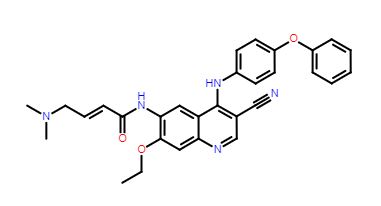
HS-10340
CAS 2156639-66-4
MF C26 H31 N7 O5
MW 521.57
1,8-Naphthyridine-1(2H)-carboxamide, N-[5-cyano-4-[[(1R)-2-methoxy-1-methylethyl]amino]-2-pyridinyl]-7-formyl-3,4-dihydro-6-[(tetrahydro-2-oxo-1,3-oxazepin-3(2H)-yl)methyl]-
(R)-N-(5-cyano-4-((1-methoxypropan-2-yl)amino)pyridin-2-yl)-7-formyl-6-((2-carbonyl)-1,3-oxazepine-3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxamide

CAS 2307670-65-9
Jiangsu Hansoh Pharmaceutical Group Co Ltd
Being investigated by Jiangsu Hansoh, Shanghai Hansoh Biomedical and Changzhou Hengbang Pharmaceutical ; in June 2018, the product was being developed as a class 1 chemical drug in China.
Useful for treating liver cancer, gastric cancer and prostate cancer.
Use for treating cancers, liver cancer, gastric cancer, prostate cancer, skin cancer, ovary cancer, lung cancer, breast cancer, colon cancer, glioma and rhabdomyosarcoma
The fibroblast growth factor receptor (FGFR) belongs to the receptor tyrosine kinase transmembrane receptor and includes four receptor subtypes, namely FGFR1, FGFR2, FGFR3 and FGFR4. FGFR regulates various functions such as cell proliferation, survival, differentiation and migration, and plays an important role in human development and adult body functions. FGFR is abnormal in a variety of human tumors, including gene amplification, mutation and overexpression, and is an important target for tumor-targeted therapeutic research.
FGFR4, a member of the FGFR receptor family, forms dimers on the cell membrane by binding to its ligand, fibroblast growth factor 19 (FGF19), and the formation of these dimers can cause critical tyrosine in FGFR4’s own cells. The phosphorylation of the amino acid residue activates multiple downstream signaling pathways in the cell, and these intracellular signaling pathways play an important role in cell proliferation, survival, and anti-apoptosis. FGFR4 is overexpressed in many cancers and is a predictor of malignant invasion of tumors. Decreasing and reducing FGFR4 expression can reduce cell proliferation and promote apoptosis. Recently, more and more studies have shown that about one-third of liver cancer patients with continuous activation of FGF19/FGFR4 signaling pathway are the main carcinogenic factors leading to liver cancer in this part of patients. At the same time, FGFR4 expression or high expression is also closely related to many other tumors, such as gastric cancer, prostate cancer, skin cancer, ovarian cancer, lung cancer, breast cancer, colon cancer and the like.
The incidence of liver cancer ranks first in the world in China, with new and dead patients accounting for about half of the total number of liver cancers worldwide each year. At present, the incidence of liver cancer in China is about 28.7/100,000. In 2012, there were 394,770 new cases, which became the third most serious malignant tumor after gastric cancer and lung cancer. The onset of primary liver cancer is a multi-factor, multi-step complex process with strong invasiveness and poor prognosis. Surgical treatments such as hepatectomy and liver transplantation can improve the survival rate of some patients, but only limited patients can undergo surgery, and most patients have a poor prognosis due to recurrence and metastasis after surgery. Sorafenib is the only liver cancer treatment drug approved on the market. It can only prolong the overall survival period of about 3 months, and the treatment effect is not satisfactory. Therefore, it is urgent to develop a liver cancer system treatment drug targeting new molecules. FGFR4 is a major carcinogenic factor in liver cancer, and its development of small molecule inhibitors has great clinical application potential.
At present, some FGFR inhibitors have entered the clinical research stage as anti-tumor drugs, but these are mainly inhibitors of FGFR1, 2 and 3, and the inhibition of FGFR4 activity is weak, and the inhibition of FGFR1-3 has hyperphosphatemia. Such as target related side effects. Highly selective inhibitor of FGFR4 can effectively treat cancer diseases caused by abnormal FGFR4 signaling pathway, and can avoid the side effects of hyperphosphatemia caused by FGFR1-3 inhibition. Highly selective small molecule inhibitors against FGFR4 in tumor targeted therapy The field has significant application prospects.
SYN
PATENT
WO2017198149
where it is claimed to be an FGFR-4 inhibitor for treating liver and prostate cancers, assigned to Jiangsu Hansoh Pharmaceutical Group Co Ltd and Shanghai Hansoh Biomedical Co Ltd .
PATENT
WO2019085860
Compound (R)-N-(5-Cyano-4-((1-methoxypropan-2-yl)amino)pyridin-2-yl)-7-formyl-6-((2-carbonyl-) 1,3-oxazepine-3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxamide (shown as Formula I). The compound of formula (I) is disclosed in Hausen Patent PCT/CN2017/084564, the compound of formula I is a fibroblast growth factor receptor inhibitor, and the fibroblast growth factor receptor (FGFR) belongs to the receptor tyrosine kinase transmembrane receptor. The body includes four receptor subtypes, namely FGFR1, FGFR2, FGFR3 and FGFR4. FGFR regulates various functions such as cell proliferation, survival, differentiation and migration, and plays an important role in human development and adult body functions. FGFR is abnormal in a variety of human tumors, including gene amplification, mutation and overexpression, and is an important target for tumor-targeted therapeutic research.
[0003]

Example 1: Preparation of a compound of formula (I)
[0048]
First step 4-(((2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl)methyl)amino)butane Preparation of 1-propanol
[0050]
2-(Dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-carbaldehyde (1.0 g, 4.2 mmol), 4-aminobutyl at room temperature l-ol (0.45g, 5.1mmol) was dissolved in DCE (15mL), stirred for 2 hours, followed by addition of NaBH (OAc) . 3 (1.35 g of, 6.4 mmol), stirred at room temperature overnight. The reaction was treated with CH 2 CI 2 was diluted (100 mL), the organic phase was washed with water (10mL) and saturated brine (15mL), and dried over anhydrous sodium sulfate, and concentrated by column chromatography to give compound 4 – (((2- ( Dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl)methyl)amino)butan-1-ol (0.9 g, 69%) .
[0051]
. 1 H NMR (400 MHz, CDCl3 . 3 ) [delta] 7.13 (S, IH), 5.17 (S, IH), 4.84 (S, IH), 3.73 (S, 2H), 3.66-3.49 (m, 2H), 3.42 ( s, 6H), 3.40-3.36 (m, 2H), 2.71 (t, J = 6.3 Hz, 2H), 2.68-2.56 (m, 2H), 1.95-1.81 (m, 2H), 1.74-1.55 (m, 4H);
[0052]
MS m/z (ESI): 310.2 [M+H] + .
[0053]
The second step is 3-((2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl)methyl)-1,3- Preparation of oxazepine-2 ketone
[0055]
4-(((2-(Dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl)methyl)amino) in an ice water bath Butan-1-ol (0.6 g, 1.94 mmol) was dissolved in DCE (15 mL), then bis(trichloromethyl) carbonate (0.22 g, 0.76 mmol) was added and triethylamine (0.78 g, 7.76) was slowly added dropwise. Methyl) and then stirred at room temperature for 3 hours. The reaction temperature was raised to 80 ° C, and the reaction was carried out at 80 ° C for 6 hours. After the reaction was cooled to room temperature, it was diluted with CH 2 Cl 2 (100 mL), and the organic phase was washed sequentially with water (10 mL) and brine (15 mL) Drying with sodium sulfate, concentration and column chromatography to give the compound 3-((2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl) )methyl)-1,3-oxazepin-2-one (0.37 g, 57%).
[0056]
MS m/z (ESI): 336.2 [M+H] + .
[0057]
The third step is phenyl 7-(dimethoxymethyl)-6-((2-carbonyl-1,3-oxazepine-3-yl)methyl)-3,4-dihydro-1, Preparation of 8-naphthyridin-1(2H)-carboxylate
[0059]
3-((2-(Dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl)methyl)-1,3-oxan -2-one (670mg, 2mmol), diphenyl carbonate (643mg, 3mmol) mixing in of THF (15 mL), N 2 in an atmosphere, cooled to -78 deg.] C, was added dropwise LiHMDS in THF (4mL, 4mmol) was Naturally, it was allowed to react to room temperature overnight. After adding saturated aqueous NH 4 Cl (100 mL), ethyl acetate (100 mL×2), EtOAc. Methyl)-6-((3-carbonylmorpholino)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxylate (432 mg, 47%) .
[0060]
. 1 H NMR (400 MHz, CDCl3 . 3 ) [delta] 7.56 (S, IH), 7.38 (m, 2H), 7.21 (m, 3H), 5.22 (S, IH), 4.77 (S, 2H), 4.16 (m, 2H), 3.95 (m, 2H), 3.39 (s, 6H), 3.25 (m, 2H), 2.84 (t, J = 6.5 Hz, 2H), 1.87 (m, 2H), 1.64 (m, 4H);
[0061]
MS m/z (ESI): 456.2 [M+H] + .
[0062]
The fourth step: (R)-N-(5-cyano-4-((1-methoxypropan-2-yl)amino)pyridin-2-yl)-7-(dimethoxymethyl) -6-((2-carbonyl-1,3-oxazepine-3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxamide synthesis
[0064]
(R)-6-Amino-4-((1-methoxypropan-2-yl)amino) nicotinenitrile (30 mg, 0.14 mmol), phenyl 7-(dimethoxymethyl)-6- ( (2-carbonyl-1,3-oxazepine-3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxylate (60 mg, 0.13 Methyl acetate was dissolved in THF (5 mL), cooled to -78 ° C under N 2atmosphere, and a solution of THF (0.3 mL, 0.3 mmol) of LiHMDS was added dropwise to the reaction mixture. After adding a saturated aqueous solution of NH 4 Cl (50 mL), EtOAc (EtOAc) (5-Cyano-4-((1-methoxypropan-2-yl)amino)pyridin-2-yl)-7-(dimethoxymethyl)-6-((2-carbonyl-1) 3-oxoheptyl-3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxamide (65 mg, 86%).
[0065]
1H NMR (400MHz, CDCl3) δ 13.70 (s, 1H), 8.18 (s, 1H), 7.60 (s, 2H), 5.41 (s, 1H), 5.12 (d, J = 7.8 Hz, 1H), 4.73 (s, 2H), 4.20-4.11 (m, 2H), 4.06-3.99 (m, 2H), 3.93 (s, 1H), 3.52-3.48 (m, 7H), 3.46-3.42 (m, 1H), 3.39 (s, 3H), 3.26-3.21 (m, 2H), 2.83 (t, J = 6.2 Hz, 2H), 2.03-1.95 (m, 2H), 1.91-1.83 (m, 2H), 1.67-1.62 (m , 2H), 1.31 (d, J = 6.6 Hz, 3H);
[0066]
MS m/z (ESI): 568.3 [M+H] + .
[0067]
Step 5: (R)-N-(5-Cyano-4-((1-methoxypropan-2-yl)amino)pyridin-2-yl)-7-formyl-6-((2) Synthesis of -carbonyl-1,3-oxoheptyl-3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxamide
[0069]
(R)-N-(5-Cyano-4-((1-methoxypropan-2-yl)amino)pyridin-2-yl)-7-(dimethoxymethyl)-6-( (2-carbonyl-1,3-oxazepine-3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxamide (65 mg, 0.12 mmol) Dissolved in THF/water (volume ratio: 11/4, 4.5 mL), concentrated HCl (0.45 mL, 5.4 mmol), and allowed to react at room temperature for 2 h. Saturated NaHC03 . 3 solution (50mL), (50mL × 2 ) and extracted with ethyl acetate, the organic phases were combined and washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated by column chromatography to give the title compound (R) -N- ( 5-cyano-4-((1-methoxypropan-2-yl)amino)pyridin-2-yl)-7-formyl-6-((2-carbonyl-1,3-oxazepine) 3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1 (2H)-carboxamide (30 mg, 51%).
[0070]
. 1 H NMR (400 MHz, CDCl3 . 3 ) [delta] 13.57 (S, IH), 10.26 (S, IH), 8.17 (S, IH), 7.71 (S, IH), 7.63 (S, IH), 5.27 (S, 1H), 4.95 (s, 2H), 4.19-4.12 (m, 2H), 4.11-4.04 (m, 2H), 3.94 (s, 1H), 3.52 (m, 1H), 3.48-3.37 (m, 4H) , 3.33 – 3.28 (m, 2H), 2.93 (t, J = 6.3 Hz, 2H), 2.04 (m, 2H), 1.93-1.85 (m, 2H), 1.73 (m, 2H), 1.39-1.28 (m , 3H);
[0071]
MS m/z (ESI): 522.2 [M+H] + .
PATENT
WO-2019085927
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019085927&tab=FULLTEXT
Novel crystalline salt (such as hydrochloride, sulfate, methane sulfonate, mesylate, besylate, ethanesulfonate, oxalate, maleate, p-toluenesulfonate) forms of FGFR4 inhibitor, particularly N-[5-cyano-4-[[(1R)-2-methoxy-1-methyl-ethyl]amino]-2-pyridyl]-7-formyl-6-[(2-oxo-1,3-oxazepan-3-yl)methyl]-3,4-dihydro-2H-1,8-naphthyridine-1-carboxamide (designated as Forms I- IX), compositions comprising them and their use as an FGFR4 inhibitor for the treatment of cancer such as liver cancer, gastric cancer, prostate cancer, skin cancer, ovarian cancer, lung cancer, breast cancer, colon cancer and glioma or rhabdomyosarcoma are claimed.
Example 1: Preparation of a compound of formula (I)
First step 4-(((2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl)methyl)amino)butane Preparation of 1-propanol
2-(Dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-carbaldehyde (1.0 g, 4.2 mmol), 4-aminobutyl at room temperature l-ol (0.45g, 5.1mmol) was dissolved in DCE (15mL), stirred for 2 hours, followed by addition of NaBH (OAc) . 3 (1.35 g of, 6.4 mmol), stirred at room temperature overnight. The reaction was treated with CH 2 CI 2 was diluted (100 mL), the organic phase was washed with water (10mL) and saturated brine (15mL), and dried over anhydrous sodium sulfate, and concentrated by column chromatography to give compound 4 – (((2- ( Dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl)methyl)amino)butan-1-ol (0.9 g, 69%) .
. 1 H NMR (400 MHz, CDCl3 . 3 ) [delta] 7.13 (S, IH), 5.17 (S, IH), 4.84 (S, IH), 3.73 (S, 2H), 3.66-3.49 (m, 2H), 3.42 ( s, 6H), 3.40-3.36 (m, 2H), 2.71 (t, J = 6.3 Hz, 2H), 2.68-2.56 (m, 2H), 1.95-1.81 (m, 2H), 1.74-1.55 (m, 4H);
MS m/z (ESI): 310.2 [M+H] + .
The second step is 3-((2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl)methyl)-1,3- Preparation of oxazepine-2 ketone
4-(((2-(Dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl)methyl)amino) in an ice water bath Butan-1-ol (0.6 g, 1.94 mmol) was dissolved in DCE (15 mL), then bis(trichloromethyl) carbonate (0.22 g, 0.76 mmol) was added and triethylamine (0.78 g, 7.76) was slowly added dropwise. Methyl) and then stirred at room temperature for 3 hours. The reaction temperature was raised to 80 ° C, and the reaction was carried out at 80 ° C for 6 hours. After the reaction was cooled to room temperature, it was diluted with CH 2 Cl 2 (100 mL), and the organic phase was washed sequentially with water (10 mL) and brine (15 mL) Drying with sodium sulfate, concentration and column chromatography to give the compound 3-((2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl) )methyl)-1,3-oxazepin-2-one (0.37 g, 57%).
MS m/z (ESI): 336.2 [M+H] + .
The third step is phenyl 7-(dimethoxymethyl)-6-((2-carbonyl-1,3-oxazepine-3-yl)methyl)-3,4-dihydro-1, Preparation of 8-naphthyridin-1(2H)-carboxylate
3-((2-(Dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl)methyl)-1,3-oxan -2-one (670mg, 2mmol), diphenyl carbonate (643mg, 3mmol) mixing in of THF (15 mL), N 2 in an atmosphere, cooled to -78 deg.] C, was added dropwise LiHMDS in THF (4mL, 4mmol) was Naturally, it was allowed to react to room temperature overnight. After adding saturated aqueous NH 4 Cl (100 mL), ethyl acetate (100 mL×2), EtOAc. Methyl)-6-((3-carbonylmorpholino)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxylate (432 mg, 47%) .
. 1 H NMR (400 MHz, CDCl3 . 3 ) [delta] 7.56 (S, IH), 7.38 (m, 2H), 7.21 (m, 3H), 5.22 (S, IH), 4.77 (S, 2H), 4.16 (m, 2H), 3.95 (m, 2H), 3.39 (s, 6H), 3.25 (m, 2H), 2.84 (t, J = 6.5 Hz, 2H), 1.87 (m, 2H), 1.64 (m, 4H);
MS m/z (ESI): 456.2 [M+H] + .
The fourth step: (R)-N-(5-cyano-4-((1-methoxypropan-2-yl)amino)pyridin-2-yl)-7-(dimethoxymethyl) -6-((2-carbonyl-1,3-oxazepine-3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxamide synthesis
(R)-6-Amino-4-((1-methoxypropan-2-yl)amino) nicotinenitrile (30 mg, 0.14 mmol), phenyl 7-(dimethoxymethyl)-6- ( (2-carbonyl-1,3-oxazepine-3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxylate (60 mg, 0.13 Methyl acetate was dissolved in THF (5 mL), cooled to -78 ° C under N 2atmosphere, and a solution of THF (0.3 mL, 0.3 mmol) of LiHMDS was added dropwise to the reaction mixture. After adding a saturated aqueous solution of NH 4 Cl (50 mL), EtOAc (EtOAc) (5-Cyano-4-((1-methoxypropan-2-yl)amino)pyridin-2-yl)-7-(dimethoxymethyl)-6-((2-carbonyl-1) 3-oxoheptyl-3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxamide (65 mg, 86%).
1H NMR (400MHz, CDCl3) δ 13.70 (s, 1H), 8.18 (s, 1H), 7.60 (s, 2H), 5.41 (s, 1H), 5.12 (d, J = 7.8 Hz, 1H), 4.73 (s, 2H), 4.20-4.11 (m, 2H), 4.06-3.99 (m, 2H), 3.93 (s, 1H), 3.52-3.48 (m, 7H), 3.46-3.42 (m, 1H), 3.39 (s, 3H), 3.26-3.21 (m, 2H), 2.83 (t, J = 6.2 Hz, 2H), 2.03-1.95 (m, 2H), 1.91-1.83 (m, 2H), 1.67-1.62 (m , 2H), 1.31 (d, J = 6.6 Hz, 3H);
MS m/z (ESI): 568.3 [M+H] + .
Step 5: (R)-N-(5-Cyano-4-((1-methoxypropan-2-yl)amino)pyridin-2-yl)-7-formyl-6-((2) Synthesis of -carbonyl-1,3-oxoheptyl-3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxamide
(R)-N-(5-Cyano-4-((1-methoxypropan-2-yl)amino)pyridin-2-yl)-7-(dimethoxymethyl)-6-( (2-carbonyl-1,3-oxazepine-3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxamide (65 mg, 0.12 mmol) Dissolved in THF/water (volume ratio: 11/4, 4.5 mL), concentrated HCl (0.45 mL, 5.4 mmol), and allowed to react at room temperature for 2 h. Saturated NaHC03 . 3 solution (50mL), (50mL × 2 ) and extracted with ethyl acetate, the organic phases were combined and washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated by column chromatography to give the title compound (R) -N- ( 5-cyano-4-((1-methoxypropan-2-yl)amino)pyridin-2-yl)-7-formyl-6-((2-carbonyl-1,3-oxazepine) 3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1 (2H)-carboxamide (30 mg, 51%).
. 1 H NMR (400 MHz, CDCl3 . 3 ) [delta] 13.57 (S, IH), 10.26 (S, IH), 8.17 (S, IH), 7.71 (S, IH), 7.63 (S, IH), 5.27 (S, 1H), 4.95 (s, 2H), 4.19-4.12 (m, 2H), 4.11-4.04 (m, 2H), 3.94 (s, 1H), 3.52 (m, 1H), 3.48-3.37 (m, 4H) , 3.33 – 3.28 (m, 2H), 2.93 (t, J = 6.3 Hz, 2H), 2.04 (m, 2H), 1.93-1.85 (m, 2H), 1.73 (m, 2H), 1.39-1.28 (m , 3H);
MS m/z (ESI): 522.2 [M+H] + .
///////////HS-10340 , HS 10340 , HS10340, CANCER, Jiangsu Hansoh, Shanghai Hansoh Biomedical, Changzhou Hengbang, CHINA, liver cancer, gastric cancer, prostate cancer, skin cancer, ovary cancer, lung cancer, breast cancer, colon cancer, glioma, rhabdomyosarcoma
C[C@H](COC)Nc1cc(ncc1C#N)NC(=O)N4CCCc3cc(CN2CCCCOC2=O)c(C=O)nc34
CCS(=O)(=O)O.C[C@H](COC)Nc1cc(ncc1C#N)NC(=O)N4CCCc3cc(CN2CCCCOC2=O)c(C=O)nc34

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader