml_) (see Table below). The reactions were stirred in parallel at 50 °C or 22 °C. Samples were taken in timely intervals for UPLC analysis.
The results of reaction monitoring of the transformations of 27 in ethanol with NaOH:

dol. = dolutegravir
Example 32:

Compound 3 (30 g, 1 10 mmol; assay 99%) was suspended in acetonitrile (450 mL), acetic acid (73 mL) and methanesulfonic acid (25 mL) were added. The reaction mixture was stirred 4 h at 70 °C. The clear red solution was cooled to 25 °C. Triethylamine (77 mL) and (S)-2-aminopropanol (17 mL) were added and the mixture was stirred at reflux temperature for 20 h. The reaction mixture was cooled to 25 °C and the insoluble product filtered, washed with 1 M HCI(aq) (60 mL), water (3 * 60 mL) and dried to give 4c (19.49 g, 67%): mp = 313-315 °C; 1H NMR (DMSO-d6): δ 1.31 (d, J = 6.3 Hz, 3H), 3.65 (dd, J = 8.6, 6.8 Hz, 1 H), 4.13 (dd, J = 1 1.7, 10.3 Hz, 1 H), 4.28 (m, 1 H), 4.39 (dd, J = 8.6, 6.8 Hz, 1 H), 4.92 (dd, J = 12.3, 4.2 Hz, 1 H), 5.45 (dd, J = 10.2, 4.1 Hz, 1 H), 7.16 (s, 1 H), 8.84 (s, 1 H), 15.74 (s, 1 H); 13C NMR (DMSO-d6) 16.5, 51.6, 52.9, 72.4, 81.6, 1 15.8, 1 18.1 , 141.5, 147.6, 153.4, 165.3, 179.0.
Example 33

Compound 4c (2.78 g) was suspended in dimethylformamide (40 mL), cooled to 0 °C, then triethylamine (3.52 mL) was added, followed by ethyl chloroformate (1 .31 mL). After 10 min there was added 2,4-difluorobenzylamine (1 .57 mL). The mixture was then stirred at 25 °C for 1 h. Water (150 mL) was added and the mixture extracted with dichloromethane (50 mL). The organic phase was separated, washed with water (2 χ 50 mL), dried over sodium sulfate and evaporated under reduced pressure. The residue (4.31 g) was treated with boiling 2-propanol (40 mL), the suspension cooled, the product filtered and dried to give the product 5c as a white powder (2.70 g, 69%): 99.80 area% by HPLC at 258 nm; mp = 222-223 °C; MS (ESI) m/z = 390 [MH]+; 1H NMR (DMSO-d6): δ 1 .30 (d, J = 6.3 Hz, 3H), 3.63 (dd, J = 8.6, 6.8 Hz, 1 H), 4.02 (m, 1 H), 4.26 (m, 1 H), 4.37 (dd, J = 8.6, 6.8 Hz, 1 H), 4.53 (d, J = 6.0 Hz, 2H), 4.84 (dd, J = 12.2, 4.2 Hz, 1 H), 5.40 (dd, J = 12.2, 4.2 Hz, 1 H), 6.91 (s, 1 H), 7.05 (m, 1 H), 7.24 (m, 1 H), 7.38 (m, 1 H), 8.62 (s, 1 H), 10.43 (t, J = 6.0 Hz, 1 H).

To a suspension of 5c (2.70 g, 6.9 mmol) in acetonitrile (32 mL) was added DABCO (39 mg, 5 mol%) and TCCA (1.01 g, 4.3 mmol). The mixture was stirred 20 h at 40 °C protected from light and then quenched with a mixture of DMSO (0.81 mL) and water (0.20 mL). The insoluble cyanuric acid was removed by filtration and washed with acetonitrile (10 mL). The filtrate was evaporated under reduced pressure to give viscous oil that was crystallized from a mixture of methanol (10 mL) and water (5 mL), by slowly cooling the solution from 60 °C to room temperature. The product 6c was filtered, washed with cold methanol (8 mL) and dried to give an off-white powder (1 .20 g, 41 %): mp = 225-227 °C; MS (ESI) m/z = 424 [MH]+; 1H NMR
(DMSO-d6): δ 1.28 (d, J = 6.3 Hz, 3H), 3.65 (dd, J = 8.6, 6.9 Hz, 1 H), 4.09 (m, 1 H), 4.26 (m, 1 H), 4.35 (dd, J = 8.6, 6.6 Hz, 1 H), 4.54 (d, J = 5.9 Hz, 2H), 4.85 (dd, J = 12.3, 3.8 Hz, 1 H), 5.42 (dd, J = 10.1 , 3.8 Hz, 1 H), 7.06 (m, 1 H), 7.24 (m, 1 H), 7.40 (m, 1 H), 8.67 (s, 1 H), 10.24 (t, J = 6.0 Hz, 1 H).
Example 35

cabotegravir
The suspension of 6c (1.00 g, 2.4 mmol) and sodium hydroxide (0.57 g, 14.2 mmol) in absolute ethanol (7 mL) was stirred at 40 °C for 16 h. The reaction was quenched with 0.5M H2S04 (15 mL), extracted with dichloromethane (20 mL), the extract washed with water (20 mL) and evaporated under reduced pressure. The residue was triturated in MTBE (10 mL), the product filtered, washed with MTBE (10 mL) and dried to give cabotegravir as an off-white solid (0.74 g, 77%): MS (ESI) m/z = 405 [MH]+.
Lek, a Sandoz company, opens the first production facility in Slovenia for drug substances for innovative medicines at its Mengeš site
Vojmir Urlep, president of Lek Board of Management

Dr Miro Cerar, the Prime Minister of the Republic of Slovenia
Lek, a Sandoz company, awarded for cooperation in practical training of students of the Faculty of Chemistry and Chemical Technology
30. 1. 2015
At a ceremony held on 22 January 2015 at the Faculty of Chemistry and Chemical Technology, University of Ljubljana, the Maks Samec awards and recognitions for 2014 were presented for the best doctoral thesis in the field of chemistry, the best doctoral thesis in the field of chemical engineering and chemical technology and for services and merits to the Faculty in the year 2014. On this occasion, the Faculty also wanted to thank all the companies and individuals who shared their knowledge and resources to help the Faculty on its education and research path.
Lek, a Sandoz company, received a plaque for taking part in the implementation of practical training, which was collected, on behalf of the company, by Samo Roš, Head of Human Resources and a Member of the Lek Board of Management. By doing so, the Faculty of Chemistry and Chemical Technology thanked all the mentors who directly transfer their expertise and valuable experience onto students, teaching them specific skills, encouraging their development, guiding them through the work process and ensuring that students become socialized in the workplace.
* * *
Lek, a Sandoz company, is one of key pillars of the second-largest generic pharmaceutical company globally. Its role within Sandoz is to act as: a leading global development center for technologically demanding products and technologies; a global manufacturing center for active pharmaceutical ingredients and medicines; a competence center for the development of vertically integrated products; a Sandoz competence center in the field of development and manufacturing of biosimilar products; and, a supply center for the markets of Central and Eastern Europe (CEE), South East Europe (SEE) and Commonwealth of Independent States (CIS), and it is responsible for sales on the Slovenian market. For further information please visit http://www.lek.si/en.
Sandoz, the generic pharmaceuticals division of Novartis, is a global leader in the generic pharmaceutical sector. Sandoz employs over 26,400 employees and its products are available in more than 160 countries, offering a broad range of high-quality, affordable products that are no longer protected by patents. With USD 9.6 billion in sales in 2014, Sandoz has a portfolio of approximately 1,100 molecules, and holds the #1 position globally in biosimilars as well as in generic injectables, ophthalmics, dermatology and antibiotics, complemented by leading positions in the cardiovascular, metabolism, central nervous system, pain, gastrointestinal, respiratory, and hormonal therapeutic areas. Sandoz develops, produces, and markets these medicines, as well as active pharmaceutical and biotechnological substances. Nearly half of Sandoz’s portfolio is in differentiated products, which are defined as products that are more difficult to scientifically develop and manufacture than standard generics. In addition to strong organic growth since consolidating its generics businesses under the Sandoz brand name in 2003, Sandoz has benefitted from strong growth of its acquisitions, which include Lek (Slovenia), Sabex (Canada), Hexal (Germany), Eon Labs (US), EBEWE Pharma (Austria), Oriel Therapeutics (US), and Fougera Pharmaceuticals (US).
Sandoz is on Twitter. Sign up to follow @Sandoz_global at http://twitter.com/Sandoz_Global.
Novartis provides innovative healthcare solutions that address the evolving needs of patients and societies. Headquartered in Basel, Switzerland, Novartis offers a diversified portfolio to best meet these needs: innovative medicines, eye care, cost-saving generic pharmaceuticals, preventive vaccines and over-the-counter products. Novartis is the only global company with leading positions in these areas. In 2014, the Group achieved net sales of USD 58.0 billion, while R&D throughout the Group amounted to approximately USD 9.9 billion (USD 9.6 billion excluding impairment and amortization charges). Novartis Group companies employ approximately 130,000 full-time-equivalent associates. Novartis products are available in more than 180 countries around the world. For more information, please visit www.novartis.com
////////////Carbotegravir, Dolutegravir, New Patent, WO 2016113372, Lek Pharmaceutical and Chemical Co DD

{kind=link}