
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
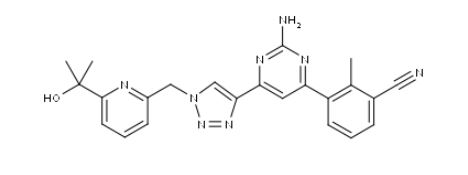
ETRUMADENANT
CAS 2239273-34-6
C23 H22 N8 O, 426.47
Benzonitrile, 3-[2-amino-6-[1-[[6-(1-hydroxy-1-methylethyl)-2-pyridinyl]methyl]-1H-1,2,3-triazol-4-yl]-4-pyrimidinyl]-2-methyl-
- 3-[2-Amino-6-[1-[[6-(1-hydroxy-1-methylethyl)-2-pyridinyl]methyl]-1H-1,2,3-triazol-4-yl]-4-pyrimidinyl]-2-methylbenzonitrile
- AB 928
Arcus Biosciences is developing etrumadenant, the lead from the small molecule adenosine (A2a/A2b) dual receptor antagonist program, for treating cancer. In November 2020, preliminary data from ARC-7 in metastatic NSCLC were expected to report in the first half of 2021.
- OriginatorArcus Biosciences
- ClassAmines; Antineoplastics; Nitriles; Pyridines; Pyrimidines; Small molecules; Triazoles
- Mechanism of ActionAdenosine A2A receptor antagonists; Adenosine A2B receptor antagonists
- Phase IINon-small cell lung cancer
- Phase I/IIProstate cancer
- Phase IBladder cancer; Breast cancer; Cancer; Colorectal cancer; Endometrial cancer; Gastrointestinal cancer; Head and neck cancer; Malignant melanoma; Merkel cell carcinoma; Oesophageal cancer; Ovarian cancer; Renal cancer
- 19 Sep 2020Updated efficacy and adverse events data from a phase I/Ib trial in Non-small cell lung cancer presented at the 45th European Society for Medical Oncology Congress (ESMO-2020)
- 06 Aug 2020Efficacy data from a phase I trial in Colorectal cancer presented at the American Association for Cancer Research Meeting (AACR-2020)
- 13 Jul 2020Arcus Biosciences and Gilead Sciences complete closing of partnership agreement to co-develop and co-promote AB 928 in USA
PAPER
Organic Process Research & Development (2020), 24(7), 1254-1261.
https://pubs.acs.org/doi/10.1021/acs.oprd.0c00124
AB928 is a potent and selective dual antagonist of the A2a and A2b receptors, which is currently in clinical trials. Here, we report the development of two scalable and practical syntheses of AB928. The first-generation synthesis was used to successfully obtain AB928 in excellent yield and purity to support our preclinical and initial clinical studies. Recently, we have developed a second-generation synthesis of AB928 featuring a palladium-free protocol to access 3-(2-amino-6-chloropyrimidin-4-yl)-2-methylbenzonitrile, a key intermediate in the AB928 synthesis. The new method is scalable, practical, and significantly more cost-effective.



PAPER
Tetrahedron Letters (2020), 61(20), 151855.
PAPENT
WO 2020018680
Example 1: Synthesis of 3-[2-amino-6-(l-{[6-(2-hydroxypropan-2-yl)pyridin-2-yl]methyl}-lH-l,2,3-triazol-4-yl)pyrimidin-4-yl]-2-methylbenzonitrile (Compound I)
[0208] Step 1 : In a 250mL round bottom flask equipped with a magnetic stir bar was successively charged the boronic ester (3.89 g, 16 mmol) and the 2-amino-4,6-dichloropyrimidine (3.67 g, 22,4 mmol). Absolute ethanol (100 mL) was added followed by a solution of KHCO3 (4.81 g, 48 mmol) in deionized water (19 mL). The resulting suspension was degassed with nitrogen for 5 minutes. PdChiPPluk (112 mg, 1 mol%) was then added and the mixture was heated to 78 °C for 3 hours under a nitrogen atmosphere. Ethanol was evaporated under reduced pressure and deionized water (150 mL) was added. The suspension was filtered and the solid was washed with additional water (100 mL). The solid was then dissolved in acetone (220 mL) and collected in a 500 mL round bottom flask. A mixture of silica and celite (1 : 1, 150 g) was added and the solvent was removed under reduced pressure. The resulting crude material was purified by flash chromatography over silica gel (dichloromethane/ethyl acetate gradient 0% to 15%). The desired product was obtained as a white solid (1.91 g, 49%). LCMS: Method A, retention time = 2.93 min, ESI MS [M+H]+ for C12H9CIN4, calcd 245.7, found 245.2
[0209] Step 2 : In a round-bottom flask 5.1 g (20.8 mmol) of chloro-pyrimidine was suspended in 42 mL of degassed THF. To this suspension was added 8.68 mL (62.4 mmol) of Et3N and 5.95 mL (25.0 mmol) of TIPS-acetylene. The reaction mixture was stirred for 5 min, followed by addition of 219 mg (0.312 mmol) of PdCl2(PPh3)2 and 119 mg (0.624 mmol) of Cul. The reaction mixture was stirred at 50 °C for 5h under N2. After cooling the reaction to room temp., solvent was removed and the crude material was resuspended in 100 mL EtOAc from which insoluble solid was filtered off. The filtrate was washed with (1 : 1) NH4CI/NH4OH (2 x 100 mL) and 10% Na2S204 (1 x 100 mL). The organic layer was dried using Na2S04, concentrated and taken to next step without further purification.
[0210] Step 3 : In a round-bottom flask the crude TIPS product from previous step was dissolved in 42 mL dry THF and cooled to 0 °C. To this was added 25 mL (25.0 mmol) of TBAF (1.0 M in THF). The reaction was stirred at 0 °C for 15 min. Saturated NH4CI (100 mL) was added to quench the reaction. The organics were extracted from the aqueous layer with EtOAc (2 x 100 mL). The combined organic layer was washed with (1 : 1) NH4CI/NH4OH (2 x 100 mL) and 10% Na2S204 (1 x 100 mL). The organic layer was dried using Na2S04, concentrated and the pure product 5 was obtained by triturating with 40% CH2Cl2/Hexane as a light brown solid. Yield: 3.71 g (76%, 2-steps).
[0211] Step 4 : To a solution of methylmagnesium bromide (3 M in Et20, 40 mL, 120 mmol, 4.0 equiv) at 0 °C under N2 was added a solution of methyl 2-(hydroxymethyl)pyridine-2-carboxylate (5.0 g, 29.9 mmol) in THF (70 mL, 0.4 M) over the course of 30 minutes. The resulting mixture was allowed to warm to room temperature and stirred for 3 h. The reaction mixture was quenched with NH4CI aq (55 mL) and EtOAc (50 mL) was added. The organic phase was separated, and the aqueous phase was extracted with EtOAc (3 x 40 mL). The combined organic extracts were washed with saturated aqueous sodium bisulfite (7 x 20 mL), then dried (Ni^SCh), filtered and concentrated in vacuo to give the title compound (3.45 g, 69% yield; 96% purity as judged by LCMS) as a pale yellow liquid. LCMS: Method A, retention time = 0.722 and 1.06 min, ESI MS [M+H]+ for C9H13NO2, calcd 167.09, found 167.2
[0212] Step 5 : To a solution of 2-hydroxymethyl-6-(l -hydroxy- 1 -methyl ethyljpyri dine (5 g,
29.9 mmol, 1.0 equiv) in PhMe (33 mL, 0.9 M) at 0 °C under N2 was added diphenylphosphoryl azide (7.73 mL, 35.9 mmol, 1.2 equiv.), followed by l,8-diazabicyclo[5.4.0]undec-7-ene (5.37 mL, 35.9 mmol, 1.2 equiv.). The resulting mixture was to warm to room temperature and stirred for 14 h. Upon completion, diluted with ethyl acetate and washed with water, the organic layer was dried (Na2S04), filtered and concentrated. The residue was dissolved in 1N aq HC1 (2 eq, 60 mmol) and extracted with MTBE in hexanes (3:7, 100 mL), the organic layer was washed with water (50 mL) and the combined aqueous layer was neutralized with 2N aqueous NaOH and extracted with ethyl acetate (3X75 mL), dried the organic layer (Na2S04), filtered through a plug of cotton and concentrated the filtrate to afford the pure compound as pale yellow color liquid (3.75 g, 75%). LCMS: Method A, retention time = 2.67 min, ESI MS [M+H]+ for C9H12N4O, calcd 193.1, found 193.2
[0213] Step 6: A mixture of azide (3.34 g, 17.4 mmol), alkyne (3.71 g, 15.8 mmol), copper(II) sulfate (39 mg; 0.158 mmol), and sodium ascorbate (156 mg, 0.790 mmol) in 2: 1 /-BuOH/EbO (158 mL) was heated at 60 °C for 13 h. The solvent was removed in vacuo, the residue dry loaded onto silica gel, and purified by silica gel chromatography (0-100% EtOAc in hexanes) to afford the desired product as an off-white solid (6.08 g, 90%). ‘H NMR (400 MHz, DMSO-cfc) d 8.69 (s, 1H), 7.90 (d, J= 7.8 Hz, 1H), 7.80 (t, J= 7.8 Hz, 1H), 7.76 (d, J= 7.8 Hz, 1H), 7.61 (d, J= 8.0 Hz, 1H), 7.51 (t, /= 7.8 Hz, 1H), 7.28 (s, 1H), 7.10 (d, J= 7.6 Hz, 2H), 6.90 (s, 2H), 5.81 (s, 2H), 5.23 (s, 1H), 2.55 (s, 3H), 1.38 (s, 6H). ESI MS [M+H]+ for C23H23N8O, calcd 427.2, found 427.3.
Example 2: Preparation of Crystalline Solid Form of 3-[2-amino-6-(l-{[6-(2-hydroxypropan-2-yl)pyridin-2-yl]methyl}-lH-l,2,3-triazol-4-yl)pyrimidin-4-yl]-2-methylbenzonitrile
[0214] The product from Example 1, Step 6 (7.53 g) was dissolved in acetone (109 mL) by heating to reflux at which point water (218 mL) was added at a rate of 10 mL/min to initiate crystallization. The mixture was cooled and the solids were collected by filtration, washed with 1 :2 acetone/water (109 mL), and dried under vacuum to afford Form I of Compound I as a white solid (7.08 g; 94%).
PATENT
WO 2019161054
PATENT
WO2020185859 , claiming method for treating a subject identified as having an oncogene driven cancer comprising an agent (eg AB-928) targeting the extracellular production of adenosine and/or antagonizing the activation by adenosine of one of its receptors.
PATENT
WO-2020247789
Processes for preparing aminopyrimidine compounds, particularly etrumadenant (AB-928).
Example 1: Trifluoroethanol Assisted Condensation of B-Ketoesters to Provide a
Hydroxypyrimidine (and Chloropyrimidine).
bromo-2-methylaniline (18.6 g, 100 mmol) dropwise so that a fine white suspension forms. The mixture was cooled to 0 °C and a solution of sodium nitrite (7.31 g, 106 mmol) in water (15.1 mL) was added dropwise. The mixture was stirred at 0 °C for 30 minutes. To the resultant homogeneous mixture at 0 °C was added sodium bicarbonate (17.8 g, 212 mmol) at such a rate to avoid excessive gas evolution. The aqueous phase of the resultant brown suspension was found to have pH ~7. This suspension was maintained at 0 °C.
[0070] In a separate flask, copper cyanide (9.85 g, 110 mmol), potassium cyanide (13.0 g, 200 mmol), and water (31 mL) were heated to 60 °C to form a homogeneous solution. To this solution at 60 °C with stirring was added the above suspension dropwise to avoid excessive gas evolution. After addition, the mixture was stirred at 100 °C for 30 minutes. The mixture was cooled, MTBE (200 mL) was added, the mixture agitated, and filtered to remove any solids, washing with MTBE. The organic phase was dried over Na2SO4 and concentrated. The resultant crude product was purified by vacuum distillation to afford the desired product as a light orange solid (13.6 g, 69%).
[0071] Step 2: In a two liter two-necked flask, aryl bromide (101.9 g, 520 mmol, 1.0 equiv.) was dissolved in THF (520 mL) under an atmosphere of N2, and the mixture was cooled in an
ice-water bath. iPrMgClLiCl (400 mL, 1.3 M in THF, 520 mmol, 1.0 equiv.) was added by cannula. Upon completion of the addition, the ice bath was removed. After four hours, the flask was cooled in an ice-water bath and dry ice (~ 230 g, 5.2 mol, 10 equiv.) was added portionwise to prevent overheating or bubbling over (note: CO2 gas can be bubbled through the solution in place of solid dry ice). When bubbling from the addition was complete, the mixture was diluted with MTBE (500 mL) and 2M HC1 (250 mL). The layers were separated, and the aqueous layer was washed with additional MTBE (500 mL). The organic layer was extracted with 10% NaOH (190 mL x 2), and the combined aqueous layers were cooled in an ice-water bath and acidified with concentrated HC1 until a white precipitate formed. The precipitate was isolated by filtration and washed with water before being dried overnight in a vacuum oven at 80° C to afford the benzoic acid as a white solid (64.1 g, 76% yield).
[0072] Step 3: The benzoic acid (50 g, 311 mmol, 1.0 equiv.) was suspended in CH2CI2, and oxalyl chloride (40 mL, 466 mmol, 1.5 equiv.) was added, followed by DMF (~ 30 drops). Off gassing was observed immediately, and the reaction flask was open to the atmosphere under positive pressure of N2. Upon complete consumption of the starting acid as determined by LCMS and visual inspection (complete dissolution of starting material), the reaction mixture was concentrated. Excess oxalyl chloride was removed by azeotropic distillation with toluene to afford the corresponding acid chloride as a tannish-brown solid.
[0073] In a separate two-necked flask equipped with an overhead stirrer, potassium ethyl malonate (66.1 g, 388 mmol, 1.25 equiv.), triethylamine (108 mL, 777 mmol, 2.5 equiv.) and MeCN (777 mL) were cooled in a salt/ice-brine bath. Solid MgCl2 (74 g, 777 mmol, 2.5 equiv.) was added, and the resulting suspension was vigorously stirred at ~ -10° C. After one hour, the solid acid chloride was added at a rate to ensure dissolution into the thick suspension. The suspension rapidly became homogenous, and the stirring rate was reduced to avoid splashing.
The ice bath was removed. Upon complete consumption of the starting material as determined by TLC analysis, the reaction mixture was cooled in an ice-water bath, and 2M HC1 (971 mL, 1.9 mol, 6.25 equiv.) was added, and the ice bath was removed. After 30 minutes, the layers were separated, and the aqueous layer was extracted with MTBE. The combined organic layers were washed with saturated NaHCO3 and brine, dried over sodium sulfate, filtered, and concentrated to afford the keto-ester as a tannish-brown solid (67 g, 93% yield).
[0074] Step 4: A round-bottom flask was charged with 42.0 g (181.8 mmol) of the b-keto-ester, 32.7 g (181.8 mmol) of guanidinium carbonate and 227 mL of trifluoroethanol. The suspension was then heated to reflux under N2 for 16 h.
[0075] Work-up: The reaction was cooled to room temperature and solvent was evaporated under reduced pressure to obtain a viscus red oil. The oil was re-dissolved in 250 mL H2O and the aqueous solution was extracted with dichloromethane (2 x 250 mL). The aqueous phase is then acidified to pH ~2-3 using 1.0 M HCl(aq ). The precipitated product was collected by filtration, washed thoroughly with H2O and dried in a vacuum oven at 70 °C. Yield 30.81 g (75%), Purity >99%.
[0076] Step 5: A round-bottom flask was charged with 50.0 g (221.2 mmol) pyrimidone from step 4 and 100.8 g (442.2 mmol) of benzyltriethylammonium chloride. The mixture was suspended in 442.2 mL of dry acetonitrile and 31.0 mL (331.8 mmol) of POCI3 was added. The suspension thus obtained was then heated to reflux under N2 for 4 h.
[0077] Work-up: The reaction was cooled to room temperature and ~200 g crushed ice was added. The mixture was then stirred for 30 min flowed by dropwise addition of ice-cold 15% aqueous NH4OH to ~ pH 10 -11. {Note: Slow addition of cold NH4OH is recommended to avoid sudden exotherm due to quenching of excess POCI3). The suspension was then stirred at room temperature for an additional 1.5 h. The precipitated product was collected by filtration, washed thoroughly with H2O and dried in a vacuum oven at 70 °C. Yield 48.2 g (89%), Purity >99%.
HPLC conditions
HPLC: Agilent 1 100
Column: YMC-HPLC Column; 250 x 4.6; S-5 pm, 20 nm; AQ20S05-2546WT; No.0425058945
Solvent: H2O / MeCN with 0.1% HCO2H
Flow Rate: 0.8 mL/min
Column Temperature: 30 °C
Method:
Example 2: Comparative Pyrimidine Coupling
[0078] The synthetic route for preparing 3-[2-amino-6-(l- {[6-(2-hydroxypropan-2-yl)pyridin-2-yl]methyl}-1H-1 ,2,3-triazol-4-yl)pyrimidin-4-yl]-2-methylbenzonitrile utilizing boronic ester benzonitrile to linked the phenyl and pyrimidine rings is shown below and is also provided in WO2018/136700.
[0079] The scheme below displays the synthetic route used to prepare the boronic ester benzonitrile used in the process above and subsequent reaction with pyrimidine to form a compound of Formula (I). Notably, the desired linkage between the pyrimidine and the phenyl provides a yield of less than 50%.
[0080] The below scheme displays the synthetic route used to prepare a compound of Formula (I) that utilized a conversion of a b-diketoester to a pyrimidine using guanidine. The route provides a 75% yield.
PATENT
WO 2018136700
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018136700
Example 1: Synthesis of 3-[2-amino-6-(1-{[6-(2-hydroxypropan-2-yl)pyridin-2-yl]methyl}-1H-1,2,3-triazol-4-yl)pyrimidin-4-yl]-2-methylbenzonitrile
[0269] Step 1: In a 250mL round bottom flask equipped with a magnetic stir bar was successively charged the boronic ester (3.89 g, 16 mmol) and the 2-amino-4,6- dichloropyrimidine (3.67 g, 22,4 mmol). Absolute ethanol (100 mL) was added followed by a solution of KHCO3 (4.81 g, 48 mmol) in deionized water (19 mL). The resulting suspension was degassed with nitrogen for 5 minutes. PdCl2(PPh3)2 (112 mg, 1 mol%) was then added and the mixture was heated to 78 °C for 3 hours under a nitrogen atmosphere. Ethanol was evaporated under reduced pressure and deionized water (150 mL) was added. The suspension was filtered and the solid was washed with additional water (100 mL). The solid was then dissolved in acetone (220 mL) and collected in a 500 mL round bottom flask. A mixture of silica and celite (1:1, 150 g) was added and the solvent was removed under reduced pressure. The resulting crude material was purified by flash chromatography over silica gel (dichloromethane/ethyl acetate gradient 0% to 15%). The desired product was obtained as a white solid (1.91 g, 49%). LCMS: Method A, retention time = 2.93 mm, ESI MS [M+H]+ for C12H9ClN4, calcd 245.7, found 245.2
[0270] Step 2: In a round-bottom flask 5.1 g (20.8 mmol) of chloro-pyrimidine was suspended in 42 mL of degassed THF. To this suspension was added 8.68 mL (62.4 mmol) of Et3Ν and 5.95 mL (25.0 mmol) of TIPS -acetylene. The reaction mixture was stirred for 5 min, followed by addition of 219 mg (0.312 mmol) of PdCl2(PPh3)2 and 119 mg (0.624 mmol) of Cul. The reaction mixture was stirred at 50 °C for 5h under N2. After cooling the reaction to room temp., solvent was removed and the crude material was resuspended in 100 mL EtOAc from which insoluble solid was filtered off. The filtrate was washed with (1:1) NH4C1/NH4OH (2 × 100 mL) and 10% Na2S2O4 (1 × 100 mL). The organic layer was dried using Na2SO4, concentrated and taken to next step without further purification.
[0271] Step 3: In a round-bottom flask the crude TIPS product from previous step was dissolved in 42 mL dry THF and cooled to 0 °C. To this was added 25 mL (25.0 mmol) of TBAF (1.0 M in THF). The reaction was stirred at 0 °C for 15 mm. Saturated NH4Cl (100 mL) was added to quench the reaction. The organics were extracted from the aqueous layer with EtOAc (2 x 100 mL). The combined organic layer was washed with (1:1) NH4Cl/NH4OH (2 x 100 mL) and 10% Na2S2O4 (1 x 100 mL). The organic layer was dried using Na2SO4, concentrated and the pure product 5 was obtained by triturating with 40% CH2Cl2/Hexane as a light brown solid. Yield: 3.71 g (76%, 2-steps).
[0272] Step 4: To a solution of methylmagnesium bromide (3 M in Et2O, 40 mL, 120 mmol, 4.0 equiv) at 0 °C under N2 was added a solution of methyl 2-(hydroxymethyl)pyridine-2-carboxylate (5.0 g, 29.9 mmol) in THF (70 mL, 0.4 M) over the course of 30 minutes. The resulting mixture was allowed to warm to room temperature and stirred for 3 h. The reaction mixture was quenched with NH4Cl aq (55 mL) and EtOAc (50 mL) was added. The organic phase was separated, and the aqueous phase was extracted with EtOAc (3 x 40 mL). The combined organic extracts were washed with saturated aqueous sodium bisulfite (7 x 20 mL), then dried (Na2SO4), filtered and concentrated in vacuo to give the title compound (3.45 g, 69% yield; 96% purity as judged by LCMS) as a pale yellow liquid. LCMS: Method A, retention time = 0.722 and 1.06 mm, ESI MS [M+H]+ for C9H13NO2, calcd 167.09, found 167.2
[0273] Step 5: To a solution of 2-hydroxymethyl-6-(1-hydroxy-1-methylethyl)pyridine (5 g, 29.9 mmol, 1.0 equiv) in PhMe (33 mL, 0.9 M) at 0 °C under N2 was added diphenylphosphoryl azide (7.73 mL, 35.9 mmol, 1.2 equiv.), followed by l,8-diazabicyclo[5.4.0]undec-7-ene (5.37 mL, 35.9 mmol, 1.2 equiv.). The resulting mixture was to warm to room temperature and stirred for 14 h. Upon completion, diluted with ethyl acetate and washed with water, the organic layer was dried (Na2SO4), filtered and concentrated. The residue was dissolved in 1N aq HCl (2 eq, 60 mmol) and extracted with MTBE in hexanes (3:7, 100 mL), the organic layer was washed with water (50 mL) and the combined aqueous layer was neutralized with 2N aqueous NaOH and extracted with ethyl acetate (3×75 mL), dried the organic layer (Na2SO4), filtered through a plug of cotton and concentrated the filtrate to afford the pure compound as pale yellow color liquid (3.75 g, 75%). LCMS: Method A, retention time = 2.67 mm, ESI MS [M+H]+ for C9H12N4O, calcd 193.1, found 193.2
[0274] Step 6: A mixture of azide (3.34 g, 17.4 mmol), alkyne (3.71 g, 15.8 mmol), copper(II) sulfate (39 mg; 0.158 mmol), and sodium ascorbate (156 mg, 0.790 mmol) in 2:1 t-BuOH/H2O (158 mL) was heated at 60 °C for 13 h. The solvent was removed in vacuo, the residue dry loaded onto silica gel, and purified by silica gel chromatography (0-100% EtOAc in hexanes) to afford the desired product as an off-white solid (6.08 g, 90%). 1H NMR (400 MHz, DMSO-d6) δ 8.69 (s, 1H), 7.90 (d, J = 7.8 Hz, 1H), 7.80 (t, J = 7.8 Hz, 1H), 7.76 (d, J = 7.8 Hz, 1H), 7.61 (d, J= 8.0 Hz, 1H), 7.51 (t, J = 7.8 Hz, 1H), 7.28 (s, 1H), 7.10 (d, J = 7.6 Hz, 2H), 6.90 (s, 2H), 5.81 (s, 2H), 5.23 (s, 1H), 2.55 (s, 3H), 1.38 (s, 6H). ESI MS [M+H]+ for C23H23N8O, calcd 427.2, found 427.3.
/////////ETRUMADENANT, AB-928, AB 928, PHASE 2

