
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

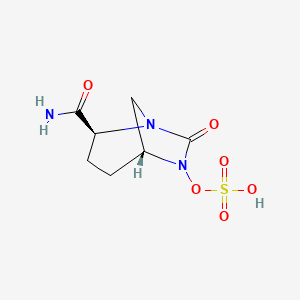
NXL-104, Avibactam
trans-7-oxo-6-(sulphooxy)-1,6-diazabicyclo[3,2,1]octane-2-carboxamide sodium salt (e.g., NXL-104)
CAS 396731-20-7, 1192491-61-4
AVE-1330
AVE-1330A
PHASE 1 a broad-spectrum intravenous beta-lactamase inhibitor, was under development for the treatment of infections due to nosocomial drug resistant Gram-negative bacteria
SANOFI INNOVATOR
Novexel holds exclusive worldwide development and commercialization rights from Sanofi.
NXL104; Avibactam; UNII-7352665165;
| Molecular Formula: | C7H11N3O6S |
|---|---|
| Molecular Weight: | 265.24374 g/mol |
CAS 1192500-31-4, 396731-14-9
[(2S,5R)-2-carbamoyl-7-oxo-1,6-diazabicyclo[3.2.1]octan-6-yl] hydrogen sulfate
(2S,5R)-7-oxo-6-(sulfooxy)-1,6-diazabicyclo[3.2.1]octane-2-carboxamide
trans-7-oxo-6-(sulfooxy)-1,6-diazabicyclo[3.2.1]octan-2-carboxamide
1,6-Diazabicyclo(3.2.1)octane-2-carboxamide, 7-oxo-6-(sulfooxy)-, (1R,2S,5R)-rel-
NXL-104/ceftazidime, Cephems (Cephalosporins), Fixed-Dose Combination

Avibactam is a non-β-lactam β-lactamase inhibitor antibiotic being developed by Actavis jointly with AstraZeneca. A new drug application for avibactam in combination with ceftazidime was approved by the FDA on February 25, 2015, for treating complicated urinary tract and complicated intra-abdominal Infections caused by antibiotic resistant-pathogens, including those caused by multi-drug resistant gram-negative bacterial pathogens.[2][3][4]
Increasing resistance to cephalosporins among Gram-(-) bacterial pathogens, especially among hospital-acquired infections, results in part from the production of beta lactamase enzymes that deactivate these antibiotics. While the co-administration of a beta lactamase inhibitor can restore antibacterial activity to the cephalorsporin, previously approved beta lactamase inhibitors such astazobactam and Clavulanic acid do not inhibit important classes of beta lactamase including Klebsiella pneumoniae carbapenemases (KPCs), metallo-beta lactamases, and AmpC. Avibactam inhibits KPCs, AmpC, and some Class D beta lactamases, but is not active aganist NDM-1.[5]
U.S. Pat. No. 7,112,592 discloses novel heterocyclic compounds and their salts, processes for making the compounds and methods of using the compounds as antibacterial agents. One such compound is sodium salt of trans-7-oxo-6-(sulphooxy)-1,6-diazabicyclo[3,2,1]octane-2-carboxamide. Application WO 02/10172 describes the production of azabicyclic compounds and salts thereof with acids and bases, and in particular, trans-7-oxo-6-sulphoxy-1,6-diazabicyclo[3.2.1]octane-2-carboxamide and its pyridinium, tetrabutylammonium and sodium salts. Application WO 03/063864 and U.S. Patent Publication No. 2005/0020572 describe the use of compounds including trans-7-oxo-6-(sulphooxy)-1,6-diazabicyclo[3,2,1]octane-2-carboxamide sodium salt, as β-lactamase inhibitors that can be administered alone or in, combination with β-lactamine antibacterial agents. These references are incorporated herein by reference, in their entirety.


| INGREDIENT | UNII | CAS | Average: 287.22
C7H10N3NaO6S |
|---|---|---|---|
| Avibactam sodium | 9V824P8TAI | 1192491-61-4 |
Avibactam, sodium (2S,5R)-2-carbamoyl-7-oxo-1,6-diazabicyclo[3.2.1]octan-6-yl sulfonate, containing a diazabicyclo[3.2.1]octane (DBO) heterocyclic core structure, is a novel diazabicyclooctane non-β-lactama β-lactamase inhibitor. It has a unique mechanism of inhibition among β-lactamase inhibitors, which is able to bind reversibly and covalently to β-lactamase.
As a new drug featured with bacterial resistance, avibactam has been widely used in clinic and its combination with Ceftazidime (Zavicefta) has recently been approved by the EMA and FDA for treatment of complicated intra-abdominal infectious (CIAI), complicated urinary tract infectious (CUTI), hospital acquired pneumonia (HAP), etc.
Moreover, compared with the three known β-lactamase inhibitors named clavulanic acid, sulbactam, and tazobactam, the efficiency of avibactam is stronger and its spectrum is also broader: avibactam is active against class A including Class A Klebsiella pneumoniae carbapenemase (KPCs) and ESBLs, class C, and some class D β-lactamases

Synthesis
SYN1

Substantial effort has been devoted to the preparation of avibactam . Initially, the Aventis infection division in Romainville (France) disclosed route A for its synthesis in the early stage of drug discovery. In this process, double-chiral piperidine derivatives were used as starting material to provide avibactam via inversion of configuration, deprotection, urea-cyclization, deprotection, and sulfonation with about 9.0% total yields. Miller et al. also prepared avibactam based on route A. This route suffers from a long synthetic procedure, low yield, and heavy laborious workups. Besides, the raw materials (double-chiral piperidine derivatives) are expensive and a number of environmentally undesirable reagents and solvents are required in this route.
-
Dubreuil, L. J.; Mahieux, S.; Neut, C.; Miossec, C.; Pace, J. Int. J. Antimicrob. Agents 2012, 39 (6) 500– 504 DOI: 10.1016/j.ijantimicag.2012.02.013
-
(a) Mangion, I. K.; Ruck, R. T.; Rivera, N.; Huffman, M. A.; Shevlin, M. Org. Lett. 2011, 13 (20) 5480– 5483 DOI: 10.1021/ol202195n
(b) Krishnamurthy, S.; Venkataprasad, J.; Vagvala, T. C.; Moriguchi, T.; Tsuge, A. RSC Advances, 5 (64), 52154– 52160.
-
(a) Aventis Pharma SA: WO2002010172, 2002.
(a) Aszodi, J.; Lampilas, M.; Fromentin, C.; Rowlands, D. A. Aventis Pharma SA, FR, 2835186 (2003) .
(c) Novexel: WO2008142285, 2008.
Baldwin, J. E.; Adlington, R. M.; Godfrey, C. R.; Gollins, D. W.; Smith, M. L.; Russel, A. T. Synlett 1993, 1993 (01) 51– 53 DOI: 10.1055/s-1993-22345
Miller, S. P.; Zhong, Y. L.; Liu, Z.; Simeone, M.; Yasuda, N.; Limanto, J.; Chen, Z.; Lynch, J.; Capodanno, V. Org. Lett. 2014, 16 (1) 174– 177DOI: 10.1021/ol4031606
The Wockhardt developed route B to obtain avibactam from l-glutamate acid or l-pyroglutamic acid. In this route, the skeleton of the target molecular diazabicyclo[3.2.1]octane heterocyclic core structure (DBO) was constructed through the steps of ring-opening, ring-closing, deoxygenization, and then by deprotection, sulfonation, and other steps to obtain avibactam with a total yield of about 11.0%. This method, producing small scale avibactam in a single batch, has some drawbacks limiting the large-scale synthesis: (a) a long synthetic procedure, (b) complicated purification process, and (c) the employment of excessive environmentally unfriendly reagents such as diphosgene.
(a) Patil, V. J.; Tadiparthi, R.; Birajdar, S.; Bhagwat, S. P. US8969334, 2015.
(b) Hecker, E. A.; Baldwin, A. B-lactamase inhibitor picoline salt: P, US9120796, 2015.
(c) Aszodi, J.; Fromentin, C.; Lampilas, M.; Rowlands, D. A. P. US7612087, 2009.
(d) Gu, Y. G.; He, Y.; Yin, N.; Alexander, D. P. WO2013149136, 2013.
(e) Hwang, Y. S.; Gu, J. Q.; Jain, A.; Garad, S.; Jacob, P. S. P. US20140275001, 2014.
Recently, AstraZeneca and Forest Laboratories have optimized the process: from commercially available Boc-benzylglutamate in only 5 isolated steps with an overall yield of 35.0% (without including the construction of DBO). Another route is based on the olefin metathesis reaction to construct the DBO skeleton (Route C)
Xiong, H.; Chen, B.; Durand-Réville, T. F.; Joubran, C.; Alelyunas, Y. W.; Wu, D.; Huynh, H. ACS Med. Chem. Lett. 2014, 5 (10) 1143– 1147DOI: 10.1021/ml500284k
Ball, M.; Boyd, A.; Ensor, G. J.; Evans, M.; Golden, M.; Linke, S. R.; Milne, D.; Murphy, R.; Telford, A.; Kalyan, Y.; Lawton, G. R.; Racha, s.; Zhou, S. H. Org. Process Res. Dev. 2016, 20 (10) 1799– 1805 DOI: 10.1021/acs.oprd.6b00268
SYN2
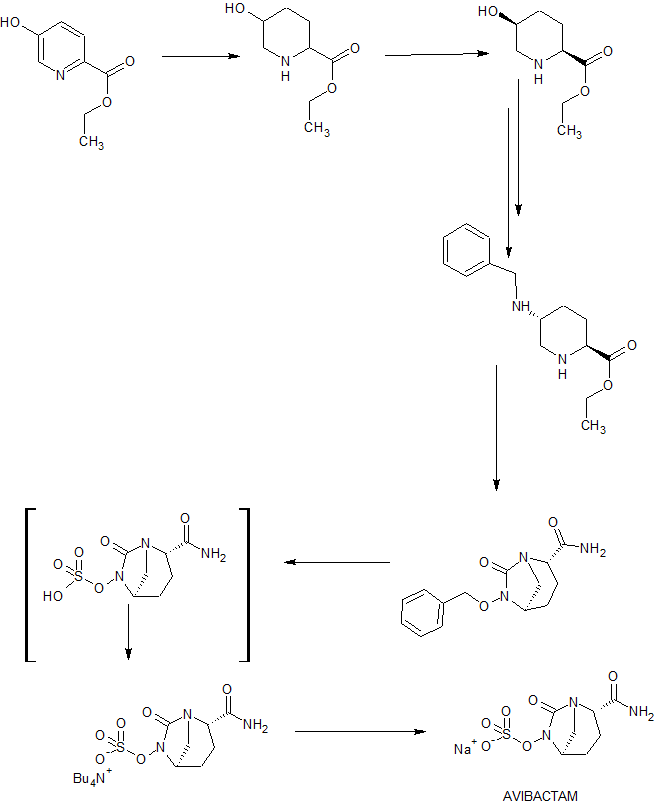
A New Synthetic Route to Avibactam: Lipase Catalytic Resolution and the Simultaneous Debenzylation/Sulfation


An efficient synthesis of avibactam starting from commercially available ethyl-5-hydroxypicolinate was completed in 10 steps and 23.9% overall yield. The synthesis features a novel lipase-catalyzed resolution, in the preparation of (2S,5S)-5-hydroxypiperidine-2-carboxylate acid, which is a valuable precusor of the key intermediate ethyl (2S,5R)-5-((benzyloxy)amino)piperidine-2-carboxylate. An optimized one-pot debenzylation/sulfation reaction, followed by cation exchange, gave the avibactam sodium salt on a 400.0 g scale.
Preparation of Avibactam Sodium Salt (1)
PATENT
In some embodiments, sulphaturamide or tetrabutylammonium salt of (1R,2S,5R)-7-oxo-6-(sulphooxy)-1,6-diazabicyclo[3.2.1]octane-2-carboxamide may be prepared by chiral resolution of its racemic precursor trans-7-oxo-6-(phenylmethoxy)-1,6-diazabicyclo[3.2.1]octane-2-carboxamide, the preparation of which is described in Example 33a Stage A in Application WO 02/10172. In exemplary embodiments, injection of 20 μl of a sample of 0.4 mg/mL of trans-7-oxo-6-(sulphooxy)-1,6-diazabicyclo[3.2.1]octane-2-carboxamide, eluted on a Chiralpak ADH column (5 25 cm×4.6 mm) with heptane-ethanol-diethylamine mobile phase 650/350/0.05 vol at 1 mL/min makes it possible to separate the (1R,2S,5R) and (1S,2R,5S) enantiomers with retention times of 17.4 minutes and 10.8 minutes respectively. The sulphaturamide is then obtained by conversion according to the conditions described in Example 33a Stage B then Stage C and finally in Example 33b of Application WO 02/10172.
In other embodiments, the sulphaturamide can be prepared from the mixture of the oxalate salt of (2S)-5-benzyloxyamino-piperidine-2-carboxylic acid, benzyl ester (mixture (2S,5R)/(2S,5S) ˜50/50) described in application FR2921060.
For example, the preparation may proceed in the following stages:

EXAMPLES Example 1 Preparation and characterization of amorphous trans-7-oxo-6-(sulphooxy)-1,6-diazabicyclo[3,2,1]octane-2-carboxamide sodium salt
Amorphous trans-7-oxo-6-(sulphooxy)-1,6-diazabicyclo[3,2,1]octane-2-carboxamide can be prepared as described in U.S. Pat. No. 7,112,592. The XRD pattern was obtained by mounting samples on a sample holder of Rigaku Miniflex X-ray diffractometer with the Kβ radiation of copper (λ=1.541 Å). The samples, without grinding, were put on a glass plate and were analyzed at ambient temperature and humidity. Data were collected at 0.05° interval, 2°/minute from 3°-40° 2θ. FIG. 1shows the X-ray diffraction (XRD) pattern for amorphous trans-7-oxo-6-(sulphooxy)-1,6-diazabicyclo[3,2,1]octane-2-carboxamide sodium salt.
A solution, in a water-acetone mixture (1-1), of the sodium salt of the racemic trans-7-oxo-6-(sulphoxy)-1,6-diazabicyclo[3.2.1]octane-2-carboxamide described in Example 33c of Application WO 02/10172 is evaporated under reduced pressure, under the conditions of concentration described in said example. The salt is obtained in crystallized form. The X-ray spectra (“XRPD diffraction patterns”) of the polymorphic Forms were compared. The diffraction pattern of the racemic form obtained according to the prior art is different from each of those of the polymorphic Forms.
Example 2 Preparation and characterization of Form I of trans-7-oxo-6-(sulphooxy)-1,6-diazabicyclo[3,2,1]octane-2-carboxamide sodium salt
Method I
A solution of the 5.067 g (10 mmoles) of the tetrabutylammonium salt of trans-7-oxo-6-(sulfooxy)-1,6-diazabicyclo[3,2,1]octane-2-carboxamide in 12.5 ml of 200 proof ethanol and 12.5 ml of 190 proof ethanol was filtered through a 1.6 μm filter and added to a 100 ml jacketed-reactor equipped with magnetic stirrer. The solution was warmed to an internal temperature of 35° C. Separately, a solution of 3.3 g (20 mmoles) of sodium 2-ethylhexanoate in 25 ml 200 proof ethanol was filtered through a 1.6 μm filter. 2.5 ml of this solution was added to the reactor and the mixture was stirred for 1 h at 35° C. Crystallization occurred during this time. The remainder of the sodium 2-ethylhexanoate solution was added over 20 min. The mixture was stirred for an additional 1 h at 35° C., followed by 12 h at 25° C. The mixture was cooled to 0° C. for 2 h. The crystals were isolated by filtration and washed with 10 ml ethanol. The crystals were dried under vacuum at 35° C. for 16 h. 2.72 g of the sodium salt of trans-7-oxo-6-(sulfooxy)-1,6-diazabicyclo[3,2,1]octane-2-carboxamide (Form I) was obtained, corresponding to a yield of 95%
PATENT
http://www.google.com/patents/WO2014135930A1?cl=en
Example -1
Preparation of sodium salt of (2S, 5R)-sulfuric acid mono-{2-carboxamido-7-oxo-l,6-diaza- bicyclo Γ3.2.11 octane
Step-1: Preparation of (2S, 5R)-2-Carboxamido-6-benzyloxy-7-oxo-l,6-diaza- bicyclo [3.2.1] octane:
Method-1:
The starting compound ((2S, 5R)-sodium 6-benzyloxy-7-oxo-l,6-diaza-bicyclo [3.2.1] octane-2-carboxylate; compound of Formula (II)) was prepared according to a procedure disclosed in Indian Patent Application No. 699/MUM/2013. To a 100 ml round bottom flask equipped with magnetic stirrer was charged (2S, 5R)-sodium 6-benzyloxy-7- oxo-l,6-diaza-bicyclo [3.2.1] octane-2-carboxylate (10.0 gm, 0.033 mol), followed by freshly prepared HOBt. ammonia complex (10.0 gm, 0.066 mol), EDC hydrochloride (9.62 gm, 0.050 mol) and 1-hydroxy benzotriazole (4.51 gm, 0.033 mol). To this mixture of solids, water (30 ml) was added at about 35°C, and stirring was started. Precipitation occurred after 30 minutes. The reaction mixture was stirred for additional 20 hours at about 35°C. Dichloro methane (150 ml) was added to the suspension and the reaction mass was allowed to stir for 10 minutes. The layers were separated. Aqueous layer was washed with additional dichloro methane (50 ml). Combined organic layer was evaporated under vacuum to provide a residue (21 gm). The residue was stirred with acetone (21 ml) for 30 minutes and filtered under suction to provide (2S, 5R)-2-carboxamido-6-benzyloxy-7-oxo-l,6-diaza- bicyclo [3.2.1] octane as a white solid in 5.5 gm quantity in 60% yield after drying under vacuum at about 45 °C.
Analysis
H!NMR (DMSO-de)
7.35 -7.45 (m, 6H), 7.25 (bs, 1H), 4.89 – 4.96 (dd, 2H), 3.68 (d, 1H), 3.62 (s, 1H), 2.90 (s, 2H), 2.04 – 2.07 (m, 1H), 1.70-1.83 (m, 1H), 1.61-1.66 (m, 2H).
MS (ES+) C14H17N3O3 = 276.1 (M+l) Purity: 93.95% as determined by HPLC Specific rotation: [a]25 D – 8.51° (c 0.5%, CHC13) Method-2:
Alternatively, the above compound was prepared by using the following process. To a 50 ml round bottom flask equipped with magnetic stirrer was charged a solution of (2S, 5R)- sodium 6-benzyloxy-7-oxo-l,6-diaza-bicyclo [3.2.1] octane-2-carboxylate (1 gm, 0.003 mol) in water (15 ml) followed by EDC hydrochloride (1 gm, 0.005 mol) and 1- hydroxybenzotriazole (0.39 gm, 0.003 mol) at 35°C under stirring. The reaction mass was stirred for 1 hour to obtain a white suspension. At this point, aqueous ammonia was added (2 ml, 40% w/v), under stirring. The reaction mixture was stirred for additional 5 hours. The suspension was filtered, washed with additional water (10 ml) to provide (2S, 5R)-2- carboxamido-6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1] after drying under vacuum at 45°C in 0.21 gm quantity.
Step-2: Preparation of Tetrabutyl ammonium salt of (2S, 5R)-2-carboxamido-6-sulfooxy-7- oxo-l,6-diaza-bicyclo [3.2.1] octane:
To a Parr shaker bottle, was charged (2S, 5R)-2-carboxamido-6-benzyloxy-7-oxo-l,6- diaza-bicyclo [3.2.1] octane (7.0 gm, 0.025 mol) followed by a 1:1 mixture of N,N- dimethylformamide and dichloro methane (35 ml: 35 ml). To the clear solution was added 10% palladium on carbon (1.75 gm) and hydrogen pressure was applied up to 50 psi. The suspension was shaken for 3 hours at 35°C. The catalyst was removed by filtering the reaction mixture over celite bed. The catalyst bed was washed with dichloro methane (30 ml). Combined filtrate was evaporated under vacuum at a temperature below 40°C to obtain an oily residue. The oily residue (4.72 gm) was dissolved in N,N-dimethylformamide (35 ml) and to the clear solution was added sulfur trioxide.DMF complex at 10°C under stirring in one lot. The mixture was allowed to stir at 35°C for additional 2 hours. As TLC showed complete conversion, 10% aqueous solution of tetrabutyl ammonium acetate (9.44 gm, 0.031 mol, in 30 ml water) was added under stirring and the reaction mixture was stirred for overnight and then subjected to high vacuum distillation on rotavapor by not exceeding temperature above 40°C to obtain a residue. Xylene (50 ml) was added to the residue and similarly evaporated to remove traces of DMF. The dry residue thus obtained was stirred with water (70 ml) and extracted with dichloro methane (70 ml x 2). Combined organic extract was dried over sodium sulfate and solvent was evaporated under vacuum below 40°C to obtain oily residue in 7 gm quantity as a crude product. It was stirred with methyl isobutyl ketone (21 ml) for 30 minutes at about 35°C to obtain a white solid in 5.9 gm quantity as a tetrabutyl ammonium salt of (2S, 5R)-2-carboxamido-6-sulfooxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane in pure form in 46% yield.
Analysis
NMR: (CDC13)
6.63 (s, 1H), 5.48 (s, 1H), 4.34 (br s, 1H), 3.90 (d, 1H), 3.27-3.40 (m, 9H), 2.84 (d, 1H), 2.38 (dd, 1H), 2.21-2.20 (m, 1H), 1.60-1.71 (m, 12H), 1.40-1.50 (m, 8H), 1.00 (t, 12H).
MS (ES-) C7H10N3O6S. N(C4H9)4 = 264.0 (M-l) as a free sulfonic acid.
Purity: 98.98% as determined by HPLC.
Specific rotation: [a]25 D – 30.99° (c 0.5%, MeOH)
Step-3: Synthesis of Sodium salt of (2S, 5R)-2-carboxamido-6-sulfooxy-7-oxo-l,6-diaza- bicyclo [3.2.1] octane
To a 100 ml round bottom flask equipped with magnetic stirrer was charged tetrabutyl ammonium salt of (2S, 5R)-2-carboxamido-6-sulfooxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane ( 5.5 gm, 0.0108 mol) followed by ethanol (28 ml) to provide a clear solution under stirring at about 35°C. To the reaction mixture was added a solution of sodium 2-ethyl hexanoate (3.6 gm, 0.021 mol) dissolved in ethanol (28 ml) in one lot under stirring to provide precipitation. The suspension was stirred for additional 2 hours to effect complete precipitation at about 35°C. The reaction mixture was filtered under suction and the wet cake was washed with acetone (30 ml x 2). The wet cake was dried at 40°C under vacuum to provide sodium salt of (2S, 5R)-2-carboxamido-6-sulfooxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane as a white solid in 2.6 gm quantity in 83% yield.
Analysis
H!NMR (DMSO-d6)
7.39 (s, 1H), 7.24 (s, 1H), 3.98 (s, 1H), 3.68 (d, 1H), 3.02 (d, 1H), 2.92 (d, 1H), 2.00- 2.10 (m, 1H), 2.80-2.90 (m, 1H), 1.55-1.70 (m, 2H).
MS (ES-) C7H10N3O6SNa = 264.0 (M-l) as a free sulfonic acid;
Purity: 97.98% as determined by HPLC
Specific rotation: [a]25 D – 49.37° (c 0.5%, water)
Powder X-ray diffractogram: (degrees 2 theta):
PATENT
http://www.google.com/patents/WO2015150941A1?cl=en
References
- “Full Prescribing Information: AVYCAZ™ (ceftazidime-avibactam) for Injection, for intravenous use”. ©2015 Actavis. All rights reserved. Retrieved 1 June 2015.
- Zhanel, GG (2013). “Ceftazidime-avibactam: a novel cephalosporin/β-lactamase inhibitor combination”. Drugs 73 (2): 159-77.doi:10.1007/s40265-013-0013-7. PMID 23371303.
- “Actavis Announces FDA Acceptance of the NDA Filing for Ceftazidime-Avibactam, a Qualified Infectious Disease Product”. Actavis—a global, integrated specialty pharmaceutical company—Actavis. Actavis plc. Retrieved 1 June 2015.
- Ehmann, DE; Jahic, H; Ross, PL; Gu, RF; Hu, J; Durand-Réville, TF; Lahiri, S; Thresher, J; Livchak, S; Gao, N; Palmer, T; Walkup, GK; Fisher, SL (2013). “Kinetics of Avibactam Inhibition against Class A, C, and D β-Lactamases”. The Journal of biological chemistry 288 (39): 27960–71. doi:10.1074/jbc.M113.485979. PMC 3784710. PMID 23913691.
- “www.accessdata.fda.gov” (PDF).
External links
- T. Edeki, J. Armstrong, J. Li. “Pharmacokinetics of Avibactam (AVI) and Ceftazidime (CAZ) Following Separate or Combined Administration in Healthy Volunteers”.

| Patent | Submitted | Granted |
|---|---|---|
| NOVEL CRYSTALLINE FORMS OF TRANS-7-OXO-6-(SULPHOOXY)-1,6-DIAZABICYCLO[3,2,1]OCTANE-2-CARBOXAMIDE SODIUM SALT [US2014349967] | 2014-08-07 | 2014-11-27 |
| PROCESS FOR PREPARING A COMPOUND USEFUL FOR PRODUCING AN OPTICALLY ACTIVE DIAZABICYCLOOCTANE COMPOUND [US2014303375] | 2014-05-27 | 2014-10-09 |
| QUICK METHOD FOR DETECTING ENYZMES AND MICROORANISMS [US2013089883] | 2011-03-01 | 2013-04-11 |
| Crystalline forms of trans-7-oxo-6-(sulphooxy)-1,6-diazabicyclo[3,2,1]octane-2-carboxamide sodium salt [US8835455] | 2013-05-24 | 2014-09-16 |
| WO2009091856A2 * | Jan 15, 2009 | Jul 23, 2009 | Merck & Co Inc | Beta-lactamase inhibitors |
| WO2012086241A1 * | Jun 30, 2011 | Jun 28, 2012 | Meiji Seika Pharma Co., Ltd. | Optically-active diazabicyclooctane derivative and method for manufacturing same |
| INMU06992013A | Title not available | |||
| US7112592 | Jul 24, 2001 | Sep 26, 2006 | Aventis Pharma S.A. | Azabicyclic compounds, preparation thereof and use as medicines, in particular as antibacterial agents |
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
[(2S,5R)-2-Carbamoyl-7-oxo-1,6-diazabicyclo[3.2.1]octan-6-yl] hydrogen sulfate
|
|
| Clinical data | |
| Trade names | Avycaz (formulated with ceftazidime) |
| Legal status |
|
| Routes of administration |
intravenous |
| Pharmacokinetic data | |
| Bioavailability | 100% (intravenous) |
| Protein binding | 5.7–8.2%[1] |
| Metabolism | nil |
| Onset of action | increases in proportion to dose |
| Excretion | Renal (97%) |
| Identifiers | |
| CAS Number | 1192500-31-4 |
| ATC code | J01 |
| PubChem | CID: 9835049 |
| ChemSpider | 8010770 |
| ChEBI | CHEBI:85984 |
| ChEMBL | CHEMBL1689063 |
| Chemical data | |
| Formula | C7H11N3O6S |
| Molecular mass | 265.24 g/mol |
SEE BACTAM SERIES…………..http://apisynthesisint.blogspot.in/p/bactam-series.html
////////
[Na+].NC(=O)[C@@H]2CC[C@@H]1CN2C(=O)N1OS([O-])(=O)=O
C1CC(N2CC1N(C2=O)OS(=O)(=O)O)C(=O)N

update…………
Development of a Manufacturing Route to Avibactam, a β-Lactamase Inhibitor
Abstract

Process development work to provide an efficient, robust, and cost-effective manufacturing route to avibactam, a β-lactamase inhibitor is presented herewith. Aspects of this optimization work include the counterintuitive introduction of a protecting group to effect a difficult urea formation and the use of controlled feed hydrogenation conditions to facilitate an elegant one pot debenzylation and sulfation reaction. Overall, the commercial process delivers avibactam in much improved yield with significant reduction in the environmental footprint.
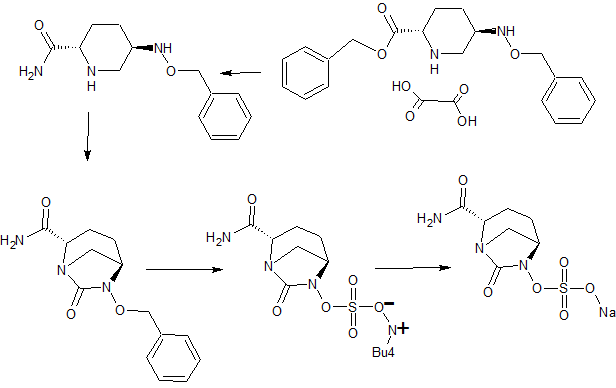
Preparation of Benzyl (2S,5R)-5-[(Benzyloxy)amino]piperidine-2-carboxylate Ethanedioate (1:1)
1H NMR (400 MHz, DMSO) δ: 1.52 (1H, m), 1.70 (1H, m), 1.94 (1H, d, J = 12.3 Hz), 2.22 (1H, dd, J = 13.8 Hz, J = 3.6 Hz), 2.79 (1H, t, J = 11.5 Hz), 3.27 (1H, m), 3.46 (1H, d, J = 11.5 Hz), 4.14 (1H, dd, J = 12.3 Hz, J = 3.2 Hz), 4.68 (2H, s), 5.24 (2H, s), 7.34 (10H, m). 13C NMR (100 MHz, DMSO) δH 25.4 (s), 26.1 (s), 46.5 (s), 54.0 (s), 56.4 (s), 67.3 (s), 76.4 (s), 128.5 (m), 135.7 (s), 138.5 (s), 164.7 (s), 167.5 (s). HRMS Calcd for C20H25N2O3: 341.1860; HRMS found [M+H]+: 341.1858.
Preparation of (2S,5R)-5-[(Benzyloxy)amino]piperidine-2-carboxamide
1H NMR (400 MHz, DMSO) δH 1.12 (1H, m), 1.27 (1H, m), 1.83 (2H, m), 2.22 (1H, dd, J = 10.1 Hz, J = 12.0 Hz), 2.76 (1H, m), 2.89 (1H, dd, J = 2.8 Hz, J = 10.9 Hz), 3.14 (1H, dd, J = 4.1 Hz, J = 12.0 Hz), 4.58 (2H, s), 6.46 (1H, d, J = 5.6 Hz), 6.91 (1H, s), 7.09 (1H, s), 7.32 (5H, m). 13C NMR (100 MHz, DMSO) δH 28.4 (s), 29.2 (s), 49.5 (s), 57.5 (s), 59.8 (s), 76.3 (s), 128.3 (m), 138.9 (s), 175.6 (s). HRMS Calcd for C13H20N3O2: 250.1550; HRMS found [M+H]+: 250.1551.
Preparation of (2S,5R)-6-(Benzyloxy)-7-oxo-1,6-diazabicyclo[3.2.1]octane-2-carboxamide
1H NMR (400 MHz, DMSO) δH 1.65 (2H, m), 1.83 (1H, m), 2.07 (1H, m), 2.91 (2H, s), 3.63 (1H, s), 3.69 (1H, d, J = 6.7 Hz), 4.92 (2H, dd, J = 18.1 Hz, J = 11.4 Hz), 7.38 (7H, m). 13C NMR (100 MHz, DMSO) δH 18.6 (s), 21.1 (s), 47.2 (s), 57.5 (s), 60.1 (s), 77.4 (s), 129.0 (m), 136.3 (s), 167.9 (s), 171.8 (s). HRMS Calcd for C14H18N3O3: 276.1343; HRMS found [M+H]+: 276.1336
Preparation of Tetrabutylammonium [(2S,5R)-2-Carbamoyl-7-oxo-1,6-diazabicyclo[3.2.1]octan-6-yl] Sulfate
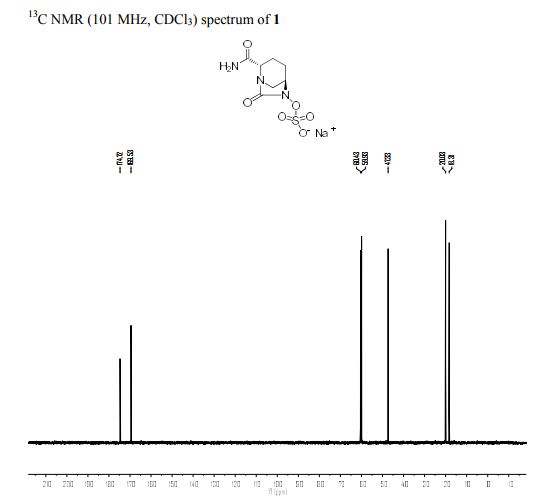
1H NMR (400 MHz, CDCl3) δH 1.00 (12H, t, J = 7.2 Hz), 1.45 (8H, m), 1.67 (9H, m), 1.87 (1H, m), 2.16 (1H, m), 2.37 (1H, dd, J = 15.0 Hz, J = 7.0 Hz), 2.87 (1H, d, J = 11.6 Hz), 3.31 (9H, m), 3.91 (1H, d, J = 7.9 Hz), 4.33 (1H, s), 5.87 (1H, s), 6.69 (1H, s). 13C NMR (100 MHz, D2O) δH 12.8 (s), 18.1 (s), 19.1 (s), 19.9 (s), 23.1 (s), 47.2 (s), 58.1 (s), 59.8 (s), 60.3 (s), 169.4 (s), 174.7 (s). HRMS Calcd for C7H10N3O6S: 264.0296; HRMS found [M–H]–: 264.0298
AVIBACTAM
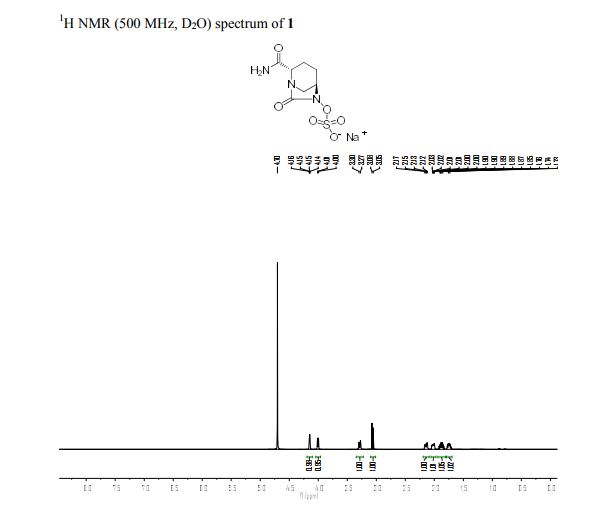
1H NMR (500 MHz, DMSO) δH 1.63 (2H, m), 1.83 (1H, m), 2.05 (1H, m), 2.90 (1H, d, J = 11.8 Hz), 3.00 (1H, d, J = 11.6 Hz), 3.67 (1H, d, J = 6.9 Hz), 3.98 (1H, s), 7.29 (1H, s), 7.44 (1H, s). 13C NMR (100 MHz, D2O) δH 18.1 (s), 19.9 (s), 47.2 (s), 59.8 (s), 60.3 (s), 169.4 (s), 174.7 (s). HRMS Calcd for C7H12N3O6S: 266.0441; HRMS found [M+H]+: 266.0441


Shifts
| Index | Name | Shift (ppm) |
|---|---|---|
| 16 | H6 | 4.573 |
| 27 | H5 | 2.416 |
| 18 | H7 | 1.742 |
| 19 | H8 | 1.870 |
| 26 | H4 | 1.949 |
| 11 | H2 | 3.497 |
| 12 | H3 | 4.412 |
| 14 | H1 | 5.267 |
///////////

