
Home » 2024 (Page 4)
Yearly Archives: 2024
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Obeldesivir

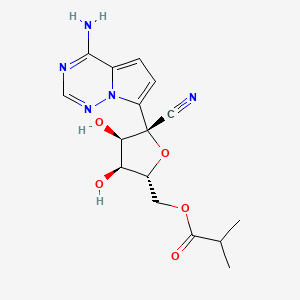
Obeldesivir,
2647441-36-7
361.35 g/mol
C16H19N5O5
[(2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxyoxolan-2-yl]methyl 2-methylpropanoate
Q55KCM7PXB; ATV006; UNII-Q55KCM7PXB
SCHEME
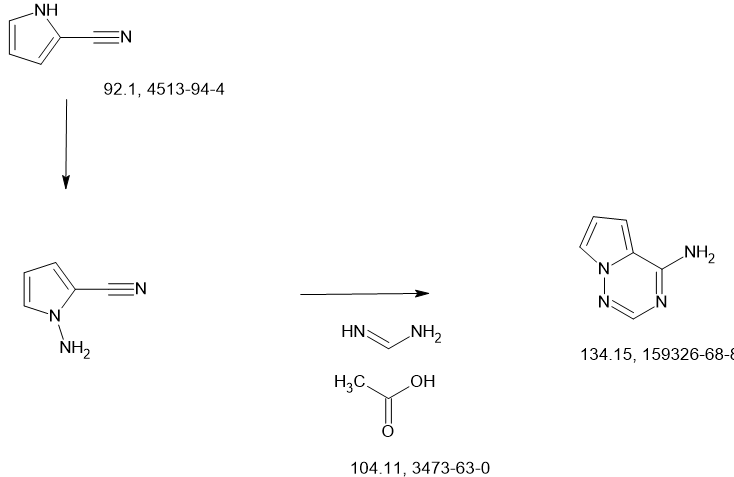
NEXT
CN11687449 68%
Journal of Medicinal Chemistry (2023), 66(17), 11701-11717 50%
WO2023167938
CN115583954 67%
WO2022047065
CN113698405 49%
https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c00750
Obeldesivir (GS-5245, ATV006) is an isobutyric ester prodrug of GS-441524 made by Gilead Sciences that is currently in Phase III trials for the outpatient treatment of COVID-19 in high risk patients.[1][2] The purpose of the isobutyric ester modification on obeldesivir is to improve the oral bioavailability of the parent nucleoside, GS-441524. Obeldesivir is hydrolyzed to its parent nucleoside, GS-441524, which is in turn converted to remdesivir-triphosphate (GS-443902) by a nucleoside kinase, adenylate kinase and nucleotide diphosphate kinase.[3][4]
GS-443902 is a bioactive ATP analogue with broad-spectrum antiviral activity [5] and is the same compound formed by remdesivir, though by a different enzymatic pathway. Unlike remdesivir, which is metabolized by enzymes that are highly expressed in the liver, GS-441524 released by obeldesivir is metabolized by enzymes that are evenly expressed throughout the body. Due to their different metabolic pathways, obeldesivir can be administered orally, whereas remdesivir must be administered intravenously for COVID-19 treatment.
The pharmacokinetic properties of obeldesivir and improved was first published by Chinese researchers in May 2022. The Chinese group pursued investigation of obeldesivir independently from Gilead Sciences. Compared to IV administered GS-441524 in rats at 5 mg/kg, orally administered obeldesivir at 25 mg/kg (referred to as “ATV006”) yielded approximately 22% bioavailability.[6] Treatment with obdeldesivir reduced viral load and prevents lung pathology in KI-hACE2 and Ad5-hACE2 mouse models of SARS-CoV-2. A patent filed by Gilead Sciences with a priority date of August 27, 2020,[7] found the bioavailability of GS-441524 after oral administration of obdeldesivir (compound 15) in mice, rats, ferrets, dogs, and cynomolgus macaques to be 41%, 63.9%, 154%, 94%, and 38%, respectively. Across all species evaluated, obeldesivir showed improved oral bioavailability compared to oral administration of the parent nucleoside, GS-441524
PAPER
https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c00750
Abstract

Remdesivir 1 is an phosphoramidate prodrug that releases the monophosphate of nucleoside GS-441524 (2) into lung cells, thereby forming the bioactive triphosphate 2-NTP. 2-NTP, an analog of ATP, inhibits the SARS-CoV-2 RNA-dependent RNA polymerase replication and transcription of viral RNA. Strong clinical results for 1 have prompted interest in oral approaches to generate 2-NTP. Here, we describe the discovery of a 5′-isobutyryl ester prodrug of 2 (GS-5245, Obeldesivir, 3) that has low cellular cytotoxicity and 3–7-fold improved oral delivery of 2 in monkeys. Prodrug 3 is cleaved presystemically to provide high systemic exposures of 2 that overcome its less efficient metabolism to 2-NTP, leading to strong SARS-CoV-2 antiviral efficacy in an African green monkey infection model. Exposure-based SARS-CoV-2 efficacy relationships resulted in an estimated clinical dose of 350–400 mg twice daily. Importantly, all SARS-CoV-2 variants remain susceptible to 2, which supports development of 3 as a promising COVID-19 treatment.
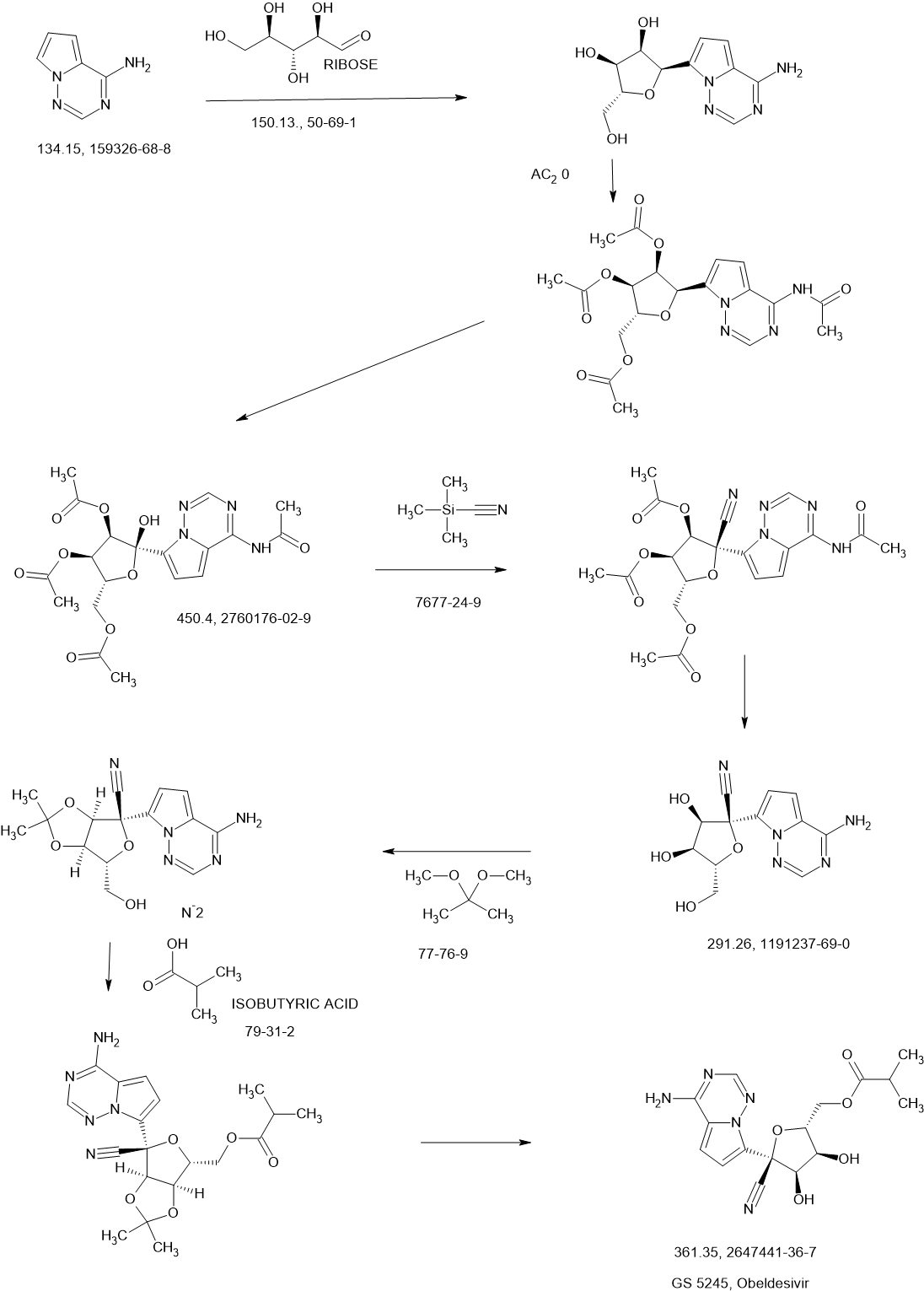
Synthesis of ((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl Isobutyrate (3)
To a solution of (3aR,4R,6R,6aR)-4-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-6-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxole-4-carbonitrile 4 (2000 mg, 6.0 mmol) (30) and isobutyric acid (638 mg, 7.2 mmol) in DMF (5 mL), N,N′-diisopropylcarbodiimide (914 mg, 7.2 mmol) was slowly added followed by 4-(dimethylamino)pyridine (737 mg, 6.0 mmol). The reaction mixture was stirred for 4 h and then diluted with ethyl acetate, washed with water and brine, dried over sodium sulfate, and concentrated under reduced pressure. The residue was subjected to silica gel chromatography, eluting with 20% MeOH in CH2Cl2 to provide the intermediate ((3aR,4R,6R,6aR)-6-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-6-cyano-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl isobutyrate. LCMS m/z = 402.2 (M + 1). To a solution of the intermediate (1500 mg) in THF (10 mL), conc. HCl (2 mL) was added, and the mixture was stirred at rt for 4 h. The reaction mixture was diluted with CH2Cl2, washed with water, saturated aqueous bicarbonate, and brine, dried over sodium sulfate, concentrated, and subjected to silica gel chromatography, eluting with 30% MeOH in CH2Cl2 to afford the title compound (660 mg, 50%). 1H NMR (400 MHz, MeOH-d4) δ 7.88 (s, 1H), 6.96–6.85 (m, 2H), 4.50–4.27 (m, 4H), 4.16 (dd, J = 6.2, 5.3 Hz, 1H), 2.56 (p, J = 7.0 Hz, 1H), 1.14 (dd, J = 7.0, 3.8 Hz, 6H). LCMS m/z: 362.1 (M + 1). 13C NMR (400 MHz, CHCl3–d3) δ 175.9, 155.6, 147.9, 123.5, 110.2, 100.8, 116.9, 116.6, 81.3, 79.0, 74.0, 70.2, 62.9, 33.2, 18.7, 18.6. HRMS m/z: 362.14615, C16H20N5O5 362.14644.

PATENT
WO-2022047065-A2
PATENT
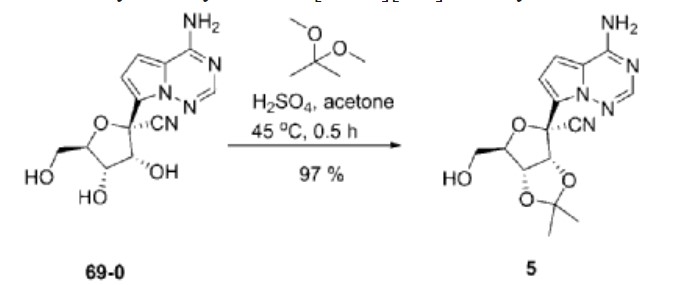
| Example 1. (3aR, 4R, 6R, 6aR)-4-(4-aminopyrrole[2,1-f][1,2,4]triazin-7-yl)-6-(hydroxymethyl- Synthesis of 2,2-dimethyltetrahydrofuran[3,4-d][1,3]dioxolyl-4-carbonitrile (compound 5) |

| Dissolve 5.62g of compound 69-0 in 30mL of acetone, then add 11.50mL of 2,2-dimethoxypropane and 1.34mL of 98% sulfuric acid, stir at 45°C for half an hour, cool to room temperature, and remove by rotary evaporation Organic solvents. Extract with 100 mL of ethyl acetate and 100 mL of saturated sodium bicarbonate solution. Repeat the extraction three times. Combine the ethyl acetate layers, add anhydrous sodium sulfate to dry, and filter to remove sodium sulfate. The organic solvent was removed by rotary evaporation, and 6.20 g of compound 5 (white solid, yield 97%) was obtained through column chromatography (eluent: petroleum ether/ethyl acetate (v/v) = 1/2). The hydrogen spectrum of the obtained compound 5 was detected, and the results are as follows: |
| 氢谱:1H NMR(400MHz,Chloroform-d)δ7.95(s,1H),7.11(d,J=4.7Hz,1H),6.69(dd,J=4.8,2.4Hz,1H),5.77(s,2H),5.42(d,J=6.6Hz,1H),5.24(dd,J=6.6,2.4Hz,1H),4.67(q,J=1.9Hz,1H),3.99(dd,J=12.5,1.9Hz,1H),3.84(dd,J=12.5,1.7Hz,1H),1.81(s,3H),1.40(s,3H)。 |
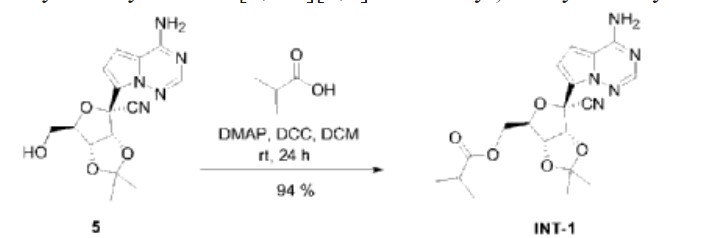
| Example 2. ((3aR,4R,6R,6aR)-6-(4-aminopyrrole[2,1-f][1,2,4]triazin-7-yl)-6-cyano-2 ,Synthesis of 2-dimethyltetrahydrofuran[3,4-d][1,3]dioxol-4-yl)methylisobutyrate (compound INT-1) |
| Dissolve 1.50g of compound 5 in 15ml of dichloromethane, then add 0.42mL of isobutyric acid and 55.40mg of 4-dimethylaminopyridine, stir for 10 minutes, add 1.02g of dicyclohexylcarbodiimide, and stir at room temperature. Stir for 24h. After column chromatography separation (eluent: petroleum ether/ethyl acetate (v/v) = 1/1), 1.71 g of compound INT-1 (white solid, yield 94%) was obtained. The hydrogen spectrum and carbon spectrum of the obtained compound INT-1 were detected, and the results are as follows: |
| Hydrogen spectrum: 1 H NMR (400MHz, CDCl 3 ,ZQF-RD01-2)δ(ppm):7.99(s,1H),6.99(d,J=4.6Hz,1H),6.62(d,J=4.6Hz,1H),5.72(br,2H),5.49(d,J=6.8Hz,1H),4.93-4.90(dd,J=6.8Hz,4.3Hz,1H),4.61-4.58(q,J=4.4Hz,1H),4.44-4.26(m,2H),2.61-2.50(m,1H),1.77(s,3H),1.42(s,3H),1.17-1.14(q,J=3.8Hz,6H)。 |
| Carbon spectrum: 13 C NMR (100MHz, CDCl 3 ,ZQF-RD01-2)δ(ppm):176.7,155.2,147.3,123.5,117.2,116.7,115.6,112.6,100.0,83.8,83.0,82.0,81.4,63.1,33.8,26.4,25.6,18.9。 |
PATENT
WO2023167938

To a solution of (3aR,4R,6R,6aR)-4-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-6-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxole-4-carbonitrile, Intermediate 1 (2000 mg, 6.0 mmol) (Siegel et. al. J. Med. Chem.2017, 60, 1648-1661) in tetrahydrofuran (THF) was added N,N-dimethylaminopyridine (DMAP) (0.03 eq). To the reaction mixture, isobutyric anhydride (1.1 eq) was added slowly. After the completion of the staring material, the reaction mixture was concentrated and purified by flash chromatography using 20% methanol in DCM as an eluant to give ((3aR,4R,6R,6aR)-6-(4- aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-6-cyano-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4- yl)methyl isobutyrate, Intermediate 1a. LCMS: MS m/z: 402.2 (M+1).
Concentrated HCl (5 eq, 1mL) was added to a solution of Intermediate 1a (1000 mg) in acetonitrile (10 mL), which was then stirred at room temperature for 2 hours. LCMS showed the product formation. The reaction was stopped after 4 hours. The reaction mixture was diluted with ethyl acetate and quenched with saturated bicarbonate. The organic layer was separated, washed with brine, dried over sodium sulphate, and concentrated. The residue was purified by flash chromatography using 30% methanol DCM as an eluant. The collected fractions were concentrated to give Intermediate 2.1H NMR (400 MHz, Methanol-d4) δ 7.88 (s, 1H), 6.96 – 6.85 (m, 2H), 4.50 – 4.27 (m, 4H), 4.16 (dd, J = 6.2, 5.3 Hz, 1H), 2.56 (p, J = 7.0 Hz, 1H), 1.14 (dd, J = 7.0, 3.8 Hz, 6H); LCMS: MS m/z: 362.1 (M+1).


AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
//////////
| Names | |
|---|---|
| IUPAC name[(2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f] [1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxyoxolan-2-yl]methyl 2-methylpropanoate | |
| Identifiers | |
| CAS Number | 2647441-36-7 |
| 3D model (JSmol) | Interactive image |
| ChEMBL | ChEMBL4863829 |
| ChemSpider | 115037299 |
| PubChemCID | 162513664 |
| UNII | Q55KCM7PXB |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C16H19N5O5 |
| Molar mass | 361.358 g·mol−1 |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
References
- ^ “Clinical Trials Register”.
- ^ “Gilead touts Veklury resilience against mutated coronavirus, plots phase 3 for new COVID oral antiviral”. 19 October 2022.
- ^ Cho, A.; Saunders, O. L.; Butler, T.; Zhang, L.; Xu, J.; Vela, J. E.; Feng, J. Y.; Ray, A. S.; Kim, C. U. (2012). “Synthesis and antiviral activity of a series of 1′-substituted 4-aza-7,9-dideazaadenosine C-nucleosides”. Bioorganic & Medicinal Chemistry Letters. 22 (8): 2705–2707. doi:10.1016/j.bmcl.2012.02.105. PMC 7126871. PMID 22446091.
- ^ Yan, Victoria C.; Muller, Florian L. (2020). “Advantages of the Parent Nucleoside GS-441524 over Remdesivir for Covid-19 Treatment”. ACS Medicinal Chemistry Letters. 11 (7): 1361–1366. doi:10.1021/acsmedchemlett.0c00316. PMC 7315846. PMID 32665809.
- ^ Gordon, C. J.; Tchesnokov, E. P.; Woolner, E.; Perry, J. K.; Feng, J. Y.; Porter, D. P.; Götte, M. (2020). “Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency”. The Journal of Biological Chemistry. 295 (20): 6785–6797. doi:10.1074/jbc.RA120.013679. PMC 7242698. PMID 32284326.
- ^ Cao, Liu; et al. (2022). “The adenosine analog prodrug ATV006 is orally bioavailable and has preclinical efficacy against parental SARS-CoV-2 and variants”. Science Translational Medicine. 14 (661): eabm7621. doi:10.1126/scitranslmed.abm7621. PMC 9161374. PMID 35579533.
- ^ “Espacenet – search results”.
//////////Obeldesivir, GS-5245, ATV006
CC(C)C(OC[C@H]1O[C@@](C#N)(C2=CC=C3C(N)=NC=NN32)[C@H](O)[C@@H]1O)=O
Oditrasertib
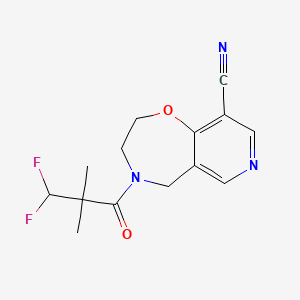
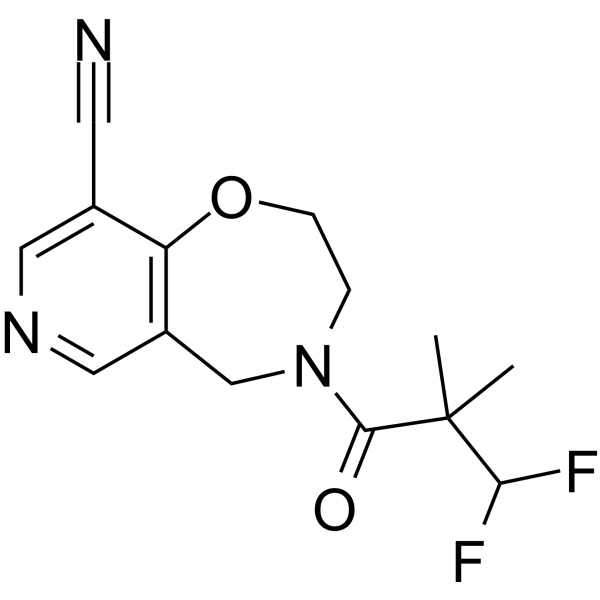
Oditrasertib
SAR443820; DNL788
| Molecular Formula | C14H15F2N3O2 |
| Molecular Weight | 295.2846 |
- UNII-I1YQT8HC89
- I1YQT8HC89
- cas 2252271-93-3
- 4-(3,3-Difluoro-2,2-dimethylpropanoyl)-3,5-dihydro-2H-pyrido(3,4-F)(1,4)oxazepine-9-carbonitrile
PYRIDO(3,4-F)-1,4-OXAZEPINE-9-CARBONITRILE, 4-(3,3-DIFLUORO-2,2-DIMETHYL-1-OXOPROPYL)-2,3,4,5-TETRAHYDRO-
Oditrasertib (SAR443820) is an orally active, BBB-penetrable and selective reversible inhibitor of RIPK1. Oditrasertib can be used in the research of chronic inflammatory central nervous system diseases, such as amyotrophic lateral sclerosis and multiple sclerosis.
Oditrasertib is a serine/threonine kinase (STK) inhibitor.
OriginatorHarvard University
DeveloperDenali Therapeutics Inc; Sanofi
ClassAntidementias; Small molecules
Mechanism of ActionRIPK1 protein inhibitors
- 24 Feb 2025Sanofi terminates its license for DNL 788 from Denali Therapeutics
- 21 Nov 2024Chemical structure information added.
- 08 Nov 2024Discontinued – Phase-II for Multiple sclerosis (In adults) in Canada, China, France, Italy, Poland, Germany, Belgium, Spain (PO)
SCHEME
WO2018213632

PATENT
[WO2018213632A1]
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018213632

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
//////////
///////////Oditrasertib,
CC(C)(C(F)F)C(=O)N1CCOC2=C(C=NC=C2C1)C#N
- [1]. Fidalgo JDV, et al. Preparation of substituted oxazepines and related compounds as RIPK1 inhibitors and uses thereof. WO2018213632A1. 2018-11-22.[2]. Hincelin-Mery A, et al. Safety, pharmacokinetics, and target engagement of a brain penetrant RIPK1 inhibitor, SAR443820 (DNL788), in healthy adult participants. Clin Transl Sci. 2024 Jan;17(1):e13690. [Content Brief][3]. Montalban X, et al. Effect of RIPK1 inhibitor, SAR443820, on serum neurofilament light levels in patients with multiple sclerosis: a phase 2 trial design (P6-3.011)[J]. Neurology, 2023, 100(17_supplement_2): 2178.
ORZILOBEN
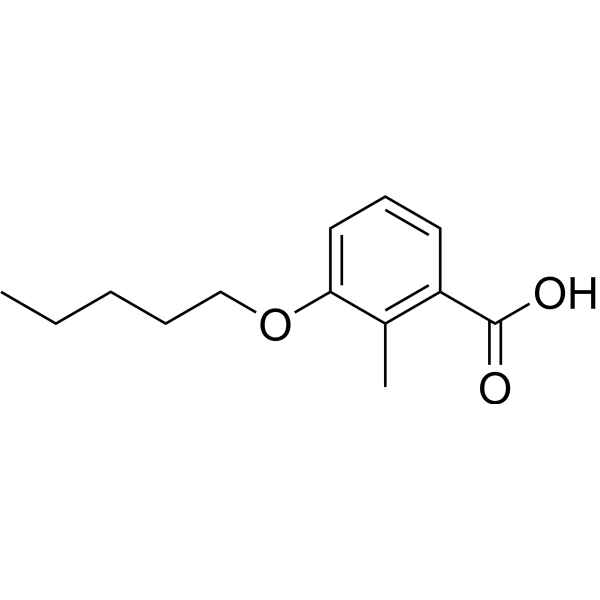

ORZILOBEN,
CAS 1555822-28-0
2-methyl-3-(pentyloxy)benzoic acid
| Molecular Weight | 222.28 |
|---|---|
| Formula | C13H18O3 |
Orziloben is a medium chain fatty acid (MCFA) analogue[1]
Phase IILiver disorders
OriginatorPronova BioPharma
DeveloperNorthSea Therapeutics
ClassAntifibrotics; Hepatoprotectants; Omega 3 fatty acids; Unsaturated fatty acids
Mechanism of ActionUndefined mechanism.
28 Jan 2025No recent reports of development identified for phase-I development in Liver-disorders(In volunteers) in United Kingdom (PO)- 24 Dec 2024Chemical structure information added.
- 23 Aug 2024NorthSea Therapeutics completes a phase I trial in healthy volunteers in the United Kingdom (PO) (ISRCTN12367117)
Patent
WO2020074964
The rising global epidemic of obesity and its comorbidities, e.g., type 2 diabetes mellitus and hyperlipidemia, is placing an enormous burden both on public health (mortality and morbidity) and on the available public health resources required to treat these conditions.
Current drugs that treat hyperlipidemia (e.g., statins, omega-3 fatty acids, fibrates) have mostly neutral effects on glycemic control, whilst drugs targeting glycemic control e.g., insulin, thiazolidinediones (TZDs), have adverse effects upon bodyweight and (for TZDs) other unwanted side-effects restricting their use.
In addition to hyperlipidemia and type 2 diabetes, a marked increase in the prevalence of non-alcoholic fatty liver disease (NAFLD) has occurred. NAFLD has become the most common chronic liver condition in Western populations in relation to the obesity and type 2 diabetes epidemics. The prevalence of non-alcoholic steatohepatitis (NASH), a form of NAFLD that is associated with hepatic inflammation and ballooning of hepatocytes, is expected to increase by 63% between 2015 and 2030 in the United States (Estes, Hepatology, 2018; 67(1): 123-133), where NASH is expected to become the leading cause of liver transplantation by 2020. As liver fibrosis, but not inflammation, is associated with mortality and morbidity in NASH patients, drugs which prevent progression/induce regression of fibrosis are also a focus of biomedical research.
The development of novel compounds that simultaneously target both hyperlipidemia and glycemic control, without the adverse side-effects (e.g., weight gain) typically associated with insulin sensitising drugs is thus a desirable goal. Such compounds would be even more attractive if they could additionally prevent the progression/reverse hepatic fibrosis and reduce hepatic steatosis. The present invention addresses these needs for new treatment methods, compounds, and pharmaceutical compositions.

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
//////////
/////////ORZILOBEN, 1555822-28-0, OBESITY, NST-6179, SEFA-6179, NST 6179, SEFA 6179
O=C(C1=C(C)C(OCCCCC)=CC=C1)O
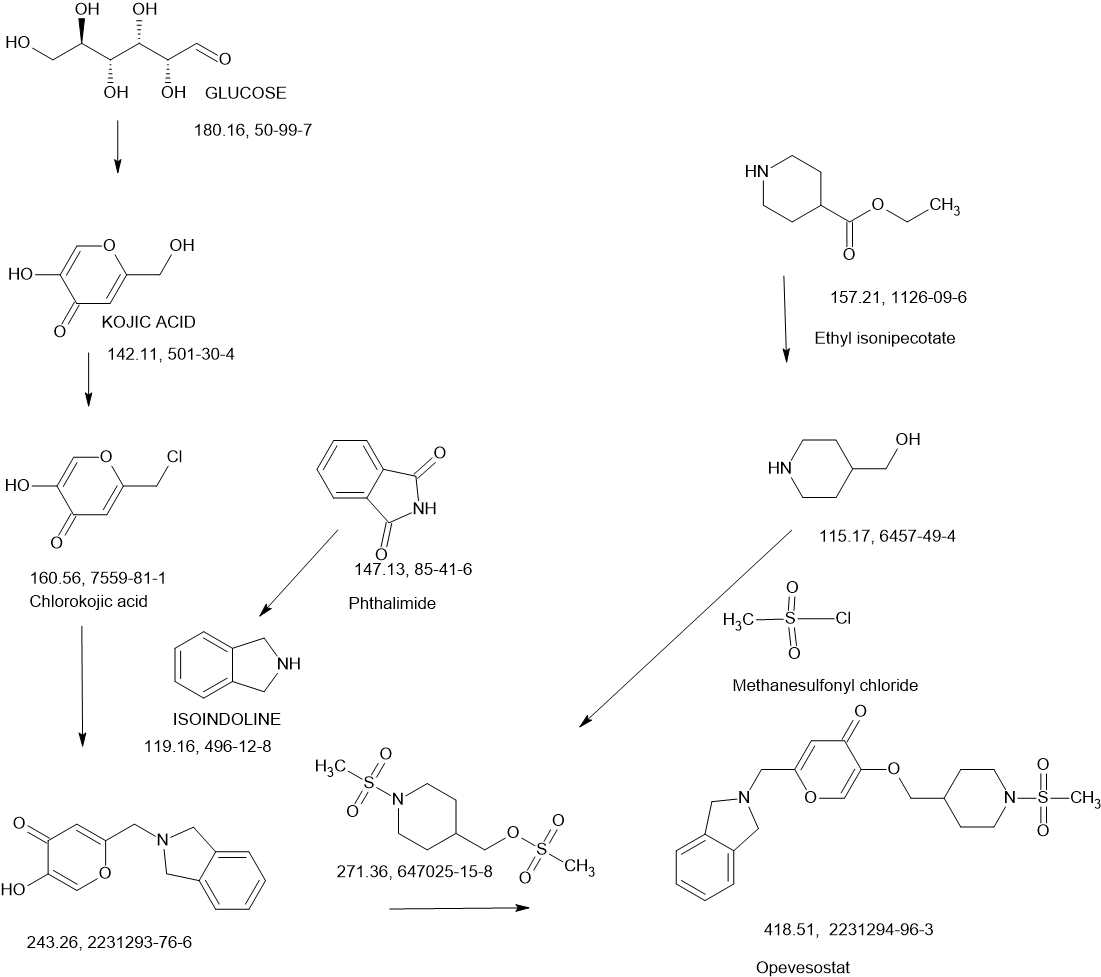
Opevesostat
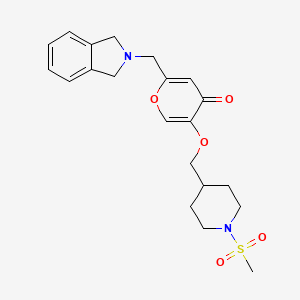


Opevesostat
- ODM208
2231294-96-3
Chemical Formula: C21H26N2O5S
MK-5684, ODM-208
Molecular Weight: 418.508
2-[(1,3-dihydro-2H-isoindol-2-yl)methyl]-5-{[1-(methanesulfonyl)piperidin-4-yl]methoxy}-4H-pyran-4-one
| Opevesostat tosylate |
4H-Pyran-4-one, 2-[(1,3-dihydro-2H-isoindol-2-yl)methyl]-5-[[1-(methylsulfonyl)-4-piperidinyl]methoxy]-, 4-methylbenzenesulfonate (1:1)
2-((1,3-DIHYDRO-2H-ISOINDOL-2-YL)METHYL)-5-((1-(METHYLSULFONYL)-4-PIPERIDINYL)METHOXY)-4H-PYRAN-4-ONE, TOSYLATE
SCHEME
SEE AT THE END OF PAGE
Opevesostat is an investigational new drug being developed for the treatment of metastatic castration-resistant prostate cancer (mCRPC).[1] It is a non-steroidal, selective inhibitor of CYP11A1 (cholesterol side-chain cleavage enzyme)[2] discovered by Orion Corporation and currently undergoing clinical development by Merck & Co., Inc. Opevesostat’s mechanism of action involves suppressing the production of steroid hormones and their precursors that may activate the androgen receptor signaling pathway, which is crucial in prostate cancer progression. As of 2024, opevesostat is being evaluated in two Phase 3 clinical trials, OMAHA1 and OMAHA2a, which are assessing its efficacy and safety in combination with hormone replacement therapy for patients with mCRPC who have failed prior treatments.[3][4][5]
useful in the treatment of a steroid receptor, in particular androgen receptor (AR), dependent conditions and diseases, and to pharmaceutical compositions containing such compounds.
Prostate cancer is worldwide the most common cancer in men. Even though the 5-year survival rate of patients with localized prostate cancer is high, the prognosis for those patients, who develop castration-resistant prostate cancer (CRPC) within that 5-year follow-up period, is poor.
The androgen receptor (AR) signalling axis is critical in all stages of prostate cancer. In the CPRC stage, disease is characterized by high AR expression, AR amplification and persistent activation of the AR signalling axis by residual tissue/tumor androgens and by other steroid hormones and intermediates of steroid biosynthesis. Thus, treatment of advanced prostate cancer involves androgen deprivation therapy (ADT) such as hormonal manipulation using gonadotropin-releasing hormone (GnRH) agonists/antagonists or surgical castration, AR antagonists or CYP17A1 inhibitors (such as abiraterone acetate in combination with prednisone).
Although therapies can initially lead to disease regression, eventually majority of the patients develop a disease that is refractory to currently available therapies. Increased progesterone levels in patients treated with abiraterone acetate has been hypothesized to be one of the resistance mechanisms. Several nonclinical and clinical studies have indicated upregulation of enzymes that catalyse steroid biosynthesis at the late stage of CRPC. Very recently it has been published that 11β-OH androstenedione can be
metabolized into 11-ketotestosterone (11-K-T) and 11-ketodehydrotestosterone (11-K-DHT) which can bind and activate AR as efficiently as testosterone and dihydrotestosterone. It has been shown that these steroids are found in high levels in plasma and tissue in prostate cancer patients, suggesting their role as AR agonists in CRPC. Furthermore, it has been addressed that prostate cancer resistance to CYP17A1 inhibition may still remain steroid dependent and responsive to therapies that can further suppress de novo intratumoral steroid synthesis upstream of CYP17A1, such as by CYP11A1 inhibition therapy (Cai, C. et al, Cancer Res., 71(20), 6503-6513, 2011).
Cytochrome P450 monooxygenase 11A1 (CYP11A1), also called cholesterol side chain cleavage enzyme, is a mitochondrial monooxygenase which catalyses the conversion of cholesterol to pregnenolone, the precursor of all steroid hormones. By inhibiting CYP11A1, the key enzyme of steroid biosynthesis upstream of CYP17A1, the total block of the whole steroid biosynthesis can be achieved. CYP11A1 inhibitors may therefore have a great potential for treating steroid hormone dependent cancers, such as prostate cancer, even in advanced stages of the disease, and especially in those patients who appear to be hormone refractory. It has been recently shown that a compound having CYP11A1 inhibitory effect significantly inhibited tumor growth in vivo in a murine CRPC xenograft model (Oksala, R. et al, Annals of Oncology, (2017) 28 (suppl.
PATENT
WO2018115591
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018115591&_cid=P20-LQXJT7-60871-1
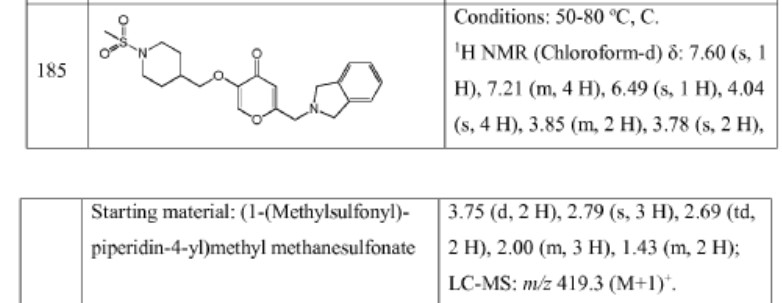

Example 4. SIMILAR
N-((4-(((6-(Isoindolin-2-ylmethyl)-4-oxo-4H-pyran-3-yl)oxy)methyl)cyclohexyl)- methyl)methanesulfonamide (Compound 173)

To a solution of 5-hydroxy-2-(isoindolin-2-ylmethyl)-4H-pyran-4-one (0.10 g, 0.41 mmol) in DMF (2 ml) were added (4-(methylsulfonamidomethyl)cyclohexyl)methyl methanesulfonate (0.14 g, 0.45 mmol) and K2CO3 (0.12 g, 0.8 mmol). The reaction mixture was heated at 80 °C for 2 h. The mixture was cooled to RT, water (10 ml) was added and the product was extracted with EtOAc. The combined extracts were washed with water, dried with Na2SO4, filtered and evaporated. The crude product was purified by column chromatography to afford the title compound (0.06 g). 1H NMR (400 MHz, Chloroform-d) δ ppm 0.92 – 1.11 (m, 4 H) 1.40 – 1.63 (m, 2 H) 1.78 – 2.00 (m, 4 H) 2.91 – 2.99 (m, 5 H) 3.65 (d, J=6.46 Hz, 2 H) 3.77 (s, 2 H) 4.03 (s, 4 H) 5.04 (br t, J=6.31 Hz, 1 H) 6.49 (s, 1 H) 7.20 (s, 4 H) 7.59 (s, 1 H).
ntermediate 58: 5-Hydroxy-2-(isoindolin-2-ylmethyl)-4H-pyran-4-one
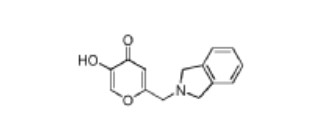
To a stirred solution of 2-(chloromethyl)-5-hydroxy-4H-pyran-4-one (2.0 g, 12.5 mmol) in acetonitrile (50 mL) were added DIPEA (3.22 mL, 25.0 mmol) and isoindoline (1.78 g, 25.0 mmol) at RT. When the reaction was complete, the precipitated solid was filtered and washed with EtOAc. The title compound was collected as pale brown solid (1.1 g). LC-MS: m/z 244.1 (M+H)+.
///////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
References
- ^ “Opevesostat – Orion”. AdisInsight.
- ^ Kim C, Jeong E, Lee YB, Kim D (July 2024). “Steroidogenic cytochrome P450 enzymes as drug target”. Toxicological Research. 40 (3): 325–333. Bibcode:2024ToxRe..40..325K. doi:10.1007/s43188-024-00237-0. PMC 11187042. PMID 38911541.
- ^ “Merck and Orion Announce Mutual Exercise of Option Providing Merck Global Exclusive Rights to Opevesostat”. StockTitan. July 2024. Retrieved 2024-11-23.
- ^ “Inside Information: Orion and MSD Announce Mutual Exercise of Option Providing MSD Global Exclusive Rights to Opevesostat”. Orion. Retrieved 2024-11-23.
- ^ “Merck and Orion Announce Mutual Exercise of Option Providing Merck Global Exclusive Rights to Opevesostat”. Merck. Retrieved 2024-11-23.
 |
|
| Clinical data | |
|---|---|
| Other names | MK-5684, ODM-208 |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C21H26N2O5S |
| Molar mass | 418.51 g·mol−1 |
| 3D model (JSmol) | |
|
show
|
|
//////////
/////////Opevesostat, ODM 208, MK-5684, ODM-208, MK 5684
O=C1C=C(CN2CC3=C(C=CC=C3)C2)OC=C1OCC4CCN(S(=O)(C)=O)CC4
SCHEME
.
Sotuletinib HCl
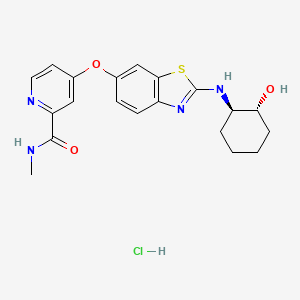

Sotuletinib HCl
CAS: 2222138-31-8 (HCl)
Chemical Formula: C20H23ClN4O3S
Molecular Weight: 434.939
7W3V82OQ0P
Synonym: Sotuletinib HCl; Sotuletinib hydrochloride, Sotuletinib monohydrochloride, BLZ945; BLZ 945; BLZ-945;
IUPAC/Chemical Name: 4-((2-(((1R,2R)-2-hydroxycyclohexyl)amino)benzo[d]thiazol-6-yl)oxy)-N-methylpicolinamide hydrochloride
| 2-PYRIDINECARBOXAMIDE, 4-((2-(((1R,2R)-2-HYDROXYCYCLOHEXYL)AMINO)-6-BENZOTHIAZOLYL)OXY)-N-METHYL- |
Sotuletinib, also known as BLZ945, is a potent and selective CSF-1R kinase inhibitor. BLZ945 showed effects of CSF1R inhibition on other tumor-infiltrating immune cells. BLZ945 attenuates the turnover rate of TAMs while increasing the number of CD8+ T cells that infiltrate cervical and breast carcinomas. BLZ945 decreases the growth of malignant cells in the mouse mammary tumor virus-driven polyomavirus middle T antigen (MMTV-PyMT) model of mammary carcinogenesis. BLZ945 prevents tumor progression in the keratin 14-expressing human papillomavirus type 16 (K14-HPV-16) transgenic model of cervical carcinogenesis.
Sotuletinib (BLZ945) is an experimental drug in development for the treatment of amyotrophic lateral sclerosis (ALS). It works as a colony-stimulating factor 1 (CSF1) receptor inhibitor.[1][2][3]
- OriginatorCelgene Corporation; Novartis
- ClassAmides; Amines; Antineoplastics; Benzothiazoles; Cyclohexanols; Ethers; Pyridines; Small molecules
- Mechanism of ActionMacrophage colony stimulating factor receptor antagonists
- Phase IIAmyotrophic lateral sclerosis
- Phase I/IISolid tumours
- 05 Dec 2022Novartis Pharmaceuticals terminates a phase I/II trials in Solid tumours (Combination therapy, Late-stage disease, Metastatic disease) in Taiwan, Japan, Israel (PO) in US, Israel, Italy, Japan, Singapore, Spain, Taiwan and Switzerland (EudraCT2015-005806-12) (NCT02829723)
- 14 Feb 2022Adverse events and pharmacodynamics data from preclinical macaque model study in brain disorders presented at the 29th Conference on Retroviruses and Opportunistic Infections
- 03 Dec 2020Chemical structure information added
An orally bioavailable inhibitor of colony stimulating factor 1 receptor (CSF-1R; CSF1R), with potential antineoplastic activity. CSF1R inhibitor BLZ945 selectively binds to CSF1R expressed on tumor-associated macrophages (TAMs), blocks the activity of CSF1R, and inhibits CSF1R-mediated signal transduction pathways. This inhibits the activity and proliferation of TAMs, and reprograms the immunosuppressive nature of existing TAMs. Altogether, this reduces TAM-mediated immune suppression in the tumor microenvironment, re-activates the immune system, and improves anti-tumor cell responses mediated by T-cells. CSF1R, also known as macrophage colony-stimulating factor receptor (M-CSFR) and CD115 (cluster of differentiation 115), is a cell-surface receptor for its ligand, colony stimulating factor 1 (CSF1); this receptor is overexpressed by TAMs in the tumor microenvironment, and plays a major role in both immune suppression and the induction of tumor cell proliferation.
Syn

WO2007/121484
WO2018069892
WO2017137958
PATENT
The free base and salts of the compound of formula (I) may be prepared for example, according to the procedures given in International Patent Application No. PCT/US2007/066898 filed on Apr. 18, 2007 and published as WO2007/121484 on Oct. 25, 2007. The compound of formula (I) has the chemical name: 4-(2-((1R,2R)-2-hydroxycyclohexylamino)benzothiazol-6-yloxy)-N-methylpicolinamide and is also known as BLZ945.






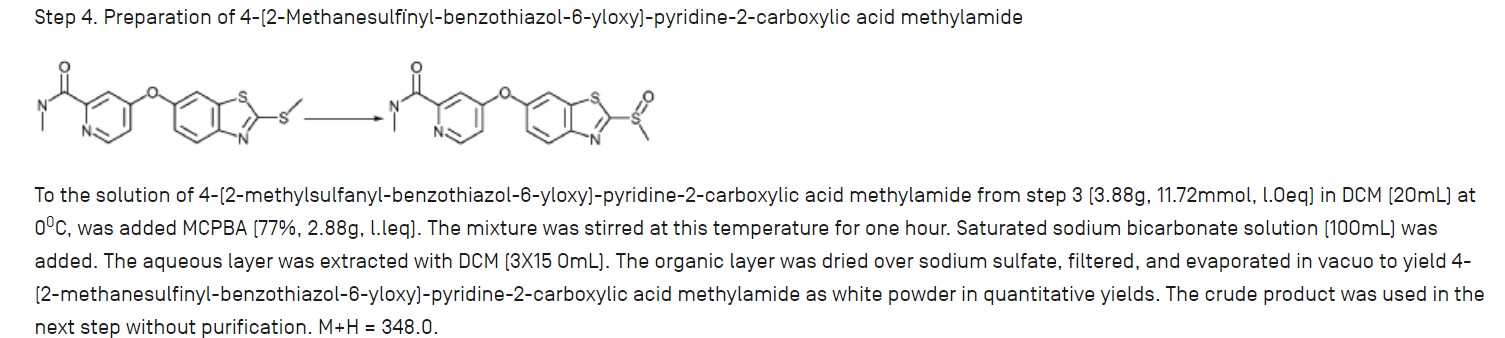
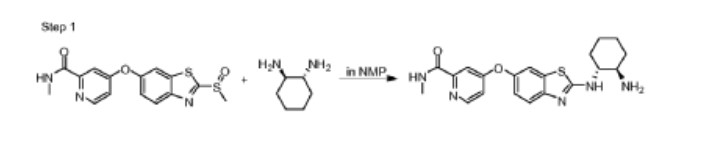
Step 1. Preparation of 4-(2-((lR,2R)-2-aminocyclohexylamino)benzo[d]thiazol-6-yloxy)-N-methylpicolinamide
To the solution of N-methyl-4-(2-(methylsulfinyl)benzo[d]thiazol-6-yloxy)picolinamide (15 mg, 43 μmole) in 400 μL of NMP was added (lR,2R)-cyclohexane-1,2-diamine (17 mg, 150 μmole). The reaction solution was stirred at 105°c for 24 hours. The crude reaction solution was purified on prep HPLC and evaporated in vaccuo to give 4-(2-((lR,2R)-2-aminocyclohexylamino)benzo[d]thiazol-6-yloxy)-N-methylpicolinamide (12 mg, 30 μmole) as white powder. ES/MS m/z 398.1(MH+).
PATENT
https://patents.google.com/patent/US20200093801A1
PATENT
CN116139135
PATENT
US20200190057
PATENT
CN110475555
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
//////////
///////////
References
- ^ Pognan, François; Buono, Chiara; Couttet, Philippe; Galarneau, Jean-René; Timsit, Yoav; Wolf, Armin (29 October 2022). “Liver enzyme delayed clearance in rat treated by CSF1 receptor specific antagonist Sotuletinib”. Current Research in Toxicology. 3: 100091. doi:10.1016/j.crtox.2022.100091. ISSN 2666-027X.
- ^ Thongchot, Suyanee; Duangkaew, Supani; Yotchai, Wasan; Maungsomboon, Sorranart; Phimolsarnti, Rapin; Asavamongkolkul, Apichat; Thuwajit, Peti; Thuwajit, Chanitra; Chandhanayingyong, Chandhanarat (2 December 2022). “Novel CSF1R-positive tenosynovial giant cell tumor cell lines and their pexidartinib (PLX3397) and sotuletinib (BLZ945)-induced apoptosis”. Human Cell. 36 (1): 456–467. doi:10.1007/s13577-022-00823-0.
- ^ Martinez-Gonzalez, Loreto; Martinez, Ana (1 February 2023). “Emerging clinical investigational drugs for the treatment of amyotrophic lateral sclerosis”. Expert Opinion on Investigational Drugs. 32 (2): 141–160. doi:10.1080/13543784.2023.2178416.
////////Sotuletinib HCl, BLZ945, BLZ 945, BLZ-945,
O=C(NC)C1=NC=CC(OC2=CC=C3N=C(N[C@H]4[C@H](O)CCCC4)SC3=C2)=C1.[H]Cl
TACACICLIB, AUR-102, AURIGENE
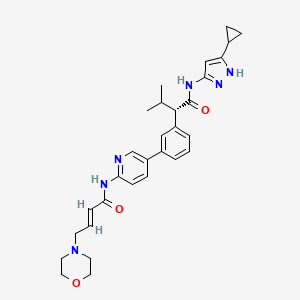
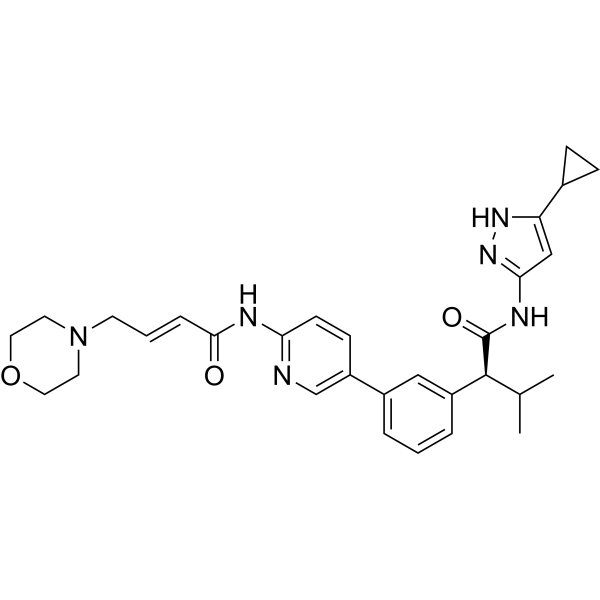
Tacaciclib
2768774-66-7
AUR-102
- Tacaciclib
- SCHEMBL24548621
- GTPL12880
- 528.6 g/mol
- C30H36N6O3
INN 12755
UNI D3G4JKK1MA
(2S)-N-(5-cyclopropyl-1H-pyrazol-3-yl)-3-methyl-2-[3-[6-[[(E)-4-morpholin-4-ylbut-2-enoyl]amino]pyridin-3-yl]phenyl]butanamide
(αS)-N-(5-Cyclopropyl-1H-pyrazol-3-yl)-α-(1-methylethyl)-3-[6-[[(2E)-4-(4-morpholinyl)-1-oxo-2-buten-1-yl]amino]-3-pyridinyl]benzeneacetamide
Benzeneacetamide, N-(5-cyclopropyl-1H-pyrazol-3-yl)-α-(1-methylethyl)-3-[6-[[(2E)-4-(4-morpholinyl)-1-oxo-2-buten-1-yl]amino]-3-pyridinyl]-, (αS)-
Tacaciclib is a CDK inhibitor, antineoplastic effect.
The present invention is directed to methods of preparation of compound of formula (I) that is useful for inhibiting Cyclin-dependent kinase 7 (CDK7) and for treating diseases or disorders mediated thereby.
CDK7, which complexes with cyclin H and RING-finger protein MAT1, phosphorylates the cell cycle CDKs in the activation of T-loop, to promote their activities (Fisher et al., Cell., Aug 26;78(4):713- 24, 1994). As such, it has been proposed that inhibiting CDK7 would provide a potent means of inhibiting cell cycle progression, which may be especially relevant given that there is compelling evidence from gene knockout studies in mice for lack of an absolute requirement for CDK2, CDK4 and CDK6 for the cell cycle at least in most cell types (M alumbres et al., Nature Cell Biology, 11, 1275 – 1276, 2009), whilst different tumors appear to require some, but they are independent of other interphase CDKs (CDK2, CDK4 , CDK6). Recent genetic and biochemical studies have confirmed the importance of CDK7 for cell cycle progression (Larochelle. et al., Mol Cell., Mar 23;25(6):839-50. 2007; Ganuza et al., EM BO J., May 30; 31(11): 2498-510, 2012).
Cyclin-dependent kinase 7 (CDK7) activates cell cycle CDKs and is a member of the general Transcription factor II Human (TFIIH). CDK7 also plays a role in transcription and possibly in DNA repair. The trimeric Cak complex CDK7/CyclinH/MATl is also a component of TFIIH, the general transcription/DNA repair factor IIH (Morgan, DO., Annu.Rev. Cell Dev. Biol. 13, 261-91, 1997). As a TFIIH subunit, CDK7 phosphorylates the CTD (Carboxy-Terminal-Domain) of the largest subunit of RNA polymerase II (pol II). The CTD of mammalian pol (II) consists of 52 heptad repeats with the consensus sequence 1 YSPTSPS 7 and the phosphorylation status of the Ser residues at positions 2 and 5 has been shown to be
important in the activation of RNAP-II indicating that it is likely to have a crucial role in the function of the CTD. CDK7, which primarily phosphorylates Ser-5 (PSS) of RNAP-II at the promoter as part of transcriptional initiation (Gomes et ah, Genes Dev. 2006 Mar 1; 20(5):601-12, 2006), in contrast with CDK9, which phosphorylates both Ser-2 and Ser-5 of the CTD heptad (Pinhero et al., Eur. J. Biochem., 271, pp. 1004-1014, 2004).
In addition to CDK7, other CDKs have been reported to phosphorylate and regulate RNA pol (II) CTD. The other CDKs include, Cdk9/ Cyclin T1 or T2 that constitute the active form of the positive transcription elongation factor (P-TEFb) (Peterlin and Price, Mol Cell., Aug 4; 23(3): 297-305,2006) and Cdkl2/Cyclin K and Cdkl3/Cyclin K as the latest members of RNAPII CTD kinases (Bartkowiak et al., Genes Dev., Oct 1 5;24(20):2303-16, 2010; Blazek et al., Genes Dev .Oct 15;25(20):2158-72, 2011).
Disruption of RNAP II CTD phosphorylation has been shown to preferentially effect proteins with short half-lives, including those of the anti-apoptotic BCL-2 family. (Konig et al., Blood, 1, 4307-4312, 1997; The transcriptional non-selective cyclin-dependent kinase inhibitor flavopiridol induces apoptosis in multiple myeloma cells through transcriptional repression and down-regulation of Mcl-1; (Gojoet al., Clin. Cancer Res. 8, 3527-3538, 2002).
This suggests that the CDK7 enzyme complexes are involved in multiple functions in the cell: cell cycle control, transcription regulation and DNA repair. It is surprising to find one kinase involved in such diverse cellular processes, some of which are even mutually exclusive. It also is puzzling that multiple attempts to find cell cycle dependent changes in CDK7 kinase activity remained unsuccessful. This is unexpected since activity and phosphorylation state of its substrate, CDC2, fluctuate during the cell cycle. In fact, it is shown that cdk7 activity is required for the activation of both Cdc2/Cyclin A and Cdc2/Cyclin B complexes, and for cell division. (Larochelle, S. et al. Genes Dev 12,370-81, 1998). Indeed, flavopiridol, a non-selective pan-CDK inhibitor that targets CTD kinases, has demonstrated efficacy for the treatment of chronic lymphocytic leukemia (CLL), but suffers from a poor toxicity profile (Lin et al.,). 27, 6012-6018, 2009; Christian et al., Clin. Lymphoma Myeloma, 9, Suppl.
3, S179-S185, 2009).
International publication WO2016193939, which is incorporated herein by reference for all purposes describes CDK7 inhibitors and processes for the preparation thereof. Inhibitors of CDK7 are currently being developed for the treatment of cancer. For drug development, it is typically advantageous to employ individual stereoisomers as they exhibit marked differences in pharmacodynamic, pharmacokinetic, and toxicological properties.
SYN
COUPLER

MAIN

Aurigene Discovery Technologies Ltd.
WO2022249141
WO2022130304
WO2022084930
WO2023224961
WO2023107861
WO2022249141
WO2022229835
WO2022130304
WO2022084930
PATENT
WO 2016/193939 COMPD 44
https://patents.google.com/patent/WO2016193939A1/en
InventorSusanta SamajdarRamulu PoddutooriChetan PanditSubhendu MUKHERJEERajeev Goswami
AURIGENE DISCOVERY TECHNOLOGIES LIMITED [IN]/[IN]
Inventors
- SAMAJDAR, Susanta
- PODDUTOORI, Ramulu
- PANDIT, Chetan
- MUKHERJEE, Subhendu
- GOSWAMI, Rajeev
PATENT
Applicants
- AURIGENE ONCOLOGY LIMITED [IN]/[IN]
Inventors
- PODDUTOORI, Ramulu
- VIJAYKUMAR BHAT, Uday
- THIMMASANDRA SEETHAPPA, Devaraja
WO2022229835
Example- 1: Preparation of compound of formula (I)
Scheme-1: Preparation of KRM-A
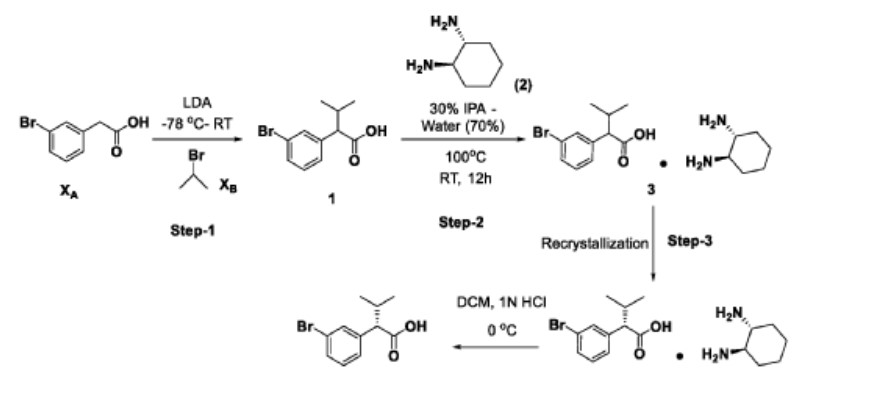

Step-4
KRM-A 4
Step-1: Preparation of 2-(3-bromophenyl)-3-methylbutanoic acid (1)
2M LDA (698 mL, 1.38mol) was added to a solution of 2-(3-bromophenyl) acetic acid (XA, 150 g, 0.69 mol) in THF (700mL) at -78 °C over a period of 30 min. The reaction mixture was stirred for 2h at -78 °C followed by a drop wise addition of isopropyl bromide (X B , 255 g, 2.07 mol) over a period of 30 min. The reaction mixture was stirred at room temperature overnight. Then, the reaction mixture was quenched with IN HC1 (pH 2) and the obtained product was extracted to ethyl acetate (500 mL x 3). The combined organic layer was washed with water followed by brine solution. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to afford the crude compound which was purified by silica column by eluting with 0-10% ethyl acetate-hexane system to afford the title compound (150 g, 83% yield) , HPLC purity-96%. The compound of formula (1) can also be prepared by the procedure described in CN 110590747.
Step-2: Preparation of Compound 3
2-(3-bromophenyl)-3-methylbutanoic acid (1, 510 g, 1.98 mol) was dissolved in 30% of IP A in water (10.2 L; 3.06 L of IPA-7.14 L of water) and ( 1L\ 2i ?)-cyclohexane-1,2-diamine (2, 113 g, 0.9 mol) was added. The reaction mixture was stirred at room temperature for 10 min until the precipitation was observed, then was heated to 100 °C until the solution became clear and stirred at the same temperature for another 30 min. The reaction mixture was allowed to slowly reach room temperature for 8-12h. The obtained solid was filtered and washed with 500 mL of 30% IPA-water mixture and dried under vacuum to afford the compound 3 (620 g, wet).
Work up (for Chiral purity): Small portion (100 mg) of compound 3 was taken in DCM (2-3 mL) and was added IN HC1 (pH 2) at 0 °C until the clear solution was observed. The compound was extracted into DCM, dried over NaiSCL and the solvent was evaporated to afford the title compound as white solid (20 mg). Chiral HPLC was recorded for this sample and 20.6% of undesired isomer was observed in chiral HPLC.
In order to improve the chiral purity of the title compound, the recrystallization method was performed as described below.
Step-3: Recrystallization
The compound 3 (619.90 g) was taken in 30% of IP A in water (12.4 L), then the mixture was heated to 100 °C until the solution became clear and was stirred at the same temperature for another 30min. The reaction mixture was allowed to reach room temperature slowly for 8-12h.
The obtained solid was filtered and washed with 500mL 30% IPA-water and dried under vacuum to afford a desired compound (360g, wet).
Work up for analysis (for Chiral purity): Small portion (100 mg) from above compound was taken in DCM (2-3mL), was added IN HC1 (pH 2) at 0 °C until the clear solution was observed and the compound was extracted to DCM, dried over NaiSCL and the solvent was evaporated to afford title compound as white solid (35 mg). Chiral HPLC was recorded for this sample and 10.3% of undesired isomer was observed in chiral HPLC.
The recrystallization method was repeated for three more times by using 30% of IPA in water as per the aforesaid procedure to obtain the purity of greater than 98.50% ee along with 0.27% other isomer to afford 286 g of compound 4.
Step-4: Preparation of (S)-2-(3-bromophenyl)-3-methylbutanoic acid (KRM-A)
The compound 4 (286 g) was taken in DCM (1.3 L), then was added IN HC1 at 0 °C until the clear solution was observed, and the compound was extracted to DCM (500 mL x 2). The organic layer was separated, washed with brine solution (500 mL) and dried over NaiSCL. The solvent was evaporated from the reaction mixture to afford title compound as white solid (148 g, 60% yield). Chiral HPLC: 98.50%
*H NMR (400MHz, DMSO-de): d 12.5 (s, 1H), 7.50-7.44 (m, 2H), 7.34-7.26 (m, 2H), 3.16 (d, 1H), 2.23-2.11 (m, 1H), 0.98 (d, 3H), 0.63 (d, 3H); Chiral HPLC: 98.50% retention time: 4,588 min.
Scheme-2: Preparation of compound of formula (I)
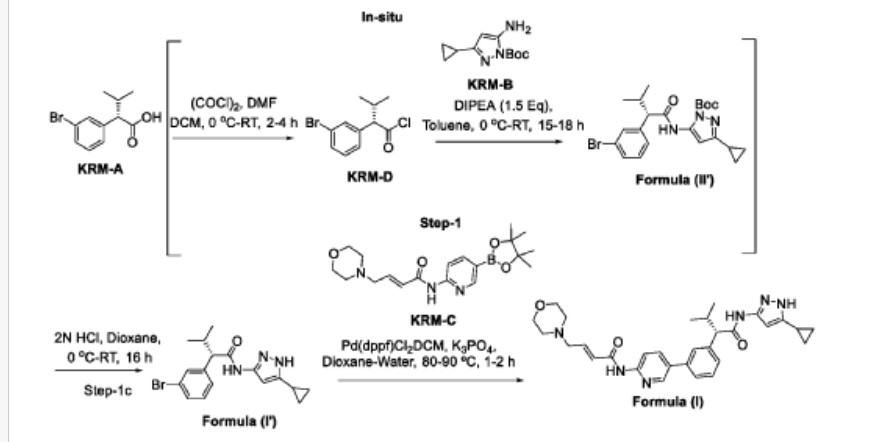

Step-1: Synthesis of (S)-2-(3-bromophenyl)-N-(5-cyclopropyl-1H-pyrazol-3-yl)-3-methylbutanamide
Step-la: Preparation ofKRM-D
To a stirred solution of KRM-A (lOOg, O.388mol) in dry DCM (600 mL, 6 vol), a catalytic amount of DMF (10 mL) was added followed by oxalyl chloride (45 mL, 0.525 mol) dropwise at 0°C over a period of 30 min. After completion of addition, the reaction mixture was stirred for 15 min at the same temperature. The reaction mixture was allowed to reach room temperature and stirred for 2 to 4h. After completion of the reaction (reaction was monitored by TLC, acid chloride formation was checked by quenching an aliquot of reaction mixture with MeOH), the reaction mixture was concentrated under vacuum at 40°C-45°C to afford crude (S)- 2-(3-bromophenyl)-3-methylbutanoyl chloride (KRM-D). The crude KRM-D was dissolved in toluene (500mL) and used for next step.
Step-lb: Preparation of compound of formula (II)
(5)-2-(3-bromophcnyl)-3-mcthylbutanoyl chloride in toluene was added slowly to a pre-cooled solution (0 to 5 °C) of ieri-butyl 3-amino-5-cyclopropyl-1H-pyrazole- l-carboxylate (KRM-B, 95.5g, 0.427 mol) and N, N-diisopropylethyl amine (100 mL, 0.583 mol) in toluene (1.2 L) at 0 °C for the period of l-2h. The reaction mixture was allowed to reach RT and stirred overnight. The reaction mixture was then cooled to 0-5°C and washed with ice-cold 1.5N HC1 (3 x 500 mL). The organic layer was washed with sodium bicarbonate solution (500 mL), brine solution (500 mL), dried over anhydrous NaiSCL , filtered and concentrated under vacuum at 45-50°C to afford crude tert-butyl (S)-5-( 2-(3-bromophenyl)-3-methylbutanamido)-3-
cyclopropyl- lH-pyrazole- 1-carboxylate (compound of formula (IG)) as light brown oil (~180g, LCMS: m/z= 461.9 (M+H) + , HPLC: 80.80%, retention time:15.89 min) . The crude product was taken as such for next step without further purification.
Step-1 c: Preparation of compound of formula (I)
To a suspension of tert-butyl (S)-5-(2-(3-bromophenyl)-3-methylbutanamido)-3-cyclopropyl-1H-pyrazole-1-carboxylate (180 g, 1,731 mol) in dioxane (360 mL ) was added 2N aqueous HC1 (360 mL) at 0 °C. The reaction mixture was stirred overnight at room temperature. After completion of the reaction, dioxane was concentrated, and the reaction mixture was diluted with water (500 mL) and basified with solid sodium bicarbonate (until pH-8). The obtained compound was extracted with DCM (700 mL x 3). The combined organic layers were washed with water (300 mL), brine solution (300 mL), and dried over anhydrous NaiSCL . The organic layer was concentrated to obtain a crude (S)-2-(3-bromophenyl)-N-(5-cyclopropyl-lH-pyrazol-3-yl)-3-methylbutanamide (Compound of formula (G)) as a semi-solid. The crude was dissolved in toluene (500 mL) and the solution was stirred for 18 h. The obtained solid was filtered and washed with toluene (100 mL) and n-heptane (200 mL). The solid was further dried under vacuum at 45-50°C for 6 h to afford a title compound (1 lOg, Yield: 78% over two steps). LCMS: m/z= 362 (M+H) + , HPLC: 97.66%, retention time: 24.10 min
Step-2: Preparation of (S, E)-N-(5-(3-(l-((5-cyclopropyl-lH-pyrazol-3-yl) amino)-3-methyl-l-oxobutan-2- yl) phenyl) pyridin-2-yl)-4-morpholinobut-2-enamide (Compound of formula (I))
To a degassed solution of (5)-2-(3-bromophcnyl)-N-(5-cyclopropyl-1 H-pyrazol-3-yl)-3-methylbutanamide (50 g, 0.138 mol) and (E)-4 -morpholino-N-(5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridin-2-yl)but-2-enamide (KRM-C, 56.6 g, 0.151 mol, 1.1 eq) (prepared according to the procedure described in W02020202001) in 1,4-dioxane (500 mL, 10 vol) and water (100 mL, 2 vol) was added K3PO4 tribasic (73.2 g, 0.345 mol, 2.5 eq) at room temperature The reaction mass was stirred for 20 min with argon purging (degassing). Pd(dppf)Ch.DCM (3.38 g, 0.0042 mol, and 0.03eq) was added to the reaction mixture and the reaction mixture was heated to 90°C for 1-2 h (The reaction was monitored by TLC using 10% methanol in DCM as solvent system).
After completion of the reaction, the reaction mass was cooled to room temperature and filtered through Celite ® bed. The bed was washed with 1, 4-dioxane (200 mL) and the filtrate was concentrated to get crude compound. The crude compound was dissolved in 5% methanol in DCM (400 mL) and washed with water (200 mL x 2). The aqueous layer was separated and
extracted with DCM (100 mL x 2). The combined organic layer was washed with brine solution, filtered and dried over sodium sulfate. The organic layer was concentrated under vacuum at 35-40°C to obtain crude title compound (~80g).
The crude compound of formula (I), (80 g) was dissolved in 700 mL of ethyl acetate. The reaction mixture was cooled to 15°C and 2N HC1 was slowly added (until pH ~1). The reaction mixture was then stirred at room temperature for 20 min and the layers were separated. The aqueous layer (containing the product) was washed with ethyl acetate (300 mL x 3). The aqueous layer was cooled to 0°C and adjusted the pH to ~8 using 20% aqueous NaiCCL solution. The product was extracted with 10% methanol in DCM (300 mL x 3). The combined organic layer was washed with water (300 mL), dried over sodium sulfate and filtered. The filtrate was treated with activated charcoal (16 g, 20% w/w with respect to crude input of 80 g), then the reaction mixture was stirred overnight at room temperature and filtered through Celite ® bed. The bed was washed with 5% methanol in DCM (~ 20 vol, until absence of product by TLC). The filtrate was concentrated under vacuum at 35°C – 40°C to afford compound of formula (I) (70g, HPLC purity: 92.70%, retention time: 15.65 min).
Work-up for improved chiral purity: The above compound of formula (I) was dissolved in ethylacetate (~30 vol, 2L) and washed with aqueous citric acid (2 times, 400 mL x 1 and 200mL x 1), aqueous NaHCCL solution (2%, 500 mL x 1) and aqueous NaCl solution (10%, 500 mL x 1). The combined organic layer was dried over sodium sulfate and filtered. The filtrate was concentrated under vacuum at 35°C – 40°C to afford compound of formula (I) (~60g).
*H NMR (400MHz, DMSO-rfe): d: 10.79 (s, 1H), 10.46 (s, 1H), 8.61 (d, 1H), 8.28 (d, 1H), 8.07-8.05 (m, 1H), 7.69 (s, 1H), 7.56 (d, 1H), 7.39 (m, 2H), 6.84-6.77 (m, 1H), 6.62 (s, 2H), 6.51 (d, 1H), 6.13 (s, 1H) , 3.62-3.59 (m, 4H), 3.35 (d, 1H), 3.15-3.13 (m, 2H), 2.42-2.39 (m, 5H), 1.80-1.77 (m, 1H), 0.98 (d, 3H) , 0.88-0.85 (m, 2H), 0.67 (d, 3H), 0.62-0.60 (m, 2H); LCMS: m/z= 529.25-free base (M+H) + , HPLC: 98.98%, retention time: 15.40 min.
Patent
PATENT
In some embodiments, the compound of formula (I) is (E)-N-(5-(3-(l-((5-cyclopropyl-lH-pyrazol-3-yl)amino)-3-methyl-l-oxobutan-2-yl)phenyl)pyridin-2-yl)-4-morpholinobut-2-enamide or a pharmaceutically acceptable salt or a stereoisomer thereof (Compound 44).
Compound 44 is disclosed in WO 2016/193939 Al, published December 8, 2016, entitled “Substituted heterocyclyl derivatives as cdk inhibitors,” the entire contents of which are incorporated herein by reference. Compound 44A can be in the form of a fumaric acid salt or cocrystal as described in WO 2022/130304 Al, published June 23, 2022, entitled “Cocrystal of a cdk inhibitor,” the entire contents of which are incorporated herein by reference.
Example 3: Synthesis of Compounds 44A & 44B via Chiral Separation
Scheme-1
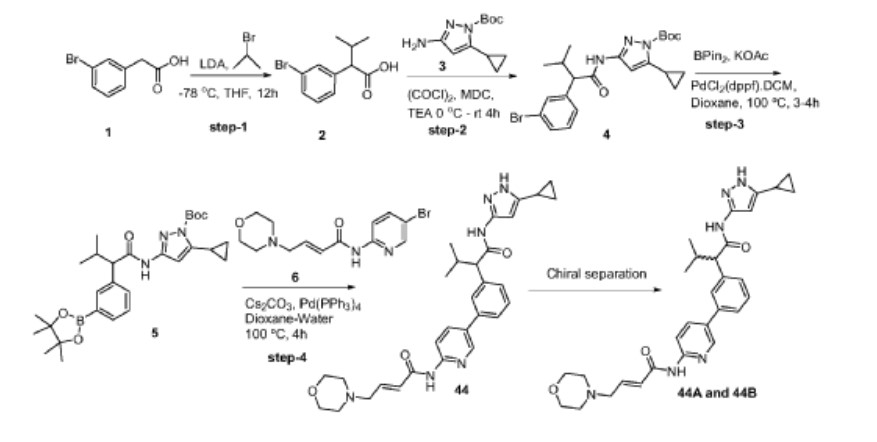

Step-1: Synthesis of 2-(3-bromophenyl)-3-methylbutanoic acid
[0352] 2M LDA (698 mL, 1.38mol) was added to a solution of 2-(3 -bromophenyl) acetic acid (reagent-1, 150g, 0.69mol) in THF (700mL) at -78 °C over a period of 30 min. The reaction mass was stirred for 2h at -78 °C followed by the drop wise addition of Isopropyl bromide (255 g, 2.07mol) over a period of 30 min at -78 °C. The reaction mass was stirred at room temperature for overnight. The reaction mass was quenched with IN HC1 (pH 2) and product extracted to ethyl acetate (500mL x 3). The combined organic layer washed with water followed by brine, dried and concentrated under reduced pressure to afford the title crude compound which was purified by silica column by eluting with 0-10% ethyl acetate -hexane system to afford the title compound 2 (150g, 83% yield). LCMS: m/z = 254.80 (M-2H)’
Step-2: Synthesis of tert-butyl 3-(2-(3-bromophenyl)-3-methylbutanamido)-5-cyclopropyl-lH-pyrazole-1 -carboxylate
[0353] 2-(3-bromophenyl)-3-methylbutanoic acid (intermediate-2, 70g, 0.0.27mol) was dissolved in dry DCM (500 mL) and added oxalyl chloride (68 mL, 0.78mol) dropwise at 0 °C followed by addition of catalytic amount of DMF (0.8mL) and maintained reaction mass at same temperature for 30min. The reaction mass was allowed to room temperature and stirred for 4h, distilled off the solvent and excess oxalyl chloride under vacuum. Re-dissolved the residue in DCM (250 mL) and added slowly to the cooled solution of tert-butyl 3 -amino-5 -cyclopropyl- 1H-pyrazole-1 -carboxylate (intermediate-3, 49g, 0.218mol) and TEA (55 mL, 0.546mol) in THF (250 mL) at 0 °C for 30min, The reaction was stirred at room temperature for 12h then the reaction mass was concentrated under reduced pressure and the residue was dissolved in DCM, washed with saturated NaHCO3 solution and brine. The organic layer was dried over anhydrous sodium sulphate and concentrated under reduced pressure, the crude was purified by silica gel column chromatography by eluting with 15% ethyl acetate-hexane to afford the title compound 4 (90g, 71% ) LCMS: m/z = 363.80 (M-Boc+2).
Step-3: Synthesis of tert-butyl 5-cyclopropyl-3-(3-methyl-2-(3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)butanamido)-l H -pyr azole- 1 -carboxylate
[0354] To a degassed solution of tert-butyl 3-(2-(3-bromophenyl)-3-methylbutanamido)-5-cyclopropyl-lH-pyrazole-1 -carboxylate (intermediate-4, 90g, 0.193mol) and 4, 4, 4′, 4′, 5, 5, 5′, 5′-octamethyl-2,2′-bi(l,3,2-dioxaborolane) (62g, 0.25 Imol) in 1,4-Dioxane (500 mL) was added potassium acetate (37.80g, 0.386mol). The reaction mass was allowed to stir for 10 min with degassing at RT and added PdC12(dppf).DCM complex (12.5g, 0.015mol). The reaction mass was heated for 3-4 h at 100 °C. Reaction mixture cooled to RT and filtered on celite bed, filtrate evaporated to get dark brown liquid. The crude material was purified by silica column chromatography by eluting with 20% ethyl acetate in hexane to afford the compound 5 (90g, 86%). LCMS: m/z = 410 (M-Boc+1)+.
Step-4: Synthesis of (E)-N-(5-(3-(l-((5-cyclopropyl-lH-pyrazol-3-yl)amino)-3-methyl-l-oxobutan-2-yl)phenyl)pyridin-2-yl)-4-morpholinobut-2-enamide
[0355] To a degassed solution of tert-butyl 5-cyclopropyl-3-(3-methyl-2-(3-(4, 4,5,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)phenyl)butanamido)- 1 H-pyrazole- 1 -carboxylate, 5 (10g, 0.019mol) and (E)-N-(5-bromopyridin-2-yl)-4-morpholinobut-2-enamide (7.7g, 0.023mol) in
1,4-Dioxane (lOOmL) and water (40mL) followed by Cs2CO3 (14.5g, 0.045mol) were added. The reaction mass was allowed to stir for 10 min with degassing and added Pd(PPh3)4 (1.1g, 0.00095mol), heated the reaction mass for 4 h at 100 °C in a sealed tube. The reaction mass was cooled and diluted with brine solution. The aqueous layer was separated and re-extracted with ethyl acetate. The combined organic layer was evaporated to dryness and crude material was purified by silica column chromatography by eluting with 10%-l 5 % methanol in DCM to get desired pure compound 44 (4.5g, 44%). LCMS: m/z = 529.15 (M+H)+; HPLC: 95.17%, rt: 6.34 min.
[0356] Racemic (E)-N-(5 -(3 -( 1 -((5 -cyclopropyl- 1 H-pyrazol-3 -yl)amino)-3 -methyl- 1 -oxobutan-2-yl)phenyl)pyridin-2-yl)-4-morpholinobut-2-enamide was separated by using chiral preparative HPLC column (Method: Column: Chiral Pak IA (20mm X 250 mm, 5 micron), Elution: isocratic (50:50), A=ACN, B= MeOH, Flow: 20mL/min ) to afford the pure Isomer- 1 and Isomer-2.
Isomer-1 (Compound 44-A):
[0357] 1HNMR (DMSO-d6, 400MHz): 5 12.02 (s, 1H), 10.78 (s, 1H), 10.44 (s, 1H), 8.61 (s, 1H), 8.28 (d, 1H), 8.07-8.05 (m, 1H), 7.68 (s, 1H), 7.57 (d, 1H), 7.41-7.37 (m, 2H), 6.81-6.78 (m, 1H), 6.49 (d, 1H), 6.13 (s, 1H), 3.61-3.58 (m, 4H), 3.36-3.34 (m, 1H), 3.12 (d, 2H), 2.41-2.32 (m, 5H), 1.82-1.76 (m, 1H), 0.97 (d, 3H), 0.88-0.85 (m, 2H), 0.67 (d, 3H), 0.62-0.59 (m, 2H); LCMS: m/z = 529.15 (M+H)+; HPLC: 96.72%, rt: 6.39 min; Chiral HPLC: 97.68%, rt: 14.47.
Isomer-2 (Compound 44B):
[0358] 1HNMR (DMSO-d6, 400MHz): 5 12.02 (s, 1H), 10.78 (s, 1H), 10.44 (s, 1H), 8.61 (s, 1H), 8.28 (d, 1H), 8.07-8.04 (m, 1H), 7.68 (s, 1H), 7.57 (d, 1H), 7.41-7.37 (m, 2H), 6.81-6.78 (m, 1H), 6.50 (d, 1H), 6.14 (s, 1H), 3.61-3.58 (m, 4H), 3.36-3.34 (m, 1H), 3.12 (d, 2H), 2.40-2.39 (m, 5H), 1.82-1.76 (m, 1H), 0.97 (d, 3H), 0.88-0.85 (m, 2H), 0.67 (d, 3H), 0.62-0.60 (m, 2H); LCMS: m/z = 529.15 (M+H)+; HPLC: 96.24%, rt: 6.39 min; Chiral HPLC: 97.92%, rt: 8.80.
Example 4: Preparation of Compound 44-A via Chiral Synthesis
Preparation of KRM-A (chemical precursor to Compound 44-A)

Step-4
KRM-A
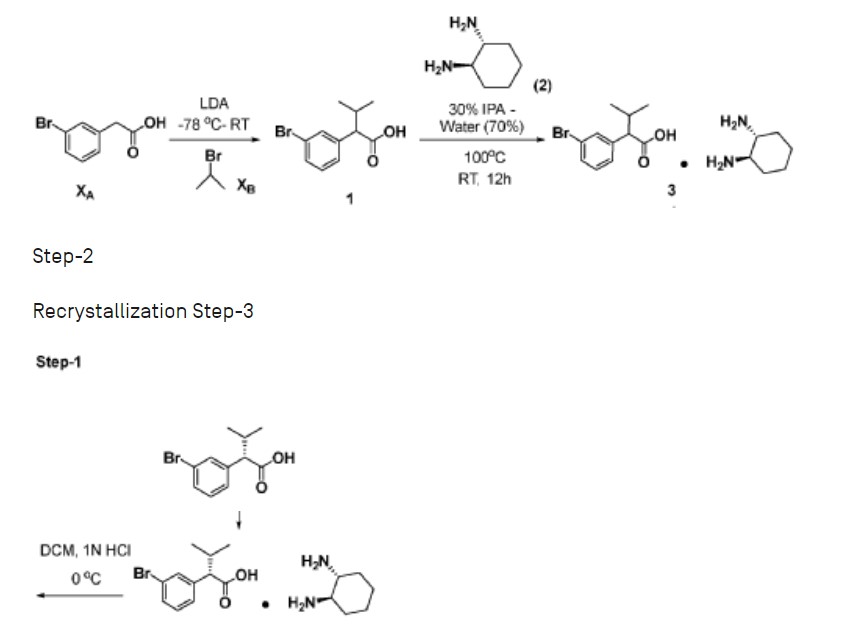
Step-1: Preparation of 2-(3-bromophenyl)-3-methylbutanoic acid (1)
[0359] 2M LDA (698 mL, 1.38mol) was added to a solution of 2-(3 -bromophenyl) acetic acid (150 g, 0.69 mol) in THF (700mL) at -78 °C over a period of 30 min. The reaction mixture was stirred for 2h at -78 °C followed by drop wise addition of isopropyl bromide (XB, 255 g, 2.07 mol) over a period of 30 min at -78 °C. The reaction mass was stirred at room temperature overnight. The reaction mass was quenched with IN HC1 (pH 2) and the obtained product was extracted to ethyl acetate (500 mL x 3). The combined organic layer was washed with water followed by brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure to afford the title crude compound which was purified by silica column by eluting with 0-10% ethyl acetate -hexane system to afford the title compound (150 g, 83% yield), HPLC purity-96%. The compound of formula (1) can also be prepared by the procedure described in CN110590747.
Step-2: Preparation of Compound 3
[0360] 2-(3-bromophenyl)-3-methylbutanoic acid (1, 510 g, 1.98 mol) was dissolved in 30% of IPA in water (10.2 L; 3.06 L of IPA-7.14 L of water) and (1R, 27?)-cyclohexane-l,2-diamine (2, 113 g, 0.9 mol) was added. The reaction mixture was stirred at room temperature for 10 min until the precipitation was observed, then heated to 100 °C till the solution becomes clear and was stirred at same temperature for another 30 min. The reaction mixture was allowed to attain room temperature slowly for 8-12h. The obtained solid was filtered and washed with 500 mL of 30% IPA-water mixture and dried under vacuum to afford the compound 3 (620 g, wet).
[0361] Work up for analysis (for Chiral purity): Small portion (100 mg) of compound 3 was taken in DCM (2-3 mL) and was added IN HC1 (pH 2) at 0 °C till the clear solution was observed. The compound was extracted into DCM, dried over Na2SC>4 and the solvent was evaporated to afford the title compound as white solid (20 mg). Chiral HPLC was recorded for this sample and 20.6% of undesired isomer was observed in chiral HPLC.
[0362] In order to improve the chiral purity of the title compound, the recrystallization method was performed as described below.
Step-3: Recrystallization
[0363] The compound 3 (619.90 g) was taken in 30% of IPA in water (12.4 L), then the mixture was heated to 100 °C till the solution becomes clear and stirred at same temperature for another 30min. The reaction mixture was allowed to attain room temperature slowly for 8-12h. The obtained solid was filtered and washed with 500mL 30% IP A- water and dried under vacuum to afford a desired compound (360g, wet).
[0364] Work up for analysis (for Chiral purity): Small portion (100 mg) from above compound was taken in DCM (2-3mL), was added IN HC1 (pH 2) at 0 °C till the clear solution was observed and the compound was extracted to DCM, dried over Na2SCL and the solvent was evaporated to afford title compound as white solid (35 mg). Chiral HPLC was recorded for this sample and 10.3% of undesired isomer was observed in chiral HPLC.
[0365] The recrystallization method was repeated for three more times by using 30% of IPA in water as described above to get the purity >98.50% ee along with 0.27% other isomer to afford 286 g of compound 4.
Step-4: Preparation of (S)-2-(3-bromophenyl)-3-methylbutanoic acid (KRM-A)
[0366] The compound 4 (286 g) was taken in DCM (1.3 L), then was added IN HC1 at 0 °C until the clear solution was observed, and the compound was extracted to DCM (500 mL x 2). The organic layer was separated and washed brine solution (500 mL) and dried over Na2SO4, the solvent was evaporated to afford title compound as white solid (148 g, 60% yield). Chiral HPLC: 98.50%
[0367] ‘H NMR (400MHz, DMSO-d6): 8 12.5 (s, 1H), 7.50-7.44 (m, 2H), 7.34-7.26 (m, 2H), 3.16 (d, 1H), 2.23-2.11 (m, 1H), 0.98 (d, 3H), 0.63 (d, 3H); Chiral HPLC: 98.50% retention time: 4.588 min.
Preparation of Compound 44-A
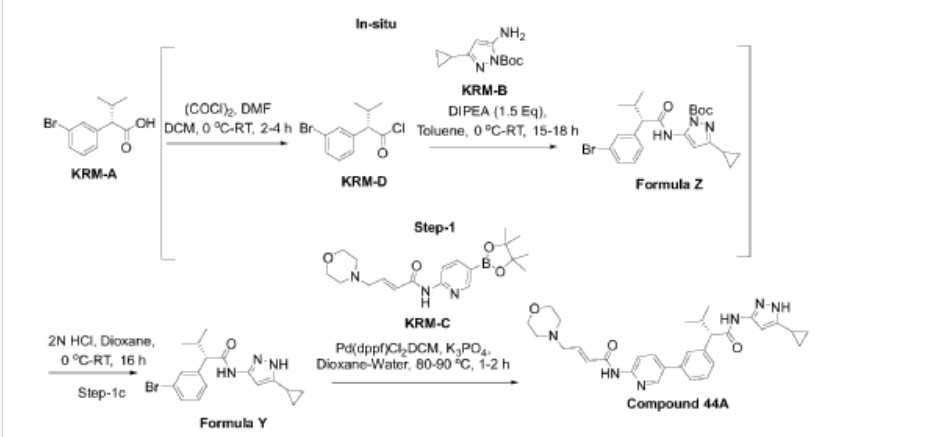

Step-1: Synthesis of (S)-2-(3-bromophenyl)-N-(5-cyclopropyl-lH-pyrazol-3-yl)-3-methylhutanamide
Step- la: Preparation of KRM-D
[0368] To a stirred solution of KRM-A (100g, 0.388mol) in dry DCM (600 mL, 6 vol), a catalytic amount of DMF (10 mL) was added followed by oxalyl chloride (45 mL, 0.525 mol) dropwise at 0 °C over a period of 30 min. After completion of addition, the reaction mixture was stirred for 15 min at the same temperature. The reaction mixture was allowed to reach room temperature and stirred for 2 to 4h. After completion of the reaction (reaction was monitored by TLC, acid chloride formation was checked by quenching an aliquot of reaction mixture with MeOH), the reaction mixture was concentrated under vacuum at 40°C-45°C to afford crude 2-(3-bromophenyl)-3 -methylbutanoyl chloride (KRM-D). The crude KRM-D was dissolved in toluene (500mL) and used for next step.
Step- lb: Preparation of compound of formula Z
[0369] (S)-2-(3-bromophenyl)-3 -methylbutanoyl chloride in toluene was added slowly to a pre-cooled solution (0 to 5 °C) of te/7-butyl 3 -amino-5 -cyclopropyl- IH-pyrazole-l -carboxylate (KRM-B, 95.5g, 0.427 mol) and N, N-diisopropylethyl amine (100 mL, 0.583 mol) in toluene (1.2 L) at 0 °C for the period of l-2h. The reaction mixture was allowed to attain RT and stirred for overnight. The reaction mixture was then cooled to 0-5°C and washed with ice-cold 1.5N HCI (3 x 500 mL). The organic layer was washed with sodium bicarbonate solution (500 mL),
brine solution (500 mL), dried over anhydrous Na2SO4, filtered and concentrated under vacuum at 45-50°C to afford crude tert-butyl (5)-5-(2-(3-bromophenyl)-3-methylbutanamido)-3-cyclopropyl-lH-pyrazole-1 -carboxylate (compound of formula Z) as light brown oil (~180g, LCMS: m/z= 461.9 (M+H)+, HPLC: 80.80%, retention time: 15.89 min). The crude product was taken as such for next step without further purification.
Step-lc: Preparation of compound of formula Y
[0370] To a suspension of tert-butyl (S)-5-(2-(3-bromophenyl)-3-methylbutanamido)-3-cyclopropyl-lH-pyrazole-1 -carboxylate (180 g, 1.731 mol) in dioxane (360 mL) was added 2N aqueous HC1 (360 mL) at 0 °C. The reaction mixture was stirred overnight at room temperature.
[0371] After completion of the reaction, dioxane was concentrated, and the reaction mixture was diluted with water (500 mL) and basified with solid sodium bicarbonate (until pH-8). The resulted compound was extracted with DCM (700 mL x 3). The combined organic layers were washed with water (300 mL) and brine solution (300 mL), and dried over anhydrous Na2SO4. The organic layer was concentrated to get a crude (<S)-2-(3-bromophenyl)-N-(5-cyclopropyl-lH-pyrazol-3-yl)-3-methylbutanamide (Compound of formula Y) as a semi solid. The crude was dissolved in toluene (500 mL) and the solution was stirred for 18 h. The solid formed was filtered and washed with toluene (100 mL) and n-heptane (200 mL). The solid was further dried under vacuum at 45-50°C for 6 h to afford a title compound (110g, Yield: 78% over two steps). LCMS: m/z= 362 (M+H)+, HPLC: 97.66%, retention time: 24.10 min
[0372] Step-2: Preparation of (S, E)-N-(5-(3-(l-((5-cyclopropyl-lH-pyrazol-3-yl) amino)-3-methyl-l-oxobutan-2-yl) phenyl) pyridin-2-yl)-4-morpholinobut-2-enamide (Compound 44A)
[0373] To a degassed solution of (<S)-2-(3-bromophenyl)-N-(5-cyclopropyl-lH-pyrazol-3-yl)-3-methylbutanamide (50 g, 0.138 mol) and (£)-4-morpholino-N-(5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridin-2-yl)but-2-enamide (KRM-C, 56.6 g, 0.151 mol, 1.1 eq) (prepared according to the procedure described in W02020202001) in 1,4-dioxane (500 mL, 10 vol) and water (100 mL, 2 vol) was added K3PO4 tribasic (73.2 g, 0.345 mol, 2.5 eq) at room temperature The reaction mass was stirred for 20 min with argon purging (degassing). Pd(dppf)C12.DCM [l,l’-Bis(diphenylphosphino)ferrocene]dichloropalladium(II) complex with dichloromethane] (3.38 g, 0.0042 mol, and 0.03eq) was added and the reaction mixture was heated to 90°C for 1-2 h (The reaction was monitored by TLC using 10% methanol in DCM as solvent system).
[0374] After completion of the reaction, the reaction mass was cooled to room temperature and filtered through Celite® bed. The bed was washed with 1, 4-dioxane (200 mL) and the filtrate was concentrated to get crude compound. The crude compound was dissolved in 5% methanol in DCM (400 mL) and washed with water (200 mL x 2). The aqueous layer was separated and extracted with DCM (100 mL x 2). The combined organic layer was washed with brine solution, filtered and dried over sodium sulphate. The organic layer was concentrated under vacuum at 35-40°C to get crude title compound (~80g).
[0375] The crude compound 44A, (80 g) was dissolved in 700 mL of ethyl acetate. The reaction mixture was cooled to 15°C and 2N HC1 was slowly added (until pH ~1). The reaction mixture was then stirred at room temperature for 20 min and the layers were separated. The aqueous layer (containing the product) was washed with ethyl acetate (300 mL x 3). The aqueous layer was cooled to 0°C and adjusted the pH to ~8 using 20 % aqueous Na2COs solution. The product was extracted with 10% methanol in DCM (300 mL x 3). The combined organic layer was washed with water (300 mL), dried over sodium sulphate and filtered. The filtrate was treated with activated charcoal (16 g, 20% w/w with respect to crude input of 80 g), stirred overnight at room temperature and filtered through Celite® bed. The bed was washed with 5% methanol in DCM (~ 20 vol, till absence of product by TLC). The filtrate was concentrated under vacuum at 35°C – 40°C to afford compound 44A (70g, HPLC purity: 92.70%, retention time: 15.65 min).
[0376] ‘ H NMR (400MHz, DMSO-^): <5: 10.79 (s, 1H), 10.46 (s, 1H), 8.61 (d, 1H), 8.28 (d, 1H), 8.07-8.05 (m, 1H), 7.69 (s, 1H), 7.56 (d, 1H), 7.39 (m, 2H), 6.84-6.77 (m, 1H), 6.62 (s, 2H), 6.51 (d, 1H), 6.13 (s, 1H), 3.62-3.59 (m, 4H), 3.35 (d, 1H), 3.15-3.13 (m, 2H), 2.42-2.39 (m, 5H), 1.80-1.77 (m, 1H), 0.98 (d, 3H), 0.88-0.85 (m, 2H), 0.67 (d, 3H), 0.62-0.60 (m, 2H);
LCMS: m/z= 529.25-free base (M+H)+, HPLC: 98.98%, retention time: 15.40 min.
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
//////////
REF
POSTER SESSION: Molecular Targeted Agents| Volume 138, SUPPLEMENT 2, S47, October 01, 2020
//////Tacaciclib, GTPL12880, AUR-102
CC(C)C(C1=CC=CC(=C1)C2=CN=C(C=C2)NC(=O)C=CCN3CCOCC3)C(=O)NC4=NNC(=C4)C5CC5

NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}