MK 2048
Molecular Formula: C21H21ClFN5O4 Molecular Weight: 461.873943
869901-69-9, 3oyl, 3oyn
Merck & Co., Inc.
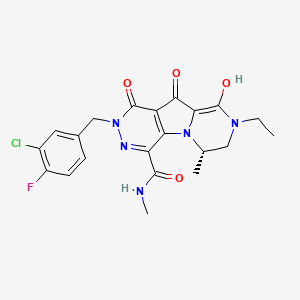
| (6S)-2-(3-chloro-4-fluorobenzyl)-8-ethyl-10-hydroxy-N,6-dimethyl-1,9-dioxo-1,2,6,7,8,9-hexahydropyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyridazine-4-carboxamide |
6(S)-2-(3-Chloro-4-fluorobenzyl)-8-ethyl-10-hydroxy-N,6-dimethyl-l,9-dioxo-l,2,6,7,8,9- hexahydropyrazino[r,2′:l,5]pyrrolo[2,3-d]pyridazine-4-carboxamide
|
|
5-27-2009
|
Hiv Integrase Inhibitors
|
MK-2048 is a second generation integrase inhibitor, intended to be used against HIV infection. It is superior to the first available integrase inhibitor,raltegravir, in that it inhibits the HIV enzyme integrase 4 times longer. It is being investigated for use as part of pre-exposure prophylaxis (PrEP). [1]
It is being developed by Merck & Co.[2]

MK-2048 is a second generation integrase inhibitor for HIV-1 integrase. MK-2048 inhibits subtype B and subtype C integrase activities. MK-2048 inhibits R263K mutants slightly more effectively than G118R mutants.
MK-2048 inhibits S217H intasome and, by contrast, MK2048 remains fully active against the N224H intasome. MK2048 displays substantially lower dissociation rates compared with raltegravir, another integrase inhibitor.
MK-2048 is active against viruses resistant to RAL and EVG. MK-2048 exposure leads to the selection of G118R as a possible novel resistance mutation after 19 weeks. MK-2048, with continued pressure, subsequently leads to an additional substitution, at position E138K, after 29 weeks, within the IN gene.
Although the G118R mutation alone confers only slight resistance to MK-2048 but not to RAL or EVG, its presence arouses a dramatic reduction in viral replication capacity compared to wild-type NL4-3. E138K both partially restores viral replication capacity and also contributes to increased levels of resistance against MK-2048.

Structure of MK-2048 with important pharmacophore highlighted


…………………..
Synthesis
WO2005110415A1
http://www.google.as/patents/WO2005110415A1?cl=en
EXAMPLE 62 6(S)-2-(3-Chloro-4-fluorobenzyl)-8-ethyl-10-hydroxy-N,6-dimethyl-l,9-dioxo-l,2,6,7,8,9- hexahydropyrazino[r,2′:l,5]pyrrolo[2,3-d]pyridazine-4-carboxamide
Step 1: te rt-Butyl[( 1 S)-2-(ethylamino)- 1 -methylethyl] carbamate To a cold (0 °C) solution of N-(tø/ -butoxycarbonyl)-L-alanine N’-methoxy-N’- methylamide (15.6 g, 67.2 mmol) in anhydrous THF (150 mL) and diethyl ether (400 mL), solid lithium aluminum hydride (5.1 g, 134.3 mmol) was added portionwise over a period of 30 minutes. The mixture was stirred at room temperature for 3 hours and cooled back to 0 °C. The reaction was treated carefully with an aqueous solution of potassium hydrogen sulfate (250 mL, 1M). The resultant mixture was diluted with diethyl ether.
The organic extract was washed successively with dilute hydrochloric acid, and brine, dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum to provide the corresponding aldehyde as colorless solid. Without further purification, a cold (0 °C), stirred solution of the intermediate aldehyde (10.7 g, 61.8 mmol) and ethylamine hydrogen chloride (10.1 g, 123.5 mmol) in methanol (72 mL) was treated with sodium triacetoxyborohydride (17.2 g, 80.9 mmol) in one portion. The mixture was allowed to warm up to room temperature.
After stirring at room temperature overnight, the solution was concentrated under vacuum. The residue was partitioned between diethyl ether and cold aqueous sodium hydroxide (1.5 M). The ethereal extract was washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum to provide the titled compound. lH NMR (400 MHz, CDCI3) δ 4.68 (br s, IH), 3.75 (br t, IH), 2.62 (m, 5 H), 1.13 (d, J = 6.7 Hz, 3H),
1.09 (t, J = 7.0 Hz, 3H). ES MS M+l = 203
Step 2: ført-Butyl { ( 1 S)-2-[(bromoacetyl)ethylamino] – 1 -methylethyl } carbamate To a cold (0 °C) stirred solution of ?ert-butyl[(lS)-2-(ethylamino)-l- methylethyl]carbamate (11.0 g, 54.6 mmol) in a mixture of ethyl acetate (107 mL) and saturated aqueous sodium bicarbonate (65 mL), bromoacetyl bromide (12.1 g, 60.0 mmol) was added portionwise under an atmosphere of nitrogen. The mixture was allowed to warm up to room temperature over a period of 3.5 hours. The organic phase was separated, washed successively with saturated aqueous sodium bicarbonate, and brine. The organic extract was dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum. The residue was concentrated as a solution in toluene under vacuum to afford the title compound. ES MS M+l = 323, 325.
Step 3: fe7 -Butyl (2S)-4-ethyl-2-methyl-5-oxopiperazine-l-carboxylate To a stirred slurry of sodium hydride (1.7 g, 69.8 mmol) in anhydrous THF (800 mL), a solution of tert-butyl{(lS)-2-[(bromoacetyl)ethylamino]-l-methylethyl}carbamate (17.4 g, 53.7 mmol) in anhydrous THF (100 mL) was added dropwise over a period of 1 hour under an atmosphere of nitrogen. The reaction mixture was stirred at room temperature for two hours, cooled in an ice-water bath, and quenched with dropwise addition of aqueous citric acid (80 mL, 1M). The mixture was concentrated under vacuum. The residue was partitioned between chloroform and saturated aqueous sodium bicarbonate. The organic extract was dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum. The residue was subjected to column chromatography on silica gel eluting with a gradient of 0-15% acetonitrile in chloroform. Collection and concentration of appropriate fractions provided the title compound. lH NMR (400 MHz, CDCI3) δ 4.46 (br s, IH), 4.24 (d, J = 18.4 Hz, 1 H), 3.78 (d, J = 18.4 Hz, 1 H),
3.64 (dd, J = 12.3, 4.2 Hz, 1 H), 3.54 (heptet, J = 7.1 Hz, 1 H), 3.38 (heptet, J = 7.1 Hz, 1 H), 2.99 (dd, J = 12.3, 1.8 Hz, 1 H), 1.47 (s, 9H), 1.21 (d, J = 6.8 Hz, 3H), 1.14 (t, J = 7.1 Hz, 3H). ES MS M+l = 243.
Step 4: (5S)-l-Ethyl-5-methylpiperazin-2-one hydrochloride Anhydrous hydrogen chloride gas was bubbled into a cold (-20 °C) solution of tert-butyl (2S)-4-ethyl-2-methyl-5-oxopiperazine-l-carboxylate (10.5 g, 43.4 mmol) in ethyl acetate (250 mL) under nitrogen. After the solution was saturated with hydrogen chloride, the reaction mixture was stirred in an ice-water bath for 30 minutes. The product mixture was purged with nitrogen, concentrated under vacuum to provide the title hydrogen chloride salt as pale yellow solid. lH NMR (400 MHz, DMSO-d6) δ 10.00 (br d, 2H), 3.72 (d, J = 16.6 Hz, 1 H), 3.62(d, J = 16.6 Hz, 1 H),
3.49-3.35 (m, 5 H), 3.29 (heptet, /= 7.3 Hz, 1 H), 1.31 (d, / = 6.6 Hz, 3H), 1.05 (t, J = 7.1 Hz, 3H).
Step 5: Ethyl (4S)-2-ethyl-8-hydroxy-4-methyl-l-oxo-l,2,3,4-tetrahydropyrrolo[l,2-a]pyrazin-7- carboxy late Anhydrous ammonia gas was bubbled into a cold (0 °C) solution of (5S)-l-Ethyl-5- methylpiperazin-2-one hydrochloride (5.8 g, 32.3 mmol) in chloroform for 30 minutes. The resultant slurry was filtered and concentrated under vacuum. The residual oil was concentrated as a solution in toluene under vacuum, redissolved in toluene (120 mL) and treated with diethyl ethoxymethylenemalonate (7.0 g, 32.3 mmol) and heated in a sealed flask in an oil bath at 100 °C overnight. The resultant solution was concentrated under vacuum. The residual oil was concentrated as a solution in toluene under vacuum to provide the corresponding diethyl { [(2S)-4-ethyl-2-methyl-5- oxopiperazin-l-yl]methylene}malonate. Without further purification, to a solution of the malonate (10.5 g, 33.5 mmol) in anhydrous THF (330 mL) warmed with an external oil bath at 65 °C under an atmosphere of nitrogen, a solution of lithium bis(trimethylsilyl)amide (35.1 mL, 1 M, 35.1 mmol) was added. The solution was heated at the same temperature for one hour and concentrated under vacuum. The residue was partitioned between dichloromethane and hydrochloric acid (1M). The organic extract was washed with brine, dried over anhydrous magnesium sulfate, filtered, and concentrated under vacuum. The residue was triturated with diethyl ether. The solid precipitated was filtered, washed with diethyl ether to provide the title compound as pale brown solid. lH NMR (400 MHz, CDCI3) δ 8.43 (s, IH), 7.11 (s, IH), 4.32 (q, J = 7.1 Hz, 2H), 4.24 (m, IH), 3.65-
3.35 (m, 4H), 1.51 (d, J = 6.4 Hz, 3H), 1.36 (t, J = 7.0 Hz, 3H), 1.19 (t, J = 7.0 Hz, 3H). ES MS M+l = 267
Step 6: Ethyl (4S)-2-ethyl-8-methoxy-4-methyl-l-oxo-l,2,3,4-tetrahydropyrrolo[l,2-a]pyrazin-7- carboxylate A mixture of ethyl (4S)-2-ethyl-8-hydroxy-4-methyl-l -oxo- 1,2,3, 4-tetrahydropyrrolo[ 1,2- a]pyrazin-7-carboxylate (6.6 g, 24.8 mmol), anhydrous potassium carbonate (13.7 g, 99.1 mmol, 325 mesh), and iodomethane (4.2 g, 29.7 mmol) in anhydrous DMF (123 mL) was stirred at room temperature overnight. The mixture was filtered and concentrated under vacuum. The residue was partitioned between chloroform and dilute hydrochloric acid. The organic extract was washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum. The residue was subjected to column chromatography on silica gel eluting with a gradient of 0-3% methanol in chloroform. Collection and concentration of appropriate fractions provided the title compound. Residual methanol was removed by concentrating from its solution in toluene under vacuum. lH NMR (400 MHz, CDCI3) δ 7.19 (s, IH), 4.29 (q, J = 7.1 Hz, 2 H), 4.24 (m, IH), 4.03 (s, 3H), 3.70-
3.32 (m, 4 H), 1.52 (d, J = 6.6 Hz, 3H), 1.35 (t, J = 7.0 Hz, 3H), 1.19 (t, J = 7.2 Hz, 3H). ES MS M+l = 281
Step 7: Ethyl (4S)-6-bromo-2-ethyl-8-methoxy-4-methyl-l-oxo-l,2,3,4-tetrahydropyrrolo[l,2- a]pyrazin-7-carboxylate To a mixture of ethyl (4S)-2-ethyl-8-(methoxy)-4-methyl-l-oxo-l,2,3,4- tetrahydropyrrolo[l,2- ]pyrazine-7-carboxylate (6.2 g, 22.1 mmol) and sodium bicarbonate (20.0 g, 238.0 mmol) in dichloromethane (500 mL) at 0 °C, a solution of bromine in dichloromethane (24.2 mmol, 0.5 M) was added over a period of 60 minutes. The reaction mixture was stirred at room temperature for 2 h, filtered, and concentrated under vacuum. The residue was subjected to column chromatography on silica gel eluted with ethyl acetate. Collection and concentration of appropriate fractions provided the corresponding bromide. Residual ethyl acetate was removed by concentrating from its solution in benzene under vacuum. lH NMR (400 MHz, CDCI3) δ 4.58 (br m, IH), 4.34 (m, IH), 3.99 (s, 3H), 3.92 (dd, J = 13.0, 4.0 Hz,
IH), 3.67 (heptet, J = 7.1 Hz, 1 H), 3.49 (heptet, J = 7.1 Hz, 1 H), 3.23 (d, J = 13.0 Hz, IH), 1.40 (d, J = 7.1 Hz, 3H), 1.38 (t, 7 = 7.0 Hz, 3H), 1.20 (t, J = 7.0 Hz, 3H). ES MS M+l = 359, 361.
Step 8: Ethyl (4S)-2-ethyl-8-(methoxy)-6-[methoxy(oxo)acetyl]-4-methyl-l-oxo-l,2,3,4- tetrahydropyrrolo[ 1 ,2- ]pyrazine-7-carboxylate To a cold (-78 °C) solution of ethyl (4S)-6-bromo-2-ethyl-8-methoxy-4-methyl-l-oxo- l,2,3,4-tetrahydropyrrolo[l,2-a]pyrazin-7-carboxylate (8.51 g, 23.7 mmol) in anhydrous THF (800 mL) under an atmosphere of dry nitrogen, a solution of n-BuLi in hexane (10.5 mL, 26.3 mmol, 2.5 M) was added. The resultant mixture was stirred at -78 °C for 20 minutes. A solution of dimethyl oxalate (6.4 g, 53.8 mmol; dried from concentration from benzene under vac) in anhydrous THF (30 mL) was added. The reaction mixture was stirred at -78 °C for 1 hour and cannulated into a mixture of aqueous sulfuric acid (240 mL, 2M) and THF (200 mL) maintained between at -5 to -35 °C. The mixture was extracted with ethyl acetate (3 times). The organic extracts were combined, washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under vacuum. The residue was subjected to column chromatography on silica gel eluted with 40 to 100% ethyl acetate- hexane gradient. Collection and concentration of appropriate fractions provided the titled compound. lH NMR (400 MHz, CDCI3) δ 5.07 (m, IH), 4.29 (q, J = 7.2 Hz, 2H), 4.00 (s, 3H), 3.99-3.93 (m, IH), 3.89 (s, 3H), 3.74-3.66 (m, IH), 3.53-3.48 (m, IH), 3.23 (dd, J = 1.3, 13.2 Hz, IH), 1.46 (d, J = 6.6 Hz, 3H), 1.36 (t, J = 7.2 Hz, 3H), 1.22 (t, 7= 7.1 Hz, 3H). ES MS M+l = 367
Step 9: (6S)-8-Ethyl-10-methoxy-6-methyl-l,9-dioxo-l,2,6,7,8,9- hexahydropyrazino[r,2′:l,5]pyrrolo[2,3-d]pyridazine-4-carbohydrazide A mixture of ethyl (4S)-2-ethyl-8-(methoxy)-6-[methoxy(oxo)acetyl]-4-methyl-l-oxo- l,2,3,4-tetrahydropyrrolo[l,2-α]pyrazine-7-carboxylate (3.3 g, 8.9 mmol) and anhydrous hydrazine (1.7 mL, 53.7 mmol) in methanol (400 mL) was stirred at room temperature for one hour. The reaction mixture was concentrated under vacuum. The residue was concentrated from toluene. The resultant gummy solid was treated with methanol (20 mL). Diethyl ether was added to the resultant slurry which was filtered to provide the title compound as white solid. lH NMR (400 MHz, CDCI3) δ 8.99 (br s, 2H), 5.54 (br m, IH), 4.12 (m, IH), 4.10 (s, 3H), 3.81 (m, IH),
3.39 (m, IH), 3.21 (d, 7 = 12.6 Hz, IH), 1.44 (d, 7 = 6.4 Hz, 3H), 1.23 (t, 7 = 7.3 Hz, 3H). ES MS M+l =
335
Step 10: (6S)-8-Ethyl-10-methoxy-N,6-dimethyl-l,9-dioxo-l,2,6,7,8,9- hexahydropyrazino[r,2′:l,5]pyrrolo[2,3-d]pyridazine-4-carboxamide To a solution of (6S)-8-ethyl-10-methoxy-6-methyl-l,9-dioxo-l,2,6,7,8,9- hexahydropyrazino[r,2′:l,5]pyrrolo[2,3-d]pyridazine-4-carbohydrazide (0.39 g, 1.2 mmol) and methylamine (5.9 mL, 11.8 mmol; 2 M in THF) in anhydrous dichloromethane (25 mL) in a water bath at room temperature, a solution of iodine (0.60 g, 2.4 mmol) in dichloromethane was added dropwise.
After the addition was completed, an aqueous solution of sodium sulfite was added and the mixture was stirred vigorously for 10 minutes. The organic phase was separated, diluted with chloroform, and washed with brine. The organic extract was dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum. The residue was triturated with a mixture of ethanol (7 mL) and diethyl ether (25 mL). The white solid precipitated was obtained by filtration and dried from its solution in toluene under vacuum. 1H NMR (400 MHz, CDCI3) δ 11.57 (s, IH), 7.38 (m, IH), 5.95 (br m, IH), 4.17 (s, 3H), 4.03 (dd, 7 =
13.4, 3.8 Hz, 1 H), 3.76 (heptet, 7 = 7.1 Hz, 1 H), 3.50 (heptet, 7 = 7.1 Hz, 1 H), 2.99 (dd, 7 = 12.9, 1.0 Hz, 1 H), 3.03 (d, 7 = 5.0 Hz, 3H), 1.44 (d, 7 = 6.6 Hz, 3H), 1.23 (t, 7 = 7.2 Hz, 3H). ES MS M+l = 334 Step 11: (6S)-2-(3-Chloro-4-fluorobenzyl)-8-ethyl-10-methoxy-N,6-dimethyl-l,9-dioxo- l,2,6,7,8,9-hexahydropyrazino[r,2′: l,5]pyrrolo[2,3-d]pyridazine-4-carboxamide To a cold (0 °C) solution of (6S)-8-ethyl-10-methoxy-N,6-dimethyl-l,9-dioxo- l,2,6,7,8,9-hexahydropyrazino[l’,2′: l,5]pyrrolo[2,3-d]pyridazine-4-carboxamide (1.58 g, 4.73 mmol) in anhydrous DMF (50 mL), a solution of lithium bis(trimethylsilyl)amide (4.97 mL, 4.97 mmol, 1 M in THF) was added. After stirring at the same temperature for 25 minutes, 3-chloro-4-fluorobenzyl bromide (1.27 g, 5.68 mmol) was added. The reaction mixture was stirred at room temperature for 10 minutes and concentrated under vacuum. The residue was partitioned between chloroform and brine. The organic extract was dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum. The residue was subjected to column chromatography on silica gel eluting with a 1-5% methanol in ethyl acetate gradient. Collection and concentration of appropriate fractions provided the title compound. lH NMR (400 MHz, CDCI3) δ 7.46 (dd, 7 = 6.9, 2.2 Hz, IH), 7.32 (m, IH), 7.09 (t, 7 = 7.6 Hz, IH), 7.03
(br signal, IH), 5.92 (m, IH), 5.32 (d, 7 = 14.1 Hz, IH), 5.26 (d, 7= 14.1 Hz, IH), 4.14 (s, 3H), 3.97 (dd, 7 = 13.2, 3.7 Hz, IH), 3.73 (heptet, 7 = 7.2 Hz, 1 H), 3.51 (heptet, 7 = 7.1 Hz, IH), 3.21 (dd, 7= 13.2, 1.7 Hz, IH), 3.03 (d, 7 = 5.0 Hz, 3H), 1.42 (d, 7 = 6.6 Hz, 3H), 1.23 (t, 7 = 7.1 Hz, 3H). ES MS M+l = 476
Step 12:
(6S)-2-(3-Chloro-4-fluorobenzyl)-8-ethyl-10-hydroxy-N,6-dimethyl-l,9-dioxo- l,2,6,7,8,9-hexahydropyrazino[r,2′:l,5]pyrrolo[2,3-d3pyridazine-4-carboxamide
To a solution of (6S)-2-(3-chloro-4-fluorobenzyl)-8-ethyl-10-methoxy-N,6-dimethyl-l,9- dioxo-l,2,6,7,8,9-hexahydropyrazino[r,2′:l,5]pyrrolo[2,3-d]pyridazine-4-carboxamide (1.15 g, 2.41 mmol) in anhydrous dichloromethane (800 mL), a solution of boron tribromide in dichloromethane (3.14 mL, 3.14 mmol; 1 M) was added. After stirring at room temperature for 5 minutes, the reaction mixture was treated with anhydrous methanol, stirred for 30 minutes, and concentrated under vacuum. The procedure was repeated twice. The residue was dissolved in a mixture of methanol and acetonitrile and treated with aqueous sodium hydroxide. The mixture was subjected to purification on preparative reverse phase high pressure column chromatography. Collection and lyophilization of appropriate fractions provided the title compound as white amorphous solid.
MK 2048
lH NMR (400 MHz, CDCI3) δ 7.48 (dd, 7 = 7.0, 2.2 Hz, IH), 7.33 (m, IH), 7.09 (t, 7 = 8.7 Hz, IH), 6.01 (m, IH), 5.33 (d, 7= 14.1 Hz, IH), 5.27 (d, 7 = 14.1 Hz, IH), 3.99 (dd, 7= 12.8, 4.0 Hz, 1 H), 3.71(heptet, 7 = 7.1 Hz, 1 H), 3.49 (heptet, 7 = 7.1 Hz, 1 H), 3.24 (dd, 7 = 13.2, 1.5 Hz, 1 H), 3.03 (d, 7 = 5.1 Hz, 3H), 1.42 (d, 7 = 6.6 Hz, 3H), 1.24 (t, 7 = 7.3 Hz, 3H). ES MS M+l = 462
The amorphous product was dissolved in boiling methanol (1.4 g/200 mL). Upon cooling in an ice-water bath, a precipitate formed which was separated by obtained by filtration to afford a white crystalline solid.
MK 2048sodium salt
The corresponding sodium salt was prepared by treatment of a solution of (6S)-2-(3- chloro-4-fluorobenzyl)-8-ethyl- 10-hydroxy-N,6-dimethyl-l ,9-dioxo- 1 ,2,6,7,8,9- hexahydropyrazino[r,2′:l,5]pyrrolo[2,3-d]pyridazine-4-carboxamide (920 mg, 1.99 mmol) in aqueous acetonitrile with aqueous sodium hydroxide (1.03 equivalent), followed by lyophilization of the resultant solution.

(5S)-1-ethyl-5-methylpiperazin-2-one
 1,5-Cyclooctadiene-iridium(I) chloride dimer, Chloro(1,5-cyclooctadiene)iridium(I) dimer, Di-μ-chlorobis[(1,2,5,6-η)-1,5-cyclooctadiene]diiridium, Iridium(I) chloride 1,5-cyclooctadiene complex dimer, [Ir(1,5-cod)Cl]2, [Ir(1,5-cod)Cl]2, [Ir(cod)Cl]2
1,5-Cyclooctadiene-iridium(I) chloride dimer, Chloro(1,5-cyclooctadiene)iridium(I) dimer, Di-μ-chlorobis[(1,2,5,6-η)-1,5-cyclooctadiene]diiridium, Iridium(I) chloride 1,5-cyclooctadiene complex dimer, [Ir(1,5-cod)Cl]2, [Ir(1,5-cod)Cl]2, [Ir(cod)Cl]2

(S)-1-[(R)-2-Di-(4-methoxy-3,5-dimethylphenyl-phosphino)ferrocenyl]-ethyl-dicyclohexylphosphine
SL-J006-2
(5S)-l-Ethyl-5-methylpiperazin-2-one was alternatively prepared as follows:
Step 1: N^rf-Butoxycarbonyl-N^ethylglycinamide Ethylamine (37 g, 0.82 mol) was condensed into a pressure vessel at 0 °C. N-(tert- butoxycarbonyl)glycine methyl ester (50 mL, 0.34 mol) was added. The vessel was sealed and the mixture was stirred at room temperature overnight. The product mixture was concentrated under vacuum and the residue was passed through a pad of silica gel eluting with ethyl acetate. The solution was concentrated under vacuum to provide the title compound as a clear oil. lH NMR (400 MHz, CDCI3) δ 6.11 (br s, IH), 5.18 (br s, IH), 3.77 (d, 7 = 5.7 Hz, 2H), 3.31 (q, 7 = 7.1
Hz, 2H), 1.15 (t, 7 = 7.1 Hz, 3H).
Step 2: l-Ethyl-5-methylpyrazin-2(lH)-one A cold (0 °C) solution of N^tø^butoxycarbonyl-N^ethylglycinamide (68.0 g, 0.33 mol) in anhydrous dichloromethane (500 mL) was saturated with anhydrous hydrogen chloride gas. After stirring at the same temperature for 1.5 hours, the solution was recharged with more hydrogen chloride gas and stirred for additional 15 minutes. The reaction mixture was concentrated under vacuum. The residue was dissolved in methanol, diluted with toluene, and concentrated under vacuum to afford the intermediate N-ethylglycinamide HCI salt.
This was stored under vacuum overnight and used without further purification. A solution of N-ethylglycinamide HCI salt (44.2 g, 0.32 mol), aqueous sodium hydroxide (640 mL, 1M), water (350 mL), pyruvic aldehyde (20.9 mL, 40% solution in water) was heated in an oil bath at 120 °C for one hour. The reaction mixture was cooled and saturated with solid sodium chloride. The mixture was extracted with chloroform (4×250 mL).
The combined organic extract was dried over anhydrous sodium sulfate, filtered, and passed through a plug of silica gel. The silica gel was rinsed successively with ethyl acetate and then 2% methanol in ethyl acetate. The eluted fractions were combined and concentrated under vacuum. The residual solid was recrystallized from diethyl ether to afford the title compound as pale yellow solid. lH NMR (400 MHz, CDCI3) δ 8.11 (s, IH), 6.92 (s, IH), 3.92 (q, 7 = 7.2 Hz, 2H), 2.28 (s, 3H), 1.37 (t, 7 = 7.2 Hz, 3H).
Step 3: (5 S)- 1 -Ethyl-5-methylpiperazin-2-one
A mixture of chloro-l,5-cyclooctadiene iridium (I) dimer (34 mg, 51 μmol) and (S)-l-[(R)-2-di-(3,5-bis(trifluoromethyl)phenyl)phosphino)ferrocenyl]ethyldicyclohexylphosphine (44 mg, 51 μmol; Solvias AG, SL-J006-2) in a mixture of 1:2 toluene and methanol (100 mL; purged with nitrogen for 15 minutes) was sonicated under an atmosphere of nitrogen for 15 minutes. To the resultant mixture, iodine (0.39 g, 1.52 mmol) and l-ethyl-5-methylpyrazin-2(lH)-one (7.0 g, 50.66 mmol) was added. The resultant mixture was heated in an oil bath at 50 °C under an atmosphere of hydrogen gas at 800 psi for 48 hours. The product mixture was filtered through a pad of Celite. The filtrate was concentrated under vacuum. The residue was treated with chloroform saturated with ammonia gas (100 mL). The resultant suspension was filtered through a pad of Celite, which was the rinsed with chloroform saturated with ammonia gas. The combined filtrate was concentrated under vacuum. The residue was concentrated as a solution in toluene for subsequent reaction. lH NMR (400 MHz, CDCI3) δ 3.58 (d, 7 = 17.2 Hz, IH), 3.53(d, 7 = 17.2 Hz, IH), 3.49-3.35 (m, 2H),
1.19 (d, 7 = 5.9 Hz, 3H), 1.14 (t, 7 = 7.2 Hz, 3H).
……………..
US 7538112
http://www.google.com/patents/US7538112
Step 12: (6S)-2-(3-Chloro-4-fluorobenzyl)-8-ethyl-10-hydroxy-N,6-dimethyl-1,9-dioxo-1,2,6,7,8,9-hexahydropyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyridazine-4-carboxamide
To a solution of (6S)-2-(3-chloro-4-fluorobenzyl)-8-ethyl-10-methoxy-N,6-dimethyl-1,9-dioxo-1,2,6,7,8,9-hexahydropyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyridazine-4-carboxamide (1.15 g, 2.41 mmol) in anhydrous dichloromethane (800 mL), a solution of boron tribromide in dichloromethane (3.14 mL, 3.14 mmol; 1 M) was added. After stirring at room temperature for 5 minutes, the reaction mixture was treated with anhydrous methanol, stirred for 30 minutes, and concentrated under vacuum. The procedure was repeated twice. The residue was dissolved in a mixture of methanol and acetonitrile and treated with aqueous sodium hydroxide. The mixture was subjected to purification on preparative reverse phase high pressure column chromatography. Collection and lyophilization of appropriate fractions provided the title compound as white amorphous solid.
1H NMR (400 MHz, CDCl3) δ 7.48 (dd, J=7.0, 2.2 Hz, 1H), 7.33 (m, 1H), 7.09 (t, J=8.7 Hz, 1H), 6.01 (m, 1H), 5.33 (d, J=14.1 Hz, 1H), 5.27 (d, J=14.1 Hz, 1H), 3.99 (dd, J=12.8, 4.0 Hz, 1 H), 3.71 (heptet, J=7.1 Hz, 1 H), 3.49 (heptet, J=7.1 Hz, 1 H), 3.24 (dd, J=13.2, 1.5 Hz, 1 H), 3.03 (d, J=5.1 Hz, 3H), 1.42 (d, J=6.6 Hz, 3H), 1.24 (t, J=7.3 Hz, 3H). ES MS M+1=462
The amorphous product was dissolved in boiling methanol (1.4 g/200 mL). Upon cooling in an ice-water bath, a precipitate formed which was separated by obtained by filtration to afford a white crystalline solid.
The corresponding sodium salt was prepared by treatment of a solution of (6S)-2-(3-chloro-4-fluorobenzyl)-8-ethyl-10-hydroxy-N,6-dimethyl-1,9-dioxo-1,2,6,7,8,9-hexahydropyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyridazine-4-carboxamide (920 mg, 1.99 mmol) in aqueous acetonitrile with aqueous sodium hydroxide (1.03 equivalent), followed by lyophilization of the resultant solution.
References
- Keith Alcorn. Ralvetgravir shows potential for use as PrEP drug AIDSmap.com. 28 April 2009. Accessed 8 Nov 2009.
- Mark Mascolini. Merck Offers Unique Perspective on Second-Generation Integrase Inhibitor. 10th International Workshop on Clinical Pharmacology of HIV Therapy, April 15–17, 2009, Amsterdam. Accessed 8 Nov 2009.
| WO2011121105A1 |
1 Apr 2011 |
6 Oct 2011 |
Tibotec Pharmaceuticals |
Macrocyclic integrase inhibitors |
| EP1756114A2 * |
3 May 2005 |
28 Feb 2007 |
Merck and Co., Inc. |
Hiv integrase inhibitors |
| US7517929 |
3 Dec 2004 |
14 Apr 2009 |
Momentive Performance Materials Inc. |
Star-branched silicone polymers as anti-mist additives for coating applications |
| US5693640 * |
6 Jun 1995 |
2 Dec 1997 |
Merck, Sharp & Dohme, Ltd. |
Pyridazino-indole derivatives |
| US5756501 * |
3 Dec 1996 |
26 May 1998 |
American Home Products Corporation |
Saturated and unsaturated pyridazino 4,5-B! indolizines useful as antidementia agents |

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....













 ACH 702
ACH 702