
Home » Posts tagged 'Rhizen Pharmaceuticals'
Tag Archives: Rhizen Pharmaceuticals
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
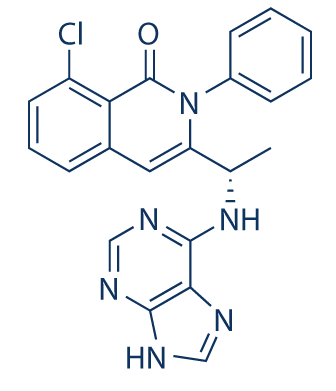
RP 12146

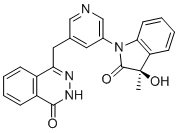

RP 12146, EX-A7910
cas 2732869-64-4
4-[[5-[(3R)-3-hydroxy-3-methyl-2-oxoindol-1-yl]pyridin-3-yl]methyl]-2H-phthalazin-1-one
| M.Wt | 398.422 | |
| Formula | C23H18N4O3 | |
RP12146 (RP12146) is a novel, selective, and potent small molecule inhibitor of PARP1/2 with IC50 of 0.6/0.5 nM, with several fold selectivity over other isoforms.
SCHEME
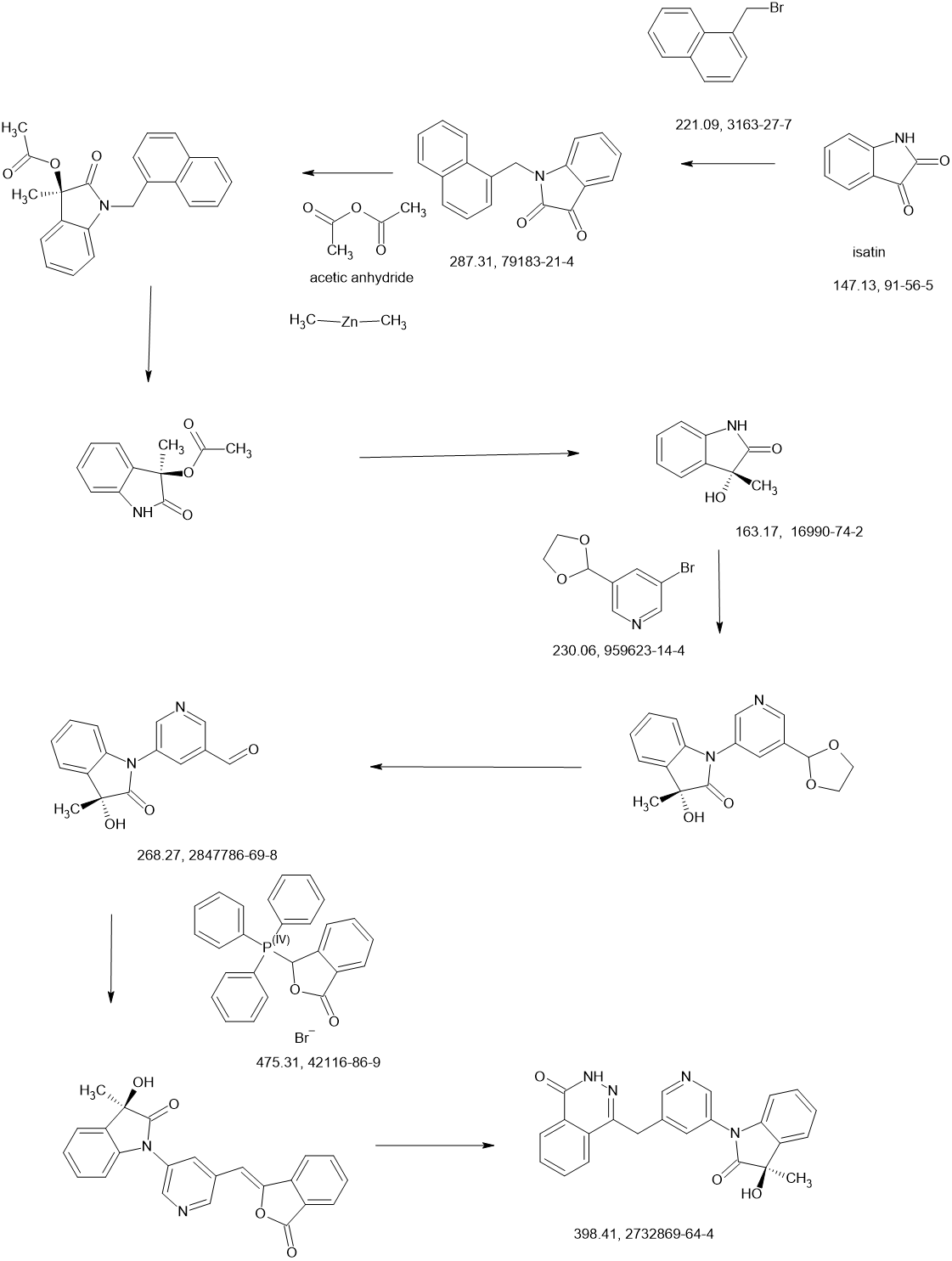
Ref
WO2021220120
Rhizen Pharmaceuticals AG
WO2022215034
Rhizen Pharmaceuticals AG; Incozen Therapeutics Pvt. Ltd.
RP-12146 is an oral poly (ADP-ribose) polymerase (PARP) inhibitor in phase I clinical development at Rhizen Pharmaceuticals for the treatment of adult patients with locally advanced or metastatic solid tumors.
Solid TumorExtensive-stage Small-cell Lung CancerLocally Advanced Breast CancerMetastatic Breast CancerPlatinum-sensitive Ovarian CancerPlatinum-Sensitive Fallopian Tube CarcinomaPlatinum-Sensitive Peritoneal Cancer
Poly(ADP-ribose) polymerase (PARP) defines a family of 17 enzymes that cleaves NAD+ to nicotinamide and ADP-ribose to form long and branched (ADP-ribose) polymers on glutamic acid residues of a number of target proteins, including PARP itself. The addition of negatively charged polymers profoundly alters the properties and functions of the acceptor proteins. Poly(ADP-ribosyl)ation is involved in the regulation of many cellular processes, such as DNA repair, gene transcription, cell cycle progression, cell death, chromatin functions and genomic stability. These functions have been mainly attributed to PARP-1 that is regarded as the best characterized member of the PARP family. However, the identification of novel genes encoding PARPs, together with the characterization of their structure and subcellular localization, have disclosed different roles for poly(ADP-ribosyl)ation in cells, including telomere replication and cellular transport.
Recently, poly(ADP-ribose) binding sites have been identified in many DNA damage checkpoint proteins, such as tumor suppressor p53, cyclin-dependent kinase inhibitor p21Cip1/waf1, DNA damage recognition factors (i.e., the nucleotide excision repair xeroderma pigmentosum group A complementing protein and the mismatch repair protein MSH6), base excision repair (BER) proteins (i.e. DNA ligase III, X-ray repair cross-complementing 1, and XRCC1), DNA-dependent protein kinase (DNA-PK), cell death and survival regulators (i.e.,
NF-kB, inducible nitric oxide synthase, and telomerase). These findings suggest that the different components of the PARP family might be involved in the DNA damage signal network, thus regulating protein-protein and protein-DNA interactions and, consequently, different types of cellular responses to genotoxic stress. In addition to its involvement in BER and single strand breaks (SSB) repair, PARP-1 appears to aid in the non-homologous end-joining (NHEJ) and homologous recombination (HR) pathways of double strand breaks (DSB) repair. See Lucio Tentori et al., Pharmacological Research, Vol. 45, No. 2, 2002, page 73-85.
PARP inhibition might be a useful therapeutic strategy not only for the treatment of BRCA mutations but also for the treatment of a wider range of tumors bearing a variety of deficiencies in the HR pathway. Further, the existing clinical data (e.g., Csaba Szabo et al., British Journal of Pharmacology (2018) 175: 192-222) also indicate that stroke, traumatic brain injury, circulatory shock and acute myocardial infarction are some of the indications where PARP activation has been demonstrated to contribute to tissue necrosis and inflammatory responses.
As of now, four PARP inhibitors, namely olaparib, talazoparib, niraparib, and rucaparib have been approved for human use by regulatory authorities around the world.
Patent literature related to PARP inhibitors includes International Publication Nos. WO 2000/42040, WO 2001/016136, WO 2002/036576, WO 2002/090334, WO2003/093261, WO 2003/106430, WO 2004/080976, WO 2004/087713, WO 2005/012305, WO 2005/012524, WO 2005/012305, WO 2005/012524, WO 2005/053662, W02006/033003, W02006/033007, WO 2006/033006, WO 2006/021801, WO 2006/067472, WO 2007/144637, WO 2007/144639, WO 2007/144652, WO 2008/047082, WO 2008/114114, WO 2009/050469, WO 2011/098971, WO 2015/108986, WO 2016/028689, WO 2016/165650, WO 2017/153958, WO 2017/191562, WO 2017/123156, WO 2017/140283, WO 2018/197463, WO 2018/038680 and WO 2018/108152, each of which is incorporated herein by reference in its entirety for all purposes.
There still remains an unmet need for new PARP inhibitors for the treatment of various diseases and disorders associated with cell proliferation, such as cancer.
PATENT
Illustration 1
CLIP
https://cancerres.aacrjournals.org/content/81/13_Supplement/1233
Abstract 1233: Preclinical profile of RP12146, a novel, selective, and potent small molecule inhibitor of PARP1/2
Srikant Viswanadha, Satyanarayana Eleswarapu, Kondababu Rasamsetti, Debnath Bhuniya, Gayatriswaroop Merikapudi, Sridhar Veeraraghavan and Swaroop VakkalankaProceedings: AACR Annual Meeting 2021; April 10-15, 2021 and May 17-21, 2021; Philadelphia, PA
Abstract
Background: Poly (ADP-ribose) polymerase (PARP) activity involves synthesis of Poly-ADP ribose (PAR) polymers that recruit host DNA repair proteins leading to correction of DNA damage and maintenance of cell viability. Upon combining with DNA damaging cytotoxic agents, PARP inhibitors have been reported to demonstrate chemo- and radio-potentiation albeit with incidences of myelosuppression. A need therefore exists for the development selective PARP1/2 inhibitors with a high therapeutic window to fully exploit their potential as a single agent or in combination with established therapy across various tumor types. Additionally, with the emerging concept of ‘synthetic lethality’, the applicability PARP inhibitors can be expanded to cancers beyond the well-defined BRCA defects. Herein, we describe the preclinical profile of RP12146, a novel and selective small molecule inhibitor of PARP1 and PARP2.
Methods: Enzymatic potency was evaluated using a PARP Chemiluminescent Activity Assay Kit (BPS biosciences). Cell growth was determined following incubation with RP12146 in BRCA1 mutant and wild-type cell lines across indications. Apoptosis was evaluated following incubation of cell lines with compound for 120 h, subsequent staining with Annexin-V-PE and 7-AAD, and analysis by flow cytometry. For cell cycle, cells were incubated with compound for 72 h, and stained with Propidium Iodide prior to analysis by flow cytometry. Expression of downstream PAR, PARP-trapping, phospho-γH2AX and cleaved PARP expression were determined in UWB1.289 (BRCA1 null) cells by Western blotting. Anti-tumor potential of RP12146 was tested in OVCAR-3 Xenograft model. Pharmacokinetic properties of the molecule were also evaluated. Results: RP12146 demonstrated equipotent inhibition of PARP1 (0.6 nM) and PARP2 (0.5 nM) with several fold selectivity over the other members of the PARP family. Compound caused a dose-dependent growth inhibition of both BRCA mutant and non-mutant cancer cell lines with GI50 in the range of 0.04 µM to 9.6 µM. Incubation of UWB1.289 cells with RP12146 caused a G2/M arrest with a corresponding dose-dependent increase in the percent of apoptotic cells. Expression of PAR was inhibited by 86% at 10 nM with a 2.3-fold increase in PARP-trapping observed at 100 nM in presence of RP12146. A four-fold increase in phospho-γH2AX and > 2-fold increase in cleaved PARP expression was observed at 3 µM of the compound. RP12146 exhibited anti-tumor potential with TGI of 28% as a single agent in OVCAR-3 xenograft model. Efficay was superior compared to Olaparib tested at an equivalent dose. Pharmacokinetic studies in rodents indicated high bioavailability with favorable plasma concentrations relevant for efficacy
Conclusions: Data demonstrate the therapeutic potential of RP12146 in BRCA mutant tumors. Testing in patients is planned in H1 2021.
Citation Format: Srikant Viswanadha, Satyanarayana Eleswarapu, Kondababu Rasamsetti, Debnath Bhuniya, Gayatriswaroop Merikapudi, Sridhar Veeraraghavan, Swaroop Vakkalanka. Preclinical profile of RP12146, a novel, selective, and potent small molecule inhibitor of PARP1/2 [abstract]. In: Proceedings of the American Association for Cancer Research Annual Meeting 2021; 2021 Apr 10-15 and May 17-21. Philadelphia (PA): AACR; Cancer Res 2021;81(13_Suppl):Abstract nr 1233.

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
CLIP
Rhizen Pharmaceuticals AG Announces First Patient Dosing in a Phase I/Ib Study of Its Novel PARP Inhibitor (RP12146) in Patients With Advanced Solid Tumors
RHIZEN’S PARP INHIBITOR EFFORTS ARE PART OF A LARGER DDR PLATFORM THAT ALSO INCLUDES AN EARLY STAGE POLθ-DIRECTED PROGRAM; PLATFORM ENABLES PROPRIETARY IN-HOUSE COMBINATIONS
- Rhizen Pharma commences dosing in a phase I/Ib trial to evaluate its novel PARP inhibitor (RP12146) in patients with advanced cancers.
- Rhizen indicated that RP12146 has comparable preclinical activity vis-à-vis approved PARP inhibitors and shows improved preclinical safety that it expects will translate in the clinic.
- The two-part multi-center phase I/Ib study is being conducted in Europe and is designed to initially determine safety, tolerability and MTD/RP2D of RP12146 and to subsequently assess its anti-tumor activity in expansion cohorts with HRR mutation-enriched ES-SCLC, ovarian and breast cancer patients.
- RP12146 is part of a larger DDR platform at Rhizen that includes a preclinical-stage Polθ inhibitor program; the DDR platform enables novel, proprietary, in-house combinations
November 01, 2021 07:24 AM Eastern Daylight Time
BASEL, Switzerland–(BUSINESS WIRE)–Rhizen Pharmaceuticals AG (Rhizen), a Switzerland-based privately held, clinical-stage oncology & inflammation-focused biopharmaceutical company, announced today that it has commenced dosing in a multi-center, phase I/Ib trial to evaluate its novel poly (ADP-ribose) polymerase (PARP) inhibitor (RP12146) in patients with advanced solid tumors. This two-part multi-center phase I/Ib study is being conducted in Europe and has been designed to initially determine safety, tolerability, maximum tolerated dose (MTD), and/or recommended phase II dose (RP2D) of RP12146 and to subsequently assess its anti-tumor activity in expansion cohorts with HRR mutation-enriched ES-SCLC, ovarian and breast cancer patients.
“Our PARP program is foundational for our DDR platform efforts and will be the backbone for several novel proprietary combinations that we hope to bring into development going forward.”
Rhizen indicated that RP12146 has shown preclinical activity and efficacy comparable to the approved PARP inhibitor Olaparib, and shows improved safety as seen in the preclinical IND-enabling toxicology studies; an advantage that Rhizen hopes will translate in the clinical studies. Rhizen also announced that its PARP program is part of a larger DNA Damage Response (DDR) platform effort, which includes a preclinical-stage polymerase theta (Polθ) inhibitor program. Rhizen expects the platform to enable novel proprietary combinations of its PARP and Polθ assets given the mechanistic synergy and opportunity across PARP resistant/refractory settings.
“PARP inhibitors are a great success story in the DNA damage response area, but they are not without safety concerns that have limited realization of their full potential. Although our novel PARP inhibitor is competing in a crowded space, we expect its superior preclinical safety to translate into the clinic which will differentiate our program and allow us to extend its application beyond the current landscape of approved indications and combinations”, said Swaroop Vakkalanka, Founder & CEO of Rhizen Pharma. Swaroop also added that “Our PARP program is foundational for our DDR platform efforts and will be the backbone for several novel proprietary combinations that we hope to bring into development going forward.”
About Rhizen Pharmaceuticals AG.:
Rhizen Pharmaceuticals is an innovative, clinical-stage biopharmaceutical company focused on the discovery and development of novel oncology & inflammation therapeutics. Since its establishment in 2008, Rhizen has created a diverse pipeline of proprietary drug candidates targeting several cancers and immune associated cellular pathways.
Rhizen has proven expertise in the PI3K modulator space with the discovery of our first PI3Kδ & CK1ε asset Umbralisib, that has been successfully developed & commercialized in MZL & FL by our licensing partner TG Therapeutics (TGTX) in USA. Beyond this, Rhizen has a deep oncology & inflammation pipeline spanning discovery to phase II clinical development stages.
Rhizen is headquartered in Basel, Switzerland.
REF
Safety, Pharmacokinetics and Anti-tumor Activity of RP12146, a PARP Inhibitor, in Patients With Locally Advanced or Metastatic Solid Tumors….https://clinicaltrials.gov/ct2/show/NCT05002868
//////////RP 12146, oral poly (ADP-ribose) polymerase (PARP) inhibitor, phase I, clinical development, INCOZEN, Rhizen Pharmaceuticals, adult patients, locally advanced, metastatic solid tumors, PARP, CANCER, EX-A7910, EX A7910

NEW DRUG APPROVALS
ONE TIME
$10.00
Umbralisib, TGR-1202, a Phosphoinositide-3 kinase delta inhibitor, Rhizen Pharmaceuticals S.A./TG Therapeutics
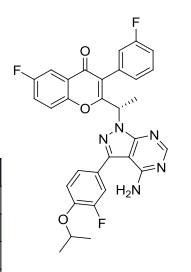

| Molecular Formula: | C31H24F3N5O3 |
|---|---|
| Molecular Weight: | 571.54917 g/mol |
RP-5307
TGR-1202
TGR-1202 PTSA
FU8XW5V3FS (UNII code)
RP-5264 (free base)
A PI3K inhibitor potentially for treatment of chronic lymphocytic leukemia, leukemia,lymphoma,B-cell
TGR‐1202, a next generation PI3K-δ delta inhibitor. TGR-1202 (RP-5264) is a highly specific, orally available, PI3K delta inhibitor, targeting the delta isoform with nanomolar potency and several fold selectivity over the alpha, beta, and gamma isoforms of PI3K.
TG Therapeutics, under license from Rhizen Pharmaceuticals, is developing TGR-1202 (structure shown; formerly RP-5264), a lead from a program of PI3K delta inhibitors, for the potential oral treatment of hematological cancers including Hodgkin lymphoma, non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), B-cell lymphoma and mantle cell lymphoma (MCL)
Incozen Therapeutics Pvt Ltd
TG Therapeutics
TGR-1202 potential to perform as the best PI3K inhibitor in its class and the possible superiority of TG-1101 over Rituxan®.
| Rhizen Pharmaceuticals S.A. | |
| Description | Phosphoinositide 3-kinase (PI3K) delta inhibitor |
Leukemia, chronic lymphocytic PHASE 3, TG Therapeutics
Orphan Drug
Umbralisib is a novel phosphatidylinositol 3-kinase delta (PI3Kdelta) inhibitor under development at TG Therapeutics in phase III clinical trials, in combination with ublituximab, for the treatment of chronic lymphocytic leukemia (CLL) and for the treatment of diffuse large B-cell lymphoma (DLBCL). The company refers to the combination regimen of ublituximab and TGR-1202 as TG-1303. The drug is also in phase II clinical development for the oral treatment of hematologic malignancies, as a single agent or in combination therapy. Phase I clinical trials are ongoing in patients with select relapsed or refractory solid tumors, such as adenocarcinoma of the pancreas, adenocarcinoma of the colon, rectum, gastric and GE junction cancer, and GI Stromal Tumor (GIST).
In 2016, orphan drug designation was assigned to the compound in the U.S. for the treatment of CLL. In 2017, additional orphan drug designation was granted in the U.S. for the treatment of CLL and DLBCL, in combination with ublituximab.
Originated by Rhizen Pharmaceuticals, the product was jointly developed by Rhizen Pharmaceuticals and TG Therapeutics since 2012. In 2014, exclusive global development and commercialization rights (excluding India) were licensed to TG Therapeutics.
CLINICAL TRIALS……….https://clinicaltrials.gov/search/intervention=TGR-1202
B-cell lymphoma; Chronic lymphocytic leukemia; Hematological neoplasm; Hodgkins disease; Mantle cell lymphoma; Non-Hodgkin lymphoma
Phosphoinositide-3 kinase delta inhibitor
SYNTHESIS


Rhizen Pharmaceuticals Announces Out-licensing Agreement for TGR-1202, a Novel Next Generation PI3K-delta Inhibitor
Rhizen to receive upfront payment of $8.0 million — Rhizen to retain global manufacturing and supply rights — Rhizen to retain development and commercialization for India
Rhizen to retain development and commercialization for India
| Source: Rhizen Pharmaceuticals SA
La Chaux-de-Fonds, Switzerland, Sept. 23, 2014 (GLOBE NEWSWIRE) — Rhizen Pharmaceuticals S.A. today announced an out-licensing agreement for TGR-1202, a novel next generation PI3K-delta inhibitor. TG Therapeutics exercised its option for early conversion to a licensing agreement from a 50:50 joint venture partnership.
In exchange for this licensing agreement, TG Therapeutics will pay Rhizen an upfront payment of $8.0 million ($4.0 million in cash and $4.0 million in TG Therapeutics common stock). In addition to the upfront payment, Rhizen will be eligible to receive regulatory filing, approval and sales based milestones in the aggregate of approximately $240 million, and tiered royalties based on net sales.
Swaroop Vakkalanka, Ph.D. and President of Rhizen stated, “We are extremely happy and take pride in discovering a novel, next generation, once-daily PI3K-delta inhibitor under active development led by TG Therapeutics. We are encouraged by the progress of TRG-1202 to date, and the speed at which TG Therapeutics is developing the asset in various hematological malignancies. We look forward to the day this novel drug reaches cancer patients in need of new and safe therapies.”
About Rhizen Pharmaceuticals S.A.:
Rhizen Pharmaceuticals is an innovative, clinical-stage biopharmaceutical company focused on the discovery and development of novel therapeutics for the treatment of cancer, immune and metabolic disorders. Since its establishment in 2008, Rhizen has created a diverse pipeline of proprietary drug candidates targeting several cancers and immune associated cellular pathways. Rhizen is headquartered in La-Chaux-de-Fonds, Switzerland. For additional information, please visit Rhizen’s website, www.rhizen.com.

TGR-1202.with Idelalisib and IPI-145 (left to right) for comparison.
IPI 145
PATENTS
WO 2011055215
http://www.google.com/patents/WO2011055215A2?cl=en

PATENT
WO 2015181728
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015181728
TGR-1202, chemically known as (S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-3-(3-fluorophenyl)-4H-chromen-4-one, has the following chemical structure:

Example 1: Preparation of the PTSA Salt of TGR-1202 (Form A)

7100 g of TGR-1202 was charged in a reactor containing 56.8 litres of acetone and stirred at ambient temperature. 4680 g of p-toluene sulphonic acid was added and the reaction mixture was heated at a temperature of 60-65° C for about 6 hours. The solvent was removed by distillation under reduced pressure to obtain a wet residue. The wet residue was degassed and allowed to cool to < 20° C. Approximately 142 litres of diethyl ether was then added and the resulting mixture was stirred overnight, then filtered to obtain a solid mass which was washed with diethyl ether and dried in vacuo to yield a solid mass. The solid mass was re-suspended in diethyl ether, stirred for 6 hours, and then filtered to yield a solid mass which was subsequently dissolved in 56.8 litres of acetone, filtered through a HiFlow bed, and concentrated under reduced pressure. The resulting residue mass was stirred with water overnight, then filtered and vacuum dried to yield 6600 g of the PTSA salt of TGR-1202. HPLC: 99.21% and chiral purity of 99.64:0.36 (S:R).
Example 2: Preparation of the PTSA Salt of TGR-1202 (Form B)

1000 g of TGR-1202 was charged in a reactor containing 8 litres of acetone and stirred at ambient temperature. 666 g of p-toluene sulphonic acid was then added and the reaction mixture was heated at a temperature of 60-65 °C for about 6 hours. The solvent was removed by distillation under reduced pressure to obtain a wet residue. The wet residue was degassed and allowed to cool to < 20° C. Approximately 20 litres of diethyl ether was added and the resulting mixture was stirred overnight, then filtered to obtain a solid mass which was washed with diethyl ether and dried in vacuo to yield a solid mass which was then vacuum dried to yield 1150 g of the PTSA salt of TGR-1202. HPLC: 99.33% and chiral purity: 99.61:0.39 (S:R).
Table 1 lists the XRPD pattern peaks and relative peak intensities for the products of Examples 1 and 2.
TABLE 1

The tablet composition comprising a PTSA salt of TGR-1202 prepared according to Example 2 exhibited a Cmax about 2.5 fold and an area under the curve (AUC) about 1.9 fold greater than that of the tablet composition comprising a PTSA salt of TGR-1202 prepared according to Example 1. The results are provided in Table 8 below.
TABLE 8

PATENT
WO 2014071125
http://www.google.com/patents/WO2014071125A1?cl=en
formula (A) that is a ΡΒΚδ selective inhibitor,

(A)
Synthesis of Compound of Formula A
Unless otherwise stated, purification implies column chromatography using silica gel as the stationary phase and a mixture of petroleum ether (boiling at 60-80°C) and ethyl acetate or dichloromethane and methanol of suitable polarity as the mobile phases. The term “RT” refers to ambient temperature (25-28°C).
Intermediate 1 : 2-( l-bromoethyl)-6-fluoro-3-f3-fluorophenyl)-4H-chromen-4-one
Step-1 [l-(5-Fluoro-2-hydroxyphenyl)-2-(3-fluorophenyl)ethanone]: 3- Fluorophenylacetic acid (7.33 g, 47.56 mmoles) was dissolved in 25 ml dichloromethane. To this mixture, oxalylchloride (7.54 g, 59.46 mmoles) and DMF (3 drops) were added at 0°C and stirred for 30 min. The solvent was evaporated and dissolved in 25 ml dichloromethane. To this mixture, 4-fluoroanisole (5.00 g, 39.64 mmoles) was added and cooled to 0°C. At 0°C A1C13 (7.95 g, 59.46 mmoles) was added and the reaction mixture was warmed to RT and stirred for 12 hours. The reaction mixture was quenched by the addition of 2N HC1, extracted with ethyl acetate, dried over sodium sulphate and concentrated. The crude product was purified by column chromatography with ethyl acetate :petroleum ether to afford the title compound as colorless solid (4.5 g, 45% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 11.34 (s, 1H), 7.75 (dd, J=9.4, 3.1 Hz, 1H), 7.42 (m, 2H), 7.12 (m, 3H), 7.05 (dd, J=9.0, 4.5 Hz, 1H), 4.47 (s, 2H).
Step-2 [2-Ethyl-6-fiuoro-3-(3-fluorophenyl)-4H-chromen-4-one]: l-(5-Fluoro-2- hydroxyphenyl)-2-(3-fluorophenyl)ethanone obtained from Step-1 (3.00 g, 12.08 mmoles) was placed in a round bottom flask and to this triethylamine (25 ml) and propionic anhydride (4.92 g, 37.82 mmoles) were added, and the mixture was refluxed for 24 hours. After cooling to RT, the reaction mixture was acidified by the addition of IN HC1 solution, extracted with ethyl acetate, washed with sodium bicarbonate solution, dried with sodium sulphate and concentrated. The crude product was purified by column chromatography with ethyl acetate :petroleum ether to afford the title compound as off-yellow solid (1.80 g, 52% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 7.80 (m, 1H), 7.76 (m, 2H), 7.51 (dd, J=8.0, 6.4 Hz), 7.22 (m, 1H), 7.18 (m, 2H), 2.56 (q, J=7.6 Hz, 2H), 1.20 (t, J=7.6 Hz, 3H).
Step-3: To a solution of 2-Ethyl-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one obtained from Step-2 (1.80 g, 6.28 mmoles) in carbon tetrachloride (20 ml), N- bromosuccinimide (1.11 g, 6.28 mmoles) was added and heated to 80°C. Azobisisobutyronitrile (10 mg) was added to the reaction mixture at 80°C. After 12 hours, the reaction mixture was cooled to RT, diluted with dichloromethane and washed with water. The organic layer was dried over sodium sulphate and concentrated under reduced pressure to afford the crude title compound as yellow solid (1.25 g, 55% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 7.91 (dd, J=9.2, 4.3 Hz, 1H), 7.81 (dt, j=8.2, 2.8 Hz, 1H), 7.74 (dd, J=8.3, 3.1 Hz, 1H), 7.57 (m, 1H), 7.32 (dt, J=8.5, 2.4 Hz, 1H), 7.19 (m, 2H), 5.00 (q, J=6.8 Hz, 1H), 1.97 (d, J=6.8 Hz, 3H).
Intermediate 2: 6-fluoro-3-f3-fluorophenyl)-2-fl-hvdroxyethyl)-4H-chromen-4-one

To a solution of Intermediate 1 (15.0 g, 40.84 mmol) in DMSO (150 ml), n-butanol (7.5 ml) was added and heated to 120°C for 3 hours. The reaction mixture was cooled to RT, quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (7.90 g, 64%). 1H-NMR (δ ppm, CDC13, 400 MHz): 7.85 (dd, J = 8.1, 3 Hz, 1H), 7.54 (dd, J = 9.2, 4.2 Hz, 1H), 7.47-7.37 (m, 2H), 7.15-6.98 (m, 3H), 4.74 (quintet, J= 6.8 Hz, 1H), 2.23 (d, J = 7.4 Hz, 1H), 1.54 (d, J = 6.6 Hz, 3H).
Intermediate 3 : 2-acetyl-6-fluoro-3-( 3-fluorophenyl)-4H-chromen-4-one

DMSO (5.60 ml, 79.14 mmol) was added to dichloromethane (40 ml), and cooled to – 78°C, followed by oxalyl chloride (3.40 ml, 39.57 mmol). After 10 min., intermediate 2 (6.00 g, 19.78 mmol) in dichloromethane (54 ml) was added dropwise and stirred for 20 min.
Triethylamine (12 ml) was added and stirred for 1 hour. The reaction mixture was quenched with water and extracted with dichloromethane. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (4.2 g, 71%) which was used as such in the next step.
Intermediate 4: fS)-6-fluoro-3-f3-fluorophenyl)-2-fl-hvdroxyethyl)-4H-chromen-4-one

To intermediate 3 (2.00 g, 6.66 mmol), R-Alpine borane (0.5 M in THF, 20 ml) was added and heated to 60°C for 20 hours. The reaction mixture quenched with 2N HC1, and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (1.51 g, 75%).
Enantiomeric excess: 94.2%, enriched in the fast eluting isomer (retention time: 8.78 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 5: fR)-l-f6-fluoro-3-f3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl 4- chlorobenzoate

To a solution of intermediate 4 (1.45 g, 4.78 mmol) in THF (15 ml), 4-chlorobenzoic acid (0.748 g, 4.78 mmol) and triphenylphosphine (1.88 g, 7.17 mmol) were added and heated to 45°C followed by diisopropylazodicarboxylate (1.4 ml, 7.17 mmol). After 1 hour, the reaction mixture was concentrated and the residue was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (1.81 g, 86%) which was used without purification in the next step. Intermediate 6: fR)-6-fluoro-3-f3-fluorophenyl)-2-fl-hvdroxyethyl)-4H-chromen-4-one

Method A
Intermediate 5 (1.75 g, 3.96 mmol) in methanol (17 ml) was cooled to 10°C, potassium carbonate (0.273 g, 1.98 mmol) was added and stirred for 30 min. The reaction mixture was concentrated, acidified with 2N HCl solution, extracted with ethyl acetate, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (1.05 g, 87% yield). Enantiomeric excess: 93.6%>, enriched in the late eluting isomer (retention time: 11.12 min.) as determined by HPLC on a chiralpak AD-H column.
Method B
Step-1 [(R)-2-(l-(benzyloxy)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one]: To l-(5-fluoro-2-hydroxyphenyl)-2-(3-fluorophenyl)ethanone (11.00 g, 44.31 mmol) in dichloromethane, HATU (33.7 g, 88.63 mmol) and R-(+)2-benzyloxypropionic acid (9.58 g, 53.17 mmol) were added and stirred for 10 min. Triethylamine (66.7 ml, 0.47 mol) was added dropwise and stirred at RT for 24 hours. The reaction mixture was quenched with water, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate:
petroleum ether to afford the title compound as a yellow solid (10.5 g, 60%> yield). 1H-NMR (δ ppm, CDCls, 400 MHz): 7.85 (dd, J = 8.1,3 Hz, 1H), 7.58 (dd, J = 9.1, 4.1 Hz, 1H), 7.47-7.39 (m, 1H), 7.39-7.34 (m, 1H), 7.28-7.20 (m, 3H), 7.20-7.14 (m, 2H), 7.16-7.07 (m, 1H), 6.99-6.89 (m, 2H), 4.50-4.31 (m, 3H), 1.56 (d, J = 6.4 Hz, 3H).
Step-2: (R)-2-(l-(benzyloxy)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one obtained in Step-1 (10.5 g, 26.69 mmol) in dichloromethane (110 ml) was cooled to 0°C, aluminium chloride (5.35 g, 40.03 mmol) was added portionwise and stirred at RT for 6 hours. The reaction mixture was quenched with 2N HCl solution, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford intermediate 6 a yellow solid (6.1 g, 76% yield). Enantiomeric excess: 97.7%, enriched in the late eluting isomer (retention time: 11.12 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 7: 4-bromo-2-fluoro-l-isopropoxybenzene

To a solution of 4-bromo-3-fluorophenol (10 g, 52.35 mmol) in THF (100ml), isopropyl alcohol (4.8 ml, 62.62 mmol) and triphenylphosphine (20.6 g, 78.52 mmol) were added and heated to 45°C followed by diisopropylazodicarboxylate (15.4 ml, 78.52 mmol). The mixture was refluxed for 1 hour, concentrated and the residue was purified by column
chromatography with ethyl acetate: petroleum ether to afford the title compound as a colorless liquid (13.1 g, 99% yield), which was used without purification in the next step.
Intermediate 8: 2-f3-fluoro-4-isopropoxyphenyl)-4,4,5.,5-tetramethyl-l,3i2-dioxaborolane

Potassium acetate (10.52 g, 107.2 mmol) and bis(pinacolato)diboron (15 g, 58.96 mmol) were added to a solution of intermediate 7 (10.52 g, 107.2 mmol) in dioxane (125 ml), and the solution was degassed for 30 min. [l, -Bis(diphenylphosphino)ferrocene]dichloro palladium(II) CH2CI2 (4.4 g, 5.36 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12 hours, the reaction mixture was filtered through celite and concentrated. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow oil (13.9g, 99%) which was used without purification in the next step.
Intermediate 9: 3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-dlpyrimidin-4-amine

To a solution of 3-iodo-lH-pyrazolo[3,4-d]pyrimidin-4-amine (11.0 g, 42.14 mmol) in DMF (110 ml), ethanol (55 ml) and water (55 ml), intermediate 8 (23.4 g, 84.28 mmol) and sodium carbonate (13.3 g, 126.42 mmol) were added and degassed for 30 min.
Tetrakis(triphenylphosphine)palladium(0) (2.4 g, 2.10 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12 hours, the reaction mixture was filtered through celite, concentrated and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was triturated with diethyl ether, filtered and dried under vacuum to afford the title compound as light brown solid (3.2 g, 26% yield) which is used as such for the next step.
(RS)- 2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
To a solution of intermediate 9 (0.080 g, 0.293 mmol) in DMF (2 ml), potassium carbonate (0.081 g, 0.587 mmol) was added and stirred at RT for 10 min. To this mixture intermediate 1 (0.215 g, 0.587 mmol) was added and stirred for 12 hours. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with methanol: dichloromethane to afford the title compound as a pale yellow solid (0.045 g). MP: 175-177°C. 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 8.20 (s, 1H), 7.85 (dd, J = 81, 3.0 Hz, 1H), 7.48-7.33 (m, 5H), 7.14 (t, J= 8.3 Hz, 1H), 7.02 (m, 2H), 6.90 (m, 1H), 6.10 (q, J = 7.1 Hz, 1H), 5.42 (s, 2H), 4.64 (quintet, J = 6.0 Hz, 1H), 1.99 (d, J = 7.1 Hz, 3H), 1.42 (d, J= 6.1 Hz, 6H).
fS)-2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (“S-isomer”)
To a solution of intermediate 9 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 6 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.15 ml, 0.749 mmol) was added heated to 45°C. After 2 hours, the reaction mixture was quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as an off-white solid (0.049 g, 20 % yield). MP: 139-142°C. Mass: 571.7 (M+). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 10.64 min.). fR)-2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-ehromen-4-one
To a solution of intermediate 8 (0.284 g, 0.989 mmol) in THF (5.0 ml), intermediate 4 (0.250 g, 0.824 mmol) and tris(4-methoxy)phenylphosphine (0.435 g, 1.23 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.25 ml, 1.23 mmol) was added stirred at RT. After 12 hours, the reaction mixture was quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate :
petroleum ether to afford the title compound as an off-white solid (0.105 g, 22 % yield). MP: 145-148°C. Mass: 571.7 (M+). Enantiomeric excess: 95.4% as determined by HPLC on a chiralpak AD-H column, enriched in the late eluting isomer (retention time = 14.83 min.).
PATENT
WO 2014006572
http://www.google.com/patents/WO2014006572A1?cl=en
 B1 IS DESIRED
B1 IS DESIRED
(S)-2- (l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-6- fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (compound-B l)
Intermediate 11
[119] Intermediate 11: 4-bromo-2-fluoro-l-isopropoxybenzene:To a solution of 4-bromo-2- fluorophenol (lOg, 52.35 mmol) in THF (100ml), isopropyl alcohol (4.8ml, 62.62 mmol) and triphenylphosphine (20.6g, 78.52 mmol) were added and heated to 45 C followed by diisopropylazodicarboxylate (15.4ml, 78 52 mmol). The mixture was refluxed for lh, concentrated and the residue was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a colourless liquid (13. lg, 99%) which was used without purification in the next step. Intermediate 12
[120] Intermediate 12: 2-(3-fluoro-4-isopropoxyphenyl)-4,4,5,5-tetramethyl- 1,3,2- dioxaborolane: Potassium acetate (10.52 g, 107.2 mmol) and bis(pinacolato)diboron (15g, 58.96 mmol) were added to a solution of intermediate 11 (10.52 g, 107.2 mmol) in dioxane (125 ml), and the solution was degassed for 30 min. [1,1 ‘- Bis(diphenylphosphino)ferrocene]dichloro palladium(II).CH2Cl2 (4.4g, 5.36 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12h the reaction mixture was filtered through celite and concentrated. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow oil (13.9g, 99%) which was used without purification in the next step.
Intermediate 13
[121] Intermediate 13: 3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-4- amine: To a solution of 3-iodo-lH-pyrazolo[3,4-d]pyrimidin-4-amine (11.0 g, 42.14 mmol) in DMF 110 ml), ethanol (55 ml) and water (55 ml), intermediate 12 (23.4 g, 84.28 mmol) and sodium carbonate (13.3 g, 126.42 mmol) were added and degassed for 30 min. Tetrakis(triphenylphosphine)palladium(0) (2.4 g, 2.10 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12h, the reaction mixture was filtered though celite, concentrated and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was triturated with diethyl ether, filtered and dried under vacuum to afford the title compound as light brown solid (3.2 g, 26% yield) which is used as such for the next step.
Example Bl
(S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
[127] To a solution of intermediate 13 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 5 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate ( 0.15 ml, 0.749 mmol) was added heated to 45°C. After 2h, the reaction mixture was quenched with with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as an off-white solid (0.049 g, 20 %). MP: 139- 142°C. Mass : 571.7 (M H-NMR (δ ppm, CDC13, 400 MHz): 8.24 (s, 1H), 7.85 (dd, J = 8.2,3.1 Hz, 1H), 7.50-7.29 (m, 5H), 7.14 (t, J = 8.4 Hz, 1H), 7.02 (m, 2H), 6.92 (d, J = 8.4 Hz, 1H), 6.11 (q, J = 7.1 Hz, 1H), 5.40 (s, 2H), 4.66 (quintet, J = 6.1 Hz, 1H), 2.00 (d, J = 7.1Hz, 3H), 1.42 (d, J = 6.1 Hz, 6H). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 10.64min.).
PATENT
US 2014/0011819 describe the synthesis of TGR-1202 (Example B l)
http://www.google.co.in/patents/US20140011819
Example B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
-
To a solution of intermediate 13 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 5 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.15 ml, 0.749 mmol) was added heated to 45° C. After 2 h, the reaction mixture was quenched with with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate:petroleum ether to afford the title compound as an off-white solid (0.049 g, 20%). MP: 139-142° C. Mass: 571.7 (M+).1H-NMR (δ ppm, CDCl3, 400 MHz): 8.24 (s, 1H), 7.85 (dd, J=8.2, 3.1 Hz, 1H), 7.50-7.29 (m, 5H), 7.14 (t, J=8.4 Hz, 1H), 7.02 (m, 2H), 6.92 (d, J=8.4 Hz, 1H), 6.11 (q, J=7.1 Hz, 1H), 5.40 (s, 2H), 4.66 (quintet, J=6.1 Hz, 1H), 2.00 (d, J=7.1 Hz, 3H), 1.42 (d, J=6.1 Hz, 6H). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=10.64 min)
4-Methylbenzenesulfonate Salt of Compound B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one 4-methylbenzenesulfonate
-

-
(S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one 4-methylbenzenesulfonate: To (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (22.7 g, 39.69 mmol) in isopropanol (600 ml), p-toluenesulphonic acid (8.30 g, 43.66 mmol) was added and refluxed for 1 h. The reaction mixture was concentrated, co-distilled with petroleum ether and dried. To the residue water (300 ml) was added and stirred for 30 min. The solid was filtered, washed with petroleum ether and dried under vacuum to afford the title compound as off-white solid (28.2 g, 95%). MP: 138-141° C. 1H-NMR (δ ppm, CDCl3, 400 MHz): 8.11 (s, 1H), 7.85 (dd, J=8.0, 3.0 Hz, 1H), 7.80 (d, J=8.2 Hz, 2H), 7.51 (dd, J=9.3, 4.3 Hz, 1H), 7.45 (dd, J=7.5, 3.1 Hz, 1H), 7.42-7.31 (m, 3H), 7.29 (m, 2H), 7.22 (d, J=8.0 Hz, 2H), 7.16 (t, J=8.3 Hz, 1H), 7.08 (dt, J=8.5, 2.5 Hz, 1H), 6.97 (br s, 1H), 6.88 (br s, 1H), 6.11 (q, J=7.2 Hz, 1H), 4.67 (quintet, J=6.0 Hz, 1H), 2.36 (s, 3H), 2.03 (d, J=7.1 Hz, 3H), 1.43 (d, J=6.0 Hz, 6H). Mass: 572.4 (M++1-PTSA). Enantiomeric excess: 93.4% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=12.35 min.)

Sulphate Salt of Compound B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one sulfate
-

-
(S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one sulphate: To (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (15.0 g, 26.24 mmol) in isopropanol (600 ml) was cooled to 0° C. To this Sulphuric acid (2.83 g, 28.86 mmol) was added and stirred at room temperature for 24 h. The reaction mass was filtered and washed with petroleum ether and dried under vacuum. To the solid, water (150 ml) was added and stirred for 30 min. The solid was filtered, washed with petroleum ether and dried under vacuum to afford the title compound as off-white solid (13.5 g, 76%). MP: 125-127° C. 1H-NMR (δ ppm, CDCl3, 400 MHz): 8.11 (s, 1H), 7.85 (dd, J=8.0, 3.0 Hz, 1H), 7.51 (dd, J=9.2, 4.2 Hz, 1H), 7.45-7.31 (m, 3H), 7.29 (m, 1H), 7.15 (t, J=8.3 Hz, 1H), 7.08 (dt, J=8.5, 2.4 Hz, 1H), 6.96 (br s, 1H), 6.88 (br s, 1H), 6.09 (q, J=7.1 Hz, 1H), 4.676 (quintet, J=6.1 Hz, 1H), 2.01 (d, J=7.1 Hz, 3H), 1.42 (d, J=6.1 Hz, 6H). Mass: 572.2 (M++1-H2SO4). Enantiomeric excess: 89.6% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=12.08 min.)
-
Various other acid addition salts of compound B1 were prepared as provided in Table 1.
-
TABLE 1 Melting Point Acid Method of preparation (° C.) Hydro- Compound B1 (1 eq.) dissolved in THF, 130-132 chloric excess HCl/Et2O was added, the clear acid solution obtained was evaporated completely. The residue obtained was washed with water. p- Compound B1 (1 eq.) dissolved in 138-141° C. Toluene- isopropyl alcohol (IPA), refluxed for sulfonic 30 min., acid (1.1 eq.) in IPA was added, acid the clear solution obtained was evaporated completely. The residue obtained was washed with water. Benzene- Compound B1 (1 eq.) dissolved in IPA, 170-172 sulphonic refluxed for 30 min., acid(1.1 eq.) in IPA acid was added, the clear solution not obtained, the residue was evaporated completely and was washed with water. Maleic Compound B1 (1 eq.) dissolved in IPA, 107-109 acid refluxed for 30 min., acid (1.1 eq.) in IPA was added, the clear solution not obtained, the residue was evaporated completely and was washed with water. Camphor Compound B1 (1 eq.) dissolved in IPA, 120-121 sulfonic refluxed for 30 min., acid (1.1 eq.) in IPA acid was added, the clear solution not obtained, the residue was evaporated completely and was washed with water. Sulphuric Compound B1 (1 eq.) dissolved in IPA, 125-127 acid refluxed for 30 min., acid(1.1 eq.) in IPA was added, the clear solution obtained was evaporated completely. The residue obtained was washed with water.

REFERENCES
WO 2014/006572 and U.S. Patent Publication No. 2014/0011819,
http://www.tgtherapeutics.com/O’ConnorTGR202Single%20AgentEHA&Lugano2015.pdf
-
TGR-1202: Phase I/II started 09/28/2015
Week in Review, Clinical StatusRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) … -
Ublituximab: Phase I/II started 09/28/2015
Week in Review, Clinical StatusLFB S.A., Les Ulis, France TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: Ublituximab (TGTX-1101, TG-1101, LFB-R603) Business: Cancer Molecular target: CD20 Description: Glycoengineered mAb against CD20 … -
The Daily Extra, Company NewsTG Therapeutics Inc. (NASDAQ:TGTX) rose $2.65 (23%) to $14.37 after the company said it received an SPA from FDA for the Phase III UNITY-CLL trial of ublituximab (TG-1101) in combination with TGR-1202 to treat chronic …
-
Targets & Mechanisms: The battle for IRAK 04/23/2015
Nimbus, Aurigene and TG Therapeutics are chasing IRAK4 inhibitors for cancerBC Innovations, Targets & MechanismsNow that Nimbus has put IRAK4 on the map for B cell lymphoma, several companies are closing in with their own inhibitors, and they’re all on track for IND-enabling studies this year. -
TGR-1202: Additional Phase I/II data 01/26/2015
Week in Review, Clinical ResultsRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) … -
Ublituximab: Additional Phase I/II data 01/26/2015
Week in Review, Clinical ResultsLFB S.A., Les Ulis, France TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Ildong Pharmaceutical Co. Ltd. (KSE:000230), Seoul, South Korea Product: Ublituximab (TGTX-1101, TG-1101, LFB-R603) Business: Cancer … -
TGR-1202: Phase I started 12/15/2014
Week in Review, Clinical StatusRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) … -
Rhizen, TG Therapeutics deal 12/08/2014
Week in Review, DealsRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Business: Cancer TG Therapeutics exercised an option under a 2012 deal to license exclusive, worldwide …
| Patent | Submitted | Granted |
|---|---|---|
| NOVEL SELECTIVE PI3K DELTA INHIBITORS [US2014011819] | 2013-07-02 | 2014-01-09 |
| Treatment Of Cancers Using PI3 Kinase Isoform Modulators [US2014377258] | 2014-05-30 | 2014-12-25 |
////////Umbralisib
CC(C)OC1=C(C=C(C=C1)C2=NN(C3=C2C(=NC=N3)N)C(C)C4=C(C(=O)C5=C(O4)C=CC(=C5)F)C6=CC(=CC=C6)F)F
