
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
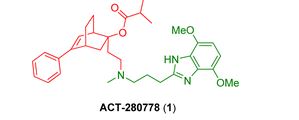
ACT-280778
(1R,2R,4R)-2-(2-((3-(4,7-Dimethoxy-1H-benzo[d]imidazol-2-yl)propyl)(methyl)amino)ethyl)-5-phenylbicyclo[2.2.2]oct-5-en-2-yl Isobutyrate
Propanoic acid, 2-methyl-, (1R,2R,4R)-2-[2-[[3-(4,7-dimethoxy-1H-benzimidazol-2-yl)propyl]methylamino]ethyl]-5-phenylbicyclo[2.2.2]oct-5-en-2-yl ester
isobutyric acid (1R,2R,4R)-2-(2-{[3-(4,7-dimethoxy-1H-benzoimidazol-2-yl)-propyl]-methyl-amino}-ethyl)-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl ester
Actelion Pharmaceuticals Ltd innovator
C33 H43 N3 O4
1075744-31-8
bis-MALEATE SALT 1537197-53-7
Chiral bicyclic benzimidazole 1 (ACT-280778) is a L/T calcium channel blocker potentially indicated for the treatment of hypertension and angina pectoris
Many cardiovascular disorders have been associated with a ‘calcium overload’ resulting from an abnormal elevated calcium influx through the plasma membrane of cardiac and vascular smooth muscle cells. There are 3 major pathways through which extracellular calcium can enter these cells: 1 ) receptor-activated calcium channels, 2) ligand-gated calcium channels and 3) voltage-operated calcium channels (VOCs). 0 VOCs have been classified into 6 main categories: L (Long-lasting), T (Transient), N (Neuronal), P (Purkinje cells), Q (after P) and R (Remaining or Resistant).
L-type calcium channels are responsible for the inward movement of calcium that initiates contraction in cardiac and smooth muscle cells suggesting a putative application for blockers of these channels in the cardiovascular field. In this view, L-type calcium channel blockers5 have been used in clinic since the early 60s and are now recommended as a first line of treatment for systolic-diastolic hypertension and angina pectoris.
T-type calcium channels are found in various tissues such as coronary and peripheral vasculature, sinoatrial node and Purkinje fibres, brain, adrenal glands and in the kidney. This broad distribution suggests a T-type channel blocker to have a putative cardiovascular0 protection, to have en effect on sleep disorders, mood disorders, depression, migraine, hyperaldosteroneemia, preterm labor, urinary incontinence, brain aging or neurodegenerative disorders such as Alzheimers disease.
Mibefradil (Posicor®), the first L-type and T-type calcium channels blocker demonstrated a superior effect over calcium channel blockers, which target the L channel predominantly. Mibefradil was used for the treatment of hypertension and angina without showing negative side-effects often seen by L channel blockers like inotropy, reflex tachycardia, vasoconstrictive hormone release or peripheral edema. Additionally, mibefradil showed a potentially cardioprotective effect (Villame, Cardiovascular Drugs and Therapy 15, 41-28, 2001 ; Ramires, J MoI Cell Cardiol 1998, 30, 475-83), a renal protective effect (Honda, Hypertension 19, 2031-37, 2001 ), and showed a positive effect in the treatment of heart failure (Clozel, Proceedings Association American Physicians 1999, 1 11 , 429-37).
Despite the enormous demand for a compound of this profile, mibefradil was withdrawn from the market in 1998 (one year after its launch), due to unacceptable CYP 3A4 drug interactions. Moreover, ECG abnormalities (i.e. QT prolongations) and interaction with the MDR-1 mediated digoxin efflux were also reported (du Souich, Clin Pharmacol Ther 67, 249- 57, 2000; Wandel, Drug Metab Dispos 2000, 28, 895-8).
It has now been found that crystalline salt forms of COMPOUND (isobutyric acid (1 R*,2R*,4R*)-2-(2-{[3-(4,7-dimethoxy-1 H-benzoimidazol-2-yl)-propyl]-methyl-amino}-ethyl)- 5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl ester) may under certain conditions be found. Said crystalline salt forms of COMPOUND are novel and may have advantageous properties, especially compared to the free base (WO2008/132679) or the di-hydrochloride salt of COMPOUND. Such advantages may include better flow properties, better solubility, less hygroscopicity, better reproducibiliy in manufacturing (for example better filtration parameters, better reproducibility of formation, better sedimentation),
………………………
http://www.google.com/patents/WO2009130679A1?cl=en
Scheme 1


……………
http://www.google.com/patents/CN102186828A?cl=en
http://www.google.com/patents/EP2344461A1?cl=en
The preparation of COMPOUND is known from WO2008/132679: Preparation of intermediates
General procedures for the preparation of key intermediates K: Key intermediates K1A and K2A which are bicyclo[2.2.2]oct-5-en-2-yl or bicyclo[3.2.2]non-8- en-6-yl derivatives are obtained as a mixture between the major racemate having the relative configuration (R*, R*, R*) (i.e. the bridge -(CH2)2– of the cyclohexene moiety is cis to the group -OR2 being hydroxy) and the minor racemate having the relative configuration (R*, S*, R*) (i.e. the bridge -(CH2)2– of the cyclohexene moiety is trans to the group -OR2 being hydroxy). The major and the minor racemates can be separated as described for key intermediate K1A in procedure A1.5. The major racemate is isolated and used in the preparation of the examples below.
K1 A: rac-(1 R*,2R*,4R*)-(2-Hydroxy-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl)-acetic acid tert. -butyl ester K1 A.1 (Procedure A1.1 ): rac-(1 R*.4R*VBicvclor2.2.21octane-2.5-dione
25 ml. of 2-(trimethylsilyloxy)-1 ,3-cyclohexadiene and 13 ml. of α-acetoxyacrylonitrile were mixed and heated at 1500C in a closed vessel for 22 h. The obtained dark orange viscous oil was dissolved in 200 ml. of MeOH. After dropwise addition of a solution of 2.2 g of sodium methoxide in 150 ml. of MeOH the reaction mixture was stirred for 3 h at rt, poured into ice/water and extracted with DCM. The organic phases were concentrated in vacuo and the crude residue was purified by CC with EtOAc-Hept (1 :2) to yield 7.9 g of rac-(1 R*,4R*)- bicyclo[2.2.2]octane-2,5-dione. LC-MS: tR = 0.44 min.
K1A.2 (Procedure A1.2): rac-(1 R*.4R*VSpirorbicvclor2.2.2loctane-2.2′-ri .3ldioxolanl-5-one To 4.0 g of rac-(1 R*,4R*)-bicyclo[2.2.2]octane-2,5-dione (intermediate K1A.1 ), dissolved in 120 ml. of toluene, 1.7 ml. of ethylene glycol and 0.27 g of TsOH were added and the solution was heated under vigorous stirring to reflux for 3.5 h. The reaction mixture was cooled to rt, quenched with saturated aq. NaHCO3, extracted with Et2O, and the organic phase was evaporated. The crude product was purified by CC with Hex-EtOAc (7:3) to yield 2.41 g of rac-(1 R*,4R*)-spiro[bicyclo[2.2.2]octane-2,2′-[1 ,3]dioxolan]-5-one as yellow oil. LC-MS: tR = 0.64 min; [M+H+CH3CN]+: 224.35. K1A.3 (Procedure A1.3): Mixture of rac-(7R*.8R*.10R*V and rac-(7R*.8S*.10R*V7.10-(1.2- Ethylen)-8-phenyl-1 ,4-dioxa-spiror4.5ldecan-8-ol
To a solution of 2.41 g of rac-(1 R*,4R*)-spiro[bicyclo[2.2.2]octane-2,2′-[1 ,3]dioxolan]-5-one
(intermediate K1A.2) in 80 ml. Et2O, 14.5 ml. phenylmagnesium bromide solution (1 M in Et2O) was added dropwise over 10 min. The reaction mixture was stirred for 4 h at rt. Then, the mixture was quenched carefully with ice, 8 ml. 2N HCI were added and the phases were separated. The organic phase was evaporated and the crude product was purified by CC with Hept-EtOAC (7:3) to give 0.37 g of 7,10-(1 ,2-ethylen)-8-phenyl-1 ,4-dioxa- spiro[4.5]decan-8-ol as colorless oil. (Separation of the diastereomers by CC is possible but was not performed here.)
LC-MS: tR = 0.84 min; [M-H2O+H]+: 243.34.
K1A.4 (Procedure A1.4): rac-(1 R*,4R*)-5-Phenyl-bicvclor2.2.2loct-5-en-2-one
To a solution of 0.54 g of 7,10-(1 ,2-ethylen)-8-phenyl-1 ,4-dioxa-spiro[4.5]decan-8-ol (intermediate K1A.3) in 20 ml. acetone was added 200 mg of TsOH and then the mixture was stirred for 2 d at rt. The reaction mixture was quenched with sat. aq. NaHCO3, extracted with EtOAC and the organic phase was evaporated. The crude product was purified by CC with Hept-EtOAC (7:3) to give 0.34 g of rac-(1 R*,4R*)-5-phenyl-bicyclo[2.2.2]oct-5-en-2-one as colorless oil. LC-MS: tR = 0.93 min; [M+H+CH3CN]+: 240.1 1. K1A.5 (Procedure A1.5): rac-(1 R*.2R*.4R*H2-Hvdroxy-5-phenyl-bicvclor2.2.2loct-5-en-2-vn- acetic acid tert.-butyl ester and rac-(1 R*,2S*,4R*H2-hvdroxy-5-phenyl-bicvclor2.2.2loct-5-en- 2-yl)-acetic acid tert.-butyl ester
To a solution of 0.51 mL of DIPA in 0.5 mL THF 2.2 mL of n-butyllithium (1.6M in Hex) were added dropwise at -200C. After 10 min, 0.5 mL of toluene were added and the solution was stirred for 30 min. The mixture was cooled to -500C, 0.73 mL of tert.-butyl acetate were added and stirring was continued for 1 h at -500C. Then 0.32 g of rac-(1 R*,4R*)-5-phenyl- bicyclo[2.2.2]oct-5-en-2-one (intermediate K1A.4) dissolved in 1 mL of THF was added and the solution was stirred at -50 to -200C over 2.5 h. The reaction mixture was poured on ice/aq. HCI, the organic phase was separated, washed and evaporated. The crude reaction product was purified by CC with Hept-EtOAc (9:1 ) to yield 0.30 g of the major racemate, rac- (1 R*,2R*,4R*)-2-hydroxy-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl)-acetic acid tert.-butyl ester, as white solid and 0.07 g of the minor racemate, rac-(1 R*,2S*,4R*)-2-hydroxy-5-phenyl- bicyclo[2.2.2]oct-5-en-2-yl)-acetic acid tert.-butyl ester, as colorless oil. LC-MS (major racemate): tR = 1.06 min; [M-(CH3)3-H2O+H]+: 241.1 1. LC-MS (minor racemate): tR = 1.05 min; [M+H]+: 315.18. K1A.6: (1 S.2S.4SV(2-Hvdroxy-5-Dhenyl-bicvclor2.2.2loct-5-en-2-vn-acetic acid tert.-butyl ester and (1 R,2R,4R)-(2-Hvdroxy-5-phenyl-bicvclor2.2.2loct-5-en-2-yl)-acetic acid tert.-butyl ester rac-(1 R*,2R*,4R*)-(2-Hydroxy-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl)-acetic acid tert.-butyl ester was separated into the respective enantiomers using prep, chiral HPLC (column: Daicel ChiralPak AD-H, 20×250 mm, 5 μm; Hex/ EtOH 95:5, flow 16 mL/min) Chiral analytic HPLC (Daicel ChiralPak AD-H, 4.6×250 mm, 5 μm; Hex/ EtOH 95:5, flow 0.8 mL/min):
(1 R,2R,4R)-(2-Hydroxy-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl)-acetic acid tert.-butyl ester: Enantiomer A: tR = 6.70 min.
(1S,2S,4S)-(2-Hydroxy-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl)-acetic acid tert.-butyl ester: Enantiomer B: tR = 7.93 min.
BB. [3-(4,7-Dimethoxy-1 H-benzoimidazol-2-yl)-propyl]-methyl-amine
BB.1 3,6-Dimethoxy-benzene-1 ,2-diamine 3, 6-Dimethoxy-benzene-1 ,2-diamine was synthesized by dissolving 6.0 g of 1 ,4-dimethoxy- 2,3-dinitro-benzene (Eur.J.Org.Chem. 2006, 2786-2794) in 220 mL EtOH, evacuating 3 times with N2 and adding 600 mg of 10wt% Pd/C. The reaction was stirred under a H2 atmosphere (balloon). Another 300 mg of 10wt% Pd/C were added after 2 days and the mixture was stirred for another 24 h. Filtration over a pad of celite and washing with EtOH and EtOAc yielded after concentration in vacuo 4.3 g of 3, 6-dimethoxy-benzene-1 ,2-diamine as black solid. LC-MS: tR = 0.48 min; [M+H]+: 169.09.
BB.2 r3-(2-Amino-3,6-dimethoxy-phenylcarbamoyl)-propyll-methyl-carbamic acid benzyl ester To a solution of 3.1 g of 4-(benzyloxycarbonyl-methyl-amino)-butyric acid in 80 mL DCM were added 6.5 mL of DIPEA, 1.8 g of HOBt, 2.6 g of EDC and 154 mg of DMAP. After stirring for 10 min, 2.1 g of 3, 6-dimethoxy-benzene-1 ,2-diamine, dissolved in 20 mL DCM, were added and the mixture was stirred at rt overnight. The reaction was quenched with sat. aq. NaHCO3, the phases were separated and the organic phase was washed with brine, dried over MgSO4 and concentrated in vacuo to yield the crude title compound as black oil. LC-MS: tR = 0.88 min; [M+H]+: 402.06.
BB.3 [3-(4,7-Dimethoxy-1 H-benzoimidazol-2-yl)-propyl1-methyl-carbamic acid benzyl ester
To a mixture of the above crude 3-(2-amino-3,6-dimethoxy-phenylcarbamoyl)-propyl]-methyl- carbamic acid benzyl ester in 16 mL toluene were added 4 mL of DMF and 1.9 g of TsOH and the reaction was heated to 1500C for 2 h in the microwave. Sat. aq. NaHCO3 was added and the phases were separated. The organic phase was washed with brine, dried over MgSO4, concentrated in vacuo, filtered over a short pad of silica gel with EtOAc and concentrated again. Purification by CC with EtOAc yielded 2.7 g of 3-(4,7-dimethoxy-1 H- benzoimidazol-2-yl)-propyl]-methyl-carbamic acid benzyl ester as brown resin. LC-MS: tR = 0.85 min; [M+H]+: 384.62.
BB.4 r3-(4,7-Dimethoxy-1 H-benzoimidazol-2-yl)-propyll-methyl-amine
A solution of 2.6 g of 3-(4,7-dimethoxy-1 H-benzoimidazol-2-yl)-propyl]-methyl-carbamic acid benzyl ester in 60 ml. EtOH was evacuated 3 times with N2 before 260 mg of 10 wt% Pd/C were added. The reaction mixture was then stirred under a H2atmosphere (balloon) for 5 h at rt. Filtration over a pad of celite and washing with EtOH yielded after concentration in vacuo 1.7 g of 3-(4,7-dimethoxy-1 H-benzoimidazol-2-yl)-propyl]-methyl-amine as brown foam. LC-MS: tR = 0.57 min; [M+H]+: 250.13.
Preparation of COMPOUND Reference Example 1A: rac-lsobutyric acid (1 R*,2R*,4R*)-2-(2-{[3-(4,7-dimethoxy-1H- benzoimidazol-2-yl)-propyl]-methyl-amino}-ethyl)-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl ester
1.1 (Procedure P1.1 V rac-(1 R*.2R*.4R*H2-Hvdroxy-5-phenyl-bicvclor2.2.2loct-5-en-2-vn- acetic acid To a solution of 4.0 g of rac-(1 R*,2R*,4R*)-(2-hydroxy-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl)- acetic acid tert.-butyl ester in 25 mL EtOH were added 2.1 g of LiOH-H2O, 8 mL H2O and 22 mL MeOH. The reaction mixture was stirred at rt for 3 d and then concentrated. The residue was partitioned between water and Et2O. The aq. layer was separated and acidified with 1 N HCI resulting in the formation of a white solid. The solid was filtrated, washed with 5 mL aq. HCI and dried in vacuo to obtain 3.2 g of rac-(1 R*,2R*,4R*)-(2-hydroxy-5-phenyl- bicyclo[2.2.2]oct-5-en-2-yl)-acetic acid as white solid. LC-MS: tR = 0.86 min; [M-H2O+H]+: 241.28.
1.2 (Procedure P1.2): rac-(1 R*,2R*,4R*)-N-r3-(4,7-Dimethoxy-1 H-benzoimidazol-2-yl)- propyl1-2-(2-hvdroxy-5-phenyl-bicvclo[2.2.21oct-5-en-2-yl)-N-methyl-acetamide To a solution of 280 mg of rac-(1 R*,2R*,4R*)-(2-hydroxy-5-phenyl-bicyclo[2.2.2]oct-5-en-2- yl)-acetic acid in 7 mL THF were added 0.58 mL of DIPEA, 175 mg of HOBt and 250 mg of EDC at rt. After stirring for 10 min, 270 mg of 3-(4,7-dimethoxy-1 H-benzoimidazol-2-yl)- propyl]-methyl-amine were added and the reaction mixture was stirred at rt overnight. The reaction mixture was quenched with sat. aq. NaHCO3, the phases were separated and the organic phase was washed with water and brine, dried over MgSO4 and concentrated in vacuo. Purification by CC using EtOAc-MeOH (5:1 to 2:1 ) yielded 475 mg of rac- (1 R*,2R*,4R*)-N-[3-(4,7-dimethoxy-1 H-benzoimidazol-2-yl)-propyl]-2-(2-hydroxy-5-phenyl- bicyclo[2.2.2]oct-5-en-2-yl)-N-methyl-acetamide as white foam. LC-MS: tR = 0.91 min; [M+H]+: 490.06.
1.3 (Procedure P1.3): rac-(1 R*.2R*.4R*V2-(2-fr3-(4.7-Dimethoxy-1 H-benzoimidazol-2-ylV propyll-methyl-amino}-ethyl)-5-phenyl-bicvclor2.2.21oct-5-en-2-ol
To a solution of 310 mg of rac-(1 R*,2R*,4R*)-N-[3-(4,7-dimethoxy-1 H-benzoimidazol-2-yl)- propyl]-2-(2-hydroxy-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl)-N-methyl-acetamide in 8 mL toluene were added dropwise 0.77 ml. of a Red-AI solution (65% in toluene) at 00C. After stirring for 10 min at 00C, the cooling bath was removed and stirring was continued for 3 h at rt. The reaction mixture was then carefully poured onto a mixture of 1 M NaOH/ice and stirred for 10 min. The aq. phase was extracted with toluene, the combined organic phases were washed with brine, dried over MgSO4 and concentrated in vacuo. Purification by CC using EtOAc-MeOH (2:1 ) yielded 230 mg of rac-(1 R*,2R*,4R*)-2-(2-{[3-(4,7-dimethoxy-1 H- benzoimidazol^-y^-propyll-methyl-aminoj-ethy^-δ-phenyl-bicyclop^^loct-δ-en^-ol as white foam. LC-MS: tR = 0.79 min; [M+H]+: 476.13. 1.4: rac-lsobutyric acid (1 R*.2R*.4R*‘)-2-(2-fr3-(4.7-dimethoxy-1 H-benzoimidazol-2-vn- propyll-methyl-amino}-ethyl)-5-phenyl-bicvclor2.2.21oct-5-en-2-yl ester
To a solution of 199 mg of rac-(1 R*,2R*,4R*)-2-(2-{[3-(4,7-dimethoxy-1 H-benzoimidazol-2- yO-propyO-methyl-aminoJ-ethy^-δ-phenyl-bicycloP^^loct-δ-en^-ol in 4 mL DCM were added 0.2 mL of NEt3 and 0.1 mL of isobutyrylchloride at 0°C. The reaction mixture was stirred overnight allowing the temperature to reach slowly rt. The reaction was quenched with sat. aq. NaHCO3, the phases were separated and the water phase was re-extracted with DCM. The combined organic phases were washed with brine, dried over MgSO4 and concentrated in vacuo. The residue was redissolved in 3 mL EtOAc, silica gel and 1.5 mL MeOH were added and the mixture was stirred vigorously for 7 d. The mixture was filtered, thouroughly washed with EtOAc-MeOH (2:1 ) and evaporated. Purification by CC using EtOAc-MeOH (5:1 to 3:1 + 0.1 % NEt3) yielded 186 mg of rac-isobutyric acid (1 R*,2R*,4R*)-2- (2-{[3-(4,7-dimethoxy-1 H-benzoimidazol-2-yl)-propyl]-methyl-amino}-ethyl)-5-phenyl- bicyclo[2.2.2]oct-5-en-2-yl ester as beige foam. LC-MS: tR = 0.90 min; [M+H]+: 546.23. Reference Example 2A: lsobutyric acid (1S,2S,4S)-2-(2-{[3-(4,7-dimethoxy-1H- benzoimidazol-2-yl)-propyl]-methyl-amino}-ethyl)-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl ester
2.1 : (1S.2S.4SV(2-Hvdroxy-5-Dhenyl-bicvclor2.2.2loct-5-en-2-vn-acetic acid Prepared according to procedure P1.1 in Reference Example 1A using enantiomer B of rac- (1 R*,2R*,4R*)-(2-hydroxy-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl)-acetic acid tert.-butyl ester (see K1A.6). LC-MS: tR = 0.91 min; [M-H2CHH]+: 241.10.
2.2: (1S.2S.4SV2-(2-fr3-(4.7-Dimethoxy-1 H-benzoimidazol-2-ylVDroDyll-methyl-amino>- ethyl)-5-phenyl-bicvclor2.2.21oct-5-en-2-ol
Prepared according to procedures P1.2 to P1.3 in Reference Example 1A using the above (2-hydroxy-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl)-acetic acid. LC-MS: tR = 0.78 min; [M+H]+: 476.09.
2.3: Isobutyric acid (1S,2S,4S)-2-(2-{[3-(4J-dimethoxy-1 H-benzoimidazol-2-yl)-propyl1- methyl-amino}-ethyl)-5-phenyl-bicvclor2.2.21oct-5-en-2-yl ester
Prepared according to procedure P1.4 in Reference Example 1A using the above 2-(2-{[3-
(4,7-dimethoxy-1 H-benzoimidazol-2-yl)-propyl]-methyl-amino}-ethyl)-5-phenyl- bicyclo[2.2.2]oct-5-en-2-ol.
LC-MS: tR = 0.89 min; [M+H]+: 546.19. Reference Example 3A: lsobutyric acid (1 R,2R,4R)-2-(2-{[3-(4,7-dimethoxy-1H- benzoimidazol-2-yl)-propyl]-methyl-amino}-ethyl)-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl ester
3.1 : (1 R.2R.4RH2-Hvdroxy-5-phenyl-bicvclor2.2.21oct-5-en-2-vn-acetic acid
Prepared according to procedure P1.1 in Reference Example 1 using enantiomer A of rac- (1 R*,2R*,4R*)-(2-hydroxy-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl)-acetic acid tert.-butyl ester (see K1A.6). LC-MS: tR = 0.91 min; [M-H2CHH]+: 241.16.
3.2: (1 R,2R,4R)-2-(2-{[3-(4,7-Dimethoxy-1 H-benzoimidazol-2-yl)-propyl1-methyl-amino}- ethvD-δ-phenyl-bicvclo^^^loct-δ-en^-ol Prepared according to procedures P1.2 to P1.3 in Reference Example 1 using the above (2- hydroxy-5-phenyl-bicyclo[2.2.2]oct-5-en-2-yl)-acetic acid. LC-MS: tR = 0.79 min; [M+H]+: 476.09. 3.3: Isobutyric acid (1 R.2R.4RV2-(2-{r3-(4.7-dimethoxy-1 H-benzoimidazol-2-ylVpropyll- methyl-amino}-ethyl)-5-phenyl-bicvclor2.2.21oct-5-en-2-yl ester
Prepared according to procedure P1.4 in Reference Example 1A using the above 2-(2-{[3- (4,7-dimethoxy-1 H-benzoimidazol-2-yl)-propyl]-methyl-amino}-ethyl)-5-phenyl- bicyclo[2.2.2]oct-5-en-2-ol.
LC-MS: tR = 0.89 min; [M+H]+: 546.11. Optical rotation: alpha D (c = 10 mg/mL EtOH) = -21.5°.
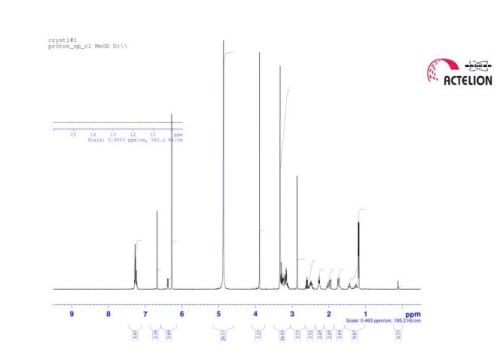
1 H NMR (MeOD, 400 MHz) δ 7.39-7.37 (m, 2H), 7.30 (t, J = 6.4 Hz, 2H), 7.24-7.20 (m, 1 H), 6.60 (s, 2 H), 6.43 (br d, J = 7.6 Hz, 1 H), 3.91 (s, 6H), 3.27-3.23 (m, 1 H), 3.18-3.15 (m, 1 H), 2.87 (t, J = 7.6 Hz, 2H), 2.54 (sept, J = 7.0 Hz, 1 H), 2.47-2.37 (m, 4H), 2.21 (s, 3H), 2.19- 2.12 (m, 1 H), 2.01-1.92 (m, 5H), 1.75-1.65 (m, 2H), 1.48-1.38 (m, 1 H), 1.27-1.19 (m, 1 H), 1.16 (d, J = 7.0 Hz, 6H).
Example S5: Preparation and characterization of the di-maleic acid salt of COMPOUND
Maleic acid (256 g, 2.2 mol, 2.1 eq), dissolved in MeOH (630 ml_, 1.1 volumes) was added to a refluxing solution of COMPOUND (682 g, 84% w/w (NMR assay), 1.05 mol) in EtOAc (6.3 L, 11 volumes). The resulting mixture was stirred under reflux for 15 minutes and was then cooled to 65-68°C within 30 minutes and seeded with 0.04% w/w of seeding crystals of di- maleic acid salt of COMPOUND (Seeding crystals were obtained after careful crystallisation using the same protocol.). The mixture was then cooled from 65-68°C to 400C within 3 h. The obtained suspension was then cooled down to 200C over 1 h, filtered under 0.2 bar of nitrogen and rinsed with EtOAc (1500 ml. 2.6 volumes). The obtained white solid was then dried under 1 atmosphere of nitrogen for 24 hours to yield 715 g (88%) of the di-maleic acid salt of COMPOUND.
Table S5: Characterisation data for the di-maleic acid salt of COMPOUND

………….
paper
A scalable access to 1 (ACT-280778), a potent L/T calcium channel blocker, has been developed. The synthesis, amenable to kilogram manufacturing, comprises 10 chemical steps from enantiomerically pure 5-phenylbicyclo[2.2.2]oct-5-en-2-one (3) and 1,4-dimethoxybenzene with a longest linear sequence of 7 steps. Key to the success of this fit-for-purpose approach are a robust and atom-efficient access to benzimidazole 4, the substrate-controlled diastereoselective enolate addition toward carboxylic acid 2 that was isolated by simple crystallization with high dr (>99:1), the convenient selective N-deacylation of intermediate 10, and the identification of a suitable solid form of 1 as the bis-maleate salt (1·2 C4H4O4). As an illustration of the robustness of this process, 14 kg of drug substance, suitable for human use, was produced with an overall yield of 38% over the longest linear sequence (7 steps).

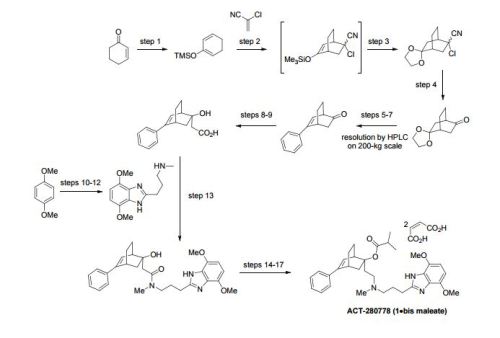


LC-MS were run using the following conditions: Finnigan Navigator with HP 1 100 Binary Pump and DAD, column: 4.6×50 mm, Zorbax SB-AQ, 5 μm, 120 A, gradient: 5-95% acetonitrile in water, 1 min, with 0.04% trifluoroacetic acid, flow: 4.5 mL/min, tR is given in min.
Compounds are purified by preparative HPLC (column: X-terra RP18, 50×19 mm, 5 μm, gradient: 10-95% acetonitrile in water containing 0.5 % of formic acid) or by column chromatography on silica gel. Racemates can be separated into their enantiomers by preparative HPLC (preferred conditions: Daicel, ChiralCel OD 20×250 mm, 10 μm, 4% ethanol in hexane, flow 10-20 mL/min).
-
this work was preliminarily disclosed: Funel, J.-A. In Practical Synthesis of L/T Calcium Channel Blocker ACT-280778, 30th SCI Process Development Symposium, Cambridge, UK, December 5–7, 2012; Funel, J.-A.; In Practical Synthesis of 5-Phenylbicyclo[2.2.2]oct-5-en-2-one toward L/T Calcium Channel Blocker ACT-280778. Application of the Diels–Alder Reaction on kg-Scale, 1st Smart Synthesis and Advanced Purification Conference, April 21–23, 2013; Lyon, FR.
-
(a) Hilpert, K., Hubler, F., and Renneberg, D. WO/2008/132679A1, 2008.
(b) Hubler, F.,Hilpert, K., and Renneberg, D. WO/2009/130679A1, 2009.
-
Funel, J.-A.; Schmidt, G.; Abele, S. Org. Process Res. Dev. 2011, 15, 1420– 1427
-
Abele, S.; Schwaninger, M.; Fierz, H.; Schmidt, G.; Funel, J.-A.; Stoessel, F. Org. Process Res. Dev. 2012, 16, 2015– 2020
-
(a) Abele, S.; Inauen, R.; Funel, J.-A.; Weller, T. Org. Process Res. Dev. 2012, 16, 129–140
(b) Abele, S.; Funel, J.-A. WO/2012/052943A1, 2012.
(c) Abele,S.; Funel, J.-A. WO/2012/052939A2, 2012.
-
Abele, S.; Inauen, R.; Spielvogel, D.; Moessner, C. J. Org. Chem. 2012, 77, 4765– 4773


Reblogged this on ORGANIC CHEMISTRY SELECT.
LikeLike