
Home » 2016 (Page 37)
Yearly Archives: 2016
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
DS 2330 by Daiichi Sankyo
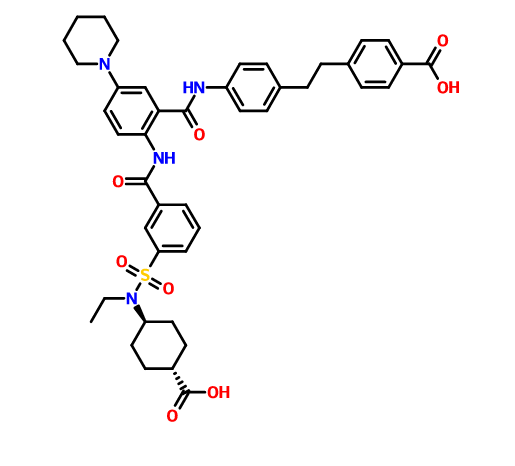
DS 2330
a trans compd
4-[2-(4-{[2-({3-[(trans-4-carboxy-cyclohexyl)(ethyl)sulfocarbamoyl]benzoyl}amino)-5-(piperidin-1-yl)benzoyl]amino}phenyl)ethyl]benzoic acid,
4- [2- (4 – {[2 – ({3 – [(trans-4-carboxy-cyclohexyl) (ethyl) sulfur carbamoyl] benzoyl} amino) -5- (piperidin-1-yl) benzoyl] amino} phenyl) ethyl] benzoate
- Originator Daiichi Sankyo Inc
- Class Hyperphosphataemia therapies
useful for treating hyperphosphatemia, DS-2330, a phosphorous lowering agent, being developed by Daiichi Sankyo, for treating hyperphosphatemia in chronic kidney disease. In April 2016, DS-2330 was reported to be in phase 1 clinical development.
- Phase IHyperphosphataemia
- 31 Oct 2015Phase-I clinical trials in Hyperphosphataemia in USA (unspecified route)
![]()
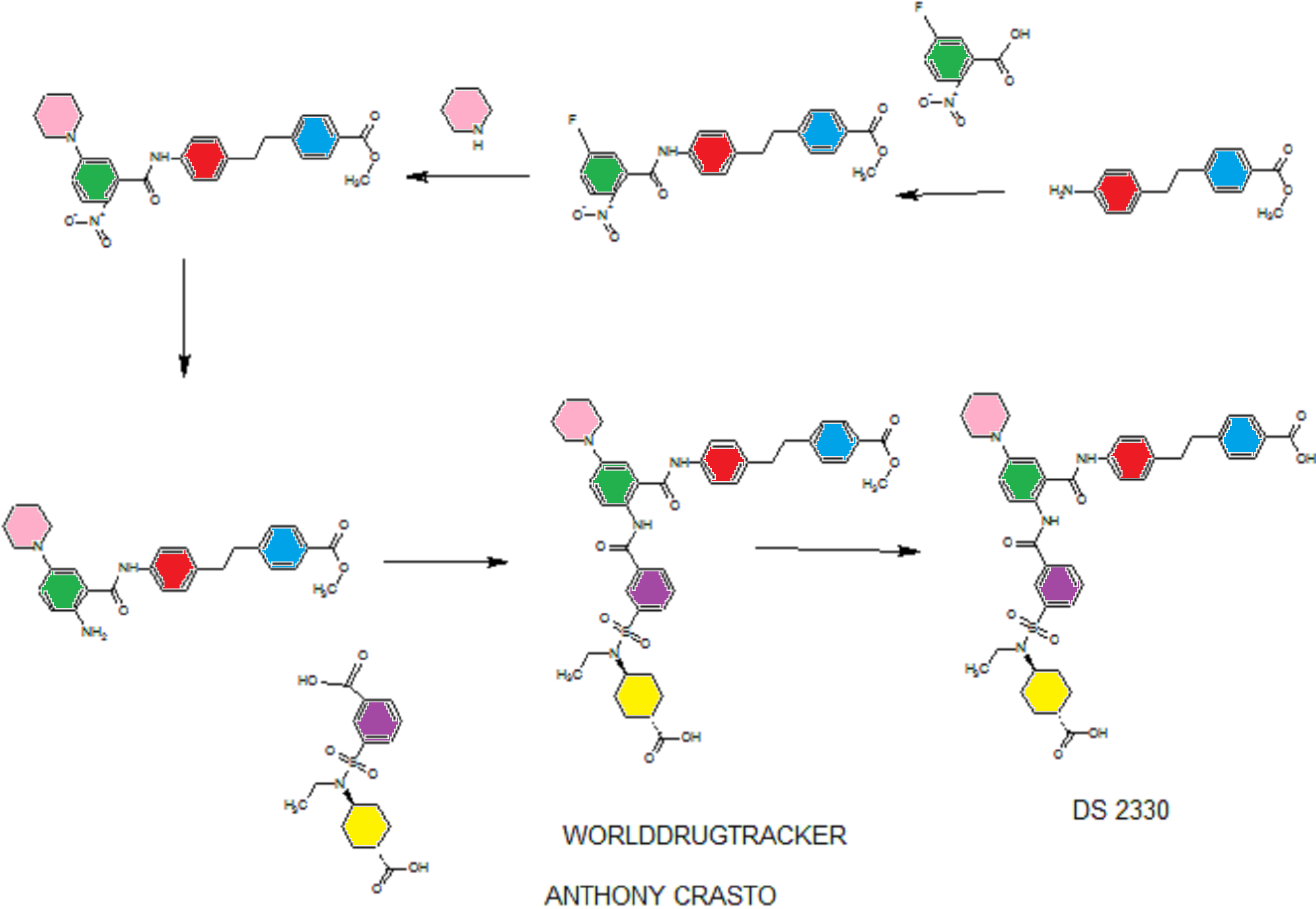
SEE WO2015108038,
PATENT
WO2014175317
http://www.google.com/patents/EP2990400A1?cl=en
PATENT
he problem is to provide a pharmaceutical for the prevention or treatment of hyperphosphatemia. The solution is a salt of a compound including formula (I), or a crystal of a hydrate thereof.
disodium 4- [2- (4 – {[2 – ({3 – [(trans-4-carboxy-cyclohexyl) (ethyl) sulfur carbamoyl] benzoyl} amino) -5- (piperidin-1-yl ) benzoyl] amino} phenyl) ethyl] benzoic acid trihydrate
Disodium 4- [2- (4 – { [2 – ({3 – [(trans-4-carboxylatocyclohexyl) (ethyl) sulfamoyl] benzoyl} amino) – 5- (piperidin-1-yl) benzoyl] amino} phenyl) ethyl] benzoate trihydrate
of α crystal
4- [2- (4 – {[2 – ({3 – [(trans-4-carboxy-cyclohexyl) (ethyl) sulfur carbamoyl] benzoyl} amino) -5- (piperidin-1-yl) benzoyl] amino} phenyl) ethyl] 1 mol / L NaOH aqueous solution to benzoic acid (1.2 g) (3.1 mL) was added and dissolved completely. After stirring at room temperature for 1 day was added acetonitrile (60 mL), at 40 ° C.
and stirred for further 1 day. The precipitated solid was collected by filtration, and 3 hours drying under reduced pressure at room temperature to give the title compound 1.1 g (85%).
in water (46.4 mL), 1-PrOH (72 mL), 4 mol / L NaOH aqueous solution (25.54 mL) was added, then filtered after stirring insolubles at room temperature, water / 1-PrOH: was washed with (3 7, 80 mL). The filtrate was heated up to 40 ℃, 1-PrOH the (160 mL) was added, and further seed crystal (α crystals, 0.2g) was added. Then the temperature was raised to 50 ℃, 1-PrOH (96 ml) was added, and the mixture was stirred overnight.Thereafter, 1-PrOH (480 ml) was added and after overnight stirring, was collected by filtration the precipitated solid was cooled to room temperature.Thereafter, and vacuum dried overnight at 40 ° C., to give the title compound 39.4 g (96%).
REFERENCES
////////////DS 2330, DS-2330, DAIICHI SANKYO, phase 1
O=C(O)[C@@H]1CC[C@H](CC1)N(CC)S(=O)(=O)c2cccc(c2)C(=O)Nc5ccc(cc5C(=O)Nc4ccc(CCc3ccc(cc3)C(=O)O)cc4)N6CCCCC6
OR
O=C(O)[C@@H]1CC[C@H](CC1)N(CC)S(=O)(=O)c2cccc(c2)C(=O)Nc5ccc(cc5C(=O)Nc4ccc(CCc3ccc(cc3)C(=O)O)cc4)N6CCCCC6
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Facebook 

 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
GDC-0084

GDC-0084
CAS#: 1382979-44-3
Chemical Formula: C18H22N8O2
Exact Mass: 382.1866
Synonym: RG7666; RG-7666; RG 7666; GDC-0084; GDC0084; GDC 0084.
IUPAC/Chemical Name: 5-(6,6-dimethyl-4-morpholino-8,9-dihydro-6H-[1,4]oxazino[4,3-e]purin-2-yl)pyrimidin-2-amine
| Latest Stage of Development | Phase I |
| Standard Indication | Brain cancer |
| Indication Details | Treat progressive or recurrent high-grade glioma |
| Regulatory Designation | |
| Partner | Genentech Inc. |
- Originator Genentech
- Class Antineoplastics; Small molecules
- Mechanism of Action 1 Phosphatidylinositol 3 kinase inhibitors
- 28 Jan 2015 Discontinued – Phase-I for Glioma in Spain (unspecified route)
- 28 Jan 2015 Discontinued – Phase-I for Glioma in USA (unspecified route)
- 01 Jan 2015 Genentech completes a phase I trial in Glioma in USA and Spain (NCT01547546)
GDC-0084, also known as RG7666, is a phosphatidylinositol 3-kinase (PI3K) inhibitor with potential antineoplastic activity. PI3K inhibitor GDC-0084 specifically inhibits PI3K in the PI3K/AKT kinase (or protein kinase B) signaling pathway, thereby inhibiting the activation of the PI3K signaling pathway. This may result in the inhibition of both cell growth and survival in susceptible tumor cell populations. Activation of the PI3K signaling pathway is frequently associated with tumorigenesis.
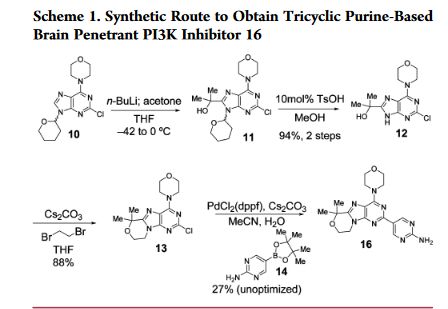
http://pubs.acs.org/doi/pdf/10.1021/acsmedchemlett.6b00005
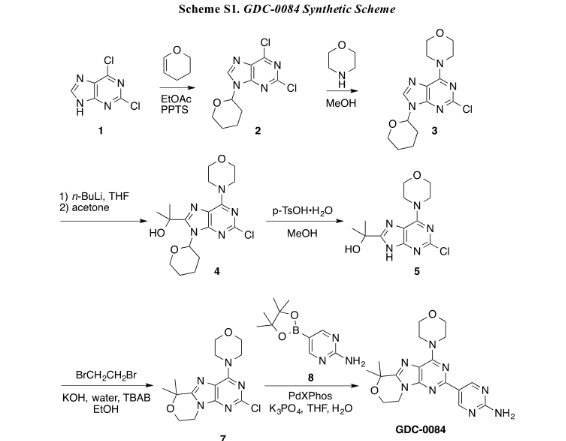

An improved, efficient process with a significantly reduced process mass intensity (PMI) led to the multikilogram synthesis of a brain penetrant PI3K inhibitor GDC-0084. Highlights of the synthesis include a phase transfer catalyzed annulation in water, an efficient Suzuki-Miyaura cross-coupling of a chloropyrimidine with an arylboronic acid using a low palladium catalyst loading, and the development of a controlled crystallization to provide the API. The process delivered GDC-0084 with low levels of both impurities and residual metals.
Development of an Efficient, Safe, and Environmentally Friendly Process for the Manufacture of GDC-0084
//////GDC-0084
NC1=NC=C(C2=NC(N3CCOCC3)=C4N=C(C(C)(C)OCC5)N5C4=N2)C=N1
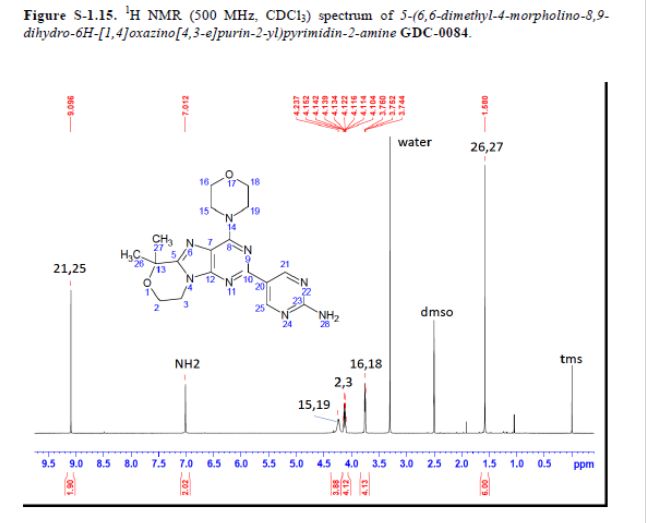
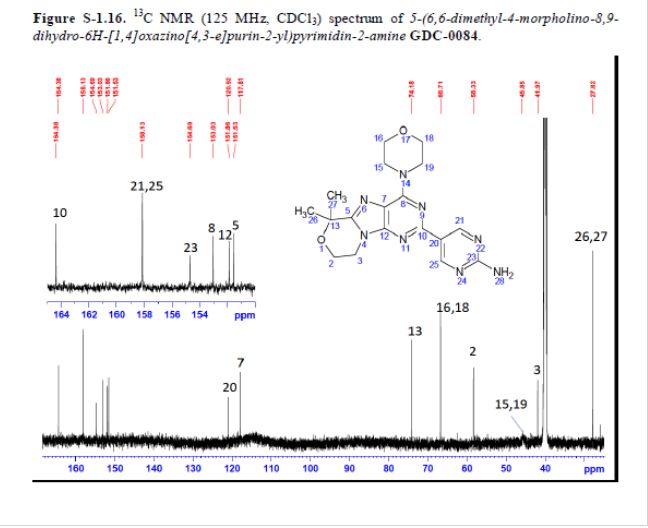
5-(6,6-Dimethyl-4-morpholino-8,9-dihydro-6H-[1,4]oxazino[4,3-e]purin-2-yl)pyrimidin-2-amine GDC-0084
mp 211 °C; 1H NMR (500 MHz, DMSO-d6) δ 9.09 (s, 2H), 7.03 (s, 2H), 4.32–4.17 (m, 4H), 4.17–4.04 (m, 4H), 3.84–3.65 (m, 4H), 1.58 (s, 6H); 13C NMR (125 MHz, DMSO-d6) δ 163.8, 157.6, 154.2, 152.5, 151.3, 151.0, 120.3, 117.3, 73.7, 66.2, 57.8, 45.2, 41.5, 27.3. HRMS [M + H]+calcd for C18H22N8O2 383.1938; found 383.1945.
-
The Discovery of Clinical Development Candidate GDC-0084, a Brain Penetrant Inhibitor of Class I Phosphoinositide 3-Kinases (PI3K) and mTOR.
Heffron, T.; Ndubaku, C.; Salphati, L.; Alicke, B.; Cheong, J.;Drobnick, J.; Edgar, K.; Gould, S.; Lee, L.; Lesnick, J.; Lewis, C.; Nonomiya, J.; Pang, j.; Plise, E.; Sideris,S.; Wallin, J.; Wang, L.; Zhang, X.; Olivero, A. ACS Med. Chem. Lett. 2016, , DOI: 10.1021/acsmedchemlett.6b00005
-
(a) Purine Derivatives Useful as PI3 Kinase Inhibitors. Goldsmith, P.; Hancox, T. C.; Hudson, A.; Pegg, N. A.; Kulagowski, J. J.; Nadin, A. J.; Price, S. PCT Int. Appl. WO 2009053716 A1 Apr 30, 2009.
(b) Preparation of Purine Derivatives with PI3K Inhibitory Activity and Methods of Use Thereof. Castanedo, G.; Chuckowree,I.; Folkes, A.; Sutherlin, D. P.; Wan, N. C. PCT Int. Appl. WO 2009146406 A1 Dec 3, 2009
The new Annex 16 “Certification by a Qualified Person and Batch Release” will become effective as of 15 April 2016.
DRUG REGULATORY AFFAIRS INTERNATIONAL
 The new Annex 16 is coming into Force
The new Annex 16 is coming into Force
The new Annex 16 “Certification by a Qualified Person and Batch Release” will become effective as of 15 April 2016. The contents will reflect the coming state of expectations regarding the batch release.
see
The new Annex 16 “Certification by a Qualified Person and Batch Release” will become effective as of 15 April 2016.
It is centrally pointed out that the main duty of a Qualified Person (QP) is the certification of batches. In this context, the QP must personally ensure that the responsibilities listed under Chapter 1.6 are fulfilled. Chapter 1.7 lists many other responsibilities to be guaranteed by the QP. However the related activities can be delegated and the QP can rely on the respective quality management systems. Yet, the “QP should have on-going assurance that this reliance is well founded” (1.7). The 21 responsibilites listed include amongst others:
- The…
View original post 406 more words
ICH Q3D implemented in the European Pharmacopoeia: Revision of Two General Monographs with Regard to Elemental Impurities
DRUG REGULATORY AFFAIRS INTERNATIONAL
 ICH Q3D implemented in the European Pharmacopoeia: Revision of Two General Monographs with Regard to Elemental Impurities
ICH Q3D implemented in the European Pharmacopoeia: Revision of Two General Monographs with Regard to Elemental Impurities
Two general monographs of the European Pharmacopoeia have been revised and published for comment in the newest “Pharmeuropa” edition. Read more about what you will have to consider in future with regard to the control of elemental impurities in pharmaceutical preparations, APIs and excipients.
see
In a press release dated 30 November 2015, the EDQM announced the revision of two general pharmacopoeial monographs: “Substances for pharmaceutical use” (2034) and “Pharmaceutical preparations” (2619). The decision was taken during the 153rd session of the European Pharmacopoeia Commission; the Commission follows its strategy for implementing the ICH Guideline Q3D “Guideline for Elemental Impurities” in the European Pharmacopoeia. A section “Elemental Impurities” has been added to both monographs which emphasizes that the provisions laid down in General Chapter 5.20 of the Pharmacopoeia (identical in wording with…
View original post 272 more words
I (Anthony Crasto) am Editorial Board member for our Journal of Analytical & Pharmaceutical Research


I am on editorial board ……… Editorial Board member for our Journal of Analytical & Pharmaceutical Research………http://medcraveonline.com/JAPLR/editorial-board
This is possible with your cooperation and support
read…….http://medcraveonline.com/JAPLR/JAPLR-02-00010.pdf
http://medcraveonline.com/JAPLR/JAPLR-02-00011.pdf
Tackling the Challenges with Poorly Soluble Drugs
http://medcraveonline.com/JAPLR/JAPLR-01-00001.pdf
BTI-320 (formerly PAZ320), Soluble mannan polysaccharides from Boston Therapeutics for the treatment of type 2 diabetes in combination with oral agents or insulin
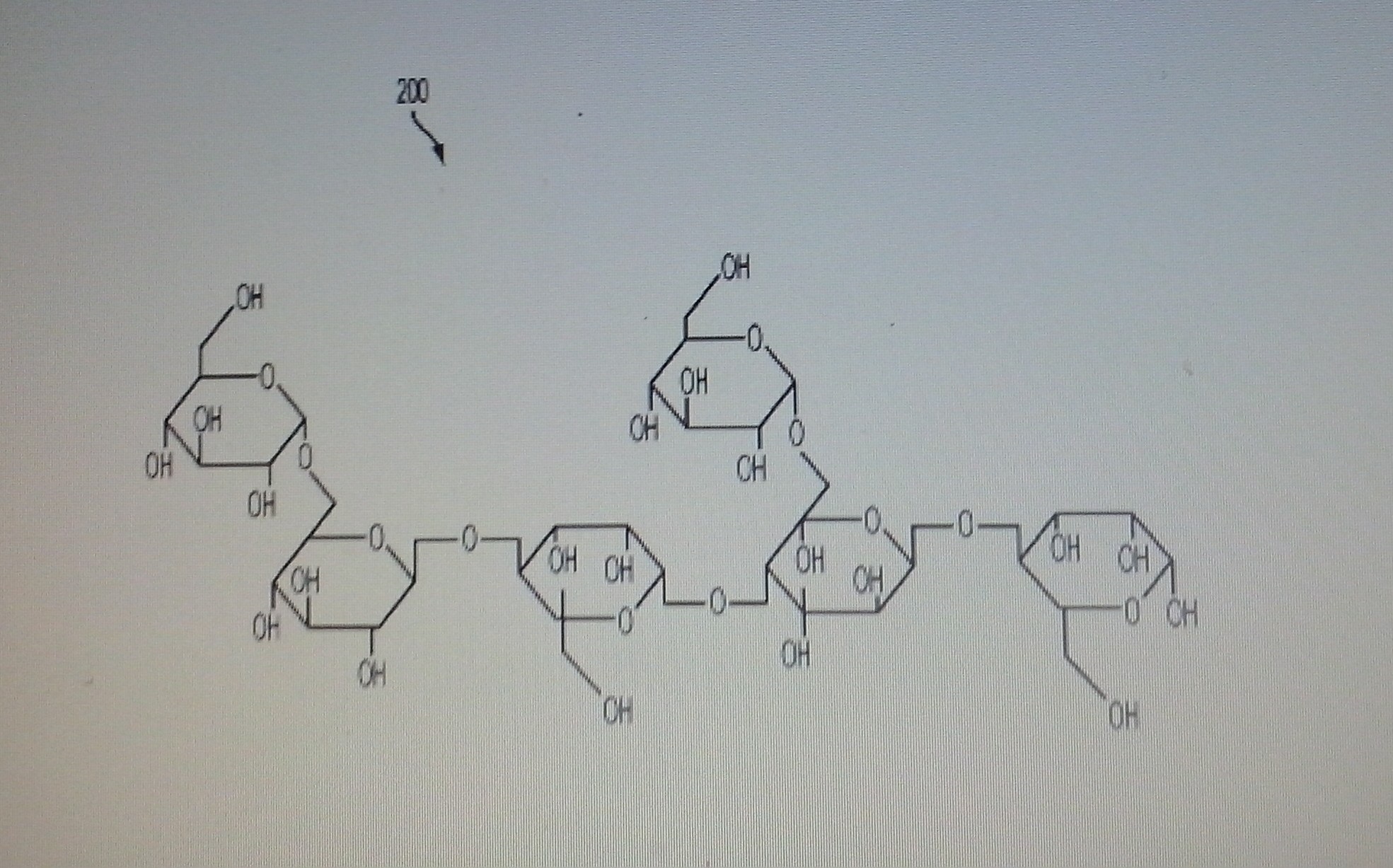
BTI-320 (formerly PAZ320)
PAZ 320
Non-insulin dependent diabetes
Alpha-glucosidase inhibitor; Hydrolase inhibitor; Sucrose alpha-glucosidase inhibitor
Composition of chemically purified (fractionation) soluble mannan polysaccharides from legume’s seeds
BTI-320 is in phase II clinical development at Boston Therapeutics for the treatment of type 2 diabetes in combination with oral agents or insulin, and also for the treatment of high-risk patients with pre-diabetes. A chewable tablet formulation is being developed. The product is already available as dietary supplement.
| Company | Boston Therapeutics Inc. |
| Description | Chewable polysaccharide that inhibits alpha glucosidase |
| Molecular Target | |
| Mechanism of Action | Alpha glucosidase inhibitor |
| Therapeutic Modality | Macromolecule: Polysaccharide |
| Latest Stage of Development | Phase II |
| Standard Indication | Diabetes |
| Indication Details | Treat Type II diabetes |


PATENT
http://www.google.co.in/patents/WO2012061675A1?cl=en
A composition of chemically purified soluble mannans from legumes’ seeds (e.g. Ceratonia siliqua, Cæsalpinia spinosa Trigonelle foenum-graecum, and Cyamopsis tetragonolobus) and their use in the assembly of palatable dietary supplements is disclosed herein. The fractionation process provides high-quality physiologically soluble, chemically modified and purified homogeneous size polysaccharide fibers, devoid of natural impurities, for example proteins, alkaloids, glycoalkaloids, and/or environmental impurities including heavy metals, agricultural residues and microbial toxins. This process provides hypoallergenic dietary fibers devoid of any potential allergens, cytotoxins, and gastrointestinal toxins. A sequential process for assembly of the soluble fibers with plurality of molecular weights to create a time controlled dissolution of the functional high and low molecular weight fibers for improving solubility and palatability with improved dietary performance in the oral and gastro-intestinal system is also disclosed herein.
Fig. 1 illustrates a block flow diagram of an embodiment of a method for recovering purified mannan polysaccharides;
Fig. 2 illustrates a chemical structure of a mannan polysaccharide;
Fig. 3 illustrates a block flow diagram of an embodiment of a method for recovering high molecular weight (HMW) purified mannan polysaccharides;
Fig. 4 illustrates a block flow diagram of an embodiment of a method for recovering low molecular weight (LMW) purified mannan polysaccharides;
REFERENCES
https://clinicaltrials.gov/show/NCT02060916
https://clinicaltrials.gov/show/NCT02358668
BTI-320, a nonsystemic novel drug to control glucose uptake into the bloodstream, functions as a competitive inhibitor of sugar hydrolyzing enzymes
75th Annu Meet Sci Sess Am Diabetes Assoc (ADA) (June 5-9, Boston) 2015, Abst 974-P
Boston Therapeutics’ Hong Kong Affiliate Advance Pharmaceutical’s BTI-320 Clinical Trial Reaches Mid-Point by Enrolling 30 Patients at the Chinese University of Hong Kong
Boston Therapeutics Press Release 2015, July 08
Insight into the molecular mechanism of action of BTI320, a non-systemic novel drug to control serum glucose levels in individuals with diabetes50th Annu Meet Eur Assoc Study Diabetes (EASD) (September 15-19, Vienna) 2014, Abst 545
////BTI-320, PAZ320, PHASE 2, BTI 320, PAZ 320, Macromolecule, Polysaccharide, Non-insulin dependent diabetes, Alpha-glucosidase inhibitor, Hydrolase inhibitor, Sucrose alpha-glucosidase inhibitor, phase II clinical development, Boston Therapeutics, Soluble mannan polysaccharides
Composition of chemically purified (fractionation) soluble mannan polysaccharides from legume’s seeds
POLYMER OF BELOW
CAS 9036-88-8, 51395-96-1
| refractive index : | 78.5 ° (C=1.4, H2O) |
Ailes;MANNAN;K-41K1;D-Mannan;NSC 174478;NSC 174479;NSC 174481;NSC 307194;NSC 174477;NSC 174473

| Chemical name: | 1,6-Anhydro-β-D-mannopyranose |
| Synonyms: | 1,6-Anhydro-D-mannose; 1,6-Anhydromannose; Mannosan; NSC 226600; |
| CAS Number: | 14168-65-1 |
| Possible CAS #: | NA |
| Molecular form.: | C₆H₁₀O₅ |
| Appearance: | White to Pale Beige Solid |
| Melting Point: | 182-184°C |
| Mol. Weight: | 162.14 |
Summary:
Mannans are major constitutents of hemicelluloses in plant tissue and are polymers composed of β(1→4)-linked mannose and glucose residues. Some contain galactopyranosyl side chains (see a galactomannan).
Slightly galactosylated mannans (4% galactose), considered as linear β(1→4)-D-mannans, have been isolated from the seed endosperm of vegetable ivory nut ( Phytelephas macrocarpa) and date ( Phoenix dactylifera) .
Glycan icon:

Child Classes: a 1,6-α-D-mannan backbone (0), a galactoglucomannan (0), a galactomannan (0), a glucomannan (0), a mannan oligosaccharide (1)
SMILES: C(O)C4(C(O[R1])C(O)C(O)C(OC3(C(O)C(O)C(OC2(C(O)C(O)C(OC1(C(O)C(O)C(O[R2])OC(CO)1))OC(CO)2))OC(CO)3))O4)
CAS:9036-88-8,
//////////
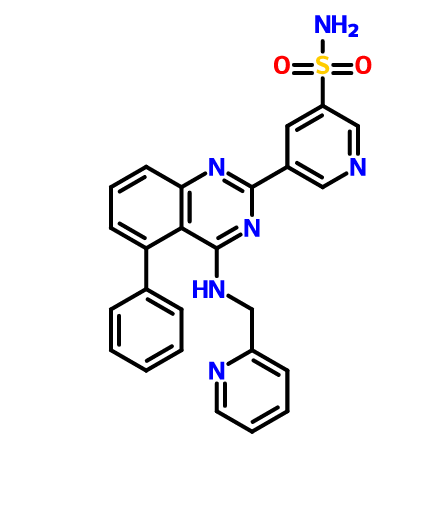
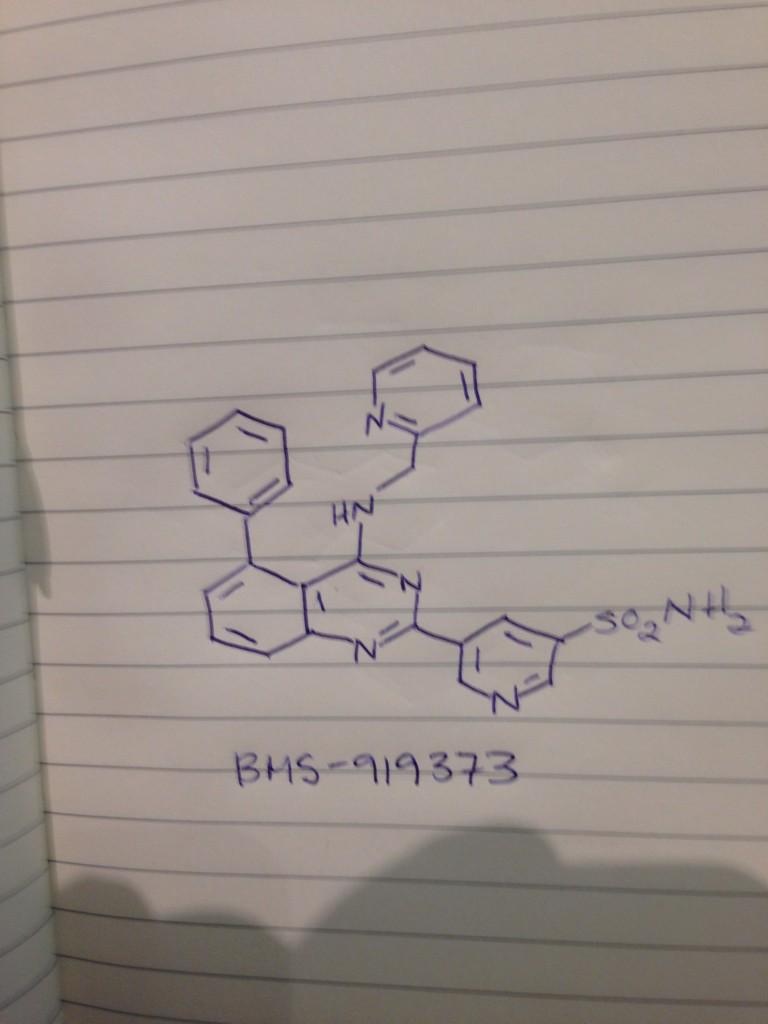
BMS 919373
 .
.
Bethany Halford on Twitter: “BMS-919373, from $BMS for …https://twitter.com/beth_halford/status/634105343719682048
Aug 19, 2015 – BMS–919373, from $BMS for atrial fibrillation #ACSBoston MEDI 1st disclosures @bmsnews pic.twitter.com/y3D4Yv2U7M.
BMS 919373
- Phase IIParoxysmal atrial fibrillation
- Phase IAcute coronary syndromes; Atrial fibrillation
-
- 01 Oct 2014Phase-I clinical trials in Atrial fibrillation in Canada (PO) (NCT02153437)
- 01 Jul 2014Phase-II clinical trials in Paroxysmal atrial fibrillation in Canada (PO) (NCT02156076)
- 01 Jul 2014Phase-II clinical trials in Paroxysmal atrial fibrillation in USA (PO)
- https://clinicaltrials.gov/ct2/show/NCT02153437
- https://clinicaltrials.gov/ct2/show/NCT02156076
- CAS HCL SALT 1272356-77-0
| Latest Stage of Development | Phase I |
| Standard Indication | Fibrillation |
| Indication Details | Treat atrial fibrillation |
Synthesis
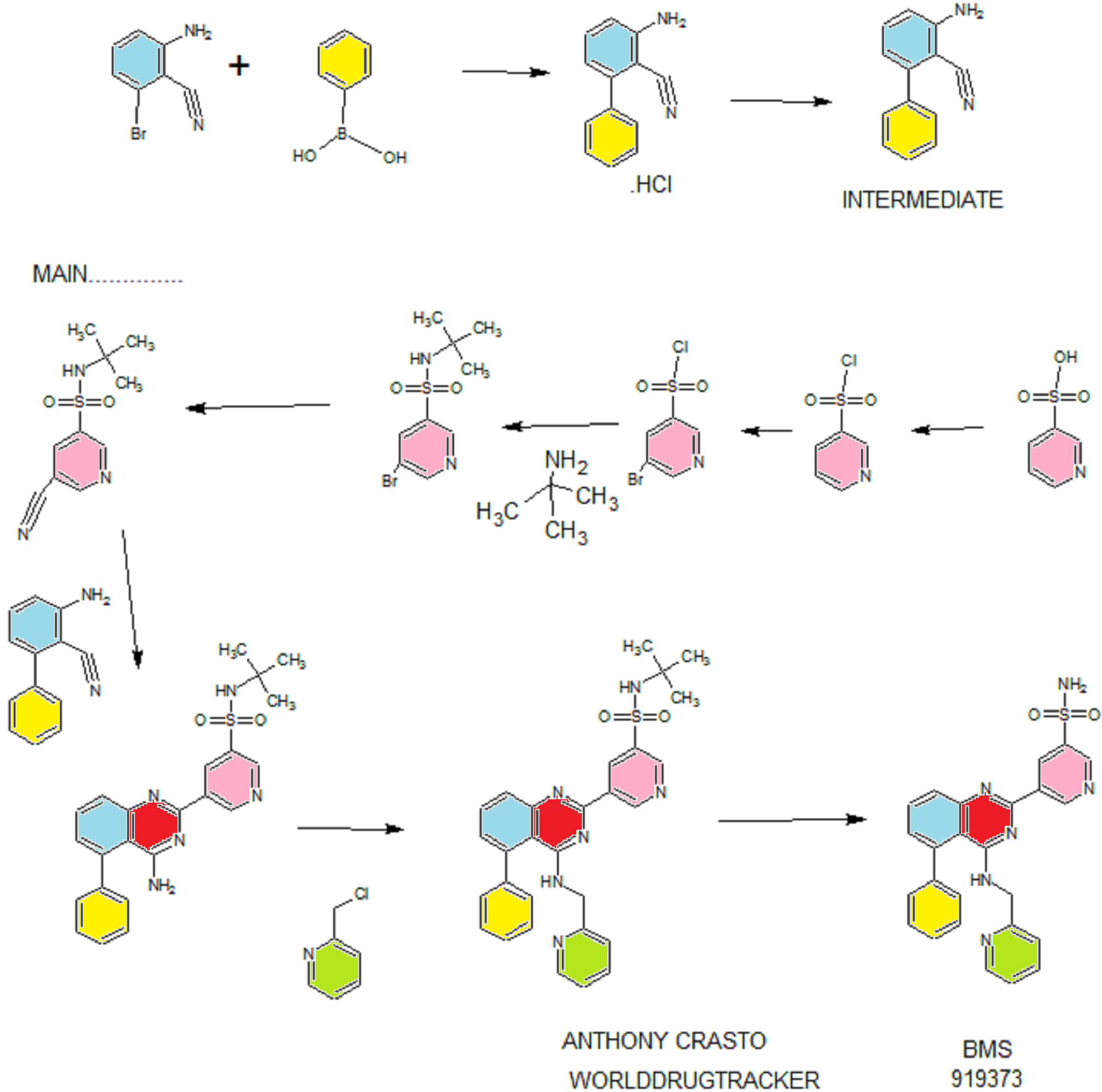
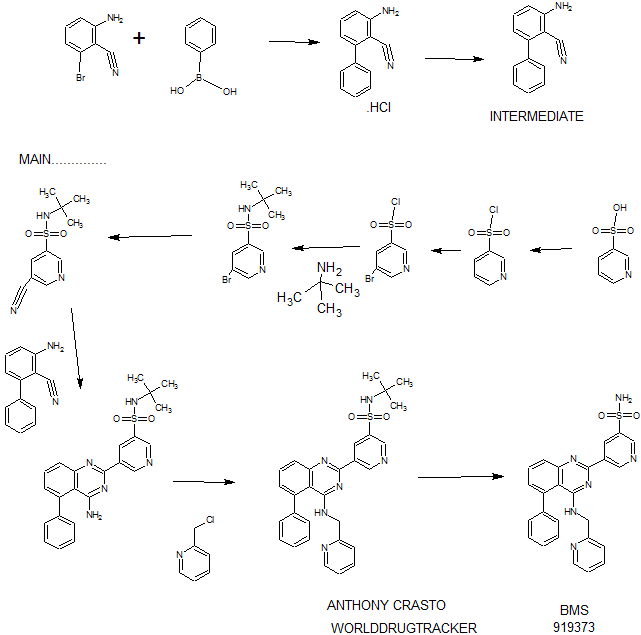
PATENT
WO 2011028741
http://www.google.co.in/patents/WO2011028741A1?cl=en
EXAMPLE 7
5-(5-Phenyl-4-(pyridin-2-ylmethylamino)quinazolin-2-yl)pyridine-3-sulfonamide

Step 1. Preparatio -Bromopyridine-3 -sulfonamide

See also U.S. Publication Nos. 2006/217387 and 2006/375834, and J. Org. Chem., 54:389 (1989). A mixture of pyridine-3 -sulfonic acid (10.3 g, 64.8 mmol), phosphorous pentachloride (20.82 g, 100 mmol) and phosphorous oxychloride (10 mL, 109 mmol) was heated to reflux where it stirred for 4h. At the conclusion of this period, the reaction mixture was allowed to cool to room temperature. Once at the prescribed temperature, the reaction mixture was evaporated to dryness under reduced pressure to yield a residue. The residue was treated with bromine (6.00 mL, 1 16 mmol) and then heated to reflux where it stirred for 14h. After this time, the reaction mixture was cooled to 0 °C and then a saturated solution of NH4OH in ¾0 (40 mL) was slowly added. The resulting mixture was allowed to warm to room temperature where it stirred for 30 min. The reaction mixture was then filtered and the filter cake was washed with hexane to afford 5 -bromopyridine-3 -sulfonamide (6.0 g) as an off- white solid. The product was used without further purification. LCMS Method Q: retention time 0.75 min; [M+l] = 237.0.
Step 2. Preparation of pyridine-3-sulfonamide-5-ylboronic acid pinacol ester

See also WO2008/150827 Al and WO2008/144463. A mixture of 5- bromopyridine-3 -sulfonamide (1.5 g, 6.33 mmol), bis(pinacolato)diboron (2.41 g, 9.5 mmol) and potassium acetate (1.86 g, 19.0 mmol) in 1,4-dioxane (15 mL) was degassed with nitrogen for 15 min then (l, l’-bis(diphenylphosphino)- ferrocene)palladium (II) chloride dichloromethane complex (232 mg, 0.317 mmol) was added and the resulting mixture was degassed again with nitrogen for 10 min. At the conclusion of this period, the reaction mixture was heated in a microwave at 120 °C for 45 min. After this time, the reaction mixture was filtered through CELITE® and the filtrate was concentrated under reduced pressure to provide pyridine-3- sulfonamide-5-ylboronic acid pinacol ester (740 mg) as a brown solid. The product was used without further purification. XH NMR (400 MHz, DMSO-d6) δ (ppm): 8.83 (s, 1H), 8.80 (s, 1H), 8.26 (s, 1H), 7.56-7.74 (bs, 2H), 1.17 (s, 12H).
Step 3. Example 7

To a solution of 2-chloro-5-phenyl-N-(pyridin-2-ylmethyl)quinazolin-4- amine (150 mg, 0.43 mmol) in 1,4-dioxane (6 mL) and ¾0 (1 mL) under nitrogen was added pyridine-3-sulfonamide-5-ylboronic acid pinacol ester (185 mg, 0.65 mmol), and potassium carbonate (119 mg, 0.86 mmol). Upon completion of addition, the mixture was degassed with nitrogen for 15 minutes and then (1, 1′- bis(diphenylphosphino)ferrocene)palladium (II) chloride dichloromethane complex (31 mg, 0.043 mmol) was added. The resulting mixture was again degassed with nitrogen for 10 min. After this time, the mixture was heated to 90 °C where it stirred for 16h. At the conclusion of this period, the reaction mixture was allowed to cool to room temperature. Once at the prescribed temperature, the reaction mixture was quenched by the addition of water and then transferred to a separation funnel. The aqueous layer was extracted with ethyl acetate. The combined organic portions were washed with water and saturated NaCl, dried over Na2S04, filtered and concentrated under reduced pressure. The resulting concentrate was purified by preparative TLC using 5% methanol in dichloromethane to afford Example 7 (50 mg) as a brown solid. ‘H NMR (400 MHz, DMSO-d6) δ (ppm): 9.81 (s, 1H), 9.17 (s, 1H), 9.09 (s, 1H), 8.24 (d, J= 4.4 Hz, 1H), 7.94 (d, J=7.2 Hz, 1H), 7.86 (t, J= 7.6 Hz, 1Η),7.75-7.72 (t, J= 7.6 Hz, 3H), 7.59-7.51 (m, 5H), 7.34 (d, J=7.2 Hz, 2H), 7.24 (t, J=6.4 Hz, 1H), 6.98 (t, J= 3.2 Hz, 1H), 4.77 (d, J= 4.0 Hz, 2H). LCMS Method Q: retention time 1.39 min; [M+l] = 469.0. HPLC Method B: purity 98.1%, retention time = 8.74 min. [00120] Alternatively, Example 7 can be synthesized as follows:
Step 1. Preparation of 5-Bromo-pyridine-3-sulfonyl chloride

PC15 (2.95 Kg, 14.16 moles) and POCl3 (2.45 Kg, 15.98 moles) were added into pyridine-3 -sulfonic acid (1.5 Kg, 9.42 mol) in 10 L RB flask equipped with mechanical stirrer under inert atmosphere. The reaction mass was heated to 120- 125°C where it stirred for 18 h. After this time, the reaction progress was monitored by HPLC, which indicated the reaction was complete. Excess POCI3 was removed under vacuum to give a residue. The residue was cooled to ambient temperature and bromine (1.2 Kg, 7.5 moles) was added. Upon completion of addition, the resulting mixture was heated to 120-125°C where it stirred for 5 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated the reaction was complete. The reaction mixture was cooled to ambient temperature and then poured into ice-water (10 L), and the resulting mixture was extracted with DCM (10.5 Lx2). The DCM extracts were combined and the solvent was removed under vacuum to yield crude product (1.8 Kg, 74.4% yield).
Step 2. Preparation of 5-bromo-N-tert-butylpyridine-3 -sulfonamide

Crude 5 -bromopyridine-3-sulfonyl chloride from step 1 above was dissolved in THF (14 L, 8 vol) and then transferred to a 20 L RB flask equipped with mechanical stirrer under inert atmosphere. The solution was cooled to 0-5°C and tert- butyl amine (1.95 Kg, 26.66 moles) was added at 0-5°C. Upon completion of addition, the reaction mixture was warmed to ambient temperature where it stirred for 2 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated that the reaction was complete. The solvent was evaporated under vacuum to give a thick residue. The residue was dissolved in ethyl acetate (18 L, 12 vol). The organic layer was separated, washed with water (9 L, 5 vol) and then concentrated under vacuum to yield a residue. Hexanes (9 L, 5 vol) were added to the residue and the product precipitated out and was collected by filtration to yield a free flowing yellow solid (1.5 Kg, 54.28% overall yield). ¾ NMR (DMSO-D6, 400 MHz, δ ppm); 8.99 (d, J = 2Hz, 1H), 8.81 (d, J= 2 Hz, 1H), 8.29 (t, J= 2Hz, 1H). [M++l] = 293. Step 3. Preparation of 5-bromo-N-tert-butylpyridine-3 -sulfonamide

5 -Bromo-N-tert-butylpyridine-3 -sulfonamide (1.5 Kg, 5.11 moles) was dissolved in dimethylformamide (7.5 L, 5 vol) and the solution was added to a 20 L glass-lined reactor equipped with mechanical stirrer. The solution was degassed with nitrogen for 30 min. After this time, potassium ferrocyanide trihydrate (867 g, 2.05 moles), sodium carbonate (1.08 Kg, 10.189 moles), copper (I) iodide (73.2 g, 0.374 moles) and dichloro-bis (triphenylphosphine) palladium (II) (71.6 g, 0.102 moles) were added. Upon completion of addition, the reaction mixture was heated to 120- 125°C where it stirred for 4 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated the reaction was complete. The reaction mixture was cooled to ambient temperature and then filtered through a celite bed. Water (18 L, 12 vol) was added into the filtrate and the resulting mixture was extracted with ethyl acetate (7.5L*2). The organic layers were combined, washed with water and then concentrated to yield a thick residue. Hexanes (7.5 L, 5 vol) were added to the residue. The product precipitated out and was collected by filtration to yield a free flowing yellow solid (1.0 Kg, 82.8% yield, 89% purity by HPLC). ¾ NMR (DMSO-D6, 400 MHz, δ ppm); 9.21 – 9.24 (d,d J= 7.2Hz, 3.2Hz, 2H), 8.70-8.71(m,lH), 7.98 (s, lH). [M++l] = 239.2.
Step 4. Preparation of 3-aminobiphenyl-2-carbonitrile

2-Amino-6-bromo-benzonitrile (1.0 Kg, 5.07 moles) and toluene (10 L, 10 vol) were added to a 20 L glass-lined reactor equipped with mechanical stirrer under inert atmosphere. Potassium acetate (996 g, 10.16 moles) and phenylboronic acid (866, 7.10 moles) were added into the solution and the solution was degassed with nitrogen for 30 min. After this time, dichloro-bis (triphenylphosphine) palladium (II) (17.8 g, 0.025 moles) was added to the reaction mixture at ambient temperature. The mixture was heated to 110°C, where it stirred for 17 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated the reaction was completed. The reaction mixture was filtered through a celite bed. The filtrate was transferred back to the reactor and concentrated hydrochloric acid (-35%, 2 L, 2 vol) was charged to the reactor at ambient temperature. The HCl salt of the title compound precipitated out from the reaction and was collected by filtration. The HCl salt was transferred into the 20 L reactor and then made basic with 10% NaOH solution (pH 8-9). The resulting product was extracted with ethyl acetate (10 L, 10 vol). The ethyl acetate layer was washed with water (5 L, 5 vol) and then the solvent was evaporated under vacuum to give a residue. Hexanes (5 L, 5 vol) were added to the residue at 35-40°C, and the resulting slurry was cooled to ambient temperature. Once at the prescribed temperature, the product was collected by filtration to provide a pale yellow solid (802 g, 81.4%, 99% by HPLC). XH NMR (DMSO-D6, 400 MHz, δ ppm); 7.43-7.52 (m, 5H), 7.33-7.37 (m, 1H), 6.83 (d, J=8Hz, 1H), 6.62 (d, J=8Hz, 1H), 6.1 (s, 2H). ES-MS: [M++l] = 194.23.
Step 5. Preparation of 5-(4-amino-5-phenylquinazolin-2-yl)-N-tert-butylpyridine-3-

3-Aminobiphenyl-2-carbonitrile (1028 g, 5.30 moles), 5-bromo-N-tert- butylpyridine-3 -sulfonamide (1440 g, 5.55 moles) and 1,4-dioxane (10 L, 10 vol) were added to a 20 L glass-lined reactor equipped with mechanical stirrer. Sodium tert-butoxide (1.275 Kg 12.870 moles) was added to the solution portion-wise at 20- 30°C. Upon completion of addition, the reaction mixture was heated to reflux where it stirred for 2 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated the reaction was complete. The reaction mixture was cooled to 30-35°C and then poured into water (40 L, 40 vol). The resulting mixture was extracted with DCM (20 L*2). The DCM layers were combined, washed with water (10 L, 10 vol) and then dried over sodium sulfate. The solvent was evaporated under vacuum to give a residue. Isopropyl alcohol (1.2 L, 1.2 vol) was added to the residue at 40°C. The resulting precipitate slurry was cooled to 10-15°C and then stirred for 2 h. After this time, the precipitate was collected by filtration and dried at 50°C for 16 h to yield the product (1.9 Kg, 82.9% yield, 99% purity by HPLC). Ή NMR (DMSO-D6, 400 MHz, δ ppm); 9.72 (s, 1H), 9.11 (s, 2H), 7.83-7.94 (m, 4H), 7.49-7.60 (m, 5H), 7.31 (d,d /=6.8Hz,1.2Hz, 1H). ES-MS: [M++l] = 433.53.
Step 6. Preparation of N-tert-butyl-5-(5-phenyl-4-(pyridin-2-ylmethylamino) quinazolin-2-yl) pyridine-3 -sulfonamide

2-(Chloromethyl) pyridine hydrochloride (564 g, 3.44 moles) and dimethyl acetamide (7L, 7 vol) were added to a 20 L RB flask- 1 equipped with mechanical stirrer under inert atmosphere. The resulting solution was cooled to 0- 5°C and triethylamine (346.3, 3.44 moles) was added at 0-5°C. 5-(4-Amino-5- phenylquinazolin-2-yl)-N-tert-butylpyridine-3-sulfonamide (1.0 Kg. 2.306 moles) and dimethylacetamide (4 L, 4 vol) were added to a separate 20 L RB flask-2 equipped with mechanical stirrer under inert atmosphere. This solution was cooled to 0-5°C and sodium tert-butoxide (884 g, 9.24 moles) was added at 0-5°C. The resulting solution was stirred to affect dissolution and then transferred to the RB flask- 1 at 0- 5°C. Upon completion of addition, the reaction mixture was stirred at 0-5°C for 2 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated that the reaction was complete. The reaction mass was poured into water (60 L, 60 vol) with stirring. The crude product was collected by filtration and dried at 60°C for 12 h. After this time, the dried material was dissolved in THF (20 L, 20 vol). Upon dissolution, 6M HC1 in isopropyl alcohol (1 L, 1 vol) was added at 20-25°C. The crude HCL salt of the product was obtained a pale-yellow free flow solid (920 g, 71% yield, 93% purity by HPLC). The crude HC1 salt (1.345 Kg, 2.56moles), methanol (6.7 L, 5 vol) and dichloromethane (13.5 L, 10 vol) were added to a 20 L glass-lined reactor equipped with mechanical stirrer. The slurry was stirred for 20-30 min at 30°C. After this time, the solvent was distilled to 4 vol with respect to input under vacuum. The resulting slurry was cooled to 20-25°C, where stirred for 2 h. At the conclusion of this period, the slurry was filtered and dried at 50°C for 6 h to yield the product (1.1 Kg, 82% yield, 98% purity by HPLC). XH NMR (DMSO- D6, 400 MHz, δ ppm); 9.72 (s, 1H), 9.10-9.14 (m, 2H), 8.39 (s, 1H), 7.92-8.03 (m, 4H), 7.56-7.58 (m, 5H), 7.43-7.49 (m, 3H), 7.1 (bs, 1H), 4.88 (s, 2H), 1.17 (2, 9H).
Step 7. Example 7

N-tert-butyl-5-(5-phenyl-4-(pyridin-2-ylmethylamino) quinazolin-2-yl) pyridine-3 -sulfonamide (1.0 Kg, 1.9 moles) and concentrated hydrochloric acid (7 L, 7 vol) were added to a 20 L glass-lined reactor equipped with mechanical stirrer. The reaction mixture was heated to 90-100°C where it stirred for 1 h. At the conclusion of this period, the reaction progress was monitored by HPLC, which indicated the reaction was complete. The reaction mixture was cooled to 5-10°C and the pH was adjusted to 1.7 to 2.0 using 12% aqueous sodium hydroxide solution. Once at the prescribed pH, the crude HC1 salt of the product was collected by filtration. The HC1 salt filter cake and ethanol (5 L, 5 vol) were added to 10 L glass-lined reactor equipped with a mechanical stirrer. The resulting mixture was made basic to pH 7-8 at 20-25°C using triethyl amine (2.25 Kg, 22.23 moles). Once at the prescribed pH, the basic mixture was stirred for 2 h. After this time, the free base of product was filtered and washed with water (10 L, 10 vol) followed by ethanol (2L, 2 vol). The resulting product was dried at 50-55°C for 8 h to yield Example 7 (644 g, 72% yield, 99.9% purity by HPLC).
XH NMR (DMSO-D6, 400 MHz, δ ppm); 9.81 (d, J=2.0Hz, 1H), 9.18 (t, J=2Hz, 1H), 9.1 1 (d, J=2Hz, 1H), 8.23 (d, J=4.4Hz, 1H), 7.92-7.94 (m, 1H), 7.83-7.87 (m, 1H), 7.78 (s, 2H), 7.70-7.72 (m, 1H), 7.50-7.59 (m, 5H), 7.31-7.34 (m, 2H), 7.22-7.25 (m, 1H), 6.95 (t, J=4Hz, 1H), 4.76 (d, J=4Hz, 2H). ES-MS: [M++l] = 469.
/////////atrial fibrillation, Potassium channel Kv1.5 (KCNA5) inhibitor, IKur antagonist, Bristol-Myers Squibb Co., BMS 919373, BMS-919373, PHASE 2
NS(=O)(=O)c1cc(cnc1)c4nc2cccc(c2c(NCc3ccccn3)n4)c5ccccc5
CRD 1152, CURADEV PHARMA PRIVATE LTD
Several candidates….one is…….CRD1152
ONE OF THEM IS CRD 1152
Kynurenine pathway regulators (solid tumors)

Compound 2
CAS1638121-21-7
N3-(3-Chloro-4- fluorophenyl) furo[2,3- c]pyridine-2,3- diamine

COMPD 190
CAS 1638118-99-6

COMPD248
7-Chloro-N3- (3-chloro-4- fluorophenyl) furo[2,3- c]pyridine-2,3- diamine, 166
DMSO-d6: δ 7.87 (d, J = 5.1 Hz, 1H), 7.25 (s, 2H), 7.16-7.10 (m, 2H), 6.88 (d, J = 5.1 Hz, 1H), 6.59 (dd, J′ = 6.2 Hz, J″ = 2.6 Hz, 1H), 6.48 (dt, J′ = 8.8 Hz, J″ = 6.7 Hz, J′′′ = 3.4 Hz, 1H) M + H] 312
OR
N3-(3,4- difluorophenyl)- 7-(pyridin-4- yl)furo[2,3- c]pyridine-2,3- diamine, 184
CD3CN: δ 8.72 (s, 2H), 8.26 (s, 3H), 7.07-7.03 (m, 2H), 6.47-6.40 (m, 2H), 5.74 (s, 1H), 5.55 (s, 2H) M + H] 339
OR

COMPD73
CAS 1638117-85-7
Several candidates………..CRD1152
 67
67
 66
66
| Company | Curadev Pharma Pvt. Ltd. |
| Description | Small molecule dual indoleamine 2,3-dioxygenase 1 (IDO1) and tryptophan 2,3-dioxygenase (TDO1; IDO) inhibitor |
| Molecular Target | Indoleamine 2,3-dioxygenase (INDO) (IDO) ; Tryptophan 2,3-dioxygenase (TDO2) (TDO) |
| Mechanism of Action | Indoleamine 2,3-dioxygenase (INDO) inhibitor |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Preclinical |
| Standard Indication | Cancer (unspecified) |
| Indication Details | Treat cancer |
| Regulatory Designation | |
| Partner | Roche |
Hoffmann-La Roche partners with Curadev Pharma Ltd. for IDO1 and TDO inhibitors (April 20, 2015)
Curadev Pharma Pvt Ltd., founded in 2010 and headquartered in New Delhi, announced that it has entered into a research collaboration and exclusive license agreement with Roche for the development and commercialization of IDO1 and TDO inhibitors to treat cancer. The agreement covers the development of CRD1152, the lead preclinical immune tolerance inhibitor and a research collaboration with Roche’s research and early development organization to further explore the IDO and TDO pathways.
IDO1 (indoleamine-2,3-dioxygenase-1) and TDO (tryptophan-2,3-dioxygenase) are enzymes that mediate cancer-induced immune suppression. This mechanism is exploited by tumor cells as well as certain type of immune cells, limiting the anti-tumor immune response. Dual inhibition of the IDO1 and TDO pathways promises to maintain the immune response, prevent local tumor immune escape and potentially avoid resistance to other immunotherapies when used in combination, and could lead to new treatment options for cancer patients. Curadev’s preclinical lead-compound, a small-molecule that shows potent inhibition of the two rate-limiting enzymes in the tryptophan to kynurenine metabolic pathways, has the potential for mono therapy as well as combination with Roche’s broad oncology pipeline and portfolio.
Under the terms of agreement, which includes a research collaboration with Roche’s research and early development organization, Curadev will receive an upfront payment of $25 million and will be eligible to receive up to $530 million in milestone payments, as well as escalating royalties potentially reaching double digits for the first product from the collaboration developed and commercialized by Roche. Curadev is also eligible for milestones and royalties on any additional products resulting from the research collaboration.




Curadev Announces Research Collaboration and Licensing Agreement to Develop Cancer Immunotherapeutic
Curadev’s dual IDO and TDO immune tolerance inhibitor – a novel approach in cancer immunotherapy
Apr 20, 2015, 06:30 ET from Curadev
NEW DELHI, India, April 20, 2015 /PRNewswire/ —
Curadev Pharma Private Ltd. today announced that it has entered into a research collaboration and exclusive license agreement with Roche for the development and commercialization of IDO1 and TDO inhibitors. The agreement covers the development of the lead preclinical immune tolerance inhibitor and a research collaboration with Roche’s research and early development organization to further explore the IDO and TDO pathways.
IDO1 (indoleamine-2, 3-dioxygenase-1) and TDO (tryptophan-2, 3-dioxygenase) are enzymes that mediate cancer-induced immune suppression. This mechanism is exploited by tumor cells as well as certain type of immune cells, limiting the anti-tumor immune response.
Dual inhibition of the IDO1 and TDO pathways promises to maintain the immune response, prevent local tumor immune escape and potentially avoid resistance to other immunotherapies when used in combination, and could lead to new treatment options for cancer patients. Curadev’s preclinical lead-compound, a small-molecule that shows potent inhibition of the two rate-limiting enzymes in the tryptophan – to kynurenine metabolic pathways, has the potential for mono therapy as well as combination with Roche’s broad oncology pipeline and portfolio.
“We are very excited to be working with the global leader in oncology with their unrivalled expertise in clinical development,” said Arjun Surya, PhD, Chief Scientific Officer, Curadev. “The collaboration acknowledges our focused research efforts on patient-critical drug targets that have yielded a drug candidate that could make a significant difference in the development of novel treatments for patients suffering from cancer.”
Under the terms of agreement, which includes a research collaboration with Roche’s research and early development organization to further extend Curadev’s findings, Curadev will receive an upfront payment of $25 million and will be eligible to receive up to $530 million in milestone payments based on achievement of certain predetermined events and sales levels as well as escalating royalties potentially reaching double digits for the first product from the collaboration developed and commercialized by Roche. Curadev would also be eligible for milestones and royalties on any additional products resulting from the research collaboration. Roche will fund future research, development, manufacturing and commercialization costs and will also provide additional research funding to Curadev for support of the research collaboration.
About Curadev
Headquartered in New Delhi, India, Curadev Pharma Private Limited was founded in 2010 by a team of professionals from the pharmaceutical and biotech sectors with the mission to improve human health and enhance the quality of human life by accelerating the discovery and delivery of new drugs. Curadev focuses on the creation and out-licensing of pre-IND assets and IND packages for drug development.
For further information:
Curadev Partnering
Manish Tandon – VP and Chief Financial Officer, manish@curadev.in
PATENT
US20160046596) INHIBITORS OF THE KYNURENINE PATHWAY
Monali Banerjee
Sandip Middya
Ritesh Shrivastava
Sushil Raina
Arjun Surya
Dharmendra B. Yadav
Veejendra K. Yadav
Kamal Kishore Kapoor
Aranapakam Venkatesan
Roger A. Smith
Scott K. Thompson
ONE ………….Example 2
Synthesis of N3-(3-Chloro-4-fluoro-phenyl)-furo[2,3-c]pyridine-2,3-diamine (Compound 2)
Step 1: 3-Methoxymethoxy-pyridine

Step 2: 3-Methoxymethoxy-pyridine-4-carbaldehyde

Step 3: 3-Hydroxy-pyridine-4-carbaldehyde

Step 4: 4-{[3-Chloro-4-fluoro-phenylimino]-methyl}-pyridin-3-ol

Step 5: N3-(3-Chloro-4-fluoro-phenyl)-furo[2,3-c]pyridine-2,3-diamine

Monali Banerjee – Director, R&D
Ms. Banerjee has more than 10 years of research experience, during which she has held positions of increasing responsibility. Her past organizations include TCG Lifesciences (Chembiotek) and Sphaera Pharma. Ms. Banerjee is a versatile scientist with a deep understanding of the fundamental issues that underlie various aspects of drug discovery. At Curadev, she has been responsible for target selection, patent analysis, pharmacophore design, assay development, ADME/PK and in vivo and in vitro pharmacology. Ms. Banerjee holds a Masters in Biochemistry and a Bachelors in Chemistry both from Kolkata University.
writeup
|
The essential amino acid Tryptophan (Trp) is catabolized through the kynurenine (KYN) pathway. The initial rate-limiting step in the kynurenine pathway is performed by heme-containing oxidoreductase enzymes, including tryptophan 2,3-dioxygenase (TDO), indoleamine 2,3-dioxygenase-1 (IDO1), and indoleamine 2,3-dioxygenase-2 (IDO2). IDO1 and IDO2 share very limited homology with TDO at the amino acid level and, despite having different molecular structures, each enzyme has the same biochemical activity in that they each catalyze tryptophan to form N-formylkynurenine. IDO1, IDO2, and/or TDO activity alter local tryptophan concentrations, and the build-up of kynurenine pathway metabolites due to the activity of these enzymes can lead to numerous conditions associated with immune suppression.
|
|
Kynurenine pathway dysregulation and IDO1 and/or TDO activity also correlate with cardiovascular risk factors, and kynurenines and IDO1 are markers for Atherosclerosis and other cardiovascular heart diseases such as coronary artery disease (Platten et al., Science, 2005, 310(5749):850-5, Wirlietner et al. Eur J Clin Invest. 2003 July; 33(7):550-4) in addition to kidney disease. The kynurenines are associated with oxidative stress, inflammation and the prevalence of cardiovascular disease in patients with end-stage renal disease (Pawlak et al., Atherosclerosis, 2009, (204)1:309-314). Studies show that kynurenine pathway metabolites are associated with endothelial dysfunction markers in the patients with chronic kidney disease (Pawlak et al., Advances in Medical Sciences, 2010, 55(2):196-203).
|
///////CRD1152, CRD-1152, CRD 1152, CURADEV PHARMA PRIVATE LTD, ROCHE, IDO1 and TDO inhibitors, COLLABORATION, CANCER, indoleamine-2,3-dioxygenase-1, Hoffmann-La Roche, kynurenine pathway regulators, solid tumors
GDC-0919; NLG-919; RG-6078

MF C18H22N2O
MW: 282.17321
GDC-0919; NLG-919; RG-6078, GDC0919; GDC-0919; GDC 0919; NLG919; NLG 919; NLG-919; RG6078; RG-6078; RG 6078.
1-cyclohexyl-2-(5H-imidazo[5,1-a]isoindol-5-yl)ethanol
CAS No.1402836-58-1
GDC-0919, also known as NLG919 and RG6078, is an orally available inhibitor of indoleamine 2,3-dioxygenase 1 (IDO1), with potential immunomodulating and antineoplastic activities. Upon administration, NLG919 targets and binds to IDO1, a cytosolic enzyme responsible for the oxidation of the essential amino acid tryptophan into kynurenine. By inhibiting IDO1 and decreasing kynurenine in tumor cells, this agent increases tryptophan levels, restores the proliferation and activation of various immune cells, including dendritic cells (DCs), natural killer (NK) cells, T-lymphocytes, and causes a reduction in tumor-associated regulatory T-cells (Tregs). Activation of the immune system, which is suppressed in many cancers, may induce a cytotoxic T-lymphocyte (CTL) response against the IDO1-expressing tumor cells

- Originator Lankenau Institute for Medical Research
- Developer Genentech; NewLink Genetics Corporation
- Class Antineoplastics; Small molecules
- Mechanism of Action Immunomodulators; Indoleamine-pyrrole 2,3-dioxygenase inhibitors
Phase I Solid tumours
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015210769 | 2015-07-30 | ANTIBODY MOLECULES TO PD-1 AND USES THEREOF |
| US2014066625 | 2014-03-06 | Fused Imidazole Derivatives Useful as IDO Inhibitors |
- 27 Sep 2015 Pharmacokinetics results from a phase-I clinical trial in Solid tumours presented at the European Cancer Congress 2015 (ECC-2015)
- 27 Sep 2015 Positive efficacy and safety results from a phase-I clinical trial in Solid tumours presented at the European Cancer Congress 2015 (ECC-2015)
- 31 Jul 2015 Phase-I clinical trials in Solid tumours (Combination therapy, Late-stage disease, Second-line therapy or greater) in USA (PO) (NCT02471846)
PATENT
http://www.google.com/patents/WO2012142237A1?cl=en

PATENT
Fused Imidazole Derivatives Useful as IDO Inhibitors
13041-cyclohexyl-2-(5H-imidazo[5,1- a]isoindol-5-yl)ethanol79 1H NMR (a mixture of diastereomers) 1.10-1.37 (m, 6H), 1.66-1.80 (m, 5H), 2.05 (m, 2H), 2.15 (m, 1H), 3.72 (m, 1H), 5.36 and 5.46 (two m, 1H), 7.16 (s, 1H), 7.25 (m, 1H), 7.34 (m, 1H), 7.43 (d, 1H, J = 7.6 Hz), 7.54 (d, 1H, J = 7.6 Hz), 7.80 (s, 1H)
| WO2011056652A1 * | Oct 27, 2010 | May 12, 2011 | Newlink Genetics | Imidazole derivatives as ido inhibitors |
| WO2012142237A1 * | Apr 12, 2012 | Oct 18, 2012 | Newlink Geneticks Corporation | Fused imidazole derivatives useful as ido inhibitors |
| WO2014159248A1 | Mar 10, 2014 | Oct 2, 2014 | Newlink Genetics Corporation | Tricyclic compounds as inhibitors of immunosuppression mediated by tryptophan metabolization |
| US8722720 | Oct 27, 2010 | May 13, 2014 | Newlink Genetics Corporation | Imidazole derivatives as IDO inhibitors |
| US9260434 | Oct 14, 2013 | Feb 16, 2016 | Newlink Genetics Corporation | Fused imidazole derivatives useful as IDO inhibitors |
| US20140066625 * | Oct 14, 2013 | Mar 6, 2014 | Newlink Genetics Corporation | Fused Imidazole Derivatives Useful as IDO Inhibitors |
| US20160002249 * | Jul 8, 2015 | Jan 7, 2016 | Newlink Genetics Corporation | Fused Imidazole Derivatives Useful as IDO Inhibitors |
REFERENCES
Nature Reviews Drug Discovery14,373(2015)doi:10.1038/nrd4658
http://www.ncbi.nlm.nih.gov/pubmed/21517759
http://www.roche.com/irp150128-annex.pdf
/////CRD1152, CRD 1152, CRD-1152, Curadev, Research Collaboration, Licensing Agreement, Develop, Cancer Immunotherapeutic, IDO1 and TDO inhibitors
OC(C1CCCCC1)CC(C2=C3C=CC=C2)N4C3=CN=C4
CFG 920, Novartis Scientists team up with Researchers at Aurigene, Bangalore, India,
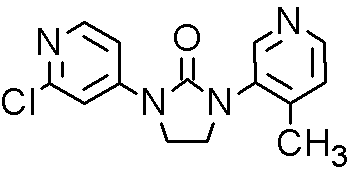
CFG920,
Inhibitor Of Prostate Cancer With Fewer Cardiac Side Effects
Cas 1260006-20-9
Novartis
Target: CYP17/CYP11B2
Disease: Castration-resistant prostate cancer
MF C14H13ClN4O
MW: 288.0778
Elemental Analysis: C, 58.24; H, 4.54; Cl, 12.28; N, 19.40; O, 5.54
Steroid 17-alpha-hydroxylase inhibitors
CFG920 is a CYP17 inhibitor, is also an orally available inhibitor of the steroid 17-alpha-hydroxylase/C17,20 lyase (CYP17A1 or CYP17), with potential antiandrogen and antineoplastic activities. Upon oral administration, CYP17 inhibitor CFG920 inhibits the enzymatic activity of CYP17A1 in both the testes and adrenal glands, thereby inhibiting androgen production. This may decrease androgen-dependent growth signaling and may inhibit cell proliferation of androgen-dependent tumor cells.
https://clinicaltrials.gov/ct2/show/NCT01647789
NCT01647789: A Study of Oral CFG920 in Patients With Castration Resistant Prostate Cancer2012
- 09 Nov 2015Adverse events, efficacy and pharmacokinetics data from the phase I part of a phase I/II trial in Prostate cancer (Metastatic disease) presented at the 27th AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics (AACR-NCI-EORTC-2015)
- 29 Jan 2013Phase-I clinical trials in Prostate cancer in Spain (PO)
- 10 Dec 2012Phase-I clinical trials in Prostate cancer in Canada (PO)
In August 2015, preclinical data were presented at the 250th ACS meeting in Boston, MA. In monkeys, treatment with CFG-920 (3 mg/kg, po) showed good bioavailability with F value of 93%, Tmax of 0.5 h, Cmax of 1382 nM.dn and AUC of 2364 nM.h, while CFG-920 (10 mg/kg, po) showed F value of 183%, Cmax of 1179 nM.dn and Tmax of 1.04 h
Bethany Halford on Twitter: “CFG920 – @Novartis CMOS for …

Novartis is developing CFG-920 (structure shown), an oral CYP17 inhibitor, for the potential treatment of metastatic castration-resistant prostate cancer. In March 2013, a phase I/II trial was initiated and at that time, the study was expected to complete in January 2015; in August 2015, clinical data were presented
2015 250th (August 19) Abs MEDI 341
Discovery of CFG920, a dual CYP17/CYP11B2 inhibitor, for the treatment of castration resistant prostate cancer
American Chemical Society National Meeting and Exposition
Christoph Gaul, Prakash Mistry, Henrik Moebitz, Mark Perrone, Bjoern Gruenenfelder, Nelson Guerreiro, Wolfgang Hackl, Peter Wessels, Estelle Berger, Mark Bock, Saumitra Sengupta, Venkateshwar Rao, Murali Ramachandra, Thomas Antony, Kishore Narayanan, Samiulla Dodheri, Aravind Basavaraju, Shekar Chelur


| Aurigene Discovery Technologies Limited, an independent subsidiary of Dr Reddy’s, CSN Murthy is chief executive officer |

WO 2010149755

Prostate cancer is the most commonly occurring cancer in men. Doctors often treat the metastatic stage of the disease by depriving the patient of sex hormones via chemical or surgical castration. But if it progresses far enough, the cancer can survive this therapy, transforming into the castration-resistant form. “Once the cancer becomes castration-resistant, the prognosis is poor,” said Novartis’s Christoph Gaul.
In recent years, CYP17, a bifunctional 17α-hydroxylase/17,20-lyase cytochrome P450 enzyme, has emerged as a target for treating castration-resistant prostate cancer. The enzyme catalyzes the biosynthesis of sex hormones, including testosterone, and blocking it can starve prostate cancer of the androgens it needs to thrive.
Johnson & Johnson’s CYP17 inhibitor, abiraterone acetate (Zytiga), a steroid that binds irreversibly to CYP17, was approved by the Food & Drug Administration in 2011. But Novartis scientists thought they could make a better CYP17 inhibitor, Gaul told C&EN. They teamed up with researchers at Aurigene, in Bangalore, India, and came up with their clinical candidate, CFG920.
Unlike abiraterone, CFG920 isn’t a steroid, and it inhibits CYP17 reversibly. It also reversibly inhibits another cytochrome P450 enzyme, CYP11B2, which is involved in the synthesis of the mineralocorticoids, hormones that regulate cardiac function.
Treating prostate cancer patients by lowering their androgen levels turns out to have negative cardiac side effects: Patients’ lipid metabolism is thrown off and their mineralocorticoid levels jump, leading to increases in blood pressure. Those changes can be stressful for the heart. “If prostate cancer patients don’t die because of the cancer, a lot of times they die because of cardiac disease,” Gaul said.
Because CFG920 also keeps mineralocorticoid levels in check, Novartis is hoping the drug candidate will ameliorate some of the cardiac side effects of inhibiting CYP17. The compound is currently in Phase I clinical trials.
PATENT
WO 2010149755
Example 58
Prύpιn”ation ofI'(2’ChIoroψ}ri(ibi-^’\l)’3’f4’metMψ}τUin’3’yl)-imiJazoliJin’2’θne (5HA)-

Using the same reaction conditions as in Example 14. 1-(4-methyl-pyridin-3-yl)- itnida/olidin-2-onc ().-.!.4b: 600 mg. 3.3898 mmol) uas reacted with 2-chloro-4-iodo- py.idine (974 mg.4.067 mmol). 1 , 4-dioxane (60 mL). copper iodide (65 mg, 0.3398 mmol), /r<w.v-1.2-diamino cycK)hexane (0.12 ml,, 1.0169 mmol) and potassium phosphate (2.15 g, 10.1694 mmol) to afford 810 mg of the product (83% yield).
1H NMR (C1DCI3. 300 Mi l/): 6 8.5-8.4 (m. 211). 8.3 (d. IH), 7.6-7.5 (m, 2H). 7.2 (S. 111). 4.1-3.9 (ni. 4H), 2.35 <s. 3H)
LCVIS puιϊt>: 90.8%. nι-7 – 289.1 (M M)
HPl C: 97.14%
REFERENCES
1: Gomez L, Kovac JR, Lamb DJ. CYP17A1 inhibitors in castration-resistant prostate cancer. Steroids. 2015 Mar;95:80-7. doi: 10.1016/j.steroids.2014.12.021. Epub 2015 Jan 3. Review. PubMed PMID: 25560485; PubMed Central PMCID: PMC4323677.
2: Yin L, Hu Q, Hartmann RW. Recent progress in pharmaceutical therapies for castration-resistant prostate cancer. Int J Mol Sci. 2013 Jul 4;14(7):13958-78. doi: 10.3390/ijms140713958. Review. PubMed PMID: 23880851; PubMed Central PMCID: PMC3742227.
///////CFG-920, CYP17 inhibitor (prostate cancer), Novartis, CFG 920, Novartis scientists, team up , researchers , Aurigene, Bangalore, India,
