
Home » Posts tagged 'Treatment'
Tag Archives: Treatment
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
ETAMICASTAT

Etamicastat HCl salt
CAS: 677773-32-9 (HCl salt)
CAS 760173-05-5 (free base).
Chemical Formula: C14H16ClF2N3OS
Molecular Weight: 347.8088
Synonym: BIA 5-453; BIA5-453; BIA-5-453; Etamicastat
IUPAC/Chemical Name: (R)-5-(2-aminoethyl)-1-(6,8-difluorochroman-3-yl)-1,3-dihydro-2H-imidazole-2-thione hydrochloride
5-(2-Aminoethyl)-1-((3R)-6,8-difluoro-3,4-dihydro-2H-chromen-3-yl)-1,3-dihydro-2h-imidazole-2-thione
R)-5-(2-aminoethyl)-1-(6,8-difluorochroman-3-yl)-1,3-dihydroimidazole-2-thione hydrochloride,
PHASE 2, Treatment of Heart Failure Therapy, Hypertension
Bial-Portela and Ca, S.A

is a novel peripherally selective dopamine β-hydroxylase (DBH) inhibitor being developed by Bial-Portela and Ca, S.A. for treatment of hypertension and congestive heart failure.(1) The compound was shown to be well tolerated in healthy volunteers.
Etamicastat, also known as BIA 5-453, is a potent, reversible, peripherally selective dopamine β-hydroxylase inhibitor (DBH inhibitor). Chronic dopamine ß-hydroxylase inhibition with etamicastat effectively decreases blood pressure, although does not prevent the development of hypertension in the spontaneously hypertensive rat.

aReagents and conditions: a) Boc2O, EtOH, rt, 2 h; b) TBDMS-Cl, Et3N, DMAP, DCM, rt, 18 h; c) Dess–Martin periodinane, DCM, rt, 1 h; d) 2, KSCN, AcOH, EtOAc, reflux, 7 h; e) 2 N HCl, EtOAc, rt, 2 h.
Paper
Development of the Asymmetric Hydrogenation Step for Multikilogram Production of Etamicastat

The asymmetric hydrogenation of methyl (6,8-difluoro-2H-chromen-3-yl)carbamate is a key step in the manufacturing route to etamicastat. A development of this step including the ruthenium or rhodium catalyst screening and the influence of the catalyst preparation (isolated, preformed in solution or in situ), solvent, temperature, pressure, additive, and concentration on the performance of the given ligand was discussed. Scale-up experiments for the best catalysts under optimized conditions were described.
PAPER
J Med Chem 2006, 49(3): 1191
in the processes .
(J?) -5- (2-Aminoethyl) -1- (6, 8-difluorochroman-3-yl) -1, 3-dihydroimidazole-2 -thione hydrochloride (the compound of formula 1, below) is a potent, non-toxic and peripherally selective inhibitor of ϋβΗ, which can be used for treatment of certain cardiovascular disorders. Compound 1 is disclosed in WO2004/033447 , along with processes for its preparation.

1
The process disclosed in WO2004/033447 involves the reaction of ( R) – 6 , 8 -difluorochroman-3 -ylamine hydrochloride (the structure of ( R) -6, 8-difluorochroman-3 -ylamine is shown below as compound QA) , [4 – ( tert-butyldimethylsilanyloxy) -3 -oxobutyl] carbamic acid tert-butyl ester and potassium thiocyanate .

QA
(R) -6 , 8-difluorochroman- 3 -ylamine (compound QA) is a key intermediate in the synthesis of compound 1. The stereochemistry at the carbon atom to which the amine is attached gives rise to the stereochemistry of compound 1, so it is advantageous that compound QA is present in as pure enantiomeric form as possible. In other words, the (R) -enantiomer of compound QA should be in predominance, with little or no (S) enantiomer present. Thus, the process for preparing compound QA will advantageously produce compound QA with as high enantiomeric excess (ee) as possible.
Advantageous processes for preparing, for example, the compound of formula QA have now been found. In one aspect, the processes involve a biotransformation step. In another aspect, the processes involve chemical transformation. The processes may also be employed in the preparation of similar precursors useful in the production of other peripherally-selective inhibitors of dopamine -β -hydroxylase .
WO2008/136695 discloses a compound of formula YA, its (R) or (S) enantiomer, a mixture of its (R) and (S) enantiomers, or pharmaceutically acceptable salts thereof.

YA
The (R) -enantiomer of the compound of formula YA has been found to be a potent dopamines-hydroxylase inhibitor having high potency and significantly reduced brain access.
As disclosed in WO2008/136695 , the compound of formula YA may be prepared by reacting the compound of formula 1 with benzaldehyde under reductive alkylation conditions. In particular, (R) -5- (2 -aminoethyl ) -1- (6 , 8-difluorochroman-3 -yl) – 1 , 3 -dihydroimidazole-2 -thione and benzaldehyde may be reacted in the presence of a solvent or mixture of solvents, and a reducing agent such as sodium cyanoborohydride or sodium triacetoxyborohydride .
The compound of formula W may be prepared using a process as disclosed herein from the nitro chromene compound M.
The compound of formula WA may also be prepared using a process comprising bromination of 2 , 4 -difluorophenol to give bromophenol, alkylation of bromophenol with 4 -chloro-3 -oxo butanoate to give ketone followed by cyclization and decarboxylation to produce compound WA.

WA
According to an aspect of the present invention, there is provided the following 2 -part synthetic route from the starting material 2 , 4 -difluorophenol to (R) -5- (2 -aminoethyl ) -1- (6 , 8-difluorochroman-3 -yl) -1 , 3 -dihydroimidazole-2 – thione
hydrochloride :
Part (1)


Preferred reagents and conditions:
a) HMTA, CF3COOH, 115°C, 18 hours
b) CH2CHCN, DABCO, DMF, water, 70°C, 16 hours
c) H2S04, AcOH, 100°C, 1 hour
d) NaClO, NaOH, MeOH, 25°C, 24 hours
e) (R) -C3 -TunePhosRu (acac) 2 S/C 3000, 30 bar H2, MeOH, 80°C, 20 hours
f) Water, 2-propanol, reflux to 20°C
g) 40% KOH, MeOH, reflux, 24 hours
h) L-tartaric acid, ethanol, water, RT, 1 hour
Part (2)

![]()

Preferred reagents and conditions
a’) methyl vinyl ketone, t-BuONa, EtOAc, EtOH, 40-50°C, 2-3 hours
Br2, MeOH, 20-25°C, 5 hours
water, reflux, 1 hour
KOH, AcOH, reflux, 1 hour
HCl, water, 2-propanol, 75 °C, 4 hours
KSCN, AcOH, 100°C, 2-4 hours
NaHC03, water, EtOH
NaBH4, 2-propanol, THF, water, 20-25°C, 16 hours
HCl, 2-propanol, water, reflux, 1-2 hours
The ( R ) -5- (2-Aminoethyl) -1- (6, 8-difluorochroman-3 -yl) -1,3-dihydroimidazole-2 – thione hydrochloride
EXAMPLES
Example 1
Nitro chromene synthesis

To 3 , 5-difluoro-2-hydroxybenzaldehyde (lOg, 63mmol, leq) , di-n-butylamine (4.1g, 32mmol, 0.5eq) , phtalic anhydride (18.7g, 126mmol, 2eq) in toluene (500mL) was added nitroethanol (5.75g, 63mmol, leq) . The round bottomed flask fitted with a dean stark apparatus was refluxed for 18h. The mixture was cooled and nitroethanol (5.75g, 63mmol, leq) was added. The resulting reaction mixture was then reflux for 12h. After cooling, the solution was evaporated down to approximately 150mL and purified over silica gel (eluent ethyl acetate : hexane 1:1) this gave several fractions that contained only the product by TLC, these was evaporated under reduced pressure to yield 1.8g which was 100% pure by HPLC aera. Several more fractions were collected containing a mixture of product and starting material. These were combined and washed with 2% NaOH solution (2x50mL) to remove starting material. The organic layer was washed with water (50mL) , dried over sodium sulfate and evaporated under reduced pressure to give 2.49g of brown solid ( 100% pure by HPLC aera) . More fractions were collected. These were combined, washed with 2% NaOH solution (3xl00mL) , water (lOOmL) and dried over sodium sulfate. This was then filtered and evaporated down in vacuum to yield 6.14g of a brown solid which was 91.3% pure by HPLC aera. 6 , 8 -difluoro-3 -nitro-2H-chromene (9.90g, 73.4%) was obtained as a brown solid.
Example 2
Nitro chromene synthesis with column purification
To a solution of isobenzofuran-1 , 3 -dione (4,68 g, 31,6 mmol) , 3 , 5-difluoro-2 -hydroxybenzaldehyde (2,5 g, 15,81 mmol) in Toluene (25 ml) was added 2 -nitroethanol (2,88 g, 31,6 mmol). The resulting mixture was heated to reflux overnight (Dean stark) .
The reaction conversion was checked by TLC (eluent PE/EtOAc 9:1) . A yellow spot was observed and corresponds to the expected product .
Reaction was cooled to room temperature and a plug of silica gel was performed. A pale brown solid (3.9g) was obtained. “””H-NMR showed presence of product and starting material. The solid was dissolved in diethylether and the organic layer was washed with aqueous sodium carbonate, dried over Na2S04, filtered and concentrated under reduced pressure. A pale brown solid (1.7g,) was obtained. The 1H-NMR was indicated no starting material but still polymer from nitroethanol and residue of phtalic anhydride. A second silica plug (eluent: PE/EtOAc 95:5) was done. A pale yellow solid (1.5g) was obtained. 1H-NMR of solid showed only product and polymer. The solid was recrystallized from methanol/water . A pale yellow solid (1.05g, 31.2%) was obtained.
Example 3
Nitro chromene synthesis without column purification
To a solution of isobenzofuran- 1 , 3 -dione (18,74 g, 127 mmol) , 3 , 5-difluoro-2 -hydroxybenzaldehyde (10 g, 63,3 mmol) in Toluene (100 ml) was added 2 -nitroethanol (6,86 ml, 95 mmol) . The resulting mixture was heated to reflux for 24h (Dean stark) .
The reaction conversion was checked by HPLC and by 1H-NMR. Only 50% conversion was obtained.
The reaction mixture was cooled to room temperature and diluted with DCM (lOOmL) and 1M NaOH solution (200mL) .
The biphasic system was stirred for 30 minutes and then separated (very difficult to see phase separation) . The aqueous layer was washed with DCM (50mL) and the combined organic layers were washed twice with water (2x50ml) , dried over sodium sulfate. The filtered organic layer was concentrated under reduced pressure. To the residue was added methanol (50mL) . The methanol was then removed by distillation under reduced pressure. A brown solution precipitated when most of the methanol was removed. More methanol was added and more solid crushed out then few drops of water was added to increase the product precipitation. The brown slurry was stirred for 30 minutes and filtered. The brown solid was washed with methanol/water (1:9, 5mL) and dried in a vacuum oven at 40°C for 12h.6, 8-difluoro-3 -nitro-2H-chroraene (4,9 g, 22,99 mmol,) was obtained as brown solid in 36.3% yield.
HPLC showed a purity of 98% and 1H-NMR confirmed the structure and purity around 95%
Example 4
Reduction of nitro chromene to nitro-alkane (racemic mixture)

To a suspension of 6 , 8 -difluoro-3 -nitro-2H-chromene (213mg, 0,999 mmol) and silica (0,8 g, 0,999 mmol) in a mixture of CHC13 (10 ml) and IPA (3,4 ml) at 0°C was added portion wise sodium borohydride (95 mg, 2,498 mmol). The resulting mixture was stirred at 0°C for 45 minutes. Reaction conversion was checked by HPLC. 1 mL of acetic acid was added at 0°C and the resulting mixture was stirred for 30 minutes at room temperature. The slurry was filtered and the silica was washed with DCM. The filtrate was diluted with ethyl acetate and water and the biphasic system was separated. The aqueous layer was back extracted with ethyl acetate. The combined organic layers were washed with brine, dried over MgS04, filtered and concentrated under reduced pressure.
6 , 8-difluoro-3 -nitrochroman (196mg, 0,911 mmol, 91 % yield) was obtained as a pale yellow oil.
Example 5
Preparation of 6 , 8 -difluorochroman-3 -one from nitro chromene

A solution of 6, 8-difluoro-3 -nitro-2H-chromene (lOOmg, 0,469 mmol) in acetic acid (0.5 ml) is added slowly to a stirred slurry of iron (262 mg, 4,69 mmol) in acetic acid (1 ml) at 60.deg. C. The reaction mixture is stirred at 60. °C for 2 hour then allowed to cool to room temperature and stirred overnight. The reaction mixture is poured onto ice-water (30 ml) and filtered through Celite. The solid was wash with dichloromethane (DCM) (50 ml) . The organic portion is separated and washed with water (2 x 30 ml) and brine (30 ml) , dried over MgS04, filtered and concentrated in vacuo to give a brown oil. 6,8-difluorochroman-3 -one (75 mg, 0,407 mmol, 87 % yield) was obtained as a brown oil.
Example 6
Preparation of 6 , 8-difluorochroman-3 -one from methyl 6,8-difluoro-2H-chromen-3 -yl-carbamate

Methanol (1000m ml) was added to a slurry of methyl fluoro-2H-chromen-3 -yl -carbamate (250 g, 1.037 mol) hydrogen chloride 6N (2000 ml, 12 mol) at room temperature. The resulting mixture was reflux and stirred for 2 hours. Reaction monitored by HPLC.
Reaction was not complete but was stopped in order to avoid degradation of the product. The yellow solution was cooled to room temperature. A slurry (two type of solid) was observed and diluted with diethyl ether (300mL) . The resulting slurry was stirred at 5°C for 1 hour then filtered. The yellow solid was washed with water. The resulting wet yellow solid was suspended in diethylether (400mL) and petroleum ether (PE) (400mL) was added. Slight yellow solid was stirred at room temperature overnight, filtered and washed with PE (300mL) , dried in a vacuum oven at 30 °C for 4h. The wet sample was checked by NMR. No starting material was detected. A pale yellow solid (72.5g, solid 1) was obtained. The mother liquors were concentrated to dryness. A yellow solid was obtained, suspended in diethyl ether and PE. The slurry was then stirred for 4 hours, filtered, washed with PE . A dark yellow solid (4.5g, solid 2) was obtained. Solid 1 (2g) was diluted in DCM and washed with water (pH =6). The organic layer was then dried over Na2S04, filtered, concentrated to dryness. A crystalline pale yellow solid (1.9g, solid 3) was obtained. NMR showed the same purity for solid 3 as for solid 1. The remaining part of solid 1 was then diluted in DCM. The resulting organic layer was washed with water, dried over Na2S04, filtered and then concentrated to dryness. Slight yellow crystalline solid (68.5g, solid 4) was obtained. NMR confirmed high quality material.
Loss on Drying (LOD) : 1.03% .
Example 7
Biotransformation: Transaminases

Codexis transaminases ATA-025, ATA-251 and ATA-P2-A07 recognized 6 , 8 -difluorochroman-3 -one as the substrate and produced the corresponding 6 , 8 -difluorochroman-3 -amine .
References
1: Igreja B, Wright LC, Soares-da-Silva P. Sustained high blood pressure reduction with etamicastat, a peripheral selective dopamine β-hydroxylase inhibitor. J Am Soc Hypertens. 2015 Dec 19. pii: S1933-1711(15)00838-4. doi: 10.1016/j.jash.2015.12.011. [Epub ahead of print] PubMed PMID: 26803288.
2: Loureiro AI, Bonifácio MJ, Fernandes-Lopes C, Pires N, Igreja B, Wright LC, Soares-da-Silva P. Role of P-glycoprotein and permeability upon the brain distribution and pharmacodynamics of etamicastat: a comparison with nepicastat. Xenobiotica. 2015;45(9):828-39. doi: 10.3109/00498254.2015.1018985. Epub 2015 Jun 10. PubMed PMID: 25915108.
3: Loureiro AI, Soares-da-Silva P. Distribution and pharmacokinetics of etamicastat and its N-acetylated metabolite (BIA 5-961) in dog and monkey. Xenobiotica. 2015;45(10):903-11. doi: 10.3109/00498254.2015.1024780. Epub 2015 Apr 14. PubMed PMID: 25869244.
4: Pires NM, Igreja B, Moura E, Wright LC, Serrão MP, Soares-da-Silva P. Blood pressure decrease in spontaneously hypertensive rats folowing renal denervation or dopamine β-hydroxylase inhibition with etamicastat. Hypertens Res. 2015 Sep;38(9):605-12. doi: 10.1038/hr.2015.50. Epub 2015 Apr 9. PubMed PMID: 25854989.
5: Bonifácio MJ, Sousa F, Neves M, Palma N, Igreja B, Pires NM, Wright LC, Soares-da-Silva P. Characterization of the interaction of the novel antihypertensive etamicastat with human dopamine-β-hydroxylase: comparison with nepicastat. Eur J Pharmacol. 2015 Mar 15;751:50-8. doi: 10.1016/j.ejphar.2015.01.034. Epub 2015 Jan 29. PubMed PMID: 25641750.
6: Pires NM, Loureiro AI, Igreja B, Lacroix P, Soares-da-Silva P. Cardiovascular safety pharmacology profile of etamicastat, a novel peripheral selective dopamine-β-hydroxylase inhibitor. Eur J Pharmacol. 2015 Mar 5;750:98-107. doi: 10.1016/j.ejphar.2015.01.035. Epub 2015 Jan 30. PubMed PMID: 25641747.
7: Igreja B, Pires NM, Bonifácio MJ, Loureiro AI, Fernandes-Lopes C, Wright LC, Soares-da-Silva P. Blood pressure-decreasing effect of etamicastat alone and in combination with antihypertensive drugs in the spontaneously hypertensive rat. Hypertens Res. 2015 Jan;38(1):30-8. doi: 10.1038/hr.2014.143. Epub 2014 Oct 9. PubMed PMID: 25298210.
8: Loureiro AI, Bonifácio MJ, Fernandes-Lopes C, Igreja B, Wright LC, Soares-da-Silva P. Etamicastat, a new dopamine-ß-hydroxylase inhibitor, pharmacodynamics and metabolism in rat. Eur J Pharmacol. 2014 Oct 5;740:285-94. doi: 10.1016/j.ejphar.2014.07.027. Epub 2014 Jul 21. PubMed PMID: 25058908.
9: Almeida L, Nunes T, Costa R, Rocha JF, Vaz-da-Silva M, Soares-da-Silva P. Etamicastat, a novel dopamine β-hydroxylase inhibitor: tolerability, pharmacokinetics, and pharmacodynamics in patients with hypertension. Clin Ther. 2013 Dec;35(12):1983-96. doi: 10.1016/j.clinthera.2013.10.012. Epub 2013 Dec 2. PubMed PMID: 24296323.
10: Loureiro AI, Rocha JF, Fernandes-Lopes C, Nunes T, Wright LC, Almeida L, Soares-da-Silva P. Human disposition, metabolism and excretion of etamicastat, a reversible, peripherally selective dopamine β-hydroxylase inhibitor. Br J Clin Pharmacol. 2014 Jun;77(6):1017-26. doi: 10.1111/bcp.12274. PubMed PMID: 24168152; PubMed Central PMCID: PMC4093927.
11: Loureiro AI, Fernandes-Lopes C, Bonifácio MJ, Wright LC, Soares-da-Silva P. N-acetylation of etamicastat, a reversible dopamine-β-hydroxylase inhibitor. Drug Metab Dispos. 2013 Dec;41(12):2081-6. doi: 10.1124/dmd.113.053736. Epub 2013 Sep 6. PubMed PMID: 24013186.
12: Nunes T, Rocha JF, Vaz-da-Silva M, Falcão A, Almeida L, Soares-da-Silva P. Pharmacokinetics and tolerability of etamicastat following single and repeated administration in elderly versus young healthy male subjects: an open-label, single-center, parallel-group study. Clin Ther. 2011 Jun;33(6):776-91. doi: 10.1016/j.clinthera.2011.05.048. PubMed PMID: 21704242.
13: Vaz-da-Silva M, Nunes T, Rocha JF, Falcão A, Almeida L, Soares-da-Silva P. Effect of food on the pharmacokinetic profile of etamicastat (BIA 5-453). Drugs R D. 2011;11(2):127-36. doi: 10.2165/11587080-000000000-00000. PubMed PMID: 21548660; PubMed Central PMCID: PMC3585837.
14: Rocha JF, Vaz-Da-Silva M, Nunes T, Igreja B, Loureiro AI, Bonifácio MJ, Wright LC, Falcão A, Almeida L, Soares-Da-Silva P. Single-dose tolerability, pharmacokinetics, and pharmacodynamics of etamicastat (BIA 5-453), a new dopamine β-hydroxylase inhibitor, in healthy subjects. J Clin Pharmacol. 2012 Feb;52(2):156-70. doi: 10.1177/0091270010390805. PubMed PMID: 21343348.
15: Nunes T, Rocha JF, Vaz-da-Silva M, Igreja B, Wright LC, Falcão A, Almeida L, Soares-da-Silva P. Safety, tolerability, and pharmacokinetics of etamicastat, a novel dopamine-β-hydroxylase inhibitor, in a rising multiple-dose study in young healthy subjects. Drugs R D. 2010;10(4):225-42. doi: 10.2165/11586310-000000000-00000. PubMed PMID: 21171669; PubMed Central PMCID: PMC3585840.
16: Beliaev A, Learmonth DA, Soares-da-Silva P. Synthesis and biological evaluation of novel, peripherally selective chromanyl imidazolethione-based inhibitors of dopamine beta-hydroxylase. J Med Chem. 2006 Feb 9;49(3):1191-7. PubMed PMID: 16451083.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO1995007284A1 * | Aug 29, 1994 | Mar 16, 1995 | Smithkline Beecham Plc | Phosphinic acid derivatives with anti-hyper glycemic and/or anti-obesity activity |
| WO2006044293A2 * | Oct 11, 2005 | Apr 27, 2006 | Pharmacopeia Drug Discovery, Inc. | Bicyclic compounds as selective melanin concentrating hormone receptor antagonists for the treatment of obesity and related disorders |
| WO2012007548A1 * | Jul 14, 2011 | Jan 19, 2012 | Dsm Ip Assets B.V. | (r)-selective amination |
| WO2013002660A2 * | Jun 29, 2012 | Jan 3, 2013 | BIAL – PORTELA & Cª, S.A. | Process |
| GR1005093B * | Title not available |
| Reference | ||
|---|---|---|
| 1 | * | AL NEIRABEYEH M. ET AL.: “Methoxy and hydroxy derivatives of 3,4-dihydro-3-(di-n-propylamino)-2H-1-benzopyrans: new synthesis and dopaminergic activity“, EUROPEAN JOURNAL OF MEDICINAL CHEMISTRY, vol. 26, no. 5, 1991, EDITIONS SCIENTIFIQUE ELSEVIER, PARIS; FR, pages 497 – 504, XP023870436, ISSN: 0223-5234, DOI: 10.1016/0223-5234(91)90145-D |
| 2 | * | BELIAEV, A. ET AL.: “Process Research for Multikilogram Production of Etamicastat: A Novel Dopamine ß-Hydroxylase Inhibitor“, ORGANIC PROCESS RESEARCH & DEVELOPMENT, no. 16, 2012, American Chemical Society, Washington; US, pages 704 – 709, XP002731798, DOI: 10.1021/op300012d |
| 3 | * | BOYE, S. ET AL.: “N,N-Disubstituted aminomethyl benzofuran derivatives: synthesis and preliminary binding evaluation“, BIOORGANIC & MEDICINAL CHEMISTRY, no. 7, 1999, ELSEVIER SCIENCE LTD; GB, pages 335 – 341, XP002731795, ISSN: 0968-0896, DOI: 10.1016/S0968-0896(98)00239-9 |
| 4 | * | COMOY, C. ET AL.: “3-Amino-3,4-dihydro-2H-1-benzopyran Derivatives as 5-HT1A Receptor Ligandsand Potential Anxiolytic Agents. 2. Synthesis and QuantitativeStructure-Activity Relationship Studies of Spiro[pyrrolidine- andpiperidine-2,3′(2’H)-benzopyrans]“, JOURNAL OF MEDICINAL CHEMISTRY., vol. 39, no. 21, 1996, AMERICAN CHEMICAL SOCIETY. WASHINGTON; US, pages 4285 – 4298, XP002731797, ISSN: 0022-2623, DOI: 10.1021/JM950861W |
| 5 | * | SHIN, C. ET AL.: “Total Synthesis of Bistratamide G, a Metabolite of the PhilippinesAscidian Lissoclinum bistratum, from Dehydrotripeptides“, CHEMISTRY LETTERS, vol. 33, no. 6, 2004, Chemical Society of Japan, Tokyo; JP, pages 664 – 665, XP002731799, ISSN: 0366-7022, DOI: 10.1246/cl.2004.664 |
| 6 | * | VASSE, J. L. ET AL.: “New efficient conditions for the reduction with NADH models“, SYNLETT, October 1998 (1998-10-01), THIEME INTERNATIONAL, STUTTGART; DE, pages 1144 – 1146, XP002731796, ISSN: 0936-5214, DOI: 10.1055/s-1998-1876 |
| 7 | * | XIAO, G.-Q. ET AL.: “3-Nitro-2H-chromenes as a New Class of Inhibitors against Thioredoxin Reductase and Proliferation of Cancer Cells“, ARCHIV DER PHARMAZIE, no. 345, 2012, VCH VERLAGSGESELLSCHAFT MBH, WEINHEIM; DE, pages 767 – 770, XP002731794, ISSN: 0365-6233, DOI: 10.1002/ardp.201200121 |
////////Etamicastat, BIA-5-453 , PHASE 2, Treatment, Heart Failure Therapy, Hypertension, Bial-Portela and Ca, S.A
SMILES Code: FC1=CC(F)=C(OC[C@H](N2C(CCN)=CNC2=S)C3)C3=C1.[H]Cl
c1c(cc(c2c1C[C@H](CO2)n3c(c[nH]c3=S)CCN)F)F
LIK 066, Licogliflozin diprolinate
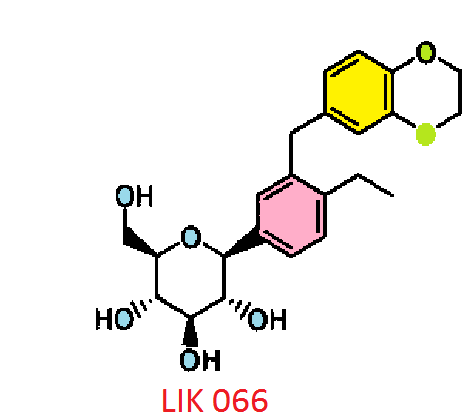
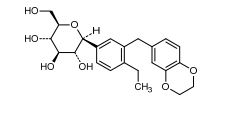
Licogliflozin
LIK 066
Licogliflozin diprolinate

LIK-066, a new flozin on the horizon
C23 H28 O7 . 2 C6 H11 N O, 642.7795, 1 :2 co-crystal of Example 62 : L-proline. A melting point 176°C…WO2011048112
CAS 1291095-45-8, (1S)-1,5-anhydro-1-C-[3-[(2,3-dihydro-1,4-benzodioxin-6-yl)methyl]-4-ethylphenyl]-D-glucitol (1:1) WITH L-Proline, compd., 1:1 Proline Co-crvstal , 1:1 Proline Co-crvstal …..…WO2011048112
CAS BASE 1291094-73-9, 416.46, C23 H28 O7
(1S)-1,5-Anhydro-1-[3-(2,3-dihydro-1,4-benzodioxin-6-ylmethyl)-4-ethylphenyl]-D-glucitol bis[1-[(2S)-pyrrolidin-2-yl]ethanone]
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethyl)-4- ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
Sodium glucose transporter-2 inhibitor
SGLT 1/2 inhibitor
Novartis Ag innovator
Clinical trial……..https://clinicaltrials.gov/ct2/show/NCT01915849
https://clinicaltrials.gov/ct2/show/NCT02470403
- 10 Jun 2015 Novartis initiates enrolment in a phase II trial for Type 2 diabetes mellitus in USA (NCT02470403)
- 02 Apr 2014 Novartis terminates a phase II trial in Type-2 diabetes mellitus in USA, Poland, Argentina, Hungary, Puerto Rico and South Africa (NCT01824264)
- 01 Jan 2014 Novartis completes a phase II trial in Type 2 diabetes mellitus in USA (NCT01915849)
Licogliflozin, a SGLT-1/2 inhibitor, is in phase II clinical development at Novartis for the treatment of metabolic disorders, for the treatment of heart failure in patients with type 2 diabetes, for the treatment of obesity and for the treatment of polycystic ovary syndrome (PCOS) in overweight and obese women. Phase II trials for the treatment of type 2 diabetes had been discontinued.
EMA/415156/2014 European Medicines Agency decision P/0183/2014 of 24 July 2014 on the agreement of a paediatric investigation plan and on the granting of a deferral and on the granting of a waiver for (S)-Pyrrolidine-2-carboxylic acid compound with (2S,3R,4R,5S,6R)-2-(3-((2,3- dihydrobenzo[b][1,4]dioxin-6-yl)methyl)-4-ethylphenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran3,4,5-triol (2:1) (LIK066) (EMEA-001527-PIP01-13) in accordance with Regulation (EC) No 1901/2006 of the European Parliament and of the Council
1. Opinion of the Paediatric Committee on the agreement of a Paediatric Investigation Plan and a deferral and a waiver. 2014, EMEA-001527-PIP01-13 (here) [ Novartis revealed the IUPAC name here].
Where name is given
http://www.who.int/medicines/publications/druginformation/issues/DrugInformation2017_Vol31-4/en/


http://www.who.int/medicines/publications/druginformation/issues/PL_118.pdf?ua=1
SEE ALSO


LIK-066 is in phase II clinical studies at Novartis for the treatment of type 2 diabetes.
In June 2014, the EMA’s PDCO adopted a positive opinion on a pediatric investigation plan (PIP) for LIK-066 for type 2 diabetes
Diabetes mellitus is a metabolic disorder characterized by recurrent or persistent hyperglycemia (high blood glucose) and other signs, as distinct from a single disease or condition. Glucose level abnormalities can result in serious long-term complications, which include cardiovascular disease, chronic renal failure, retinal damage, nerve damage (of several kinds), microvascular damage and obesity.
Type 1 diabetes, also known as Insulin Dependent Diabetes Mellitus (IDDM), is characterized by loss of the insulin-producing β-cells of the islets of Langerhans of the pancreas leading to a deficiency of insulin. Type-2 diabetes previously known as adult- onset diabetes, maturity-onset diabetes, or Non-Insulin Dependent Diabetes Mellitus (NIDDM) – is due to a combination of increased hepatic glucose output, defective insulin secretion, and insulin resistance or reduced insulin sensitivity (defective responsiveness of tissues to insulin). Chronic hyperglycemia can also lead to onset or progression of glucose toxicity characterized by decrease in insulin secretion from β-cell, insulin sensitivity; as a result diabetes mellitus is self-exacerbated [Diabetes Care, 1990, 13, 610].
Chronic elevation of blood glucose level also leads to damage of blood vessels. In diabetes, the resultant problems are grouped under “microvascular disease” (due to damage of small blood vessels) and “macro vascular disease” (due to damage of the arteries). Examples of microvascular disease include diabetic retinopathy, neuropathy and nephropathy, while examples of macrovascular disease include coronary artery disease, stroke, peripheral vascular disease, and diabetic myonecrosis.
Diabetic retinopathy, characterized by the growth of weakened blood vessels in the retina as well as macular edema (swelling of the macula), can lead to severe vision loss or blindness. Retinal damage (from microangiopathy) makes it the most common cause of blindness among non-elderly adults in the US. Diabetic neuropathy is characterized by compromised nerve function in the lower extremities. When combined with damaged blood vessels, diabetic neuropathy can lead to diabetic foot. Other forms of diabetic neuropathy may present as mononeuritis or autonomic neuropathy. Diabetic nephropathy is characterized by damage to the kidney, which can lead to chronic renal failure, eventually requiring dialysis. Diabetes mellitus is the most common cause of l adult kidney failure worldwide. A high glycemic diet (i.e., a diet that consists of meals that give high postprandial blood sugar) is known to be one of the causative factors contributing to the development of obesity.
Type 2 diabetes is characterized by insulin resistance and/or inadequate insulin secretion in response to elevated glucose level. Therapies for type 2 diabetes are targeted towards increasing insulin sensitivity (such as TZDs), hepatic glucose utilization (such as biguanides), directly modifying insulin levels (such as insulin, insulin analogs, and insulin secretagogues), increasing increttn hormone action (such as exenatide and sitagliptin), or inhibiting glucose absorption from the diet (such as alpha glucosidase inhibitors) [Nature 2001 , 414, 821-827],
Glucose is unable to diffuse across the cell membrane and requires transport proteins. The transport of glucose into epithelial cells is mediated by a secondary active cotransport system, the sodium-D-glucose co-transporter (SGLT), driven by a sodium- gradient generated by the Na+/K+-ATPase. Glucose accumulated in the epithelial cell is further transported into the blood across the membrane by facilitated diffusion through GLUT transporters [Kidney International 2007, 72, S27-S35].
SGLT belongs to the sodium/glucose co-transporter family SLCA5. Two different SGLT isoforms, SGLT1 and SGLT2, have been identified to mediate renal tubular glucose reabsorption in humans [Curr. Opinon in Investigational Drugs (2007): 8(4), 285-292 and references cited herein]. Both of them are characterized by their different substrate affinity. Although both of them show 59% homology in their amino acid sequence, they are functionally different. SGLT1 transports glucose as well as galactose, and is expressed both in the kidney and in the intestine, while SGLT2 is found exclusively in the S1 and S2 segments of the renal proximal tubule.
As a consequence, glucose filtered in the glomerulus is reabsorbed into the renal proximal tubular epithelial cells by SGLT2, a low-affinity/high-capacity system, residing on the surface of epithelial cell lining in S1 and S2 tubular segments. Much smaller amounts of glucose are recovered by SGLT1 , as a high-affinity/low-capacity system, on the more distal segment of the proximal tubule. In healthy human, more than 99% of plasma glucose that is filtered in the kidney glomerulus is reabsorbed, resulting in less than 1 % of the total filtered glucose being excreted in urine. It is estimated that 90% of total renal glucose absorption is facilitated by SGLT2; remaining 10 % is likely mediated by SGLT1 [J. Parenter. Enteral Nutr. 2004, 28, 364-371].
SGLT2 was cloned as a candidate sodium glucose co-transporter, and its tissue distribution, substrate specificity, and affinities are reportedly very similar to those of the low-affinity sodium glucose co-transporter in the renal proximal tubule. A drug with a mode of action of SGLT2 inhibition will be a novel and complementary approach to existing classes of medication for diabetes and its associated diseases to meet the patient’s needs for both blood glucose control, while preserving insulin secretion. In addition, SGLT2 inhibitors which lead to loss of excess glucose (and thereby excess calories) may have additional potential for the treatment of obesity.
Indeed small molecule SGLT2 inhibitors have been discovered and the anti-diabetic therapeutic potential of such molecules has been reported in literature [T-1095 (Diabetes, 1999, 48, 1794-1800, Dapagliflozin (Diabetes, 2008, 57, 1723-1729)].
SYNTHESIS
PATENT
WO 2011048112
https://www.google.com/patents/WO2011048112A1?cl=en
Gregory Raymond Bebernitz, Mark G. Bock, Dumbala Srinivas Reddy, Atul Kashinath Hajare, Vinod Vyavahare, Sandeep Bhausaheb Bhosale, Suresh Eknath Kurhade, Videsh Salunkhe, Nadim S. Shaikh, Debnath Bhuniya, P. Venkata Palle, Lili Feng, Jessica Liang,
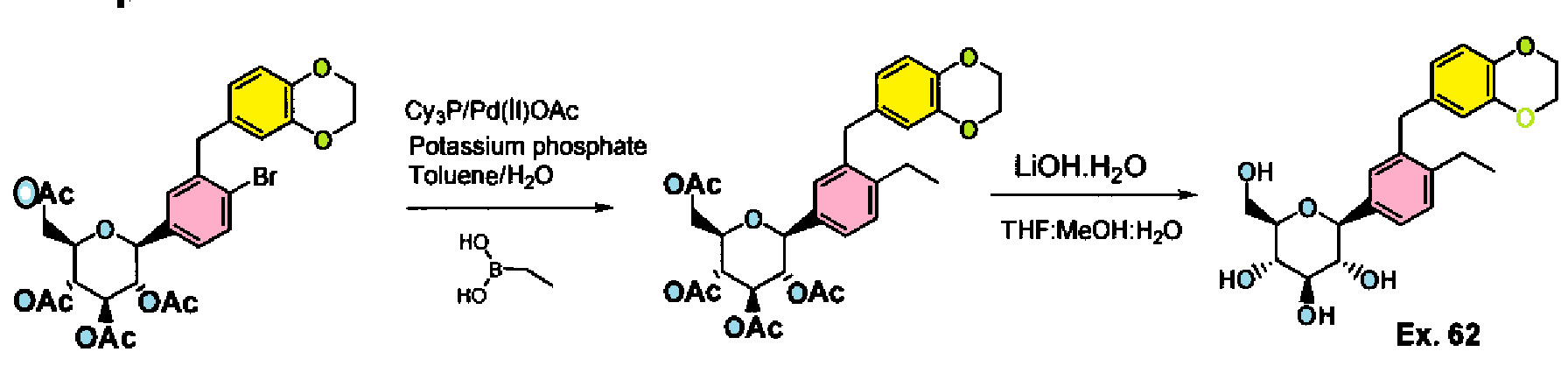
Example 61-62:

Ex. 61
Example 61 : Acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester
Step I: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3- (2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester (10.0 g, 15.74 mmol) in toluene (200 mL) was added tricyclohexylphosphine (1.76 g, 6.29 mmol), a solution of potassium phosphate tribasic (13.3 g, 62.9 mmol) in water (15 mL), and ethylboronic acid (3.4 g, 47.2 mmol). The reaction mixture was degassed for 45 min then palladium (II) acetate (529 mg, 2.3 mmol) was added. After refluxing overnight, the reaction mixture was cooled to room temperature, and water was added. The resulting mixture was extracted with ethyl acetate, (2 X 200 mL), washed with water and brine, then dried over sodium sulfate, concentrated and purified by column chromatography to furnish acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (5.4 g).
Example 62: (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1,4]dioxin-6-ylmethyl)-4- ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
Step II: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (9.3 g, 15.9 mmol) in methanol:THF:water 3:2:1 (170 mL) was added lithium hydroxide (764 mg, 19.1 mmol). After stirring for 2 h at room temperature, the volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate (150 mL) and washed with brine (75 mL), brine containing 5 mL of 5% aqueous KHS04 (75 mL), and brine (20 mL) again, then dried over sodium sulfate and concentrated to furnish (2S,3R,4R,5S,6R)-2-[4-Cyclopropyl-3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (6.59)
H NMR (400 MHz, CD3OD): δ 1.07 (t, J = 7.6 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.34- 3.50 (m, 4H), 3.68 (dd, J = 12.0, 5.6 Hz, 1 H), 3.85-3.91 (m, 3H), 4.08 (d, J = 9.6 Hz, 1 H), 4.17 (s, 4H), 6.53-6.58 (m, 2H), 6.68 (d, J – 8.4 Hz, 1 H), 7.15-7.25 (m, 3H).
MS (ES) m z 434.2 (M+18).
PICK UP IDEAS FROM HERE
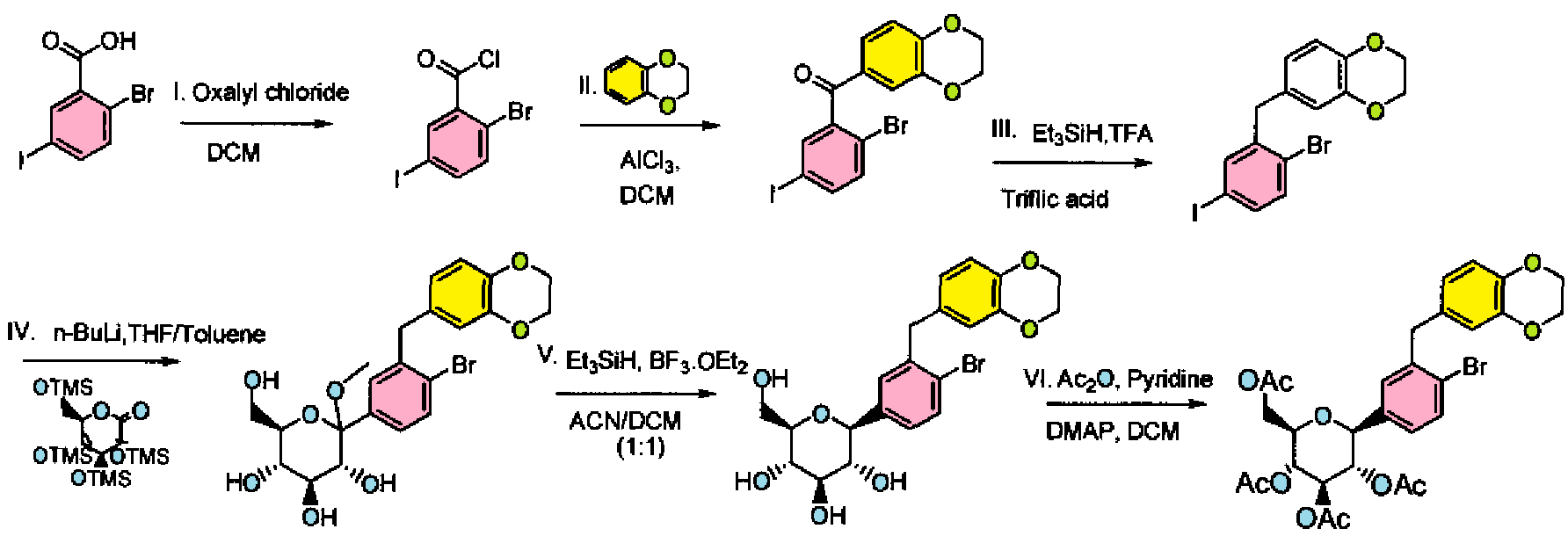
Examples 57-58:

Ex. 57 Ex. 58
Step I: To a stirred solution of 2-bromo-5-iodobenzoic acid (25.0 g, 76.48 mmol) in dichloromethane (200 mL) was added oxalyl chloride (10.3 mL, 114.74 mmol) at 0 °C followed by D F (0.9 mL). After complete addition, the reaction mixture was stirred at room temperature for 3h. Volatiles were evaporated under reduced pressure to furnish 2-bromo-5-iodo-benzoyl chloride (26.4 g). The crude product was used for the next step immediately.
Step II: To a stirred solution of 2-bromo-5-iodo-benzoyl chloride (26.4 g, 76.56 mmol) in dichloromethane (250 mL) was added benzo(1 ,4)-dioxane (10.41 g, 76.26 mmol) at 0 °C. To this reaction mixture, AICI3 (40.78 g, 305.47 mmol) was added in portions. After stirring overnight at room temperature, the reaction mixture was poured into crushed ice. The resulting mixture was extracted with dichloromethane (500 mL X 2). The dichloromethane layers were combined and washed with water (200 mL), saturated aqueous sodium bicarbonate solution (200 mL X 2), and brine (200 mL), then dried over sodium sulfate and concentrated. The solid product was triturated with hexanes, and the triturated product was dried under vacuum to furnish (2-bromo-5-iodo-phenyl)-(2,3- dihydro-benzo[1 ,4]dioxin-6-yl)-methanone (30 g).
1H N R (400 MHz, DMSO-D6): δ 4.29-4.37 (m, 4H), 7.02 (d, J = 8.4 Hz, 1 H), 7.16 (d, J = 2.4 Hz, 1 H), 7.18-7.19 (m, 1 H), 7.53 (d, J = 8.4 Hz, 1 H), 7.77-7.81 (m, 1 H), 7.82 (d, J = 2.0 Hz, 1 H).
Step III: To a stirred solution of (2-bromo-5-iodo-phenyl)-(2,3-dihydro-benzo[1 ,4]dioxin- 6-yl)-methanone (30.0 g, 67.4 mmol) in trifluoroacetic acid (100 mL) was added triethylsilane (86.2 mL, 539.3 mmol) followed by triflic acid (6.0 mL, 67.42 mmol ) at room temperature. After stirring for 25 min at room temperature, volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate and washed with saturated aqueous sodium bicarbonate solution (200 mL X 2), water (200 mL), and brine (200 mL), then dried over sodium sulfate, concentrated and purified by silica gel column chromatography to furnish 6-(2-bromo-5-iodo-benzyl)-2,3- dihydro-benzo[1 ,4]dioxine (26.5 g). H NMR (400 MHz, DMSO-D6): δ 3.90 (s, 4H), 4.2 (s, 2H), 6.65 (dd, J = 8.4 Hz, J = 2.0 Hz, H), 6.68 (d, J = 2.0 Hz, 1 H), 6.77 (d, J = 8.4 Hz, H), 7.39 (d, J = 8.4 Hz, 1 H), 7.50 (dd, J = 8.4 Hz, J = 2.4 Hz 1 H), 7.67 (d, J = 2.8 Hz, 1 H).
Step IV: To a stirred solution of 6-(2-bromo-5-iodo-benzyl)-2,3-dihydro- benzo[1 ,4]dioxine (26.5 g, 61.47 mmol) in THF:toluene 2:1 (300 mL) was added 1.6 M solution of n-BuLi in hexanes (42.3 mL, 67.62 mmol) at -78 °C. The reaction mixture was stirred for 1 h, and then transferred to a stirred solution of 2,3,4,6-tetrakis-O- (trimethylsilyl)-D-glucopyranone (28.69 g, 61.47 mmol) in toluene (100 mL) at -78 °C. After stirring for 1 h, 0.6 N methanesulfonic acid in methanol (265 mL) was added dropwise and stirred the reaction mixture for 16 h at room temperature. Reaction was quenched by the addition of aq. NaHC03 solution (~75 mL) and extracted with ethyl acetate (250 mL X 3), dried over sodium sulfate, concentrated and purified by silica gel column chromatography to furnish (3R,4S,5S,6R)-2-[4-Bromo-3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl-2-methoxy-tetrahydro-pyran- 3,4,5-triol (28.4 g)
Example 57: [(2R,3R,4R,5S,6S)-3,4,5-triacetoxy-6-[4-bromo-3-(2,3-dihydro-1 ,4- benzodioxin-6-ylmethyl)phenyl]tetrahydropyran-2-yl]methyl acetate
Step V: To a stirred solution of (3R,4S,5S,6R)-2-[4-bromo-3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl-2-methoxy-tetrahydro-pyran-3,4,5- triol (28.4 g, 57.1 mmol) in acetonitrile-dichloromethane 1 :1 (250 mL) was added triethylsilane (36.5 mL, 228.4 mmol) and boron trifluoride diethyletharate complex (14.1 mL, 114.2 mmol) at 10 °C. After stirring for 4 h at 10°C, the reaction was quenched with saturated aqueous sodium bicarbonate (~ 100 mL). The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (3 X 150 mL). The organic layers were combined and dried over sodium sulfate, concentrated to furnish (3R,4R,5S,6R)-2- [4-bromo-3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-6-hydroxymethyl- tetrahydro-pyran-3,4,5-triol (28.4 g). Crude product was used for next reaction without purification. Example 58: [(2R,3R,4R,5S,6S)-3,4,5-triacetoxy-6-[4-bromo-3-(2!3-dihydro-1,4- benzodioxin-6-ylmethyl)phenyl]tetrahydropyran-2-yl]methyl acetate Step V: To a stirred solution of (3R,4R,5S,6R)-2-[4-Bromo-3-(2,3-dihydro- benzo[ 1 ,4]dioxin-6-yl methyl)-phenyl]-6-hydroxymethyl-tetrahyd ro-pyran-3,4 , 5-triol (28.4 g, 60.81 mmol) in dichloromethane (300 mL) was added pyridine (40 mL, 486.5 mmol), acetic anhydride (50 mL, 486.5 mmol) and DMAP (740 mg, 6.08 mmol) at room temperature. After stirring for 2 h, volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate (500ml) and washed with 1 N HCI (200 mL X 2) followed by brine (200ml), then dried over sodium sulfate and
concentrated. The resulting crude compound was dissolved in ethanol (320 mL) at 65 °C and allowed to cool to room temperature while stirring. Light yellow solid formed was filtered and washed with cold ethanol (150 mL) followed by hexane (200 mL) to get acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3-(2,3-dihydro-benzo[1 ,4]dioxin- 6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester powder (22.5 g, purity 98%).
COCRYSTAL
Example 75: 1:1 Proline Co-crvstal with f2S.3R.4R.5S.6R¾-2-r3-f2.3-Dihvdro- benzori.41dioxin-6-ylmethyl)-4-ethyl-phenvn-6-hvdroxymethyl-tetrahydro-pyran- 3.4.5-triol
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Example 62) was completely amorphous initially but formed a crystalline complex with proline. This was confirmed by powder X-ray diffraction (PXRD) analysis. The stiochiometry of Example 62 and L- proline in the co-crystal prepared by method 1 was found to be 1 :1 by NMR
spectroscopy & HPLC. Characterization data for co-crystals of Example 62 and proline prepared by method 1 is shown in Table 3. Relative intensities of the most prominent powder x-ray diffraction peaks for co-crystals of Example 62 and proline are shown in Table 3A.
Table 3

Table 3A

3.70 15.78 18.36 25.18
9.68 10.68 18.88 36.33
11.07 21.21 20.42 69.29
14.26 14.81 21.18 27.94
14.80 22.97 22.50 12.25
15.40 4 98 23.78 33.08
16.12 8.45 24.56 6.92
16.59 18.78 25.79 21.69
17.31 100.0 27.46 8.90
17.60 20.35 31.97 7.65
17.98 47.20 32.46 5.98
1:1 Proline Co-crvstal
Example 77: 1:1 Proline Co-crvstal with (2S.3R.4R.5S.6Ri-2-f3-(2.3-Dihvdro- benzoh .41dioxin-6-ylmethvh-4-ethyl-phenvn-6-hvdroxymethyl-tetrahvdro-pyran- 3.4.5-triol
Method 2:
1 :1 Co-Crvstals of Example 62 with L-Proline
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]- 6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Example 62, 1500mg,3.6mmol), L- proline (415mg, 3.6mmol) and ethanol (23 ml_) were added to a 50 mL 3-neck round bottom flask equipped with nitrogen purging, magnetic stirring bar,
thermometer pocket & calcium chloride guard tube and the mixture was stirred at 25-30°C for 30 min., then heat to reflux. A clear solution was observed which was refluxed for 30 min., then slowly cool to 25-30°C causing percipitation. Di- isopropyl ether (DIPE, 23 mL) was added while maintaining the mixture at 25-30°C and stirring continuously for additional one to two hours at the same temperature. The precipitate was collected by filtration using vacuum (Nitrogen atmosphere), and the filter cake was washed with ethanol-DIPE mixture (1 :1 v/v, 10ml) followed by DIPE (23 mL). The product was vacuum dried at 65-70°C for 5-6 hrs.
1:1 Proline Co-crvstal (ΔΗ 53 J/g) was observed by differential scanning calorimetry (DSC) and is shown in Fig. 1. A powder X-ray diffraction (PXRD) spectrum is shown in Fig. 2.
2:1 Proline Co-crvstal
Example 78: 2:1 Proline Co-crvstal with f2S.3R.4R.5S.6R>-2-r3-f2.3-Pihvdro-benzof1.41dioxin-6-ylmethvH-4-ethyl-phenvn-6-hvdroxymethyl-tetrahvdro-pyran- 3.4.5-triol
Method 3: 1 :2 Co-Crvstals of Example 62 with L-Proline
(2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Example 62, 1 kg) was added to 15 L of ethanol with agitation while maintaining the mixture at 20-25 °C. The mixture was stirred for 10 min at 20-25 °C, then L-proline (537 gm) was added while maintaining the mixture at 20-25 °C. The mixture was stirred at this temperature for 30 min., then heated to reflux and refluxed for 30 min. The mixture was slowly cooled to 25-30°C then stired for 1 hr. DIPE (15 L) was added while maintaining the temperature at 25-30 °C and the mixture was stirred at this temperature for 1 hr. The precipitated product was collected by filtration and the product was washed with DIPE (5 L). The product was air dried at 65-70 °C to yield 1.22 kg
(79%) of a 1 :2 co-crystal of Example 62 : L-proline. A melting point 176°C (ΔΗ 85 J/g) was observed by differential scanning calorimetry (DSC) and is shown in Fig.
3. A powder X-ray diffraction (PXRD) spectrum is shown in Fig. 4. Relative
intensities of the most prominent powder x-ray diffraction peaks for the 1 :2 co- crystals of Example 62 and proline are shown in Table 5.
Table 5

PATENT
WO 2012140597
http://www.google.co.in/patents/WO2012140597A1?cl=en
. TABLE 2:

Intermediate 2: (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-

Intermediate 2
Intermediate 1
Step I: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[4-bromo-3- (2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-phenyl]-tetrahydro-pyran-2-ylmethyl ester (Intermediate 1 , 10.0 g, 15.74 mmol) in toluene (200 mL) was added
tricyclohexylphosphine (1.76 g, 6.29 mmol), a solution of potassium phosphate tribasic (13.3 g, 62.9 mmol) in water (15 mL), and ethylboronic acid (3.4 g, 47.2 mmol). The reaction mixture was degassed for 45 min then palladium (II) acetate (529 mg, 2.3 mmol) was added. After refluxing overnight, the reaction mixture was cooled to room temperature, and water was added. The resulting mixture was extracted with ethyl acetate, (2 X 200 ml_), washed with water and brine, then dried over sodium sulfate, concentrated and purified by column chromatography to furnish acetic acid
(2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-tetrahydro-pyran-2-ylmethyl ester (5.4 g).
Step II: To a stirred solution of acetic acid (2R,3R,4R,5S)-3,4,5-triacetoxy-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-tetrahydro-pyran-2-ylmethyl ester (9.3 g, 15.9 mmol) in methanol:THF:water 3:2:1 (170 ml.) was added lithium hydroxide (764 mg, 19.1 mmol). After stirring for 2 h at room temperature, the volatiles were evaporated under reduced pressure. The resulting residue was taken up in ethyl acetate (150 ml.) and washed with brine (75 ml_), brine containing 5 ml. of 5% aqueous KHS04 (75 ml_), and brine (20 ml.) again, then dried over sodium sulfate and concentrated to furnish (2S,3R,4R,5S,6R)-2-[4-Cyclopropyl-3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)- phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (6.5 g)
1H NMR (400 MHz, CD3OD): δ 1.07 (t, J = 7.6 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.34- 3.50 (m, 4H), 3.68 (dd, J = 12.0, 5.6 Hz, 1 H), 3.85-3.91 (m, 3H), 4.08 (d, J = 9.6 Hz, 1 H), 4.17 (s, 4H), 6.53-6.58 (m, 2H), 6.68 (d, J = 8.4 Hz, 1 H), 7.15-7.25 (m, 3H).
MS (ES) m/z 434.2 (M+18).
Example 3: Synthesis of phosphoric acid (2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl ester diethyl ester

To a stirred solution of (2S,3R,4R,5S,6R)-2-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)- 4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Intermediate 2, 500 mg, 1.2 mmol) in pyridine (5 ml) was added diethylchlorophosphate (0.27 ml, 1 .9 mmol) at -40°C. After stirring for 1 h at same temperature, reaction was quenched with the addition of 1 N HCI and extracted with ethyl acetate (2 X 10 ml). Combined organic layers were washed with brine (10 ml), dried over sodium sulfate, concentrated and purified by preparative HPLC to give 220 mg of phosphoric acid (2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl ester diethyl ester as a white solid. 1H NMR (400 MHz, CD3OD): δ 1.07 (t, J = 7.6 Hz, 3H), 1.15 (td J = 7.2, 1.2 Hz, 3H), 1.22 (td, J = 6.8, 0.8 Hz, 3H), 2.57 (q, J = 7.6 Hz, 2H), 3.36-3.46 (m, 3H), 3.53-3.55 (m, 1 H),3.89 (s, 2H), 3.96-4.11 (m, 5H), 4.17 (s, 4H), 4.18-4.22 (m 1 H), 4.30-4.34 (m, 1 H), 6.52 (d, J = 2.0 Hz, 1 H),6.57 (dd, J = 8.4, 2.4 Hz, 1 H), 6.68 (d, J = 8.4 Hz, 1 H), 7.15- 7.22(m, 3H). MS (ES) m/z 553.3 (M+1 ).
Example 4: Synthesis of disodium salt of phosphoric acid mono- {(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]- 3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl} ester

To a stirred solution of (2S,3R,4R,5S,6R)-2-[3-(2,3-Dihydro-benzo[1 ,4]dioxin-6- ylmethyl)-4-ethyl-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol (Intermediate 2, 1.0 g, 2.4 mmol) in THF (15 ml) was added a solution of Diethyl-phosphoramidic acid di- tert-butyl ester (780 mg, 3.12 mmol) in THF (5 ml) at 0°C followed by a solution of tetrazole (435 mg, 6.2 mmol) in DCM (12.5 ml). After stirring for 5 min at same temperature, it was stirred at room temperature for 20 min. Reaction mixture was cooled to -40 °C and added a solution of m-CPBA (830 mg, 4.8 mmol) in DCM (5 ml). The reaction mixture was stirred at same temperature for 5 min and then at room temperature for 2 h. Reaction mixture was cooled to 0°C and quenched by the addition of 10% sodium bisulfite solution (5 ml). This was extracted with ether (3 X 10 ml). Combined organic layer was washed with brine (5 ml), dried over sodium sulfate and concentrated to give 700 mg of phosphoric acid di-tert-butyl ester (2R,3S,4R,5R,6S)-6- [3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro- pyran-2-ylmethyl ester.
To the stirred solution of phosphoric acid di-tert-butyl ester (2R,3S,4R,5R,6S)-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl ester (500 mg) in methanol (20 ml) was added amberlyst 15 ion exchange resin (250 mg) and refluxed for overnight. Reaction mixture was cooled to room temperature, filtered through celite bed and filtrate was concentrated to give 300 mg of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl- phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2-ylmethyl} ester. The crude material was taken up for next reaction.
To a solution of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3-dihydro- benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl} ester (300 mg, 0.6 mmol) in methanol (5 ml) was added 1 N sodium bicarbonate solution (80 mg, 0.7 mmol) in water. After stirring at room temperature for 2 h, the volatiles were evaporated under reduced pressure. The resulting solid was triturated with diethyl ether. The resulting residue was purified by preparative HPLC to give 95 mg of disodium salt of phosphoric acid mono-{(2R,3S,4R,5R,6S)-6-[3-(2,3- dihydro-benzo[1 ,4]dioxin-6-ylmethyl)-4-ethyl-phenyl]-3,4,5-trihydroxy-tetrahydro-pyran-2- ylmethyl} ester.
1H NMR (400 MHz, CD3OD): δ 1.06 (t, J = 7.4 Hz, 3H), 2.56 ( q, J = 7.3 Hz, 2H), 3.34- 3.41 (m, 2H), 3.49 (t, J = 8.8 Hz, 1 H), 3.81-3.88 (m, ,3H), 3.92-3.99 (m, 1 H), 4.05 (d, J = 9.3 Hz, 1 H), 4.16 (s, 4H), 4.20-4.25 (m, 1 H), 6.54 (m, 2H), 6.67 (d, J = 7.8 Hz, 1 H), 7.12-7.21 (m, 3H). MS (ES) m/z 497.1 (M+1 ) for phosphoric acid.
![]()
PATENT
SEE INDIAN PATENT
IN 2009DE02173
Glycoside derivatives and uses thereof
REFERENCES
Pediatric investigation plan (PIP) decision: (S)-Pyrrolidine-2-carboxylic acid compound with (2S,3R,4R,5S,6R)-2-(3-((2,3-dihydrobenzo[b][1,4]dioxin-6-yl)methyl)-4-ethylphenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (2:1) ( LIK066) (EMEA-001527-PIP01-13)
European Medicines Agency (EMA) Web Site 2014, July 24
Safety, tolerability, pharmacokinetics (PK) and pharmacodynamics (PD) assessment of LIK066 in healthy subjects and in patients with type 2 diabetes mellitus (T2DM) (NCT01407003)
ClinicalTrials.gov Web Site 2011, August 07
IN 2009DE02173
| WO2001016147A1 | 24 Aug 2000 | 8 Mar 2001 | Kissei Pharmaceutical | Glucopyranosyloxypyrazole derivatives, medicinal compositions containing the same and intermediates in the production thereof |
| WO2001027128A1 | 2 Oct 2000 | 19 Apr 2001 | Bruce Ellsworth | C-aryl glucoside sglt2 inhibitors |
| WO2001068660A1 | 15 Mar 2001 | 20 Sep 2001 | Hideki Fujikura | Glucopyranosyloxy benzylbenzene derivatives, medicinal compositions containing the same and intermediates for the preparation of the derivatives |
| WO2001074834A1 | 29 Mar 2001 | 11 Oct 2001 | Squibb Bristol Myers Co | O-aryl glucoside sglt2 inhibitors and method |
| WO2003020737A1 | 5 Sep 2002 | 13 Mar 2003 | Squibb Bristol Myers Co | O-pyrazole glucoside sglt2 inhibitors and method of use |
| WO2003043985A1 | 20 Nov 2002 | 30 May 2003 | Andrew Thomas Bach | Heterocyclic compounds and methods of use |
| WO2004018491A1 | 21 Aug 2003 | 4 Mar 2004 | Nobuhiko Fushimi | Pyrazole derivatives, medicinal composition containing the same, medicinal use thereof, and intermediate for production thereof |
| WO2004078163A2 | 26 Feb 2004 | 16 Sep 2004 | Oern Almarsson | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| WO2004080990A1 | 12 Mar 2004 | 23 Sep 2004 | Kazuhiro Ikegai | C-glycoside derivatives and salts thereof |
| WO2004099230A1 | 30 Apr 2004 | 18 Nov 2004 | Eikyu Yoshiteru | Monosaccharide compounds |
| WO2004103995A1 | 19 May 2004 | 2 Dec 2004 | Gary Michael Ksander | N-acyl nitrogen heterocycles as ligands of peroxisome proliferator-activated receptors |
| WO2005011592A2 | 29 Jul 2004 | 10 Feb 2005 | Janssen Pharmaceutica Nv | Substituted indazole-o-glucosides |
| WO2005021566A2 | 20 Aug 2004 | 10 Mar 2005 | Barsoumian Edward Leon | Glucopyranosyloxy- pirazoles, drugs containing said compounds the use and production method thereof |
| WO2005085237A1 | 3 Mar 2005 | 15 Sep 2005 | Kissei Pharmaceutical | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| WO2005085265A1 | 3 Mar 2005 | 15 Sep 2005 | Kissei Pharmaceutical | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| WO2006011502A1 | 27 Jul 2005 | 2 Feb 2006 | Chugai Pharmaceutical Co Ltd | Novel glucitol derivative, prodrug thereof and salt thereof, and therapeutic agent containing the same for diabetes |
| WO2006054629A1 | 17 Nov 2005 | 26 May 2006 | Kissei Pharmaceutical | 1-SUBSTITUTED-3-β-D-GLUCOPYRANOSYLATED NITROGENOUS HETERO- CYCLIC COMPOUNDS AND MEDICINES CONTAINING THE SAME |
| WO2008016132A1 | 3 Aug 2007 | 7 Feb 2008 | Daiichi Sankyo Co Ltd | Benzyl phenyl glucopyranoside derivative |
| WO2011048112A1 * | 19 Oct 2010 | 28 Apr 2011 | Novartis Ag | Glycoside derivatives and uses thereof |
| US20030114390 * | 4 Oct 2002 | 19 Jun 2003 | Washburn William N. | C-aryl glucoside SGLT2 inhibitors and method |
| US20040018998 | 21 Sep 2001 | 29 Jan 2004 | Hideki Fujikura | Glucopyranosyloxybenzylbenzene derivatives and medicinal compositions containing the same |
| US20060009400 | 28 Jun 2005 | 12 Jan 2006 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060019948 | 15 Jul 2005 | 26 Jan 2006 | Boehringer Ingelheim International Gmbh | Methylidene-D-xylopyranosyl- and oxo-D-xylopyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060025349 | 27 Jul 2005 | 2 Feb 2006 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-phenyl-substituted cycles, medicaments containing such compounds, their use and process for their manufacture |
| US20060035841 | 9 Aug 2005 | 16 Feb 2006 | Boehringer Ingelheim International Gmbh | D-xylopyranosyl-phenyl-substituted cycles, medicaments containing such compounds, their use and process for their manufacture |
| US20060074031 | 30 Sep 2005 | 6 Apr 2006 | Boehringer Ingelheim International Gmbh | D-pyranosyl-substituted phenyl derivatives, medicaments containing such compounds, their use and process for their manufacture |
| US20060293252 | 14 Aug 2006 | 28 Dec 2006 | Sanofi-Aventis Deutschland Gmbh | Novel Thiophene Glycoside Derivatives, Processes for The Preparation, Medicaments Comprising These Compounds, and The Use Thereof |
| US20080027014 | 26 Jul 2007 | 31 Jan 2008 | Tanabe Seiyaku Co., Ltd. | Novel SGLT inhibitors |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2015032272A1 * | 19 Aug 2014 | 12 Mar 2015 | Jiangsu Hansoh Pharmaceutical Co., Ltd. | C-aryl glucoside derivative, preparation method for same, and medical applications thereof |
| US9034921 | 1 Jun 2012 | 19 May 2015 | Green Cross Corporation | Diphenylmethane derivatives as SGLT2 inhibitors |
INVENTORS OF LIK 066
Gregory Raymond Bebernitz, Mark G. Bock, Dumbala Srinivas Reddy, Atul Kashinath Hajare, Vinod Vyavahare, Sandeep Bhausaheb Bhosale, Suresh Eknath Kurhade, Videsh Salunkhe, Nadim S. Shaikh, Debnath Bhuniya, P. Venkata Palle, Lili Feng, Jessica Liang,
| BEBERNITZ, Gregory, Raymond; (US). BOCK, Mark, G.; (US). REDDY, Dumbala Srinivas; (IN). HAJARE, Atul Kashinath; (IN). VYAVAHARE, Vinod; (IN). BHOSALE, Sandeep Bhausaheb; (IN). KURHADE, Suresh Eknath; (IN). SALUNKHE, Videsh; (IN). SHAIKH, Nadim, S.; (IN). BHUNIYA, Debnath; (IN). PALLE, P., Venkata; (IN). FENG, Lili; (US). LIANG, Jessica; (US) |
 Mark G Bock
Mark G Bock
BEBERNITZ, Gregory, Raymond….PIC NOT AVAILABLE

Dr. Srinivasa Reddy
 NADEEM SHAIKH
NADEEM SHAIKH
 Venkata Palle
Venkata Palle
ONLY FEW…………………….
//////Licogliflozin diprolinate
see……..http://medcheminternational.blogspot.in/2015/11/lik-066-novartis-for-treatment-of-type.html
FDA approves new treatment for HIV

November 5, 2015
Release
The U.S. Food and Drug Administration today approved Genvoya (a fixed-dose combination tablet containing elvitegravir, cobicistat, emtricitabine, and tenofovir alafenamide) as a complete regimen for the treatment of HIV-1 infection in adults and pediatric patients 12 years of age and older.
The CDC estimates that 1.2 million persons ages 13 years and older are living with HIV infection, and that more than another 150,000 persons in this age range have HIV but are unaware of their infection. Over the past decade, the number of people living with HIV has increased, while the annual number of new HIV infections has remained relatively stable.
“Today’s approval of a fixed dose combination containing a new form of tenofovir provides another effective, once daily complete regimen for patients with HIV-1 infection,” said Edward Cox, M.D., director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research.
Genvoya is approved for use in HIV-infected adults and children ages 12 years and older weighing at least 35 kilograms (77 pounds) who have never taken HIV therapy (treatment-naïve) and HIV-infected adults whose HIV-1 virus is currently suppressed. While Genvoya is not recommended for patients with severe renal impairment, those with moderate renal impairment can take Genvoya.
Genvoya’s safety and efficacy in adults were evaluated in 3,171 participants enrolled in four clinical trials. Depending on the trial, participants were randomly assigned to receive Genvoya or another FDA approved HIV treatment. Results showed Genvoya was effective in reducing viral loads and comparable to the other treatment regimens.
Genvoya contains a new form of tenofovir that has not been previously approved. This new form of tenofovir provides lower levels of drug in the bloodstream, but higher levels within the cells where HIV-1 replicates. It was developed to help reduce some drug side effects. Genvoya appears to be associated with less kidney toxicity and decreases in bone density than previously approved tenofovir containing regimens based on laboratory measures. Patients receiving Genvoya experienced greater increases in serum lipids (total cholesterol and low-density lipoprotein) than patients receiving other treatment regimens in the studies.
Genvoya carries a Boxed Warning alerting patients and health care providers that the drug can cause a buildup of lactic acid in the blood and severe liver problems, both of which can be fatal. The Boxed Warning also states that Genvoya is not approved to treat chronic hepatitis B virus infection. The most common side effect associated with Genvoya is nausea. Serious side effects include new or worsening kidney problems, decreased bone mineral density, fat redistribution and changes in the immune system (immune reconstitution syndrome). Health care providers are advised to monitor patients for kidney and bone side effects. Genvoya should not be given with other antiretroviral products and may have drug interactions with a number of other commonly used medications.
Genvoya is marketed by Gilead Sciences Inc. based in Foster City, California.
/////////
BMS-582949 in phase 2 for Treatment of Antipsoriatics , Rheumatoid arthritis
BMS 582949, PS-540446
UNII-CR743OME9E
CAS 623152-17-0
4-[5-(N-Cyclopropylcarbamoyl)-2-methylphenylamino]-5-methyl-N-propylpyrrolo[2,1-f][1,2,4]triazine-6-carboxamide
4-(5-(Cyclopropylcarbamoyl)-2-methylphenylamino)-5-methyl-N-propylpyrrolo[1,2-f][1,2,4]triazine-6-carboxamide
Bristol-Myers Squibb Company
M.Wt: 406.48
Cas : 623152-17-0 Formula: C22H26N6O2
BMS-582949 had been in phase II clinical trials at Bristol-Myers Squibb for the oral treatment of moderate to severe psoriasis and for the treatment of rheumatoid arthritis (RA) in combination with methotrexate and for the treatment of inflammation in atherosclerotic plaque. However, no recent development has been reported for this research.
…………………..
http://www.google.com/patents/WO2012031057A1?cl=en
The present invention generally relates to a method of treating resistant rheumatic disease, such as refractory rheumatoid arthritis, with a therapeutically effective amount of a dual action p38 inhibitor that is safe and well-tolerated. A dual action p38 kinase inhibitor is a compound that inhibits both activation of p38 kinase and p38 kinase activity in cells.
A large number of cytokines participate in the inflammatory response, including IL- 1 , IL-6, IL-8 and TNF-a. Overproduction of cytokines such as IL-1 and TNF-a are implicated in a wide variety of diseases, including inflammatory bowel disease, rheumatoid arthritis, psoriasis, multiple sclerosis, endotoxin shock, osteoporosis, Alzheimer’s disease, and congestive heart failure, among others. See e.g., Henry et al., Drugs Fut. , 24: 1345- 1354 ( 1999); Salituro et al., Curr. Med. Ckem., 6:807-823 (1999)]. Important mediators of proinflammatory cytokines such as TNFct and IL-1 β,. as well as cellular responses to such cytokines production, are the mitogen-activated protein (MAP) kinases, and in particular, p38 kinase. See e.g., Schieven, G.L., “The biology of p38 kinase: a central role in inflammation”, Current Topics in Medicinal Chemistry, 5 :921 – 928 (2005). Accordingly, modulation of p38 kinase may be useful in the treatment of inflammatory disease including rheumatic diseases such as rheumatoid arthritis (RA).
Compounds that reportedly inhibit p38 kinase and cytokines such as IL-1 and TNF-a for use in treating inflammatory diseases are disclosed in U.S. Patent Nos.
6,277,989 and 6, 130,235 to Scios, Inc; U.S. Patent. Nos. 6, 147,080 and 5,945,41 8 to Vertex Pharmaceuticals Inc; U.S. Patent Nos. 6,251 ,914, 5,977, 103 and 5,658,903 to Smith-Kline Beecham Corp.; U.S. Patent Nos. 5,932,576 and 6,087,496 to G.D. Searle & Co.; WO 00/56738 and WO 01 /27089 to Astra Zeneca; WO 01/34605 to Johnson & Johnson; WO 00/12497 (quinazoHne derivatives as p38 kinase inhibitors); WO 00/56738 (pyridine and pyrimidine derivatives for the same purpose); WO 00/12497 (discusses the relationship between p38 kinase inhibitors); and WO 00/12074 (piperazine and piperidine compounds useful as p38 inhibitors). Other compounds that inhibit p38 kinase are pyrrolotriazine aniline compounds, information on these compounds is disclosed in U.S. Patent Nos. 6,670,357; 6,867,300; 7,034, 151 ; 7, 160,883; 7,21 1,666; 7,253, 167; and U.S. Publication Nos. 2003/023283 1 (published Dec. 18, 2003); 2004/0229877 (published Nov. 1 8, 2004); 2005/0043306 (published Feb. 24, 2005; 2006/0003967 (published Jan. 5, 2006); 2006/0030708 (published Feb. 9, 2006); 2006/0041 124 (published Feb. 23, 2006); 2006/0229449 (published Oct. 12, 2006); 2006/0235020 (published Oct. 19, 2006); and 2007/0213300 (published Sept 13, 2007).
In particular, WO 2003/090912 (U.S. Patent Nos. 7, 160,883, 7,388,009, p38 inhibitor, BMS-582949 (Example 7,

including processes of making and uses thereof.
……………………
http://www.google.com/patents/WO2003090912A9?cl=en
Examples 4-22

Compounds having the formula (Id), above, wherein R4 has the values listed in the following Table, were prepared following the same procedure described for Example 3, using the appropriate amine in place of ra-butylamine.



…………………………
WO 2006020904
http://www.google.com.br/patents/WO2006020904A1?cl=en
EXAMPLE IA St


Part a.
A solution of Example 1 (0.86 g, 2.20 mmol, 1.0 eq.) in THF (4.0 mL) and 1 N aqueous NaOH (9.0 mL, 4.1 eq.) was stirred at 6O0C overnight. After cooling to RT, the reaction mixture was concentrated in vacuo but not to dryness. To the solution at O0C was added 1 N aqueous hydrochloric acid until it was acidic and the precipitate was collected and dried to afford crude Example IA acid (0.51 g, 64.0 % yield). HPLC Ret. t. = 2.400 min.; LC/MS (M+H) + = 366.06+. The filtrate was then extracted with EtOAc (3x) and the organic layers were combined, dried over sodium sulfate, and concentrated in vacuo to give Example IA acid (0.035 g, 4.4 % yield). Part b.

A solution of Part a. acid (0.026 g, 0.071 mmol, 1.0 eq.), EDC (0.021 g, 0.11 mmol, 1.5 eq.), HOBt (0.015 g, 0.11 mmol, 1.5 eq), ^-propylamine (0.015 mL, 0.15 mmol, 2.1 eq.) and DIPEA (0.040 mL, 0.23 mmol, 3.2 eq.) in DMF (0.20 mL) was shaken at RT overnight. Water (1 mL) was added and the precipitate collected by filtration, washed with water, and dried to give Example IA amide (0.021 g, 70% yield); HPLC Ret. t. = 2.883 min.; LC/MS (M+H)+ = 421.18 +.
EJiAMPLE 2 Direct Aminolysis Procedure

n-Buli/THF
Ester Compound I or Hexyllithium/THF
-^
,NH9

1. Aminolysis with hexyllithium
To a dried 100 ml flask was added THF (10 ml) under nitrogen, which was then cooled to -100C. Hexyllithium (2.3 M in hexane, 6.5 ml, 15.0 mmol) was added slowly (exothermic, temperature was up to 5°C), followed by dropwise addition of propylamine (1.01 g, 1.4 ml, 17.1 mmol) at such a rate to maintain the temperature below 5°C. The resulting mixture was stirred at O0C for 20 minutes. A suspension of ester compound I (1.0 g, 2.5 mmol) in THF (12 ml) was added over a 10 minute period (exothermic, T<5°C). After being stirred at 00C for 20 minutes, the mixture was allowed to warm to room temperature and stirred for 5 hours. Ester compound I was <0.1 AP at this point by HPLC analysis. The mixture was cooled to -50C. Acetic acid (2 ml) was added slowly to maintain the temperature <10°C. The resulting thick slurry was stirred at room temperature for 20 minutes, and then solvents were exchanged with DMF (15 ml) on a rotavapor. To the resulting yellow slurry, water (15 ml) was added slowly to keep T<25°C. During the addition of water, the slurry became a clear solution, and a new slurry was formed. The slurry was stirred at room temperature for overnight. In the morning the slurry was filtered and the solid was washed with DMF/water (1:1, 5 ml), water (5 ml) and acetone (5 ml). The cake was dried under vacuum at 55°C for 24 hours to afford 0.90 g of amide product II (yield: 87.2%) as a white solid. HPLC: 99.70 AP.
2. Aminolysis with n-butyllithium
To a dried 100 ml of flask was added THF (10 ml) under nitrogen and then cooled to -100C. n-Butyllithium (2.5 M in hexane, 6.0 ml, 15.0 mmol) was added slowly, followed by dropwise addition of propylamine (0.98 g, 16.5 mmol) at such a rate to keep the temperature below 00C. The resulting mixture was stirred at O0C for 20 minutes. A suspension of ester compound I (1.0 g, 2.5 mmol) in THF (12 ml) was added over a 10 minute period (T<5°C). After being stirred at O0C for 30 minutes, the mixture was allowed to warm to room temperature and stirred for overnight (~22h, Note 1). Compound I was not detected at this point by HPLC analysis. The mixture was cooled to -7°C. Acetic acid (2 ml) was added dropwise to maintain the temperature <10°C. The resulting thick slurry was stirred at 50C for 2 hours and at room temperature for 20 minutes, followed by evaporation on a rotavapor to give a wet yellow solid. To this solid was added acetone (10 ml) and water (20 ml). The slurry was stirred at room temperature for one and half hours. Filtration gave a white solid. This solid was washed with 35% acetone in water (10 ml), water (5 ml) and acetone (5 ml). The cake was dried under vacuum at 55°C for the weekend to afford 0.94g of amide product II (yield: 91.0%) as a white solid. HPLC: 99.76 AP. Note 1: Compound I was -0.056 AP at 2.5 hours.
……………………
WO 2003090912
http://www.google.com/patents/WO2003090912A1?cl=en
……………………..
Discovery of 4-(5-(Cyclopropylcarbamoyl)-2-methylphenylamino)-5-methyl-N-propylpyrrolo[1,2-f][1,2,4]triazine-6-carboxamide (BMS-582949), a clinical p38a MAP kinase inhibitor for the treatment of inflammatory diseases
J Med Chem 2010, 53(18): 6629
http://pubs.acs.org/doi/abs/10.1021/jm100540x
The discovery and characterization of 7k (BMS-582949), a highly selective p38α MAP kinase inhibitor that is currently in phase II clinical trials for the treatment of rheumatoid arthritis, is described. A key to the discovery was the rational substitution of N-cyclopropyl for N-methoxy in 1a, a previously reported clinical candidate p38α inhibitor. Unlike alkyl and other cycloalkyls, the sp2 character of the cyclopropyl group can confer improved H-bonding characteristics to the directly substituted amide NH. Inhibitor 7k is slightly less active than 1a in the p38α enzymatic assay but displays a superior pharmacokinetic profile and, as such, was more effective in both the acute murine model of inflammation and pseudoestablished rat AA model. The binding mode of 7k with p38α was confirmed by X-ray crystallographic analysis.

EXAMPLE 3

Direct Aminolysis
Ester Compound I

Amide Product II
Method A:
A solution of n-propylamine (6.5 eq) in THF (20 ml/g of ester compound I) was cooled to — 5°C and was slowly treated with 2.5 M solution of n-butyllithium (6.1 eq). The mixture was stirred for 10 minutes. At the end of the period, a slurry of ester compound I (1 eq) in THF (14 ml/g of ester compound I) was cannulated into the performed Li-NHPr solution. The reaction mixture was warmed to 25°C and stirred till all of ester compound I was consumed (~ 3 hours). After the reaction was judged to be completed by HPLC, the reaction mixture was cooled to ~0°C and was slowly treated with acetic acid (5 ml/g of ester compound I). The slurry was then warmed to -2O0C and was stirred for 1 hour. At the end of the period, the solvent was distilled under vacuum to the minimum volume and the concentrated slurry was diluted with a solution of acetone (10 ml/g of ester compound I) and water (20 ml/g of ester compound I). The slurry was stirred for 1 hour and was cooled to ~5°C. The slurry was filtered and the cake was washed with acetone (5 ml/g of ester compound I). The cake was dried to give the amide product II (typically in 85% yield and 99 AP).
Method B:
A solution of n-propylamine (20 eq) in 2,2,2-trifmoroethanol (10 ml/g of ester compound I) was slowly treated with 2.5 M solution of n-butyllithium (1.5 eq). The mixture was stirred for 5 minutes. At the end of the period, the starting material, ester compound I, was added and the reaction mixture was warmed to 900C. The reaction mixture was held at 900C for 24 hours and was allowed to cool to ~20°C. The reaction mixture was then analyzed by HPLC. Typically, analysis indicated there was only 1.57 AP of starting material left.
Method C:
A solution of n-propylamine (2 eq) in methylene chloride (10 ml/g of ester compound I) at 200C was slowly treated with 2.0 M solution of trimethylaluminum (4 eq) in hexanes. The mixture was stirred for 15 minutes. At the end of the period, the starting material, ester compound 1 (1 eq), was added and the reaction mixture was warmed to 600C. The reaction mixture was held at 600C for 24 hours and was allowed to cool to ~20°C. The reaction mixture was then slowly quenched with aqueous HCl solution and analyzed by HPLC. Typically, analysis indicated there was 96.8AP of amide compound II product with 0.03 AP of the dipropylamide impurity.
…………………………………….
| WO2003090912A1 * | 15 abr. 2003 | 6 nov. 2003 | Squibb Bristol Myers Co | Pyrrolo-triazine aniline compounds useful as kinase inhibitors |
Liu C, Lin J, Everlof G, Gesenberg C, Zhang H, Marathe PH, Malley M, Galella MA, McKinnon M, Dodd JH, Barrish JC, Schieven GL, Leftheris K.
Bioorg Med Chem Lett. 2013 May 15;23(10):3028-33. doi: 10.1016/j.bmcl.2013.03.022. Epub 2013 Mar 15.
Freebern WJ, Bigwarfe TJ, Price KD, Haggerty HG.
J Immunotoxicol. 2013 Jan-Mar;10(1):106-17. doi: 10.3109/1547691X.2012.736427. Epub 2012 Nov 23.
Liu C, Lin J, Wrobleski ST, Lin S, Hynes J, Wu H, Dyckman AJ, Li T, Wityak J, Gillooly KM, Pitt S, Shen DR, Zhang RF, McIntyre KW, Salter-Cid L, Shuster DJ, Zhang H, Marathe PH, Doweyko AM, Sack JS, Kiefer SE, Kish KF, Newitt JA, McKinnon M, Dodd JH, Barrish JC, Schieven GL, Leftheris K.
J Med Chem. 2010 Sep 23;53(18):6629-39. doi: 10.1021/jm100540x.
BMS-582949: crystalline form of a p38alpha inhibitor? WO2008079857.
Norman P.
Expert Opin Ther Pat. 2009 Aug;19(8):1165-8. doi: 10.1517/13543770902816160.
| WO2000012074A2 | Aug 27, 1999 | Mar 9, 2000 | Sarvajit Chakravarty | Use of piperidines and/or piperazines as inhibitors of p38-alpha kinase | |
| WO2000012497A2 | Aug 27, 1999 | Mar 9, 2000 | Sarvajit Chakravarty | Quinazoline derivatives as medicaments | |
| WO2000056738A1 | Mar 17, 2000 | Sep 28, 2000 | Astrazeneca Ab | Pyridine and pyrimidine derivatives and their use as inhibitors of cytokine mediated disease | |
| WO2001027089A1 | Oct 10, 2000 | Apr 19, 2001 | Astrazeneca Ab | Pyrimidine derivatives | |
| WO2001034605A1 | Oct 27, 2000 | May 17, 2001 | Ortho Mcneil Pharm Inc | SUBSTITUTED 2-ARYL-3-(HETEROARYL)-IMIDAZO[1,2-a]PYRIMIDINES, AND RELATED PHARMACEUTICAL COMPOSITIONS AND METHODS | |
| WO2003090912A1 | Apr 15, 2003 | Nov 6, 2003 | Squibb Bristol Myers Co | Pyrrolo-triazine aniline compounds useful as kinase inhibitors | |
| US4200750 | Dec 8, 1977 | Apr 29, 1980 | Westwood Pharmaceuticals Inc. | 4-Substituted imidazo [1,2-a]quinoxalines | |
| US5658903 | Jun 3, 1996 | Aug 19, 1997 | Smithkline Beecham Corporation | Cytokine inhibitors | |
| US5932576 | May 22, 1998 | Aug 3, 1999 | G. D. Searle & Company | 3(5)-heteroaryl substituted pyrazoles as p38 kinase inhibitors | |
| US5945418 | Mar 20, 1997 | Aug 31, 1999 | Vertex Pharmaceuticals Incorporated | Administering to the mammal to inhibit a mammalian protein kinase p38 which causes cell proliferation, cell death and response to extracellular stimuli | |
| US5977103 | Jan 10, 1997 | Nov 2, 1999 | Smithkline Beecham Corporation | Substituted imidazole compounds | |
| US6087496 | Apr 1, 1999 | Jul 11, 2000 | G. D. Searle & Co. | Enzyme inhibitors | |
| US6130235 | Aug 3, 1998 | Oct 10, 2000 | Scios Inc. | Piperidine moieties coupled to indole, benzimidazole or benzotriazole. | |
| US6147080 | Jun 10, 1997 | Nov 14, 2000 | Vertex Pharmaceuticals Incorporated | Inhibitors of p38 | |
| US6251914 | Jul 1, 1998 | Jun 26, 2001 | Smithkline Beecham Corporation | Treating cytokine mediated diseases | |
| US6277989 | Mar 14, 2000 | Aug 21, 2001 | Scios, Inc. | Quinazoline derivatives as medicaments | |
| US6670357 | Nov 7, 2001 | Dec 30, 2003 | Bristol-Myers Squibb Company | Antiinflammatory agents | |
| US6867300 | Nov 6, 2002 | Mar 15, 2005 | Bristol-Myers Squibb Company | Methods for the preparation of pyrrolotriazine compounds useful as kinase inhibitors | |
| US7034151 | Feb 5, 2004 | Apr 25, 2006 | Bristol-Myers Squibb Company | 1,4-dihydro-4-oxo-pyrrolo[2,1-f][1,2,4]triazine-6-carboxylates; novel approach to the formation of the bicyclic heterocyclic ring system | |
| US7041501 | Oct 31, 2002 | May 9, 2006 | Bristol-Myers Squibb Company | Methods of screening for toxicity of test compounds | |
| US7160883 | Apr 22, 2003 | Jan 9, 2007 | Bristol-Myers-Squibb Company | Pyrrolo-triazine aniline compounds useful as kinase inhibitors | |
| US7211666 | Dec 22, 2004 | May 1, 2007 | Bristol-Myers Squibb Company | N-Cyclopropyl-4-[[5-[(methoxyamino)carbonyl]-2-methylphenyl]amino]-5-methylpyrrolo[2,1-f][1,2,4]triazine-6-carboxamide; aminating with chloramine to produce a pyrrole with a Nitrogen nitrogen bond; reacting with formamide, cyclizing to form the pyrrolotriazine core; kinase inhibitors | |
| US7253167 | Jun 29, 2005 | Aug 7, 2007 | Bristol-Myers Squibb Company | Tricyclic-heteroaryl compounds useful as kinase inhibitors | |
| US7388009 | Oct 3, 2003 | Jun 17, 2008 | Bristol-Myers Squibb Company | Heterocyclic drugs as enzyme inhibitors for Kinase enzymes or prodrugs | |
| US7462616 | Oct 24, 2006 | Dec 9, 2008 | Bristol-Myers Squibb Company | Pyrrolo-triazine aniline compounds useful as kinase inhibitors | |
| US7759343 | Oct 28, 2008 | Jul 20, 2010 | Bristol-Myers Squibb Company | Pyrrolo-triazine aniline compounds useful as kinase inhibitors | |
| US61379001 | Title not available | ||||
| US20030232831 | Apr 22, 2003 | Dec 18, 2003 | Alaric Dyckman | Aryl ketone pyrrolo-triazine compounds useful as kinase inhibitors | |
| US20040229877 | Oct 29, 2003 | Nov 18, 2004 | Katerina Leftheris | Administering pyrrolotriazine carboxamide and benzamide compounds for therapy of p38 kinase-associated conditions | |
| US20050043306 | Oct 3, 2003 | Feb 24, 2005 | Katerina Leftheris | Heterocyclic drugs as enzyme inhibitors for Kinase enzymes or prodrugs | |
| US20060003967 | Jun 28, 2005 | Jan 5, 2006 | Zhongping Shi | Method for preparing pyrrolotriazine compounds | |
| US20060030708 | Aug 5, 2005 | Feb 9, 2006 | Lobben Paul C | Methods for the preparation of pyrrolotriazine compounds | |
| US20060041124 | Oct 14, 2005 | Feb 23, 2006 | Bang-Chi Chen | Process for preparing pyrrolotriazine kinase inhibitors | |
| US20060229449 | Apr 3, 2006 | Oct 12, 2006 | Apurba Bhattacharya | Reacting with chloramine in presence of aqueous base, phase transfer catalyst; anti-cancer agents, kinase inhibitors | |
| US20060235020 | Apr 4, 2006 | Oct 19, 2006 | Soojin Kim | Process for preparing salts of 4-[[5-[(cyclopropylamino)carbonyl]-2-methylphenyl]amino]-5-methyl-N-propylpyrrolo[2,1-f][1,2,4]triazine-6-carboxamide and novel stable forms produced therein | |
| US20070213300 | Mar 6, 2007 | Sep 13, 2007 | Bristol-Myers Squibb Company | Pyrrolotriazine aniline prodrug compounds useful as kinase inhibitors |
Animation of nanoparticles for drug delivery in cancer treatment

Animation of nanoparticles for drug delivery in cancer treatment
read at
FDA Approves Perjeta for Neoadjuvant Breast Cancer Treatment

The structure of HER2 and pertuzumab
pertuzumab
Sept. 30, 2013 — The U.S. Food and Drug Administration today granted accelerated approval to Perjeta (pertuzumab) as part of a complete treatment regimen for patients with early stage breast cancer before surgery (neoadjuvant setting). Perjeta is the first FDA-approved drug for the neoadjuvant treatment of breast cancer.
Perjeta was approved in 2012 for the treatment of patients with advanced or late-stage (metastatic) HER2-positive breast cancer. HER2-positive breast cancers have increased amounts of the HER2 protein that contributes to cancer cell growth and survival
cut paste of my old article
he European Medicines Agency (EMA) has approved Roche’s PERJETA (pertuzumab) for patients with previously untreated HER2-positive metastatic breast cancer (mBC)
MARCH 5, 2013 8:59 AM / 4 COMMENTS /
The structure of HER2 and pertuzumab
march 4, 2013
Pertuzumab (also called 2C4, trade name Perjeta) is a monoclonal antibody. The first of its class in a line of agents called “HER dimerization inhibitors”. By binding to HER2, it inhibits the dimerization of HER2 with other HER receptors, which is hypothesized to result in slowed tumor growth.[1] Pertuzumab received US FDA approval for the treatment of HER2-positive metastatic breast cancer on June 8, 2012.[2] Pertuzumab was developed at Genentech and is now owned by Roche which acquired Genentech in 2009.
Clinical trials
Early clinical trials of pertuzumab in prostate, breast, and ovarian cancers have been met with limited success.[3]
The dosage of pertuzumab used in the pivotal phase III CLEOPATRA (Clinical Evaluation of Pertuzumab and Trastuzumab) trial was as follows: IV 840 mg loading dose followed by IV 420 mg every three weeks.[4]
The pharmacokinetics of intravenous pertuzumab appear to be unaffected by age and no drug-drug interaction has been reported with docetaxel. The pharmacokinetics and pharmacodynamics of pertuzumab were summarized in a Feb 2012 review by Gillian Keating.[4]
The combination of pertuzumab plus trastuzumab plus docetaxel, as compared with placebo plus trastuzumab plus docetaxel, when used as first-line treatment for HER2-positive metastatic breast cancer, significantly prolonged progression-free survival, with no increase in cardiac toxic effects in the randomized, double-blind, multinational, phase III CLEOPATRA trial.[5]
Intravenous pertuzumab is currently being evaluated in patients with breast cancer in the following trials: MARIANNE (advanced breast cancer), NEOSPHERE (early breast cancer), TRYPHAENA (HER2-positive stage II/III breast cancer) and APHINITY (HER2-positive nonmetastatic breast cancer).[4]
References
- de Bono, Johann S.; Bellmunt, J; Attard, G; Droz, JP; Miller, K; Flechon, A; Sternberg, C; Parker, C et al. (20 January 2007). “Open-Label Phase II Study Evaluating the Efficacy and Safety of Two Doses of Pertuzumab in Castrate Chemotherapy-Naive Patients With Hormone-Refractory Prostate Cancer”. Journal of Clinical Oncology 25 (3): 257–262.doi:10.1200/JCO.2006.07.0888. PMID 17235043.
- “FDA Approves Perjeta (Pertuzumab) for People With HER2-Positive Metastatic Breast Cancer” (Press release). Genentech. Retrieved 2012-06-09.
- Genentech press release – May 15, 2005
- Keating GM. Pertuzumab: in the first-line treatment of HER2-positive metastatic breast cancer. Drugs 2012 Feb 12; 72 (3): 353-60.Link text
- Baselga J, Cortés J, Kim SB, and the CLEOPATRA Study Group. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med 2012 Jan 12; 366 (2): 109-19. Link text
Simeprevir has been approved in Japan for the treatment of genotype 1 chronic hepatitis C infection

simeprevir
| CAS number | 923604-59-5 | ||
| Formula | C38H47N5O7S2 | ||
| Weight | 749.93908 |
Stockholm, Sweden — Medivir AB (OMX: MVIR) today reports that Janssen Pharmaceutical R&D Ireland (Janssen) has been informed by the Japanese Ministry of Health, Labour and Welfare (MHLW) that simeprevir has been approved for the treatment of genotype 1 chronic hepatitis C virus (HCV) infection.
read all at
http://www.pharmalive.com/japan-approves-simeprevir
Hepatitis C virus (HCV) infections affect approximately 3 percent of the worldwide population and often lead to cirrhosis and hepatocellular carcinoma. The standard therapy of pegylated- interferon and ribavirin induces serious side effects and provides viral eradication in less than 50% of patients. Combination therapy of HCV including ribavirin and interferonare currently is the approved therapy for HCV. Unfortunately, such combination therapy also produces side effects and is often poorly tolerated, resulting in major clinical challenges in a significant proportion of patients. Numerous direct acting agents (DAAs) have been or are being developed for treatment of HCV, such as telaprevir and boceprevir (both received MA approved in 2011 for use with interferon and ribavirin based therapy), however direct acting agents are linked to increased toxicity of treatment, the emergence of resistance, and to date do not provide a standard of care which is interferon free. The combination of direct acting agents can also result in drug-drug interactions. To date, no HCV therapy has been approved which is interferon free. There is therefore a need for new combination therapies which have reduced side effects, and interferon free, have a reduced emergence of resistance, reduced treatment periods and/or and enhanced cure rates.
Simeprevir (formerly TMC435) is an experimental drug candidate for the treatment of hepatitis C. It is being developed byMedivir and Johnson & Johnson‘s pharmaceutical division Janssen Pharmaceutica and is currently in Phase III clinical trials.[1]
Simeprevir is a hepatitis C virus protease inhibitor.[2]
Simeprevir is being tested in combination regimens with pegylated interferon alfa-2a and ribavirin,[3] and in interferon-free regimens with other direct-acting antiviral agents including daclatasvir[4] and sofosbuvir [5]
Food and Drug Administration (FDA) has granted Priority Review to the New Drug Application (NDA) for simeprevir (TMC435). Simeprevir is an investigational NS3/4A protease inhibitor taken orally (150 mg capsule) once a day along with pegylated interferon and ribavirin for genotype 1 chronic hepatitis C virus (HCV) infection in adult patients with compensated liver disease (meaning the liver is heavily scarred but still functional).
“Hepatitis C is a complex disease and Janssen is committed to working with the HCV community, caregivers, and health care systems to address this global epidemic,” said Gaston Picchio, Hepatitis Disease Area Leader, Janssen Research & Development. “We are pleased that the FDA has granted simeprevir Priority Review, as it is a significant step forward in making this therapy available to physicians and their hepatitis C patients.”
The FDA grants Priority Review to medicines that may offer major advances in care or provide a treatment option where no adequate therapy exists. Under the Prescription Drug User Fee Act, FDA review will begin approximately 60 days after receipt of the application and will aim to be completed within six months from when the review period begins.
The regulatory submission for simeprevir is supported in part by data from three pivotal Phase 3 studies: QUEST-1 and QUEST-2 in treatment-naïve patients and PROMISE in patients who have relapsed after prior interferon-based treatment. Janssen also recently submitted simeprevir for marketing authorization to regulatory authorities in Japan and Europe.

- “Medivir Announces That Simeprevir (TMC435) Data Will Be Presented at the Upcoming AASLD Meeting”. Yahoo News. October 1, 2012. Retrieved November 6, 2012.
- Lin, TI; Lenz, O; Fanning, G; Verbinnen, T; Delouvroy, F; Scholliers, A; Vermeiren, K; Rosenquist, A et al. (2009). “In vitro activity and preclinical profile of TMC435350, a potent hepatitis C virus protease inhibitor”. Antimicrobial agents and chemotherapy 53 (4): 1377–85. doi:10.1128/AAC.01058-08. PMC 2663092. PMID 19171797.
|displayauthors=suggested (help) - “Phase 3 Studies Show Simeprevir plus Interferon/Ribavirin Cures Most Patients in 24 Weeks”. hivandhepatitis.com. December 27, 2012.
- Medivir announces TMC435 in an expanded clinical collaboration. Medivir. 18 April 2012.
- Results from a phase IIa study evaluating Simeprevir and Sofosbuvir in prior null responder Hepatitis C patients have been presented at CROI. 6 March 2013.
IUPAC standard name
(1R, 4R, 6S, 15R, 17R)-N-(cyclopropanesulfonyl) -17 – ({7-methoxy-8-methyl-2-[4 – (propan-2-yl) -1,3-thiazol-2 -yl] quinolin-4-yl} oxy)-13-methyl-2 ,14-dioxo-3 ,13-diazatricyclo [13.3.0.0 4 , 6 ] octadec-7-ene-4-carboxamide
IUPAC traditional name
(1R, 4R, 6S, 15R, 17R)-N-(cyclopropanesulfonyl) -17 – {[2 – (4-isopropyl-1 ,3-thiazol-2-yl)-7-methoxy-8-methylquinolin-4- yl] oxy}-13-methyl-2 ,14-dioxo-3 ,13-diazatricyclo [13.3.0.0 4 , 6 ] octadec-7-ene-4-carboxamide
Aliases
TMC435
TMC435350

,,,,,,,,,,,,,
NS3/4A protease inhibitors
Ciluprevir (BILN 2061) Boehringer Ingelheim
Boceprevir (SCH503034) Merck
Telaprevir (VX-950) Vertex
Danoprevir (RG7227) Roche
simeprevir /TMC435 Tibotec / Medivir
Vaniprevir (MK-7009) Merck
Bl 201335 Boehringer Ingelheim
BMS-650032 Bristol-Myers Squibb
GS-9256 Gilead
ABT-450 Abbott / Enanta
Narlaprevir (SCH900518) Merck
PHX1766 Phenomix
ACH-1625 Achillion
IDX320 Idenix
MK-5172 Merck
VX-985 Vertex Drug name Company
GS-9451 Gilead
Telaprevir
Accordin to http://en.wikipedia.Org/wiki/File:Telaprevir.svg, Teaprevir has the structure

Systematic lUPAC Name: (1 S,3aR,6aS)-2-[(2S)-2-[[(2S)-2-Cyclohexyl-2-(pyrazine-2- carbonylamino)acetyl]amino]-3,3-dimethylbutanoyl]-/\/-[(3S)-1-(cyclopropylamino)-1 ,2- dioxohexan-3-yl]-3,3a,4,5,6,6a-hexahydro-1/-/-cyclopenta[c]pyrrole-1-carboxamide
Telaprevir may be administered in a unit dose of, for example between about 250 and about l OOOmg, such as about 750mg/kg. Typically once, twice, three or four times daily, such as three times daily for the duration of the pre-treatment period and/or combination treatment period.
Boceprevir
Accordin to http://en.wikipedia.0rg/wiki/File:B0ceprevir.svg, Boceprevir has the structure:

Systematic lUPAC Name: (1 R,2S,5S)-N-[(2≡)-4-amino-1-cyclobutyl-3,4-dioxobutan-2-yl)]- 3-{(2S)-2-[(tert-butylcarbamoyl)amino]-3,3-dimethylbutanoyl}- 6,6-dimethyl-3- azabicyclo[3.1.0]hexane-2-carboxamide
Boceprevir may be administered in a unit dose of, for example between about 250 and about 1000mg, such as about 800mg/kg. Typically once, twice, three or four times daily, such as three times daily for the duration of the pre-treatment period and/or combination treatment period.
Compound 1: miR-122 inhibitor
As reported in Young et al., JACS 2010, 132, 7976-7981) (hereby incorporated by reference), it is possible to assay for small molecule inhibitors of miR122 and small molecule are known, such as those illustrated below:



» {7.02 ± 1.40) If (4. S3 * 0.45)

The numerical values refer to luciferase expression due to miR-122 deprepression, and values greater than 1 indicate miR-122 inhibition.
Merck Announces FDA Acceptance of New Drug Application for Investigational Fertility Treatment

corifollitropin alfa
WHITEHOUSE STATION, N.J.–(BUSINESS WIRE)–Merck (NYSE:MRK), known as MSD outside the United States and Canada, today announced that the New Drug Application (NDA) for its investigational fertility treatment, corifollitropin alfa, has been accepted for standard review by the U.S. Food and Drug Administration (FDA). Merck is seeking FDA approval of corifollitropin alfa for Controlled Ovarian Stimulation (COS) in women participating in assisted reproductive technology.
If approved, corifollitropin alfa would be the first sustained follicular stimulant for use in a fertility treatment regimen.
read all at
http://www.pharmalive.com/fda-accepts-mercks-fertility-treatment-nda

Corifollitropin alfa
Merck received approval on February 15, 2010 from the European Commission for ELONVA (corifollitropin alfa) a long lasting single injection fusion protein lacking LH activity. Only one injection is required for the first seven days, replacing the first seven daily injections of conventional FSH. Initial results demonstrates similar pregnancy rates as daily recombinant FSH injections.[7][8]
- ref 7 N. P. Koper, R. Boostanfar, P. Devroey, B. C. Fauser, P. C. IJzerman-Boon, B. M. J. L. Mannaerts. Global ClinicalDevelopment, Organon, Part of Schering-Plough Corporation, Oss, Netherlands; Huntington Reproductive Center, Tarzana, CA; Center of Reproductive Medicine, Dutch-speaking Free University, Brussels, Belgium; University Medical Center Utrecht, Utrecht, Netherlands; Biometrics, NV Organon, Part of Schering-Plough Corporation, Oss, Netherlands. “Corifollitropin alfa demonstrates similar pregnancy rates as compared to daily recombinant FSH treatment in a controlled ovarian stimulation regimen for IVF/ICSI.” Fertility and Sterility, 90:page S75.
- ref 8 ^ Devroey P, Boostanfar R, Koper NP, Mannaerts BM, Ijzerman-Boon PC, Fauser BC, 2009. “A double-blind, non-inferiority RCT comparing corifollitropin alfa and recombinant FSH during the first seven days of ovarian stimulation using a GnRH antagonist protocol.” Human Reproduction, 2009, August 14, [Epub ahead of print]. PMID 19684043.
In May2013, MSD launched ELONVA® (corifollitropin alfa injection) – a new treatment for fertility, – in Singapore. Approved for controlled ovarian stimulation in combination with a GnRH antagonist for the development of multiple follicles, Corifollitropin alfa injection is the first sustained follicle stimulant. A single subcutaneous injection of the recommended dose of corifollitropin alfa injection may replace the first seven injections of any
Findings showed that other failed repeated treatments may lead to depression, anxiety, sexua conventional daily recombinant follicle stimulating hormone (rFSH) preparation in a controlled ovarian stimulation treatment cycle. Simplified fertility treatment with Elonva not only helps to reduce the emotional and physical burden of fertility, it may also reduce dropout rates and potentially improve the overall chances of pregnancy.
l anxiety/difficulty, relationship problems with partner, family and friends, increased sense of self-blame and guilt, particularly for the partner experiencing fertility problem. ”By reducing the number of daily injections, the
availability of corifollitropin alfa injection is a positive step towards helping reduce the burden of fertility treatment for women experiencing difficulty conceiving. Simplifying fertility treatment with new modalities of treatment and new medication may encourage more infertile couple to embark
on treatment earlier when the wife’s age is younger and ovarian reserve better.” said Dr Loh Seong Feei, Medical Director of Thomson Fertility Centre

{kind=link}
{kind=link}