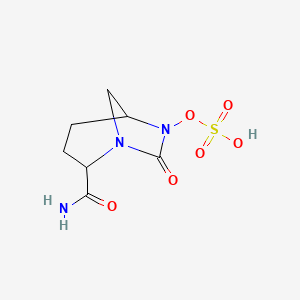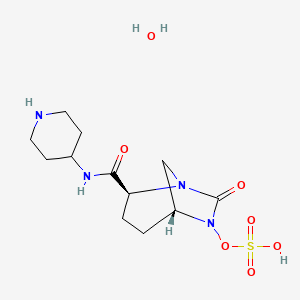
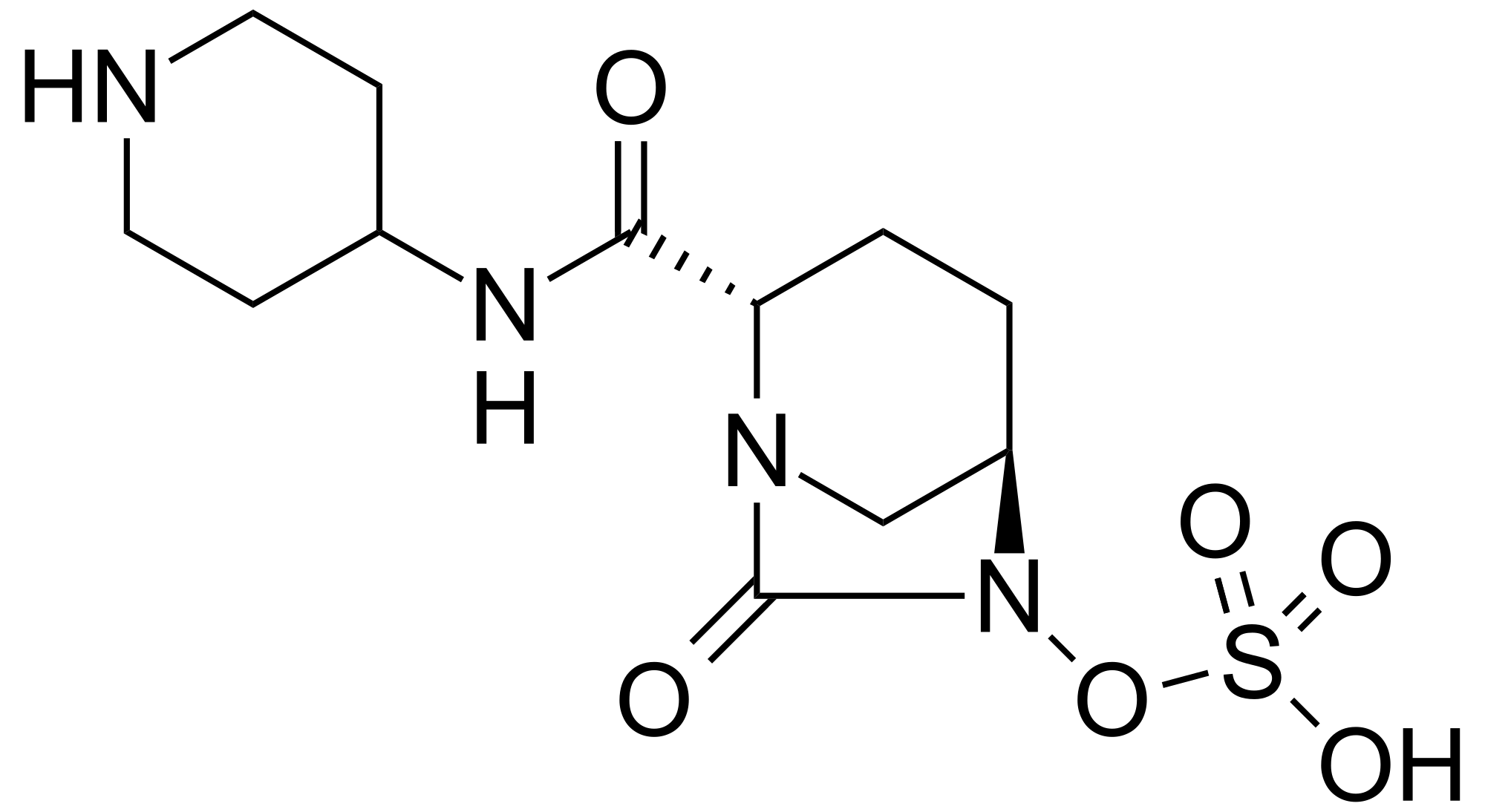
MK 7655, RELEBACTAM
(1R,2S,5R)-7-Oxo-N-(4-piperidinyl)-6-(sulfooxy)-1,6-diazabicyclo[3.2.1]octane-2-carboxamide
(1R,2S,5R)-7-oxo-2-((piperidin-4-yl)carbamoyl)-1,6-diazabicyclo(3.2.1)octan-6-yl hydrogen sulfate monohydrate
Sulfuric acid, mono((1R,2S,5R)-7-oxo-2-((4-piperidinylamino)carbonyl)-1,6-diazabicyclo(3.2.1)oct-6-yl) ester, hydrate (1:1)
CAS 1174020-13-3
β-Lactamase inhibitor
MK-7655 is a beta-lactamase inhibitor in phase III clinical studies at Merck & Co for the treatment of serious bacterial infections…….See clinicaltrials.gov, trial identifier numbers NCT01505634 and NCT01506271.
In 2014, Qualified Infectious Disease Product (QIDP) and Fast Track designations were assigned by the FDA for the treatment of complicated urinary tract infections, complicated intra-abdominal infections and hospital-acquired bacterial pneumonia/ventilator-associated bacterial pneumonia.

PAPER
A concise synthesis of a beta-lactamase inhibitor
Org Lett 2011, 13(20): 5480
http://pubs.acs.org/doi/abs/10.1021/ol202195n
http://pubs.acs.org/doi/suppl/10.1021/ol202195n/suppl_file/ol202195n_si_001.pdf
MK-7655 (1) is a β-lactamase inhibitor in clinical trials as a combination therapy for the treatment of bacterial infection resistant to β-lactam antibiotics. Its unusual structural challenges have inspired a rapid synthesis featuring an iridium-catalyzed N–H insertion and a series of late stage transformations designed around the reactivity of the labile bicyclo[3.2.1]urea at the core of the target.
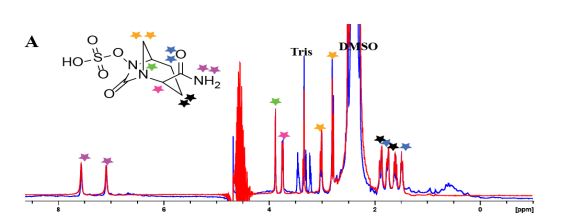
H NMR (400 MHz, DMSO-d6): δ 8.30 (br s, 2H), 8.20 (d, J = 7.8 Hz, 1H), 4.01 (s, 1H), 3.97-3.85 (m, 1H), 3.75 (d, J = 6.5 Hz, 1H), 3.28 (dd, J = 12.9, 2.5 Hz, 2H), 3.05-2.93 (m, 4H), 2.08-1.97 (m, 1H), 1.95-1.79 (m, 3H), 1.73-1.59 (m, 4H);
13C NMR (DMSO-d6, 100 MHz) δ 169.7, 166.9, 59.8, 58.3, 46.9, 44.3, 42.9, 28.5, 28.3, 20.8, 18.9;
HRMS calculated for C12H20N4O6S (M+H): 349.1182, found: 349.1183.
[α]D 25 = -23.3 (c = 1.0, CHCl3)


PATENT
WO 2009091856
http://www.google.com/patents/WO2009091856A2?cl=en
EXAMPLE IA
(2S ,5 R)-7-Oxo-N-piperidin-4-yl-6-(sulfooxy)- 1 ,6-diazabicyclo [3.2.1 ]octane-2-carboxamide
Step 1 : tert-butyl 4-({[(2S,5R)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1]oct-2- yljcarbonyl } amino)piperidine- 1 -carboxylate : To a solution of (2S,5R)-6-(phenylmethoxy)-7-oxo-l,6-diazabicyclot3.2.1]octane-
2-carboxylic acid (1.484 g, 5.37 mmol) in dry dichloromethane (60 ml) was added triethylamine (1.88 ml, 13.49 mmol), 2-chloro-l-methylpyridinium iodide (1.60 g, 6.26 mmol), and 4-amino-l- BOC-piperidine (1.30 g, 6.49 mmol) sequentially at room temperature under nitrogen. The reaction was then heated to 500C for 1 hour. The reaction mixture was concentrated under vacuum and purified by silica gel chromatography on an Isco Combiflash (40 g silica gel, 40 mL/min, 254 nM, 15% to 100% EtOAc/hexane over 14 column volumes then 100% EtOAc for 4 column volumes; title compuond eluted at 65% ethyl acetate/hexane) to afford the title compound as a pale orange solid.
Step 2: tert-butyl 4-({[(2S,5R)-6-hydroxy-7-oxo-l ,6-diazabicyclo[3.2.1]oct-2- yl] carbonyl } amino)piρeridine- 1 -carboxylate:
Palladium on carbon (394 mg; 10% Pd/C) was added to a solution of the product of step 1 (1.81 g, 3.95 mmol) in methanol (50.6 mL) and the resulting mixture was stirred under hydrogen (balloon) overnight. LC/MS analysis indicated the reaction was not complete. Acetic acid (6 drops) and additional catalyst (159 mg of 10% Pd/C) were added to the reaction and the resulting mixture was stirred under hydrogen (balloon) for an additional 90 minutes. Additional catalyst (0.2085 g of 10% Pd/C) was added to the reaction and stirring under hydrogen was continued for an additional 2.5 hours at which time the reaction was judged complete by LC-MS analysis. The reaction was filtered through a celite pad and the collected solid was washed well wtih MeOH. The filtrate was concentrated under vacuum to afford the title compound as a colorless oil which was used without purification in the next step.
Step 3 : tert-butyl-4-({ [(2S,5R)-7-oxo-6-(sulfooxy)- 1 ,6-diazabicyclo[3.2.1 ]oct-2- yl] carbonyl } amino)ρiperidine- 1 -carboxylate:
To a solution of the product of step 2 (1.455 g, 3.95 mmol; theoretical yield of step 2) in dry pyridine (30 mL) was added sulfur trioxide pyridine complex (3.2 g, 20.11 mmol) at room temperature under nitrogen. The resulting thick mixture was stirred over the weekend.
The reaction was filtered and the white insoluble solids were washed well with dichloromethane. The filtrate was concentrated in vacuo. The residue was further azeotroped with toluene to remove excess pyridine to afford the title compound which was used without purification in the next step.
Step 4: (2S,5R)-7-oxo-N-piperidin-4-yl-6-(sulfooxy)-l,6-diazabicyclo[3.2.1]octane-2- carboxamide:
To a mixture of the product of step 3 (1.772 g, 3.95 mmol; theoretical yield of step 3) in dry dichloromethane (30 ml) at 00C under nitrogen was slowly added trifluoroacetic acid (6.1 ml, 79 mmol). Immediately the reaction became a solution. After 1 hour, additional trifluoroacetic acid (8 ml) was added to the reaction. The reaction was stirred at 00C until judged complete by LC-MS analysis then concentrated in vacuo. The residue was triturated with ether (3X) to remove excess TFA and organic impurities. The resulting white insoluble solid was collected via centrifugation, dried in vacuo, then purified by preparative HPLC (250X21.2 mm Phenomenex Synergi Polar-RP 80A column; 10 micron; 35 mL/min.; 210 nM; 0% to 30% methanol/water over 15 minutes; title compound eluted at 10% methanol/water). Fractions containing the title compound were combined and Iyophilized overnight to afford the title compound as a white solid. LC-MS (negative ionization mode) m/e 347 (M-H).
PAPER
Discovery of MK-7655, a beta-lactamase inhibitor for combination with Primaxin
Bioorg Med Chem Lett 2014, 24(3): 780
http://www.sciencedirect.com/science/article/pii/S0960894X13014856

PATENT
WO 2014200786
http://www.google.dj/patents/WO2014200786A1?cl=en







Exemplary Scheme

– 50% isolated yield overall from 1 to 5

O via crystallization
XAMPLE 1
(2S,5R)-7-oxo-N-piperidin-4-yl-6-(sulfooxy)- 1 ,6-diazabicyclo[3.2.1 ]octane-2-carboxamide 
Preparation of (15′,45)-5-((2-nitrophenyl)sulfonyl)-2-oxa-5-azabicyclo[2.2.2]octan-3 one (2)
To a reactor (R-1) equipped with an additional funnel, nitrogen inlet and agitator was charged (2S,5S)-5-hydroxypiperidine-2-carboxylic acid (77.3 wt%) (50.0 g, 344 mmol), and water (150 mL). Agitation was begun, the pH adjusted to 10-11 by addition of 10 N NaOH (~ 46.5 mL) and the reactor charged with acetone (50.0 mL).
In a separate reactor (R-2) equipped with an agitator and nitrogen inlet was charged 2-nitrobenzene-l-sulfonyl chloride (97%) (106.0 g, 478 mmol) and acetone (80 mL). The contents of R-2 were transferred to R-1 at 23-30 °C while the pH of the solution was maintained at 10-11 by simultaneously addition of 10 N NaOH. After 15 to 30 min, the pH was adjusted to about 6 by addition of 12 N HC1. The solution was charged with EtOAc (500 mL) and the pH adjusted to 3.0 by addition of 12 N HC1. The layers were separated and the aqueous back-extracted with EtOAc (150 mL x 2).
To a separate reactor (R-3) was charged product la in the combined organic layers, 2-nitrobenzene-l-sulfonyl chloride (73.0 g, 329 mmol), and triethylamine (130 mL). The batch in R-3 was agitated at 20-28°C for 30 min. The solution was charged with water (100 mL), the layers separated, and the aqueous back extracted with EtOAc (150 mL x 2). The combined EtOAc layer was washed with 10% NaHC03 (100 mL) and brine (100 mL). The organic phase was concentrated to 150 mL upon which a crystalline slurry was formed. The concentrated solution was agitated at 13-18°C for 2-3 hours followed by filtration of crystalline solids. The resulting wet cake was washed with EtOAc (60 mL) and then dried under vacuum oven at 25-30°C to afford 2 (65.6 g, 79% yield), m.p. 126.0-126.7 °C. 1H NMR (CDC13, 400 MHz) δ: 8.02 (m, 1 H), 7.80-7.71 (m, 2 H), 7.66 (m, 1 H), 4.88 (m, 1 H), 4.55 (dd, J= 3.8, 2.7 Hz, 1 H), 3.78 (dt, J= 11.2, 3.0 Hz, 1 H), 3.66 (dd, J = 11.2, 1.1 Hz, 1 H), 2.44 (m, 1 H), 2.11 (m, 2 H), 1.91 (m, 1 H); 13C NMR (CDC13, 100 MHz) δ: 168.4, 148.3, 134.4, 132.1, 131.0, 130.7, 124.2, 73.5, 51.4, 48.0, 25.1, 23.2
Preparation oftert-butyl 4-((25*,55)-l-((2-nitrophenyl)sulfonyl)-5-(((2- nitrophenyl)sulfony l)oxy)piperidine-2-carboxamido)piperidine- 1 -carboxylate (3)
To a reactor (R-l) was charged lactone 2 (65.5 g, 210 mmol), THF (131 mL) and tert-butyl 4-aminopiperidine-l -carboxylate (44.5 g, 222 mmol). The stirred solution was heated to reflux (typical temperature 72 °C) for ~18 hr. The reaction was cooled to 25-35 °C and then charged with THF (325 mL) and 4-dimethylaminopyridine (40.1 g, 328 mmol) followed by agitation for 30 minutes.
To a separate reactor (R-2) was charged 2-nitrobenzene-l-sulfonyl chloride (60.9 g,
275 mmol) and THF (200 mL). The contents of R-2 were added to R-l over the course of 45 to 75 minutes maintaining batch temperature of 20 to 30°C. The batch in R-l was agitated for 2 to 4 hours at a temperature of 20 to 30°C.
To a separate reactor (R-3) was charged water (600 mL) and methanol (600 mL). The contents of R-3 were charged to the main batch over the course of 45 to 75 minutes with agitation while maintaining temperature of 20 to 30°C. The batch was cooled to 5 to -5°C and then agitated at 5 to -5°C for at least 4 hours. The solids were filtered and then washed twice with methanol (130 mL x 2). The wet cake was dried in a vacuum oven at 40 to 50°C to afford 3 (144.0 g, 98% yield), m.p. 131.8-133.1 °C. 1H NMR (CDC13, 400 MHz) δ: 8.14 (m, 2 H), 7.83-7.74 (m, 6 H), 6.50 (d, J= 7.9 Hz, 1 H), 4.69 (m, 1 H), 4.43 (s, 1H), 4.11 (dd, , J= 13.7, 4.9 Hz, 1H), 3.95 (m, 2H), 3.83 (m, 1H), 3.47 (s, 1H), 3.10 (dd, J= 13.7, 11.0 Hz, 1H), 2.81 (m, 2H), 2.51 (m, 1H), 2.12 (m, 1H), 1.85-1.72 (m, 4H), 1.45 (s, 9H), 1.26 (m, 1H); 13C NMR (CDC13, 100 MHz) δ: 166.9, 154.6, 148.2, 147.6, 135.2, 134.8, 132.6, 132.5, 131.9, 131.6, 131.4, 129.7, 124.9, 124.7, 79.8, 76.5, 55.0, 47.1, 46.0, 31.8, 31.5, 28.4, 27.3, 24.4.
Preparation of N-4-nitrobenzene sulfonyl-O-benzylhydroxylamine
To a reactor (R-l) was charged O-benzylhydroxylamine hydrochloride (61.0g, 382 mmol) and pyridine (400 mL). The solution cooled to 5 to -5°C.
To a separate reactor (R-2) was charged 4-nitrobenzenesulfonyl chloride (89.0 g, 402 mmol) and pyridine (200 mL). The contents of R-2 were transferred to R-l at a rate to maintain temperature range of -5 to -5°C. The batch in R-l was agitated at 5 to -5 °C for 15 to 45 minutes then warmed to 20 to 30°C for 45 to 75 minutes. Water (250 mL) was then added at a rate to maintain 20 to 30°C and agitated 5 to 15 minutes. The solids were filtered and the wet cake washed with water (100 mL x 3). The wet cake was dried in vacuum oven at 50°C to afford N-4-nitrobenzenesulfonyl-O-benzylhydroxylamine (113.3 g, 96% yield), m.p. 128.4-130.0 °C. 1H NMR (CDCls, 400 MHz) δ: 8.36 (d, J = 8.9 Hz, 2 H), 8.11 (d, J = 8.9 Hz, 2 H), 7.36 (m, 5H), 7.11 (s, 1H), 5.02 (s, 2H); 13C NMR (CDC13, 100 MHz) δ: 151.0, 142.5, 134.9, 130.2, 129.7, 129.3, 128.9, 124.5, 80.2.
Step C. Preparation of tert-butyl 4-((2S,5R)-5-((benzyloxy)amino)piperidine -2-carboxamido)piperidine- 1 -carboxylate (4)
Boc
To a reactor (R-l) was charged tert-butyl 4-((2R,5R)-l-((2-nitrophenyl)sulfonyl)-5-(((2-nitrophenyl)sulfonyl)oxy)piperidine-2-carboxamido)piperidine-l -carboxylate (3) (110 g, 158 mmol), N-4-nitrobenzene sulfonyl-O-benzylhydroxylamine (58 g, 188 mmol), potassium carbonate (25.9 g, 187 mmol) and dimethylacetamide (440 mL). The stirred solution was heated to 60 to 70°C for 24 – 32 hours. The batch was cooled to 20 to 30°C and charged with toluene (660 mL). The batch was extracted with 1 N sodium hydroxide (3×220 mL) then washed with water (220 mL).
The toluene solution was azotropically distilled at ~50°C to about 1/3 volume. The solution was solvent-switched to MeOH at 45-55°C, adjusted to 237 mL.
The batch was cooled to 20-25°C, charged with thioglycolic acid (57.9 g, 629 mmol) at 10 °C, and then charged with K2CO3 anhydrous (172.0 g, 1225 mmol). The batch was agitated at 10-15°C for 0.5 h, warmed to 20-25°C, agitated at 20-25°C for 10-15 h, and heated at 48-53°C for 3-6 h.
The batch was charged with 10 wt% sodium chloride (1.10 L) and toluene (880 mL) at about 40°C. The layers were separated and the aq. layer back-extracted with toluene (3 x440 mL). The combined organic layer was washed with 10% NaHC03 (2 x220 mL). The batch was concentrated at 40-50°C to 165 mL, then cooled to 35-40°C. The batch was charged with seed (50 mg) and agitated for 1 h at 35-40°C. The batch was charged with heptanes (110 mL) at 35-40°C over 1 h, then slowly cooled to 15-20°C over 1 h. The batch was agitated for 3 h and the solids filtered. The wet cake was washed with toluene/heptanes (137.5 mL) then dried in vacuum oven at 30 °C for 3-8 h to affored 4. (47.3 g, 70% overall yield from 3), m.p. 117.5-118.0 °C. 1H NMR (CDC13, 500 MHz) δ: 7.37-7.29 (m, 5 H), 6.64 (d, J= 8.2 Hz, 1 H), 5.36 (brs, 1 H), 4.67 (s, 2 H), 4.00 (m, 2 H), 3.90 (m, 1 H), 3.28 (ddd, J= 11.8, 4.0, 1.7 Hz, 1 H), 3.12 (dd, J= 10.2, 3.2 Hz, 1 H), 2.95 (m, 1 H), 2.86 (m, 2 H), 2.46 (dd, J= 11.8, 9.5 Hz, 1 H), 2.10 (m, 1 H), 1.93-1.83 (m, 3 H), 1.58 (brs, 1 H), 1.45 (s, 9 H), 1.41 (m, 1 H), 1.35-1.23 (m, 3 H); 13C NMR (CDC13, 125 MHz) δ: 172.8, 154.7, 137.7, 128.4 (4 C), 127.9, 79.6, 76.9, 59.8, 57.0, 49.2, 46.1, 42.8 (br, 2 C), 32.0 (2 C), 28.4 (3 C), 28.3, 27.2.
Step D: Preparation of tert-butyl 4-((lR,2S,5R)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1 ]octane-2-carboxamido)piperidine- 1 -carboxylate (5)
To a reactor (R-l) was charged tert-butyl 4-((2S,5R)-5-((benzyloxy)amino)piperidine-2-carboxamido)piperidine-l-carboxylate (4) (46.3 g, 107 mmol), dichloromethane (463 mL), and Hunig’s base (58.0 mL). The batch was cooled to -18°C and then charged with triphosgene in four portions (25.1 g total; 85 mmol) at <-8°C. The batch was agitated at -5 to 0°C for 0.5 h then charged with 11.4 wt% aqueous H3P04 at -5 to 0 °C (347 g, 3541 mmol). The batch was agitated at 20-25°C for 15-20 h then phase cut. The aqueous layer was back-extracted with dichloromethane (138 mL). The combined organic layer was washed with 10% NaHC03 (115 mL), then water (115 mL). The organic solution was concentrated at atmospheric pressure to ~80
mL, then charged with MTBE (347 mL) at 35-45 °C over 0.5 h, then concentrated at 35-45 °C to 231 mL two times to form a slurry.
The slurry was charged with heptanes (139 mL) at 35-45 °C over 2 h, then slowly cooled to 15-20°C over 1 h. The batch was agitated at 15-20°C for 6-8 h. Solids were filtered and the wet cake washed with MTBE/heptanes (1.4 : 1 , 185 mL) then dried under vacuum at 25-30°C for 5-10 hours to afford 5 (43.7 g, 92% yield), m.p. 161.3-161.8 °C. 1H NMR (CDC13, 500 MHz) δ: 7.45-7.32 (m, 5 H), 6.55 (d, J= 8.2 Hz, 1 H), 5.05 (d, J= 11.6 Hz, 1 H), 4.90 (d, J= 11.6 Hz, 1 H), 4.02 (m, 2 H), 3.90 (m, 2 H), 3.30 (m, 1 H), 2.99 (dt, J= 11.7, 1.1 Hz, 1 H), 2.86 (m, 2 H), 2.64 (d, J = 11.7 Hz, 1 H), 2.37 (dd, J= 14.6, 6.9 Hz, 1 H), 2.04-1.82 (m, 4 H), 1.58 (m, 1 H), 1.45 (s, 9 H), 1.30 (m, 2 H); 13C NMR (CDC13, 125 MHz) δ: 168.3, 167.5, 154.7, 135.6, 129.2 (2 C), 128.8, 128.6 (2 C), 79.7, 78.3, 60.4, 57.8, 47.5, 46.8, 42.5 (br, 2 C), 32.0, 31.7, 28.4 (3 C), 20.8, 17.2.
Step E: Preparation of tert-butyl 4-((2S,5R)-6-hydroxy-7-oxo-l,6-diazabicyclo[3.2.1“|octane- 2-carboxamido) iperidine- 1 -carboxylate
tert-butyl 4-((2S,5R)-6-hydroxy-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxamido)piperidine-l -carboxylate (9.2 g, 20.1 mmol) was charged to a glass bottle, and the solids were dissolved in THF (150 mL). The solution was then charged to a hydrogenation reactor along with Pd/Al203 (10 wt%, 1.5 g). The reaction was purged three times with hydrogen and then set to a hydrogen pressure of 50 psi. The reaction temperature was adjusted to 25°C and the reaction was allowed to agitate for 22 hours. After the reaction was complete as determined by HPLC analysis, the solution was filtered through SOLKA-FLOC® (Interational Fiber Corporation, North Tonawanda, NY) to remove the catalyst and the filter cake was washed with THF. The filtrate and washes were then solvent switched by vacuum distillation to iPrOAc to a final volume of 40 mL. The resulting iPrOAc slurry was aged at room temperature for 1 hour. The solids were then filtered and washed with iPrOAc (20 mL) and dried under vacuum and N2 at 40°C to afford the title product (6.62 g., 17.97 mmol, 90% isolated yield). Spectral data matched the reference compound.
Preparation of (2S,5R)-7-oxo-N-piperidin-4-yl-6-(sulfooxy)- 1 ,6-diazabicyclo[3.2.1 ]octane-2-carboxamide
tert-butyl 4-((2S,5R)-6-hydroxy-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxamido)piperidine-l-carboxylate (20 g, 54.3 mmol), THF (200 mL), 2-picoline (10.9 mL, 309 mmol) and pyridine-S03 complex (30.2 g, 190 mmol) were charged to a flask under nitrogen. The heterogeneous mixture was allowed to stir overnight (~15 h). The reaction mixture was cooled to -10°C then DCM (200 mL) was added. 0.5 M K2HP04 (168 mL, 84 mmol) was added over 10 minutes. Bu4NHS04 (19.4 g, 57 mmol) was then added over 10 minutes. The biphasic mixture was stirred for 30 minutes, phase cut and the water layer was back extracted with 40 ml of DCM. The combined DCM solution was washed with water (120 ml), phase cut and the organic solution was solvent-switched to MeCN (320 ml) by vacuum distillation with 3 bed volumes of MeCN (total 1.0 L) and used as is in the next step. The solution of Bu4N+ OSO3 salt 7 in MeCN solution was used with an assumed yield of 100% (37.5 g, 54.3 mmol). The reaction mixture was cooled in an ice bath, and TMSI (10.26 ml, 70.7 mmol) was added via addition funnel over 30 minutes between 0°C and 5°C. The resulting mixture was agitated for 1-2 h and then quenched with H20:MeCN (1 :1, 6 ml) to afford a slurry. The slurry was warmed to room temperature and agitated for 12 h and after this time the pH of the supernatant was about 3.0. Tetrabutylammonium acetate (13.6 ml, 13.59 mmol) was slowly added over 30 min. The slurry was agitated for 1 h and pH of the supernatant was about 4.0. Solids were collected by filtration. The solid was washed with 60 mL of aqueous MeCN to afford 19.5 g of the crude product 8 in a 93% isolated yield from compound 6 .
At this stage, all byproducts (including hydro lyzation products of TMS-carbonate) and impurities were soluble in the organic phase.
The product was dissolved back into 140 ml of MeCN:H20 (1 :2) at room temperature. 1-Butanol (390 ml) as antisolvent was slowly added into the solution to afford a slurry. The slurry was agitated overnight. The white crystalline solid was filtered and washed with 3:1 IPA: water (40 ml) and dried under vacuum and nitrogen at room temperature to afford the title product in the form of a crystalline hydrate. (Yield = 16.3 g, 82%). Spectral data matched reference compound.
Preparation of (2S,5R)-7-oxo-2-(piperidin- 1 -ium-4-ylcarbamoyl)- 1 ,6-diazabicyclo[3.2.1 ]octan-6-yl sulfate (1).
tert-Butyl 4-( {[(25*,5i?)-6-hydroxy-7-oxo- 1 ,6-diazabicyclo[3.2.1 ]oct-2-yl]carbonyl}amino)piperidine-l-carboxylate 16 (0.54 g, 1.5 mmol), THF (5.4 mL), 2-picoline (0.29 mL, 2.9 mmol) and pyridine-S03 complex (0.70 g, 4.4 mmol) were charged to a vial under nitrogen. The heterogeneous mixture was allowed to stir overnight (~15 hr). The reaction mixture was cooled to -10°C then dichloromethane (5.4 mL) was added. 0.5 M K2HPO4 (4.5 mL, 2.3 mmol) was added over 10 minutes. BU4NHSO4 (0.53 g, 1.54 mmol) was then added over 10 min. The biphasic mixture was stirred for 30 min, phase cut and the water layer was back extracted with 1 ml of DCM. The combined DCM solution was washed with water (2.0 mL), phase cut and the organic solution was solvent-switched to MeCN (3.2 mL) by vacuum distillation with 3 bed volumes of MeCN. The product was used as is in the next step (water content less than 1000 ppm).
The solution of Bu4N+S04~~ salt 8 in MeCN solution was used with an assumed yield of 100% (1.0 g, 1.47 mmol). The reaction mixture was cooled in an ice bath, and Ν,Ο-bis(trimethylsilyl)trifluoroacetamide (BSTFA) (0.4 lg, 1.59 mmol) was added into the reaction and was allowed to stir for 10 min. TMSI (0.06g, 0.27 mmol) was added between 0°C and 5°C. The resulting mixture was allowed to agitate for 2 hr and then quenched with H2O (0.07g, 4.1 mmol) and acetic acid (0.08g, 1.5 mmol) to afford a slurry. The slurry was warmed to room temperature and agitated for 12 hr. Filter to collect the solid. The solid was washed with MeCN/water (94:6, 1 mL X 4) to afford the crystalline product 1 (0.38 g) in a 75% yield.
If NO-bis(trimethylsilyl)acetamide (BSA) (0.32g, 1.59 mmol) was applied, the reaction needed 24 hr to achieve full conversion.
Patent
WO2015033191
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015033191&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=PCTDescription
Scheme 1.

Formula (V)
Formula (VI)

Formula (I)
Scheme – 1
Example -1
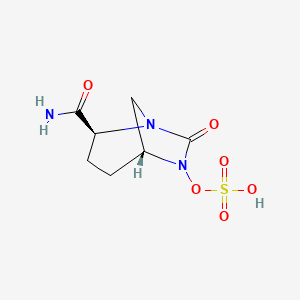
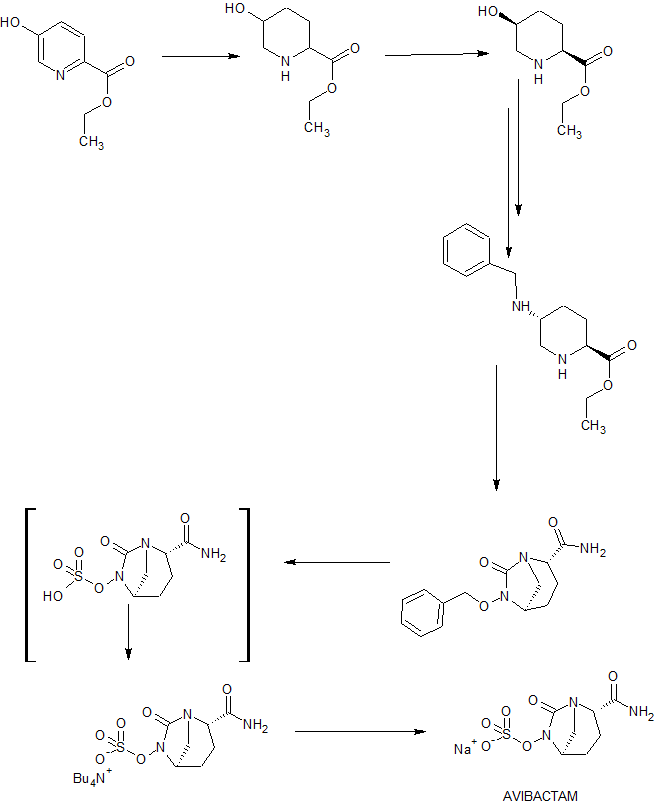
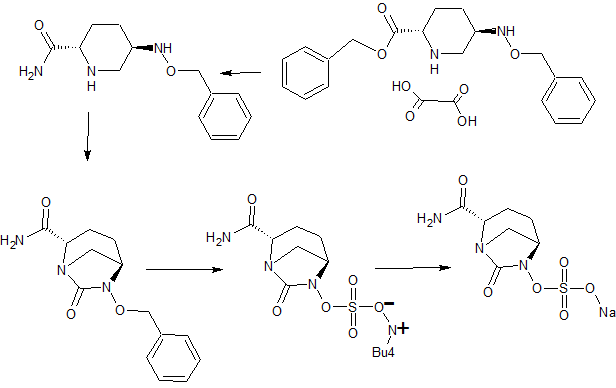
Preparation of (2S, 5R)-Sulfuric acid mono-{2-[N’-(4-aminopiperidinyl)-carbonyl]-7-oxo- l,6-diaza-bicyclo[3.2.1]oct-6-yl} ester (I).
Step-1: Preparation of (2S, 5R)-tert-butyl { (6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate (IV):
To a 250 ml round bottom flask equipped with magnetic stirrer was charged a solution of (2S, 5R)-sodium 6-benzyloxy-7-oxo-l,6-diaza-bicyclo [3.2.1] octane-2-carboxylate (11.1 gm, 0.037 mol, prepared using a method disclosed in Indian Patent Application No 699/MUM/2013) in water (180 ml) followed by l-tert-butoxycarbonyl-4-amino-piperidine (7.8 gm, 0.039 mol), EDC hydrochloride (11 gm, 0.055 mol) and 1 -hydro ybenzotriazole (4.8 gm, 0.037 mol) at 30°C successively under stirring. The reaction mixture was stirred for 24 hours at 30°C to provide a suspension. The suspension was filtered under suction and washed with 45°C warm water (40 ml) to provide (2S, 5R)-tert-butyl { (6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate in 12.7 gm quantity in 74% yield after drying under vacuum.
Analysis
NMR: (CDC13,) = 7.36-7.44 (m, 5H), 6.56 (d,lH), 5.06 (d,lH), 4.91 (d, 1H), 4.03 (br s, 1H), 3.88-3.97 (m, 2H), 3.29 (s, 1H), 3.00 (d, 1H), 2.86 (t, 2H), 2.64 (d, 1H), 2.37 (dd, 1H), 1.85-2.01 (m, 4H), 1.54-1.62 (m, 2H), 1.45 (s, 9H), 1.25-1.36 (m, 2H).
MS (ES+) C24H34N405 = 459.5 (M+l).
Step-2: Preparation of (2S, 5R)-tert-butyl { (6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate (V):
To a 100 ml single neck round bottom flask equipped with magnetic stirrer was charged a solution of (2S, 5R)-tert-butyl { (6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate (9 g, 19.5 mmol) in methanol (90 ml) followed by 10% palladium on carbon (2.7 g) at 35°C. The reaction mixture was stirred under 1 atm hydrogen pressure at 35°C for 2 hours. The catalyst was removed by filtering the reaction mixture under suction over a celite bed. The celite bed was washed with dichloromethane (50 ml). The combined filtrate was evaporated under vacuum below 35°C to provide (2S, 5R)-tert-butyl {(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate in 8.45 g quantity; it was used as such for the next reaction.
Analysis
NMR: (CDC13,) = 6.60 (d, 1H), 3.88-4.10 (m, 4H), 3.78 (s, 1H), 3.20 (d, 1H), 3.90 (t, 2H), 2.80 (d, 1H), 2.46 (dd, 1H), 2.1-2.2 (m, 1H), 2.85-2.20 (m, 4H), 1.70-1.80 (m, 1H), 2.47 (s, 9H), 1.30-1.41 (m, 3H).
MS (ES+) C17H28N405 = 369.4 (M+l).
Step-3: Preparation of Tetrabutyl ammonium salt of (2S, 5R)-tert-butyl {(6-sulfooxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate (VI):
To a 100 ml single neck round bottom flask equipped with magnetic stirrer was charged a solution of (2S, 5R)-tert-butyl {(6-hydroxy-7-oxo-l,6-diaza-bicyclo [3.2.1 ]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate (6.40 g, 7.6 mmol) in dichloromethane (90 ml), triethyl amine (9.3 ml), followed by pyridine – sulfur trioxide complex (5.4 g, 34.2 mmol) at 35°C under stirring. The reaction mixture was stirred for additional 4 hours at 35°C. The solvent was evaporated under vacuum below 40°C to provide a residue. The residue was stirred with 0.5N aqueous potassium dihydrogen phosphate solution (90 ml) for 1 hour. The resulting solution was extracted with dichloromethane (2 x 100 ml) to remove impurities. To the aqueous layer was added tetrabutyl ammonium hydrogen sulfate (6.9 g, 20.52 mmol) and the reaction mixture was stirred for 14 hours at 35°C. It was extracted with dichloromethane (3 x 30 ml). Combined organic layer was dried over sodium sulfate and evaporated under vacuum to provide tetrabutyl ammonium salt of (2S, 5R)-tert-butyl {(6-sulfooxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate in 8.0 g quantity in 62% yield.
Analysis
NMR: (CDC13,) – 6.64 (d, 1H), 4.36 (br s, 1H), 4.05(br s, 2H), 3.90-4.00 (m, 1H), 3.87 (d, 1H), 2.28-3.34 (m, 10H), 3.80-3.95 (m, 2H), 3.74 (d, 1H), 2.42 (dd, 1H), 2.15-2.24 (m, 1H), 1.82-1.97 (m, 4H), 1.61-1.74 (m, 14 H), 1.41-1.52 (m, 10 H), 1.02 (t, 12H).
MS (ES-) C17H27N408S. N(C4H9)4 = 447.4 (M-l) as a free sulfonic acid.
Step-4: Synthesis of (2S, 5R)- Sulfuric acid mono-{ [(4-aminopiperidin-4-yl) carbonyl]-7-oxo-l,6-diaza-bicyclo[3.2.1]-oct-6-yl} ester (I):
To a 100 ml round bottom flask equipped with magnetic stirrer was charged a solution of tetrabutyl ammonium salt of (2S, 5R)-tert-butyl {(6-sulfooxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl-carbonyl) amino} piperidine-l-carboxylate (6.0 g) in dichloromethane (15 ml). The solution was cooled to -10°C under stirring and to it was added trifluoro acetic acid (15 ml) drop wise. The reaction mixture was stirred at -10°C for 1 hour. Solvents were evaporated under vacuum below 30°C to its 1/3 volume to provide a thick residue. The thick residue was stirred twice with diethyl ether (60 ml each time) to provide a precipitation. The solid obtained was filtered at suction and suspended in acetone (90 ml). To the suspension was added 10% solution of sodium-2-ethyl-hexanoate in acetone to adjust pH between 4.5 to 5.5. The suspension was stirred for 10 minutes and filtered under suction. The wet cake was washed with acetone and dried under vacuum below 40°C to provide 3 gm crude compound. The crude compound was stirred with aqueous isopropanol (3ml water: 21 ml iospropanol) for overnight to purify further. The resulting suspension was filtered under suction and washed with aqueous isopropanol (1 ml water: 7 ml IPA mixture). Finally the cake was dried under vacuum below 40°C to provide the title compound as a off-white solid in 1.8 g quantity in 65% yield.
Analysis
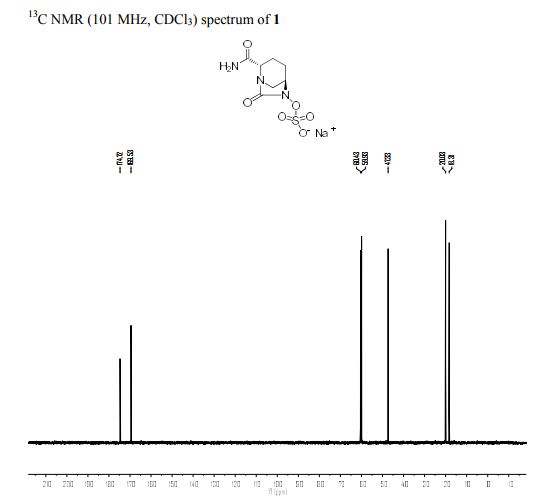
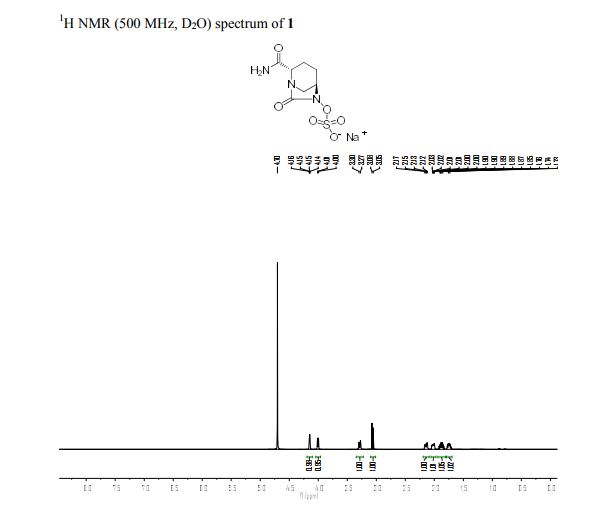
H1NMR (DMSO-d6, D20 exchange) = 8.19 (d, exchanges with D20), 3.99 (s, 1H), 3.82-3.92 (m, 1H), 3.72 (d, 1H), 2.24 (br d, 3H), 2.90-3.04 (m, 5H), 1.96-2.06 (m, 1H), 1.80-1.94 (m, 3H), 1.58-1.72 (m, 4H).
MS (ES+) C12H20N4O6S = 349.2 (M+l) as a free sulfonic acid;
Purity by HPLC: 99.2%
Specific rotation: [a] D -45.25 °, (c 0.3%, water)

SEE BACTAM SERIES…………..http://apisynthesisint.blogspot.in/p/bactam-series.html
//////
C1CC(N2CC1N(C2=O)OS(=O)(=O)O)C(=O)NC3CCNCC3.O
UPDATE,,,,,,,,,,
Improved Preparation of a Key Hydroxylamine Intermediate for Relebactam: Rate Enhancement of Benzyl Ether Hydrogenolysis with DABCO
Process R&D Department, MRL, Merck & Co., Inc., Rahway, New Jersey 07065, United States
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.7b00381
Publication Date (Web): February 1, 2018
Copyright © 2018 American Chemical Society
Previous methods to prepare a bicyclic N-hydroxyl urea intermediate in the synthesis of the potent β-lactamase inhibitor relebactam were effective, but deemed unsuitable for long-term use. Therefore, we developed an in situ protection protocol during hydrogenolysis and a robust deprotection/isolation sequence of this unstable intermediate employing a reactive crystallization. During the hydrogenation studies, we discovered a significant rate enhancement of O-benzyl ether hydrogenolysis in the presence of organic amine bases, especially DABCO. The broader utility of the application of organic bases on the hydrogenolysis of a range of O– and N-benzyl-containing substrates was demonstrated.

5 could be isolated by concentrating the filtrate and storing the solution at 5 °C overnight. 1H NMR (500 MHz, CDCl3): δ 6.58 (d, J = 7.9 Hz, 1H), 4.10–3.86 (m, 4H), 3.55 (bs, 1H), 3.14 (bd, J = 11.5 Hz, 1H), 2.86 (bt, J = 12.0 Hz, 2H), 2.76 (d, J = 11.5 Hz, 1H), 2.36 (dd, J = 15.1, 7.1 Hz, 1H), 2.12 (m, 1H), 2.00–1.82 (m, 3H), 1.66 (m, 1H), 1.44 (s, 9H), 1.31 (m, 2H), 0.25 (S, 9H). 13C NMR (125 MHz, CDCl3): δ 169.2, 168.3, 154.8, 79.8, 60.7, 60.0, 47.3, 46.9, 42.6 (br, 2C), 32.2, 31.9, 28.5 (3C), 20.5, 17.5, −0.75 (3C). (+)-ESI HRMS: calcd for C20H36N4NaO3Si (M + Na)+, 463.2347; found, 463.2348.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....







































































 .
.