

| Patent | Submitted | Granted |
|---|---|---|
| Compound useful for the treatment of degenerative and inflammatory diseases [US8088764] | 2010-12-30 | 2012-01-03 |
| NOVEL COMPOUNDS USEFUL FOR THE TREATMENT OF DEGENERATIVE AND INFLAMMATORY DISEASES [US2011190260] | 2011-08-04 |
Home » Posts tagged 'rheumatoid arthritis' (Page 2)
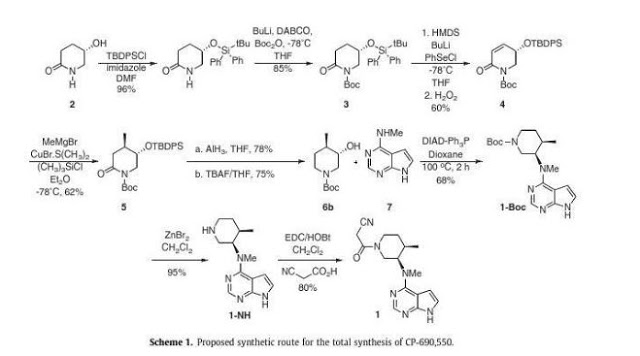
Tag Archives: rheumatoid arthritis
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
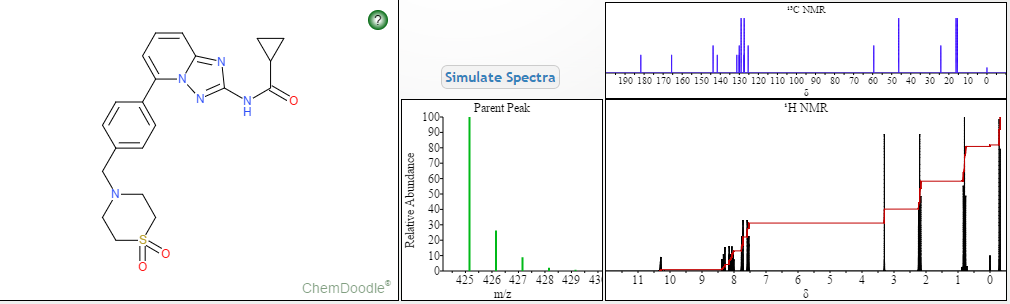
Filgotinib

Filgotinib
EU APPROVED 2020/9/24, JYSELECA
JAPAN APPROVED2020/9/25
- C21H23N5O3S
- MW425.504
- Elemental Analysis: C, 59.28; H, 5.45; N, 16.46; O, 11.28; S, 7.54
1206161-97-8
Cyclopropanecarboxamide, N-[5-[4-[(1,1-dioxido-4-thiomorpholinyl)methyl]phenyl][1,2,4]triazolo[1,5-a]pyridin-2-yl]-
G146034
GLPG0634
N-(5-(4-((1,1-dioxidothiomorpholino)methyl)phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl)cyclopropanecarboxamide
Galapagos Nv INNOVATOR
PHASE 3, Crohn’s disease, Rheumatoid arthritis, Ulcerative colitis
Filgotinib is an orally available inhibitor of JAK1/JAK2 and TYK2 in phase III clinical development at Galapagos and Gilead for the treatment of rheumatoid arthritis, moderate or severe Crohn’s disease and ulcerative colitis
IL-6 antagonist; Jak1 tyrosine kinase inhibitor; Tyk2 tyrosine kinase inhibitor; Jak3 tyrosine kinase inhibitor; Jak2 tyrosine kinase inhibitor
Autoimmune disease; Cancer; Colitis; Crohns disease; Inflammatory disease; Neoplasm; Rheumatoid arthritis; Transplant rejection
In 2017, orphan drug designation was assigned to the compound in the U.S. for the treatment of pediatric Crohn’s disease and pediatric ulcerative colitis.
GlaxoSmithKline had been developing filgotinib preclinically for the treatment of rheumatoid arthritis pursuant to a license; however, in 2010, the compound was re-acquired by Galapagos. In 2012, the product was licensed to Abbott for development and marketing. In January 2013, Abbott spun-off its research-based pharmaceutical business into a newly-formed company AbbVie. The license agreement between Galapagos and Abbott was terminated in September 2015, Galapagos regaining all rights to the product. The same year, Galapagos and Gilead entered into a global partnership and Gilead obtained the global rights of codevelopment and commercialization for the treatment of inflammatory diseases
Filgotinib (GLPG0634), by the Belgian biotech company Galápagos NV, is a drug which is currently under investigation for the treatment of rheumatoid arthritis and Crohn’s disease.
Filgotinib (GLPG0634) is an orally-available, selective inhibitor of JAK1 (Janus kinase 1) for the treatment of rheumatoid arthritis and potentially other inflammatory diseases. Filgotinib (GLPG0634) dose-dependently inhibited Th1 and Th2 differentiation and to a lesser extent the differentiation of Th17 cells in vitro. GLPG0634 was well exposed in rodents upon oral dosing, and exposure levels correlated with repression of Mx2 expression in leukocytes. The JAK1 selective inhibitor GLPG0634 (Filgotinib) is a promising novel therapeutic with potential for oral treatment of rheumatoid arthritis and possibly other immune-inflammatory diseases. Filgotinib (GLPG0634) is currently in a Phase 2 study in Crohn’s disease.

Mechanism of action
Filgotinib is a Janus kinase inhibitor with selectivity for subtype JAK1 of this enzyme. It is considered a promising agent as it inhibits JAK1 selectively. Less selective JAK inhibitors (e.g. tofacitinib) are already being marketed. They show long-term efficacy in the treatment of various inflammatory diseases. However, their lack of selectivity leads to dose-limiting side effects.[1] It is thought that inhibition of all JAK isoenzymes is beneficial in rheumatoid arthritis. However, pan-JAK inhibition might also lead to unwanted side effects that might not outweigh its benefits. This is the rationale for the development of newer and more selective inhibitors like filgotinib.
The signal transmission of large numbers of proinflammatory cytokines is dependent on JAK1. Inhibition of JAK2 may also contribute to the efficacy against RA. Nonetheless it is thought that JAK2 inhibition might lead to anemia and thrombopenia by interference witherythropoietin and thrombopoietin and granulocyte-macrophage colony-stimulating factor. Therefore one might prefer to choose a more selective JAK1 inhibitor as a primary therapeutic option. Filgotinib exerts a 30-fold selectivity for JAK1 compared to JAK2.[2] It is however still to be seen to what extent JAK2 inhibition should be avoided.
Novel crystalline forms of filgotinib salts, particularly hydrochloride salt, useful for treating JAK-mediated diseases eg inflammatory diseases, autoimmune diseases, proliferative diseases, allergy and transplant rejection. Galapagos and licensee AbbVie are developing filgotinib, a selective JAK-1 inhibitor, for treating rheumatoid arthritis (RA) and Crohn’s disease (CD). In August 2015, the drug was reported to be in phase 2 clinical development for treating RA and CD. The drug is also being investigated for the treatment of colitis and was discovered as part of the company’s arthritis alliance with GSK; however in August 2010 Galapagos reacquired the full rights. See WO2013189771, claiming use of filgotinib analog for treating inflammatory diseases. Also see WO2010010190 (co-assigned with GSK and Abbott) and WO2010149769 (assigned to Galapagos) claiming filgotinib, generically and specifically, respectively.
Clinical trials and approval
The efficacy of filgotinib is currently studied in a phase2b program (DARWIN trial 1, 2) with involvement of 886 rheumatoid arthritis patients and 180 Crohn’s disease patients.
Phase 1 study
It was shown in phase 1 studies that the pharmacokinetics of filgotinib metabolism is independent of hepatic CYP450 enzymatic degradation. The drug metabolism is however mediated by carboxylesterases. There is no interference reported with the metabolism of methotrexate nor with any of the investigated transport proteins.[3]
Phase 2 study: Proof of concept (2011)
In november 2011 Galápagos released the results of their phase 2 study (identification: NCT01384422, Eudract: 2010-022953-40) in which 36 patients were treated who showed a suboptimal clinical response to methotrexate treatment. Three groups of twelve patients were treated either with 200 mg filgotinib in a single dose, 200 mg divided in two doses or placebo. The primary end-point was the ACR20 score, which monitors improvements in the symptomatology of the patient. After the scheduled 4 weeks of treatment, 83% of the respondents showed an improved ACR20-score. Half of the treated patients showed a complete (or near complete) remission of the disease. There were no reports ofanemia nor changes in lipidemia. The company stated in their press release that filgotinib is the first selective JAK1 inhibitor that shows clinical efficacy. As a result of this study, the company stated that “GLPG0634 shows one of the highest initial response rates ever reported for rheumatoid arthritis treatments”.[4]
DARWIN 1 trial
The DARWIN 1 trial is a 24 week double blind placebo-controlled trial with 599 rheumatoid arthritis patients enrolled. All participants have moderate to severe RA and showed an insufficient response to standard methotrexate treatment. The trial compares three dosages of filgotinib as a once or twice per day regimen. During the trial all participants remain on their methotrexate treatment. According to the company, the results of this trial are expected in July 2015.[5]
DARWIN 2 trial
The DARWIN 2 trial is a double blind placebo-controlled trial with 280 rheumatoid arthritis patients enrolled who show an insufficient response to standard methotrexate treatment. This trial, in contrast to the previous DARWIN 1 trial, methotrexate is discontinued. Therefore, this trial investigates filgotinib as a monotherapy.[6] The recruitment of DARWIN trial 2b ended in november 2014.[7] Preliminary results are expected in the second quarter of 2015 and a full completion of the study is expected in the third quarter of 2015.
DARWIN 3 trial
Patients who complete DARWIN 1 and 2 will be eligible for DARWIN 3.
COSY PREDICT
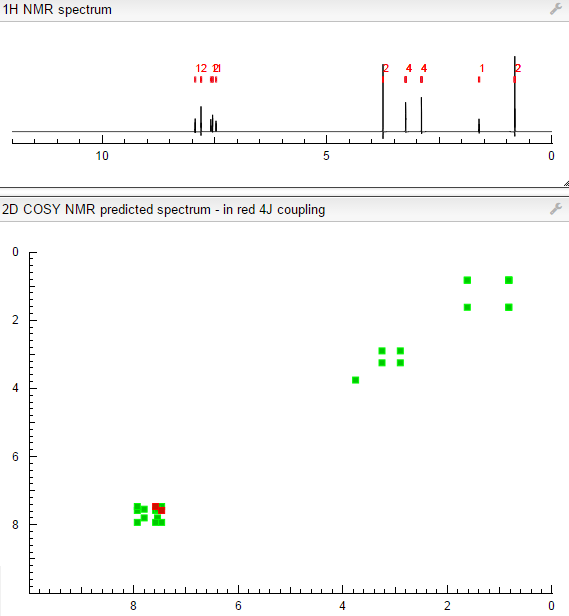
Time line
- june 2011: results of first phase 2 trial
- november 2014: initiation of DARWIN 1 and 2 trials
- april 2015: expected date of DARWIN 1 trial results
- june 2015: expected date of DARWIN 2 trial results
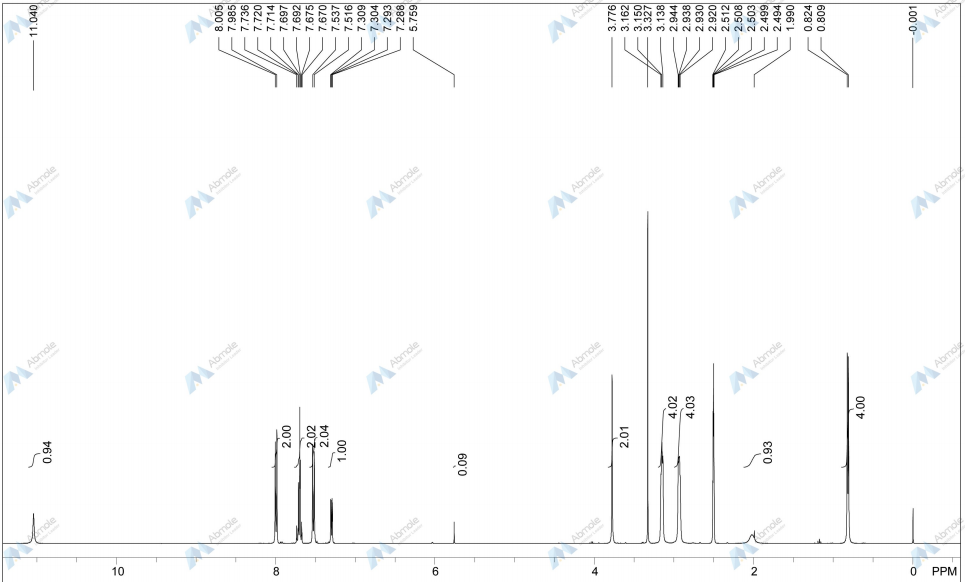
NMR FROM NET….ABMOLE, DMSOD6
NMR MEDKOO DMSOD6

CHEMIETEK
1H NMR PREDICT


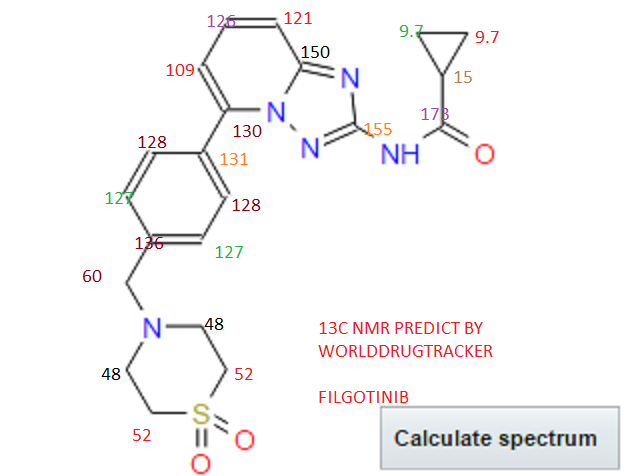
13C NMR PREDICT
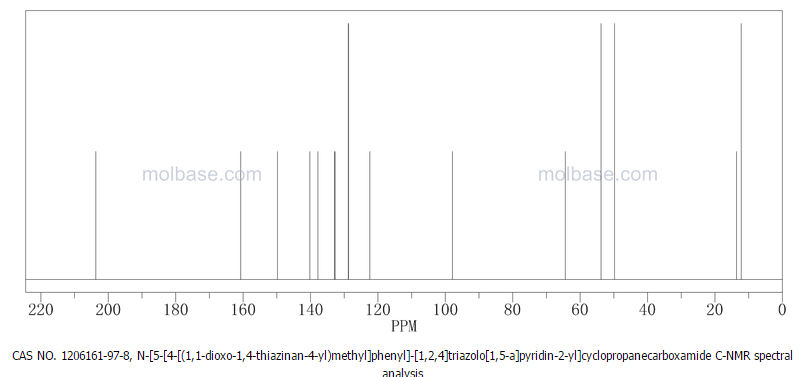

……………………
MORE PREDICTS
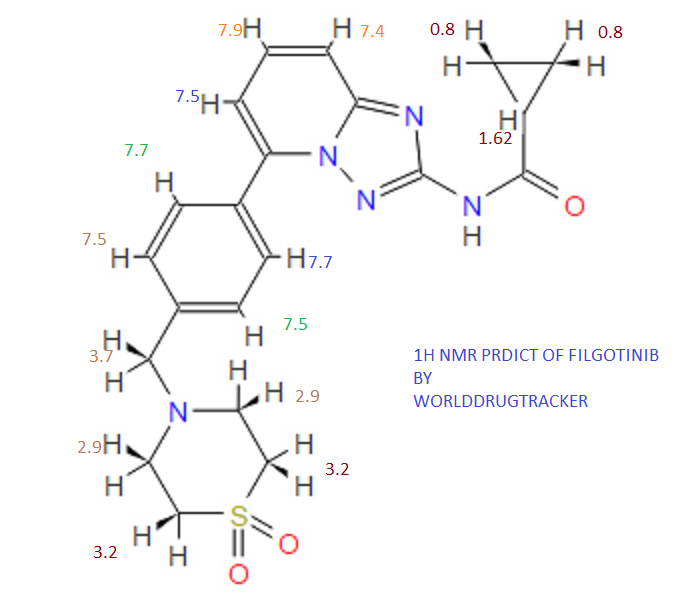
1H NMR PREDICT
13C NMR PREDICT
PRODUCT PATENT
http://www.google.com/patents/WO2010149769A1?cl=en
| Applicants: | GALAPAGOS NV [BE/BE]; Generaal De Wittelaan L11/A3 B-2800 Mechelen (BE) (For All Designated States Except US). MENET, Christel Jeanne Marie [FR/BE]; (BE) (For US Only). SMITS, Koen Kurt [BE/BE]; (BE) (For US Only) |
| Inventors: | MENET, Christel Jeanne Marie; (BE). SMITS, Koen Kurt; (BE) |
| International Filing Date: | 25.06.2010 |
ESTIMATED EXP 2030
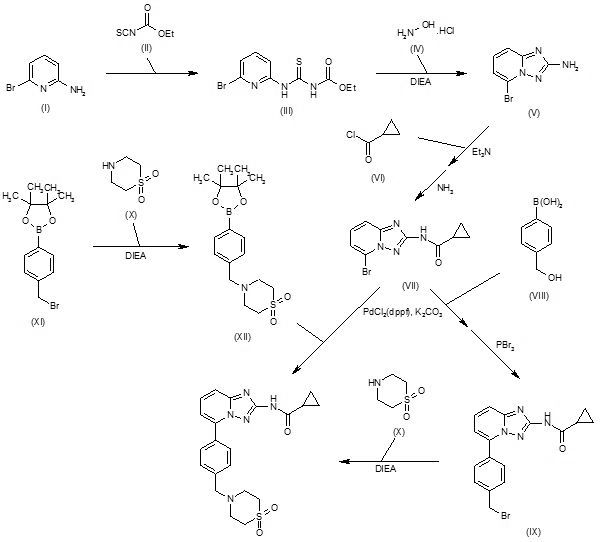
Condensation of 2-amino-6-bromopyridine (I) with ethoxycarbonyl isothiocyanate (II) in CH2Cl2 gives 1-(6-bromopyridin-2-yl)-3-carboethoxythiourea (III), which upon cyclization with hydroxylamine hydrochloride (IV) in the presence of DIEA in EtOH/MeOH yields 2-amino-5-bromo[1,2,4]triazolo[1,5-a]pyridine (V). N-Acylation of amine (V) with cyclopropanecarbonyl chloride (VI) using Et3N in acetonitrile, and subsequent treatment with methanolic ammonia furnishes the carboxamide (VII) (1-3), which upon Suzuki coupling with 4-(hydroxymethyl)phenylboronic acid (VIII) in the presence of PdCl2(dppf) and K2CO3 in dioxane/H2O at 90 °C, followed by bromination with PBr3 in CHCl3 affords intermediate (IX). Condensation of benzyl bromide derivative (IX) with thiomorpholine-1,1-dioxide (X) using DIEA in CH2Cl2/MeOH yields filgotinib (1,2). Alternatively, condensation of (4-bromomethylphenyl)-4,4,5,5-tetramethyl-[1,3,2]dioxaborolane (XI) with thiomorpholine 1,1-dioxide (X) in the presence of DIEA in CH2Cl2/MeOH gives intermediate (XII), which undergoes Suzuki coupling with aryl bromide (VII) in the presence of PdCl2(dppf) and K2CO3 in dioxane/H2O at 90 °C to afford the target filgotinib
The present invention is based on the discovery that the compound of the invention is able to act as an inhibitor of JAK and that it is useful for the treatment of inflammatory conditions, autoimmune diseases, proliferative diseases, transplantation rejection, diseases involving impairment of cartilage turnover, congenital cartilage malformations, and/or diseases associated with hypersecretion of IL6. In a specific aspect the compound is an inhibitor of JAKl and JAK2. The present invention also provides methods for the production of this compound, a pharmaceutical composition comprising this compound and methods for treating inflammatory conditions, autoimmune diseases, proliferative diseases, transplantation rejection, diseases involving impairment of cartilage turnover, congenital cartilage malformations, and/or diseases associated with hypersecretion of IL6 by administering the compound of the invention.
Accordingly, in a first aspect of the invention, a compound of the invention is provided having a formula (I):

[0017] The compound of the invention is a novel inhibitor of JAK that appears to exhibit a dramatically improved in vivo potency as compared to structurally similar compounds. In a particular embodiment the compound of the invention is an inhibitor of JAKl and JAK2. In particular it appears to exhibit this increase in potency at lower in vivo exposure levels compared to structurally similar compounds. The use of a compound with these improvements is expected to result in a lower dosage requirement (and therefore an improved dosing schedule).
General Synthetic Method Scheme 1

1. RCOCI, Et3N 2. NH3 / MeOH CH3CN, 20 0C 2O 0C

wherein Ar represents phenyl-Ll-heterocycloalkyl, where Ll is a bond, -CH2– or -CO- and the heterocycloalkyl group is optionally substituted.
General
1.1.1 l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2)

(2)
[00117] To a solution of 2-amino-6-bromopyridine (1) (253.8 g, 1.467 mol) in DCM (2.5 L) cooled to 5 0C is added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction mixture is then allowed to warm to room temp. (20 0C) and stirred for 16 h. Evaporation in vacuo gives a solid which may be collected by filtration, thoroughly washed with petrol (3×600 mL) and air-dried to afford (2). The thiourea may be used as such for the next step without any purification. 1H (400 MHz, CDCl3) δ 12.03 (IH, br s, NH), 8.81 (IH, d, J 7.8 Hz, H-3), 8.15 (IH, br s, NH), 7.60 (IH, t, J 8.0 Hz, H-4), 7.32 (IH, dd, J 7.7 and 0.6 Hz, H-5), 4.31 (2H, q, J 7.1 Hz, CH2), 1.35 (3H, t, J 7.1 Hz, CH3).
7.7.2 5-Bromo-[l, 2, 4]triazolo[l, 5-a]pyridin-2-ylamine (3)

[00118] To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH
(1 :1, 900 mL) is added N,N-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture is stirred at room temp. (20 0C) for 1 h. l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) is then added and the mixture slowly heated to reflux (Note: bleach scrubber is required to quench H2S evolved). After 3 h at reflux, the mixture is allowed to cool and filtered to collect the precipitated solid. Further product is collected by evaporation in vacuo of the filtrate, addition Of H2O (250 mL) and filtration. The combined solids are washed successively with H2O (250 mL), EtOH/MeOH (1 : 1, 250 mL) and Et2O (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound may be used as such for the next step without any purification. 1H (400 MHz, DMSO-t/β) δ 7.43-7.34 (2H, m, 2 x aromatic-H), 7.24 (IH, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1 :1, M+H+, 100%).
7.7.3 General procedure for mono-acylation to afford intermediate (4):

[00119] To a solution of the 2-amino-triazolopyridine (3) (7.10 g, 33.3 mmol) in dry CH3CN
(150 mL) at 5 0C is added Et3N (11.6 mL, 83.3 mmol) followed by cyclopropanecarbonyl chloride (83.3 mmol). The reaction mixture is then allowed to warm to ambient temperature and stirred until all starting material (3) is consumed. If required, further Et3N (4.64 mL, 33.3 mmol) and cyclopropanecarbonyl chloride (33.3 mmol) is added to ensure complete reaction. Following solvent evaporation in vacuo the resultant residue is treated with 7 N methanolic ammonia solution (50 mL) and stirred at ambient temp, (for 1-16 h) to hydro lyse any bis-acylated product. Product isolation is made by removal of volatiles in vacuo followed by trituration with Et2O (50 mL). The solids are collected by filtration, washed with H2O (2x50mL), acetone (50 mL) and Et2O (50 mL), then dried in vacuo to give the required bromo intermediate (4).
Method A
Preparation of compounds of the invention via Suzuki coupling (5):
[00120] An appropriate boronic acid (2eq.) is added to a solution of bromo intermediate (4) in
1 ,4-dioxane/water (5:1). K2CO3 (2 eq.) and PdCl2dppf (5%) are added to the solution. The resulting mixture is then heated in a microwave at 140 0C for 30 min (this reaction can also be carried out by traditional heating in an oil bath at 900C for 16h under N2). Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhyd. MgSθ4 and evaporated in vacuo. The final compound is obtained after purification by flash chromatography or preparative HPLC. HPLC: Waters
XBridge Prep Cl 8 5μm ODB 19mm ID x 100mm L (Part No.186002978). All the methods are using
MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Method B

Bl. 4 4-[2-(Cyclopropanecarbonyl-amino)-[ 1 , 2, 4]triazolo[l, 5-a] pyridin-5-yl] -benzoyl chloride

[00121] 2 Drops of DMF are added to a solution of 4-[2-(cyclopropanecarbonyl-amino)- [l,2,4]triazolo[l,5-a]pyridin-5-yl]-benzoic acid (1 eq) obtained by Method A using 4-carboxyphenylboronic acid in DCM under N2 atmosphere. Then oxalyl chloride (2 eq) is added dropwise to this resulting solution (gas release). The mixture is stirred at room temperature for 2 hours. After completion of the reaction by LCMS, the solvent is removed. The crude acid chloride is used without further purification in next step.
B2. Amide formation (General Method)

[00122] An appropriate amine (1.1 eq) and Et3N (5 eq) are dissolved in DCM under N2 atmosphere and cooled at 00C. The acid chloride (Bl, 1 eq) dissolved in DCM is added dropwise to this solution. The reaction is stirred at room temperature for 16 h. After this time, reaction is complete. The compound is extracted with EtOAc and water, washed with brine and dried over anhyd. MgSO4. Organic layers are filtered and evaporated. The final compound is isolated by preparative HPLC. Preparative HPLC: Waters XBridge Prep C18 5μm ODB 19mm ID x 100mm L (Part No.186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Method C

Wherein R3a or R3b together with the nitrogen atom to which they are attached, may form a heterocycloalkyl.
Reductive alkylation (general method)
[00123] An appropriate amine (2 eq.), cyclopropanecarboxylic acid (for example cyclopropanecarboxylic acid [5-(4-formyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridine-2-yl]-amide) prepared by method A (1 eq.) and Ti(OPr)4 are mixed and stirred at room temperature for 3 hrs. The mixture is diluted in ethanol and Na(CN)BH3 (leq.) is added. The resulting solution is stirred at room temperature for 16 hrs. The mixture is diluted in water and filtered. The filtrate is washed with ethanol. The combined solvent phases are evaporated under vacuum. The final compound is isolated by preparative HPLC.
Method D 
wherein R1 and R2 together with the Nitrogen atom to which they are attached, may form a heterocycloalkyl.
Reaction ofalkylation

[00124] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (leq) and Et3N (2 eq) (or AgCO3) are dissolved in DCM/MeOH (4:1 v:v) under N2 and an amine (2 eq) is added dropwise. The resulting solution is stirred at room temperature for 16h. After this time, the reaction is complete. The solvent is evaporated. The compound is extracted with EtOAc and water, washed with brine and dried over anhyd. MgSθ4. Organic layers are filtered and evaporated. The final compound is isolated by flash chromatography.
Suzuki coupling

[00125] The obtained boronic acid (2eq.) is added to a solution of cyclopropanecarboxylic acid
(5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide (4) in 1 ,4-dioxane/water (5:1). K2CO3 (2 eq.) and PdCl2dppf (5%) are added to the solution. The resulting mixture is then heated in a microwave at 140 0C for 30 min (This reaction can also be carried out by traditional heating in an oil bath at 900C for 16h under N2). Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhyd. MgSθ4 and evaporated in vacuo. The final compound is obtained after purification by flash chromatography or preparative HPLC. HPLC: Waters XBridge Prep C18 5μm ODB 19mm ID x 100mm L (Part No.186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Synthesis of the compound of the invention and comparative examples
Compound l(the compound of the invention)
Step 1:

[00126] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (leq) and DIPEA
(2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (2 eq) was added portionwise. The resulting solution was stirred at room temperature for 16h. After this time, the reaction was complete. The solvent was evaporated. The compound was extracted with EtOAc and water, washed with brine and dried over anhyd. MgS O4. Organic layers were filtered and evaporated. The final compound was isolated without further purification.
Step 2: Suzuki coupling

[00127] 4-[4-(4,4,5,5-Tetramethyl-[l,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-l,l-dioxide
(l.leq.) was added to a solution of cyclopropanecarboxylic acid (5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 900C for 16h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSθ4 and evaporated in vacuo. The final compound was obtained after purification by flash chromatography.
[00128] Alternatively, after completion of the reaction, a palladium scavenger such as 1,2-bis(diphenylphosphino)ethane, is added, the reaction mixture is allowed to cooled down and a filtration is performed. The filter cake is reslurried in a suitable solvent (e.g. acetone), the solid is separated by filtration, washed with more acetone, and dried. The resulting solid is resuspended in water, aqueous HCl is added, and after stirring at RT, the resulting solution is filtered on celite (Celpure P300). Aqueous NaOH is then added to the filtrate, and the resulting suspension is stirred at RT, the solid is separated by filtration, washed with water and dried by suction. Finally the cake is re-solubilised in a mixture of THF/H2O, treated with a palladium scavenger (e.g. SMOPEX 234) at 500C, the suspension is filtered, the organic solvents are removed by evaporation, and the resulting slurry is washed with water and methanol, dried and sieved, to obtain the title compound as a free base.
Alternative route to Compound l(the compound of the invention):
Step 1:

[00129] 4-(Hydroxymethyl)phenylboronic acid (l.leq.) was added to a s o luti o n o f cyclopropanecarboxylic acid (5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 900C for 16h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSθ4 and evaporated in vacuo. The resulting mixture was used without further purification.
Step 2:

[00130] To a solution of cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)- [l,2,4]triazolo[l,5-a]pyridin-2-yl]-amide (1.0 eq) in chloroform was slowly added phosphorus tribromide (1.0 equiv.). The reaction mixture was stirred at room temperature for 20 hours, quenched with ice and water (20 mL) and extracted with dichloromethane. The organic layer was dried over anhyd. MgSθ4, filtered and concentrated to dryness. The resulting white residue was triturated in dichloromethane/diethyl ether 2:1 to afford the expected product as a white solid.
Step 3:

[00131] Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridin- 2-yl]-amide (leq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (1.1 eq) was added dropwise. The resulting solution was stirred at room temperature for 16h. After this time, the reaction was complete. The solvent was evaporated. The compound was dissolved in DCM, washed with water and dried over anhyd. MgSO^ Organic layers were filtered and evaporated. The final compound was isolated by column chromatography using EtOAc to afford the desired product.
PATENT
WO 2010010190
WO 2013173506
WO 2013189771
WO 2015117980
WO 2015117981
POLYMORPH
CN 105061420
https://encrypted.google.com/patents/CN105061420A?cl=en
JAK inhibitor N-(5-(4-(1,1-dioxothiomorpholinyl)methyl)phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl)cyclopropanecarboxamide, and methods for preparing the four crystal forms, wherein the four crystal forms respectively are a crystal form H1, a crystal form H2, a crystal form H3 and a crystal form H4,
POLYMORPH
E CRYSTAL
CN 105111206
D CRYSTAL
CN 105111207
H CRYSYAL
CN 105198876
G
CN 105198877
F CN 105198878
C CN 105198880
POLYMORPH
WO 2016105453
POLYMORPH
Preparation of solid forms of filgotinib free base
WO 2017012773
The present invention relates to cryst. filgotinib free base (I), a method of its prepn. and a pharmaceutical compn. comprising the same. Thus, reacting the compd. II with thiomorpholine dioxide afforded 92% I (purity of 98.6%) which was then converted into its hydrochloride salt. Pharmaceutical compn. comprising compd. I was disclosed.
POLYMORPH
CN 105669669
The present invention provides a crystal form A, B, D, G and M of N-[5-[4-[(1,1-dioxido-4-thiomorpholinyl)methyl]phenyl][1,2,4]triazolo[1,5-a]pyridin-2-yl]cyclopropanecarboxamide hydrochloride.
PAPER
Future Medicinal Chemistry (2015), 7(2), 203-235. | Language: English, Database: CAPLUSA review. The discovery of the JAK-STAT pathway was a landmark in cell biol. The identification of these pathways has changed the landscape of treatment of rheumatoid arthritis and other autoimmune diseases. The two first (unselective) JAK inhibitors have recently been approved by the US FDA for the treatment of myelofibrosis and rheumatoid arthritis and many other JAK inhibitors are currently in clin. development or at the discovery stage. Research groups have demonstrated the different roles of JAK member and the therapeutic potential of targeting them selectively. ………..
https://www.future-science.com/doi/10.4155/fmc.14.149
PAPER
Journal of Pharmaceutical Sciences (Philadelphia, PA, United States) (2018), 107(6), 1624-1632.
PATENT
US2010/331319 A1, ; Page/Page column 13-14
http://www.google.com/patents/US20100331319
Synthetic Preparation of the Compound of the Invention and Comparative Examples
The compound of the invention and the comparative examples can be produced according to the following scheme.

wherein Ar represents phenyl-L1-heterocycloalkyl, where L1 is a bond, —CH2— or —CO— and the heterocycloalkyl group is optionally substituted.
General 1.1.1 1-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2)

To a solution of 2-amino-6-bromopyridine (1) (253.8 g, 1.467 mol) in DCM (2.5 L) cooled to 5° C. is added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction mixture is then allowed to warm to room temp. (20° C.) and stirred for 16 h. Evaporation in vacuo gives a solid which may be collected by filtration, thoroughly washed with petrol (3×600 mL) and air-dried to afford (2). The thiourea may be used as such for the next step without any purification. 1H (400 MHz, CDCl3) δ 12.03 (1H, br s, NH), 8.81 (1H, d, J=7.8 Hz, H-3), 8.15 (1H, br s, NH), 7.60 (1H, t, J=8.0 Hz, H-4), 7.32 (1H, dd, J 7.7 and 0.6 Hz, H-5), 4.31 (2H, q, J 7.1 Hz, CH2), 1.35 (3H, t, J 7.1 Hz, CH3).
1.1.2 5-Bromo-[1,2,4]triazolo[1,5-a]pyridin-2-ylamine (3)

To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH (1:1, 900 mL) is added N,N-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture is stirred at room temp. (20° C.) for 1 h. 1-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) is then added and the mixture slowly heated to reflux (Note: bleach scrubber is required to quench H2S evolved). After 3 h at reflux, the mixture is allowed to cool and filtered to collect the precipitated solid. Further product is collected by evaporation in vacuo of the filtrate, addition of H2O (250 mL) and filtration. The combined solids are washed successively with H2O (250 mL), EtOH/MeOH (1:1, 250 mL) and Et2O (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound may be used as such for the next step without any purification. 1H (400 MHz, DMSO-d6) δ 7.43-7.34 (2H, m, 2×aromatic-H), 7.24 (1H, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1:1, M+H+, 100%).
1.1.3 General Procedure for Mono-Acylation to Afford Intermediate (4)

To a solution of the 2-amino-triazolopyridine (3) (7.10 g, 33.3 mmol) in dry CH3CN (150 mL) at 5° C. is added Et3N (11.6 mL, 83.3 mmol) followed by cyclopropanecarbonyl chloride (83.3 mmol). The reaction mixture is then allowed to warm to ambient temperature and stirred until all starting material (3) is consumed. If required, further Et3N (4.64 mL, 33.3 mmol) and cyclopropanecarbonyl chloride (33.3 mmol) is added to ensure complete reaction. Following solvent evaporation in vacuo the resultant residue is treated with 7 N methanolic ammonia solution (50 mL) and stirred at ambient temp. (for 1-16 h) to hydrolyse any bis-acylated product. Product isolation is made by removal of volatiles in vacuo followed by trituration with Et2O (50 mL). The solids are collected by filtration, washed with H2O (2×50 mL), acetone (50 mL) and Et2O (50 mL), then dried in vacuo to give the required bromo intermediate (4).
Method A Preparation of Compounds of the Invention Via Suzuki Coupling (5):
An appropriate boronic acid (2 eq.) is added to a solution of bromo intermediate (4) in 1,4-dioxane/water (5:1). K2CO3 (2 eq.) and PdCl2dppf (5%) are added to the solution. The resulting mixture is then heated in a microwave at 140° C. for 30 min (this reaction can also be carried out by traditional heating in an oil bath at 90° C. for 16 h under N2). Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhyd. MgSO4 and evaporated in vacuo. The final compound is obtained after purification by flash chromatography or preparative HPLC. HPLC: Waters XBridge Prep C18 5 μm ODB 19 mm ID×100 mm L (Part No. 186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Method B

B1. 4 4-[2-(Cyclopropanecarbonyl-amino)-[1,2,4]triazolo[1,5-a]pyridin-5-yl]-benzoyl chloride

2 Drops of DMF are added to a solution of 4-[2-(cyclopropanecarbonyl-amino)-[1,2,4]triazolo[1,5-a]pyridin-5-yl]-benzoic acid (1 eq) obtained by Method A using 4-carboxyphenylboronic acid in DCM under N2 atmosphere. Then oxalyl chloride (2 eq) is added dropwise to this resulting solution (gas release). The mixture is stirred at room temperature for 2 hours. After completion of the reaction by LCMS, the solvent is removed. The crude acid chloride is used without further purification in next step.
B2. Amide Formation (General Method)

An appropriate amine (1.1 eq) and Et3N (5 eq) are dissolved in DCM under N2 atmosphere and cooled at 0° C. The acid chloride (B1, 1 eq) dissolved in DCM is added dropwise to this solution. The reaction is stirred at room temperature for 16 h. After this time, reaction is complete. The compound is extracted with EtOAc and water, washed with brine and dried over anhyd. MgSO4. Organic layers are filtered and evaporated. The final compound is isolated by preparative HPLC. Preparative HPLC: Waters XBridge Prep C18 5 μm ODB 19 mm ID×100 mm L (Part No. 186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
…
Synthesis of the Compound of the Invention and Comparative Examples Compound 1 (the Compound of the Invention) Step 1:

2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[1,3,2]dioxaborolane (1 eq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1-dioxide (2 eq) was added portionwise. The resulting solution was stirred at room temperature for 16 h. After this time, the reaction was complete. The solvent was evaporated. The compound was extracted with EtOAc and water, washed with brine and dried over anhyd. MgSO4. Organic layers were filtered and evaporated. The final compound was isolated without further purification.
STEP 2: Suzuki coupling

4-[4-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-1,1-dioxide (1.1 eq.) was added to a solution of cyclopropanecarboxylic acid (5-bromo-[1,2,4]triazolo[1,5-a]pyridin-2-yl)-amide in 1,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 90° C. for 16 h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSO4 and evaporated in vacuo. The final compound was obtained after purification by flash chromatography.
Alternatively, after completion of the reaction, a palladium scavenger such as 1,2-bis(diphenylphosphino)ethane, is added, the reaction mixture is allowed to cooled down and a filtration is performed. The filter cake is reslurried in a suitable solvent (e.g. acetone), the solid is separated by filtration, washed with more acetone, and dried. The resulting solid is resuspended in water, aqueous HCl is added, and after stirring at RT, the resulting solution is filtered on celite (Celpure P300). Aqueous NaOH is then added to the filtrate, and the resulting suspension is stirred at RT, the solid is separated by filtration, washed with water and dried by suction. Finally the cake is re-solubilised in a mixture of THF/H2O, treated with a palladium scavenger (e.g. SMOPEX 234) at 50° C., the suspension is filtered, the organic solvents are removed by evaporation, and the resulting slurry is washed with water and methanol, dried and sieved, to obtain the title compound as a free base.
Alternative Route to Compound 1 (the Compound of the Invention): Step 1:

4-(Hydroxymethyl)phenylboronic acid (1.1 eq.) was added to a solution of cyclopropanecarboxylic acid (5-bromo-[1,2,4]triazolo[1,5-a]pyridin-2-yl)-amide in 1,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 90° C. for 16 h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSO4 and evaporated in vacuo. The resulting mixture was used without further purification.
Step 2:

To a solution of cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl]-amide (1.0 eq) in chloroform was slowly added phosphorus tribromide (1.0 equiv.). The reaction mixture was stirred at room temperature for 20 hours, quenched with ice and water (20 mL) and extracted with dichloromethane. The organic layer was dried over anhyd. MgSO4, filtered and concentrated to dryness. The resulting white residue was triturated in dichloromethane/diethyl ether 2:1 to afford the expected product as a white solid.
Step 3:

Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl]-amide (1 eq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1-dioxide (1.1 eq) was added dropwise. The resulting solution was stirred at room temperature for 16 h. After this time, the reaction was complete. The solvent was evaporated. The compound was dissolved in DCM, washed with water and dried over anhyd. MgSO4. Organic layers were filtered and evaporated. The final compound was isolated by column chromatography using EtOAc to afford the desired product.
…………………….
PATENT
Novel salts and pharmaceutical compositions thereof for the treatment of inflammatory disorders
Also claims a method for preparing filgotinib hydrochloride trihydrate. The present filing forms a pair with this week’s filing, WO2015117980, claiming a tablet composition comprising filgotinib hydrochloride.
The compound cyclopropanecarboxylic acid {5-[4-(l,l-dioxo-thiomorpholin-4-ylmethyl)-phenyl]-[l,2,4]triazolo[l,5-a]pyridin-2-yl -amide (Compound 1), which has the chemical structure:

is disclosed in our earlier application WO 2010/149769 (Menet C. J., 2010) as being an inhibitor of JAK and as being useful in the treatment of inflammatory conditions, autoimmune diseases, proliferative diseases, allergy, transplant rejection, diseases involving impairment of cartilage turnover, congenital cartilage malformations, and/or diseases associated with hypersecretion of IL6 or interferons. Hereafter this compound is named Compound 1. The data presented in WO 2010/149769 demonstrate that despite similar in vitro activities, Compound 1 has unexpectedly high in vivo potency compared with structurally similar compounds.
Example 1. Preparation of Compound 1
1.1. Route 1
1.1.1. 4-[4-(4,4,5,5-Tetramethyl-[l,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-l,l-dioxide

[00205] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (1 eq) and DIPEA (2 eq) are dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (2 eq) is added portionwise. The resulting solution is stirred at room temperature for 16h. After this time, the reaction is complete. The solvent is evaporated. The compound is extracted with EtOAc and water, washed with brine and dried over anhydrous MgSO i. Organic layers are filtered and evaporated. The final compound is isolated without further purification.
1.1.2. Cyclopropanecarboxylic acid (5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide

1.1.2.1. Step i): l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea
[00206] To a solution of 2-amino-6-bromopyridine (1) (253.8 g, 1.467 mol) in DCM (2.5 L) cooled to 5°C is added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction
mixture is then allowed to warm to room temp. (20 °C) and stirred for 16 h. Evaporation in vacuo gives a solid which may be collected by filtration, thoroughly washed with petrol (3 x 600 niL) and air-dried to afford the desired product. The thiourea may be used as such for the next step without any purification. lH (400 MHz, CDC13) δ 12.03 (1H, br s), 8.81 (1H, d), 8.15 (1H, br s), 7.60 (1H, t), 7.32 (1H, dd), 4.31 (2H, q), 1.35 (3H, t).
1.1.2.2. Step ii): 5-Bromo-[l,2,4]triazolo[l,5-a]pyridin-2-ylamine
[00207] To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH (1 : 1, 900 mL) is added NN-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture is stirred at room temp. (20 °C) for 1 h. l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) is then added and the mixture slowly heated to reflux (Note: bleach scrubber is required to quench H2S evolved). After 3h at reflux, the mixture is allowed to cool and filtered to collect the precipitated solid. Further product is collected by evaporation in vacuo of the filtrate, addition of H20 (250 mL) and filtration. The combined solids are washed successively with H20 (250 mL), EtOH/MeOH (1 : 1, 250 mL) and Et20 (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound may be used as such for the next step without any purification. lH (400 MHz, DMSO-i¼) δ 7.43-7.34 (2H, m, 2 x aromatic-H), 7.24 (1H, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1 : 1, M+H+, 100%).
1.1.2.3. Step Hi): Cyclopropanecarboxylic acid (5-bromo-[l ,2,4]triazolo[l ,5-a]pyridin-2-yl)-amide
[00208] To a solution of the 2-amino-triazolopyridine obtained in the previous step (7.10 g, 33.3 mmol) in dry MeCN (150 mL) at 5°C is added Et3N (11.6 mL, 83.3 mmol) followed by cyclopropanecarbonyl chloride (83.3 mmol). The reaction mixture is then allowed to warm to ambient temperature and stirred until all starting material is consumed. If required, further Et3N (4.64 mL, 33.3 mmol) and cyclopropanecarbonyl chloride (33.3 mmol) is added to ensure complete reaction. Following solvent evaporation in vacuo the resultant residue is treated with 7 N methanolic ammonia solution (50 mL) and stirred at ambient temp, (for 1-16 h) to hydro lyse any bis-acylated product. Product isolation is made by removal of volatiles in vacuo followed by trituration with Et20 (50 mL). The solids are collected by filtration, washed with H20 (2x50mL), acetone (50 mL) and Et20 (50 mL), then dried in vacuo to give the desired compound.
1.1.3. Compound 1

[00209] 4-[4-(4,4,5,5-Tetramethyl-[l ,3,2]dioxaborolan-2-yl)-benzyl] hiomoφholine , l -dioxide (l . l eq.) is added to a solution of cyclopropanecarboxylic acid (5-bromo-[l ,2,4]triazolo[l ,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water (4: 1). K2CO3 (2 eq.) and PdC^dppf (0.03 eq.) are added to the solution. The resulting mixture is then heated in an oil bath at 90°C for 16h under N2. Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhydrous MgS04 and evaporated in vacuo.
[00210] The final compound is obtained after purification by flash chromatography.
[00211] Alternatively, after completion of the reaction, a palladium scavenger such as 1 ,2-bis(diphenylphosphino)ethane, is added, the reaction mixture is allowed to cool down and a filtration is performed. The filter cake is reslurried in a suitable solvent (e.g. acetone), the solid is separated by filtration, washed with more acetone, and dried. The resulting solid is resuspended in water, aqueous HC1 is added, and after stirring at room temperature, the resulting solution is filtered on celite (Celpure P300). Aqueous NaOH is then added to the filtrate, and the resulting suspension is stirred at room temperature, the solid is separated by filtration, washed with water and dried by suction. Finally the cake is re-solubilised in a mixture of THF/H20, treated with a palladium scavenger (e.g. SMOPEX 234) at 50°C, the suspension is filtered, the organic solvents are removed by evaporation, and the resulting slurry is washed with water and methanol, dried and sieved, to obtain the desired compound as a free base.
1.2. Route 2
1.2.1. Step 1: cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)-[l,2, 4]triazolo[l, 5- a] pyridin-2-yl] -amide

[00212] 4-(Hydroxymethyl)phenylboronic acid (l . l eq.) is added to a solution of cyclopropanecarboxylic acid (5-bromo-[l ,2,4]triazolo[l ,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water
(4:1). K2CO3 (2 eq.) and PdC^dppf (0.03 eq.) are added to the solution. The resulting mixture is then heated in an oil bath at 90°C for 16h under N2. Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhydrous MgS04 and evaporated in vacuo. The resulting mixture is used without further purification.
1.2.2. Step 2: Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[l,2,4]triazolo[l,5- a Jpyridin-2-ylJ -amide

[00213] To a solution of cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridin-2-yl] -amide (1.0 eq) in chloroform is slowly added phosphorus tribromide (1.0 eq.). The reaction mixture is stirred at room temperature for 20 h, quenched with ice and water (20 mL) and extracted with dichloromethane. The organic layer is dried over anhydrous MgSO i, filtered and concentrated to dryness. The resulting white residue is triturated in dichloromethane/diethyl ether 2:1 to afford the desired product.
1.2.3. Step 3:

[00214] Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridin-2-yl]-amide (l eq) and DIPEA (2 eq) are dissolved in DCM/MeOH (5: 1 v:v) under N2 and thiomorpho line 1,1-dioxide (1.1 eq) is added dropwise. The resulting solution is stirred at room temperature for 16h. After this time, the reaction is complete. The solvent is evaporated. The compound is dissolved in DCM, washed with water and dried over anhydrous MgSO i. Organic layers are filtered and evaporated. The final compound is isolated by column chromatography using EtOAc to afford the desired product.
…………………
PATENT
http://www.google.co.in/patents/WO2013189771A1?cl=en
Example 1. Synthesis of the compounds
1.1. Route 1
1.1.1. Synthesis of 5-Bromo-[l,2,4]triazolo[l,5-a]pyridin-2-ylamine (Intermediate 3)

led to 5 °C was added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction mixture was then allowed to warm to room temp. (20 °C) and stirred for 16 h. Evaporation in vacuo gave a solid which was collected by filtration, thoroughly washed with petrol (3×600 mL) and air-dried to afford (2). The thiourea was used as such in the next step without any purification.
[00157] lH (400 MHz, CDC13) δ 12.03 (IH, br s, NH), 8.81 (IH, d, J 7.8 Hz, H-3), 8.15 (IH, br s, NH), 7.60 (IH, t, J 8.0 Hz, H-4), 7.32 (IH, dd, J 7.7 and 0.6 Hz, H-5), 4.31 (2H, q, J 7.1 Hz, CH2), 1.35 (3H, t, J 7.1 Hz, CH3).
1.1.1.2. 5-Bromo-f 1,2, 4]triazolo[ 1 ,5-a] pyridin-2-ylamine (3)
[00158] To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH (1 : 1, 900 mL) was added NN-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture was stirred at room temp. (20 °C) for 1 h. l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) was then added and the mixture slowly heated to reflux (Note: bleach scrubber was required to quench H2S evolved). After 3 h at reflux, the mixture was allowed to cool and filtered to collect the precipitated solid. Further product was collected by evaporation in vacuo of the filtrate, addition of H20 (250 mL) and filtration. The combined solids were washed successively with H20 (250 mL), EtOH/MeOH (1 : 1, 250 mL) and Et20 (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound was used as such in the next step without any purification.
[00159] lH (400 MHz, DMSO-i¼) δ 7.43-7.34 (2H, m, 2 x aromatic-H), 7.24 (1H, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1 : 1, M+H+, 100%).
1.1.2. Synthesis of 4-[ 4-(4, 4, 5, 5-Tetramethyl-f 1, 3,2] ‘ dioxaborolan-2-yl) -benzyl] ‘- thiomor holine- 1, 1 -dioxide (Intermediate 4)

[00160] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (1 eq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (2 eq) was added portion wise. The resulting solution was stirred at room temperature for 16h. After this time, the reaction was complete. The solvent was evaporated. The compound was extracted with EtOAc and water, washed with brine and dried over anhydrous MgSO i. Organic layers were filtered and evaporated. The final compound was isolated without further purification.
1.1.3. Synthesis of 5-[4-(l, l-Dioxothiomorpholin-4-ylmethyl)-phenyl]-[l,2,4]triazolo[l,5- a ridin-2-ylamine (Formula I)

[00161] 4-[4-(4,4,5,5-Tetramethyl-[l,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-l,l-dioxide (l .leq.) was added to a solution of 5-bromo-[l,2,4]triazolo[l,5-a]pyrid in-2-ylamine (4: 1). K2CO3 (2 eq.) and PdC^dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 90°C for 16h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhydrous MgSC>4 and evaporated in vacuo. The final compound was obtained after purification by flash chromatography.
[00162] lH (400 MHz, CDC13) δ 7.94-7.92 (d, 2H), 7.52-7.48 (m, 3H), 7.37-7.34 (m, 1H), 7.02-7.00 (m, 1H), 6.00 (d, 2H), 3.76 (d, 2H), 3.15-3.13 (m, 4H), 2.93-2.91 (m, 4H).
[00163] m/z 358.2 (M+H+, 100%). 1.2. Route 2
1.2.1. Cyclopropanecarboxylic acid {5-[4-(l, l-dioxo-thiomorpholin-4-ylmethyl)-phenylJ- [l,2,4]triazolo[l,5-a]pyridin-2-yl}-amide (Formula II)
[00164] The compound according to Formula II may be synthesized according to the procedure described in WO 2010/149769.
1.2.2. Synthesis of 5-[4-(l, l-Dioxothiomorpholin-4-ylmethyl)-phenyl]-[l,2,4]triazolo[l,5- aJpyridin-2-ylamine (Formula I)
[00165] The compound according to Formula I can also be produced by hydrolysis of the compound accor ing to Formula II:

[00166] Hydrochloric acid 30% aq (12.06 kg; 3.9 rel. volumes) was added to a slurry of the compound according to Formula II (3.45 kg; 1.0 equiv.) in demineralized water (10.0 kg; 3.0 rel. volumes). Subsequently, a line rinse was performed with demineralized water (3.4 kg; 1.0 rel. volumes). The reaction mixture was heated to 80±5°C for 14.5 h. After completion of the reaction (conversion > 99%>), the reaction mixture was cooled to 20±5°C. The reaction mixture was diluted with demineralized water (6.8 kg; 2.0 rel. volumes) and sodium hydroxide 33%> aq (9.52 kg; 3.7 rel volumes) was dosed at such a rate that the temperature of the reactor contents remained below 35°C. An additional amount of sodium hydroxide 33%> aq (2.55 kg; 1.0 rel. volumes) was needed to get the pH > 10. The product was filtered off, washed twice with demineralized water (1.5 rel. volumes) and dried under vacuum for 1 h, thus yielding the crude compound according to Formula I.
[00167] The crude compound according to Formula I (5.70 kg) was re-slurried in demineralized water (23.0 kg; 8.5 rel. volumes). Hydrochloric acid 30%> aq (1.65 kg; 0.7 rel. volumes) and demineralized water (4.3 kg; 1.6 rel. volumes) were added and the reaction mixture was stirred at 20±5°C for 45 min. As the compound according to Formula I was not dissolved completely, the reaction mixture was stirred at 45±5°C for 1 h. The reaction mixture was filtered and the residue was washed with demineralized water (2.0 kg 0.75 rel. volumes). Sodium hydroxide 33%> aq (1.12 kg; 0.6 rel volumes) was added to the filtrate. An additional amount of sodium hydroxide 33%> aq (1.01 kg) was needed to get the pH > 10. The resulting reaction mixture was stirred at 20±5°C for about 3 h. The product was filtered off, washed twice with demineralized water (4.1 kg; 1.5 rel. volumes), and twice with methyl tert-butyl ether (MTBE; 3.0 kg; 1.5 rel. volumes) and dried under vacuum for 15.5 h on the filter. The product was further dried in a vacuum oven at 40±5°C for 202 h, thus affording the desired compound according to Formula I.
Update
Scheme 1: General S nthesis of Compounds of Formula I or A

Formula A
Scheme 7.

(16) (17) (18)
(18a): R3a=R3b=R2a=R (18b): R3a=R3b=D; R2a 18c): R3a=R3b=H; R2a

References
- Namour, Florence; Diderichsen, Paul Matthias; Cox, Eugène; Vayssière, Béatrice; Van der Aa, Annegret; Tasset, Chantal; Van’t Klooster, Gerben (2015-02-14). “Pharmacokinetics and Pharmacokinetic/Pharmacodynamic Modeling of Filgotinib (GLPG0634), a Selective JAK1 Inhibitor, in Support of Phase IIB Dose Selection”. Clin Pharmacokinet. Epub ahead of print.doi:10.1007/s40262-015-0240-z.
- Van Rompaey, L; Galien, R; Van der Aar, E; Clement-Lacroix, P; Van der Aar, E; Nelles, L; Smets, B; Lepescheux, L; Cristophe, T; Conrath, K; Vandeghinste, N; Vayssiere, B; De Vos, S; Fletcher, S; Brys, R; Van’t Klooster, G; Feyen, J; Menet, C (2013-10-01). “Preclinical characterization of GLPG0634, a selective inhibitor of JAK1 for the treatment of inflammatory diseases”. J Immunol. 191(7). doi:10.4049/jimmunol.1201348.
- http://acrabstracts.org/abstracts/phase-1-and-phase-2-data-confirm-that-glpg0634-a-selective-jak1-inhibitor-has-a-low-potential-for-drug-drug-interactions/
- “Galapagos’ GLPG0634 shows excellent efficacy and safety in rheumatoid arthritis Phase II study” (PDF) (Press release). Retrieved 2015-02-26.
- “Galapagos reports that the last patient in DARWIN 1 has completed 12 weeks of treatment” (PDF) (Press release). Retrieved 2015-02-26.
- “Galapagos completes recruitment for Darwin 1 study with GLPG0634 (filgotinib) in RA”. EuroInvestor. Retrieved 2015-02-26.
- NASDAQ OMX Corporate Solutions. “Galapagos completes recruitment for Darwin 2 monotherapy study with GLPG0634 (filgotinib) in RA”. Yahoo Finance. Retrieved 2015-02-26.
| US8551980 | Nov 17, 2010 | Oct 8, 2013 | Bayer Intellectual Property Gmbh | Substituted triazolopyridines |
| US8796457 | Jun 25, 2010 | Aug 5, 2014 | Galapagos Nv | Compound useful for the treatment of degenerative and inflammatory diseases |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-[5-[4-[(1,1-dioxo-1,4-thiazinan-4-yl)methyl]phenyl]-[1,2,4]triazolo[1,5-a]pyridin-2-yl]cyclopropanecarboxamide
|
|
| Clinical data | |
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Biological half-life | 6 hours[1] |
| Identifiers | |
| CAS Registry Number | 1206161-97-8 |
| ATC code | L01XE18 |
| IUPHAR/BPS | 7913 |
| ChemSpider | 28189566 |
| UNII | 3XVL385Q0M |
| ChEMBL | CHEMBL3301607 |
| Chemical data | |
| Formula | C21H23N5O3S |
| Molecular mass | 425.50402 g/mol |
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


Join me on twitter
 LIONEL MY SON
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL
/////////Galapagos, GLPG0634, Filgotinib, PHASE 2, orphan drug designation, PHASE 3, Crohn’s disease, Rheumatoid arthritis, Ulceraticolitis
ve SMILES code: O=C(C1CC1)NC2=NN3C(C4=CC=C(CN5CCS(CC5)(=O)=O)C=C4)=CC=CC3=N2
TOFACITINIB 的合成, トファシチニブ, Тофацитиниб, توفاسيتين يب SPECTRAL VISIT
Tofacitinib Citrate, 的合成
托法替布, トファシチニブクエン酸塩, Тофацитиниба Цитрат
3-{(3R,4R)-4-methyl-3-[methyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3-oxo-propionitrile citrate salt
CAS : 540737-29-9
ROTATION +
Tofacitinib; Tasocitinib;
477600-75-2 base ; CP-690550;
3-((3R,4R)-4-methyl-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)-3-oxopropanenitrile;
3-{(3R,4R)-4-methyl-3-rmethyl-(7H-pyrrolor2,3-dlpyrimidin-4-yl)-amino1- piperidin-1-yl}-3-oxo-propionitrile mono citrate salt
CP 690550 Tofacitinib; CP-690550; CP-690550-10; Xeljanz; Jakvinus; Tofacitinib citrate
Trademarks: Xeljanz; Jakvinus
MF: C16H20N6O
CAS : 477600-75-2 BASE ; 540737-29-9(citrate) 3-[(3R,4R)-4-methyl-3-[methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]piperidin-1-yl]-3-oxopropanenitrile
Molecular Weight: 312.369
SMILES: C[C@@H]1CCN(C[C@@H]1N(C)C2=NC=NC3=C2C=CN3)C(=O)CC#N
Activity: Treatment of Rheumatoid Arthritis; RA Treatment, JAK Inhibitor; Protein Kinase Inhibitor; JAK3 Inhibitor; Janus Kinase 3 Inhibitor; JAK-STAT Signaling Pathway; JAK1 Kinase Inhibitor; Selective Immunosuppressants
Status: Launched 2012
Originator: Pfizer 
Pfizer Inc’s oral JAK inhibitor tofacitinib was approved on November 6, 2012 by US FDA for the treatment of rheumatoid arthritis.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
Tofacitinib (trade names Xeljanz and Jakvinus, formerly tasocitinib,[1] CP-690550[2]) is a drug of the janus kinase (JAK) inhibitor class, discovered and developed by Pfizer. It is currently approved for the treatment of rheumatoid arthritis (RA) in the United States,Russia, Japan and many other countries, is being studied for treatment of psoriasis, inflammatory bowel disease, and other immunological diseases, as well as for the prevention of organ transplant rejection. 

An Improved and Efficient Process for the Preparation of Tofacitinib Citrate
Yogesh S. Patil, Nilesh L. Bonde, Ankush S. Kekan, Dhananjay G. Sathe*1, and Arijit Das
Publication Date (Web): November 17, 2014 (Article)
DOI: 10.1021/op500274j
MS m/z 313 (M+ + 1);
mp 201–202 °C;
|
1H NMR (CDCl3) δ 8.34 (s, 1H), δ 7.38 (d, 1H, J = 2.4 Hz), δ 6.93 (d, 1H, J = 2.4 Hz), δ 4.97 (m, 1H), δ 3.93–4.03 (m, 4H), δ 3.66 (m, 1H), δ 3.50 (m, 4H), δ 2.91 (d, 2H, J = 15.6 Hz), δ 2.80 (t, 2H, J = 12.8 Hz), δ 2.55 (m, 1H), δ 1.99 (m, 1H), δ 1.77 (m, 1H), δ 1.13–1.18 (m, 3H).
|



TEAMWORK
Part of the Pfizer group responsible for Xeljanz: Front row, from left: Sally Gut Ruggeri, Chakrapani Subramanyam, Eileen Elliott Mueller, and Frank Busch. Second row, from left: Matthew Brown, Mark Flanagan, and Robert Dugger. Back row, from left: Elizabeth Kudlacz and Douglas Ball.
Credit: Pfizer
Mark Flanagan, who was on the team at Pfizer that discovered Xeljanz, (tofacitinib citrate), an oral treatment for rheumatoid arthritis, remembers testing the drug in a rat model and seeing the drug decrease the level of inflammation in the rats’ footpads. “What we look for is physical measurements of the size of the joint. In the control animals, there was quite a bit of inflammation in the joints, whereas animals treated with different doses of the drug showed a dose-dependent decrease in the size of the joint. “Tofacitinib showed robust efficacy in the first such study run. I can remember the excitement that this data generated on the team,” he says.
Tofacitinib, chemically known as (3R,4R)-4-methyl-3-(methyl-7H-pyrrolo [2,3- d]pyrimidin-4-ylamino)-B-oxo-l -piperidinepi panenitrile, is represented Formula I. Tofacitinib citrate, a janus kinase inhibitor, is approved as XELJANZ® tablets for treatment .of rheumatoid arthritis.

Various intermediates and processes for preparation of tofacitinib are disclosed in patents like US7301 023 and US8232394.

Formula I or isomers or a mixture of isomers thereof by following any method provided in the prior art, for example, by following Example 14 of U.S. Patent No. RE41,783 or by following Example 6 of U.S. Patent No. 7,301,023. Tofacitinib of Formula I or isomers of tofacitinib or a mixture of isomers thereof may be converted into a salt by following any method provided in the prior art, for example, by following Example 1 of U.S. Patent No. 6,965,027 or by following Example 1 or Example 8 of PCT Publication No. WO 2012/135338. The potential significance of JAK3 inhibition was first discovered in the laboratory of John O’Shea, an immunologist at the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health (NIH).[5] In 1994, Pfizer was approached by the NIH to form a public-private partnership in order to evaluate and bring to market experimental compounds based on this research.[5] Pfizer initially declined the partnership but agreed in 1996, after the elimination of an NIH policy dictating that the market price of a product resulting from such a partnership would need to be commensurate with the investment of public taxpayer revenue and the “health and safety needs of the public.”[5] The drug discovery, preclinical development, and clinical development of tofacitinib took place exclusively at Pfizer.[6] In November 2012, the U.S. Food and Drug Administration (FDA) approved tofacitinib for treatment of rheumatoid arthritis. Once on the market, rheumatologists complained that the $2,055 a month wholesale price was too expensive, though the price is 7% less than related treatments.[6] A 2014 study showed that tofacitinib treatment was able to convert white fat tissues into more metabolically active brown fat, suggesting it may have potential applications in the treatment of obesity.[7] It is an inhibitor of the enzyme janus kinase 1 (JAK1) and janus kinase 3 (JAK 3) , which means that it interferes with the JAK-STAT signaling pathway, which transmits extracellular information into the cell nucleus, influencing DNA transcription.[3] Recently it has been shown in a murine model of established arthritis that tofacitinib rapidly improved disease by inhibiting the production of inflammatory mediators and suppressing STAT1-dependent genes in joint tissue. This efficacy in this disease model correlated with the inhibition of both JAK1 and 3 signaling pathways, suggesting that tofacitinib may exert therapeutic benefit via pathways that are not exclusive to inhibition of JAK3.[4]
Preparation of 3-{(3R,4R)-4-methyl-3-[methyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3-oxo-propionitrile citrate salt (Tofacitinib citrate, Xeljanz, CP-690550-10)
To a round-bottomed flask fitted with a temperature probe, condenser, nitrogen source, and heating mantle, methyl-[(3R,4R)-4-methyl-piperidin-3-yl]-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amine (5.0 g, 20.4 mmol) was added followed by 1-butanol (15 mL), ethyl cyanoacetate (4.6 g, 40.8 mmol), and DBU (1.6 g, 10.2 mmol). The resulting amber solution was stirred at 40 °C for 20 h. Upon reaction completion, citric acid monohydrate (8.57 g, 40.8 mmol) was added followed by water (7.5 mL) and 1-butanol (39.5 mL). The mixture was heated to 81 °C and held at that temperature for 30 min. The mixture was then cooled slowly to 22 ºC and stirred for 2 h. The slurry was filtered and washed with 1-butanol (20 mL). The filter cake was dried in a vacuum oven at 80 °C to afford 9.6 g (93%) of tofacitinib citrate as an off-white solid.
1H NMR (500 MHz, d6-DMSO): δ 8.14 (s, 1H), 7.11 (d, J=3.6 Hz, 1H), 6.57 (d, J=3.6 Hz, 1H), 4.96 (q, J=6.0 Hz, 1H), 4.00-3.90 (m, 2H), 3.80 (m, 2H), 3.51 (m, 1H), 3.32 (s, 3H), 2.80 (Abq, J=15.6 Hz, 2H), 2.71 (Abq, J=15.6 Hz, 2H), 2.52-2.50 (m, 1H), 2.45-2.41 (m, 1H), 1.81 (m, 1H), 1.69-1.65 (m, 1H), 1.04 (d, J=6.9 Hz, 3H).
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
PAPER
3-((3R,4R)-4-Methyl-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)-3-oxopropanenitrile (1) Monocitrate
J. Med. Chem., 2010, 53 (24), pp 8468–8484
DOI: 10.1021/jm1004286
1monocitrate as a white crystalline solid (mp = 201 dec).
LRMS: m/z 313.2 (MH+).
1H NMR (400 MHz) (D2O) δ HOD: 0.92 (2 H, d, J = 7.2 Hz), 0.96 (1 H, d, J = 7.6 Hz), 1.66 (1 H, m), 1.80 (1 H, m), 2.37 (1 H, m), 2.58 (2 H, 1/2 ABq, J = 15.4 Hz), 2.70 (2 H, 1/2 ABq, J = 15.4 Hz), 3.23 (2 H, s), 3.25 (1 H, s), 3.33 (1 H, m), 3.46 (1 H, m), 3.81 (4 H, m), 4.55 (1 H, m), 6.65 (1 H, d, J = 3.2 Hz), 7.20 (1 H, t, J = 3.2 Hz), 8.09 (1 H, m).
Anal. Calcd for C22H28N6O8: C, 52.38; H, 5.59; N, 16.66. Found: C, 52.32; H, 5.83; N, 16.30. For additional characterization of the monocitrate salt of 1 see WO 03/048162.
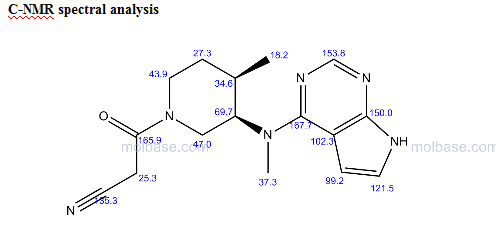
NMR PREDICT



References:
Weiling Cai, James L. Colony,Heather Frost, James P. Hudspeth, Peter M. Kendall, Ashwin M. Krishnan,Teresa Makowski, Duane J. Mazur, James Phillips, David H. Brown Ripin, Sally Gut Ruggeri, Jay F. Stearns, and Timothy D. White; Investigation of Practical Routes for the Kilogram-Scale Production of cis-3-Methylamino-4-methylpiperidines; Organic Process Research & Development 2005, 9, 51−56
Ripin, D. H.B.; 3-amino-piperidine derivatives and methods of manufacture, US patent application publication, US 2004/0102627 A1
Ruggeri, Sally, Gut;Hawkins, Joel, Michael; Makowski, Teresa, Margaret; Rutherford, Jennifer, Lea; Urban,Frank,John;Pyrrolo[2,3-d]pyrimidine derivatives: their intermediates and synthesis, PCT pub. No. WO 2007/012953 A 2, US20120259115 A1, United States Patent US8232393. Patent Issue Date: July 31, 2012
Kristin E. Price, Claude Larrive´e-Aboussafy, Brett M. Lillie, Robert W. McLaughlin, Jason Mustakis, Kevin W. Hettenbach, Joel M. Hawkins, and Rajappa Vaidyanathan; Mild and Efficient DBU-Catalyzed Amidation of Cyanoacetates, Organic Letters, 2009, vol.11, No.9, 2003-2006
MORE NMR PREDICT

Tofacitinib 
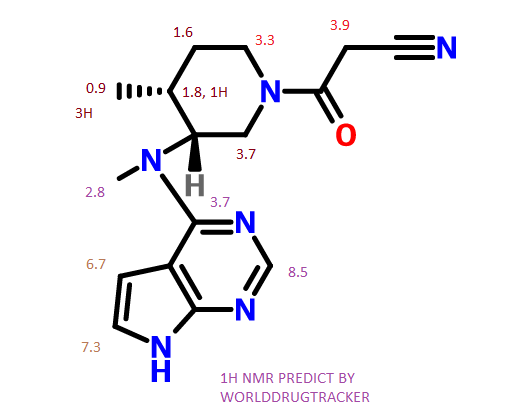
13C NMR PREDICT 

SEE…….https://newdrugapprovals.org/2015/07/24/tofacitinib-%E7%9A%84%E5%90%88%E6%88%90-spectral-visit/
COSY PREDICT 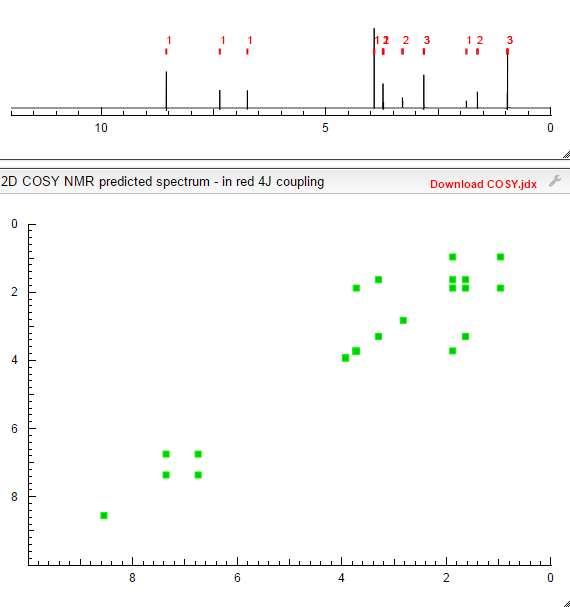 सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
SEE………http://orgspectroscopyint.blogspot.in/2014/12/tofacitinib-citrate.html
NMR PICTURE FROM THE NET

PAPER
Volume 54, Issue 37, 11 September 2013, Pages 5096–5098

Asymmetric total synthesis of Tofacitinib
- a Laboratory of Asymmetric Synthesis, Chemistry Institute of Natural Resources, University of Talca, P.O. Box 747, Talca, Chile
- b Laboratory of Natural Products, Department of Chemistry, University of Antofagasta, P.O. Box 170, Antofagasta, Chile
http://dx.doi.org/10.1016/j.tetlet.2013.07.042
Abstract
A novel stereoselective synthesis of Tofacitinib (CP-690,550), a Janus tyrosine kinase (JAK3) specific inhibitor, has been achieved starting from (5S)-5-hydroxypiperidin-2-one in 10 steps from 2 with a 9.5% overall yield. The potentiality of this synthetic route is the obtention of tert-butyl-(3S,4R)-3-hydroxy-4-methylpiperidine-1-carboxylate (6b) as a new chiral precursor involved in the synthesis of CP690,550, in a three-step reaction, without epimerizations, rather than the 5 or more steps used in described reactions to achieve this compound from analogues of 6b.
Graphical abstract

…………………. Tofacitinib synthesis: US2001053782A1

Tofacitinib synthesis: WO2002096909A1

Tofacitinib synthesis: Org Process Res Dev 2014, 18(12), 1714-1720 (also from a chinese publication, same procedure just slight changes in reagents/conditions)
References:
1. Blumenkopf, T. A.; et. al. Pyrrolo[2,3-d]pyrimidine compounds. US2001053782A1
2. Flanagan, M. E.; et. al. Optical resolution of (1-benzyl-4-methylpiperidin-3-yl) -methylamine and the use thereof for the preparation of pyrrolo 2,3-pyrimidine derivatives as protein kinases inhibitors. WO2002096909A1
3. Das, A.; et. al. An Improved and Efficient Process for the Preparation of Tofacitinib Citrate. Org Process Res Dev2014, 18(12), 1714-1720.
Synthesis of Tofacitinib Citrate

PATENT https://www.google.co.in/patents/WO2003048162A1?cl=en The crystalline form of the compound of this invention 3-{4-methyl-3-[methyl- (7H-pyrrolot2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3-oxo-propionitrile mono citrate salt is prepared as described below. Scheme 1


Scheme 2




Example 1 3-{(3R,4R)-4-methyl-3-rmethyl-(7H-pyrrolor2,3-dlpyrimidin-4-yl)-amino1- piperidin-1-yl}-3-oxo-propionitrile mono citrate salt Ethanol (13 liters), (3R, 4R)-methyl-(4-methyl-piperidin-3-yl)-(7H-pyrrolo[2,3- d]pyrimidin-4-yl)-amine (1.3 kg), cyano-acetic acid 2,5-dioxo-pyrrolidin-1-yl ester (1.5 kg), and triethylamine (1.5 liters) were combined and stirred at ambient temperature. Upon reaction completion (determined by High Pressure Liquid Chromotography (HPLC) analysis, approximately 30 minutes), the solution was filtered, concentrated and azeotroped with 15 liters of methylene chloride. The reaction mixture was washed sequentially with 12 liters of 0.5 N sodium hydroxide solution, 12 liters of brine and 12 liters of water. The organic layer was concentrated and azeotroped with 3 liters of acetone (final pot temperature was 42°C). The resulting solution was cooled to 20°C to 25°C followed by addition of 10 liters of acetone. This solution was filtered and then aqueous citric acid (0.8 kg in 4 liters of water) added via in-line filter. The reaction mixture was allowed to granulate. The slurry was cooled before collecting the solids by filtration. The solids were dried to yield 1.9 kg (71 %) (3R, 4R)- 3-{4-Methyl-3-[methyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3-oxo- propionitrile mono citrate. This material was then combined with 15 liters of a 1:1 ratio of ethanol/water and the slurry was agitated overnight. The solids were filtered and dried to afford 1.7 kg (63% from (3R, 4R)-methyl-(4-methyl-piperidin-3-yl)-(7H- pyrrolo[2,3-d]pyrimidin-4-yl)-amine) of the title compound as a white crystalline solid. 1H NMR (400 MH2)(D20) δ HOD: 0.92 (2H, d, J = 7.2 Hz), 0.96 (1H, d, J = 7.6 Hz), 1.66 (1H, m), 1.80 (1H, m), 2.37 (1H, m), 2.58 (2H, 1/2 ABq, J = 15.4 Hz), 2.70 (2H, 3 ABq, J = 154 Hz), 3.23 (2H, s), 3.25 (1H, s), 3.33 (1H, m), 3.46 (1H, m), 3.81 (4H, m), 4.55 (1 H, m), 6.65 (1 H, d, J = 3.2 Hz), 7.20 (1 H, t, J = 3.2 Hz), 8.09 (1 H, m).
Patent
http://www.google.co.in/patents/EP1913000A2?cl=en Example 10 Preparation of methyl-[(3R, 4R)-4-methyl-piperidin-3-yl]-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amine:
KEY INTERMEDIATE
To a clean, dry, nitrogen-purged 2 L hydrogenation reactor were charged 20 wt% Pd(OH)2/C (24.0 g, 50% water wet), water (160 ml), isopropanol (640 ml), (1-benzyl-4-methyl-piperidin-3-yI)-methyi- (7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amine (160.0 g, 0.48 mol), and acetic acid (28.65 g, 0.48 mol). The reactor was purged with three times at 50 psi with nitrogen and three times at 50 psi with hydrogen. Once purging was complete, the reactor was heated to 45-55°C and pressurized to 50 psi with hydrogen through a continuous feed. The hydrogen uptake was monitored until no hydrogen was consumed for 1 hour. The reactor was cooled to 20-300C and purged three times at 50 psi with nitrogen. The reaction mixture was filtered through wet Celite and the filtrate was sent to a clean, dry, nitrogen-purged vessel. A solution of sodium hydroxide (39.33 g) in water (290 ml) was charged and the mixture was stirred for a minimum of 1 hour then heated to 75-900C. The isopropanol was removed by distillation. The reaction mixture was cooled to 20-30°C and 2-methyltetrahydrofuran (1.6 L) was added. The aqueous layer was drained off and the 2-methyltetrahydrofuran was displaced with toluene (1.6 L). The distillation was continued until the final volume was 800 ml. The slurry was cooled to 20-30°C and held for a minimum of 7 hours. The resulting solids were isolated by filtration and washed with toluene (480 ml). After drying under vacuum between 40-50DC for a minimum of 24 hours with a slight nitrogen bleed 102.3 g (87.3%) of the title compound were isolated. Mp 158.6-159.8°C. 1H NMR (400 MHz, CDCI3): δ 11.38 (bs, 1H), 8.30 (s, 1H), 7.05 (d, J=3.5 Hz, 1H), 6.54 (d, J=3.5 Hz, 1H), 4.89-4.87 (m, 1H), 3.39 (s, 3H), 3.27 (dd, J=12.0, 9.3 Hz, 1 H), 3.04 (dd, J=12.0, 3.9 Hz, 1H), 2.94 (td, J=12.6, 3.1 Hz, 1H0, 2.84 (dt, J=12.6, 4.3 Hz, 1H), 2.51-2.48 (m, 1H), 2.12 (bs, 2H), 1.89 (ddt, J=13.7, 10.6, 4 Hz, 1 H), 1.62 (dq, J=13.7, 4Hz, 1 H), 1.07 (d, J=7.3 Hz, 3H). 13C NMR (400 MHz, CDCI3): δ 157.9, 152.0, 151.0, 120.0, 103.0, 102.5, 56.3, 46.2, 42.4, 34.7, 33.4, 32.4, 14.3.  KEY INT
KEY INT
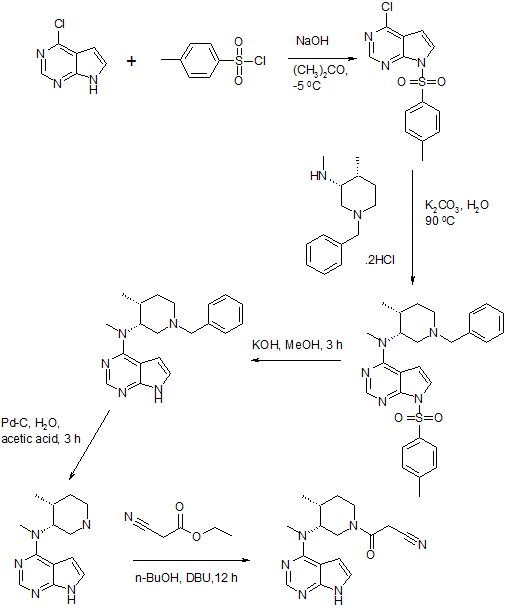
Example 11 Preparation of 3-{(3R, 4R)-4-methyl-3-[methyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3- oxo-propionitrile….TOFACITINIB BASE
To a clean, dry, nitrogen-purged 1.0 L reactor were charged methyl-(4-methyl-piperidin-3-yI)-(7H- pyrroIo[2,3-d]pyrimidin-4-yl)-amine (32.0 g, 0.130 mol), toluene (160 ml), ethyl cyanoacetate (88.53 g, 0.783 mol) and triethyl amine (26.4 g, 0.261 mol). The reaction was heated to 1000C and held for 24 hours. The reaction was washed with water (160 ml). The organic layer concentrated to a volume of 10 ml and water (20 ml) was added. The residual toluene was removed by distillation and the mixture was cooled to room temperature. Acetone (224 ml) was added followed by citric acid (27.57 g, 0.144 mol) in water (76 ml). The resulting slurry was stirred for 7 hours. The solids were isolate by filtration, washed with acetone (96 ml), and dried under vacuum to afford 42.85 g (65.3%) of the title compound. Example 13 Preparation of 3-{(3R, 4R)~4-methyl-3-[methyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3-oxo- propionitrile citrate salt:…………..TOFACITINIB CITRATE To a clean, dry, nitrogen-purged 500 ml reactor were charged methyl-(4-methyl-piperidin-3-yl)-(7H- pyrrolo[2,3-d]pyrimidin-4-yl)-amine (25.0 g, 0.102 mol) and methylene chloride (250 ml). The mixture was stirred at room temperature for a minimum of 2.5 hours. To a clean, dry, nitrogen-purged 1 L reactor were charged cyanoacetic acid (18.2 g, 0.214 mol), methylene chloride (375 ml), and triethyl amine (30.1 ml, 0.214 mol). The mixture was cooled to -15.0— 5.00C over one hour and trimethylacetyl chloride (25.6 ml, 0.204 mol) was added at a rate to maintain the temperature below O0C. The reaction was held for a minimum of 2.5 hours, then the solution of the amine was added at a rate that maintained the temperature below O0C. After stirring for 1 hour, the mixture was warmed to room temperature and 1 M sodium hydroxide (125 ml) was added. The organic layer was washed with water (125 ml) The methylene chloride solution.was displaced with acetone until a volume of 500 ml and a temperature of 55-650C had been achieved. Water (75 ml) was charged to the mixture while maintaining the temperature at 55-65°C. A solution of citric acid (20.76 g, 0.107 mol) in water (25.0) was charged and the mixture was cooled to room temperature. The reactor was stirred for a minimum of 5 hours and then the resulting solids were isolated by filtration and washed with acetone (2×75 ml), which was sent to the filter. The salt was charged into a clean, dry, nitrogen-purged 1L reactor with 2B ethanol (190 ml) and water (190 ml). The slurry was heated to 75-850C for a minimum of 4 hours. The mixture was cooled to 20-300C and stirred for an additional 4 hours. The solids were isolated by filtration and washed with 2B ethanol (190 ml). After drying in a vacuum oven at 500C with a slight nitrogen bleed, 34.6 g (67.3%) of the title compound were isolated. 1H NMR (500 MHz, CZ6-DMSO): δ 8.14 (s, 1 H), 7.11 (d, J=3.6 Hz, 1 H), 6.57 (d, J=3.6 Hz, 1 H), 4.96 (q, J=6.0 Hz, 1 H), 4.00-3.90 (m, 2H), 3.80 (m, 2H), 3.51 (m, 1 H), 3.32 (s, 3H), 2.80 (Abq, J=15.6 Hz, 2H), 2.71 (Abq, J=15.6 Hz, 2H), 2.52-2.50 (m, 1 H), 2.45-2.41 (m, 1 H), 1.81 (m, 1 H), 1.69-1.65 (m, 1 H), 1.04 (d, J=6.9 Hz, 3H)
PAPER
Org. Lett., 2009, 11 (9), pp 2003–2006
DOI: 10.1021/ol900435t
http://pubs.acs.org/doi/full/10.1021/ol900435t 
PATENT
http://www.omicsonline.org/open-access/advances-in-the-inhibitors-of-janus-kinase-2161-0444.1000540.php?aid=29799  ……………..
……………..

 सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
Clinical trials
Rheumatoid arthritis
Phase II clinical trials tested the drug in rheumatoid arthritis patients that had not responded to DMARD therapy. In a tofacitinib monotherapy study, the ACR score improved by at least 20% (ACR-20) in 67% of patients versus 25% who received placebo; and a study that combined the drug with methotrexate achieved ACR-20 in 59% of patients versus 35% who received methotrexate alone. In a psoriasis study, the PASI score improved by at least 75% in between 25 and 67% of patients, depending on the dose, versus 2% in the placebo group.[8] The most important side effects in Phase II studies were increased blood cholesterol levels (12 to 25 mg/dl LDL and 8 to 10 mg/dl HDL at medium dosage levels) andneutropenia.[8] Phase III trials testing the drug in rheumatoid arthritis started in 2007 and are scheduled to run until January 2015.[9] In April 2011, four patients died after beginning clinical trials with tofacitinib. According to Pfizer, only one of the four deaths was related to tofacitinib.[10] By April 2011, three phase III trials for RA had reported positive results.[11] In November 2012, the U.S. FDA approved tofacitinib “to treat adults with moderately to severely active rheumatoid arthritis who have had an inadequate response to, or who are intolerant of, methotrexate.”[12]
Psoriasis
As of April 2011 a phase III trial for psoriasis is under way.[11]
Alopecia
In June 2014, scientists at Yale successfully treated a male patient afflicted with alopecia universalis. The patient was able to grow a full head of hair, eyebrows, eyelashes, facial, armpit, genitalia and other hair. No side effects were reported in the study.[13]
Ulcerative colitis
The OCTAVE study of Tofacitinib in Ulcerative Colitis started in 2012. It is currently enrolling patients, though the NIH trials page states that they expect the trial to close in June 2015.[14]
Vitiligo
In a June 2015 study, a 53-year-old woman with vitiligo showed noticeable improvement after taking tofacitinib for five months.[15]
Development of Safe, Robust, Environmentally Responsible Processes for New Chemical Entities
– Dr. V. Rajappa, Director & Head-Process R&D, Bristol-Myers Squibb, India
A PRESENTATION
 The presentation will load below
The presentation will load below
![]()
Scroll with mouse to view 76 pages


- Herper, Matthew (2 March 2011). “Why Pfizer’s Biggest Experimental Drug Got A Name Change”. Forbes. Retrieved 3 March 2011.
- Kremer, J. M.; Bloom, B. J.; Breedveld, F. C.; Coombs, J. H.; Fletcher, M. P.; Gruben, D.; Krishnaswami, S.; Burgos-Vargas, R. N.; Wilkinson, B.; Zerbini, C. A. F.; Zwillich, S. H. (2009). “The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis: Results of a double-blind, placebo-controlled phase IIa trial of three dosage levels of CP-690,550 versus placebo”. Arthritis & Rheumatism 60 (7): 1895–1905. doi:10.1002/art.24567. PMID 19565475.
- “Tasocitinib”. Drugs in R&D 10 (4): 271–284. 2010. doi:10.2165/11588080-000000000-00000. PMC 3585773. PMID 21171673.
- Ghoreschi, K.; Jesson, M. I.; Li, X.; Lee, J. L.; Ghosh, S.; Alsup, J. W.; Warner, J. D.; Tanaka, M.; Steward-Tharp, S. M.; Gadina, M.; Thomas, C. J.; Minnerly, J. C.; Storer, C. E.; Labranche, T. P.; Radi, Z. A.; Dowty, M. E.; Head, R. D.; Meyer, D. M.; Kishore, N.; O’Shea, J. J. (2011). “Modulation of Innate and Adaptive Immune Responses by Tofacitinib (CP-690,550)”. J Immunol. 186 (7): 4234–4243. doi:10.4049/jimmunol.1003668. PMC 3108067. PMID 21383241.
- ^ Jump up to:a b c “Seeking Profit for Taxpayers in Potential of New Drug”, Jonathan Weisman, New York Times, March 18, 2013
- Ken Garber (9 January 2013). “Pfizer’s first-in-class JAK inhibitor pricey for rheumatoid arthritis market”. Nature Biotechnology 31 (1): 3–4. doi:10.1038/nbt0113-3. PMID 23302910.
- Jump up^ Moisan A, et al. White-to-brown metabolic conversion of human adipocytes by JAK inhibition. Nature Cell Biology, 8 December 2014. DOI 10.1038/ncb3075
- “EULAR: JAK Inhibitor Effective in RA But Safety Worries Remain”. MedPage Today. June 2009. Retrieved 9 February 2011.
- Clinical trial number NCT00413699 for “Long-Term Effectiveness And Safety Of CP-690,550 For The Treatment Of Rheumatoid Arthritis” at ClinicalTrials.gov
- Matthew Herper. “Pfizer’s Key Drug Walks A Tightrope”. Forbes.
- “Two Phase III Studies Confirm Benefits of Pfizer’s Tofacitinib Against Active RA”. 28 Apr 2011.
- “FDA approves Xeljanz for rheumatoid arthritis”. 6 Nov 2012.
- “Hairless man grows full head of hair in yale arthritis drug trial”. 19 Jun 2014.
- https://clinicaltrials.gov/ct2/show/NCT01465763?term=A3921094&rank=1
- “This Drug Brought Pigment Back for Woman with Vitiligo”. TIME. June 27, 2015. Retrieved June 29, 2015.
- Nordqvist, Christian (27 April 2013). “Pfizer’s Arthritis Drug Xeljanz (tofacitinib) Receives A Negative Opinion In Europe”. Medical News Today. Retrieved 2 August 2013.
- “”XALEJANZ PRESCRIBING INFORMATION @ Labeling.Pfizer.com””.
SEE………http://orgspectroscopyint.blogspot.in/2014/12/tofacitinib-citrate.html
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
3-[(3R,4R)-4-methyl-3-[methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]piperidin-1-yl]-3-oxopropanenitrile
|
|
| Clinical data | |
| Trade names | Xeljanz, Jakvinus |
| AHFS/Drugs.com | entry |
| Licence data | US FDA:link |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration | Oral |
| Pharmacokinetic data | |
| Bioavailability | 74% |
| Protein binding | 40% |
| Metabolism | Hepatic (via CYP3A4 andCYP2C19) |
| Biological half-life | 3 hours |
| Excretion | Urine |
| Identifiers | |
| CAS Registry Number | 477600-75-2 |
| ATC code | L04AA29 |
| PubChem | CID: 9926791 |
| IUPHAR/BPS | 5677 |
| DrugBank | DB08183 |
| ChemSpider | 8102425 |
| UNII | 87LA6FU830 |
| ChEBI | CHEBI:71200 |
| ChEMBL | CHEMBL221959 |
| Synonyms | CP-690550 |
| Chemical data | |
| Formula | C16H20N6O |
| Molecular mass | 312.369 g/mol |
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.

सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
LIONEL MY SONHe was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
//////
INCB-039110, Janus kinase-1 (JAK-1) inhibitor……..for the treatment of rheumatoid arthritis, myelofibrosis, rheumatoid arthritis and plaque psoriasis.
 INCB-39110,
INCB-39110,
CAS 1334298-90-6
INCB-039110, Jak1 tyrosine kinase inhibitor
- 3-Azetidineacetonitrile, 1-[1-[[3-fluoro-2-(trifluoromethyl)-4-pyridinyl]carbonyl]-4-piperidinyl]-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]-
C26H23F4N9O (MW, 553.51)
{ l- { l-[3-fluoro-2- (trifluoromethyl)isonicotinoyl]piperidin-4-yl}-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4- yl)-lH-pyrazol-l-yl]azetidin-3-yl}acetonitrile
2-(3-(4-(7H-pyrrolo[2,3-( Jpyrimidin-4-yl)-lH- pyrazol- 1 -yl)- 1 -( 1 -(3 -fluoro-2-(trifluoromethyl)isonicotinoyl)piperidin-4-yl)azetidin- 3-yl)acetonitrile
2-(3-(4-(7H- Pyrrolo[2,3 -i/]pyrimidin-4-yl)- lH-pyrazol- 1 -yl)- 1 -(1 -(3 -fluoro-2- (trifluoromethyl)isonicotinoyl)piperidin-4-yl)azetidin-3-yl)acetonitrile adipate MAY BE THE DRUG… HAS CAS 1334302-63-4
 ADIPATE OF INCB-39110
ADIPATE OF INCB-39110
ALSO/OR

3-Azetidineacetonitrile, 1-[1-(3-fluorobenzoyl)-4-methyl-4-piperidinyl]-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]-, 2,2,2-trifluoroacetateMAY BE THE DRUG ????… HAS CAS 1334300-52-5
US 2011/0224190 is the pdt patent

| Incyte Corporation |
Clinical trials
IN PHASE 2 for the treatment of rheumatoid arthritis, myelofibrosis, rheumatoid arthritis and plaque psoriasis.
SEE
http://clinicaltrials.gov/show/NCT01633372
Jak2 tyrosine kinase inhibitor; Jak1 tyrosine kinase inhibitor
Breast tumor; Chronic obstructive pulmonary disease; Crohns disease; Inflammatory bowel disease; Influenza virus infection; Insulin dependent diabetes; Liver tumor; Multiple sclerosis; Prostate tumor; Rheumatoid arthritis; SARS coronavirus infection
Used for treating cancers (eg prostate cancer, hepatic cancer and pancreatic cancer) and autoimmune diseases. Follows on from WO2013036611, claiming the process for preparing the same JAK inhibitor. Incyte is developing INCB-39110 (phase II, September 2014), for the oral treatment of myelofibrosis, hematological neoplasm and non-small cell lung cancer.
INCB-039110 is a Jak1 inhibitor in phase II clinical studies at Incyte for the treatment of rheumatoid arthritis, myelofibrosis, rheumatoid arthritis and plaque psoriasis. The company is also conducting a phase I clinical study for the treatment of advanced or metastatic solid tumors.
Protein kinases (PKs) regulate divINCB-039110 is a Jak1 inhibitor in phase II clinical studies at Incyte for the treatment of rheumatoid arthritis, myelofibrosis, rheumatoid arthritis and plaque psoriasis. The company is also conducting a phase I clinical study for the treatment of advanced or metastatic solid tumors.erse biological processes including cell growth, survival, differentiation, organ formation, morphogenesis, neovascularization, tissue repair, and regeneration, among others. Protein kinases also play specialized roles in a host of human diseases including cancer. Cytokines, low-molecular weight polypeptides or glycoproteins, regulate many pathways involved in the host
inflammatory response to sepsis. Cytokines influence cell differentiation,
proliferation and activation, and can modulate both pro-inflammatory and antiinflammatory responses to allow the host to react appropriately to pathogens.
Signaling of a wide range of cytokines involves the Janus kinase family (JAKs) of protein tyrosine kinases and Signal Transducers and Activators of Transcription
(STATs). There are four known mammalian JAKs: JAK1 (Janus kinase-1), JAK2, JAK3 (also known as Janus kinase, leukocyte; JAKL; and L-JAK), and TYK2
(protein-tyros ine kinase 2).
Cytokine-stimulated immune and inflammatory responses contribute to pathogenesis of diseases: pathologies such as severe combined immunodeficiency (SCID) arise from suppression of the immune system, while a hyperactive or inappropriate immune/inflammatory response contributes to the pathology of autoimmune diseases (e.g., asthma, systemic lupus erythematosus, thyroiditis, 20443-0253WO1 (INCY0124-WO1) PATENT myocarditis), and illnesses such as scleroderma and osteoarthritis (Ortmann, R. A., T. Cheng, et al. (2000) Arthritis Res 2(1): 16-32).
Deficiencies in expression of JAKs are associated with many disease states. For example, Jakl-/- mice are runted at birth, fail to nurse, and die perinatally (Rodig, S. J., M. A. Meraz, et al. (1998) Cell 93(3): 373-83). Jak2-/- mouse embryos are anemic and die around day 12.5 postcoitum due to the absence of definitive
erythropoiesis.
The JAK/STAT pathway, and in particular all four JAKs, are believed to play a role in the pathogenesis of asthmatic response, chronic obstructive pulmonary disease, bronchitis, and other related inflammatory diseases of the lower respiratory tract. Multiple cytokines that signal through JAKs have been linked to inflammatory diseases/conditions of the upper respiratory tract, such as those affecting the nose and sinuses (e.g., rhinitis and sinusitis) whether classically allergic reactions or not. The JAK/STAT pathway has also been implicated in inflammatory diseases/conditions of the eye and chronic allergic responses.
Activation of JAK/STAT in cancers may occur by cytokine stimulation (e.g. IL-6 or GM-CSF) or by a reduction in the endogenous suppressors of JAK signaling such as SOCS (suppressor or cytokine signaling) or PIAS (protein inhibitor of activated STAT) (Boudny, V., and Kovarik, J., Neoplasm. 49:349-355, 2002).
Activation of STAT signaling, as well as other pathways downstream of JAKs (e.g., Akt), has been correlated with poor prognosis in many cancer types (Bowman, T., et al. Oncogene 19:2474-2488, 2000). Elevated levels of circulating cytokines that signal through JAK/STAT play a causal role in cachexia and/or chronic fatigue. As such, JAK inhibition may be beneficial to cancer patients for reasons that extend beyond potential anti-tumor activity.
JAK2 tyrosine kinase can be beneficial for patients with myeloproliferative disorders, e.g., polycythemia vera (PV), essential thrombocythemia (ET), myeloid metaplasia with myelofibrosis (MMM) (Levin, et al, Cancer Cell, vol. 7, 2005: 387- 397). Inhibition of the JAK2V617F kinase decreases proliferation of hematopoietic cells, suggesting JAK2 as a potential target for pharmacologic inhibition in patients with PV, ET, and MMM. 20443-0253WO1 (INCY0124-WO1) PATENT
Inhibition of the JAKs may benefit patients suffering from skin immune disorders such as psoriasis, and skin sensitization. The maintenance of psoriasis is believed to depend on a number of inflammatory cytokines in addition to various chemokines and growth factors (JCI, 1 13 : 1664-1675), many of which signal through JAKs (Adv Pharmacol. 2000;47: 113-74).
JAKl plays a central role in a number of cytokine and growth factor signaling pathways that, when dysregulated, can result in or contribute to disease states. For example, IL-6 levels are elevated in rheumatoid arthritis, a disease in which it has been suggested to have detrimental effects (Fonesca, J.E. et al, Autoimmunity
Reviews, 8:538-42, 2009). Because IL-6 signals, at least in part, through JAKl, antagonizing IL-6 directly or indirectly through JAKl inhibition is expected to provide clinical benefit (Guschin, D., N., et al Embo J 14: 1421, 1995; Smolen, J. S., et al. Lancet 371 :987, 2008). Moreover, in some cancers JAKl is mutated resulting in constitutive undesirable tumor cell growth and survival (Mullighan CG, Proc Natl Acad Sci U S A.106:9414-8, 2009; Flex E., et al.J Exp Med. 205:751-8, 2008). In other autoimmune diseases and cancers elevated systemic levels of inflammatory cytokines that activate JAKl may also contribute to the disease and/or associated symptoms. Therefore, patients with such diseases may benefit from JAKl inhibition. Selective inhibitors of JAKl may be efficacious while avoiding unnecessary and potentially undesirable effects of inhibiting other JAK kinases.
Selective inhibitors of JAKl, relative to other JAK kinases, may have multiple therapeutic advantages over less selective inhibitors. With respect to selectivity against JAK2, a number of important cytokines and growth factors signal through JAK2 including, for example, erythropoietin (Epo) and thrombopoietin (Tpo)
(Parganas E, et al. Cell. 93:385-95, 1998). Epo is a key growth factor for red blood cells production; hence a paucity of Epo-dependent signaling can result in reduced numbers of red blood cells and anemia (Kaushansky K, NEJM 354:2034-45, 2006). Tpo, another example of a JAK2-dependent growth factor, plays a central role in controlling the proliferation and maturation of megakaryocytes – the cells from which platelets are produced (Kaushansky K, NEJM 354:2034-45, 2006). As such, reduced Tpo signaling would decrease megakaryocyte numbers (megakaryocytopenia) and lower circulating platelet counts (thrombocytopenia). This can result in undesirable 20443-0253WO1 (INCY0124-WO1) PATENT and/or uncontrollable bleeding. Reduced inhibition of other JAKs, such as JAK3 and Tyk2, may also be desirable as humans lacking functional version of these kinases have been shown to suffer from numerous maladies such as severe-combined immunodeficiency or hyperimmunoglobulin E syndrome (Minegishi, Y, et al.
Immunity 25:745-55, 2006; Macchi P, et al. Nature. 377:65-8, 1995). Therefore a JAK1 inhibitor with reduced affinity for other JAKs would have significant
advantages over a less-selective inhibitor with respect to reduced side effects involving immune suppression, anemia and thrombocytopenia.

……………………….
http://www.google.com/patents/US20110224190
EXAMPLESThe example compounds below containing one or more chiral centers were obtained in enantiomerically pure form or as scalemic mixtures, unless otherwise specified.Unless otherwise indicated, the example compounds were purified by preparativeHPLC using acidic conditions (method A) and were obtained as a TFA salt or using basic conditions (method B) and were obtained as a free base.Method A:Column: Waters Sun Fire C18, 5 μm particle size, 30×100 mm;
Mobile phase: water (0.1% TFA)/acetonitrile
Flow rate: 60 mL/min
Gradient: 5 min or 12 min from 5% acetonitrile/95% water to 100% acetonitrileMethod B:Column: Waters X Bridge C18, 5 μm particle size, 30×100 mm;
Mobile phase: water (0.15% NH4OH)/acetonitrileMethod C:Column: C18 column, 5 μm OBD
Mobile phase: water+0.05% NH4OH (A), CH3CN+0.05% NH4OH (B)Gradient: 5% B to 100% B in 15 minFlow rate: 60 mL/minExample 1
{1-{1-[3-Fluoro-2-(trifluoromethyl)isonicotinoyl]piperidin-4-yl}-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]azetidin-3-yl}acetonitrile 










Mobile phase: water (0.1% TFA)/acetonitrile
Flow rate: 60 mL/min
Gradient: 5 min or 12 min from 5% acetonitrile/95% water to 100% acetonitrileMethod B:Column: Waters X Bridge C18, 5 μm particle size, 30×100 mm;
Mobile phase: water (0.15% NH4OH)/acetonitrileMethod C:Column: C18 column, 5 μm OBD
Mobile phase: water+0.05% NH4OH (A), CH3CN+0.05% NH4OH (B)Gradient: 5% B to 100% B in 15 minFlow rate: 60 mL/minExample 1
{1-{1-[3-Fluoro-2-(trifluoromethyl)isonicotinoyl]piperidin-4-yl}-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]azetidin-3-yl}acetonitrile

Step A: tert-Butyl 3-Oxoazetidine-1-carboxylate

To a mixture of tert-butyl 3-hydroxyazetidine-1-carboxylate (10.0 g, 57.7 mmol), dimethyl sulfoxide (24.0 mL, 338 mmol), triethylamine (40 mL, 300 mmol) and methylene chloride (2.0 mL) was added sulfur trioxide-pyridine complex (40 g, 200 mmol) portionwise at 0° C. The mixture was stirred for 3 hours, quenched with brine, and extracted with methylene chloride. The combined extracts were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column (0-6% ethyl acetate (EtOAc) in hexanes) to give tert-butyl 3-oxoazetidine-1-carboxylate (5.1 g, 52% yield).
Step B: tert-Butyl 3-(Cyanomethylene)azetidine-1-carboxylate

An oven-dried 1 L 4-neck round bottom flask fitted with stir bar, septa, nitrogen inlet, 250 ml addition funnel and thermocouple was charged with sodium hydride (5.6 g, 0.14 mol) and tetrahydrofuran (THF) (140 mL) under a nitrogen atmosphere. The mixture was chilled to 3° C., and then charged with diethyl cyanomethylphosphonate (22.4 mL, 0.138 mol) dropwise via a syringe over 20 minutes. The solution became a light yellow slurry. The reaction was then stirred for 75 minutes while warming to 18.2° C. A solution of tert-butyl 3-oxoazetidine-1-carboxylate (20 g, 0.1 mol) in tetrahydrofuran (280 mL) was prepared in an oven-dried round bottom, charged to the addition funnel via canula, then added to the reaction mixture dropwise over 25 minutes. The reaction solution became red in color. The reaction was allowed to stir overnight. The reaction was checked after 24 hours by TLC (70% hexane/EtOAc) and found to be complete. The reaction was diluted with 200 mL of 20% brine and 250 mL of EtOAc. The solution was partitioned and the aqueous phase was extracted with 250 mL of EtOAc. The combined organic phase was dried over MgSO4 and filtered, evaporated under reduced pressure, and purified by flash chromatography (0% to 20% EtOAc/hexanes, 150 g flash column) to give the desired product, tert-butyl 3-(cyanomethylene)azetidine-1-carboxylate (15 g, 66.1% yield).
Step C: 4-Chloro-7-{[2-(trimethylsilyl)ethoxy]methyl}-7H-pyrrolo[2,3-d]pyrimidine

To a suspension of sodium hydride (36.141 g, 903.62 mmol) in N,N-dimethylacetamide (118 mL) at −5° C. (ice/salt bath) was added a dark solution of 4-chloropyrrolo[2,3-d]pyrimidine (119.37 g, 777.30 mmol) in N,N-dimethylacetamide (237 mL) slowly. The flask and addition funnel were rinsed with N,N-dimethylacetamide (30 mL). A large amount of gas was evolved immediately. The mixture became a slightly cloudy orange mixture. The mixture was stirred at 0° C. for 60 min to give a light brown turbid mixture. To the mixture was slowly added [2-(trimethylsilyl)ethoxy]methyl chloride (152.40 g, 914.11 mmol) and the reaction was stirred at 0° C. for 1 h. The reaction was quenched by addition of 12 mL of H2O slowly. More water (120 mL) was added followed by methyl tert-butyl ether (MTBE) (120 mL). The mixture was stirred for 10 min. The organic layer was separated. The aqueous layer was extracted with another portion of MTBE (120 mL). The organic extracts were combined, washed with brine (120 mL×2) and concentrated under reduced pressure to give the crude product 4-chloro-7-{[2-(trimethylsilyl)ethoxy]methyl}-7H-pyrrolo[2,3-d]pyrimidine as a dark oil. Yield: 85.07 g (97%); LC-MS: 284.1 (M+H)+. It was carried to the next reaction without purification.
Step D: 4-(1H-Pyrazol-4-yl)-7-{[2-(trimethylsilyl)ethoxy]methyl}-7H-pyrrolo[2,3-d]pyrimidine

A 1000 mL round bottom flask was charged with 4-chloro-7-{[2-(trimethylsilyl)ethoxy]methyl}-7H-pyrrolo[2,3-d]pyrimidine (10.00 g, 35.23 mmol), 1-butanol (25.0 mL), 1-(1-ethoxyethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (15.66 g, 52.85 mmol), water (25.0 mL) and potassium carbonate (12.17 g, 88.08 mmol). This solution was degased 4 times, filling with nitrogen each time. To the solution was added tetrakis(triphenylphosphine)palladium(0) (4.071 g, 3.523 mmol). The solution was degased 4 times, filling with nitrogen each time. The mixture was stirred overnight at 100° C. After being cooled to room temperature, the mixture was filtered through a bed of celite and the celite was rinsed with ethyl acetate (42 mL). The filtrate was combined, and the organic layer was separated. The aqueous layer was extracted with ethyl acetate. The organic extracts were combined and concentrated under vacuum with a bath temperature of 30-70° C. to give the final compound 4-(1H-pyrazol-4-yl)-7-{[2-(trimethylsilyl)ethoxy]methyl}-7H-pyrrolo[2,3-d]pyrimidine. Yield: 78%. LC-MS: 316.2 (M+H)+.
Step E: tert-Butyl 3-(Cyanomethyl)-3-[4-(7-{[2-(trimethylsilyl)ethoxy]methyl}-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]azetidine-1-carboxylate

A 2 L round bottom flask fitted with overhead stirring, septa and nitrogen inlet was charged with tert-butyl 3-(cyanomethylene)azetidine-1-carboxylate (9.17 g, 0.0472 mol), 4-(1H-pyrazol-4-yl)-7-{[2-(trimethylsilyl)ethoxy]methyl}-7H-pyrrolo[2,3-d]pyrimidine (14.9 g, 0.0472 mol) and acetonitrile (300 mL). The resulting solution was heterogeneous. To the solution was added 1,8-diazabicyclo[5.4.0]undec-7-ene (8.48 mL, 0.0567 mol) portionwise via syringe over 3 min at room temperature. The solution slowly became homogeneous and yellow in color. The reaction was allowed to stir at room temperature for 3 h. The reaction was complete by HPLC and LC/MS and was concentrated by rotary evaporation to remove acetonitrile (˜150 mL). EtOAc (100 mL) was added followed by 100 ml of 20% brine. The two phases were partitioned. The aqueous phase was extracted with 150 mL of EtOAC. The combine organic phases were dried over MgSO4, filtered and concentrated to yield an orange oil. Purification by flash chromatography (150 grams silica, 60% EtOAc/hexanes, loaded with CH2Cl2) yielded the title compound tert-butyl 3-(cyanomethyl)-3-[4-(7-{[2-(trimethylsilyl)ethoxy]methyl}-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]azetidine-1-carboxylate as a yellow oil (21.1 g, 88% yield). LC-MS: [M+H]+=510.3.
Step F: {3-[4-(7-{[2-(Trimethylsilyl)ethoxy]methyl}-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]azetidin-3-yl}acetonitrile dihydrochloride

To a solution of tert-butyl 3-(cyanomethyl)-3-[4-(7-{[2-(trimethylsilyl)ethoxy]methyl}-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]azetidine-1-carboxylate (2 g, 3.9 mmol) in 10 mL of THF was added 10 mL of 4 N HCl in dioxane. The solution was stirred at room temperature for 1 hour and concentrated in vacuo to provide 1.9 g (99%) of the title compound as a white powder solid, which was used for the next reaction without purification. LC-MS: [M+H]+=410.3.
Step G: tert-Butyl 4-{3-(Cyanomethyl)-3-[4-(7-{[2-(trimethylsilyl)ethoxy]methyl}-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]azetidin-1-yl}piperidine-1-carboxylate

Into the solution of {3-[4-(7-{[2-(trimethylsilyl)ethoxy]methyl}-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]azetidin-3-yl}acetonitrile dihydrochloride (2.6 g, 6.3 mmol), tert-butyl 4-oxo-1-piperidinecarboxylate (1.3 g, 6.3 mmol) in THF (30 mL) were added N,N-diisopropylethylamine (4.4 mL, 25 mmol) and sodium triacetoxyborohydride (2.2 g, 10 mmol). The mixture was stirred at room temperature overnight. After adding 20 mL of brine, the solution was extracted with EtOAc. The extract was dried over anhydrous Na2SO4 and concentrated. The residue was purified by combiflash column eluting with 30-80% EtOAc in hexanes to give the desired product, tert-butyl 4-{3-(cyanomethyl)-3-[4-(7-{[2-(trimethylsilyl)ethoxy]methyl}-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]azetidin-1-yl}piperidine-1-carboxylate. Yield: 3.2 g (86%); LC-MS: [M+H]+=593.3.
Step H: {1-Piperidin-4-yl-3-[4-(7-{[2-(trimethylsilyl)ethoxy]methyl}-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]azetidin-3-yl}acetonitrile trihydrochloride

To a solution of tert-butyl 4-{3-(cyanomethyl)-3-[4-(7-{[2-(trimethylsilyl)ethoxy]methyl}-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]azetidin-1-yl}piperidine-1-carboxylate (3.2 g, 5.4 mmol) in 10 mL of THF was added 10 mL of 4 N HCl in dioxane. The reaction mixture was stirred at room temperature for 2 hours. Removing solvents under reduced pressure yielded 3.25 g (100%) of {1-piperidin-4-yl-3-[4-(7-{[2-(trimethylsilyl)ethoxy]methyl}-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]azetidin-3-yl}acetonitrile trihydrochloride as a white powder solid, which was used directly in the next reaction. LC-MS: [M+H]+=493.3. 1H NMR (400 MHz, DMSO-d6): δ 9.42 (s 1H), 9.21 (s, 1H), 8.89 (s, 1H), 8.69 (s, 1H), 7.97 (s, 1H), 7.39 (d, 1H), 5.68 (s, 2H), 4.96 (d, 2H), 4.56 (m, 2H), 4.02-3.63 (m, 2H), 3.55 (s, 2H), 3.53 (t, 2H), 3.49-3.31 (3, 3H), 2.81 (m, 2H), 2.12 (d, 2H), 1.79 (m, 2H), 0.83 (t, 2H), −0.10 (s, 9H).
Step I: {1-{1-[3-Fluoro-2-(trifluoromethyl)isonicotinoyl]piperidin-4-yl}-3-[4-(7-{[2-(trimethylsilyl)ethoxy]methyl}-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]azetidin-3-yl}acetonitrile

A mixture of {1-piperidin-4-yl-3-[4-(7-{[2-(trimethylsilyl)ethoxy]methyl}-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]azetidin-3-yl}acetonitrile trihydrochloride (1.22 g, 2.03 mmol), 3-fluoro-2-(trifluoromethyl)isonicotinic acid (460 mg, 2.2 mmol), benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate (1.07 g, 2.42 mmol), and triethylamine (2.0 mL, 14 mmol) in dimethylformamide (DMF) (20.0 mL) was stirred at room temperature overnight. LS-MS showed the reaction was complete. EtOAc (60 mL) and saturated NaHCO3 aqueous solution (60 mL) were added to the reaction mixture. After stirring at room temperature for 10 minutes, the organic phase was separated and the aqueous layer was extracted with EtOAc three times. The combined organic phase was washed with brine, dried over anhydrous Na2SO4, filtered and evaporated under reduced pressure. Purification by flash chromatography provided the desired product {1-{1-[3-fluoro-2-(trifluoromethyl)isonicotinoyl]piperidin-4-yl}-3-[4-(7-{[2-(trimethylsilyl)ethoxy]methyl}-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]azetidin-3-yl}acetonitrile. LC-MS: 684.3 (M+H)+.
Step J: {1-{1-[3-Fluoro-2-(trifluoromethyl)isonicotinoyl]piperidin-4-yl}-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]azetidin-3-yl}acetonitrile

Into a solution of {1-{1-[3-fluoro-2-(trifluoromethyl)isonicotinoyl]piperidin-4-yl}-3-[4-(7-{[2-(trimethylsilyl)ethoxy]methyl}-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]azetidin-3-yl}acetonitrile (56 mg, 0.1 mmol) in methylene chloride (1.5 mL) was added trifluoroacetic acid (1.5 mL). The mixture was stirred at room temperature for 2 hours. After removing the solvents in vacuum, the residue was dissolved in a methanol solution containing 20% ethylenediamine. After being stirred at room temperature for 1 hour, the solution was purified by HPLC (method B) to give the title compound. LC-MS: 554.3 (M+H)+; 1H NMR (400 MHz, CDCl3): 9.71 (s, 1H), 8.82 (s, 1H), 8.55 (d, J=4.6 Hz, 1H), 8.39 (s, 1H), 8.30 (s, 1H), 7.52 (t, J=4.6 Hz, 1H), 7.39 (dd, J1=3.4 Hz, J2=1.5 Hz, 1H), 6.77 (dd, J1=3.6 Hz, J2=0.7 Hz, 1H), 4.18 (m, 1H), 3.75 (m, 2H), 3.63 (dd, J1=7.8 Hz, J2=3.7 Hz, 2H), 3.45 (m, 2H), 3.38 (s, 2H), 3.11 (m, 1H), 2.57 (m, 1H), 1.72 (m, 1H), 1.60 (m, 1H), 1.48 (m, 1H), 1.40 (m, 1H).

………………………..
http://www.google.com/patents/US20130060026
Example 1Synthesis of 4-(1H-pyrazol-4-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine (5)

Step 1. 4-Chloro-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine (3)
To a flask equipped with a nitrogen inlet, an addition funnel, a thermowell, and the mechanical stirrer was added 4-chloro-7H-pyrrolo[2,3-d]pyrimidine (1, 600 g, 3.91 mol) and N,N-dimethylacetimide (DMAC, 9.6 L) at room temperature. The mixture was cooled to 0-5° C. in an ice/brine bath before solid sodium hydride (NaH, 60 wt %, 174 g, 4.35 mol, 1.1 equiv) was added in portions at 0-5° C. The reaction mixture turned into a dark solution after 15 minutes. Trimethylsilylethoxymethyl chloride (2, SEM-Cl, 763 mL, 4.31 mol, 1.1 equiv) was then added slowly via an addition funnel at a rate that the internal reaction temperature did not exceed 5° C. The reaction mixture was then stirred at 0-5° C. for 30 minutes. When the reaction was deemed complete determined by TLC and HPLC, the reaction mixture was quenched by water (1 L). The mixture was then diluted with water (12 L) and methyl tert-butyl ether (MTBE) (8 L). The two layers were separated and the aqueous layer was extracted with MTBE (8 L). The combined organic layers were washed with water (2×4 L) and brine (4 L) and solvent switched to 1-butanol. The solution of crude product (3) in 1-butanol was used in the subsequent Suzuki coupling reaction without further purification. Alternatively, the organic solution of the crude product (3) in MTBE was dried over sodium sulfate (Na2SO4). The solvents were removed under reduced pressure. The residue was then dissolved in heptane (2 L), filtered and loaded onto a silica gel (SiO2, 3.5 Kg) column eluting with heptane (6 L), 95% heptane/ethyl acetate (12 L), 90% heptane/ethyl acetate (10 L), and finally 80% heptane/ethyl acetate (10 L). The fractions containing the pure desired product were combined and concentrated under reduced pressure to give 4-chloro-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine (3, 987 g, 1109.8 g theoretical, 88.9% yield) as a pale yellow oil which partially solidified to an oily solid on standing at room temperature. For 3: 1H NMR (DMSO-d6, 300 MHz) δ 8.67 (s, 1H), 7.87 (d, 1H, J=3.8 Hz), 6.71 (d, 1H, J=3.6 Hz), 5.63 (s, 2H), 3.50 (t, 2H, J=7.9 Hz), 0.80 (t, 2H, J=8.1 Hz), 1.24 (s, 9H) ppm; 13C NMR (DMSO-d6, 100 MHz) δ 151.3, 150.8, 150.7, 131.5, 116.9, 99.3, 72.9, 65.8, 17.1, −1.48 ppm; C12H18ClN3OSi (MW 283.83), LCMS (EI) m/e 284/286 (M++H).
Step 2. 4-(1H-Pyrazol-4-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine (5)
To a reactor equipped with the overhead stirrer, a condenser, a thermowell, and a nitrogen inlet was charged water (H2O, 9.0 L), solid potassium carbonate (K2CO3, 4461 g, 32.28 mol, 2.42 equiv), 4-chloro-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine (3, 3597 g, 12.67 mol), 1-(1-ethoxyethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (4, 3550 g, 13.34 mol, 1.05 equiv), and 1-butanol (27 L) at room temperature. The resulting reaction mixture was degassed three timed backfilling with nitrogen each time before being treated with tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4, 46 g, 0.040 mol, 0.003 equiv) at room temperature. The resulting reaction mixture was heated to gentle reflux (about 90° C.) for 1-4 hours. When the reaction was deemed complete determined by HPLC, the reaction mixture was gradually cooled down to room temperature before being filtered through a Celite bed. The Celite bed was washed with ethyl acetate (2×2 L) before the filtrates and washing solution were combined. The two layers were separated, and the aqueous layer was extracted with ethyl acetate (12 L). The combined organic layers were concentrated under reduced pressure to remove solvents, and the crude 4-(1-(1-ethoxyethyl)-1H-pyrazol-4-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine (6) was directly charged back to the reactor with tetrahydrofuran (THF, 4.2 L) for the subsequent acid-promoted de-protection reaction without further purification.
To a suspension of crude 4-(1-(1-ethoxyethyl)-1H-pyrazol-4-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine (6), made as described above, in tetrahydrofuran (THF, 4.2 L) in the reactor was charged water (H2O, 20.8 L), and a 10% aqueous HCl solution (16.2 L, 45.89 mol, 3.44 equiv) at room temperature. The resulting reaction mixture was stirred at 16-30° C. for 2-5 hours. When the reaction was deemed complete by HPLC analysis, the reaction mixture was treated with a 30% aqueous sodium hydroxide (NaOH) solution (4 L, 50.42 mol, 3.78 equiv) at room temperature. The resulting reaction mixture was stirred at room temperature for 1-2 hours. The solids were collected by filtration and washed with water (2×5 L). The wet cake was charged back to the reactor with acetonitrile (21.6 L), and resulting suspension was heated to gentle reflux for 1-2 hours. The clear solution was then gradually cooled down to room temperature with stirring, and solids were precipitated out from the solution with cooling. The mixture was stirred at room temperature for an additional 1-2 hours. The solids were collected by filtration, washed with acetonitrile (2×3.5 L), and dried in oven under reduced pressure at 45-55° C. to constant weight to afford 4-(1H-pyrazol-4-yl)-7-(2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine (5, 3281.7 g, 3996.8 g theoretical, 82.1% yield) as white crystalline solids (99.5 area % by HPLC). For 5: 1H NMR (DMSO-d6, 400 MHz) δ 13.41 (br. s, 1H), 8.74 (s, 1H), 8.67 (br. s, 1H), 8.35 (br. s, 1H), 7.72 (d, 1H, J=3.7 Hz), 7.10 (d, 1H, J=3.7 Hz), 5.61 (s, 2H), 3.51 (t, 2H, J=8.2 Hz), 0.81 (t, 2H, J=8.2 Hz), 0.13 (s, 9H) ppm; C15H21N5OSi (MW, 315.45), LCMS (EI) m/e 316 (M++H).
Example 2tert-Butyl 3-(cyanomethylene)azetidine-1-carboxylate (13)

Step 1. 1-Benzhydrylazetidin-3-ol hydrochloride (9)
A solution of diphenylmethanamine (7, 2737 g, 15.0 mol, 1.04 equiv) in methanol (MeOH, 6 L) was treated with 2-(chloromethyl)oxirane (8, 1330 g, 14.5 mol) from an addition funnel at room temperature. During the initial addition a slight endotherm was noticed. The resulting reaction mixture was stirred at room temperature for 3 days before being warmed to reflux for an additional 3 days. When TLC showed that the reaction was deemed complete, the reaction mixture was first cooled down to room temperature and then to 0-5° C. in an ice bath. The solids were collected by filtration and washed with acetone (4 L) to give the first crop of the crude desired product (9, 1516 g). The filtrate was concentrated under reduced pressure and the resulting semisolid was diluted with acetone (1 L). This solid was then collected by filtration to give the second crop of the crude desired product (9, 221 g). The crude product, 1-benzhydrylazetidin-3-ol hydrochloride (9, 1737 g, 3998.7 g theoretical, 43.4% yield), was found to be sufficiently pure to be used in the subsequent reaction without further purification. For 9: 1H NMR (DMSO-d6, 300 MHz), δ 12.28 (br. d, 1H), 7.7 (m, 5H), 7.49 (m, 5H), 6.38 (d, 1H), 4.72 (br. s, 1H), 4.46 (m, 1H), 4.12 (m, 2H), 3.85 (m, 2H) ppm; C16H18ClNO (free base of 9, C16K7NO MW, 239.31), LCMS (EI) m/e 240 (M++H).
Step 2. tert-Butyl 3-hydroxyazetidine-1-carboxylate (10)
A suspension of 1-benzhydrylazetidin-3-ol hydrochloride (9, 625 g, 2.27 mol) in a 10% solution of aqueous sodium carbonate (Na2CO3, 5 L) and dichloromethane (CH2Cl2, 5 L) was stirred at room temperature until all solids were dissolved. The two layers were separated, and the aqueous layer was extracted with dichloromethane (CH2Cl2, 2 L). The combined organics extracts were dried over sodium sulfate (Na2SO4) and concentrated under reduced pressure. This resulting crude free base of 9 was then dissolved in THF (6 L) and the solution was placed into a large Parr bomb. Di-tert-butyl dicarbonate (BOC2O, 545 g, 2.5 mol, 1.1 equiv) and 20% palladium (Pd) on carbon (125 g, 50% wet) were added to the Parr bomb. The vessel was charged to 30 psi with hydrogen gas (H2) and stirred under steady hydrogen atmosphere (vessel was recharged three times to maintain the pressure at 30 psi) at room temperature for 18 h. When HPLC showed that the reaction was complete (when no more hydrogen was taken up), the reaction mixture was filtered through a Celite pad and the Celite pad was washed with THF (4 L). The filtrates were concentrated under reduced pressure to remove the solvent and the residue was loaded onto a Biotage 150 column with a minimum amount of dichloromethane (CH2Cl2). The column was eluted with 20-50% ethyl acetate in heptane and the fractions containing the pure desired product (10) were collected and combined. The solvents were removed under reduced pressure to afford tert-butyl 3-hydroxyazetidine-1-carboxylate (10, 357 g, 393.2 g theoretical, 90.8% yield) as colorless oil, which solidified upon standing at room temperature in vacuum. For 10: 1HNMR (CDCl3, 300 MHz), δ 4.56 (m 1H), 4.13 (m, 2H), 3.81 (m, 2H), 1.43 (s, 9H) ppm.
Step 3. tert-Butyl 3-oxoazetidine-1-carboxylate (11)
A solution of tert-butyl 3-hydroxyazetidine-1-carboxylate (10, 50 g, 289 mmol) in ethyl acetate (400 mL) was cooled to 0° C. The resulting solution was then treated with solid TEMPO (0.5 g, 3.2 mmol, 0.011 equiv) and a solution of potassium bromide (KBr, 3.9 g, 33.2 mmol, 0.115 equiv) in water (60 mL) at 0-5° C. While keeping the reaction temperature between 0-5° C. a solution of saturated aqueous sodium bicarbonate (NaHCO3, 450 mL) and an aqueous sodium hypochlorite solution (NaClO, 10-13% available chlorine, 450 mL) were added. Once the solution of sodium hypochlorite was added, the color of the reaction mixture was changed immediately. When additional amount of sodium hypochlorite solution was added, the color of the reaction mixture was gradually faded. When TLC showed that all of the starting material was consumed, the color of the reaction mixture was no longer changed. The reaction mixture was then diluted with ethyl acetate (EtOAc, 500 mL) and two layers were separated. The organic layer was washed with water (500 mL) and the saturated aqueous sodium chloride solution (500 mL) and dried over sodium sulfate (Na2SO4). The solvent was then removed under reduced pressure to give the crude product, tert-butyl 3-oxoazetidine-1-carboxylate (11, 48 g, 49.47 g theoretical, 97% yield), which was found to be sufficiently pure and was used directly in the subsequent reaction without further purification. For crude 11: 1HNMR (CDCl3, 300 MHz), δ 4.65 (s, 4H), 1.42 (s, 9H) ppm.
Step 4. tert-Butyl 3-(cyanomethylene)azetidine-1-carboxylate (13)
Diethyl cyanomethyl phosphate (12, 745 g, 4.20 mol, 1.20 equiv) and anhydrous tetrahydrofuran (THF, 9 L) was added to a four-neck flask equipped with a thermowell, an addition funnel and the nitrogen protection tube at room temperature. The solution was cooled with an ice-methanol bath to −14° C. and a 1.0 M solution of potassium tert-butoxide (t-BuOK) in anhydrous tetrahydrofuran (THF, 3.85 L, 3.85 mol, 1.1 equiv) was added over 20 minutes keeping the reaction temperature below −5° C. The resulting reaction mixture was stirred for 3 hours at −10° C. and a solution of 1-tert-butoxycarbonyl-3-azetidinone (11, 600 g, 3.50 mol) in anhydrous tetrahydrofuran (THF, 2 L) was added over 2 h keeping the internal temperature below −5° C. The reaction mixture was stirred at −5 to −10° C. over 1 hour and then slowly warmed up to room temperature and stirred at room temperature for overnight. The reaction mixture was then diluted with water (4.5 L) and saturated aqueous sodium chloride solution (NaCl, 4.5 L) and extracted with ethyl acetate (EtOAc, 2×9 L). The combined organic layers were washed with brine (6 L) and dried over anhydrous sodium sulfate (Na2SO4). The organic solvent was removed under reduced pressure and the residue was diluted with dichloromethane (CH2Cl2, 4 L) before being absorbed onto silica gel (SiO2, 1.5 Kg). The crude product, which was absorbed on silica gel, was purified by flash column chromatography (SiO2, 3.5 Kg, 0-25% EtOAc/hexanes gradient elution) to afford tert-butyl 3-(cyanomethylene)azetidine-1-carboxylate (13, 414.7 g, 679.8 g theoretical, 61% yield) as white solid. For 13: 1H NMR (CDCl3, 300 MHz), δ 5.40 (m, 1H), 4.70 (m, 2H), 4.61 (m, 2H), 1.46 (s, 9H) ppm; C10H14N2O2 (MW, 194.23), LCMS (EI) m/e 217 (M′+Na).
Example 3(3-Fluoro-2-(trifluoromethyl)pyridin-4-yl)(1,4-dioxa-8-azaspiro[4,5]decan-8-yl)methanone (17)

Step 1. 1,4-Dioxa-8-azaspiro[4.5]decane (15)
To a 30 L reactor equipped with a mechanic stirrer, an addition funnel and a septum was charged sodium hydroxide (NaOH, 1.4 kg, 35 mol) and water (7 L, 3.13 kg, 17.43 mol). To the solution thus obtained was added 1,4-dioxa-8-azaspiro[4.5]decane hydrochloric acid (14, 3.13 kg, 17.43 mol). The mixture was stirred at 25° C. for 30 minutes. Then the solution was saturated with sodium chloride (1.3 kg) and extracted with 2-methyl-tetrahydrofuran (3×7 L). The combined organic layer was dried with anhydrous sodium sulfate (1.3 kg), filtered and concentrated under reduced pressure (70 mmHg) at 50° C. The yellow oil thus obtained was distilled under reduced pressure (80 mmHg, bp: 115° C. to 120° C.) to give compound 15 (2.34 kg, 16.36 mol, 93.8%) as a clear oil, which was used directly in the subsequent coupling reaction.
Step 2. (3-Fluoro-2-(trifluoromethyl)pyridin-4-yl)(1,4-dioxa-8-azaspiro[4,5]decan-8-yl)methanone (17)
To a dried 100 L reactor equipped with a mechanic stirrer, an addition funnel, a thermometer and a vacuum outlet were placed 3-fluoro-2-(trifluoromethyl)isonicotinic acid (16, 3.0 kg, 14.35 mol), benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate (BOP reagent, 7.6 kg, 17.2 mol, 1.20 equiv) in dimethylformamide (DMF, 18 L). To the resulting solution was added 1,4-dioxa-8-azaspiro[4.5]decane (15, 2.34 kg, 16.36 mol, 1.14 equiv) with stirring over 20 minutes. Triethylamine (Et3N, 4 L, 28.67 mol, 2.00 equiv) was then added over 1 hour. The temperature was kept between 5° C. and 10° C. during the additions. The dark brown solution thus obtained was stirred for 12 hours at 20° C. and then chilled to 10° C. With vigorous stirring, 18 L of saturated sodium bicarbonate solution and 36 L of water were sequentially added and the temperature was kept under 15° C. The precipitation (filter cake) thus obtained was collected by filtration. The aqueous phase was then saturated with 12 kg of solid sodium chloride and extracted with EtOAc (2×18 L). The combined organic layer was washed with saturated sodium bicarbonate solution (18 L), and water (2×18 L) in sequence. The filter cake from the previous filtration was dissolved back in the organic phase. The dark brown solution thus obtained was washed twice with 18 L of water each and then concentrated under reduced pressure (40-50° C., 30 mm Hg) to give 5.0 kg of the crude product as viscous brown oil. The crude product 17 obtained above was dissolved in EtOH (8.15 L) at 50° C. Water (16.3 L) was added over 30 minutes. The brown solution was seeded, cooled to 20° C. over 3 hours with stirring and stirred at 20° C. for 12 h. The precipitate formed was filtered, washed with a mixture of EtOH and water (EtOH:H2O=1:20, 2 L) and dried under reduced pressure (50 mmHg) at 60° C. for 24 hours to afford (3-fluoro-2-(trifluoromethyl)pyridin-4-yl)(1,4-dioxa-8-azaspiro[4,5]decan-8-yl)methanone (17, 3.98 kg, 11.92 mol, 83.1%) as a white powder. For 17: 1H NMR (300 MHz, (CD3)2SO) δ 8.64 (d, 3JHH=4.68 Hz, 1H, NCH in pyridine), 7.92 (dd, 3JHH=4.68 Hz, 4JHF=4.68 Hz, 1H, NCCH in pyridine), 3.87-3.91 (m, 4H, OCH2CH2O), 3.70 (br s, 2H, one of NCH2 in piperidine rine, one of another NCH2 in piperidine ring, both in axial position), 3.26 (t, 3JHH=5.86 Hz, 2H, one of NCH2 in piperidine rine, one of another NCH2 in piperidine ring, both in equatorial position), 1.67 (d, 3JHH=5.86 Hz, 2H, one of NCCH2 in piperidine ring, one of another NCCH2 in piperidine ring, both in equatorial position), 1.58 (br s, 2H, one of NCCH2 in piperidine ring, one of another NCCH2 in piperidine ring, both in axial position) ppm; 13C NMR (75 MHz, (CD3)2SO) δ 161.03 (N—C═O), 151.16 (d, 1JCF=266.03 Hz, C—F), 146.85 (d, 4JCF=4.32 Hz, NCH in pyridine), 135.24 (d, 2JCF=11.51 Hz, C—C═O), 135.02 (quartet, 2JCF=34.57 Hz, NCCF3), 128.24 (d, 4JCF=7.48 Hz, NCCH in pyridine), 119.43 (d×quartet, 1JCF=274.38 Hz, 3JCF=4.89 Hz, CF3), 106.74 (OCO), 64.60 (OCCO), 45.34 (NC in piperidine ring), 39.62 (NC in piperidine ring), 34.79 (NCC in piperidine ring), 34.10 (NCC in piperidine ring) ppm; 19F NMR (282 MHz, (CD3)2SO) δ-64.69 (d, 4JFF=15.85 Hz, F3C), −129.26 (d×quartet, 4JFF=15.85 Hz, 4JFH=3.96 Hz, FC) ppm; C14H14F4N2O3 (MW, 334.27), LCMS (EI) m/e 335.1 (M++H).
Example 4(3-Fluoro-2-(trifluoromethyl)pyridin-4-yl) (1,4-dioxa-8-azaspiro[4,5]decan-8-yl)methanone (18)

In a 5 L 4-necked round bottom flask equipped with a mechanical stirrer, a thermocouple, an addition funnel and a nitrogen inlet was placed (3-fluoro-2-(trifluoromethyl)pyridin-4-yl)(1,4-dioxa-8-azaspiro[4,5]decan-8-yl)methanone (17, 100 g, 0.299 mol) in acetonitrile (ACN, 400 mL) at room temperature. The resultant solution was cooled to below 10° C. To the reaction mixture was added 6.0 N aqueous hydrochloric acid (HCl, 450 mL, 2.70 mol, 9.0 equiv), while the internal temperature was kept below 10° C. The resulting reaction mixture was then warmed to room temperature and an additional amount of 6.0 N aqueous hydrochloric acid (HCl, 1050 mL, 6.30 mol, 21.0 equiv) was slowly introduced to the reaction mixture at room temperature in 8 hours via the addition funnel. The reaction mixture was then cooled to 0° C. before being treated with 30% aqueous sodium hydroxide (NaOH, 860 mL, 8.57 mmol, 28.6 equiv) while the internal temperature was kept at below 10° C. The resulting reaction mixture was subsequently warmed to room temperature prior to addition of solid sodium bicarbonate (NaHCO3, 85.0 g, 1.01 mol, 3.37 equiv) in 1 hour. The mixture was then extracted with EtOAc (2×1.2 L), and the combined organic phase was washed with 16% aqueous sodium chloride solution (2×800 mL) and concentrated to approximately 1.0 L by vacuum distillation. Heptane (2.1 L) was added to the residue, and the resulting mixture was concentrated to 1.0 L by vacuum distillation. To the concentrated mixture was added heptane (2.1 L). The resulting white slurry was then concentrated to 1.0 L by vacuum distillation. To the white slurry was then added methyl tert-butyl ether (MTBE, 1.94 L). The white turbid was heated to 40° C. to obtain a clear solution. The resulting solution was concentrated to about 1.0 L by vacuum distillation. The mixture was stirred at room temperature for 1 hour. The white precipitate was collected by filtration with pulling vacuum. The filter cake was washed with heptane (400 mL) and dried on the filter under nitrogen with pulling vacuum to provide compound 18 (78.3 g, 90.1%) as an off-white solid. For 18: 1H NMR (300 MHz, (CD3)2SO) δ 8.68 (d, 3JHH=4.69 Hz, 1H, NCH in pyridine), 7.97 (dd, 3JHH=4.69 Hz, 4JHF=4.69 Hz, 1H, NCCH in pyridine), 3.92 (br s, 2H, one of NCH2 in piperidine rine, one of another NCH2 in piperidine ring, both in axial position), 3.54 (t, 3JHH=6.15 Hz, 2H, one of NCH2 in piperidine rine, one of another NCH2 in piperidine ring, both in equatorial position), 2.48 (t, 3JHH=6.44 Hz, 2H, NCCH2), 2.34 (t, 3JHE=6.15 Hz, 2H, NCCH2) ppm; 13C NMR (75 MHz, (CD3)2SO) δ 207.17 (C═O), 161.66 (N—C═O), 151.26 (d, 1JCF=266.89 Hz, C—F), 146.90 (d, 4JCF=6.05 Hz, NCH in pyridine), 135.56 (C—C═O), 134.78-135.56 (m, NCCF3), 128.27 (d, 3JCF=7.19 Hz, NCCH in pyridine), 119.52 (d×quartet, 1JCF=274.38 Hz, 3JCF=4.89 Hz, CF3), 45.10 (NC in piperidine ring) ppm, one carbon (NCC in piperidine ring) missing due to overlap with (CD3)2SO; 19F NMR (282 MHz, (CD3)2SO) δ-64.58 (d, 4JFF=15.85 Hz, F3C), −128.90 (d×quartet, 4JFF=15.85 Hz, 4JFH=4.05 Hz, FC) ppm; C12H10F4N2O2 (MW, 290.21), LCMS (EI) m/e 291.1 (M++H).
Example 53-[4-(7-{[2-(Trimethylsilyl)ethoxy]methyl}-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]azetidin-3-yl}acetonitrile dihydrochloride (20)

Step 1. tent-Butyl 3-(cyanomethyl)-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)azetidine-1-carboxylate (19)
In a dried 30 L reactor equipped with a mechanic stirrer, a thermometer, an addition funnel and a vacuum outlet were placed 4-(1H-pyrazol-4-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidine (5, 4.50 kg, 14.28 mol), tert-butyl 3-(cyanomethylene)azetidine-1-carboxylate (13, 3.12 kg, 16.08 mol, 1.126 equiv) in acetonitrile (9 L) at 20±5° C. To the resultant pink suspension was added 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 225 mL, 1.48 mol, 0.10 equiv) over 40 minutes. The batch temperature was kept between 10° C. and 20° C. during addition. The brown solution obtained was stirred at 20° C. for 3 hours. After the reaction was complete, water (18 L) was added with stirring over 80 minutes at 20° C. The mixture was seeded and the seeded mixture was stirred at room temperature for 12 hours. The solids were collected by filtration and the filter cake was washed with a mixture of acetonitrile and water (1:2, 9 L) and dried in a vacuum oven with nitrogen purge for 12 hours at 60° C. to provide the crude product (19, 7.34 kg) as a light yellow powder. The crude product obtained above was dissolved in methyl tert-butyl ether (MTBE, 22 L) at 60° C. in a 50 L reactor equipped with a mechanic stirrer, a thermometer, an addition funnel and a septum. Hexanes (22 L) was added over 1 hour at 60° C. The solution was then seeded, cooled to 20° C. over 3 hours and stirred at 20° C. for 12 hours. The precipitation was collected by filtration. The resultant cake was washed with a mixture of MTBE and hexane (1:15, 3 L) and dried in a vacuum oven for 10 hours at 50° C. to provide the compound 19 (6.83 kg, 13.42 mol, 94.0%) as a white powder. For 19: 1H NMR (400 MHz, CDCl3) δ 8.87 (s, 1H), 8.46 (d, J=0.6 Hz, 1H), 8.36 (d, J=0.7 Hz, 1H), 7.44 (d, J=3.7 Hz, 1H), 6.82 (d, J=3.7 Hz, 1H), 5.69 (s, 2H), 4.57 (d, J=9.6 Hz, 2H), 4.32 (d, J=9.5 Hz, 2H), 3.59-3.49 (m, 2H), 3.35 (s, 2H), 1.49 (s, 9H), 0.96-0.87 (m, 2H), −0.03-−0.10 (s, 9H) ppm; 13C NMR (101 MHz, CDCl3) δ 157.22, 153.67, 153.24, 151.62, 142.13, 130.16, 129.67, 124.47, 116.72, 115.79, 102.12, 82.54, 74.23, 68.01, 60.25, 58.23, 29.65, 29.52, 19.15, −0.26 ppm; C25H35N7O3Si (MW, 509.68), LCMS (EI) m/e 510.1 (M++H).
Step 2. 3-[4-(7-{[2-(Trimethylsilyl)ethoxy]methyl}-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]azetidin-3-yl}acetonitrile dihydrochloride (20)
In a 2 L 4-necked round bottom flask equipped with a mechanical stirrer, a thermocouple, an addition funnel and a nitrogen inlet was added compound 19 (55.0 g, 0.108 mol) and methanol (MeOH, 440 mL) at 20±5° C. The resulting white turbid was stirred for 20 minutes at room temperature to provide a light yellow solution. A solution of hydrochloric acid (HCl) in isopropanol (5.25 M, 165 mL, 0.866 mol, 8.02 equiv) was then added to the reaction mixture via the addition funnel in 5 minutes. The resulting reaction mixture was then heated to 40° C. by a heating mantle. After 2 hours at 40° C., water (165 mL, 9.17 mol, 84.8 equiv) was added to the reaction mixture via the addition funnel to provide a light green solution at 40° C. Methyl tert-butyl ether (MTBE, 440 mL) was added to the resulting mixture via the addition funnel at 40° C. The resulting mixture was slowly cooled to 10° C. The solids were collected by filtration and washed with MTBE (2×220 mL). The white solids were dried in the filter under nitrogen with a pulling vacuum for 18 hours to afford compound 20 (52.2 g, KF water content 5.42%, yield 94.9%). For 20: 1H NMR (400 MHz, (CD3)2SO) δ 10.39 (brs, 1H), 10.16 (brs, 1H), 9.61 (s, 1H), 9.12 (s, 1H), 9.02 (s, 1H), 8.27-8.21 (d, J=3.8 Hz, 1H), 7.72-7.66 (d, J=3.8 Hz, 1H), 5.82 (s, 2H), 4.88-4.77 (m, 2H), 4.53-4.44 (m, 2H), 4.12 (s, 2H), 3.69-3.60 (m, 2H), 0.98-0.89 (m, 2H), 0.01 (s, 9H) ppm; 13C NMR (101 MHz, (CD3)2SO) δ 151.25, 146.45, 145.09, 140.75, 133.38, 132.44, 116.20, 116.09, 112.79, 102.88, 73.07, 66.14, 59.16, 53.69, 26.44, 17.15, −1.36 ppm; C20H29Cl2N7OSi (free base of 20, C20H27N7OSi, MW 409.56), LCMS (EI) m/e 410.2 (M++H).
Example 62-(1-(1-(3-Fluoro-2-(trifluoromethyl)isonicotinoyl)piperidin-4-yl)-3-(4-(7-(2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)azetidin-3-yl)acetonitrile (21)

In a 100 L dried reactor equipped with a mechanical stirrer, a thermocouple, a condenser, and a nitrogen inlet was added (20, 3.24 kg, 6.715 mol) and dichloromethane (32 L) at 20±5° C. The mixture was stirred at room temperature for 10 minutes before being treated with triethylamine (TEA, 1.36 kg, 13.44 mol, 2.00 equiv) at an addition rate which keeping the internal temperature at 15-30° C. Compound 18 (2.01 kg, 6.926 mol, 1.03 equiv) was then added to the reactor at room temperature. After 10 minutes, sodium triacetoxyborohydride (NaBH(OAc)3, 2.28 kg, 10.75 mol, 1.60 equiv) was added portion wise to the reactor in 1 hour while the internal temperature was kept at 15-30° C. The resulting reaction mixture was stirred at 15-30° C. for an additional one hour. Once the reductive amination reaction is deemed complete, the reaction mixture was treated with a 4% aqueous sodium bicarbonate solution (NaHCO3, 32 L) to adjust the pH to 7-8. After stirring for 30 minutes at room temperature, the two phases were separated. The aqueous phase was extracted with dichloromethane (29 L). The combined organic phase was sequentially washed with 0.1 N aqueous hydrochloric acid solution (16 L), 4% aqueous sodium bicarbonate solution (16 L), 8% aqueous sodium chloride solution (2×16 L). The resultant organic phase was partially concentrated and filtered. The filtrate was subjected to solvent exchange by gradually adding acetonitrile (65 L) under vacuum. The white solids were collected by filtration, washed with acetonitrile (10 L) and dried at 40-50° C. in a vacuum oven with nitrogen purge to afford compound 21 (4.26 kg, 6.23 mol, 92.9%). For 21: 1H NMR (500 MHz, (CD3)2SO) δ 8.84 (s, 1H), 8.76 (s, 1H), 8.66 (d, J=4.7 Hz, 1H), 8.43 (s, 1H), 7.90 (t, J=4.7 Hz, 1H), 7.78 (d, J=3.7 Hz, 1H), 7.17 (d, J=3.7 Hz, 1H), 5.63 (s, 2H), 4.07 (dt, J=11.1, 4.9 Hz, 1H), 3.75 (d, J=7.8 Hz, 2H), 3.57 (dd, J=10.2, 7.8 Hz, 2H), 3.55 (s, 2h), 3.52 (dd, J=8.5, 7.4 Hz, 2H), 3.41 (dq, J=13.3, 4.3 Hz, 1H), 3.26 (t, J=10.0 Hz, 1H), 3.07 (ddd, J=13.1, 9.4, 3.2 Hz, 1H), 2.56 (dt, J=8.5, 4.7 Hz, 1H), 1.81-1.73 (m, 1H), 1.63 (m, 1H), 1.29 (m, 1H), 1.21 (m, 1H), 0.82 (dd, J=8.5, 7.4 Hz, 2H), −0.12 (s, 9H) ppm; 13C NMR (101 MHz, (CD3)2SO) δ 161.68, (154.91, 152.27), 153.08, 152.69, 151.53, 147.69, 140.96, (136.19, 136.02), (136.48, 136.36, 136.13, 136.0, 135.78, 135.66, 135.43, 135.32), 131.43, 130.84, 129.03, (126.17, 123.42, 120.69), 117.99, 122.77, 118.78, 114.71, 102.02, 73.73, 67.04, 62.86, 61.88, 58.51, 45.63, 30.03, 29.30, 28.60, 18.52, 0.00 ppm; C32H37F4N9O2Si (MW, 683.77), LCMS (EI) m/e 684.2 (M++H).
Example 72-(3-(4-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-1-(1-(3-fluoro-2-(trifluoromethyl)isonicotinoyl)piperidin-4-yl)azetidin-3-yl)acetonitrile (22)
 BASE OF INCB 39110
BASE OF INCB 39110To a 250 mL 4-necked round bottom flask equipped with a mechanical stirrer, a thermocouple, an addition funnel and a nitrogen inlet was added compound 21 (9.25 g, 13.52 mmol, KF water content 3.50%) and acetonitrile (74 mL) at 20±5° C. The resulting white slurry was cooled to below 5° C. Boron trifluoride diethyl etherate (BF3.OEt2, 6.46 mL, 51.37 mmol, 3.80 equiv) was then added at a rate while the internal temperature was kept at below 5.0° C. The reaction mixture was then warmed to 20±5° C. After stirring at 20±5° C. for 18 hours, the reaction mixture was cooled to 0-5° C. and an additional amount of BF3.OEt2 (0.34 mL, 2.70 mmol, 0.2 equiv) was introduced to the reaction mixture at below 5.0° C. The resulting reaction mixture was warmed to 20±5° C., and kept stirring at room temperature for an additional 5 hours. The reaction mixture was then cooled to 0-5° C. before water (12.17 mL, 0.676 mol, 50 equiv) was added. The internal temperature was kept at below 5.0° C. during addition of water. The resultant mixture was warmed to 20±5° C. and kept stirring at room temperature for 2 hours. The reaction mixture was then cooled to 0-5° C. and aqueous ammonium hydroxide (NH4OH, 5 N, 121.7 mmol, 9.0 equiv) was added. During addition of aqueous ammonium hydroxide solution, the internal temperature was kept at below 5.0° C. The resulting reaction mixture was warmed to 20±5° C. and stirred at room temperature for 20 hours. Once the SEM-deprotection was deemed complete, the reaction mixture was filtered, and the solids were washed with EtOAc (9.25 mL). The filtrates were combined and diluted with EtOAc (74 mL). The diluted organic solution was washed with 13% aqueous sodium chloride solution (46.2 mL). The organic phase was then diluted with EtOAc (55.5 mL) before being concentrated to a minimum volume under reduced pressure. EtOAc (120 mL) was added to the residue, and the resulting solution was stirred at 20±5° C. for 30 minutes. The solution was then washed with 7% aqueous sodium bicarbonate solution (2×46 mL) and 13% aqueous sodium bicarbonate solution (46 mL). The resultant organic phase was diluted with EtOAc (46 mL) and treated with water (64 mL) at 50±5° C. for 30 minutes. The mixture was cooled to 20±5° C. and the two phases were separated. The organic phase was treated with water (64 mL) at 50±5° C. for 30 minutes for the second time. The mixture was cooled to 20±5° C. and the two phases were separated. The resultant organic phase was concentrated to afford crude compound 22 (free base), which was further purified by column chromatography (SiO2, 330 g, gradient elution with 0-10% of MeOH in EtOAc) to afford analytically pure free base (22, 7.00 g, 93.5%) as an off-white solid. For 22:
1H NMR (400 MHz, (CD3)2SO) δ 12.17 (d, J=2.8 Hz, 1H), 8.85 (s, 1H), 8.70 (m, 2H), 8.45 (s, 1H), 7.93 (t, J=4.7 Hz, 1H), 7.63 (dd, J=3.6, 2.3 Hz, 1H), 7.09 (dd, J=3.6, 1.7 Hz, 1H), 4.10 (m, 1H), 3.78 (d, J=7.9 Hz, 2H), 3.61 (t, J=7.9 Hz, 1H), 3.58 (s, 2H), 3.46 (m, 1H), 3.28 (t, J=10.5 Hz, 1H), 3.09 (ddd, J=13.2, 9.5, 3.1 Hz, 1H), 2.58 (m, 1H), 1.83-1.75 (m, 1H), 1.70-1.63 (m, 1H), 1.35-1.21 (m, 2H) ppm;
13C NMR (101 MHz, (CD3)2SO) δ 160.28, (153.51, 150.86), 152.20, 150.94, 149.62, (146.30, 146.25), 139.48, (134.78, 134.61), (135.04, 134.92, 134.72, 134.60, 134.38, 134.26, 134.03, 133.92), 129.22, 127.62, 126.84, 121.99, 122.04, (124.77, 122.02, 119.19, 116.52), 117.39, 113.00, 99.99, 61.47, 60.49, 57.05, 44.23, 28.62, 27.88, 27.19 ppm;
C26H23F4N9O (MW, 553.51), LCMS (EI) m/e 554.1 (M′+H).
ADIPATE
Example 8
2-(3-(4-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-1-(1-(3-fluoro-2-(trifluoromethyl)isonicotinoyl)piperidin-4-yl)azetidin-3-yl)acetonitrile adipate (25)

Step 1. 2-(3-(4-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-1-(1-(3-fluoro-2-(trifluoromethyl)isonicotinoyl)piperidin-4-yl)azetidin-3-yl)acetonitrile adipate crude salt (24)
The process of making compound 22 in Example 7 was followed, except that the final organic phase was concentrated by vacuum distillation to the minimum volume to afford crude compound 22, which was not isolated but was directly used in subsequent adipate salt formation process. To the concentrated residue which containing crude compound 22 was added methanol (200 mL) at room temperature. The mixture was the concentrated by vacuum distillation to a minimum volume. The residue was then added methanol (75 mL) and the resulting solution was heated to reflux for 2 hours. Methyl isobutyl ketone (MIBK, 75 mL) was added to the solution and the resulting mixture was distilled under vacuum to about 30 mL while the internal temperature was kept at 40-50° C. Methanol (75 mL) was added and the resulting mixture was heated to reflux for 2 hours. To the solution was added MIBK (75 mL). The mixture was distilled again under vacuum to about 30 mL while the internal temperature was kept at 40-50° C. To the solution was added a solution of adipic acid (23, 2.15 g, 14.77 mmol) in methanol (75 mL). The resultant solution was then heated to reflux for 2 hours. MIBK (75 mL) was added. The mixture was distilled under vacuum to about 60 mL while the internal temperature was kept at 40-50° C. Heating was stopped and heptane (52.5 mL) was added over 1-2 hours. The resultant mixture was stirred at 20±5° C. for 3-4 hours. The white precipitates were collected by filtration, and the filter cake was washed with heptane (2×15 mL). The solid was dried on the filter under nitrogen with a pulling vacuum at 20±5° C. for 12 hours to provide compound 24 (crude adipate salt, 8.98 g, 12.84 mmol., 95.0%). For 24: 1H NMR (400 MHz, (CD3)2SO) δ 12.16 (s, 1H), 12.05 (brs, 2H), 8.85 (s, 1H), 8.72 (s, 1H), 8.69 (d, J=4.7 Hz, 1H), 8.45 (s, 1H), 7.93 (t, J=4.7 Hz, 1H), 7.63 (dd, J=3.6, 2.3 Hz, 1H), 7.09 (dd, J=3.6, 1.7 Hz, 1H), δ 4.11 (dt, J=11.0, 4.4 Hz, 1H), 3.77 (d, J=7.8 Hz, 2H), 3.60 (t, J=7.8 Hz, 2H), 3.58 (s, 2H), 3.44 (dt, J=14.4, 4.6 Hz, 1H), 3.28 (t, J=10.4 Hz, 1H), 3.09 (ddd, J=13.2, 9.6, 3.2 Hz, 1H), 2.58 (tt, J=8.6, 3.5 Hz, 1H), 2.28-2.17 (m, 4H), 1.83-1.74 (m, 1H), 1.67 (d, J=11.0 Hz, 1H), 1.59-1.46 (m, 4H), 1.37-1.21 (m, 2H) ppm; 13C NMR (101 MHz, (CD3)2SO) δ 174.38, 160.29, (153.52, 150.87), 152.20, 150.94, 149.63, (146.30, 146.25), 139.48, (134.79, 134.62), (135.08, 134.97, 134.74, 134.62, 134.38, 134.28, 134.04, 133.93), 129.21, 127.62, 126.84, 122.05, (124.75, 122.02, 119.29, 116.54), 117.39, 113.01, 99.99, 61.47, 60.50, 57.06, 44.24, 33.42, 30.70, 28.63, 27.89, 27.20, 24.07 ppm; C32H33F4N9O5 (Mol. Wt: 699.66; 24: C26H23F4N9O, MW 553.51), LCMS (EI) m/e 554.0 (M++H).
Step 2.
2-(3-(4-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-1-(1-(3-fluoro-2-(trifluoromethyl)isonicotinoyl)piperidin-4-yl)azetidin-3-yl)acetonitrile adipate (25)
In a 100 L dried reactor equipped with a mechanical stirrer, a thermocouple, an addition funnel and a nitrogen inlet was added compound 24 (3.40 kg, 4.86 mol) and acetone (23.8 L). The resulting white turbid was heated to 55-60° C. to provide a clear solution. The resultant solution was filtered through an in-line filter to another 100 L reactor. Heptane (23.8 L) was filtered through an in-line filter to a separated 50 L reactor. The filtered heptane was then charged to the acetone solution in the 100 L reactor at a rate while the internal temperature was kept at 55-60° C. The reaction mixture in the 100 L reactor was then cooled to 20±5° C. and stirred at 20±5° C. for 16 hours. The white precipitates were collected by filtration and the cake was washed with heptane (2×5.1 L) and dried on the filter under nitrogen with a pulling vacuum. The solid was further dried in a vacuum oven at 55-65° C. with nitrogen purge to provide compound 25 (3.11 kg, 92.2%) as white to off-white powder. For 25:
ADIPATE OF INCB 39110
1H NMR (400 MHz, (CD3)2SO) δ 12.16 (s, 1H), 12.05 (brs, 2H), 8.85 (s, 1H), 8.72 (s, 1H), 8.69 (d, J=4.7 Hz, 1H), 8.45 (s, 1H), 7.93 (t, J=4.7 Hz, 1H), 7.63 (dd, J=3.6, 2.3 Hz, 1H), 7.09 (dd, J=3.6, 1.7 Hz, 1H), δ 4.11 (dt, J=11.0, 4.4 Hz, 1H), 3.77 (d, J=7.8 Hz, 2H), 3.60 (t, J=7.8 Hz, 2H), 3.58 (s, 2H), 3.44 (dt, J=14.4, 4.6 Hz, 1H), 3.28 (t, J=10.4 Hz, 1H), 3.09 (ddd, J=13.2, 9.6, 3.2 Hz, 1H), 2.58 (tt, J=8.6, 3.5 Hz, 1H), 2.28-2.17 (m, 4H), 1.83-1.74 (m, 1H), 1.67 (d, J=11.0 Hz, 1H), 1.59-1.46 (m, 4H), 1.37-1.21 (m, 2H) ppm;
13C NMR (101 MHz, (CD3)2SO) δ 174.38, 160.29, (153.52, 150.87), 152.20, 150.94, 149.63, (146.30, 146.25), 139.48, (134.79, 134.62), (135.08, 134.97, 134.74, 134.62, 134.38, 134.28, 134.04, 133.93), 129.21, 127.62, 126.84, 122.05, (124.75, 122.02, 119.29, 116.54), 117.39, 113.01, 99.99, 61.47, 60.50, 57.06, 44.24, 33.42, 30.70, 28.63, 27.89, 27.20, 24.07 ppm;
C32H33F4N9O5 (Mol. Wt: 699.66; free base: C26H23F4N9O (MW, 553.51), LCMS (EI) m/e 554.0 (M++H).

…………………………
http://www.google.com/patents/WO2014138168A1?cl=en
Processes for preparing JAK inhibitor (preferably INCB-39110) comprising the reaction of a substituted 1H-pyrazole compound with 4-chloro-7H-pyrrolo[2,3-d]pyrimidine in the presence of a base (eg cesium fluoride) and a solvent under Suzuki coupling conditions ([1,1′- bis(dicyclohexylphosphino)ferrocene]dichloropalladium (II)), followed by deprotection and then reaction with a piperidine derivative, and salt synthesis are claimed. Also claimed are novel intermediates and processes for their preparation. The compound is disclosed to be useful for treating disease mediated by JAK activity (targeting JAK-1 and 2), such as multiple sclerosis, rheumatoid arthritis, type I diabetes, inflammatory bowel disease, Crohn’s disease, COPD, prostate cancer, hepatic cancer, breast cancer, influenza, and SARS.
Example 1. Synthesis of 2-(3-(4-(7H-Pyrrolo[2,3-< ]pyrimidin-4-yl)-lH-pyrazol-l- yl)-l-(l-(3-fluoro-2-(trifluoromethyl)isonicotinoyl)piperidin-4-yl)azetidin-3- yl)acetonitrile Adipate (9)20443-0253WO1 (INCY0124-WO1) PATENT

tert-Butyl 3-(cyanomethyl)-3-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH- pyrazol-l-yl)azetidine-l-carboxylate (3). To a 1-L flask equipped with a nitrogen inlet, a thermocouple, and a mechanical stirrer were sequentially added isopropanol (IP A, 200 mL), l,8-diazabicyclo[5,4,0]undec-ene (DBU, 9.8 g, 64.4 mmol, 0.125 equiv), 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (1, 101 g, 520.51 mmol, 1.01 equiv) and tert-butyl 3-(cyanomethylene)azetidine-l-carboxylate (2, 100 g, 514.85 mmol) at ambient temperature to generate a reaction mixture as a
suspension. The resulting reaction mixture was heated to reflux in 30 minutes to provide a homogenous solution and the mixture was maintained at reflux for an additional 2 – 3 hours. After the reaction was complete as monitored by HPLC, n- heptane (400 mL) was gradually added to the reaction mixture in 45 minutes while maintaining the mixture at reflux. Solids were precipitated out during the w-heptane addition. Once w-heptane addition was complete, the mixture was gradually cooled to ambient temperature and stirred at ambient temperature for an additional 1 hour. The solids were collected by filtration, washed with w-heptane (200 mL), and dried under vacuum at 50 °C with nitrogen sweeping to constant weight to afford tert-butyl 3- 20443-0253WO1 (INCY0124-WO1) PATENT
(cyanomethyl)-3-(4-(4,4,5,5-tetramethyl- 1 ,3,2-dioxaborolan-2-yl)- IH-pyrazol- 1 – yl)azetidine-l -carboxylate (3, 181 g, 199.9 g theoretical, 90.5%) as a white to pale yellow solid. For 3: XH NMR (400 MHz, DMSO-i¾) δ 8.31 (s, 1H), 7.74 (s, 1H), 4.45 – 4.23 (m, 2H), 4.23 – 4.03 (m, 2H), 3.56 (s, 2H), 1.38 (s, 9H), 1.25 (s, 12H) ppm; 13C NMR (101 MHz, DMSO-i/6) δ 155.34, 145.50, 135.88, 1 16.88, 107.08 (br), 83.15, 79.36, 58.74 (br), 56.28, 27.96, 26.59, 24.63 ppm; Ci9H29B 404 (MW 388.27),
LCMS (EI) mle 389 (M+ + H). teri-Butyl 3-(4-(7H-pyrrolo[2,3-< |pyrimidin-4-yl)-lH-pyrazol-l-yl)-3- (cyanomethyl)-azetidine-l-carboxylate (5). To a 1-L flask equipped with a nitrogen inlet, a thermocouple, and a mechanical stirrer were added 4-chloro-7H-pyrrolo[2,3- i/]pyrimidine (4, 39.6 g, 257.6 mmol), tert-butyl 3-(cyanomethyl)-3-(4-(4,4,5,5- tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- IH-pyrazol- 1 -yl)azetidine- 1 -carboxylate (3, 100 g, 257.6 mmol, 1.0 equiv), cesium fluoride (136.9 g, 901.4 mmol, 3.5 equiv), tert- butanol (250 mL), water (250 mL), and [l, l’-bis(di- cyclohexylphosphino)ferrocene]dichloropalladium(II) (Pd-127, 351.4 mg, 0.46 mmol, 0.0018 equiv) at ambient temperature. The resulting reaction mixture was de-gassed and refilled with nitrogen for 3 times before being heated to reflux and maintained at reflux under nitrogen for 20 – 24 hours. When HPLC showed the reaction was complete, the reaction mixture was cooled to 45 – 55 °C in 30 minutes, the two phases were separated, and the aqueous phase was discarded. To the organic phase was added w-heptane (125 mL) in 30 minutes at 45 – 55 °C. The resulting mixture was slowly cooled to ambient temperature in one hour and stirred at ambient temperature for an additional 2 hours. The solids were collected by filtration, washed with n- heptane (100 mL), and dried under vacuum at 50 °C with nitrogen sweeping to constant weight to afford tert-butyl 3-(4-(7H-pyrrolo[2,3-<i]pyrimidin-4-yl)-lH- pyrazol-l-yl)-3-(cyanomethyl)-azetidine-l -carboxylate (5, 96.8 g, 97.7 g theoretical, 99%) as a pale yellow solid. For 5: XH NMR (400 MHz, DMSO-i¾) δ 8.89 (s, 1H), 8.68 (s, 1H), 8.44 (s, 1H), 7.60 (d, J= 3.5 Hz, 1H), 7.06 (d, J= 3.6 Hz, 1H), 4.62 – 4.41 (m, 2H), 4.31 – 4.12 (m, 2H), 3.67 (s, 2H), 1.39 (s, 9H) ppm; 13C NMR (101 MHz, DMSO-i¾) δ 155.40, 152.60, 150.63, 149.15, 139.76, 129.53, 127.65, 122.25, 20443-0253WO1 (INCY0124-WO1) PATENT
116.92, 113.21, 99.71, 79.45, 58.34 (br), 56.80, 27.99, 26.83 ppm; Ci9H21 702 (MW 379.4), LCMS (EI) mle 380 (M+ + H).
2- (3-(4-(7H-Pyrrolo[2,3-< |pyrimidin-4-yl)-lH-pyrazol-l-yl)azetidin-3- yl)acetonitrile dihydrochloride salt (6). To a 0.5-L flask equipped with a nitrogen inlet, a thermocouple, an additional funnel, and a mechanical stirrer were added tert- butyl 3 -(4-(7H-pyrrolo [2,3 -<i]pyrimidin-4-yl)- lH-pyrazol- 1 -yl)-3 – (cyanomethyl)azetidine-l-carboxylate (5, 15 g, 39.5 mmol), water (7.5 mL, 416 mmol) and dichloromethane (75 mL) at room temperature. The mixture was stirred at room temperature to generate a suspension. To the suspension was added a solution of 5 M hydrogen chloride (HQ) in isopropanol (55 mL, 275 mmol, 7.0 equiv) in 5 minutes. The resulting reaction mixture was then heated to gentle reflux and
maitained at reflux for 3-4 hours. After the reaction was completed as mornitored by HPLC, tert-butyl methyl ether (TBME, 45 mL) was added to the reaction suspension. The mixture was gradually cooled to room temperature, and stirred for an additional one hour. The solids were collected by filtration, washed with tert-butyl methyl ether (TBME, 45 mL) and dried under vacuum at 50 °C with nitrogen sweeping to constant weight to afford 2-(3-(4-(7H-pyrrolo[2,3-i/]pyrimidin-4-yl)-lH-pyrazol-l-yl)azetidin-
3- yl)acetonitrile dihydrochloride salt (6, 13.6 g, 13.9 g theoretical, 98%) as an off- white to light yellow solid. For 6: XH NMR (400 MHz, D20) δ 8.96 (s, 1H), 8.81 (s, 1H), 8.49 (s, 1H), 7.78 (d, J= 3.8 Hz, 1H), 7.09 (d, J= 3.7 Hz, 1H), 4.93 (d, J= 12.8 Hz, 2H), 4.74 (d, J= 12.5 Hz, 2H), 3.74 (s, 2H) ppm; 13C NMR (101 MHz, D20) δ 151.35, 143.75, 143.33, 141.33, 132.03, 131.97, 115.90, 114.54, 113.85, 103.18, 59.72, 54.45 (2C), 27.02 ppm; Ci4H15Cl2N7 (Ci4H13N7 for free base, MW 279.30), LCMS (EI) mle 280 (M+ + H).
2-(3-(4-(7H-Pyrrolo[2,3-< |pyrimidin-4-yl)-lH-pyrazol-l-yl)-l-(l-(3-fluoro-2- (trifluoromethyl)isonicotinoyl)piperidin-4-yl)azetidin-3-yl)acetonitrile (8, Free Base). To a 0.5-L flask equipped with a nitrogen inlet, a thermocouple, an additional funnel, and a mechanical stirrer were added 2-(3-(4-(7H-pyrrolo[2,3-<i]pyrimidin-4- yl)-lH-pyrazol-l-yl)azetidin-3-yl)acetonitrile dihydrochloride salt (6, 20 g, 56.78 mmol), dichloromethane (200 mL) and triethylamine (TEA, 16.62 mL, 119.2 mmol, 20443-0253WO1 (INCY0124-WO1) PATENT
2.1 equiv) at ambient temperature. The mixture was stired at ambient temperature for 30 minutes before l-(3-fluoro-2-(trifluoromethyl)-isonicotinoyl)piperidin-4-one (7, 17.15 g, 57.91 mmol, 1.02 equiv) was added to the mixture. The mixture was then treated with sodium triacetoxyborohydride (25.34 g, 1 13.6 mmol, 2.0 equiv) in 5 minutes at ambient temperature (below 26 °C). The resulting reaction mixture was stirred at ambient temperature for 2 hours. After the reaction was complete as mornitored by HPLC, the reaction mixture was quenched with saturated aHC03 aqueous solution (200 mL). The two phases were separated and the aqueous phase was extracted with methylene chloride (200 mL). The combined organic phase was washed with 4% brine (100 mL) followed by solvent switch of methylene chloride to acetone by distillation. The resulting solution of the desired crude product (8) in acetone was directly used for the subsequent adipate salt formation. A small portion of solution was purified by column chromatography (S1O2, 0 – 10% of MeOH in EtOAc gradient elution) to afford the analytically pure 2-(3-(4-(7H-pyrrolo[2,3- i/]pyrimidin-4-yl)- lH-pyrazol- 1 -yl)- 1 -( 1 -(3 -fluoro-2-
(trifluoromethyl)isonicotinoyl)piperidin-4-yl)azetidin-3-yl)acetonitrile (8 free base) as an off-white solid. For 8: ¾ NMR (400 MHz, DMSO-i¾) δ 12.17 (d, J= 2.8 Hz, 1H), 8.85 (s, 1H), 8.70 (m, 2H), 8.45 (s, 1H), 7.93 (t, J= A J Hz, 1H), 7.63 (dd, J= 3.6, 2.3 Hz, 1H), 7.09 (dd, J= 3.6, 1.7 Hz, 1H), 4.10 (m, 1H), 3.78 (d, J= 7.9 Hz, 2H), 3.61 (t, J= 7.9 Hz, 1H), 3.58 (s, 2H), 3.46 (m, 1H), 3.28 (t, J= 10.5 Hz, 1H), 3.09 (ddd, J = 13.2, 9.5, 3.1 Hz, 1H), 2.58 (m, 1H), 1.83 – 1.75 (m, 1H), 1.70 – 1.63 (m, 1H), 1.35 – 1.21 (m, 2H) ppm; 13C MR (101 MHz, DMSO-i/6) δ 160.28, (153.51, 150.86), 152.20, 150.94, 149.62, (146.30, 146.25), 139.48, (134.78, 134.61), (135.04, 134.92, 134.72, 134.60, 134.38, 134.26, 134.03, 133.92), 129.22, 127.62, 126.84, 121.99, 122.04, (124.77, 122.02, 1 19.19, 1 16.52), 117.39, 113.00, 99.99, 61.47, 60.49, 57.05, 44.23, 28.62, 27.88, 27.19 ppm;
(MW, 553.51), LCMS (EI) mle 554.1 (M+ + H).
2-(3-(4-(7H-Pyrrolo[2,3-< |pyrimidin-4-yl)-lH-pyrazol-l-yl)-l-(l-(3-fluoro-2- (trifluoromethyl)isonicotinoyl)piperidin-4-yl)azetidin-3-yl)acetonitrile Adipate (9). To a 0.5-L flask equipped with a mechanical stirrer, a thermocouple, an addition funnel, and a nitrogen inlet was added a solution of crude 2-(3-(4-(7H-pyrrolo[2,3- 20443-0253WO1 (INCY0124-WO1) PATENT i/]pyrimidin-4-yl)- lH-pyrazol- 1 -yl)- 1 -( 1 -(3 -fluoro-2-
(trifluoromethyl)isonicotinoyl)piperidin-4-yl)azetidin-3-yl)acetonitrile (8 free base, 31.38 g, 56.7 mmol) in acetone (220 mL) and adipic acid (8.7 g, 59.53 mmol, 1.05 equiv) at ambient temperature. The reaction mixture was then heated to reflux to give a solution. w-Heptane (220 mL) was gradually added to the reaction mixture at 40 – 50 °C in one hour. The resulting mixture was gradually cooled to ambient temperature in one hour and stirred at ambient temperature for an additional 16 hours. The solids were collected by filtration, washed with w-heptane (2 X 60 mL), and dried under vacuum at 50 °C with nitrogen sweeping to constant weight to afford 2-(3-(4-(7H- Pyrrolo[2,3 -i/]pyrimidin-4-yl)- lH-pyrazol- 1 -yl)- 1 -(1 -(3 -fluoro-2- (trifluoromethyl)isonicotinoyl)piperidin-4-yl)azetidin-3-yl)acetonitrile adipate (9,34.0 g, 39.7 g theoretical, 85.6% for two steps) as a white to off-white solid. 9:
XH NMR (400 MHz, DMSO-i/6) δ 12.16 (s, 1H), 12.05 (brs, 2H), 8.85 (s, 1H), 8.72 (s, 1H), 8.69 (d, J= A J Hz, 1H), 8.45 (s, 1H), 7.93 (t, J= A J Hz, 1H), 7.63 (dd, J= 3.6, 2.3 Hz, 1H), 7.09 (dd, J= 3.6, 1.7 Hz, 1H), 5 4.1 1 (dt, J= 1 1.0, 4.4 Hz, 1H), 3.77 (d, J= 7.8 Hz, 2H), 3.60 (t, J= 7.8 Hz, 2H), 3.58 (s, 2H), 3.44 (dt, J= 14.4, 4.6 Hz, 1H), 3.28 (t, J= 10.4 Hz, 1H), 3.09 (ddd, J= 13.2, 9.6, 3.2 Hz, 1H), 2.58 (tt, J= 8.6, 3.5 Hz, lH), 2.28 – 2.17 (m, 4H), 1.83 – 1.74 (m, 1H), 1.67 (d, J= 11.0 Hz, 1H), 1.59 – 1.46 (m, 4H), 1.37 – 1.21 (m, 2H) ppm;
13C MR (101 MHz, DMSO-i/6) δ 174.38, 160.29, (153.52, 150.87), 152.20, 150.94, 149.63, (146.30, 146.25), 139.48, (134.79, 134.62), (135.08, 134.97, 134.74, 134.62, 134.38, 134.28, 134.04, 133.93), 129.21, 127.62, 126.84, 122.05, (124.75, 122.02, 1 19.29, 1 16.54), 117.39, 113.01, 99.99, 61.47, 60.50, 57.06, 44.24, 33.42, 30.70, 28.63, 27.89, 27.20, 24.07 ppm;
C32H33F4N9O5 ( MW 699.66;![]() for free base, MW, 553.51), LCMS (EI) mle 554.0 (M+ + H).
for free base, MW, 553.51), LCMS (EI) mle 554.0 (M+ + H).
Example 2: Alternative Synthesis of 2-(3-(4-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)- lH-pyrazol-l-yl)-l-(l-(3-fluoro-2-(trifluoromethyl)isonicotinoyl)piperidin-4- yl)azetidin-3-yl)acetonitrile 20443-0253WO1 (INCY0124-WO1) PATENT
Scheme II

………………………………..COMPD11……………………………………………………………………………………………………..COMPD 8 BASE
C26H3i BF4N603 C26H23F4N9O Mol. Wt: 562.37 Mol. Wt: 553.51
2- (Azetidin-3-ylidene)acetonitrile hydrochloride (2a). To a 0.5-L flask equipped with a nitrogen inlet, a thermocouple, and a mechanical stirrer were added tert-butyl
3- (cyanomethylene)azetidine-l-carboxylate (2, 30 g, 154.46 mmol) and
methylenechloride (300 mL) at ambient temperature. The solution was then treated with a solution of 5 M hydrogen chloride (HQ) in isopropanol solution (294.2 mL, 1.54 mol, 10 equiv) at ambient temperature and the resulting reaction mixture was stirred at ambient temperature for 18 hours. After the reaction was complete as monitored by HPLC, the suspension was added tert-butyl methyl ether (TBME, 150 mL), and the mixture was stirred at ambient temperature for 2 hours. The solids was collected by filtration, washed with w-heptane (2 X 100 mL), and dried on the filtration funnel at ambient temperature for 3 hours to afford 2-(azetidin-3- ylidene)acetonitrile hydrochloride (2a, 13.7 g, 20.2 g theoretical, 67.8 %) as a white solid. For 2a: XH NMR (500 MHz, DMSO-i¾) δ 9.99 (s, 2H), 5.94 (p, J= 2.5 Hz, 1H), 20443-0253WO1 (INCY0124-WO1) PATENT
4.85 – 4.80 (m, 2H), 4.77 – 4.71 (m, 2H) ppm; C NMR (126 MHz, DMSO-i¾) δ 155.65, 114.54, 94.78, 55.26, 54.63 ppm; C5H7C1N2 (MW 130.58; C5H6N2 for free base, MW 94.11), LCMS (EI) mle 95 (M+ + H).
2-(l-(l-(3-Fluoro-2-(trifluoromethyl)isonicotinoyl)piperidin-4-yl)azetidin-3- ylidene)acetonitrile (10). To a 0.25-L flask equipped with a nitrogen inlet, a thermocouple, and a magnetic stirrer were added 2-(azetidin-3-ylidene)acetonitrile hydrochloride (2a, 4.5 g, 34.46 mmol), l-(3-fluoro-2-
(trifluoromethyl)isonicotinoyl)piperidin-4-one (7, 10 g, 34.46 mmol, 1.0 equiv), and methylenechloride (100 mL) at ambient temperqature and the resulting mixture was then treated with sodium triacetoxyborohydride (14.6 g, 68.93 mmol, 2.0 equiv) at ambient temperature. The reaction mixture was stirred at ambient temperature for 2 hours before being quenched with saturated sodium bicarbonate (NaHCOs) aqueous solution (50 mL). The two phases were separated and the aqueous phase was extracted with dichloromethane (200 mL). The combined organic phase was washed with water (50 mL) and brine (50 mL) and concentrated under reduced pressure to afford the crude desired product (10), which was purified by column chromatography (S1O2, 0 – 10 % of ethyl acetate in hexane gradient elution) to afford 2-(l-(l-(3- fluoro-2-(trifluoromethyl)isonicotinoyl)piperidin-4-yl)azetidin-3-ylidene)acetonitrile (10, 9.5 g, 12.7 g theoretical, 74.8 %) as a white solid. For 10: XH NMR (400 MHz, CDCI3) δ 8.57 (d, J= A J Hz, 1H), 7.54 (t, J= 4.6 Hz, 1H), 5.29 (p, J= 2.4 Hz, 1H), 4.18 – 4.08 (m, 1H), 4.08 – 4.03 (m, 2H), 3.98 – 3.94 (m, 2H), 3.57 – 3.39 (m, 2H), 3.17 – 3.04 (m, 1H), 2.56 (tt, J= 7.4, 3.5 Hz, 1H), 1.86 – 1.77 (m, 1H), 1.75 – 1.64 (m, 1H), 1.54 – 1.43 (m, 1H), 1.43 – 1.31 (m, lH) ppm; 13C MR (101 MHz, CDC13) δ 161.34, 160.73, 152.62 (d, J= 269.1 Hz), 145.75 (d, J= 6.1 Hz), 136.73 (qd, J = 36.1, 12.0 Hz), 134.56 (d, J= 16.9 Hz), 126.89, 120.58 (qd, J= 275.0, 4.9 Hz),
115.11, 92.04, 62.05, 60.57 (2C), 44.47, 39.42, 29.38, 28.47 ppm; Ci7H16F4N40 (MW 368.33), LCMS (EI) mle 369 (M++ H).
2-(l-(l-(3-Fluoro-2-(trifluoromethyl)isonicotinoyl)piperidin-4-yl)-3-(4-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol-l-yl)azetidin-3-yl)acetonitrile (11). To a 25 mL flask equipped with a nitrogen inlet, a thermocouple, and a magnetic 20443-0253WO1 (INCY0124-WO1) PATENT stirrer were added 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (1, 210 mg, 1.08 mmol, 1.08 equiv), 2-(l-(l-(3-fluoro-2-
(trifluoromethyl)isonicotinoyl)piperidin-4-yl)azetidin-3 -ylidene)acetonitrile (10, 370 mg, 1.0 mmol) and acetonitrile (3 mL) at ambient temperature. The solution was then treated with l,8-diazabicyclo[5,4,0]undec-ene (DBU, 173 mg, 0.17 mL, 1.12 mmol, 1.12 equiv) at ambient temperature and the resulting reaction mixture was warmed to 50 °C and stirred at 50 °C for overnight. When the reaction was complete as
monitored by HPLC, the reaction mixture was directly load on a solica gel (S1O2) column for chromatographic purification (0 – 2.5 % MeOH in ethyl acetate gradient elution) to afford 2-(l-(l-(3-fluoro-2-(trifluoromethyl)isonicotinoyl)piperidin-4-yl)-3- (4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol-l-yl)azetidin-3- yl)acetonitrile
 COMPD 11
COMPD 11
(11, 263 mg, 562.4 mg theoretical, 46.7 %) as a white solid.
For 11: ΧΗ NMR (400 MHz, DMSO-i/6) δ 8.64 (d, J= 4.7 Hz, 1H), 8.22 (d, J= 0.6 Hz, 1H), 7.88 (dd, J= A J Hz, 1H), 7.69 (s, 1H), 4.10 – 3.99 (m, 1H), 3.58 (d, J= 7.8 Hz, 2H), 3.52 – 3.42 (m, 2H), 3.44 (s, 2H), 3.41 – 3.33 (m, 1H), 3.28 – 3.15 (m, 1H), 3.03 (ddd, J= 12.9, 9.2, 3.2 Hz, 1H), 2.51 – 2.44 (m, 1H), 1.77 – 1.66 (m, 1H), 1.64 – 1.54 (m, 1H), 1.28 – 1.17 (m, 2H), 1.24 (s, 12H) ppm;
13C MR (101 MHz, DMSO-i/6) δ 160.22, 152.13 (d, J= 265.8 Hz), 146.23 (d, J= 5.7 Hz), 145.12, 135.41, 134.66 (d, J= 16.9 Hz), 134.43 (qd, J= 35.0, 1 1.7 Hz), 127.58, 120.61 (qd, J= 274.4, 4.6 Hz), 117.35, 106.59 (br), 83.10, 61.40, 60.53 (2C), 56.49, 44.17, 38.99, 28.55, 27.82, 27.02, 24.63 ppm; C26H3iBF4 603 (MW 562.37), LCMS (EI) mle 563 (M+ + H).
2-(3-(4-(7H-Pyrrolo[2,3-< |pyrimidin-4-yl)-lH-pyrazol-l-yl)-l-(l-(3-fluoro-2- (trifluoromethyl)isonicotinoyl)piperidin-4-yl)azetidin-3-yl)acetonitrile (8). To a
25-mL flask equipped with a nitrogen inlet, a thermocouple, an additional funnel, and a magnetic stirrer were added 2-(l-(l-(3-fluoro-2-(trifluoromethyl)- isonicotinoyl)piperidin-4-yl)-3-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH- pyrazol-l-yl)azetidin-3-yl)acetonitrile (11, 307 mg, 0.546 mmol), 4-chloro-7H- pyrrolo[2,3-if|pyrimidine (4, 84.8 mg, 0.548 mmol, 1.0 equiv), sodium bicarbonate (NaHC03, 229 mg, 2.72 mmol, 5.0 equiv), water (1.6 mL), and 1,4-dioxane (1.6 mL) at ambient temperature. The mixture was then teated with
tetrakis(triphenylphosphine)palladium(0) (12.8 mg, 0.011 mmol, 0.02 equiv) at 20443-0253WO1 (INCY0124-WO1) PATENT ambient temperature and the resulting reaction mixture was de-gassed and refilled with nitrogen for 3 times before being heated to 85 °C. The reaction mixture was stired at 85 °C under nitrogen for overnight. When the reaction was complete as monitored by HPLC, the reaction mixture was concentrated to dryness under reduced pressure and the desired product, 2-(3-(4-(7H-pyrrolo[2,3-( Jpyrimidin-4-yl)-lH- pyrazol- 1 -yl)- 1 -( 1 -(3 -fluoro-2-(trifluoromethyl)isonicotinoyl)piperidin-4-yl)azetidin- 3-yl)acetonitrile (8 free base, 135 mg, 302.2 mg theoretical, 44.6 %), was obtained as off- white solids by direct silica gel (S1O2) cloumn chromatography (0 – 10% of ethyl acetate in hexane gradient elution) purification of the dried reaction mixture. The compound obtained by this synthetic approach is identical in every comparable aspect to the compound 8 manufactured by the synthetic method as described above inExample 1.

……………………………………………….
A Double-Blind, Placebo-Controlled Study Exploring the Safety, Tolerability, and Efficacy of a 28 Day Course of INCB-039110 in Subjects With Active Rheumatoid Arthritis (NCT01626573)
ClinicalTrials.gov Web Site 2012, June 25
A double-blind, placebo-controlled study exploring the safety, tolerability, and efficacy of a 28-day course of escalating doses of an oral JAK 1 inhibitor (INCB039110) in subjects with stable, chronic plaque psoriasis
22nd Congr Eur Acad Dermatol Venereol (EADV) (October 3-6, Istanbul) 2013, Abst FC01.6
A randomized, dose-ranging, placebo-controlled, 84-day study of INCB039110, a selective janus kinase-1 inhibitor, in patients with active rheumatoid arthritis
77th Annu Sci Meet Am Coll Rheumatol (October 26-30, San Diego) 2013, Abst 1797
Safety Study of INCB-039110 in Combination With Gemcitabine and Nab-Paclitaxel in Subjects With Advanced Solid Tumors (NCT01858883)
ClinicalTrials.gov Web Site 2013, May
An Open-Label, Phase II Study Of The JAK1 Inhibitor INCB039110 In Patients With Myelofibrosis
55th Annu Meet Am Soc Hematol (December 7-10, New Orleans) 2013, Abst 663
| WO2013036611A1 * | Sep 6, 2012 | Mar 14, 2013 | Incyte Corporation | Processes and intermediates for making a jak inhibitor |
| WO2013043962A1 * | Sep 21, 2012 | Mar 28, 2013 | Merck Sharp & Dohme Corp. | Cyanomethylpyrazole carboxamides as janus kinase inhibitors |
Decernotinib … JAK inhibitor for the treatment of autoimmune and inflammatory diseases, including rheumatoid arthritis.

Decernotinib
Decernotinib
N2-[2-(1H-Pyrrolo[2,3-b]pyridin-3-yl)pyrimidin-4-yl]-N-(2,2,2-trifluoroethyl)-D-isovalinamide
(R)-2-(2-(lH-pyrrolo[2,3-b]pyridin-3-yl)pyrimidin-4-ylamino)-2-methyl-N-(2,2,2- trifluoroethyl)butanamide
Vertex Pharmaceuticals Inc
UNII-MZK2GP0RHK, VX-509, VRT-831509, cas 944842-54-0
Molecular Formula: C18H19F3N6O
Molecular Weight: 392.37827
In phase 3 for the treatment of autoimmune and inflammatory diseases, including rheumatoid arthritis.
 DECERNOTINIB
DECERNOTINIB
The Janus kinases (JAK) are a family of tyrosine kinases consisting of JAK1, JAK2, JAK3, and TYK2. The JAKs play a critical role in cytokine signaling. The down-stream substrates of the JAK family of kinases include the signal transducer and activator of transcription (STAT) proteins. JAK/STAT signaling has been implicated in the mediation of many abnormal immune responses such as psoriasis. Moreover, JAK kinases represent an established therapeutic target for this disease.
For example, JAK kinases are an established therapeutic target for treating psoriasis. Stump K. L., et al., Arthritis Res. Ther. (201 1) 13:R68; Fridman J.S., et al., J Immunol. (2010) 184:5298-5307; West K., Curr. Op. Investig. Drugs (2009) 10:491-504; Kremer J. M. et al., Arthritis Rheumatism (2009) 60(7):1895- 1905; Xiong, W. et al., Ther Adv Musculoskelet Dis. (201 1) 3(5): 255-266; Panes, J. et al. 19th Ann. Eur. Gastroenterology Week (Oct 22-26, 2011) Stockholm, SE, PI 456; and Drugs in R & D “Tofacitinib” (2010) 10(4):271-84.
Compounds described as kinase inhibitors, particularly the JAK family kinases, are disclosed in WO 2005/095400 and WO 2007/084557. Also disclosed in these publications are processes and intermediates for preparing these compounds
Decernotinib ( VX-509 ) is an oral selective JAK3 inhibitor being evaluated for the treatment of rheumatoid arthritis ( RA ). This was a 24-week, randomized, placebo-controlled, double-blind, phase 2 study of four dosing regimens of Decernotinib, administered to patients with RA with inadequate response to Methotrexate ( MTX ).
The aim of the study was to assess the efficacy and safety of four dosing regimens of VX-509 administered to patients with rheumatoid arthritis on stable background Methotrexate therapy.
Patients with active rheumatoid arthritis ( C-reactive protein [ CRP ] greater than ULN, greater than or equal to 6 swollen joints [ of 66 ], and greater than or equal to 6 tender joints [ of 68 ] ) taking stable doses of MTX were randomized 1:1:1:1:1 to receive placebo or one of four dosing regimens of Decernotinib ( 100 mg QD, 150 mg QD, 200 mg QD, or 100 mg BID ) for a duration of 24 weeks.
The primary efficacy endpoints at week 12 were met and have previously been reported; 24-week efficacy and safety results are now reported.
A total of 358 patients were randomized and received greater than or equal to 1 dose of study drug; 81% of patients were female, with a mean age of 53 years.
At baseline, the mean tender joint count was 23.8, the mean swollen joint count was 16.1, and the average disease duration was 7.3 years.
After 24 weeks of treatment the proportion of patients achieving ACR20, ACR50, ACR70, DAS28 ( CRP ) less than 2.6 and DAS28 ( ESR ) less than 2.6 and the decrease from baseline in DAS28 ( CRP ) were statistically significantly greater in each of the Decernotinib dose groups than in the placebo group.
Over 24 weeks, the percentage of patients with any adverse event was higher in the Decernotinib group ( all Decernotinib dose groups combined ) ( 59.9% ) relative to placebo ( 42.3% ) and led to study discontinuation in 9.1% and 8.5% of patients in the Decernotinib and placebo groups, respectively.
The most common adverse reactions in the Decernotinib group were headache ( 8.7% ), hypercholesterolemia ( 5.2% ), and diarrhea ( 4.5% ).
Serious adverse reactions occurred in similar proportions of patients receiving Decernotinib ( 7.3% ) or placebo ( 5.6% ), but there were more serious infections in the Decernotinib group ( 3.5% ) compared with placebo ( 1.4% ).
Through 24 weeks there were two serious adverse effects that resulted in death; one was cardiac failure in the Decernotinib 100 mg BID group ( previously reported ) and one was pancytopenia in a patient with pneumonia in the Decernotinib 200 mg QD group.
Elevations in transaminase levels and decreases in median neutrophil and lymphocyte counts were observed in the Decernotinib groups and were generally mild.
Safety profiles were comparable across groups receiving Decernotinib.
In conclusion, all tested doses of Decernotinib significantly improved signs and symptoms of rheumatoid arthritis versus placebo when administered in combination with stable background Methotrexate therapy for 24 weeks.
Decernotinib was associated with small increases in adverse reactions rates, serious infections, and mostly minor laboratory abnormalities. ( Xagena )
Source: EULAR Meeting – van Vollenhoven R et al, Ann Rheum Dis 2014;73(Suppl2)
see
WO 2007084557
http://www.google.com/patents/WO2007084557A2?cl=en
………………………………………
WO 2013006634
http://www.google.com/patents/WO2013006634A2?cl=en

Formula I is:
The present invention provides a process for preparing (R)-2-(2-(lH-pyrrolo[2,3- b]pyridin-3-yl)pyrimidin-4-ylamino)-2-methyl-N-(2,2,2-trifluoroethyl)butanamide of Formula la:

la
comprising the steps of:
ivb) reacting lH-pyrrolo[2,3-b]pyridine (5a) with p-toluenesulfonyl chloride in the presence of an organic solvent to generate l-tosyl-lH-pyrrolo[2,3-b]pyridine (9a)

5a 9a
vb) reacting l-tosyl-lH-pyrrolo[2,3-b]pyridine (9a) in an organic solvent with N-bromosuccinimide to generate 3-bromo-l-tosyl-lH-pyrrolo[2,3-b]pyridine (7a)

vi) reacting 3-bromo-l-tosyl-lH-pyrrolo[2,3-b]pyridine (7a) with triisopropyl borate in the presence of a strong lithium base in an organic solvent to generate
l-tosyl-lH-pyrrolo[2,3-b]pyridin-3-ylboronic acid (8a) 0H

8a
vii) esterifying l-tosyl-lH-pyrrolo[2,3-b]pyridin-3-ylboronic acid (8a) with pinacolate alcohol in an organic solvent to generate
3 -(4,4,5 ,5 -tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 -tosyl- 1 H-pyrrolo[2,3 -bjpyridine (la) :

viiib) reacting 2,4-dichloropyrimidine (11a) with a hydrochloride salt of D-isovaline (15a) under coupling condition to generate a compound of Formula 2a

11a 2a
ixb) reacting the compound of Formula 2a with HC1 to generate the hydrochloride salt of the compound of Formula 2a;
i) reacting the compound of Formula la with the compound of Formula 2a with in the presence of water, an organic solvent, an inorganic base, and a transition metal catalyst to generate a compound of Formula 3a,

ii) deprotecting the compound of Formula 3a under basic conditions to generate a compound of Formula 4a

4a ; and iii) reacting the compound of Formula 4a with 2,2,2-trifluoroethylamine in the presence of a coupling agent and an organic solvent to generate the compound of Formula la.


– l13C4, 15N2]


……………………………………………………………….
WO 2013070606
http://www.google.com/patents/WO2013070606A1?cl=en
………………………………………………….
patent WO2014074471
WO2014074471 claiming use of heterocyclic compound (preferably decernotinib) for treating psoriasis. Vertex is developing decernotinib, an oral JAK 3 inhibitor, for the treatment of autoimmune and inflammatory diseases, including rheumatoid arthritis. As of July 2014, the drug is Phase 3 trials.
http://www.google.com/patents/WO2014074471A1?cl=en
Table 1:
COMPD 1 IS DECERNOTINIB


Example 1: Analytical Methods Used
[0260] (A) HPLC on C18 column. Mobile phase was acetonitrile/water/TFA (60:40:0.1). Flow rate was 1.0 mL/min. Detection at wavelength of 230 nm. Run time was 25-26 minutes.
[0261] (B) HPLC on C18 column. Mobile phase was acetonitrile/water/TFA (90: 10:0.1). Flow rate was 1.0 mL/min. Detection at wavelength of 230 nm.
[0262] (C) HPLC on a Waters XBridge Phenyl column, 4.6 x 150 mm, 3.5 μπι. Mobile phase A was water/1 M ammonium formate, pH 4.0 (99: 1). Mobile phase B was
acetonitrile/water/ 1M ammonium formate, pH 4.0 (90:9:1). Gradient 5 % to 90 % B in 15 minutes. Total run time 22 minutes. Flow rate 1.5 mL/min. Detection at UV, 245 nm.
T = 25 °C.
[0263] (D) HPLC on a Waters XBridge Phenyl column, 4.6 x 150 mm, 3.5 μπι. Mobile phase A was water/1 M ammonium formate, pH 4.0 (99: 1). Mobile phase B was
acetonitrile/water/ 1M ammonium formate, pH 4.0 (90:9: 1). Gradient 15% to 90 % B in 15 minutes. Total run time 22 minutes. Flow rate 1.5 mL/min. Detection at UV, 220 nm.
T = 35 °C.
[0264] Example 2: Preparation of Compounds of Formula I [0265] General Synthetic Scheme

[0266] The Boc-protected amino acid starting material (1) undergoes amidation in the presence of an activating agent, a coupling reagent, and the acid salt of the amine HNR7R17 to generate the Boc-protected amide intermediate (2). The amide intermediate (2) is
deprotected under acidic conditions and reacted with the halogenated heteroaryl (3) to generate the aminoheteroaryl intermediate (4). Boronated azaindole (5) is coupled with the aminoheteroaryl intermediate (4) under cross-coupling condition to generate the compound of Formula I.
………………………………………………………………………….
Patent
http://www.google.com/patents/US8163917
| 346 | M+H393.20 | RT 1.60 | (DMSO-d6, 300 MHz) 11.95 (bs, 1H), 8.7 (d, |
| 1H), 8.25 (m, 2H), 8.12 (d, 1H), 8.02 (d, 1H), | |||
| 7.28 (s, 1H), 7.13 (dd, 1H), 6.38 (bd, 1H), 3.75 | |||
| (m, 2H), 2.06 (m, 1H), 1.83 (m, 1H), 1.46 (s, | |||
| 3H), 0.8 (t, 3H); |
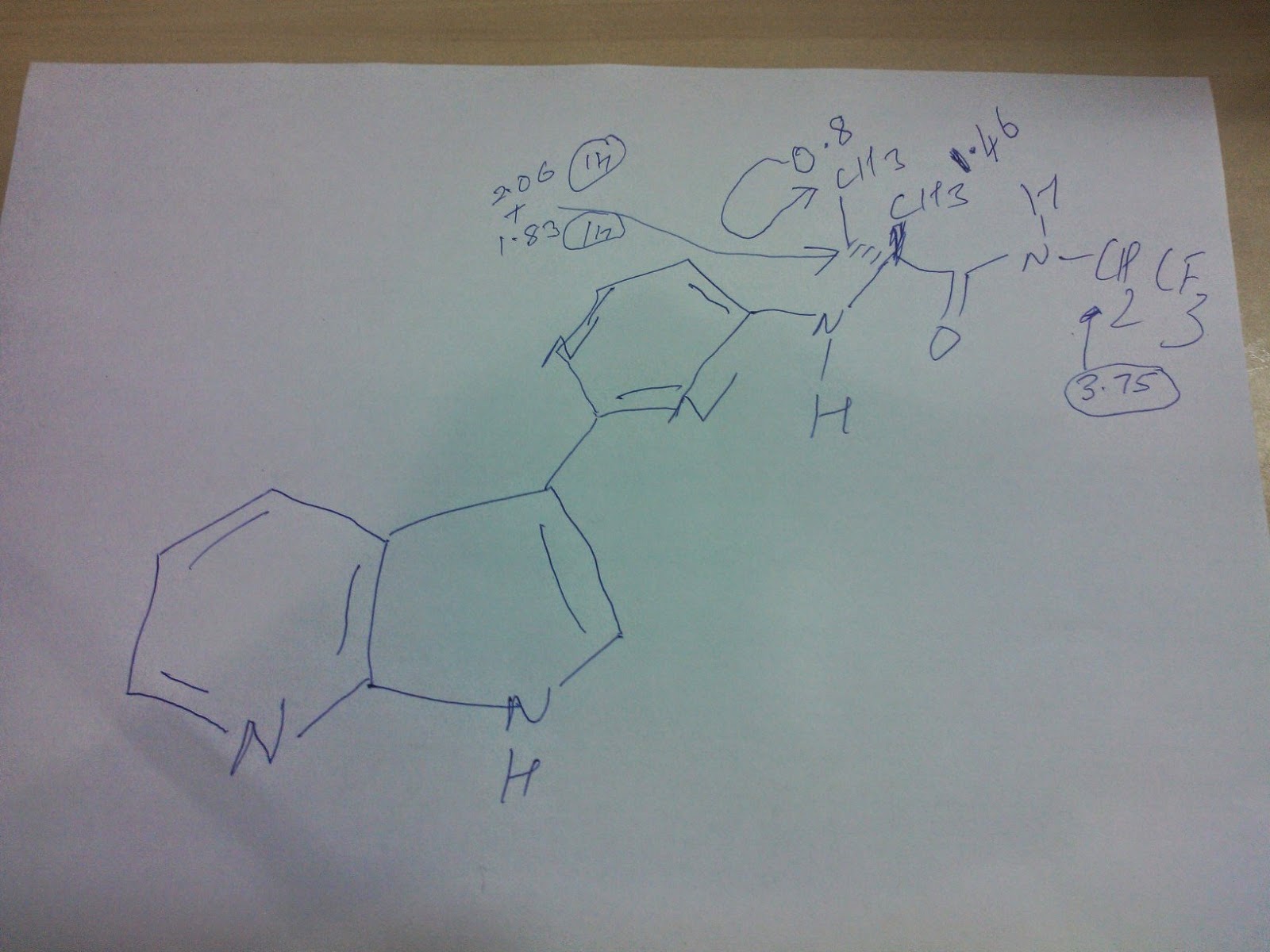
| 346 |
|
|
Example 1 Preparation of Compounds of the Invention
General Synthetic Scheme

Step 1
To a stirred solution of Boc-valine (1; R1 is Me; 3.8 g, 0.02 mol), EDC (4.63 g, 0.024 mol), HOBt (4.0 g, 0.026 mol), DIEA (10.5 mL, 0.06 mol) in 100 mL of DCM is added trifluoroethylamine HCl (2.92 g, 0.022 mol). The reaction mixture is stirred for 16 h. It is concentrated to dryness and redissolved in EtOAc, washed successively with 0.5N HCl, saturated aqueous solution of NaHCO3 and brine. The organic layer is dried (Na2SO4) and concentrated in vacuo to give 5.4 g (98%) of 2 as a white solid.
Step 2
Compound 2 (5.32 g, 0.0197 mol) is deprotected with a 1:1 mixture of DCM/TFA at rt for 45 min. Concentration to dryness gives the intermediate amine that is used directly for the next step. A mixture of 5-fluoro-2,4-dichloropyrimidine (3; R is F; 3.28 g, 0.0197 mol), the crude amine TFA salt (5.25 g, 0.0197 mol) and DIEA (10.27 mL, 0.059 mol) are stirred in isopropanol at rt for 16 h. The reaction mixture is concentrated in vacuo and redissolved in EtOAc, washed successively with 0.5N HCl, saturated aqueous solution of NaHCO3 and brine. The organic layer is dried (Na2SO4) and concentrated in vacuo to give a crude oil that is subjected to chromatography (50% EtOAc/50% hexanes) to yield the desired compound 4.
Step 3
A mixture of 5 (30 mg, 0.075 mmol; prepared according to WO 2005/095400), 4 (23 mg, 0.075 mmol), Pd (Ph3P)4 (9 mg, 0.0078 mmol) and sodium carbonate 2M (115 uL, 0.23 mmol) in 1 mL of DME is microwaved at 150° C. for 10 minutes. The reaction mixture is filtered through a short pad of silica gel with 30% EtOAc-70% hexanes as eluent to provide, after concentration to dryness, the crude intermediate that is used directly for the next step.
The crude intermediate is dissolved in 1 mL of dry methanol and 200 uL of sodium methoxide in methanol 25% was added. The reaction mixture is stirred at 60° C. for 1 h and quenched with 6N HCl (154 uL). The mixture is dried under a flow of nitrogen and purified by reverse phase HPLC (10-60 MeCN/water w/0.5% TFA) to provide the desired material of formula 6a.
Compounds of formulae 6b and 6c may be prepared in an analogous manner using the appropriate starting reagents. For instance, a compound of formula 6b may generally be made by substituting Cert-butyl 2-(2,2,2-trifluoroethylcarbamoyl)pyrrolidine-1-carboxylate for compound 1, while a compound of formula 6c may generally be made by substituting tert-butyl 2-(2,2,2-trifluoroethylcarbamoyl)propan-2-ylcarbamate for compound 1.
Example 2 Analytical Results
Tables 4, 5 and 6 below depicts exemplary 1H-NMR data (NMR) and liquid chromatographic mass spectral data, reported as mass plus proton (M+H), as determined by electrospray, and retention time (RT) for certain compounds of the present invention, wherein compound numbers in Tables 4, 5 and 6 correspond to the compounds depicted in Tables 1, 2 and 3, respectively (empty cells indicate that the test was not performed):
PATENTS
|
4-25-2012
|
Azaindoles Useful as Inhibitors of Janus Kinases
|
|
|
8-4-2010
|
Azaindoles useful as inhibitors of janus kinases
|
new patent
WO-2014110259
| US8450489 * | Mar 1, 2012 | May 28, 2013 | Vertex Pharmaceuticals Incorporated | Azaindoles useful as inhibitors of janus kinases |
| US8530489 * | May 22, 2012 | Sep 10, 2013 | Vertex Pharmaceuticals Incorporated | 5-cyano-4-(pyrrolo [2,3B] pyridine-3-yl)-pyrimidine derivatives useful as protein kinase inhibitors |
| US8686143 * | Oct 25, 2011 | Apr 1, 2014 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of Janus kinases |
| US20120157429 * | Oct 25, 2011 | Jun 21, 2012 | Wannamaker Marion W | Compounds useful as inhibitors of janus kinases |
| US20120165307 * | Mar 1, 2012 | Jun 28, 2012 | Vertex Pharmaceuticals Incorporated | Azaindoles useful as inhibitors of janus kinases |
| US20120309963 * | May 22, 2012 | Dec 6, 2012 | Vertex Pharmaceuticals Incorporated | 5-cyano-4- (pyrrolo [2,3b] pyridine-3-yl) -pyrimidine derivatives useful as protein kinase inhibitors |
| US20130237516 * | Apr 25, 2013 | Sep 12, 2013 | Vertex Pharmaceuticals Incorporated | Azaindoles useful as inhibitors of janus kinases |
| WO2013173506A2 | May 15, 2013 | Nov 21, 2013 | Rigel Pharmaceuticals, Inc. | Method of treating muscular degradation |
| WO2005095400A1 | Mar 30, 2005 | Oct 13, 2005 | Vertex Pharma | Azaindoles useful as inhibitors of jak and other protein kinases |
| WO2007084557A2 | Jan 17, 2007 | Jul 26, 2007 | Vertex Pharma | Azaindoles useful as inhibitors of janus kinases |
| WO2013070606A1 * | Nov 6, 2012 | May 16, 2013 | Vertex Pharmaceuticals Incorporated | Methods for treating inflammatory diseases and pharmaceutical combinations useful therefor |
BMS-582949 in phase 2 for Treatment of Antipsoriatics , Rheumatoid arthritis
BMS 582949, PS-540446
UNII-CR743OME9E
CAS 623152-17-0
4-[5-(N-Cyclopropylcarbamoyl)-2-methylphenylamino]-5-methyl-N-propylpyrrolo[2,1-f][1,2,4]triazine-6-carboxamide
4-(5-(Cyclopropylcarbamoyl)-2-methylphenylamino)-5-methyl-N-propylpyrrolo[1,2-f][1,2,4]triazine-6-carboxamide
Bristol-Myers Squibb Company
M.Wt: 406.48
Cas : 623152-17-0 Formula: C22H26N6O2
BMS-582949 had been in phase II clinical trials at Bristol-Myers Squibb for the oral treatment of moderate to severe psoriasis and for the treatment of rheumatoid arthritis (RA) in combination with methotrexate and for the treatment of inflammation in atherosclerotic plaque. However, no recent development has been reported for this research.
…………………..
http://www.google.com/patents/WO2012031057A1?cl=en
The present invention generally relates to a method of treating resistant rheumatic disease, such as refractory rheumatoid arthritis, with a therapeutically effective amount of a dual action p38 inhibitor that is safe and well-tolerated. A dual action p38 kinase inhibitor is a compound that inhibits both activation of p38 kinase and p38 kinase activity in cells.
A large number of cytokines participate in the inflammatory response, including IL- 1 , IL-6, IL-8 and TNF-a. Overproduction of cytokines such as IL-1 and TNF-a are implicated in a wide variety of diseases, including inflammatory bowel disease, rheumatoid arthritis, psoriasis, multiple sclerosis, endotoxin shock, osteoporosis, Alzheimer’s disease, and congestive heart failure, among others. See e.g., Henry et al., Drugs Fut. , 24: 1345- 1354 ( 1999); Salituro et al., Curr. Med. Ckem., 6:807-823 (1999)]. Important mediators of proinflammatory cytokines such as TNFct and IL-1 β,. as well as cellular responses to such cytokines production, are the mitogen-activated protein (MAP) kinases, and in particular, p38 kinase. See e.g., Schieven, G.L., “The biology of p38 kinase: a central role in inflammation”, Current Topics in Medicinal Chemistry, 5 :921 – 928 (2005). Accordingly, modulation of p38 kinase may be useful in the treatment of inflammatory disease including rheumatic diseases such as rheumatoid arthritis (RA).
Compounds that reportedly inhibit p38 kinase and cytokines such as IL-1 and TNF-a for use in treating inflammatory diseases are disclosed in U.S. Patent Nos.
6,277,989 and 6, 130,235 to Scios, Inc; U.S. Patent. Nos. 6, 147,080 and 5,945,41 8 to Vertex Pharmaceuticals Inc; U.S. Patent Nos. 6,251 ,914, 5,977, 103 and 5,658,903 to Smith-Kline Beecham Corp.; U.S. Patent Nos. 5,932,576 and 6,087,496 to G.D. Searle & Co.; WO 00/56738 and WO 01 /27089 to Astra Zeneca; WO 01/34605 to Johnson & Johnson; WO 00/12497 (quinazoHne derivatives as p38 kinase inhibitors); WO 00/56738 (pyridine and pyrimidine derivatives for the same purpose); WO 00/12497 (discusses the relationship between p38 kinase inhibitors); and WO 00/12074 (piperazine and piperidine compounds useful as p38 inhibitors). Other compounds that inhibit p38 kinase are pyrrolotriazine aniline compounds, information on these compounds is disclosed in U.S. Patent Nos. 6,670,357; 6,867,300; 7,034, 151 ; 7, 160,883; 7,21 1,666; 7,253, 167; and U.S. Publication Nos. 2003/023283 1 (published Dec. 18, 2003); 2004/0229877 (published Nov. 1 8, 2004); 2005/0043306 (published Feb. 24, 2005; 2006/0003967 (published Jan. 5, 2006); 2006/0030708 (published Feb. 9, 2006); 2006/0041 124 (published Feb. 23, 2006); 2006/0229449 (published Oct. 12, 2006); 2006/0235020 (published Oct. 19, 2006); and 2007/0213300 (published Sept 13, 2007).
In particular, WO 2003/090912 (U.S. Patent Nos. 7, 160,883, 7,388,009, p38 inhibitor, BMS-582949 (Example 7,

including processes of making and uses thereof.
……………………
http://www.google.com/patents/WO2003090912A9?cl=en
Examples 4-22

Compounds having the formula (Id), above, wherein R4 has the values listed in the following Table, were prepared following the same procedure described for Example 3, using the appropriate amine in place of ra-butylamine.



…………………………
WO 2006020904
http://www.google.com.br/patents/WO2006020904A1?cl=en
EXAMPLE IA St


Part a.
A solution of Example 1 (0.86 g, 2.20 mmol, 1.0 eq.) in THF (4.0 mL) and 1 N aqueous NaOH (9.0 mL, 4.1 eq.) was stirred at 6O0C overnight. After cooling to RT, the reaction mixture was concentrated in vacuo but not to dryness. To the solution at O0C was added 1 N aqueous hydrochloric acid until it was acidic and the precipitate was collected and dried to afford crude Example IA acid (0.51 g, 64.0 % yield). HPLC Ret. t. = 2.400 min.; LC/MS (M+H) + = 366.06+. The filtrate was then extracted with EtOAc (3x) and the organic layers were combined, dried over sodium sulfate, and concentrated in vacuo to give Example IA acid (0.035 g, 4.4 % yield). Part b.

A solution of Part a. acid (0.026 g, 0.071 mmol, 1.0 eq.), EDC (0.021 g, 0.11 mmol, 1.5 eq.), HOBt (0.015 g, 0.11 mmol, 1.5 eq), ^-propylamine (0.015 mL, 0.15 mmol, 2.1 eq.) and DIPEA (0.040 mL, 0.23 mmol, 3.2 eq.) in DMF (0.20 mL) was shaken at RT overnight. Water (1 mL) was added and the precipitate collected by filtration, washed with water, and dried to give Example IA amide (0.021 g, 70% yield); HPLC Ret. t. = 2.883 min.; LC/MS (M+H)+ = 421.18 +.
EJiAMPLE 2 Direct Aminolysis Procedure

n-Buli/THF
Ester Compound I or Hexyllithium/THF
-^
,NH9

1. Aminolysis with hexyllithium
To a dried 100 ml flask was added THF (10 ml) under nitrogen, which was then cooled to -100C. Hexyllithium (2.3 M in hexane, 6.5 ml, 15.0 mmol) was added slowly (exothermic, temperature was up to 5°C), followed by dropwise addition of propylamine (1.01 g, 1.4 ml, 17.1 mmol) at such a rate to maintain the temperature below 5°C. The resulting mixture was stirred at O0C for 20 minutes. A suspension of ester compound I (1.0 g, 2.5 mmol) in THF (12 ml) was added over a 10 minute period (exothermic, T<5°C). After being stirred at 00C for 20 minutes, the mixture was allowed to warm to room temperature and stirred for 5 hours. Ester compound I was <0.1 AP at this point by HPLC analysis. The mixture was cooled to -50C. Acetic acid (2 ml) was added slowly to maintain the temperature <10°C. The resulting thick slurry was stirred at room temperature for 20 minutes, and then solvents were exchanged with DMF (15 ml) on a rotavapor. To the resulting yellow slurry, water (15 ml) was added slowly to keep T<25°C. During the addition of water, the slurry became a clear solution, and a new slurry was formed. The slurry was stirred at room temperature for overnight. In the morning the slurry was filtered and the solid was washed with DMF/water (1:1, 5 ml), water (5 ml) and acetone (5 ml). The cake was dried under vacuum at 55°C for 24 hours to afford 0.90 g of amide product II (yield: 87.2%) as a white solid. HPLC: 99.70 AP.
2. Aminolysis with n-butyllithium
To a dried 100 ml of flask was added THF (10 ml) under nitrogen and then cooled to -100C. n-Butyllithium (2.5 M in hexane, 6.0 ml, 15.0 mmol) was added slowly, followed by dropwise addition of propylamine (0.98 g, 16.5 mmol) at such a rate to keep the temperature below 00C. The resulting mixture was stirred at O0C for 20 minutes. A suspension of ester compound I (1.0 g, 2.5 mmol) in THF (12 ml) was added over a 10 minute period (T<5°C). After being stirred at O0C for 30 minutes, the mixture was allowed to warm to room temperature and stirred for overnight (~22h, Note 1). Compound I was not detected at this point by HPLC analysis. The mixture was cooled to -7°C. Acetic acid (2 ml) was added dropwise to maintain the temperature <10°C. The resulting thick slurry was stirred at 50C for 2 hours and at room temperature for 20 minutes, followed by evaporation on a rotavapor to give a wet yellow solid. To this solid was added acetone (10 ml) and water (20 ml). The slurry was stirred at room temperature for one and half hours. Filtration gave a white solid. This solid was washed with 35% acetone in water (10 ml), water (5 ml) and acetone (5 ml). The cake was dried under vacuum at 55°C for the weekend to afford 0.94g of amide product II (yield: 91.0%) as a white solid. HPLC: 99.76 AP. Note 1: Compound I was -0.056 AP at 2.5 hours.
……………………
WO 2003090912
http://www.google.com/patents/WO2003090912A1?cl=en
……………………..
Discovery of 4-(5-(Cyclopropylcarbamoyl)-2-methylphenylamino)-5-methyl-N-propylpyrrolo[1,2-f][1,2,4]triazine-6-carboxamide (BMS-582949), a clinical p38a MAP kinase inhibitor for the treatment of inflammatory diseases
J Med Chem 2010, 53(18): 6629
http://pubs.acs.org/doi/abs/10.1021/jm100540x
The discovery and characterization of 7k (BMS-582949), a highly selective p38α MAP kinase inhibitor that is currently in phase II clinical trials for the treatment of rheumatoid arthritis, is described. A key to the discovery was the rational substitution of N-cyclopropyl for N-methoxy in 1a, a previously reported clinical candidate p38α inhibitor. Unlike alkyl and other cycloalkyls, the sp2 character of the cyclopropyl group can confer improved H-bonding characteristics to the directly substituted amide NH. Inhibitor 7k is slightly less active than 1a in the p38α enzymatic assay but displays a superior pharmacokinetic profile and, as such, was more effective in both the acute murine model of inflammation and pseudoestablished rat AA model. The binding mode of 7k with p38α was confirmed by X-ray crystallographic analysis.

4-(5-(Cyclopropylcarbamoyl)-2-methylphenylamino)-5-methyl-N-propylpyrrolo[1,2-f][1,2,4]triazine-6-carboxamide (7k)
A mixture of 4-(5-(cyclopropylcarbamoyl)-2-methylphenylamino)-5-methylpyrrolo[1,2-f][1,2,4]triazine-6-carboxylic acid (6b) (2.16 g, 5.91 mmol), n-propylamine (1.0 mL, 12.2 mmol), BOP (3.40 g, 7.69 mmol), and N-methylmorpholine (2.5 mL, 22.7 mmol) in DMF (10 mL) was stirred at 50 °C for 3 h. The mixture was poured into a mixture prepared from saturated NaHCO3 solution (60 mL) and water (60 mL). The precipitating product was collected by suction filtration was washed with water. This crude product was suspended into ethyl acetate (100 mL) and stirred at 70 °C for 1 h. Upon cooling to rt, the title compound (2.07 g, 86% yield) was collected as a white solid by suction filtration; 98% purity by HPLC. LCMS (EI)
m/z Calcd for C22H26N6O2 (M + H)+ = 407.21. Found: 407.22.
1H NMR (500 MHz, DMSO-d6) δ 8.49 (d, J = 3.6 Hz, 1H), 8.23 (s, 1H), 8.21 (s, 1H), 7.86 (s, 1H), 7.80 (s, 1H), 7.77 (d, J = 7.8 Hz, 1H), 7.42 (d, J = 7.8 Hz, 1H), 3.20 (m, 2H), 2.87 (m, 1H), 2.82 (s, 3H), 2.25 (s, 3H), 1.54 (m, 2H), 0.91 (t, J = 7.4 Hz, 3H), 0.68 (m, 2H), 0.59 (m, 2H).
13C NMR (125 MHz, DMSO-d6) δ 167.3, 164.45, 155.3, 148.7, 138.8, 137.1, 133.0, 130.6, 127.2, 125.8, 119.6, 118.8, 114.4, 113.3, 41.0, 23.6, 23.1, 18.5, 12.1, 12.0, 6.2.
EXAMPLE 3

Direct Aminolysis
Ester Compound I

Amide Product II
Method A:
A solution of n-propylamine (6.5 eq) in THF (20 ml/g of ester compound I) was cooled to — 5°C and was slowly treated with 2.5 M solution of n-butyllithium (6.1 eq). The mixture was stirred for 10 minutes. At the end of the period, a slurry of ester compound I (1 eq) in THF (14 ml/g of ester compound I) was cannulated into the performed Li-NHPr solution. The reaction mixture was warmed to 25°C and stirred till all of ester compound I was consumed (~ 3 hours). After the reaction was judged to be completed by HPLC, the reaction mixture was cooled to ~0°C and was slowly treated with acetic acid (5 ml/g of ester compound I). The slurry was then warmed to -2O0C and was stirred for 1 hour. At the end of the period, the solvent was distilled under vacuum to the minimum volume and the concentrated slurry was diluted with a solution of acetone (10 ml/g of ester compound I) and water (20 ml/g of ester compound I). The slurry was stirred for 1 hour and was cooled to ~5°C. The slurry was filtered and the cake was washed with acetone (5 ml/g of ester compound I). The cake was dried to give the amide product II (typically in 85% yield and 99 AP).
Method B:
A solution of n-propylamine (20 eq) in 2,2,2-trifmoroethanol (10 ml/g of ester compound I) was slowly treated with 2.5 M solution of n-butyllithium (1.5 eq). The mixture was stirred for 5 minutes. At the end of the period, the starting material, ester compound I, was added and the reaction mixture was warmed to 900C. The reaction mixture was held at 900C for 24 hours and was allowed to cool to ~20°C. The reaction mixture was then analyzed by HPLC. Typically, analysis indicated there was only 1.57 AP of starting material left.
Method C:
A solution of n-propylamine (2 eq) in methylene chloride (10 ml/g of ester compound I) at 200C was slowly treated with 2.0 M solution of trimethylaluminum (4 eq) in hexanes. The mixture was stirred for 15 minutes. At the end of the period, the starting material, ester compound 1 (1 eq), was added and the reaction mixture was warmed to 600C. The reaction mixture was held at 600C for 24 hours and was allowed to cool to ~20°C. The reaction mixture was then slowly quenched with aqueous HCl solution and analyzed by HPLC. Typically, analysis indicated there was 96.8AP of amide compound II product with 0.03 AP of the dipropylamide impurity.
…………………………………….
| WO2003090912A1 * | 15 abr. 2003 | 6 nov. 2003 | Squibb Bristol Myers Co | Pyrrolo-triazine aniline compounds useful as kinase inhibitors |
Liu C, Lin J, Everlof G, Gesenberg C, Zhang H, Marathe PH, Malley M, Galella MA, McKinnon M, Dodd JH, Barrish JC, Schieven GL, Leftheris K.
Bioorg Med Chem Lett. 2013 May 15;23(10):3028-33. doi: 10.1016/j.bmcl.2013.03.022. Epub 2013 Mar 15.
Freebern WJ, Bigwarfe TJ, Price KD, Haggerty HG.
J Immunotoxicol. 2013 Jan-Mar;10(1):106-17. doi: 10.3109/1547691X.2012.736427. Epub 2012 Nov 23.
Liu C, Lin J, Wrobleski ST, Lin S, Hynes J, Wu H, Dyckman AJ, Li T, Wityak J, Gillooly KM, Pitt S, Shen DR, Zhang RF, McIntyre KW, Salter-Cid L, Shuster DJ, Zhang H, Marathe PH, Doweyko AM, Sack JS, Kiefer SE, Kish KF, Newitt JA, McKinnon M, Dodd JH, Barrish JC, Schieven GL, Leftheris K.
J Med Chem. 2010 Sep 23;53(18):6629-39. doi: 10.1021/jm100540x.
BMS-582949: crystalline form of a p38alpha inhibitor? WO2008079857.
Norman P.
Expert Opin Ther Pat. 2009 Aug;19(8):1165-8. doi: 10.1517/13543770902816160.
| WO2000012074A2 | Aug 27, 1999 | Mar 9, 2000 | Sarvajit Chakravarty | Use of piperidines and/or piperazines as inhibitors of p38-alpha kinase | |
| WO2000012497A2 | Aug 27, 1999 | Mar 9, 2000 | Sarvajit Chakravarty | Quinazoline derivatives as medicaments | |
| WO2000056738A1 | Mar 17, 2000 | Sep 28, 2000 | Astrazeneca Ab | Pyridine and pyrimidine derivatives and their use as inhibitors of cytokine mediated disease | |
| WO2001027089A1 | Oct 10, 2000 | Apr 19, 2001 | Astrazeneca Ab | Pyrimidine derivatives | |
| WO2001034605A1 | Oct 27, 2000 | May 17, 2001 | Ortho Mcneil Pharm Inc | SUBSTITUTED 2-ARYL-3-(HETEROARYL)-IMIDAZO[1,2-a]PYRIMIDINES, AND RELATED PHARMACEUTICAL COMPOSITIONS AND METHODS | |
| WO2003090912A1 | Apr 15, 2003 | Nov 6, 2003 | Squibb Bristol Myers Co | Pyrrolo-triazine aniline compounds useful as kinase inhibitors | |
| US4200750 | Dec 8, 1977 | Apr 29, 1980 | Westwood Pharmaceuticals Inc. | 4-Substituted imidazo [1,2-a]quinoxalines | |
| US5658903 | Jun 3, 1996 | Aug 19, 1997 | Smithkline Beecham Corporation | Cytokine inhibitors | |
| US5932576 | May 22, 1998 | Aug 3, 1999 | G. D. Searle & Company | 3(5)-heteroaryl substituted pyrazoles as p38 kinase inhibitors | |
| US5945418 | Mar 20, 1997 | Aug 31, 1999 | Vertex Pharmaceuticals Incorporated | Administering to the mammal to inhibit a mammalian protein kinase p38 which causes cell proliferation, cell death and response to extracellular stimuli | |
| US5977103 | Jan 10, 1997 | Nov 2, 1999 | Smithkline Beecham Corporation | Substituted imidazole compounds | |
| US6087496 | Apr 1, 1999 | Jul 11, 2000 | G. D. Searle & Co. | Enzyme inhibitors | |
| US6130235 | Aug 3, 1998 | Oct 10, 2000 | Scios Inc. | Piperidine moieties coupled to indole, benzimidazole or benzotriazole. | |
| US6147080 | Jun 10, 1997 | Nov 14, 2000 | Vertex Pharmaceuticals Incorporated | Inhibitors of p38 | |
| US6251914 | Jul 1, 1998 | Jun 26, 2001 | Smithkline Beecham Corporation | Treating cytokine mediated diseases | |
| US6277989 | Mar 14, 2000 | Aug 21, 2001 | Scios, Inc. | Quinazoline derivatives as medicaments | |
| US6670357 | Nov 7, 2001 | Dec 30, 2003 | Bristol-Myers Squibb Company | Antiinflammatory agents | |
| US6867300 | Nov 6, 2002 | Mar 15, 2005 | Bristol-Myers Squibb Company | Methods for the preparation of pyrrolotriazine compounds useful as kinase inhibitors | |
| US7034151 | Feb 5, 2004 | Apr 25, 2006 | Bristol-Myers Squibb Company | 1,4-dihydro-4-oxo-pyrrolo[2,1-f][1,2,4]triazine-6-carboxylates; novel approach to the formation of the bicyclic heterocyclic ring system | |
| US7041501 | Oct 31, 2002 | May 9, 2006 | Bristol-Myers Squibb Company | Methods of screening for toxicity of test compounds | |
| US7160883 | Apr 22, 2003 | Jan 9, 2007 | Bristol-Myers-Squibb Company | Pyrrolo-triazine aniline compounds useful as kinase inhibitors | |
| US7211666 | Dec 22, 2004 | May 1, 2007 | Bristol-Myers Squibb Company | N-Cyclopropyl-4-[[5-[(methoxyamino)carbonyl]-2-methylphenyl]amino]-5-methylpyrrolo[2,1-f][1,2,4]triazine-6-carboxamide; aminating with chloramine to produce a pyrrole with a Nitrogen nitrogen bond; reacting with formamide, cyclizing to form the pyrrolotriazine core; kinase inhibitors | |
| US7253167 | Jun 29, 2005 | Aug 7, 2007 | Bristol-Myers Squibb Company | Tricyclic-heteroaryl compounds useful as kinase inhibitors | |
| US7388009 | Oct 3, 2003 | Jun 17, 2008 | Bristol-Myers Squibb Company | Heterocyclic drugs as enzyme inhibitors for Kinase enzymes or prodrugs | |
| US7462616 | Oct 24, 2006 | Dec 9, 2008 | Bristol-Myers Squibb Company | Pyrrolo-triazine aniline compounds useful as kinase inhibitors | |
| US7759343 | Oct 28, 2008 | Jul 20, 2010 | Bristol-Myers Squibb Company | Pyrrolo-triazine aniline compounds useful as kinase inhibitors | |
| US61379001 | Title not available | ||||
| US20030232831 | Apr 22, 2003 | Dec 18, 2003 | Alaric Dyckman | Aryl ketone pyrrolo-triazine compounds useful as kinase inhibitors | |
| US20040229877 | Oct 29, 2003 | Nov 18, 2004 | Katerina Leftheris | Administering pyrrolotriazine carboxamide and benzamide compounds for therapy of p38 kinase-associated conditions | |
| US20050043306 | Oct 3, 2003 | Feb 24, 2005 | Katerina Leftheris | Heterocyclic drugs as enzyme inhibitors for Kinase enzymes or prodrugs | |
| US20060003967 | Jun 28, 2005 | Jan 5, 2006 | Zhongping Shi | Method for preparing pyrrolotriazine compounds | |
| US20060030708 | Aug 5, 2005 | Feb 9, 2006 | Lobben Paul C | Methods for the preparation of pyrrolotriazine compounds | |
| US20060041124 | Oct 14, 2005 | Feb 23, 2006 | Bang-Chi Chen | Process for preparing pyrrolotriazine kinase inhibitors | |
| US20060229449 | Apr 3, 2006 | Oct 12, 2006 | Apurba Bhattacharya | Reacting with chloramine in presence of aqueous base, phase transfer catalyst; anti-cancer agents, kinase inhibitors | |
| US20060235020 | Apr 4, 2006 | Oct 19, 2006 | Soojin Kim | Process for preparing salts of 4-[[5-[(cyclopropylamino)carbonyl]-2-methylphenyl]amino]-5-methyl-N-propylpyrrolo[2,1-f][1,2,4]triazine-6-carboxamide and novel stable forms produced therein | |
| US20070213300 | Mar 6, 2007 | Sep 13, 2007 | Bristol-Myers Squibb Company | Pyrrolotriazine aniline prodrug compounds useful as kinase inhibitors |
World’s first biosimilar antibody is approved in Korea

celltrion Worlds first biosimilar antibody* is approved in Korea
South Korean biosimilar manufacturer Celltrion, today declared that, Korean Food and Drug Administration approved its first biosimilar monoclonal antibody, Remsima.
Remsima is a biosimilar version of Remicade, the blockbuster in Rheumatoid Arthitis (RA). Korean FDA approved the product in several indications including RA, ankylosing spondylitis, ulcerative colitis, psoriasis and Crohn’s disease.
http://www.biosimilarnews.com/worlds-first-biosimilar-antibody-is-approved-in-korea
Molecular imaging gets to the root of rheumatoid arthritis
Rheumatoid arthritis causes chronic pain for almost half of adults by the time they retire, but a new molecular imaging technique can visualize inflammation in the joints, giving doctors a clear read on chronic pain and possible joint destruction, say researchers at the Society of Nuclear Medicine and Molecular Imaging’s 2014 Annual Meeting.
In order to image arthritis inside the joints, researchers used multiple molecular imagingsystems, positron emission tomography (PET) and single photon emission tomography (SPECT), both of which image physiological processes with the help of specialized detectors that pick up signals from injected radionuclide imaging agents. In this case researchers evaluated anti-fibroblast activation protein (FAP) antibodies involved in the inflammation associated with rheumatoid arthritis. This was made possible with radiotracers that combine the molecular compound 28H1, which can bind to FAP in the body, with the radionuclides In-111, used in conjunction with SPECT imaging systems, and Zr-89…
View original post 322 more words
Chinese Herb Beats Drug At Treating Rheumatoid Arthritis
A Chinese herb called thunder god vine works better than a widely-prescribed pharmaceutical drug at easing rheumatoid arthritis, a new study has found.

The herb has long been used in China to treat this potentially crippling autoimmune disease, which typically strikes hand and foot joints. It is known in Mandarin as ‘lei gong teng’ and to botanists as Tripterygium wilfordii Hook F.
Extracts of the herb have already fired the interest of drug laboratories as they contain hundreds of compounds, including intriguing molecules called diterpenoids which are believed to ease inflammation and immune response.
In a study published in the journal Annals of the Rheumatic Diseases, Chinese researchers recruited 207 patients with rheumatoid arthritis and gave them either the herb; the drug methotrexate; or a combination of the two.
After six months, the patients were given a doctor’s assessment and were also asked if they felt…
View original post 279 more words
Bristol-Myers Squibb announced promising results from phase 2b study of its rheumatoid arthritis drug clazakizumab
clazakizumab. BMS-945429, ALD518
Bristol-Myers Squibb, phase 2b study, rheumatoid arthritis,
NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL
CLAZAKIZUMAB
PRONUNCIATION klaz” a kiz’ ue mab
THERAPEUTIC CLAIM Autoimmune diseases, rheumatoid arthritis
CHEMICAL NAMES
1. Immunoglobulin G1, anti-(human interleukin 6) (human-Oryctolagus cuniculus monoclonal BMS-945429/ALD518 heavy chain), disulfide with human-Oryctolagus cuniculus monoclonal BMS-945429/ALD518 κ-chain, dimer
2. Immunoglobulin G1, anti-(human interleukin-6 (B-cell stimulatory factor 2, CTL differentiation factor, hybridoma growth factor, interferon beta-2)); humanized rabbit monoclonal BMS-945429/ALD518 [300-alanine(CH2-N67>A67)]1 heavy chain (223-217′)-disulfide with humanized rabbit monoclonal BMS-945429/ALD518 light chain dimer (229-229”:232-232”)-bisdisulfide, O-glycosylated
MOLECULAR FORMULA C6426H9972N1724O2032S42
MOLECULAR WEIGHT 145.2 kDa
SPONSOR Bristol-Myers Squibb
CODE DESIGNATION BMS-945429, ALD518
CAS REGISTRY NUMBER 1236278-28-6
Monoclonal antibody
Type Whole antibody
Source Humanized
Target IL6
CAS number 1236278-28-6
Clazakizumab is a humanized monoclonal antibody designed for the treatment of rheumatoid arthritis.[1]
Clazakizumab was developed by Alder Biopharmaceuticals and Bristol-Myers Squibb.
Bristol-Myers Squibbalong with Alder Biopharmaceuticals, announced the presentation of efficacy and safety data from a Phase 2b dose-ranging study of subcutaneous (SC) clazakizumab in adults with moderate-to-severe rheumatoid arthritis (RA) and an inadequate response to methotrexate (MTX). Clazakizumab is a humanized anti-IL-6 monoclonal antibody that is directed against the IL-6 cytokine rather than its receptor.
In the Phase IIb study clazakizumab doses ranging from 25-200 mg monotherapy and in combination with MTX were studied vs. MTX alone. Adalimumab in combination with MTX was included as an active reference arm. All clazakizumab treatment arms, alone or in combination with MTX, demonstrated efficacy in controlling the signs and symptoms of RA, and met the predefined primary endpoint of a higher ACR20 response rate vs. MTX alone after 12 weeks of treatment. All clazakizumab treatment groups were also associated with improved ACR 20/50/70 response rates and HAQ-DI scores vs. MTX at week 24. Rates of low disease activity and remission with clazakizumab plus MTX, as measured by DAS28 CRP, CDAI and SDAI criteria were numerically greater for clazakizumab at 12 and 24 weeks than the active comparator.
The adverse event (AE) rates were similar across all clazakizumab arms. The most frequent AE for clazakizumab was dose-related injection site reactions. The most frequent reason for discontinuation due to AE in clazakizumab treated patients was laboratory abnormality, predominantly transaminase elevations, more frequent in MTX-containing arms. The most frequent serious adverse events (SAEs) were serious infections. Rates of serious infections were generally comparable for clazakizumab and adalimumab combination arms and were numerically greater than MTX alone.
“There is a great need for additional disease-modifying therapies that can provide more patients with deep and sustainable remission, helping preserve function and limit further joint damage,” said Paul Emery, MD, director of MSK Biomedical Unit at the Leeds Teaching Hospitals Trust in the United Kingdom. “Currently, less than 30% of RA patients experience sustained remission as defined by ACR criteria. Clazakizumab is an investigational therapy that neutralizes IL-6 signaling by blocking the IL-6 cytokine, and provides promising remission data that will need to be further investigated.”
Bristol-Myers Squibb has exclusive worldwide rights to develop and commercialize clazakizumab for all indications outside of cancer under a collaboration agreement with its discoverer, Alder Biopharmaceuticals.
clazakizumab
immunoglobulin G1-kappa, anti-[Homo sapiens IL6 (interleukin 6, IL-6)], humanized monoclonal antibody;
gamma1 heavy chain (1-450) [humanized VH (Homo sapiens IGHV3-66*01 (83.50%) -(IGHD)-IGHJ3*02 M123>L (115)) [8.8.14] (1-120) -Homo sapiens IGHG1*03 CH
gamma1 heavy chain (1-450) [humanized VH (Homo sapiens IGHV3-66*01 (83.50%) -(IGHD)-IGHJ3*02 M123>L (115)) [8.8.14] (1-120) -Homo sapiens IGHG1*03 CH

BMS and Simcere will co-develop and co-commercialize the subcutaneous formulation of Orencia® for the treatment of rheumatoid arthritis in China.

AsianScientist (Jun. 17, 2013) – Bristol-Myers Squibb Company and Simcere Pharmaceutical Group announced this week a new collaboration to co-develop and commercialize the subcutaneous (SC) formulation of BMS’s Orencia® (abatacept) for the treatment of rheumatoid arthritis in China.
Orencia SC is already on the market for the treatment of rheumatoid arthritis in the U.S., Europe, and Japan.
http://www.asianscientist.com/tech-pharma/bms-simcere-pharma-co-develop-orencia-sc-china-2013/
