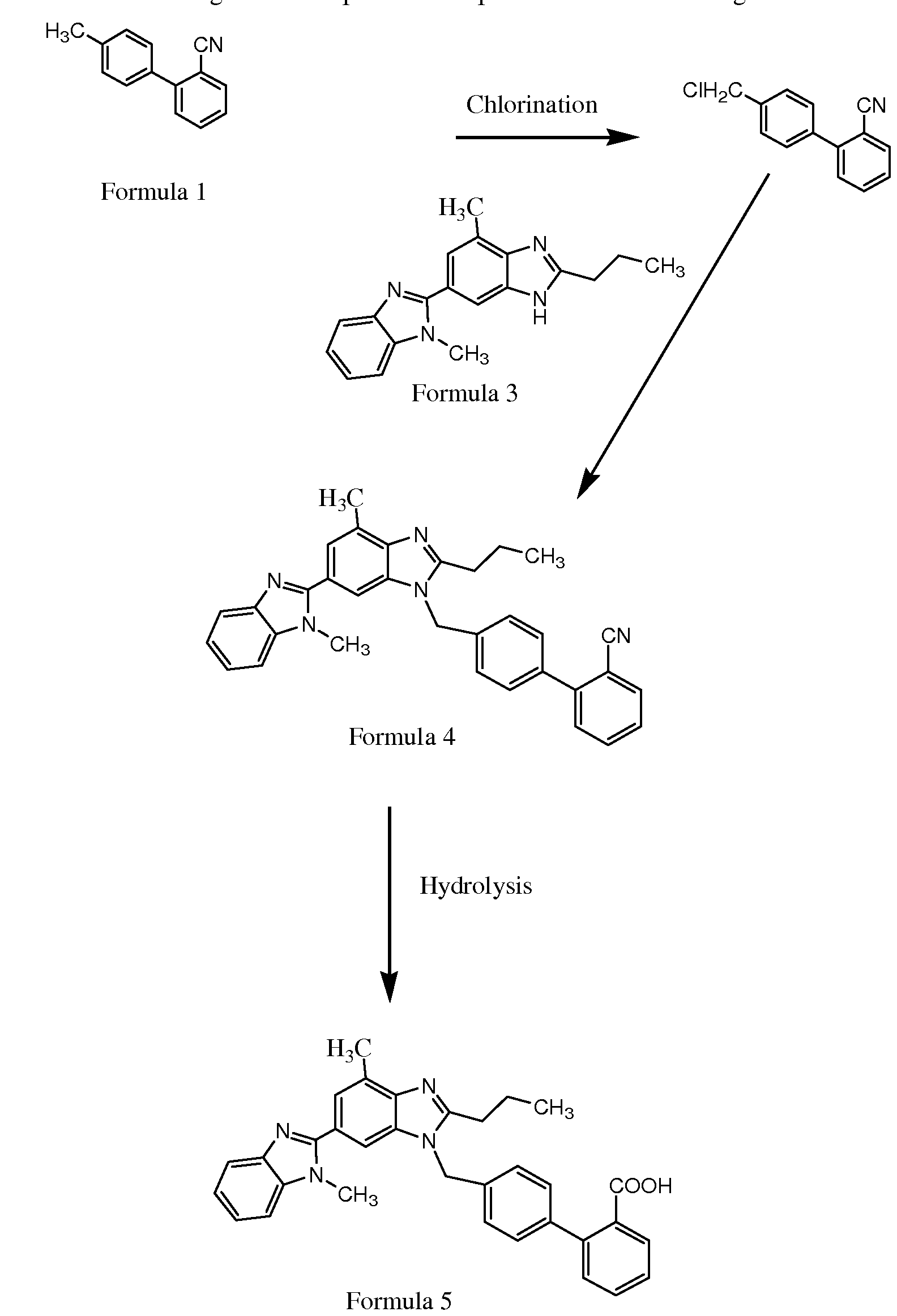
4′-[[4-methyl-6-(1-methyl-1h-benzimidazol-2-yl)-2-propyl-1h-benzimidazol-1yl]methyl]biphenyl-2-carboxylic acid (telmisartan)
PART 1……..http://orgspectroscopyint.blogspot.in/2015/04/telmisartan-part-13.html
PART 2……..http://orgspectroscopyint.blogspot.in/2015/04/telmisartan-part-23.html
OR https://newdrugapprovals.org/2015/04/06/telmisartan-part-23/
PART3…… http://orgspectroscopyint.blogspot.in/2015/04/telmisartan-part-33.html
GENERAL DESCRIPTION
Telmisartan is currently available as oral tablets in 20, 40, and 80 mg strengths for use in the treatment of hypertension. It is also marketed as Micardis® HCT which is a fixed dose combination with Hydrochlorothiazide (HCTZ) in 40/12.5, 80/12.5, 80/25 mg/mg strengths, and Twynsta® its fixed dose combination with Amlodipine in 40/5, 80/5, 40/10, 80/10 mg/mg strengths.
In 2009, Boehringer Ingelheim (Boehringer) gained approval to extend the market authorised indication of the Telmisartan 80 mg strength to include reducing the risk of myocardial infarction, stroke or death from cardiovascular disorders.
The Telmisartan molecule was discovered and developed by Boehringer, and was launched in Europe and the US in 1998. Boehringer has co-marketing agreements with Bayer Schering Pharma and GlaxoSmithKline in certain countries.

Telmisartan (1) is an angiotensin II receptor antagonist useful in the treatment of hypertension, heart diseases, heart strokes, and bladder diseases.1 Telmisartan (1) is currently available in the market as an antihypertensive drug2 under the brand name of MICARDIS. The first reported synthetic method3 for this molecule consists of 8 steps (Scheme 1) involving condensation of 4-amino-3-methyl benzoic acid methyl ester (2) with butyryl chloride (3) in chlorobenzene to yield 4. Nitration of 4 followed by reduction of the resulting 5-substituted nitro compound 5 over Pd-C in methanol yielded amine 6. Cyclisation of 6 in acetic acid reflux affords the monobenzimidazole derivative 7, which upon further hydrolysis yielded an acid intermediate 8 by a saponification process. Condensation of compound 8 with diamine derivative 9 in polyphosphoric acid yielded the dibenzimidazole compound 10, which was further alkylated with 4′-bromomethyl-biphenyl-2-carboxylic acid tert-butyl ester (11)4 to afford product 12. Finally, hydrolysis of ester12 in trifluoracetic acid yielded telmisartan (1) in an overall yield of around 21% with several impurities. This process suffers from disadvantages such as (a) a multistep synthesis for compound 8 (3 steps from compound 5); (b) the solvents dimethyl formamide (DMF) or dimethylsulfoxide (DMSO) used in the penultimate stage are unrecoverable, while the use of potassium tert-butoxide resulted in high organic volatile impurities (OVI) in telmisartan; (c) deprotection of the tert-butyl group using trifluoroacetic acid in DMF lead to the formation of several byproducts; (d) residue on ignition (ROI) in API obtained from this process is always >1.0% (ICH limit <0.1%), and there is no specified process mentioned in the literature to control the ash content. This is mainly due to very poor solubility of the telmisartan in most of the solvents including water; and (e) the overall yield (21%) of this process is discouraging, which makes the process less viable for commercial production.
(1) (a) Battershill, A. J.; Scott, L. J. Drugs 2006, 66 (1), 51-83. (b) Norbert, H.; Berthold, N.; Uwe, R.; Jacobus, C. A.; Van, M.; Wolfgang, W.; Michael, E. U.S. Patent 5,591,762, 1997. (c) Ruth, R. W.; William, J. C.; John, D. I.; Michael, R. C.; Kristine, P.; Ronald, D. S.; Pieter, B. M. W. M. T. J. Med. Chem. 1996, 39 (3), 625-656.
(2) http://www.rxlist.com/cgi/generic2/telmisartan.htm.
(3) (a) Uwe, J. R.; Gerhard, B. N.; Kai, M. H.; Helmut, W.; Michael, E.; Jacobus, C. A.; Van, M.; Wolfgang, W.; Norbert, H. H. J. Med. Chem. 1993, 36, 4040-4051. (b) Merlo
(4) Carini, D. J.; Dunicia, J. V. Eu. Patent 2,53,310, 1988. (5) Venkataraman, S.; Mathad, V. T.; Kikkuru, S. R.; Neti, S.; Chinta, R. R.; Arunagiri, M.; Routhu, L. K PCT WO 06/044754A2, 2006.
(6) The intermediate 9 is prepared via monomethylation of o-nitroaniline (15) using dimethylsulfate followed by hydrogenation over Pd-C catalyst in methanol with 75% of overall yield. Of the several methylating agents such as CH3I, DMS, HCOOH, and H2CO explored
(7) Structures of these impurities were tentatively proposed based on MS-MS data and a probable reaction mechanism and then synthesized as shown in Scheme 3. These impurities were characterized by NMR, mass, and IR techniques and further confirmed to be present in the sample by HPLC coinjection and spiking methods (0.1%). (8) Shen, J.; Li, J.; Yan, T.; Li, H.; Ji, R. CN 1,344,712, 2002.
(9) Several brominating agents such as molecular bromine, N-bromosuccinimide (NBS), and 1,3-dibromo-5,5-dimethylhydantoin (DBDMH) resulted in 13 along with the dibromo impurity 26. The formation of the dibromo impurity 26 is varying from 20-45% by HPLC. The content of 26 is nearly 45% in the case of NBS bromination, whereas the same is in the range of 15%- 20% in the case of DBDMH. Hence, DMDBH has been utilized as the brominating agent in the process. However impurity 26 did not participated in the next step and was easily washed out to a nondetected level during the isolation of 14 in the condensation step.
(10) Robert, E. D.; Peter, S.; Herbert, N.; Kenneth, S.; William, I. F. D. J. Pharm. Sci. 2000, 89 (11), 1465-1479.
Whilst patent protection for Telmisartan molecule, DE4103492A, has expired in Canada, it is still in force in the US until January 2014, receiving the longer term based on 17 years from the issue date for patents filed prior to June 8 1995. The equivalent European patent, EP0502314 (‘314), has been extended by SPC in France, Germany, Spain and the UK until December 2013 (see Figure 3).
Boehringer, seeking to protect its Telmisartan franchise, has also filed SPC applications for its Telmisartan-HCTZ and Telmisartan-Amlodipine products, for the basic patent ‘314, in France, Germany, Spain and the UK, potentially extending protection until January 2017 (see Figure 3). GenericsWeb’s proprietary SPC analyser has identified the basic patent as a ‘C3’ category, suggesting the claims of the basic patent do not protect the combinations and therefore the SPC may be invalid. The response by the national IPOs in respect to the invalidity of SPCs for the Telmisartan combinations has varied. The French SPC application (FR02C0028) for Telmisartan-HCTZ was initially rejected by the Institut National de la Propriété Industrielle (INPI) in December 2010, finding the claims of the basic patent did not protect a medicine comprising Telmisartan in association with HCTZ. The Paris Court of Appeal upheld INPI’s decision in June 2012, denying Boehringer’s request for appeal. Similarly, on June 2012, the Juzgado de lo Mercantil de Pamplona (the Court) held the Spanish SPC (C20020018) for Telmisartan-HCTZ invalid following a revocation suit filed by Cinfa and Actavis against Boehringer in April 2010. The Court’s decision relied on the ECJ’s findings in the ‘Medeva’ decision relating to SPCs for combination products, which concluded that to satisfy article 3(a) of SPC regulation 469/2009 the wording of the claims of the basic patent had to specify all active ingredients. Therefore, the Court found the SPC to be invalid on the grounds of article 15.1(a) in regard to 3(a), finding ‘314 did not specify a composition of Telmisartan in association with HCTZ. In February 2013, revocation proceedings were filed in the Bundespatentgericht for the German SPC (DE10299029) for Telmisartan-HCTZ. This raises the question of whether the SPC will prevent a generic Telmisartan-HCTZ product in Germany until conclusion of the revocation proceedings or will generic companies launch their products ‘at risk’ upon expiry of the SPC for Telmisartan, therefore assuming invalidity based on the ‘Medeva’ decision and similar findings by other PTOs and Courts in the matter.
The French (FR11C0008), German (DE122011000013) and Spanish (C201100010) SPCs for the Telmisartan-Amlodipine combination have been withdrawn. However, the UKIPO has granted the SPCs for both combination products (see Figure 3). No litigation proceedings have been detected in the UK. This may be due to amendments, under section 27 of the Patents Act 1977, of the specification for the UK designation of ‘314, in 2004 and 2011. The amendments were in the form of amended claim pages which included a pharmaceutical composition comprising HCTZ or a calcium channel blocker. Patents in the family with priority GB9722026A protect authorised indicated uses of the 80 mg dosage form of Telmisartan for reducing cardiac tissue damage associated with myocardial infarction and prevention or treatment of stroke, so are considered to be a constraint only for those indicated uses.
The family with the priority DE19901921A protects the crystalline form used in the commercially available product but are not considered to be a constraint to generic competition because the protected technology is likely to be circumvented. Families AU2002242676A, DE10301371A and EP04026234A protect Telmisartan combination products (see Figure 2). AU2002242676A and EP04026234A claim bilayer tablets comprising Telmisartan and HCTZ or Amlodipine, respectively. They are not considered to be a constraint to generic competition because the protected technologies are likely to be circumvented by generic reformulation. However, patents in the family DE19901921A expiring in July 2024, claiming composition of Telmisartan and several other drugs, including Amlodipine, are considered to be a constraint to generic competition for the Telmisartan-Amlodipine product. The family was deemed key due to its Canadian member 2534006 being listed on Health Canada’s patent register. Equivalent patents in the US have not been granted yet, but claims listed in the image wrapper in USPTO appear to limit the claims to a currently unauthorised use of Telmisartan and Amlodipine, therefore may not be a constraint for generic entry in the US.
Amongst the US approvals, Watson is the only company to have obtained tentative market approvals for all dosage strengths for the Telmisartan tablets and the fixed dose combination of Telmisartan and HCTZ. Lupin has gained a tentative approval for the Telmisartan and Amlodipine fixed dose combination. No generics are currently on the market in the UK due to unexpired patent protection, however several companies including Egis, Sandoz and Glenmark have obtained market authorisation for Telmisartan tablets in all dosage strengths. Actavis and Teva have obtained market authorisations via the centralised procedure. Dr Reddy’s Lab and Krka hold generic authorisations for both Telmisartan and Telmisartan-HCTZ fixed dose combination tablets. These generic approvals are suggestive of the competition Micardis® will face across Europe upon molecule patent expiry. Currently no generic Telmisartan-Amlodipine approvals have been identified in Europe. This is due to data exclusivity previsions in Europe, preventing the filing of generic market authorisation until October 2018, and a further 2 year market exclusivity period could prevent the launch of a generic equivalent until October 2020. In Canada, Mylan was one of the generic competitors to launch Telmisartan and Telmisartan-HCTZ following molecule patent expiry. This is likely to be mirrored in other territories upon expiry of the molecule patent.
Figure 4: Marketing Authorisations for products containing Telmisartan in Key Countries

In summary, patent protection remains a significant barrier to generic entry for the Telmisartan products in most major markets due to the molecule patent being in force. Boehringer’s lifecycle management attempts to maintain a monopoly for their blockbuster drug, including combination products and extensions of indications. Patent protection for its products, apart from the molecule patent, include a ‘use’ patent and combination patents which may pose a barrier to generic competition and may see Boehringer retain some of their market share. SPCs for the Telmisartan-HCTZ combination have been the subject of litigation in France and Spain, resulting in their invalidation, a revocation proceeding is on-going in Germany. Data exclusivity provisions in Europe will prevent the launch of a generic Telmisartan-Amlodipine fixed dose combination. In Canada, generic competition for Telmisartan and Telmisartan-HCTZ entered the market shortly after the expiry of the molecule patent. This is likely to be mirrored in other territories with generic companies already holding market authorisations for both products.
……………………………………..
PATENT
WO 2010018441
http://www.google.im/patents/WO2010018441A2?cl=en
Telmisartan is chemically named as 4′-[(1,4′-Dimethyl-2I-propyl[2l6′-bi-1H- benzimidazol]-1′-yl)methyl][1 ,1′-biphenyl]-2-carboxylic acid; or 4′-[[4-methyl-6-(1-methyl-2- benzimidazolyO^-propyl-i-benzimidazolyllmethyll^-biphenylcarboxylic acid.
The key raw material used to prepare Telmisartan is Bltyl, chemically named as 1,7′- dimethyl-2′-propyl-2,5′-bi-1H-benzimidazole, also known by other names, i.e 2-Propyl-4- methyl-6-(1 -methylbenzimidazol-2-yl)benzimidazole; 4-Methyl-6-(1 -methyl benzimidazol-2- yl)-2-propylbenzimidazole, and the structure shown as below:
BIM WO2006136916 describes substantially pure micronized particles of Telmisartan or a pharmaceutically acceptable salt, ester or derivative. The “substantially pure” is further defined as “Telmisartan or pharmaceutically acceptable salt, ester or derivative thereof having a purity of greater than or equal to about 98%, preferably a purity of greater than or equal to about 99% and more preferably a purity of greater than or equal to about 99.5%.° The substantially pure Telmisartan or a pharmaceutically acceptable salt, ester or derivative has an effective average particle size of less than about 300 microns.
A Journal of http://www.IP.com (2005), 5(7B), 4 – describes a process for purification of 4′-(2-propyl-4-methyl-6-(1-methylbenzimidazol-2-yl)benzimidazol-1-ylmethyl)biphenyl-2- carboxylic acid (Telmisartan). The pure compound was isolated by filtration under reduced pressure.
US20060276525 claims Telmisartan form A having HPLC purity > 99.5 %. It further provides a process for preparing Telmisartan form A by crystallization from a polar organic solvent selected from the group consisting of dimethyl sulfoxide, DMF, N.N-dimethyl acetamide, N-methyl 2-pyrrolidone, water and mixtures thereof. The process provides Telmisartan with a limit of DMSO at a level of < 1000 ppm. The process uses high boiling solvent in the last step for getting required purity, and which is also an extra purification step, which limits its commercial application.
US5591762 ( column-37,38 ) described the general process for the preparation of compound of formula-V
wherein bromine in structure IV is leaving group. There are several other leaving groups such as chlorine, iodine, a substituted sulphonyloxy group, e.g. a methane sulphonyloxy, phenylsulphonyloxy or p-toluenesulphonyloxy group are reported.
US5591762 describes preparation of Telmisartan from Telmisartan tert. butyl ester using trifluoroacetic acid in DMF as a solvent in 63.9 % yield. (Example-9) The resulting product had a melting point of 261-2630C.
The process for the preparation of tert. Butyl ester of Telmisartan is not commercially viable and deprotection involving the use of trifluoro acetic acid is not eco-friendly.
US 6385986 describes polymorphs of 4′-[2-n-propyl-4-methyl-6-(1-methylbenzimid- azol-2-yl) benzimidazol-1-ylmethyl] biphenyl-2-carboxylic acid (Telmisartan) i.e. polymorphic form B, mixtures of the polymorphs. The processes for preparing Telmisartan containing form B and the use for preparing a pharmaceutical composition. US ‘986 further describes that Telmisartan obtained process of as described in EP502314B1 to give a solid in the form of long needles which is difficult to filter, wash and isolate. It is further characterized that it requires a long time for drying due to the presence of solvent which forms large and hard fragments during the drying process. The fragments on grinding produce a dry powder which exhibits strong tendency to electrostatic charging and is virtually impossible to pour. The polymorphic form B of Telmisartan shows virtually no tendency to electrostatic charging and easy for suction filtration, centrifuge, washing, drying and is free-flowing even without being ground up.
Therefore, as a consequence of the alleged unsuitability of Telmisartan form A for pharmaceutical use, only a mixture of crystalline Telmisartan form A and form B is claimed in the ‘986 patent, wherein Telmisartan form A is characterized by having an endothermic maximum at 269±2°C, and Telmisartan form B is characterized by having an endothermic maximum at 183±2°C.
Apparently Telmisartan form A is similar to the original form characterized by its’ melting point in the ‘762 patent. The differences between the DSC value and the measured melting point may be attributed to the different methodologies used-the DSC maxima can be slightly different than the visually observed melting point.
Hence, the prior art teaches a lengthy, complicated and industrially disadvantageous process for obtaining crystalline Telmisartan form A. The need to further reprocess the re- crystallized Telmisartan, as taught in the examples of the ‘986 patent, shows that the product was not highly-pure and/or that it contained residual solvents, because the solvents used therein have high boiling point. JMC-1993, vol-36, No25 pg-4040-4051 describes preparation of Telmisartan tert. butyl ester using BIM and 2-(4’bromomethyl phenyl) tert. butyl benzoate using pot. Tert butoxide as a base in DMSO as solvent.
Formula 6
The preparative details for compound of formula-VII on page-4049, coloumn-3, compound 33, paragraph-4; line1-4 reads as follows.
Potassium tert-butoxide was added to the solution of BIM in DMSO at room temperature followed by the addition of the compound of formula Vl. Upon stirring for 14 hrs, the mixture was poured into water and extracted with ethyl acetate, the combined extract was dried on MgSO4 and evaporated. Residue was purified by silica gel column chromatography to give compound of formula-VII. The above mentioned process uses chromatographic purification, which is generally cumbersome and time consuming process and also requires solvents in high volume.
US20060094883 describes a process for the preparing Telmisartan, wherein Telmisartan alkyl ester – a
compound of formula-ll is prepared , comprising the steps of :
(a) combining i.y-dimethyl^’-propyl-IH.S’H-p.S1 ] bibenzimidazole (referred to as BIM) of formula III,
Formula 3 with 4′-bromomethyl-biphenyl-2-carboxylic acid alkyl ester (referred to as BMBP alkyl ester) of formula IV1
Formula 4 an inorganic base and a low boiling point organic solvent, to obtain a mixture;
(b) heating the mixture obtained in step (a) to a temperature of about 55°C. to about 1200C;
(c) maintaining the mixture obtained in step (b) for about 1 hour to about 8 hours, to obtain Telmisartan alkyl ester of formula II; and
(d) recovering Telmisartan alkyl ester of formula II, wherein, R is a straight or branched chain C1-C4 alkyl.
WO2005108375 describes process for the preparation of Telmisartan, characterized in that 1H-Benzimidazole-2-n-propyl-4-methyl-6-(1 ‘-methyl benzimidazole- 2’yl) of formula (II) and methyl-4- (bromo methyl)biphenyl 2-carboxylate of formula (III) are subjected to
WO 2007/010558 describes a method for the preparation of Telmisartan involving
Telmisartan dihydrochloride which comprises, i) condensing 4-Methyl-2-n-propyl-IH- benzimidazole-6-carboxylic acid with N-Methyl- O-phenylene diamine dihydrochloride to yields 4-methyl-6 (1 -methyl benzimidazol-2- yl)-2-n-propyl IH- benzimidazole, ii) treating 4- methyl-6-(l -methyl benzimidazol-2-yl)-2-n-propyl-IH-benzimidazole with
4*– (bromomethyl)-2-biphenyl-2-carboxylate in presence of a base in an organic solvent and isolating the ester as acid addition salt, iii) converting ester acid addition salt to Telmisartan dihydrochloride and iv) converting Telmisartan dihydrochloride to Telmisartan. CN1344712 describes method comprising reaction of 4-methyl-6-(1-methyl-2(1H)- benzimidazolyl)-1H-benzimidazole with 4′-bromomethyl-biphenyl-2-carboxylic acid alkyl ester [wherein alkyl is methyl or ethyl] in solvent i.e. DMF, DMSO, THF, dioxane, chloroform, dichloroethane, etc. in the presence of base [such as Na alcoholate, triethylamine, tributylamine, tripropylamine, KOH, NaOH, CsOH, Ba(OH)2 etc.] as acid capturer at 20- 1000C for 8-10 hrs, and then hydrolyzing with acid (such as H2SO4, HCI, HBr, HOAc, etc) at room temp, to reflux temp, or with base in Ci-5 alc.-water at 20-1600C for 1-10 hour. WO 2006/125592 describes a new process for the preparation of saltans 2-butyl-3- [[2″-[1 -(triphenylmethyl)-i H- tetrazol-5-yl][1 , 1 ‘-biphenyl]-4-yl]methyl]-1 ,3-diazaspiro[4.4] non- 1-en-4-one is disclosed, which proceeds via novel intermediate, 4-[(2-butyl-4-oxo-1 ,3- diazaspiro[4.4]non-1-en-3-yl)methyl]phenylboronic acid (Formula (H)) or its analogs. Compound (II) reacts with 5-(2-bromophenyl)-1-(triphenylmethyl)-1H-tetrazole (III) in the presence of catalyst, using conditions of Suzuki reaction, to give trityl irbesartan (I), whereas analogs to compound (II) may give candesartan, valsartan, Telmisartan, losartan and olmesartan.
WO 2006/050509 describes the amorphous form of Telmisartan sodium and the preparation thereof. Also provided are the Telmisartan sodium polymorph crystal Forms 0 to
XIII and XV to XX and preparations thereof. Also provided are pharmaceutical composition of amorphous and polymorphic forms of Telmisartan sodium or mixtures thereof, and methods of treatment of a mammal in need thereof.
WO 2006/044754 describes a process for preparing Telmisartan and intermediates formed in the process.
WO 2004/087676 describes a novel method for the production of Telmisartan by reacting 2-n-propyl-4-methyl-6-(1′-methylbenzimidazol-2’-yl)-benzimidazol with a compound of general formula (IV)1 in which Z is a leaving group, wherein the compound 2-cyano-4′-[2″- n-propyl-4″-methyl-6″-(1 ‘”-methylbenzimidazol-2l“-yl)benzimidazol-1 “-ylmethyl]biphenyl is obtained, and subsequently conducting hydrolysis of the nitrile to acid function.
WO2000/043370 describes polymorphs of 4′-[2-n-propyl-4-methyl-6(1 -methyl benzimidazol -2-yl) benzimidazol -1-ylmethyl] biphenyl-2-carboxylic acid (INN: Telmisartan), and in particular the polymorphous form B of formula (I), characterized by an endothermic peak at 183 ± 2°C during thermal analysis by differential scanning calorimetry. The invention also relates to mixtures of said polymorphs, methods for producing Telmisartan containing form B and to the use thereof in the preparation of a medicament.
Example-5 : Preparation of 4′-[[2-n-propyl-4methyl-6-(1-methylbenzimidazol-2-yl)- benzimidazol-1-yl]-methyl] biphenyl carboxylic acid [Telmisartan]
90 gm of ethyl-4′-[[2-n-propyl-4-methyl-6-(1-methylbenzimidazol-2-yl)-benzimidazol- 1-yl]-methyl] biphenyl carboxylate was stirred with 810 ml aq. HCI [32-35 % wt/ vol] at 95±2°C for about 8-10 hours. The reaction mixture was cooled to 25-300C. 180 ml of Dichloromethane and 1350 ml of water were added, pH of the reaction mixture was adjusted to -9.0 to 10.0 using 20 % aq. NaOH. The reaction mixture was stirred at 30-350C for about 30 minutes and the layer was allowed to separate. 1800 ml of MDC was added to aqueous phase at 25-300C. pH of the solution was adjusted to ~3 to 3.5 with acetic acid. The mixture was stirred for about 20 minutes and the layer was allow to separate. The aqueous layer was extracted with 900 ml DCM and organic layer was separated and washed with 2 X 900 ml water. The organic phase was dried over anhy. Sodium sulfate and charcoalized followed by distillation to remove about 80-85 % of DCM at 40-420C. The reaction mixture was slowly cooled to 80C and stirred at 8-120C for about 1Hr. 2700 ml of acetone (100C was slowly added and temperature is maintained at 8-12°C.The reaction mixture was stirred for 2 hours with slow RPM. The mixture was filtered at 8-120C and washed with 2×180 ml of acetone. The product was obtained through suction drying for 30-45 minutes, and under vacuum at 85-900C. 70.0 gm of Telmisartan is obtained having purity of 99.84%.
…………………………
PATENT
WO2009006860A2
http://www.google.im/patents/WO2009006860A2?cl=en
Telmisartan (I) is produced in accordance with the original patent of Boehringer Ingelheim (US 5 591 762) from telmisartan tert-butyl ester (II). The hydrolysis is carried out using of trifluoroacetic acid in the toxic solvent N,N-dimethylformamide.
According to another patent applied by the same company (US 2004 236113) the manufacture was problematic and this is why this procedure was replaced with hydrolysis of the corresponding nitrile (III). However, during the hydrolysis, which is carried out with potassium hydroxide in ethylene glycol, a high temperature (160 0C) is used, which causes browning of the product, which must be subsequently purified by means of activated carbon. Also, the energy demands of several-ton production would be considerably high.

In a newer application of Cipla (WO 2005/10837) the last two synthetic steps (iii+iv) are combined and telmisartan is isolated after alkaline hydrolysis by acidifying of the reaction mixture in water or extraction with dichloromethane and precipitation with acetone. Both the ways of isolation are unsuitable for industrial production. In the case of telmisartan of crystalline form A its isolation from water or aqueous solutions of organic solvents is very difficult since a hardly filterable product is formed. Extraction of the product with dichloromethane and precipitation with acetone brings a well-filterable product, but the use of dichloromethane is virtually impossible from the point of view of environment protection.

Another method has been described by Dr. Reddy (WO 2006/044754), which starts from telmisartan methylester hydrochloride, which is hydrolyzed to produce the potassium salt of termisartan, which is further acidified in aqueous acetonitrile; after isolation it crystallizes from a dichloromethane/methanol mixture and finally from methanol alone, and wherein a pressure apparatus is used for the dissolution in methanol at a temperature above its boiling point (80 °C). The result of this complex procedure, which manifests the already above mentioned shortcomings, is a low yield of the product.
The method of Teva (WO 2006/044648) is in many aspects similar to the above mentioned procedure of Cipla, wherein the last two steps of the synthesis are also combined. The method comprises phase separations, which lead to low yields (69 % – 80 %) besides increased tediousness. Matrix starts from telmisartan tert-butyl ester (II), which is first converted to telmisartan dihydrochloride, which in turn, by action of aqueous ammonia in methanol, provides telmisartan with a low total yield of 73%.
http://www.google.im/patents/WO2009006860A2?cl=en
Example 1
4′-[[4-methyl-6-(l-methyl-lH“-benzimidazol-2-yl)-2-proρyl-lH-benzimidazol- lyl]methyl]biphenyl-2-carboxylic acid (telmisartan)
Telmisartan methylester (VI) (40 g) was refluxed in methanol (440 ml) with potassium hydroxide (14.9 g) for 24 hours. To the boiling solution, methanol (240 ml) and then acetic acid (45.5 g) were added. While boiling, the mixture was stirred for another 1 hour, after cooling to 4°C the product was aspirated within 1 hour and washed with methanol (2 x 80 ml). After drying at the laboratory temperature (24 h) 35.18 g (90 %) of the product were obtained.
Analytic assessment: HPLC purity: 99.90 %,
Content of residual solvents: methanol (below the detection limit) acetic acid (360 ppm) Titration content: 100.9 % Sulfate ash content: 0.04 % DSC: form A
Example 2
4′-[[4-Methyl-6-(l-methyl-lH-benzimidazol-2-yl)-2-propyl-lH-benzimidazol- lyl]methyl]biphenyl-2-carboxylic acid (telmisartan)
Telmisartan methylester (VI) (20 g) was refluxed in methanol (300 ml) with potassium hydroxide (7 g ) for 24 h. After addition of formic acid (17 g) and after cooling to 4 °C the product was aspirated within 1 hour and washed with methanol (2 x 80 ml). After drying at the laboratory temperature (24 h) 18.7 g (96 %) of the product were obtained.
Example 3
4′-[[4-methyl-6-(l-methyl-lH-benzimidazol-2-yl)-2-propyl-l/J-benzimidazol- lyl]methyl]biρhenyl-2-carboxylic acid (telmisartan)
Telmisartan methylester (VT) (20 kg) was refluxed in methanol (400 1) with potassium hydroxide (7 kg) for 24 h. After addition of acetic acid (20 kg) and cooling to 4 °C the product was aspirated within 1 hour and washed with methanol (2 x 80 1). After drying at the laboratory temperature (24 h) 18.5 kg (95 %) of the product were obtained. Example 4
4′-[[4-methyl-6-(l-methyl-lH-benzimidazol-2-yl)-2-propyl-lH‘-benzimidazol- lyl]methyl]biphenyl-2-carboxylic acid (telmisartan)
Telmisartan methylester (40 g) was refluxed in methanol (240 ml) with potassium hydroxide (14.9 g) for 24 h. To the boiling solution methanol (240 ml) and then acetic acid (45.5 g) were added. After cooling to 4 °C the product was aspirated within 1 hour and washed with methanol (2 x 80 ml). After drying at the laboratory temperature (24 h) 36 g (92%) of the product were obtained.
…………………………..
PAPER

Org. Process Res. Dev., 2007, 11 (1), pp 81–85
DOI: 10.1021/op060200g
Telmisartan (1), a substituted dibenzimidazole derivative, is an antihypertensive drug, essentially used to control blood pressure. An improved, cost-effective, and impurity-free process for telmisartan (1) suitable for large-scale production is described here by addressing various process development issues. The overall yield obtained from this newly developed process is around 50% (over five steps) compared to the literature reported process (21%, over eight steps).
4′-[(1,4′-Dimethyl-2′-propyl-[2,6′-bi-1H-benzimidazol]- 1′-yl)methyl]-[1,1′-biphenyl]-2-carboxylic Acid (1).
Telmisartan (1) as a white crystalline powder. Yield 7 g (77%); purity by HPLC 99.9%; mp 260- 262 °C; Pd content not detected; Heavy metals <10 ppm; MS m/z 515 (M+ + H);
1 H NMR (CDCl3) δ 12.8 (s, 1H), 7.05-7.5 (m, 14H), 5.60 (s, 2H), 3.82 (s, 3H), 2.97 (t, J ) 7.5, 2H), 2.63 (s, 3H), 1.88 (q, J ) 7.3, 2H), 1.04 (t, J ) 7.3, 3H);
13C NMR (DMSO-d6) δ 13.5, 16.7, 20.6, 27.6, 32.7, 47.1, 51.7, 112.0, 112.7, 114.7, 118.6, 125.3, 125.7, 125.8, 127.0, 127.4, 128.6, 129.3, 130.4, 130.6, 131.5, 132.3, 133.1, 133.7, 134.5, 140.2, 140.5, 150.2, 157.3, 168.1.
Anal. Calcd for C33H30N4O2: C, 77.02; H, 5.88; N, 10.89; O, 6.22. Found: C, 77.0; H, 5.82; N, 10.89; O, 6.20.
………………….
PATENT
EP1719766A2
http://www.google.im/patents/EP1719766A2?cl=en
he present invention provides a process for the preparation of a compound of formula (I) or a salt thereof
comprising the reaction of a compound of formula (II) or a salt thereof
with a synthon of formula (III) or a salt thereof
prepared by reaction of a compound of formula (IV)
with a compound of formula (V)
-
Telmisartan, 4′-[(1,7′ -dimethyl-2′ -propyl[2,5′ -bis-l H-benzimidazol]-3′-yl)methyl][1,1′-biphenyl]-2-carboxylic acid is a known ACE inhibitor useful in therapy as antihypertensive agent. Its preparation is disclosed inEP 502314 and comprises the alkylation of 4-methyl-6-(1-methyl-benzimidazol-2-yl)-2-propylbenzimidazole (A) with t-butyl 4′-(bromomethyl)biphenyl-2-carboxylate (B)
-
However, compound (B) is not commercially available and its synthesis requires a number of steps, among them the protection of the carboxylic function which is finally removed by hydrolysis. There is therefore the need for an alternative synthesis for the industrial preparation of telmisartan, which makes use of commercially available or easy to prepare intermediates and which, if possible, avoids the additional steps of protection and deprotection of the carboxylic function.
Example 4. 4′-[[4-Methyl-6-(1-methyl-2-benzimidazolyl)-2-propyl-1-benzimidazolyl]methyl]-2-biphenylcarboxylic acid (telmisartan)
-
(4′-Methyl-2′-propyl-1H-benzimidazol-6′-yl)-1-methyl benzimidazole (3.0 g, 9.8 mmol), 4-(4,4,5,5-tetramethyl[1,3,2]dioxaborolan-2-yl)benzyl methanesulfonate (3.12 g, 10 mmol), tetrahydrofuran (15 ml) and potassium carbonate (1.38 g, 10 mmol) are loaded into a round-bottom flask equipped with magnetic stirrer, condenser and under nitrogen atmosphere. The mixture is stirred at room temperature for 8 hours, then 10% hydrochloric acid is added to pH=2.
-
THF is evaporated off, which causes precipitation of boronic acid. After recrystallization from ethyl acetate, 4.2 g of product are obtained.
-
The boronic acid (3.5 g, 8.0 mmol), ethyl 2-bromoacetate (1.83 g, 8.0 mmol), sodium hydroxide (1.28 g, 32 mmol), water (5 ml), tetrahydrofuran (20 ml), triphenylphosphine (315 mg, 1.2 mmol) and palladium acetate (90 mg, 0.4 mmol) are loaded into a round-bottom flask equipped with magnetic stirrer and condenser. All the residual air is removed with nitrogen and then the mixture is heated at 60°C for 18 hours, thereafter is cooled, added with water (30 ml) and tetrahydrofuran is evaporated off. Ethyl acetate is added (30 ml) and the mixture is acidified with acetic acid to pH=5. The product is filtered and washed with water, to obtain 2.8 g of crude telmisartan, which is purified by dissolution in concentrated ammonia (2 ml), addition of acetone and reprecipitation with acetic acid.
……………………
PATENT
http://www.google.im/patents/EP2305650A1?cl=en
-
Telmisartan and its physiologically acceptable salts have valuable pharmacological properties. Telmisartan is an angiotensin-II-antagonist, which may be used to treat hypertension and cardiac insufficiency, ischaemic peripheral circulatory disorders, myocardial ischaemia (angina). Furthermore, Telmisartan may be used to prevent the progression of cardiac insufficiency after myocardial infarct, to treat diabetic neuropathy, glaucoma, gastrointestinal diseases and bladder diseases. Telmisartan is also suitable for treating pulmonary diseases, e. g. lung oedema and chronic bronchitis, for preventing arterial restenosis after angioplasty, for preventing thickening of blood vessel walls after vascular operations, and for preventing arteriosclerosis and diabetic angiopathy. In view of the effects of angiotensin on the release of acetyl-choline and dopamine in the brain, Telmisartan is also suitable for alleviating central nervous system disorders, e. g. depression, Alzheimer’s disease, Parkinson syndrome, bulimia and disorders of cognitive function.
-
Telmisartan is a compound of formula (I)
chemically known as 4′-((1,7′-dimethyl-2′-propyl-1H,3′H-2,5′,-bibenzo[d]imidazol-3′-yl)methyl)biphenyl-2-carboxylic acid, which is disclosed in EP 502314 B1 and marketed under the trade name Micardis®.
-
Several methods have been used to prepare Telmisartan.
-
The process described inEP 502314 B1 comprises the alkylation of 4-methyl-6-(1-methyl-benzimidazol-2-yl)-2-propylbenzimidazole (III)
with t-butyl 4′-(bromomethyl)biphenyl-2-carboxylate and subsequently hydrolysis to Telmisartan. t-Butyl 4′-(bromomethyl)biphenyl-2-carboxylate is not commercially available and its synthesis requires a number of steps, among them the protection of the carboxylic function which is finally removed by hydrolysis.
-
The patent application
WO 2006044648 relates to a method for the production of Telmisartan by reacting 4-methyl-6-(1-methyl-benzimidazol-2-yl)-2-propylbenzimidazole (III) with 4′-(bromomethyl)biphenyl-2-carboxylic acid alkyl ester and subsequently hydrolysis.
-
The patent application
WO 2004087676 relates to a method for the production of Telmisartan by reacting 4-methyl-6-(1-methyl-benzimidazol-2-yl)-2-propylbenzimidazole (III) with 4-bromomethyl-2′-cyanobiphenyl and subsequently hydrolysis of the nitrile to the acid function.
-
The patent application
EP 1719766 relates to a method for the production of Telmisartan, by coupling with a Suzuki reaction the
N-4-bromobenzyl derivative of the compound of formula (III) with 2-carboxylphenyl boronic acid. As described in
EP 1878735 , 2-carboxyphenyl boronic acid requires a very laborious process to separate it, since it is extremely soluble in water, making the process unattractive for an industrial application. Thus, the active substance prepared by the process known up till now can only be obtained in a satisfactory quality after running through a number of process steps, wherein additional steps of protection and deprotection of the carboxylic function or additional steps to obtain the carboxylic function are often present.
Example 2 4′-((1,7′-dimethyl-2′-propyl-1H,3′H-2,5′-bibenzo[d]imidazol-3′-yl)methyl)biphenyl-2-carboxylic acid (I)
-
-
A 2L four-necked glass reactor, fitted with mechanical stirrer, thermometer, dropping funnel, under nitrogen atmosphere, was charged with sodium hydride (60% in mineral oil) (12.5 g, 312 mmol) and toluene (450 mL). The suspension was stirred and trimethylsilanol (31 g, 343 mmol) was added dropwise. After stirring for 15 minutes, methyl 4′-((1,7′-dimethyl-2′-propyl-1H, 3′H-2,5′-bibenzo[d]imidazol-3′-yl)methyl)biphenyl-2-carboxylate (V) (145 g, 274 mmol) was added, the mixture was stirred for 5 hours at about 100°C and monitored by quantitative TLC (elution with 5% MeOH in EtOAc) until complete conversion. The mixture was then cooled at room temperature, water (130 mL) was added, and the mixture was brought at 50°C. The phases were separated and the aqueous phase was stripped under vacuum to remove residual toluene.
350 g of an aqueous solution were obtained and used as such in the next step.
-
A 1L four-necked glass reactor, fitted with mechanical stirrer, thermometer, dropping funnel, under nitrogen atmosphere, was charged with the aqueous solution in MeOH (600 mL). The mixture was heated under stirring at 40°C until dissolution and charcoal (7 g) was added. The suspension was stirred at 40°C for 30 minutes, filtered through a pad of Celite and the resulting solid was washed with a mixture of MeOH/water 4/1 (100 mL). The filtrate and the washings were combined, the resulting solution was heated to reflux temperature and acetic acid (17.7 g, 295 mmol) was added dropwise over 1 hour. The suspension was then cooled, filtered and the solid was washed with a mixture MeOH/water 4/1 (3 x 50 mL). The collected solid was then dried at 55°C under reduced pressure affording the title compound (130 g) as a white solid.
………………………
PATENT
WO2014067237A1
http://www.google.im/patents/WO2014067237A1?cl=en
Telmisartan is a novel non-peptide angiotensin Π (ΑΤ Π) receptor antagonist, for the clinical treatment of hypertension, its chemical name is 4 ‘- [(1,4′-dimethyl – 2′-propyl [2,6’- two -1Η- benzoimidazol] -Γ–yl) methyl] biphenyl] -2-carboxylic acid, knot
Telmisartan
Telmisartan synthetic route has mainly 3-methyl-4-amino-benzoic acid methyl ester as the starting material by N- acylation, nitration, reduction, cyclization, ester hydrolysis and condensation reaction intermediates 2-n-propyl-4-methyl – 6 – (1 – methyl-benzimidazol-2-yl) benzimidazole-α), Ϊ with 4′-bromomethyl-biphenyl-2-carboxylate (V) via nucleophilic substitution, hydrolysis reaction to give the final product two Bu telmisartan (reaction formula 1) (J Med Chem, 1993, 36: 4040-4051).
Reaction Scheme 1
After has been reported by 4′-bromomethyl-biphenyl-2-carboxylic acid methyl ester (or ethyl ester) (VI) or 4′-bromomethyl-biphenyl-2-carbonitrile (VII) Preparation of telmisartan (CN01126367 .9, CN01131915.1).


Example 16: Preparation of telmisartan
The title compound of Example 15 (III, R = CN) (24.8g, 0.05mol) was added propylene glycol (100ml) and water (100ml) (or other previously described embodiments will be an aqueous mixed solvent :), potassium hydroxide (or e.g. prior to said other inorganic bases) (0.2mol), was refluxed for 10 hours. After no starting material by TLC was cooled to room temperature, concentrated under reduced pressure to a small volume, was added dropwise hydrochloric acid adjusted to pH 5 to 6, the precipitated solid was filtered, washed with water to obtain telmisartan.
Telmisartan Preparation: 17 Examples
The title compound I (30.4g, 0.10moi>, embodiments of Example 14 4′-chloro-methyl-biphenyl-2-carbonitrile (0.12mol), sodium ethoxide (or other organic bases as previously described) (0.3mol) and DMF (or other solvent as previously described) (200ml) mixed, 65 ° C for about 5 hours. TLC detected no starting material, was added ethylene glycol (100ml and water (50ml) (or other aqueous solvent), and heated to 160 ° C. TLC detected no starting material, concentrated hydrochloric acid under ice cooling to adjust pH to 5-6, to precipitate a solid, the resulting solid was filtered, washed with water to give crude telmisartan, by recrystallization in telmisartan.
Examples 18 to 24: Preparation of telmisartan reference method of Example 8, the title compound of Example 6 to the compound of formula I (10g, leq) and implemented as a reactant, with NaH as a base, the reaction temperature under different conditions the reaction, the reaction solution was subjected to phase detection by conventional post-treatment to give telmisartan (crude), yield was calculated, and the purity of the liquid phase detection telmisartan. The test results are shown in Table 2.
Table 2 compares the reaction conditions
……………………..
PATENT
WO2011077444A1

1 Telmisartan ……………………………….2 Impurity B
Table 1 : Preparation of Telmisartan and 2 with reported synthetic schemes

process for the preparation of telmisartan, comprising: condensation of -n-propyl-4-methyl-6-(l’-methylbenzimidazol-2′-yl)benzimidazole (I)

with a compound of general formula II)

wherein Z denotes a leaving group such as a halogen atom, for example, a chlorine, bromine, or iodine atom to obtain the compound 2-cyano-4′-[2″-n-propyl-4″-methyl-6″-( 1 “‘-methylbenzimidazol-2″‘-yl)benzimidazol- 1 “-ylmethyl]biphenyl (III), and subsequent

hydrolysis of nitrile in the presence of excess base and solvent followed by acid/base purification to obtain pure telmisartan.

Scheme-I


EXAMPLES:
Experiment-1: Preparation of 2-cyano-4-[2-n-propyl-4-methyI-6-(l- methylbenzimidazol-2-yl) bnzimidazol-l-ylmethyl] biphenyl.
Add 2-n-propyl-4-methyl-6-(l ‘-methylbenzimidazol-2′-yl) benzimidazole 100 g in 1000ml of acetone and of potassium hydroxide 22.0 g with stirring at 20-25°C. Then of 4-bromomethyl-2′-cyanobiphenyl 92g is added at 20-25°C. Monitor the reaction on thin layer chromatography, after compilation reaction, the crystals are suction filtered, washed with chilled acetone, water, and then dried in a air drying cupboard at 80° C. Yield: 135.0 g (82.82% of theory); melting point: 196° C.-197° C; HPLC: 99.30%. N-3 isomer: 0.08%.
Experiment-2: Preparation of 2-cyano-4-[2-n-propyl-4-methyl-6-(l- methylbenzimidazol-2-yl) benzimidazol-l-ylmethyl] biphenyl.
Add 2-n-propyl-4-methyl-6-( -methylbenzimidazol-2′-yl) benzimidazole 100 g in 1000ml of acetone and of potassium hydroxide 22.0 g with stirring at 20-25°C. Then of 4-bromomethyl-2′-cyanobiphenyl 92g is added at 20-25 °C. Monitor the reaction on thin layer chromatography, after the reaction is completed, cooled to 0 to 5.0° C. and stirred for another hour at this temperature. The material is filtered, washed with chilled acetone, then wash with water, and then dried in a air drying cupboard at 80° C. Yield: 141.50 g (87.73% of theory); melting point: 196° C.-197° C; HPLC: 99.50%. N-3 isomer: 0.16%
Experiment-3: Preparation of Telmisartan.
Add potassium hydroxide 80g in 500ml of ethylene glycol then add 2-cyano-4′- [2-n-propyl-4-methyl-6-( 1 -methyl benzimidazol-2-yl) benzimidazol- 1 -ylmethyl] biphenyl lOOgm at room temperature. Stir the reaction mixture and raise temperature to 150- 155° C. The mixture is stirred for 15 to 18 hours at this temperature and monitor reaction mass by HPLC. After compilation of reaction cool to 30 to 35°C then diluted with 800 ml methanol then telmisartan precipitates by adding of acetic acid at 25 to 30°C and further diluted with water. Then stirred for further 90min at 25 to 30°C. After the crystals have been suction filtered. The wet material dissolve in 500ml methanol with 12gm potassium hydroxide then after treatment of charcoal crystallize the telmisartan to adjusting of pH 6.0 to 6.4 by acetic acid then dilute with 400ml water. Filtered and then dried in a vacuum tray drier at 85°C. Yield: 90g (87.37% of theory); HPLC: 99.91%.
Experiment-4: Preparation of Telmisartan.
Add potassium hydroxide lOOg in 500ml of ethylene glycol then add 2-cyano- 4’-[2-n-propyl-4-methyl-6-(l -methyl benzimidazol-2-yl) benzimidazol- 1 -ylmethyl] biphenyl 1 OOgm at room temperature. Stir the reaction mixture and raise temperature to 150-155° C. The mixture is stirred for 15 to 18 hours at this temperature and monitor reaction mass by HPLC. After compilation of reaction cool to 30 to 35°C then diluted with 800ml methanol then telmisartan crystallize by adding of acetic acid at 25 to 30°C then dilute with 300ml water. Stir for further 90min at 25 to 30°C. Filter and then dried in a vacuum drying cupboard at 85°C. Yield: 101 g (1.03% of theory); HPLC: 99.90%.
Experiment-5: Preparation of pure Telmisartan.
Crude telmisartan 101 g (from example 4) & activated carbon lOg is added in methanol 100ml , dichloromethane 500ml at 25 to 30°C. Stir the reaction mixture then the brown solution is filtered through hyflow bed, Completely distilled out filtrate below 50°C then add 800ml water at that temperature and stir for lhr. The telmisartan is hot filtered and washed with water. The telmisartan is dried at 80° C. in a vacuum drying cupboard. Yield: 90 g (87.37% of theory); HPLC: >99.95%.
Experiment-6: Preparation of Telmisartan.
2-cyano-4′- [2-n-propyl-4-methyl-6-( 1 -methyl benzimidazol-2-yl) benzimidazol- 1 – ylmethyl] biphenyl lOOgm is added in 500ml of ethylene glycol with lOOg of potassium hydroxide powder at 20-25°C. Stir and raise temperature to 160° C. to 165° C. The mixture is stirred for 15 to 18 hours at this temperature and monitor reaction mass by HPLC. After compilation of reaction cool to 70 to 75°C then diluted with methanol and water then telmisartan crystallize by adding of acetic acid to adjust the pH 5.5 to 6.0 at 25 to 30°C. Stir for further 60min at 25 to 30°C. After the crystals have been suction filtered. The wet material dissolve in methanol with potassium hydroxide 12gm then after treatment of charcoal crystallize the telmisartan by adding of acetic acid by adjusting of pH 6.0 to 6.4 then stir for further 60min. The material is filtered and dried in a vacuum drying cupboard at 85°C. Yield: 86.56g (84.03% of theory); HPLC: >99.96%.
Experiment-7: Preparation of Telmisartan.
of 2-n-propyl-4-methyl-6-(r-methylbenzimidazol-2′-yl) benzimidazole 100 g is add in 1000 ml of acetone, and of potassium hydroxide 22 gm with stirring at 20-25° C and then 90.0 g of 4-bromomethyl-2′-cyanobiphenyl is added at 20-25°C. The temperature of the reaction mixture is maintained at 20 to 25° C. Stir for a further 6.0 to 8.0 hours at 20 to 25° C. Monitor the reaction on thin layer chromatography, after compilation reaction distil out acetone. Add ethylene glycol 500ml and potassium hydroxide lOOgm to residue Stir the reaction mixture and raise temperature to 150° C. to 155° C. The mixture is stirred for 15 to 18 hours at this temperature and monitor reaction mass by HPLC. After compilation of reaction cool to 30 to 35°C. Reaction mass diluted with methanol and stir for 30min then telmisartan precipitated by adding of acetic acid to adjust the pH 6.0 to 6.5 at 25 to 30°C. Then dilute with water and filter, wash with of methanol. Wet telmisartan is dissolved in methanolic potassium hydroxide, filtered to remove un dissolved material. Acetic acid is added to adjust the pH 6.0 to 6.4 , water added for complete precipitation of material. Finally telmisartan is suction filter and wash with water at 40 to 45 °C. The telmisartan is dried at 80° C. in a vacuum drying cupboard. Yield: 130g
HPLC: 99.4%.
1H NMR (DMSO-d6) δ 1.0 (t,3H), 1.9 (q, 2H), 2.95 (t, 2H), 2.4 (s, 3H), 3.95 (s, 3H), 5.8 (s, 2H), 7.28 (s,lH),7.80 (s,lH), 7.75 (d, 2H), 7.25 (t, 2H), 7.10 (d, 2H), 7.30 (d, 2H), 7.40 (d, 1H), 7.40 (t, 1H), 7.30 (t, 1H), 7.45 (d, 1H). 12.9 (s, 1H).
m/z 514.7 [ M + H]+.
…………………
PATENT
US 6358986
EXAMPLE
3185 kg of recrystallised telmisartan (recrystallised from dimethylformamide or dimethylacetamide), 5.6 kg of activated charcoal, 185 l of water, 190.4 kg of formic acid (99-100%) and 185 l of methylethylketone are placed in a 1200 l stirring apparatus. The mixture is stirred for about 1 h at 60-70° C. and then filtered into another 1200 l stirring apparatus and washed with a mixture of 74 l of methylethylketone and 8.3 kg of formic acid (99-100%). About 278 l of solvent are distilled off at 80-100° C. whilst simultaneously 278 l of water are added. The mixture is then cooled to 20-30° C. and precipitated by the metered addition of 281.5 kg of 25% ammonia solution. The product precipitated is centrifuged, washed with water and dried at 120-125° C. Yield: 178 kg of telmisartan (96.2% of theory)
Comparison Example
150 kg of telmisartan (recrystallised from dimethylformamide or dimethylacetamide), 7.5 kg of activated charcoal, 750 l of ethanol and 30 kg of 25% aqueous ammonia solution are placed in a 1200 l stirring apparatus. The mixture is stirred for about 1 h and then filtered into another 1200 l stirring apparatus and washed with 150 l of ethanol. The mixture is heated to 70-80° C., 35 kg of glacial acetic acid are added and the mixture is stirred for a further 1.5-2 h at 75-80° C. The mixture is then cooled to 0-10° C. and stirred for a further 2 h. The product precipitated is centrifuged, washed with 300 l of ethanol and with 300 l of water and dried at 70-90° C. Yield: 135 kg of telmisartan (90% of theory) pure form AIn the preparation process according to the invention, as a result of the partial conversion of the polymorphic form B into the polymorphic form A during the drying process, telmisartan occurs as a pure substance in a mixture of two polymorphic forms. However, this does not affect the properties of the pharmaceutical composition, as in the course of the manufacture of telmisartan tablets, for example, the mixture of the polymorphic forms A and B is dissolved in 0.1 N NaOH solution and converted by spray drying into a homogeneous and totally amorphous granulate which is then subjected to the other tablet making steps. For more detailed information on the use of the products according to the invention for preparing a pharmaceutical composition, cf. EP 502314 B1, the contents of which are hereby referred to.
…………………………..
PATENT
WO2009006860A2
Telmisartan (I) is produced in accordance with the original patent of Boehringer Ingelheim (US 5 591 762) from telmisartan tert-butyl ester (II). The hydrolysis is carried out using of trifluoroacetic acid in the toxic solvent N,N-dimethylformamide.
According to another patent applied by the same company (US 2004 236113) the manufacture was problematic and this is why this procedure was replaced with hydrolysis of the corresponding nitrile (III). However, during the hydrolysis, which is carried out with potassium hydroxide in ethylene glycol, a high temperature (160 0C) is used, which causes browning of the product, which must be subsequently purified by means of activated carbon. Also, the energy demands of several-ton production would be considerably high.
In a newer application of Cipla (WO 2005/10837) the last two synthetic steps (iii+iv) are combined and telmisartan is isolated after alkaline hydrolysis by acidifying of the reaction mixture in water or extraction with dichloromethane and precipitation with acetone. Both the ways of isolation are unsuitable for industrial production. In the case of telmisartan of crystalline form A its isolation from water or aqueous solutions of organic solvents is very difficult since a hardly filterable product is formed. Extraction of the product with dichloromethane and precipitation with acetone brings a well-filterable product, but the use of dichloromethane is virtually impossible from the point of view of environment protection.
Another method has been described by Dr. Reddy (WO 2006/044754), which starts from telmisartan methylester hydrochloride, which is hydrolyzed to produce the potassium salt of termisartan, which is further acidified in aqueous acetonitrile; after isolation it crystallizes from a dichloromethane/methanol mixture and finally from methanol alone, and wherein a pressure apparatus is used for the dissolution in methanol at a temperature above its boiling point (80 °C). The result of this complex procedure, which manifests the already above mentioned shortcomings, is a low yield of the product.
The method of Teva (WO 2006/044648) is in many aspects similar to the above mentioned procedure of Cipla, wherein the last two steps of the synthesis are also combined. The method comprises phase separations, which lead to low yields (69 % – 80 %) besides increased tediousness. Matrix starts from telmisartan tert-butyl ester (II), which is first converted to telmisartan dihydrochloride, which in turn, by action of aqueous ammonia in methanol, provides telmisartan with a low total yield of 73%.
WO2009006860A2
Example 3
4′-[[4-methyl-6-(l-methyl-lH-benzimidazol-2-yl)-2-propyl-l/J-benzimidazol- lyl]methyl]biρhenyl-2-carboxylic acid (telmisartan)
Telmisartan methylester (VT) (20 kg) was refluxed in methanol (400 1) with potassium hydroxide (7 kg) for 24 h. After addition of acetic acid (20 kg) and cooling to 4 °C the product was aspirated within 1 hour and washed with methanol (2 x 80 1). After drying at the laboratory temperature (24 h) 18.5 kg (95 %) of the product were obtained.
Example 4
4′-[[4-methyl-6-(l-methyl-lH-benzimidazol-2-yl)-2-propyl-lH‘-benzimidazol- lyl]methyl]biphenyl-2-carboxylic acid (telmisartan)
Telmisartan methylester (40 g) was refluxed in methanol (240 ml) with potassium hydroxide (14.9 g) for 24 h. To the boiling solution methanol (240 ml) and then acetic acid (45.5 g) were added. After cooling to 4 °C the product was aspirated within 1 hour and washed with methanol (2 x 80 ml). After drying at the laboratory temperature (24 h) 36 g (92%) of the product were obtained.
……………………………….
PATENT
WO2010004385
Telmisartan was first disclosed in US 5,591,762. US 5,591,762 also discloses a process for the preparation of Telmisartan by reacting l,4′-dimethyl-2′-propyl[2,6′-bi-lH- benzimidazole (II) with 4′-(bromomethyl)[l,r-biphenyl]-2-carboxylic acid 1,1- dimethylethyl ester (III) in a solvent optionally in the presence of an acid binding agent to produce the intermediate 4′-[(l,4′-dimethyl-2′-propyl[2,6l-bi-lH-benzimidazol]-l- yl)methyl]-[l,l’-biphenyl]-2-carboxylic acid 1,1-dimethylethyl ester (IV), which is further hydrolysed to produce crude Telmisartan. The crude product obtained is purified over a silica gel column and finally crystallized from acetone. The process is shown in Scheme 1 :
Hydrolysis
(I)
The disadvantage with the above process is the use of column chromatography in the purification of Telmisartan. Employing column chromatography technique is tedious and laborious and also involves use of large quantities of solvents, and hence is not suitable for industrial scale operations.
US 6,358,986 describes two crystalline forms of Telmisartan donated as Form A, Form B. In US 6,358,986, the process for preparing crystalline Telmisartan Form A comprises mixing the Telmisartan with ethanol, adding activated charcoal and aqueous ammonia and mixing for one hour, then filtering to another stirring apparatus and washing with ethanol. Resulting solution is heated to 70~80°C, adding glacial acetic acid and stirring for further 1.5-2 hours at the same temperature, cooling to 0-10°C, stirring for further 2 hours, isolating the product by centrifugation, washing with ethanol then with water and drying at 70-90°C. According to the detailed description given in the US ‘986 patent, in addition to the disadvantageously prolonged drying process of the Telmisartan Form A, very hard particles are obtained. The grinding process of these particles produces a dry powder, which has strong tendency to electrostatic charging and which is virtually impossible to pour and manipulate for pharmaceutical preparations. On the other hand, Telmisartan Form B is free from the above-mentioned limitations. However, the inventors of the US ‘986 patent could not obtain pure, dry Form B because upon drying, some of Form B transformed into Form A. According to the teachings of the US ‘986 patent, mixtures of Telmisartan Form A and Form B ranging from 90:10 to 60:40 are suitable for industrial scaling-up, and even a content of 10% of Form B is sufficient to ensure that the product will have the positive qualities required for large-scale production.
US 2006/0276525 Al describes a process for the preparation of crystalline solid of Telmisartan Form A by dissolving Telmisartan in a polar solvent such as dimethylsulfoxide (DMSO), N,N-dimethylformamide (DMF), ΛζiV-dimethylacetamide (DMA)5 iV-methyl-2-pyrrolidone (ΝMP) and cooling the solution for sufficient time to produce Telmisartan Form A crystals, which are filtered and dried.
EXAMPLE-8
PREPARATION OF 4′-[[4-METHYL-O-(I-METHYL-Z-BENZIMIDAZOLYL) ^- PROPYL-1-BENZIMIDAZOLYL] METHYL]-Z-BIPHENYLCARBOXYLIC
ACID [TELMISARTAN]
Powdered sodium hydroxide (6.83 g) was added in N,N~dimethylformamide (175 ml) at 4°C followed by 4-methyl-6-(l-methyl-2-benzimidazolyl)-2 -propyl- 1- benzimidazole monohydrate (50 g) and stirred for 5 min. Thereafter, methyl-2-[4′- (bromomethylphenyl)]benzoate (54.76 g) was added at 0°C and stirred to the reaction mass till completion of the reaction. Methylene chloride (250 ml) was added, followed by water (500 ml) at 20C and stirred for 10 min. The aqueous layer was separated and extracted with methylene chloride (50 ml). The combined organic extract was washed with water (250 ml) to obtain 380 ml of the organic solution containing Telmisartan methyl ester. 320 ml of this organic layer was concentrated at ambient pressure to collect 210 ml of the distillate. Methanol (120 ml) was added to the concentrated mass and distilled to collect 96 ml of the distillate. The concentrated mass was diluted with 160 ml of methanol at 5O0C. Thereafter, aqueous sodium hydroxide solution (17.4 g of NaOH in 40 ml of water) was added at 5O0C and heated to reflux at 69-7O0C and stirred at reflux temperature till completion of hydrolysis reaction. Thereafter, the reaction mass was concentrated under reduced pressure at 60-650C till no more solvent distils. Water (600 ml) and methylene chloride (200 ml) was added to this solution. pH was adjusted to 4 with hydrochloric acid (22 ml, 35% w/w) at 27-28°C. The aqueous layer was separated and extracted with methylene chloride (40 ml). The combined organic layer was washed with water (80 ml) to obtain 280 ml of the organic solution. This is divided in to four parts and taken for isolation of Telmisartan as given below.
Part-1 The organic layer (70 ml) as obtained above was diluted with N,N-dimethylformamide (500 ml) at 27°C and seeded with Telmisartan form-A. The solution was left on standing without stirring for 30 min. The resulting suspension was stirred at 27-28°C for 30 min at this temperature. Solid was filtered, washed with precooled N5N- dimethylformamide (15 ml, -5°C) followed by precooled ethanol (10 ml, -2°C) and dried at 85-900C under reduced pressure to afford 10.1 g of Telmisartan.
Part-2
The organic layer (70 ml) as obtained above was diluted with N,N-dimethylformamide (50 ml) at 27°C and seeded with Telmisartan form-A. The solution was left on standing without stirring for 30 min. The resulting suspension was concentrated under reduced pressure at 65-700C to collect 30 ml of the distillate. Thereafter, the concentrated mass was cooled to -5°C and stirred for 30 min at this temperature. Product was filtered, washed with precooled N,N-dimethylformamide (15 ml, -3°C) followed by precooled ethanol (10 ml, -2°C) and dried at 85-900C under reduced pressure to afford 11.4 g of Telmisartan.
Part-3
The organic layer (70 ml) as obtained above was diluted with N,N-dimethylformamide (60 ml) at 27°C and seeded with Telmisartan form-A. The solution was left on standing without stirring for 30 min. The resulting suspension was concentrated under reduced pressure at 65-70°C to collect 50 ml of the distillate. Thereafter, stirred at 30°C for 15 min, cooled to -5°C and stirred for 30 min at this temperature. Product was filtered, washed with precooled N,N-dimethylformamide (15 ml, -5°C) followed by precooled ethanol (10 ml, -20C) and dried at 85-900C under reduced pressure to afford 11.7 g of Telmisartan.
Part-4
The organic layer (70 ml) as obtained above was diluted with N,N-dimethylformamide (40 ml) at 27°C arid seeded with Telmisartan form-A. The solution was left on standing without stirring for 30 min. The resulting suspension was concentrated under reduced pressure at 65-700C to collect 45 ml of the distillate. Thereafter, stirred at 300C for 15 min, cooled to -5°C and stirred for 30 min at this temperature. Product was filtered, washed with precooled N,N-dimethylformamide (15 ml, -5°C) followed by precooled ethanol (10 ml, -20C) and dried at 85-900C under reduced pressure to afford 12.3 g of Telmisartan.
…………………………………
|
DATABASE CA [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; LIU, LI-YAN: “Study on the optimization synthesis of telmisartan” XP002548425 retrieved from STN Database accession no. 2007:587025 & HUAXUE GONGCHENGSHI , 21(3), 60-61 CODEN: HGUOAP; ISSN: 1002-1124, 2007, |
| WO2006044754A2 * |
18 Oct 2005 |
27 Apr 2006 |
Muthulingam Arunagiri |
Process for preparing telmisartan |
| WO2009006860A2 * |
8 Jul 2008 |
15 Jan 2009 |
Zentiva As |
A method of manufacturing 4′-[[4-methyl-6-(1-methyl-1h-benzimidazol-2-yl)-2-propyl-1h-benzimidazol-1yl]methyl]biphenyl-2-carboxylic acid (telmisartan) |
| EP0502314A1 * |
31 Jan 1992 |
9 Sep 1992 |
Dr. Karl Thomae GmbH |
Benzimidazol, medicaments containing them and process for their preparation |
| US20060211866 * |
21 Mar 2006 |
21 Sep 2006 |
Glenmark Pharmaceuticals Limited |
Process for the preparation of angiotensin receptor blockers and intermediates thereof |
| US6737432 |
30 Oct 2002 |
18 May 2004 |
Boehringer Ingelheim Pharma Kg |
Crystalline form of telmisartan sodium |
| US7193089 |
17 Mar 2004 |
20 Mar 2007 |
Boehringer Ingelheim International Gmbh |
Process for manufacture of telmisartan |
| US7501448 |
13 Oct 2005 |
10 Mar 2009 |
Teva Pharmaceutical Industries, Ltd. |
high yields, low cost process; easy solvent extraction; nontoxic, safe, environmentally friendly, low boiling point organic solvents; 1,7′-dimethyl-2′-propyl-1H,3’H-[2,5′]bibenzoimidazolyl is reacted with 4’bromomethyl-biphenyl-2-carboxylic acid alkyl ester; industrial scale; hydrolysis |
| US7511145 |
30 Jul 2004 |
31 Mar 2009 |
Genelabs Technologies, Inc. |
Bicyclic heteroaryl derivatives |
| US7943781 |
18 Oct 2005 |
17 May 2011 |
Dr. Reddy’s Laboratories Limited |
Process for preparing telmisartan |
| US8541593 * |
20 Jun 2008 |
24 Sep 2013 |
Amgen Inc. |
Process for making substituted 2-amino-thiazolones |
| US8691999 |
10 May 2005 |
8 Apr 2014 |
Cipla Limited |
Process for the preparation of telmisartan |
| US20100222402 * |
8 Jul 2008 |
2 Sep 2010 |
Jan Stach |
Method of manufacturing 4′-[[4-methyl-6-(1-methyl-1h-benzimidazol-2-yl)-2-propyl-1h-benzimidazol-1yl]methyl]biphenyl-2-carboxylic acid (telmisartan) |
| US20100280255 * |
20 Jun 2008 |
4 Nov 2010 |
Amgen Inc. |
Process for making substituted 2-amino-thiazolones |
| US20120196896 * |
17 Sep 2010 |
2 Aug 2012 |
Georgetown University |
Treatment for Oxidative Stress and/or Hypertension |
| EP2420231A2 |
4 Oct 2007 |
22 Feb 2012 |
Boehringer Ingelheim Vetmedica GmbH |
Angiotensin II receptor antagonist for the prevention or treatment of cardiovascular disease in cats |
| EP2420232A2 |
4 Oct 2007 |
22 Feb 2012 |
Boehringer Ingelheim Vetmedica GmbH |
Angiotensin II receptor antagonist for the prevention or treatment of cardiovascular diseases in cats |
| EP2420233A2 |
4 Oct 2007 |
22 Feb 2012 |
Boehringer Ingelheim Vetmedica GmbH |
Angiotensin II receptor antagonist for the prevention or treatment of systemic diseases in cats |
| WO2005108375A1 * |
10 May 2005 |
17 Nov 2005 |
Cipla Ltd |
Process for the preparation of telmisartan |
| WO2009006860A2 * |
8 Jul 2008 |
15 Jan 2009 |
Zentiva As |
A method of manufacturing 4′-[[4-methyl-6-(1-methyl-1h-benzimidazol-2-yl)-2-propyl-1h-benzimidazol-1yl]methyl]biphenyl-2-carboxylic acid (telmisartan) |
| WO2010075347A2 |
22 Dec 2009 |
1 Jul 2010 |
Takeda Pharmaceutical Company Limited |
Methods of treating hypertension with at least one angiotensin ii receptor blocker and chlorthalidone |
| EP0502314B1 |
31 Jan 1992 |
20 May 1998 |
Dr. Karl Thomae GmbH |
Benzimidazol derivatives, medicaments containing them and process for their preparation |
| WO2010018441A2 * |
10 Aug 2009 |
18 Feb 2010 |
Cadila Pharmaceuticals Ltd. |
An improved process for the preparation of substantially pure telmisartan |
| WO2010146187A2 * |
21 Jun 2010 |
23 Dec 2010 |
Krka, Tovarna Zdravil, D.D., Novo Mesto |
Process for the preparation of telmisartan |
| WO2011077444A1 * |
28 May 2010 |
30 Jun 2011 |
Inogent Laboratories Private Limited |
A new process for the preparation of pure telmisartan |
| WO2012028925A2 * |
29 Aug 2011 |
8 Mar 2012 |
Ogene Systems (I) Pvt Ltd |
An improved process for the preparation of telmisartan |
| CN1768044A * |
26 Mar 2004 |
3 May 2006 |
贝林格尔·英格海姆国际有限公司 |
Process for manufacture of telmisartan |
| CN102731407A * |
4 Jul 2012 |
17 Oct 2012 |
宁波九胜创新医药科技有限公司 |
Method for preparing telmisartan |
| EP0627433A1 * |
7 Dec 1993 |
7 Dec 1994 |
Eisai Co., Ltd. |
Process for producing imidazopyridine derivative and intermediate |
| EP2123648A1 * |
20 May 2008 |
25 Nov 2009 |
Chemo Ibérica, S.A. |
A process for the preparation of Telmisartan. |
| EP2305650A1 * |
21 Sep 2009 |
6 Apr 2011 |
Chemo Ibérica, S.A. |
Novel process for the preparation of telmisartan |
| EP0502314B1 |
31 Jan 1992 |
20 May 1998 |
Dr. Karl Thomae GmbH |
Benzimidazol derivatives, medicaments containing them and process for their preparation |
| EP1719766A2 |
18 Apr 2006 |
8 Nov 2006 |
Dipharma S.p.A. |
A process for the preparation of telmisartan |
| EP1878735A1 |
28 Jun 2007 |
16 Jan 2008 |
Dipharma Francis S.r.l. |
Process for the preparation of boronic acids and derivatives thereof |
| WO2004087676A1 |
26 Mar 2004 |
14 Oct 2004 |
Boehringer Ingelheim Int |
Method for the production of telmisartan |
| WO2006044648A1 |
13 Oct 2005 |
27 Apr 2006 |
Teva Pharma |
Process for preparing telmisartan |
| WO2006136916A2 * |
20 Jun 2006 |
28 Dec 2006 |
Glenmark Pharmaceuticals Ltd |
Substantially pure micronized particles of telmisartan and pharmaceutical compositions containing same |
| EP1878735A1 * |
28 Jun 2007 |
16 Jan 2008 |
Dipharma Francis S.r.l. |
Process for the preparation of boronic acids and derivatives thereof |
| EP2103609A1 * |
20 Mar 2008 |
23 Sep 2009 |
Lek Pharmaceuticals D.D. |
Catalyzed carbonylation in the synthesis of angiotensin ii antagonists |
| EP2123648A1 |
20 May 2008 |
25 Nov 2009 |
Chemo Ibérica, S.A. |
A process for the preparation of Telmisartan. |
| EP2149566A1 |
15 Jul 2008 |
3 Feb 2010 |
Chemo Ibérica, S.A. |
A process for the preparation of telmisartan |
| EP2305650A1 |
21 Sep 2009 |
6 Apr 2011 |
Chemo Ibérica, S.A. |
Novel process for the preparation of telmisartan |
| CN102015690B |
19 Mar 2009 |
30 Apr 2014 |
力奇制药公司 |
Catalyzed carbonylation in the synthesis of angiotensin II antagonists |
| US8410285 |
19 Mar 2009 |
2 Apr 2013 |
Lek Pharmaceuticals D.D. |
2′-halobiphenyl-4-yl intermediates in the synthesis of angiotensin II antagonists |
| US8445693 |
19 Mar 2009 |
21 May 2013 |
Lek Pharmaceuticals D.D. |
Catalyzed carbonylation in the synthesis of angiotensin II antagonists |
| WO2009115585A1 * |
19 Mar 2009 |
24 Sep 2009 |
Lek Pharmaceuticals D.D. |
Catalyzed carbonylation in the synthesis of angiotensin ii antagonists |
| WO2009123483A1 |
30 Mar 2009 |
8 Oct 2009 |
Zaklady Farmaceutyczne Polpharma Sa |
Process for preparation of telmisartan |
| WO2010018441A2 * |
10 Aug 2009 |
18 Feb 2010 |
Cadila Pharmaceuticals Ltd. |
An improved process for the preparation of substantially pure telmisartan |
| WO2010146187A2 |
21 Jun 2010 |
23 Dec 2010 |
Krka, Tovarna Zdravil, D.D., Novo Mesto |
Process for the preparation of telmisartan |
| EP0502314A1 * |
31 Jan 1992 |
9 Sep 1992 |
Dr. Karl Thomae GmbH |
Benzimidazol, medicaments containing them and process for their preparation |
| DE4142366A1 * |
20 Dec 1991 |
24 Jun 1993 |
Thomae Gmbh Dr K |
New phenylalkyl derivs. – are angiotensin II antagonists used to treat hypertension, coronary insufficiency, angina, cns disorders etc. |
| US20040162327 * |
12 Feb 2004 |
19 Aug 2004 |
Boehringer Ingelheim Pharma Kg |
Treatment of hypertension and cardiac insufficiency, ischaemic peripheral circulatory disorders, diabetic neuropathy, glaucoma, gastrointestinal diseases and bladder diseases |
| WO2004087676A1 * |
26 Mar 2004 |
14 Oct 2004 |
Boehringer Ingelheim Int |
Method for the production of telmisartan |
| WO2005108375A1 |
10 May 2005 |
17 Nov 2005 |
Cipla Ltd |
Process for the preparation of telmisartan |
| WO2006044648A1 * |
13 Oct 2005 |
27 Apr 2006 |
Teva Pharma |
Process for preparing telmisartan |
| WO2006044754A2 * |
18 Oct 2005 |
27 Apr 2006 |
Muthulingam Arunagiri |
Process for preparing telmisartan |
| US6358986 * |
10 Jan 2000 |
19 Mar 2002 |
Boehringer Ingelheim Pharma Kg |
Polymorphs of telmisartan |
| US20040236113 |
17 Mar 2004 |
25 Nov 2004 |
Boehringer Ingelheim International Gmbh |
Process for manufacture of telmisartan |
| WO2010004385A1 * |
15 Jun 2009 |
14 Jan 2010 |
Aurobindo Pharma Limited |
Process for the preparation of pure 4′-[4-methyl-6-(1-methyl-2-benzimidazolyl)-2-propyl-1-benzimidazolyl]methyl]-2-biphenylcarboxylic acid |
| WO2010018441A2 * |
10 Aug 2009 |
18 Feb 2010 |
Cadila Pharmaceuticals Ltd. |
An improved process for the preparation of substantially pure telmisartan |
| WO2012055941A1 |
26 Oct 2011 |
3 May 2012 |
Krka,Tovarna Zdravil, D. D., Novo Mesto |
Multilayer pharmaceutical composition comprising telmisartan and amlodipine |
| WO2005108375A1 * |
10 May 2005 |
17 Nov 2005 |
Cipla Ltd |
Process for the preparation of telmisartan |
| WO2006044754A2 * |
18 Oct 2005 |
27 Apr 2006 |
Muthulingam Arunagiri |
Process for preparing telmisartan |
| WO2009006860A2 * |
8 Jul 2008 |
15 Jan 2009 |
Zentiva As |
A method of manufacturing 4′-[[4-methyl-6-(1-methyl-1h-benzimidazol-2-yl)-2-propyl-1h-benzimidazol-1yl]methyl]biphenyl-2-carboxylic acid (telmisartan) |
| EP1719766A2 * |
18 Apr 2006 |
8 Nov 2006 |
Dipharma S.p.A. |
A process for the preparation of telmisartan |
| US20060211866 * |
21 Mar 2006 |
21 Sep 2006 |
Glenmark Pharmaceuticals Limited |
Process for the preparation of angiotensin receptor blockers and intermediates thereof |
| US20060276525 * |
17 May 2006 |
7 Dec 2006 |
Itai Adin |
Processes of preparing highly pure telmisartan form A, suitable for pharmaceutical compositions |

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....






























 COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR

























 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..
















 Russian Journal of Bioorganic Chemistry, , vol. 31, # 4 p. 378 – 382
Russian Journal of Bioorganic Chemistry, , vol. 31, # 4 p. 378 – 382 US5605897 A1, ;
US5605897 A1, ;