
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
New comprehensive GMP Inspection Database available
DRUG REGULATORY AFFAIRS INTERNATIONAL
Just recently a so called “inspection tracker” was launched by Health Canada. Now, the agency offers an additional database which contains 3,821 inspections (per March 2015) which have been performed since 2012 – many of them outside Canada, e.g. in Europe or Asia. The information is available in an online database. The use of the database is very easy and search results are excellent.
By using the database even inspections in progress can be displayed. This is a service no other agency can provide. The database also offers further information about past inspections at the same production site. No further search is necessary because the information about past inspections will be displayed in the search result for a given production site. The rating of the inspection is also provided, and in case of GMP non compliance a detailed and very structured information about the findings is provided. The quality…
View original post 157 more words
The “Industry Coalition” gives practical advice for the control of elemental impurities in active substances and excipients
DRUG REGULATORY AFFAIRS INTERNATIONAL
.jpg)
The requirements of the “Guideline for Elemental Impurities ICH Q3D” published in December of last year mean a considerable expense for the affected pharmaceutical companies and drug manufacturers in terms of laboratory and personnel upgrading (see also our news about “ICH Q3D – Elemental Impurities” of 07 January 2015). In addition, the deadlines for the implementation of this guideline are quite tight. (June 2016 for newly approved drugs and December 2017 for already approved drugs, see our news “CHMP adopts ICH Q3D Guideline as “Scientific Guideline” of 21 January 2015).
In the March issue of “Pharmaceutical Technology Europe”, an article of the “Industry Coalition” has been published with the title “Implementation of ICH Q3D Elemental Impurities Guideline: Challenges and Opportunities“, which is intended to support the efffected companies with a number of pragmatic pieces of advice in the implementation of these requirements.
View original post 275 more words
What is SMU-B?

cas 1253286-89-3
Spiro[3H-indole-3,4′-piperidin]-2(1H)-one, 5-[6-amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-3-pyridinyl]-1′-methyl-
SMU-B
or is it
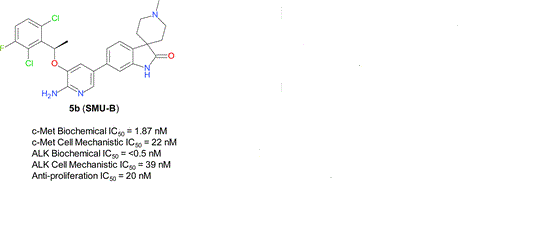
1253286-90-6
Spiro[3H-indole-3,4′-piperidin]-2(1H)-one, 6-[6-amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-3-pyridinyl]-1′-methyl-
SMU-B

A series of novel aminopyridyl/pyrazinyl-substituted spiro[indoline-3,4′-piperidine]-2-ones were designed, synthesized, and tested in various in vitro/in vivo pharmacological and antitumor assays. 6-[6-Amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-3-pyridyl]-1′-methylspiro[indoline-3,4′-piperidine]-2-one (compound 5b or SMU-B) was identified as a potent, highly selective, well-tolerated, and orally efficacious c-Met/ALK dual inhibitor, which showed pharmacodynamics effect by inhibiting c-Met phosphorylation in vivo and significant tumor growth inhibitions (>50%) in GTL-16 human gastric carcinoma xenograft models.
see..http://pubs.acs.org/doi/abs/10.1021/ml400203d
cas 1253286-90-6
6-[6-Amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-3-pyridyl]-1′-methylspiro[indoline-3,4′-piperidine]-2-one (compound 5b or SMU-B)
SEE

1_4,3_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] -2-nitro-approved P set

obtained in Step 1-3 (IS) -I- (2,6- dichloro-3-fluorophenyl) ethanol (2. 09g, IOmmol) was dissolved in dry THF (80 ml). Then, at room temperature under a nitrogen atmosphere, a solution of 3-hydroxy-2-nitro-pyridine (1.54g, llmmol) and triphenylphosphine (3. 409g, 13mmol), and so is completely dissolved, cooled to 0 ° C, was added Diisopropyl azodicarboxylate (DIAD, 2.63g, 13mmol), After the addition, the mixture was stirred at 0 ° C for 16 hours, the solvent was removed by rotary evaporation and the oily residue was purified by silica gel column chromatography (petroleum ether / ethyl acetate : 4/1) to give the desired product as a white solid (3. 046g, yield: 92%) o 1H-NMR (CDClySOOMHz): 8 (ppm) I. 86 (d, J = 6. 4Hz, 3H), 6 . 10 (q, J = 6. 4Hz, 1H), 7. 09 (dd, J = 7. 6,8. 8Hz, 1H), 7. 21 (dd, J = 8. 4, I. 2Hz, 1H ), 7. 31 (dd, J = 4. 8,8. 8Hz, 1H),
7. 37 (dd, J = 4. 8,8. OHz, 1H), 8. 04 (dd, J = L 6,4. 4Hz, 1H). Mass spectrum m / z:. 330 94 [M + H, 35C1,35Cl], 332. 92 [M + H, 35Cl, 37Cl].
1_5,3_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -2_ atmosphere based grant given P

to take steps 1-4 to get the 3 – [(lR) _l- (2,6_ dichloro-3-fluorophenyl) ethoxy] -2_ nitro than Li Jie (2. 649g, 8mmol) was dissolved in ethanol (15mL) was added iron powder (3. 575g, 64mmol) were mixed under nitrogen with vigorous stirring at 90 ° C oil bath, was added via syringe 0.8mL IM HCl (aq), after 10 minutes, was added 0. 8mL IMHCl (aq). Stirring was continued for 30 minutes, TLC showed the reaction. Cooled to room temperature, filtered through Celite, the filter residue washed with ethanol (3X IOmL). The combined organic phase was removed by rotary evaporation of the solvent gave the desired product as a light brown solid (2. 41g, yield: 100%) o 1H-Nmr (Cdci3JOOmHz): 8 (ppm) I. 81 (d, J = 6. 8Hz, 3H ), 5. 03 (s, br, 2H), 6. 01 (q, J = 6. 8Hz, 1H), 6. 47 (dd, J = 4. 8,7. 6Hz, 1H), 6. 70 (d, J = 8. OHz, 1H), 7. 05 (t, J = 8. 8Hz, 1H), 7. 28 (dd, J = 4. 0,8. 0Hz, 1H), 7. 57 ( d, J = 5.2Hz, lH). Mass spectrum m / z:. 301 00 [M + H, 35Cl, 35Cl], 302. 77 [M + H, 35Cl, 37Cl].
l-6,5_ desert _3_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -2_ atmosphere base than Li Jie

The steps 1-5 obtained 3_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] -2-yl atmosphere than Li Jie (1.506g, 5mmol) dissolved in acetonitrile (20mL) in. Then, at 0 ° C and the degree of stirring in added portionwise N- bromosuccinimide (0.908g, 5. Lmmol), After the addition, stirring was continued for 30 minutes. The solvent was removed by rotary evaporation, the crude product was obtained as a white solid was the desired product (1.045g, yield: 55%) was purified by column chromatography on silica gel. 1H-NMR (⑶Cl3,500MHz): 8 (ppm) I. 81 (d, J = 6. 8Hz, 3H), 4 85 (s, br, 2H), 6 98 (q, J = 6. 8Hz.. , 1H), 6. 82 (d, J =
2. 0Hz, 1H), 7. 08 (t, J = 8. 4Hz, 1H), 7. 31 (dd, J = 4. 8,8. 8Hz, 1H), 7. 65 (d, J = 2 . OHz, 1H). Mass spectrum m / z:… 378 84 [M + H, 35Cl, 35Cl, 79Br], 380 82 [M + H, 35Cl, 35Cl, 81Br or 35Cl, 37Cl, 79Br], 382 80 [M + H, 35Cl , 37Cl, 81Bror 37Cl, 37Cl, 79Br].
Step 2, I ‘- methyl-5- (4,4,5,5-tetramethyl -I, 3,2- dioxolane boron-2-yl) spiro [indoline Spray – 3,4 ‘- piperidin] -2_ one

2-1,5- bromo -I ‘- methyl-spiro [indoline-3,4’ – piperidin] _2_ one

[0300] 5-bromo – indol-2-one (I. 272g, 6mmol) was suspended THF (15mL) at, and cooled to -78 ° C, added dropwise with stirring IM NaN (SiMe3) THF solution of 2 (30mL, 30mmol). After the addition was stirred at _78 ° C 30 min, then 2-chloro -N- (2- chloro-ethyl) -N- methyl-ethylamine hydrochloride solid (I. 155g, 6mmol). After the addition stirring was continued for 30 minutes, then warmed to room temperature and stirred for two days. TLC showed the reaction was completed, to the pink suspension was carefully added aqueous 4M hydrochloric acid (IOmL), and then adjusted with concentrated aqueous ammonia to pH ^ 9, and extracted with DCM (3 X 80mL). The organic phases were combined, dried (Na2SO4), and concentrated to give the crude product was purified by silica gel column chromatography (7M NH3 in methanol solution / DCM: 5/95) to give the desired product (I. 38g, yield: 78%) was purified. 1H-NMR (CD3ODjOOMHz):. 8 (ppm) I. 86-1 92 (m, 2H), I 94-1 98 (m, 2H), 2 44 (s, 3H), 2 62-…. 2. 68 (m, 2H), 2. 86-2. 91 (m, 2H), 6. 76 (d, J = 7. 6Hz, 1H), 7. 33 (dd, J = I. 2,7 . 6Hz, 1H), 7. 44 (d, J = I. 6Hz, 1H), 7. 81 (s, br, 1H). Mass spectrum m / z:. 294 99 [M + H, 79Br], 296 82 [M + H, 81Br]..
2-2, V – methyl-5- (4,4,5,5-tetramethyl–1,3,2_ dioxolane Borane _2_ yl) spiro [indoline – 3,4 ‘- piperidin] -2_ one

Under nitrogen, obtained in Step 2-1 to 5-bromo -I ‘- methyl-spiro [indoline-_3,4’ – piperidin] _2_ one (147. 6mg, 0. 5mmol) , the United pinacols drop acid unitary purpose (140mg, 0. 55mmol) and acetic acid Bell (147mg, I. 5mmol) in DMSO (0. 2ml) was added in PdCl2 (dppf) • CH2Cl2 (20. 4mg, 0. 025mmol ), to the resulting solution was bubbled with nitrogen for 2 minutes, and then stirred at 80 ° C of 16 hours. LC-MS showed completion of the reaction, after cooling to room temperature, water (2mL), extracted with DCM (3X5mL). The organic phases were combined, dried (Na2SO4), and concentrated to give the desired product (170mg, yield: 100%) o MS m / z:. 342 07 [M + H], 343. 08 [M + H, 100%], 344. 11 [M + H].
Step 3,5_ [6_ atmosphere base _5_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -3_ batch P fixed base] -I ‘- A group spiro [indoline-3,4 ‘- piperidin] -2_ one
The steps 1-6 5_ desert obtained _3_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] -2-yl batch atmosphere pyridine (75. 8mg , 0. 2mmol), I’- step 2_2 obtained methyl 5- (4,4,5,5-tetramethyl-l, 3,2-dioxolane Borane 2-yl) spiro [ indoline-3,4′-piperidin] -2-one (82mg, 0. 24mmol) and potassium carbonate (82. 9mg, 0. 6mmol) was dissolved in DME / water mixture solution (4 / 1,2. Oml ). Then, under nitrogen, was added Pd (PPh3) 4 (II. 6mg, 0. Olmmol), to the resulting mixture was bubbled with nitrogen for 2 minutes, and then stirred at 80 ° C of 18 hours. LC-MS showed completion of the reaction, after cooling to room temperature, water (5mL), extracted (3 X IOmL) with DCM. The organic phases were combined, dried (Na2SO4), and concentrated to give the crude product was purified by silica gel column chromatography (7M NH3 in methanol solution / DCM: 5/95) to give the desired product (88. 6mg, yield: 86%) was purified. 1H-Nmr (Cdci3JOOmHz): 8 (ppm) I. 86 (d, J = 6. 4Hz, 3H), I 93-2 02 (m, 4H), 2 44 (s, 3H),…
2. 66-2. 72 (m, 2H), 2. 89-2. 93 (m, 2H), 4. 87 (s, br, 2H), 6. ll (q, J = 6. 4Hz, 1H ), 6. 88 (d, J =
8. OHz, 1H), 6. 94 (d, J = I. 2Hz, 1H), 7. 06 (t, J = 8. 4Hz, 1H), 7. 19 (dd, J = I. 2,8 . OHz, 1H),
7. 31 (m, 1H), 7. 36 (s, 1H), 7. 66 (s, br, 1H), 7. 80 (d, J = 2. OHz, 1H). Mass spectrum m / z:.. 515 05 [M + H, 35Cl, 35Cl], 517 03 [M + H, 35Cl, 37Cl].
Example 2: 6_ [6_ atmosphere base _5_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -3_ than Li Jie base] -I ‘ – methyl-spiro [indoline-3,4 ‘- piperidin] -2_ one

Step I, I ‘- methyl-6- (4,4,5,5-tetramethyl–I, 3,2- dioxolane boron-2-yl) spiro [indoline Spray – 3,4 ‘- piperidin] -2_ one
1-1,6- bromo -I ‘- methyl-spiro [indoline-3,4’ – piperidin] -2_ one

As described in Example I steps 2-1 of the method from the commercially available 6-bromo – indol-2-one was prepared, Yield: 82%. Analysis of the data obtained the desired product are = 1H-Nmr (Cd3OdJOOmHz): 8 (ppm) 1.90-1.98 (m, 4H),
2. 44 (s, 3H), 2. 64-2. 68 (m, 2H), 2. 86-2. 92 (m, 2H), 7. 05 (d, J = 2. 0Hz, 1H), 7. 16-7. 21 (m, 2H), 7. 91 (s, br, 1H). Mass spectrum m / z: 295 00 [M + H, 79Br], 296 78 [M + H, 81Br]… [0312] 1-2, 1 ‘- methyl-6- (4,4,5,5-tetramethyl-_1,3,2_ dioxolane Borane _2_ yl) spiro [indoline – 3,4 ‘- piperidin] -2_ one

In the step 1-1 of the obtained 6-bromo -I ‘- methyl-spiro [indoline-_3,4’ – piperidin] -2_ ketone and commercially available linking pinacol boronic ester material, the method of Example I was prepared in accordance with steps 2-2, Yield: 95%. Analysis of the data obtained of the target product are as follows: Mass spectrum m / z:. 342 06 [M + H], 343 04 [M + H, 100%], 344. 12 [M + H]..
Step 2,6_ [6_ atmosphere base _5_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -3 ratio Li Jie base] -I ‘- methyl-spiro [indoline-3,4 ‘- piperidin] -2_ one
Example I steps 1-6 to obtain 5-bromo -3 – [(IR) -I- (2,6- dichloro-3-fluorophenyl) ethoxy] -2-amino- pyridine, I obtained in Example 1-2 of the present embodiment in step ‘- methyl-6- (4,4,5,5-tetramethyl-l, 3,2-dioxolane-2-yl borane) spiro [indoline-_3,4 ‘- piperidin] -2-one, prepared as in Example I Step 3. Yield: 82%. 1H-Nmr (Cdci3JOOmHz): 8 (ppm) I. 86 (d, J = 6. 4Hz, 3H), I. 91-1 95 (m, 2H), I 97-2 03 (m, 2H… ), 2. 45 (s, 3H), 2. 65-2. 72 (m, 2H), 2. 89-2. 95 (m, 2H), 5. 12 (s, hr, 2H),
6. 12 (q, J = 6. 4Hz, 1H), 6. 94-7. 00 (m, 3H), 7. 06 (t, J = 8. 4Hz, 1H), 7. 31 (m, 1H ), 7. 35 (d, J = 7. 2Hz, 1H), 7. 90 (d, J = 2. 0Hz, 1H), 9. 28 (s, br, 1H). Mass spectrum m / z:.. 515 05 [M + H, 35Cl, 35Cl], 517 03 [M + H, 35Cl, 37Cl].
5- [6-amino-5 – [(2,6-dichloro-3-fluorophenyl) methoxy] _3_ pyridinyl] -I’–methyl-spiro [indole: 3 [0317] Example morpholine-3,4 ‘- piperidin] -2-one
H2N N


Step I, 5_ desert _3_ (2,6_ two gas -3- integrity oxy) _2_ atmosphere based grant given P
1-1,2,6_ two gas acid gas _3_
Cl OF
Sodium hydroxide (13g, 325mmol) in water (IlOmL) was cooled to _5 ° C was added dropwise under vigorous stirring of liquid bromine (12. 5g, 78. 2mmol), added after the addition of pre-cooled to 10 ° C dioxane (75mL). The above mixture under vigorous stirring was added dropwise a pre-cooled to 5 ° C of I- (2,6- dichloro-3-fluorophenyl) ethanone (5g, 21. 2mmol) in dioxane (330mL) and water (90mL) was added. After the addition, at room temperature for 2 hours Lan Xiang, Xiang Lan then 90 C for 30 minutes. TLC was not shown with the S starting material disappeared, and was acidified with concentrated hydrochloric acid to PH~9. The resulting mixture was rotary evaporated to dryness, added water (20mL), and extracted with diethyl ether (2X80mL), the organic phases were combined, dried (Na2SO4), and concentrated to give an oily product solidified after cooling to a transparent, slightly yellow solid (3. 4g, Yield: 67%). 1H-Nmr (Cdci3AOOmHz):. 8 (ppm) 7. 21 (. Dd, J = 8. 0,8 8Hz, 1H), 7 35 (. Dd, J = 4. 4,9 2Hz, 1H), 9 . 79 (s, br, 1H). Mass spectrum m / z (ES “:. 207 11 [M_H, 35Clj35Cl], 209 10 [MH, 35Cl, 37Cl]..
1-2,2,6–dichloro-3-fluoro-benzyl alcohol
^ Coh
F
[0325] To be filled with 2,6-dichloro-3-fluoro benzoic acid (3g, 14. 35mmol) added dropwise to the flask IM BH3. THF (43mL, 43mmol), added after the mixture was stirred under reflux for 24 hours. TLC showed the reaction was complete, methanol (50mL) to destroy excess borane, and the solvent was distilled off under reduced pressure and the resulting trimethyl borate, the process is repeated twice more to give a viscous product 2. I g, yield: 75% . 1H-Nmr (Cdci3JOOmHz): 8 (ppm) 2. 09 (t, J = 6. 4Hz, 1H), 4. 97 (d, J = 6. 4Hz, 2H), 7 09 (t, J = 8. . 8Hz, 1H), 7. 32 (dd, J = 4. 8,9. 1Hz, 1H). Mass spectrum m / z (ES-):.. 193 08 [M_H, 35Cl, 35Cl], 195 12 [MH, 35Cl, 37Cl].
1-3,3_ (2,6-gas _3_ integrity oxy) _2_ nitro grant given P

Following the procedure of steps 1-4 of Example I, was prepared from 2,6-dichloro-3-fluoro-benzyl alcohol and 3-hydroxy-2-nitropyridine prepared in yield (in this example embodiment steps 1_2) : 90%. 1H-Nmr (Cdci3AOOmHz): 8 (ppm) 5. 45 (s, 2H), 7 20, 7 37 (dd, J = 4. 8. (Dd, J = 8. 0,9 2Hz, 1H.). , 9. 2Hz, 1H), 7. 59 (dd, J = 4. 4,8. 4Hz, 1H),
7. 74 (dd, J = L 2,8. 4Hz, 1H), 8. 17 (dd, J = L 6,4. 4Hz, 1H). Mass spectrum m / z:. 316 89 [M + H, 35Cl, 35Cl], 318. 89 [M + H, 35Cl, 37Cl].
1_4,3_ (2,6-gas _3_ integrity oxy) _2_ atmosphere based grant given P

The method according to Example I step 1_5 from 3- (2,6-gas -3- integrity oxy) _2_ nitro Jie ratio 唳 preparation (in this case, steps 1-3), that Yield: 95% o 1H-Nmr (Cdci3JOOmHz):. 8 (ppm) 4 65 (s, br, 2H), 5 31 (s, 2H), 6 66 (dd, J = 5. 2,8.. . 0Hz, 1H), 7. 14 (dd, J = I. 2,8. 0Hz, 1H), 7. 18 (dd, J =
8. 4,9. 2Hz, 1H), 7. 37 (dd, J = 4. 8,8. 8Hz, 1H), 7. 73 (dd, J = I. 6,5. 6Hz, 1H). Mass spectrum m / z:. 286 95 [M + H, 35Cl, 35Cl], 288 85 [M + H, 35Cl, 37Cl]..
1-5,5_ desert -3- (2,6-gas -3_ integrity oxy) ~ 2 ~ atmosphere based grant given P

Following the procedure of Example I step 1_6 embodiment, starting from 3- (2,6-gas _3_ integrity yloxy) _2_ atmosphere group given the preparation of the batch P (in the example of the present embodiment in step 1-4), Yield: 60% o 1H-Nmr (Cdci3JOOmHz):. 8 (ppm) 4 68 (s, br, 2H), 5 28 (s, 2H), 7 21 (dd, J = 8. 0,8.. . 8Hz, lH), 7. 24 (dd, J = 2. OHz, 1H), 7. 39 (dd, J = 4. 8,
9. 2Hz, 1H), 7. 78 (d, J = 2. OHz, 1H). Mass spectrum m / z:. 364 83 [M + H, 35Cl, 36Cl, 79Br], 366 77 [M + H], 368 69 [M + H]…
Step 2,5_ [6_ atmosphere base _5_ [(2,6_ two gas -3- gas) methoxy] -3_ than Li Jie base] -I-methyl-spiro [indoline _ 3,4 ‘- piperidin] -2-one
The present embodiment 5_ desert steps 1_5 obtained _3_ (2,6_ two gas _3_ integrity yloxy) pyridine ~ 2 ~ atmosphere, Examples 2-2 obtained in step I I ‘- methyl-5- (4,4,5,5-tetramethyl-borane _1,3,2- dioxolane-2-yl) spiro [indoline-_3,4’ – piperidine ] -2-one, prepared as in Example I Step 3. Yield: 85 V0o 1H-Nmr (Cdci3JOOmHz):.. 8 (ppm) I. 92-2 02 (m, 4H), 2. 43 (s, 3H), 2. 65-2 71 (m, 2H) , 2. 90-2. 91 (m, 2H), 4. 92 (s, br, 2H), 5. 52 (s, 2H), 6. 89 (d, J = 8. 4Hz, 1H), 6 . 90 (d, J = L 2Hz, 1H), 7. 06 (t, J = 8. OHz, 1H), 7. 21 (dd, J = L 2,8. OHz, 1H), 7. 31 ( m, 1H),
7. 37 (s, 1H), 7. 79 (s, br, 1H), 7. 80 (d, J = 2.0Hz, lH). MS m / z:. 501 06 [M + H, 35Cl, 35Cl], 503 04 [M + H, 35Cl, 37Cl]..
6- [6-amino-5 – [(2,6-dichloro-3-fluorophenyl) methoxy] _3_ pyridinyl] -I’- methyl-spiro [indole: 4 [0337] Example morpholine _3,4 ‘- piperidin] -2-one



H2N N
Following the procedure in Example I step of Example 3, the procedure of Example 3 to give 5-bromo-1-5 _3_ (2,6-dichloro-3-fluoro-benzyloxy) -2-amino-pyridine and Step 2 in Example I to give the embodiment 1-2 ‘- methyl-6- (4,4,5,5-tetramethyl-1,3,2-dioxolane Borane 2-yl) spiro [ indoline-3,4 ‘- piperidine] _2_ ester -one, yield:. 78 V0o 1H-Nmr (Cdci3JOOmHz): 8 (ppm) I. 96-2 00 (m, 2H), 2. 01 -2. 12 (m, 2H), 2. 46 (s, 3H), 2. 66-2. 73 (m, 2H), 2. 90-2. 96 (m, 2H), 5. 30 (s , hr, 2H), 6. 94-7. 01 (m, 3H), 7. 07 (t, J =
8. 4Hz, 1H), 7. 30 (m, 1H), 7. 34 (d, J = 7. 2Hz, 1H), 7. 89 (d, J = 2. OHz, 1H), 8. 56 ( s, br, 1H). MS m / z:. 501 06 [M + H, 35Cl, 35Cl], 503 04 [M + H, 35Cl, 37Cl]..
Example 5: 5_ [5_ atmosphere base -6- [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] Batch-2-yl] -I ‘ – methyl-spiro [indoline-3,4 ‘- piperidin] -2-one
J0A = o
. | J: too
[0342] Step 1,5_ desert _2_ atmosphere base _3_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] Jie than exposure
Cl 6, / ISL / Br
xy
H2N N
In at 0 ° C, NaH (80mg of NaH in mineral oil, 2mmol) force the mouth (1R) -1_ (2,6- dichloro-3-fluorophenyl) ethanol (418mg, 2mmol. See example Example I Step 1_3) in anhydrous THF (6mL) and stirred for half an hour, a solution of 2-amino-3,5-dibromo-pyrazine (506mg, 2mmol) in THF (6mL) was added. The resulting mixture was warmed to room temperature, heated under reflux for 20 hours. TLC showed the reaction was substantially complete. After cooling to room temperature, water was added (IOmL), the mixture was extracted three times with ethyl acetate (3x20mL), the organic phases were combined, dried, concentrated, and the residue to give 594mg product was purified by column chromatography (l-3Me0H inhexanes), yield: 78%. 1H-NMR (O) Cl3, 500MHz):. 8 (ppm) I. 83 (d, J = 7. 2Hz, 3H), 5. 12 (s, br, 2H), 6 73 (q, J = 6 . 8Hz, 1H), 7. 05 (t, J = 8. OHz, 1H), 7. 28 (dd, J = 4. 8,
8. 8Hz, 1H), 7. 58 (s, 1H). Mass spectrum m / z:. 379 83 [M + H, 35Cl, 35Cl, 79Br], 381. 81 [M + H, 35Cl, 35Cl, 81Br], 383 79 [M + H, 35Cl, 37Cl, 81Br]..
Step 2,5_ [5_ atmosphere base _6_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] Batch-2-yl] -I ‘- A group spiro [indoline-3,4 ‘- piperidin] -2-one
5_ bromide present embodiment obtained in step I _2_ amino _3_ [(IR) -I- (2,6_ dichloro _3_ fluorophenyl) ethoxy] pyrazine, implemented I’- methyl step 2-2 obtained in Example I-5 (4,4,5,5-tetramethyl -I, 3,2- dioxolane boron
2-yl) spiro [indoline-3,4 ‘- piperidin] -2-one, prepared as in Example I Step 3. Yield: 54%. 1H-NMR (CD3ODjOOMHz): 8 (ppm) I. 85 (d, J = 6. 8Hz, 3H), I 85-1 88 (m, 2H), I 97-2 04 (m, 2H…. ), 2. 46 (s, 3H), 2. 76-2. 82 (m, 2H), 2. 97-3. 02 (m, 2H), 6. 74 (q, J = 6. 4Hz, 1H ), 6. 85 (d, J = 8. OHz, 1H), 7. 15 (t, J = 8. 4Hz, 1H), 7. 41 (dd, J = 4. 8,9. 2Hz, lH) , 7. 54 (dd, J = I. 6,
8. OHz, 1H), 7. 69 (d, J = I. 8Hz, 1H), 7. 81 (dt, J = 2. 0,8. 0Hz, 1H), 7. 87 (s, 1H). Mass spectrum m / z:. 515 92 [M + H, 35Cl, 35Cl], 517. 90 [M + H, 35Cl, 37Cl].
Example 6: 6- [5-amino -6 – [(lR) -l_ (2,6- dichloro _3_ fluorophenyl) ethoxy] pyrazin-2-yl] -I ‘ – methyl-spiro [indoline-3,4 ‘- piperidin] -2-one

The embodiment of Example 5, 5_ bromo obtained in step I _2_ amino _3_ [(IR) -I- (2,6_ dichloro _3_ fluorophenyl) ethoxy] pyrazine, Example I’- methyl-2 obtained in steps 1-2 6- (4,4,5,5-tetramethyl–I, 3,2- dioxolane boron-2-yl) spiro [indole morpholine -3,4’_ piperidin] -2-one, prepared as in Example I Step 3. Yield: 67% 0
1H-NMR (CD3ODjOOMHz): 8 (ppm) I. 85 (d, J = 6. 8Hz, 3H), I 88-1 96 (m, 4H), 2 48 (s… , 3H), 2. 76-2. 82 (m, 2H), 2. 98-3. 05 (m, 2H), 6. 75 (q, J = 6. 4Hz, 1H), 7. 16 (t , J = 8. 8Hz, 1H), 7. 31 (d, J = 2. OHz, 1H), 7. 36-7. 43 (m, 3H), 7. 88 (s, 1H).
Mass spectrum m / z:. 515 99 [M + H, 35Clj35Cl], 517 90 [M + H, 35Cl, 37Cl]..
SEE
Bioorganic & Medicinal Chemistry Letters (2014), 24(16), 3673-3682.
School of Pharmaceutical Sciences, Southern Medical University,



 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


 amcrasto@gmail.com
amcrasto@gmail.com





 Stockholm, Sweden
Stockholm, Sweden Despite the cold weather, public came and enjoyed different activities. The famous chef, Paul Svensson who works in one of the fanciest and most famous …
Despite the cold weather, public came and enjoyed different activities. The famous chef, Paul Svensson who works in one of the fanciest and most famous …
Amgen receives FDA approval for chronic heart failure medicine Corlanor
Amgen receives FDA approval for chronic heart failure medicine Corlanor
Amgen has received approval from the US Food and Drug Administration (FDA) for its Corlanor (ivabradine) to treat patients with chronic heart failure.

keep watching
will be updated………………..
1.5 lakh views on Top Organic Spectroscopy blog in the world
ORGANIC SPECTROSCOPY INTERNATIONAL
1.5 lakh views on this blog………http://orgspectroscopyint.blogspot.in/
Techniques for Organic Compounds ie NMR, MASS, IR, UV Etc. Starters,
Learners, advanced, all alike, contains content which is basic or advanced,
by Dr Anthony Melvin Crasto, Worlddrugtracker,
email me ……….. amcrasto@gmail.com, call +91 9323115463

1.5 lakh views on this blog………http://orgspectroscopyint.blogspot.in/
FRANCE
.jpg)
Republican march, Place de la République, Paris.



The St. Bartholomew’s Day massacre (1572) was the climax of theFrench Wars of Religion, which were brought to an end by the Edict of Nantes (1598).

One of the Lascaux paintings: a horse – Dordogne, approximately 18,000 BC

With Clovis‘ conversion to Catholicism in 498, theFrankish monarchy, electiveand secular until then, became hereditary and ofdivine right.

Frankish expansion from 481 to 843/870.

The Storming of the Bastille on 14 July 1789 was the starting event of theFrench Revolution.

Louis XIV, the “sun king” was the absolute monarch of France and made France the leading European power.

Napoleon, Emperor of the French, and his Grande Armée built a vast Empire across Europe. He helped spread the French revolutionary ideals and his legal reforms had a major influence worldwide.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
COCK WILL TEACH YOU NMRCOCK SAYS MOM CAN TEACH YOU NMR
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE amcrasto@gmail.com
FDA approves first generic Copaxone to treat multiple sclerosis

April 16, 2015
The U.S. Food and Drug Administration today approved the first generic version of Copaxone (glatiramer acetate injection), used to treat patients with relapsing forms of multiple sclerosis (MS).
Sandoz has received FDA approval to market generic glatiramer acetate in a 20 mg/1 ml daily injection.
“Health care professionals and patients can be assured that FDA-approved generic drugs have met the same rigorous standards of quality as the brand-name drug,” said Janet Woodcock, M.D., director of the FDA’s Center for Drug Evaluation and Research. “Before approving this generic product, given its complexity, we reviewed additional information to make sure that the generic product is as safe and effective as the brand name product.”
The FDA applies the same rigorous and reliable standards to evaluate all generic drug products. As needed, the agency requires appropriate information to demonstrate sameness for complex active ingredients, such as glatiramer acetate. For this approval, FDA scientists established a thorough scientific approach for demonstrating active ingredient sameness that takes into consideration the complexity of glatiramer acetate.

MS is a chronic, inflammatory, autoimmune disease of the central nervous system that disrupts communication between the brain and other parts of the body. It is among the most common causes of neurological disability in young adults and occurs more frequently in women than men. For most people with MS, episodes of worsening function (relapses) are initially followed by recovery periods (remissions). Over time, recovery periods may be incomplete, leading to progressive decline in function and increased disability. MS patients often experience muscle weakness and difficulty with coordination and balance. Most people experience their first symptoms of MS between the ages of 20 and 40.
In the clinical trials for Copaxone, the most common adverse reactions reported by those taking Copaxone were skin problems at the injection site (redness, pain, swelling and itching), flushing (vasodilation), rash, shortness of breath and chest pain.
BLOG STATS OF NEW DRUG APPROVALS
MOSCOW


COCK WILL TEACH YOU NMRCOCK SAYS MOM CAN TEACH YOU NMRDRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE amcrasto@gmail.com
Zydus-Cadila is developing ZYH-7, a PPAR alpha modulator for the potential treatment of dyslipidemia


Zydus-Cadila is developing ZYH-7, a PPAR alpha modulator for the potential treatment of dyslipidemia .
By January 2012, phase II trials had begun ; in January 2014, the drug was still listed as being in phase II development
By January 2012 phase II trials had begun for Diabetes type 2 Lipoprotein disorders
Obesity
In August 2007, an IND was filed , and by March 2008, a phase I trial was underway ; by April 2011, the trial had been completed
| Zydus Cadila has filed an Investigational New Drug (NID) application for seeking DCGI’s permission for conducting clinical trials for its New Molecular Entity (NME) ZYH7. |
 |
| According to a company release, it claims that ZYH7 is a novel drug candidate for treating dyslipidemia and metabolic disorders. The company inform that ZYH7 had been conceptualised and developed by its scientists from Zydus Research Centre. |
| The company has its in-house research centre and it had recently concluded pre-clinical studies on ZYH7, which have reported interesting and encouraging finding which indicate a novel molecule to treat dyslipidemia and associated metabolic disorders. |
| Commenting on the new development, Pankaj Patel, chairman and managing director, Zydus Cadila said, “We have been building a promising pipeline of new molecular entities at the Zydus Research Centre and ZYH7 is an important step in this direction”. |
| Starting with its first IND filing in 2005, Zydus today has four INDs in various stages of clinical trials. NME – ZYH1 for treating dyslipidemia and ZYI1 for treating pain and inflammation are undergoing Phase II trials. ZYH2 for treating diabetes and the novel CB-1 antagonist, ZYO1 for treating obesity, are undergoing Phase I trials. |
| Diabetes, a worldwide health problem, affects more than 150 million people, a number expected to double to 300 million by 2025. People with diabetes are at especially high risk for dyslipidemia, particularly high triglyceride levels and low HDL levels. |
| Dyslipidemia is also a key independent risk factor for cardiovascular disease (CVD), which is the largest therapeutic segment in the world pharmaceutical market. |
| With an increasing correlation between several endocrine and metabolic disorders, there has been considerable emphasis in recent times on metabolic syndrome. The metabolic components of cardiovascular disease, diabetes and obesity, are linked in numerous ways with each having an impact on the other. |
| For instance, it is also well known that patients with Type 2 diabetes have a two to four-fold excess risk of coronary heart disease and that these patients very often have increased cardiovascular risk factors even before the onset of their diabetes. |
Dyslipidemia is an abnormal amount of lipids (e.g. cholesterol and/or fat) in the blood. In developed countries, most dyslipidemias are hyperlipidemias; that is, an elevation of lipids in the blood. This is often due to diet and lifestyle. Prolonged elevation of insulin levels can also lead to dyslipidemia. Likewise, increased levels of O-GlcNAc transferase (OGT) may cause dyslipidemia.
| Dyslipidemia | |
|---|---|
| Classification and external resources | |
| ICD–10 | E78 |
| ICD–9 | 272 |
| DiseasesDB | 33452 |
| MeSH | D050171 |
Classification
Physicians and basic researchers classify dyslipidemias in two distinct ways:
- Phenotype, or the presentation in the body (including the specific type of lipid that is increased)
- Etiology, or the reason for the condition (genetic, or secondary to another condition). This classification can be problematic, because most conditions involve the intersection of genetics and lifestyle issues. However, there are a few well-defined genetic conditions that are usually easy to identify.
Fredrickson Classification:[1]
| Phenotype | I | IIa | IIb | III | IV | V |
|---|---|---|---|---|---|---|
| Elevated Lipoprotein | Chylomicron | LDL | LDL and VLDL | IDL | VLDL | VLDL and chylomicrons |
WO 2008035359
https://www.google.com/patents/WO2008035359A2?cl=en
Scheme 1 below which comprises:

Scheme 2 below which comprises

| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2009021740A2 | Aug 14, 2008 | Feb 19, 2009 | Sanofis Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2010049946A2 * | Oct 22, 2009 | May 6, 2010 | Cadila Healthcare Limited | Thyroid receptor ligands |
| WO2010084512A1 * | Dec 22, 2009 | Jul 29, 2010 | Cadila Healthcare Limited | Novel oxime derivatives |
| WO2010110479A1 * | Mar 24, 2010 | Sep 30, 2010 | Nippon Chemiphar Co., Ltd. | Activator for peroxisome proliferator-activated receptor |
| WO2011157827A1 | Jun 17, 2011 | Dec 22, 2011 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2013037390A1 | Sep 12, 2011 | Mar 21, 2013 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2014192023A1 * | May 20, 2014 | Dec 4, 2014 | Cadila Healthcare Limited | Novel compounds suitable for the treatment of dyslipidemia |
| EP2567959A1 | Sep 12, 2011 | Mar 13, 2013 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US8742117 | Dec 22, 2009 | Jun 3, 2014 | Cadila Healthcare Limited | Oxime derivatives |
| US8822414 * | Dec 26, 2011 | Sep 2, 2014 | Cadila Healthcare Limited | Heterocyclic compounds suitable for the treatment of dyslipidemia |
………….
PARIS

Myself recovering from leg swelling

DR ANTHONY CRASTO at Metro hospital Manpada Thane, India
13-16 Apr, 2015
I am back
vladivostok







COCK WILL TEACH YOU NMRCOCK SAYS MOM CAN TEACH YOU NMRDRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE amcrasto@gmail.com
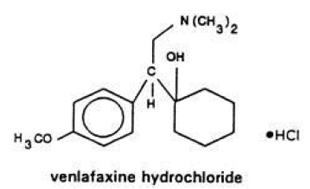
VENLAFAXINE PART 1/3


 SEE
SEEpart 1………http://orgspectroscopyint.blogspot.in/2015/04/venlafaxine.html / https://newdrugapprovals.org/2015/04/09/venlafaxine-part-12/
part 2……..https://newdrugapprovals.org/2015/04/09/venlafaxine-22/
PART 3…..http://orgspectroscopyint.blogspot.in/2015/04/venlafaxine-part-33.html
SEE ALSO
http://www.google.com/patents/WO2008059525A2?cl=en
WILL BE UPDATED………..
Chemistry

 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (RS)-1-[2-dimethylamino-1-(4-methoxyphenyl)-ethyl]cyclohexanol | |
| Clinical data | |
| Trade names | Effexor XR, Effexor, Trevilor |
| AHFS/Drugs.com | monograph |
| Licence data | US Daily Med:link |
|
|
|
|
| Oral | |
| Pharmacokinetic data | |
| Bioavailability | 42±15%[1] |
| Protein binding | 27±2% (parent compound), 30±12% (active metabolite,desvenlafaxine)[2] |
| Metabolism | Hepatic (~50% of the parent compound is metabolised on first pass through the liver)[1][2] |
| Half-life | 5±2 h (parent compound for immediate release preparations), 15±6 h (parent compound for extended release preparations), 11±2 h (active metabolite)[1][2] |
| Excretion | Renal (87%; 5% as unchanged drug; 29% asdesvenlafaxine and 53% as other metabolites)[1][2] |
| Identifiers | |
| 93413-69-5 |
|
| N06AX16 | |
| PubChem | CID 5656 |
| DrugBank | DB00285 |
| ChemSpider | 5454 |
| UNII | GRZ5RCB1QG |
| ChEBI | CHEBI:9943 |
| ChEMBL | CHEMBL637 |
| Chemical data | |
| Formula | C17H27NO2 |
| 277.402 g/mol | |

HSQC

1H NMR PREDICT OF HCL

13C NMR PREDICT OF HCL


BASE


External links[Drug information
- U.S. Food and Drug Administration information on Effexor
- Efexor patient information leaflet (UK)
- Effexor XR prescribing information for healthcare professionals (pdf) (USA only) Archived from the original on 17 September 2006.
- Detailed Patient/Parent Information on Effexor
- List of international brand names for Venlafaxine
- U.S. National Library of Medicine: Drug Information Portal -Venlafaxine
Diagnostic tools
Patient experiences
: The views expressed are my personal and in no-way suggest the views
of the professional body or the company that I represent.
: The views expressed are my personal and in no-way suggest the views
of the professional body or the company that I represent.
: The views expressed are my personal and in no-way suggest the views
of the professional body or the company that I represent.
COCK WILL TEACH YOU NMRCOCK SAYS MOM CAN TEACH YOU NMR
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE amcrasto@gmail.com
VENLAFAXINE PART 2/3

part 1………http://orgspectroscopyint.blogspot.in/2015/04/venlafaxine.html / https://newdrugapprovals.org/2015/04/09/venlafaxine-part-12/
part 2……..https://newdrugapprovals.org/2015/04/09/venlafaxine-22/
PART 3…..http://orgspectroscopyint.blogspot.in/2015/04/venlafaxine-part-33.html
http://www.google.com/patents/WO2008059525A2?cl=en
WILL BE UPDATED………..
………………….

……………………….
PAPER
DOI: 10.1039/C4RA00840E
http://pubs.rsc.org/en/content/articlelanding/2014/ra/c4ra00840e#!divAbstract
A protecting group free asymmetric total synthesis of (−)-venlafaxine is reported. The strategy employs Sharpless epoxidation and regio-selective epoxide ring opening by an in situgenerated Gilman reagent as key steps. This paper reports a 53% overall yield in 6 steps for total synthesis of (−)-venlafaxine.


…………………
http://www.google.com/patents/EP2181982B1?cl=en
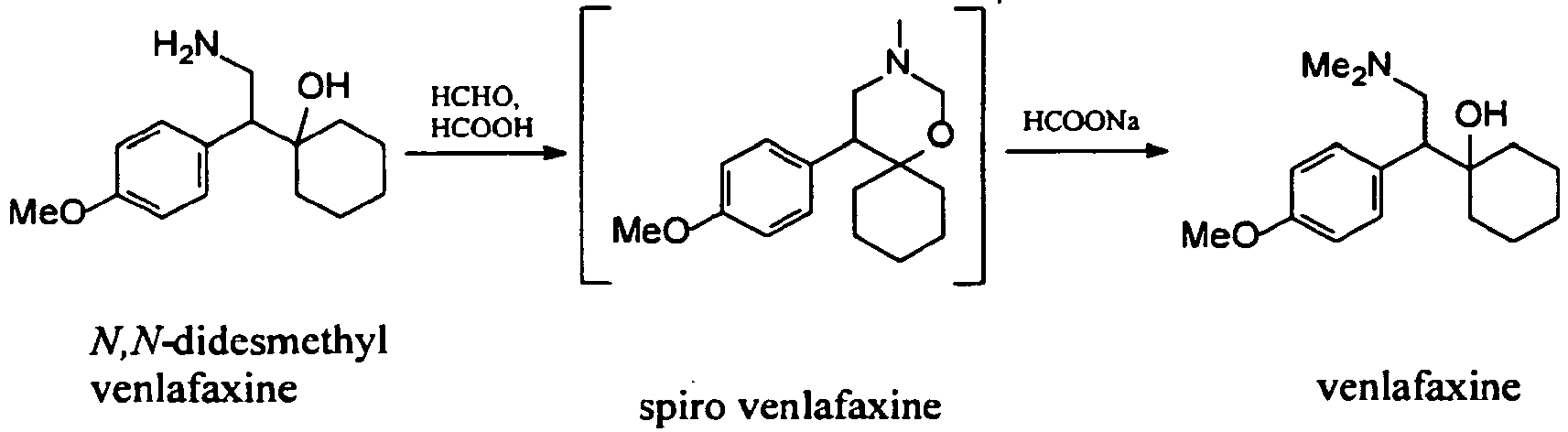
Examples:Example 1 – Preparation of venlafaxine from N,N-didesmethyl venlafaxine hydrochloride
-
A 50 % aqueous NaOH solution (4 ml, 74 mmol) was added to a stirred solution of N,N-didesmethyl venlafaxine hydrochloride (5.72 g, 20 mmol) in water (16 ml) at room temperature. Formic acid (98 %, 11.5 ml, 305 mmol) and 37 % aqueous solution of formaldehyde (8.4 ml, 113 mmol) were added to this mixture. The mixture was stirred under reflux temperature and the conversion was completed in 5 h (HPLC: 98.67 area %). Then the solution was cooled to room temperature and adjusted with 50 % aequous NaOH to pH 12. The mixture was extracted twice with 66 ml of isopropyl acetate. The collected organic phases were washed three times with water (66 ml). The isolated solution of venlafaxine base was very pure (HPLC: 98.9 area%).
Example 2 – Preparation of venlafaxine hydrochloride form I from the solution of venlafaxine base in isopropyl acetate
-
To the solution of venlafaxine base in isopropyl acetate from example 1 (66 ml, 10 mmol) 5 ml of 2 M aqueous HCl were added. The mixture was heated and water was removed by azeotropic distillation using a Dean-Starck trap. When all water was removed from the mixture, the product began slowly to crystallize. The obtained suspension was heated under reflux temperature for 1.5 h, then cooled and filtered. 2.75 g (88 % from N,N-didesmethyl venlafaxine hydrochloride) of pure venlafaxine hydrochloride form I (HPLC: 99.65 area %) were obtained.
Example 3 (exemplary) – Preparation of venlafaxine hydrochloride form I from the solution of venlafaxine base in isopropyl acetate
-
The solution of venlafaxine in isopropyl acetate from example 1 (66 ml, 10 mmol) was concentrated to ½ of the volume. Then 10 to 50 mg of venlafaxine hydrochloride form I was added to the solution. Subsequently, a 2.5 M solution of HCl in ethanol (4.0 ml) was slowly added within 30 min. After the whole amount of acid was added, the obtained suspension was stirred for another 2 h. Then the mixture was filtered and the product was washed with isopropyl acetate and dried. We obtained 2.69 g (86 % from N,N-didesmethyl venlafaxine hydrochloride) of pure venlafaxine hydrochloride form I (HPLC: 99.65 area %).
…………………..
PATENT
http://www.google.com/patents/WO2006035457A1?cl=en
Venlafaxine is known by the chemical name 1-[2-dimethylamino-1-(4 methoxyphenyl ethyl Cyclohexanol hydrochloride and structure of formula (V).

(V)
Venlafaxine is a useful pharmaceutical agent as an antidepressant. Venlafaxine, the intermediates in the manufacture of Venlafaxine, the process of preparing said Venlafaxine and their intermediates are well known from US Patents 4,535,186, US Patent No. 6,350,912, and CN 1225356.
Further International Publication No. WO03/050074 discloses the manufacture of Venlafaxine Hydrochloride and crystalline polymorphs Form I, Form II, Form III and optically pure (R) and (S) enantiomers exhibiting different crystalline structures of Venlafaxine hydrochloride. The preparation of all the forms of Venlafaxine and their inter-conversion are also described in said WO03/0500074 publication. U.S. Patents 4535186, 4761501 disclose a process for manufacture of 1-[2- amino-i-(p-methoxyphenyl) ethyljcyclohexanol (free base of formula IV), an intermediate produced during the preparation of Venlafaxine in two stages by reacting p-methoxyphenyl acetonitrile with cyclohexanone in the presence of n- butyl lithium (Stage 1) to form 1-[cyano(p-methoxyphenyl) methyl] Cyclohexanol of formula III

(111)
This process is commonly used for the preparation of formula III. The US Patent 4,535,186 produces a yield of about 30% based on p-methoxyphenyl acetonitrile.
WO/03/050074 suggests an alternate way of preparing compound of formula III without using butyl lithium i.e. by reacting p-methoxyphenyl acetonitrile with cyclohexanone in the presence of alkali metal hydroxide in a mixture of toluene and hexane. The publication WO/03/050074 also suggests a material yield of 74% based upon p-methoxyphenyl acetonitrile and purity.
The drop wise addition of butyl lithium to p-methoxyphenyl acetonitrile is hazardous and hence it requires skill and safety measures to be taken by the person skilled in the art for handling butyl lithium over the addition period to avoid any accidents during the preparation process.
The second stage i.e. conversion of compound of formula III to formula IV described in US patent US 4,535,186 is by hydrogenating compound of formula III using Rhodium on alumina. The catalyst Rhodium is recycled by filtering and washing the catalyst with ethanol and the combined filtrate evaporated and dried under vacuum yielding free base as an oil. However, the cost of Rhodium catalyst is very high and hence the catalyst has to be recovered.
WO/02/500017 suggests the use of a Nickel or cobalt catalyst for the hydrogenation, which is highly economical when compared with the Rhodium catalyst as suggested by US Patent No. 4,535,186. The International Publication WO/02/500017 teaches that the hydrogenation reaction of Stage Il may be carried out in the presence of an organic solvent preferably an alcohol. The international publication also suggests the pretreatment of the catalyst with ethanol.
The US Patent 4,535,186 describes the third stage in the process of preparing Venlafaxine i.e. conversion of compound of formula IV (free base) to compound V i.e. Venlafaxine by methylating the compound of formula IV (free base) with a mixture of formaldehyde and formic acid in water.

(IV)
US Patent Publication No. 2005/0033088 describes a process for preparing phenylethylamine derivative, an intermediate of Venlafaxine hydrochloride; said process comprising steps of reduction of compound of formula III with palladium on charcoal in an organic acid selected from formic acid, acetic acid or propionic acid, preferably acetic acid in an autoclave at a pressure of 5 to 25 kg/cm2 preferably 10 to 15 kg/cm2 at a temperature in the range of 30 to 75°C, preferably at 50 to 55°C till the hydrogenation substantially complete, filtering the palladium catalyst and evaporating the filtrate. Extracting the filtrate with halogenated hydrocarbon solvent and purifying the same. The process also describes the preparation of Venlafaxine hydrochloride without isolation of freebase.
The route of synthesis for Venlafaxine (formula V) and intermediate of Venlafaxine (formula IV) is depicted in the following scheme:
Step -I

(I) (II) (III) Step-ll

…………..(III) ……………………(IV)
Step-ll I

(IV) (V)
Accordingly, the present invention relates to an improved process for the preparation of compound of formula IV

(IV)
comprising the step of hydrogenating a compound of formula

(Hi)
in the presence of toluene, water, and a catalyst wherein the said process yields 66% formula (IV) with 99% HPLC purity.
The compound of formula IV is further methylated using formaldehyde and formic acid mixture to form Venlafaxine (formula V) followed by the treatment with HCL gas dissolved in Isopropanol.

(IV) (V)
According to the preferred embodiment of the present invention, nickel catalyst, preferably Raney nickel catalyst is used. The catalyst is washed in water to remove the alkali. No pretreatment of the nickel catalyst is required.

(HI) (IV)
According to another embodiment of the present invention, compound of formula IV is prepared by hydrogenating compound of formula III in the presence of Raney Nickel and water. According to another embodiment of the present invention, compound of formula III is prepared by charging p-methoxyphenyl acetonitrile into butyl lithium at -70 to -75°C and tetrahydrofuran; cooling the reaction mixture to about -50°C to -750C; adding cyclohexanone at a temperature below -500C quenching with ice and saturated ammonium chloride solution below 0°C; and stirring and filtering the product of formula (III) wherein the said process yields 89% of compound of formula III with 99.8% purity. The reaction scheme is depicted as follows:

…………(I) …………(H)………………………………. (III)
Example 1 :
Preparation of 1-[cyano-(4-methoxyphenyl) methyl Icvclohexanol.
In a 2 ltr 4 necked round bottom flask equipped with a overhead stirrer, thermometer and dropping funnel, 100 ml dry THF followed by 210 ml Butylithium (1.6 M solution in Hexane) was charged. The reaction mixture was cooled to – 700C. Added gradually a solution of 50 gm p-methoxyphenyl acetonitrile dissolved in 50 ml dry THF at -70 to -75°C. After 30 min, added solution of 33.1 gm Cyclohexanone in 50 ml THF. After the addition, maintained at -65 to -700C and monitored by TLC. After 4 hrs, reaction mixture was gradually added over mixture of ice and 150 ml saturated ammonium chloride solution below 0°C and adjusted pH to 7 with dilute Hydrochloric acid. Stirred for 1 hr and filtered the product. Washed the product with 200 ml hexane and dried to obtain 74.3 gm. (The yield based on p-methoxyphenyl acetonitrile 89%, Melting range 123- 125°C, HPLC purity of 99.8%).
Example 2:
Preparation of 1-[2-amino-(4-methoxyphenyl) ethyl] Cyclohexanol acetate
In an autoclave are charged 100 gm 1-[cyano(4- methoxyphenyl)ethyl]cyclohexanol, 100 ml toluene and 400 ml water at RT. Stirred and cooled to 10°C. Charged 20 gm Raney Nickel (which was prewashed with water to make it free of Alkali) and 100 ml liquor ammonia (20%). Then pressurized the autoclave with hydrogen to 4 – 5 kg pressure and maintained for 120 minutes below 120C. Then the reaction temperature slowly raised to bout 500C along with the increase in the hydrogen pressure to 7 to 8 kg. Maintained between 45 – 50°C for 8 hr. After the completion of the reaction, cooled the reaction to RT, released the hydrogen pressure and charged 400 ml toluene. Filtered the catalyst and washed bed with 100 ml toluene. Separated the organic layer from the filtrate. The organic layer was washed with 10% Sodium chloride solution. To the organic layer was added 40 ml methanol followed by 10 ml acetic acid. Stirred for 15 minutes and then again charged 10 ml acetic acid. Then heated to 75-8O0C and maintained for 15 minutes. Cooled to 0 – 50C. Filtered the product. Washed the product with 100 ml ethyl acetate and dried: 83.5 gm (Yield 66%, Melting range 164-166°C, HPLC purity 99%).
Example 3:
Preparation of H2-dimethylamino-1-(4-methoxyphenyl) ethvH Cyclohexanol Hydrochloride
To a stirred solution of 100 gm of 1-[2-amino-(4-methoxyphenyl) ethyl] Cyclohexanol acetate in 300 ml water was added 117 gm of formic acid (88%) and 91 gm of formaldehyde (40% solution). The solution was heated to 98°C and maintained for 20 hrs. Reaction mixture was cooled to about 100C and added 500 ml ethyl acetate. The pH was adjusted to about 7 with sodium hydroxide solution and further to 10 – 10.5 with ammonium hydroxide solution. Layers were separated. Aqueous layer was extracted with ethyl acetate. Combined organic layers were washed with water. Combined organic extract was stirred with activated carbon (5 gm) and filtered. Filtrate was concentrated in vacuum to completely remove ethyl acetate. Residue was dissolved in isopropanol (300 ml) and acidified at 300C (pH 1-1.5) with the solution of HCI in isopropanol. Temperature was then raised to 600C and maintained for 60 to 90 min. The reaction mass was cooled under agitation to 10°C and maintained under agitation at 10°C for 60 min. Product was isolated by filtration. Finally it was washed with isopropanol and dried at 60°C.
Dry wt. : 85 gm (84% yield, HPLC 99.9% purity with all individual impurities below 0.1% concentration). This material exhibited following characteristic x-ray powder diffraction pattern with characteristic peaks expressed in d-values (A) at.
(The abbreviations in brackets mean : (vs) = Relative intensity above 80%; (s) = 30% – 80%; (m) = 15% – 30%; ![]() = 8% to 15% and (vw) = below 8%.) 2.23 (VW), 2.29(VW), 2.32 (VW)1 2.35(VW), 2.38(VW), 2.43(VW), 2.46(VW), -2.48(VW)12.55(M), 2.64(W), 2.69(W), 2.73(VW), 2.8(W), 2.83(W), 2.88(W), 2.93(VW), 3.09(VW), 3.12(M), 3.26(VM), 3.31(VM), 3.38(W), 3.45(VW), 3.5(VW), 3.55(M), 3.69(VW), 3.87(VW), 3.99(VW), 4.07(M), 4.18(S), 4.35(VS), 4.48(VW), 4.68(M), 5.1 (VW), 5.27(W), 5.42(VW), 5.55(VW), 5.63(M), 5.68(M), 5.76(VW), 6.5(S), 6.95(VS), 8.65(VW), 10.56(M), 13.06(M).
= 8% to 15% and (vw) = below 8%.) 2.23 (VW), 2.29(VW), 2.32 (VW)1 2.35(VW), 2.38(VW), 2.43(VW), 2.46(VW), -2.48(VW)12.55(M), 2.64(W), 2.69(W), 2.73(VW), 2.8(W), 2.83(W), 2.88(W), 2.93(VW), 3.09(VW), 3.12(M), 3.26(VM), 3.31(VM), 3.38(W), 3.45(VW), 3.5(VW), 3.55(M), 3.69(VW), 3.87(VW), 3.99(VW), 4.07(M), 4.18(S), 4.35(VS), 4.48(VW), 4.68(M), 5.1 (VW), 5.27(W), 5.42(VW), 5.55(VW), 5.63(M), 5.68(M), 5.76(VW), 6.5(S), 6.95(VS), 8.65(VW), 10.56(M), 13.06(M).
Example 4 :
Preparation of 1-f2-amino-(4-methoxyphenyl) ethyl] Cyclohexanol acetate (IV)
In an autoclave charged 150 gm 1-[cyano(4-methoxyphenyl)ethyl]cyclohexanol, and 675 ml water at RT. Stirred and cooled to 1O0C. Charged 30 gm Raney Nickel (prewashed with water to make it free of Alkali) and 150 ml liquor Ammonia (20%). Then pressurized the autoclave with hydrogen to 4 – 5 kg pressure and maintaind for 120 minutes below 12°C. After completion of 120 minutes slowly raised the temperature to about 500C along with the increase in the hydrogen pressure to 7 to 8 kg. Maintained between 45 – 5O0C for about 20 hrs. Monitored reaction by TLC to ensure disappearance of starting material. After the completion of the reaction cooled the reaction to RT, released the hydrogen pressure and filtered through celite bed. Washed bed with 300 ml toluene. To the filtrate added 300 ml toluene. Shaken well and separated the organic layer. The organic layer was washed with 5% Sodium chloride solution. To the organic layer was added 45 ml methanol and 15 ml acetic acid. Stirred for 15 minutes and then again charged 15 ml acetic acid. Then heated to 75-8O0C and maintained for 15 mins. Cooled to 0 – 5°C. Filtered the product. Washed the product with 100 ml ethyl acetate and dried: 104 gm (Yield 53%, Melting range 152-153°C, HPLC purity 90%).
……………….
PATENT
http://www.google.com/patents/WO2008059525A2?cl=en
Venlafaxine acts by inhibiting re-uptake of norepinephrine and serotonin. It has been reported that its (-) enantiomer is a more potent inhibitor of norepinephrine synaptosomal uptake while its (+) enantiomer is more selective in inhibiting serotonin uptake (J. Med. Chem. 1990, 33(10), 2899-2905) In humans, venlafaxine is transformed by a metabolic pathway into two minor metabolites, N-desmethylvenlafaxine of formula II, N,O-di- desmethylvenlafaxine of formula IV and one major metabolite, O-desmethylvenlafaxine of formula III.

formula I formula II

formula III formula IV
In the literature there are several processes reported for the synthesis of venlafaxine of formula I and venlafaxine hydrochloride of formula Ia.

formula Ia
The synthesis of venlafaxine from 2-(l-hydroxycyclohexyl)-2-(4-methoxyphenyl)acetonitrile (hereinafter called as cyano-intermediate and represented by formula V) involving two step synthesis is known in the prior art.

formula V
US 4,535,186 discloses the preparation of venlafaxine of formula I by the reaction of p- methoxyphenylacetonitrile with cyclohexanone at -78 0C in the presence of n-butyllithium as a base which yields 2-(l -hydroxy cyclohexyl)-2-(4-methoxyphenyl)acetonitrile of formula V. Reduction of the cyano-intermediate under hydrogen pressure with rhodium on alumina catalyst gives l-[2-amino-l-(4-methoxyphenyl)ethyl]cyclohexanol. N-Methylation of the amino compound is accomplished employing formaldehyde and formic acid (Eschweiler- Clarke reaction) to give venlafaxine of formula I.
The reaction is as shown in the Scheme- 1.

Reduction


Scheme-1
Another prior art reference, Zhou Jinpei et.al, J. China Pharm. University, 1999, 30(4), 249- 50) discloses the preparation of venlafaxine starting from anisole. Anisole is acylated to the chloroacetyl derivative, which is then animated using N,N-dirnethylamine. The carbonyl group of this compound is reduced to the alcohol using KBH4 and is converted to the bromo- derivative using PBr3 which in turn when reacted with Mg and cyclohexanone undergoes a Grignard reaction to provide venlafaxine of formula I.
The reaction is as shown in the Scheme-2.

Scheme-2
US 2005033088 discloses a two step process for venlafaxine starting from the cyano- intermediate. The cyano-intermediate is reduced in the presence of palladium on charcoal in acetic acid at a hydrogen pressure of 5-25 kg/cm2 at a temperature in the range of 30-75 0C. The product of step 1 is N-methylated using formic acid, formaldehyde solution at a temperature of 90-98 0C for 19 hrs to yield venlafaxine, which is then converted to its hydrochloride salt.
WO2006035457 also discloses a process of making venlafaxine and its intermediates. The process comprises the step of hydrogenating the cyano-intermediate in the presence of toluene, water, and Raney nickel where in the said process yields 66% of an intermediate with 99% HPLC purity. This reaction is carried out at 10-12 0C and at 4-5 kg/cm2 of hydrogen pressure for 2 hrs and further at 50 0C at 7-8 kg/cm2 for 7-8 hrs. This intermediate is N-methylated using formaldehyde and formic acid mixture to form venlafaxine, which is treated with IPA/HC1 to get venlafaxine hydrochloride.
US 6,350,912 discloses a one pot process for the preparation of venlafaxine in 15-28 % yield from the cyano-intermediate. In the said patent venlafaxine has been prepared by the reduction of cyano-intermediate in the presence of Raney nickel and without isolation of intermediate l-[2-amino-l-(4-methoxyphenyl) ethyl] cyclohexanol
CN 1850781 discloses a process for the preparation of venlafaxine by following steps: (1) carrying out condensation of 4-methoxyphenylacetonitrile and cyclohexanone in presence of base to obtain 2-(l-hydroxycyclohexyl)-2-(4-methoxyphenyl)acetonitrile, (2) reacting 2-(l- hydroxycyclohexyl)-2-(4-methoxyphenyl)acetonitrile with cuprous chloride and dimethylamine to obtain 2-(l -hydroxy cyclohexyl)-2-(4-methoxyphenyl)-N,N- dimethylacetimidamide, and (3) reacting 2-(l-hydroxycyclohexyl)-2-(4-methoxyphenyl)- N,N-dimethylacetimidamide with KBH4 to obtain 1 – [2-dimethylamino)- 1 -(4-methoxyphenyl) ethyl)cyclohexanol (venlafaxine).
The reaction is as shown in the Scheme-3.

Scheme-3
The processes disclosed in the prior art have many disadvantages. Most of the prior art processes employ formaldehyde as a reactant for N-methylation step which is known to be a carcinogen. Acute exposure of the same is highly irritating to the eyes, nose and throat. Ingestion of formaldehyde is fatal and long term exposure causes respiratory problems and skin irritation.
Another disadvantage is the formation of an impurity (represented by formula VI), which is formed during N-methylation step using formaldehyde as a reagent.

formula VI
As may be appreciated, all the above well-known processes share the same strategy of synthesis, consisting of two steps for the synthesis of venlafaxine from cyano-intermediate or are prepared in one pot with poor yield.
Yet another drawback of the processes disclosed in the prior art is the use of expensive catalysts like rhodium on alumina and use OfBF3 etherate which is highly corrosive.
The prior art references disclose the synthesis of alkoxyphenylethyldimethylamine from alkoxyphenylacetonitrile using excess dimethylamine and palladium catalyst in methanol solution which is firstly reported by Kindler and Hensse. (1. Kindler and Hesse; Arch. Pharm., 1933, 271, 439. 2. Johannes S. Buck, Richard Baltzly and Walter S. Ide; J. Am. Chem. Soc. 1938, 60(8), 1789-1792; 3. Albert J. Schuster and Eugene R. Wagner; J. Labelled Compounds and Radiopharmaceuticals 1992, XXXIII(3), 213-217).
The reaction is as shown in the Scheme-5.

Scheme-5
According to an aspect of the invention there is provided a novel single step process for the synthesis of venlafaxine of formula I and N-desmethylvenlafaxine of formula II from 2-(l- hydroxycyclohexyl)-2-(4-methoxyphenyl)acetonitrile of formula V comprising reaction of 2- (l-hydroxycyclohexyl)-2-(4-methoxyphenyl)acetonitrile with an alkylamine and/or its salt in a solvent in the presence of a transition metal catalyst, under hydrogen atmosphere.

formula I formula II formula V DETAILED DESCRIPTION OF THE INVENTION
The present invention describes a single step process for venlafaxine and its analog starting from the cyano-intermediate. The present invention circumvents the difficulties encountered in the prior art and is an economically viable process.
The present invention particularly relates to a single step synthesis of venlafaxine of formula I, and N-desmethylvenlafaxine of formula II from the cyano-intermediate of formula V.
The reaction of the present invention is as shown in Scheme-4:

formula V formula L R = CH3 (Venlafaxine) formula II. R = H (N-desmethylvenlafaxine)

formula Ia. R = CH3, X = Cl formula Ha. R = H, X = Cl
Example-1:
General procedure for synthesis of venlafaxine and N-desmethylvenlafaxine To the, stirred solution of 2-(l -hydroxy cyclohexyl)-2-(4-methoxyphenyl)acetonitrile (1.0 equiv.) in methanol (10-20 volumes), alkylamine (3-5 equiv.) was added and the mixture was stirred for 5-10 minutes to get a clear solution. Palladium catalyst (10-50 wt %) was added under nitrogen atmosphere to the above reaction mixture. The reaction mixture was purged with hydrogen gas (three times) and allowed to stir under hydrogen (1-2 atmospheric pressure.) at room temperature for 5 to 40 hrs. The progress of the reaction was monitored by TLC and HPLC. After completion of the reaction, the catalyst was filtered through celite and washed with methanol. The combined filtrate was concentrated to dryness under reduced pressure and the residue was poured in water. The aqueous layer was basifϊed with 10% aq. NaOH to pH 8-10 and extracted with ethyl acetate (3 times). The combined ethyl acetate layers were washed with brine and dried over sodium sulphate. Ethyl acetate was evaporated under vacuum to obtain the title compound.
Example-2: Venlafaxine
To the stirred solution of 2-(l-hydroxycyclohexyl)-2-(4-methoxyphenyl)acetonitrile (20.0 g, 1 equiv.) in methanol (460.0 ml), dimethylamine hydrochloride (26.6 g, 4 equiv.) was added and the mixture was stirred for 5-10 minutes at room temperature to obtain a clear solution. 5% Palladium on alumina (4.0 g, 20 wt %) was added under nitrogen to the above clear solution. The reaction mixture was purged with hydrogen gas (three times) and allowed to stir under hydrogen (1-2 atmospheric pressure.) at room temperature for 20 hrs. After completion of the reaction product was isolated by the procedure as described in Example- 1 above to obtain light yellow viscous liquid (18.Og) which was directly converted to its hydrochloride salt using IPA/HC1. HPLC purity of the crude reaction mixture (18.Og) = 81.64%.
Venlafaxine-free base:
1H NMR in CDCl3 (300 MHz): δ 7.05 (d, 2H), 6.81 (d, 2H), 3.79 (s, 3H), 3.27 (t, IH), 2.94 , (dd, IH), 2.31 (s, 6H), 2.28 (dd, IH), 1.71-0.94 (m, 10H). 13C NMR in CDCl3: δ 158.16,’ 132.65, 130.01, 113.20, 74.14, 61.14, 55.05, 51.52, 45.37, 37.99, 31.07, 25.91, 21.52, 21.23.
IR (KBr): 3152, 2980, 2941, 2895, 1728, 1607, 1512, 1462, 1439, 1358, 1279, 1204, 1186, 1177, 1146, 1103, 1040, 1011, 968, 851 cm“1. HPLC Purity: 99.37 % (area %). GC-MS: 178 (M+H+).
Venlafaxine hydrochloride salt:
1H NMR in D2O (300 MHz): δ 7.23 (d, 2H), 6.91 (d, 2H), 3.71 (s, 3H), 3.54 (t, IH), 3.48 (dd, IH), 2.97 (dd, IH), 2.69 (s, 6H), 1.41-1.0 (m, 10H).
13C NMR in D2O: δ 158.52, 130.86, 128.16, 114.25, 73.23, 58.26, 55.25, 50.38, 44.87, 41.41, 35.07, 33.34, 24.76, 21.08, 20.86.
IR (KBr): 3321, 2941, 2928, 2675, 2644, 2623, 2611, 2587, 2521, 2482, 2359, 2330, 1512, 1441, 1242, 1179, 1038, 829 cm“1. HPLC Purity: 99.64 % (area %). GC-MS: 178 (M+H+).
Example-3: N-Desmethyl venlafaxine To the stirred solution of 2-(l -hydroxy cyclohexyl)-2-(4-methoxyphenyl)acetonitrile (20.0 g, 1 equiv.) in methanol (350.0 ml), monomethylamine hydrochloride (27.7 g, 4 equiv.) was added and the mixture was stirred for 5-10 minutes at room temperature to obtain a clear solution. 5% Palladium on alumina (4.0 g, 20 wt %) was added under nitrogen to the above clear solution. The reaction mixture was purged with hydrogen gas (three times) and allowed to stir under hydrogen (1-2 atmospheric pressure.) at room temperature for 24 hrs. After completion of the reaction product was isolated by the procedure as described in Example- 1 above to obtain light yellow viscous liquid (18.5g) which was directly converted to its hydrochloride salt using IPA/HCl.
HPLC conversion to N-desmethylvenlafaxine (in the crude reaction mixture = 18.5g): 35.46%. N-Desmethyl venlafaxine-free base:
1H NMR in CDCl3 (300 MHz): δ 7.23 (d, 2H), £91 (d, 2H), 3.71 (s, 3H), 3.54 (t, IH), 3.48 (dd, IH), 2.97 (dd, IH), 2.69 (s, 3H), 1.41-1.0 (m, 10H)
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2003050074A1 * | Mar 19, 2002 | Jun 19, 2003 | Cadila Healthcare Ltd | Manufacture of venlafaxine hydrochloride and crystalline polymorphs thereof |
| US4535186 * | Oct 26, 1983 | Aug 13, 1985 | American Home Products Corporation | 2-Phenyl-2-(1-hydroxycycloalkyl or 1-hydroxycycloalk-2-enyl)ethylamine derivatives |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2008059525A2 * | Oct 1, 2007 | May 22, 2008 | Calyx Chemicals And Pharmaceut | An improved process for the preparation of venlafaxine and its analogs |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2006035457A1 * | Sep 16, 2005 | Apr 6, 2006 | Amoli Organics Ltd | A process for the manufacture of venlafaxine and intermediates thereof |
| US4535186 * | Oct 26, 1983 | Aug 13, 1985 | American Home Products Corporation | 2-Phenyl-2-(1-hydroxycycloalkyl or 1-hydroxycycloalk-2-enyl)ethylamine derivatives |
| US6350912 * | Feb 28, 2001 | Feb 26, 2002 | Council Of Scientific And Industrial Research | One pot process for the preparation of 1-[2-dimethylamino-(4-methoxyphenyl)-ethyl]cyclohexanol |
| US20050033088 * | Jun 7, 2004 | Feb 10, 2005 | Dr. Reddy’s Laboratories Limited | Catalytic hydrogenation of phenylacetonitrile using palladium on carbon supports |
| Reference | ||
|---|---|---|
| 1 | * | CHAVAN S P ET AL: “An efficient and green protocol for the preparation of cycloalkanols: a practical synthesis of venlafaxine” TETRAHEDRON LETTERS, ELSEVIER, AMSTERDAM, NL, vol. 45, no. 39, 20 September 2004 (2004-09-20), pages 7291-7295, XP004558985 ISSN: 0040-4039 |
| 2 | * | J.S. BUCK ET AL: “beta-Phenylethylamine Derivatives. Tertiary and quaternary salts” J.AM.CHEM.SOC., 1938, pages 1789-1792, XP002478651 cited in the application |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2010100520A1 * | Mar 4, 2009 | Sep 10, 2010 | Hikal Limited | A process for preparation of phenethylamine derivative |
| WO2011124190A2 | Apr 6, 2011 | Oct 13, 2011 | Zentiva, K.S. | Method of producing 4-(2-(substituted)-1-(1-hydroxycyclohexyl)ethyl)phenols by o- demethylation of their methylethers by means of inodorous aromatic thiols |



Chernivtsi central square

KIEV

Independence Square (Maidan Nezalezhnosty)

DONETSK
COCK WILL TEACH YOU NMRCOCK SAYS MOM CAN TEACH YOU NMRDRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE amcrasto@gmail.com
: The views expressed are my personal and in no-way suggest the views
of the professional body or the company that I represent.
: The views expressed are my personal and in no-way suggest the views
of the professional body or the company that I represent.
: The views expressed are my personal and in no-way suggest the views
of the professional body or the company that I represent.
COCK WILL TEACH YOU NMRCOCK SAYS MOM CAN TEACH YOU NMR
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE