
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
New EU GMP Annex 15 Revision published – Valid as of 1 October 2015
DRUG REGULATORY AFFAIRS INTERNATIONAL
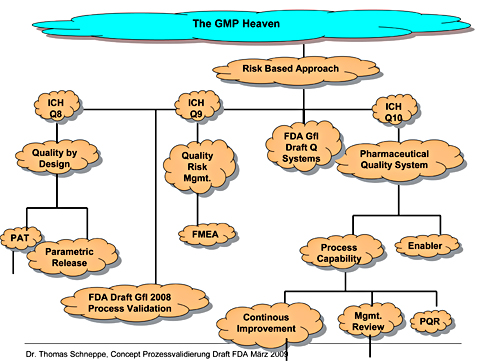
In February 2014 the draft for the revision of Annex 15 was published. Compared with the currently valid version the changes were partly significant. Now the draft was published as final document and will be valid as of 1 October 2015. Read more about the Changes in Annex 15.
In February 2014 the draft for the revision of EU GMP Annex 15 was published (see the GMP-News from 11 February 2014 “Revision of the EU GMP Annex 15 for Qualification and Validation published“). Compared with the currently valid version the changes were significant in some parts (see also the GMP-News from 21 March 2014 “Detailed Analysis of Annex 15 Draft“. Now the draft was published as final document and will be valid as of 1 October 2015.
What will change? Following you will find an overview about the changes.
With 16 pages the document is much…
View original post 1,474 more words
Overview about API manufacturing for the European market
DRUG REGULATORY AFFAIRS INTERNATIONAL
![]()
EudraGMDP provides some interesting information about the API manufacturing sites as well as about importers, distributors of APIs to be used as starting material in Medicinal Products for human use in Europe. Please read more about the API registrations in EudraGMDP.
EudraGMDP provides some interesting information about the API manufacturing sites as well as about importers, distributors of APIs to be used as starting material in Medicinal Products for human use in Europe. Although the database is still not complete (not all competent authorities in Europe have established a system to make sure that all registration data will be entered into EudraGMDP in a timely manner) the current information is already very interesting.
Currently (as per 19 March 2015) the database counts 3.275 API manufacturing sites, importers or distributors located outside Europe. On the other side 936 API manufacturing sites, importers or distributors are located in EEA countries (EU Member states…
View original post 143 more words
A surprising source of serotonin could affect antidepressant activity

This schematic drawing of a serotonergic neuron shows exocytotic release of serotonin from vesicles (red arrow) and the nonexocytotic release described by Mlinar and colleagues (blue arrow). Reuptake of serotonin (green arrow) is blocked by SSRI antidepressants, increasing the extracellular serotonin concentration. Credit: Adell 2015
Depression affects an estimated 350 million people worldwide and poses a major public health challenge, according to the World Health Organization. Researchers have discovered an unconventional way that serotonin is released from neurons that could play an important role in the mechanism through which antidepressant drugs work. The Journal of General Physiology study is highlighted in the April issue.
Serotonin is a chemical in the brain that plays a key role in regulating various emotions and behaviors. Like other neurotransmitters, which relay signals between neurons, serotonin is stored in small sacs called vesicles in the presynaptic terminal of one neuron and released into the synapse…
View original post 202 more words
LINEZOLID
 LINEZOLID
LINEZOLID

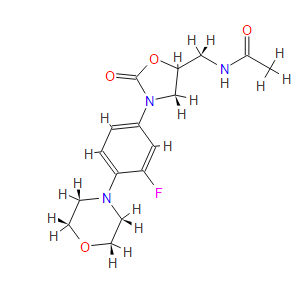
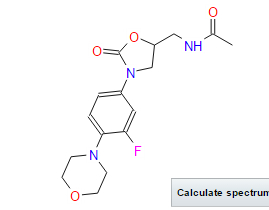
(S)-N-[[3-(3-fluoro-4-morpholinylphenyl)-2-oxo-5-oxazolidinyl]methyl] acetamide.
| N-[[(5s)-3-(3-fluoro-4-morpholin-4-ylphenyl)-2-oxo-1,3-oxazolidin-5-yl]methyl]acetamide PRODUCT PATENTUS5688792 (1997 to Pharmacia & Upjohn) |
|
| CAS No.: | 165800-03-3 |
|---|---|
| Synonyms: | |
| Formula: | C16H20FN3O4 |
| Exact Mass: | 337.14400 |

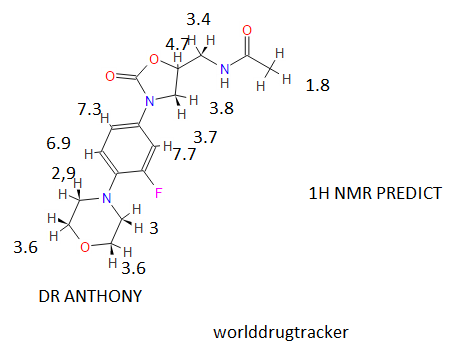
13C
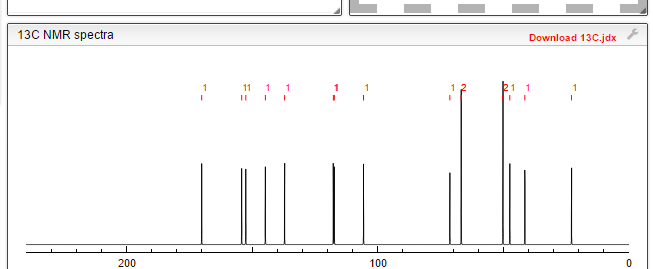
1H NMR AND 13C PREDICT
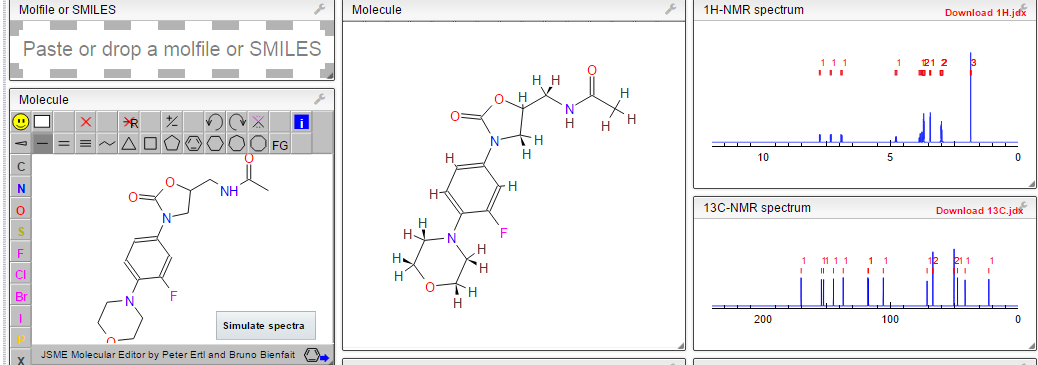

1H NMR PREDICT
![N-[[(5S)-3-(3-fluoro-4-morpholin-4-ylphenyl)-2-oxo-1,3-oxazolidin-5-yl]methyl]acetamide NMR spectra analysis, Chemical CAS NO. 165800-03-3 NMR spectral analysis, N-[[(5S)-3-(3-fluoro-4-morpholin-4-ylphenyl)-2-oxo-1,3-oxazolidin-5-yl]methyl]acetamide H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-19/000/030/236/165800-03-3-1h.png)
13C NMR PREDICT
![N-[[(5S)-3-(3-fluoro-4-morpholin-4-ylphenyl)-2-oxo-1,3-oxazolidin-5-yl]methyl]acetamide NMR spectra analysis, Chemical CAS NO. 165800-03-3 NMR spectral analysis, N-[[(5S)-3-(3-fluoro-4-morpholin-4-ylphenyl)-2-oxo-1,3-oxazolidin-5-yl]methyl]acetamide C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-19/000/030/236/165800-03-3-13c.png)
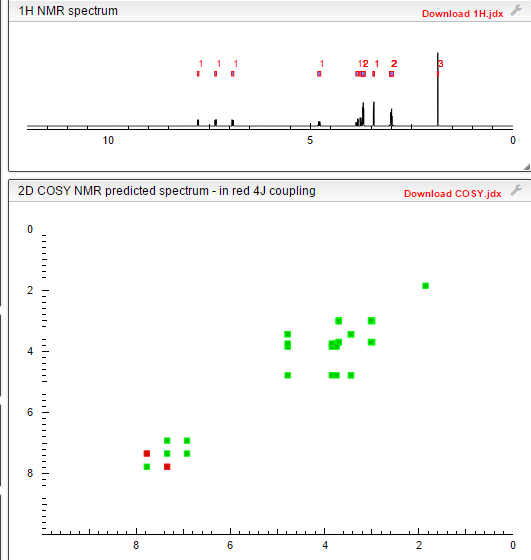
COSY
PREDICT
HMBC

ORIGINAL 1H NMR…………...http://www.selleckchem.com/products/Linezolid(Zyvox).html

INTERMEDIATES USED
Arkivoc, , vol. 2012, # 6 p. 45 – 56
WO2011/137222 A1, ;

Union Quimico Farmaceutica, S.A. (UQUIFA) Patent: EP2163547 A1, 2010 ; Location in patent: Page/Page column 11 ;

THE REGENTS OF THE UNIVERSITY OF CALIFORNIA; GARG, Neil K.; RAMGREN, Stephen D.; SILBERSTEIN, Amanda L.; QUASDORF, Kyle W. Patent: WO2012/94622 A2, 2012 ; Location in patent: Page/Page column 31-32 ;

Lianhe Chemical Technology Co., Ltd. Patent: EP2388251 A1, 2011 ; Location in patent: Page/Page column 11 ;
Tammana, Rajesh; Vemula, Kiran Kumar; Guruvindapalli, Ramadasu; Yanamandr, Ramesh; Gutta, Madhusudhan Arkivoc, 2012 , vol. 2012, # 6 p. 45 – 56

Union Quimico Farmaceutica, S.A. (UQUIFA) Patent: EP2163547 A1, 2010 ; Location in patent: Page/Page column 10 ;

Song, Lirong; Chen, Xiaobei; Zhang, Shilei; Zhang, Haoyi; Li, Ping; Luo, Guangshun; Liu, Wenjing; Duan, Wenhu; Wang, Wei Organic Letters, 2008 , vol. 10, # 23 p. 5489 – 5492

Union Quimico Farmaceutica, S.A. (UQUIFA) Patent: EP2163547 A1, 2010 ; Location in patent: Page/Page column 10 ;

JUBILANT LIFE SCIENCES LIMITED; BISWAS, Sujay; PANDA, Atulya, Kumar; GUPTA, Ashish, Kumar; SINGH, Shishupal; TIWARI, Praveen; VIR, Dharam; THOMAS, Saji Patent: WO2013/111048 A1, 2013 ; Location in patent: Page/Page column 24; 25 ;

Perrault, William R.; Pearlman, Bruce A.; Godrej, Delara B.; Jeganathan, Azhwarsamy; Yamagata, Koji; Chen, Jiong J.; Lu, Cuong V.; Herrinton, Paul M.; Gadwood, Robert C.; Chan, Lai; Lyster, Mark A.; Maloney, Mark T.; Moeslein, Jeffery A.; Greene, Meredith L.; Barbachyn, Michael R. Organic Process Research and Development, 2003 , vol. 7, # 4 p. 533 – 546

US6362334 B1, ; Example 13 ;

NMR OF INTERMEDTIATES






-
Linezolid is a pharmaceutically active compound useful as an antibacterial agent, e.g. for the treatment of diabetic food infections caused by Gram-positive bacteria. It is represented by the formula (I),

-
[0003]The marketed pharmaceutical compositions are a sterile isotonic solution for an i.v. infusion, a tablet for oral administration and an aqueous suspension for oral administration. They are marketed, i.e., under brand name ZYVOX by Pfizer.
-
[0004]The molecule of linezolid has one asymmetric carbon in the molecule allowing for 2 enantiomers; the marketed compound is the (S)-enantiomer. In the above-marketed compositions, linezolid is present as a free base.
-
[0005]Hereinunder, the name linezolid will be used as the generic name for N-(3-(3-fluoro-4-(morpholin-4-yl)phenyl)-2-oxooxazolidin-5(S)-ylmethyl)acetamide, unless indicated to the contrary.
-
[0006]Linezolid was first disclosed in WO 95/07271 ( EP 0717738 , US 5,688,792 ) of the Upjohn Company.
-
[0007]Various processes for making linezolid are known in the art. In particular, the important ones are these, the final step of which comprises acetylation of an amine precursor of the formula (II) with an acetylhalide or acetic anhydride (see, e.g., WO 2005 099353 ),

-
[0008]This amine precursor (II) may be made from various starting materials, e.g.:
- a) By a reduction of an azide compound of formula (III) by a suitable reductant ( WO2006/091731 , WO 95/07271 , US 5837870 , WO2009/063505 , US 7291614 ),

The starting compound (III) may be made from the corresponding tosylate or chloride of general formula (VII) below ( WO 2005/099353 ).
- b) By a decomposition of a phthalimide compound of formula (IV), e.g. by methylamine ( WO95/07271 ) or by hydrazine ( US 5837870 ),

The starting compound (IV) may be made from the same tosylate or chloride as sub a) ( WO2005/099353 ) or by a cyclization of the oxazolidine ring ( WO 99/24393 , WO2006/008754 ).
- c) From a sulfonate compound of formula (V),

by treatment with ammonium hydroxide in isopropanol or THF ( WO 95/07271 ) or by treatment with ammonia under enhanced pressure ( WO 97/37980 ).
- d) By a reduction of an imine (VI),

wherein R2 is a chlorophenyl, bromophenyl or 2,4,-dichlorophenyl moiety ( WO 2007/116284 ).
- a) By a reduction of an azide compound of formula (III) by a suitable reductant ( WO2006/091731 , WO 95/07271 , US 5837870 , WO2009/063505 , US 7291614 ),
-
[0009]Except of the imine (VI), each of the preceded synthetic approaches is based on a step of converting a starting material of the general formula (VII),

wherein L is a suitable leaving group, for instance a halogen or an alkyl-or aryl sulfonyloxy group,
by a reaction with a nitrogen nucleophile (an azide salt, phthalimide salt, ammonia or ammonium hydroxide), followed, if necessary, by a next step of conversion of the formed reaction intermediate (e.g., compound (III) or compound (IV)) into the amino/compound (II). Apparently, making the starting amine-compound (II) in a good yield and purity is the key aspect of commercial success of any of the above synthetic routes yielding linezolid. However, the known approaches have various drawbacks, for instance serious toxicity and explosion hazard of the azide salts, long reaction times and hazardous agents (hydrazine, methyl amine) in using the phthalimide intermediate, low yields and many side products at the ammonium hydroxide approach, or harsh reaction conditions in reaction with ammonia.










is reacted without isolation with acetic anhydride as an oily product, or in solution, to produce the acetamide, linezolid (1). This is followed by procedures for isolating the linezolid (1) such as those described in U.S. Pat. No. 5,688,792, at col. 15, 11. 22-28 (chromatography and separation of the desired fraction, followed by evaporation and trituration of the product to obtain pure linezolid (1)).

is reduced to its corresponding amine, S-N-(4-morpholinyl-3-fluorophenyl)-2-oxo-5-oxazolidinyl-methyl amine (2) in the solvent ethyl acetate by hydrogenation using hydrogen gas and a palladium/carbon catalyst. These reaction conditions lead to the production of an undesirable level of reaction by-products, and, following the acetylation of the intermediate amine (2) to linezolid (1), to undesirably high levels of bis-linezolid (4)

http://www.google.com/patents/US20060252932



A Novel Synthesis of Oxazolidinone Derivatives (A Key Intermediate of Linezolid)
2Center for Pharmaceutical sciences, Jawaharlal Nehru Technological University, Kukatpally, Hyderabad, India

N-[[(5S)-3-[3-fluoro-4-(4-morpholinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide (7a):
IR (KBr, cm-1): 3338 (N-H stretching), 3117, 3066 (aromatic C-H stretching), 2971, 2863, 2818 (aliphatic C-H stretching), 1738, 1662 (C=O stretching), 1545, 1516,1453 (aromatic C=C stretching), 1425 (C-N stretching), 1381 (aliphatic C-H bending), 1334 (C-F stretching), 1274 (C-O stretching), 1198, 1177 (C-N bending), 1117, 1081 (aromatic C-H bending).
1H NMR (CDCl3) δ ppm: 7.44 (m, 1H), 7.26 (m, 1H), 6.99 (m, 1H), 6.01 (t,1H), 4.76 (m, 1H), 4.02 (m, 2H), 3.80 (m, 4H), 3.61(m, 2H), 3.05 (m, 4H), 2.02 (t, 3H):
C13NMR(CDCl3) δppm: 171.33, 156.87, 154.44, 136.40, 132.84, 118.67, 113.81, 107.52, 71.96, 66.76, 50.79, 47.46, 41.68, 22.81. MS: 338 (M++H);
……………………………………………………………………….
ARKIVOC 2012 (vi) 45-56 Page 45 ©ARKAT-USA, Inc.
An expeditious construction of 3-aryl-5-(substituted methyl)-2- oxazolidinones: a short and efficient synthesis of Linezolid
Rajesh Tammana,a,b Kiran Kumar Vemula,a Ramadasu Guruvindapalli,a Ramesh Yanamandra,c and Madhusudhan Gutta* a
aDepartment of Research & Development, Inogent Laboratories Pvt. Ltd.,
A GVK BIO Company, 28A, IDA, Nacharam, Hyderabad 500 076, Andhra Pradesh, India
bCentre for Pharmaceutical Sciences, Institute of Science and Technology, Jawaharlal Nehru Technological University, Hyderabad 500 072, Andhra Pradesh, India
cDepartment of Analytical Research & Development, GVK Biosciences Pvt. Ltd., 28A, IDA, Nacharam, Hyderabad 500 076, Andhra Pradesh, India
E-mail: madhusudhan.gutta@inogent.com
http://www.arkat-usa.org/get-file/42622/
N-(((S)-3-(3-fluoro-4-morpholinophenyl)-2-oxooxazolidin-5-yl)methyl)acetamide 1 (Linezolid) 1 was prepared according to the method described in literature.12,15
Mp 182-183 °C, (lit.12a 181.5- 182.5 °C); enantiomeric purity 99.9% (by chiral HPLC);
IR (KBr): ν 3343 (NH), 3075 (Ar-H), 2967 (CH), 1741 (C═O), 1660 (C═O) cm-1 ;
1H NMR (CDCl3): δ 2.03 (s, 3H), 3.04-3.07 (t, 4H), 3.56-3.77 (m, 3H), 3.86-3.89 (t, 4H), 4.00-4.06 (t, 1H), 4.74-4.79 (m, 1H), 5.96 (s, 1H), 6.90- 6.96 (t, 1H), 7.06-7.10 (d, 1H), 7.43-7.48 (d, 1H).
13C NMR (DMSO-d6): δ 22.4, 41.4, 47.3, 50.6, 66.1, 71.5, 106.4, 114.0, 119.1, 133.3, 135.5, 154.0, 156.2, 170.0;
ESI-MS (C16H20FN3O4): m/z (%) 338.18 (100, M+ +1).
12. (a) Brickner, S. J.; Hutchinson, D. K.; Barbachyn, M. R.; Manninen, P. R.; Ulanowicz, D. A.;
Garmon, S. A.; Grega, K. C.; Hendges, S. K.; Toops, D. S.; Ford, C. W.; Zurenko, G. E. J.
Med. Chem. 1996, 39, 673. (b) Barbachyn, M. R.; Brickner, S. J.; Hutchinson, D. K. U.S.
patent 5688792; 1997; Chem. Abstr. 1995, 123, 256742. (c) Dhananjay, G. S.; Nandu, B. B.;
Avinash, V. N.; Kamlesh, D. S.; Anindya, S. B.; Tushar, A. N. PCT Int. Appl. 063505, 2009;
Chem. Abstr. 2009, 150, 515152.
13. (a) Imbordino, R. J.; Perrault, W. R.; Reeder, M. R. PCT Int. Appl. 116284, 2007; Chem.
Abstr. 2007, 147, 469356. (b) Pearlman, B. A.; Perrault, W. R.; Barbachyn, M. R.;
Manninen, P. R.; Toops, D. S.; Houser, D. J.; Fleck, T. J. U.S. Patent 5837870, 1998; Chem.
Abstr. 1998, 130, 25061. (c) Perrault, W. R.; Pearlman, B. A.; Godrej, D. B.; Jeganathan, A.;
Yamagata, K.; Chen, J. J.; Lu, C. V.; Herrinton, P. M.; Gadwood, R. C.; Chan, L.; Lyster, M.
A.; Maloney, M. T.; Moeslein, J. A.; Greene, M. L.; Barbachyn, M. R. Org. Proc. Res. Dev.
2003, 7, 533.
14. (a) Yu, D. S.; Huang, L.; Liang, H.; Gong, P. Chin. Chem. Lett. 2005, 16, 875. (b) Pearlman,
B. A. PCT Int. Appl. 9924393, 1999; Chem. Abstr. 1999, 130, 338099. (c) Weigert, F. J. J.
Org. Chem. 1973, 38, 1316.
15. (a) Wang, M.; Tong, H. CN patent 101220001, 2008. (b) Mohan Rao, D.; Krishna Reddy, P.
PCT Int. Appl. 099353, 2005; Chem. Abstr. 2005, 143, 440395. (c) Mohan Rao, D.; Krishna
Reddy, P. PCT Int. Appl. 008754, 2006; Chem. Abstr. 2006, 144, 170978.
……………………………………
Organic Process Research and Development, 2003 , vol. 7, # 4 p. 533 – 546
http://pubs.acs.org/doi/abs/10.1021/op034028h


(S)-N-[[3-[3-Fluoro-4-(4-morpholinyl)phenyl]-2-oxo- 5-oxazolidinyl]methyl]acetamide: Linezolid: Zyvox
HPLC analyses showed the first and second crops to be 98.9 and 94.6 wt % linezolid, respectively, with <0.2% enantiomer in each; also, an additional 9.7% yield of linezolid was detected in the filtrate by external standard HPLC (total ) 80.6%). Analysis data for 1st crop material: mp ) 73-76 °C;
1 H NMR (CDCl3, 400 MHz)
δ 7.43 (dd, J ) 14.4, 2.4 Hz, 1H), 7.07 (dd, J ) 8.8, 2.0 Hz, 1H), 6.91 (t, J ) 8.8 Hz, 1H), 6.43 (br t, 1H), 4.77 (m, 1H), 4.02 (t, J ) 9.2 Hz, 1H), 3.86 (t, J ) 4.4 Hz, 4H), 3.76 (dd, J ) 8.8, 6.8 Hz, 1H), 3.66 (m, 2H), 3.05 (t, J ) 4.8 Hz, 4H), 2.02 (s, 3H);
13C NMR (CDCl3, 100 MHz)
δ 23.07 (q), 41.93 (t), 47.66 (t), 51.00 (t), 66.95 (t), 71.99 (d), 107.56 (dd, JC-F ) 26.16 Hz), 113.97 (dd, JC-F ) 3.02 Hz), 118.85 (dd, JC-F ) 4.03 Hz), 132.90 (sd, JC-F ) 4.03 Hz), 136.58 (sd, JC-F ) 9.06 Hz), 154.42 (s), 155.50(sd, JC-F ) 246.53 Hz), 171.19 (s)
MS (EI) m/z (relative intensity) 337 (90), 293 (81), 209 (100);
[R]25D ) -16 (c ) 1.05, ethanol).
Anal. Calcd for C16H20FN3O4: C, 56.97; H, 5.97; N, 12.46; found: C, 56.86; H, 6.05; N, 12.44
HPLC (99.0 wt %, 98.9 area % linezolid, tR 1.60 min) conditions: InertsilODS-2 5.0 µm 150 mm × 4.6 mm, flow rate ) 2.0 mL/ min, gradient elution from 40:60 A:B to 80:20 A:B over 10 min; A ) acetonitrile; B ) water. External standard HPLC analysis of the filtrate showed
d 12.9% and 7.6% yield of linezolid and 8, respectively.
SEE HPLC AT http://file.selleckchem.com/downloads/hplc/S140801-Linezolid-Zyvox-HPLC-Selleck.pdf
………………………….
http://www.google.com/patents/WO2007064818A1?cl=en

Linezolid



Desfluoro Linezolid


HTTP://WWW.GOOGLE.COM/PATENTS/US6559305
HTTP://WWW.GOOGLE.COM/PATENTS/US7989618
……………………………………….
http://www.google.com/patents/EP2690100A1?cl=en
Example 3
-
[0034]To a 25 ml, round-bottomed flask equipped with a magnetic stirring bar was charged “amine” (0.49 g) followed by water (8.30 ml). A heterogeneous mixture was stirred and hydrochloric acid (0.12 mL, 35 %) was added. A homogenous solution was obtained. The solution was cooled down in an ice-water bath to 0°C. Acetic anhydride (0.31 mL) was added followed by sodium bicarbonate (0.45 g). Carbon dioxide was immediately released and a formation of white precipitate was observed. The precipitate was filtered off and the filter cake was washed with water (10 ml). The filter cake was collected and dried (100 mbar) at 70°C overnight. An off-white solid linezolid (0.26 g) was isolated.
…………………………..
PATENT
http://www.google.com/patents/WO2007116284A1?cl=en



Example 4 Trituration (convert linezolid crystalline Form I to linezolid crystalline Form E) The product from Example (89.18 g) is transferred to a 3L round bottom flask equipped with a mechanical stirrer, thermocouple and heating mantel. Ethyl acetate (2.23 L, 15 mL/g) is added and seeded with Linezolid form II crystals and the slurry is heated to ca. 500C. A slight exotherm of 30C is observed. After 30 minutes of heating the form change is observable as the solid is changing to long needles. Stirring is continued for 2 hours at 500C, at which time the contents are cooled to ambient temperature and stirred for an additional 30 minutes. The contents are then cooled to 30C for 1.5 hours, filtered and washed with cold ethyl acetate (300 mL total). The resultant solids are dried under vacuum at 50°C for 18 hours to give Linezolid (78.12 g) Form II by XRD, 99.8 wt%, 99.9% ee. HPLC conditions: YMC 5μ ODS-AM 150 nm X 4.6 nm column, etuting with CH3CN /water + 0.1% TFA from 20% CH3CN to 80% CH3CN in 8 min at 0.5 mL/min, detecting at 254nm. TR (Linezolid) = 4.4 min; HPLC conditions: Chiralcel OJ-H 250 nm X 4.6 nm column, eluting with 90% CO2/ 10%MeOH at 3.0 mL/min, detecting at 255 nm. TR [title compound] = 3.6 min; TR (enantiomer of title compound) = 4.1 rain
……………………………………..
http://www.google.com/patents/EP2516408A1?cl=en
The polymorphic form obtained by following process disclosed in U.S. Pat. No. 5,688,792 is designated as Form I. Figure- 1 depicts the PXRD graph of Form I obtained by following prior art process. [15] Disadvantage of the process disclosed in U.S. Pat. No. 5,688,792 is that it involves use of n-butyl lithium. Due to its explosive nature it is difficult to handle at plant scale. Also, the said reaction is carried out at temperature of -78°C, which is difficult to attain during commercial production. Further the intermediate obtained requires purification by column chromatography. Column chromatography is a cumbersome technique and difficult to practice during commercial scale production.
The process for the preparation of Linezolid is also disclosed in Journal of Medicinal Chemistry (1996), 39(3), 673-9, U.S. Pat. Nos. 6,492,555, 5,837,870, 6,887,995, 7,307,163, 7,429,661, etc.
Linezolid was first disclosed in U.S. Pat. No. 5,688,792. The process for synthesis is as disclosed in Scheme-I


………………………………………..
https://acs.confex.com/acs/green08/techprogram/P52019.HTM
Wednesday, June 25, 2008 – 2:00 PM
New York (Capital Hilton)
128
Convergent Green Synthesis of Linezolid (Zyvox)
………………………………….

……………………………………..

……………………………………….
http://www.google.com/patents/EP2072505A2?cl=en
-
WO 95/07271 , which specifically describes the synthesis of linezolid, namely [(S)-N-[[3-(3-fluoro-4-morpholinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide], according to the following scheme:

-
[0003]Other synthetic routes for the preparation of linezolid are reported for example in US 6107519 and in Tetrahedron Letters, Vol 37, N° 44, pages 7937-7940, wherein the chiral compound shown below is used instead of glycidyl butyrate as a synthon containing the molecule stereocenter.

-
[0004]It should be appreciated that all of the known approaches to the preparation of linezolid make use of chiral synthons for the construction of the stereocenter. These are small molecules characterized by a high cost, therefore they are not suitable for the production of the compound on an industrial scale.
-
[0005]There is therefore the need for an alternative synthesis which provides oxazolidinone derivatives, linezolid included, from inexpensive starting materials, and which does not require a chiral synthon for the construction of the molecule, so that it can be used for the industrial preparation of such derivatives.


………………………………….
http://pubs.rsc.org/en/content/articlelanding/2010/md/c0md00015a/unauth

………………………………….

…………………………………………

…………………………………
DOI: 10.1039/C3RA45186K
http://pubs.rsc.org/en/content/articlelanding/2013/ra/c3ra45186k#!divAbstract


A new asymmetric synthesis of the antibiotic Linezolid was performed through a copper-catalyzed Henry reaction as the key step. The use of camphor-derived aminopyridine ligands helped to improve the yields of the chiral precursor and to obtain Linezolid in good overall yield and enantiomeric excess.
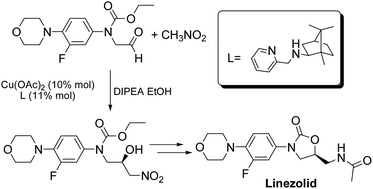
Linezolid 1. Mp: 181–182 C [lit. 181.5–182.5 C];
1 H-NMR (300 MHz; CDCl3) d 2.02 (s, 3H), 3.06 (t, J ¼ 4.7 Hz, 4H), 3.61– 3.78 (m, 3H), 3.87 (t, J ¼ 4.7 Hz, 4H), 4.03 (t, J ¼ 9.0 Hz, 1H), 4.72–4.82 (m, 1H), 6.17 (bt, 1H, exch. with D2O), 6.93 (t, J ¼ 9.0 Hz, 1H), 7.08 (dd, J1 ¼ 9.0 Hz, J2 ¼ 2.5 Hz, 1H), 7.44 (dd, J1 ¼ 14.4 Hz, J2 ¼ 2.5 Hz, 1H); ee ¼ 71%;
HPLC (Daicel CHIRALPAK-IA, hexane/i-PrOH ¼ 70 : 30, ow rate 0.8 mL min 1 , l ¼ 254 nm); tR (major) ¼ 14.1 min; tR (minor) ¼ 16.4 min. A true sample of (S)-Linezolid (ee > 98%) under the same HPLC conditions gave a tR ¼ 14.1 min.
………………………………..
http://www.slideshare.net/vishwajeeta/introduction-new-ppt
………………………………


http://www.slideshare.net/pushechnikov/linezolid-case-study
…………………………………
http://pubs.rsc.org/en/content/articlelanding/2011/cc/c1cc15503b#!divAbstract

…………………………………
http://www.mdpi.com/1424-8247/3/7/1988/htm

………………………………

Numbered structure of linezolid, showing the pharmacophore required for good activity (in blue) and desirable structural features (in orange).
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (S)-N-({3-[3-fluoro-4-(morpholin-4-yl)phenyl]-2-oxo-1,3-oxazolidin-5-yl}methyl)acetamide | |
| Clinical data | |
| Trade names | Zyvox, Zyvoxam, Zyvoxid |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a602004 |
| Licence data | US FDA:link |
|
|
|
|
| Intravenous infusion, oral | |
| Pharmacokinetic data | |
| Bioavailability | ~100% (oral) |
| Protein binding | Low (31%) |
| Metabolism | Hepatic (50–70%, CYPnot involved) |
| Half-life | 4.2–5.4 hours (shorter in children) |
| Excretion | Nonrenal, renal, and fecal |
| Identifiers | |
| 165800-03-3 |
|
| J01XX08 | |
| PubChem | CID 441401 |
| DrugBank | DB00601 |
| ChemSpider | 390139 |
| UNII | ISQ9I6J12J |
| KEGG | D00947 |
| ChEMBL | CHEMBL126 |
| NIAID ChemDB | 070944 |
| Chemical data | |
| Formula | C16H20FN3O4 |
| 337.346 g/mol | |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO1995007271A1 * | Aug 16, 1994 | Mar 16, 1995 | Michael R Barbachyn | Substituted oxazine and thiazine oxazolidinone antimicrobials |
| AU2001100437A4 * | Title not available | |||
| EP0963980A2 * | Mar 10, 1999 | Dec 15, 1999 | The Wellcome Foundation Limited | 1,2,4-Triazine derivative, its preparation and its use as reference marker for testing purity and stability of “lamotrigine” |
| Reference | ||
|---|---|---|
| 1 | * | [Online] August 2002 (2002-08), XP002388488 Retrieved from the Internet: URL:www.emea.eu.int/pdfs/human/ich/273799e n.pdf> [retrieved on 2006-07-03] |
| 2 | * | [Online] June 1995 (1995-06), XP002388489 Retrieved from the Internet: URL:www.emea.eu.int/pdfs/human/ich/38195en .pdf> [retrieved on 2006-07-03] |
| 3 | * | DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; LIU, JUN ET AL: “Preparation of oxazolidone derivatives as antibacterial agents” XP002429969 retrieved from STN Database accession no. 2003:576097 -& CN 1 355 165 A (INSTITUTE OF MEDICAL AND BIOLOGICAL TECHNOLOGY, CHINESE ACADEMY OF MED) 26 June 2002 (2002-06-26) |
| 4 | * | GLEAVE D M ET AL: “Synthesis and antibacterial activity of [6,5,5] and [6,6,5] tricyclic fused oxazolidinones” BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, OXFORD, GB, vol. 8, no. 10, 19 May 1998 (1998-05-19), pages 1231-1236, XP004137053 ISSN: 0960-894X |
| 5 | * | REDDY K V S R K ET AL: “Isolation and characterization of process related impurities in linezolid” JOURNAL OF PHARMACEUTICAL AN BIOMEDICAL ANALYSIS, vol. 30, no. 3, 15 October 2003 (2003-10-15), pages 635-642, XP002388486 |
| WO2001057035A1 * | Jan 29, 2001 | Aug 9, 2001 | Upjohn Co | Linezolid-crystal form ii |
| WO2002032857A1 * | Oct 17, 2001 | Apr 25, 2002 | Robert C Gadwood | Methods of producing oxazolidinone compounds |
| WO2002085849A2 * | Apr 15, 2002 | Oct 31, 2002 | Delara B Godrej | Process to prepare oxazolidinones |
| WO2005099353A2 * | Apr 19, 2004 | Oct 27, 2005 | Reddy Pingili Krishna | A novel process for the preparation of linezolid and related compounds |
| WO2006008754A1 | Jul 20, 2004 | Jan 26, 2006 | Reddy Pingili Krishna | Novel intermediates for linezolid and related compounds |
| WO2006031179A1 * | Sep 12, 2005 | Mar 23, 2006 | Astrazeneca Ab | Process for preparation of phtalimide |
| WO2007116284A1 * | Mar 26, 2007 | Oct 18, 2007 | Pfizer Prod Inc | Process for preparing linezolid |
| WO2010081404A1 | Jan 8, 2010 | Jul 22, 2010 | Lianhe Chemical Technology Co., Ltd. | Method for preparing linezolid and intermediates thereof |
| WO2012019632A1 | Aug 11, 2010 | Feb 16, 2012 | Synthon B.V. | Process for making linezolid |
| WO2012019862A1 | Jul 14, 2011 | Feb 16, 2012 | Synthon B.V. | Process for making linezolid |
| WO2012114354A1 | Feb 21, 2012 | Aug 30, 2012 | Lee Pharma Limited | Anhydrous linezolid crystalline form-ii |
| WO2013072923A1 | Sep 18, 2012 | May 23, 2013 | Cadila Healthcare Limited | Process for the preparation of crystalline linezolid |
| WO2013111048A1 | Jan 22, 2013 | Aug 1, 2013 | Jubilant Life Sciences Limited | Improved process for the preparation of stable crystalline form-i of linezolid, substantially free of residual solvent |
| WO2014071990A1 | Nov 9, 2012 | May 15, 2014 | Synthon Bv | Process for making linezolid |
| EP1403267A1 * | Sep 25, 2003 | Mar 31, 2004 | Daiso Co., Ltd. | Process for preparing glycidylphthalimide |
| EP1564215A1 * | Sep 25, 2003 | Aug 17, 2005 | Daiso Co., Ltd. | Process for preparing glycidylphthalimide |
| EP2100884A1 | Oct 16, 2003 | Sep 16, 2009 | Symed Labs Limited | Crystalline form of linezolid |
| EP2690100A1 | Jul 14, 2011 | Jan 29, 2014 | Synhton B.V. | Process for making linezolid |
| US6444813 | Jan 29, 2001 | Sep 3, 2002 | Pharmacia & Upjohn Company | Mixing linezolid of an >98% enantomeric purity in a solvent at >80 degrees; separating a crystal (ii) of >99% purity; analysis by the powder x-ray diffraction spectrum/infrared spectrum as a mineral oil mull; bactericides; stability |
| US6514529 | Mar 15, 2001 | Feb 4, 2003 | Pharmacia & Upjohn Company | A compressed tablet of antibacterial oxazolidinone selected from the group consisting of linezolid, eperezolid and (S)-N-((3-(3-fluoro-4-(tetrahydro-2H-thiopyran-4-yl)phenyl-2-o xo-5-oxazolidinylmethyl)acetamide S,S-dioxide |
| US6544991 | Jun 21, 2001 | Apr 8, 2003 | Pharmacia & Upjohn Company | Compositions and methods for treating bacterial infections |
| US6559305 | May 23, 2002 | May 6, 2003 | Pharmacia & Upjohn Company | Linezolid—crystal form II |
| US6617339 | Jun 3, 1999 | Sep 9, 2003 | Syngenta Limited | Oxazolidinone derivatives, process for their preparation and pharmaceutical compositions containing them |
| US6796975 | Mar 15, 2001 | Sep 28, 2004 | Pharmacia & Upjohn Company | Container for linezolid intravenous solution |
| US6833453 | Oct 17, 2001 | Dec 21, 2004 | Pharmacia & Upjohn Company | As an example, manufacturing a 5-(tert-butylcarbamoyl)-amino-methyl-oxazolidinone by condensing a carbamate with a tert-butylcarbamoyl protected derivative of glycidylamine or a 3-amino-1-halopropanol |
| US6875875 | Sep 25, 2003 | Apr 5, 2005 | Daiso Co., Ltd. | Process for preparing glycidylphthalimide |
| US6887995 | Apr 15, 2002 | May 3, 2005 | Pharmacia & Upjohn Company | Reacting N-aryl-O-alkylcarbamate with an amide derivative in the presence of a lithium cation, a base, and a nucleophile |
| US6989381 | Aug 20, 2001 | Jan 24, 2006 | Pharmacia Corporation | Containing s cyclodextrin compound in a concentration sufficient to maintain the drug in solution at such a drug concentration. |
| US7087784 | Mar 25, 2004 | Aug 8, 2006 | Pharmacia & Upjohn | Process to prepare oxazolidinones |
| US7128928 | Feb 20, 2003 | Oct 31, 2006 | Pharmacia Corporation | Ophthalmic formulation with novel gum composition |
| US7135576 | Jan 7, 2005 | Nov 14, 2006 | Daiso Co., Ltd. | Process for preparing glycidylphthalimide |
| US7307163 | Apr 19, 2004 | Dec 11, 2007 | Symed Labs Limited | Process for the preparation of linezolid and related compounds |
| US7351824 | Oct 8, 2007 | Apr 1, 2008 | Symed Labs Limited | Intermediates for oxazolidinone antibacterials; N-[3-Chloro-2-(R)-hydroxypropyl]-3-fluoro-4-morpholinyl aniline |
| US7429661 | Jul 20, 2004 | Sep 30, 2008 | Symed Labs Limited | Intermediates for linezolid and related compounds |
| US7524954 | Oct 8, 2007 | Apr 28, 2009 | Symed Labs Limited | Reacting 3-fluoro-4-morpholinyl aniline derivative with epichlorohydrin; converting chloromethyl oxazolidinone to aminomethyl oxazolidinone; carbonylation ; reacting with potassium phthalimide, hydrazine hydrate, and acetic anhydride; cyclization, carbamylation |
| US7714128 | Oct 16, 2003 | May 11, 2010 | Symed Labs Limited | crystalline linezolid form III (N-[[(5S)-3-[3-fluoro-4-(4-morpholinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide) an antibacterial agent; thermal stability |
| US7718799 | Sep 26, 2007 | May 18, 2010 | Symed Labs Limited | Crystalline form of linezolid |
| US7718800 | Sep 26, 2007 | May 18, 2010 | Symed Labs Limited | Prepared by mixing linezolid with solvent or mixture of solvents, cooling contents to below 15 degrees C., optionally seeding contents with linezolid form III, stirring, and collecting linezolid form III crystals by filtration or centrifugation; antibacterial agent; thermally stable |
| US7732597 | Sep 26, 2007 | Jun 8, 2010 | Symed Labs Limited | Prepared by acetylating (S)-N-[[3-[3-fluoro-4-[4-morpholinyl]phenyl]-2-oxo-5-oxazolidinyl]methyl]amine in a solvent, optionally in presence of an organic base to form linezolid, seeding reaction mixture, and isolating linezolid form III; antibacterial agent; thermally stable |
| US7741480 | Oct 8, 2007 | Jun 22, 2010 | Symed Labs Limited | Process for the preparation of linezolid and related compounds |
| US8658789 | Jan 8, 2010 | Feb 25, 2014 | Lianhe Chemical Technology Co., Ltd. | Method for preparing linezolid and intermediates thereof |
| US5837870 | Mar 28, 1997 | Nov 17, 1998 | Pharmacia & Upjohn Company | Process to prepare oxazolidinones |
| US6107519 | Oct 13, 1998 | Aug 22, 2000 | Pharmacia & Upjohn Company | Amido-substituted secondary alcohol intermediates and preparation thereof |
| US6444813 | Jan 29, 2001 | Sep 3, 2002 | Pharmacia & Upjohn Company | Mixing linezolid of an >98% enantomeric purity in a solvent at >80 degrees; separating a crystal (ii) of >99% purity; analysis by the powder x-ray diffraction spectrum/infrared spectrum as a mineral oil mull; bactericides; stability |
| US6492555 | Jan 15, 2002 | Dec 10, 2002 | Pharmacia & Upjohn Company | Reaction of a carbamate with either a (s)-secondary alcohol or (s)-epoxide or (s)-ester; bactericides |
| US6559305 | May 23, 2002 | May 6, 2003 | Pharmacia & Upjohn Company | Linezolid—crystal form II |
| US6716980 | Jun 27, 2003 | Apr 6, 2004 | Pharmacia & Upjohn Company | Cyclization and acylation of carbamate |
| US6740754 | Apr 24, 2003 | May 25, 2004 | Pharmacia & Upjohn Company | Process to produce oxazolidinones |
| US6833453 | Oct 17, 2001 | Dec 21, 2004 | Pharmacia & Upjohn Company | As an example, manufacturing a 5-(tert-butylcarbamoyl)-amino-methyl-oxazolidinone by condensing a carbamate with a tert-butylcarbamoyl protected derivative of glycidylamine or a 3-amino-1-halopropanol |
| US6887995 | Apr 15, 2002 | May 3, 2005 | Pharmacia & Upjohn Company | Reacting N-aryl-O-alkylcarbamate with an amide derivative in the presence of a lithium cation, a base, and a nucleophile |
| US7649096 * | Jul 17, 2006 | Jan 19, 2010 | Glenmark Pharmaceuticals Limited | crystallization of linezolid antibacterial agent in solvent and antisolvent |
| US20060111350 | Jun 29, 2005 | May 25, 2006 | Judith Aronhime | Solid forms of linezolid and processes for preparation thereof |
| US20060142283 | Jun 29, 2005 | Jun 29, 2006 | Judith Aronhime | Crystalline form IV of linezolid |
| US20090156806 | Dec 11, 2008 | Jun 18, 2009 | Dipharma Francis S.R.I. | Process for the Preparation of Oxazolidinone Derivatives |
| WO1995007271A1 | Aug 16, 1994 | Mar 16, 1995 | Michael R Barbachyn | Substituted oxazine and thiazine oxazolidinone antimicrobials |
| WO2005035530A1 | Oct 16, 2003 | Apr 21, 2005 | Reddy Pingili Krishna | A novel crystalline form of linezolid |
| WO2007026369A1 | Aug 29, 2005 | Mar 8, 2007 | Reddy Pingili Krishna | A novel amorphous form of linezolid |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2009032294A2 * | Sep 5, 2008 | Mar 12, 2009 | Teva Pharma | Processes for the preparation of a linezolid intermediate, linezolid hydroxide |
| WO2011076678A1 * | Dec 17, 2010 | Jun 30, 2011 | F. Hoffmann-La Roche Ag | Substituted benzamide derivatives |
……………
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.

 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR


 amcrasto@gmail.com
amcrasto@gmail.com
Methyl (S)-aminobutyrate hydrochloride…..Levetiracetam intermediate
Methyl (S)-aminobutyrate hydrochloride…..Levetiracetam intermediate
 |
- (s)-2-aminobutyric Acid Methyl Ester
- CAS No.: 15399-22-1
http://www.google.im/patents/WO2003014080A2?cl=en





 COCK WILL TEACH YOU NMRCOCK SAYS MOM CAN TEACH YOU NMR
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
COCK WILL TEACH YOU NMRCOCK SAYS MOM CAN TEACH YOU NMR
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE amcrasto@gmail.com
JALGAON, MAHARASHTRA, INDIA

.




 .
.



.
 MANUDEVI
MANUDEVI

(S)-2-amino-butanamide hydrochloride………. Key intermediate of Levetiracetam
(S)-2-amino-butanamide hydrochloride………. Key intermediate of Levetiracetam
(S)-2-amino-butanamide hydrochloride
Key intermediate of Levetiracetam

-
CAS Number 7682-20-4
-
Linear Formula CH3CH2CH(NH2)CONH2 · HCl

Into the above (S)-2-aminobutyric acid methyl ester hydrochloride is added Isopropanol is then added, followed by the introduction of ammonia gas at a pressure about 60 psi (413 kPa) until the reaction is complete. After filtering to remove formed ammonium chloride, the solvent is partially evaporated and isopropanol hydrochloride is added. The mixture is stirred while solid product forms, then the solid is separated by filtration and washed with isopropanol.

………..
13C NMR PREDICT

COCK WILL TEACH YOU NMRCOCK SAYS MOM CAN TEACH YOU NMR amcrasto@gmail.com
TIRUPATI, INDIA
.
| Tirupati తిరుపతి |
|
|---|---|
| City | |

Clockwise from top: Tirumala Venkateswara Temple, Tirumala ghat road, City skyline and Chandragiri fort
|
|

Tirupati
Location in Andhra Pradesh, India |
|
| Coordinates: 13.65°N 79.42°ECoordinates: 13.65°N 79.42°E | |
| Country | India |
| State | Andhra Pradesh |
| Region | Rayalaseema |
| District | Chittoor |
| Government | |
| • Member of Parliament | Varaprasad Rao Velagapalli |
| Area | |
| • City | 24 km2 (9 sq mi) |
| Elevation | 161 m (528 ft) |
| Population (2011)[1] | |
| • City | 287,035 |
| • Density | 12,000/km2 (31,000/sq mi) |
| • Metro[2] | 459,985 |
| Languages | |
| • Official | Telugu |
| Time zone | IST (UTC+5:30) |
| PIN | 517501 |
| Telephone code | +91–877 |
| Vehicle registration | AP 03 |
| Website | Tirupati Mucnicipal Corporation |

 .
.
 .
.
Kapila Theertham in Tirupati








Food Service During Tirumala Tirupati Devastanam’s ‘Srinivasa Kalyanam Utsavam’ at MARG Swarnabhoomi



(2S)-2- Oxopyrrolidin-1-yl)butanoic acid………….Key Levetiracetam intermediate
(2S)-2- Oxopyrrolidin-1-yl)butanoic acid………….Key Levetiracetam intermediate

| (s)-2-(2-oxopyrrolidin-1-yl)butanoic Acid | |
| CAS No.: | 102849-49-0 |
|---|---|
| Synonyms: | |
| Formula: | C8H13NO3 |
| Exact Mass: | 171.09000 |
1H NMR PREDICT
1H NMR (CDCl3, 400 MHz): δ 0.93 (t, J = 7.7 Hz, 3H), 1.67–1.76 (m, 1H), 1.99–2.13 (m, 3H), 2.49 (t, J = 7.7 Hz, 2H), 3.37 (m, J = 8.7, 5.8 Hz, 1H), 3.52-3.58 (m, 1H), 4.64 (dd, J = 10.6, 4.8 Hz, 1H);
Journal of Chemical and Pharmaceutical Research, 2012, 4(12):4988-4994

Journal of Chemical and Pharmaceutical Research, 2012, 4(12):4988-4994

Cosy predict.BELOW
SYNTHESIS AS IN PAPER
Asymmetric synthesis of chiral amines by highly diastereoselective 1,2-additions of organometallic reagents to N-tert-Butanesulfinyl Imines
Chandra Babu K1,2*, Buchi Reddy R3 , Mukkanti K2 , Madhusudhan G1 and Srinivasulu P1
1 Inogent Laboratories (A GVK BIO Company), 28A, IDA, Nacharam, Hyderabad 500 076, India 2Centre for Pharmaceutical Sciences, JNT University, Kukatpally, Hyderabad 500 072, India
3Orchid Chemicals & Pharmaceuticals Ltd, 476/14, R&D Centre, Chennai -600 119, India __________________________________________________________________________
http://jocpr.com/vol4-iss12-2012/JCPR-2012-4-12-4988-4994.pdf
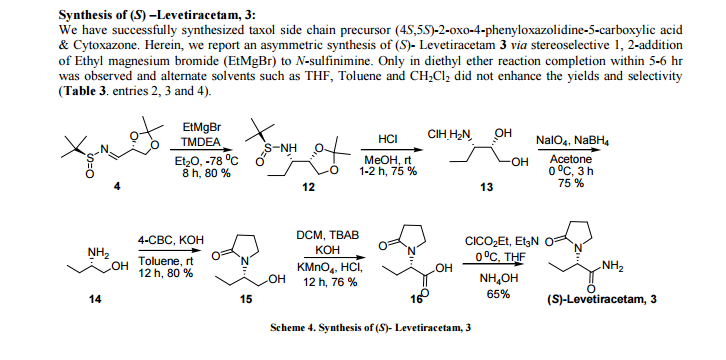
ABSTRACT We report an asymmetric synthesis of chiral amines (4S,5S)-Cytoxazone, Taxol side chain moiety and (S)- Levetiracetam starting from versatile new chiral N- sulfinimine (4). The key step, stereoselective 1,2-addition of Grignard reagent to chiral N-sulfinimine derived from (R)-glyceraldehyde acetonide and (S)-t-BSA gave the corresponding sulfonamide in high diastereoselectivity. Subsequent reactions yielded the targeted biological active and pharmaceutical important compounds with high purity (>99%) and yield
Journal of Chemical and Pharmaceutical Research, 2012, 4(12):4988-4994
(S)-2-(2-oxopyrrolidin-1-yl)butanoic acid, 16 Potassium hydroxide (1.0 g, 0.017 mol)) was dissolved into water (18.0 ml). Tetra-n-butyl ammonium bromide (0.2 g, 0.0062 mol)) and (S)-15 (1.0 g, 0.0063 mol)) in methylene chloride (10 ml) were charged in 30 min. charged Potassium permanganate (1.5 g, 0.094 mol)). After completion of reaction filtered through a celite bed and washed with water (10.0 ml). The aqueous layer pH was adjusted to 3 using hydrochloric acid (2 ml). Added sodium phosphate (2.5 g, 0.0152 mol) and toluene (25.0 ml). The reaction mixture extracted with dichloromethane (5 x 25 ml). The organic solution was dried with (Na2SO4) distilled under vacuo to give compound 16 as oil. To the residue toluene (10 ml) was added and stirred at 0 °C for about 30 min. The solid was filtered and washed with toluene (5 ml) afford the pure compound 16 (0.83g, 76%);
Mp: 124–125 °C; [α] 25 D = – 24.3 (c l.0, acetone);
1H NMR (CDCl3, 400 MHz): δ 0.93 (t, J = 7.7 Hz, 3H), 1.67–1.76 (m, 1H), 1.99–2.13 (m, 3H), 2.49 (t, J = 7.7 Hz, 2H), 3.37 (m, J = 8.7, 5.8 Hz, 1H), 3.52-3.58 (m, 1H), 4.64 (dd, J = 10.6, 4.8 Hz, 1H);
13C NMR (CDCl3, 125 MHz) : δ 10.8, 18.2, 21.9, 30.8, 43.9, 55.4, 173.7, 177.2;
IR (CHCl3) ν max : 2975, 1731, 1620 cm–1; ESI-MS: m/z 170.0 [M- +1].
Orchid Chemicals & Pharmaceuticals Ltd



Centre for Pharmaceutical Sciences, JNT University


Inogent Laboratories (A GVK BIO Company)
![]()

COCK WILL TEACH YOU NMRCOCK SAYS MOM CAN TEACH YOU NMR amcrasto@gmail.com
Levetiracetam industrial process
Levetiracetam industrial process


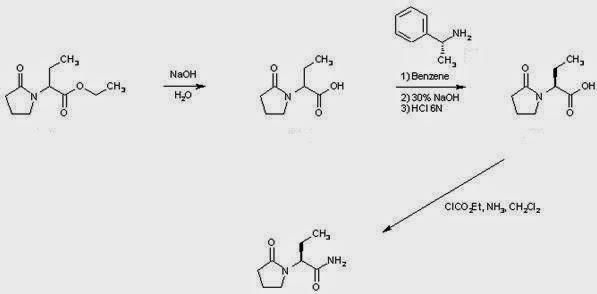
EXAMPLE 1 (a) Preparation of the (R)-alpha-methyl-benzylamine salt of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid
(b) Preparation of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid
EXAMPLE 2 (a) Preparation of ethyl (S)-4-[[1-(aminocarbonyl)propyl]amino]butyrate
(b) Preparation of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetamide
EXAMPLE 3 (a) Preparation of (S)-N-[1(aminocarbonyl)propyl]-4-chlorobutanamide
(b) Preparation of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetamide
EXAMPLE 4 Preparation of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetamide……levetiracetam
—
(±)-(R,S)-alpha-ethyl-2- oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide a key levetiracetam intermediate

(±)-(R,S)-alpha-ethyl-2- oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide
methyl (±)-(R,S)-alpha-ethyl-2-oxo-l -pyrrolidine acetate with (+)-(R)-(l-phenylethyl)- amine in toluene in the presence of a base such as sodium hydride or methoxide; crystallization- induced dynamic resolution of the resultant (±)-(R,S)-alpha-ethyl-2- oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide
(R)-(+)-1-Phenylethylamine
33978-83-5
1-Pyrrolidineacetic acid, α-ethyl-2-oxo-, methyl ester

1004767-60-5
1-Pyrrolidineacetamide, α-ethyl-2-oxo-N-[(1R)-1-phenylethyl]-
(±)-(R.S)-alpha-ethyl-2-oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide
Example 1
(±)-(R,S)-alpha-ethyl-2-oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide.
In a 100 ml reactor equipped with mechanical stirring, thermometer and bubble condenser, 13.4 g of (±)-(R,S)-alpha-ethyl-2-oxo-l-pyrrolidineacetic acid methyl ester (71.6 mmol), 8.8 g of (+)-(R)-(l-phenylethyl)-amine (72.5 mmol) and 45 ml of tetrahydrofuran were charged. 3.4 g of NaH (60% dispersion in mineral oil, 85.6 mmol) was added in small portions under nitrogen atmosphere. Reaction mixture was maintained at room temperature for about 2 h. Then, it was heated up to 350C and kept under stirring overnight. Reaction was controlled by TLC (Rf = 0.5, AcOEt/silica gel).
7.35-7.19 (1OH, m),
6.49 (2H, br s),
5.09-5.00 (2H, m),
4.41 (IH, dd, J = 8.3, 7.4 Hz),
4.36 (IH, dd, J = 8.6, 7.1 Hz),
3.49 (IH, ddd, J = 9.8, 7.7, 6.6 Hz),
3.41 (IH, ddd, J = 9.8, 7.7, 6.2 Hz),
3.30 (IH, ddd, J = 9.6, 8.3, 5.5 Hz),
3.13 (IH, ddd, 9.7, 8.5, 6.1 Hz), 2.47-2.38 (2H, m), 2.41 (IH, ddd, J = 17.0, 9.6, 6.3 Hz), 2.26 (IH, ddd, 17.0, 9.5, 6.6 Hz), 2.10-1.98 (2H, m), 2.01-1.89 (IH, m), 1.99-1.88 (IH, m), 1.98-1.85 (IH, m), 1.88-1.78 (IH, m), 1.75- 1.62 (IH, m), 1.72-1.59 (IH, m), 1.45 (3H, d, J = 7.1 Hz), 1.44 (3H, d, J = 7.1 Hz), 0.90 (3H, t, J = 7.4 Hz), 0.86 (3H, t, J = 7.4 Hz).


13C NMR (100.62 MHz, CDCl3, 25 0C): δ (ppm, TMS)
176.05 (CO), 176.00 (CO), 169.08 (CO),
168.81 (CO), 143.59 (Cquat),
143.02 (Cquat), 128.66 (2 x CH), 128.55 (2 x CH),
127.33 (CH), 127.19 (CH), 126.05 (2 x CH),
125.80 (2 x CH), 56.98 (CH), 56.61 (CH),
48.90 (CH), 48.84 (CH), 44.08 (CH2),
43.71 (CH2), 31.19 (CH2), 31.07 (CH2), 22.08 (CH3),
22.04 (CH3), 21.21 (CH2), 20.68 (CH2),
18.28 (CH2), 18.08 (CH2), 10.50 (CH3), 10.45 (CH3).


Example 2 (±)-(R.S)-alpha-ethyl-2-oxo-l-pyrrolidineacet-N-(+)-(R)-(l-phenylethyl)-amide (alternative 1).
At reaction completed, reaction mixture was cooled and when room temperature was reached, 100 ml of water was slowly charged. Aqueous phases were separated and extracted with toluene (2 x 75 ml). Collected organic phases were treated with acid water till neuter pH. Solvent was evaporated and residue was suspended in about 100 ml of heptane for about 30 minutes. Product was isolated by filtration and dried in oven at 400C temperature under vacuum overnight to give 45.2 g of the title compound (164.54 mmol, 83.2% yield, d.e. 0.0%) as white dusty solid.
Example 3
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
COCK WILL TEACH YOU NMRCOCK SAYS MOM CAN TEACH YOU NMR
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE amcrasto@gmail.com
updated
US 7902380, Levetiracetam
 US 7902380, Levetiracetamhttp://www.google.im/patents/US7902380
US 7902380, Levetiracetamhttp://www.google.im/patents/US7902380


Suitable resolving agents include optically pure bases such as alpha-methylbenzylamine and dehydroabietylamine, of which alpha-methylbenzylamine is preferred. (S)-2 can be prepared by forming the salt with (R)-alpha-methylbenzylamine and the (R)-2 can be prepared by forming the salt with (S)-alpha-methylbenzylamine.
NOTE……R)-alpha-methylbenzylamine is desired agent to get levetiracetam



EXAMPLE 1
Preparation of (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetamide from (S)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid
EXAMPLE 2
Preparation of (R)-alpha-ethyl-2-oxo-1-pyrrolidineacetamide from (R)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid
EXAMPLE 3
Preparation of (S)-alpha-Ethyl-2-oxo-1-pyrrolidineacetic acid (R)-alpha-methylbenzylamine salt
EXAMPLE 4
Recovery and Epimerization of (R)-alpha-ethyl-2-oxo-1-pyrrolidineacetic acid from the Mother Liquor
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on twitter
Join me on google plus 
 amcrasto@gmail.com
amcrasto@gmail.com

vietnam
.

 dalat city
dalat city
 hanoi
hanoi

Levetiracetam Green process construction

Dr. Rakeshwar Bandichhorl Director API – R&D,
Dr Reddys
LEVETIRACETAM GREEN PROCESS
Ravikumar Mylavarapu a , Ramasamy Vijaya Anand a , Golla China Mala Kondaiah a , Lekkala
Amarnath Reddy a , Gade Srinivas Reddy a , Arnab Roy a , Apurba Bhattacharya a , Kagga
Mukkanti b & Rakeshwar Bandichhor a
a Innovation Plaza, IPDO, R&D , Dr. Reddy’s Laboratories Ltd. , Survey Nos. 42, 45,46 & 54,
Bachupally, Qutubullapur, 500073, R.R. Dist, Andhra Pradesh, India
b Center for Environmental Science, Institute of Science and Technology , J.N.T. University ,
Kukatpally, Hyderabad, 500 072, Andhra Pradesh, India
Vol. 3, No. 3, September 2010, 225230
Gade Srinivas Reddy , Arnab Roy , Apurba Bhattacharya , Kagga Mukkanti & Rakeshwar Bandichhor (2010)
synthesis of levetiracetam, Green Chemistry Letters and Reviews, 3:3, 225-230, DOI: 10.1080/17518251003716568
To link to this article: http://dx.doi.org/10.1080/17518251003716568
– See more at: http://organicsynthesisinternational.blogspot.in/#sthash.ruewyXXk.dpuf
Dr Rakeshwar Bandichhor

| Rakeshwar Bandichhor Associate Director, API, R&D Dr. Reddy’s Laboratories India |
 |
|||||||||||
 |
||||||||||||
| BiographyRakeshwar Bandichhor holds a doctorate in Chemistry from University of Lucknow/University of Regensburg, Germany and worked as Postdoctoral Fellow at University of Regensburg, Germany, University of Pennsylvania and Texas A&M University. Dr. Rakeshwar has more than 150 papers including patents and book chapters published/accepted in various International Journals and contributed to more than 60 academic national and international conferences. He has won the various awards in his career | ||||||||||||
| Dr. Rakeshwar has more than 80 papers including patents and book chapters published/accepted in various International Journals. Notably, in the area of Organic Chemistry, Dr. Rakeshwar has coauthored a chapter in the book entitled “Green Chemistry in Pharmaceutical industry”. He has won the various awards in his career e.g. Chairman Excellence Award in the category of individual functional excellence, Best Cost Leadership Award for the development of Lopinavir, Ritonavir & their components and Anveshan Award at Dr. Reddy’s. As a part of organizational building efforts, he also supervises master’s & Ph.D. students in their dissertations. He has been invited in several conferences e.g. IIT-Mumbai, IGCW-2009, BIT-Ranchi, BITS Pilani, 9th Heterocyclic Conference, University of Florida, JNTU-Hyderabad, ISCB-2011, Apollo Hospitals Educational & Research Foundation, Hyderabad etc. to deliver lectures. He is also currently acting as an Associate Editor of GERF Bulletin of Bioscience. Recently, he has become a member National Advisory Board of Indian Society of Chemists and Biologists. |
||||||||||||
|
||||||||||||

Innovation Plaza, IPDO, R&D , Dr. Reddy’s Laboratories Ltd.


 .
.
MAHABALIPURAM, INDIA
Mahabalipuram – Wikipedia, the free encyclopedia
Mahabalipuram, also known as Mamallapuram is a town in Kancheepuram district in the Indian state of Tamil Nadu. It is around 60 km south from the city of …Shore Temple – Seven Pagodas – Pancha Rathas –
.
.



Krishna’s Butter Ball in Mahabalipuram, India. The surface below the rock is …




http://www.weather-forecast.com/locations/Mamallapuram
 Come to Mahabalipuram (also known as Mammallapuram), an enchanting beach that is located on the east coast of India.
Come to Mahabalipuram (also known as Mammallapuram), an enchanting beach that is located on the east coast of India.
 Moonraikers Restaurant, Mamallapuram
Moonraikers Restaurant, Mamallapuram


 Hotel Mamalla Bhavan – Mahabalipuram Chennai – Food, drink and entertainment
Hotel Mamalla Bhavan – Mahabalipuram Chennai – Food, drink and entertainment
 .
.
A carving at the Varaha Temple, Mahabalipuram

/////////////
The 10-Hydroxy-2-Decenoic Acid (10-2-HDA) content in Royal Jelly, is said to possess strong inhibition of malignant cell growth, namely transferable AKR leukemia, TA3 breast malignancy

Royal jelly is a honey bee secretion that is used in the nutrition of larvae, as well as adult queens.[1] It is secreted from the glands in the hypopharynx of worker bees, and fed to all larvae in the colony, regardless of sex or caste.[2]
When worker bees decide to make a new queen, because the old one is either weakening or dead, they choose several small larvae and feed them with copious amounts of royal jelly in specially constructed queen cells. This type of feeding triggers the development of queen morphology, including the fully developed ovaries needed to lay eggs.[3]
Other Common Names: Apilak, Gelée Royale, Queen Bee Jelly
Royal Jelly has been called the “Crown Jewel” of the beehive that has become extremely popular since the 1950s as a wonderful source of energy and natural way to increase stamina; perhaps that is the reason why the Queen Bee is so strong and enduring. It is also thought to be a great nutritional source of enzymes, proteins, sugars and amino acids, but there is no scientific proof to verify the supplement’s efficacy for its use as an overall health tonic. You’ll have to decide.

History:
Royal Jelly is a thick, milky material that is secreted from the hypopharyngea- salivary glands in the heads of the young nurse bees between the sixth and twelfth days of life, and when honey and pollen are combined and refined within the nurse bee, Royal Jelly is naturally created. While all larvæ in a colony are fed Royal Jelly, it is the only food that is fed to the Queen Bee throughout her life; other adult bees do not consume it at all. All female eggs may produce a Queen Bee, but this occurs only when – during the whole development of the larvæ – she is cared for and fed by this material – in large quantities. As a result of this special nutrition, the Queen develops reproductive organs (while the worker bee develops traits that relate only to work, i.e., stronger mandibles, brood food, wax glands and pollen baskets). The Queen develops in about fifteen days, while the workers require twenty-one; and finally, the Queen endures for several years, while workers survive only a few months. “10-2 HDA,” thought to be the principle active substance in Royal Jelly, makes the Queen Bee fifty percent larger than the other female worker bees and gives her incredible stamina, ovulation ability and longevity, living four to five years longer than worker bees who only live forty or more days. Perhaps this is the reason why so many positive qualities have been attributed to Royal Jelly as a truly rare gift of nature, but it should be noted that there is no clinical evidence to support the claims. There is even great controversy as to the constituents included in the supplement. Most researchers claim that it includes all the B-vitamins and vitamins A, C, D and E; some disagree. It does contain proteins, sugars, lipids (essential fatty acids), many essential amino acids, collagen, lecithin, enzymes and minerals, in addition to the very valuable
10-2-HDA (10-Hydroxy-2-Decenoic Acid). It is said that Royal Jelly may be most effective when combined with honey. You can decide whether any improvements you derive from Royal Jelly’s use are purely coincidental, but if (and when) you feel better when using it, just enjoy the benefits.

10-2-HDA (10-Hydroxy-2-Decenoic Acid)
Beneficial Uses:
Many fans claim that Royal Jelly is a great way to increase energy, as well as a remarkable stamina booster. In addition, it is also considered a means to enhance the immune system and maintain overall health.
Royal Jelly is said to alleviate a variety of problems, such as exhaustion, anxiety, mild depression, insomnia and lack of energy and stamina. Royal Jelly is also believed to have a calming effect on the nervous system.
Some people maintain that Royal Jelly has helped to improve skin disorders and has slowed down the ageing process. Royal Jelly’s collagen, lecithin and vitamins A, C, D and E all benefit the skin, helping to moisturize dry skin and soothe dermatitis.
In 1977, scientists at the Beijing Medical University reported that when Royal Jelly was administered to male and female neurasthenia patients, all patients reported very effective (86%) or effective (14%) improvement. Insomnia was eliminated, quality of sleeping increased and headache and dizziness were alleviated. It was also said that physical and mental abilities, appetite and working efficiency were improved.
The 10-Hydroxy-2-Decenoic Acid (10-2-HDA) content in Royal Jelly, is said to possess strong inhibition of malignant cell growth, namely transferable AKR leukemia, TA3 breast malignancy, etc., and recent studies indicated immuno-regulation and anti-malignancy activities. It can promote the growth of T-lymphocyte subsets, Interleukin-2 and the generation of tumor necrosis factor. Much research is being conducted on this valuable active constituent, which has exhibited positive physiological and pharmacological effects including vasodilative and hypotensive activities, antihypercholesterolemic activity and anti-inflammatory functions. In addition to these activities, the 10-HDA in Royal Jelly has been suggested to improve menopausal symptoms.
Other benefits attributed to the qualities of Royal Jelly include relief of bronchial asthma, liver, pancreatic and kidney ailments, stomach ulcers and bone fractures.
Contraindications:
Royal Jelly Nutritional Supplement is a natural bee product and may induce allergic reactions in some people and should, therefore, be tested in very small amounts before continued use. Symptoms of allergy include breathing problems or tightness in your throat or chest, chest pain, skin hives, rash or itchy or swollen skin.
Cultivation
Royal jelly is secreted from the glands in the heads of worker bees, and is fed to all bee larvae, whether they are destined to become drones (males), workers (sterile females), or queens (fertile females). After three days, the drone and worker larvae are no longer fed with royal jelly, but queen larvae continue to be fed this special substance throughout their development. It is harvested by humans by stimulating colonies with movable frame hives to produce queen bees. Royal jelly is collected from each individual queen cell (honey comb) when the queen larvae are about four days old. It is collected from queen cells because these are the only cells in which large amounts are deposited; when royal jelly is fed to worker larvae, it is fed directly to them, and they consume it as it is produced, while the cells of queen larvae are “stocked” with royal jelly much faster than the larvae can consume it. Therefore, only in queen cells is the harvest of royal jelly practical. A well-managed hive during a season of 5–6 months can produce approximately 500 g of royal jelly. Since the product is perishable, producers must have immediate access to proper cold storage (e.g., a household refrigerator or freezer) in which the royal jelly is stored until it is sold or conveyed to a collection center. Sometimes honey or beeswax are added to the royal jelly, which is thought to aid its preservation.
Composition
The overall composition of royal jelly is 67% water, 12.5% crude protein, including small amounts of many different amino acids, and 11% simple sugars (monosaccharides), also including a relatively high amount (5%) of fatty acids. The main acid is the 10-hydroxy-2-decenoic acid or 10-HDA (about 2 – 3%).It also contains many trace minerals, some enzymes, antibacterial and antibiotic components, pantothenic acid (vitamin B5), vitamin B6 (pyridoxine) and trace amounts of vitamin C,[2] but none of the fat-soluble vitamins, A, D, E and K.[4]
Royalactin
The component of royal jelly that causes a bee to develop into a queen appears to be a single protein that has been called royalactin. Jelly which had been rendered inactive by prolonged storage had a fresh addition of each of the components subject to decay and was fed to bees; only jelly laced with royalactin caused the larvae to become queens.[5] Royalactin also induces similar phenotypical change in the fruitfly (Drosophila melanogaster), marked by increased body size and ovary development.
Epigenetic effects
The honey bee queens and workers represent one of the most striking examples of environmentally controlled phenotypic polymorphism. In spite of their identical clonal nature at the DNA level, they are strongly differentiated across a wide range of characteristics including anatomical and physiological differences, longevity of the queen, and reproductive capacity.[6] Queens constitute the sexual caste and have large active ovaries, whereas workers have only rudimentary, inactive ovaries and are functionally sterile. The queen/worker developmental divide is controlled epigenetically by differential feeding with royal jelly; this appears to be due specifically to the protein royalactin. A female larva destined to become a queen is fed large quantities of royal jelly; this triggers a cascade of molecular events resulting in development of a queen.[3] It has been shown that this phenomenon is mediated by an epigenetic modification of DNA known as CpG methylation.[7] Silencing the expression of an enzyme that methylates DNA in newly hatched larvae led to a royal jelly-like effect on the larval developmental trajectory; the majority of individuals with reduced DNA methylation levels emerged as queens with fully developed ovaries. This finding suggests that DNA methylation in honey bees allows the expression of epigenetic information to be differentially altered by nutritional input.
Uses
Citing various potential health benefits seen in lab studies, royal jelly is collected and sold as a dietary supplement for humans, but the European Food Safety Authority has rejected these claims stating that the current evidence does not support consuming royal jelly will give health benefits in humans.[8] In the United States, both the Federal Trade Commission and the Food and Drug Administration have taken legal action against companies that have used unfounded claims of health benefits to market royal jelly products.[9][10][11][12]
Adverse effects
Royal jelly may cause allergic reactions in humans ranging from hives, asthma, to even fatal anaphylaxis.[13][14][15][16][17][18] The incidence of allergic side effect in people who consume royal jelly is unknown. The risk of having an allergy to royal jelly is higher in people who have other allergies.[13]

The benefits of Royal Jelly are truly extensive. The list of benefits is so extensive that it may actually appear to be ‘too good to be true’ to many of us, myself included. I’m still amazed every time I scan the many studies done on this amazing substance.Royal Jelly is one of the naturally occurring miraculous super foods on the planet that gets very little press! It packs a powerful health punch and here’s why:Royal Jelly is a substance produced by worker honey bees. Bee colonies function on a hierarchical system: Bees all start out as unisex larvae, blank slate bee babies if you will. Then they break off into 1 of 3 roles within their colony. The worker bees (females), the drones (males used for reproduction) and The Queen Bee.The workers and drones have a typical life span of 3-4 months, whereas The Queen Been can live for up to 7 years!

Cancer-fighting properties – According to a study published in a 2009 edition of the BMC Complementary and Alternative Medicine, royal jelly fights cancer by suppressing the blood supply to tumors. When the Japanese researchers tested various royal jelly types on umbilical vein tissue cultures, all of them inhibited the formation of blood vessels, especially those richest in caffeic acid, a compound responsible for the greatest suppressive levels. Moreover, since the fatty components of royal jelly contain estrogenic effects – as proved by a study published in the December 2010 edition of PLoS One – it is possible that royal jelly can treat breast and cervical cancer.
Improves blood health – A study published in the November 2008 edition of the Biological and Pharmaceutical Bulletin showed that royal jelly can improve insulin resistance and blood pressure. The researchers fed the jelly to rats suffering from high blood pressure and insulin resistance due to a high-fructose diet. After two months, the rats demonstrated noticeably fewer instances of blood vessel constriction, which resulted in lower triglyceride and insulin levels.
Skincare properties – Although Royal jelly is best-known as a health supplement, it is often used in skincare products because it contains DNA and gelatin, two ingredients that aid collagen production (and thus anti-aging activity). For this reason, many people like to apply royal jelly topically and allow it to nourish and invigorate their skin.
Antibacterial components – According to a study published in the July 1990 edition of the Journal of Biological Chemistry, a protein found in royal jelly – unofficially named royalisin – provides numerous antibacterial and antimicrobial properties, and is effective at dealing with certain bacterial cultures at lower levels.
Rich in nutrients – As with other bee products such as bee pollen and propolis, royal jelly’s biggest attraction is probably its impressive concentration of vitamins and minerals. Indeed, an average serving of royal jelly contains seventeen different amino acids (including all eight essential amino acids, making it a complete protein), most of the B-vitamins (which are used for the production and synthesis of energy), and respectable levels of iron and calcium, which are essential for superior blood and bone Health. Royal jelly also contains vitamins A, C, and E, which are important antioxidants that can neutralize free radical activity, thus guarding us from degenerative diseases.
Infertility treatment – It is not a coincidence that worker bees are infertile, while queen bees can lay up to 2,000 eggs per day throughout their extensive 4 to 6 year lifespan. This is because royal jelly stimulates estrogen production, thereby stabilizing menstrual cycles in women, improving sperm morphology in men, and increasing the libido of both sexes.
- royal jelly acid
- Bee propolis
- 3-Hydroxydecanoic acid
- 3,10-Dihydroxydecanoic acid
- 3,11-Dihydroxydodecanoic acid
Notes
- ^ Jung-Hoffmann L: Die Determination von Königin und Arbeiterin der Honigbiene. Z Bienenforsch 1966, 8:296-322.
- ^ a b Graham, J. (ed.) (1992) The Hive and the Honey Bee (Revised Edition). Dadant & Sons.
- ^ a b Maleszka, R, Epigenetic integration of environmental and genomic signals in honey bees: the critical interplay of nutritional, brain and reproductive networks. Epigenetics. 2008, 3, 188-192.
- ^ “Value-added products from beekeeping. Chapter 6.”.
- ^ Kamakura, M. (2011). “Royalactin induces queen differentiation in honeybees”. Nature 473 (7348): 478–483. doi:10.1038/nature10093. PMID 21516106.
- ^ Winston, M, The Biology of the Honey Bee, 1987, Harvard University Press
- ^ Kucharski R, Maleszka, J, Foret, S, Maleszka, R (2008). “Nutritional Control of Reproductive Status in Honeybees via DNA Methylation”. Science 319 (5871): 1827–1833. doi:10.1126/science.1153069.
- ^ “Scientific Opinion”. EFSA Journal 9 (4): 2083. 2011.
- ^ “QVC to Pay $7.5 Million to Settle Charges that It Aired Deceptive Claims”. Federal Trade Commission. March 19, 2009.
- ^ “Complaint in the Matter of CC Pollen Company et al.”. Federal Trade Commission. March 16, 1993.
- ^ “Federal Government Seizes Dozens of Misbranded Drug Products: FDA warned company about making medical claims for bee-derived products”. Food and Drug Administration. Apr 5, 2010.
- ^ “Inspections, Compliance, Enforcement, and Criminal Investigations: Beehive Botanicals, Inc”. Food and Drug Administration. March 2, 2007.
- ^ a b Leung, R; Ho, A; Chan, J; Choy, D; Lai, CK (March 1997). “Royal jelly consumption and hypersensitivity in the community”. Clin. Exp. Allergy 27 (3): 333–6. doi:10.1111/j.1365-2222.1997.tb00712.x. PMID 9088660.
- ^ Takahama H, Shimazu T (2006). “Food-induced anaphylaxis caused by ingestion of royal jelly”. J Dermatol. 33 (6): 424–426. doi:10.1111/j.1346-8138.2006.00100.x. PMID 16700835.
- ^ Lombardi C, Senna GE, Gatti B, Feligioni M, Riva G, Bonadonna P, Dama AR, Canonica GW, Passalacqua G (1998). “Allergic reactions to honey and royal jelly and their relationship with sensitization to compositae”. Allergol Immunopathol (Madr). 26 (6): 288–290.
- ^ Thien FC, Leung R, Baldo BA, Weiner JA, Plomley R, Czarny D (1996). “Asthma and anaphylaxis induced by royal jelly”. Clin Exp Allergy 26 (2): 216–222. doi:10.1111/j.1365-2222.1996.tb00082.x. PMID 8835130.
- ^ >Leung R, Thien FC, Baldo B, Czarny D (1995). “Royal jelly-induced asthma and anaphylaxis: clinical characteristics and immunologic correlations”. J Allergy Clin Immunol 96 (6 Pt 1): 1004–1007. doi:10.1016/S0091-6749(95)70242-3. PMID 8543734.
- ^ Bullock RJ, Rohan A, Straatmans JA (1994). “Fatal royal jelly-induced asthma”. Med J Aust 160 (1): 44.
References
- Balch, Phyllis A.; Balch, James F. (2000). Prescription for Nutritional Healing, Third Edition. New York: Avery. ISBN 1-58333-077-1.
- Ammon, R. and Zoch, E. (1957) Zur Biochemie des Futtersaftes der Bienenkoenigin. Arzneimittel Forschung 7: 699-702
- Blum, M.S., Novak A.F. and Taber III, 5. (1959). 10-Hydroxy-decenoic acid, an antibiotic found in royal jelly. Science, 130 : 452-453
- Bonomi, A. (1983) Acquisizioni in tema di composizione chimica e di attivita’ biologica della pappa reale. Apitalia, 10 (15): 7-13.
- Braines, L.N. (1959). Royal jelly I. Inform. Bull. Inst. Pchelovodstva, 31 pp (with various articles)
- Braines, L.N. (1960). Royal jelly II. Inform. Bull. Inst. Pchelovodstva, 40 pp.
- Braines, L.N. (1962). Royal jelly III. Inform. Bull. Inst. Pchelovodstva, 40
- Chauvin, R. and Louveaux, 1. (1956) Etdue macroscopique et microscopique de lagelee royale. L’apiculteur.
- Cho, Y.T. (1977). Studies on royal jelly and abnormal cholesterol and triglycerides. Amer. Bee 1., 117 : 36-38
- De Belfever, B. (1958) La gelee royale des abeilles. Maloine, Paris.
- Destrem, H. (1956) Experimentation de la gelee royale d’abeille en pratique geriatrique (134 cas). Rev. Franc. Geront, 3.
- Giordani, G. (1961). [Effect of royal jelly on chickens.] Avicoltura 30 (6): 114-120
- Hattori N, Nomoto H, Fukumitsu H, Mishima S, Furukawa S. [Royal jelly and its unique fatty acid, 10-hydroxy-trans-2-decenoic acid, promote neurogenesis by neural stem/progenitor cells in vitro.] Biomed Res. 2007 Oct;28(5):261-6.
- Hashimoto M, Kanda M, Ikeno K, Hayashi Y, Nakamura T, Ogawa Y, Fukumitsu H, Nomoto H, Furukawa S. (2005) Oral administration of royal jelly facilitates mRNA expression of glial cell line-derived neurotrophic factor and neurofilament H in the hippocampus of the adult mouse brain. Biosci Biotechnol Biochem. 2005 Apr;69(4):800-5.
- Inoue, T. (1986). The use and utilization of royal jelly and the evaluation of the medical efficacy of royal jelly in Japan. Proceeding sof the XXXth International Congress of Apiculture, Nagoya, 1985, Apimondia, 444-447
- Jean, E. (1956). A process of royal jelly absorption for its incorporation into assimilable substances. Fr. Pat., 1,118,123
- Jacoli, G. (1956) Ricerche sperimentali su alcune proprieta’ biologiche della gelatina reale. Apicoltore d’Italia, 23 (9-10): 211-214.
- Jung-Hoffmann L: Die Determination von Königin und Arbeiterin der Honigbiene. Z Bienenforsch 1966, 8:296-322.
- Karaali, A., Meydanoglu, F. and Eke, D. (1988) Studies on composition, freeze drying and storage of Turkish royal jelly. J. Apic. Res., 27 (3): 182-185.
- Kucharski R, Maleszka, J, Foret, S, Maleszka, R, Nutritional Control of Reproductive Status in Honeybees via DNA Methylation. Science. 2008 Mar 28;319(5871):1827-3
- Lercker, G., Capella, P., Conte, L.S., Ruini, F. and Giordani, G. (1982) Components of royal jelly: II. The lipid fraction, hydrocarbons and sterolds. J. Apic. Res. 21(3):178-184.
- Lercker, G., Vecchi, M.A., Sabatini, A.G. and Nanetti, A. 1984. Controllo chimicoanalitico della gelatina reale. Riv. Merceol. 23 (1): 83-94.
- Lercker, G., Savioli, S., Vecchi, M.A., Sabatini, A.G., Nanetti, A. and Piana, L. (1986) Carbohydrate Determination of Royal Jelly by High Resolution Gas Chromatography (HRGC). Food Chemistry, 19: 255-264.
- Lercker, G., Caboni, M.F., Vecchi, M.A., Sabatini, A.G. and Nanetti, A. (1992) Caratterizzazione dei principali costituenti della gelatina reale. Apicoltura 8:11-21.
- Maleszka, R, Epigenetic integration of environmental and genomic signals in honey bees: the critical interplay of nutritional, brain and reproductive networks. Epigenetics. 2008, 3, 188-192.
- Nakamura, T. (1986) Quality standards of royal jelly for medical use. proceedings of the XXXth International Congress of Apiculture, Nagoya, 1985 Apimondia (1986) 462-464.
- Rembold, H. (1965) Biologically active substances in royal jelly. Vitamins and hormones 23:359-382.
- Salama, A., Mogawer, H.H. and El-Tohamy, M. 1977 Royal jelly a revelation or a fable. Egyptian Journal of Veterinary Science 14 (2): 95-102.
- Takenaka, T. Nitrogen components and carboxylic acids of royal jelly. In Chemistry and biology of social insects (edited by Eder, J., Rembold, H.). Munich, German Federal Republic, Verlag J. Papemy (1987): 162-163.
- Wagner, H., Dobler, I., Thiem, I. Effect of royal jelly on the peirpheral blood and survival rate of mice after irradiation of the entire body with X-rays. Radiobiologia Radiotherapia (1970) 11(3): 323-328.
- Winston, M, The Biology of the Honey Bee, 1987, Harvard University Press
Disclaimer:
The information presented herein by this post is intended for educational purposes only. These statements have not been evaluated by the FDA and are not intended to diagnose, cure, treat or prevent disease. Individual results may vary, and before using any supplements, it is always advisable to consult with your own health care provider.
