





( Angew. Chem., Int. Ed. 2015,54, 4945−4948).
Volume 54, Issue 16April 13, 2015 Pages 4945–4948
WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
Home » 2016 (Page 23)
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |

MIGLUSTAT
![]()
Gauchers disease type I; Niemann Pick disease type C
EP-03031800, Process for the preparation of high purity miglustat
Navinta, LLC ; Shah, Shrenik K. ; Kharatkar, Raju Mahadev ; Bhatt, Chiragkumar Anilkumar ; Kevat, Jitendra Bhagwandas
The present invention provides a process for the preparation and isolation of crystalline miglustat without the use of a column chromatography or ion exchange purification. The crystalline miglustat has a high purity and a melting point of 128 °C and an endothermic peak is 133 °C.
Process for preparing and isolating crystalline form of miglustat with a high purity is claimed. Represents a first PCT filing from the inventors on miglustat. Actelion, under license from Oxford GlycoSciences (OGS; then Celltech, now UCB), which licensed the compound from GD Searle & Co, has developed and launched miglustat.
Product patent WO9426714, will expire in the US in 2018.
Kharatkar is affiliated with Sterling Biotech, Bhatt is affiliated with Intas and Kevat is affiliated with Orchid Chemicals & Pharmaceuticals.
INVENTORS Shah, Shrenik K.; Kharatkar, Raju Mahadev; Bhatt, Chiragkumar Anilkumar; Kevat, Jitendra Bhagwandas
About Navinta
Navinta, LLC in Ewing, N.J. is a technology driven Pharmaceutical Company that focuses on novel routes of synthesis of new and existing drug molecules, complex pharmaceutical ingredients, novel formulations of liquid dosage form, novel oral dosage form, novel injectable dosage form and implantable drug delivery devices. Navinta has currently at least fifteen (15) patents granted or pending with the United States Patent and Trademark Office.
![]()
EP-03031800 LINK EMBEDDED
|
Miglustat is a potent inhibitor of glycosyltransferase. It is primarily used in the treatment of Gaucher’s disease. Miglustat is chemically known as N-butyl-1,5-dideoxy-1,5-imino-D-glucitol of formula (I) and is sometimes referred as N-butyl-1-deoxynojirimycin. Miglustat is a white to off-white crystalline solid with a melting point of 125-126° C. Its empirical formula is C10H21NO4 and has a molecular weight of 219.28 g/mol.
|
Example 1
Synthesis of 2, 3, 4, 6-tetra-O-benzyl-N-butyl-1-deoxynojirimycin hydrochloride of Formula (VI)
To a solution of 2, 3, 4, 6-tetra-O-benzyl-1-deoxynojirimycin hydrochloride (V) (prepared as in Organic Process Research & Development, 2008, 12, 414-423) (45 g, 0.08 mol) in 1575 mL of methanol, n-butyraldehyde (21.6 g, 0.24 mol) and sodium cyanoborohydride (25.2 g, 0.4 mol) were added and stirred. The reaction was maintained under stirring at a temperature from about 25.degree. C. to about 30.degree. C. After the completion of the reaction, the reaction was quenched by adding 765 ml of 10% HCl in methanol, while keeping the temperature between 25.degree. C. to 30.degree. C. The reaction mass was cooled to 0.degree. C. to 5.degree. C. and the resulting precipitate solids were filtered. The filtrate was treated with aqueous HCl and the solid formed was filtered, suspended in 1 N HCl, stirred for 1 hour and filtered. The collected solid was washed with diisopropylether and dried under vacuum to furnish 46.2 g of compound (IV) (46.2 g, 0.075 mol, 94% yield) of high chemical purity based on HPLC analysis (>99.0%).
Example 2
Synthesis of Miglustat Hydrochloride of Formula (III)
A solution of 2, 3, 4, 6-tetra-O-benzyl-N-butyl-1-deoxynojirimycin hydrochloride (VI) (100 g, 0.16 mol) in methanol (1000 mL), 10% HCl solution in methanol (100 mL), and 10% Pd/C (50% wet) (10 g) were mixed and stirred under hydrogen atmosphere at a temperature of about 25.degree. C. to about 30.degree. C. until completion of the reaction. The reaction mass was filtered and the solvent was removed from the filtrate by rotary evaporation. Ethyl acetate (1000 mL) was added to the residue from the rotary evaporation to precipitate the solid. The solid was filtered and dried to isolate Miglustat hydrochloride (III) (42 g, 0.16 mol, 100% yield) of >99.5% purity as measured by HPLC analysis. The DSC thermogram of this product is provided as FIG. 3, and the FTIR spectrum of this product is provided as FIG. 4.
Example 3
Synthesis of Miglustat of Formula (I)
Miglustat hydrochloride (III) (42 g, 0.16 mol) obtained from Example 2 was dissolved in 420 mL of methanol and DBU (1,8-diazabicycloundec-7-ene) (34.1 mL) was added. The reaction mass was warmed slightly and stirred for about 2 hours. The reaction was concentrated by removal of methanol. Dichloromethane (900 mL) was added to the residue. The resulting solid was filtered and dried to obtain crystalline miglustat (I) (27 g, 0.12 mol, 75% yield) of >99.5% purity as measured by HPLC analysis. The melting point of the crystalline miglustat (I) is 128.degree. C. The DSC thermogram and FTIR spectrum of the product are provided as FIG. 1 and FIG. 2, respectively. The crystalline miglustat (I) contained <0.05% of the 5R isomer (IV) as measured by HPLC.

WO-2016092561, Ivacaftor, NEW PATENT
https://www.google.com/patents/WO2016092561A2?cl=en
Novel polymorphs of ivacaftor, process for its preparation and pharmaceutical composition thereof
Laurus Labs Pvt Ltd
LAURUS LABS PRIVATE LIMITED [IN/IN]; Plot No. DS1, IKP Knowledge Park, Genome Valley Turkapally, Shameerpet Mandal, Ranga District Hyderabad 500078 (IN)

| Ram Thaimattam, Venkata Srinivasa Rao DAMA, Venkata Sunil Kumar Indukuri, Seeta Rama Anjaneyulu GORANTLA,Satyanarayana Chava, | |
| Applicant | Laurus Labs Private Limited |
THAIMATTAM, Ram; (IN).
DAMA, Venkata Srinivasa Rao; (IN).
INDUKURI, Venkata Sunil Kumar; (IN).
GORANTLA, Seeta Rama Anjaneyulu; (IN).
CHAVA, Satyanarayana; (IN)

Novel crystalline forms of ivacaftor (designated as forms L1 to L14), processes for their preparation and composition comprising them are claimed.
Vertex, in research collaboration with Cystic Fibrosis Foundation Therapeutics, had developed and launched ivacaftor.
Ivacaftor, also known as N-(2,4-di-tert-butyl-5-hydroxyphenyl)-l,4-dihydro-4-oxoquinoline-3-carboxamide, having the following Formula I:

Formula I
Ivacaftor was approved by FDA and marketed by Vertex pharma for the treatment of cystic fibrosis under the brand name KALYDECO® in the form of 150 mg oral tablets.
WO2006/002421 publication discloses modulators of ATP-binding cassette transporters such as ivacaftor. This patent generally discloses a process for the preparation of modulators of ATP-binding cassette transporters such as quinoline compounds; however, specific process for the preparation of ivacaftor and its solid state details were not specifically disclosed.
WO2007/079139 publication discloses Form A, Form B and amorphous form of ivacaftor characterized by PXRD, DSC and TGA and process for their preparation. Further this publication discloses ethanol crystalate of ivacaftor in example part.
WO2009/038683 publication discloses the solid forms of ivacaftor, which are designated as Form-I (2-methylbutyric acid), Form-II (propylene glycol), Form-HI (PEG400.KOAc), Form-IV (lactic acid), Form-V (isobutyric acid), Form-VI (propionic
acid), Form- VII (ethanol), Form- VIII (2-propanol), Form-IX (monohydrate), Form-X (besylate Form A), Form-XI (besylate Form B), Form-XII (besylate Form D), Form-XIII (besylate Form E), Form-XIV (besylate Form F), Form-XV (besylate (2: 1)), Form-XVI (besylate mono hydrate). This publication also discloses the characterization details like PXRD, DSC and TGA for the above forms and process for their preparation.
WO201 1/1 16397 publication discloses crystalline Form C of ivacaftor, process for its preparation and pharmaceutical composition comprising the same. Also discloses characterization details of Form C, such as PXRD, IR, DSC and 13CSSNMR.
WO2013/158121 publication discloses solvated forms of ivacaftor, which are designated as Form D (acetonitrile or acetonitrile/water (75/25) solvate), Form E (Methyl ethyl ketone (MEK), MEK/water (90/1), MEK/water (90/10), MEK/water (80/20) solvate), Form F (acetonitrile/water (75/25) solvate), Form G (isopropyl acetate solvate), Form H (isopropyl acetate/water (95/5) solvate), Form I (MEK solvate), Form J (MEK/water (99/1) solvate), Form K (MEK or MEK/water (99/1) or MEK/water (90/10) or MEK/water (80/20) solvate), Form L (isopropyl acetate/water (95/5) solvate), Form M (MEK or MEK/water (99/1) solvate), Form N (MEK water (90/10) or MEK/water (80/20) solvate), Form O (MEK or MEK/water (99/1) solvate), Form P (MEK water (90/10) or MEK water (80/20) solvate), Form Q (MEK/water (80/20) solvate), Form R (acetonitrile solvate), Form S (MEK/water (80/20) solvate), Form T (isopropyl acetate/water (95/5) solvate), Form W (acetonitrile/water (90/10) solvate), Form XX (from 10% water/ acetonitrile) and hydrate B (hydrated form). This patent further discloses characterization details like PXRD and TGA for the above forms and process for their preparation.
WO2014/118805 publication discloses crystalline forms of ivacaftor designated as Form D, Form E, Form El, Form G and Form G’; amorphous ivacaftor designated as Form I and Form II; crystalline ivacaftor solvates such as n-butanol solvate, methanol solvate, propylene glycol solvate, DMF solvate, THF solvate, DMF:ethylacetate solvate. This publication further discloses the process for the preparation of said forms along with their characterization details.
WO2015/070336 publication discloses polymorphic form APO-I and MIBK solvate of ivacaftor along with its characteristic PXRD details, process for its preparation and pharmaceutical composition comprising them.
CN 104725314A publication discloses ivacaftor new polymorph D, which is obtained by crystallization of ivacaftor from acetonitrile/water. This publication further discloses characteristic details such PXRD, IR and DSC of ivacaftor new polymorph D.
Polymorphism is the occurrence of different crystalline forms of a single compound and it is a property of some compounds and complexes. Thus, polymorphs are distinct solids sharing the same molecular formula, yet each polymorph may have distinct physical properties. Therefore, a single compound may give rise to a variety of polymorphic forms where each form has different and distinct physical properties, such as different solubility profiles, different melting point temperatures and/or different x-ray diffraction peaks. Since the solubility of each polymorph may vary, identifying the existence of pharmaceutical polymorphs is essential for providing pharmaceuticals with predictable solubility profiles. It is desirable to investigate all solid state forms of a drug, including all polymorphic forms and solvates, and to determine the stability, dissolution and flow properties of each polymorphic form.
Polymorphic forms and solvates of a compound can be distinguished in a laboratory by X-ray diffraction spectroscopy and by other methods such as, infrared spectrometry. Additionally, polymorphic forms and solvates of the same drug substance or active pharmaceutical ingredient, can be administered by itself or formulated as a drug product (also known as the final or finished dosage form), and are well known in the pharmaceutical art to affect, for example, the solubility, stability, flowability, tractability and compressibility of drug substances and the safety and efficacy of drug products.
The discovery of new polymorphic forms and solvates of a pharmaceutically useful compound, like ivacaftor, may provide a new opportunity to improve the performance characteristics of a pharmaceutical product. It also adds to the material that a formulation scientist has available for designing, for example, a pharmaceutical dosage form of a drug with a targeted release profile or other desired characteristic. New polymorphic forms of the ivacaftor have now been discovered and have been designated as ivacaftor Form-Ll, Form-L2, Form-L3, Form-L4, Form-L5, Form-L6, Form-L7, Form-L8, Form-L9, Form-LlO, Form-Ll 1, Form-Ll 2 A, Form-Ll 2B, Form-Ll 3 and Form-Ll 4.
EXAMPLE 1 : Preparation of Ivacaftor Form-Ll
A suspension of ivacaftor ethanolate (5 g) in n-heptane (200 mL) was heated to 95-100°C and stirred for 5 hrs at the same temperature. Then the reaction mixture was cooled to 25-35°C and stirred for an hour. The solid obtained was filtered, washed with n-heptane and suck dried. The wet solid was further dried at 60-65°C for 16 hrs under vacuum yielded ivacaftor Form-Ll . The XRPD is set forth in Figure- 1.
In a similar manner, ivacaftor Form-Ll was prepared from different solvates of ivacaftor in place of ivacaftor ethanolate as input using the following conditions;

Ivacaftor cyclopentyl methyl ether (0.5 g) n-heptane (20 mL) 50°C/8 hr
Ivacaftor methyltertiarybutyl ether (0.5 g) n-heptane (20 mL) 50°C/8 hr

When Dr Satyanarayana Chava started Laurus Labs in 2007, he invested nearly Rs 60 crore of his own money into it. His confidence in its success was neither bravado nor bluster, but defined by his knowledge of the pharmaceutical industry. Eight years on, the Hyderabad-based company is on track to reach revenues of Rs 2,000 crore by the end of FY2016.
Chava, now 52, has more than two decades of experience in the pharmaceutical industry; in his last job, he was chief operating officer (COO) of the successful startup, Matrix Laboratories. Of his 10 years there, he says with pride, “I never skipped a promotion and got to work in all departments.” His dedication, coupled with a sound understanding of what it takes to start a pharmaceutical company, is what makes Laurus Labs among the hottest startups in this sector.
Initially, Chava planned the business around research and development (R&D). He wanted Laurus Labs to focus on contract research and make money from royalties. “In India, companies start with manufacturing and then get into R&D,” he explains. “I did it the other way round.” He focussed his fledgling company’s resources on developing formulations for medicines, and licensed them to other pharmaceutical players. In the early months, Laurus Labs had 10 people in manufacturing and 300 in R&D.

In June 2007, Aptuit, a US-based contract research organisation (CRO), signed it on for a $20 million (then Rs 80 crore) contract. But despite this injection of funds, Chava was unable to sustain his original idea of developing technologies for other companies. At the time of the Aptuit deal, Laurus Labs’s annual revenues were not even $20,000 (Rs 8 lakh at the time). In 2008, Chava decided to start manufacturing active pharmaceutical ingredients (API), which, as the name suggests, are chemicals or key ingredients in drugs required to make the medication work. His early investment into R&D benefitted Laurus Labs; it maintains a large repository of research-based knowledge that forms the bedrock of any successful pharmaceutical business.
Today, it is a key manufacturer supplier of APIs and holds its own against better-known competitors like US generic drug giant Mylan, which, incidentally, acquired a controlling stake in Matrix around the time Chava founded Laurus Labs. It has also carved a niche for itself by supplying antiretroviral or ARVs (used to fight infections caused by retroviruses like HIV) and oncology drugs. And despite being a relatively new player, its clients include giants like Pfizer, Teva Pharmaceutical Industries and Merck.

The person behind it
A Master’s degree in chemistry was never on the cards for Chava. In the early 1980s, the best students usually studied physics, and he had planned to do the same. But when he went to his college in Amravati (Andhra Pradesh) to enroll, his elder sister’s friend suggested he study chemistry too. Chava took up the subject on a whim. He ended up liking chemistry so much so that in his final year he topped his batch despite not having written one out of the four required papers. He went on to complete his PhD in the subject in 1991.
Upon graduating, he was hired by Ranbaxy Laboratories in Delhi as a researcher. In those early years itself Chava knew he’d spend a lifetime in the industry. He enjoyed the work and gained valuable experience as a young researcher in what was then India’s finest pharmaceutical company.
But through his years in the industry, Chava was conscious of the fact that he needed to broaden his experience outside of research. His stint at Matrix Laboratories afforded him that opportunity. As it was a startup, he was able to rise through the ranks quickly and got the opportunity to work in key departments from sales and marketing to finance and accounts. Within eight years of joining Matrix, he became its COO.

This experience was to come in handy when, due to differences with the board—he refused to elaborate on this—he decided to leave Matrix and set up Laurus Labs. And though he is the company’s chief executive officer (CEO), Chava remains true to his calling as a chemist. He has strived to build an organisation that is not very hierarchical. It is not uncommon to see him interacting with the chemists in the company and discussing formulations with them—something unheard of in an industry where most CEOs are from a sales and marketing background.


Chandrakanth Chereddi
VP Synthesis Business Unit
Prior to his current assignment at Laurus Labs India, Chandra headed the Project Management division for all scientific projects at the Laurus R&D center. Chandra previously worked for McKinsey & Company in India as a member of the healthcare practice and at Google Inc. as a software engineer in Google’s Mountain View, CA office. Chandra holds a BE from the College of Engineering, Osmania University, Hyderabad, and MS from University of Illinois at Urbana-Champaign, and an MBA from Indian School of Business, Hyderabad.
///////WO 2016092561, Ivacaftor, New patent, Laurus Labs Pvt Ltd

Continuous production is a flow production method used to manufacture, produce, or process materials without interruption. Continuous production is called a continuous process or a continuous flow process because the materials, either dry bulk or fluids that are being processed are continuously in motion, undergoing chemical reactions or subject to mechanical or heat treatment. Continuous processing is contrasted with batch production.
Continuous usually means operating 24 hours per day, seven days per week with infrequent maintenance shutdowns, such as semi-annual or annual. Some chemical plants can operate for more than one or two years without a shutdown. Blast furnaces can run four to ten years without stopping.[1]
Production workers in continuous production commonly work in rotating shifts.
Processes are operated continuously for practical as well as economic reasons. Most of these industries are very capital intensive and the management is therefore very concerned about lost operating time.
Shutting down and starting up many continuous processes typically results in off quality product that must be reprocessed or disposed of. Many tanks, vessels and pipes cannot be left full of materials because of unwanted chemical reactions, settling of suspended materials or crystallization or hardening of materials. Also, cycling temperatures and pressures from starting up and shutting down certain processes (line kilns, boilers, blast furnaces, pressure vessels, etc.) may cause metal fatigue or other wear from pressure or thermal cycling.

In the more complex operations there are sequential shut down and start up procedures that must be carefully followed in order to protect personnel and equipment. Typically a start up or shut down will take several hours.
Continuous processes use process control to automate and control operational variables such as flow rates, tank levels, pressures, temperatures and machine speeds.[2]
Many processes such as assembly lines and light manufacturing that can be easily shut down and restarted are today considered semi-continuous. These can be operated for one or two shifts if necessary.
The oldest continuous flow processes is the blast furnace for producing pig iron. The blast furnace is intermittently charged with ore, fuel and flux and intermittently tapped for molten pig iron and slag; however, the chemical reaction of reducing the iron and silicon and later oxidizing the silicon is continuous.
Semi-continuous processes, such as machine manufacturing of cigarettes, were called “continuous” when they appeared.
Many truly continuous processes of today were originally batch operations.
The Fourdrinier paper machine, patented in 1799, was one of the earliest of the industrial revolution era continuous manufacturing processes. It produced a continuous web of paper that was formed, pressed, dried and reeled up in a roll. Previously paper had been made in individual sheets.
Another early continuous processes was Oliver Evans‘es flour mill (ca. 1785), which was fully automated.
Early chemical production and oil refining was done in batches until process control was sufficiently developed to allow remote control and automation for continuous processing. Processes began to operate continuously during the 19th century. By the early 20th century continuous processes were common.
In addition to performing maintenance, shut downs are also when process modifications are performed. These include installing new equipment in the main process flow or tying-in or making provisions to tie-in sub-processes or equipment that can be installed while the process is operating.
Shut-downs of complicated processes may take weeks or months of planning. Typically a series of meetings takes place for co-ordination and planning. These typically involve the various departments such as maintenance, power, engineering, safety and operating units.
All work is done according to a carefully sequenced schedule that incorporates the various trades involved, such as pipe-fitters, millwrights, mechanics, laborers, etc., and the necessary equipment (cranes, mobile equipment, air compressors, welding machines, scaffolding, etc.) and all supplies (spare parts, steel, pipe, wiring, nuts and bolts) and provisions for power in case power will also be off as part of the outage. Often one or more outside contractors perform some of the work, especially if new equipment is installed.
Safety meetings are typically held before and during shutdowns. Other safety measures include providing adequate ventilation to hot areas or areas where oxygen may become depleted or toxic gases may be present and checking vessels and other enclosed areas for adequate levels of oxygen and insure absence of toxic or explosive gases. Any machines that are going to be worked on must be electrically disconnected, usually through the motor starter, so that it cannot operate. It is common practice to put a padlock on the motor starter, which can only be unlocked by the person or persons who is or are endangered by performing the work. Other disconnect means include removing couplings between the motor and the equipment or by using mechanical means to keep the equipment from moving. Valves on pipes connected to vessels that workers will enter are chained and locked closed, unless some other means is taken to insure that nothing will come through the pipes.
Continuous Production can be supplemented using a Continuous Processor. Continuous Processors are designed to mix viscous products on a continuous basis by utilizing a combination of mixing and conveying action. The Paddles within the mixing chamber (barrel) are mounted on two co-rotating shafts that are responsible for mixing the material. The barrels and paddles are contoured in such a way that the paddles create a self-wiping action between themselves minimizing buildup of product except for the normal operating clearances of the moving parts. Barrels may also be heated or cooled to optimize the mixing cycle. Unlike an extruder, the Continuous Processor void volume mixing area is consistent the entire length of the barrel ensuring better mixing and little to no pressure build up. The Continuous Processor works by metering powders, granules, liquids, etc. into the mixing chamber of the machine. Several variables allow the Continuous Processor to be versatile for a wide variety of mixing operations:[3]
Continuous Processors are used in the following processes:
The Continuous Processor has an unlimited material mixing capabilities but, it has proven its ability to mix:

In the development of a new route to bendamustine hydrochloride, the API in Treanda, the key benzimidazole intermediate 5 was generated via catalytic heterogeneous hydrogenation of an aromatic nitro compound using a batch reactor. Because of safety concerns and a site limitation on hydrogenation at scale, a continuous flow hydrogenation for the reaction was investigated at lab scale using the commercially available H-Cube. The process was then scaled successfully, generating kilogram quantities on the H-Cube Midi. This flow process eliminated the safety concerns about the use of hydrogen gas and pyrophoric catalysts and also showed 1200-fold increase in space–time yield versus the batch processing.


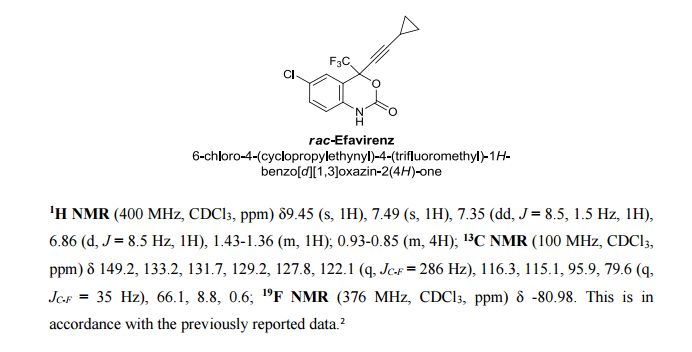
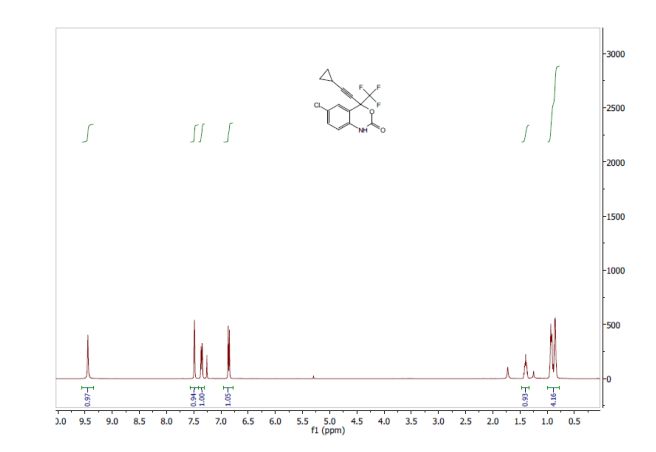
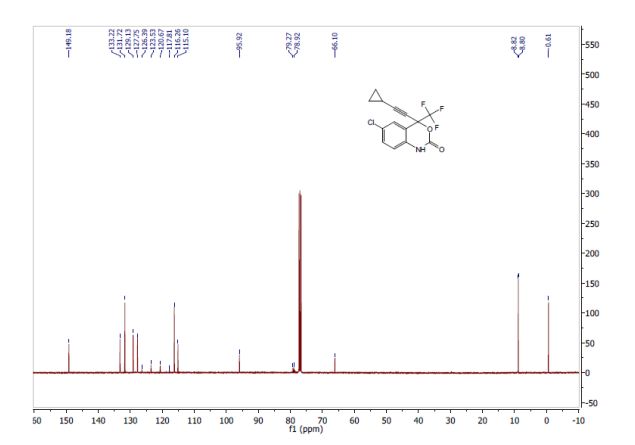
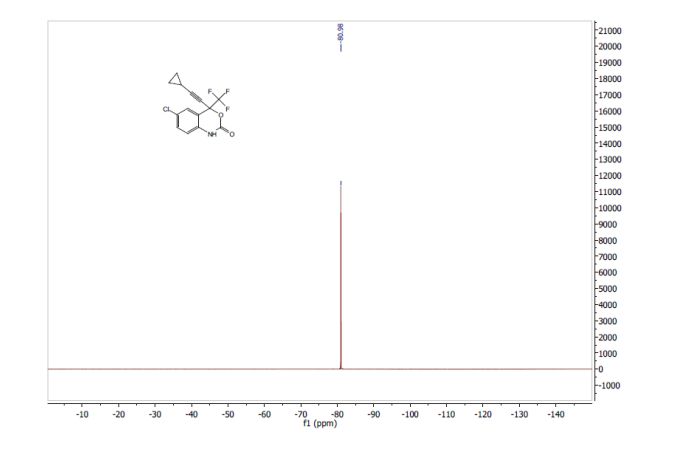

Volume 54, Issue 16April 13, 2015 Pages 4945–4948


The synthesis of 2-(2′-hydroxy-5′-methylphenyl)benzotriazole from 2-nitro-2′-hydroxy-5′-methylazobenzene over Pd/γ-Al2O3 in a fixed-bed reactor was investigated. Pd/γ-Al2O3 catalysts were prepared by two methods and characterized by XRD, TEM, H2-TPR, and N2 adsorption–desorption. Employed in the above reaction, the palladium catalyst impregnated in hydrochloric acid exhibited much better catalytic performance than that impregnated in ammonia–water, which was possibly attributed to the better dispersion of palladium crystals on γ-Al2O3. This result demonstrated that the preparation process of the catalyst was very important. Furthermore, the reaction parameters were optimized. Under the optimized conditions (toluene, NAB/triethylamine molar ratio 1:2, 60 °C, 2.5 MPa hydrogen pressure, 0.23 h–1 liquid hourly space velocity), about 90% yield of 2-(2′-hydroxy-5′-methylphenyl)benzotriazole was obtained. Finally, the time on stream performance of the catalyst was evaluated, and the reaction could proceed effectively over 200 h without deactivation of the catalyst.
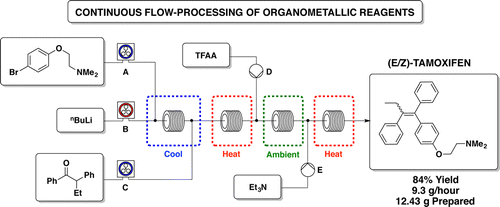
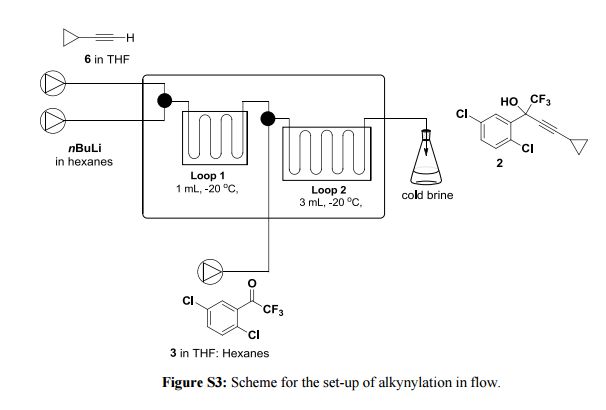
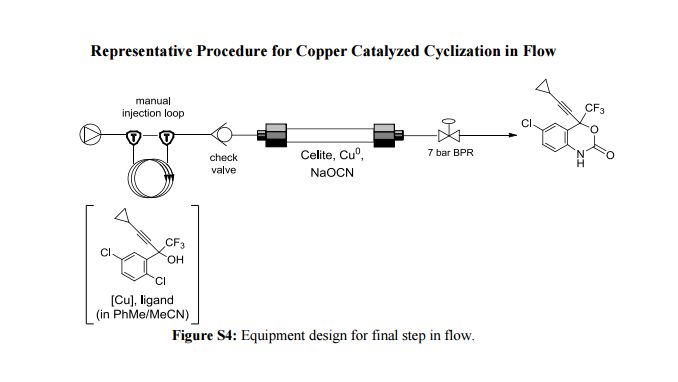
A new enabling technology for the pumping of organometallic reagents such as n-butyllithium, Grignard reagents, and DIBAL-H is reported, which utilises a newly developed, chemically resistant, peristaltic pumping system. Several representative examples of its use in common transformations using these reagents, including metal–halogen exchange, addition, addition–elimination, conjugate addition, and partial reduction, are reported along with examples of telescoping of the anionic reaction products. This platform allows for truly continuous pumping of these highly reactive substances (and examples are demonstrated over periods of several hours) to generate multigram quantities of products. This work culminates in an approach to the telescoped synthesis of (E/Z)-tamoxifen using continuous-flow organometallic reagent-mediated transformations.

A versatile multi-step continuous flow synthesis for the preparation of substituted pyrazoles is presented.
The automated synthesis utilises a metal-free ascorbic acid mediated reduction of diazonium salts prepared from aniline starting materials followed by hydrolysis of the intermediate hydazide and cyclo-condensation with various 1,3-dicarbonyl equivalents to afford good yields of isolated functionalised pyrazole products.
The synthesis of the COX-2 selective NSAID was demonstrated using this approach.

Continuous flow methodologyhas been used to enhance several steps in the synthesis of a precursor to Sacubitril.
In particular, a key carboethoxyallylation benefited from a reducedprocessing time and improved reproducibility, the latter attributable toavoiding the use of a slurry as in the batch procedure. Moreover, in batchexothermic formation of the organozinc species resulted in the formation ofside products, whereas this could be avoided in flow because heat dissipationfrom a narrow packed column of zinc was more efficient
Cyclohexaneperoxycarboxylic acid (6, has been developed as a safe, inexpensive oxidant, with demonstrated utility in a Baeyer−Villiger rearrangement.34 Solutions of cyclohexanecarboxylic acid in hexane and 50% aqueous H2O2 were continuously added to 45% H2SO4 at 50−70 °C and slightly reduced pressure. The byproduct H2O was removed azeotropically, and the residence time in the reactor was 3 h. Processing was adjusted to maintain a concentration of 6 at 17−19%, below the detonable level, and the product was kept as a stable solution in hexane. These operations enhanced the safety margin in preparing 6.

Scheme . Generation of cyclohexaneperoxycarboxylic acid

The conversion of a batch process to continuous (flow) operation has been investigated. The manufacture of 4,d-erythronolactone at kilogram scale was used as an example. Fully continuousprocessing was found to be impracticable with the available plant because of the difficulty in carrying out a multiphase isolation step continuously, so hybrid batch–continuous options were explored. It was found that very little additional laboratory or process safety work other than that required for the batch process was required to develop the hybrid process. A hybrid process was chosen because of the difficulty caused by the precipitation of solid byproduct during the isolation stage. While the project was a technical success, the performance benefits of the hybrid process over the batch were not seen as commercially significant for this system.




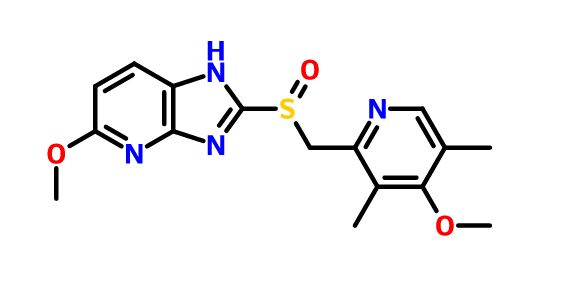
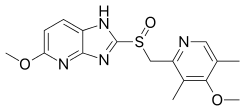
Tenatoprazole
テナトプラゾール
泰妥拉唑
| Tenatoprazole; 113712-98-4; Ulsacare; Protop; TU 199; TU-199; | |
| Molecular Formula: | C16H18N4O3S |
|---|---|
| Molecular Weight: | 346.40412 g/mol |
5-methoxy-2-[(4-methoxy-3,5-dimethylpyridin-2-yl)methylsulfinyl]-1H-imidazo[4,5-b]pyridine
2-[2-(3,5-Dimethyl)pyridylmethylsulfinyl]-5-methoxyimidazo[4,5-b]pyridine
Phase I
PHASE 1 FOR ………..A proton pump inhibitor potentially for the treatment of gastroesophageal reflux disease.
![]()
Research Code TU-199
CAS No. 113712-98-4
Mitsubishi Tanabe Pharma and was licensed to Negma Laboratories

Tenatoprazole is a proton pump inhibitor drug candidate that was undergoing clinical testing as a potential treatment for refluxoesophagitis and peptic ulcer as far back as 2003.[1] The compound was invented by Mitsubishi Tanabe Pharma and was licensed to Negma Laboratories (part of Wockhardt as of 2007[2]).[3]:22
Mitsubishi reported that tenatoprazole was still in Phase I clinical trials in 2007[4]:27 and again in 2012.[3]:17
Tenatoprazole has an imidazopyridine ring in place of the benzimidazole moiety found in other proton pump inhibitors, and has a half-life about seven times longer than other PPIs.[5]

Tenatoprazole, or (+)-5-methoxy-2-{[(4-methoxy-3,5-dimethyl-2-pyridyl) methyl] sulfinyl} imidazo-[4,5-b] pyridine, is described in Patent No. EP 254,588. It belongs to the group of drugs considered as proton pump inhibitors, which inhibit the secretion of gastric acid and are useful in the treatment of gastric and duodenal ulcers. It can also be used to treat gastro-oesophageal reflux, digestive bleeding and dyspepsia, because of its relatively long elimination half-life, as described in the application for French patent No. FR 02. 13113.
The first known derivative of this series of proton pump inhibitors was omeprazole, described in Patent No. EP 001,529, which is endowed with properties which inhibit the secretion of gastric acid and is widely employed as an anti-ulcerative in human therapeutics.
In addition to omeprazole, other proton pump inhibitors are well known, and particular mention can be made of rabeprazole, pantoprazole and lansoprazole, which all exhibit structural analogy and lansoprazole, which all exhibit structural analogy and belong to the group of pyridinyl methyl sulfinyl benzimidazoles. These compounds are sulfoxides presenting with asymmetry at the level of the sulphur atom, and therefore generally take the form of a racemic mixture of two enantiomers.
Like omeprazole and other sulfoxide with an analogue structure, tenatoprazole has an asymmetric structure and may therefore be present in the form of a racemic mixture or of its enantiomers. Thus it may exist in the form of its two enantiomers with R and S configurations, or (+) or (−), respectively.
Recent studies have shown that, unlike all the other proton pump inhibitors such as, for example, omeprazole or lansoprazole, and unexpectedly, tenatoprazole is endowed with a markedly prolonged duration of action, resulting from a plasma half-life which is about seven times longer. Thus the clinical data collected have shown that tenatoprazole enables a degree of symptom relief and healing of gastric lesions which is superior to that achieved by other drugs belonging to the same therapeutic category of proton pump inhibitors, which thus allows its effective use in the treatment of atypical and oesophageal symptoms of gastro-oesophageal reflux, digestive bleeding and dyspepsia, as indicated above.
Studies performed by the application have made it possible to show that the two enantiomers contribute differently to the properties of tenatoprazole, and that the two enantiomers, (+) and (−) exhibit significantly different pharmacokinetic properties. Thus it is possible to prepare medicinal products with specific activity by isolating the enantiomers, and these enantiomers themselves exhibit a different pharmacokinetic profile from that of the known racemic mixture. It then becomes possible to use each of these enantiomers more effectively in precise indications for the treatment of perfectly identified pathologies.
Anti-ulcer drug
tenatoprazole (tenatoprazole) is a new proton pump inhibitor, by the Japanese company Tokyo Tanabe, Japan’s Mitsubishi Corporation and Japan’s Hokuriku pharmaceutical companies jointly developed, has passed Phase II clinical trials. It acts on gastric parietal cells, reducing treatment of gastric ulcer, duodenal ulcer, reflux wall cell H + / K + -ATP activity, inhibition of gastric acid secretion, and H. pylori antibacterial activity, mainly for esophagitis and Zhuo – Ellison syndrome and gastric acid secretion disorders related diseases. Compared with the same types of drugs, Tenatoprazole suppress H + / K + -ATP enzyme activity is stronger, more stable, its efficacy than similar products currently widely used in clinical omeprazole strong 7 times. It has not been in the domestic market, nor ratified the production, with broad market prospects and development potential.
Proton pump inhibitors (proton pump inhibitors) for the treatment of acid-related diseases, the past ten years a wide range of clinical applications, better effect of the drug. It can quickly pass through the stomach wall membrane, gathered in a strongly acidic secretory tubules, and H + / K + -ATP enzyme (proton pump) thiol groups covalently bonded to form a disulfide bond, proton pump inactivation, inhibition of the enzyme H + / K + transport, so as to achieve the effect of acid suppression. Proton pump inhibitors and conventional clinical application of gastric acid suppression drugs H2 receptor antagonists compared with different sites of action and have different characteristics, namely acid-suppressing effect at night is good, rapid onset of acid inhibition strong and long time, easy to take these drugs can quickly and efficiently inhibit gastric acid secretion and clearance of Helicobacter pylori, it is widely used gastric ulcer, duodenal ulcer, reflux esophagitis and Zhuo – Ellison syndrome and other diseases treatment. Currently, proton pump inhibitors has been listed on the main omeprazole, lansoprazole, pantoprazole, rabeprazole and esomeprazole.Physical and
chemical properties ofwhite or white crystalline powder, melting point 174 ~ 175 ℃. Soluble in chloroform, insoluble in alcohol and water.
This product and other proton pump inhibitors as compared to chemically stable. China had 34 omeprazole preparations from Portugal, Brazil, India, China and other 13 countries, the stability of the measurements were made. The results showed that only six products (18%) during the trial showing good physical and chemical stability of. 27 products (79%) less (including Chinese product), the active ingredient a significant chemical decomposition, color and physical properties such as dissolution, are also a corresponding change. The results of a stability test designed to compare the various proton pump inhibitors show investigated eight days at 60 ℃, relative humidity of 75%, after omeprazole decomposition only 3% of the active ingredient, the tenatoprazole 77% of the data, said Alpha pantoprazole stability far superior to omeprazole, is already developed similar products in the most promising products.
Matsuishi, N.; Takeda, H.; Iizumi, K.; Murakami, K.; Hisamitsu, A. US Patent 4,808,596, 1989
Synthesis of Tenatoprazole 1 commences with the coupling of 2-mercapto-5-methoxyimidazo[4,5-b]pyridine 2 with 2-chloromethyl-4-methoxy-3,5-dimethyl pyridine hydrochloride 3 in the presence of potassium hydroxide affords 4 with 73% yield in ethanol and chloroform. The oxidation of the penultimate sulfide intermediate4 with m-CPBA in chloroform (100 vol) afforded 1
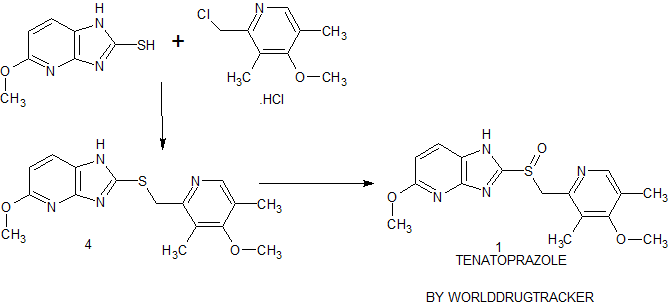
Syn 2
synthesis of 1 begins with the solvent-free condensation of 2-mercapto-5-methoxyimidazo[4,5-b]pyridine 2 with 2-chloromethyl-4-methoxy-3,5-dimethyl pyridine hydrochloride 3 to deliver the sulfide intermediate4 with 98% yield.
The final step of the synthesis is the oxidation of the sulfide intermediate with m-CPBA to form tenatoprazole 1. The sulfide intermediate 4 on treatment with 0.9 equiv of m-chloroperbenzoic acid (m-CPBA) at −10 to −15 °C afforded the crude tenatoprazole which was isolated as its sodium salt. The sodium salt of tenatoprazole 5 was purified by recrystallsation using dimethyl formamide and ethyl acetate (2:1 ratio) to yield the pure crystalline tenatoprazole sodium 5. Treatment of tenatoprazole sodium 5 with dil. HCl in the presence of acetone and water afforded the pure tenatoprazole 1
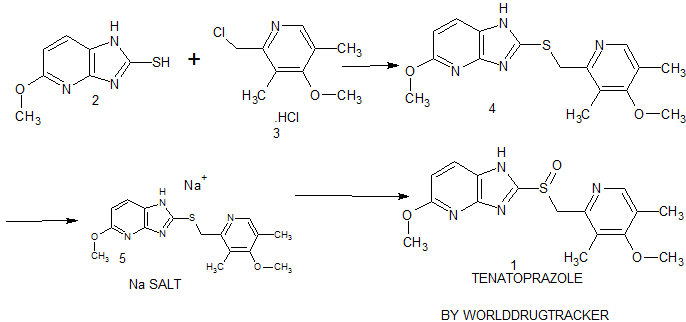
PATENT
CN 1861600
CN 1982311
WO 2009116072
CN 101429192
WO 2010043601
IN 2010CH00462
IN 251400
CN 102304127
WO 2012004802
CN 102703922
IN 2009DE01392
WO 2014111957
IN 2013MU00181
IN 2014CH01419
PAPER
Dai, Liyan; Synthetic Communications 2008, V38(4), P576-582
Advanced Materials Research (Durnten-Zurich, Switzerland) (2011), 233-235(Pt. 1, Fundamental of Chemical Engineering), 160-164.
Organic Process Research & Development (2013), 17(10), 1293-1299
Enantiomeric separation of proton pump inhibitors on new generation chiral columns using LC and supercritical fluid chromatography
Journal of Separation Science (2013), 36, (18), 3004-3010………http://onlinelibrary.wiley.com/doi/10.1002/jssc.201300419/abstract
PATENT
CN 102304127
https://www.google.com/patents/CN102304127A?cl=en
Tenatoprazole is a new type of gastric H + / K + -ATP enzyme inhibitors (proton pump inhibitor PPI), the chemical name 5-methoxy-2- (4-methoxy-3, 5-dimethyl-2-methylsulfinyl) imidazole and W, 5-b] pyridine, useful in the treatment of gastric ulcer, duodenal ulcer, reflux esophagitis and Zhuo – Ai syndrome and gastric acid secretion disorders related diseases. The drug was developed by Japan’s Tokyo Tanabe, Japan’s Mitsubishi Corporation and Japan’s Hokuriku pharmaceutical companies. Compared with other varieties of the same type, which inhibit H + / K + -ATP enzyme activity is stronger, ulcers of various tests are effective, and significantly improve the stability compared with other proton pump inhibitors.
US patent US4808596 “hidazo [4,5_b] pyridine compounds and pharmaceutical compositions containing same)) synthesis process disclosed Tenatoprazole the below formula:

By The route of 2-chloro-3,5-dimethyl-4-methoxypyridine hydrochloride with 2-mercapto-5-methoxy-imidazole, 5-b] pyridine under basic conditions condensation of Intermediate 2- [2- (3,5_-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazo W, 5-b] pyridine, and then oxidizing the Thai duly omeprazole. This route for the synthesis of pull azole classic line, many pull azoles such as omeprazole can be synthesized by a similar route, this route mild condition, simple operation. But the route condensation, oxidation treatment after use of large amounts of toxic solvent chloroform, is not conducive to industrial scale; lower oxidation yields, the resulting Tenatoprazole containing unreacted starting materials 2- [2_ (3,5 – dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazo W, 5-b] pyridine, further comprising a sulfone by-product, N- oxide, N- oxide sulfone, These by-products may interfere with purification of tenatoprazole.
Japanese Patent invention Wo 丨 J JP05222038 “5_methoxy-2- [[(4_methoxy-3, 5-dimethyl-2-pyridyl) methyl] thio] imidazo [4,5 ~ b] pyridine and intermediates)) male
Synthesis open Tenatoprazole the below formula:

4-chloro-2-chloromethyl-3,5-dimethylpyridine -N- oxide 2_ mercapto _5_ methoxy-imidazo – [4, 5-b] pyridine in alkaline under condensation of Intermediate 5-Methoxy-2- (4-chloro-3,5-dimethyl-2-methylthio Bi) imidazo W, 5-b] pyridine-oxide -N- ( yield 82%), then refluxed in a solution of sodium methoxide in methanol to give 5-methoxy-2- (4-oxo-3,5-dimethyl-2-methyl sulfide) imidazo W , 5-b] pyridine -N- oxide (income ¥ 71%), and then at room temperature in methylene chloride, phosphorus trichloride treated with deoxy (yield 95%), and finally oxidation in Tenatoprazole (income Rate not reported). The novel synthetic route, mild reaction conditions, simple operation, the yield of each step is higher, but the route is too long resulting in a total yield is not high, prolonged and rising production costs.
Reaction route is as follows:

Example 1:
] a) 2- [2- (3,5-dimethyl) -4-methoxy-picolyl thioether _5_ methoxy] imidazo [4,5_b] pyridine:
To a reaction flask was added 2-mercapto-5-methoxy-imidazole, 5-b] pyridine 18. lg, 12g of sodium hydroxide and water 144. 8g, stirred and dissolved at 25 ° C, was added dropwise within Ih 20g of the 2-chloromethyl-dimethyl-4-methoxy _3,5- pyridine hydrochloride and 60g of water were mixed solution dropwise at 25 ° C the reaction 2h, the reaction is completed, filtered, washed with water 144. 8g, 36. 2mL ethanol and washed to obtain a wet powder; wet powder was dried at 50 ° C in vacuo to constant weight to give 2- [2_ (3,5-dimethyl) -4-methoxy-pyridylmethyl sulfide -5 – methoxy] imidazo [4,5-b] pyridine 32. Og;
2) Preparation of tenatoprazole lithium salt: To a reaction flask was added 2- [2- (3,5_-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazo W, 5-b] pyridine 30g, dichloromethane 300g, methanol 15g, and dissolved with stirring; cooled to -10 ° C, was added dropwise the 15g and 485g m-chloroperbenzoic acid in methylene chloride mixed solution, dropwise addition the reaction temperature was controlled at -10 ° C, the dropping time of the pool; the dropwise addition, the temperature control at -10 ° C, the reaction 30min; completion of the reaction, at 10 ° C by the dropwise addition of lithium hydroxide and 135g water 15g mixed solution, drip complete, insulation stirred Ih; filtered cake was washed with acetone 60mL, get wet powder; wet powder was dried at 35 ° C under vacuum to constant weight to give Tenatoprazole lithium salt ^ g;
3) Preparation Tenatoprazole: To a reaction flask 加入泰 pantoprazole lithium salt 25g, acetone 63mL, water IOOmL, cooling M0 ° C, dropping lmol within lh / L hydrochloric pH7 0, drops. Albert, stirring 30min; the filter cake washed with water 50mL, washed with acetone and 50mL, wet powder was dried at 35 ° C under vacuum to constant weight to give Tenatoprazole 19. Sg.
Example 2:
a) 2- [2- (3,5-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazo W, 5_b] pyridine (4) Preparation: To the reaction flask was added 2-mercapto-5-methoxy-imidazo 44,5-b] pyridine 18. lg, 11. 2g of potassium hydroxide and water 217mL, stirred and dissolved at! 35 ° C, was added dropwise within 2h by the 33. 3g of 2-chloro-3,5-dimethyl-4-methoxypyridine hydrochloride and 133. 2mL water mixed solution, dropwise at 35 ° C the reaction 4h, the reaction is completed, filtration, water 217mL, 72. 4mL ethanol and washed to obtain a wet powder; wet powder was dried at 60 ° C in vacuo to constant weight to give 2- [2- (3,5-dimethyl) -4-methoxy-pyridylmethyl sulfide -5-methoxy-yl] imidazo W, 5-b] pyridine 33. Ig;
2) Preparation of tenatoprazole lithium salt: To a reaction flask was added 2- [2- (3,5_-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazole and W, 5-b] pyridine 30g, dichloromethane 400mL, methanol 50mL, stirring to dissolve; cooled to _15 ° C, was added drop by the m-chloroperoxybenzoic acid 16g of mixed solution of dichloromethane and 400mL , the process reactor temperature control was added dropwise at -20 ° C, the dropping time 2. 5h; the dropwise addition, the temperature control _15 ° C, the reaction 35min; completion of the reaction, at 15 ° C by the dropwise addition of 20g of hydrogen Lithium oxide and 200mL water mixed solution, drip completed, insulation mixing 1. 5h; filtration, the filter cake washed with acetone 90mL, get wet powder; wet powder was dried at 40 ° C under vacuum to constant weight to give Tenatoprazole lithium salt 28. 6g;
3) Preparation Tenatoprazole: To a reaction flask 加入泰 pantoprazole lithium salt 25g, ethanol 75mL, water 150mL, cooled to 10 ° C, dropping 6mol / L hydrochloric pH8 0 within 2h,. drops Albert, stirring 40min; the filter cake washed with water 100mL, washed with acetone IOOmL, wet powder was dried at 40 ° C under vacuum to constant weight, yield powder was Tenatoprazole 19. 5g.
Example 3:
a) 2- [2- (3,5-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazo W, 5_b] pyridine (4) Preparation: To the reaction flask was added 2-mercapto-5-methoxy-imidazo 44,5-b] pyridine 18. lg, 8.4g of lithium hydroxide and water 180ml, stirred and dissolved at 30 ° C, was added dropwise within 1. 5h by the Guang .6g 2-chloro-3,5-dimethyl-4-methoxy-pyridine hydrochloride and 90mL water mixed solution, drop end at 30 ° C reaction 3h, the reaction is complete, filtration, water 217mL , washed with 85mL ethanol to obtain a wet powder; wet powder was dried at 55 ° C in vacuo to constant weight to give 2- [2- (3,5-dimethyl) -4-methoxy-5-pyridylmethyl sulfide oxy] imidazo [4,5-b] pyridine 32. 4g;
2) Preparation of tenatoprazole lithium salt: To a reaction flask was added 2- [2- (3,5_-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazole and W, 5-b] pyridine 30g, dichloromethane 600mL, methanol 60mL, stirring to dissolve; cooled to -20 ° C, was added drop by the m-chloroperoxybenzoic acid 18g of mixed solution of dichloromethane and 600mL , dropwise addition the reaction temperature is controlled at _20 ° C, the dropping time of the pool; the dropwise addition, the temperature control at _20 ° C, the reaction 40min; completion of the reaction, at 20 ° C by the dropwise addition of lithium hydroxide and 300mL 30g water mixed solution, drip complete insulation mixing tank; filter, the filter cake washed with acetone and 120mL, get wet powder; wet powder was dried at 40 ° C under vacuum to constant weight to give Tenatoprazole lithium salt 28. 7g;
3) Preparation Tenatoprazole: To a reaction flask 加入泰 pantoprazole lithium salt 25g, methanol 75mL, water 120mL, cooled to 5 ° C, dropping dilute hydrochloric acid within 1 5h tune pH7 5,.. drops Albert, stirring 35min; the filter cake washed with water 75mL, 75mL acetone washed, wet powder was dried at 40 ° C under vacuum to constant weight, yield powder was Tenatoprazole 19. 6g.
Example 4:
a) 2- [2- (3,5-dimethyl) -4-methoxy-picolyl thioether _5_ methoxy] imidazo [4,5_b] pyridine ⑷ Preparation of: To a solution The reaction flask was added 2-mercapto-5-methoxy imidazole -½, 5-b] pyridine 18. lg, IOg sodium hydroxide and water 150ml, stirred and dissolved at 30 ° C, the 1. 5h dropwise added from 21 . 5g of 2-chloro-3,5-dimethyl-4-methoxypyridine hydrochloride and 90mL water mixed solution, dropwise at 30 ° C the reaction 3h, completion of the reaction, was filtered, washed with water 217mL, The wet powder was washed with ethanol to give 85mL; wet powder was dried at 55 ° C in vacuo to constant weight to give 2- [2- (3,5_-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide ] imidazo [4,5-b] pyridine 32. 3g;
2) Preparation of tenatoprazole lithium salt: To a reaction flask was added 2- [2- (3,5_-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazole and W, 5-b] pyridine 30g, dichloromethane 500mL, methanol 60mL, stirring to dissolve; cooled to -20 ° C, was added drop by the m-chloroperoxybenzoic acid 18g of mixed solution of dichloromethane and 500mL , the process reactor temperature control was added dropwise at -20 ° C, the dropping time pool; the dropwise addition, the temperature control in -20 ° C, the reaction 40min; completion of the reaction, at 20 ° C by the dropwise addition of lithium hydroxide 30g and 300mL water mixed solution, drip complete insulation mixing tank; filter, the filter cake washed with acetone and 120mL, get wet powder; wet powder was dried at 40 ° C under vacuum to constant weight to give Tenatoprazole lithium salt 28. 6g;
3) Preparation Tenatoprazole: To a reaction flask 加入泰 pantoprazole lithium salt 25g, isopropanol 75mL, water 120mL, cooled to 5 ° C, dropping 3mol / L hydrochloric within 1 5h. . pH7 5, drops Albert, stirring 35min; the filter cake washed with water 75mL, 75mL acetone washed, wet powder was dried at 40 ° C under vacuum to constant weight, yield powder was Tenatoprazole 19. 7g.
PAPER
http://pubs.acs.org/doi/full/10.1021/op800173u

An efficient, cost-effective and multikilogram-scale process for the synthesis of tenatoprazole 1, an antiulcerative drug, is described. The key steps in this synthesis involve the coupling of 2-mercapto-5-methoxyimidazo[4,5-b]pyridine 2 with 2-chloromethyl-4-methoxy-3,5-dimethyl pyridine hydrochloride 3 to yield 4 and its subsequent oxidation with m-CPBA to produce sulfoxide 1. The process has been scaled up for the multikilogram-scale of compound 1 with an overall yield of 72%. The new process requires no purification process and affords the target compound 1 with 99.8% purity by HPLC.
2-[2-(3,5-dimethyl)pyridylmethylsulfinyl]-5-methoxyimidazo[4,5-b]pyridine (1, 15.5 kg, 74%). Purity by HPLC 99.8%; 1H NMR (200 MHz, DMSO) δ 2.2 (s, 6H), 3.8 (s, 6H), 4.8 (s, 2H), 6.6 (d, 1H), 7.8 (d, 1H), 8.2 (s, 1H), 13.0 (s, 1H).
http://www.google.co.in/patents/US7507746
the (+) enantiomer of tenatoplazole can be obtained by using chloroform, an industrially acceptable solvent, in accordance with the method proposed by Umemura et al. (J. Org. Chem. 1993, 58, 4592) as follows:

Example 1 (−)-5-methoxy-2-{(4-methoxy-3,5-dimethyl-2-pyridyl)methyl]sulfinyl}-1H-imidazo[4,5-b]pyridineThe conditions for preparative chromatography, shown as an example, are as follows:
Column: 265×110 mm ChiralPak®
Chiral Stationary Phase selector of the Amylose tris type [(S)-a methylbenzylcarbamate]
Flow rate: 570 ml/min
Detection: UV 240 nm
Temperature: Ambient temperature
These conditions are implemented on a liquid preparative chromatography apparatus.
Introduce approximately 2 g of the racemic mixture if tenatoprazole exhibiting purity higher than 99.5%. The (−) enantiomer is identified by measuring the angle of optical rotation, which must be laevogyre. This measurement can be performed directly on the column, the product being dissolved in the solvent (acetonitrile).
Example 2 (+)-5-methoxy-2-{(4-methoxy-3, 5-dimethyl-2-pyridyl)methyl]sulfinyl}-1H-imidazo[4,5-b]pyridine(R)-(+)-binaphthol 85 g (0.311 mol, 0.2 equivalence), ortho titanic acid isopropyl 42 g (0.148 mol, 0.1 equivalence), water 55 g (3.06 mol) and chloroform 7.5 L were stirred for 1 hour at room temperature. To the resultant, 5-methoxy-2-{(4-methoxy-3,5-dimethyl-2-pyridyl)methyl]thio}imidazo[4,5-b]pyridine (MPI), 0.5 kg, was added and stirred for 0.5 hours at room temperature. The thus-prepared mixture was cooled to 5° C. and then 70% aqueous solution of tert-butylhydroperoxide, 0.4 L (approx. 3.0 mol, 2.0 equivalence) was added and stirred for 72 hours at the same temperature as above. After the reaction endpoint was confirmed by HPLC, an aqueous solution of sodium hydroxide was added thereto to separate the aqueous layer, thus removing foreign matter. Then, the resultant was concentrated. Ethyl acetate was added to concentrated residues, which were then heated and suspended. The thus-prepared crude crystalline substances were dissolved in water and neutralized to pH 6.8 with a diluted sulfuric acid solution which was chilled with ice. Deposited crystals were filtered, dried and recrystallized by addition of ethanol to obtain (+)-5-methoxy-2-{(4-methoxy-3,5-dimethyl-2-pyridyl)methyl]sulfinyl}-1H-imidazo[4,5-b]pyridine {(+)-TU-199}
Yield: 77%
Optical purity: 96.6% ee
Chemical purity: 94.5%
Melting point: 135° C.
Optical rotation: +184° (conditions: C=1.0, N,N-dimethylformaldehyde solution)
Ultraviolet absorption spectrum: (10 μg/mL)λmax (nm): 316, 273, 206
When measurements were carried out, for a solubility of (+), (−) forms and a racemic form (±) of tenatoprazole in relation to water, it was found that the (+) form dissolved almost 3 times greater than the racemic body and (−) form dissolved over 2 times greater than the racemic form, exhibiting favorable physical properties in preparing drugs (refer to Table 2 below).
| TABLE 2 | |||
| (+) form | (−) form | (±)racemic form | |
| Solubility (water) μg/mL | 93.0 | 74.4 | 34.6 |

Tenatoprazole is a pyridinylmethylsulfinyl imidazopyridine compound, which is a weak base. This compound has three pKas. One is the pyridine pKa of pyridinylmethyl moiety and the others are the imidazole pKa and the pyridine pKa of the imidazopyridine moiety. The pyridine pKa1 enables tenatoprazole accumulation in the acidic canaliculus of the parietal cell. Protonation of the imidazopyridine ring enhances electron deficiency at the C-2 position, allowing intramolecular rearrangement to the active form. The active form is the sulfenic acid and/or cyclic sulfonamide, and reacts with luminal cysteine thiols of the enzyme to inhibit the enzyme activity

| US4808596 * | 24 Jul 1987 | 28 Feb 1989 | Tokyo Tanabe Company, Ltd. | Imidazo[4,5-b]pyridine compounds and pharmaceutical compositions containing same |
| US5753265 * | 7 Jun 1995 | 19 May 1998 | Astra Aktiebolag | Multiple unit pharmaceutical preparation |
| US5798120 * | 6 Oct 1994 | 25 Aug 1998 | Tokyo Tanabe Company Limited | Enteric granule-containing tablets |
| EP0124495A2 | 28 Feb 1984 | 7 Nov 1984 | Aktiebolaget Hässle | Omeprazole salts |
| EP0254588A1 | 24 Jul 1987 | 27 Jan 1988 | Tokyo Tanabe Company Limited | Imidazo[4,5-b] pyridine compounds, process for preparing same and pharmaceutical compositions containing same |
| Reference | ||
|---|---|---|
| 1 | * | Andersson et al., Pharmacology & Therapeutics, 2005, vol. 108, pp. 294-307. |
| 2 | * | Anon et al., Drugs in R&D, 2002, vol. 3, pp. 276-277. |
| 3 | Kakinoki et al., Methods and Findings in Experimental and Clinical Pharmacology, 21(3): 179-187 (1999). | |
| 4 | Komatsu et al., J. Org. Chem., 58(17): 4529-4533 (1993). | |
| 5 | Uchiyama et al., Journal of Pharmacy and Pharmacology, 51(4): 457-464 (1999). | |
| 6 | Uchiyama et al., Methods and Findings in Experimental and Clinical Pharmacology, 21(2): 115-122 (1999). | |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US20120220623 * | 30 Aug 2012 | Mitsubishi Tanabe Pharma Corporation | The enantiomer of tenatoprazole and the use thereof in therapy |
| CN1453278A * | May 10, 2002 | Nov 5, 2003 | 中国人民解放军军事医学科学院放射医学研究所 | Omprazole compound and its prepn and application |
| CN1861600A * | Jun 14, 2006 | Nov 15, 2006 | 浙江大学 | Preparation process of taytrolazole |
| Reference | ||
|---|---|---|
| 1 | * | 《Organic Process Research & Development》 20081112 Somaiah Sripathi et al. An Improved Synthesis of Antiulcerative Drug:Tenatoprazole 第804-806页 1-6 第13卷, |
| 2 | * | 《Synthetic Communication》 20080101 Liyan Dai et al. Improved Synthetic Approach to Tenatoprazole 第576-582页 1-6 第38卷, |
| 3 | * | 《中国药物化学杂志》 20061231 赵冬梅等 抗溃疡药泰妥拉唑的合成 第360-362页 1-6 第16卷, 第6期 |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
5-methoxy-2-[(4-methoxy-3,5-dimethylpyridin-2-yl)methylsulfinyl]-1H-imidazo[4,5-b]pyridine
|
|
| Clinical data | |
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Metabolism | Hepatic (CYP2C19-mediated) |
| Biological half-life | 4.8 to 7.7 hours |
| Identifiers | |
| CAS Number | 113712-98-4 |
| ATC code | none |
| PubChem | CID 636411 |
| ChemSpider | 552196 |
| UNII | RE0689TX2K |
| Chemical data | |
| Formula | C16H18N4O3S |
| Molar mass | 346.405 g/mol |
| Chirality | Racemic mixture |
テナトプラゾール
Tenatoprazole

C16H18N4O3S : 346.4
[113712-98-4]
/////////////Tenatoprazole, 113712-98-4, TU-199, proton pump inhibitor, treatment of gastroesophageal reflux disease, Mitsubishi Tanabe Pharma, Negma Laboratories, PHASE 1, テナトプラゾール
CC1=CN=C(C(=C1OC)C)CS(=O)C2=NC3=C(N2)C=CC(=N3)OC
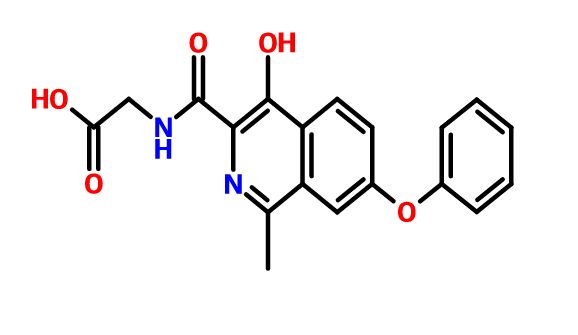

ROXADUSTAT
ASP1517; ASP 1517; ASP-1517; FG-4592; FG 4592; FG4592; Roxadustat.
CAS 808118-40-3
Chemical Formula: C19H16N2O5
Exact Mass: 352.10592
THERAPEUTIC CLAIM, Treatment of anemia
CHEMICAL NAMES
(4-hydroxy-1-methyl-7-phenoxyisoquinoline-3-carbonyl)glycine
1. Glycine, N-[(4-hydroxy-1-methyl-7-phenoxy-3-isoquinolinyl)carbonyl]-
2. N-[(4-hydroxy-1-methyl-7-phenoxyisoquinolin-3-yl)carbonyl]glycine
MF C19H16N2O5
MW 352.3
SPONSOR FibroGen
CODE FG-4592; ASP1517
CAS 808118-40-3
WHO NUMBER 9717
Roxadustat, also known as ASP1517 and FG-4592, is an HIF α prolyl hydroxylase inhibitor in a cell-free assay. It stabilizes HIF-2 and induces EPO production and stimulates erythropoiesis. Roxadustat transiently and moderately increased endogenous erythropoietin and reduced hepcidin
|
FG-4592 (also known as ASP1517), 2-(4-hydroxy-1-methyl-7-phenoxyisoquinoline-3-carboxamido)acetic acid,
is a potent small molecule inhibitor of hypoxia-inducible factor prolyl hydroxylase (HIF-PH),
an enzyme up-regulating the expression of endogenous human erythropoietin (Epo). It is currently being investigated as an oral treatment for anemia associated with chronic kidney disease (CKD). Unlike other anemia treating agents, erythropoiesis-stimulating agents (ESAs),
FG-4592 inhibits HIF, through a distinctive mechanism, by stabilization of HIF. According to previous studies,
FG-4592 is capable of correcting and maintaining hemoglobin levels in CKD patients not
receiving dialysis and in patients of end-stage renal disease who receives dialysis but do not need intravenous iron supplement.
Reference
1. Luis Borges. Different modalities of erythropoiesis stimulating agents.
Port J Nephrol Hypert 2010; 24(2): 137-145 2. “FibroGen and Astellas announce initiation of phase 3 trial of FG-4592/ASP1517 for treatment of anemia of chronic kidney disease” Fibrogen Press Release. Dec 11 2012
3. “FibroGen announces initiation of phase 2b studies of FG-4592, an oral HIF prolyl hydroxylase inhibitor, for treatment of anemia”
|
![]()

Roxadustat (FG-4592) is a novel new-generation oral hypoxia-induciblefactor (HIF) prolyl hydroxylase inhibitor (PHI) for the treatment of ane-mia in patients with chronic kidney disease (CKD). HIF is a cytosolic tran-scription factor that induces the natural physiological response to lowoxygen conditions, by stimulating erythropoiesis and other protectivepathways. Roxadustat has been shown to stabilize HIF and induce ery-thropoiesis. Consequently, it corrects anemia and maintains hemoglo-bin levels without the need for intravenous iron supplementation in CKDpatients not yet receiving dialysis and in end-stage renal disease pa-tients receiving dialysis. There are many concerns about the use of ery-thropoiesis-stimulating agents (ESA) to treat anemia as they causesupra-physiologic circulating erythropoietin (EPO) levels and are asso-ciated with adverse cardiovascular effects and mortality. Available clin-ical data show that modest and transient increases of endogenous EPOinduced by HIF-PHI (10- to 40-fold lower than ESA levels) are sufficientto mediate erythropoiesis in CKD patients. Evidence suggests that rox-adustat is well tolerated and, to date, no increased risk of cardiovascu-lar events has been found. This suggests that roxadustat provides adistinct pharmacological and clinical profile that may provide a saferand more convenient treatment of CKD anemia
FG-4592 is a new-generation hypoxia-inducible factor prolyl hydroxylase inhibitor in early clinical trials at FibroGen for the oral treatment of iron deficiency anemia and renal failure anemia. Preclinical studies are ongoing for the treatment of sickle cell anemia.
The investigational therapy is designed to restore balance to the body’s natural process of erythropoiesis through mechanisms including: natural EPO production, suppression of the effects of inflammation, downregulation of the iron sequestration hormone hepcidin, and an upregulation of other iron genes, ensuring efficient mobilization and utilization of the body’s own iron stores. In April 2006, FG-4592 was licensed to Astellas Pharma by originator FibroGen in Asia, Europe and South Africa for the treatment of anemia. FibroGen retains rights in the rest of the world. In 2007, the FDA put the trial on clinical hold due to one case of death by fulminant hepatitis during a phase II clinical trial for patients with anemia associated with chronic kidney disease and not requiring dialysis. However, in 2008, the FDA informed the company that clinical trials could be resumed. Phase II/III clinical trials for this indication resumed in 2012. In 2013, the compound was licensed to AstraZeneca by FibroGen for development and marketing in US, CN and all major markets excluding JP, Europe, the Commonwealth of Independent States, the Middle East and South Africa, for the treatment of anemia associated with chronic kidney disease (CKD) and end-stage renal disease (ESRD).
PATENTS
WO 2004108681
WO 2008042800
WO 2009058403
WO 2009075822
WO 2009075824
WO 2012037212
WO 2013013609
WO 2013070908
CN 104892509
MACHINE TRANSLATED
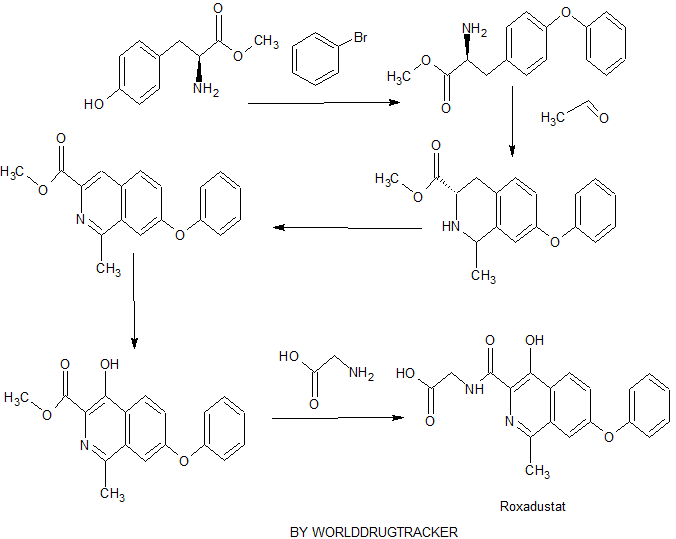
Connaught orlistat (Roxadustat) by the US company Phibro root (FibroGen) R & D, Astellas AstraZeneca and licensed by a hypoxia-inducible factor (HIF) prolyl hydroxylase small molecule inhibitors, codenamed FG-4592.As a first new oral drug, FG-4592 is currently in Phase III clinical testing stage, for the treatment of chronic kidney disease and end-stage renal disease related anemia. Because the drug does not have a standard Chinese translation, so the applicant where it is transliterated as “Connaught Secretary him.”
Connaught orlistat (Roxadustat, I) the chemical name: N_ [(4- hydroxy-1-methyl-7-phenoxy-3-isoquinolinyl) carbonyl] glycine, its structural formula is:

The original research company’s international patent W02004108681 Division provides a promise he was prepared from the intermediate and intermediate Connaught Secretary for his synthetic route:

Zhejiang Beida company’s international patent W02013013609 preparation and acylation of core intermediate was further optimized synthesis route is:

n PhO. eight XOOH
original research company’s international patent W02014014834 and W02014014835 also provides another synthetic route he Connaught Secretary prepared:

Analysis of the above synthetic route, although he continued to Connaught Division to improve and optimize the synthesis, but its essence rings manner that different form quinoline ring is basically the same mother. Especially methyl isoquinoline replaced either by way of introducing the Suzuki reaction catalyzed by a noble metal element, either through amine reduction achieved. Moreover, the above reaction scheme revelation raw materials are readily available, many times during the reaction need to be protected and then deprotected. Clearly, the preparation process is relatively complicated, high cost, industrial production has brought some difficulties.

Example One:
tyrosine was added to the reaction flask and dried (18. lg, 0.1 mmol) and methanol 250mL, cooling to ice bath 0_5 ° C, was added dropwise over 1 hour a percentage by weight of 98% concentrated sulfuric acid 10g. Drops Albert, heating to reflux. The reaction was stirred for 16-20 hours, TLC the reaction was complete. Concentrated under atmosphere pressure, the residue was added water 100mL, using 10% by weight sodium hydroxide to adjust the pH to 6. 5-7.0, precipitated solid was filtered, washed with methanol and water chloro cake (I: 1) and dried in vacuo tyrosine methyl ester as a white solid (11) 15.38, yield 78.5% out 1–] \ ^ 111/2: 196 [] \ 1 + 1] +!.
Example Two:
[0041] a nitrogen atmosphere and ice bath, was added to the reaction flask tyrosine methyl ester (II) (9. 8g, 50mmol), potassium methoxide (3. 5g, 50mmol) and methanol 50mL, until no gas generation after, was heated to reflux, the reaction was stirred for 2 hours. Concentrated under atmosphere pressure to remove the solvent, the residue was added dimethylsulfoxide 25mL, freshly prepared copper powder (0.2g, 3. Lmmol), was slowly warmed to 150-155 ° C, for about half an hour later, a solution of bromobenzene ( 7. 9g, 50mmol), continue to heat up to 170-175 ° C, the reaction was stirred for 3 hours, TLC detection of the end of the reaction. Was cooled to 60 ° C, and methanol was added to keep micro-boiling, filtered while hot, the filter cake washed three times with hot ethanol, and the combined organic phases, was cooled to square ° C, filtered, and dried in vacuo to give a white solid of 2-amino-3- ( 4-phenoxyphenyl) propanoate (111) 8 11.5, yield 84.9% as 1 -] \ ^ 111/2:! 272 [] \ 1 + 1] +.
Example Three:
in the reaction flask was added 2-amino-3- (4-phenoxyphenyl) propionic acid methyl ester (III) (10. 8g, 40mmol), 40% by weight acetaldehyde (20g, 0. 2mol ) and the percentage by weight of 35% concentrated hydrochloric acid 50mL, refluxed for 1 hour. Continue 40% by weight was added acetaldehyde (10g, 0.1mol), and the percentage by weight of 35% concentrated hydrochloric acid 25mL, and then the reaction was refluxed for 3-5 hours. Was cooled to 4-7 ° C, ethyl acetate was added, and extracted layers were separated. The aqueous layer was adjusted with sodium hydroxide solution to pH 11-12, extracted three times with ethyl acetate. The combined organic phase was dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give a white solid of 1-methyl-3-carboxylate -7- phenoxy-1,2,3,4-tetrahydroisoquinoline (IV) 8 4g, 70.7% yield; Mass spectrum (EI): EI-MS m / z: 298 [M + H] + .
Example Four:
Under ice bath, the reaction flask was added methyl 3-carboxylate I- -7- phenoxy-1,2, 3,4-tetrahydro-isoquinoline (IV) (5. 9g, 20mmol) and dichloromethane 100mL, 0 ° C and under stirring added potassium carbonate (13. 8g, 0. lmol), p-toluenesulfonyl chloride (11. 4g, 60mmol), the addition was completed, the ice bath was removed and stirred at room temperature 3 hour. Water was added 30mL, after stirring standing layer, the organic phase was washed with dilute hydrochloric acid, water and saturated brine, and concentrated, the resulting product was added a 30% by weight sodium hydroxide solution (8. 0g, 60mmol) and dimethyl sulfoxide 60mL, gradually warming to 120-130 ° C, the reaction was stirred for 2-4 hours to complete the reaction by TLC. Cooled to room temperature, water was added lOOmL, extracted three times with ethyl acetate, the combined organic phase was successively washed with water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated, the resulting oil was treated with ethyl acetate and n-hexane (1: 3) recrystallization, vacuum dried to give an off-white solid 1-methyl-3-carboxylate 7-phenoxyheptanoic isoquinoline (V) 5. 25g, yield 89. 6%; EI-MS m / z: 294 [M + H] VH NMR (DMS0-d6) δ 2. 85 (s, 3H), 3 · 97 (s, 3H), 7 · 16-7. 24 (m, 3H), 7 · 49-7. 60 (m, 4Η), 8 · 35 (d, J = 9 · 0,1Η), 8 · 94 (s, 1Η).
Example five:
[0047] added 1-methyl-3-carboxylic acid methyl ester 7-phenoxyheptanoic isoquinoline (V) (2. 93g, IOmmol) and glacial acetic acid 50mL reaction flask, stirring solution of 30% by weight hydrogen peroxide 5mL, warmed to 60-70 ° C, was slowly added dropwise within 10 hours the percentage by weight of a mixture of 30% hydrogen peroxide 2mL and 12mL of glacial acetic acid, a dropping was completed, the reaction was continued for 20-24 hours. Concentrated under reduced pressure, ethanol was added, distillation is continued to be divisible remaining glacial acetic acid. The residue was dissolved with dichloromethane, washed with 5% by weight of sodium bicarbonate, the organic phase was separated, dried over anhydrous sodium sulfate. Filtered and the resulting solution was added p-toluenesulfonyl chloride (3. 8g, 20mmol), was heated to reflux, the reaction was stirred for 3-4 hours, TLC detection completion of the reaction. The solvent was distilled off under reduced pressure, cooled to room temperature, methanol was added, the precipitated solid, cooled to square ° C, allowed to stand overnight. Filtered, the filter cake washed twice with cold methanol and vacuum dried to give an off-white solid 1- methyl-3-methyl-4-hydroxy-phenoxy-isoquinoline -7- (VI) I. 86g, yield 60.2 %; EI-MS m / z:.. 310 [M + H] +, 1H NMR (DMS0-d6) δ 2.90 (s, 3H), 4.05 (s, 3H), 7 17-7 26 (m, 3H ), 7. 49-7. 61 (m, 4H), 8. 38 (d, J = 9. 0,1H), 11. 7 (s, 1H) 〇
Example VI:
in the reaction flask with magnetic stirring and pressure to join I- methyl-3-methyl-4-hydroxy-7-phenoxyheptanoate isoquinoline (VI) (1.55g, 5mmol), glycine (I. 13g, 15mmol) and sodium methoxide (3. 25g, 6mmol) in methanol (30mL).Sealed, slowly heated to 120 ° C, the reaction was stirred for 8-10 hours to complete the reaction by TLC. Cooled to room temperature, solid precipitated. Filtration, and the resulting solid was recrystallized from methanol, acetone and then beating the resulting solid was dried under vacuum to give a white solid Connaught orlistat 1.40g, yield 79.5%;
EI-MS m / z: 353 [M + H] +,
1H NMR (DMS0-d6) S2.72 (s, 3H), 3 · 99 (d, J = 6 · 0, 2H), 7 · 18-7. 28 (m, 3H), 7 · 49-7. 63 (m, 4H), 8 · 31 (d, J = 8 · 8,1H), 9 · 08 (s, lH), 13.41 (brs, lH).
PATENT

Example 10. Preparation of Compound A
a) 5-Phenoxyphthalide

[0200] A reactor was charged with DMF (68 Kg), and stirring was initiated. The reactor was then charged with phenol (51 Kg), acetylacetone (8 Kg), 5-bromophthalide (85 Kg), copper bromide (9 Kg), and potassium carbonate (77 Kg). The mixture was heated above 85 °C and maintained until reaction completion and then cooled. Water was added. Solid was filtered and washed with water. Solid was dissolved in dichloromethane, and washed with aqueous HCl and then with water. Solvent was removed under pressure and methanol was added. The mixture was stirred and filtered. Solid was washed with methanol and dried in an oven giving 5- phenoxyphthalide (Yield: 72%, HPLC: 99.6%). b) 2-Chloromethyl-4-phenoxybenzoic acid methyl ester

[0201] A reactor was charged with toluene (24 Kg), and stirring was initiated. The reactor was then charged with 5-phenoxyphthalide (56 Kg), thionyl chloride (41 Kg), trimethyl borate (1
Kg), dichlorotriphenylphosphorane (2.5 Kg), and potassium carbonate (77 Kg). The mixture was heated to reflux until reaction completion and solvent was removed leaving 2-chloromethyl-4- phenoxybenzoyl chloride. Methanol was charged and the mixture was heated above 50 °C until reaction completion. Solvent was removed and replaced with DMF. This solution of the product methyl 2-chloromethyl-4-phenoxybenzoic acid methyl ester in DMF was used directly in the next step (HPLC: 85%). c) 4-Hydroxy-7-phenoxyisoquinoline-3-carboxylic acid methyl ester (la)

[0202] A reactor was charged with a solution of 2-chloromethyl-4-phenoxybenzoic acid methyl ester (~68 Kg) in DMF, and stirring was initiated. The reactor was then charged with p- toluenesulfonylglycine methyl ester (66 Kg), potassium carbonate (60 Kg), and sodium iodide (4 Kg). The mixture was heated to at least 50 °C until reaction completion. The mixture was cooled. Sodium methoxide in methanol was charged and the mixture was stirred until reaction completion. Acetic acid and water were added, and the mixture was stirred, filtered and washed with water. Solid was purified by acetone trituration and dried in an oven giving la (Yield from step b): 58%; HPLC: 99.4%). 1H NMR (200 MHz, DMSO-d6) δ 11.60 (s, 1 H), 8.74 (s, 1H),
8.32 (d, J = 9.0 Hz, 1 H), 7.60 (dd, J = 2.3 & 9.0 Hz, 1H), 7.49 (m, 3 H), 7.24 (m, 3 H), 3.96 (s, 3 H); MS-(+)-ion M+l = 296.09 d) Methyl l-((dimethylamino)methyl)-4-hydroxy-7-phenoxyisoquinoline-3-carboxylate
(lb)

[0203] A flask was charged with la (29.5 g) and acetic acid (44.3 g ± 5%), and then stirred. Bis-dimethylaminomethane (12.8 g ± 2%) was slowly added. The mixture was heated to 55 ± 5 °C and maintained until reaction completion. The reaction product was evaluated by MS, HPLC and 1H NMR. 1H NMR (200 MHz, DMSO-d6) δ 11.7 (s, 1 H), 8.38 (d, J = 9.0 Hz, 1 H), 7.61 (dd, J = 9.0, 2.7 Hz, 1 H), 7.49 (m, 3 H), 7.21 (m, 3 H), 5.34 (s, 2 H), 3.97 (s, 3 H), 1.98 (s, 3 H); MS-(+)-ion M+l = 368.12. e) Methyl l-((acetoxy)methyl)-4-hydroxy-7-phenoxyisoquinoline-3-carboxylate (lc)

[0204] The solution of lb from a) above was cooled below 25 °C, at which time acetic anhydride (28.6 g ± 3.5 %) was added to maintain temperature below 50 °C. The resulting mixture was heated to 100 ± 5 °C until reaction completion.
[0205] The solution of lc and Id from above was cooled to less than 65 ± 5 °C. Water (250 mL) was slowly added. The mixture was then cooled to below 20 ± 5 °C and filtered. The wet cake was washed with water (3 x 50 mL) and added to a new flask. Dichloromethane (90 mL) and water (30 mL) were added, and the resulting mixture was stirred. The dichloromethane layer was separated and evaluated by HPLC.
[0206] The organic layer was added to a flask and cooled 5 ± 5 °C. Morpholine was added and the mixture was stirred until reaction completion. Solvent was replaced with acetone/methanol mixture. After cooling, compound lc precipitated and was filtered, washed and dried in an oven (Yield: 81%, HPLC: >99.7%). 1H NMR (200 MHz, DMSO-d6) δ 11.6 (S, 1 H), 8.31 (d, J = 9.0 Hz, 1 H), 7.87 (d, J = 2.3 Hz, 1 H), 7.49 (m, 3 H), 7.24 (m, 3 H), 3.95 (s, 3 H), 3.68 (s, 2H), 2.08 (s, 6 H); MS-(+)-ion M+l = 357.17. f) Methyl 4-hydroxy-l-methyl-7-phenoxyisoquinoline-3-carboxylate (le)

[0207] A reactor was charged with lc (16.0 g), Pd/C (2.08 g), anhydrous Na2C03 (2.56 g) and ethyl acetate (120 mL). The flask was vacuum-purged with nitrogen (3X) and vacuum-purged with hydrogen (3X). The flask was then pressurized with hydrogen and stirred at about 60 °C until completion of reaction. The flask was cooled to 20-25 °C, the pressure released to ambient, the head space purged with nitrogen three times and mixture was filtered. The filtrate was concentrated. Methanol was added. The mixture was stirred and then cooled. Product precipitated and was filtered and dried in an oven (Yield: 90%, HPLC: 99.7%). g) [(4-Hydroxy-l-methyl-7-phenoxy-isoquinoline-3-carbonyl)-amino]-acetic acid
(Compound A)
[0208] A pressure flask was charged with le (30.92 g), glycine (22.52 g), methanol (155 mL), sodium methoxide solution (64.81 g) and sealed (as an alternative, sodium glycinate was used in place of glycine and sodium methoxide). The reaction was heated to about 110 °C until reaction was complete. The mixture was cooled, filtered, washed with methanol, dried under vacuum, dissolved in water and washed with ethyl acetate. The ethyl acetate was removed and to the resulting aqueous layer an acetic acid (18.0 g) solution was added. The suspension was stirred at room temperature, filtered, and the solid washed with water (3 x 30 mL), cold acetone (5-10 °C, 2 x 20 mL), and dried under vacuum to obtain Compound A (Yield: 86.1%, HPLC: 99.8%). Example 11. Biological Testing
[0209] The solid forms provided herein can be used for inhibiting HIF hydroxylase activity, thereby increasing the stability and/or activity of hypoxia inducible factor (HIF), and can be used to treat and prevent HIF-associated conditions and disorders (see, e.g., U.S. Patent No. 7,323,475, U.S. Patent Application Publication No. 2007/0004627, U.S. Patent Application Publication No. 2006/0276477, and U.S. Patent Application Publication No. 2007/0259960, incorporated by reference herein).

Condensation of 5-bromophthalide (I) with phenol (II) in the presence of K2CO3, CuBr and acetylacetone in DMF gives 5-phenoxyphthalide (III), which upon lactone ring opening using SOCl2, Ph3PCl2, B(OMe)3 and K2CO3 in refluxing toluene yields 2-chloromethyl-4-phenoxybenzoyl chloride (IV). Esterification of acid chloride (IV) with MeOH at 50 °C furnishes the methyl ester (V), which is then condensed with methyl N-tosylglycinate (VI) in the presence of K2CO3 and NaI in DMF at 50 °C to afford N-substituted aminoester (VII). Cyclization of the intermediate diester (VII) using NaOMe in MeOH leads to methyl 4-hydroxy-7-phenoxyisoquinoline-3-carboxylate (VIII), which is submitted to Mannich reaction with bis-dimethylaminomethane (IX) in the presence of AcOH at 57 °C to provide the dimethylaminomethyl compound (X). Treatment of amine (X) with Ac2O at 103 °C, followed by selective hydrolysis of the phenolic acetate with morpholine leads to methyl 1-acetoxymethyl-4-hydroxy-7-phenoxyisoquinoline-3-carboxylate (XI). Hydrogenolysis of the benzylic acetate (XII) in the presence of Pd/C and Na2CO3 in EtOAc yields methyl 4-hydroxy-1-methyl-7-phenoxyisoquinoline-3-carboylate (XII), which finally couples with glycine (XIII) in the presence of NaOMe in MeOH at 110 °C to afford the target roxadustat (1-3).

Cyclization of 4-phenoxyphthalic acid (I) with glycine (II) at 215 °C gives the phthalimide (III), which upon esterification with MeOH and H2SO4 at reflux yields methyl ester (IV). Subsequent rearrangement of phthalimidoacetate (IV) by means of Na in BuOH at 97 °C, followed by flash chromatography provides the isoquinoline-2-carboxylate (V). Bromination of intermediate (V) using POBr3 and NaHCO3 in acetonitrile leads to butyl 8-bromo-3-hydroxy-6-phenoxy-isoquinoline-2-carboxylate (VI), which upon hydrolysis with NaOH in refluxing H2O/EtOH furnishes carboxylic acid (VII). Substitution of bromine in intermediate (VII) using MeI and BuLi in THF at -78 °C, followed by alkylation with PhCH2Br in the presence of K2CO3 in refluxing acetone affords the 2-methyl isoquinoline (VIII). Ester hydrolysis in intermediate (VIII) using KOH in MeOH gives the corresponding carboxylic acid (IX), which is then activated with i-BuOCOCl and Et3N in CH2Cl2, followed by coupling with benzyl glycinate hydrochloride (X) to yield benzylated roxadustat (XI). Finally, debenzylation of intermediate (XI) with H2 over Pd/C in EtOAc/MeOH provides the title compound (1).

Condensation of 4-nitro-ortho-phthalonitrile (I) with phenol (II) in the presence of K2CO3 in DMSO gives 4-phenoxy-ortho-phthalonitrile (III) (1), which upon hydrolysis with NaOH (1) or KOH (2) in refluxing MeOH yields 4-phenoxyphthalic acid (IV) (1,2). Dehydration of dicarboxylic acid (IV) using Ac2O and AcOH at reflux furnishes the phthalic anhydride (V), which is then condensed with methyl 2-isocyanoacetate (VI) using DBU in THF to provide oxazole derivative (VII). Rearrangement of intermediate (VII) with HCl in MeOH at 60 °C leads to isoquinoline derivative (VIII), which is partially chlorinated by means of POCl3 at 70 °C to afford 1-chloro-isoquinoline derivative (IX). Substitution of chlorine in intermediate (IX) using Me3B, Pd(PPh3)4 and K2CO3 in refluxing dioxane gives methyl 4-hydroxy-1-methyl-7-phenoxy-3-carboxylate (X), which is then hydrolyzed with aqueous NaOH in refluxing EtOH to yield the carboxylic acid (XI). Coupling of carboxylic acid (XI) with methyl glycinate hydrochloride (XII) by means of PyBOP, (i-Pr)2NH and Et3N in CH2Cl2 yields roxadustat methyl ester (XII), which is finally hydrolyzed with aqueous NaOH in THF to afford the target roxadustat (1).
CLIPS

1: Besarab A, Provenzano R, Hertel J, Zabaneh R, Klaus SJ, Lee T, Leong R, Hemmerich S, Yu KH, Neff TB. Randomized placebo-controlled dose-ranging and pharmacodynamics study of roxadustat (FG-4592) to treat anemia in nondialysis-dependent chronic kidney disease (NDD-CKD) patients. Nephrol Dial Transplant. 2015 Oct;30(10):1665-73. doi: 10.1093/ndt/gfv302. Epub 2015 Aug 3. PubMed PMID: 26238121; PubMed Central PMCID: PMC4569392.
2: Forristal CE, Levesque JP. Targeting the hypoxia-sensing pathway in clinical hematology. Stem Cells Transl Med. 2014 Feb;3(2):135-40. doi: 10.5966/sctm.2013-0134. Epub 2013 Dec 26. PubMed PMID: 24371328; PubMed Central PMCID: PMC3925058.
3: Bouchie A. First-in-class anemia drug takes aim at Amgen’s dominion. Nat Biotechnol. 2013 Nov;31(11):948-9. doi: 10.1038/nbt1113-948b. PubMed PMID: 24213751.
4: Flight MH. Deal watch: AstraZeneca bets on FibroGen’s anaemia drug. Nat Rev Drug Discov. 2013 Oct;12(10):730. doi: 10.1038/nrd4135. PubMed PMID: 24080688.
5: Beuck S, Schänzer W, Thevis M. Hypoxia-inducible factor stabilizers and other small-molecule erythropoiesis-stimulating agents in current and preventive doping analysis. Drug Test Anal. 2012 Nov;4(11):830-45. doi: 10.1002/dta.390. Epub 2012 Feb 24. Review. PubMed PMID: 22362605.
6: Cases A. The latest advances in kidney diseases and related disorders. Drug News Perspect. 2007 Dec;20(10):647-54. PubMed PMID: 18301799.
///////////////
http://zliming2004.lofter.com/post/1cc9dc55_79ad5d8

FG-4592 is a new-generation hypoxia-inducible factor prolyl hydroxylase inhibitor in early clinical trials at FibroGen for the oral treatment of iron deficiency anemia and renal failure anemia. Preclinical studies are ongoing for the treatment of sickle cell anemia.
The investigational therapy is designed to restore balance to the body’s natural process of erythropoiesis through mechanisms including: natural EPO production, suppression of the effects of inflammation, downregulation of the iron sequestration hormone hepcidin, and an upregulation of other iron genes, ensuring efficient mobilization and utilization of the body’s own iron stores. In April 2006, FG-4592 was licensed to Astellas Pharma by originator FibroGen in Asia, Europe and South Africa for the treatment of anemia. FibroGen retains rights in the rest of the world. In 2007, the FDA put the trial on clinical hold due to one case of death by fulminant hepatitis during a phase II clinical trial for patients with anemia associated with chronic kidney disease and not requiring dialysis. However, in 2008, the FDA informed the company that clinical trials could be resumed. Phase II/III clinical trials for this indication resumed in 2012. In 2013, the compound was licensed to AstraZeneca by FibroGen for development and marketing in US, CN and all major markets excluding JP, Europe, the Commonwealth of Independent States, the Middle East and South Africa, for the treatment of anemia associated with chronic kidney disease (CKD) and end-stage renal disease (ESRD).
Phase Organization ConditionPhase IIIAstellas Pharma
AstraZeneca
FibroGenAnemia, renal failure
Condensation of 5-bromophthalide (I) with phenol (II) in the presence of K2CO3, CuBr and acetylacetone in DMF gives 5-phenoxyphthalide (III), which upon lactone ring opening using SOCl2, Ph3PCl2, B(OMe)3 and K2CO3 in refluxing toluene yields 2-chloromethyl-4-phenoxybenzoyl chloride (IV). Esterification of acid chloride (IV) with MeOH at 50 °C furnishes the methyl ester (V), which is then condensed with methyl N-tosylglycinate (VI) in the presence of K2CO3 and NaI in DMF at 50 °C to afford N-substituted aminoester (VII). Cyclization of the intermediate diester (VII) using NaOMe in MeOH leads to methyl 4-hydroxy-7-phenoxyisoquinoline-3-carboxylate (VIII), which is submitted to Mannich reaction with bis-dimethylaminomethane (IX) in the presence of AcOH at 57 °C to provide the dimethylaminomethyl compound (X). Treatment of amine (X) with Ac2O at 103 °C, followed by selective hydrolysis of the phenolic acetate with morpholine leads to methyl 1-acetoxymethyl-4-hydroxy-7-phenoxyisoquinoline-3-carboxylate (XI). Hydrogenolysis of the benzylic acetate (XII) in the presence of Pd/C and Na2CO3 in EtOAc yields methyl 4-hydroxy-1-methyl-7-phenoxyisoquinoline-3-carboylate (XII), which finally couples with glycine (XIII) in the presence of NaOMe in MeOH at 110 °C to afford the target roxadustat (1-3).
Cyclization of 4-phenoxyphthalic acid (I) with glycine (II) at 215 °C gives the phthalimide (III), which upon esterification with MeOH and H2SO4 at reflux yields methyl ester (IV). Subsequent rearrangement of phthalimidoacetate (IV) by means of Na in BuOH at 97 °C, followed by flash chromatography provides the isoquinoline-2-carboxylate (V). Bromination of intermediate (V) using POBr3 and NaHCO3 in acetonitrile leads to butyl 8-bromo-3-hydroxy-6-phenoxy-isoquinoline-2-carboxylate (VI), which upon hydrolysis with NaOH in refluxing H2O/EtOH furnishes carboxylic acid (VII). Substitution of bromine in intermediate (VII) using MeI and BuLi in THF at -78 °C, followed by alkylation with PhCH2Br in the presence of K2CO3 in refluxing acetone affords the 2-methyl isoquinoline (VIII). Ester hydrolysis in intermediate (VIII) using KOH in MeOH gives the corresponding carboxylic acid (IX), which is then activated with i-BuOCOCl and Et3N in CH2Cl2, followed by coupling with benzyl glycinate hydrochloride (X) to yield benzylated roxadustat (XI). Finally, debenzylation of intermediate (XI) with H2 over Pd/C in EtOAc/MeOH provides the title compound (1).
Condensation of 4-nitro-ortho-phthalonitrile (I) with phenol (II) in the presence of K2CO3 in DMSO gives 4-phenoxy-ortho-phthalonitrile (III) (1), which upon hydrolysis with NaOH (1) or KOH (2) in refluxing MeOH yields 4-phenoxyphthalic acid (IV) (1,2). Dehydration of dicarboxylic acid (IV) using Ac2O and AcOH at reflux furnishes the phthalic anhydride (V), which is then condensed with methyl 2-isocyanoacetate (VI) using DBU in THF to provide oxazole derivative (VII). Rearrangement of intermediate (VII) with HCl in MeOH at 60 °C leads to isoquinoline derivative (VIII), which is partially chlorinated by means of POCl3 at 70 °C to afford 1-chloro-isoquinoline derivative (IX). Substitution of chlorine in intermediate (IX) using Me3B, Pd(PPh3)4 and K2CO3 in refluxing dioxane gives methyl 4-hydroxy-1-methyl-7-phenoxy-3-carboxylate (X), which is then hydrolyzed with aqueous NaOH in refluxing EtOH to yield the carboxylic acid (XI). Coupling of carboxylic acid (XI) with methyl glycinate hydrochloride (XII) by means of PyBOP, (i-Pr)2NH and Et3N in CH2Cl2 yields roxadustat methyl ester (XII), which is finally hydrolyzed with aqueous NaOH in THF to afford the target roxadustat (1).
EP 1644336
US 8278325
JP 2006527200
JP 2010111697
CN 102977015
US 2014343094
CN 102977016
US 7323475
US 8765956
US 2013310565
CN 103145616
US 2013178417
CN 102718708
WO 2004108681
US 2004254215
JP 2011148810
EP 2357175
US 8017625
US 2012029011
US 8916585
Drug Substances
WO 2013013609
EP 2734504
CN 104024227
US 2015031721

Polymorphs
Drug Substances
WO 2014014834
CN 103435546
Synthesis
Synthesis Intermediates
CN 103539735
US 2014024676
WO 2014014835
US 2014303202
US 2015038529
EP 2872488
Drug Substances
Polymorphs
US 2014024675
US 8883823
KR 2015058147
US 2015175550
Polymorphs
Drug Substances
EP 1644336
US 8278325
JP 2006527200
JP 2010111697
CN 102977015
US 2014343094
CN 102977016
US 7323475
US 8765956
US 2013310565
CN 103145616
US 2013178417
CN 102718708
WO 2004108681
US 2004254215
JP 2011148810
EP 2357175
US 8017625
US 2012029011
US 8916585
Product patent
WO 2004108681
https://patents.google.com/patent/WO2004108681A1/pt
WO 2013013609
https://patents.google.com/patent/WO2013013609A1

polymorph scan
[(4-Hydroxy- 1 -methyl-7-phenoxy-isoquinoline-3-carbonyl)-amino] -acetic acid (hereinafter, Compound A) is a potent inhibitor of hypoxia inducible factor (HIF) prolyl hydroxylase, as described in U.S. Patent No. 7,323,475. HIF prolyl hydroxylase inhibitors are useful for increasing the stability and/or activity of HIF, and useful for, inter alia, treating and preventing disorders associated with HIF, including anemia, ischemia, and hypoxia.
Innovator
WO2004108681
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2004108681
Example D-81
e) [(4-Hydroxy-l-methyl-7-phenoxy-isoquinoline-3-carbonyl)-amino]-acetic acid
[0604] Synthesized from [(4-benzyloxy- l-methyl-7-phenoxy-isoquinoline-3-carbonyl)-amino]-acetic acid benzyl ester in analogy to Example D-78 d); MS-(-)-ion: M-l = 351.1.
Example D-78
d) [(4-Hydroxy-l-methoxymethyl-isoquinoline-3-carbonyl)-amino]-acetic acid [0590] A mixture of [(4-benzyloxy- 1 -methoxymethyl-isoquinoline-3 -carbonyl)-amino] -acetic acid benzyl ester (134 mg, 0.285 mmol), Pd/C (100 mg, 10 wt% Pd), EtOAc (10 ml) and MeOH (50 ml) was stirred under a Hj-atmosphere at ambient pressure and temperature for 18 h. Then the mixture was filtered through a pad of celite. Celite and filter cake were washed thoroughly with EtOAc and the combined organic phases were concentrated in vacuo to give the title compound as a tan solid (74 mg); MS-(-)-ion: M-l = 289.2
WO 2014014835
https://patents.google.com/patent/WO2014014835A2

Compound A.
In one embodiment, the pharmaceutical composition comprises a compound selected from the group consisting of: Compound A Form A, Compound A Form B, Compound A Form C, Compound A Form D, Compound A sodium salt, Compound A L-arginine salt, Compound A L-lysine salt, Compound A ethanolamine salt, Compound A diethanolamine salt, Compound A tromethamine salt, amorphous Compound A, and Compound A potassium salt, as described generally above, and at least one pharmaceutically acceptable excipient.
Solid Forms of Compound A
[0073] As described generally above, the present disclosure provides solid forms of [(4- hydroxy-l-methyl-7-phenoxy-isoquinoline-3-carbonyl)-amino] -acetic acid (Compound A).
[0074] Compound A Form A is characterized by its X-ray powder diffractogram that comprises peaks at 8.5, 16.2, and 27.4 °2Θ ± 0.2 °2Θ. The diffractogram comprises additional peaks at 12.8, 21.6, and 22.9 °2Θ ± 0.2 °2Θ. Form A also is characterized by its full X-ray powder diffractogram as substantially shown in Figure 1.
[0075] In some embodiments, Form A is characterized by its differential scanning calorimetry (DSC) curve that comprises an endotherm at about 223 °C. Form A also is characterized by its full DSC curve as substantially as shown in Figure 2.
[0076] Compound A Form B is characterized by its X-ray powder diffractogram that comprises peaks at 4.2, 8.3, and 16.6 °2Θ ± 0.2 °2Θ. The diffractogram comprises additional peaks at 12.5, 14.1, and 17.4 °2Θ ± 0.2 °2Θ. Form B also is characterized by its full X-ray powder
diffractogram as substantially shown in Figure 3.
[0077] In some embodiments, Form B is characterized by its differential scanning calorimetry (DSC) curve that comprises an endotherm at about 222 °C. Form B also is characterized by its full DSC curve as substantially as shown in Figure 4.
[0078] Compound A Form C is characterized by its X-ray powder diffractogram that comprises peaks at 4.5, 13.7, and 16.4 °2Θ ± 0.2 °2Θ. The diffractogram comprises additional peaks at 15.4, 15.5, and 20.6 °2Θ ± 0.2 °2Θ. Form C also is characterized by its full X-ray powder diffractogram as substantially shown in Figure 5.
[0079] In some embodiments, Form C is characterized by its differential scanning calorimetry (DSC) curve that comprises an endotherm at about 222 °C. Form C also is characterized by its full DSC curve as substantially as shown in Figure 6.
[0080] Compound A Form D is characterized by its X-ray powder diffractogram that comprises peaks at 8.4, 8.5, and 16.8 °2Θ ± 0.2 °2Θ. The diffractogram comprises additional peaks at 4.2, 12.6, and 28.4 °2Θ ± 0.2 °2Θ. Form D also is characterized by its full X-ray powder diffractogram as substantially shown in Figure 7. [0081] In some embodiments, Form D is characterized by its differential scanning calorimetry (DSC) curve that comprises an endotherm at about 222 °C. Form D also is characterized by its full DSC curve as substantially as shown in Figure 8.
Example 10. Preparation of Compound A
a) 5-Phenoxyphthalide

[0200] A reactor was charged with DMF (68 Kg), and stirring was initiated. The reactor was then charged with phenol (51 Kg), acetylacetone (8 Kg), 5-bromophthalide (85 Kg), copper bromide (9 Kg), and potassium carbonate (77 Kg). The mixture was heated above 85 °C and maintained until reaction completion and then cooled. Water was added. Solid was filtered and washed with water. Solid was dissolved in dichloromethane, and washed with aqueous HCl and then with water. Solvent was removed under pressure and methanol was added. The mixture was stirred and filtered. Solid was washed with methanol and dried in an oven giving 5- phenoxyphthalide (Yield: 72%, HPLC: 99.6%). b) 2-Chloromethyl-4-phenoxybenzoic acid methyl ester
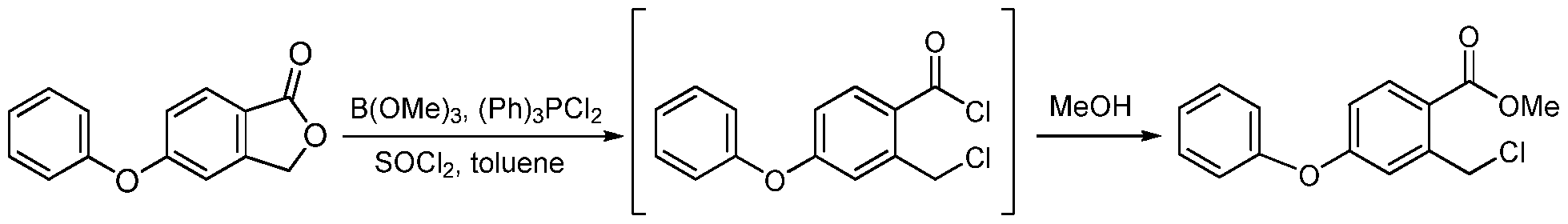
[0201] A reactor was charged with toluene (24 Kg), and stirring was initiated. The reactor was then charged with 5-phenoxyphthalide (56 Kg), thionyl chloride (41 Kg), trimethyl borate (1
Kg), dichlorotriphenylphosphorane (2.5 Kg), and potassium carbonate (77 Kg). The mixture was heated to reflux until reaction completion and solvent was removed leaving 2-chloromethyl-4- phenoxybenzoyl chloride. Methanol was charged and the mixture was heated above 50 °C until reaction completion. Solvent was removed and replaced with DMF. This solution of the product methyl 2-chloromethyl-4-phenoxybenzoic acid methyl ester in DMF was used directly in the next step (HPLC: 85%). c) 4-Hydroxy-7-phenoxyisoquinoline-3-carboxylic acid methyl ester (la)

[0202] A reactor was charged with a solution of 2-chloromethyl-4-phenoxybenzoic acid methyl ester (~68 Kg) in DMF, and stirring was initiated. The reactor was then charged with p- toluenesulfonylglycine methyl ester (66 Kg), potassium carbonate (60 Kg), and sodium iodide (4 Kg). The mixture was heated to at least 50 °C until reaction completion. The mixture was cooled. Sodium methoxide in methanol was charged and the mixture was stirred until reaction completion. Acetic acid and water were added, and the mixture was stirred, filtered and washed with water. Solid was purified by acetone trituration and dried in an oven giving la (Yield from step b): 58%; HPLC: 99.4%). 1H NMR (200 MHz, DMSO-d6) δ 11.60 (s, 1 H), 8.74 (s, 1H),
8.32 (d, J = 9.0 Hz, 1 H), 7.60 (dd, J = 2.3 & 9.0 Hz, 1H), 7.49 (m, 3 H), 7.24 (m, 3 H), 3.96 (s, 3 H); MS-(+)-ion M+l = 296.09 d) Methyl l-((dimethylamino)methyl)-4-hydroxy-7-phenoxyisoquinoline-3-carboxylate
(lb)

[0203] A flask was charged with la (29.5 g) and acetic acid (44.3 g ± 5%), and then stirred. Bis-dimethylaminomethane (12.8 g ± 2%) was slowly added. The mixture was heated to 55 ± 5 °C and maintained until reaction completion. The reaction product was evaluated by MS, HPLC and 1H NMR. 1H NMR (200 MHz, DMSO-d6) δ 11.7 (s, 1 H), 8.38 (d, J = 9.0 Hz, 1 H), 7.61 (dd, J = 9.0, 2.7 Hz, 1 H), 7.49 (m, 3 H), 7.21 (m, 3 H), 5.34 (s, 2 H), 3.97 (s, 3 H), 1.98 (s, 3 H); MS-(+)-ion M+l = 368.12. e) Methyl l-((acetoxy)methyl)-4-hydroxy-7-phenoxyisoquinoline-3-carboxylate (lc)
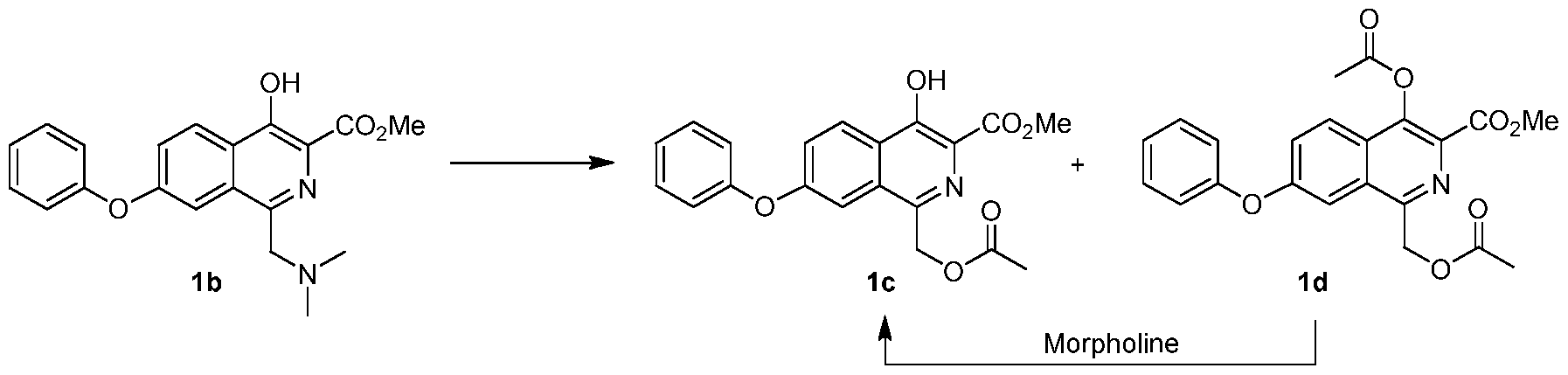
[0204] The solution of lb from a) above was cooled below 25 °C, at which time acetic anhydride (28.6 g ± 3.5 %) was added to maintain temperature below 50 °C. The resulting mixture was heated to 100 ± 5 °C until reaction completion.
[0205] The solution of lc and Id from above was cooled to less than 65 ± 5 °C. Water (250 mL) was slowly added. The mixture was then cooled to below 20 ± 5 °C and filtered. The wet cake was washed with water (3 x 50 mL) and added to a new flask. Dichloromethane (90 mL) and water (30 mL) were added, and the resulting mixture was stirred. The dichloromethane layer was separated and evaluated by HPLC.
[0206] The organic layer was added to a flask and cooled 5 ± 5 °C. Morpholine was added and the mixture was stirred until reaction completion. Solvent was replaced with acetone/methanol mixture. After cooling, compound lc precipitated and was filtered, washed and dried in an oven (Yield: 81%, HPLC: >99.7%). 1H NMR (200 MHz, DMSO-d6) δ 11.6 (S, 1 H), 8.31 (d, J = 9.0 Hz, 1 H), 7.87 (d, J = 2.3 Hz, 1 H), 7.49 (m, 3 H), 7.24 (m, 3 H), 3.95 (s, 3 H), 3.68 (s, 2H), 2.08 (s, 6 H); MS-(+)-ion M+l = 357.17. f) Methyl 4-hydroxy-l-methyl-7-phenoxyisoquinoline-3-carboxylate (le)

[0207] A reactor was charged with lc (16.0 g), Pd/C (2.08 g), anhydrous Na2C03(2.56 g) and ethyl acetate (120 mL). The flask was vacuum-purged with nitrogen (3X) and vacuum-purged with hydrogen (3X). The flask was then pressurized with hydrogen and stirred at about 60 °C until completion of reaction. The flask was cooled to 20-25 °C, the pressure released to ambient, the head space purged with nitrogen three times and mixture was filtered. The filtrate was concentrated. Methanol was added. The mixture was stirred and then cooled. Product precipitated and was filtered and dried in an oven (Yield: 90%, HPLC: 99.7%). g) [(4-Hydroxy-l-methyl-7-phenoxy-isoquinoline-3-carbonyl)-amino]-acetic acid
(Compound A)

[0208] A pressure flask was charged with le (30.92 g), glycine (22.52 g), methanol (155 mL), sodium methoxide solution (64.81 g) and sealed (as an alternative, sodium glycinate was used in place of glycine and sodium methoxide). The reaction was heated to about 110 °C until reaction was complete. The mixture was cooled, filtered, washed with methanol, dried under vacuum, dissolved in water and washed with ethyl acetate. The ethyl acetate was removed and to the resulting aqueous layer an acetic acid (18.0 g) solution was added. The suspension was stirred at room temperature, filtered, and the solid washed with water (3 x 30 mL), cold acetone (5-10 °C, 2 x 20 mL), and dried under vacuum to obtain Compound A (Yield: 86.1%, HPLC: 99.8%). Example 11. Biological Testing
[0209] The solid forms provided herein can be used for inhibiting HIF hydroxylase activity, thereby increasing the stability and/or activity of hypoxia inducible factor (HIF), and can be used to treat and prevent HIF-associated conditions and disorders (see, e.g., U.S. Patent No. 7,323,475, U.S. Patent Application Publication No. 2007/0004627, U.S. Patent Application Publication No. 2006/0276477, and U.S. Patent Application Publication No. 2007/0259960, incorporated by reference herein).
[0210] The biological activity of the solid forms provided herein may be assessed using any conventionally known method. In particular embodiments, cells derived from animal tissues, preferably human tissues, capable of expressing erythropoietin when stimulated by compounds of the invention are cultured for the in vitro production of endogenous proteins. Cells contemplated for use in such methods include, but are not limited to, cells derived from hepatic, hematopoietic, renal, and neural tissues.
[0211] Cell culture techniques are generally available in the art and include any method that maintains cell viability and facilitates expression of endogenous proteins. Cells are typically cultured in a growth medium optimized for cell growth, viability, and protein production. Cells may be in suspension or attached to a substrate, and medium may be supplied in batch feed or continuous flow-through regimens. Compounds of the invention are added to the culture medium at levels that stimulate erythropoietin production without compromising cell viability. Erythropoietin produced by the cells is secreted into the culture medium. The medium is then collected and the erythropoietin is purified using methods known to those of skill in the art. (See, e.g., Lai et al. (1987) U.S. Pat. No. 4,667,016; and Egrie (1985) U.S. Pat. No. 4,558,006.)
PATENT
https://patents.google.com/patent/CN104892509A/en
Connaught orlistat (Roxadustat, I) has the chemical name: N_ [(4- hydroxy-l-methyl-3-isoquinolinyl) carbonyl] glycine, having the formula:
[0004]

[0005] The originator’s International Patent W02004108681 provides a Connaught orlistat prepared from the intermediates and orlistat Connaught intermediate synthetic route:
[0006]

[0007] Zhejiang International Patent W02013013609 Beida’s preparation and acylation of the intermediate core is further optimized, which is a synthetic route:
[0008]
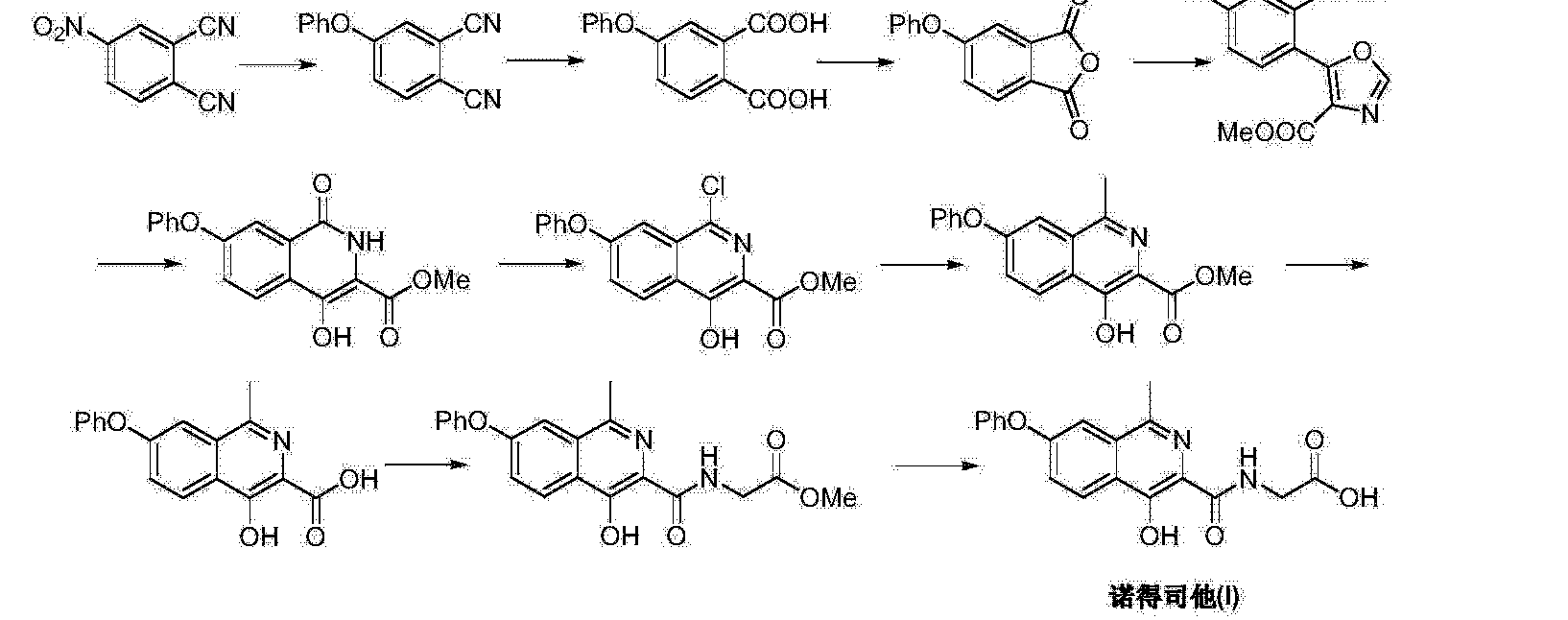
n PhO. eight XOOH
[0009] The originator’s International Patent W02014014834 and W02014014835 also provides another synthetic route Division Connaught his prepared:
[0010]

[0011]

PATENT
https://patents.google.com/patent/US9617218B2/en
PATENT
WO 2013013609
https://patents.google.com/patent/WO2013013609A1
Zhejiang Beta Pharma Incorporation
The present invention relates to approximately pure crystalline polymorphs, wherein these polymorphs are the polymorphs of the compound of Formula I, and/or a hydrate thereof, and/or a solvate thereof.

Formula I .
The compound of Formula I of the present invention exists in one or more crystal forms. The inventors designated these crystal forms Form I, Form II, Form III, Form IV, Form V, Form VI and Form VII.

8, 9, 10
Synthesis of Compound 1
Under inert gas (N?), 4~Mtro~o~phth.ak>nitrile (9.2 g), phenol (5.0 g), .2CO3 (7.3 g) and DMSO (40 mL) were added into a flask, and were stirred and reacted at room temperature for 48 hrs., then heated to 60 °C and reacted for 2 hrs. After cooled down, the reaction mixture was filtered and the resulted yellow solid was dried to obtain 11.6 g of Compound 1.
Synthesis of Compound 2
50 % of NaOH solution (25 mL) was added into the methanol solution of Compound 1 (11.3 g). The solution was heated to reflux for 48 hr until the reaction was complete. Concentrated HCl was then added to adjust the pH value to 3. The precipitate was filtered and dried to obtain 10.5 g of Compound 2.
Synthesis of Compound 3
Compound 2 (6.0 g) was dissolved in glacial acetic acid (60 mL) and acetic anhydride (60 mL) and heated to reflux for 3 hrs. The solvent was removed on a rotary evaporator to obtain Compound
3.
Synthesis of Compound 4
Compounds 3 (6.0 g) and methyl isocyanoacetate (2.65 g) were dissolved in THF (60 mL). 3.54 g of DBU (CAS No. 6674-22-2) was added in drop-wise at room temperature and stirred for 1 hr. at room temperature. After extracted with ethyl acetate under alkaline conditions to remove the impurities, the pH value of the aqueous phase was adjusted to 3 with diluted HCL Extracted with ethyl acetate, washed with water and dried with anhydrous Na2S04 and fi ltered, the resulting organic phase was distilled on a rotary evaporator to obtain 9.0g of Compound 4. Synthesis of Compound 5
Compound 4 (9.0 g) in CH3OH was added in concentrated HC1 and heated to 60 °C for 4 hrs. The resulted precipitation was filtered to obtain 5.8 g of crude product. The product was further purified by chromatography to obtain 1.85 g of Compound 5.
Synthesis of Compound 6
Compound 5 (1.77 g) in POCI3 (10 mL) was heated to about 70 and reacted for 3 hrs., then cooled dow and poured into ice. After POCU was completely decomposed, the resulting precipitate was filtered and washed with water, to obtain 1.45 g of Compound 6.
Synthesis of Compound 7
Under N2 atmosphere, Compound 6 (1 .41 g), dioxane (20 mL), rdj Pii ).Π )φ (0.49 g), .2CO3 (1.78 g) and trimethyl borane (0.54 g) were stirred mixed and heated to reflux for 3 hrs., then stirred at room temperature for 48 hrs. After concentration, the resulting mixture was extracted with ethyl acetate, washed with water, dried and filtered, then distilled on a rotary evaporator, followed by further purification through chromatography, to obtain 0.42 g of Compound 7.
Synthesis of Compound 8
Compound 7 (1.02 g) was added into the mixture of etiianol (10 mL) and 2N of NaOH (10 mL), and refluxed for 1.5 hrs. After removing the impurities by filtration, the resulting mixture was distilled to remove ethanol on a rotary evaporator. The resulting pale yellow precipitate was then filtered, washed with water, and dried to obtain 0.5 g of Compound 8.
Synthesis of Compound 9
Compound 8 (0.37 g), glycine methyl ester hydrochloride (0.44 g) and 1.00 g of PyBOP (CAS No. 128625-52-5) were added into dichloromethane (15 mL). and then added triethyiamine (0.74 mL) and bis(isopropyl)eth.y I amine ( 1.0 mL), stirred and reacted at room temperature for 3 hrs. After filtration, the organic phase was washed with water, dried and filtered, followed by a rotary evaporation, and further purification by a silica gel column, to obtain 0.29 g of Compound 9.
Synthesis of Compound 10, the compound of Formula I
Compound 9 (0.28 g) in THF was added in 1 NaOH (5 mL) and stirred and reacted for 1 hr. at room temperature. After remo ving THF by a rotary evaporation, the pFi value of the residue was adjusted to about 3 by diluted HQ, washed further by ethyl acetate, filtered and dried, to obtain 0.21 g of Compound 10, the compound of Formula I. Example 2
Preparation of Crystalline Form I of the compound of Formula I
The compound of Formula I prepared from the method disclosed in Example 1 above, was dissolved in the mixed solvent of methanol/MTBE (methyl tertbutyl ether) at room temperature, followed by a spontaneous precipitation to obtain the desired Polymorph Form I, with the melting point of 174-177 °C.
Example 2
Preparation of Crystalline Form I of the compound of Formula I
The compound of Formula I prepared from the method disclosed in Example 1 above, was dissolved in the mixed solvent of methanol/MTBE (methyl tertbutyl ether) at room temperature, followed by a spontaneous precipitation to obtain the desired Polymorph Form I, with the melting point of 174-177 °C.
Example 3
Preparation of Crystalline Form II of the compound of Formula I
A slurry suspension of excess amount of the compound of Formula I prepared from the method disclosed in Example 1 above, was stirred in the mixed solvent of H20/acetonitrile (3: 1) or H20/ethanol at room temperature or 50°C at least 48 hrs., or in the mixed solvent of methanol/H20 at room temperature over 48 hr, to obtain the desired Crystalline Form II, with the melting point of 209-212 °C .
Example 4
Preparation of Crystalline Form III of the compound of Formula I
The compound of Formula I prepared from the method disclosed in Example 1 above, was dissolved in the mixed solvent of methanol/acetonitrile at room temperature, followed by a spontaneous precipitation to obtain the desired Crystalline Form III.
Or, a slurry suspension of excess amount of the compound of Formula I prepared from the method disclosed in Example 1 above, was stirred in H20, CH2C12, isopropyl acetate (IPAc), ethyl acetate (EtOAc), or the mixed solvent of IPAc/heptane or H20/acetone at 50 °C over 48 hrs., to obtain the desired Crystalline Form III, with the melting point of 198-200 °C ..
Example 5
Preparation of Crystalline Form IV of the compound of Formula I
A slurry suspension of excess amount of the compound of Formula I prepared from the method disclosed in Example 1 above, was stirred in MTBE, or the mixed solvent of MTBE/heptane, IPAc/heptane, ethyl acetate/heptane or H20/acetone at room temperature over 48 hrs., to obtain the desired Crystalline Form IV.
Or, a slurry suspension of excess amount of the compound of Formula I prepared from the method disclosed in Example 1 above, was stirred in the mixed solvent of ethyl acetate/heptane at 50 °C over 48 hrs., to obtain the desired Ciystalline Form IV.
Or, a slurry suspension of excess amount of the Crystalline Form III as prepared in Example 4 was stirred in the mixed solvent of FFiO/acetone at 50°C for 12-14 days, to obtain the desired Crystalline Form IV, with the melting point of 204-207 °C .
Example 6
Preparation of Crystalline Form V of the compound of Formula I
A slurry suspension of excess amount of the compound of Formula I prepared from the method disclosed in Example 1 above, was stirred in the mixed solvent of MTBE/heptane at 50 C over 48 hr, to obtain she desired Crystalline Form V; or, water was added as anti-solvent into the methanol solution of the compound of Formula I, to obtain the desired Ciystalline Form V, with the melting point of 190-193 °C .
Example 7
Preparation of Crystalline Form VI of the compound of Formula I
A slurry suspension of excess amount of the compound of Formula I prepared from the method disclosed in Example 1 above, was stirred in the mixed solvent of acetonitrile/FFiO (1 : 1) or THF/H2O at room temperature over 48 hrs, to obtain she desired Crystalline Form VI.
Or, the compound of Formula I prepared from the method disclosed in Example 1 above, was dissolved in the mixed solvent of methanol/ethy! acetate at room temperature, followed by a spontaneous precipitation using Ciystalline Form IV as prepared in Example 5 as crystal seeds to obtain the desired Crystalline Form VI, with the melting point of 200-203 °C =
Example 8
Preparation of Crystalline Form VH of the compound of Formula I
Ciystalline Form V prepared from the method of Example 6 was heated to 180 °C, to obtain the desired Crystalline Form VH.
PATENT
IN 201641016266
REDDYS AMORPHOUS FORM
The US patent number 7323475 B2, Example D-81 (e), by referring Example D-78 (d), discloses a process for isolation of roxadustat by concentration of organic phases (EtOAc/Methanol) in vacuo.
The US patent number 8883823 B2 discloses amorphous, different polymorphic Forms, solvates and salts of roxadustat.
The US patent number 9206134 B2 discloses different crystalline Forms of roxadustat.
PATENT
CN 106187888
PATENT
US 2014024675
PATENT
https://patents.google.com/patent/US9206134B2/en
BEIJING BETTA PHARMACEUTICALS CO.
The present invention relates to approximately pure crystalline polymorphs, wherein these polymorphs are the polymorphs of the compound of Formula and/or a hydrate thereof, and/or a solvate thereof.

The compound of Formula I of the present invention exists in one or more crystal forms. The inventors designated these crystal forms Form I, Form II, Form III, Form IV, Form V, Form VI and Form VII.
////////////

ROXADUSTAT
ASP1517; ASP 1517; ASP-1517; FG-4592; FG 4592; FG4592; Roxadustat.
CAS 808118-40-3
Chemical Formula: C19H16N2O5
Exact Mass: 352.10592
THERAPEUTIC CLAIM
Treatment of anemia
CHEMICAL NAMES
(4-hydroxy-1-methyl-7-phenoxyisoquinoline-3-carbonyl)glycine
1. Glycine, N-[(4-hydroxy-1-methyl-7-phenoxy-3-isoquinolinyl)carbonyl]-
2. N-[(4-hydroxy-1-methyl-7-phenoxyisoquinolin-3-yl)carbonyl]glycine
MF C19H16N2O5
MW 352.3
SPONSOR FibroGen
CODE FG-4592; ASP1517
CAS 808118-40-3
WHO NUMBER 9717
Roxadustat, also known as ASP1517 and FG-4592, is an HIF α prolyl hydroxylase inhibitor in a cell-free assay. It stabilizes HIF-2 and induces EPO production and stimulates erythropoiesis. Roxadustat transiently and moderately increased endogenous erythropoietin and reduced hepcidin
|
FG-4592 (also known as ASP1517), 2-(4-hydroxy-1-methyl-7-phenoxyisoquinoline-3-carboxamido)acetic acid,
is a potent small molecule inhibitor of hypoxia-inducible factor prolyl hydroxylase (HIF-PH),
an enzyme up-regulating the expression of endogenous human erythropoietin (Epo). It is currently being investigated as an oral treatment for anemia associated with chronic kidney disease (CKD). Unlike other anemia treating agents, erythropoiesis-stimulating agents (ESAs),
FG-4592 inhibits HIF, through a distinctive mechanism, by stabilization of HIF. According to previous studies,
FG-4592 is capable of correcting and maintaining hemoglobin levels in CKD patients not
receiving dialysis and in patients of end-stage renal disease who receives dialysis but do not need intravenous iron supplement.
Reference
1. Luis Borges. Different modalities of erythropoiesis stimulating agents.
Port J Nephrol Hypert 2010; 24(2): 137-145 2. “FibroGen and Astellas announce initiation of phase 3 trial of FG-4592/ASP1517 for treatment of anemia of chronic kidney disease” Fibrogen Press Release. Dec 11 2012
3. “FibroGen announces initiation of phase 2b studies of FG-4592, an oral HIF prolyl hydroxylase inhibitor, for treatment of anemia”
|
![]()
Roxadustat (FG-4592) is a novel new-generation oral hypoxia-induciblefactor (HIF) prolyl hydroxylase inhibitor (PHI) for the treatment of ane-mia in patients with chronic kidney disease (CKD). HIF is a cytosolic tran-scription factor that induces the natural physiological response to lowoxygen conditions, by stimulating erythropoiesis and other protectivepathways. Roxadustat has been shown to stabilize HIF and induce ery-thropoiesis. Consequently, it corrects anemia and maintains hemoglo-bin levels without the need for intravenous iron supplementation in CKDpatients not yet receiving dialysis and in end-stage renal disease pa-tients receiving dialysis. There are many concerns about the use of ery-thropoiesis-stimulating agents (ESA) to treat anemia as they causesupra-physiologic circulating erythropoietin (EPO) levels and are asso-ciated with adverse cardiovascular effects and mortality. Available clin-ical data show that modest and transient increases of endogenous EPOinduced by HIF-PHI (10- to 40-fold lower than ESA levels) are sufficientto mediate erythropoiesis in CKD patients. Evidence suggests that rox-adustat is well tolerated and, to date, no increased risk of cardiovascu-lar events has been found. This suggests that roxadustat provides adistinct pharmacological and clinical profile that may provide a saferand more convenient treatment of CKD anemia
FG-4592 is a new-generation hypoxia-inducible factor prolyl hydroxylase inhibitor in early clinical trials at FibroGen for the oral treatment of iron deficiency anemia and renal failure anemia. Preclinical studies are ongoing for the treatment of sickle cell anemia.
The investigational therapy is designed to restore balance to the body’s natural process of erythropoiesis through mechanisms including: natural EPO production, suppression of the effects of inflammation, downregulation of the iron sequestration hormone hepcidin, and an upregulation of other iron genes, ensuring efficient mobilization and utilization of the body’s own iron stores. In April 2006, FG-4592 was licensed to Astellas Pharma by originator FibroGen in Asia, Europe and South Africa for the treatment of anemia. FibroGen retains rights in the rest of the world. In 2007, the FDA put the trial on clinical hold due to one case of death by fulminant hepatitis during a phase II clinical trial for patients with anemia associated with chronic kidney disease and not requiring dialysis. However, in 2008, the FDA informed the company that clinical trials could be resumed. Phase II/III clinical trials for this indication resumed in 2012. In 2013, the compound was licensed to AstraZeneca by FibroGen for development and marketing in US, CN and all major markets excluding JP, Europe, the Commonwealth of Independent States, the Middle East and South Africa, for the treatment of anemia associated with chronic kidney disease (CKD) and end-stage renal disease (ESRD).
PATENTS
WO 2004108681
WO 2008042800
WO 2009058403
WO 2009075822
WO 2009075824
WO 2012037212
WO 2013013609
WO 2013070908
CN 104892509
MACHINE TRANSLATED
Connaught orlistat (Roxadustat) by the US company Phibro root (FibroGen) R & D, Astellas AstraZeneca and licensed by a hypoxia-inducible factor (HIF) prolyl hydroxylase small molecule inhibitors, codenamed FG-4592.As a first new oral drug, FG-4592 is currently in Phase III clinical testing stage, for the treatment of chronic kidney disease and end-stage renal disease related anemia. Because the drug does not have a standard Chinese translation, so the applicant where it is transliterated as “Connaught Secretary him.”
Connaught orlistat (Roxadustat, I) the chemical name: N_ [(4- hydroxy-1-methyl-7-phenoxy-3-isoquinolinyl) carbonyl] glycine, its structural formula is:
The original research company’s international patent W02004108681 Division provides a promise he was prepared from the intermediate and intermediate Connaught Secretary for his synthetic route:
Zhejiang Beida company’s international patent W02013013609 preparation and acylation of core intermediate was further optimized synthesis route is:
n PhO. eight XOOH
original research company’s international patent W02014014834 and W02014014835 also provides another synthetic route he Connaught Secretary prepared:
Analysis of the above synthetic route, although he continued to Connaught Division to improve and optimize the synthesis, but its essence rings manner that different form quinoline ring is basically the same mother. Especially methyl isoquinoline replaced either by way of introducing the Suzuki reaction catalyzed by a noble metal element, either through amine reduction achieved. Moreover, the above reaction scheme revelation raw materials are readily available, many times during the reaction need to be protected and then deprotected. Clearly, the preparation process is relatively complicated, high cost, industrial production has brought some difficulties.
Example One:
tyrosine was added to the reaction flask and dried (18. lg, 0.1 mmol) and methanol 250mL, cooling to ice bath 0_5 ° C, was added dropwise over 1 hour a percentage by weight of 98% concentrated sulfuric acid 10g. Drops Albert, heating to reflux. The reaction was stirred for 16-20 hours, TLC the reaction was complete. Concentrated under atmosphere pressure, the residue was added water 100mL, using 10% by weight sodium hydroxide to adjust the pH to 6. 5-7.0, precipitated solid was filtered, washed with methanol and water chloro cake (I: 1) and dried in vacuo tyrosine methyl ester as a white solid (11) 15.38, yield 78.5% out 1–] \ ^ 111/2: 196 [] \ 1 + 1] +!.
Example Two:
[0041] a nitrogen atmosphere and ice bath, was added to the reaction flask tyrosine methyl ester (II) (9. 8g, 50mmol), potassium methoxide (3. 5g, 50mmol) and methanol 50mL, until no gas generation after, was heated to reflux, the reaction was stirred for 2 hours. Concentrated under atmosphere pressure to remove the solvent, the residue was added dimethylsulfoxide 25mL, freshly prepared copper powder (0.2g, 3. Lmmol), was slowly warmed to 150-155 ° C, for about half an hour later, a solution of bromobenzene ( 7. 9g, 50mmol), continue to heat up to 170-175 ° C, the reaction was stirred for 3 hours, TLC detection of the end of the reaction. Was cooled to 60 ° C, and methanol was added to keep micro-boiling, filtered while hot, the filter cake washed three times with hot ethanol, and the combined organic phases, was cooled to square ° C, filtered, and dried in vacuo to give a white solid of 2-amino-3- ( 4-phenoxyphenyl) propanoate (111) 8 11.5, yield 84.9% as 1 -] \ ^ 111/2:! 272 [] \ 1 + 1] +.
Example Three:
in the reaction flask was added 2-amino-3- (4-phenoxyphenyl) propionic acid methyl ester (III) (10. 8g, 40mmol), 40% by weight acetaldehyde (20g, 0. 2mol ) and the percentage by weight of 35% concentrated hydrochloric acid 50mL, refluxed for 1 hour. Continue 40% by weight was added acetaldehyde (10g, 0.1mol), and the percentage by weight of 35% concentrated hydrochloric acid 25mL, and then the reaction was refluxed for 3-5 hours. Was cooled to 4-7 ° C, ethyl acetate was added, and extracted layers were separated. The aqueous layer was adjusted with sodium hydroxide solution to pH 11-12, extracted three times with ethyl acetate. The combined organic phase was dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give a white solid of 1-methyl-3-carboxylate -7- phenoxy-1,2,3,4-tetrahydroisoquinoline (IV) 8 4g, 70.7% yield; Mass spectrum (EI): EI-MS m / z: 298 [M + H] + .
Example Four:
Under ice bath, the reaction flask was added methyl 3-carboxylate I- -7- phenoxy-1,2, 3,4-tetrahydro-isoquinoline (IV) (5. 9g, 20mmol) and dichloromethane 100mL, 0 ° C and under stirring added potassium carbonate (13. 8g, 0. lmol), p-toluenesulfonyl chloride (11. 4g, 60mmol), the addition was completed, the ice bath was removed and stirred at room temperature 3 hour. Water was added 30mL, after stirring standing layer, the organic phase was washed with dilute hydrochloric acid, water and saturated brine, and concentrated, the resulting product was added a 30% by weight sodium hydroxide solution (8. 0g, 60mmol) and dimethyl sulfoxide 60mL, gradually warming to 120-130 ° C, the reaction was stirred for 2-4 hours to complete the reaction by TLC. Cooled to room temperature, water was added lOOmL, extracted three times with ethyl acetate, the combined organic phase was successively washed with water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated, the resulting oil was treated with ethyl acetate and n-hexane (1: 3) recrystallization, vacuum dried to give an off-white solid 1-methyl-3-carboxylate 7-phenoxyheptanoic isoquinoline (V) 5. 25g, yield 89. 6%; EI-MS m / z: 294 [M + H] VH NMR (DMS0-d6) δ 2. 85 (s, 3H), 3 · 97 (s, 3H), 7 · 16-7. 24 (m, 3H), 7 · 49-7. 60 (m, 4Η), 8 · 35 (d, J = 9 · 0,1Η), 8 · 94 (s, 1Η).
Example five:
[0047] added 1-methyl-3-carboxylic acid methyl ester 7-phenoxyheptanoic isoquinoline (V) (2. 93g, IOmmol) and glacial acetic acid 50mL reaction flask, stirring solution of 30% by weight hydrogen peroxide 5mL, warmed to 60-70 ° C, was slowly added dropwise within 10 hours the percentage by weight of a mixture of 30% hydrogen peroxide 2mL and 12mL of glacial acetic acid, a dropping was completed, the reaction was continued for 20-24 hours. Concentrated under reduced pressure, ethanol was added, distillation is continued to be divisible remaining glacial acetic acid. The residue was dissolved with dichloromethane, washed with 5% by weight of sodium bicarbonate, the organic phase was separated, dried over anhydrous sodium sulfate. Filtered and the resulting solution was added p-toluenesulfonyl chloride (3. 8g, 20mmol), was heated to reflux, the reaction was stirred for 3-4 hours, TLC detection completion of the reaction. The solvent was distilled off under reduced pressure, cooled to room temperature, methanol was added, the precipitated solid, cooled to square ° C, allowed to stand overnight. Filtered, the filter cake washed twice with cold methanol and vacuum dried to give an off-white solid 1- methyl-3-methyl-4-hydroxy-phenoxy-isoquinoline -7- (VI) I. 86g, yield 60.2 %; EI-MS m / z:.. 310 [M + H] +, 1H NMR (DMS0-d6) δ 2.90 (s, 3H), 4.05 (s, 3H), 7 17-7 26 (m, 3H ), 7. 49-7. 61 (m, 4H), 8. 38 (d, J = 9. 0,1H), 11. 7 (s, 1H) 〇
Example VI:
in the reaction flask with magnetic stirring and pressure to join I- methyl-3-methyl-4-hydroxy-7-phenoxyheptanoate isoquinoline (VI) (1.55g, 5mmol), glycine (I. 13g, 15mmol) and sodium methoxide (3. 25g, 6mmol) in methanol (30mL).Sealed, slowly heated to 120 ° C, the reaction was stirred for 8-10 hours to complete the reaction by TLC. Cooled to room temperature, solid precipitated. Filtration, and the resulting solid was recrystallized from methanol, acetone and then beating the resulting solid was dried under vacuum to give a white solid Connaught orlistat 1.40g, yield 79.5%;
EI-MS m / z: 353 [M + H] +,
1H NMR (DMS0-d6) S2.72 (s, 3H), 3 · 99 (d, J = 6 · 0, 2H), 7 · 18-7. 28 (m, 3H), 7 · 49-7. 63 (m, 4H), 8 · 31 (d, J = 8 · 8,1H), 9 · 08 (s, lH), 13.41 (brs, lH).
PATENT
Example 10. Preparation of Compound A
a) 5-Phenoxyphthalide
[0200] A reactor was charged with DMF (68 Kg), and stirring was initiated. The reactor was then charged with phenol (51 Kg), acetylacetone (8 Kg), 5-bromophthalide (85 Kg), copper bromide (9 Kg), and potassium carbonate (77 Kg). The mixture was heated above 85 °C and maintained until reaction completion and then cooled. Water was added. Solid was filtered and washed with water. Solid was dissolved in dichloromethane, and washed with aqueous HCl and then with water. Solvent was removed under pressure and methanol was added. The mixture was stirred and filtered. Solid was washed with methanol and dried in an oven giving 5- phenoxyphthalide (Yield: 72%, HPLC: 99.6%). b) 2-Chloromethyl-4-phenoxybenzoic acid methyl ester
[0201] A reactor was charged with toluene (24 Kg), and stirring was initiated. The reactor was then charged with 5-phenoxyphthalide (56 Kg), thionyl chloride (41 Kg), trimethyl borate (1
Kg), dichlorotriphenylphosphorane (2.5 Kg), and potassium carbonate (77 Kg). The mixture was heated to reflux until reaction completion and solvent was removed leaving 2-chloromethyl-4- phenoxybenzoyl chloride. Methanol was charged and the mixture was heated above 50 °C until reaction completion. Solvent was removed and replaced with DMF. This solution of the product methyl 2-chloromethyl-4-phenoxybenzoic acid methyl ester in DMF was used directly in the next step (HPLC: 85%). c) 4-Hydroxy-7-phenoxyisoquinoline-3-carboxylic acid methyl ester (la)
[0202] A reactor was charged with a solution of 2-chloromethyl-4-phenoxybenzoic acid methyl ester (~68 Kg) in DMF, and stirring was initiated. The reactor was then charged with p- toluenesulfonylglycine methyl ester (66 Kg), potassium carbonate (60 Kg), and sodium iodide (4 Kg). The mixture was heated to at least 50 °C until reaction completion. The mixture was cooled. Sodium methoxide in methanol was charged and the mixture was stirred until reaction completion. Acetic acid and water were added, and the mixture was stirred, filtered and washed with water. Solid was purified by acetone trituration and dried in an oven giving la (Yield from step b): 58%; HPLC: 99.4%). 1H NMR (200 MHz, DMSO-d6) δ 11.60 (s, 1 H), 8.74 (s, 1H),
8.32 (d, J = 9.0 Hz, 1 H), 7.60 (dd, J = 2.3 & 9.0 Hz, 1H), 7.49 (m, 3 H), 7.24 (m, 3 H), 3.96 (s, 3 H); MS-(+)-ion M+l = 296.09 d) Methyl l-((dimethylamino)methyl)-4-hydroxy-7-phenoxyisoquinoline-3-carboxylate
(lb)
[0203] A flask was charged with la (29.5 g) and acetic acid (44.3 g ± 5%), and then stirred. Bis-dimethylaminomethane (12.8 g ± 2%) was slowly added. The mixture was heated to 55 ± 5 °C and maintained until reaction completion. The reaction product was evaluated by MS, HPLC and 1H NMR. 1H NMR (200 MHz, DMSO-d6) δ 11.7 (s, 1 H), 8.38 (d, J = 9.0 Hz, 1 H), 7.61 (dd, J = 9.0, 2.7 Hz, 1 H), 7.49 (m, 3 H), 7.21 (m, 3 H), 5.34 (s, 2 H), 3.97 (s, 3 H), 1.98 (s, 3 H); MS-(+)-ion M+l = 368.12. e) Methyl l-((acetoxy)methyl)-4-hydroxy-7-phenoxyisoquinoline-3-carboxylate (lc)
[0204] The solution of lb from a) above was cooled below 25 °C, at which time acetic anhydride (28.6 g ± 3.5 %) was added to maintain temperature below 50 °C. The resulting mixture was heated to 100 ± 5 °C until reaction completion.
[0205] The solution of lc and Id from above was cooled to less than 65 ± 5 °C. Water (250 mL) was slowly added. The mixture was then cooled to below 20 ± 5 °C and filtered. The wet cake was washed with water (3 x 50 mL) and added to a new flask. Dichloromethane (90 mL) and water (30 mL) were added, and the resulting mixture was stirred. The dichloromethane layer was separated and evaluated by HPLC.
[0206] The organic layer was added to a flask and cooled 5 ± 5 °C. Morpholine was added and the mixture was stirred until reaction completion. Solvent was replaced with acetone/methanol mixture. After cooling, compound lc precipitated and was filtered, washed and dried in an oven (Yield: 81%, HPLC: >99.7%). 1H NMR (200 MHz, DMSO-d6) δ 11.6 (S, 1 H), 8.31 (d, J = 9.0 Hz, 1 H), 7.87 (d, J = 2.3 Hz, 1 H), 7.49 (m, 3 H), 7.24 (m, 3 H), 3.95 (s, 3 H), 3.68 (s, 2H), 2.08 (s, 6 H); MS-(+)-ion M+l = 357.17. f) Methyl 4-hydroxy-l-methyl-7-phenoxyisoquinoline-3-carboxylate (le)
[0207] A reactor was charged with lc (16.0 g), Pd/C (2.08 g), anhydrous Na2C03 (2.56 g) and ethyl acetate (120 mL). The flask was vacuum-purged with nitrogen (3X) and vacuum-purged with hydrogen (3X). The flask was then pressurized with hydrogen and stirred at about 60 °C until completion of reaction. The flask was cooled to 20-25 °C, the pressure released to ambient, the head space purged with nitrogen three times and mixture was filtered. The filtrate was concentrated. Methanol was added. The mixture was stirred and then cooled. Product precipitated and was filtered and dried in an oven (Yield: 90%, HPLC: 99.7%). g) [(4-Hydroxy-l-methyl-7-phenoxy-isoquinoline-3-carbonyl)-amino]-acetic acid
(Compound A)
[0208] A pressure flask was charged with le (30.92 g), glycine (22.52 g), methanol (155 mL), sodium methoxide solution (64.81 g) and sealed (as an alternative, sodium glycinate was used in place of glycine and sodium methoxide). The reaction was heated to about 110 °C until reaction was complete. The mixture was cooled, filtered, washed with methanol, dried under vacuum, dissolved in water and washed with ethyl acetate. The ethyl acetate was removed and to the resulting aqueous layer an acetic acid (18.0 g) solution was added. The suspension was stirred at room temperature, filtered, and the solid washed with water (3 x 30 mL), cold acetone (5-10 °C, 2 x 20 mL), and dried under vacuum to obtain Compound A (Yield: 86.1%, HPLC: 99.8%). Example 11. Biological Testing
[0209] The solid forms provided herein can be used for inhibiting HIF hydroxylase activity, thereby increasing the stability and/or activity of hypoxia inducible factor (HIF), and can be used to treat and prevent HIF-associated conditions and disorders (see, e.g., U.S. Patent No. 7,323,475, U.S. Patent Application Publication No. 2007/0004627, U.S. Patent Application Publication No. 2006/0276477, and U.S. Patent Application Publication No. 2007/0259960, incorporated by reference herein).
SYNTHESIS……..http://zliming2004.lofter.com/post/1cc9dc55_79ad5d8
Condensation of 5-bromophthalide (I) with phenol (II) in the presence of K2CO3, CuBr and acetylacetone in DMF gives 5-phenoxyphthalide (III), which upon lactone ring opening using SOCl2, Ph3PCl2, B(OMe)3 and K2CO3 in refluxing toluene yields 2-chloromethyl-4-phenoxybenzoyl chloride (IV). Esterification of acid chloride (IV) with MeOH at 50 °C furnishes the methyl ester (V), which is then condensed with methyl N-tosylglycinate (VI) in the presence of K2CO3 and NaI in DMF at 50 °C to afford N-substituted aminoester (VII). Cyclization of the intermediate diester (VII) using NaOMe in MeOH leads to methyl 4-hydroxy-7-phenoxyisoquinoline-3-carboxylate (VIII), which is submitted to Mannich reaction with bis-dimethylaminomethane (IX) in the presence of AcOH at 57 °C to provide the dimethylaminomethyl compound (X). Treatment of amine (X) with Ac2O at 103 °C, followed by selective hydrolysis of the phenolic acetate with morpholine leads to methyl 1-acetoxymethyl-4-hydroxy-7-phenoxyisoquinoline-3-carboxylate (XI). Hydrogenolysis of the benzylic acetate (XII) in the presence of Pd/C and Na2CO3 in EtOAc yields methyl 4-hydroxy-1-methyl-7-phenoxyisoquinoline-3-carboylate (XII), which finally couples with glycine (XIII) in the presence of NaOMe in MeOH at 110 °C to afford the target roxadustat (1-3).
Cyclization of 4-phenoxyphthalic acid (I) with glycine (II) at 215 °C gives the phthalimide (III), which upon esterification with MeOH and H2SO4 at reflux yields methyl ester (IV). Subsequent rearrangement of phthalimidoacetate (IV) by means of Na in BuOH at 97 °C, followed by flash chromatography provides the isoquinoline-2-carboxylate (V). Bromination of intermediate (V) using POBr3 and NaHCO3 in acetonitrile leads to butyl 8-bromo-3-hydroxy-6-phenoxy-isoquinoline-2-carboxylate (VI), which upon hydrolysis with NaOH in refluxing H2O/EtOH furnishes carboxylic acid (VII). Substitution of bromine in intermediate (VII) using MeI and BuLi in THF at -78 °C, followed by alkylation with PhCH2Br in the presence of K2CO3 in refluxing acetone affords the 2-methyl isoquinoline (VIII). Ester hydrolysis in intermediate (VIII) using KOH in MeOH gives the corresponding carboxylic acid (IX), which is then activated with i-BuOCOCl and Et3N in CH2Cl2, followed by coupling with benzyl glycinate hydrochloride (X) to yield benzylated roxadustat (XI). Finally, debenzylation of intermediate (XI) with H2 over Pd/C in EtOAc/MeOH provides the title compound (1).
Condensation of 4-nitro-ortho-phthalonitrile (I) with phenol (II) in the presence of K2CO3 in DMSO gives 4-phenoxy-ortho-phthalonitrile (III) (1), which upon hydrolysis with NaOH (1) or KOH (2) in refluxing MeOH yields 4-phenoxyphthalic acid (IV) (1,2). Dehydration of dicarboxylic acid (IV) using Ac2O and AcOH at reflux furnishes the phthalic anhydride (V), which is then condensed with methyl 2-isocyanoacetate (VI) using DBU in THF to provide oxazole derivative (VII). Rearrangement of intermediate (VII) with HCl in MeOH at 60 °C leads to isoquinoline derivative (VIII), which is partially chlorinated by means of POCl3 at 70 °C to afford 1-chloro-isoquinoline derivative (IX). Substitution of chlorine in intermediate (IX) using Me3B, Pd(PPh3)4 and K2CO3 in refluxing dioxane gives methyl 4-hydroxy-1-methyl-7-phenoxy-3-carboxylate (X), which is then hydrolyzed with aqueous NaOH in refluxing EtOH to yield the carboxylic acid (XI). Coupling of carboxylic acid (XI) with methyl glycinate hydrochloride (XII) by means of PyBOP, (i-Pr)2NH and Et3N in CH2Cl2 yields roxadustat methyl ester (XII), which is finally hydrolyzed with aqueous NaOH in THF to afford the target roxadustat (1).
CLIPS
1: Besarab A, Provenzano R, Hertel J, Zabaneh R, Klaus SJ, Lee T, Leong R, Hemmerich S, Yu KH, Neff TB. Randomized placebo-controlled dose-ranging and pharmacodynamics study of roxadustat (FG-4592) to treat anemia in nondialysis-dependent chronic kidney disease (NDD-CKD) patients. Nephrol Dial Transplant. 2015 Oct;30(10):1665-73. doi: 10.1093/ndt/gfv302. Epub 2015 Aug 3. PubMed PMID: 26238121; PubMed Central PMCID: PMC4569392.
2: Forristal CE, Levesque JP. Targeting the hypoxia-sensing pathway in clinical hematology. Stem Cells Transl Med. 2014 Feb;3(2):135-40. doi: 10.5966/sctm.2013-0134. Epub 2013 Dec 26. PubMed PMID: 24371328; PubMed Central PMCID: PMC3925058.
3: Bouchie A. First-in-class anemia drug takes aim at Amgen’s dominion. Nat Biotechnol. 2013 Nov;31(11):948-9. doi: 10.1038/nbt1113-948b. PubMed PMID: 24213751.
4: Flight MH. Deal watch: AstraZeneca bets on FibroGen’s anaemia drug. Nat Rev Drug Discov. 2013 Oct;12(10):730. doi: 10.1038/nrd4135. PubMed PMID: 24080688.
5: Beuck S, Schänzer W, Thevis M. Hypoxia-inducible factor stabilizers and other small-molecule erythropoiesis-stimulating agents in current and preventive doping analysis. Drug Test Anal. 2012 Nov;4(11):830-45. doi: 10.1002/dta.390. Epub 2012 Feb 24. Review. PubMed PMID: 22362605.
6: Cases A. The latest advances in kidney diseases and related disorders. Drug News Perspect. 2007 Dec;20(10):647-54. PubMed PMID: 18301799.

UCT’s H3D is a center of excellence for research and innovation with an already strong track record in malaria drug discovery. The vision of H3D is to be the leading organization for integrated drug discovery and development on the African continent.

H3D is Africa’s first integrated drug discovery and development centre. The Centre was founded at the University of Cape Town in April 2011 and pioneers world-class drug discovery in Africa.
To be the leading organisation for integrated drug discovery and development from Africa, addressing global unmet medical needs.
To discover and develop innovative medicines for unmet medical needs on the African continent and beyond, by performing state-of-the-art research and development and bridging the gap between basic science and clinical studies.
We embrace partnerships with local and international governments, pharmaceutical companies, academia, and the private sector, as well as not-for-profit and philanthropic organisations, while training scientists to be world experts in the field.
The H3D collaboration with the Medicines for Malaria Venture (MMV) focuses on delivering potential agents against malaria that will be affordable and safe to use. In line with the global aim to eradicate malaria, projects are pursued that not only eliminates blood-stage Plasmodium falciparum and Plasmodium vivax infection, but also acts against liver stages and blocks transmission of the infection. The projects embrace multidisciplinary activities to optimise hit compounds from screening libraries through the drug discovery pipeline and deliver clinical candidates.
Merck Serono Announces Recipients of the Second Annual €1 Million Grant for Multiple Sclerosis Innovation
Merck signed a research agreement with the University of Cape Town (UCT), South Africa, to co-develop a new R&D platform. It aims at identifying new lead programs for potential treatments against malaria, with the potential to expand it to other tropical diseases. It combines Merck’s R&D expertise and the drug discovery capabilities of the UCT Drug Discovery and Development Centre, H3D.
UCT’s H3D is a center of excellence for research and innovation with an already strong track record in malaria drug discovery. The vision of H3D is to be the leading organization for integrated drug discovery and development on the African continent. They say that working with partners like Merck is critical to build up a comprehensive pipeline to tackle malaria and related infectious diseases.
Physical Address:
Department of Chemistry
7.32 H3D Lab Suite, PD Hahn Building, Level 7
North Lane off Ring Road
Upper Campus, University of Cape Town
Rondebosch, 7700, South Africa
Postal Address:
University of Cape Town
Private Bag X3
Rondebosch 7701
South Africa


![]()

//////H3D, Africa, integrated drug discovery and development centre, University of Cape Town
CAS 89647-87-0
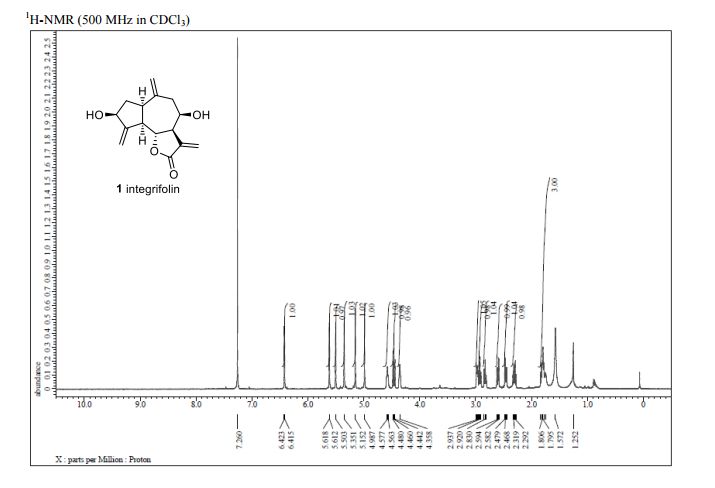
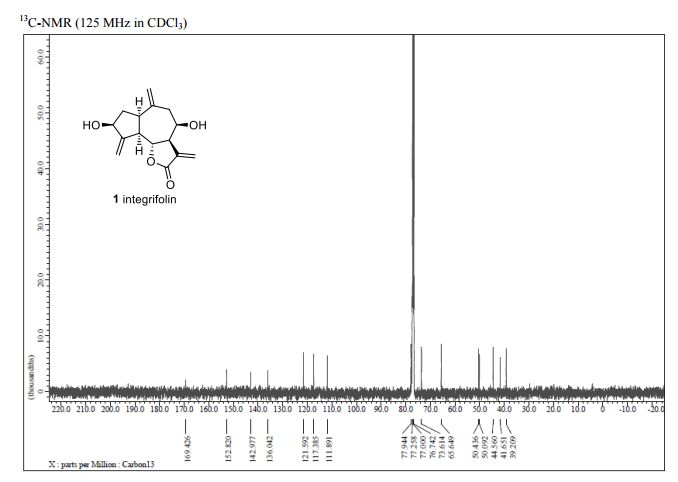
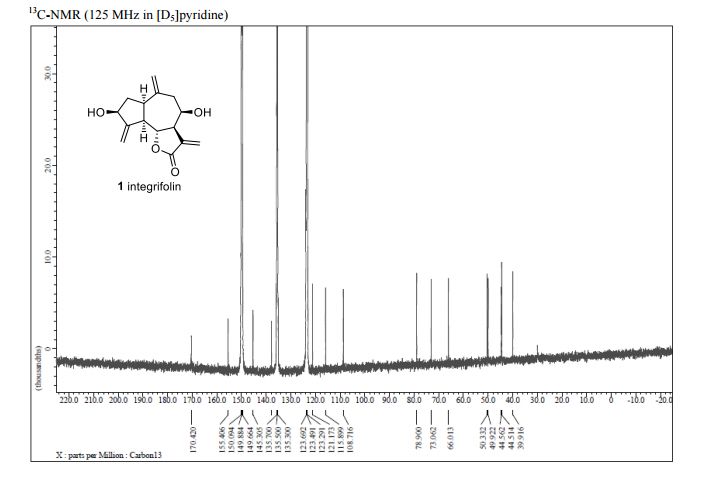
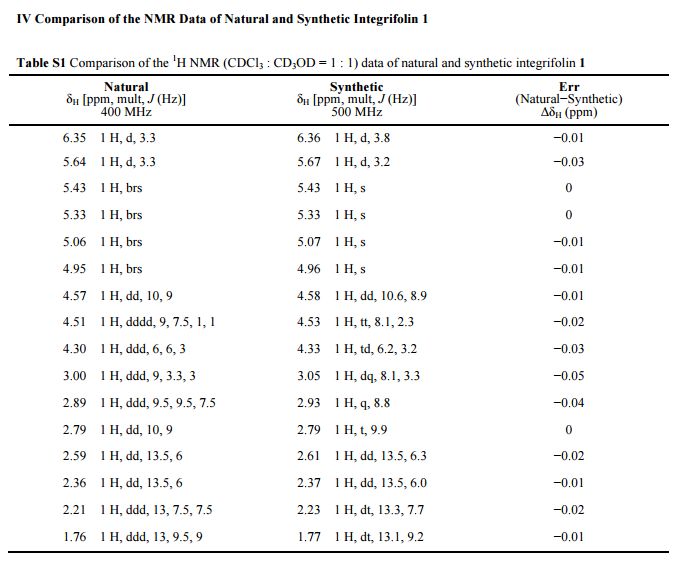
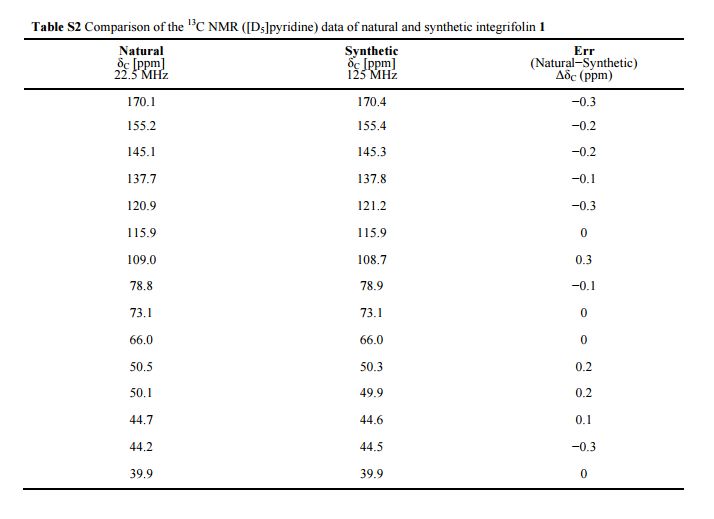
PATENT

Paper
 Journal title : Bulletin of the Korean Chemical Society Volume 33, Issue 1, 2012, pp.337-340 Publisher : Korean Chemical Society DOI : 10.5012/bkcs.2012.33.1.337
Journal title : Bulletin of the Korean Chemical Society Volume 33, Issue 1, 2012, pp.337-340 Publisher : Korean Chemical Society DOI : 10.5012/bkcs.2012.33.1.337BLOG POST FROM CHEMISTRY VIEWS, WILEY

Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
Compound from plants keeps human cancer cells from multipying
Read more at Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
Weight control is an important concern of human beings, both for medical (pharmaceutical and/or nutraceutical) as well as non-therapeutic, e.g. cosmetic, reasons. More importantly, excessive accumulation of body fat (i.e. obesity (= adiposity), especially with excessive fat in the ventral region and surrounding the viscera) can be dangerous and has been linked to health problems such as type II diabetes, hypertension, heart disease, atherosclerosis (where more than two of the preceding disorders are present, the condition is often called “Metabolic Syndrome” or “syndrome X”), hyperlipidemia, coronary heart disease, stroke, breast and colon cancer, sleep apnoea, gallbladder disease, reproductive disorders such as polycystic ovarian syndrome, gastroesophageal reflux disease, increased incidence of complications of general anesthesia, fatty liver, gout or thromboembolism (see, e.g., Kopelman, Nature 404: 635-43 (2000)). Obesity reduces life-span and carries a serious risk of the co-morbidities listed above, as well disorders such as infections, varicose veins,
acanthosis nigricans, eczema, exercise intolerance, insulin resistance, hypertension hypercholesterolemia, cholelithiasis, orthopedic injury, and thromboembolic disease (Rissanen et al, Br. Med. J. 301 : 835-7 (1990)). Obesity is one of the main factors in the development of cardiovascular diseases. As a side effect the levels of cholesterol, blood pressure, blood sugar and uric acid in obese people are usually higher than those of persons of normal weight. The morbidity from coronary heart disease among the overweight people is increased as well. Among the people aged 40-50, mortality will rise about 1% when body weight increases by 0.5 kg and the death rate will increase 74% when body weight exceeds 25% of the standard. The prevalence of obesity in the United States has more than doubled since the turn of the last century (whole population) and more than tripled within the last 30 years among children aged from 6 to 11. This problem more and more becomes a disease risk also in Europe. In Germany, particularly many people have been found to suffer from overweight recently, already 25% of the young people, children and adolescents there are affected by obesity and related disorders. Furthermore, being overweight is considered by the majority of the Western population as unattractive.
Overweight and obesity result from an imbalance between the calories consumed and the calories used by the body. When the calories consumed exceed the calories burned, the body is in positive energy balance and over time weight gain will occur. The excess calories are stored in the fat cells. When the calories burned exceed the calories consumed, the body is in negative energy balance and over time weight loss will occur.
Determinants of obesity include social factors, psychological factors, genetic factors, developmental factors and decreased physical activity. Some components of a comprehensive weight loss programs include medical assessment, behavioural and dietary modification, nutrition education, mental and cognitive restructuring, increased physical activity, and long term follow-up.
An increasing interest by consumers in the maintenance or reduction of their body weight can be found. This leads to a demand for products useful for these purposes. Preferred are such food products which can conveniently be consumed as part of the daily diet, for example meal replacer products, such as meal replacer bars and beverages. These are usually designed for use as a single-serving food product to replace one or two meals a day.
An issue is that often a saturating effect is missed when such products are consumed, resulting in hunger feelings only a relatively short time after consummation or even in the lack of a saturation feeling already directly after consummation.
Summing up, there remains a need for new safe and effective compositions for promoting weight loss and/or loss of body fat in subjects such as humans. The problem to be solved by the present invention is therefore to find compositions or compounds useful in the treatment of obesity; and/or for improving the total cholesterol HDIJLDL ratio.
Phytochemistry provides a large pool of compounds and compositions to be looked at whether they are able to solve this problem.
The present invention provides methods and compositions useful in the control, treatment and prevention of obesity and obesity-related conditions, disorders, and diseases; and/or and/or for improving the total cholesterol HDL/LDL ratio.
Rosinski, G., et al., Endocrinological Frontiers in Phyiological Insect Ecology, Wroclow Technical University Press, Wroclow 1989, describe that certain tricyclic sequiterpene lactones, such as grossheimin and repin, showed inhibition of larval growth and antifeeding activity in Mealworm (Tenebrio σιοΐϊίοή. Grossheimin shows no anti-feeding but little decrease of absorption of digested food constituents and a little decrease in efficiency in digesting. Repin exhibit low effects at all. Both compounds show no effect on lipid levels in blood.
Shimoda, H., et al, Bioinorganic & Medicinal Chemistry Letters 13 (2003), 223-228, describe that methanolic extracts from Artichoke (Cynara sclolymus L.) with cynaropicrin, aguerin B and grossheimin as components and certain sesquiterpene glycosides suppress serum triglyceride elevation in olive oil-loaded mice. Some of these compounds exhibit a moderate short term (2 hours after olive oil administration) anti-hyperlipidemic activity presented as a lowering of the serum triglyceride (serum TG) concentrations, the long term (6 hours) show in the case of cynaropicrin and aguerine B an increase of the serum TG. Furthermore the authors present data of the gastric emptying (GE) of a methanolic ectract of artichoke. They determine a significantly inhibited GE. However, as shown below, this mechanism is not an explanation for the anti obesity effect shown in the present invention (see Example 1 ).
Fritzsche, J., et al., Eur. Food Res. Technol. 215, 149-157 (2002) describe the effect of certain isolated artichoke leaflet extract components with cholesterol lowering potential. Ahn, E.M-., et al, Arch Pharm. res. 29(1 1 ), 937-941 , 2006, shows ACAT inhibitory activity for two sesquiterpene lactones. KR 20040070985 also shows an effect of certain sesquiterpene lactone derivatives on cholesterol biosynthesis involved enzymes. Gebhard, R., Phytother. Res. 16, 368-372 (2002) and J. Pharmacol. Exp. Ther. 286(3), 1 122-1 128 (1998), shows
enforcement of cholesterol biosynthesis inhibition in HepG2 cells by artichoke extracts. WO 2007/006391 also claims reduction in cholesterol by certain Cynara scolymus variety extracts.
Other reported activities of tricyclic sesquiterpene lactones are antioxidant activity (European Food Research & Technology (2002), 215(2): 149-157), inhibitors of NF kb (Food Style 21 (2007), 1 1 (6): 54-56; JP 2006-206532), serum triglyceride increase-inhibitory effect (Kagaku Kogyo (2006), 57(10): 740-745), hypoglycaemic effect (J. Trad. Med. (2003), 20(2): 57-61), bitter taste (DE 2654184). Any beneficial effects are included in this invention by reference.
None of the documents suggest that a control and treatment of obesity and body fat in warmblooded animals might be possible.
Cynaropicrin, a tricyclic sesquiterpene lactone causes in vivo a strong weight loss. More surprisingly it was found that this effect is not correlated to a decrease in food intake. The weight balance is not affected by reduction of assimilation efficiency; the decrease of body fat and body weight is presumably caused by effects on energy metabolism. Surprisingly, it was found in addition that cynaropicrin also allows for improving the total cholesterol HDL7LDL ratio
Tricyclic sequiterpene lactones or known ingredients of plants of the subclass Asterides, especially from the family of Asteraceae, more specifically from species of the genera of the list consisting of Achilea, Acroptilon, Agranthus, Ainsliaea, Ajania, Amberboa, Andryala, Artemisia, Aster, Bisphopanthus, Brachylaena, Calea, Calycocorsus, Cartolepsis, Centaurea, Cheirolophus, Chrysanthemum, Cousinia, Crepis, Cynara, Eupatorium, Greenmaniella, Grossheimia, Hemistaptia, Ixeris, Jurinea, Lapsana, Lasiolaena, Liatris, Lychnophora, Macroclinidium, Mikania, Otanthus, Pleiotaxis, Prenanthes, Pseudostifftia, Ptilostemon,
Rhaponticum, Santolina, Saussurea, Serratula, Sonchus, Stevia, Taeckholmia, Tanacetum, Tricholepis, Vernonia, Volutarella, Zaluzania; even more specifically from species of the list consisting of Achillea clypeolata, Achillea collina, Acroptilon repens, Agrianthus pungens, Ainsliaea fragrans, Ajania fastigiata, Ajania fruticulosa, Amberboa lippi, Amberboa muricata, Amberboa ramose**, Amberboa tubuliflora and other Amberboa spp.*, Andryala integrifolia, Andryala pinnatifida, Artemisia absinthium, Artemisia cana, Artemisia douglasiana, Artemisia fastigiata, Artemisia franserioides, Artemisia montana, Artemisia sylvatica, Artemisia
tripartita, Aster auriculatus, Bishopanthus soliceps, Brachylaena nereifolia, Brachylaena perrieri, Calea jamaicensis, Calea solidaginea, Calycocorsus stipitatus, Cartolepsis intermedia, Centaurea babylonica, Centaurea bella, Centaurea canariensis*, Centaurea clementei, Centaurea conicum, Centaurea dealbata, Centaurea declinata, Centaurea glastifolia, Centaurea hermanii, Centaurea hyrcanica, Centaurea intermedia, Centaurea janeri, Centaurea kalscyi, Centaurea kandavanensis, Centaurea kotschyi, Centaurea linifolia, Centaurea macrocephala, Centaurea musimomum, Centaurea nicolai, Centaurea pabotii, Centaurea pseudosinaica, Centaurea repens, Centaurea salonitana, Centaurea scoparia, Centaurea sinaica, Centaurea solstitialis, Centaurea tweediei and other Centaurea spp. *, Cheirolophus uliginosus, Chrysanthemum boreale, Cousin ia canescens, Cousinia conifera, Cousinia picheriana, Cousinia piptocephala, Crepis capillaris, Crepis conyzifolia, Crepis crocea, Crepis japonica, Crepis pyrenaica, Crepis tectorum, Crepis virens, Crepis zacintha, Cynara alba, Cynara algarbiensis, Cynara auranitica, Cynara baetica, Cynara cardunculus, Cynara cornigera, Cynara cyrenaica, Cynara humilis, Cynara hystrix, Cynara syriaca, Cynara scolymus**, Cynara sibthorpiana and other Cynara spp.*, Eupatorium anomalum,
Eupatorium chinense, Eupatorium lindleyanum, Eupatorium mohrii, Eupatorium
rotundifolium, Eupatorium semialatum, Greenmaniella resinosa, Grossheimia
macrocephala** and other Grossheimia spp. *, Hemisteptia lyrata, Ixeris chinensis, Ixeris debilis, Ixeris dentata, Ixeris repens, Ixeris stolonifera, Jurinea carduiformis, Jurinea derderioides, Jurinea maxima, Lapsana capillaris, Lapsana communis, Lasiolaena morii, Lasiolaena santosii, Liatris chapmanii, Liatris gracilis, Liatris pycnostachya, Lychnophora blanchetii, Macroclinidium trilobum, Mikania hoehnei, Otanthus maritimus, Pleiotaxis rugosa, Prenanthes acerifolia, Pseudostifftia kingii, Ptilostemon diacanthus, Ptilostemon
gnaphaloides, Rhaponticum serratuloides, Santolina jamaicensis, Saussurea affinis,
Saussurea elegans, Saussurea involucrata, Saussurea laniceps, Saussurea neopulchella** and other Sauusurea spp. *, Serratula strangulata, Sonchus arborea, Stevia sanguinea, Taeckholmia arborea, Taeckholmia pinnata, Tanacetum fruticulosum, Tanacetum
parthenium, Tricholepis glaberrima** and other Tricholepsis spp. *, Vernonia arkansana, Vernonia nitidula, Vernonia noveboracensis, Vernonia profuga, Vernonia sublutea,
Volutarella divaricata, Zaiuzania resinosa; and can potentially be isolated from any part of the plants. Those genera and/or species marked with an asterisk (*) and especially those species marked with two asterisks (**) are especially preferred.
Appropriate plant material can be obtained from various sources, e.g. from:
Alfred Galke GmbH, Gittelde/Harz, Germany; Miiggenburg Pflanzliche Rohstoffe, Bad Bramstedt, Germany; Friedrich Nature Discovery, Euskirchen, Germany; VitaPlant AG, Uttwil, Switzerland; Amoros Nature SL, Hostalric, Spain.
(±)-Integrifolin

Banksia integrifolia
Coast Banksia
Family: Proteaceae
Banksia integrifolia is a tall shrub or small tree 6 – 16m tall. It is common in sandy coastal areas, but also grows in the forests of tablelands. The light grey bark is hard and rough.
Mature leaves 5 -10 cm long, are stiff, entire (untoothed), dull dark green above and hairy-white underneath. They are generally lanceolate. Younger leaves are irregularly toothed and shorter than the mature leaves. The species name ‘integrifolia’ means whole-leaved.
The pale yellow flower spikes of Banksia integrifolia range from 7-14cm long and 7cm wide. The bent styles emerge from individual flowers on the spike, straightening and spreading.
A short time after flowering, the seed pods protrude cleanly from the woody cone and open to shed black, papery, winged seeds.
Banksia integrifolia flowers from January to June.
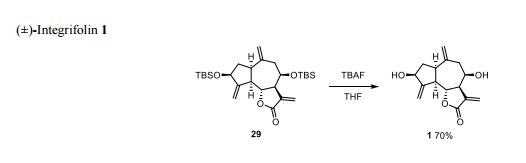

https://www.jstage.jst.go.jp/article/cpb1958/33/8/33_8_3361/_pdf
PAPER
http://onlinelibrary.wiley.com/doi/10.1002/chem.201601275/abstract
///////(±)-Integrifolin, human cancer cells, multipying
C=C1C(=O)O[C@@H]2[C@H]3C(=C)[C@@H](O)C[C@H]3C(=C)C[C@@H](O)[C@@H]12


(R)-3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide
3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide
(3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide)
(R)-3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide
cas 866772-52-3
LBX-192
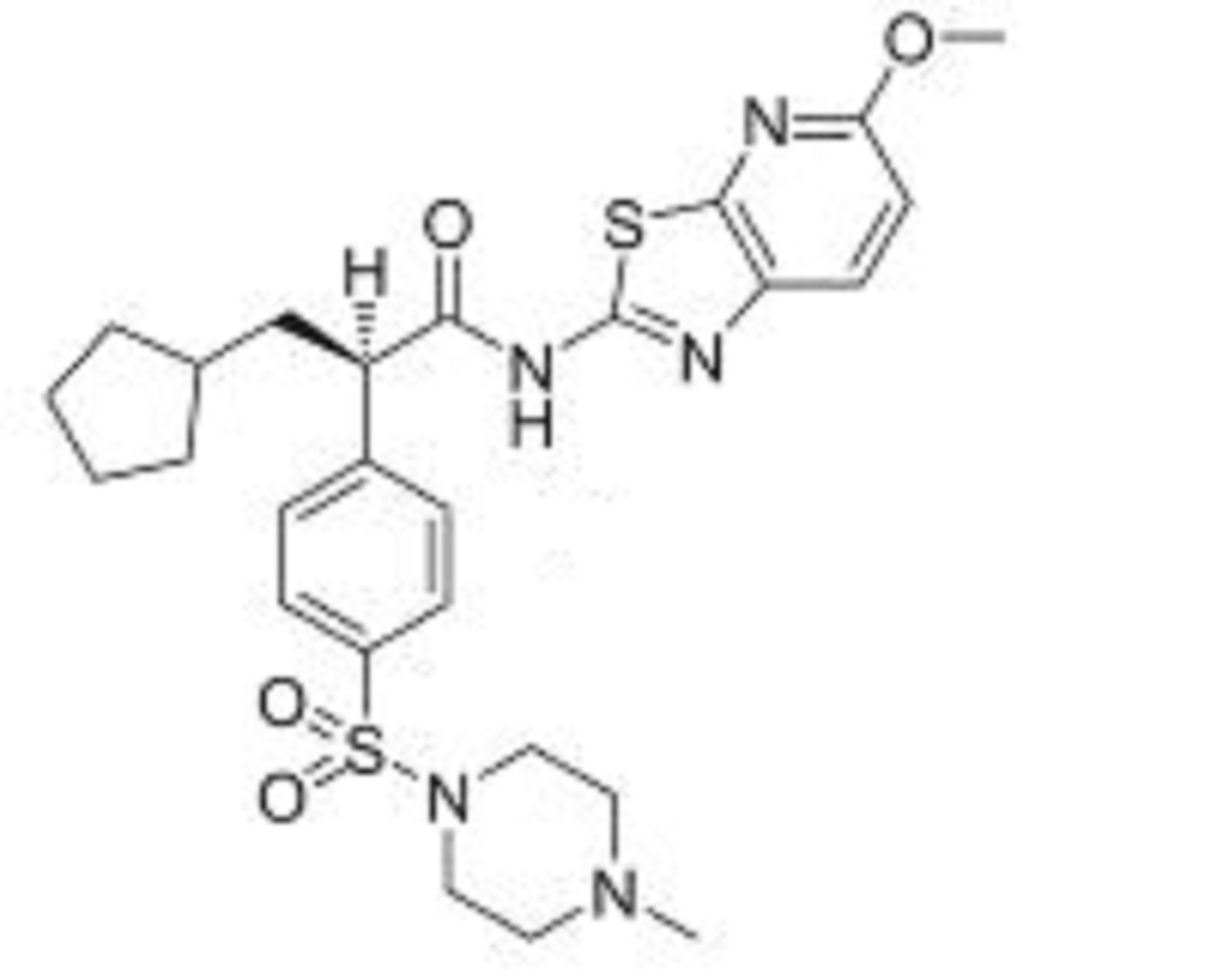
R(−) 3-cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide
R(−)17c BELOW

| Inventors | Gregory Raymond Bebernitz, Ramesh Chandra Gupta, Vikrant Vijaykumar Jagtap, Appaji Baburao Mandhare, Davinder Tuli, |
| Original Assignee | Novartis Ag
|
| Molecular Formula: | C26H33N5O4S2 |
|---|---|
| Molecular Weight: | 543.70132 g/mol |




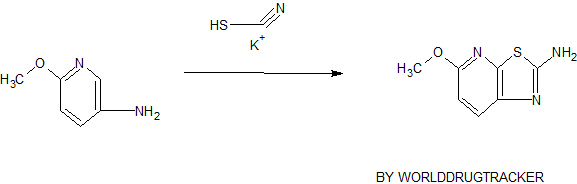
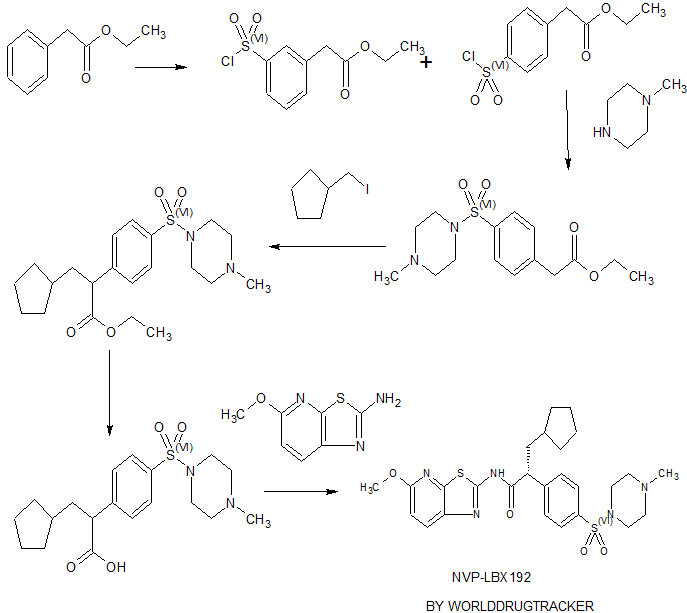
https://acs.confex.com/acs/nerm09/webprogram/Paper75087.html
Sulfonamide-Thiazolpyridine Derivatives, Glucokinase Activators, Treatment Of Type 2 Diabetes
2009 52 (19) 6142 – 6152
Investigation of functionally liver selective glucokinase activators for the treatment of type 2 diabetes
Journal of Medicinal Chemistry
Bebernitz GR, Beaulieu V, Dale BA, Deacon R, Duttaroy A, Gao JP, Grondine MS, Gupta RC, Kakmak M, Kavana M, Kirman LC, Liang JS, Maniara WM, Munshi S, Nadkarni SS, Schuster HF, Stams T, Denny IS, Taslimi PM, Vash B, Caplan SL
2010 240th (August 22) Medi-198
Glucokinase activators with improved physicochemicalproperties and off target effects
American Chemical Society National Meeting and Exposition
Kirman LC, Schuster HF, Grondine MS et al
2010 240th (August 22) Medi-197
Investigation of functionally liver selective glucokinase activators
American Chemical Society National Meeting and Exposition
Schuster HF, Kirman LC, Bebernitz GC et al
PATENT
http://www.google.com/patents/US7750020
EXAMPLE 1 3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide

A. Phenylacetic Acid Ethyl Ester
A solution of phenylacetic acid (50 g, 0.36 mol) in ethanol (150 mL) is treated with catalytic amount of sulfuric acid (4 mL). The reaction mixture is refluxed for 4 h. The reaction is then concentrated in vacuo. The residue is dissolved in diethyl ether (300 mL) and washed with saturated aqueous sodium bicarbonate solution (2×50 mL) and water (1×100 mL). The organic layer dried over sodium sulfate filtered and concentrated in vacuo to give phenylacetic acid ethyl ester as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 1.2 (t, J=7.2, 3H), 3.6 (s, 2H), 4.1 (q, J=7.2, 2H), 7.3 (m, 5H); MS 165 [M+1]+.
B. (4-Chlorosulfonyl-phenyl)-acetic acid ethyl ester
To a cooled chlorosulfonic acid (83.83 g, 48 mL, 0.71 mol) under nitrogen is added the title A compound, phenylacetic acid ethyl ester (59 g, 0.35 mol) over a period of 1 h. Reaction temperature is brought to RT (28° C.), then heated to 70° C., maintaining it at this temperature for 1 h while stirring. Reaction is cooled to RT and poured over saturated aqueous sodium chloride solution (200 mL) followed by extraction with DCM (2×200 mL). The organic layer is washed with water (5×100 mL), followed by saturated aqueous sodium chloride solution (1×150 mL). The organic layer dried over sodium sulfate, filtered and concentrated in vacuo to give crude (4-chlorosulfonyl-phenyl)acetic acid ethyl ester. Further column chromatography over silica gel (60-120 mesh), using 100% hexane afforded pure (4-chlorosulfonyl-phenyl)-acetic acid ethyl ester as a colorless oil.
C. [4-(4-Methyl-piperazine-1-sulfonyl)-phenyl]-acetic acid ethyl ester
A solution of N-methylpiperazine (9.23 g, 10.21 ml, 0.092 mol), DIEA (13 g, 17.4 mL, 0.10 mol) and DCM 80 mL is cooled to 0° C., and to this is added a solution of the title B compound, (4-chlorosulfonyl-phenyl)-acetic acid ethyl ester (22 g, 0.083 mol) in 50 mL of DCM within 30 min. Reaction mixture stirred at 0° C. for 2 h, and the reaction mixture is washed with water (100 mL), followed by 0.1 N aqueous hydrochloric acid solution (1×200 mL). The organic layer dried over sodium sulfate, filtered and concentrated under vacuo to give crude [4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-acetic acid ethyl ester. Column chromatography over silicagel (60-120 mesh), using ethyl acetate afforded pure [4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-acetic acid ethyl ester as white crystalline solid: 1H NMR (400 MHz, CDCl3) δ 1.3 (t, J=7.4, 3H), 2.3 (s, 3H), 2.5 (m, 4H), 3.0 (br s, 4H), 3.7 (s, 2H), 4.2 (q, J=7.4, 2H), 7.4 (d, J=8.3, 2H), 7.7 (d, J=7.3, 2H); MS 327 [M+1]+.
D. 3-Cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid ethyl ester
A solution of the title C compound, [4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-acetic acid ethyl ester (15 g, 0.046 mol) in a mixture of THF (60 mL) and DMTP (10 mL) is cooled to −78° C. under nitrogen. The resulting solution is stirred at −78° C. for 45 min and to this is added LDA (25.6 mL, 6.40 g, 0.059 mol, 25% solution in THF/Hexane). A solution of iodomethylcyclopentane (11.60 g, 0.055 mol) in a mixture of DMTP (12 mL) and THF (20 mL) is added over a period of 15 min at −78° C. and reaction mixture stirred at −78° C. for 3 h further, followed by stirring at 25° C. for 12 h. The reaction mixture is then quenched by the dropwise addition of saturated aqueous ammonium chloride solution (50 mL) and is concentrated in vacuo. The residue is diluted with water (50 mL) and extracted with ethyl acetate (3×100 mL). The organic solution is washed with a saturated aqueous sodium chloride (2×150 mL), dried over sodium sulfate, filtered and concentrated in vacuo. Column chromatography over silica gel (60-120 mesh), using 50% ethyl acetate in hexane as an eluent to afford 3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid ethyl ester as a white solid: 1H NMR (400 MHz, CDCl3) δ 0.9-2.1 (m, 11H), 1.2 (t, J=7.1, 3H), 2.3 (s, 3H), 2.5 (br s, 4H), 3.0 (br s, 4H), 3.6 (m, 1H), 4.1 (q, J=7.1, 2H), 7.5 (d, J=8.3, 2H), 7.7 (d, J=8.3, 2H); MS 409 [M+1]+.
E. 3-Cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid
A solution of the title D compound, 3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid ethyl ester (14 g, 0.034 mol) in methanol:water (30 mL:10 mL) and sodium hydroxide (4.11 g, 0.10 mol) is stirred at 60° C. for 8 h in an oil bath. The methanol is then removed in vacuo at 45-50° C. The residue is diluted with water (25 mL) and extracted with ether (1×40 mL). The aqueous layer is acidified to pH 5 with 3 N aqueous hydrochloric acid solution. The precipitated solid is collected by vacuum filtration, washed with water (20 mL), followed by isopropyl alcohol (20 mL). Finally, solid cake is washed with 100 mL of hexane and dried under vacuum at 40° C. for 6 h to give 3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid as a white solid: 1H NMR (400 MHz, CDCl3) δ 1.1-2.0 (m, 11H), 2.4 (s, 3H), 2.7 (br s, 4H), 3.1 (br s, 4H), 3.6 (m, 1H), 7.5 (d, J=8.3, 2H), 7.6 (d, J=8.3, 2H); MS 381 [M+l]+.
F. 5-Methoxy-thiazolo[5,4-b]pyridin-2-ylamine
A solution of 6-methoxy-pyridin-3-ylamine (5.0 g, 0.0403 mol) in 10 mL of acetic acid is added slowly to a solution of potassium thiocyanate (20 g, 0.205 mol) in 100 mL of acetic acid at 0° C. followed by a solution of bromine (2.5 mL, 0.0488 mol) in 5 mL of acetic acid. The reaction is stirred for 2 h at 0° C. and then allowed to warm to RT. The resulting solid is collected by filtration and washed with acetic acid, then partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The insoluble material is removed by filtration and the organic layer is evaporated and dried to afford 5-methoxy-thiazolo[5,4-b]pyridin-2-ylamine as a tan solid.
G. 3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide
A solution of the title E compound, 3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid (5 g, 0.013 mol) in DCM (250 mL) is cooled to 0° C. and then charged HOBt hydrate (2.66 g, 0.019 mol), followed by EDCI hydrochloride (6 g, 0.031 mol). The reaction mixture is stirred at 0° C. for 5 h. After that the solution of the title F compound, 5-methoxy-thiazolo[5,4-b]pyridin-2-ylamine (2.36 g, 0.013 mol) and D1EA (8 mL, 0.046 mol) in a mixture of DCM (60 mL) and DMF (20 mL) is added dropwise over 30 min. Reaction temperature is maintained at 0° C. for 3 h, then at RT (28° C.) for 3 days. Reaction is diluted with (60 mL) of water and the organic layer is separated and washed with saturated sodium bicarbonate solution (2×50 mL) followed by water washing (2×50 mL) and saturated sodium chloride aqueous solution (1×150 mL). Finally the organic layer is dried over sodium sulfate, filtered, and evaporated under vacuo. The crude product is purified using column chromatography over silica gel (60-120 mesh), using 40% ethyl acetate in hexane as an eluent to afford 3-cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide as a white solid: 1H NMR (400 MHz, CDCl3) δ 0.9-2.1 (m, 11H), 2.2 (s, 3H), 2.5 (br s, 4H), 3.1 (br s, 4H), 3.7 (m, 1H), 4.0 (s, 3H), 6.8 (d, J=8.8, 1H), 7.5 (d, J=8.3, 2H), 7.7 (d, J=8.3, 2H), 7.8 (d, J=8.8, 1H), 8.6 (s, 1H); MS 617 [M+1]+.
H. 3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide dihydrochloride
The title G compound, 3-cyclopentyl-2-(4-methyl piperazinyl sulfonyl)phenyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)propionamide (2.8 g, 0.0051 mol) is added to a cooled solution of 10% hydrochloric acid in isopropanol (3.75 mL). The reaction mixture is stirred at 0° C. for 1 h and then at RT for 2 h. The solid is separated, triturated with 10 mL of isopropanol and collected by vacuum filtration and washed with 50 mL of hexane. The solid is dried at 70° C. for 48 h to afford 3-cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide dihydrochloride as an off white solid.
EXAMPLE 2 (R)-3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide

The title compound is obtained analogously to Example 1 by employing the following additional resolution step:
The racemic title E compound of Example 1,3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid (10 g, 0.026 mol) in 1,4-dioxane (500 mL) is treated in a three necked 1 liter flask, equipped with heating mantle, water condenser, calcium chloride guard tube and mechanical stirrer with 3.18 g (0.026 mol) of (R)-(+)-1-phenylethylamine. This reaction mixture is then refluxed at 100° C. for 1 h. The clear reaction solution is cooled to RT (27° C.) and stirred for 10 h. The crystallized salt is collected by filtration under vacuum, washed with 5 mL of hexane and dried under vacuum to afford salt A.
The salt A is dissolved in 1,4-dioxane (500 mL) and heated at 100° C. for 1 h. The clear reaction solution is cooled to RT (27° C.) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 50 mL of hexane, and dried under vacuum to afford salt B.
The salt B is dissolved in 1,4-dioxane (290 mL) and heated at 100° C. for 1 h. The clear reaction solution is cooled to RT (27° C.) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 30 mL of hexane, and dried under vacuum to afford salt C.
The salt C is dissolved in 1,4-dioxane (100 mL) and heated at 100° C. for 1 h. The clear reaction solution is cooled to RT (27° C.) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 30 ml of hexane, and dried under vacuum to afford salt D.
The salt D is treated with aqueous hydrochloric acid solution (20 mL, 1 mL of concentrated hydrochloric acid diluted with 100 mL of water) and stirred for 5 min. The white solid precipitates out and is collected by vacuum filtration, washed with 10 mL of cold water, 5 mL of isopropanol and 20 mL of hexane, and dried under vacuum to yield the hydrochloride salt of (R)-(−)-3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid, salt E.
The salt E is neutralized by stirring with aqueous sodium bicarbonate solution (10 mL, 1 g of sodium bicarbonate dissolved in 120 mL of water) for 5 min. The precipitated solid is collected by filtration, washed with 10 mL of cold water, 100 mL of hexane, and dried to afford (R)-(−)-3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid: m.p. 202.2-203.4° C.
Alternatively, the title compound may be obtained by the resolution of the racemic title compound of Example 1 using the following preparative chiral HPLC method:
EXAMPLE 3 (S)-3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide

The title compound is prepared analogously to Example 2.
J MED CHEM 2009, 52, 6142-52
http://pubs.acs.org/doi/abs/10.1021/jm900839k
Type 2 diabetes is a polygenic disease which afflicts nearly 200 million people worldwide and is expected to increase to near epidemic levels over the next 10−15 years. Glucokinase (GK) activators are currently under investigation by a number of pharmaceutical companies with only a few reaching early clinical evaluation. A GK activator has the promise of potentially affecting both the β-cells of the pancreas, by improving glucose sensitive insulin secretion, as well as the liver, by reducing uncontrolled glucose output and restoring post-prandial glucose uptake and storage as glycogen. Herein, we report our efforts on a sulfonamide chemotype with the aim to generate liver selective GK activators which culminated in the discovery of 3-cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide (17c). This compound activated the GK enzyme (αKa = 39 nM) in vitro at low nanomolar concentrations and significantly reduced glucose levels during an oral glucose tolerance test in normal mice.


PATENT
EP-1735322-B1
Example 2(R)-3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide
The title compound is obtained analogously to Example 1 by employing the following additional resolution step:
The racemic title E compound of Example 1, 3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid (10 g, 0.026 mol) in 1,4-dioxane (500 mL) is treated in a three necked 1 liter flask, equipped with heating mantle, water condenser, calcium chloride guard tube and mechanical stirrer with 3.18 g (0.026 mol) of (R)-(+)-1-phenylethylamine. This reaction mixture is then refluxed at 100°C for 1 h. The clear reaction solution is cooled to RT (27°C) and stirred for 10 h. The crystallized salt is collected by filtration under vacuum, washed with 5 mL of hexane and dried under vacuum to afford salt A.
The salt A is dissolved in 1,4-dioxane (500 mL) and heated at 100°C for 1 h. The clear reaction solution is cooled to RT (27°C) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 50 mL of hexane, and dried under vacuum to afford salt B.
The salt B is dissolved in 1,4-dioxane (290 mL) and heated at 100°C for 1 h. The clear reaction solution is cooled to RT (27°C) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 30 mL of hexane, and dried under vacuum to afford salt C.
The salt C is dissolved in 1,4-dioxane (100 mL) and heated at 100°C for 1 h. The clear reaction solution is cooled to RT (27°C) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 30ml of hexane, and dried under vacuum to afford salt D.
The salt D is treated with aqueous hydrochloric acid solution (20 mL, 1 mL of concentrated hydrochloric acid diluted with 100 mL of water) and stirred for 5 min. The white solid precipitates out and is collected by vacuum filtration, washed with 10 mL of cold water, 5 mL of isopropanol and 20 mL of hexane, and dried under vacuum to yield the hydrochloride salt of (R)-(-)-3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid, salt E.
The salt E is neutralized by stirring with aqueous sodium bicarbonate solution (10 mL, 1 g of sodium bicarbonate dissolved in 120 mL of water) for 5 min. The precipitated solid is collected by filtration, washed with 10 mL of cold water, 100 mL of hexane, and dried to afford (R)-(-)-3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid: m.p. 202.2-203.4°C.
Alternatively, the title compound may be obtained by the resolution of the racemic title compound of Example 1 using the following preparative chiral HPLC method:
REFERENCES
US 7750020
WO-2005095418-A1
US-20080103167-A1
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015218151 | 2015-08-06 | NOVEL PHENYLACETAMIDE COMPOUND AND PHARMACEUTICAL CONTAINING SAME |
| US7750020 | 2010-07-06 | Sulfonamide-Thiazolpyridine Derivatives As Glucokinase Activators Useful The Treatment Of Type 2 Diabetes |
Type 2 diabetes is a polygenic disease which afflicts nearly 200 million people worldwide and is expected to increase to near epidemic levels over the next 10−15 years. Glucokinase (GK) activators are currently under investigation by a number of pharmaceutical companies with only a few reaching early clinical evaluation. A GK activator has the promise of potentially affecting both the β-cells of the pancreas, by improving glucose sensitive insulin secretion, as well as the liver, by reducing uncontrolled glucose output and restoring post-prandial glucose uptake and storage as glycogen. Herein, we report our efforts on a sulfonamide chemotype with the aim to generate liver selective GK activators which culminated in the discovery of 3-cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide (17c). This compound activated the GK enzyme (αKa = 39 nM) in vitro at low nanomolar concentrations and significantly reduced glucose levels during an oral glucose tolerance test in normal mice.
https://www.google.com/patents/US7750020
EXAMPLE 2 (R)-3-Cyclopentyl-N-(5-methoxy-thiazolo[5,4-b]pyridin-2-yl)-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionamide

The title compound is obtained analogously to Example 1 by employing the following additional resolution step:
The racemic title E compound of Example 1,3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid (10 g, 0.026 mol) in 1,4-dioxane (500 mL) is treated in a three necked 1 liter flask, equipped with heating mantle, water condenser, calcium chloride guard tube and mechanical stirrer with 3.18 g (0.026 mol) of (R)-(+)-1-phenylethylamine. This reaction mixture is then refluxed at 100° C. for 1 h. The clear reaction solution is cooled to RT (27° C.) and stirred for 10 h. The crystallized salt is collected by filtration under vacuum, washed with 5 mL of hexane and dried under vacuum to afford salt A.
The salt A is dissolved in 1,4-dioxane (500 mL) and heated at 100° C. for 1 h. The clear reaction solution is cooled to RT (27° C.) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 50 mL of hexane, and dried under vacuum to afford salt B.
The salt B is dissolved in 1,4-dioxane (290 mL) and heated at 100° C. for 1 h. The clear reaction solution is cooled to RT (27° C.) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 30 mL of hexane, and dried under vacuum to afford salt C.
The salt C is dissolved in 1,4-dioxane (100 mL) and heated at 100° C. for 1 h. The clear reaction solution is cooled to RT (27° C.) and stirred for 10 h. The crystallized product is collected by filtration under vacuum, washed with 30 ml of hexane, and dried under vacuum to afford salt D.
The salt D is treated with aqueous hydrochloric acid solution (20 mL, 1 mL of concentrated hydrochloric acid diluted with 100 mL of water) and stirred for 5 min. The white solid precipitates out and is collected by vacuum filtration, washed with 10 mL of cold water, 5 mL of isopropanol and 20 mL of hexane, and dried under vacuum to yield the hydrochloride salt of (R)-(−)-3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid, salt E.
The salt E is neutralized by stirring with aqueous sodium bicarbonate solution (10 mL, 1 g of sodium bicarbonate dissolved in 120 mL of water) for 5 min. The precipitated solid is collected by filtration, washed with 10 mL of cold water, 100 mL of hexane, and dried to afford (R)-(−)-3-cyclopentyl-2-[4-(4-methyl-piperazine-1-sulfonyl)-phenyl]-propionic acid: m.p. 202.2-203.4° C.
Alternatively, the title compound may be obtained by the resolution of the racemic title compound of Example 1 using the following preparative chiral HPLC method:
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015218151 | 2015-08-06 | NOVEL PHENYLACETAMIDE COMPOUND AND PHARMACEUTICAL CONTAINING SAME |
| US7750020 | 2010-07-06 | Sulfonamide-Thiazolpyridine Derivatives As Glucokinase Activators Useful The Treatment Of Type 2 Diabetes |
Torrent Research Centre, Village Bhat, Gujarat, India




Mr. Samir Mehta, 52, is the Vice Chairman of the USD 2.75 billion Torrent Group and Chairman of Torrent Pharma

Shri Sudhir Mehta – Chairman Emeritus ::
| Dr. Chaitanya Dutt – Director (Research & Development) :: |
 Born in the year 1950, Dr. Chaitanya Dutt holds an MD in Medicine. He practiced as a consulting physician before joining the company in 1982. Since then he has been associated with the Company. His rich experience spans in the areas of Pharma R&D, clinical research, manufacturing, quality assurance, etc. He is one of the key professionals in the top management team of the Company. He has been instrumental in setting up the Torrent Research Centre (TRC), the research wing of the Company. Under his prudent guidance and leadership, TRC has achieved tremendous progress in the areas of discovery research as well as development work on formulations. He does not hold any directorship in any other company. Born in the year 1950, Dr. Chaitanya Dutt holds an MD in Medicine. He practiced as a consulting physician before joining the company in 1982. Since then he has been associated with the Company. His rich experience spans in the areas of Pharma R&D, clinical research, manufacturing, quality assurance, etc. He is one of the key professionals in the top management team of the Company. He has been instrumental in setting up the Torrent Research Centre (TRC), the research wing of the Company. Under his prudent guidance and leadership, TRC has achieved tremendous progress in the areas of discovery research as well as development work on formulations. He does not hold any directorship in any other company. |
///NOVARTIS, DIABETES, Sulfonamide-Thiazolpyridine Derivatives, Glucokinase Activators, Treatment Of Type 2 Diabetes, 866772-52-3, Novartis Molecule, functionally liver selective glucokinase activators, treatment of type 2 diabetes , NVP-LBX192, LBX-192
c1(sc2nc(ccc2n1)OC)NC(C(c3ccc(cc3)S(=O)(=O)N4CCN(CC4)C)CC5CCCC5)=O

| Diabetes Nephropathy, a chronic metabolic complication of diabetes mellitus, is characterized by elevated levels of serum glucose,creatinine, urea and uric acid in addition to abnormal histopathological changes in kidney. In the recent past, many antidiabetic agents are introduced; still the diabetes and the related nephropathy complication continue to be a major medical problem, not only in developed countries but also in developing countries. Not with standing much research work, the diabetic kidney damages are increasing rapidly and patients with diabetes kidney failure undergo either painful dialysis or kidney transplantation [1] which is both costly and harmful. More and more interest is now growing about plant use as an alternative therapy for protecting kidney damage in patients with diabetes mellitus. Reactive oxygen species (ROS) have been widely implicated in the pathogenicity of diabetes mellitus and its nephropathy. A number of clinical studies suggest that the antioxidants in medicinal plants are key factors in reducing the incidence of diabetic nephropathy. Traditional medicines and extracts from medicinal plants with antioxidant potential have been extensively used as alternative medicine for better control and management of diabetes nephropathy [2]. However, searching for new antidiabetic drugs with nephroprotective properties from natural plants is currently very important. |
| Amaranthus hybridus L. (Amaranthaceae) commonly known as ‘Cheera’ in Malayalam, is an erect branched annual herb distributed throughout tropical and temperate regions of India as a common weed in the agricultural fields and wastelands. In traditional medicinal system different parts of the plant Amaranthus hybridus (A. hybridus) have been mentioned to be useful in a variety of diseases. Traditionally, the plant has been used in treating dysentery, diarrhoea, ulcers and hemorrhage of the bowel due to its astringent property [3–5]. In southern India, the leaves are used in folk medicine for the treatment of diabetes. Leaves possess antibacterial effect, cleansing effect and also help to reduce tissue swelling [5]. In Nigeria, A. hybridus leaves combined with condiments are used to prepare soup [6–8]. In Congo, their leaves are eaten as spinach or green vegetables [6,9]. These leaves boiled and mixed with a groundnut sauce are eaten as salad in Mozambique and in West Africa [10,11]. The Amaranthus species contains amaranthine, quercetin, and kaempferol glycosides [12].A. hybridus leaves are used as an antidote for snake and scorpion bite [13,14]. |
| Amaranthus species were of great importance in pre-Colombian American people’s diets [15] and A. cruentus and A. hybridus have a high nutritional value [16] (Fernand et al.). The consumption of A. cruentus products is advised for patients with celiac disease and, therefore, also for diabetic persons [17]. A. hybridus has been used traditionally for the treatment of liver infections and knee pain and for its laxative, diuretic, and cicatrisation properties [16]. |
| Furthermore, recent studies established theantihyperglycemic activities of other species of Amaranthus genus as A. spinosus [18] and A. viridis [19,20]. However, based on the literature survey, there is no scientific report proving the anti-hyperglycemic efficacy of this particular species. Therefore, the current study was designed to evaluate the nephroprotective activity of Amaranthus hybridus in STZ induced diabetic rats. |
| Balasubramanian T* and Karthikeyan M | |
| Department of Pharmacology, Al Shifa College of Pharmacy, Kerala, India | |
| Corresponding Author : | Dr. Thirumalaiswamy Balasubramanian Department of Pharmacology Al Shifa College of Pharmacy Poonthavanam Post, Kizhattur Village Perinthalmanna, Malappuram Dist Kerala-679 325, India Tel: +919544496752 E-mail: tbaluanandhi@gmail.com |
| Received December 29, 2015; Accepted January 07, 2016; Published January 14, 2016 | |
| Citation: Balasubramanian T and Karthikeyan M (2016) Therapeutic Effect of Amaranthus hybridus on Diabetic Nephropathy. J Develop Drugs 5:147.doi:10.4172/2329-6631.1000147 | |
SEE

Dr. T. Balasubramanian

Karthikeyan M
http://alshifacollegeofpharmacy.com/teaching-faculty.html
![]()







////////Therapeutic Effect, Amaranthus hybridus, Diabetic Nephropathy, AYURVEDA

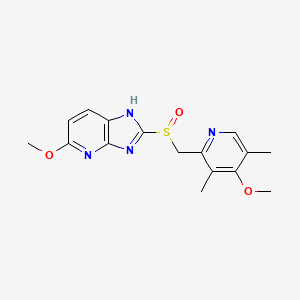
 Integrifolin
Integrifolin
