
Home » Posts tagged 'zydus cadila'
Tag Archives: zydus cadila
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
ZY 19489, MMV 253
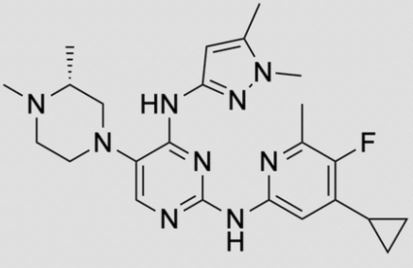

![2-N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-5-[(3R)-3,4-dimethylpiperazin-1-yl]-4-N-(1,5-dimethylpyrazol-3-yl)pyrimidine-2,4-diamine.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=92045019&t=l)
ZY 19489, MMV 253
C24 H32 FN9, 465.5
CAS 1821293-40-6
MMV253, GTPL10024, MMV674253
N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-5-((3R)-2-((1,5-dimethyl-1H-pyrazol-3-yl)amino)-3,4-dimethylpiperazin-1-yl)pyrimidin-2-amine
2-N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-5-[(3R)-3,4-dimethylpiperazin-1-yl]-4-N-(1,5-dimethylpyrazol-3-yl)pyrimidine-2,4-diamine
- N2-(4-Cyclopropyl-5-fluoro-6-methyl-2-pyridinyl)-5-[(3R)-3,4-dimethyl-1-piperazinyl]-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-2,4-pyrimidinediamine
- (R)-N2-(4-Cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine

SYN
IN 201721031453
The invention relates to triaminopyrimidine compd. of formula I, pharmaceutically acceptable salts thereof, hydrates, solvates, polymorphs, optically active forms thereof, in solid state forms useful for preventing or treating malaria. The invention also relates to a process for prepn. of triaminopyrimidine compd. and intermediates thereof. Compd. I was prepd. by condensation of 5-bromouracil with tert-Bu (R)-2-methylpiperazine-1-carboxylate to give tert-Bu (R)-4-(2,4-dichloropyrimidin-5-yl)-2-methylpiperazine-1-carboxylate, which underwent chlorination followed by condensation with 1,5-dimethyl-1H-pyrazol-3-amine followed by condensation with 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride to give (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3-methylpiperazin-1-yl)pyrimidine-2,4-diamine, which underwent Boc-deprotection followed by methylation to give I.
SYN
WO 2019049021
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019049021
Malaria is caused by protozoan parasites of the genus Plasmodium that infect and destroy red blood cells, leading to fever, severe anemia, cerebral malaria and, if untreated, death.
International (PCT) Publication No. WO 2015/165660 (the WO ‘660) discloses triaminopyrimidine compounds, intermediates, pharmaceutical compositions and methods for use for preventing or treating malaria. The WO ‘660 discloses a process for preparation of 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine (compound 5) as depicted in scheme-1.
Scheme 1
WO ‘660 discloses a process for preparation of triaminopyrimidine compounds depicted in scheme-2.

WO ‘660 discloses the preparation of compounds 8 and 4 by using microwave technique using Biotage microwave vial. WO ‘660 in example- 13, discloses the isolation of compound 1 by concentration of reaction mixture to obtain crude product, which was purified through reverse phase HPLC GILSON instrument to obtain pure solid compound 1 in 40.8% yield, without providing the purity of the solid compound 1. The process disclosed in WO ‘660 is not industrially advantageous as it requires microwave conditions as well as chromatographic purification and provides compound 1 with lower yields. The compound 1 prepared may not be suitable for pharmaceutical preparations based on various regulatory requirements.
Polymorphism, the occurrence of different crystalline forms, is a property of some molecules. A single molecule can exist in different crystalline forms having distinct physical properties like melting point, thermal behaviors (e.g. measured by thermogravimetric analysis – TGA, or different scanning calorimetry – DSC, Powder x-ray diffraction pattern – PXRD, infrared absorption – IR). One or more these techniques may be used to distinguish different polymorphic forms of a compound.
Different salts and solid states (e.g. solvates, hydrates) of an active pharmaceutical ingredient may possess different physio-chemical properties. Such variation in the properties of different salts and solid states forms may provide a basis for improving formulation, for example, by facilitating better processing or handling characteristics, changing the dissolution profile in a favorable direction, or improving stability (both chemical and polymorph) and shelf-life. These variations in the properties of different salts and solid states forms may offer improvements to the final dosage form for example, to improve bioavailability. Different salts and solid state forms of an active pharmaceutical ingredient may also give rise to a variety of polymorphs or crystalline forms or amorphous form, which may in turn provide additional opportunities to assess variations in the properties and characteristics of an active pharmaceutical ingredient.
In view of the above, the present invention provides a process for the preparation of triaminopyrimidine compound 1 or pharmaceutically acceptable salts thereof or hydrates or solvates or polymorphs or optically active forms thereof, which is industrially scalable, environment friendly and efficient so as to obtain compounds of the invention in higher yields and purity.
The process for the preparation of triaminopyrimidine compound 1 or intermediates thereof of the present invention, takes the advantage by using appropriate solvent systems and isolation techniques as well as purification techniques, thereby to overcome problems of lower yields, chromatography purifications and microwave reactions of the prior art.
SUMMARY OF THE INVENTION
The present invention provides solid state forms of triaminopyrimidine compound
1,
1
Examples: Preparation of Intermediates
Example-1: Preparation of 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine
In a 250 mL 4N round bottom flask, process water (30 ml) and cyclopropanecarboxylic acid (14.19 g, 164.88 mmol) were added at 25 to 35°C and started stirring. Sulphuric acid (4.4 ml, 82.44 mmol) was charged to the reaction mixture. Silver nitrate (4.18 g, 24.73 mmol), 6-Chloro-3-fluoro-2-methylpyridine (6 g, 41.22 mmol) were charged to the reaction mixture. Aqueous solution of ammonium persulphate (65.85 g, 288.54 mmol in 90 mL water) was added to the reaction mixture in 30 to 60 min at temperature NMT 60 °C. After the completion of the reaction as monitored by HPLC, toluene (30 ml) was added to the reaction mixture and stirred for 15 min. The reaction mixture filtered, separated layers from filtrate and extracted aqueous layer using toluene (30 mL). The organic layer was washed with aqueous sodium carbonate solution (30 mL) and water. The organic layer was distilled completely under vacuum at 60 °C to obtain 3.37 g syrupy mass as titled compound.
Example-2: Preparation of 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine
In a suitable glass assembly, process water (7.5 L) and cyclopropanecarboxylic acid (3.55 Kg, 41.24 mol) were added at 25 to 35 °C and stirred. Sulphuric acid (2.02 Kg, 20.59 mol), silver nitrate (1.05 Kg, 6.21 mol), 6-chloro-3-fluoro-2-methylpyridine (1.5 Kg, 10.3 mol) were added to the reaction mixture. Aqueous solution of ammonium persulphate (16.46 g, 72.13 mmol in 22.5 L water) was added to the reaction mixture at 55 to 60 °C and maintained. After the completion of the reaction as monitored by HPLC, toluene (7.5 L) was added to the reaction mixture and stirred for 15 min. The reaction mixture was filtered, organic layer was separated and aqueous layer was extracted using toluene (6 L), filtered the reaction mixture and washed the solid with toluene (1.5 L). The combined organic layer was washed with 20% sodium carbonate solution (9 L) and water. The organic layer was concentrated completely under vacuum at 60 °C to obtain 880 g (86.50%) syrupy mass of titled compound.
Example-3: Preparation of N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenyl-methanimine
In a 100 mL 3N round bottom flask, 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine (2.69 g, 14.48 mmol) and toluene (30 mL) were added at 25 to 35 °C. Diphenylmethanimine (3.15 g, 17.38 mmol) was charged to the reaction mixture and stirred for 5-10 min under nitrogen purging. Racemic BINAP (270 mg, 0.43 mmol) and palladium acetate (98 mg, 0.43 mmol) were added to the reaction mixture. Sodium-ie/ -butoxide (2.78 g, 28.96 mmol) was added to the reaction mixture and heated to 100 to 110° C under nitrogen. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C and filtered over hyflo bed and washed with toluene. The filtrate containing titled compound was preserved for next stage of reaction.
Example-4: Preparation of N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenyl-methanimine
In a suitable assembly, 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine (880) and toluene (7.5 L) were added at 25 to 35 °C. Diphenylmethanimine (787 g, 4.34 mmol) and BOC anhydride (237 g, 1.086 mol) was added to the reaction mixture and stirred for 5-10 min under nitrogen purging. Racemic BINAP (67.6 g, 0.108 mmol) and palladium acetate (24.4 g, 0.108 mol) were added to the reaction mixture. S odium- ieri-butoxide (870 g, 9.05 mol) was added to the reaction mixture and heated to 100 to 110 °C under nitrogen. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C, water (6 L) was added. The reaction mixture was filtered over hyflo bed and washed with toluene. The filtrate containing titled compound was preserved for next stage of reaction.
Example-5: Preparation of 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride monohydrate
In a 100 mL 3N round bottom flask, N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenylmethanimine in toluene as obtained in example-3 was added water (25 mL) at 25 to 35° C. The cone. HCl (3 mL) was charged to the reaction mixture and heated to 40 to 50 °C. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C. Layers were separated. The aqueous layer was treated with methylene dichloride and pH was adjusted to 7.5 to 8.5 using sodium carbonate solution, stirred for 15 min and layers were separated. Aqueous layer was extracted with methylene dichloride, charcoaled and acidified to pH 3 to 4 with aqueous HCl. The solvent was distilled completely and acetonitrile (9 mL) and ethyl acetate (9 mL) was added. The reaction mixture was stirred for 1 hour at 25 to 35° C. The product was filtered and washed with ethyl acetate. The product was dried at 50° C for 4 hours under vacuum to obtain 1.62 g title compound as monohydrate yellow crystalline solid having 99.51% HPLC purity.
Example-6: Preparation of 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride monohydrate
In a suitable glass assembly, N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenylmethanimine in toluene as obtained in example-4 was added water (6 L) at 25 to 35° C. The cone. HCl (750 mL) was charged to the reaction mixture and heated to 40 to 50 °C. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C. Layers were separated. The aqueous layer was treated with methylene dichloride (3 L) and pH was adjusted to 7.5 to 8.5 using sodium carbonate solution, stirred for 15 min and layers were separated. Aqueous layer was extracted with methylene dichloride (3 L), charcoaled and acidified to pH 3 to 4 with aqueous HCl. The solvent was distilled completely and acetonitrile (1.5 L) and ethyl acetate (1.5 L) were added. The reaction mixture was stirred for 1 hour at 25 to 35° C. The product was filtered and washed with ethyl acetate. The product was dried at 50° C for 4 hours under vacuum to obtain 489 g (96.80%) title compound as monohydrate yellow crystalline solid having 99.51% HPLC purity. The crystalline compound is characterized by Powder x-ray diffraction pattern (FIG.5), Differential scanning calorimetry (FIG.6) and Thermogravimetric analysis (FIG.7).
Example 7: Preparation of 2,3-dibromobutanenitrile
In a 2 L round bottom flask, dichloromethane (550 mL) and 2-butenenitrile 110 g
(1.64 mol) were cooled to 20 to 25 °C. A solution of bromine 275 g (1.72 mol) in dichloromethane (220 mL) was dropwise added at 20 to 25 °C. Hydrobromic acid 1.43 ml (0.0082 mol) in acetic acid (33%) solution was added into the reaction mixture and stirred for 4 hours. After the completion of reaction, Na2S203 (550 mL) 4% aqueous solution was added and the reaction mixture was stirred for 15 min. The separated organic layer was distilled under vacuum completely to obtain 364.2 g (97.9%) of title compound as an oil.
Example 8: Preparation of l,5-dimethyl-lH-pyrazol-3-amine
In a 5 L round bottom flask, water (1. 36 L), sodium hydroxide 340 g (8.99 mol) were added and the reaction mixture was cooled to 0 to 5°C. A solution of methyl hydrazine sulphate 237.8 g (1.65 mol) in 680 mL water was added dropwise to the reaction mixture and stirred below 10 °C. 2,3-dibromobutanenitrile 340 g (1.5 mol) prepared in example-7 was added and the reaction mixture was stirred below 10 °C for 2 hours. After the completion of reaction, toluene (630 mL) was added and the reaction mixture was stirred for 15 min. The aqueous layer was separated and the organic layer was removed. The aqueous layer was extracted with dichloromethane (5.1 L). The combined organic layer was distilled completely under vacuum to obtain residue. Diisopropyl ether (680 mL) was added and the reaction mixture was stirred at 0 to 5 °C for 1 hour. The reaction mixture was filtered, washed with diisopropyl ether and dried to obtained 121.5 g (72.93%) of title compound having 95.63% purity.
Examples: Preparation of triaminopyrimidine compounds
Example-9: Preparation of tert-butyl (R)-4-(2,4-dioxo-l,2,3,4-tetrahydro- pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate
In 2 L four neck round bottom flask, 1.25 Kg (6.545 mol) 5-bromouracil, 1.87 Kg (9.360 mol) tert-butyl (R)-2-methylpiperazine-l-carboxylate and 5L pyridine were added at 25 to 35° C. The reaction mass was stirred for 15 hours at 115 to 120°C. After completion, the reaction mass was cooled to 25 to 35°C. 12.5 L water was added and stirred for 1 hour. The reaction mass was filtered, washed with 2.5 L water and dried to obtain 1.37 Kg (67.4%) of title compound.
Example-10: Preparation of tert-butyl (R)-4-(2,4-dichloropyrimidin-5-yl)-2-methylpiperazine- 1 -carboxylate
In 20 L four neck round bottom flask, 1.36 Kg (4.382 mmol) tert-butyl (R)-4-(2,4-dioxo-1, 2,3, 4-tetrahydropyrimidin-5-yl)-2-methylpiperazine-l -carboxylate and 6.8 L phosphorus oxychloride were added at 25 to 35° C. 26.5 mL pyridine (0.329 mol) was added and the reaction mass was heated to 105 to 110 °C and stirred for 4 hours. After the completion of the reaction, phosphorus oxychloride was distilled completely at atmospheric pressure. 2.72 L acetone was added and the reaction mixture was quenched into 4.08 L water. Acetone was removed by distillation under vacuum. 20% sodium carbonate solution was added to adjust pH 7.5-8.5 of the reaction mixture. 1.14 Kg (5.258 mol) di-tert-butyl dicarbonate and 9.52 L ethyl acetate were added and stirred for 2 hours at 25 to 35 °C. After the completion of the reaction, the organic layer was separated and aqueous layer was extracted with 6.8 L ethyl acetate. The combined ethyl layers were distilled to remove ethyl acetate completely under vacuum to obtain residue. 1.36 L isopropyl alcohol was added to the residue and isopropyl alcohol was removed completely. 4.08 L isopropyl alcohol and 6.8 L water were added to the residue and stirred for 1 hour. The reaction mass was filtered, washed with water and dried to obtain 1.25 Kg of title compound.
Example-11: Preparation of tert-butyl (R)-4-(2-chloro-4-[(l,5-dimethyl-lH-pyrazol-3-yl)amino)pyrimidin-5-yl]-2-methylpiperazine-l-carboxylate
In 20 L round bottom flask, 640 g (1.843 mol) tert-butyl (R)-4-(2, 4-dichloropyrimidin-5-yl)-2-methylpiperazine-l -carboxylate, 225.3 g (2.027 g) 1,5-dimethyl-lH-pyrazol-3-amine and 9.6L toluene were added at 25 to 35°C. 1.2 Kg (3.686 mol) cesium carbonate was added. The reaction mixture was purged for 15 min under nitrogen. 12.41 g (0.0553 mol) palladium acetate and 34.43 g (0.0553 mol) racemic 2,2′-bis(diphenylphosphino)-l,l’-binaphthyl were added and the reaction mass was maintained for 16 hours at 110 to 115 °C under nitrogen. After the completion of the reaction, the reaction mixture was filtered through a celite bed and washed the bed with 1.28 L toluene. Toluene was distilled completely and 2.56 L dichlromethane was added. The compound was adsorbed by 1.92 Kg silica gel (60-120 mesh). The dichloromethane was distilled completely under vacuum and 12.8 L mixture of ethyl acetate and hexane was added to the residue and stirred for 2 hours. The silica gel was filtered and the filtrate was distilled completely under vacuum to obtain 595 g title compound.
Example-12: Preparation of tert-butyl (R)-4-(2-((4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)amino)-4-((l,5-dimethyl-lH-pyrazol-3-yl)amino) pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate
In 20 L round bottom flask, 595 g (1.40 mol) tert-butyl (R)- 4-(2-chloro-4-[(l,5-dimethyl-lH-pyrazol-3-yl)amino)pyrimidin-5-yl]-2-methylpiperazine-l-carboxylate, 305 g (1.38 mol) 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride and 11.5 L toluene were added at 25 to 35°C. 1.08 Kg (3.32 mol) cesium carbonate was added. The reaction mixture was purged for 15 min under nitrogen. 17.21 g (27.6 mmol) palladium acetate and 6.21 g (27.6 mmol) racemic 2,2′-bis(diphenylphosphino)-l, -binaphthyl were added. The reaction mass was stirred for 6 hours at 110 tol l5 °C under nitrogen. After the completion of the reaction, the reaction mixture was filtered through a celite bed and washed with toluene. The filtrate was used as such in the next step without further treatment.
Example-13: Preparation of tert-butyl (R)-4-(2-((4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)amino)-4-((l,5-dimethyl-lH-pyrazol-3-yl)amino) pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate
In 500 mL four neck round bottom flask, 7.5 g (17.77 mmol) (R)-tert-butyl 4-(2-chloro-4-[(l,5-dimethyl-lH-pyrazol-3-yl)amino)pyrimidin-5-yl]-2-methylpiperazine-l-carboxylate, 3.92 g (17.77 mmol) 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride compound and 150 mL toluene were added at 25 to 35 °C. 20 g (61.3 mmol) cesium carbonate was added. The reaction mixture was purged for 15 min under nitrogen. Then, 130 mg (0.58 mmol) palladium acetate and 360 mg (0.58 mmol) racemic 2,2′-bis(diphenylphosphino)-l,l’-binaphthyl were added. The reaction mass was stirred for 18 hours at 110 to 115° C under nitrogen. After completion, the reaction mixture was filtered through a celite bed and washed with toluene. The filtrate was used as such in the next step without further treatment.
2 4
Example-14: (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N -(1, 5-dimethyl-lH-pyrazol-3-yl)-5-(3-methylpiperazin-l-yl)pyrimidine-2,4-diamine
In 50 L glass assembly, the filtrate containing tert-butyl (R)-4-(2-((4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)amino)-4-((l,5-dimethyl-lH-pyrazol-3-yl)amino) pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate from example 13 was taken. 11.5 L water and 1.28 L Cone. HC1 were added at 25 to 35 °C. The reaction mass was stirred for 2 hours at 50 to 55 °C. After the completion of the reaction, reaction mixture was cooled to room temperature and filtered over celite bed and washed with water. The separated the aqueous layer from filtrate was basified by using 20% sodium carbonate solution and extracted with 12.8 L methylene dichloride. The organic layer was distilled completely under vacuum to obtain residue. 9.6 L acetonitrile was added to the residue and heated to reflux for 30 min. The reaction mixture was cooled and stirred at 25 to 35 °C for 1 hour. The reaction mixture was filtered, washed with 640 mL acetonitrile and dried to obtain 360 g titled compound.
2 4
Example-15: (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N -(1,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine
In 250 mL four neck round bottom flask, 4.7 g (10.4 mmol) (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine was dissolved in 56 mL ethanol. 1.89 g (23.32 mmol) formaldehyde and 1.44 g (22.90 mmol) sodium cyanoborohydride were added. Adjusted pH 5-6 using acetic acid and stirred the reaction mass at 25 to 35 °C for 2 hours. After completion, ethanol was distilled completely under vacuum. 47 mL water was added to the residue. The reaction mass was basified by 20% sodium carbonate solution and extracted with methylene dichloride. Both the organic layers were combined and distilled completely under vacuum. 94 mL acetonitrile was added to the residue and heated to reflux for 15 min. The reaction mass was cooled to 25 to 35° C and stirred for 1 hour. The reaction mass was filtered, washed with 5 mL acetonitrile and dried to obtain 3.7 g title compound as crystalline solid, having HPLC purity of about 99.61%.
2 4
Example-16: (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N -(1,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine
In 20 L round bottom flask, 725 g (1.60 mol) (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazine-l-yl)pyrimidine-2,4-diamine was dissolved in 6.52 L dichloromethane. 261.5 g (3.2 mol) formaldehyde and 510.4 g (2.4 mol) sodium triacetoxyborohydride were added and stirred the reaction mixture at 25 to 35 °C for 2 hours. After the completion of the reaction, 3.63 L water was added into the reaction mixture. The reaction mixture was basified by 20% sodium carbonate solution and the organic layer was separated. The aqueous layer was extracted with 1.45 L methylene dichloride. The combined organic layers were distilled completely under vacuum. 14.5 L acetonitrile was added to the residue and heated to reflux for 15 min. The reaction mixture was cooled to 25 to 35° C and stirred for 1 hour. The reaction mass was filtered, washed with 1.45 L acetonitrile and dried to obtain 632 g of title compound as crystalline solid having 99.01% HPLC purity. The crystalline compound is characterized by Powder x-ray diffraction pattern (FIG.l) and Differential Scanning Calorimetry (FIG.2).
2 4
Example-17: Preparation of (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N -(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine In a 10 mL round bottom flask, 300 mg (0.644 mmol) (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine, 2.7 mL acetonitrile and 0.3 mL water were added and the reaction mixture was heated to reflux for 15 min. The reaction mixture was cooled to 25 to 35 °C and stirred for 1 hour. The reaction mass was filtered, washed with acetonitrile and dried to obtain 201 mg (67%) title compound as crystalline solid. The crystalline compound is characterized by Powder x-ray diffraction pattern (FIG.3) and Differential Scanning Calorimetry (FIG.4).
SYN
WO 2015165660
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015165660
Example 13
Synthetic scheme 1
Synthetic scheme 2
(R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine
In a 50 mL round-bottomed flask (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3-methylpiperazin-1-yl)pyrimidine-2,4-diamine hydrochloride (190 mg, 0.42 mmol, Example 2) was taken in DCM (2 mL) to give a yellow suspension. To this Hunig’s Base (0.184 mL, 1.05 mmol) was added and the suspension turned clear. After 10 minutes, it turned into a white suspension. After another 10 minutes, the mixture was concentrated to dryness. Resultant residue was dissolved in ethanol (absolute, 99.5%) (3 mL) and formaldehyde (0.042 mL, 0.63 mmol) was added and stirred for 10 minutes. White suspension slowly cleared to yellow solution. To this clear solution sodium cyanoborohydride (26.4 mg, 0.42 mmol) was added in one portion to get white suspension. After 30 minutes LCMS showed completion of reaction. The reaction mixture was concentrated and the crude was purified through reverse phase HPLC GILSON instrument to get the pure solid of (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine (80 mg, 40.8 %).1H NMR (300
MHz, DMSO-d6) δ ppm 0.67 – 0.78 (m, 2 H) 1.00 (d, J=6.22 Hz, 3 H) 1.02 – 1.08 (m, 2 H) 1.96 – 2.10 (m, 1 H) 2.23 (s, 7 H) 2.30 – 2.38 (m, 4 H) 2.73 – 2.96 (m, 4 H) 3.33 (s, 3 H) 6.83 (s, 1 H) 7.67 (d, J=5.09 Hz, 1 H) 8.00 (s, 1 H) 8.03 (s, 1 H) 9.26 (s,1 H) MS (ES+), (M+H)+ = 466.45 for C21H32FN9.
SYN
Nature Communications (2015), 6, 6715.
https://www.nature.com/articles/ncomms7715
Hameed P., S., Solapure, S., Patil, V. et al. Triaminopyrimidine is a fast-killing and long-acting antimalarial clinical candidate. Nat Commun 6, 6715 (2015). https://doi.org/10.1038/ncomms7715
The widespread emergence of Plasmodium falciparum (Pf) strains resistant to frontline agents has fuelled the search for fast-acting agents with novel mechanism of action. Here, we report the discovery and optimization of novel antimalarial compounds, the triaminopyrimidines (TAPs), which emerged from a phenotypic screen against the blood stages of Pf. The clinical candidate (compound 12) is efficacious in a mouse model of Pf malaria with an ED99 <30 mg kg−1 and displays good in vivo safety margins in guinea pigs and rats. With a predicted half-life of 36 h in humans, a single dose of 260 mg might be sufficient to maintain therapeutic blood concentration for 4–5 days. Whole-genome sequencing of resistant mutants implicates the vacuolar ATP synthase as a genetic determinant of resistance to TAPs. Our studies highlight the potential of TAPs for single-dose treatment of Pf malaria in combination with other agents in clinical development.

(A) Pyridine, microwave, 150 °C, 45 min. (B) (i) POCl3, reflux, 6 h (ii) sodium carbonate, di-tert-butyl dicarbonate, room temperature, 16 h. (C) N,N-Diisopropylethylamine (DIPEA), ethanol, microwave, 110 °C, 1 h. (D) (i) Potassium tert-butoxide, 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP), pd2(dba)3, toluene, reflux, 12 h. (E) HCl (4 N) in dioxane, 15–30 min. (F) Compound 9, DIPEA, dichloromethane, formaldehyde (HCHO), sodium cyanoborohydride, 15 min.
Synthesis of (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1, 5-dimethyl-1H-pyrazol-3-yl)-5-(3, 4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine (12). (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3-methylpiperazin-1-yl)pyrimidine-2,4-diamine hydrochloride (compound 9, 190 mg, 0.42 mmol) was taken in dichloromethane (2 ml) to give a yellow suspension. To this Hunig’s Base (0.184 ml, 1.05 mmol) was added and the suspension turned clear. After 10 min of stirring, reaction mixture turned into a white suspension and then it was concentrated to dryness. Resultant residue was dissolved in ethanol (absolute, 99.5%) (3 ml), and formaldehyde (0.042 ml, 0.63 mmol) was added and stirred for 10 min. To this clear solution, sodium cyanoborohydride (26.4 mg, 0.42 mmol) was added in one portion to get a white suspension. The reaction mixture was concentrated and the crude product was purified through reverse-phase chromatography to get the pure off-white solid of (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1, 5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine (80 mg, 40.8%). Yield: 40.8%, purity: >95% by HPLC (ultraviolet at 220 and 254 nm). 1H NMR (300 MHz, DMSO-d6) δ 9.26 (s,1H), 8.03 (s, 1H) 8.00 (s, 1H) 7.67 (d, J=5.1 Hz, 1H) 6.83 (s, 1H) 3.33 (s, 3H) 2.96–2.73 (m, 4H) 2.75–2.50 (m, 1H) 2.38–2.30 (m, 4H) 2.23 (s, 7H) 2.10–1.96 (m, 1H),1.08–1.02 (m, 2H) 1.00 (d, J=6.2 Hz, 3H) 0.78–0.67 (m, 2H). 13C-NMR (126 MHz, DMO-d6) δ 155.30, 154.67, 152.10, 150.93, 148.98, 146.81. 145.29, 141.95, 140.31, 138.81, 124.91, 106.20, 97.07, 58.78, 51.87, 42.16, 35.28, 17.23. 10.99 and 8.77, HRMS (ESI): m/z calculated for C24H32FN9+H [M+H]: 466.2765. Found: 466. 2838. Traces of LC-MS, HRMS, 1H NMR and 13C-NMR of compound 12 are shown in Supplementary Figs 1–3.


| Product vision |
|
| MoA |
|
| Key features |
|
| Challenges |
|
| Status |
|
| Next milestone |
|
| Previously |
|
Zydus receives Orphan Drug Designation from USFDA for ZY-19489, a novel compound to treat malaria;
ZY19489 is a novel antimalarial compound active against all current clinical strains of P. falciparum and P. vivax, including drug-resistant strains.
Zydus Cadila listed as Cadila Healthcare Limited announced that its antimalarial compound ZY19489 (MMV253), currently in development together with Medicines for Malaria Venture (MMV), a leading product development partnership (PDP) in antimalarial drug research, has received Orphan Drug Designation from the USFDA.
Orphan drug designation provides eligibility for certain development incentives, including tax credits for qualified clinical testing, prescription drug user fee exemptions, and seven-year marketing exclusivity upon FDA approval.
The company said that the Phase I study of ZY19489 has demonstrated a long half-life and potential for a single-dose cure for malaria. In a separate malaria challenge trial, potent antimalarial activity has been demonstrated following single-dose oral administration of ZY19489.
“As a global community facing threats from rapidly mutating malaria strains and the rise in artemisinin resistance cases, we have to be prepared with novel therapeutic drugs. ZY-19489 is a potential single dose radical cure for P. falciparum and P. vivax malaria which is a major global health risk today,” Pankaj R. Patel, Chairman, Zydus Group, said.
“ZY19489 is a potent, first in class molecule, originally discovered and elaborated in India” said Dr. Timothy Wells, Chief Scientific Officer, MMV. “It has tremendous potential as part of a new generation of treatments and is fully active against drug resistant strains of malaria which are increasingly a concern.”
Artemisinin resistance is seen as a mounting challenge to the global fight against malaria. ZY19489 is being developed to provide an effective alternative to the current front-line antimalarial drugs for the treatment of P. falciparum and P. vivax malaria, as artemisinin-based combination therapies (ACTs) are under threat of resistance.
As per the World Malaria Report 2021, there were an estimated 241 million cases of malaria worldwide and the estimated number of malaria deaths stood at 627,000 in 2020. A major health concern, it is estimated that a child dies from malaria every minute. About 96% of malaria deaths globally were in 29 countries. India accounted for about 82% of all malaria deaths in the WHO South-East Asia Region.
////////////ZY 19489, MMV 253, Orphan Drug Designation, PHASE 1, ZYDUS CADILA, ANTIMALARIAL
Cn1nc(Nc2nc(Nc3cc(C4CC4)c(F)c(C)n3)ncc2N2C[C@@H](C)N(C)CC2)cc1C
CC1CN(CCN1C)C2=CN=C(N=C2NC3=NN(C(=C3)C)C)NC4=NC(=C(C(=C4)C5CC5)F)C
Desidustat


Ranjit Desai
DESIDUSTAT
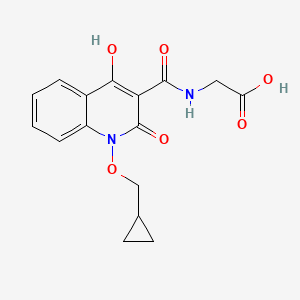
2-(1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carboxamido)acetic acid
desidustat
Glycine, N-((1-(cyclopropylmethoxy)-1,2-dihydro-4-hydroxy-2-oxo-3-quinolinyl)carbonyl)-
N-(1-(Cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl)glycine
ZYAN1 compound
(1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl) glycine in 98% yield, as a solid. MS (ESI-MS): m/z 333.05 (M+H) +. 1H NMR (DMSO-d 6): 0.44-0.38 (m, 2H), 0.62-0.53 (m, 2H), 1.34-1.24 (m, 1H), 4.06-4.04 (d, 2H), 4.14-4.13 (d, 2H), 7.43-7.39 (t, 1H), 7.72-7.70 (d, 1H), 7.89-7.85 (m, 1H), 8.11-8.09 (dd, 1H), 10.27-10.24 (t, 1H), 12.97 (bs, 1H), 16.99 (s, 1H). HPLC Purity: 99.85%
Oxemia (Desidustat) has received approval from the Drug Controller General of India. This was an incredible team effort by Zydans across the organization and I am so proud of what we have accomplished. Oxemia is a breakthrough treatment for Anemia associated with Chronic Kidney Disease in Patients either on Dialysis or Not on Dialysis, and will help improve quality of life for CKD patients. Team #zydus , on to our next effort!

Desidustat (INN, also known as ZYAN1) is a drug for the treatment of anemia of chronic kidney disease. This drug with the brand name Oxemia is discovered and developed by Zydus Life Sciences.[1] The subject expert committee of CDSCO has recommended the grant of permission for manufacturing and marketing of Desidustat 25 mg and 50 mg tablets in India,based on some conditions related to package insert, phase 4 protocols, prescription details, and GCP.[2] Clinical trials on desidustat have been done in India and Australia.[3] In a Phase 2, randomized, double-blind, 6-week, placebo-controlled, dose-ranging, safety and efficacy study, a mean hemoglobin increase of 1.57, 2.22, and 2.92 g/dL in desidustat 100, 150, and 200 mg arms, respectively, was observed.[4] The Phase 3 clinical trials were conducted at additional lower doses as of 2019.[5] Desidustat is developed for the treatment of anemia as an oral tablet, where currently injections of erythropoietin and its analogues are drugs of choice. Desidustat is a HIF prolyl-hydroxylase inhibitor. In preclinical studies, effects of desidustat was assessed in normal and nephrectomized rats, and in chemotherapy-induced anemia. Desidustat demonstrated hematinic potential by combined effects on endogenous erythropoietin release and efficient iron utilization.[6][7] Desidustat can also be useful in treatment of anemia of inflammation since it causes efficient erythropoiesis and hepcidin downregulation.[8] In January 2020, Zydus entered into licensing agreement with China Medical System (CMS) Holdings for development and commercialization of desidustat in Greater China. Under the license agreement, CMS will pay Zydus an initial upfront payment, regulatory milestones, sales milestones and royalties on net sales of the product. CMS will be responsible for development, registration and commercialization of desidustat in Greater China.[9] It has been observed that desidustat protects against acute and chronic kidney injury by reducing inflammatory cytokines like IL-6 and oxidative stress [10] A clinical trial to evaluate the efficacy and safety of desidustat tablet for the management of Covid-19 patients is ongoing in Mexico, wherein desidustat has shown to prevent acute respiratory distress syndrome (ARDS) by inhibiting IL-6.[11] Zydus has also received approval from the US FDA to initiate clinical trials of desidustat in chemotherapy Induced anemia (CIA).[12]. Desidustat has met the primary endpoints in the phase 3 clinical trials and Zydus had filed the New Drug Application (NDA) to DCGI in November, 2021.[13]\
CLIP
Zydus receives DCGI approval for new drug Oxemia; what you need to know
The new drug is an oral, small molecule hypoxia-inducible factor-prolyl hydroxylase (HIF-PH) inhibitor, Zydus said in a statement.
Gujarat-based pharma company Zydus Lifesciences on Monday received the Drugs Controller General of India (DCGI) approval for its new drug application for a first-of-its-kind oral treatment for anemia associated with Chronic Kidney Disease (CKD) – Oxemia (Desidustat).
The new drug is an oral, small molecule hypoxia-inducible factor-prolyl hydroxylase (HIF-PH) inhibitor, the drug firm said in a statement.
Desidustat showed good safety profile, improved iron mobilization and LDL-C reduction in CKD patients in DREAM-D and DREAM-ND Phase III clinical trials, conducted in approximately 1,200 subjects. Desidustat provides CKD patients with an oral convenient therapeutic option for the treatment of anemia. The pharma major did not, however, declare the cost per dose if the drug is available in the market.
“After more than a decade of research and development into the science of HIF-PH inhibitors, results have demonstrated that Oxemia addresses this unmet need and additionally reduces hepcidin, inflammation and enables better iron mobilization. This advancement offers ease of convenience for the patient and will also reduce the disease burden by providing treatment at an affordable cost, thereby improving the quality of life for patients suffering from Chronic Kidney Disease,” Chairman of Zydus Lifesciences Pankaj Patel said.
Chronic Kidney Disease (CKD) is a progressive medical condition characterised by a gradual loss of kidney function and is accompanied by comorbidities like anemia, cardiovascular diseases (hypertension, heart failure and stroke), diabetes mellitus, eventually leading to kidney failure.
PATENT
|
Scheme 3:
|
Step 1′a Process for Preparation of ethyl 2-iodobenzoate (XI-a)
Step-2 Process for the Preparation of ethyl 2-((tert-butoxycarbonyl)(cyclopropylmethoxy)aminolbenzoate (XII-a)
Step 3 Process for the Preparation of ethyl 2-((cyclopropylmethoxy)amino)benzoate (XIII-a)
Step 4 Process for the Preparation of ethyl 24N-(cyclopropylinethoxy)-3-ethoxy-3-oxopropanamido)benzoate (XIV-a)
Step 5: Process for the Preparation of ethyl 1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2 dihydroquinolline-3-carboxylate (XY-a)
Purification
Step 6 Process for the Preparation of ethyl (1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl)glycinate (XVI-a)
Purification
Step 7: Process for the Preparation of (1-(cyclopropylmethoxy)-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbonyl)glycine (I-a)
Polymorphic Data (XRPD):
References[edit]
- ^ “Zydus receives DCGI approval for new drug Oxemia; what you need to know”.
- ^ CDSCO, SEC Committee. “SEC meeting to examine IND proposals, dated 29.12.2021”. CDSCO website Govt of India. CDSCO. Retrieved 19 January 2022.
- ^ Kansagra KA, Parmar D, Jani RH, Srinivas NR, Lickliter J, Patel HV, et al. (January 2018). “Phase I Clinical Study of ZYAN1, A Novel Prolyl-Hydroxylase (PHD) Inhibitor to Evaluate the Safety, Tolerability, and Pharmacokinetics Following Oral Administration in Healthy Volunteers”. Clinical Pharmacokinetics. 57 (1): 87–102. doi:10.1007/s40262-017-0551-3. PMC 5766731. PMID 28508936.
- ^ Parmar DV, Kansagra KA, Patel JC, Joshi SN, Sharma NS, Shelat AD, Patel NB, Nakrani VB, Shaikh FA, Patel HV; on behalf of the ZYAN1 Trial Investigators. Outcomes of Desidustat Treatment in People with Anemia and Chronic Kidney Disease: A Phase 2 Study. Am J Nephrol. 2019 May 21;49(6):470-478. doi: 10.1159/000500232.
- ^ “Zydus Cadila announces phase III clinical trials of Desidustat”. 17 April 2019. Retrieved 20 April 2019 – via The Hindu BusinessLine.
- ^ Jain MR, Joharapurkar AA, Pandya V, Patel V, Joshi J, Kshirsagar S, et al. (February 2016). “Pharmacological Characterization of ZYAN1, a Novel Prolyl Hydroxylase Inhibitor for the Treatment of Anemia”. Drug Research. 66 (2): 107–12. doi:10.1055/s-0035-1554630. PMID 26367279.
- ^ Joharapurkar AA, Pandya VB, Patel VJ, Desai RC, Jain MR (August 2018). “Prolyl Hydroxylase Inhibitors: A Breakthrough in the Therapy of Anemia Associated with Chronic Diseases”. Journal of Medicinal Chemistry. 61 (16): 6964–6982. doi:10.1021/acs.jmedchem.7b01686. PMID 29712435.
- ^ Jain M, Joharapurkar A, Patel V, Kshirsagar S, Sutariya B, Patel M, et al. (January 2019). “Pharmacological inhibition of prolyl hydroxylase protects against inflammation-induced anemia via efficient erythropoiesis and hepcidin downregulation”. European Journal of Pharmacology. 843: 113–120. doi:10.1016/j.ejphar.2018.11.023. PMID 30458168. S2CID 53943666.
- ^ Market, Capital (20 January 2020). “Zydus enters into licensing agreement with China Medical System Holdings”. Business Standard India. Retrieved 20 January 2020 – via Business Standard.
- ^ Joharapurkar, Amit; Patel, Vishal; Kshirsagar, Samadhan; Patel, Maulik; Savsani, Hardikkumar; Jain, Mukul (22 January 2021). “Prolyl hydroxylase inhibitor desidustat protects against acute and chronic kidney injury by reducing inflammatory cytokines and oxidative stress”. Drug Development Research. 82 (6): 852–860. doi:10.1002/ddr.21792. PMID 33480036. S2CID 231680317.
- ^ “Zydus’ trials of Desidustat shows positive results for Covid-19 management”. The Hindu Business Line. The Hindu. Retrieved 25 January 2021.
- ^ “Zydus receives approval from USFDA to initiate clinical trials of Desidustat in cancer patients receiving chemotherapy”. PipelineReview.com. La Merie Publishing. Retrieved 22 January 2021.
- ^ “Stock Share Price | Get Quote | BSE”.
|
WO – 30.04.2020
|
|
2.WO/2020/058882METHODS OF PRODUCING VENOUS ANGIOBLASTS AND SINUSOIDAL ENDOTHELIAL CELL-LIKE CELLS AND COMPOSITIONS THEREOF
WO – 26.03.2020
|
|
3.110876806APPLICATION OF HIF2ALPHA AGONIST AND ACER2 AGONIST IN PREPARATION OF MEDICINE FOR TREATING ATHEROSCLEROSIS
CN – 13.03.2020
|
|
US – 28.11.2019
|
|
WO – 06.09.2019
|
|
EA – 30.10.2015
Настоящее изобретение относится к новым соединениям общей формулы (I), фармацевтическим композициям, содержащим указанные соединения, применению этих соединений для лечения состояний, опосредованных пролилгидроксилазой HIF, и к способу лечения анемии, включающему введение заявленных соединений |
|
EP – 28.10.2015
|
|
US – 22.10.2015
The present invention relates to novel compounds of the general formula (I), their tautomeric forms, their stereoisomers, their pharmaceutically acceptable salts, pharmaceutical compositions containing them, methods for their preparation, use of these compounds in medicine and the intermediates involved in their preparation. |
|
WO – 03.07.2014
|
 |
|
| Clinical data | |
|---|---|
| Other names | ZYAN1 |
| Identifiers | |
| CAS Number | |
| UNII | |
| Chemical and physical data | |
| Formula | C16H16N2O6 |
| Molar mass | 332.312 g·mol−1 |
| 3D model (JSmol) | |
Date
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT04215120 | Desidustat in the Treatment of Anemia in CKD on Dialysis Patients | Phase 3 | Recruiting | 2020-01-02 |
| NCT04012957 | Desidustat in the Treatment of Anemia in CKD | Phase 3 | Recruiting | 2019-12-24 |
////////// DESIDUSTAT, ZYDUS CADILA, COVID 19, CORONA VIRUS, PHASE 3, ZYAN 1, OXEMIA, APPROVALS 2022, INDIA 2022

Zydus Cadila to launch India’s 1st Tetravalent Inactivated Influenza vaccine – VaxiFlu – 4

Zydus Cadila to launch India’s 1st Tetravalent Inactivated Influenza vaccine – VaxiFlu – 4
Ahmedabad, February 24, 2017
Zydus Cadila, a research-driven, global healthcare provider has received approvals from the Drug Controller General of India (DCGI), Central Drugs Standard Control Organization (CDSCO) and the Central Drug Laboratory (CDL) to market the Tetravalent Inactivated Influenza vaccine for seasonal flu, VaxiFlu – 4. With this, Zydus Cadila will become the first Indian pharma company and second in the world to launch a Tetravalent Inactivated Influenza vaccine. The vaccine provides protection from the four influenza viruses- H1N1, H3N2, Type B (Brisbane) and Type B (Phuket).

VaxiFlu – 4 will be marketed by Zydus Vaxxicare – a division of the group focussing on preventives. The Tetravalent Inactivated Influenza vaccine has been developed at the Vaccine Technology Centre (VTC) in Ahmedabad which has proven capabilities in researching, developing, and manufacturing of safe and efficacious vaccines. The group was also the first to indigenously develop, manufacture and launch India’s first vaccine against H1N1 – Vaxiflu-S.
VTC further plans to develop a wide spectrum of vaccines against bacterial, viral and protozoal infections and has a robust pipeline of vaccines like Pentavalent (DTP-Hib-HepB), Conjugated Typhoid Vaccine, HPV, MMRV, Malaria and Hepatitis B vaccines. The group also markets the anti-rabies vaccine and the typhoid vaccine.
Speaking on the development Mr. Pankaj R. Patel, Chairman and Managing Director, Zydus Cadila said, “Disease prevention is the key to public health in both the developing and the developed world and vaccines have the potential to improve the quality of life in both spectrums. In countries such as India, there is a pressing need for low cost, high quality vaccines that can address healthcare challenges. With the launch of vaccines like VaxiFlu – 4 we are serving the cause of public health and meeting the twin challenge of affordability and accessibility.”
Influenza, or the “flu” as it is commonly called, is an infection of the respiratory tract. It is a dreaded disease and the morbidity and mortality rates associated with influenza are especially high during pandemics. Annually it is estimated that it attacks 5-10% of adults and 20-30% of children globally and causes significant levels of illness, hospitalization and death. In India, the 2009 swine flu pandemic infected more than 10 million people and resulted in more than 18000 deaths worldwide.
The last major outbreak in India occurred in 2015 with more than 33000 registered cases of influenza and over 2000 deaths. There are different strains of influenza viruses that infect human beings, the predominant ones being influenza A and influenza B. The common subtypes of influenza A found in general circulation amongst people are H1N1 (which was responsible for the devastating swine flu pandemic) and H3N2.
The subtypes of influenza B commonly found in circulation are influenza B (Brisbane – Victoria lineage) and influenza B (Phuket – Yamagata lineage). Vaccination against influenza is the most effective way to protect oneself against the dangers of influenza. Majority of the influenza vaccines available in India are inactivated trivalent influenza vaccines.
These vaccines provide protection against 2 strains of influenza A and 1 strain of influenza B. Protection against only 1 subtype of influenza B often leads to a vaccine mismatch i.e. the antigen of influenza B present in the trivalent vaccine may not match the influenza B subtype circulating during the season, leading to suboptimal protection. A quadrivalent vaccine, by virtue of having a comprehensive coverage against 2 strains of both influenza A and influenza B, provides a broader protection and significantly reduces the risk of vaccine mismatch. Vaxiflu – 4 is the first quadrivalent influenza vaccine in india.
About Zydus Zydus Cadila is an innovative, global pharmaceutical company that discovers, develops, manufactures and markets a broad range of healthcare therapies, including small molecule drugs, biologic therapeutics and vaccines. The group employs over 19,500 people worldwide, including 1200 scientists engaged in R & D, and is dedicated to creating healthier communities globally. For more information, please visit http://www.zyduscadila.com
Zydus’ vaccine research programme The Vaccine Technology Centre (VTC) is the vaccine research centre of the Zydus Group. The group has two state-of-the-art R & D Centers, one located in Catania, Italy and the other in Ahmedabad, in the western part of India. The goup has been developing vaccines for the basic vaccine programmes such as Diphtheria, Pertussis, Tetanus, Haemophilus Influenzae type B, Hepatitis B, Measles, Mumps, Rubella, Varicella, Influenza and Typhoid fever. In addition, it is developing new vaccines such as Human Papilloma Virus, Leishmaniasis, Malaria, Haemorrhagic Congo Fever, Ebola and Japanese Encephalitis.
Ref
Zydus Cadila to launch India’s 1st Tetravalent Inactivated Influenza vaccine – VaxiFlu – 4 Read more: https://goo.gl/xuSTfK #ZydusAnnouncement

///////////Zydus Cadila, Tetravalent Inactivated, Influenza vaccine, VaxiFlu – 4
Identification of an Orally Efficacious GPR40/FFAR1 Receptor Agonist from Zydus Cadila



The compounds of theese type lower blood glucose, regulate peripheral satiety, lower or modulate triglyceride levels and/or cholesterol levels and/or low-density lipoproteins (LDL) and raises the high-density l ipoproteins (HDL) plasma levels and hence are useful in combating different medical conditions, where such lowering (and raising) is beneficial. Thus, it could be used in the treatment and/or prophylaxis of obesity, hyperlipidemia, hypercholesteremia, hypertension, atherosclerotic disease events, vascular restenosis, diabetes and many other related conditions.
The compounds of are useful to prevent or reduce the risk of developing atherosclerosis, which leads to diseases and conditions such as arteriosclerotic cardiovascular diseases, stroke, coronary heart diseases, cerebrovascular diseases, peripheral vessel diseases and related disorders. -These compounds are useful for the treatment and/or prophylaxis of metabolic disorders loosely defined as Syndrome X. The characteristic features of Syndrome X include initial insulin resistance followed by hyperinsulinemia, dyslipidemia and impaired glucose tolerance. The glucose intolerance can lead to non-insulin dependent diabetes mel litus (N I DDM, Type 2 diabetes), which is characterized by hyperglycemia, which if not controlled may lead to diabetic complications or metabolic disorders caused by insulin resistance. Diabetes is no longer considered to be associated only with glucose metabol ism, but it affects anatomical and physiological parameters, the intensity of which vary • depending upon stages/duration and severity of the diabetic state. The compounds of this invention are also useful in prevention, halting or slowing progression or reducing the risk of the above mentioned disorders along with the resulting secondary diseases such as cardiovascular diseases, l ike arteriosclerosis, atherosclerosis; diabetic retinopathy, diabetic neuropathy and renal disease including diabetic nephropathy, glomerulonephritis, glomerular sclerosis, nephrotic syndrome, hypertensive nephrosclerosis and end stage renal diseases, like microalbuminuria and albuminuria, which may be result of hyperglycemia or hyperinsulinemia.
Diabetes mellitus is a serious disease affl icting over 1 00 mi l lion people worldwide. In the United States, there are more than 12 mill ion diabetics, with 600,000 new cases diagnosed each year.
Diabetes mellitus is a diagnostic term for a group of disorders characterized by abnormal glucose homeostasis resulting in elevated blood sugar. There are many- types of diabetes, but the two most common are Type 1 (also referred to as insulin- dependent diabetes mellitus or IDDM) and Type II (also referred to as non- insulin-dependent diabetes mellitus or NIDDM).
The etiology of the different types of diabetes is not the same; however, everyone with diabetes has two things in common: overproduction of glucose by the liver and little or no ability to move glucose out of the blood, into the cells where it becomes the body’s primary fuel.
People who do not have diabetes rely on insulin, a hormone made in the pancreas, to move glucose from the blood into the cells of the body. However, people who have diabetes either don’t produce insulin or can’t efficiently use the insulin they produce; therefore, they can’t move glucose into their cells. Glucose accumulates in the blood creating a condition called hyperglycemia, and over time, can cause serious health problems.
Diabetes is a syndrome with interrelated metabolic, vascular, and neuropathic components. The metabolic syndrome, generally characterized by hyperglycemia, comprises alterations in carbohydrate, fat and protein metabolism caused by absent or markedly reduced insulin secretion and/or ineffective insulin action. The vascular syndrome consists of abnormalities in the blood vessels leading to cardiovascular, retinal and renal complications. Abnormal ities in the peripheral and autonomic nervous systems are also part of the diabetic syndrome.
About 5% to 10% of the people who have diabetes have IDDM. These individuals don’t produce insulin and therefore must inject insulin to keep their blood glucose levels normal . IDDM is characterized by low or undetectable levels of endogenous insulin production caused by destruction of the insulin-producing β cells of the pancreas, the characteristic that most readily distinguishes IDDM from NIDDM. IDDM, once termed juvenile-onset diabetes, strikes young and older adults alike.
Approximately 90 to 95% of people with diabetes have Type II (or NIDDM). NIDDM subjects produce insulin, but the cells in their bodies are insulin resistant: the cells don’t respond properly to the hormone, so glucose accumulates i n their blood. NIDDM is characterized by a relative disparity between endogenous insulin production and insulin requirements, leading to elevated blood glucose levels. In contrast to IDDM, there is always some endogenous insulin production in NIDDM; many NIDDM patients have normal or even elevated blood insul in levels, whi le other NIDDM patients have inadequate insul in production ( otwein, R. et al. N. Engl. J. Med. 308, 65-71 ( 1983)). Most people diagnosed with NIDDM are age 30 or older, and half of all new cases are age 55 and older. Compared with whites and Asians, NIDDM is more common among Native Americans, African-Americans, Latinos, and Hispanics. In addition, the onset can be insidious or even clinically non-apparent, making diagnosis difficult.
The primary pathogenic lesion on NIDDM has remained elusive. Many have suggested that primary insulin resistance of the peripheral tissues is the initial event. Genetic epidemiological studies have supported this view. Similarly, insulin secretion abnormalities have been argued as the primary defect in NIDDM. It is l ikely that both phenomena are important contributors to the disease process (Rimoin, D. L., et. al. Emery and Rimoin’s Principles and Practice of Medical Genetics 3rd Ed. 1 : 1401 – 1402 ( 1996)).
Many people with NIDDM have sedentary lifestyles and are obese; they weigh approximately 20% more than the recommended weight for their height and build. Furthermore, obesity is characterized by hyperinsul inemia and insul in resistance, a feature shared with NIDDM, hypertension and atherosclerosis.
The G-protein -coupled receptor GPR 40 functions as a receptor for long-chain free fatty acids (FFAs) in the body and as such is impl icated in a large number of metabolic conditions in the body. For example it has been alleged that a GPR 40 agonist promotes insulin secretion whilst a GPR 40 antagonist inhibits insulin secretion and so depending upon the circumstances the agonist and antagonist may be useful as therapeutic agents for the number of insul in related conditions such as type 2 diabetes, obesity, impaired glucose tolerance, insul in resistance, neurodegenerative diseases and the like.
There is increasing evidences that lipids can also serve as extracel lular l igands for a specific class of receptors and thus act as “nutritional sensors” (Nolan CJ et al. J. Clinic. Invest., 2006, 1 1 6, 1 802- 1 812The free fatty acids can regulate cell function. Free fatty acids have demonstrated as ligands for orphan G protein-coupled receptors (GPCRs) and have been proposed to play a critical role in physiological glucose homeostasis.
GPR40, GPR 120, GPR41 and GPR43 exemplify a growing number of GPCRs that have been shown to be activated by free fatty acids. GPR40 and GPR 120 are activated by medium to long-chain free fatty acids whereas GPR 41 and GPR 43 are activated by short-chain fatty acid (Brown AJ et al, 2003).
GPR 40 is highly expressed on pancreatic β-cells, and enhances glucose- stimulated insulin secretion {Nature, 2003, 422, 1 73- 1 76, J. Bio. Chem. 2003, 278, 1 1303- 1 13 1 1 , Biochem. Biophys. Res. Commun. 2003, 301, 406-4 10).
Free fatty acids regulate insulin secretion from pancreatic β cells through GPR40 is reported {Lett, to Nature 2003, 422, 1 73- 1 76).
GlaxoSmith line Research and Development, US published an article in Bioorg. Med. Chem. Lett. 2006, 16, 1840- 1 845 titled Synthesis and activity of small molecule GPR40 agonists. (Does this describe GW9508?)Another article titled Pharmacological regulation of insul in secretion in ΜΓΝ6 cells through the fatty – acid receptor GPR40: Identification of agonist and antagonist small molecules is reported in
Br. J. Pharmacol. 2006, 148, 619-928 from GlaxoSmithKl i ne. USA (Does this describe GW9508?) ‘

GW 9508.
Solid phase synthesis and SAR of small molecule agonists for the. GPR 40 receptor is published in Bioorg. Med. Chem. Lett. 2007, 16, 1 840- 1 845 by Glaxo Smith line Res. 8c Dev. USA, including those with the following structures.

Johnson & Johnson Pharmaceutical Research and development , USA published
Synthesis and Biological Evaluation of 3-Aryl-3-(4-phenoxy)-propanoic acid as a Novel Series of G-protein -coupled receptor 40 agonists J. Med. Chem. 2007,
76, 2807-2817)
National Institutes of Health, Bethesda, Maryland publ ished “Bidirectional Iterative Approach to the Structural Delineation of the Functional Chemo print in GPR 40 for agonist Recognition (J. Med. Chem. 2007. 50, 298 1 -2990).
Discov roglucinols of the following formula

as a new class of GPR40 (FFAR 1 ) agonists has been publ ished by Piramal Li fe Sciences, Ltd. in Bioorg. Med. Chem. Lett. 2008, 1 8, 6357-6361
Synthesis and SAR of 1 ,2,3,4-tctrahydroisoquinoline- l -ones as novel G-protein coupled receptor40(GPR40) antagonists of the following formula has been published in Bioorg. Med. Chem. Lett. 2009, 79, 2400-2403 by Pfizer

Piramal Life Sciences Ltd. published “Progress in the discovery and development of small molecule modulators of G-protei n coupled receptor 40(GPR40/FFA 1 /FFAR1 ), an emerging target for type 2 diabetes” in Exp. Opin. Therapeutic Patents 2009, 19(2), 237 -264.
There was a report published in Zhonggno Bingli Shengli ^Zazhi 2009, 25(7), 1376- 1380 from Sun Yat. Sen University, Guangzhou, which mentions the role GPR 40 on lipoapoptosis.
A novel class of antagonists for the FFA’s receptor GPR 40 was published in Biochem. Biophy. Res. Commun. 2009 390, 557-563.

N41 (DC260126)
Merck Res. Laboratories published “Discovery of 5-aryloxy-2,4-thiazolidinediones as potent GPR40 agonists” having the following formula in Bioorg. Med. Chem. Lett. 2010 20, 1298- 1 301

Discovery of TA -875, a potent, selective, and oral ly bioavai lable G PR 40 agonist is reported by Takeda Pharmaceutical Ltd. ACS Med. Chem. Lett. 2010,
7(6), 290-294

In another report from University of Southern Denmark” Structure -Activity of Dihydrocinnamic acids and discovery of potent FFA l (GPR40) agonist TUG-469″ is reported in ACS Me -349.

The free fatty acid 1 receptor (FFAR 1 or GPR40), which is highly expressed on pancreatic β-cells and amplifies glucose-stimulated insul in secretion, has emerged as an attractive target for the treatment of type 2 diabetes (ACS Med. Chem. Lett. 2010, 1 (6), 290-294).
G-protein coupled receptor (GPR40) expression and its regulation in human pancreatic islets: The role of type 2 diabetes and fatty acids is reported in Nutrition Metabolism & Cardiovascular diseases 2010, 2(9( 1 ), 22-25
Ranbaxy reported “Identification of Berberine as a novel agonist of fatty acid receptor GPR40” in Phytother Res. 2010, 24, 1260-63.
The following substituted 3-(4-aryloxyaryI)-propanoic acids as GPR40 agonists are reported by Merck Res. Lab. in Bioorg. ed. Chem. Lett. 201 1 , 21, 3390-3394

4 EC50=0.970 μΜ 5. EC50=2.484 μΜ
CoMSIA study on substituted aryl alkanoic acid analogs as GPR 40 agonists is reported Chem. Bio. Drug. Des. 201 1 , 77, 361 -372
Takeda further published “Design, Synthesis and biological activity of potential and orally available G-protein coupled receptor 40 agonists” in J. Med. Chem. 201 1 , 54(5), 1365- 1 378.
Amgen disclosed a potent oral ly bioavai lable GPR 40 agonist AMG-837 in Bioorg. Med. Chem. Lett.

Discovery of phenylpropanoic acid derivatives containing polar functional ities as Potent and orally bioavailable G protein-coupled receptor 40 Agonist for the treatment of type 2 Diabetes is reported in J. Med. Chem. 2012, 55, 3756-3776 by Takeda.
Discovery of AM- 1638: A potent and orally bioavailable GPR40/FFA 1 full agonist is reported in ACS Med. Chem. Lett. 2012, 3(9), 726-730.


Sameer Agarwal
Cadila Healthcare Ltd., India
Sameer Agarwal has obtained Master’s in Chemistry from IIT, Delhi and was awarded DAAD (German Govt. Scholarship) fellowship to purse research project at Karlsruhe University, Germany. He has received PhD degree from Technical University, Dresden, Germany in the field of Synthetic and bio-organic chemistry under direction of Prof. Dr. Hans-Joachim Knölker, FRSC, a well-known scientist of present times for his contribution towards Alkaloid Chemistry. He worked as Research Scientist (Post-Doc), JADO Technologies, (collaboration with Max Planck Institute (MPI) of Molecular Cell Biology and Genetics and Chemsitry Department, Technical University), Germany. He then decided to return to his home country and working with Zydus Research Centre, Cadila Healthcare Ltd., Ahmedabad as Principal Scientist / Group Leader in the area of basic drug discovery and his research interest includes discovery of cardio metabolic, anti-inflammatory and oncology drugs. He has large number of publications in international journals and patents and is a reviewer of many prestigious journals including American Chemical Society.
Paper
Identification of an Orally Efficacious GPR40/ FFAR1 Receptor Agonist

GPR40/FFAR1 is a G protein-coupled receptor predominantly expressed in pancreatic β-cells and activated by long-chain free fatty acids, mediating enhancement of glucose-stimulated insulin secretion. A novel series of substituted 3-(4-aryloxyaryl)propanoic acid derivatives were prepared and evaluated for their activities as GPR40 agonists, leading to the identification of compound 5, which is highly potent in in vitro assays and exhibits robust glucose lowering effects during an oral glucose tolerance test in nSTZ Wistar rat model of diabetes (ED50 = 0.8 mg/kg; ED90 = 3.1 mg/kg) with excellent pharmacokinetic profile, and devoid of cytochromes P450 isoform inhibitory activity
Synthesis of compound 5 is depicted in Scheme 1a.
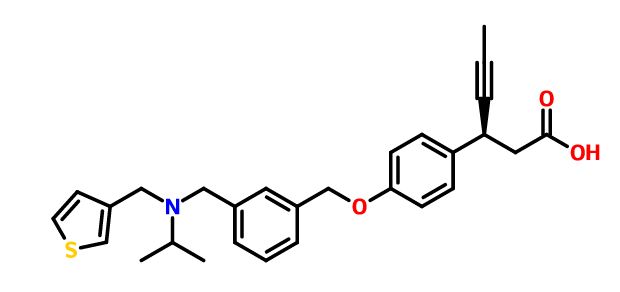
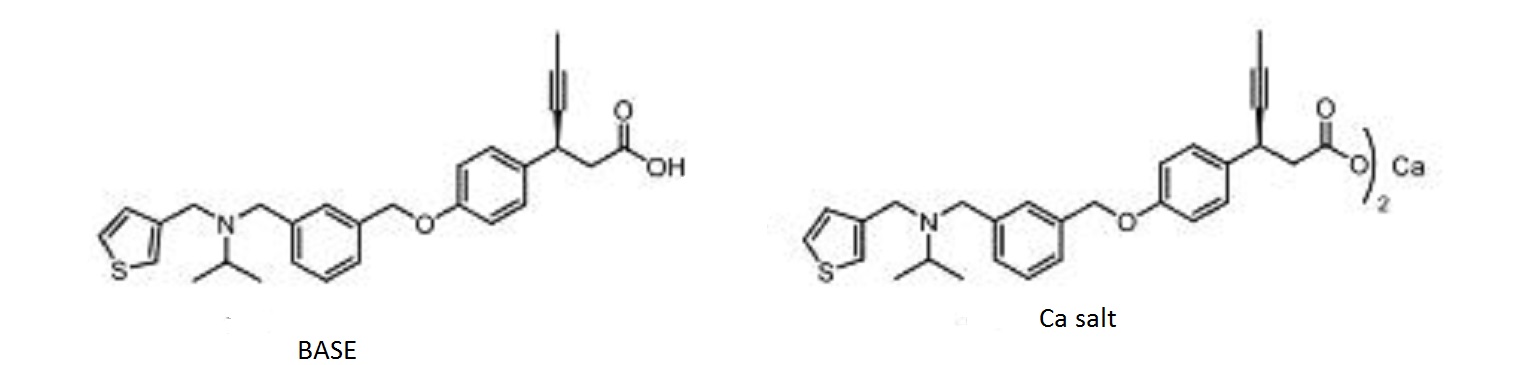
The reductive amination1 of commercially available 3-thiophene-aldehyde (3) and isopropyl amine using sodium triacetoxyborohydride resulted in secondary amine intermediate 4. Compound 4 on further reductive amination under similar conditions with aldehyde intermediate, (S)-3-(4-((3-formylbenzyl)oxy)phenyl)hex-4-ynoic acid (8), afforded 2d in high yields. The aldehyde intermediate, 8 was obtained from (S)-3-(4-hydroxyphenyl)hex-4-ynoic acid (6) as shown in Scheme 1b. Acid 6 was synthesized via 5-step reported procedure using commercially available 4-hydroxybenzaldehyde and Meldrum’s acid.2 Resolution of racemic acid 6 was accomplished via diastereomeric salt formation with (1S,2R)-1-amino-2-indanol followed by salt break with aqueous acid to furnish compound 6. Treatment of 6 with of 40% aqueous tetrabutylphosphonium hydroxide (nBu4POH) in THF, followed by addition of 3-formyl benzyl bromide (7), afforded aldehyde intermediate 8. Compound 2d was further converted to its corresponding calcium salt (5) in two-step sequence with excellent chemical purity.

Scheme 1a. Synthesis of Compounds 2d and 5. Reagent and Conditions: (a) CH(CH3)2NH2, NaB(OAc)3H, CH3COOH, dry THF, 0 ᵒC to r.t., 16 h; (b) Comp 8, NaB(OAc)3H, CH3COOH, dry THF, 0 ᵒC to r.t., 16 h; (c) NaOH, MeCN/H2O, r.t., 3 h; (d) CaCl2, MeOH/H2O, r.t., 16 h.
BASE
(S)-3-(4-((3-((isopropyl(thiophen-3- ylmethyl)amino)methyl)benzyl)oxy)phenyl)hex-4-ynoic acid (1.557 g, 3.34 mmol, 43.0 % yield) as wax solid.
1H NMR (400 MHz, DMSO-d6): δ = 12.35 (br s, 1H), 7.44 (q, J = 3.2 Hz, 2H), 7.32 – 7.24 (m, 6H), 7.04 (d, J = 4.8 Hz, 1H), 6.94 (d, J = 8.4 Hz, 2H), 5.06 (s, 2H), 3.93 (d, J = 2.4 Hz, 1H), 3.51 (d, J = 8.8 Hz, 4H), 2.84 (sept, J = 6.4 Hz, 1H), 2.57 (d, J = 8 Hz, 2H), 1.77 (d, J = 2.4 Hz, 3H), 1.01 (d, J = 6.4 Hz, 6H);
13C NMR and DEPT: DMSO-d6, 100MHz):- δ = 172.35 (C), 157.63 (C), 142.13 (C), 141.44 (C), 137.42 (C), 133.93 (C), 128.73 (CH), 128.64 (CH), 128.43 (CH), 127.99 (CH), 127.73 (CH), 126.28 (CH), 122.21 (CH), 115.10 (CH), 81.16 (C), 78.52 (C), 69.69 (CH2), 52.90 (CH2), 48.64 (CH), 48.49 (CH2), 43.44 (CH2), 33.15 (CH), 17.92 (CH3), 3.66 (CH3);
MS (EI): m/z (%) = 462.35 (100) (M+H) + ;
IR (KBr): ν = 3433, 2960, 2918, 2810, 1712, 1608, 1510, 1383, 1240, 1174, 1109, 1018 cm-1 .
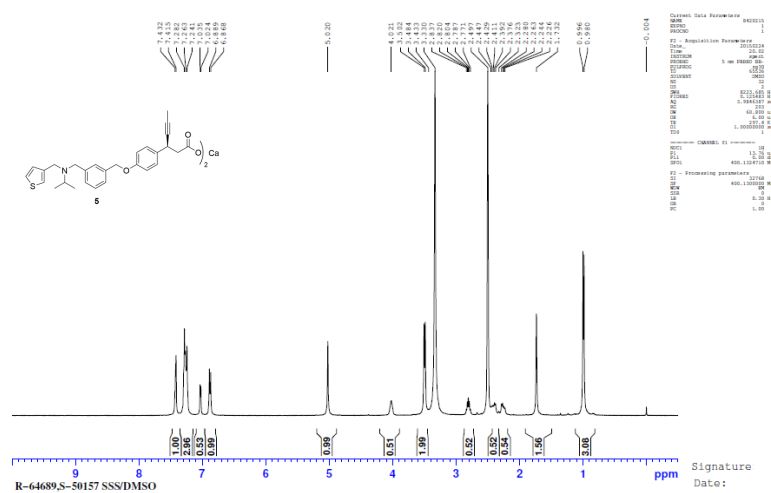
CA SALT
calcium (S)-3-(4-((3-((isopropyl(thiophen-3-yl methyl)amino)methyl)benzyl)oxy)phenyl)hex-4-ynoate (1.51 g, 1.536 mmol, 46% yield) as white powder. mp: 124.5 o C;
1H NMR (400 MHz, DMSO-d6): δ = 7.43 – 7.42 (m, 2H), 7.28 – 7.24 (m, 6H), 7.04 (d, J = 4.4 Hz, 1H), 6.89 (d, J = 8.4 Hz, 2H), 5.02 (s, 2H), 4.02 (s, 1H), 3.50 (d, J = 7.2 Hz, 4H), 2.84 – 2.77 (sept, J = 6.4 Hz, 1H), 2.43 (dd, J1 = 6.8 Hz, J2 = 7.2 Hz, 1H), 2.28 (dd, J1 = 6.8 Hz, J2 = 7.2 Hz, 1H), 1.73 (s, 3H), 0.99 (d, J = 6.4 Hz, 6H);
13C NMR and DEPT (100 MHz, DMSO-d6): δ = 177.78 (C), 157.23 (C), 142.11 (C), 141.4 (C), 137.46 (C), 135.81 (C), 128.83 (CH), 128.62 (CH), 128.40 (CH), 127.94 (CH), 127.69 (CH), 126.26 (CH), 122.18 (CH), 114.77 (CH), 83.18 (C), 77.32 (C), 69.66 (CH2), 52.89 (CH2), 48.59 (CH), 48.48 (CH2), 46.86 (CH2), 33.52 (CH), 17.88 (CH3), 3.78 (CH3);
MS (EI): m/z (%) = 462.05 (100) (M+H)+ ;
ESI-Q-TOF-MS: m/z [M+H]+ calcd for [C28H31NO3S + H]+ : 462.6280; found: 462.4988;
IR (KBr): ν = 3435, 2960, 2918, 2868, 2818, 1608, 1550, 1508, 1440, 1383, 1359, 1240 cm-1 ;
HPLC (% Purity) = 99.38%; Calcium Content (C56H60CaN2O6S2) Calcd.: 4.17%. Found: 3.99%.

COMPD Ca salt
Calcium (S)-3-(4-((3-((isopropyl(thiophen-3-yl methyl)amino)methyl)benzyl)oxy)phenyl)hex-4-ynoate



Identification of an Orally Efficacious GPR40/FFAR1 Receptor Agonist

Sr Vice President, Head Chemistry
Zydus Cadila
2012 – Present (4 years)Zydus Research Centre, Ahmedabad, India



Senior Vice President at Zydus Research Centre

Prashant Deshmukh
Research Officer at Zydus Cadila

Dr. Poonam Giri
Principal Scientist at Zydus Research Centre

Bhadresh Rami

Debdutta Bandyopadhyay
Senior General manager at Zydus Research Centre

Suresh Giri
Research Scientist



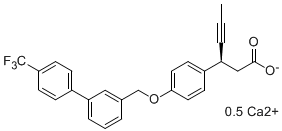
Early process development and salt selection for AMG 837, a novel GPR40 receptor agonist, is described. The synthetic route to AMG 837 involved the convergent synthesis and coupling of two key fragments, (S)-3-(4-hydroxyphenyl)hex-4-ynoic acid (1) and 3-(bromomethyl)-4′-(trifluoromethyl)biphenyl (2). The chiral β-alkynyl acid 1 was prepared in 35% overall yield via classical resolution of the corresponding racemic acid (±)-1. An efficient and scalable synthesis of (±)-1 was achieved via a telescoped sequence of reactions including the conjugate alkynylation of an in situ protected Meldrum’s acid derived acceptor prepared from 3. The biaryl bromide 2 was prepared in 86% yield via a 2-step Suzuki−Miyaura coupling−bromination sequence. Chemoselective phenol alkylation mediated by tetrabutylphosphonium hydroxide allowed direct coupling of 1 and 2 to afford AMG 837. Due to the poor physiochemical stability of the free acid form of the drug substance, a sodium salt form was selected for early development, and a more stable, crystalline hemicalcium salt dihydrate form was subsequently developed. Overall, the original 12-step synthesis of AMG 837 was replaced by a robust 9-step route affording the target in 25% yield.


Zydus Cadila’s, Lipaglyn (Saroglitazar) won a lot of support at the 75th Anniversary Conference of the American Diabetes Association
Lipaglyn (Saroglitazar) won a lot of support at the 75th Anniversary Conference of the American Diabetes Association. Lipaglyn is currently under Phase III clinical development for treatment of Non Alcoholic SteatoHepatitis (NASH), a serious liver disease and an unmet healthcare need, globally. There is currently no drug approved for treating NASH. Lipaglyn is already approved in India for the treatment of diabetic dyslipidemia


Speaking on the development, Mr. Pankaj R. Patel, Chairman and Managing Director, Zydus Cadila said, “These new robust scientific data on the safety and efficacy of Lipaglyn
(Saroglitazar) being presented at the 75th Annual Scientific Sessions of the American Diabetes Association (ADA) reflect our continued commitment to millions of patients living with Diabetes, Dyslipidemia, Non-alcoholic fatty liver disease (NAFLD) and Non-alcoholic steatohepatitis (NASH).”
Zydus Cadila, a leading global healthcare provider, today announced that new scientific and clinical data on Saroglitazar will be presented at the 75th Annual Scientific Sessions of the American Diabetes Association (ADA) in Boston, Massachusetts, USA from 5thto 9th June, 2015. Several analyses of real-world patient data of Saroglitazar will also be presented. The abstracts are available on theADA website.
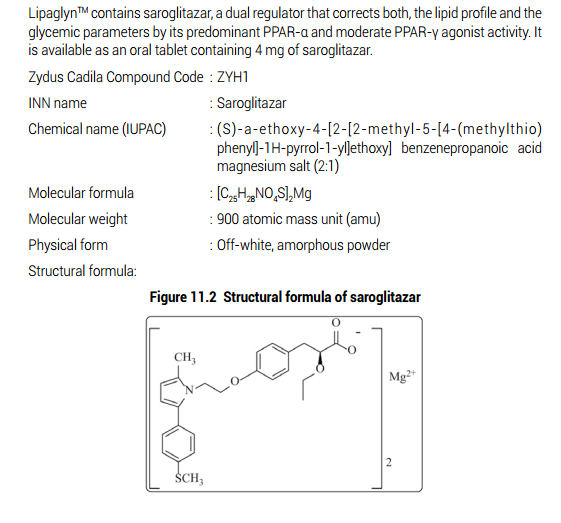
Lipaglyn – The world’s first drug for treating Diabetic Dyslipidemia combines lipid and glucose lowering effects in one single molecule.

Pankaj Patel, chairman and MD, Cadila Healthcare Ltd
Zydus is an innovation-led global healthcare provider that discovers, manufactures and markets a broad range of healthcare therapies. The group employs over 19,000 people worldwide including over 1200 scientists engaged in research and is dedicated to creating healthier communities globally.
With a strong research pipeline of NCEs, biologics and vaccines, the group became India’s first pharmaceutical company to launch its own indigenously researched therapy Lipaglyn which is also the world’s first approved therapy for diabetic dyslipidaemia. Exemptia, the world’s first biosimilar of Adalimumab is also a product of Zydus innovation. Zydus also collaborates with partners to support and make therapies affordable and accessible to communities across the world.
As a leading healthcare provider, it aims to become a global research-based pharmaceutical company by 2020.




Pankaj R. Patel (left), Chairman & Managing Director, Zybus Cadila,

Ganesh Nayak, Chief Operating Officer and Executive Director, Zydus Cadila


Zydus Cadila has announced a breakthrough in the anti-diabetic drug Lipaglyn. Lipaglyn – The world’s first drug for treating Diabetic Dyslipidemia combines lipid and glucose lowering effects in one single molecule.
The Zydus Group announced a breakthrough in its research efforts with Lipaglyn (Saroglilazar), a novel drug targeted at bridging an unmet healthcare need for treating Diabetic Dyslipidemia or Hypertriglyeeridemia in Type II diabetes, not controlled by statins alone. The drug has been approved for launch in India by the Drug Controller General of India (DCGI). With a novel action that offers lipid and glucose lowering effects in one molecule, Lipaglyn is the first Glitazar to be approved anywhere in the world.
“Lipaglyn provides patients suffering from diabetic dyslipidemia the option of a once-daily oral therapy that has a beneficial effect on both lipid parameters as well as glycemic control,” said Pankaj R. Fatel, Chairman and Managing Director, Zydus Cadila. “It has always been our dream to take a molecule right from the concept stage up to its launch. Today, we have realized this dream. It is an important breakthrough and I would like to dedicate this to all the Indian research scientists in the Held of drug discovery,” Patel added,
Diabetic Dyslipidemia is a condition where a person is diabetic and has elevated levels of the total cholesterol, the “bad” low-density lipoprotein (LDL) cholesterol and the triglycerides and a decrease in the “good” high-density lipoprotein (HDL) cholesterol concentration in the blood. Optimal LDL cholesterol levels ibr adults with diabetes are less than 100 mg/dh, optimal HDL cholesterol levels are equal to or greater than 40 mg/dL, and desirable triglycerides levels are less than 150 mg/dLT LipaglynrM, a non-thiazoKdinedione, is the first therapy to be approved for this condition,
World over, it is estimated that 30% of all deaths occur due lo cardiovascular diseases (CVD). In India, one out of every five persons is at serious risk of developing CVD, Research has shown that diabetes is one of the major risk factors of CVD. India has a population of nearly 65 million diabetics and 77 million prc-diabctics, 85 – 97% of the diabetes patients suffer from dyslipidemia or lipid abnormalities. Hence, addressing the problem of diabetes and dyslipidemia is crucial in tackling the health risk posed by CVD.
Discovered by the Zydus Research Centre, the dedicated NCE research arm of the Zydus group, LipaglynrM is a best-in-class innovation, designed to have a unique cellular mechanism of action following an extensive structure-activity relationship study initiated in the year 2000, Lipaglyn1M has a predominant affinity to PPAR alpha isoform and moderate affinity to PPAR gamma isoform of PPAR nuclear receptor subfamily. The molecule has shown beneficial effects on lipids and glyeemic control without side effects. This molecule underwent extensive pre-clinical characterisation and the I.ND was submitted in the year 2004,
As a part of the clinical development programme, extensive Phase-I, Phase-II and Phase-Ill clinical trials were conducted to evaluate the phamacokinetics, pharmacodynamics, efficacy and safety of Lipaglyn. The new drug application for Lipaglyn1 was based on a comprehensive clinical development programme spanning eight years.
Results from the first Phase III programme with Pioglitazone as a comparator drug in diabetes patients showed that the 4 mg dose of Lipaglyn led to a reduction of triglycerides and LDL (bad) cholesterol, and an increase in HDL (good) cholesterol and also showed a reduction in Fasting Plasma Glucose and glycosylated haemoglobin (HbAlc) thereby confirming its beneficial effects of both lipid and glyeemic control in diabetic patients,
In the second Phase III study, Lipaglyn was studied in diabetic dyslipidemic patients insufficiently controlled with statin therapy. The results from this study confirmed that Lipaglyn had a pronounced beneficial effect on both the lipid and glyeemic parameters in these subjects.
In both the studies, Lipaglyn was well tolerated and had a better safety profile than the comparators. Importantly Lipaglyn1 M has a non-renal route of elimination, and did not show adverse events like edema, weight gain, myopathies or derangement of liver and/or kidney functions, thus making it sale and efficacious. LipaglynIM is recommended for once daily administration as 4 mg tablets.
Zydus will offer a dedicated LipaglynIM support programme to patients and earegivers, The programme shall provide important support and information regarding access, adherence, education and thereby help patients to start and appropriately manage their disease and therapy over time.
About Lipaglyn
Lipaglyn[TM] (Saroglitazar) was launched in September 2013 in India, for treating Hypertriglyceridemia and Diabetic Dyslipidemia in Patients with Type 2 Diabetes not controlled by statins. Since then, more than 80,000 patients are availing this drug with a prescriber base over 3500 diabetologists, cardiologists and physicians. Lipaglyn[TM] helps in a reduction of triglycerides and LDL (bad) cholesterol, and an increase in HDL (good) cholesterol and has also shown a reduction in Fasting Plasma Glucose and glycosylated haemoglobin (HbA1c), thereby confirming its beneficial effects on both lipid and glycemic control in diabetic patients. Lipaglyn[TM] is a prescription medicine, and can be taken only under the advice and guidance of a registered medical practitioner.
About Zydus
Zydus Cadila is an innovative, global pharmaceutical company that discovers, manufactures and markets a broad range of healthcare therapies, including small molecule drugs, biologic therapeutics and vaccines. The group employs over 16,500 people worldwide including over 1200 scientists engaged in R & D and is dedicated to creating healthier communities globally. As a leading healthcare provider, it aims to become a global research based pharmaceutical company by 2020.
References
Zydus to present new scientific data on Lipaglyn in the US
New Delhi, Jun 8 (UNI) Healthcare services provider, Zydus Cadila today said the new scientific and clinical data on Lipaglyn (Saroglitazar) will be presented at the 75th annual scientific sessions of the American Diabetes Association (ADA) in Boston, Massachusetts, US from 5th to 9th June,2015.
Read more at http://www.uniindia.com/news/business-economy/zydus-to-present-new-scientific-data-on-lipaglyn-in-the-us/84440.html
READ …..https://newdrugapprovals.org/2013/06/07/cadila-banks-on-diabetes-druglipaglynsaroglitazar/
http://lipaglyn.com/downloads/Lipaglyn_Product_Monograph.pdf
http://www.ijpcs.net/sites/default/files/IJPCS_3_1_02_0.pdf
http://onlinelibrary.wiley.com/doi/10.1002/prp2.136/pdf
//////
Zydus Cadila, New Patent,US 20160039759, PERAMPANEL

PERAMPANEL

Zydus Cadila, New Patent,US 20160039759, PERAMPANEL
(US20160039759) PROCESS FOR THE PREPARATION OF PERAMPANEL
CADILA HEALTHCARE LIMITED
Sanjay Jagdish DESAI
Jayprakash Ajitsingh Parihar
Kuldeep Natwarlal Jain
Sachin Ashokrao Patil

Perampanel, a non-competitive AMPA receptor antagonist, is the active ingredient of FYCOMPA® tablets (U.S) which is approved as an adjunctive therapy for the treatment of partial on-set seizures with or without secondarily generalized seizures in patients with aged 12 years and older. Chemically, Perampanel is 5′-(2-cyanophenyl)-1′-phenyl-2,3′-bipyridinyl-6′(1′H)-one, with an empirical formula C23H15N30 and molecular weight 349.384 g/mol which is represented by Formula (I).

U.S. Pat. No. 6,949,571 B2 discloses perampanel and its various processes for preparation thereof.
U.S. Pat. No. 7,759,367 B2 discloses the pharmaceutical composition of perampanel and an immunoregulatory agent and their uses.
U.S. Pat. No. 8,304,548 B2 discloses the reaction of 5′-bromo-1′-phenyl-[2,3′-bipyridin]-6′(1′H)-one with 2-(1,3,2-dioxaborinan-yl)benzonitrile in the presence of palladium compound, a copper compound, a phosphorus compound and a base to form perampanel of Formula (I). Also discloses the crystalline hydrate, anhydrous crystal Form I, anhydrous crystal Form III, & anhydrous crystal Form V of perampanel of Formula (I).
U.S. Pat. No. 7,803,818 B2 discloses an amorphous form of perampanel. U.S. Pat. No. 7,718,807 B2 discloses salts of perampanel. International (PCT) publication No. WO 2013/102897 A1 discloses anhydrous crystalline Form III, V & VII of perampanel.
U.S. PG-Pub. No. 2013/109862 A1 discloses the method for preparing 2-alkoxy-5-(pyridin-2-yl)pyridine, which is an intermediate for preparing perampanel key starting material 5-(2′-pyridyl)-2-pyridone.
U.S. Pat. No. 7,524,967 B2 discloses the preparation of 5-(2′-pyridyl)-2-pyridone, an intermediate in the preparation perampanel.
International (PCT) publication No. WO 2014/023576 A1 discloses the preparation of cyanophenyl boronic acid, an intermediate in the preparation perampanel.
The prior-art processes suffer with problems of poor yield and requirement of chromatographic purification or series of crystallizations which further reduces the overall yield of the final product, which is overcome by the process of the present invention.









 Pankaj Patel, chairman, Zydus Cadila
Pankaj Patel, chairman, Zydus Cadila
EXAMPLES
The present invention is further illustrated by the following examples which is provided merely to be exemplary of the invention and do not limit the scope of the invention. Certain modification and equivalents will be apparent to those skilled in the art and are intended to be included within the scope of the present invention.
Example-A: Preparation of 5-(2-pyridyl)-1,2-dihydropyridin-2-one In a 500 mL round bottom flask, equipped with a mechanical stirrer, thermometer and an addition funnel, a solution of 188.80 g 5-bromo-2-methoxypyridine in 190 mL tetrahydrofuran and 12.92 g pyridine-2-yl boronic acid were added and refluxed. The reaction mixture was cooled to 25-30° C. and aqueous solution of hydrochloric acid was added and stirred for 1 hour. The reaction mixture was neutralized with aqueous sodium hydroxide and extracted with tetrahydrofuran.
The organic layer was washed with saline water, dried over anhydrous magnesium sulfate, and then evaporated to obtain the titled compound.
Example-1
Preparation of 3-bromo-5-(2-pyridyl)-1,2-dihydropyridin-2-one
In a 2 L round bottom flask, equipped with a mechanical stirrer, thermometer and an addition funnel, 201.5 g 5-(2-pyridyl)-1,2-dihydropyridin-2-one, 208.3 g N-bromosuccinimide and 1300 mL N,N-dimethylforamide were stirred at 25-30° C. for 2-3 hours. After completion of the reaction, the reaction mixture was poured into water and stirred for 30 min. The precipitate was filtered, washed with N,N-dimethylforamide and dried at 50° C. to obtain 230 g title compound.
Example-2
Preparation of 3-bromo-5-2-pyridyl)-1-phenyl-1,2-dihydropyridine-2-one
In a 500 mL round bottom flask, equipped with a mechanical stirrer, thermometer and an addition funnel, a solution of 18.75 g 3-bromo-5-(2-pyridyl)-1,2-dihydropyridin-2-one in 300 mL methylene dichloride, 18.36 g 1-phenyl boronic acid, 3.47 g palladium triphenylphosphine and 10 mL triethyl amine were added and the reaction mixture was stirred for 1 hour at 25-35° C. The reaction mixture was filtered and the filtrate was evaporated to dryness. The residue was crystallised from ethyl acetate to obtain the title compound.
Example-3
Preparation of Perampanel
In a 1 L round bottom flask, equipped with a mechanical stirrer, thermometer and an addition funnel, a suspension of 188 g 3-bromo-5-(2-pyridyl)-1-phenyl-1,2-dihydropyridine-2-one, 161.2 g 2-(1,3,2-dioxaborinan-2-yl)benzonitrile, 3.0 g tetrakis(triphenylphosphine)-palladium(0), 10 mL triethylamine (10 mL) in 300 mL methylene dichloride were stirred at 25-30° C. for 12 hours. To the reaction mixture was added 5 mL conc. aqueous ammonia, 10 mL water and 40 mL ethyl acetate. The separated organic layer was washed with water and saturated saline solution and dried over magnesium sulfate. The solvent was removed under vacuum. Ethyl acetate was added to the residue and heated obtain clear solution. n-hexane was added to this solution and cooled to 25-30° C. The obtained solid was filtered and washed with ethyl acetate and dried to obtain perampanel.
Example-4
Preparation of 3-Bromo-5-(2-pyridyl)-1,2-dihydropyridin-2-one
In a 2 L round bottom flask, equipped with a mechanical stirrer, thermometer and an addition funnel, 100 g 5-(2-pyridyl)-1,2-dihydropyridin-2-one, 108.5 g N-bromosuccinimide and 500 mL N,N-dimethylforamide were stirred at 30-35° C. for 3 hours. 100 mL water was added to the reaction mixture at 5-15° C. and stirred at 30-35° C. for 1 hour. The solid obtained was filtered, washed with water and dried to obtain 129 g 3-bromo-5-(2-pyridyl)-1,2-dihydropyridin-2-one.
Example-5
Preparation of 3-bromo-5-(2-pyridyl)-1-phenyl-1,2-dihydropyridine-2-one
In a 2 L round bottom flask, equipped with a mechanical stirrer, thermometer and an addition funnel, 100 g 3-bromo-5-(2-pyridyl)-1,2-dihydropyridin-2-one, 72.8 g phenylboronic acid and 500 mL N,N-dimethylformamide were added at 30-35° C. and stirred. 11.9 g copper acetate and 15.7 g pyridine were added and air was purged into the reaction mixture and stirred for 16 hours at 30-35° C. After the completion of the reaction, the reaction mixture was poured into 1200 mL aqueous ammonia at 10-15° C. and stirred for 2 hours at 30-35° C. The obtained solid was filtered, washed with water and dried to obtain 120 g 3-bromo-5-(2-pyridyl)-1-phenyl-1,2-dihydropyridine-2-one.
Example-6
Purification of 3-bromo-5-(2-pyridyl)-1-phenyl-1,2-dihydropyridine-2-one
In a 1 L round bottom flask, equipped with a mechanical stirrer, thermometer and an addition funnel, 100 g 3-bromo-5-(2-pyridyl)-1-phenyl-1,2-dihydropyridine-2-one and 500 mL isopropyl alcohol were stirred at 60-65° C. for 30 min. The reaction mixture was cooled to 20-25° C. and stirred for 30 min. The reaction mixture was filtered, washed with isopropanol and dried to obtain 96 g pure 3-bromo-5-(2-pyridyl)-1-phenyl-1,2-dihydropyridine-2-one.
Example-7
Preparation of Perampanel
In a 1 L round bottom flask, equipped with a mechanical stirrer, thermometer and an addition funnel, 100 g 3-bromo-5-(2-pyridyl)-1-phenyl-1,2-dihydropyridine-2-one and 125 g 2-(1,3,2-dioxaborinan-2-yl)benzonitrile and 1500 mL N,N-dimethylformamide were added under inert atmosphere. 44 g potassium carbonate and 4.2 g palladium tetrakis were added and stirred at 115-125° C. for 3 hours. The solvent was removed under vacuum. Ethyl acetate was added to the residue and the organic layer was distilled off to obtain perampanel (78 g).

////////Zydus Cadila, New Patent,US 20160039759, PERAMPANEL
New Patent from Zydus Cadila, Canagliflozin, US 20160002275
CADILA HEALTHCARE LIMITED [IN]

(2S,3R,4R,5S,6R)-2-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol is also known as Canagliflozin, is an inhibitor of subtype 2 sodium-glucose transport protein (SGLT2) which is chemically represented as compound of Formula (I).
U.S. Pat. No. 7,943,788 B2 discloses canagliflozin and a process for its preparation.
U.S. Pat. No. 7,943,582 B2 (the ‘582 patent) discloses crystalline form of 1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene hemihydrate and process for preparation thereof.
U.S. PG-Pub. No. 2011/0212905 discloses crystalline form of 1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene hemihydrate and process for preparation thereof.
U.S. PG-Pub. Nos. 2009/0233874, 2010/099883 and 2008/0146515 discloses similar process for the preparation of canagliflozin substantially as same as shown in scheme-1 below.
International (PCT) Publication No. WO 2011/079772 discloses a process for the preparation of canagliflozin by reduction of keto group of acetyl protected compound followed by hydrolysis.
U.S. PG-Publication No. 2014/0128595 discloses a process for the preparation of canagliflozin from anhydroglucopyranose derivative substantially as same as shown in scheme-2 below.
The prior-art processes requires sequence of protection/deprotection of canagliflozin obtained in the course of the reactions and further purification or crystallization to obtain canagliflozin in reasonably pure form. This sequences of processes results in high amount of yield loss.
In view of the above prior art, there is provided a novel, efficient and convenient process for preparation of canagliflozin which is at least a useful alternative to the prior art as well as an efficient and convenient method for purification of canagliflozin without sequence of protection and deprotection.
Scheme-3.

Ahmedabad-based pharma giant Cadila Healthcare’s chairman and managing director, Pankaj Patel,
EXAMPLES
Example-1Preparation of (3R,4S,5S,6R)-2-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (III)
In 500 mL three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel were added 2-(5-bromo-2-methylbenzyl)-5-(4-fluorophenyl)thiophene (Va) (5 g) and 150 mL toluene at 25° C. 1.5 mL (1.6M) n-butyl lithium in hexane was added dropwise at room temperature and the solution was stirred for 30 minutes. This solution was cooled to −78° C. and added dropwise to a solution of 3,4,5-tris((trimethylsilyl)oxy)-6-(((trimethylsilyl)oxy)methyl)tetrahydro-2H-pyran-2-one (IV) (6.4 g) in 100 mL toluene and the mixture was stirred for 3 hours. The reaction mixture was treated with 2.5 g methanesulfonic acid in 100 mL methanol and stirred for 1 hour. The reaction mass was warmed to 25° C. and then added to pre-cool saturated sodium bicarbonate solution and resulting mass was extracted with ethyl acetate. The extract was washed with brine, dried over Na2SO4 and evaporated under reduced pressure to obtain compound of Formula (III).
Example-1APreparation of (3R,4S,5S,6R)-2-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (III)
In 500 mL three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel were added 2-(5-bromo-2-methylbenzyl)-5-(4-fluorophenyl)thiophene (Va) (5 g) and 150 mL toluene at 25° C. 1.5 mL (1.6M) n-butyl lithium in hexane was added dropwise at room temperature and the solution was stirred for 30 minutes. This solution was cooled to −78° C. and added dropwise to a solution of 3,4,5-tris((trimethylsilyl)oxy)-6-(((trimethylsilyl)oxy)methyl)tetrahydro-2H-pyran-2-one (IV) (6.4 g) in 100 mL toluene and the mixture was stirred for 3 hours. The reaction mixture was treated with 2.5 g methanesulfonic acid in 100 mL methanol and stirred for 1 hour. The reaction mixture warmed to room temperature and stirred for 8 hours. Saturated sodium bicarbonate solution was added to the reaction mixture and the separated aqueous layer was extracted with toluene. The organic layer was distilled to remove toluene and the residue was dissolved in 50 mL methylene dichloride, washed with brine, dried over Na2SO4 and evaporated under reduced pressure to obtain residue. The residue was treated with 150 mL diisopropyl ether and stirred at 55° C. for 30 min, cooled, filtered and washed withdiisopropyl ether to obtain compound of Formula (III).
Example-1BPreparation of (3R,4S,5S,6R)-2-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (III)
In 5 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel were added 100 g 2-(5-iodo-2-methylbenzyl)-5-(4-fluorophenyl)thiophene (Vb), 114.35 g 3,4,5-tris((trimethylsilyl)oxy)-6-(((tri-methylsilyl)oxy)methyl)tetrahydro-2H-pyran-2-one (IV), 2 L toluene and 1 Ltetrahydrofuran at 30° C. The reaction mixture was cooled to −78° C. and 171.45 mL n-butyl lithium in hexane (1.6M) was added and the solution was stirred for 3 hours. The reaction mixture was treated with 94.16 g methanesulfonic acid in 1500 mL methanol and stirred for 1 hour. The reaction mixture warmed to 25° C. and stirred for 8 hours. The reaction mixture was cooled to 5° C. and saturated sodium bicarbonate solution was added to the reaction mixture and stirred for 30 min. The separated aqueous layer was extracted with toluene. The organic layer was distilled to remove toluene and the residue was dissolved in 300 mL methylene dichloride and 200 g silica gel of 60-120 mesh was added. The reaction mixture was stirred for 30 min at 30° C., washed with brine, dried over Na2SO4 and evaporated under reduced pressure to obtain residue. The residue was treated with 1 L diisopropyl ether and stirred at 55° C. for 30 min, cooled, filtered and washed with diisopropyl ether to obtain compound of Formula (III).
Example-2APreparation of (3R,4S,5S,6R)-2-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-2-methoxy-6-(((trimethylsilyl)oxy)methyl)tetrahydro-2H-pyran-3,4,5-triol (IIa1)
In 500 mL three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel were added 10 g compound of Formula (III), 80 mL methylene dichloride and 4.3 g N-methylmorpholine at −5 to 5° C. 2.7 g trimethylsilyl chloride was added slowly and stirred for 1 hour. After confirming the reaction completion TLC, 30 mL pre-cool water was slowly added, stirred and layers were separated. The separated aqueous layer was extracted with methylene dichloride and the combined organic layers were washed with 20% sodium dihydrogen phosphate dihydrate solution, water and brine. The organic layer was evaporated under reduced pressure to obtain compound of Formula (IIa).
Example-2BPreparation of (3R,4S,5S,6R)-2-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-2-methoxy-6-(((trimethylsilyl)oxy)methyl)tetrahydro-2H-pyran-3,4,5-triol (IIa1)
In 1 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel were added 100 g compound of Formula (III) and 900 mL methanol at 30° C. and stirred for 1 hour. The reaction mixture was filtered to remove silica gel and washed with methanol. The filtrate was distilled under vacuum to remove methanol completely, 350 mL methylene dichloride and 42.63 g N-methylmorpholine were added to the residue and cooled to at −5 to 5° C., 34.34 g trimethylsilyl chloride was lot-wise added and stirred for 45 min. After confirming the reaction completion TLC, 300 mL pre-cool water was slowly added, stirred and layers were separated. The separated aqueous layer was extracted with methylene dichloride and the combined organic layers were washed with 20% sodium dihydrogen phosphate dihydrate solution, water and brine. The separated organic layer was dried over sodium sulfate and filtered to obtain compound of Formula (IIa1).
Example-3APreparation of Canagliflozin of Formula (I)
In 1 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel was added solution of compound (IIa) prepared in example-2B and cooled to −70° C. 8 mL triethylsilane and 5.5 mL boron trifluoridediethyl etherate were added dropwise within 1 hour maintaining the reaction temperature between −70° C. The reaction was warmed to −30° C. and stirred for 30 min. The reaction mixture was then added to freshly preparedsodium bicarbonate solution at 5° C. and then allowed to warm to room temperature and stirred for 20 mints to adjust the pH of 7-8. The reaction mass was then slowly added to cold water. The resulting mass was extracted with ethyl acetate. The combined organic layers were washed with saturated bicarbonatesolution, dried over Na2SO4 and evaporated under reduced pressure to obtain canagliflozin having purity 86% by HPLC.
Example-3BPreparation of Canagliflozin of Formula (I)
In 2 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel was added the solution of compound (IIa) prepared in example-2B and cooled to −70° C. 67.38 g triethylsilane and 83.08 g boron trifluoridediethyl etherate were added dropwise within 1 hour maintaining the reaction temperature between −70° C. The reaction was warmed to −30° C. and stirred for 3 hours. The reaction mixture was then added to freshly prepared sodium bicarbonate solution at 5° C. and then allowed to warm to room temperature and stirred for 20 mints to adjust the pH of 7-8. The reaction mixture was then slowly added to cold water. The separated aqueous layer was extracted with 200 mL methylene dichloride. The combined organic layer was washed with 300 mL water and distilled completely to remove methylene dichloride. The resulting residue extracted with 500 mL ethyl acetate and stirred to obtain clear solution. The reaction mixture was treated with brine and saturated bicarbonate solution to separate the layers. The separated organic layer was dried over sodium sulfate, charcoalized and filtered. The filtrate is distilled to remove ethyl acetate completely under vacuum. The residue was dissolved in 300 mL methylene dichloride and 200 g silica gel of 60-120 mesh was added. The reaction mixture was stirred for 30 min at 30° C. and distilled completely under reduced pressure to obtain residue. The residue was treated with 500 L diisopropyl ether and stirred at 55° C. for 30 min, cooled, filtered and washed with diisopropyl ether to obtain canagliflozin (I) having purity 87% by HPLC.
Example-4Preparation of (3R,4S,5R,6R)-2-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-2-methoxy-6-(((trimethylsilyl)oxy)methyl)tetrahydro-2H-pyran-3,4,5-triyl)tris(oxy)tris(trimethylsilane) (IIb1)
In 500 mL three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel were added 10 g compound of Formula (III), 100 mL methylene dichloride and 15 g N-methylmorpholine at 0 to 5° C. 12.7 g trimethylsilyl chloride was added slowly and stirred for 1 hour. After confirming the reaction completion by TLC, 300 mL pre-cool water was slowly added, stirred and layers were separated. The separated aqueous layer was extracted with methylene dichloride and the combined organic layers were washed with 20% sodium dihydrogen phosphate dihydrate solution, water and brine. The organic layer was evaporated under reduced pressure to obtain compound of Formula (IIb1).
Example-5Preparation of Canagliflozin
In 500 mL three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel was added 20 g compound (IIb1) prepared in example-4 and 100 mL methylene dichloride at −25° C. to −30° C. 11 mL triethylsilane and 7.8 mL boron trifluoridediethyl etherate was added drop wise within 1-2 hours maintaining the reaction temperature between −25° C. to −30° C. The reaction was stirred for 30 min and then allowed to warn to room temperature and stirred for 1.5-2 hours. The reaction mixture was then slowly added to cold water. The reaction mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated bicarbonate solution, dried over sodium sulfate and evaporated under reduced pressure to obtain canagliflozin having purity 86% by HPLC.
Example-6Purification of Canagliflozin
In 250 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel 10 g canagliflozin (purity 85%) and 100 mLtoluene were stirred to obtain a clear solution. 10 g Polyvinylpyrrolidone was added to the solution and stirred for 2-3 hours. The reaction mixture was filtered and washed with toluene. The solid was stirred in ethyl acetate and water mixture for 30 min. The separated ethyl acetate layer was evaporated to dryness to obtain pure canagliflozin. (7.1 g. Purity 96.55% by HPLC).
Example-7Purification of Canagliflozin
In 250 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel 10 g canagliflozin (purity 87%) and 100 mLtoluene were stirred in in a round bottom flask to obtain a clear solution. 10 g β-cyclodextrin was added to the solution and stirred for 2-3 hours. The reaction mixture was filtered and washed with toluene. The solid was stirred in ethyl acetate and water mixture for 30 min. The separated ethyl acetate layer was treated with activated carbon, filtered and evaporated to dryness to obtain pure canagliflozin. (7.9 g, Purity 98.93% by HPLC).
Example-8Purification of Canagliflozin
In 250 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel 10 g canagliflozin (purity 87%) and 0.25 g activated carbon were stirred in 100 mL toluene for 15-20 min and filtered. 10 g β-cyclodextrin was added to the filtrate and stirred for 2-3 hours. The reaction mixture was filtered and washed with toluene. The solid was stirred in isopropyl acetate and water mixture for 30 min. The separated isopropyl acetate layer was evaporated to dryness to obtain pure canagliflozin. (7.7 g, Purity 99.12% by HPLC).
Example-9Purification of Canagliflozin
In 250 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel, 10 g canagliflozin (purity 87%) and 100 mLtoluene were stirred to obtain a clear solution. 10 g hydroxy propyl methyl cellulose was added to the solution and stirred for 2-3 hour. The reaction mixture was filtered, washed with toluene. The solid was stirred in isopropyl acetate and water mixture for 30 min and dried to obtain pure canagliflozin. (Purity 97-98% by HPLC).
Example-10Purification of Canagliflozin
In 2 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel, 100 g canagliflozin (purity 87%) obtained in example-3B and 900 mL methanol were stirred for 45 min at 30° C. The reaction mixture was filtered to remove silica gel. The filtrate was distilled under vacuum completely below 45° C. 400 mL toluene was added and heated to 55° C. to obtain a clear solution. The reaction mixture was filtered and the filtrate was added 100 g β-cyclodextrine. The reaction mixture was heated at 75° C. for 30 min and cooled to 30° C. and further stirred for 30 min. 5 g canagliflozin β-cyclodextrin complex was added to the solution and further cooled to 5° C. The reaction mixture was stirred for 3 hours and filtered. The wet-cake was treated with 300 mL isopropyl acetate and heated at 75° C. for 30 min. The reaction mixture was cooled to 30° C. and stirred for 6 hours and further cooled to 5° C. and stirred for 3 hours. The reaction mixture was filtered and washed with isopropyl acetate and dried at 30° C. to obtain crystalline canagliflozin β-cyclodextrine complex having 40 g pure canagliflozin with 99% purity by HPLC.
Example-11Preparation of Amorphous Canagliflozin
In 1 L three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel, 100 g canagliflozin β-cyclodextrine (purity 98%) obtained in example-10 and 400 mL acetone were stirred for 30 min at 30° C. The reaction mixture was filtered to remove β-cyclodextrine. The filtrate was distilled under vacuum completely below 45° C. 400 mL acetone was added to the residue to get clear solution at 30° C. 5 g activated charcoal was added and stirred for 20 min. The reaction mixture was filtered and the filtrate was spray dried using JISL Mini spray drier LSD-48 keeping feed pump at 30 rpm, inlet temperature at 60° C., outlet temperature at 40° C. and 2 Kg/cm2 hot air supply. The product was collected from cyclone and is further dried at 40° C.±5° C. under vacuum for 12 hours to get 80 g of amorphous canagliflozin having 99.6% purity by HPLC.
WO2014195966
https://www.google.co.in/patents/WO2014195966A2?cl=un
Canagliflozin is inhibitor of sodium dependent glucose transporter inhibitor (SGLT) which is chemically represented as (25′,3i?,4/?,55,,6 ?)-2-{3-[5-[4-Fluoro-phenyl]-thiophen-2-ylmethyl]-4-methyl-phenyl}-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol having (I).

Formula (I)
U.S. Patent No, 7,943,788 B2 (the ‘788 patent) discloses canagliflozin or salts thereof and the process for its preparation.
U.S. Patent Nos. 7,943,582 B2 and 8,513,202 B2 discloses crystalline form of 1 -(P-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl] benzene hemihydrate and process for preparation thereof. The US ‘582 B2 and US ‘202 B2 further discloses that preparation of the crystalline form of hemi-hydrate canagliflozin typically involves dissolving in a good solvent (e.g. ketones or esters) crude or amorphous compound prepared in accordance with the procedures described in WO 2005/012326 pamphlet, and adding water and a poor solvent (e.g. alkanes or ethers) to the resulting solution, followed by filtration.
U.S. PG-Pub. No. 2013/0237487 Al (the US ‘487 Al) discloses amorphous dapagliflozin and amorphous canagliflozin. The US ‘487 Al also discloses 1:1 crystalline complex of canagliflozin with L-proline (Form CS1), ethanol solvate of a 1: 1 crystalline complex of canagliflozin with D-proline (Form CS2), 1 :1 crystalline complex of canagliflozin with L-phenylalanine (Form CS3), 1:1 crystalline complex of canagliflozin with D-proline (Form CS4).
The US ‘487 Al discloses preparation of amorphous canagliflozin by adding its heated toluene solution into n-heptane. After drying in vacuo the product was obtained as a white solid of with melting point of 54.7°C to 72.0°C. However, upon repetition of the said experiment, the obtained amorphous canagliflozin was having higher amount of residual solvents. Therefore, the amorphous canagliflozin obtained by process as disclosed in US ‘487 Al is not suitable for pharmaceutical preparations.
The US ‘487 Al further discloses that amorphous canagliflozin obtained by the above process is hygroscopic in nature which was confirmed by Dynamic vapor sorption (DVS) analysis. Further, it was observed that the amorphous form underwent a physical change between the sorption/desorption cycle, making the sorption/desorption behavior different between the two cycles. The physical change that occurred was determined to be a conversion or partial conversion from the amorphous state to a crystalline state. This change was supported by a change in the overall appearance of the sample as the humidity increased from 70% to 90% RH.
The canagliflozin assessment report EMA/718531/2013 published by EMEA discloses that Canagliflozin hemihydrate is a white to off-white powder^ practically insoluble in water and freely soluble in ethanol and non-hygroscopic. Polymorphism has been observed for canagliflozin and the manufactured Form I is a hemihydrate, and an unstable amorphous Form II. Form I is consistently produced by the proposed commercial synthesis process.
Therefore, it is evident from the prior art that the reported amorphous form of canagliflozin is unstable and hygroscopic as well as not suitable for pharmaceutical preparations due to higher amount of residual solvents above the ICH acceptable limits.
Hence, there is a need to provide a stable amorphous form of canagliflozin which is suitable for pharmaceutical preparations.
Crystalline solids normally require a significant amount of energy for dissolution due to their highly organized, lattice like structures. For example, the energy required for a drug molecule to escape from a crystal is more than from an amorphous or a non-crystalline form. It is known that the amorphous forms in a number of drugs exhibit different dissolution characteristics and in some cases different bioavailability patterns compared to the crystalline form (Econno T., Chem. Pharm. Bull., 1990; 38: 2003-2007). For some therapeutic indications, one bioavailability pattern may be favoured over another.
An amorphous form of some of the drugs exhibit much higher bioavailability than the crystalline forms, which leads to the selection of the amorphous form as the final drug substance for pharmaceutical dosage from development. Additionally, the aqueous solubility of crystalline form is lower than its amorphous form in some of the drugs, which may resulted in the difference in their in vivo bioavailability. Therefore, it is desirable to have amorphous forms of drugs with high purity to meet the needs of regulatory agencies and also highly reproducible processes for their preparation.
In view of the above, it is therefore, desirable to provide canagliflozin amorphous form as well as an efficient, economical and eco-friendly process for the preparation of highly pure canagliflozin amorphous form.
Example-l:
Preparation of amorphous form of Canagliflozin
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel, 25.0 g of canagliflozin was dissolved in 250.0 mL of methanol mixture at 25°C to 30°C. The content was stirred for 30 minutes at 25°C to 30°C. To this, 1.0 g charcoal was added and stirred for 30 minutes at 25°C to 30°C. The content was filtered through Hyflo-supercel, and the Hyflo-supercel pad was washed with 50.0 mL methanol. The filtrate was concentrated under vacuum below 45°C followed by spray drying in JISL Mini spray drier LSD-48 under the below conditions. The product was collected from cyclone and is further dried at 55°C±5°C under vacuum for 16 hours to get 19.0 g of amorphous canagliflozin.

The spray-dried canagliflozin is amorphous in nature. The obtained product contains residual solvent well within ICH limit.
The obtained solid was amorphous canagliflozin as is shown by the X-ray diffraction pattern shown in FIG.1.
Example-2:
Preparation of amorphous form of Canagliflozin
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel, 25.0 g of canagliflozin was dissolved in 250.0 mL of acetone mixture at 25°C to 3O°C. The content was stirred for 30 minutes at 25°C to 30°C. To this, 1.0 g charcoal was added and stirred for 30 minutes at 25°C to 30°C. The content was filtered through Hyflo-supercel, and the Hyflo-supercel pad was washed with 50.0 mL acetone. The filtrate was concentrated under vacuum below 45°C followed by spray drying in JISL Mini spray drier LSD-48 under the below conditions. The product was collected from cyclone and is further dried at 55°C±5°C under vacuum for 16 hours to get 20.0 g of amorphous canagliflozin.

The spray-dried canagliflozin is amorphous in nature. The compound is having residual acetone less than 0.5% by GC.
The obtained solid was amorphous canagliflozin as is shown by the X-ray diffraction pattern shown in FIG.2.
Example-3:
Preparation of amorphous form of canagliflozin
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel, 10 g of canagliflozin was dissolved in 125 mL methanol and heated to obtain clear solution at 65°C. The solution was distilled to remove methanol completely. The compound thus obtained was amorphous canagliflozin.
Example-4:
Preparation of amorphous form of canagiiflozin
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel, 10 g of canagiiflozin was dissolved in 125 mL acetone and heated to obtain clear solution at 65°C. The solution was distilled to remove acetone completely. The compound thus obtained was amorphous canagiiflozin. The compound is having residual acetone less than 0.5% by GC.
Example 5:
Preparation of amorphous form of canagiiflozin
In 100 ml three necked round bottom flask equipped with mechanical stirrer, thermometer and an addition funnel, canagiiflozin (0.5 gm, 1.02 mmol), PVP K-30 (4 gm, 8 times) and 88% methanol in water (12.5ml, 25V) were heated to 65-70°C to get clear solution. The reaction mixture was stirred for 1 hour, concentrated under vacuum (1.5 mbar) at 65-70°C and degassed under vacuum (1.5 mbar) for 1 hour at 70°C to obtain the title compound in amorphous form.
Example 6:
Preparation of amorphous form of canagiiflozin
In 100 ml three necked round bottom flask equipped with mechanical stirrer, thermometer and an addition funnel, canagiiflozin (0.5 gm, 1.02 mmol), HPMC-AS (1 gm, 2 times) in 88% methanol in water (12.5 ml, 25V) were heated at 65 to 70°C to get clear solution. The reaction mixture was stirred for 2 hours, concentrated under vacuum (1.5 mbar) at 70°C and degassed under vacuum (1.5 mbar) for lhr at 70°C to obtain the title compound in amorphous form.
Example-7:
Preparation of canagliflozin-L-Proline crystalline complex
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel; 25.0 g of canagiiflozin, 6.06 g L-proline and 250 mL ethanol were heated to 75-80°C, stirred for 15 min and then cooled down to 25-30°C. The mass was filtered and washed with ethanol to obtain canagliflozin-L-proline crystalline complex.
Example-8:
Preparation of amorphous canagliflozin from canagliflozin-L-proline crystaUine complex
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel 25.0 g of Canagliflozin-L-Proline Crystalline Complex and 250 mL of ethyl acetate were stirred to get a clear solution, washed with 2×150 mL of water and the organic layer was distilled. To the residue 100 mL of isopropyl acetate and 2.5 mL of water was added and heated to 75-80°C, stirred for 15 min and cooled down to 25-30°C. The mass filtered and washed with isopropyl acetate to obtain canagliflozin. The obtained canagliflozin was subjected to spray dyring under conditions of example-2 using acetone solvent to obtain amorphous canagliflozin. Purity > 99.5% by HPLC. The compound is having residual acetone less than 0.5% by GC.
The obtained solid was amorphous canagliflozin as shown by the X-ray diffraction pattern shown in FIG.2.
HPLC Purity of amorphous canagliflozin was measured by using following chromatographic conditions:
Equipment: Shimadzu LC2010C HPLC system equipped with a dual
wavelength UV-VIS detector or equivalent
Column: romasil C-8 (250mmx4.6 mm, 5 μπι) or equivalent
Flow rate: 1.5 mL/minute
Column oven temp.: 30°C
Wavelength: 210 nm
Injection Volume: 10 μΐ, .
Diluent: Mobile Phase A: Mobile Phase B (30:70)
Mobile Phase A: Buffer:Acetonitrile:Methanol (60:30: 10)
Mobile Phase B: Acetonitrile: Methanol (80:20)
Example-9:
Preparation of amorphous form of Canagliflozin as per Example-2 of US ‘487 Al In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel 25.0 g of canagliflozin and 150 mL of ethyl acetate were stirred to get clear solution. 100 mL of n-heptane was added to the solution and the reaction mixture was filtered and dried to obtain amorphous canagliflozin. The obtained amorphous canagliflozin were dried at 65°C under vacuum for 72 hours. The residual n-heptane was 44000 ppm by GC after 72 hours drying.
Example-10:
Replacing toluene with ethyl acetate in above example-9
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel 25.0 g of canagliflozin and 150 mL of ethyl acetate were stirred to obtain clear solution. 100 mL of n-heptane was added to the solution and the reaction mixture was filtered and dried to obtain amorphous canagliflozin. The obtained amorphous canagliflozin were dried at 65°C under vacuum for 72 hours. The residual n-heptane was -44000 ppm by GC after 72 hours drying.
Example-11:
Replacing n-heptane with cyclohexane in above example-9
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel 25.0 g of canagliflozin and 150 mL of ethyl acetate were stirred to obtain clear solution. 100 mL of cyclohexane was added to the solution and the reaction mixture was filtered and dried to obtain amorphous canagliflozin. The obtained amorphous canagliflozin were dried at 55°C under vacuum for 72 hours. The residual cyclohexane was >5000 ppm by GC after 72 hours drying.
Example-12:
Preparation of amorphous form of Canagliflozin
In 100 ml three necked round bottomed flask equipped with mechanical stirrer, thermometer and addition funnel; 25.0 g of canagliflozin and 250 mL of ethyl acetate were stirred to get clear solution and then ethyl acetate was removed under reduced pressure to obtain 20.0 g of amorphous canagliflozin. The obtained amorphous canagliflozin were dried at 55°C under vacuum for 72 hours. The residual ethyl acetate was -8450 ppm by GC after 72 hours drying.
///////////////New Patent, Zydus Cadila, Canagliflozin, US 20160002275
Zydus gets USFDA nod for clinical trials of Saroglitazar

November 19, 2015
New Delhi: Zydus Cadila has received US health regulator’s nod to initiate phase II clinical trials of Saroglitazar, its new drug for treating high fat levels in body due to diabetes, obesity, and sedentary habits.
“United States Food and Drug Administration (USFDA) has endorsed company’s plan to initiate a phase II clinical trial of Saroglitazar in patients with severe hypertriglyceridemia,” Zydus Cadila said in a statement.
http://www.medicaldialogues.in/zydus-gets-usfda-nod-for-clinical-trials-of-sarolitazar/

//////////////
Zydus Cadila’s new 2-phenyl-5-heterocyclyl-tetrahydro-2h-pyran-3-amine compounds in pipeline for diabetes type 2
List of compounds as DPP-IV inhibitors


Watch out on this post as I get to correct structure………..
![]()
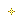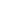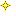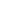

2-phenyl-5-heterocyclyl-tetrahydro-2h-pyran-3-amine compounds

One Example of 2-phenyl-5-heterocyclyl-tetrahydro-2h-pyran-3-amine compounds
CAS 1601479-87-1
(2R, 3S, 5R)-2-(2, 5-difluorophenyl)-5-(5-(methylsulfonyl)-5, 6- dihydropyrrolo [ 3, 4-c]pyrrol-2(lH, 3H, 4H)-yl)tetrahydro-2H-pyran-3-amine
(2R,3S,5R)-2-(2,5-Difluorophenyl)-5-[5-(methylsulfonyl)-3,4,5,6-tetrahydropyrrolo[3,4-c]pyrrol-2(1H)-yl]tetrahydro-2H-pyran-3-amine
MW 399.45, C18 H23 F2 N3 O3 S
INTRODUCTION
Dipeptidyl peptidase IV , CD26; DPP-IV; DP-IV inhibitors acting as glucose lowering agents reported to be useful for the treatment of type 2 diabetes. compound inhibited human DPP-IV enzyme activity (IC50 < 10 nM) in fluorescence based assays.
It lowered glucose levels (with -49.10% glucose change) when administered to C57BL/6J mice at 0.3 mg/kg p.o. in oral glucose tolerance test (OGTT).
Compound displayed the following pharmacokinetic parameters in Wistar rats at 2 mg/kg p.o.: Cmax = 459.04 ng/ml, t1/2 = 59.48 h and AUC = 4751.59 h·ng/ml.
Dipeptidyl peptidase 4 (DPP-IV) inhibitor that inhibited human DPP-IV enzyme activity with an IC50 of < 10 nM in a fluorescence based assay.
Watch out on this post as I get to correct structure………..![]()

PATENT
http://www.google.com/patents/WO2014061031A1?cl=en
Compound 8: (2R, 3S, 5R)-2-(2, 5-difluorophenyl)-5-(5-(methylsulfonyl)-5, 6- dihydropyrrolo [ 3, 4-c]pyrrol-2(lH, 3H, 4H)-yl)tetrahydro-2H-pyran-3-amine
1H NMR: (CD3OD, 400 MHz): 7.32-7.28 (m, IH), 7.26-7.23 (m, 2H), 4.77 (d, IH, J= 10Hz), 4.32(dd, IH, J,= 2.0Hz, J2= 10.8Hz), 4.19 (s, 4H), 3.89-3.83 (m, 4H), 3.70- 3.65 (m, IH), 3.61 (t, IH, J= 11.6Hz), 3.53-3.46 (m, IH), 3.04 (s, 3H), 2.65-2.62 (dd, IH, Ji= 1.2Hz, J2= 12Hz), 1.84 (q, IH, J = 12 Hz); ESI-MS: (+ve mode) 400.0 (M+H)+ (100 %); HPLC: 99.4 %.
Compound 4: (2R, 3S, 5R)-2-(2, 5-difluorophenyl)-5-(hexahydropyrrolo[3, 4-c Jpyrrol- 2(lH)-yl)tetrahydro-2H-pyran-3-amine
1H NMR: (CD3OD, 400 MHz):

.23-7.20 (m, 2H), 4.64 (d, IH, J= 10.4 Hz), 4.38-4.35 (dd, IH, J,= 2.4Hz, J2= 10.4Hz), 3.69 (t, IH, J= 11Hz), 3.57-3.53 (m, 4H), 3.34-3.30 (m, 8H), 2.68-2.65 (m, IH), 2.04 (q, IH, J = 1 1.6 Hz); ESI-MS: (+ve mode) 323.9 (M+H)+ (100 %), 345.9 (M+Na)+ (20%); HPLC: 98.6 %

PATENT
IN 2012MU03030
“NOVEL DPP-IV INHIBITORS”
3030/MUM/2012
Abstract:
The present invention relates to novel compounds of the general formula (I) their tautomeric forms, their enantiomers, their diastereoisomers, their pharmaceutically accepted salts, or pro-drugs thereof, which are useful for the treatment or prevention of diabetes mellitus (DM), obesity and other metabolic disorders. The invention also relates to process for the manufacture of said compounds, and pharmaceutical compositions containing them and their use.

Pankaj R. Patel (right), Chairman and Managing Director,
////////////2-phenyl-5-heterocyclyl-tetrahydro-2h-pyran-3-amine compounds, DPP-IV inhibitors
ZYD 1/ZYDPLA 1 From Zydus Cadila, a New NCE in Gliptin class of Antidiabetic agents.

GENERAL STRUCTURE

3-[4-(5-methyl-1,3,4-oxadiazol-2-yl)phenoxy]-5-[[(3R)-1-methyl-2-oxo-3-pyrrolidinyl]oxy]-N-2-thiazolyl- Benzamide
3-(4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl-2-oxopyrrolidin-3- yloxy)-iV-(thiazol-2-yl)benzainide
(S)-3-(4-(5-Methyl-l,3,4-oxadiazol-2-yI)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-N-(thiazol-2-yl)benzamide……S CONF…..WO2011013141A2
(Λ)-3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-Λ’-(thiazol-2-yl)benzamide…..R CONF…..WO2011013141A2
CAS 1263402-84-1 R CONF
CAS 1263402-76-1 S CONF
ZYD 1/ZYDPLA 1……….Probable Representative structure only, I will modify it as per available info
Watch out on this post as I get to correct structure………..![]()
ZYDPLA1 is an orally active, small molecule NCE, discovered and developed by the Zydus Research Centre, the NCE research wing of Zydus. ZYDPLA1 is a novel compound in the Gliptin class of antidiabetic agents. It works by blocking the enzyme Dipeptidyl Peptidase-4 (DPP-4), which inactivates the Incretin hormone GLP-1.
By increasing the GLP-1 levels, ZYDPLA1 glucose-dependently increases insulin secretion and lowers glucagon secretion. This results in an overall improvement in the glucose homoeostasis, including reduction in HbA1c and blood sugar levels.
In October 2013, Zydus received IND approval from the US FDA to initiate a phase I trial in type II diabetes
Clinical trials..Type 2 Diabetes Mellitus
NCT01972893; ZYD1/1001;
CTRI/2011/04/001684;
ZYD1
ZYD1/1001
ZYD1 is a novel GLP-1 receptor agonist. The ZYD1 exhibits increased stability to proteolytic cleavage, especially against dipeptidyl peptidase-4 (DPP-IV).ZYD1 is a potent antidiabetic agent without gastrointestinal side-effects. A first in human (FIH) Phase I study intends to evaluate the safety, tolerability, pharmacokinetics and pharmacodynamics of ZYD1 in normal healthy adult volunteers……..https://clinicaltrials.gov/show/NCT01972893
A randomized, double blind, placebo controlled Phase I clinical study to evaluate the safety, tolerability and pharmacokinetics of ZYD1, a selective GLP-1 agonist, following the subcutaneous administrations in healthy volunteers …………http://www.ctri.nic.in/Clinicaltrials/pdf_generate.php?trialid=2263&EncHid=&modid=&compid=%27,%272263det%27
Some clippings I found

ONE MORE……………

Zydus announces data presentations on ZYDPLA1 “A once-weekly small molecule DPP-IV inhibitor for treating diabetes”, at the ENDO conference in Chicago, Illinois, USA. Ahmedabad, India June 9, 2014 The Zydus group will be presenting data on its molecule ZYDPLA1 a novel compound in the Gliptin class of anti-diabetic agents during the joint meeting of the International Society of Endocrinology and the Endocrine Society: ICE/ENDO 2014 to be held from June 21-24, 2014 in Chicago, Illinois.
ZYDPLA1, currently in Phase I clinical evaluation in USA, is an orally active, small molecule NCE, discovered and developed by the Zydus Research Centre. ZYDPLA1 works by blocking the enzyme Dipeptidyl Peptidase-4 (DPP-4), which inactivates the Incretin hormone GLP-1. By increasing the GLP- 1 levels, ZYDPLA1 glucose-dependently increases insulin secretion. This results in an overall improvement in the glucose homoeostasis, including reduction in HbA1c and blood sugar levels.
The Chairman & Managing Director of Zydus, Mr. Pankaj R. Patel said, “Currently, all available DPP-4 inhibitors are dosed once-daily. ZYDPLA1 with a once-a-week dosing regimen would provide diabetic patients with a more convenient treatment alternative. ZYDPLA1 will offer sustained action, which will result in an improved efficacy profile.”
The abstract of Poster Number: LB-PP02-4 can also be viewed on the ENDO web program at https://endo.confex.com/endo/2014endo/webprogram/authora.html. The Poster Preview is scheduled on Sunday, June 22, 2014 at McCormick Place West.
The number of diabetics in the world is estimated to be over 360 million. In 2025 nearly half of the world’s diabetic population will be from India, China, Brazil, Russia and Turkey. The sales of the DPP IV inhibitors is expected to peak at almost $14 billion by 2022. Research in the field of anti-diabetic therapy seeks to address the problems of hypoglycemia, GI side effects, lactic acidosis, weight gain, CV risks, edema, potential immunogenicity etc., which pose a major challenge in the treatment of diabetes.
About Zydus
Headquartered in Ahmedabad, India, Zydus Cadila is an innovative, global pharmaceutical company that discovers, manufactures and markets a broad range of healthcare therapies. The group employs over 16,000 people worldwide including over 1100 scientists engaged in R & D and is dedicated to creating healthier communities globally. As a leading healthcare provider, it aims to become a global researchbased pharmaceutical company by 2020. The group has a strong research pipeline of NCEs, biologics and vaccines which are in various stages of clinical trials including late stage.
About Zydus Research Centre
The Zydus Research Centre has over 20 discovery programmes in the areas of cardio-metabolic disorders, pain, inflammation and oncology. Zydus has in-house capabilities to conduct discovery research from concept to IND-enabling pre-clinical development and human proof-of-concept clinical trials. The Zydus Research group had identified and developed Lipaglyn™ (Saroglitazar) which has now become India’s first NCE to reach the market. Lipaglyn™ is a breakthrough therapy in the treatment of diabetic dyslipidemia and Hypertriglyceridemia. The company recently announced the commencement of Phase III trials of LipaglynTM (Saroglitazar) in patients suffering from Lipodystrophy.
PATENT
http://www.google.com/patents/WO2011013141A2?cl=en
Rajendra Kharul, Mukul R. Jain, Pankaj R. Patel
Substituted benzamide derivatives as glucokinase (gk) activators

Scheme 2:

Scheme 3:

Scheme 4A:


Scheme 4B.
] Scheme 5 A:

Scheme 5B:

Scheme 6:

Example 1
3-(4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl-2-oxopyrrolidin-3- yloxy)-iV-(thiazol-2-yl)benzainide
4-(Dimethylamino)pyridine (DMAP) (0.149 g), N-(3-Dimethylaminopropyl)-N’- ethylcarbodiimide hydrochloride (EDCI.HC1) (0.524 g) were added to a solution of 3-
( 1 -Methoxypropan-2-yloxy)-5-(4-(5 -methyl- 1 ,3,4-oxadiazol-2-yl) phenoxy) benzoic acid (0.5 g) (Intermediate 1) in dry DCM under nitrogen at 0-5 0C. 2-Aminothiazole (0.134 g) was added and the mixture was stirred for 16 h at room temperature. It was diluted with commercially available DCM. Organic phase was washed with dil HCl, saturated solution of NaHCO3, water, brine, dried over Na2SO4, filtered and concentrated in vacuo to get the crude residue. The residue was chromatographed using silica gel as stationary phase and MeOH: CHCl3 gradient as mobile phase up to yield the product (0.3 g) as a white solid.
1H NMR (DMSO-<4, 400 MHz) δ ppm: 1.92-2.01 (m, 1 H), 2.59 (s, 3 H), 2.60-2.65 (m,
I H), 2.79 (s, 3 H), 3.31-3.34 (m, 1 H), 3.36-3.44 (m ,1 H), 5.15 (t, J = 7.6 Hz, 1 H),
7.08 (s, 1 H), 7.24 (d, J= 8.8 Hz, 2 H), 7.27-7.29 (m, 1 H), 7.40 (s, 1 H), 7.54 (s, 1 H),
7.62 (s, 1 H), 7.99 (d, J = 8.8 Hz, 2 H), 12.60 (bs, 1 H); ESI-MS mix (relative intensities): 492.03 (M+H)+ (100 %), 514.02 (M+Na)+(15 %); UPLC Purity: 93.59 %, Rettime: 3.59 min.
Intermediate 1: 3-(4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl-2-oxo pyrrolidin -3-yloxy)benzoic acid
A solution of Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl- 2-oxopyrrolidin-3-yloxy)benzoate (7 g) (Intermediate 2) in a mixture of THF and methanol (1 :1 ratio) was treated with a solution of sodium hydroxide (2 g) in water and the reaction mixture was stirred for 1 h at room temperature. The resulting solution was concentrated under vacuum to remove THF and methanol, diluted with water, and washed with EtOAc. The aqueous phase was cooled and acidified with 0.1 N HCl and extracted with DCM, combined organic extracts washed with brine, dried over Na2SO4 and concentrated in vacuo to give the product (3.5 g) as white solid.
1H NMR (CDCl3, 400 MHz) δ ppm: 2.20-2.27 (m, 1 H), 2.59-2.67 (m, 1 H), 2.77 (s, 3 H), 2.95 (s, 3 H), 3.38-3.44 (m, 1 H), 3.49-3.54 (m, 1 H), 4.96 (t, J = 7.2 Hz, 1 H), 6.93-6.95 (m, 1 H), 7.07 (d, J= 8.8 Hz, 2 H), 7.32-7.34 (m, 1 H), 7.52 (d, J= 8.8 Hz, 2 H), 9.96-9.98 (m, 2 H); ESI-MS (relative intensities): 431.9 (M+ Na)+ (70%).
Intermediate 2: Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-(l-methyl-2- oxo- pyrrolidin-3-yloxy)benzoate
To a stirred mixture of Methyl 3-hydroxy-5-(l-methyl-2-oxopyrrolidin-3-yloxy) benzoate (15 g) (Intermediate 3), N,N-dimethylglycine hydrochloride (2.3 g), copper (II) iodide (1 g) in dry 1,4-dioxane was added 2-(4-iodophenyl)-5 -methyl- 1,3,4- oxadiazole (15.4 g) (Intermediate 4) under nitrogen. The reaction mixture was refluxed for 24 h. The reaction mixture was cooled, quenched with water and extracted with DCM. Combined organic washings were washed with water, brine, dried over Na2SO4, filtered and concentrated in vacuo to get the crude product. The crude product was purified by column chromatography using silica gel as stationary phase and ethyl acetate: petroleum ether (9:1) as mobile phase to give the product (7 g) as thick liquid. 1H NMR (DMSO-<4, 400 MHz) δ ppm: 1.91-1.98 (m, 1 H), 2.49-2.54 (m, 1 H), 2.56 (s, 3 H), 2.77 (s, 3 H), 3.34-3.41 (m, 2 H), 3.81 (s, 3 H), 5.12 (t, J= 7.6 Hz, 1 H), 7.13- 7.15 (m, 2 H), 7.22 (d, J = 8.8 Hz, 2 H), 7.42 (s, 1 H), 7.97 (d, J = 8.8 Hz, 2 H); ESI- MS (relative intensities): 423.9 (M+H)+ (100%), 446.2 (M+ Na)+ (30%).
Intermediate 3: Methyl 3-hydroxy-5-(l-methyl-2-oxopyrrolidin-3-yloxy)benzoate
To a stirred solution of Methyl 3, 5-dihydroxybenzoate (20 g) [CAS No. 2150- 44-9] in dry DMF was added potassium carbonate (48 g) and the suspension stirred at ambient temperature under nitrogen. To this 3-Bromo-l-methyl-pyrrolidin-2-one (4Og) (Intermediate 5) [J. Med. Chem., 1987, 30, 1995-98] was added in three equal portions in 4 h intervals at room temperature and stirred overnight at ambient temperature. It was then quenched with water. The aqueous suspension was extracted with DCM. The combined extracts were washed with water, brine, dried over Na2SO4, and filtered, concentrated under reduced pressure to get the thick liquid residue. The crude product was purified by column chromatography using silica gel as stationary phase and ethyl acetate: petroleum ether as a mobile phase to yield the product as white solid (15 g).1H NMR (CDCl3, 400 MHz) δ ppm: 2.08-2.10 (m, 1 H), 2.60-2.67 (m, 1 H), 3.04 (s, 3 H), 3.40-
3.43 (m, 1 H), 3.48-3.51 (m, 1 H), 3.87 (s, 3 H), 4.91 (t, J = 7.2 Hz, 1 H), 6.59- 6.61 (m, 1 H), 7.07-7.09 (m, 1 H), 7.09-7.13 (m, 1 H), 8.02 (s, 1 H); ESI-MS (relative intensities): 287.9 (M+ Na)+ (30%).
Example 68…. S CONFIGURATION
(S)-3-(4-(5-Methyl-l,3,4-oxadiazol-2-yI)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-N-(thiazol-2-yl)benzamide
To a stirring solution of S-(-)-3-[4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5- [(l-methyl-2-oxo-pyrrolidin-3-yl)oxy]benzoic acid (3.5 g) (Intermediate 13) in dry DCM in single necked round bottomed flask fitted with stop cock with N2(g) balloon, 4- (dimethylamino)pyridine (2.24 g) followed by N-(3-Dimethy lam inopropy I)-N5– ethylcarbodiimide hydrochloride (EDCI. HCl) (3.3 g) were added at room temperature. After stirring at the same temperature for 15 min, 2-aminothiazole (0.94 g) was added and stirring was continued for 16 h. Progress of reaction was monitored by TLC. After completion, reaction mixture was diluted with DCM (200 mL), washed with dil HCl (20 mL, 0.05 Ν), saturated sodium bicarbonate solution, water and brine, dried over anhydrous sodium sulphate, filtered and concentrated under vacuum to get crude brown solid (3.5 g). The crude brown solid was purified by solvent trituration.
1H ΝMR (CDCl3, 400 MHz) δ ppm: 2.13-2.22 (m, 1 H), 2.62 (s, 3 H), 2.56-2.64 (m, 1 H), 2.93 (s, 3 H), 3.39-3.43 (m, 1 H), 3.48-3.53 (m ,1 H), 4.92 (t, J= 7.2 Hz, 1 H), 7.01 (s, 1 H), 7.04 (t, J= 2 Hz, 1 H), 7.21 (d, J = 8.8 Hz, 2 H), 7.26 (s, 1 H), 7.36 (s, 1 H), 7.44 (s, 1 H), 7.99 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 492.1 (M+H)+ (100 %), 513.8 (M+Νa)+ (10 %); UPLC Purity: 98.13 %, Ret. time: 3.577 min. Chiral Purity by HPLC: 97.31 %, Ret. time: 22.93 min. % ee: 94.62 %
Intermediate 13: S-(-)-3-[4-(5-Methyl-l, 3, 4-oxadiazol-2-yl)phenoxy]-5-[(l-methyl-2- oxo-pyrro- lidin-3-yl)oxy] benzoic acid
Sodium hydroxide (pallets, 1.5 g) was added to a stirring mixture of (.S)-(-)-Methyl 3- [4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-[(l-methyl-2-oxo-pyrrolidin-3-yl)oxy] benzoate (5.3g) (Intermediate 14) in MeOH:H2O (1:1) at room temperature. The reaction was monitored by TLC. After completion, methanol was evaporated from the reaction mixture and water was added. The aqueous layer was washed with EtOAc, acidified with dil. HCl (0.05 N) to obtain solid. The solid obtained was filtered, washed with water, dried under suction or vacuum to get pure white solid (3.5 g).
1H NMR (CDCl3, 400 MHz) δ ppm: 2.17-2.22 (m, 1 H), 2.62 (s, 3 H), 2.58-2.66 (m, 1 H), 2.93 (s, 3 H), 3.39-3.43 (m, 1 H), 3.48-3.53 (m ,1 H), 4.99 (t, J= 7.2 Hz, 1 H), 6.89 (t, J = 2.4 Hz, 1 H), 7.07 (d, J = 8.8 Hz, 2 H), 7.28 (s, 1 H), 7.53 (s, 1 H), 7.95 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 410 (M+H)+ (100 %); UPLC Purity: 97.85 %, Ret. time: 3.136 min. Chiral Purity by HPLC: 99.59 %, Ret. Time: 57.46 min. % ee: 99.18 %
Intermediate 14: (S) -(-) -Methyl 3-[4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-[(l- methyl-2-oxo- pyrrolidin-3-yl) oxyjbenzoate
Sodium hydride suspension (0.71 g, 50 %) was added to a stirring solution of (£)-(-)- methyl 3 -(4-(5 -methyl- 1 ,3,4-oxadiazol-2-yl)phenoxy)-5-((2-oxopyrrolidin-3- yl)oxy)benzoate (5.5 g) (Intermediate 15) in dry DMF taken in a round bottomed flask fitted with anhydrous CaCl2 guard tube at room temperature. The reaction mixture was stirred at the same temperature for 15 min. Methyl iodide (0.91 mL) was added and stirred till the reaction completion. The reaction mixture was quenched with ice-water, extracted with DCM. All organic layers were combined, washed with water, brine, dried over sodium sulphate, filtered and concentrated in vaccuo to get the thick liquid product. The liquid was triturated with EtOAc: hexane to get the white solid product (5.3 g).
1H NMR (CDCl3, 400 MHz) δ ppm: 2.14-2.21 (m, 1 H), 2.58-2.63 (m, 1 H), 2.64 (s, 3 H), 2.93 (s, 3 H), 3.39-3.43 (m, 1 H), 3.48-3.53 (m , 1 H), 3.89 (s, 3 H), 4.99 (t, J = 7.2 Hz, 1 H), 6.99 (t, J = 2 Hz, 1 H), 7.07 (d, J= 8.8 Hz, 2 H), 7.35 (s, 1 H), 7.53 (s, 1 H), 7.99 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 424.1 (M+H)+ (100 %); UPLC Purity: 96.1 1 %, Ret. time: 3.68 min. Chiral Purity by HPLC: 92.05 %, Ret. Time: 39.33 min.
Intermediate 15: (S) -(-) -Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((2- oxo pyrrolidin-3-yl)oxy) benzoate
To a stirring mixture of Methyl 3-hydroxy-5-[4-(5-methyl-l,3,4-oxadiazol-2- yl)phenoxy] benzoate (7 g) (Intermediate 7) and (/?)-(+)-3-hydroxy-2-pyrrolidinone (Intermediate 16) (2.4g) in dry THF (200 mL) taken in round bottomed flask fitted with anhydrous CaCl2 guard tube, triphenyl phosphine (1 1.3 g) was added. Diisopropyl azodicarboxylate (DIAD) (6.2 mL) in dry THF (10 mL) was added drop wise to the above reaction mixture. The reaction was stirred at room temperature. Reaction was monitored by TLC for completion. After completion, reaction mixture was concentrated under vacuum to remove the solvents. Diluted with DCM and coated over silica gel and chromatographed to furnish the product as white solid (6 g). 1H NMR (CDCl3, 400 MHz) δ ppm: 2.26-2.33 (m, 1 H), 2.62 (s, 3 H), 2.64-2.71 (m, 1 H), 3.40-3.47 (m, 1 H), 3.51-3.55 (m, 1 H), 3.89 (s, 3 H), 4.89 (t, J= 7.6 Hz, 1 H), 6.07 (bs, 1 H), 6.99 (t, J= 2.4 Hz, 1 H), 7.11 (d, J= 8.8 Hz, 2 H), 7.36 (s, 1 H), 7.51 (s, 1 H), 8.03 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 410.1 (M+H)+ (100 %); UPLC Purity: 98.35 %, Ret. time: 3.47 min. Chiral Purity by HPLC: 95.31 %, Ret. Time: 47.97 min. ee: 90.62 %.
Intermediate 16: (R)-(+)-3-Hydroxy-2-pyrrolidinone
To a stirring mixture of 4-Nitrobenzoic acid (21.5 g) and (5)-(-)-3-hydroxy-2- pyrrolidinone (11.8 g) (Intermediate 17) in dry THF (360 mL) taken in a round bottomed flask fitted with anhydrous CaCl2 guard tube, triphenyl phosphine (61.2 g) was added. To this reaction mixture, diisopropyl diazodicarboxylate (DIAD) (34 mL) was added drop wise in three portions at room temperature. The reaction was stirred at room temperature. The progress of the reaction was monitored by TLC (developing agents: UV, I2, as well as aqueous acidic KMnO4). After completion, reaction mixture was concentrated under vacuum to obtain residue. Methanol (360 mL) was added to the residue followed by potassium carbonate (10 g) at room temperature. The reaction was stirred at room temperature. The progress of the reaction was monitored by TLC (developing agents: UV, I2, as well as aqueous acidic KMnO4). After completion, reaction mixture was diluted with CHCl3 and filtered through celite. Celite bed was successively washed with 1 % MeOH:CHCl3. The filtrates were combined and concentrated to dryness to remove solvents. The residues were partitioned between EtOAc: dil. HCl (200 mL, 9:1) and stirred for 15 min. Layers were separated, aq. layer was washed with EtOAc thrice until all organic impurities were washed out. The aq. Layer was concentrated to dryness to remove the water and solid residues were obtained. The residues obtained were washed with 1-2 % MeOH: CHCl3 (3 x 100 mL), dried over sodium sulfate, filtered trough cotton, concentrated to get brown thick liquid product.
1U NMR (CDCl3, 400 MHz) δ ppm: 2.03-2.13 (m, 1 H), 2.46-2.54 (m, 1 H), 3.28-3.35 (m, IH), 3.38-3.48 (m, 1 H), 4.50 (t, J = 8.4 Hz, 1 H), 4.55 (bs, 1 H), 7.02 (bs, 1 H); [α]D25: + 68, c = l, CHCl3
Intermediate 17: (S)-(-)-3-hydroxy-2-pyrrolidinone
Cone. H2SO4 (14.8 g, 8 mL) was added drop wise over 5 min to the stirring solution of (5)-(-)-4-Amino-2-hydroxybutyric acid (15 g) [CAS No. 40371-51-5] in MeOH (95 rnL) under dry conditions using anhydrous CaCl2 guard tube. After refluxing for 4 h, the reaction mixture was allowed to cool to room temperature and diluted with water (15 mL). Potassium carbonate (24 g) was added in portions to the reaction mixture and stirred overnight (20 h). Reaction mixture was diluted with CHCl3, filtered through celite. Celite bed was thoroughly washed with 1 % MeOHiCHCl3. The filtrates were combined and evaporated to dryness to obtain thick liquid residue. The residue was subjected to aging using 1-2 % MeOHiCHCl3 and then filtered. Organic layers were combined, dried over anhydrous sodium sulphate, filtered and concentrated to obtain the white solid. (1 1.8 g)
1H NMR (CDCl3, 400 MHz) δ ppm: 2.03-2.13 (m, 1 H), 2.48-2.55 (m, 1 H), 3.30-3.35
(m, IH), 3.36-3.50 (m, 1 H), 4.34 (t, J = 8.4 Hz, 1 H), 6.51 (bs, 1 H); [α]D25: + 98, c =
1, CHCl3
Following examples (Example 70-76) were prepared by using similar procedure as that of example lwith suitable modifications as are well within the scope of a skilled person
Example 77 R CONFIGURATION
(Λ)-3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-Λ’-(thiazol-2-yl)benzamide
CORRECTED AS (R)-3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((l-methyl-2-oxopyrrolidin-3- yl) oxy)-N-(thiazol-2-yl)benzamide
To a stirring solution of (/?j-(+)-3-[4-(5-Methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-
[(l-methyl-2-oxo-pyrrolidin-3-yl)oxy]benzoic acid (0.2 g) (Intermediate 18) in dry DCM in single necked round bottomed flask fitted with stop cock with N2(g) balloon, N.ΛP-dimethylamino pyridine (0.060 g) followed by EDCI. HCl (0.23 g) were added at room temperature. After stirring at the same temperature for 15 min, 2-aminothiazole (0.054 g) was added and stirring was continued for 16 h. Progress of reaction was monitored by TLC. After completion, reaction mixture was diluted with DCM (20 mL), washed with dil HCl (5 mL, 0.05 Ν), saturated sodium bicarbonate solution, water and brine, dried over anhydrous sodium sulphate, filtered and concentrated under vacuum to get crude brown solid (0.080 g). The crude brown solid was purified by solvent trituration.
1H NMR (CDCl3, 400 MHz) δ ppm: 2.15-2.20 (m, 1 H), 2.55-2.60 (m, 1 H), 2.62 (s, 3 H), 2.93 (s, 3 H), 3.38-3.43 (m, 1 H), 3.47-3.53 (m, 1 H), 4.91 (t, J= 6.8 Hz, 1 H), 6.99 (d, J= 8.8 Hz, 2 H), 7.10-7.14 (m, 2 H), 7.23-7.26 (m, 1 H), 7.36 (s, 1 H), 7.43 (s, 1 H), 8.03 (d, J = 8.8 Hz, 2 H), 10.75 (bs, 1 H); ESI MS m/z (relative intensities): 492.1 (M+H)+ (100 %), 514.0 (M+Na)+ (20 %); UPLC Purity: 95.25 %, Ret.time: 3.578 min. Chiral Purity by HPLC: 95.93 %, Ret.time: 14.17min. % ee: 91.86 %
Intermediate 18: (R)-(+)-3-[4-(5-Methyl-l, 3, 4-oxadiazol-2-yl)phenoxy]-5-[(l-methyl- 2-oxo- pyrrolidin-3-yl)oxy] benzoic acid
Sodium hydroxide (pallets, 0.35 g) was added To a stirring mixture of (/?)-(+)-Methyl 3-[4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-[(l-methyl-2-oxo- pyrrolidin-3-yl) oxyjbenzoate (1.1 g) (Intermediate 19) in MeOH:H2O (1:1) at room temperature. The reaction was monitored by TLC. After completion, methanol was evaporated from the reaction mixture and water was added. The aqueous layer was washed with EtOAc, acidified with dil. HCl (0.05 N) to obtain solid. The solid obtained was filtered, washed with water, dried under suction or vacuum to get pure white solid (0.76 g).
1H NMR (DMSO-J6, 400 MHz) δ ppm: 1.92-1.99 (m, 1 H), 2.62 (s, 3 H), 2.58-2.66 (m, 1 H), 3.31 (s, 3 H), 3.32-3.40 (m, 2 H), 5.12 (t, J = 7.2 Hz, 1 H), 7.08 (s, 1 H), 7.14 (s, 1 H), 7.23 (d, J= 8.8 Hz, 2 H), 7.40 (s, 1 H), 7.99 (d, J= 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 410.1 (M+H)+ (65 %), 410.1 (M+H)+ (100 %); UPLC Purity: 96.95 %, Ret. time: 3.12 min. Chiral Purity by HPLC: 89.04 %, Ret. Time: 48.15 min. Intermediate 19: (R)-(+)-Methyl 3-[4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy]-5-[(l- methyl-2-oxo- pyrrolidin-3-yl) oxyjbenzoate:
Sodium hydride suspension (0.16 g, 50 %) was added to a stirring solution of (R)- (+)-Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((2-oxopyrrolidin-3- yl)oxy)benzoate (1.5 g) (Intermediate 20) in dry DMF taken in a round bottomed flask fitted with anhydrous CaCl2 guard tube, at room temperature. The reaction mixture was stirred at the same temperature for 15 min. Methyl iodide (0.20 mL) was added and stirred till the reaction completed. The reaction mixture was quenched with ice-water, extracted with DCM. All organic layers were combined, washed with water, brine, dried over sodium sulphate, filtered and concentrated in vacuum to get the thick liquid product. The liquid was triturated with EtOAc: hexane to get the white solid product
(1.2 g).
1U NMR (DMSO-J6, 400 MHz) δ ppm: 1.95-1.98 (m, 1 H), 2.51-2.55 (m, 1 H), 2.56 (s, 3 H), 2.88 (s, 3 H), 3.29-3.34 (m, 1 H), 3.37-3.40 (m ,1 H), 3.81 (s, 3 H), 5.12 (t, J = 7.2 Hz, 1 H), 7.13-7.17 (m, 2 H), 7.24 (d, J= 8.8 Hz, 2 H), 7.41 (s, 1 H), 7.99 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 423.9 (M+H)+ (100 %); UPLC Purity: 90.38 %, Ret. time: 3.68 min.
Intermediate 20: (R)-(+)-Methyl 3-(4-(5-methyl-l,3,4-oxadiazol-2-yl)phenoxy)-5-((2- oxopyrrolidin -3-yl)oxy)benzoate
To a stirring mixture of Methyl 3-hydroxy-5-[4-(5-methyl-l,3,4-oxadiazol-2- yl)phenoxy] benzoate (2.5 g) (Intermediate 7) and (5)-(-)-3-hydroxy-2-pyrrolidinone (Intermediate 17) (0.8 g) in dry THF (70 mL) taken in round bottomed flask fitted with anhydrous CaCl2 guard tube, triphenyl phosphine (3.77 g) was added. Diisopropyl azodicarboxylate (DIAD) (2.1 mL) in dry THF (2 mL) was added drop wise to the above reaction mixture. The reaction was stirred at room temperature. Reaction was monitored by TLC for completion. After completion, reaction mixture was concentrated under vacuum to remove the solvents. Diluted with DCM and coated over silica gel and chromatographed to furnish the product as white solid (2 g).
1H NMR (CDCl3, 400 MHz) δ ppm: 2.23-2.30 (m, 1 H); 2.62 (s, 3 H), 2.64-2.71 (m, 1 H), 3.40-3.46 (m, 1 H), 3.50-3.55 (m, 1 H), 3.89 (s, 3 H), 4.89 (t, J= 7.6 Hz, 1 H), 6.99 (t, J= 2.4 Hz, 1 H), 7.11 (d, J= 8.8 Hz, 2 H), 7.36 (s, 1 H), 7.51 (s, 1 H), 8.03 (d, J = 8.8 Hz, 2 H); ESI MS m/z (relative intensities): 410.1 (M+H)+ (45 %); UPLC Purity: 96.40 %, Ret. time: 3.48 min. Chiral Purity by HPLC: 90.92 %, Ret. Time: 48.36 min.


http://zyduscadila.com/wp-content/uploads/2015/09/ZYDPLA1-a-Novel-LongActing-DPP-4-Inhibitor.pdf
http://zyduscadila.com/wp-content/uploads/2015/05/PressNote23-10-13.pdf
http://zyduscadila.com/wp-content/uploads/2015/07/annual_report_14-15.pdf
http://pharmaxchange.info/press/2012/08/glucokinase-activators-gkas-in-diabetes-management/
LB-PP02-4 ZYDPLA1, a novel long-acting DPP-4 inhibitor
Jt Int Congr Endocrinol Annu Meet Endocr Soc (ICE/ENDO) (June 21-24, Chicago) 2014, Abst LBSU-1075
LB-PP02-4 ZYDPLA1, a Novel Long-Acting DPP-4 Inhibitor
Session: LBSU 1074-1087-Diabetes & Obesity
Translational
Disclosure: MRJ: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. AAJ: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. RB: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. HP: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. SK: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. PJ: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. VP: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. KP: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. VKR: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India. PRP: Chairman, Cadila Healthcare Limited, Ahmedabad, India. RD: Employee, Zydus Research Centre, Cadila Healthcare Limited, Ahmedabad, India.

////////Dipeptidyl Peptidase IV, CD26, DPP-IV, DP-IV, Inhibitors
