
Home » Posts tagged 'organic chemistry' (Page 3)
Tag Archives: organic chemistry
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
NETUPITANT

NETUPITANT
- Ro 67-3189/000
- UNII-7732P08TIR
- Ro-67-3189
- Netupitant, an NK-1 antagonist is under development for the treatment of overactive bladder. HELSINN GROUP
CAS: 290297-26-6
290296-54-7 (di HCl)
U.S. Pat. Nos. 6,303,790, 6,531,597, 6,297,375 and 6,479,483, 6,719,996 and 6,593,472 to Hoffmann La Roche(originator).
IUPAC/Chemical name:
2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethyl-N-(6-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridin-3-yl)propanamide
Chemical Formula: C30H32F6N4O
Exact Mass: 578.24803
Molecular Weight: 578.59
Elemental Analysis: C, 62.28; H, 5.57; F, 19.70; N, 9.68; O, 2.77
Netupitant is another selective NKi receptor antagonist under development by Helsinn Healthcare, having the formula 2-[3,5-bis(trifluoromethyl)phenyl]-N,2-dimethyl-N-[4-(2- methylphenyl)-6-(4-methylpiperazin- l-yl)pyridin-3-yl]propanamide, or Benzeneacetamide, N,a,a-trimethyl-N-[4-(2-methylphenyl)-6-(4-methyl-l-piperazinyl)-3-pyridinyl]-3,5- bis(trifluoromethyl)-, and the below chemical structure:

Netupitant is a tachykinin NK-1 antagonist which had been in phase III clinical trials at Helsinn for the prophylaxis of chemotherapy-induced nausea and vomiting and in phase II clinical studies for the treatment of overactive bladder. However, no recent development has been reported for this research.
NK-1 receptor antagonists work by blocking the action of neurokinin-1 (Substance P), a naturally-occurring neurotransmitter in the brain that causes emesis. Netupitant was originally developed at Roche. In June 2005, Helsinn and Roche signed a licensing agreement granting Helsinn worldwide rights to the drug candidate.
Methods of synthesizing and formulating netupitant and its prodrugs are described in U.S. Patent Nos. 6,297,375, 6,719,996 and 6,593,472 to Hoffmann La Roche.
Netupitant is a highly selective NK1 receptor antagonist, which is thought to work by blocking the action of substance P, an endogenous neurotransmitter contained in high concentrations in the vomiting center of the brainstem that can stimulate the vomiting reflex. Netupitant is currently under phase III trials.
Chemotherapy is one of the treatment options utilized by oncologists in treating different types of cancers. Nausea and vomiting are the most common side-effects experienced by cancer patients when administered with chemotherapy. Netupitant-palonosetron, which is currently in Phase III trials helps in preventing CINV. The blockage of P/NK1 receptors by Netupitant in the central nervous system inhibits the binding of endogenous tachykinin neuropeptide substance and this result in preventing the chemotherapy-induced nausea and vomiting. Moreover, Palonosetron helps in the blockage of serotonin at 5-hydroxytryptamine type 3 (5-HT3) receptors and it also helps in the chemotherapy-induced nausea and vomiting.

Netupitant-Palonosetron FDC is estimated to answer significant unmet needs of the CINV market post its launch that is expected to be commercialized in 2014, as it would overcome the problems associated with current treatment with 5-HT3 receptor antagonists. Similar to Emend, Netupitant-Palonosetron FDC would gain considerable patient pool after its estimated launch in 2014, and subsequently match the patient share of Aloxi by 2018. Netupitant-Palonosetron FDC sales are expected to reach an estimated USD 515.0 million USD by 2018. FDC combination of 5-HT3 receptor antagonist and neurokinin-1 (NK1) receptor antagonist have shown better efficacy results in Phase II clinical trials for CINV patients and would thus lead to high uptake due to shifting physician and patient preference pattern towards better treatment for CINV.

Neurokinin 1 receptor antagonists are being developed for the treatment of a number of physiological disorders associated with an excess or imbalance of tachykinin, in particular substance P. Examples of conditions in which substance P has been implicated include disorders of the central nervous system such as anxiety, depression and psychosis (WO 95/16679, WO 95/18124 and WO 95/23798).
The neurokinin-1 receptor antagonists are further useful for the treatment of motion sickness and for treatment induced vomiting. The New England Journal of Medicine, Vol. 340, No. 3 190-195, 1999 has been described the reduction of cisplatin-induced emesis by a selective neurokinin-l-receptor antagonist. US5,972,938 describes a method for treating a psychoimmunologic or a psychosomatic disorder by administration of a tachykinin receptor, such as NK-1 receptor antagonist.
With the development of the 5-HT3 antagonist in the early 1990s, there emerged new strategies in the medical community to better control nausea and vomiting caused by various medical procedures, including chemotherapy (CINV), surgery (PONV), and radiation therapy (RINV). When added to steroids such as dexamethasone, several 5-HT3 antagonists have been demonstrated to significantly improve the standard of life for patients undergoing emetogenic medical procedures. Examples of 5-HT3 antagonists include ondansetron, marketed by
GlaxoSmithKline, and palonosetron, developed by Helsinn Healthcare.
Netupitant is another selective NKi receptor antagonist under development by Helsinn Healthcare, having the formula 2-[3,5-bis(trifluoromethyl)phenyl]-N,2-dimethyl-N-[4-(2- methylphenyl)-6-(4-methylpiperazin- l-yl)pyridin-3-yl]propanamide, or Benzeneacetamide, N,a,a-trimethyl-N-[4-(2-methylphenyl)-6-(4-methyl-l-piperazinyl)-3-pyridinyl]-3,5- bis(trifluoromethyl)-, and the below chemical structure:
Methods of synthesizing and formulating netupitant and its prodrugs are described in U.S. Patent Nos. 6,297,375, 6,719,996 and 6,593,472 to Hoffmann La Roche.
Other representative NKi antagonists include ZD4974 (developed by AstraZeneca), CGP49823 (developed by Ciba-Geigy), Lanepitant and LY686017 (developed by Eli Lilly), FK888 (developed by Fujisawa), Vofopitant, Vestipitant and Orvepitant (developed by
GlaxoSmithKline), Befetupitant (developed by Hoffmann-La Roche), Rl 16031 (developed by Janssen), L-733060 and L-736281 (developed by Merck), TKA731, NKP608 and DNK333 (developed by Novartis), CP-96345, CP-99994, CP- 122721, CJ-17493, CJ-11974 and CJ-11972 (developed by Pfizer), RP67580 and Dapitant (developed by Rhone-Poulenc Rorer),
Nolpitantium and SSR240600 (developed by Sanofi-Aventis), SCH388714 and Rolapitant (developed by Schering-Plough), TAK637 (developed by Takeda), HSP117 (developed by Hisamitsu), KRP103 (developed by Kyorin Pharm) and SLV317 (developed by Solvay).
Chemical structures of the above-mentioned NKi antagonists are shown below and discussion of those compounds as well as other NKi antagonists is present in Expert Opin. Ther. Patents (2010) 20(8), pp 1019- 1045 by Huang et al.
………………………………………………
WO 2013057554
WO 2011061622
WO 2010119347
WO 2003006016
WO 2006002860///
WO 2002085458
US 2002091265…….
…………………………………………………..
http://pubs.acs.org/doi/full/10.1021/jo0523666
…………………………………………..
https://www.google.co.in/patents/US6297375
(2-(3,5-bis-trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide) which has the formula Ib

and to pharmaceutically acceptable acid addition salts thereof.
The compound of formula Ib and its salts is also characterized by valuable therapeutic properties as a highly selective antagonist of the Neurokinin 1 (NK-1, substance P) The present compound of formula lb and its pharmaceutically acceptable salts can be prepared by methods known in the art, for example, by processes described below, which process comprises
a) reacting the compound of formula

with the compound of formula

to the compound of formula



EXAMPLE 14
2-(3,5-Bis-trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide hydrochloride (1:2)
a) 1-Methyl-4-(5-nitro-pyridin-2-yl)-piperazine
To a solution of 20 g (126 mmol) of 2-chloro-5-nitropyridine in 200 ml tetrahydrofuran were added dropwise 35 ml (315 mmol) 1-methylpiperazine within 10 min. The reaction mixture was refluxed for additional 1.5 h. After cooling to room temperature, the solvent was removed in vacuo and the residue was re-dissolved in 200 ml ethyl acetate. The organic phase was washed with 200 ml 1 N sodium bicarbonate solution, dried (magnesium sulfate) and evaporated to give 27.9 g (quantitative) of the title compound as a yellow solid.
MS m/e (%):223 (M+H+, 100).
b)2,2-Dimethyl-N-[6-(4-methyl-piperazin-1-yl)-pyridin-3-yl]-propionamide
To a solution of 27.9 g (125.5 mmol) of 1-methyl-4-(5-nitro-pyridin-2-yl)-piperazine in 400 ml methanol were added 2.6 g of 10% of palladium on activated charcoal. The reaction mixture was hydrogenated (room temperature to ca. 45° C., 1 bar) until the theoretical amount of hydrogen was taken up (about 2 h). The catalyst was filtered off and was washed twice with 100 ml portions of methanol. The filtrate was evaporated in vacuo to give 28 g of a purple oil which consisted to ca. 90% of the desired aniline derivative according to analysis by thin layer chromatography.
This crude product was dissolved in a mixture of 400 ml tetrahydrofuran and 100 ml diethyl ether. After cooling to 0° C., 30 ml (215 mmol) of triethylamine were added in one portion. Stirring was continued while 26 g (215 mmol) of pivaloyl chloride were added dropwise within a period of 10 min. The ice bath was removed and the reaction mixture was stirred for 1 h at room temperature. Then, the solvent was removed in vacuo and the residue was suspended in 200 ml 1 N sodium bicarbonate solution. The product was extracted three times with 200 ml portions of dichloromethane, dried (sodium sulfate) and purified by flash chromatography to give 30 g (86%) of the title compound as pink crystals.
MS m/e (%):277 (M+H+, 100).
c) N-[4-Iodo-6-(4-methyl-piperazin-1-yl)-pyridin-3-yl]-2,2-dimethyl-propionamide
A solution of 30 g (108 mmol) 2,2-dimethyl-N-[6-(4-methyl-piperazin-1-yl)-pyridin-3-yl]-propionamide and 58 ml (380 mmol) N,N,N′,N′-tetramethylethylenediamine under argon in 650 ml tetrahydrofuran was cooled in a dry ice bath to −78° C. Within lh, 239 ml (380 mmol) of a 1.6 N n-butyllithium solution in hexane were added dropwise. The reaction mixture was allowed to warm up to −30° C. overnight. After cooling again to −78° C., 43.6 g (170 mmol) iodine dissolved in 60 ml tetrahydrofuran were added dropwise during 15 min. The dry ice bath was replaced by an ice bath and a solution of 90 g (363 mmol) sodium thiosulfate pentahydrate in 250 ml water were added within 10 min when the temperature of the reaction mixture had reached 0° C. Then, 1000 ml diethyl ether were added and the organic layer was separated. The aqueous layer was extracted twice with 500 ml dichloromethane and the combined organic layers were dried (magnesium sulfate) and evaporated. Flash chromatography gave 18.5 g (42%) of the title compound as a light brown oil which crystallized upon standing at room temperature.
MS m/e (%): 403 (M+H+, 100).
d) 2,2-Dimethyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-propionamide
A mixture of 54 g (134 mmol) N-[4-iodo-6-(4-methyl-piperazin-1-yl)-pyridin-3-yl]-2,2-dimethyl-propionamide, 420 ml toluene, 150 ml 2 N sodium carbonate solution, 4.63 g (3.9 mmol) tetrakis(triphenylphosphine)palladium(0) and 20.16 g (147 mmol) o-tolylboronic acid was heated under argon at 80° C. for 12 h. After cooling to room temperature, the aqueous phase was separated and washed twice with toluene. The combined organic layers were washed with 50 ml brine, dried (sodium sulfate), evaporated and dried in vacuo to yield 49 g (quantitative) of the title compound as a brown oil.
MS m/e (%): 367 (M+H+, 100).
e) 6-(4-Methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-ylamine
A suspension of 56 g (152 mmol) 2,2-dimethyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-propionamide in 1300 ml 3 N hydrochloric acid solution was heated to 90-95° C. overnight. The reaction mixture was cooled to room temperature, washed with three 500 ml portions diethyl ether and filtered over celite. The filtrate was diluted with 500 ml water and was adjusted to pH 7-8 by addition of 28% sodium hydroxide solution under ice cooling. The product was extracted with four 1000 ml portions of dichloromethane. The combined organic layers were washed with 500 ml brine, dried (magnesium sulfate) and evaporated to give 35 g (82%) of the title compound as a light brown oil.
MS m/e (%):283 (M+H+, 100).
f) Methyl-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-amine
A solution of 35 g (124 mmol) 6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-ylamine in 270 ml trimethyl orthoformate and 8 drops trifluoroacetic acid was heated for 3 h at 130° C. The reaction mixture was evaporated and dried in vacuo for 30 min. The residual oil was dissolved in 100 ml tetrahydrofuran and was added dropwise under ice cooling to 9.4 g (248 mmol) lithium aluminum hydride in 300 ml tetrahydrofuran. The reaction mixture was stirred for lh at room temperature, cooled to 0° C. again and acidified (pH 1-2) by addition of 28% hydrochloric acid solution. After stirring for 5 min, 28% sodium hydroxide solution was added to reach pH 10. The solution was filtered over celite, evaporated and purified by flash chromatography to give 23.6 g (64%) of the title compound as a light brown oil.
MS m/e (%):297 (M+H+, 100).
g) 2-(3,5-Bis-trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide
A solution of 20 g (67.5 mmol) methyl-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-amine and 17.5 ml (101 mmol) N-ethyldiisopropylamine in 200 ml dichloromethane was cooled in an ice bath and a solution of 24 g (75 mmol)2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionyl chloride in 50 ml dichloromethane was added dropwise. The reaction mixture was warmed to 35-40° C. for 3 h, cooled to room temperature again and was stirred with 250 ml saturated sodium bicarbonate solution. The organic layer was separated and the aqueous phase was extracted with dichloromethane. The combined organic layers were dried (magnesium sulfate) and evaporated. The residue was purified by flash chromatography to give 31.6 g (81%) of the title compound as white crystals. M.p. 155-157° C.
MS m/e (%): 579 (M+H+, 100).
h)2-(3,5-Bis-trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide hydrochloride (1:2)
To a solution of 31.6 g (54.6 mmol)2-(3,5-bis-trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide in 250 ml diethyl ether were added under ice cooling 60 ml 3 N hydrochloric acid solution in diethyl ether. After stirring for 15 min at 0° C., the suspension was evaporated to dryness, re-suspended in 100 ml diethyl ether, filtered and dried in vacuo to give 34.8 g (98%) of the title compound as white crystals. M.p. 235-238° C.
MS m/e (%): 579 (M+H+, 100).
……………………………….
2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethyl-N-(6-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridin-3-yl)propanamide (Netupitant)

Other general procedures of preparing similar compounds to intermediate 1 of Scheme 1 are also disclosed in U.S. Pat. Nos. 6,303,790, 6,531,597, 6,297,375 and 6,479,483, the entirety of which are incorporated herein by reference.
Synthesis of methyl-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-amine

Step 1:
13.0 g (82.5 mMol) 6-Chloro-nicotinic acid in 65 ml THF were cooled to 0° C. and 206.3 ml (206.3 mMol) o-tolylmagnesium chloride solution (1M in THF) were added over 45 minutes. The solution obtained was further stirred 3 hours at 0° C. and overnight at room temperature. It was cooled to −60° C. and 103.8 ml (1.8 Mol) acetic acid were added, followed by 35 ml THF and 44.24 g (165 mMol) manganese(III) acetate dihydrate. After 30 minutes at −60° C. and one hour at room temperature, the reaction mixture was filtered and THF removed under reduced pressure. The residue was partitioned between water and dichloromethane and extracted. The crude product was filtered on silica gel (eluent: ethyl acetate/toluene/formic acid 20:75:5) then partitioned between 200 ml aqueous half-saturated sodium carbonate solution and 100 ml dichloromethane. The organic phase was washed with 50 ml aqueous half-saturated sodium carbonate solution, The combined aqueous phases were acidified with 25 ml aqueous HCl 25% and extracted with dichloromethane. The organic extracts were dried (Na2SO4) and concentrated under reduced pressure to yield 10.4 g (51%) of 6-chloro-4-o-tolyl-nicotinic acid as a yellow foam. MS (ISN): 246 (M−H, 100), 202 (M-CO2H, 85), 166 (36).
Step 2:
To a solution of 8.0 g (32.3 mMol) 6-chloro-4-o-tolyl-nicotinic acid in 48.0 ml THF were added 3.1 ml (42.0 mMol) thionylchloride and 143 .mu.l (1.8 mMol) DMF. After 2 hours at 50° C., the reaction mixture was cooled to room temperature and added to a solution of 72.5 ml aqueous ammonium hydroxide 25% and 96 ml water cooled to 0″C. After 30 minutes at 0° C., THF was removed under reduced pressure and the aqueous layer was extracted with ethyl acetate. Removal of the solvent yielded 7.8 g (98%) 6-chloro-4-o-tolyl-nicotinamide as a beige crystalline foam. MS (ISP): 247 (M+H30 , 100).
Step 3:
1.0 g (4.05 mMol) 6-Chloro-4-o-tolyl-nicotinamidein 9.0 ml 1-methyl-piperazine was heated to 100° C. for 2 hours. The excess N-methyl-piperazine was removed under high vacuum and the residue was filtered on silica gel (eluent: dichloromethane) to yield 1.2 g (95%) 6-(4-methyl-piperazin-1yl)-4-o-tolyl-nicotinamide as a light yellow crystalline foam. MS (ISP): 311 (M+H+, 100), 254 (62).
Step 4:
A solution of 0.2 g (0.6 mMol) 6-(4-methyl-piperazin-1-yl)-4-o-tolyl-nicotinamide in 1.0 ml methanol was added to a solution of 103 mg (2.6 mMol) sodium hydroxide in 1.47 ml (3.2 mMol) NaOCl (13%) and heated for 2 hours at 70° C. After removal of methanol, the aqueous layer was extracted with ethyl acetate. The combined. organic extracts were dried (Na2SO4), concentrated under reduced pressure and the residue filtered on silica gel (eluent: dichloromethane/methanol 4:1) to yield 100 mg (70%) 6-(4-methyl-piperazine-1-yl)-4o-tolyl-pyridin-3-ylamine as a brown resin. MS (ISP): 283 (M+H+, 100), 226 (42).
Step 5:
2.15 ml (11.6 mMol) Sodium methoxide in methanol were added over 30 minutes to a suspension of 0.85 g (4.6 mMol) N-bromosuccinimide in 5.0 ml dichloromethane cooled to −5° C. The reaction mixture was stirred 16 hours at −5° C. Still at this temperature, a solution of 1.0 g (3.1 mMol) 6-(4-methyl-piperazin-1-yl)-4-o-tolyl-nicotinamide in 5.0 ml methanol was added over 20 minutes and stirred for 5 hours. 7.1 ml (7.1 mMol) Aqueous HCl 1N and 20 ml dichloromethane were added. The phases were separated and the organic phase was washed with deionized water. The aqueous phases were extracted with dichloromethane, brought to pH=8 with aqueous NaOH 1N and further extracted with dichloromethane. The latter organic, extracts were combined, dried (Na2SO4) and concentrated to yield 1.08 g (quant.) [6-(4-methyl-piperazin-1yl)-4-o-tolyl-pyridin-3-yl]-carbamic acid methyl ester as a grey foam. MS (ISP): 341 (M+H+, 100), 284 (35).
Step 6:
A solution of 0.5 g (1.4 mMol) [6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-carbamic acid methyl ester in 3.0 ml dichloromethane was added over 10 minutes to a solution of 1.98 ml (6.9 mMol) Red-Al.RTM. (70% in toluene) and 2.5 ml toluene (exothermic, cool with a water bath to avoid temperature to go >50° C.). The reaction mixture was stirred 2 hours at 50° C. in CH2Cl2, extracted with ethyl acetate and cooled to 0° C. 4 ml Aqueous NaOH 1N were carefully (exothermic) added over 15 minutes, followed by 20 ml ethyl acetate. The phases were separated and the aqueous phase was extracted with ethyl acetate. The combined organic extracts were washed with deionized water and brine, dried (Na2SO4) and concentrated under reduced pressure to yield 0.37 g (89%) methyl-[6-4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-amine as an orange resin. MS (ISP): 297 (M+H+, 100).
Synthesis of 2-(3,5-bis-Trifluoromethyl-phenyl)-2-methyl-propionyl Chloride

15.0 g (50 mmol) 2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionic acid were dissolved in 127.5 ml dichloromethane in the presence of 0.75 ml DMF. 8.76 ml (2 eq.) Oxalyl chloride were added and after 4.5 hours, the solution was rotary evaporated to dryness. 9 ml Toluene were added and the resulting solution was again rotary evaporated, then dried under high vacuum yielding 16.25 g (quant.) of 2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionyl chloride as a yellow oil of 86% purity according to HPLC analysis. NMR (250 MHz, CDCl3): 7.86 (br s, 1H); 7.77, (br s, 2H, 3 Harom); 1.77 (s, 6H, 2 CH3).
Synthesis of 2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethyl-N-(6-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridin-3-yl)propanamide (Netupitant)

A solution of 20 g (67.5 mmol) methyl-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-amine and 17.5 ml (101 mmol) N-ethyldiisopropylamine in 200 ml dichloromethane was cooled in an ice bath and a solution of 24 g (75 mmol)2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionyl chloride in 50 ml dichloromethane was added dropwise. The reaction mixture was warmed to 35-40° C. for 3 h, cooled to room temperature again and was stirred with 250 ml saturated sodium bicarbonate solution. The organic layer was separated and the aqueous phase was extracted with dichloromethane, The combined organic layers were dried (magnesium sulfate) and evaporated. The residue was purified by flash chromatography to give 31.6 g (81%) of 2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethyl-N-(6-(4-methylpiperazin-1yl)-4-(o-tolyl)pyridin-3yl)propanamide as white crystals. M.P. 155-157° C.; MS m/e (%): 579 (M+H+, 100).
…………………………………..
http://www.google.com/patents/US20130231315
N OXIDE SYNTHESIS
Synthesis of 5-(2-(3,5-bis(trifluoromethyl)phenyl-N,2-dimethylpropanamido)2-(4-methylpiperazin-1yl)-4-(o-tolyl)pyridine 1-oxide

Step 1:
The solution of 6-chloropyridin-3-amine (115 g, 0.898 mol) and (Boc)2O (215.4 g, 0.988 mol) in 900 mL of dioxane was refluxed overnight. The resulting solution was poured into 1500 mL of water. The resulting solid was collected, washed with water and re-crystallized from EtOAc to afford 160 g tert-butyl (6-chloropyridin-3yl)carbamate as a white solid (Yield: 78.2%).
Step 2:
To the solution of tert-butyl (6-chloropyridin-3-yl)carbamate (160 g, 0.7 mol) in 1 L of anhydrous THF was added n-BuLi (600 mL, L5 ml) at −78° C. under N2 atmosphere. After the addition was finished, the solution was stirred at −78° C. for 30 min, and the solution of I2 (177.68 g, 0.7 mol) in 800 mL of anhydrous THF was added. Then the solution was stirred at −78° C. for 4 hrs, TLC indicated the reaction was over. Water was added for quench, and EtOAc was added to extract twice. The combined organic phases were washed with brine, dried over Na2SO4, filtered and purified by flash chromatography to afford 80 g of tert-butyl (6-chloro-4-iodopyridin-3-yl)carbamate as a yellow solid (32.3%).
Step 3:
To the solution of tert-butyl (6-chloro-4-iodopyridin-3-yl)carbamate (61 g, 0.172 mol) in 300 of anhydrous THF was added 60% NaH (7.6 g, 0.189 mol) at 0° C. under N2 atmosphere. After the addition was finished, the solution was stirred for 30 min, and then the solution of MeI (26.92 g, 0.189 mol) in 100 mL of dry THF was added. Then the solution was stirred at 0° C. for 3 hrs. TLC indicated the reaction was over. Water was added for quench, and EtOAc was added to extract twice. The combined organic phases were washed with brine, dried over Na2SO4, filtered and concentrated to afford 63 g of crude tert-butyl (6-chloro-4-iodopyridin-3-yl)methyl)carbamate used into the following de-protection without the further purification.
Step 4:
To the solution of tert-butyl (6-chloro-4-iodopyridin-3-yl)(methyl)carbamate (62.5 g, 0.172 mol) in 500 mL of anhydrous DCM was added 180 mL of TFA. Then the solution was stirred at room temperature for 4 hrs. Concentrated to remove the solvent, and purified by flash chromatography to afford 45.1 g 6-chloro-4-iodo-N-methylpyridin-3-amine as a yellow solid (Yield: 97.3%).
Step 5:
To the solution of 6-chloro-4-iodo-N-methylpyridin-3-amine (40.3 g, 0.15 mol) and 2-methylbenzene boric acid (24.5 g, 0.18 mol) in 600 mL of anhydrous toluene was added 400 mL of 2 N aq. Na2CO3 solution, Pd(OAc)2 (3.36 g, 15 mmol) and PPh3(7.87 g, 0.03 mmol), The solution was stirred at 100° C. for 2 hrs. Cooled to room temperature, and diluted with water. EtOAc was added to extract twice. The combined organic phases were washed with water and brine consecutively, dried over Na2SO4, concentrated and purified by flash chromatography to afford 19 g 6-chloro-N-methyl-4-(o-tolyl)pyridin-3-amine as a white solid (Yield: 54.6%).
Step 6:
To the solution of 6-chloro-N-methyl-4-(o-tolyl)pyridin-3-amine (18.87 g, 81.3 mmol) and DMAP (29.8 g, 243.9 mmol) in 200 mL of anhydrous toluene was added the solution of 2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionyl chloride (28.5 g, 89.4 mmol) in toluene under N2 atmosphere. The solution was heated at 120° C. for 23 hrs. Cooled to room temperature, poured into 1 L of 5% aq. NaHCO3 solution, and extracted with EtOAc twice. The combined organic phases were washed by water and brine consecutively, dried. over Na2SO4, filtered and purified by flash chromatography to afford 35 g 2-(3,5-bis(trifluoromethyl)phenyl)-N-(6-chloro-4-(4-tolyl)pyridin-3-yl)-N,2-dimethylpropanamide as a white solid (Yield: 83.9%).
Step 7:
To the solution of 2-(3,5-bis(trifluoromethyl)phenyl)-N-(6-chloro-4-(o-tolyl)pyridin-3-yl)-N,2-dimethylpropanamide (5.14 g, 10 mmol) in 60 mL of DCM was added m-CPBA (6.92 g, 40 mmol) at 0° C. under N2 atmosphere. Then the solution was stirred overnight at room temperature. 1 N aq. NaOH solution was added to wash twice for removing the excess m-CPBA. and a side product. The organic phase was washed by brine, dried over Na2SO4, filtered and concentrated to afford 5.11 g of crude 5-(2-(3,5-bis(trifluoromethyl)phenyl-N,2-dimethylpropanamido)-2-chloro-4(o-tolyl)pyridine 1-oxide as a white solid (Yield: 96.4%).
Step 8:
To the solution of crude 5-(2-(3,5-bis(trifluoromethyl)phenyl)-N,2-dimethylpropanamido)-2-chloro-4-(o-tolyl)pyridine 1-oxide (5.1 g, 9.62 mmol) in 80 mL of n-BuOH was added N-methylpiperazine (7.41 g, 74.1 mmol) under N2 atmosphere. Then the solution was stirred at 80° C. overnight. Concentrated and purified by flash chromatography to afford 4.98 g 5-(2-(3,5-bis(trifluoromethyl)phenyl-N,2-dimethylpropanamido)-2-(4-methylpiperazin-1-yl)-4-(o-tolyl)pyridine 1-oxide as a white solid (Yield: 87.2%), 1HNMR (CDCl3, 400 MHz) δ 8.15 (s, 1H), 7.93 (s, 1H), 7.78 (s, 2H), 7.38 (m, 2H), 7.28 (m, 1H), 7.17 (m, 1H), 7.07 (s, 1H), 5.50 (s, 3H), 2.72 (d, J=4.4 Hz, 4H), 2.57 (m, 3H), 2.40 (s, 3H), 2.23 (s, 3H), 1.45-1.20 (m, 6H).
………………………………….
https://www.google.co.in/patents/US6479483


EXAMPLE 14 2-(3,5-bis-Trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperan-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide Hydrochloride (1:2)
a) 1-Methyl-4-(5-nitro-pyridin-2-yl)-piperazine
To a solution of 20 g (126 mmol) of 2-chloro-5-nitropyridine in 200 ml tetrahydrofuran were added dropwise 35 ml (315 mmol) 1-methylpiperazine within 10 min. The reaction mixture was refluxed for additional 1.5 h. After cooling to room temperature, the solvent was removed in vacuo and the residue was re-dissolved in 200 ml ethyl acetate. The organic phase was washed with 200 ml 1 N sodium bicarbonate solution, dried (magnesium sulfate) and evaporated to give 27.9 g (quantitative) of the title compound as a yellow solid.
MS m/e (%): 223 (M+H+, 100).
b) 2,2-Dimethyl-N-[6-(4-methyl-piperazin-1-yl)-pyridin-3-yl-propionamide
To a solution of 27.9 g (125.5 mmol) of 1-methyl-4-(5-nitro-pyridin-2-yl)-piperazine in 400 ml methanol were added 2.6 g of 10% of palladium on activated charcoal. The reaction mixture was hydrogenated (room temperature to ca. 45° C., 1 bar) until the theoretical amount of hydrogen was taken up (about 2 h). The catalyst was filtered off and was washed twice with 100 ml portions of methanol. The filtrate was evaporated in vacuo to give 28 g of a purple oil which consisted to ca. 90% of the desired aniline derivative according to analysis by thin layer chromatography.
This crude product was dissolved in a mixture of 400 ml tetrahydrofuran and 100 ml diethyl ether. After cooling to 0° C., 30 ml (215 mmol) of triethylamine were added in one portion. Stirring was continued while 26 g (215 mmol) of pivaloyl chloride were added dropwise within a period of 10 min. The ice bath was removed and the reaction mixture was stirred for 1 h at room temperature. Then, the solvent was removed in vacuo and the residue was suspended in 200 ml 1 N sodium bicarbonate solution. The product was extracted three times with 200 ml portions of dichloromethane, dried (sodium sulfate) and purified by flash chromatography to give 30 g (86%) of the title compound as pink crystals.
MS m/e (%): 277 (M+H+, 100).
c) N-[4-Iodo-6-(4-methyl-piperazin-1-yl)-pyridin-3-yl]-2,2-dimethyl-propionamide
A solution of 30 g (108 mmol) 2,2-dimethyl-N-[6-(4-methyl-piperazin-1-yl)-pyridin-3-yl]-propionamide and 58 ml (380 mmol) N,N,N′,N′-tetramethylethylenediamine under argon in 650 ml tetrahydrofuran was cooled in a dry ice bath to −78° C. Within 1 h, 239 ml (380 mmol) of a 1.6 N n-butyllithium solution in hexane were added dropwise. The reaction mixture was allowed to warm up to −30° C. overnight. After cooling again to −78° C., 43.6 g (170 mmol) iodine dissolved in 60 ml tetrahydrofuran were added dropwise during 15 min. The dry ice bath was replaced by an ice bath and a solution of 90 g (363 mmol) sodium thiosulfate pentahydrate in 250 ml water were added within 10 min when the temperature of the reaction mixture had reached 0° C. Then, 1000 ml diethyl ether were added and the organic layer was separated. The aqueous layer was extracted twice with 500 ml dichloromethane and the combined organic layers were dried (magnesium sulfate) and evaporated. Flash chromatography gave 18.5 g (42%) of the tide compound as a light brown oil which crystallized upon standing at room temperature.
MS m/e (%): 403 (M+H+, 100).
d) 2,2-Dimethyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-propionamide
A mixture of 54 g (134 mmol) N-[4-iodo-6-(4-methyl-piperazin-1-yl)-pyridin-3-yl]-2,2-dimethyl-propionamide, 420 ml toluene, 150 ml 2 N sodium carbonate solution, 4.63 g (3.9 mmol) tetrakis(triphenylphosphine)palladium(0) and 20.16 g (147 mmol) o-tolylboronic acid was heated under argon at 80° C. for 12 h. After cooling to room temperature, the aqueous phase was separated and washed twice with toluene. The combined organic layers were washed with 50 ml brine, dried (sodium sulfate), evaporated and dried in vacuo to yield 49 g (quantitative) of the title compound as a brown oil.
MS m/e (%): 367 (M+H+, 100).
e) 6-(4-Methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-ylamine
A suspension of 56 g (152 mmol) 2,2-dimethyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-propionamide in 1300 ml 3 N hydrochloric acid solution was heated to 90-95° C. overnight. The reaction mixture was cooled to room temperature, washed with three 500 ml portions diethyl ether and filtered over celite. The filtrate was diluted with 500 ml water and was adjusted to pH 7-8 by addition of 28% sodium hydroxide solution under ice cooling. The product was extracted with four 1000 ml portions of dichloromethane. The combined organic layers were washed with 500 ml brine, dried (magnesium sulfate) and evaporated to give 35 g (82%) of the title compound as a light brown oil.
MS m/e (%):283 (M+H+, 100).
f) Methyl-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-amine
A solution of 35 g (124 mmol) 6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-ylamine in 270 ml trimethyl orthoformate and 8 drops trifluoroacetic acid was heated for 3 h at 130° C. The reaction mixture was evaporated and dried in vacuo for 30 min. The residual oil was dissolved in 100 ml tetrahydrofuran and was added dropwise under ice cooling to 9.4 g (248 mmol) lithium aluminum hydride in 300 ml tetrahydrofuran. The reaction mixture was stirred for 1 h at room temperature, cooled to 0° C. again and acidified (pH 1-2) by addition of 28% hydrochloric acid solution. After stirring for 5 min, 28% sodium hydroxide solution was added to reach pH 10. The solution was filtered over celite, evaporated and purified by flash chromatography to give 23.6 g (64%) of the title compound as a light brown oil.
MS m/e (%): 297 (M+H+, 100).
g) 2-(3,5-bis-Trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide
A solution of 20 g (67.5 mmol) methyl-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]amine and 17.5 ml (101 mmol) N-ethyldiisopropylamine in 200 ml dichloromethane was cooled in an ice bath and a solution of 24 g (75 mmol) 2-(3,5-bis-trifluoromethyl-phenyl)-2-methyl-propionyl chloride in 50 ml dichloromethane was added dropwise. The reaction mixture was warmed to 35-40° C. for 3 h, cooled to room temperature again and was stirred with 250 ml saturated sodium bicarbonate solution. The organic layer was separated and the aqueous phase was extracted with dichloromethane. The combined organic layers were dried (magnesium sulfate) and evaporated. The residue was purified by flash chromatography to give 31.6 g (81%) of the title compound as white crystals. M.p. 155-157° C.
MS m/e (%): 579 (M+H+, 100).
h) 2-(3,5-bis-Trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide Hydrochloride (1:2)
To a solution of 31.6 g (54.6 mmol) 2-(3,5-bis-trifluoromethyl-phenyl)-N-methyl-N-[6-(4-methyl-piperazin-1-yl)-4-o-tolyl-pyridin-3-yl]-isobutyramide in 250 ml diethyl ether were added under ice cooling 60 ml 3 N hydrochloric acid solution in diethyl ether. After stirring for 15 min at 0° C., the suspension was evaporated to dryness, re-suspended in 100 ml diethyl ether, filtered and dried in vacuo to give 34.8 g (98%) of the title compound as white crystals. M.p. 235-238° C.
MS m/e (%): 579 (M+H+, 100).
…………………………….
Research and development of an efficient process for the construction of the 2,4,5-substituted pyridines of NK-1 receptor antagonists
Org Process Res Dev 2006, 10(6): 1157
Navari RM.
Drugs. 2013 Mar;73(3):249-62. doi: 10.1007/s40265-013-0019-1. Review.
-
Hoffmann-Emery F, Hilpert H, Scalone M, Waldmeier P.
J Org Chem. 2006 Mar 3;71(5):2000-8.
Hoffmann T, Bös M, Stadler H, Schnider P, Hunkeler W, Godel T, Galley G, Ballard TM, Higgins GA, Poli SM, Sleight AJ.
Bioorg Med Chem Lett. 2006 Mar 1;16(5):1362-5. Epub 2005 Dec 5.
http://www.sciencedirect.com/science/article/pii/S0960894X05014824
…………………………………….……………………………………………………….
| US6897226 * | 9 Jul 2003 | 24 May 2005 | Hoffmann-La Roche Inc. | NK-1 receptor active amine oxide prodrugs |
| US7211579 * | 15 Mar 2006 | 1 May 2007 | Hoffmann-La Roche Inc. | NK-1 receptor antagonists |
| US8426450 | 23 May 2012 | 23 Apr 2013 | Helsinn Healthcare Sa | Substituted 4-phenyl pyridines having anti-emetic effect |
| WO2011061622A1 | 18 Nov 2010 | 26 May 2011 | Helsinn Healthcare S.A. | Compositions for treating centrally mediated nausea and vomiting |
| WO2013057554A2 | 10 Oct 2012 | 25 Apr 2013 | Helsinn Healthcare Sa | Therapeutic combinations of netupitant and palonosetron |
| US8426450 | 23 May 2012 | 23 Apr 2013 | Helsinn Healthcare Sa | Substituted 4-phenyl pyridines having anti-emetic effect |
| WO2011061622A1 | 18 Nov 2010 | 26 May 2011 | Helsinn Healthcare S.A. | Compositions for treating centrally mediated nausea and vomiting |
| WO2013057554A2 | 10 Oct 2012 | 25 Apr 2013 | Helsinn Healthcare Sa | Therapeutic combinations of netupitant and palonosetron |
……………………………………………………………………………………….


THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007
Palbociclib
PALBOCICLIB
Mechanism of action: selective inhibitor of the cyclin-dependent kinases CDK4 and CDK6
Indication: Estrogen receptor-positive (ER+), HER2-negative (HER2 -) breast cancer
Current Status: Phase III (US, UK, EU), (US Clinical trials numbers NCT01864746,NCT01740427, NCT01942135)
Expected Launch Date: 2015
Potential Sales(peak):$5 billion
Company:Pfizer
CHEMICAL NAMES
1. Pyrido[2,3-d]pyrimidin-7(8H)-one, 6-acetyl-8-cyclopentyl-5-methyl-2-[[5-(1-
piperazinyl)-2-pyridinyl]amino]-
2. 6-acetyl-8-cyclopentyl-5-methyl-2-{[5-(piperazin-1-yl)pyridin-2-
yl]amino}pyrido[2,3-d]pyrimidin-7(8H)-one
MOLECULAR FORMULA C24H29N7O2
MOLECULAR WEIGHT 447.5
TRADEMARK None as yet
SPONSOR Pfizer Inc.
CODE DESIGNATION PD-0332991
CAS#: 571190-30-2 (PD0332991); 827022-32-2 (PD0332991 HCl salt) 827022-33-3 (palbociclib isethionate)
http://www.ama-assn.org/resources/doc/usan/palbociclib.pdf FOR STRUCTURE AND DETAILS
recent studies have identified a number of selective CDK4 inhibitors that, as discussed above, may prove useful in treating cancer—either as anti-cancer agents or as chemoprotective agents—and in treating cardiovascular disorders, such as restenosis and atherosclerosis, diseases caused by infectious agents, and autoimmune disorders, including rheumatoid arthritis. For a disclosure of these selective CDK4 inhibitors, see commonly assigned International Patent Application PCT/IB03/00059, filed Jan. 10, 2003 (the ‘059 application), which is herein incorporated by reference in its entirety for all purposes.
The ‘059 application discloses a particularly potent and selective CDK4 inhibitor, 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one:

In standard enzyme assays the compound of Formula 1 exhibits IC50 concentrations for CDK4 and CDK2 inhibition (at 25° C.) of 0.011 μM and >5 μM, respectively. For a discussion of standard CDK4 and CDK2 assays for IC50 determinations, see D. W. Fry et al., J. Biol. Chem. (2001) 16617-16623.
Though the compound of Formula 1 is a potent and selective CDK4 inhibitor, its use in pharmaceutical products presents challenges. For example, the free base has poor water solubility (9 μg/mL) and exhibits low bioavailability in animal studies. A di-HCl salt of the compound of Formula 1 appears to exhibit adequate water solubility. However, moisture uptake studies reveal that, even at low relative humidity (10% RH), the di-HCl salt absorbs water in an amount greater than about 2% of its mass, making it unsuitable for use in a solid drug product. A mono-HCl salt of the compound of Formula 1 is marginally hygroscopic, absorbing more than 2% of its mass at a relative humidity above 80%. However, the process for preparing the mono-HCl salt yields partially crystalline drug substance, indicating potential problems with process scale-up. Other salt forms of the compound of Formula 1 are thus needed.
Pfizer’s breast cancer drug Palbociclib (PD-0332991), a first in the class oral inhibitor of cyclin-dependent kinases (CDK) 4 and 6, is widely seen by investors as Pfizer’s most valuable compound in late-stage development. The FDA awarded Palbociclib “breakthrough therapy designation” in April 2013 based on the preliminary phase 2 data showing palbociclib, combined with Novartis’ drug,Femara (Letrozole), stopped breast tumors progression for more than two years as compared with 7.5 months with letrozole alone. The phase 3 trial started in February 2013 and estimated final completion date is March 2016. Leerink Swann analyst Seamus Fernandez forecasts palbociclib could become a $5 billion drug, with potential for $3 billion in first-line metastatic breast cancer alone.
Palbociclib, also known as PD0332991, is an orally available pyridopyrimidine-derived cyclin-dependent kinase (CDK) inhibitor with potential antineoplastic activity. PD-0332991 selectively inhibits cyclin-dependent kinases (particularly Cdk4/cyclin D1 kinase), which may inhibit retinoblastoma (Rb) protein phosphorylation; inhibition of Rb phosphorylation prevents Rb-positive tumor cells from entering the S phase of the cell cycle (arrest in the G1 phase), resulting in suppression of DNA replication and decreased tumor cell proliferation. PD 0332991 is a highly specific inhibitor of cyclin-dependent kinase 4 (Cdk4) (IC50 = 0.011 μmol/L) and Cdk6 (IC50 = 0.016 μmol/L), having no activity against a panel of 36 additional protein kinases.
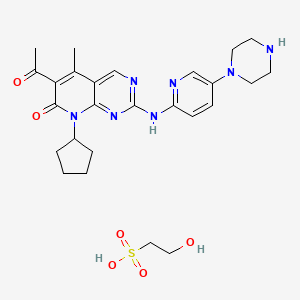
6-Acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one hydrochloride (also referred to as “Compound 1”),

as well as its intermediates. Compound 1 is described in U.S. Pat. No. 6,936,612, the disclosure of which is hereby incorporated in its entirety. This compound is a protein kinase inhibitor and represents a synthetic, small molecule inhibitor capable of modulating cell cycle control.
A method of preparing Compound 1 is disclosed as Example 36 of U.S. patent application Ser. No. 6,936,612. Methods of preparing the isethionate salt forms of Compound 1 are disclosed in Examples 1-13 of WO 2005/005426. These methods are for synthesis of small quantities of the salt forms of Compound 1 and are not designed for commercial scale-up. Therefore, a preparation of the salt forms for CDK inhibitor 6-Acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one hydrochloride which is cost-efficient, scaleable and productive is highly desirable.


USAN (zz-153)
PALBOCICLIB ISETHIONATE
THERAPEUTIC CLAIM Antineoplastic
CHEMICAL NAMES
1. Ethanesulfonic acid, 2-hydroxy-, compd. with 6-acetyl-8-cyclopentyl-5-methyl-
2-[[5-(1-piperazinyl)-2-pyridinyl]amino]pyrido[2,3-d]pyrimidin-7(8H)-one (1:1)
2. 6-acetyl-8-cyclopentyl-5-methyl-2-{[5-(piperazin-1-yl)pyridin-2-
yl]amino}pyrido[2,3-d]pyrimidin-7(8H)-one mono(2-hydroxyethanesulfonate)
MOLECULAR FORMULA C24H29N7O2 . C2H6O4S
MOLECULAR WEIGHT 573.7
SPONSOR Pfizer, Inc.
CODE DESIGNATIONS PD 0332991-0054, PF-00080665-73
CAS REGISTRY NUMBER 827022-33-3
- PD 0332991-0054
- PF-00080665-73
- UNII-W1NYL2IRDR

SYNTHESIS

:WO2008032157
……………………………….
http://www.google.com/patents/US7781583



COMPARATIVE EXAMPLE 1A Preparation of 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester
A suspension of 6-bromo-8-cyclopentyl-2-methansulfinyl-5-methyl-8H-pyrido[2,3-d]pyrimidin-7-one (10.00 g, 0.027 mol, prepared as in Example 6 of WO 01/707041, which is incorporated herein by reference) and 10.37 g (0.0373 mol) of 4-(6-amino-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester in toluene (100 mL) was heated under nitrogen in an oil bath for 7 hours. Thin layer chromatography (SiO2, 10% MeOH/DCM) indicated the presence of both starting materials. The suspension was heated under reflux for an additional 18 hours. The resulting suspension was cooled to RT and filtered to give 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester (5.93 g, 38%). Melting point>250° C. MS (APCI) M++1: calc’d, 584.2, found, 584.2.
COMPARATIVE EXAMPLE 1B Preparation of 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester
A suspension of 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester (5.93 g, 0.010 mol, prepared as in Example 1A), tetrakis(triphenylphosphine)palladium(0) (1.40 g, 0.00121 mol), and tributyl(1-ethoxyvinyl)tin (5.32 mL, 0.0157 mol) in toluene (30 mL) was heated under reflux for 3.5 hours. The mixture was cooled and filtered to give a solid. Purification of the solid by silica gel chromatography using a gradient of 5%-66% ethyl acetate/hexane over 15 minutes gave 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester as a yellow foam (4.50 g, 78%). MS (APCI) M++1: calc’d 576.2, found, 576.3.
COMPARATIVE EXAMPLE 1C Preparation of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one hydrochloride
Hydrogen chloride gas was bubbled into an ice-bath cooled solution of 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester (4.50 g, 0.00783 mol, prepared as in 2005-0059670A1) in DCM (100 mL). The resulting suspension was stoppered and stirred at RT overnight, then diluted with diethyl ether (200 mL). The solid was collected by filtration, washed with diethyl ether, and dried to give the hydrochloride salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one as a yellow solid (4.01 g, 92%). Melting point 200° C. HPLC, C18 reverse phase, 10%-95% gradient of 0.1% TFA/CH3CN in 0.1% TFA/H2O during 22 minutes: 99.0% at 11.04 minutes. MS (APCI) M++1: calc’d, 448.2, found, 448.3. Anal. calc’d for C24H29N7O2.2.4H2O.1.85 HCl: C, 51.64; H, 6.44; N, 17.56, Cl (total), 11.75. Found: C, 51.31; H, 6.41; N, 17.20; Cl (total), 12.11.
EXAMPLE 2 Preparation of 4-(6-Nitro-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester

EXAMPLE 2A Preparation of 4-(6-Nitro-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester
To 1.0 kg (5 mol) 5-bromo-2-nitropyridine was added 1.2 kg (6.4 mol) boc piperazine (tert-Butyl piperazine-1-carboxylate) in 2.6 L DMSO and 0.5 kg triethylamine under nitrogen. The mixture was heated to 65-70° C. and held for 30 hours after which some solids precipitated. Water was added and the reaction cooled to 25° C. over 2 hrs. The resulting slurry was filtered, washed and dried at 45° C. to give 1.2 kg (79% crude yield) of canary yellow solid intermediate (2A), which was used without further purification in the subsequent step.
EXAMPLE 2 Preparation of 4-(6-Nitro-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester (2)
60.0 g of 20% Pd(OH)2/C, 1213.1 g (3.9 moles) of intermediate 2a, and isopropanol were charged and stirred in a Parr reactor, then purged under gas, followed by removal of the catalyst under pressure. The filtrates were concentrated in vacuo at ˜20° C. leaving 917 g of dry brown powder (crude yield ˜84%).
EXAMPLE 3 Preparation of 2-Chloro-8-cyclopentyl-5-methyl-8H-pyrido[2,3-d]pyrimidin-7-one

EXAMPLE 3A Preparation of 5-bromo-2-chloro-4-cyclopentyl-aminopyrimidine
To 1 g (0.004 mol) of 5-bromo-2,4-dichloropyrimidine in ethanol was added 1.5 kg (0.018 mol) cyclopentylamine under nitrogen. The mixture was stirred at 25° C. for 2 hrs. Water was added to precipitate the product, and the solid was recrystallized using hexane 4:1 to give a white crystalline product (3A).
EXAMPLE 3 Preparation of 2-Chloro-8-Cyclopentyl-5-methyl-8H-pyrido[2,3-d]pyrimidin-7-one
41.5 g (0.15 mol) of 5-bromo-2-chloro-4-cyclopentylaminopyrimidine 3a and 32.3 g (0.375 mol) of crotonic acid were mixed in 100 L of THF and 105 ml (1.6 mol) diisopropyl ethylamine under nitrogen. The slurry was stirred, evacuated and refilled with nitrogen three times, after which 860 mg (0.0022 mol) palladium dichloride dibenzonitrile complex and 685 mg (0.0022 mol) tri-ortho-tolylphosphine were added and the resulting slurry degassed an additional three times. The mixture was then heated and stirred at 70° C. for 16 hrs, after which 35 ml acetic anhydride was added and the mixture stirred for an additional 1.5 hrs. The mixture was cooled and diluted with 100 ml MTBE and then extracted with 1NHCl, then aqueous sodium bicarbonate and brine. The organic phase was dried over magnesium sulfate, filtered, concentrated in vacuo, and recrystallized from IPA to yield 31.2 g (68%) of crude product (3).
EXAMPLE 4 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester

EXAMPLE 4A Preparation of 2-chloro-8-cyclopentyl-5-methyl-8H-pyrido[2,3-d]pyrimidine-7-one
10 g (0.04 mol) of intermediate 3 and 13 g (0.16 mol) of sodium acetate were mixed with 50 ml of glacial acetic acid and 12 g (0.08 mol) bromine under nitrogen. The solution was heated to 50° C. and stirred for 35 hrs, then cooled to room temperature. Sodium bisulfite solids were added until the bromine color disappeared, then quenched, filtered and washed to provide a solid which was subsequently dissolved in 500 ml hot IPA, filtered hot, and cooled. The resulting crystals were further filtered, and dried in vacuo at 65° C. to yield 8 g (61%) of crude product (4A).
EXAMPLE 4 Preparation of 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester
3.78 g (2.10 equiv; 13.6 mmoles) of intermediate 1, 25 ml toluene and lithium bis(trimethylsilyl)amide in 1 M THF (13.6 mmoles; 13.6 mL; 12.1 g) were mixed for 10 min under nitrogen to form a dark solution. In a separate beaker the intermediate 4a (1.00 equiv, 6.47 mmoles; 2.50 g) was slurried in toluene then added to the mixture containing 1 and stirred for 30 min, after which the combined mixture was quenched with 25 ml 1 M sodium bicarbonate and then filtered. Alternatively, the combined mixture can be quenched with ammonium chloride. The filter cake was washed with toluene, then acetone, then water and dried at 60° C. to give 3.5 g (92%) of a grey-yellow solid 4.
EXAMPLE 5 Preparation of 4-{6-[6-(1-butoxy-vinyl)-8-cycloentyl-5-methyl-7-oxo-7,8-dihydropyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester

768 g (1.3 mol) of intermediate 4, was mixed with 395 g (3.9 mol) of butyl vinyl ether, 4.7 L of n-butanol, and 275 ml (1.6 mol) diisopropyl ethylamine under nitrogen. The slurry was stirred and placed under ca. 50 tore vacuum and then refilled with nitrogen; this was repeated 2 more times. To this degassed solution was added 22 g (0.03 mol) Bis-(diphenylphosphinoferrocene)palladium dichloride dichloromethane complex and the resulting slurry was degassed an additional three times as described above. The mixture was then heated and stirred at 95° C. for 20 hrs. The resulting thin red slurry was diluted with 4 L branched octane’s and cooled to about 5° C. after which 1 L saturated aq. potassium carbonate was added and the mixture was filtered and rinsed with 500 ml branched octanes. After drying for 16 hrs at 45° C., 664 g (83%) of gray-solid product (5) was obtained. In addition, column chromatography can be used to further purify the crude product.
EXAMPLE 6 Preparation of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one

11.6 g (1.00 eq, 19.2 mmol) of intermediate 5, water (10.1 equiv; 193 mmoles; 3.48 mL; 3.48 g) and methanol (3.62 moles; 146 mL; 116 g) were combined and heated to 55-60° C. Isethionic acid was added slowly until a clear solution was obtained; 3.3 g isethionic acid solution was necessary to reach this end point. The resulting clear orange solution was filtered through paper and rinsed through with 20 ml methanol, after which the filtrate was reheated to 55-60° C. and the remaining isethionic acid was added (a total of 9.93 g was added). The reaction mixture precipitated and thickened for 6 hours, after which it was cooled and held at 30-35° C. while triethylamine (2.92 g; 28.8 mmoles) was added slowly as a 10% solution in methanol over 12 hrs. About halfway through the addition of triethylamine, desired polymorphic seeds were added to help formation of the desired polymorph. The resulting slurry was cooled and held at 5° C. for 15 minutes and the crystals were filtered and washed with methanol. The solid product was dried in vacuo at 55° C. to obtain 11 g of yellow crystals of the title compound.

……………………………………………………………………
http://www.google.com/patents/US7345171
EXAMPLES
The following examples are intended to be illustrative and non-limiting, and represent specific embodiments of the present invention.
Example 1 Preparation of 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester
A suspension of 6-bromo-8-cyclopentyl-2-methansulfinyl-5-methyl-8H-pyrido[2,3-d]pyrimidin-7-one (10.00 g, 0.027 mol, prepared as in Example 6 of WO 01/707041, which is incorporated herein by reference) and 10.37 g (0.0373 mol) of 4-(6-amino-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester in toluene (100 mL) was heated under nitrogen in an oil bath for 7 hours. Thin layer chromatography (SiO2, 10% MeOH/DCM) indicated the presence of both starting materials. The suspension was heated under reflux for an additional 18 hours. The resulting suspension was cooled to RT and filtered to give 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester (5.93 g, 38%). Melting point>250° C. MS (APCI) M++1: calc’d, 584.2, found, 584.2.
Example 2 Preparation of 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2.3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester
A suspension of 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester (5.93 g, 0.010 mol, prepared as in Example 1), tetrakis(triphenylphosphine)palladium(0) (1.40 g, 0.00121 mol), and tributyl(1-ethoxyvinyl)tin (5.32 mL, 0.0157 mol) in toluene (30 mL) was heated under reflux for 3.5 hours. The mixture was cooled and filtered to give a solid. Purification of the solid by silica gel chromatography using a gradient of 5%-66% ethyl acetate/hexane over 15 minutes gave 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester as a yellow foam (4.50 g, 78%). MS (APCI) M++1: calc’d 576.2, found, 576.3.
Example 3 Preparation of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one hydrochloride
Hydrogen chloride gas was bubbled into an ice-bath cooled solution of 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester (4.50 g, 0.00783 mol, prepared as in Example 2) in DCM (100 mL). The resulting suspension was stoppered and stirred at RT overnight, then diluted with diethyl ether (200 mL). The solid was collected by filtration, washed with diethyl ether, and dried to give the hydrochloride salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one as a yellow solid (4.01 g, 92%). Melting point 200° C. HPLC, C18 reverse phase, 10%-95% gradient of 0.1% TFA/CH3CN in 0.1% TFA/H2O during 22 minutes: 99.0% at 11.04 minutes. MS (APCI) M++1: calc’d, 448.2, found, 448.3. Anal. calc’d for C24H29N7O2.2.4H2O.1.85 HCl: C, 51.64; H, 6.44; N, 17.56, Cl (total), 11.75. Found: C, 51.31; H, 6.41; N, 17.20; Cl (total), 12.11.
Example 4 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2.3-d]pyrimidin-7-one (Form B)
To a slurry of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one (7.0 g, 15.64 mmol, prepared as in Example 3 following contact with NaOH) dispersed in 250 mL of water was added drop-wise 30 mL of a 0.52 M solution of isethionic acid in MeOH (15.64 mmol) to a pH of 5.2. The solution was filtered through a glass filter (fine) and the clear solution was freeze-dried to give 9.4 g of the amorphous salt. The amorphous salt (3.16 g) was mixed with 25 mL of MeOH and after almost complete dissolution a new precipitate formed. Another 25 mL of MeOH was added and the mixture was stirred at 46° C. to 49° C. for four hours. The mixture was slowly cooled to 32° C. and put in a cold room (+4° C.) overnight. A sample was taken for PXRD, which indicated formation of Form B. The mixture was filtered and the precipitate was dried overnight at 50° C. in a vacuum oven. This furnished 2.92 g of the mono-isethionate salt of the compound of Formula 1 in 92% yield. HPLC-99.25%, PXRD-Form B, CHNS, H-NMR were consistent with the structure.
Example 5 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2.3-d]pyrimidin-7-one (Form B)
MeOH (100 mL) was placed in a 250 mL flask equipped with a mechanical stirrer, thermocouple/controller, condenser, and heating mantle and preheated to 35° C. An amorphous isethionate salt (2 g, prepared as in Example 4) was slowly added in three even portions with a 25 min to 30 min interval between the additions. The reaction mixture was stirred overnight at 35° C. and subsequently cooled. A sample was filtered and examined by PXRD. It was pure Form B. The whole reaction mixture was then used as Form B seeds in a larger scale experiment.
Example 6 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one (Form B)
MeOH (50 mL) was placed in a 250 mL flask equipped with a magnetic stirrer, condenser, thermocouple/controller, and heating mantle, and preheated to 40° C. An amorphous isethionate salt (1 g, prepared as in Example 4) was slowly added in three even portions with 30 min interval between the portions and then stirred overnight at 40° C. The reaction was monitored by in-situ Raman spectroscopy. The sample was taken, filtered and analyzed by PXRD. It was pure Form B by PXRD and Raman spectroscopy. The mixture was cooled to 25° C. at a rate of 3° C./h, cooled to −10° C., filtered, and vacuum dried to furnish 0.85 g of the Form B crystalline product.
Example 7 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one (Form B)
The free base (Formula 1, 0.895 mg, 2 mmol) was mixed with 10 mL of MeOH and seeded with 33 mg of a mono-isethionate salt of the compound of Formula 1 (Form B). Then 5.6 mL of a 0.375 M solution of isethionic acid in MeOH (2.1 mmol) was added in 10 even portions over 75 min time period. The mixture was stirred for an additional hour and a sample was taken for PXRD analysis. It confirmed formation of crystalline Form B. The mixture was stirred at RT overnight and another PXRD was taken. There was no change in the crystal form. The mixture was cooled in a refrigerator at −8° C. overnight, filtered, and dried at 50° C. in a vacuum oven to give 1.053 g (91.8% of theory) of the above-named compound (Form B). HPLC—99.8%, CHNS, H-NMR, IR are consistent with the structure, PXRD-Form B.
Example 8 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2.3-d]pyrimidin-7-one (Form A)
An amorphous isethionate salt (47 mg, prepared as in Example 4) was mixed with 4 mL of EtOH in a 15 mL flask equipped with a magnetic stirrer, thermocouple and condenser. The mixture was heated to reflux, which resulted in the formation of a nearly clear solution. After refluxing for 10-15 min, the mixture became cloudy. It was slowly cooled to 50° C. and was seeded at 69° C. with Form A. The mixture was held at 50° C. for 5 h and was allowed to cool to RT overnight. The mixture was subsequently cooled to 1° C. with an ice bath, held for 1.5 h, filtered, washed with 0.5 mL of cold EtOH, air-dried, and then dried in a vacuum oven at 70° C. overnight to furnished 38.2 mg of a fine crystalline material. The crystalline material was found to be mono-isethionate salt Form A by PXRD. H-NMR was consistent for the mono-isethionate salt and indicated the presence of residual EtOH ca. 5.9 mol % or 0.6 wt %.
Example 9 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one (Form D)
An amorphous isethionate salt (9.0 g, prepared as in Example 4) was mixed with 300 mL of MeOH, stirred and heated to 63.8° C. (at reflux). To the slightly cloudy mixture was added two 50-mL portions of MeOH. The hot mixture was filtered into a 2-L flask equipped with a mechanical stirrer. The mixture was briefly heated to reflux and then cooled to 60° C. IPA (100 mL) was added to the mixture. The mixture was again heated to 60° C. and an additional 110 mL of IPA was added. A precipitate started to form at 59.7° C. The mixture was reheated to 67.5° C., cooled to 50° C., and held overnight. A sample was taken the next morning for PXRD analysis. The mixture was cooled to 25° C. at a rate of 3° C./h and another PXRD sample was taken when the mixture reached 28° C. The mixture was allowed to cool to RT overnight. A precipitate was collected and dried in a vacuum oven at 65° C. and 30 Torr. The procedure produced 7.45 g (82.8% yield) of the crystalline compound (Form D by PXRD analysis). Previously analyzed samples were also Form D. HPLC showed 98.82% purity and CHNS microanalysis was within +/−0.4%. A slurry of isethionate salt Form A, B, and D in MeOH yielded substantially pure Form B in less than three days.
Example 10 Preparation of isethionic acid (2-hydroxy-ethanesulfonic acid)
A 5-L, four-necked, round-bottomed flask, equipped with mechanical stirrer, thermocouple, gas sparger, and an atmosphere vent through a water trap was charged with 748 g (5.05 mol) of sodium isethionate (ALDRICH), and 4 L of IPA. The slurry was stirred at RT. An ice bath was used to keep the internal temperature below 50° C. as 925 g (25.4 mol) of hydrogen chloride gas (ALDRICH) was sparged into the system at a rate such that it dissolved as fast as it was added (as noted by lack of bubbling through the water trap). Sufficient HCl gas was added until the system was saturated (as noted by the start of bubbling through the water trap). During the addition of HCl, the temperature rose to 45° C. The slurry was cooled to RT and filtered over a coarse-fritted filter. The cake was washed with 100 mL of IPA and the cloudy filtrate was filtered through a 10-20μ filter. The resulting clear, colorless filtrate was concentrated under reduced pressure on a rotary evaporator, while keeping the bath temperature below 50° C. The resulting 1.07 kg of clear, light yellow oil was diluted with 50 mL of tap water and 400 mL of toluene and concentrated under reduced pressure on a rotary evaporator for three days, while keeping the bath temperature below 50° C. The resulting 800 g of clear, light yellow oil was diluted with 500 mL of toluene and 250 mL of IPA and concentrated under reduced pressure on a rotary evaporator for 11 days, keeping the bath temperature below 50° C. The resulting 713 g of clear, light yellow oil was titrated at 81 wt % (580 g, 91.1% yield) containing 7.9 wt % water and 7.5 wt % IPA.
Example 11 Preparation of 4-{6-[6-(1-butoxy-vinyl)-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester
A 5-L, three-necked, round-bottomed flask, equipped with a mechanical stirrer, a thermocouple, and a nitrogen inlet/outlet vented through a silicone oil bubbler was placed under a nitrogen atmosphere and charged with 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester (300 g, 0.51 mol, prepared as in Example 2), butyl vinyl ether (154 g, 1.54 mol, ALDRICH), n-butanol (1.5 L, ALDRICH), and diisopropyl ethylamine (107 mL, 0.62 mol, ALDRICH). The slurry was placed under approximately 50 Torr vacuum and then refilled with nitrogen 3 times. To this was added 8.3 g (0.01 mol) bis-(diphenylphosphinoferrocene) palladium dichloride dichloromethane (JOHNSON MATTHEY, Lot 077598001) and the resulting slurry was purged an additional three times as described above. The mixture was then heated to 95° C. and stirred for 20 h. The resulting thin red slurry was diluted with 2 L of heptane and cooled to approximately 5° C. At this temperature, 400 mL saturated aqueous potassium carbonate was added and the mixture was filtered and rinsed with 250 mL of heptane. After drying in an oven for 16 h at 45° C., 231.7 g (75% yield) of the title compound was obtained as a yellow solid.
Example 12 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-Pyrido[2,3-d]pyrimidin-7-one (Form B)
A 22-L, three-necked, round-bottomed flask, equipped with a mechanical stirrer, a thermocouple, and a nitrogen inlet/outlet vented through a silicone oil bubbler was placed under a nitrogen atmosphere and charged with 4-{6-[6-(1-butoxy-vinyl)-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester (725 g, 1.20 mol, prepared as in Example 11) and MeOH (14 L). The slurry was stirred at RT as it was charged with a solution of isethionic acid (530 g, 4.20 mol, prepared as in Example 10), MeOH (1.5 L), and water (70 mL, 3.89 mol). The resulting slurry was heated to 55° C. over 30 minutes and then stirred at 55° C. for 30 minutes. A solution of 175 g (1.73 mol) of Et3N (ALDRICH) in 200 mL of MeOH was charged to the slurry as it was cooled to 30° C. The slurry was held at 30° C. as a solution of 128 g (1.26 mol) of Et3N in 2 L of MeOH was added dropwise over 6 hours. The resulting slurry was sampled to determine crystal form (Form B). The slurry was cooled and held at 5° C. for 15 minutes and was subsequently filtered through a coarse-fritted filter. The resulting filter cake was washed with multiple washes of 200 mL of cold MeOH. The solid product was dried at 55° C. under vacuum to yield 710 g (91% yield) of the title compound as yellow crystals.

1)Peter L. Toogood, Patricia J. Harvey, Joseph T. Repine, Derek J. Sheehan, Scott N. VanderWel, Hairong Zhou, Paul R. Keller, Dennis J. McNamara, Debra Sherry, Tong Zhu, Joanne Brodfuehrer, Chung Choi, Mark R. Barvian, and David W. Fry;Discovery of a Potent and Selective Inhibitor of Cyclin-Dependent Kinase 4/6; Journal of Medicinal Chemistry, 2005, 48(7),2388-2406;
2)Scott N. VanderWel, Patricia J. Harvey, Dennis J. McNamara, Joseph T. Repine, Paul R. Keller, John Quin III, R. John Booth, William L. Elliott, Ellen M. Dobrusin, David W. Fry, and Peter L. Toogood; Pyrido[2,3-d]pyrimidin-7-ones as Specific Inhibitors of Cyclin-Dependent Kinase 4; Journal of Medicinal Chemistry,2005,48(7),2371-2387;
3)Erdman, David Thomas et al;Preparation of 2-(pyridin-2-ylamino)-pyrido[2,3-d]pyrimidin-7-ones;PCT Int. Appl., WO2008032157
4)Sharpless, Norman E. et al;Hematopoietic protection against chemotherapeutic compounds using selective cyclin-dependent kinase 4/6 inhibitors;PCT Int. Appl., WO2010039997
5)Dirocco, Derek Paul et al;Protection of renal tissues from schema through inhibition of the proliferative kinases CDK4 and CDK6;PCT Int. Appl., WO2012068381
6)Logan, Joshua E.et al.;PD- 0332991, a potent and selective inhibitor of cyclin-dependent kinase 4/6, demonstrates inhibition of proliferation in renal cell carcinoma at nanomolar concentrations and molecular markers predict for sensitivity; Anticancer Research (2013), 33(8), 2997-3004.
7)Phase III Study Evaluating Palbociclib (PD-0332991), a Cyclin-Dependent Kinase (CDK) 4/6 Inhibitor in Patients With Hormone-receptor-positive, HER2-normal Primary Breast Cancer With High Relapse Risk After Neoadjuvant Chemotherapy “PENELOPEB”;ClinicalTrials.gov number:NCT01864746;currently recruiting participants(as of January 2, 2013)
8)A Randomized, Multicenter, Double-Blind Phase 3 Study Of PD-0332991 (Oral CDK 4/6 Inhibitor) Plus Letrozole Versus Placebo Plus Letrozole For The Treatment Of Postmenopausal Women With ER (+), HER2 (-) Breast Cancer Who Have Not Received Any Prior Systemic Anti Cancer Treatment For Advanced Disease;ClinicalTrials.gov number:NCT01740427;currently recruiting participants(as of January 2, 2013)
9)Multicenter, Randomized, Double-Blind, Placebo-Controlled, Phase 3 Trial Of Fulvestrant (Faslodex®) With Or Without PD-0332991 (Palbociclib) +/- Goserelin In Women With Hormone Receptor-Positive, HER2-Negative Metastatic Breast Cancer Whose Disease Progressed After Prior Endocrine Therapy;ClinicalTrials.gov number:NCT01942135;currently recruiting participants(as of January 2, 2013)
| US6936612 | Jan 16, 2003 | Aug 30, 2005 | Warner-Lambert Company | 2-(Pyridin-2-ylamino)-pyrido[2,3-d]pyrimidin-7-ones |
| WO2005005426A1 | Jun 28, 2004 | Jan 20, 2005 | Vladimir Genukh Beylin | Isethionate salt of a selective cdk4 inhibitor |
| US20030229026 * | Dec 18, 2000 | Dec 11, 2003 | Al-Awar Rima Salim | Agents and methods for the treatment of proliferative diseases |
| US20040006074 * | Dec 2, 2002 | Jan 8, 2004 | The Government Of The United States Of America | Cyclin dependent kinase (CDK)4 inhibitors and their use for treating cancer |
| US20040048915 * | Sep 24, 2001 | Mar 11, 2004 | Engler Thomas Albert | Methods and compounds for treating proliferative diseases |
| US20050222163 * | Mar 30, 2005 | Oct 6, 2005 | Pfizer Inc | Combinations of signal transduction inhibitors |
| US20070027147 * | Dec 3, 2004 | Feb 1, 2007 | Takashi Hayama | Biarylurea derivatives |
| WO2008032157A2 * | Aug 27, 2007 | Mar 20, 2008 | David Thomas Erdman | Synthesis of 2-(pyridin-2-ylamino)-pyrido[2,3-d]pyrimidin-7-ones |
| WO2010075074A1 | Dec 15, 2009 | Jul 1, 2010 | Eli Lilly And Company | Protein kinase inhibitors |
| WO2012098387A1 | Jan 17, 2012 | Jul 26, 2012 | Centro Nacional De Investigaciones Oncológicas (Cnio) | 6, 7-ring-fused triazolo [4, 3 – b] pyridazine derivatives as pim inhibitors |
| US7781583 | Sep 10, 2007 | Aug 24, 2010 | Pfizer Inc | Synthesis of 2-(pyridin-2-ylamino)-pyrido[2,3-d] pryimidin-7-ones |
| US7855211 | Dec 15, 2009 | Dec 21, 2010 | Eli Lilly And Company | Protein kinase inhibitors |
| US8247408 * | Oct 9, 2006 | Aug 21, 2012 | Exelixis, Inc. | Pyridopyrimidinone inhibitors of PI3Kα for the treatment of cancer |
| US8273755 | Feb 9, 2010 | Sep 25, 2012 | Pfizer Inc | 4-methylpyridopyrimidinone compounds |

old info
Date: April 10, 2013
Pfizer Inc. said that its experimental pill for advanced, often deadly breast cancer has been designated as a breakthrough therapy by the Food and Drug Administration.
The breakthrough designation, created under legislation enacted last summer to fund and improve operations of the FDA, is meant to speed up development and review of experimental treatments that are seen as big advances over existing therapies for serious diseases. Pfizer is working with the agency to determine exactly what research results it will need to apply for approval of the drug.
Palbociclib is being evaluated as an initial treatment for the biggest subgroup of postmenopausal women whose breast cancer is locally advanced or has spread elsewhere in the body. About 60% of women with such advanced breast cancer have tumors classified as ER+, or estrogen-receptor positive, but HER2-, or lacking an excess of the growth-promoting protein HER2.
Estrogen-receptor positive tumors have proteins inside and on the surface of their cells to which the estrogen hormone can attach and then fuel growth of cells. These tumors tend to grow slowly and can be fought with drugs that block estrogen’s effects.
Meanwhile, about 80% of breast cancer tumor cells are HER2 negative. That means that unlike HER2 positive tumors, they don’t produce too much of the HER2 protein, which makes tumors grow and spread more aggressively than in other breast cancer types.
New York-based Pfizer is currently running a late-stage study of palbociclib at multiple centers, comparing its effects when used in combination with letrozole with the effects of letrozole alone.
Letrozole, sold under the brand name Femara for about the past 15 years, is a pill that works by inhibiting aromatase. That’s an enzyme in the adrenal glands that makes estrogen.
According to Pfizer, palbociclib targets enzymes called cyclin dependent kinases 4 and 6. By inhibiting those enzymes, the drug has been shown in laboratory studies to block cell growth and suppress copying of the DNA of the cancer cells.
Pfizer, which has made research on cancer medicines a priority in recent years, also is testing palbociclib as a treatment for other cancers.
| Highlight of recent study using PD-0332991 |
Phase I study of PD-0332991: Forty-one patients were enrolled. DLTs were observed in five patients (12%) overall; at the 75, 125, and 150 mg once daily dose levels. The MTD and recommended phase II dose of PD 0332991 was 125 mg once daily. Neutropenia was the only dose-limiting effect. After cycle 1, grade 3 neutropenia, anemia, and leukopenia occurred in five (12%), three (7%), and one (2%) patient(s), respectively. The most common non-hematologic adverse events included fatigue, nausea, and diarrhea. Thirty-seven patients were evaluable for tumor response; 10 (27%) had stable disease for ≥4 cycles of whom six derived prolonged benefit (≥10 cycles). PD 0332991 was slowly absorbed (median T(max), 5.5 hours), and slowly eliminated (mean half-life was 25.9 hours) with a large volume of distribution (mean, 2,793 L). The area under the concentration-time curve increased linearly with dose. Using an E(max) model, neutropenia was shown to be proportional to exposure. CONCLUSIONS:
PD 0332991 warrants phase II testing at 125 mg once daily, at which dose neutropenia was the sole significant toxicity. (Source: Clin Cancer Res; 18(2); 568-76.)
Phase I study of PD-0332991 in 3-week cycles (Schedule 2/1): Six patients had DLTs (18%; four receiving 200 mg QD; two receiving 225 mg QD); the MTD was 200 mg QD. Treatment-related, non-haematological adverse events occurred in 29 patients (88%) during cycle 1 and 27 patients (82%) thereafter. Adverse events were generally mild-moderate. Of 31 evaluable patients, one with testicular cancer achieved a partial response; nine had stable disease (≥10 cycles in three cases). PD 0332991 was slowly absorbed (mean T(max) 4.2 h) and eliminated (mean half-life 26.7 h). Volume of distribution was large (mean 3241 l) with dose-proportional exposure. Using a maximum effective concentration model, neutropenia was proportional to exposure. CONCLUSION: PD 0332991 was generally well tolerated, with DLTs related mainly to myelosuppression. The MTD, 200 mg QD, is recommended for phase II study. (source: Br J Cancer. 2011 Jun 7;104(12):1862-8)

| References |
1: Flaherty KT, Lorusso PM, Demichele A, Abramson VG, Courtney R, Randolph SS, Shaik MN, Wilner KD, O’Dwyer PJ, Schwartz GK. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin Cancer Res. 2012 Jan 15;18(2):568-76. doi: 10.1158/1078-0432.CCR-11-0509. Epub 2011 Nov 16. PubMed PMID: 22090362.
2: Smith D, Tella M, Rahavendran SV, Shen Z. Quantitative analysis of PD 0332991 in mouse plasma using automated micro-sample processing and microbore liquid chromatography coupled with tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2011 Oct 1;879(27):2860-5. doi: 10.1016/j.jchromb.2011.08.009. Epub 2011 Aug 16. PubMed PMID: 21889427.
3: Katsumi Y, Iehara T, Miyachi M, Yagyu S, Tsubai-Shimizu S, Kikuchi K, Tamura S, Kuwahara Y, Tsuchiya K, Kuroda H, Sugimoto T, Houghton PJ, Hosoi H. Sensitivity of malignant rhabdoid tumor cell lines to PD 0332991 is inversely correlated with p16 expression. Biochem Biophys Res Commun. 2011 Sep 16;413(1):62-8. doi: 10.1016/j.bbrc.2011.08.047. Epub 2011 Aug 17. PubMed PMID: 21871868; PubMed Central PMCID: PMC3214763.
4: Schwartz GK, LoRusso PM, Dickson MA, Randolph SS, Shaik MN, Wilner KD, Courtney R, O’Dwyer PJ. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1). Br J Cancer. 2011 Jun 7;104(12):1862-8. doi: 10.1038/bjc.2011.177. Epub 2011 May 24. PubMed PMID: 21610706; PubMed Central PMCID: PMC3111206.
5: Nguyen L, Zhong WZ, Painter CL, Zhang C, Rahavendran SV, Shen Z. Quantitative analysis of PD 0332991 in xenograft mouse tumor tissue by a 96-well supported liquid extraction format and liquid chromatography/mass spectrometry. J Pharm Biomed Anal. 2010 Nov 2;53(3):228-34. doi: 10.1016/j.jpba.2010.02.031. Epub 2010 Feb 26. PubMed PMID: 20236782.
6: Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, Los G, Slamon DJ. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11(5):R77. doi: 10.1186/bcr2419. PubMed PMID: 19874578; PubMed Central PMCID: PMC2790859.
7: Menu E, Garcia J, Huang X, Di Liberto M, Toogood PL, Chen I, Vanderkerken K, Chen-Kiang S. A novel therapeutic combination using PD 0332991 and bortezomib: study in the 5T33MM myeloma model. Cancer Res. 2008 Jul 15;68(14):5519-23. doi: 10.1158/0008-5472.CAN-07-6404. PubMed PMID: 18632601.
8: Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, Toogood PL. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004 Nov;3(11):1427-38. PubMed PMID: 15542782.

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family
TEDIZOLID (torezolid)
TEDIZOLID PHOSPHATE
[(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl]phenyl}-2-oxo-5-oxazolidinyl]methyl]phosphate,
DA 7157
THERAPEUTIC CLAIM Treatment of complicated skin and skin structure infections
CHEMICAL NAMES
1. 2-Oxazolidinone, 3-[3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)-3-pyridinyl]phenyl]-5- [(phosphonooxy)methyl]-, (5R)-
2. [(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl]phenyl}-2-oxooxazolidin-5- yl]methyl hydrogen phosphate
http://www.ama-assn.org/resources/doc/usan/tedizolid-phosphate.pdf
MOLECULAR FORMULA C17H16FN6O6P
MOLECULAR WEIGHT 450.3
TRADEMARK None as yet
SPONSOR Trius Therapeutics
CODE DESIGNATION TR-701 FA
CAS REGISTRY NUMBER 856867-55-5
Note: This adoption statement supersedes the USAN torezolid phosphate (N09/81), which is hereby rescinded and replaced by the USAN tedizolid phosphate (N10/118).\
……………………………..
Tedizolid, 856866-72-3
(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl]phenyl}-5-(hydroxymethyl)-1,3-oxazolidin-2-one
(5R)-3-[3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)-3-pyridinyl]phenyl]-5-(hydroxymethyl)-2-oxazolidinone,
TR 700
- Molecular Formula: C17H15FN6O3
- Average mass: 370.337799
Torezolid (also known as TR-701 and now tedizolid[1]) is an oxazolidinone drug being developed by Trius Therapeutics (originator Dong-A Pharmaceuticals) for complicated skin and skin-structure infections (cSSSI), including those caused by Methicillin-resistantStaphylococcus aureus (MRSA).[2]
As of July 2012, tedizolid had completed one phase III trial, with another one under way. [3]Both trials compare a six-day regimen of tedizolid 200mg once-daily against a ten-day regimen of Zyvox (linezolid) 600mg twice-daily.
The prodrug of tedizolid is called “TR-701″, while the active ingredient is called “TR-700″.[4][5]

Trius Therapeutics will soon be reporting data from its second phase III trial (ESTABLILSH-2) and the recently announced publication of the data from its first phase III trial (ESTABLISH-1) in the Journal of the American Medical Association (JAMA)
- “Trius grows as lead antibiotic moves forward”. 31 Oct 2011.
- “Trius Completes Enrollment In Phase 2 Clinical Trial Evaluating Torezolid (TR-701) In Patients With Complicated Skin And Skin Structure Infections”. Jan 2009.
- http://clinicaltrials.gov/ct2/results?flds=Xf&flds=a&flds=b&term=tedizolid&phase=2&fund=2&show_flds=Y
- PMID 19528279 In vitro activity of TR-700, the active ingredient of the antibacterial prodrug TR-701, a novel oxazolidinone antibacterial agent.
- PMID 19218276 TR-700 in vitro activity against and resistance mutation frequencies among Gram-positive pathogens.
…………………………………………………….
Emergence of bacterial resistance to known antibacterial agents is becoming a major challenge in treating bacterial infections. One way forward to treat bacterial infections, and especially those caused by resistant bacteria, is to develop newer antibacterial agents that can overcome the bacterial resistance. Coates et al. (Br. J. Pharmacol. 2007; 152(8), 1147-1154.) have reviewed novel approaches to developing new antibiotics. However, the development of new antibacterial agents is a challenging task. For example, Gwynn et al. (Annals of the New York Academy of Sciences, 2010, 1213: 5-19) have reviewed the challenges in the discovery of antibacterial agents.
Several antibacterial agents have been described in the prior art (for example, see PCT International Application Nos. PCT/US2010/060923, PCT/EP2010/067647, PCT/US2010/052109, PCT/US2010/048109, PCT/GB2009/050609, PCT/EP2009/056178 and PCT/US2009/041200). However, there remains a need for potent antibacterial agents for preventing and/or treating bacterial infections, including those caused by bacteria that are resistant to known antibacterial agents.
Various oxazolidinone-containing compounds have been disclosed for use asantibiotics. For example, oxazolidinone-containing compounds have been described in U.S. patent application Ser. No. 10/596,412 (filed Dec. 17, 2004), and WO 04/048350, WO 03/022824 and WO 01/94342, which are incorporated herein by reference.
U.S. patent application Ser. No. 12/577,089 (filed Oct. 9, 2009) and U.S. patent application Ser. No. 12/699,864 (filed Feb. 3, 2010), which are assigned to the same assignee as in the present application, disclose phosphate dimer impurities made during the process of making of the compounds disclosed therein. Surprisingly, it has been found that compounds containing at least two phosphates binding two oxazolidinone-containing moieties, such as dimers of oxazolidinone-containing compounds have antibacterial activity similar to their dihydrogen monophosphate analog
active drug of Formula I is (5R)-3-[3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)-3-pyridinyl]phenyl]-5-(hydroxymethyl)-2-oxazolidinone, i.e.,

These active compounds have been disclosed in WO 05/058886 and US Patent Publication No. 20070155798, while processes for making these and related compounds have been disclosed in U.S. patent application Ser. No. 12/577,089 (filed Oct. 9, 2009), and a crystalline form of the phosphate ester and related salts of the above compound has been disclosed in U.S. patent application Ser. No. 12/699,864 (filed Feb. 3, 2010).
US Patent Publication No. 20070155798, recently disclosed a series of potently anti-bacterial oxazolidinones including

wherein R═H, PO(OH)2, and PO(ONa)2.
Cubist Announces Submission of New Drug Application for Investigational Antibiotic Tedizolid for Treatment of Serious Skin Infections
LEXINGTON, Mass.–(BUSINESS WIRE)– Cubist Pharmaceuticals, Inc. today announced that it has submitted a New Drug Application (NDA) to the U.S. Food and Drug Administration (FDA) for approval of its investigational antibiotic tedizolid phosphate (TR-701). Cubist is seeking approval of tedizolid phosphate for the treatment of acute bacterial skin and skin structure infections (ABSSSI). Tedizolid phosphate is a once daily oxazolidinone being developed for both intravenous (I.V.) and oral administration for the treatment of serious Gram-positive infections, including those caused by methicillin-resistant Staphylococcus aureus (MRSA).
http://www.drugs.com/nda/tedizolid_131023.html
…………………………………………………………..
Espinoza-González NA, Welsh O, de Torres NW, Cavazos-Rocha N, Ocampo-Candiani J, Said-Fernandez S, Lozano-Garza G, Choi SH, Vera-Cabrera L.
Molecules. 2008 Jan 11;13(1):31-40.
…………………………………………..
imp patents
|
12-3-2010
|
OXAZOLIDINONE CONTAINING DIMER COMPOUNDS, COMPOSITIONS AND METHODS TO MAKE AND USE
|
|
|
10-20-2010
|
Oxazolidinone derivatives
|
|
|
7-31-2009
|
NOVEL OXAZOLIDINONE DERIVATIVES
|
…………………………………………..
TEDIZOLID disodium salt
59 nos in
http://www.google.com/patents/US20130102523

 38 nos
38 nos
Tedizolid (formerly known as torezolid or TR-700) is the active hydroxymethyl oxazolidinone having the following formula:

Pharmaceutical prodrugs such as tedizolid phosphate (also referred to as TR-701, torezolid phosphate, and TR-701 “free acid” or FA) have the following formula:

The disodium salt of tedizolid phosphate, has the following structure:

Example 1 Preparation of the Phosphate Monohydrogen Diester, Formula III
In this and the following Examples, “Formula III” refers to a compound wherein Z is
Figure US20100305069A1-20101202-C00024
and M=OH.
A 1-L, three-neck round-bottom flask equipped with a magnetic stirrer, nitrogen inlet/outlet and thermocouple was charged with the compound of Formula Ia below (16.0 g, 0.0499 mol], THF (320 mL, 20 vol) and Et3N (21.9 g, 0.216 mol, 5.0 equiv.).
Figure US20100305069A1-20101202-C00025
POCl3 (3.31 g, 0.0216 mol, 0.5 equiv.) was added dropwise via syringe over 5 minutes. The reaction temperature was maintained below 25° C. The batch was aged for 16 hours at room temperature at which point HPLC analysis (XBridge, C18) indicated that the reaction was complete. The reaction vessel was then immersed in an ice-water bath and a 500-mL addition funnel charged with 320 mL of H2O was attached to the reaction vessel. When the temperature of the reaction reached 2.7° C., H2O was added drop wise over 30 minutes. The temperature of the reaction was maintained below 10° C. Upon completion of the H2O addition, the ice-water bath was removed and the batch was aged for 3 hours. The solution was transferred to a 2-L round-bottom flask and concentrated under reduced pressure on a rotary evaporator. After removal of most of the THF from the solution, the aqueous mixture was extracted with 5 1-L portions of CH2Cl2:MeOH (9:1). The CH2Cl2 layers were combined and concentrated to a dark oil. This crude material was purified on 200 g of silica gel, eluting with 10% MeOH/CH2Cl2 to 20% 2 N NH3 in MeOH/CH2Cl2. Fractions containing mostly the bis-ester (as judged by TLC Rf=0.3 eluting with 20% 2 N NH3 in MeOH/CH2Cl2) were combined and concentrated under reduced pressure on a rotary evaporator, during which time a white precipitate was observed. The flask containing the slurry was removed from the rotary evaporator and equipped with a magnetic stir bar and allowed to stir while cooling to room temperature over 3 hours, during which time the slurry thickened. The solid was filtered and dried in a vacuum oven at 45° C. for 16 hours to give 3.55 g of bis-ester as an off-white solid (20% yield). HPLC analysis (Method A): 99.0% (AUC), tR=16.3 min. This reaction was repeated and the combined lots of the compound of Formula III (6.7 g) were slurried in 100 mL of MeOH (15 vol). The slurry was heated to 40° C. for 30 minutes and then allowed to cool to room temperature over 1 hour. The off-white solid was filtered and dried in a vacuum oven at 40° C. for 16 hours to give 6.15 g of the compound of Formula III (92% yield). The 1H NMR analysis of the product was consistent with the assigned structure. HPLC analysis (Method A): 99.0% (AUC), tR=16.3 min.
Example 2 Preparation of the Diphosphate Dihydrogen Diester, Formula IV
In Examples 2-5, “Formula IV” refers to a compound wherein Z is
Figure US20100305069A1-20101202-C00026
n=0 and M=O-imidazolium salt.
A 250-mL 3-neck round-bottom flask equipped with a magnetic stirrer, nitrogen inlet/outlet and thermocouple was charged with the compound of Formula IIa below (5.0 g, 11.1 mmol), carbonyldiimidazole (890 mg, 5.55 mmol, 0.5 equiv.) and DMF (100 mL, 20 vol).
Figure US20100305069A1-20101202-C00027
The suspension was heated to 50° C. and held at that temperature for 4 hours at which point HPLC analysis (XBridge, C18) indicated that the reaction was complete. The reaction was filtered at 50° C. and dried in a vacuum oven at 50° C. for 24 hours to give 5.15 g of the imidazolium salt (i.e., the compound of Formula IV) as an off-white solid (98% yield). The 1H NMR analysis of the product was consistent with the assigned structure. HPLC analysis (Method A): 94.5% (AUC), tR=14.6 min.
TABLE 1
Method A (Waters XBridge C18 Column)
Time (min) Flow (mL/min) % A % B
0.0 1.0 98.0 2.0
15.0 1.0 5.0 95.0
25.0 1.0 5.0 95.0
27.0 1.0 98.0 2.0
30.0 1.0 98.0 2.0
A = 87% 25 mM ammonium bicarbonate solution in water/13% Acetonitrile
B = Acetonitrile
Wavelength = 300 nm
 disodium salt is TR 701
disodium salt is TR 701
……………………………………
Various oxazolidinone-containing compounds have been disclosed for use as antibiotics. For example, oxazolidinone-containing compounds have been described in U.S. patent application Ser. No. 10/596,412 (filed Dec. 17, 2004), and WO 04/048350, WO 03/022824 and WO 01/94342, which are incorporated herein by reference.
U.S. patent application Ser. No. 12/577,089 (filed Oct. 9, 2009) and U.S. patent application Ser. No. 12/699,864 (filed Feb. 3, 2010), which are assigned to the same assignee as in the present application, disclose phosphate dimer impurities made during the process of making of the compounds disclosed therein. Surprisingly, it has been found that compounds containing at least two phosphates binding two oxazolidinone-containing moieties, such as dimers of oxazolidinone-containing compounds have antibacterial activity similar to their dihydrogen monophosphate analog,
These active compounds have been disclosed in WO 05/058886 and US Patent Publication No. 20070155798, while processes for making these and related compounds have been disclosed in U.S. patent application Ser. No. 12/577,089 (filed Oct. 9, 2009), and a crystalline form of the phosphate ester and related salts of the above compound has been disclosed in U.S. patent application Ser. No. 12/699,864 (filed Feb. 3, 2010). The latter two applications are assigned to the same assignee as in the present application
………………………………………………………………………………………………………………………..
SYNTHESIS

DESCRIPTION OF COMPDS
10,
(R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-on (compound 10)

………………………………………………………………………………………………………………………….
18
Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-fluoromethyl oxazolidin-2-on (compound 18)
………………………………………………………………………………………………………………………………………………………….
33
(R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-methoxymethyl oxazolidin-2-on (compound 33)
…………………………………………………………………………………………………………………………………………..
59
(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl disodiumphosphate (compound 59)

………………………………………………………………………………………………………………………………………………………
72
mono-[(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl]phosphate (compound 72)

COMPLETE SYNTHESIS
Example 5
Preparation of 2-cyano-5-bromopyridine
In 1 L of dimethylformamide was dissolved 100 g of 2,5-dibromopyridine, 32 g of cupper cyanide and 17.8 g of sodium cyanide were added to the solution at room temperature and the solution was stirred at the temperature of 150° C. for 7 hours for reaction. After being cooled to room temperature, the reaction mixture was added with water and extracted with ethyl acetate. The organic layer was washed with brine, dehydrated, filtered and concentrated in vacuo. The title compound 54 g was obtained. Yield 70%.
1HNMR(CDCl3) δ 8.76(s,1H), 7.98(dd,1H), 7.58(dd,1H)
Example 6
Preparation of 2-(tetrazol-5-yl)-5-bromopyridine
10 g of 2-cyano-5-bromopyridine prepared in the Preparation example 5 was dissolved in 100 ml of dimethylformamide, 5.33 g of sodiumazide, and 4.4 g of ammonium chloride were added to the solution at room temperature, and the solution was stirred at the temperature of 110° C. for 3 hours for reaction. The reaction mixture was added with water and then was extracted with ethyl acetate. The organic layer, thus separated, was washed with brine, dehydrated, filtrated and concentrated in vacuo thereby to obtain 10.5 g of the title compound. Yield 85%.
Preparation Example 7 Preparation of 2-(1-methyltetrazol-5-yl)-5-bromopyridine and 2-(2-methyltetrazol-5-yl)-5-bromopyridine
10.5 g of 2-(tetrazol-5-yl)-5-bromopyridine prepared in the Preparation example 6 was dissolved in 100 ml of dimethylformamide. And then 6.5 g of sodium hydroxide was added to the solution and 9.3 g of iodomethane was slowly added to the solution at the temperature of 0° C. The solution was stirred for 6 hours at room temperature, added with water, extracted with ethyl acetate. And then the organic layer was washed with brine, dehydrated, filtrated, concentrated in vacuo and purified by column chromatography to obtain 4 g of 2-(1-methyltetrazol-5-yl)-5-bromopyridine and 5 g of 2-(2-methyltetrazol-5-yl)-5-bromopyridine.
1) 2-(1-methyltetrazol-5-yl)-5-bromopyridine
1HNMR(CDCl3) δ 8.77(t,1H), 8.23(dd,1H), 8.04(dd,1H), 4.46(s,3H)
2) 2-(2-methyltetrazol-5-yl)-5-bromopyridine
1HNMR(CDCl3) δ 8.80(t,1H), 8.13(dd,1H), 7.98(dd,1H), 4.42(s,3H)
Example 1
Preparation of N-Carbobenzyloxy-3-fluoroaniline
3-fluoroaniline 100 g was dissolved in 1 L of tetrahydrofuran (THF) and the solution was added with 150 g (1.8 mol) of sodium bicarbonate (NaHCO3). After being cooled to 0° C., the solution was slowly added with 154 ml of N-carbobenzyloxy chloride (CbzCl) for reaction. While the temperature was maintained at 0° C., the reaction mixture was let to react for 2 hours with stirring. Afterwards, the reaction was extracted with 0.5 L of ethyl acetate. The organic layer, after being separated, was washed with brine, dried over anhydrous magnesium sulfate (MgSO4) and concentrated in vacuo. The residue was washed twice with n-hexane to afford the title compound as white crystal. 132 g. Yield 85%.
Example 2
Preparation of (R)-3-(3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol
132 g of N-carbobenzyloxy-3-fluoroaniline 132 g prepared in the Preparation example 1 was dissolved in 1.3 L of tetrahydrofuran and the solution was cooled to −78° C. 370 ml of n-buthyllitium (n-BuLi, 1.6M/n-hexane) was slowly added to the solution in a nitrogen atmosphere, followed by stirring for 10 min. And 84 ml of (R)-(−)-glycidylbuthylate was slowly added to the reaction mixture, stirred at the same temperature for 2 hours and allowed to react for 24 hours at room temperature. After completion of the reaction, the solution was added with ammonium chloride (HH4Cl) solution and extracted with 0.5 L of ethyl acetate at room temperature. The organic layer, thus separated, was washed with brine, dried over anhydrous magnesium sulfate and concentrated in vacuo. The residue was dissolved in 100 ml of ethyl acetate and washed with n-hexane to give white crystals, which were purified to the title compound. 80 g. Yield 70%.
1H NMR (DMSO-d6) δ 7.85(t,1H), 7.58(dd,1H), 7.23(dd,1H), 4.69(m,1H), 4.02 (t,1H), 3.80(dd,1H), 3.60(br dd,2H).
Example 3
Preparation of (R)-3-(4-iodo-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol
In 300 ml of acetonitryl was dissolved 30 g of (R)-3-(3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol prepared in the Preparation example 2, and 46 g of trifluoroacetic acid silver salt (CF3COOAg) and 43 g of iodide were added to the solution. After being stirred for one day at room temperature, the solution was added with water and was extracted with ethyl acetate. The organic layer, thus separated, was washed with brine and dehydrated. And then the residue was filtered, concentrated in vacuo and dried thereby to form the title compound 44 g. Yield 94%.
1H NMR (DMSO-d6) δ 7.77(t,1H), 7.56(dd,1H), 7.20(dd,1H), 5.20(m,1H), 4.70 (m,1H), 4.07(t,1H), 3.80(m,1H), 3.67(m,2H), 3.56(m,3H)
Example 4
Preparation of (R)-3-(4-tributhylstannyl-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol
In 660 ml of 1,4-dioxan was dissolved 50 g of (R)-3-(4-iodo-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol prepared in the Preparation example 3, 52 g of hexabutylditin ((Bu3Sn)2) and 9.3 g of dichlorobistriphenylphosphinpalladium were added into the solution, and stirred for 2 hours. The solution was filtered using celite and concentrated in vacuo. The residue was purified by column chromatography and 45 g of the title compound was formed.
1H NMR (DMSO-d6) δ 7.74(m,3H), 5.20(t,1H), 4.71(m,1H), 4.08(t,1H), 3.82(dd,1H), 3.68(m,1H), 3.52(m,1H), 1.48(m, 6H), 1.24(m, 6H), 1.06(m,6H), 0.83(t,9H)
COMPD 10
Example 1 Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-on (compound 10)
In 150 ml of 1-methyl-2-pyrrolidone was dissolved 37 g of (R)-3-(4-tributhylstannyl-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol. The solution was added with 19.7 g of 2-(2-methyltetrazol-5-yl)-5-bromopyridine, 10.44 g of lithium chloride and 2.9 g of dichlorobistriphenylphospine palladium(II) at room temperature and then stirred at the temperature of 120° C. for 4 hours. The reaction mixture was added with water and then extracted with ethyl acetate. The organic layer, thus separated, was washed with brine, dehydrated, filtrated, concentrated in vacuo and purified by column chromatography to provide 8 g of the title compound. Yield 26%.
1H NMR (DMSO-d6) δ 8.90(s,1H), 8.18(m,2H), 7.70(m,2H), 7.49(dd,1H), 5.25(t,1H), 4.74(m,1H), 4.46(s,3H), 4.14(t,1H), 3.88(dd,1H), 3.68(m,1H), 3.58 (m,1H)
COMPD 18
Example 28 Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-fluoromethyl oxazolidin-2-on (compound 18)
In 5 ml of methylenchloride was dissolved 100 mg of the compound 10. The solution was added with 43 mg of diethylaminosulfurtrifloride (DAST) and 0.078 ml of triethylamine and then stirred for 24 hours. After being concentrating, the reaction mixture was purified by column chromatography to obtain the title compound 75 mg. Yield 75%.
1H NMR (DMSO-d6) δ 8.91(s,1H), 8.19(m,2H), 7.74(t,1H), 7.66(dd,1H) 7.49 (dd,1H), 5.06(m,1H), 4.89(m,2H), 4.46(s,3H), 4.23(t,1H), 3.95(dd,1H)
COMPD 33
Example 37 Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-methoxymethyl oxazolidin-2-on (compound 33)
In 10 ml of methanol was dissolved 400 mg of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-methansulfonyloxymethyl oxazolidin-2-on prepared in the secondary step of the Example 24. The solution was added with 90 mg of sodium methoxide at room temperature and then stirred for one day at room temperature. The solution was extracted with ethyl acetate and the organic layer, thus separated, was washed with water and brine. The organic layer was dehydrated, filtered, concentrated in vacuo and purified by column chromatography to provide the title compound 200 mg. Yield 58%.
1H NMR(CDCl3) δ 8.90(s,1H), 8.29(d,1H), 8.04(d,1H), 7.61(dd,1H), 7.58 (t,1H), 7.38(dd,1H), 4.80(m,1H), 4.45(s,3H), 4.08(t,1H), 3.96(dd,1H), 3.67 (m,2H), 3.43(s,3H)
COMPD 59
Example 58 Preparation of mono-[(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl]phosphate (compound 72) and (R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl disodiumphosphate (compound 59)
1. The Primary Step
In 10 ml of mixture solvent (tetrahydrofuran:methylenchloride=1:1) was dissolved 1 g of compound 10. The solution was added with 0.6 g of tetrazole and 2.3 g of di-tetrabutyl diisoprophylphosphoamidite and stirred for 15 hours at room temperature. The reaction mixture was refrigerated to −78° C., added with 0.7 g of metachloroperbenzoic acid and stirred for 2 hours. After being cooling to −78° C., the reaction mixture was added with metachloroperbenzoic acid (0.7 g). When the reaction mixture was stirred for 2 hours, the temperature of the reaction mixture was raised to room temperature. The reaction mixture was then added with ethyl acetate. The organic layer, thus separated, was washed with sodium bisulfate, sodium bicarbonate and brine, dehydrated, filtered and concentrated in vacuo, followed by purification with column chromatography thereby to provide (R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl phosphoric acid ditetrabuthylester (0.71 g, 71%).
1H NMR (DMSO-d6) δ 8.90(s,1H), 8.18(m,2H), 7.74(t,1H), 7.68 (dd,1H), 7.49(dd,1H), 4.98(m,1H), 4.46(s,3H), 4.23(t,1H), 4.18(m,1H), 4.09(m,1H), 3.89 (dd,1H), 1.39(s,9H), 1.38(s,9H)
The crystal prepared the above method was dissolved in a mixture of methanol and chloroform. And then the solution added with 3.4 ml of sodium methoxide (0.3M methanol solution) at the room temperature and stirred for 10 hours. The reaction mixture was concentrated to prepare the residue. The residue was crystallized and filtered thereby to obtain the title compound (compound 59) 300 mg.
1H NMR (D2O) δ 8.27(s,1H), 7.56(dd,2H), 7.06(m,2H), 6.90(m,1H), 4.79 (m,1H), 4.63(s,3H), 3.90(m,4H)
COMPD 72
The Secondary Step
In 30 ml of methylenchloride was dissolved the compound (0.7 g) in the Primary Step. The solution was added with 15 ml of trifluoroacetic acid and then stirred for 1 hour at room temperature. The reaction mixture was concentrated in vacuo to prepare the residue. The residue was crystallized with ethanol and ethyl ether to obtain mono-[(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl]phosphate (compound 72) 400 mg.
1H NMR (DMSO-d6) δ 8.92(s,1H), 8.20(m,2H), 7.74(t,1H), 7.66(dd,1H), 7.500(dd,1H), 4.95 (m,1H), 4.46(s,3H), 4.21(t,1H), 4.05(m,2H), 3.91(dd,1H)
………………………………………………………
IMPURITIES
| Organic Impurities in TR-701 FA Drug Substance | |
| Impurity | |
| ‘Name’ | Structure and Chemical Name |
| Rx600013 ‘Des-methyl TR- 701’ |
 |
| dihydrogen ((5R)-3-{3-fluoro-4-[6-(2H-1,2,3,4-tetrazol-5- | |
| yl)-3-pyridinyl]phenyl}-2-oxo-1,3-oxazolan-5-yl)methyl | |
| phosphate | |
| Rx600024 ‘Pyrophosphate’ |
 |
| trihydrogen ((5R)-3-{3-fluoro-4-[6-(1-methyl-1H-1,2,3,4- | |
| tetraazol-5-yl)-3-pyridinyl]phenyl}-2-oxo-1,3-oxazolan-5- | |
| yl)methyl pyrophosphate | |
| Rx600014 ‘Ring opened’ |
 |
| dihydrogen 3-{3-fluoro-4-[6-(2-methyl-2H-1,2,3,4-tetraazol-5- | |
| yl)-3-pyridinyl]aniline}-2-hydroxypropyl phosphate | |
| Rx600023 ‘Me-isomer’ |
 |
| dihydrogen ((5R)-3-{3-fluoro-4-[6-(1-methyl-1H-1,2,3,4- | |
| tetraazol-5-yl)-3-pyridinyl]phenyl}-2-oxo-1,3-oxazolan-5- | |
| yl)methyl phosphate | |
| Rx600025 ‘Overalkylated- phosphorylated impurity’ |
  |
| (R)-1-((3-(3-fluoro-4-(6-(2-methyl-2H-tetrazol-5- | |
| yl)pyridin-3-yl)phenyl)-2-oxooxazolidin-5-yl)methoxy)-3- | |
| hydroxypropan-2-yl dihydrogen phosphate; | |
| (R)-3-((3-(3-fluoro-4-(6-(2-methyl-2H-tetrazol-5- | |
| yl)pyridin-3-yl)phenyl)-2-oxooxazolidin-5-yl)methoxy)-2- | |
| hydroxypropyl dihydrogen phosphate | |
| Rx600020 ‘Dimer impurity’ |
 |
| dihydrogen bis-O-O′-[(5R)-3-{3-fluoro-4-[6-(2-methyl- | |
| 2H-1,2,3,4-tetrazol-5-yl)-3-pyridinyl]phenyl}-2-oxo-1,3- | |
| oxazolidin-5-yl]methyl pyrophosphate | |
| Rx600026 “Chloro” |
 |
| (R)-5-(chloromethyl)-3-(3-fluoro-4-(6-(2-methyl-2H- | |
| tetrazol-5-yl)pyridin-3-yl)phenyl)oxazolidin-2-one | |
| Rx600001 TR-700 |
 |
| 5R)-3-{3-Fluoro-4-[6-(2-methyl-2H-1,2,3,4-tetrazol-5-yl)- | |
| pyridin-3-yl]-phenyl}-5-hydroxymethyl-1,3-oxazolidin-2-one | |
| Rx600022 ‘Bis phosphate’ |
 |
| hydrogen bis-O-O′-[(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-1,2,3,4- | |
| tetrazol-5-yl)-3-pyridinyl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methyl | |
| phosphate | |
| Rx600042 |
 |
| 3-{[(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3- | |
| yl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methoxy}-2-hydroxypropyl | |
| [(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3- | |
| yl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methyl hydrogen phosphate | |
| Rx600043 |
 |
| 2-{[(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3- | |
| yl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methoxy}-1-hydroxyethyl | |
| [(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3- | |
| yl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methyl hydrogen phosphate | |
……………………………………………..
| US4128654 | 10 Feb 1978 | 5 Dec 1978 | E. I. Du Pont De Nemours And Company | 5-Halomethyl-3-phenyl-2-oxazolidinones |
| US4250318 | 9 Aug 1978 | 10 Feb 1981 | Delalande S.A. | Novel 5-hydroxymethyl oxazolidinones, the method of preparing them and their application in therapeutics |
| US4340606 | 23 Oct 1980 | 20 Jul 1982 | E. I. Du Pont De Nemours And Company | 3-(p-Alkylsulfonylphenyl)oxazolidinone derivatives as antibacterial agents |
| US4461773 | 5 Jan 1984 | 24 Jul 1984 | E. I. Dupont De Nemours And Company | P-Oxooxazolidinylbenzene compounds as antibacterial agents |
| US4476136 | 24 Feb 1982 | 9 Oct 1984 | Delalande S.A. | Aminomethyl-5 oxazolidinic derivatives and therapeutic use thereof |
| US4948801 | 29 Jul 1988 | 14 Aug 1990 | E. I. Du Pont De Nemours And Company | Aminomethyloxooxazolidinyl arylbenzene derivatives useful as antibacterial agents |
| US5523403 | 22 May 1995 | 4 Jun 1996 | The Upjohn Company | Tropone-substituted phenyloxazolidinone antibacterial agents |
| US5565571 | 28 Apr 1994 | 15 Oct 1996 | The Upjohn Company | Substituted aryl- and heteroaryl-phenyloxazolidinones |
| US5652238 | 27 Sep 1994 | 29 Jul 1997 | Pharmacia & Upjohn Company | Esters of substituted-hydroxyacetyl piperazine phenyl oxazolidinones |
| US5688792 | 16 Aug 1994 | 18 Nov 1997 | Pharmacia & Upjohn Company | Substituted oxazine and thiazine oxazolidinone antimicrobials |
| US6365751 | 17 Apr 2001 | 2 Apr 2002 | Zeneca Ltd. | Antibiotic oxazolidinone derivatives |
| US6627646 * | 17 Jul 2001 | 30 Sep 2003 | Sepracor Inc. | Norastemizole polymorphs |
| US6689779 | 18 May 2001 | 10 Feb 2004 | Dong A Pharm. Co., Ltd. | Oxazolidinone derivatives and a process for the preparation thereof |
| US7129259 | 1 Dec 2004 | 31 Oct 2006 | Rib-X Pharmaceuticals, Inc. | Halogenated biaryl heterocyclic compounds and methods of making and using the same |
| US7141583 | 23 Apr 2001 | 28 Nov 2006 | Astrazeneca Ab | Oxazolidinone derivatives with antibiotic activity |
| US7144911 | 24 Dec 2003 | 5 Dec 2006 | Deciphera Pharmaceuticals Llc | Anti-inflammatory medicaments |
| US7202257 | 6 Jul 2004 | 10 Apr 2007 | Deciphera Pharmaceuticals, Llc | Anti-inflammatory medicaments |
| US7396847 | 9 Sep 2002 | 8 Jul 2008 | Astrazeneca Ab | Oxazolidinone and/or isoxazoline as antibacterial agents |
| US7462633 | 29 Jun 2004 | 9 Dec 2008 | Merck & Co., Inc. | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| US7473699 | 25 Feb 2003 | 6 Jan 2009 | Astrazeneca Ab | 3-cyclyl-5-(nitrogen-containing 5-membered ring)methyl-oxazolidinone derivatives and their use as antibacterial agents |
| US7498350 | 24 Nov 2003 | 3 Mar 2009 | Astrazeneca Ab | Oxazolidinones as antibacterial agents |
| US7816379 | 17 Dec 2004 | 19 Oct 2010 | Dong-A Pharm. Co., Ltd. | Oxazolidinone derivatives |
| US20020115669 | 29 Aug 2001 | 22 Aug 2002 | Wiedeman Paul E. | Oxazolidinone chemotherapeutic agents |
| US20030166620 | 18 May 2001 | 4 Sep 2003 | Jae-Gul Lee | Novel oxazolidinone derivatives and a process for the preparation thereof |
| US20040180906 | 24 Dec 2003 | 16 Sep 2004 | Flynn Daniel L | Anti-inflammatory medicaments |
| US20050038092 | 29 Jun 2004 | 17 Feb 2005 | Yasumichi Fukuda | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| US20050107435 | 9 Sep 2002 | 19 May 2005 | Gravestock Michael B. | Oxazolidinone and/or isoxazoline as antibacterial agents |
| US20050288286 | 6 Jul 2004 | 29 Dec 2005 | Flynn Daniel L | Anti-inflammatory medicaments |
| US20060116386 | 24 Nov 2003 | 1 Jun 2006 | Astrazeneca Ab | Oxazolidinones as antibacterial agents |
| US20060116400 | 24 Nov 2003 | 1 Jun 2006 | Astrazeneca Ab | Oxazolidinone and/or isoxazoline derivatives as antibacterial agents |
| US20060270637 | 24 Feb 2004 | 30 Nov 2006 | Astrazeneca Ab | Hydroxymethyl substituted dihydroisoxazole derivatives useful as antibiotic agents |
| US20070155798 | 17 Dec 2004 | 5 Jul 2007 | Dong-A Pharm. Co., Ltd. | Novel oxazolidinone derivatives |
| US20070185132 | 29 Jun 2004 | 9 Aug 2007 | Yasumichi Fukuda | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereo |
| US20070191336 | 23 Dec 2004 | 16 Aug 2007 | Flynn Daniel L | Anti-inflammatory medicaments |
| US20070203187 | 22 Jan 2007 | 30 Aug 2007 | Merck & Co., Inc. | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| US20070208062 | 24 May 2005 | 6 Sep 2007 | Astrazeneca Ab | 3-(4-(2-dihydroisoxazol-3-ylpyridin-5-yl)phenyl)-5-triazol-1-ylmethyloxazolidin-2-one derivatives as mao inhibitors for the treatment of bacterial infections |
| US20080021012 | 24 May 2005 | 24 Jan 2008 | Astrazeneca Ab | 3-[4-{6-Substituted Alkanoyl Pyridin-3-Yl}-3-Phenyl]-5-(1H-1,2,3-Triazol-1-Ylmethyl)-1,3-Oxazolidin-2-Ones As Antibacterial Agents |
| US20080021071 | 24 May 2005 | 24 Jan 2008 | Astrazeneca Ab | 3-{4-(Pyridin-3-Yl) Phenyl}-5-(1H-1,2,3-Triazol-1-Ylmethyl)-1,3-Oxazolidin-2-Ones as Antibacterial Agents |
| US20080064689 | 24 May 2004 | 13 Mar 2008 | Astrazeneca Ab | 3-[4-(6-Pyridin-3-Yl)-3-Phenyl] -5-(1H-1,2,3-Triazol-1-Ylmethyl)-1,3-Oxazolidin-2-Ones as Antibacterial Agents |
| US20090018123 | 19 Jun 2006 | 15 Jan 2009 | Milind D Sindkhedkar | Oxazolidinones Bearing Antimicrobial Activity Composition and Methods of Preparation |
| US20090192197 | 16 Sep 2008 | 30 Jul 2009 | Dong-A Pharm. Co., Ltd. | Novel oxazolidinone derivatives |
| US20100093669 | 9 Oct 2009 | 15 Apr 2010 | Trius Therapeutics | Methods for preparing oxazolidinones and compositions containing them |
| US20100227839 | 3 Feb 2010 | 9 Sep 2010 | Trius Therapeutics | Crystalline form of r)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin- 5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate |
| AU2004299413A1 | Title not available | |||
| AU2009200606A1 | Title not available | |||
| CA2549062A1 | 17 Dec 2004 | 30 Jun 2005 | Chong Hwan Cho | Novel oxazolidinone derivatives |
| CN101982468A | 17 Dec 2004 | 2 Mar 2011 | 东亚制药株式会社 | Novel oxazolidinone derivatives and pharmaceutical compositions comprising the derivatives |
| EP0312000A1 | 12 Oct 1988 | 19 Apr 1989 | The Du Pont Merck Pharmaceutical Company | Aminomethyl oxooxazolidinyl aroylbenzene derivatives useful as antibacterial agents |
| EP0352781A2 | 27 Jul 1989 | 31 Jan 1990 | The Du Pont Merck Pharmaceutical Company | Aminomethyloxooxazolidinyl arylbenzene derivatives useful as antibacterial agents |
| EP1699784A1 | 17 Dec 2004 | 13 Sep 2006 | Dong-A Pharmaceutical Co., Ltd. | Novel oxazolidinone derivatives |
| EP2305657A2 | 17 Dec 2004 | 6 Apr 2011 | Dong-A Pharmaceutical Co., Ltd. | Oxazolidinone derivatives |
| EP2435051A1 | 27 May 2010 | 4 Apr 2012 | Trius Therapeutics | Oxazolidinone containing dimer compounds, compositions and methods to make and use |
| IN236862A1 | Title not available | |||
| JPS5799576A | Title not available | |||
| KR20110071107A | Title not available | |||
| NZ547928A | Title not available | |||
| NZ575842A | Title not available | |||
| WO1993009103A1 | 5 Oct 1992 | 13 May 1993 | Upjohn Co | Substituted aryl- and heteroarylphenyloxazolidinones useful as antibacterial agents |
| WO1993023384A1 | 21 Apr 1993 | 25 Nov 1993 | Michael Robert Barbachyn | Oxazolidinones containing a substituted diazine moiety and their use as antimicrobials |
| WO1995007271A1 | 16 Aug 1994 | 16 Mar 1995 | Michael R Barbachyn | Substituted oxazine and thiazine oxazolidinone antimicrobials |
| WO1995014684A1 | 27 Sep 1994 | 1 Jun 1995 | Michel R Barbachyn | Esters of substituted-hydroxyacetyl piperazine phenyl oxazolidinones |
| WO2001094342A1 | 18 May 2001 | 13 Dec 2001 | Cho Jong Hwan | Novel oxazolidinone derivatives and a process for the preparation thereof |
| WO2002081470A1 | 3 Apr 2002 | 17 Oct 2002 | Astrazeneca Ab | Oxazolidinones containing a sulfonimid group as antibiotics |
| WO2003022824A1 | 9 Sep 2002 | 20 Mar 2003 | Astrazeneca Ab | Oxazolidinone and/or isoxazoline as antibacterial agents |
| WO2003035648A1 | 23 Oct 2002 | 1 May 2003 | Astrazeneca Ab | Aryl substituted oxazolidinones with antibacterial activity |
| WO2003047358A1 | 2 Dec 2002 | 12 Jun 2003 | Vaughan Leslie Crow | Cheese flavour ingredient and method of its production |
| WO2003072575A1 | 25 Feb 2003 | 4 Sep 2003 | Astrazeneca Ab | 3-cyclyl-5-(nitrogen-containing 5-membered ring) methyl-oxazolidinone derivatives and their use as antibacterial agents |
| WO2003072576A2 | 25 Feb 2003 | 4 Sep 2003 | Astrazeneca Ab | Oxazolidinone derivatives, processes for their preparation, and pharmaceutical compositions containing them |
| WO2004048350A2 | 24 Nov 2003 | 10 Jun 2004 | Astrazeneca Ab | Oxazolidinones as antibacterial agents |
| WO2004083205A1 | 16 Mar 2004 | 30 Sep 2004 | Astrazeneca Ab | Antibacterial 1, 3- oxazolidin -2- one derivatives |
| WO2005005398A2 | 29 Jun 2004 | 20 Jan 2005 | Yasumichi Fukuda | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| WO2005051933A1 | 23 Nov 2004 | 9 Jun 2005 | Vijay Kumar Kaul | An improved process for the synthesis of 4-(4-benzyloxy-carbonylamino-2-fluorophenyl)-piperazine-1-carboxylic acid tert-butyl ester, a key intermediate for oxazolidinone antimicrobials and compounds prepared thereby |
| WO2005058886A1 | 17 Dec 2004 | 30 Jun 2005 | Dong A Pharm Co Ltd | Novel oxazolidinone derivatives |
| WO2005116017A1 | 24 May 2005 | 8 Dec 2005 | Astrazeneca Ab | Process for the preparation of aryl substituted oxazolidinones as intermediates for antibacterial agents |
| WO2006038100A1 | 6 Oct 2005 | 13 Apr 2006 | Ranbaxy Lab Ltd | Oxazolidinone derivatives as antimicrobials |
| WO2007023507A2 | 19 Jun 2006 | 1 Mar 2007 | Milind D Sindkhedkar | Oxazolidinones bearing antimicrobial activity composition and methods of preparation |
| WO2007138381A2 | 13 Oct 2006 | 6 Dec 2007 | Delorme Daniel | Phosphonated oxazolidinones and uses thereof for the prevention and treatment of bone and joint infections |
| WO2010042887A2 | 9 Oct 2009 | 15 Apr 2010 | Trius Therapeutics | Methods for preparing oxazolidinones and compositions containing them |
| WO2010091131A1 | 3 Feb 2010 | 12 Aug 2010 | Trius Therapeutics | Crystalline form of r)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate |
| WO2010138649A1 | 27 May 2010 | 2 Dec 2010 | Trius Therapeutics, Inc. | Oxazolidinone containing dimer compounds, compositions and methods to make and use |
……………………………………………………………………………………….
THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007
TOSEDOSTAT ….An aminopeptidase inhibitor with antineoplastic activity.

TOSEDOSTAT
An aminopeptidase inhibitor with antineoplastic activity.
- CHR 2797
- CHR-2797
- Tosedostat
- UNII-KZK563J2UW
- BB-76163Vernalis (Originator)
| CAS No. | 238750-77-1 |
| Chemical Name: | Tosedostat |
| Synonyms: | BB-76163;Chr-2797;tosedostat;CHR2797 (Tosedostat);Tosedostat (CHR2797);α-[[(2R)-2-[(1S)-1-Hydroxy-2-(hydroxyamino)-2-oxoethyl]-4-methyl-1-oxopentyl]amino]-benzeneaceticacidcyclopentlyester;alpha-[[(2R)-2-[(1S)-1-Hydroxy-2-(hydroxyamino)-2-oxoethyl]-4-methyl-1-oxopentyl]amino]benzeneacetic acid cyclopentyl ester;Benzeneacetic acid, alpha-(((2R)-2-((1S)-1-hydroxy-2-(hydroxyamino)-2-oxoethyl)-4-methyl-1-oxopentyl)amino)-, cyclopentyl ester, (alphas)- |
| Molecular Formula: | C21H30N2O6 |
| Formula Weight: | 406.47 |
CHR-2797 is an oral, once-daily experimental cancer therapy in phase II clinical development at Chroma Therapeutics for the oral treatment of refractory acute myeloid leukemia in elderly patients. It is also in early clinical development for the treatment of refractory solid tumors alone or in combination with chemotherapy.
No recent development has been reported for phase I/II studies evaluating CHR-2797 as monotherapy in hematologic/blood cancer. A phase I/II clinical trial of the compound in combination with erlotinib for non-small cell lung cancer was terminated in 2010 due to very poor recruitment of patients to the study.
Cell Therapeutics is also conducting phase II clinical trials of the compound for the treatment of myelodysplasia and acute myeloid leukemia.
CHR- 2797 is an inhibitor of aminopeptidases and has demonstrated strong preclinical efficacy as monotherapy in addition to demonstrating strong synergy with a number of leading cancer therapies in a range of cancer cells. It was originally licensed from Vernalis, where it was being evaluated for its potential in treating multiple sclerosis; however development in this indication has been discontinued.
In 2008, orphan drug designation was assigned to CHR-2797 in the U.S. for the treatment of acute myeloid leukemia. In 2011, the compound was licensed to Cell Therapeutics by Chroma Therapeutics in Central America, North America and South America for exclusive marketing and codevelopment for the oral treatment of blood-related cancers and other cancers.
In corporate news, biopharmaceutical company Cell Therapeutics, Inc. (CTIC) was up more than 6% and near 52 week highs after saying Thursday that the U.S. FDA has removed the partial clinical hold on tosedostat and all studies underway have been allowed to continue. Tosedostat is under development for the treatment of blood-related cancers. It is currently being studied in Phase 2 trials in elderly patients with newly diagnosed and relapsed acute myeloid leukemia and high-risk myelodysplastic syndromes.

Tosedostat is a proprietary orally bioavailable inhibitor of the M1 family of aminopeptidases with potential antineoplastic activity.
Tosedostat is converted intracellularly into a poorly membrane-permeable active metabolite (CHR-79888) which inhibits the M1 family of aminopeptidases, particularly puromycin-sensitive aminopeptidase (PuSA), and leukotriene A4 (LTA4) hydrolase; inhibition of these aminopeptidases in tumor cells may result in amino acid deprivation, inhibition of protein synthesis due to a decrease in the intracellular free amino acid pool, an increase in the level of the proapoptotic protein Noxa, and cell death.
Noxa is a member of the BH3 (Bcl-2 homology 3)-only subgroup of the proapoptotic Bcl-2 (B-cell CLL/lymphoma 2) protein family
Cell Therapeutics announced that it has received notification from the U.S. Food and Drug Administration (FDA) that the partial clinical hold on tosedostat (IND 075503) has been removed and all studies underway may continue. Tosedostat is a first-in-class selective inhibitor of aminopeptidases, which are required by tumor cells to provide amino acids necessary for growth and tumor cell survival, and is under development for the treatment of blood-related cancers.
Tosedostat is currently being studied in the United States and European Union in investigator-sponsored and cooperative group-sponsored Phase 2 trials in elderly patients with newly diagnosed and relapsed acute myeloid leukemia (AML) and high-risk myelodysplastic syndromes (MDS).
“We are pleased that the FDA has responded favorably to the tosedostat clinical trial data provided and removed the partial clinical hold to allow further development of tosedostat in ongoing and future studies,” said John Pagel, MD, PhD, Associate Member, Clinical Research Division, Fred Hutchinson Cancer Research Center; Associate Professor, Medical Oncology Division, University of Washington School of Medicine; and Principal Investigator in the tosedostat first-line AML/MDS trial.
Recently, WO 93/20047 disclosed a class of hydroxamic acid based MMP inhibitors which also are active in inhibiting TNF production.
As mentioned above, MMP inhibitors have been proposed with hydroxamic acid or carboxylic acid zinc binding groups. The following patent publications disclose hydroxamic acid-based MMP inhibitors:
US 4599361 (Searle) EP-A-0236872 (Roche) EP-A-0274453 (Bellon) WO 90/05716 (British Bio-technology) WO 90/05719 (British Bio-technology) WO 91/02716 (British Bio-technology) EP-A-0489577 (Celltech) EP-A-0489579 (Celltech) EP-A-0497192 (Roche) WO 92/13831 (British Bio-technology) WO 92/17460 (SmithKline Beecham) WO 92/22523 – (Research Corporation Technologies) WO 93/09090 (Yamanouchi) WO 93/09097 (Sankyo) WO 93/20047 (British Bio-technology) WO 93/24449 (Celltech) WO 93/24475 (Celltech) EP-A-0574758 (Roche) The following patent publications disclose carboxylic acid-based MMP inhibitors:
EP-A-0489577 (Celltech) EP-A-0489579 (Celltech) WO 93/24449 (Celltech) WO 93/24475 (Celltech)
|
|
|
TOSEDOSTAT
| WO1996033166A1 * | 17 Apr 1996 | 24 Oct 1996 | Du Pont Merck Pharma | Hydroxamic and carboxylic acids as metalloprotease inhibitors |
| WO1998011063A1 * | 8 Sep 1997 | 19 Mar 1998 | British Biotech Pharm | Cytostatic hydroxamic acid derivatives |
| GB2268934A * | Title not available |
| US5652262 * | 14 mar 1994 | 29 lug 1997 | British Biotech Pharmaceutical, Ltd. | Hydroxamic acid derivatives as metalloproteinase inhibitors |
| US5821262 * | 4 ott 1994 | 13 ott 1998 | British Biotech Pharmaceuticals Limited | Hydroxamic acid derivatives as inhibitors of cytokine production |
| US5861436 * | 29 apr 1997 | 19 gen 1999 | British Biotech Pharmaceuticals Limited | Hydroxamic acid derivatives as metalloproteinase inhibitors |
| EP0423943A2 | 19 set 1990 | 24 apr 1991 | Beecham Group p.l.c. | Use of collagenase inhibitors in the treatment of demyelinating diseases, in particular multiple sclerosis |
| JPH03157372A | Titolo non disponibile | |||
| WO1997049674A1 | 20 giu 1997 | 31 dic 1997 | Francesca Abrate | Matrix metalloproteinase inhibitors |
| WO1998011063A1 | 8 set 1997 | 19 mar 1998 | British Biotech Pharm | Cytostatic hydroxamic acid derivatives |
| WO1999040910A1 | 27 gen 1999 | 19 ago 1999 | Andrew Paul Ayscough | Anti-inflammatory agents |
| WO1999044602A1 | 5 mar 1999 | 10 set 1999 | British Biotech Pharm | Inflammatory cell inhibitors |
| WO1999046241A1 | 12 mar 1998 | 16 set 1999 | British Biotech Pharm | Cytostatic agents |
| WO2000044373A1 * | Jan 27, 2000 | Aug 3, 2000 | Raymond Paul Beckett | Antibacterial hydroxamic acid derivatives |
| US6545051 | Jan 27, 2000 | Apr 8, 2003 | British Biotech Pharmaceuticals, Ltd. | Antibacterial hydroxamic acid derivatives |
Drugs Fut 2009, 34(2): 115
PLoS One (2013), 8(2), e57641.
WO 1999046241
WO 1995019956
WO 1998011063
US 6462023
US 20100260674
WO 2000044373
WO 9940910
NMR
http://file.selleckchem.com/downloads/nmr/S152202-CHR-2797-NMR-Selleck.pdf
Anti-Metastatic and Anti-Invasive Agents Compounds which have the property of inhibiting the action of the metalioproteinase enzymes involved in connective tissue breakdown and remodelling, such as fibroblast collagenase (Type 1 ), PMN-collagenase, 72 kDa-gelatinase, 92 kDa- gelatinase, stromelysin, stromelysin-2 and PUMP-1 (known as “matrix metalloproteinases”, and herein referred to as MMPs) have been proposed and are being tested in the clinic for the treatment of solid tumours. Cancer cells are particularly adept at utilising the MMPs to achieve rapid remodelling of the extracellular matrix, thereby providing space for tumour expansion and permitting metastasis. MMP inhibitors should minimise these processes and thus slow or prevent cancer progression.
In view of the rapid emergence of multidrug-resistant bacteria, the development of antibacterial agents with novel modes of action that are effective against the growing number of resistant bacteria, particularly the vancomycin resistant enterococci and β-lactam antibiotic-resistant bacteria, such as methicillin-resistant Staphylocccus aureus, is of utmost importance.
The natural antibiotic actinonin (see for example J. C. S Perkin I, 1975, 819) is a hydroxamic acid derivative of Structure (A):

In ddition to actinonin, various structural analogues of actinonin have also been shown to have antibacterial activity (see for example Broughton et al. (Devlin et al. Journal of the Chemical Society. Perkin Transactions 1 (9):830-841, 1975; Broughton et al. Journal of the Chemical Society. Perkin Transactions 1 (9):857-860, 1975).
The matlystatin group of compounds, share a number of structural similarities with actinonin. Both are peptidic molecules with functional hydroxamic acid metal binding groups (Ogita et al., J. Antibiotics. 45(11):1723-1732; Tanzawa et al., J. Antibiotics. 45(11):1733-1737; Haruyama et al., J. Antibiotics. 47(12):1473-1480; Tamaki et al., J. Antibiotics. 47(12):1481-1492).
………………………………………………………….
EXAMPLE 44 2-[2R-(S-Hydroxy-hydroxycarbamoyl-methyl-pentanoylamine]-2-phenyl-ethanoic acid cyclopentyl ester

The above compound was prepared using procedures similar to those described in example 8 using phenylglycine cyclopentyl ester.
Diastereoisomer A
1H-NMR; δ (MeOD), 7.4-7.29 (5H, m), 5.43 (1H, s), 5.2-5.14 (1H, m), 4.02 (1H, d, J=6.9 Hz), 2.94-2.85 (1H, m), 1.91-1.34 (10H, bm), 1.25-1.14 (1H, m) and 0.86 (6H, dd, J=6.5, 11 5 Hz).
13C-NMR; δ (MeOD), 175.6, 171.8, 171.4, 137.8, 129.8, 129.4, 128.6, 80.0, 73.2, 58.5, 49.2, 39.1, 33.3, 33.3, 26.8, 24.5, 24.4, 23.7 and 22.1.
Diastereoisomer B
1H-NMR; 8 (MeOD), 7.33-7.19 (5H, m), 5.3 (1H, s), 5.11-5.06 (1H, m), 3.81 (1H, d, J=7.3 Hz), 2.83-2.74 (lH, m), 1.83-1.45 (10H, bm), 1.12-1.03 (lH, m) and 0.88-0.81 (6H, dd, J=6.4, 12.3 Hz). 13C-NMR; δ (MeOD), 175.8, 171.8, 171.5, 137.3, 129.8, 129.5, 128.8, 79.9, 73.3, 58.7, 48.9, 39.2, 33.3, 33.3, 26.7, 24.5, 24.5, 24.0 and 22.2
Example 1
2-[2R-(S-Hydroxy-hydroxycarbamoyl-methyl)-4-methyl-pentanoylamine]-2-phenyl- ethanoic acid cyclopentyl ester

HO Ξ CONHOH
Prepared using procedures similar to those described in Preparative Example A using phenylglycine cyclopentyl ester.
Diastereoisomer A
Η-NMR; δ (MeOD), 7.4-7.29 (5H, m), 5.43 (1 H, s), 5.2-5.14 (1 H, m), 4.02 (1 H, d,
J=6.9Hz), 2.94-2.85 (1 H, m), 1.91-1.34 (10H, bm), 1.25-1.14 (1 H, m) and 0.86 (6H, 14 dd, J=6.5, 11.5Hz).
13C-NMR; δ (MeOD), 175.6, 171.8, 171.4, 137.8, 129.8, 129.4, 128.6, 80.0, 73.2,
58.5, 49.2, 39.1 , 33.3, 33.3, 26.8, 24.5, 24.4, 23.7 and 22.1.
Diastereoisomer B
Η-NMR; δ (MeOD), 7.33-7.19 (5H, m), 5.3 (1 H, s), 5.11-5.06 (1 H, m), 3.81 (1 H, d, J=7.3Hz), 2.83-2.74 (1 H, m), 1.83-1.45 (10H, bm), 1.12-1.03 (1 H, m) and 0.88-0.81 (6H, dd, J=6.4, 12.3Hz). 13C-NMR; δ (MeOD), 175.8, 171.8, 171.5, 137.3, 129.8, 129.5, 128.8, 79.9, 73.3, 58.7, 48.9, 39.2, 33.3, 33.3, 26.7, 24.5, 24.5, 24.0 and 22.2.

tosedostat
http://www.google.it/patents/US6545051

42
| WO98/11063 | WO99/46241 ex 1b | WO 98/11063 analogy ex 8 |

43
| WO98/11063 | WO99/46241 ex 1a | WO 98/11063 analogy ex 8 |
……………………………………………………………………
entry 65 in http://www.google.com/patents/WO2000044373A1
……………………………………………………………………………………………………….
http://www.google.com/patents/WO1999044602A1
Example 43
2-[2R-(S-Hydroxy-hydroxycarbamoyl-methyl)-4-methyl-pentanoylamine]-2-phenyl- ethanoic acid cyclopentyl ester
TC

HO Ξ CONHOH
Prepared using procedures similar to those described in example 8 of WO 98/11063, using phenylglycine cyclopentyl ester.
Diastereoisomer A
1H-NMR; δ (MeOD), 7.4-7.29 (5H, m), 5.43 (1 H, s), 5.2-5.14 (1 H, m), 4.02 (1 H, d, 34
J=6.9Hz), 2.94-2.85 (1 H, m), 1.91-1.34 (10H, bm), 1.25-1.14 (1 H, m) and 0.86 (6H, dd, J=6.5, 11.5Hz).
13C-NMR; δ (MeOD), 175.6, 171.8, 171.4, 137.8, 129.8, 129.4, 128.6, 80.0, 73.2, 58.5, 49.2, 39.1 , 33.3, 33.3, 26.8, 24.5, 24.4, 23.7 and 22.1.
Diastereoisomer B
1H-NMR; δ (MeOD), 7.33-7.19 (5H, m), 5.3 (1 H, s), 5.11-5.06 (1 H, m), 3.81 (1 H, d,
J=7.3Hz), 2.83-2.74 (1 H, m), 1.83-1.45 (10H, bm), 1.12-1.03 (1 H, m) and
0.88-0.81 (6H, dd, J=6.4, 12.3Hz). 13C-NMR; δ (MeOD), 175.8, 171.8, 171.5, 137.3,
129.8, 129.5, 128.8, 79.9, 73.3, 58.7, 48.9, 39.2, 33.3, 33.3, 26.7, 24.5, 24.5, 24.0 and 22.2.
……………………………..
3R-isobutyl-4S-methoxy-dihydrofuran-2,5-dione (WO 97/02239)
…………………………………………………………………………..
2(S)-Amino(phenyl)ethanoic acid cyclopentyl ester

…………………………………………………………………..
2(R)-[2,2-Dimethyl-5-oxo-1,3-dioxolan-4(S)-yl]-4-methylpentanoic acid pentafluorophenyl ester

…………………………………………………………..
intermediates
238750-91-9
α-amino-, cyclopentyl ester Benzeneacetic acid,
……………….
cas 240489-34-3
2-[2R-(S-Hydroxy-hydroxycarbamoyl-methyl)-4-methyl-pentanoylamine]-2-phenyl- ethanoic acid cyclopentyl ester

…………………..
will be updated very soon… keep watching
THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family

RAMOSETRON

Ramosetron (INN),(1-methylindol-3-yl)-[(5R)-4,5,6,7-tetrahydro-3H-benzimidazol-5-yl]methanone, 132036-88-5 cas no
| C17H17N3O | |
| 279.33 g/mol |
(1-methyl-1H-indol-3-yl)[(5R)-4,5,6,7-tetrahydro-1H-benzimidazol-5-yl]methanone
YM060
- Nasea
- Nor-YM 060
- Ramosetron
- UNII-7ZRO0SC54Y
…………………………………………………………………………………..
HYDROCHLORIDE SALT
hyrochloride salt, cas no 132907-72-3

(−)-(R)-5-[(1-methyl-1H-indol-3-yl)carbonyl]-4,5,6,7-tetrahydro-1H-benzimidazole monohydrochloride (yield 78.8%, 99.5% e.e.). FAB-MS (m/z): 280 [M+H+]
1H NMR (DMSO-d6, 30° C.): δ ppm (TMS internal standard): 1.82-1.95 (1H, m), 2.12-2.22 (1H, m), 2.66-2.94 (4H, m), 3.63-3.72 (1H, m), 3.88 (3H, s), 7.24 (1H, t, J=8.0 Hz), 7.30 (1H, t, J=8.0 Hz), 7.56 (1H, d, J=8.0 Hz), 8.22 (1H, d, J=8.0 Hz), 8.53 (1H, s), 8.90 (1H, s), 14.42 (1H, br)
…………………………………………………………………………………….
Ramosetron (INN) is a serotonin 5-HT3 receptor antagonist for the treatment of nausea and vomiting.[1] Ramosetron is also indicated for a treatment of “diarrhea-predominant irritable bowel syndrome in males”.[2] In India it is marketed under the brand name of“IBset”.
It is only licensed for use in Japan and selected Southeast Asian countries. In Japan it is sold under the tradename Iribo (イリボー). [3] Elsewhere it is commonly sold under the tradename Nasea and in India as Nozia (300 mcg/ml Inj. & 100 mcg Tab.) [4]
- Fujii Y, Saitoh Y, Tanaka H, Toyooka H (February 2000). “Ramosetron for preventing postoperative nausea and vomiting in women undergoing gynecological surgery”.Anesth. Analg. 90 (2): 472–5. doi:10.1097/00000539-200002000-00043.PMID 10648342.
- http://www.astellas.com/en/corporate/news/detail/astellas-launches-irribow-for.html
- Summary in Japanese. Retrieved on September 4, 2012.
- Abridged prescribing information – Nasea (MIMS Philippines). Retrieved on June 13, 2008.
- Synthesis and 5-HT3 antagonistic activities of 4,5,6, 7-tetrahydrobenzimidazole derivatives
200th ACS Natl Meet (August 26-31, Washington DC) 1990, Abst MEDI 39
|
1-27-2010
|
Process for producing ramosetron or its salt
|
|
|
11-20-1996
|
Intrabuccally dissolving compressed moldings and production process thereof
|
|
|
3-6-1996
|
5-substituted tetrahydrobenzimidazole compounds
|
|
|
11-15-1995
|
Intrabuccally disintegrating preparation and production thereof
|
|
|
9-7-1994
|
Tetrahydrobenzimidazole derivatives and pharmaceutical compositions containing same
|
|
|
6-24-1994
|
NEW USE OF 5-HT3 RECEPTOR ANTAGONISTS
|
AU 9048890; EP 0381422; JP 1991223278; US 5344927
| CN1696128A | Nov 2, 2004 | Nov 16, 2005 | 天津康鸿医药科技发展有限公司 | New method for synthesizing Ramosetron Hydrochloride |
| CN1765896A | Oct 28, 2004 | May 3, 2006 | 北京博尔达生物技术开发有限公司 | Novel preparation method of ramosetron hydrochloride |
| US5496942 * | 14 Feb 1994 | 5 Mar 1996 | Yamanouchi Pharmaceutical Co., Ltd. | 5-substituted tetrahydrobenzimidazole compounds |
| US5677326 * | 30 Sep 1994 | 14 Oct 1997 | Tokyo Tanabe Company Limited | Indoline compound and 5-HT.sub.3 receptor antagonist containing the same as active ingredient |
| US7358270 | 28 Jan 2005 | 15 Apr 2008 | Astellas Pharma Inc. | Treating agent for irritable bowel syndrome |
| US7683090 | 18 Oct 2006 | 23 Mar 2010 | Astellas Pharma Inc. | Treating agent for irritable bowel syndrome |
| US7794748 | 27 Aug 2004 | 14 Sep 2010 | Yamanouchi Pharmaceutical Co., Ltd. | Stable oral solid drug composition |
WO 2010024306
WO 2013005760
WO 2013100701
WO 2011001954
The chemical name of ramosetron is (−)-(R)-5-[(1-methyl-1H-indol-3-yl)carbonyl]-4,5,6,7-tetrahydro-1H-benzimidazole, and it has the structure represented by the formula (II).

It is known that ramosetron or a salt thereof has a potent 5-HT3 receptor antagonism (Patent Reference 1, Non-patent references 1 and 2), and it is on the market as a preventive or therapeutic agent for digestive symptoms (nausea, emesis) caused by administration of an anti-malignant tumor agent (cisplatin or the like). In addition, a possibility has been reported that ramosetron or a salt thereof may be useful as an agent for treating diarrheal-type irritable bowel syndrome or an agent for improving diarrheal symptoms of irritable bowel syndrome (Patent Reference 1), and its clinical trials are now in progress as an agent for treating diarrheal-type irritable bowel syndrome or an agent for improving diarrheal symptoms of irritable bowel syndrome.
As a process for producing ramosetron or a salt thereof, the following production methods are known.
Patent Reference 1 describes a production method shown by the following Production method A, namely a method for producing a tetrahydrobenzimidazole derivative (V) by allowing a heterocyclic compound (III) to react with a carboxylic acid represented by a formula (IV) or its reactive derivative.
(Production Method A)

(In the formula, X2 is a single bond and binds to a carbon atom on the heterocyclic ring represented by Het.)
As an illustrative production method of ramosetron, Patent Reference 1 describes a production method (Production method A-1) in which racemic ramosetron are obtained by using 1-methyl-1H-indole as the compound (III), and N,N-diethyl-4,5,6,7-tetrahydrobenzimidazole-5-carboxamide or N-[(4,5,6,7-tetrahydrobenzimidazol-5-yl)carbonyl]pyrrolidine, which are acid amides, as the reactive derivative of compound (IV), and allowing them to undergo treatment with phosphorus oxychloride (Vilsmeyer reaction), and then their optical resolution is carried out by fractional crystallization using (+)-dibenzoyltartaric acid.
In addition, the Patent Reference 1 exemplifies an acid halide as one of the reactive derivatives of the compound (IV), and also describes another production method of the compound (V) (Production method A-2) in which the heterocyclic compound (III) is condensed with an acid halide of the compound (IV) by the Friedel-Crafts acylation reaction using a Lewis acid as the catalyst. However, illustrative production example of ramosetron by the Friedel-Crafts acylation reaction is not described therein.
Also, a method similar to the Production example A-1 is described in Non-patent References 1 and 2 as a production method of ramosetron.
In addition, Non-patent Reference 3 describes a method for producing ramosetron labeled with 11C, represented by a Production method B. However, it discloses only the methylation step, and does not disclose a production method of nor-YM060 as the starting material.
(Production Method B)

(In the formula, nor-YM060 means (R)-5-[(1H-indol-3-yl)carbonyl]-4,5,6,7-tetrahydro-1H-benzimidazole which was provided by the present applicant, DMF means dimethylformamide.)
- Non-patent Reference 1: Chemical & Pharmaceutical Bulletin, 1996, vol. 44, no. 9, p. 1707-1716
- Non-patent Reference 2: Drugs of the Future, 1992, vol. 17, no. 1, p. 28-29
- Non-patent Reference 3: Applied Radiation and Isotopes, 1995, vol. 46, no. 9, p. 907-910
- Patent Reference 1: JP-B-6-25153

LIU Qing-wen, XU Hao, TIAN Hua, ZHENG Liang-yu, ZHANG Suo-qin
Chemoenzymatic Synthesis of Ramosetron Hydrochloride
2012 Vol. 28 (1): 70-72 [Abstract] ( 1143 ) [HTML 1KB] [PDF 206KB] ( 1052 )
doi:http://www.cjcu.jlu.edu.cn/hxyj/EN/abstract/abstract13356.shtml
…………………………………………………………………………..

The Vilsmeier-type reaction of 1-methylindole (I) with 5 – (1-pyrrolidinocarbonyl) -4,5,6,7-1 H-tetrahydrobenzimidazole hydrochloride (II) and phosphorous oxychloride in 1,2-dichloroethane gives (-5? -. [(1-methyl-3-indolyl) carbonyl] -4,5,6,7-tetrahydro-1H-benzimidazol e (III) Optical resolution of (III) with (+)-dibenzoyltartaric acid (DIBTA) in DMF -H2O, followed by exchange of the salt affords YM060.
………………………………………………….
Ondansetron: 1,2,3 ,9-Tetrahydro-9-methyl-3-[(2-methyl1-H-imidazole-1-yl)methyl]-4H-carbazol-4-one

Granisetron: Endo-1-methyl-N-(9-methyl-9-azabicyclo[3.3.1]non-3-yl)-1H-indazole-3-carboxamide

Tropisetron: Endo-1H-indole-3-carbocylic acid8-methyl-8-azabicyclo[3.2.1]oct-3-yl ester

Dolasetron: 1H-Indole-3 -carboxylic acid (2a, 6a, 8a, 9up)-octahydro-3-oxo-2,6-methano-2H-quinolizin-8-yl Ester

Azasetron: (±)-N-Azabicyclo[2.2.2]oct-3-yl-6-chloro-3,4-dihydro-4-methyl-3-oxo-1,4-benzoxazine-8-carboxamide

Alosetron: 2,3,4,5-Tetrahydro-5-methyl-2-[(5-methyl- 1H-imidazol-4-yl)methyl]-1H-pyrido[4,3-b]indol-1-one

Ramosetron

GRAZOPREVIR, MK 5172
- Grazoprevir hydrate
- UNII-4O2AB118LA
- MK 5172
MW804.99
quinoxalinyl)pentyl]cyclopropyl]oxy]carbonyl]-3-methyl-L-valyl-(4R)-4-hydroxy-L-prolyl-1-
amino-N-(cyclopropylsulfonyl)-2-ethenyl-, cyclic (1→2)-ether, hydrate (1 :1) (1R,2S)-
ethenylcyclopropyl}-5-(1,1-dimethylethyl)-14-methoxy-3,6-dioxo-
1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-
methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-
carboxamide hydrate
MOLECULAR WEIGHT 784.92
9857
ACS Med Chem Lett 2012, 3(4): 332DOI: 10.1021/ml300017p
Org Lett 2013, 15(16): 4174
[1]. Steven Harper , John A. McCauley , Michael T. Discovery of MK-5172, a Macrocyclic Hepatitis C Virus NS3/4a Protease Inhibitor. ACS Med. Chem. Lett., 2012, 3 (4), pp 332-336[2]. Summa V, Ludmerer SW, McCauley JA, MK-5172, a selective inhibitor of hepatitis C virus NS3/4a protease with broad activity across genotypes and resistant variants. Antimicrob Agents Chemother. 2012 Aug;56(8):4161-7.
IC50 Value: 7.4nM and 7nM for genotype1b and 1a respectively, in replicon system [1]
MK-5172 is a novel P2-P4 quinoxaline macrocyclic HCV NS3/4a protease inhibitor currently in clinical development.
in vitro: In biochemical assays, MK-5172 was effective against a panel of major genotypes and variants engineered with common resistant mutations observed in clinical studies with other NS3/4a protease inhibitors. In the replicon assay, MK-5172 demonstrated subnanomolar to low-nanomolar EC50s against genotypes 1a, 1b, and 2a [2].
in vivo: In rats, MK-5172 showed a plasma clearance of 28 ml/min/kg and plasma half-life of 1.4 hr. When dosed p.o. at 5 mg/kg, the plasma exposure of MK-5172 was good with an AUC of 0.7 uM.hr. The liver exposure of the compound was quite good (23 uM at 4 hr), and MK-5172 remained in liver 24 hr after a single p.o. 5 mg/kg dose. At 24 hr, the liver concentration of MK-5172 was 0.2 uM, which was over 25-fold higher than the IC50 in the replicon assay with 50% NHS. When dosed to dogs, MK-5172 showed low clearance of 5 ml/min/kg and a 3 hr half-life after i.v. 2 mg/kg dosing and had good plasma exposure (AUC=0.4 uM.hr) after a p.o. 1 mg/kg dose [1].
Clinical trial: Evaluation of Hepatic Pharmacokinetics for MK-5172 in Participants With Chronic Hepatitis C . Phase1


1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-
methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxalin-8-
yl]carbonyl}amino)-2-ethenylcyclopropyl]carbonyl}(cyclopropylsulfonyl)azanide (15 K-salt).
8.3 Hz, 1 H), 7.15 (m, 1 H), 7.04 (m, 1 H), 5.97 (m, 1 H), 5.73 (br s, 1 H), 4.96 (m, 1 H), 4.79 (apparent q, J = 9.3 Hz, 1 H), 4.26 (dd, J = 9.7, 7.7 Hz, 1 H), 4.20 (d, J = 11.3 Hz, 1 H), 4.14 (d, J = 8.8 Hz, 1 H), 3.90 (dd, J = 11.1, 3.2 Hz, 1 H), 3.86 (s, 3 H), 3.62 (m, 1 H), 2.86-2.60 (m, 3 H), 2.38 (m, 1 H), 2.21 (m, 1 H), 1.80-1.48 (m, 6 H), 1.42 (m, 5 H), 1.14 (m, 1 H), 0.95 (m, 10 H), 0.81 (m, 2 H), 0.72-0.50 (m, 3 H), 0.41 (m, 1 H) ppm.http://pubs.acs.org/doi/suppl/10.1021/ml300017p/suppl_file/ml300017p_si_001.pdf
vinylcyclopropyl)-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-
7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-
carboxamide (MK-5172, 15).
OD) δ 7.79 (dd, J = 9.6, 1.8 Hz, 1 H), 7.23 (s, 1 H), 7.22 (m, 1 H), 7.10 (d, J = 9.6 Hz, 1 H), 6.01 (apparent t, J = 3.6 Hz, 1 H), 5.74 (m, 1 H), 5.24 (dd, J = 17.0 Hz, 1.6 Hz, 1 H), 5.11 (dd, J = 10.4 Hz, 1.6 Hz, 1 H), 4.49 (d, J = 11.2 Hz, 1 H), 4.40 (m, 2 H), 4.13 (dd, J = 12.0 Hz, 4.0 Hz, 1 H), 3.92 (s, 3 H), 3.76 (m, 1 H), 2.92 (m, 2 H), 2.85 (m, 1 H), 2.55 (dd, J = 13.6 Hz, 6.4 Hz, 1 H), 2.28 (m, 1 H), 2.18 (apparent q, J =8.8 Hz, 1 H), 1.85 (dd, J = 8.0 Hz, 5.6 Hz, 1 H), 1.73 (m, 2 H), 1.5 (m, 2 H), 1.40 (dd, J = 9.6 Hz, 5.6 Hz, 1 H), 1.3 (m, 2 H), 1.23 (m, 4 H), 1.08 (s, 9 H), 0.99 (m, 2 H), 0.89 (m, 3 H), 0.73 (m, 1 H), 0.49 (m, 1 H) ppm; HRMS (ESI) m/z 767.3411 [(M+H)+; calcd for C38H51N6O9S: 767.3433].http://pubs.acs.org/doi/suppl/10.1021/ml300017p/suppl_file/ml300017p_si_001.pdf
| Intermediate # | Structure | Name | Lit. Reference |
| A1 |  |
(1R,2S)-1-Amino-N- (cyclopropylsulfonyl)-2- vinylcyclopropanecarboxamide hydrochloride | Wang et al., U.S. Pat. No. 6,995,174 |

















Step 1: Quinoxaline Hydroxyproline Methyl Ester HCl Salt

A 250-ml RB, equipped with magnetic stirrer and N2 bubbler, was charged with chloroquinoxaline BOC hydroxyproline adduct in MeOH (100 ml), and the mixture was cooled in an ice bath. Acetyl chloride (17.9 g) was then added, and the mixture was stirred at RT for 2 h. The batch was diluted with IP Ac (80 ml). Solids were filtered off and washed with IPAc (20 ml). The washed solids were dried under vacuum for 3 d, to provide 48.9 g (100% yield ). Part of this material was used in the next step.
Example 17: Preparation of Compound A, Method A

Macrocyclic acid hemihydrate, the product of Example 15 (10.16 g, 18.03 mmol) was dissolved in THF (50 mL to 90 mL). The solution was azetropically dried at a final volume of 100 mL. Sulfonamide pTSA salt (7.98 g, 1.983 mmol) followed by DMAc (15 mL) was added at RT. The batch was cooled to 0°C to 10°C, and pyridine (10 mL) was added dropwise. Then, EDC HCl (4.49 g, 23.44 mmol) was added in portions or one portion at 0°C to 10°C. The reaction mixture was aged at 0°C to 10°C for 1 h, and then warmed to 15°C to 20°C for 2 h to 4 h. MeOAc (100 mL) followed by 15 wt% citric acid in 5% NaCl in water (50 mL) was added, while the internal temperature was maintained to < 25°C with external cooling. The separated organic phase was washed with 15 wt% citric acid in 5% NaCl in water (50 mL) followed by 5% NaCl (50 mL). The organic phase was solvent-switched to acetone at a final volume of ~80 mL. Water (10 mL) was added dropwise at 35°C to 40°C. The batch was seeded with Compound A monohydrate form III (~10 mg) and aged for 0.5 h tol h at 35°C to 40°C. Additional water (22 mL) was added dropwise over 2 h to 4 h at 35°C to 40°C. The slurry was aged at 20°C for 2 h to 4 h before filtration. The wet cake was displacement washed with 60% acetone in water (2x 40 mL). Suction drying at RT gave Compound A monohydrate form III as a white solid.
XH NMR (400 MHz, CDC13) δ 9.95 (s, br, 1 H), 7.81 (d, J = 9.1 Hz, 1 H), 7.18 (dd, J = 9.1, 2.7 Hz, 1 H), 7.16 (s, br, 1 H), 7.13 (d, J = 2.7 Hz, 1 H), 5.96 (t, J = 3.8 Hz, 1 H), 5.72 (m, 1 H), 5.68 (d, J = 10.1 Hz, 1 H), 5.19 (d, J = 17.1 Hz, 1 H), 5.07 (d, J = 10.1 Hz, 1 H), 4.52 (d, J = 11.4 Hz, 1 H), 4.45 (d, J = 9.8 Hz, 1 H), 4.36 (d, J = 10.5, 6.9 Hz, 1 H), 4.05 (dd, J = 11.5, 3.9 Hz, 1 H), 3.93 (s, 3 H), 3.78 (m, 1 H), 2.90 (m, 1 H), 2.82 (tt, J = 8.0, 4.8 Hz, 1 H), 2.74 (dt, J = 13.2, 4.8 Hz, 1 H), 2.59 (dd, J = 14.0, 6.7 Hz, 1 H), 2.40 (ddd, J = 14.0, 10.6, 4.0 Hz, 1 H), 2.10 (dd, J = 17.7, 8.7 Hz, 1 H), 1.98 (2 H, mono hydrate H20), 1.88 (dd, J 8.2, 5.9 Hz, 1 HO, 1.74 (m, 3 H), 1.61 (m, 1 H), 1.50 (m, 3 H), 1.42 (dd, J = 9.6, 5.8 Hz, 1 H), 1.22 (m, 2 H), 1.07 (s, 9 H), 0.95 (m, 4 H), 0.69 (m, 1 H), 0.47 (m, 1 H).
1 C NMR (100 MHz, CDC13) δ 173.5, 172.1, 169.1, 160.4, 157.7, 154.9, 148.4, 141.0, 134.3, 132.7, 129.1, 118.8, 118.7, 106.5, 74.4, 59.6, 59.4, 55.8, 55.5, 54.9, 41.8, 35.4, 35.3, 35.2, 34.3,. 31.2, 30.7, 29.5, 28.6, 28.2, 26.6, 22.6, 18.7, 11.2, 6.31, 6.17.
HPLC conditions: Ascentis Express Column, 10 cm x 4.6 mm, 2.7 μηι; Column temperature of 40°C; Flow rate of 1.8 mL/min; and Wavelength of 215 nm.
Gradiant: mm 0.1% ¾PO4
0 20 80
5 55 45
15 55 45
25 95 5
27 95 5
27.1 20 80
32 20 80
Retention time: mm.
Compound A 14.50
Example 18: Preparation of Compound A, Method B

To a 50-L flask equipped with overhead stirring was added macrocyclic acid (1.06 kg crude, 1.00 eq), amine-pTSA (862 g crude, 1.12 eq) and MeCN (7.42 L) at 19°C. The slurry was cooled in a water bath, pyridine (2.12 L, 13.8 eq) was added, aged 15 min, and then added EDC (586 g, 1.60 eq) in one portion, aged 1.5 h, while it turned into a clear homogeneous solution.
The solution cooled in a water bath, then quenched with 2 N HC1 (1.7 L), and seeded (9.2 g), aged 15 min, and the rest of the aqueous HC1 was added over 2.5 h. A yellow slurry was formed. The slurry was aged overnight at RT, filtered, washed with MeCN/water (1 : 1 v/v, 8 L), to obtain Compound A (Hydrate II).
Compound A was dissolved in acetone (4 L) at RT, filtered and transferred to a
12-L round-bottom flask with overhead stirring, rinsed with extra acetone (1 L), heated to 50°C, water (0.9 L) was added, seeded with Compound A monohydrate form III (-10 mg), and aged 15 min, and then added water (0.8 L) over 2.5 h, extra water 3.3 v over 2.5 h was added, stopped heating, cooled to RT, aged at RT overnight, filtered, washed with water/acetone (1 : 1 v/v, 4 L), and dried in air under vacuum. Compound A Hydrate III, 670 g, was obtained as an off-white solid.
Example 19: Preparation of Compound A, Method C

Macrocyclic acid hemihydrate from Example 15 (10.16 g, 18.03 mmol) was dissolved in THF (50 ml to 90 mL). The solution was azetropically dried at a final volume of 100 mL. Sulfonamide pTSA salt (7.98 g, 19.83 mmol) was added, followed by DMAc (15 mL), at RT. The batch was cooled to 0° to 10°C, and pyridine (10 mL) was added dropwise. Then, EDC HC1 (4.49 g, 23.44 mmol) was added (in portions or one portion) at 0°C to 10°C. The reaction mixture was aged at 0°C to 10°C for 1 h, and then warmed to 15°C to 20°C for 2 h to 4 h. THF (50 mL) was added, followed by 15 wt% citric acid in 15 wt% aq. NaCl (50 mL), while the internal temperature was maintained at < 25°C with external cooling. The separated organic phase was washed with 15 wt% citric acid in 1 % aq. NaCl (40 mL), followed by 15% NaCl (40 mL). The organic phase was solvent-switched to acetone at a final volume of ~75 mL Water (1 1 mL to 12 mL) was added dropwise at 35°C to 40°C. The batch was seeded with Compound A monohydrate form III (~20 mg) and aged for 0.5 h to 1 h at 35°C to 40°C.
Additional water (22 mL) was added dropwise over 2 h to 4 h at 35°C to 40°C. The slurry was aged at 20°C for 2 h to 4 h before filtration. The wet cake was displacement washed with 60% acetone in water (40 mL x 2). Suction drying at RT or vacuum-oven drying at 45°C gave Compound A monohydrate form III as a white solid.
Example 20: Preparation of Compound A, Method D

Macrocyclic acid hemihydrate from Example 12 (10 g, 98.4wt%, 17.74 mmol) was dissolved in THF (70 mL). The solution was azetropically dried at a final volume of ~60 mL. Sulfonamide pTSA salt (7.53 g, 18.7 mmol) was added at RT. The batch was cooled to 0°C to 5°C, and pyridine (1 1.4 mL) was added dropwise. Then, EDC HC1 (4.26 g, 22.2 mmol) was added in portions at 0°C to 15°C. The reaction mixture was aged at 10°C to 15°C for 2 h to 4 h. 35 wt% Citric acid in 10 wt% aq. NaCl (80 mL) was added, while the internal temperature was maintained at < 25°C with external cooling. The separated organic phase was solvent-switched to acetone at a final volume of ~75 mL. Water (12 mL) was added dropwise at 50°C. The batch was seeded with Compound A monohydrate form III (-300 mg) and aged for 0.5 h to 1 h at 50°C. Additional water (25 mL) was added dropwise over 6 h at 35°C to 40°C. The slurry was aged at 20°C for 2 h to 4 h before filtration. The wet cake was displacement washed with 65%) acetone in water (40 mL). Suction drying at RT or vacuum-oven drying at 45°C gave Compound A monohydrate form III as a white solid.
Example 24: Ring Closing Metathesis

To a 50 mL 2-neck RB flask with reflux condenser and needle for N2 bubbling was charged the product of Example 20 (1.034 g, 0.869 mmol, 1.0 eq), toluene (20.68 ml, 20X), and the resulting solution was degassed with N2. Hoveyda-Grubbs 2nd generation catalyst (10.90 mg, 0.017 mmol) was charged to the pot, and the system was heated to 80°C with constant sparge of N2, with color change from green to reddish. The reaction was sampled (5 h) and assay by HPLC to be approximately 80% converted. The system was removed from the heat and allowed to stir at RT overnight under N2. The reaction was again assayed and deemed complete by HPLC. Toluene was removed by concentration and the resulting red oil was purified by gradient silica gel chromatography (50 g BlOTAGE SNAP Si gel column; loaded with DCM; eluted with 0 to 10% EtOAc in DCM over 10 column volumes; then 10 to 20% EtOAc in DCM over 3 column volumes; then hold; detect by TLC-UV) to yield a yellow solid, which was further slurried in EtOAc (3 mL) and hexanes (6 mL). The resulting slurry was filtered and washed with 25% EtOAc in hexanes (6 mL) to yield the product (445 mg, 0.754 mmol, 87% yield) as a white solid.


Merck reported interim data from the Phase 2 C-WORTHY study in April 2014 at the International Liver Congress (ILC) in London that evaluated the efficacy and safety of its two-drug regimen based on NS3/4A protease inhibitor MK-5172 and NS5A replication complex inhibitor MK-8742, given with or without ribavirin, in GT1 HCV patients with cirrhosis. The once-daily single pill (without ribavirin) showed a 98% SVR12 (12-week sustained virologic response) in genotype-1, treatment-naive patients. Merck will start the phase III clinical trials (NCT02105688, NCT02105662, NCT02105467 andNCT02105701) for the combination in June 2014.
Development of a Practical, Asymmetric Synthesis of the Hepatitis C Virus Protease Inhibitor MK-5172.Org. Lett. 2013;
15: 4174-4177

MK-5172 is a hepatitis C virus protease inhibitor. Key steps in the synthesis depicted are (1) the regioselective SNAr reaction of dichloroquinoxaline A with prolinol derivative B and (2) construction of the 18-membered macrocycle using a macrolactamization (F → G).
Comment
The medicinal chemistry route to MK-5172 is based on a ring-closing metathesis strategy (S. Harper et al.ACS Med. Chem. Lett. 2012, 3, 332). The best regioselectivity (20:1) and minimization of double substitution in the SNAr reaction of A with B was achieved using 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as the base in polar solvents such as DMSO, NMP, or DMAc.
SYNTHESIS, THESIS PROCEDURES, NMR see………..http://www.allfordrugs.com/2015/07/31/mk-5172-grazoprevir/
/////////////http://www.allfordrugs.com/2015/07/31/mk-5172-grazoprevir/
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
Biota Reports That Laninamivir Octanoate is Approved for the Prevention of Influenza in Japan

Laninamivir
(4S,5R,6R)-5-acetamido-4-carbamimidamido-6-[(1R,2R)-3-hydroxy-2-methoxypropyl]-5,6-dihydro-4H-pyran-2-carboxylic acid
| Formula | C13H22N4O7 |
|---|---|
| Mol. mass | 346.33638 g/mol |
cas 203120-17-6,
Laninamivir (L174000) prodrug; a novel long-acting neuraminidase inhibitor.
laninamivir octanoate
 472.53254, C21H36N4O8, cas no 203120-46-1, R-125489, CS-8958
472.53254, C21H36N4O8, cas no 203120-46-1, R-125489, CS-8958
Daiichi Sankyo (Originator)
R-118958 is a potent, long-acting neuraminidase inhibitor (LANI) approved and launched in 2010 in Japan as an inhalable formulation for the treatment of influenza A and influenza B in adults and pediatric patients. In 2013 the product was approved in Japan for the prevention of influenza A and influenza B.
| 5-(Acetylamino)-4-[(aminoiminomethyl)amino]-2,6-anhydro-3,4,5-trideoxy-7-O-methyl-D-glycero-D-galacto-non-2-enonic Acid 9-Octanoate |
| (2R,3R,4S)-3-Acetamido-4-guanidino-2-[(1R,2R)-2-hydroxy-1-methoxy-3-(octanoyloxy)propyl]-3,4-dihydro-2H-pyran-6-carboxylic Acid |
| (4S,5R,6R)-5-Acetamido-4-guanidino-6-[(1R,2R)-2-hydroxy-1-methoxy-3-(octanoyloxy)propyl]-5,6-dihydro-4H-pyran-2-carboxylic Acid |
| CS 8958 |
ATLANTA, Dec. 20, 2013 (GLOBE NEWSWIRE) — Biota Pharmaceuticals, Inc.
(Nasdaq:BOTA) (“Biota” or the “Company”) today reported that Daiichi Sankyo Company, Limited (“Daiichi Sankyo”) has been granted regulatory approval in Japan to manufacture and market Inavir(R) Dry Powder Inhaler 20mg (generic name laninamivir octanoate) for the prevention of influenza A and B. Inavir(R) was successfully developed and launched by Daiichi Sankyo in Japan for treatment of influenza A and B viruses in October, 2010. Biota is developing laninamivir octanoate outside of Japan for the treatment of influenza, and is currently conducting a large, multi-national Phase 2 trial of laninamivir octanoate in adults infected with influenza. In 2003, the Company and Daiichi Sankyo entered into a collaboration and license agreement to develop long-acting neuraminidase inhibitors, including laninamivir octanoate, and in March 2009, the parties entered into a commercialization agreement, pursuant to which Daiichi Sankyo obtained exclusive marketing rights to laninamivir octanoate in Japan.http://www.pharmalive.com/biota-flu-drug-okd-in-japan

Laninamivir (CS-8958) is a neuraminidase inhibitor which is being researched for the treatment and prophylaxis of Influenzavirus A and Influenzavirus B.[1] It is currently in Phase III clinical trials. [2]
Laninamivir was approved for influenza treatment in Japan in 2010 and is currently marketed under the name “Inavir” by Daiichi Sankyo. Biota Pharmaceuticals [3] and Daiichi Sankyo co-own Laninamivir. On 1st April 2011, BARDA awarded up to an estimated U$231m to Biota Pharmaceuticals (Formerly Biota Holdings Ltd) wholly owned subsidiary, Biota Scientific Management Pty Ltd, for the advanced development of Laninamivir in the US. [4]
patent
|
8-13-2010
|
DRUG FOR TREATMENT OF INFLUENZA
|
The recent flu scares – first H5N1 bird flu and then H1N1 swine flu – transformed Roche’s neuraminidase inhibitor Tamiflu (oseltamivir) into a household name, along with GSK’s Relenza (zanamivir). Both of these require twice-daily dosing, and the orally available oseltamivir is the first choice, but resistance is starting to appear.
A new neuraminidase inhibitor, laninamivir, is being developed by Daiichi Sankyo.5 When administered as the octanoate prodrug form, it appears that a single dose might be sufficient to treat influenza, weekly doses could be preventative, and it is active against extremely pathogenic H5N1 strains.
Laninamivir octanoateIn a double blind, randomised, placebo-controlled Phase I study in 76 healthy male volunteers, subjects were given inhaled single doses of 5, 10, 20, 40, 80 or 120mg of the prodrug, or twice-daily doses of 20 or 40mg for three days.6 No adverse events were observed, and while the prodrug disappeared from the plasma with a half-life of about two hours, the laninamivir itself was much more slowly eliminated, with a half-life of the order of three days, suggesting the potential for giving long-lasting activity against influenza.
In another Phase I trial, a total of 20 healthy subjects with renal function ranging from normal to severely impaired were given single inhaled 20mg doses of the prodrug.7 The degree of renal impairment did not affect the maximum concentration or the time to achieve it, but the half-life increased as renal function reduced. This indicates that the rate-limiting step for elimination is drug release rate to plasma from tissues rather than renal excretion. It was well tolerated, but systemic exposure increased with increasing renal impairment.
It has also been compared with oseltamivir in patients with influenza. A total of 186 children under 10 who had had febrile influenza symptoms for no longer than 36 hours were randomised to receive 20 or 40mg of laninamivir octanoate as a single inhalation or 2mg/kg oseltamivir orally twice a day for five days.8
The new drug gave a significant reduction, of 61 hours for the 40mg group and 66 for the 20mg group, in median time to illness alleviation compared with oseltamivir in those with oseltamivir-resistant H1N1 influenza A. However, there was no significant difference in the time to alleviation of illness with H3N2 influenza A, or influenza B.
The most common side-effects were gastrointestinal problems.
In a Phase III trial, a total of 1,003 adult patients with febrile influenza symptoms for no more than 36 hours were given similar doses to those in the trial in children.9 Median time to alleviation of illness was 73h for 40mg, 86h for 20mg, and 74h for oseltamivir, and the proportion of patients shedding virus at day 3 was significantly lower in the 40mg group than for those given oseltamivir.
- Yamashita M, Tomozawa T, Kakuta M, Tokumitsu A, Nasu H, Kubo S (January 2009).“CS-8958, a prodrug of the new neuraminidase inhibitor R-125489, shows long-acting anti-influenza virus activity”. Antimicrobial Agents and Chemotherapy53 (1): 186–92.doi:10.1128/AAC.00333-08. PMC2612152. PMID18955520.
- Hayden F (January 2009). “Developing new antiviral agents for influenza treatment: what does the future hold?”. Clinical Infectious Diseases. 48. Suppl 1 (S1): S3–13.doi:10.1086/591851. PMID19067613.
- http://www.biotapharma.com
- http://www.biotapharma.com/?page=1021001&subpage=1021019
5. T. Honda et al. Synthesis and in vivo influenza virus-inhibitory effect of ester prodrug of 4-guanidino-7-O-methyl-Neu5Ac2en, Bioorg Med Chem Lett 2009, 19(11): 2938
6. H. Ishizuka et al. J. Clin. Pharmacol. 2010, 50, 1319
7. H. Ishizuka et al. J. Clin. Pharmacol. 2010, epub ahead of print, doi 10.1177/0091270010361914
8. N. Sugaya and Y. Ohashi, Antimicrob. Ag. Chemother. 2010, 54, 2575
9 A. Watanabe et al. Clin. Inf. Dis. 2010, 51, 1167
A new route toward 2-acetamido-4-O-methyl-2-deoxy-D-mannopyranose from a Ferrier derivative of tri-O-acetyl-D-glucal, which contributes to aldolase-catalyzed synthesis of laninamivir (CS-8958)
Tetrahedron 2013, 39(37): 7931

Infection of poultry with H5N1 avian influenza virus has been expanding since 2003 in wide areas including Asia, Europe and Africa. Humans infected with this virus have been found not only in Asia but also in Middle East and Africa. If a new type of H5N1 influenza virus has appeared and its infection has started, it is believed that the infection will rapidly expand to cause a worldwide spread (i.e., influenza pandemic) because most people do not possess immunity against that virus and influenza viruses spread via droplet infection and airborne infection. More than half of human patients infected with H5N1 influenza virus have died so far. Thus, the virus is highly pathogenic. It is known that three influenza pandemics, the Spanish Flu, the Asian Flu and the Hong Kong Flu, occurred in the 20th century. In the Spanish Flu which caused the largest number of patients, it is estimated that about 20-40 million people died in the world and about 0.5 million people in Japan.
According to a report from Japanese Ministry of Health, Labour and Welfare made in November, 2005, if a new type influenza virus has spread, the number of patients who will consult medical doctors in Japan as a result of infection with that virus is estimated about 18-25 million. Further, when the pathogenicity of that new type influenza virus is severe, the number of inpatients is estimated about 0.2 million while the number of dead is estimated about 0.64 million. Therefore, not only health hazard but also significant influences upon social activities are feared.
Thus, a new type influenza can cause a highly severe disease. Early development of effective therapeutics is demanded.
Although it is reported that zanamivir (in particular, zanamivir hydrate) and oseltamivir (in particular, oseltamivir phosphate or oseltamivir carboxylate) which are influenza therapeutics with neuraminidase inhibitory activity show an inhibitory activity against H5N1 influenza virus, compounds with more excellent activity are desired (Non-Patent Document 1 or 2). Further, H5N1 influenza virus strains against which oseltamivir does not show any inhibitory activity (i.e., oseltamivir resistant virus strains) have been reported. Compounds which possess an inhibitory activity against these oseltamivir resistant H5N1 influenza virus strains are desired (Non-Patent Document 1 or 2).
Compounds represented by formula (I) are known to be useful as influenza therapeutics with neuraminidase inhibitory activity (Patent Documents 1 to 3). However, it has not been reported that these compounds have an inhibitory activity against H5N1 influenza virus. Further, the structures of the compounds represented by formula (I) resemble the structure of zanamivir but are completely different from the structure of oseltamivir.
Non-Patent Document 1: Nature, 2005, vol. 437, p. 1108
Non-Patent Document 2: N. Engl. J. Med., 2005, vol. 353, (25):2667-72
Patent Document 1: U.S. Pat. No. 6,340,702 (Japanese Patent No. 3209946)
Patent Document 2: U.S. Pat. No. 6,451,766 (Japanese Patent Publication No. Hei 10-109981)
Patent Document 3: U.S. Pat. No. 6,844,363 (Japanese Patent Publication No. 2002-012590)

………………………
Preparation Example 1 5-Acetamido-4-guanidino-9-O-octanoyl-2,3,4,5-tetradeoxy-7-O-methyl-D-glycero-D-galacto-non-2-enopyranosoic acid

(1) Diphenylmethyl 5-acetamido-4-(N,N-bis-t-butyloxycarbonyl)guanidino-9-O-octanoyl-2,3,4,5-tetradeoxy-7-O-methyl-D-glycero-D-galacto-non-2-enopyranosoate (3.46 g, 4.1 mmol) disclosed in Example 35 (i) of U.S. Pat. No. 6,340,702 (Japanese Patent No. 3209946) was dissolved in methylene chloride (27 ml) and trifluoroacetic acid (14 ml). The resultant solution was stirred at room temperature overnight. The reaction solution was concentrated to dryness under reduced pressure, followed by three cycles of azeotropic distillation to dryness with toluene (5 ml). The resultant oily material was dissolved in ethyl acetate (10 ml). The solution was poured into a saturated aqueous solution of sodium hydrogencarbonate (50 ml). The pH of the resultant solution was adjusted to 8.5 by addition of 20% aqueous solution of sodium carbonate. Then, the solution was stirred at room temperature for 3 hr and its pH was adjusted to 5.0 with hydrochloric acid (3 ml), followed by stirring at room temperature for another 1 hr. The solution was further stirred for 1 hr while ice-cooling. Subsequently, precipitating crystals were suction filtered and vacuum dried for 10 hr at an external temperature of 50° C. The resultant crystals were left in the air for one day to thereby yield the subject compound as a hydrate crystal (0.97 g; yield 51%).
Infrared Absorption Spectrum (KBr) ν max cm−1: 3412, 2929, 2856, 1676, 1401, 1320, 1285, 1205, 1137, 722.
1H Nuclear Magnetic Resonance Spectrum (400 MHz, CD3OD) δ ppm: 5.88 (1H, d, J=2.5 Hz), 4.45 (3H, m), 4.27 (1H, dd, J=10.0 Hz, 10.0 Hz), 4.15 (1H, m), 3.47 (21-1, m), 3.42 (3H, s), 2.37 (2H, t, J=7.4 Hz), 2.10 (3H, s), 1.31 (2H, m), 1.20-1.40 (8H, m), 0.85-0.95 (3H, m).
13C Nuclear Magnetic Resonance Spectrum (100 MHz, CD3OD) δ ppm: 176.5, 173.7, 164.7, 158.9, 146.7, 108.7, 80.1, 78.0, 69.3, 66.8, 61.4, 52.4, 35.1, 32.8, 30.2, 30.1, 26.0, 23.7, 22.8, 14.4.
(2) The subject compound was also obtained by the method described below.
5-Acetamido-4-guanidino-9-O-octanoyl-2,3,4,5-tetradeoxy-7-O-methyl-D-glycero-D-galacto-non-2-enopyranosoic acid trifluoroacetic acid salt (3.0 g, 5.1 mmol) disclosed in Example 35 (ii) of U.S. Pat. No. 6,340,702 (Japanese Patent No. 3209946) was subjected to reversed phase column chromatography [Cosmosil 75C 18PREP (nacalai tesque), 100 g] and eluted with methanol/water (0/1-1/1-2/1). Fractions containing the compound of interest were vacuum concentrated. The precipitating crystals were suction filtered and vacuum dried. The resultant crystals were left in the air for one day to thereby yield the subject compound as a hydrate crystal (1.2 g; yield 49%). The property data of the resultant compound were consistent with those of the compound obtained in (1) above.
Preparation Example 2 5-Acetamido-4-guanidino-2,3,4,5-tetradeoxy-7-O-methyl-D-glycero-D-galacto-non-2-enopyranosoic acid

5-Acetamido-4-guanidino-2,3,4,5-tetradeoxy-7-O-methyl-D-glycero-D-galacto-non-2-enopyranosoic acid trifluoroacetic acid salt (3.0 g, 5.1 mmol) disclosed in Example 28 (viii) of U.S. Pat. No. 6,340,702 (Japanese Patent No. 3209946) was purified in an ion-exchange resin column [Dowex-50X; (i) water and (ii) 5% aqueous ammonium solution] and further purified by reversed phase column chromatography [Cosmosil 75C 18PREP (nacalai tesque); water]. Fractions containing the compound of interest were vacuum concentrated. The resultant solid was washed with methanol, filtered and dried to thereby yield the subject compound (1.43 g) as a colorless solid.
1H Nuclear Magnetic Resonance Spectrum (400 MHz, CD3OD) δ ppm: 5.64 (1H, d, J=2.0 Hz), 4.43 (2H, m), 4.22 (1H, dd, J=10.0 Hz, 10.0 Hz), 4.00 (1H, m), 3.85 (1H, dd, J=10.0 Hz, 2.4 Hz), 3.68 (1H, dd, J=10.0 Hz, 5.5 Hz), 3.58 (1H, m), 3.43 (3H, s).
………………………….


…………………………..
Process W is known as a method for manufacturing a compound represented by the formula (Ia), which is embraced in a compound represented by the formula (I) or a pharmacologically acceptable salt thereof, (hereinafter also referred to as “compound (Ia)”; the same shall be applied with respect to other (Patent Document 1). In Process W, n-Hep represents a 1-heptyl group.


Process X is known as a method for manufacturing compound (Ib), which is embraced in compound (I) or a pharmacologically acceptable salt thereof (Patent Document 2). Compound (IVk) is a synthetic intermediate in Process W. In Process X, n-Hep represents a 1-heptyl group.

Process Y is known as a method for manufacturing compound (IIIa), which is a trifluoroacetic acid salt of compound (III) (Non-patent Document 1). The procedures from compound (IVc) to compound (IVe) and from compound (IVf) to compound (IVh) in Process Y are the same as in Process W.


Process Z is known as a method for manufacturing compound (IIIa), which is a trifluoroacetic acid salt of compound (III) (Non-patent Document 2). In Process Z, the procedure from compound (IVf) to compound (IVh) is the same as in Process W, and the procedure from compound (IVh) to compound (IIIa) is the same as in Process Y.
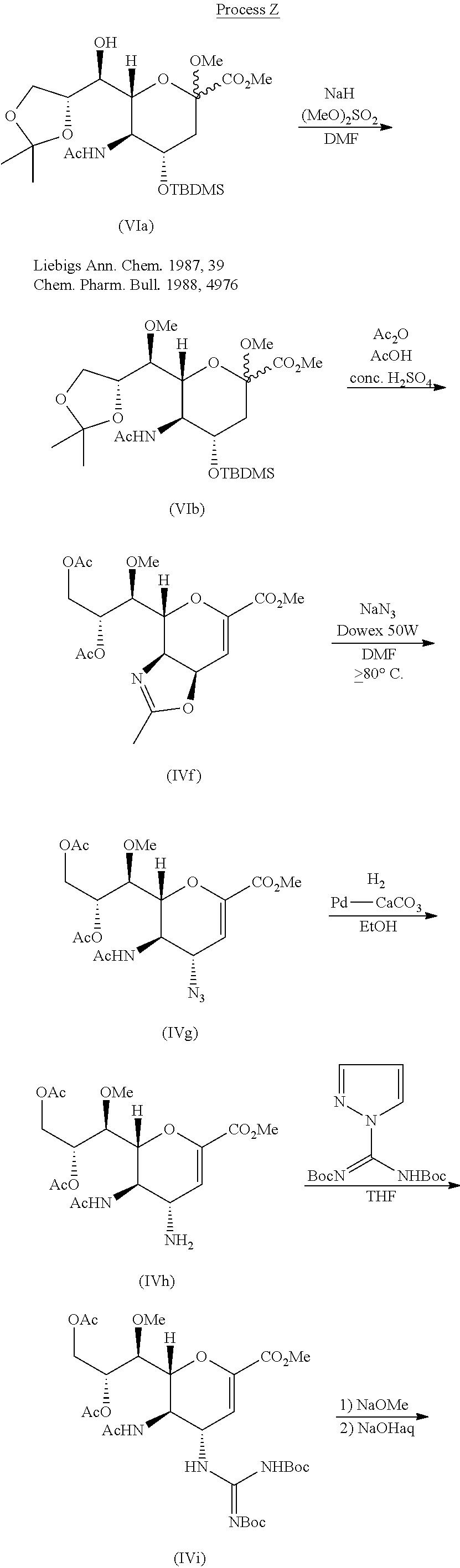

From the viewpoint of industrial production, the aforementioned Process W, Process Y, or Process Z could be improved in points such as the following:
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
update…………

:ACIE 10.1002/anie.201408138
Scheme zanamivir and Lanimiwei is based on N- acetylneuraminic acid as a starting material, the price is more expensive (ca.13000RMB / kg). Ma recently from Shanghai Institute of Organic Chemistry greatly researcher on ACIE published zanamivir, Lanimiwei and CS-8958 is more simple synthetic route. References: ACIE 10.1002 / anie.201408138
DARUNAVIR


DARUNAVIR
206361-99-1 CAS NO
[(1S,2R)-3-[[(4-Aminophenyl)sulfonyl] (2-methylpropyl)amino]-2-hydroxy-1-(phenylmethyl)propyl]carbamic acid (3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl ester
M. P.:- 72-74 °C (dec)
MW: 547.66
Darunavir and processes for its preparation are disclosed in EP0715618, W09967417, EP1725566 and Bioorganic & Medicinal Chemistry Letters (2004), 14(4), 959-963.
J Med Chem. 2013 May 23;56(10):4017-27. doi: 10.1021/jm400231v
US20050250845 discloses various pseudopolymorphs of darunavir and processes for their preparation. According to this application, “pseudopolymorph” is defined as a crystalline form of a compound in which solvent molecules are incorporated in the lattice structure. The Form B disclosed in the patent application is a pseudopolymorph wherein water is used as solvent. The thermogravimetric experiments of the Form B shows weight loss of 3.4% in the temperature range 25-78°C (water), 5.1% in the temperature range 25-1 10°C (ethanol and water) and further 1.1% weight loss (ethanol) in temperature range 110-200° C. Further at the drying step the Form B showed about 5.6% weight loss. The obtained dried product was hygroscopic and it adsorbed up to 6.8% water at high relative humidity. Amorphous form of darunavir is disclosed in US20050250845 and the publication in J.Org. Chem. 2004, 69, 7822 – 7829.
US 7700645 patent disclosed amorphous Darunavir, various solvates of Darunavir including ethanolate and method for their preparation as well as their use as a medicament. Journal of Organic Chemistry 2004, 69, 7822-7829 disclosed amorphous Darunavir is obtained by purification with column chromatography in 2% methanol in chloroform as eluent. PCT publication WO2010086844A1 disclosed crystalline dimethylsulfoxide solvate and crystalline tetrahydrofuran solvate of darunavir. The publication also disclosed the amorphous darunavir having the IR spectrum with characteristic peaks at about 1454 and 1365 cm“1
PCT publication WO201 1083287A2 disclosed crystalline darunavir hydrate substantially free of any non aqueous solvent.
Drug information:- Darunavir is an Anti-microbial drug further classified as anti-viral agent of the class protease inhibitor. It is used either single or in combination with other drugs for the treatment of human immunodeficiency virus.
Darunavir (brand name Prezista, formerly known as TMC114) is a drug used to treat HIV infection. It is in the protease inhibitor class. Prezista is an OARAC recommended treatment option for treatment-naïve and treatment-experienced adults and adolescents.Developed by pharmaceutical company Tibotec, darunavir is named after Arun K. Ghosh, the chemist who discovered the molecule at the University of Illinois at Chicago. It was approved by the Food and Drug Administration (FDA) on June 23, 2006.[2]
Darunavir is a second-generation protease inhibitor (PIs), designed specifically to overcome problems with the older agents in this class, such as indinavir. Early PIs often have severe side effects and drug toxicities, require a high therapeutic dose, are costly to manufacture, and show a disturbing susceptibility to drug resistant mutations. Such mutations can develop in as little as a year of use, and effectively render the drugs useless.
Darunavir was designed to form robust interactions with the protease enzyme from many strains of HIV, including strains from treatment-experienced patients with multiple resistance mutations to PIs.
Darunavir received much attention at the time of its release, as it represents an important treatment option for patients with drug-resistant HIV. Patient advocacy groups pressured developer Tibotec not to follow the previous trend of releasing new drugs at prices higher than existing drugs in the same class. Darunavir was priced to match other common PIs already in use, such as the fixed-dose combination drug lopinavir/ritonavir.
PREZISTA (darunavir) is an inhibitor of the human immunodeficiency virus (HIV-1) protease.
PREZISTA (darunavir), in the form of darunavir ethanolate, has the following chemical name: [(1S,2R)-3-[[(4-aminophenyl)sulfonyl](2-methylpropyl)amino]-2-hydroxy-1-(phenylmethyl)propyl]-carbamic acid (3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl ester monoethanolate. Its molecular formula is C27H37N3O7S • C2H5OH and its molecular weight is 593.73. Darunavir ethanolate has the following structural formula:
 |
Darunavir ethanolate is a white to off-white powder with a solubility of approximately 0.15 mg/mL in water at 20°C.
|
|
4-11-2012
|
METHODS FOR THE PREPARATION OF HEXAHYDROFURO[2,3-b]FURAN-3-OL
|
|
|
12-28-2011
|
Substituted Aminophenylsulfonamide Compounds as Hiv Protease Inhibitor
|
|
|
12-23-2011
|
POLYMORPHS OF DARUNAVIR
|
|
|
12-14-2011
|
METHODS FOR THE PREPARATION OF N-ISOBUTYL-N-(2-HYDROXY-3-AMINO-4-PHENYLBUTYL)-P-NITROBENZENESULFONYLAMIDE DERIVATIVES
|
|
|
11-30-2011
|
Protease inhibitor precursor synthesis
|
|
|
6-31-2011
|
PROCESS FOR THE PREPARATION OF (3R,3AS,6AR)-HEXAHYDROFURO [2,3-B] FURAN-3-YL (1S,2R)-3-[[(4-AMINOPHENYL) SULFONYL] (ISOBUTYL) AMINO]-1-BENZYL-2-HYDROXYPROPYLCARBAMATE
|
|
|
9-29-2010
|
Aminophenylsulfonamide Derivatives as Hiv Protease Inhibitor
|
|
|
8-11-2010
|
Process for the preparation of (3R,3aS,6aR)-hexahydrofuro [2,3-b] furan-3-yl (1S,2R)-3[[(4-aminophenyl) sulfonyl] (isobutyl) amino]-1-benzyl-2-hydroxypropylcarbamate
|
|
|
7-30-2010
|
RELATING TO ANTI-HIV TABLET FORMULATIONS
|
|
|
7-30-2010
|
COMBINATION FORMULATIONS
|
|
7-2-2010
|
METHODS AND INTERMEDIATES USEFUL IN THE SYNTHESIS OF HEXAHYDROFURO [2,3-B]FURAN-3-OL
|
|
|
5-7-2010
|
METHODS AND COMPOSITIONS FOR TREATING HIV INFECTIONS
|
|
|
4-21-2010
|
Pseudopolymorphic forms of a hiv protease inhibitor
|
|
|
9-21-2007
|
Immunoassays, Haptens, Immunogens and Antibodies for Anti-HIV Therapeutics
|
|
|
6-23-2006
|
Method for treating HIV infection through co-administration of tipranavir and darunavir
|
|
|
6-3-2005
|
Combination of cytochome p450 dependent protease inhibitors
|
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2010086844A1 | Dec 8, 2009 | Aug 5, 2010 | Mapi Pharma Hk Limited | Polymorphs of darunavir |
| WO2011048604A2 * | Sep 16, 2010 | Apr 28, 2011 | Matrix Laboratories Limited | An improved process for the preparation of darunavir |
| WO2011083287A2 | Oct 6, 2010 | Jul 14, 2011 | Cipla Limited | Darunavir polymorph and process for preparation thereof |
| CN102584844A * | Jan 11, 2011 | Jul 18, 2012 | 浙江九洲药业股份有限公司 | Darunavir crystal form and method for preparing same |
| US6248775 | Apr 8, 1999 | Jun 19, 2001 | G. D. Searle & Co. | α- and β-amino acid hydroxyethylamino sulfonamides useful as retroviral protease inhibitors |
| US7700645 | May 16, 2003 | Apr 20, 2010 | Tibotec Pharmaceuticals Ltd. | Pseudopolymorphic forms of a HIV protease inhibitor |
| Reference | ||
|---|---|---|
| 1 | JOURNAL OF ORGANIC CHEMISTRY vol. 69, 2004, pages 7822 – 7829 | |
| 2 | * | VAN GYSEGHEM E ET AL: “Solid state characterization of the anti-HIV drug TMC114: Interconversion of amorphous TMC114, TMC114 ethanolate and hydrate“, EUROPEAN JOURNAL OF PHARMACEUTICAL SCIENCES, ELSEVIER, AMSTERDAM, NL, vol. 38, no. 5, 8 December 2009 (2009-12-08), pages 489-497, XP026764329, ISSN: 0928-0987, DOI: 10.1016/J.EJPS.2009.09.013 [retrieved on 2009-09-24] |
Virus-encoded proteases, which are essential for viral replication, are required for the processing of viral protein precursors. Interference with the processing of protein precursors inhibits the formation of infectious virions. Accordingly, inhibitors of viral proteases may be used to prevent or treat chronic and acute viral infections. Darunavir has HIV protease inhibitory activity and is particularly well suited for inhibiting HIV-I and HIV -2 viruses. Darunavir, chemically (1 S^R.S’R.S’aS.e’aRJ-fS’he ahydrofuro^.S-b ]furanyl-[3-( 4-aminobenzenesulfonyl)isobutylamino [- 1-benzyl-zhydroxypropyl]carbamate. Darunavir is represented by the following structure:
Darunavir and its pharmaceutically acceptable salts were disclosed in US 6248775 patent, wherein Darunavir is prepared by condensing 2R-hydroxy-3-[[(4-aminophenyl)sulfonyl](2- methylpropyl)amino]-1S(phenylmethyl)propylamine with hexahydro-furo[2,3-b]furan-3-ol in anhydrous acetonitrile in the presence of anhydrous pyridine and Ν,Ν’-disuccinimidyl carbonate at ambient temperature.
US 7700645 patent disclosed amorphous Darunavir, various solvates of Darunavir including ethanolate and method for their preparation as well as their use as a medicament. Journal of Organic Chemistry 2004, 69, 7822-7829 disclosed amorphous Darunavir is obtained by purification with column chromatography in 2% methanol in chloroform as eluent. PCT publication WO2010086844A1 disclosed crystalline dimethylsulfoxide solvate and crystalline tetrahydrofuran solvate of darunavir. The publication also disclosed the amorphous darunavir having the IR spectrum with characteristic peaks at about 1454 and 1365 cm“1
PCT publication WO201 1083287A2 disclosed crystalline darunavir hydrate substantially free of any non aqueous solvent.
Darunavir Ethanolate, has the chemical name: [(1 S, 2R)-3-[[(4-aminophenyl) sulfonyl](2- methylpropyl)amino]-2-hydroxy-1-(phenylmethyl)propyl]carbamic acid (3/?, 3aS, 6a/?)- hexahydrofuro[2,3-i>]furan-3-yl ester monoethanolate and has the following structural formula:

Darunavir and its process are first disclosed in US 6248775, wherein 2 ?-hydroxy-3-[[(4- aminophenyl)sulfonyl](2-methylpropyl)amino]-1 S(phenylmethyl) propylamine (4) is reacted with (3R, 3aS, 6aR)-hexahydrofuro[2,3- >]furan-3-ol in anhydrous acetonitrile in the presence of N, W-disuccinimidyl carbonate, anhydrous pyridine at ambient temperature followed by workup to get Darunavir (Scheme A).
Scheme A

Darunavir
US 20050250845 disclosed the various solvates of Darunavir including ethanolate and method for their preparation as well as their use as a medicament. The same application disclosed the amorphous Darunavir by Raman spectra without process details.
WO 2005063770 discloses process for the preparation of Darunavir ethanolate, wherein 2R-hydroxy-3-[[(4-aminophenyl)sulfonyl](2-methylpropyl)amino]-1 S-(phenylmethyl)propyl amine (4) is reacted with (3R, 3aS, 6a ?)-hexahydrofuro[2,3-b]furan-3-ol in the presence of N, /V-disuccinimidyl carbonate, triethylamine, 41% methylamine in ethanol in a mixture of ethyl acetate and acetonitrile followed by workup and crystallization from ethanol to get Darunavir ethanolate (Scheme B).
Scheme B

In the prior art process, compound of formula 4 condensed with (3/?, 3aS, 6aR)- hexahydrofuro[2,3-6]furan-3-ol in large excess of solvent or solvent mixture containing large excess of base or mixture of bases to get Darunavir. Further, the obtained products by the processes described in the prior art are not satisfactory, from purity point of view. We have repeated the Darunavir synthetic procedures as described in the prior art and found that relatively large amounts of impurities were obtained along with Darunavir (Table-1) which need repeated crystallizations in different solvents to get desired quality of the final product resulting in poor yields. Among other impurities, the carbonic acid [(1/?,2S)-1-{((4-amino-benzenesulfonyl)-isobutyl-amino)-methyl}-2-((3R,3aSI6aR)- hexahydro-furot2,3-/3]furan-3-yloxycarbonylamino)-3-phenyl-propylester (3R,3aS,6aR)- hexahydro-furo[2,3-ft]furan-3-yl ester (difuranyl impurity of formula 1) is identified.


Conditions:-
i. Phenyl magnesium bromide, Cuprous cyanide, tetrahydrofuran, 23 °C, 1 h,
ii. t-Butyl hydroperoxide, titanium tetraisopropoxide, diethyl D-tartrate, dichloromethane, -22 °C, 24 h,
iii. Azidotrimethylsilane, titanium tetraisopropoxide, Benzene, reflux, 25 min,
iv. 2-Acetoxyisobutyryl chloride, Chloroform, 23 °C, 8 h,
v. Isobutyl amine, isopropanol, 80 °C, 12 h,
vi 4-aminobenzenesulfonyl chloride, aq. Sodium bicarbonate, dichloromethane, 23 °C, 12 h,
vii. 10% palladium on carbon, hydrogen gas (50 psi), methanol, acetic acid, tetrahydrofuran, room temperature, 2 h,
viii. [3R, 3aS,6aS]-3-hydroxyhexahydrofuro[2,3-b]-furan, disuccanamidyl carbonate, triethylamine, acetonitrile, 23 °C, 12 h
Schematic Representation for Synthesis of Darunavir
Preparation of Darunavir is described in US patent 05,158,713, and also in WO9967417 and WO9967254. Accordingly, 2-vinyloxirane 1 on reacting with phenyl magnesium bromide in presence of tetrahydrofuran solvent and cuprous cyanide catalyst give 4-phenylbut-2-ene-1-ol 2. Oxidizing 2 with t-Butyl hydroperoxide in presence of titanium tetraisopropoxide and diethyl D-tartrate using dichloromethane as solvent give [(3S)-3-benzyloxiran-2-yl]methanol 3.
Heating 3 with azidotrimethylsilane in presence of titanium tetraisopropoxide using benzene as solvent give (2S,3S)-3-azido-4-phenyl-butane-1,2-diol 4. The 1,2-dipl compound 4 underwent cyclization when treated with 2-acetoxyisobutyryl chloride in chloroform give (2S)-2-[(1S)-1-azido-2-phenyl-ethyl]oxirane 5, which was further heating with isobutylamine and isopropanol at higher temperature give (2R,3S)-3-azido-1-(isobutylamino)-4-phenyl-butan-2-ol 6. Compound 6 was reacted with 4-aminobenzenesulfonyl chloride in presence of aq. Sodium bicarbonate as base and dichloromethane as solvent resulting in to 4-amino-N-[(2R,3S)-3-azido-2-hydroxy-4-phenyl-butyl]-N-isobutyl-benzenesulfonamide 7.
Hydrogenating 7 with 10% palladium on carbon catalyst using hydrogen gas (50 psi) in methanol and tetrahydrofuran solvent in presence of small amount of acetic acid at ambient temperature resulted in to 4-amino-N-[(2R,3S)-3-amino-2-hydroxy-4-phenyl-butyl]-N-isobutyl-benzenesulfonamide 8. The final step involves reacting 8 with [3R,3aS,6aS]-3-hydroxyhexahydrofuro[2,3-b]-furan and disuccanamidyl carbonate in presence of triethylamine base and acetonitrile as solvent afford [(1S,2R)-3-[[(4-Aminophenyl)sulfonyl] (2-methylpropyl)amino]-2-hydroxy-1-(phenylmethyl)propyl]carbamic acid (3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl ester also called Darunavir 9.
…………………………
http://www.google.com/patents/WO2013114382A1?cl=en
process for the preparation of amorphous Darunavir is as

Process for the preparation of intermediate 2 is as shown in below scheme.

Examples
Example -1 : Preparation of [(1S, 2S)-3-chloro-2-hydroxy-1-(phenyl methyl) propyl] carbamic acid tert-butyl ester (5).
The solution of (3S)-3-(tert-butoxycarbonyl) amino-1-chloro-4-phenyl-2-butanone (Chloromethyl ketone 6,100 g) and aluminium isopropoxide (35 g) in isoprpylalcohol was heated to mild reflux and maintained for 3 hours. After completion of reaction distilled off isopropyl alcohol up to 50 % under vacuum and the resultant mass was cooled to 25-35°C. Water was added to the distillate, pH was adjusted to 3.0-4.0 with acetic acid and maintained the stirring for 2 hours at 25-35°C. The obtained solid was filtered and washed with water. The wet cake was taken into isopropyl alcohol (400mL) and heated to reflux for 60minutes, the mass was cooled to 25-35°C again maintain the stirring for 60minutes, the obtained solid was filtered and washed with isopropyl alcohol. The wet product was dried under normal drying to get title compound 5 (yield 80 g). Example -2: Preparation of [(1 S, 2R)-3-[(2-methylpropyl) amino]-2-hydroxy-1- (phenylmethyl) propyl] carbamic acid tert-butyl ester (4).
The mixture of [(1S, 2S)-3-chloro-2-hydroxy-1-(phenylmethyl) propyl] carbamic acid tert-butyl ester (5,100 g), isobutyl amine (294 g), sodium carbonate (31.3 g) and water was heated to 60 – 65°C and maintained for 3hours. After completion of reaction water (200 mL) was added and distilled out excess isobutyl amine under vacuum at below 75°C. Water (800 mL) was added to the distillate, cooled to 25-35°C and stirred for 2 hours. The obtained solid was filtered and washed with water to get title compound 4 (yield 105 g).
Example -3: Preparation of [(1S, 2R)-3-[[(4-nitrophenyl) sulfonyl] (2-methylpropyl) amino]- 2-hydroxy-1-(phenylmethyl) propyl] carbmic acid tert-butylester (3).
[(1 S, 2R)-3-[(2-methylpropyl) amino]-2-hydroxy-1 -(phenyl methyl) propyl] carbamic acid tert-butyl ester (4, 100 gm) and triethylamine (39.04 g) was added to methylenedichloride (1200 mL) and the temperature was raised to 40°C. p-nitro benzene sulfonyl chloride solution (72.3g of p-NBSC dissolve in 300mL methylenedichloride) was added slowly at 40-45°C for 2-3 hrs. The reaction was maintained for 3hours at 40 – 45°C. After completion of the reaction, water (500 mL) was added, separated the organic layer and distilled out methylene dichloride at atmospheric pressure. Finally, strip out the methylene dichloride by using isopropyl alcohol (200 mL). Isopropyl alcohol (1000 mL) was added to the distillate and maintained the stirring for 60 minutes at 70- 80°C. Cooled the mass to 30 – 35°C, filtered and washed with Isopropyl alcohol to get title compound 3 (yield 145 g). Example – 4: Preparation of 4-Amino-N-(2R, 3S) (3-amino-2-hydroxy-4-phenylbutyl)-N- isobutyl-benzene sulfonamide (1).
(1S, 2R)-{1-benzyl-2-hydroxy-3-[isobutyl-(4-nitro-benzenesulfonyl)-amino]-propyl}-carbamic acid tert-butyl ester (3, 100g), 10% palladium carbon (10gm) and triethanolamine (2gm) were suspended in isopropyl alcohol. The reaction was heated to 40 – 45°C and maintained under 4 – 6kg/cm2 of hydrogen pressure for 3 hours. After completion of reaction, the mass was filtered and hydrochloric acid (70mL) was added to the filtered mass. The solution was heated to reflux and maintained for 2-3hours. After completion of reaction the mass was cooled to 25-35°C, the reaction mass pH was adjusted to 6.0 – 7.0 with 20% sodium hydroxide solution and distilled out isopropyl alcohol under vacuum at below 55°C. Ethanol (200mL) and water (400mL) was added to the distillate, the mass pH was adjusted to 9.0 – 10.0 with 20% sodium hydroxide solution at 25-35°C and maintained the stirring for 2 hours at 25-35°C. The mass was cooled to 0 – 5°C, filtered and wash with water. The wet product was taken into ethanol (350mL), maintained the stirring for 30minutes at reflux temperature. The mass was cooled to 2 – 4°C, stirred for 2 hours, filtered and washed with ethanol (50 mL). The wet product was dried under normal drying to get title compound 1 (Yield 60 g).
Example-5: Preparation of ethyl-2-(4,5-dihydrofuran-3-yl)-2-oxoacetate (VI).
2, 3-Dihydrofuran (250 g) was taken in toluene (2000 mL) and triethyl amine (505 g) was added to above solution. Ethyl oxalyl chloride (536.5 g) was slowly added to the above mixture by maintaining temperature at 25-30°C and maintained the stirring for 5 hours. After completion of reaction separated the organic layer, washed the organic layer with 8% sodium bicarbonate solution (2x500mL). Organic layer was distilled completely under vacuum to get title compound VI (Yield 560g).
1 H NMR : 1.38 (t, 3H), 2.93 (t, 2H), 4.34 (q, 2H), 4.63 (t, 2H), 8.02 (s, 1 H).
Example-6: Preparation of ethyl-2-(3-bromo-2-ethoxytetrahydrofuran-3-yl)-2-oxoacetate (V).
Ethyl-2-(4,5-dihydrofuran-3-yl)-2-oxoacetate (Vl, 100g) was dissolved in dichloromethane (500ml) and Ethanol (150mL) was added. The reaction mass was cooled to 5 to 10°C. N- bromosuccinimide (1 15 g) was added lot wise by maintaining temp below 10°C. Reaction mass was then stirred at 20-30°C till completion of reaction. Reaction mass was washed with sodium bicarbonate solution (2%, 3x400mL) and the organic layer was used for the next step.
Example-7: Preparation of hexahydrofuro [2, 3-b] furan-3-ol (IV).
To the solution of Ethyl-2-(3-bromo-2-ethoxy tetra hydrofuran-3-yl)-2-oxoacetate in dichloromethane (V, 500mL) as prepared in above example, sodium sulphite solution (225g was dissolved in 1700mL of water) was added at 25-35°C. Reaction mass was stirred for 5-8hours at the same temperature and separated the organic and aqueous layers. Organic layer was washed with water (340mL). Distilled out the solvent completely get ethyl-2-(2-ethoxy tetra hydrofuran-3- yl)-2-oxoacetate. Sodium borohydride (35.5g)was dissolved in ethanol (400mL) under nitrogen atmosphere, ethyl-2-(2-ethoxytetra hydrofuran-3-yl)-2-oxoacetate was dissolved in ethanol (100mL) and slowly added to above solution at 15-30°C. Reaction mass was heated to 30-45X, maintained for 5-8 hours, the reaction mass temperature was raised to 55°C and stirred for 8 hours. The reaction mass was cooled to 20-30°C, ammonium chloride solution (1 5g in 200mL water) was slowly added and stirred for 1-2hours. The reaction mass was filtered and filtrate was distilled out under vacuum to get residue. Dichloromethane (600mL) was added to residue and cooled to -10°C. Hydrochloric acid (85mL) was added slowly drop wise in 2 hours by maintaining temp -5 to 0°C, reaction mass was stirred for 60minutes at -5 to 0°C and distilled the solvent completely. The obtained residue was stripped out with isopropyl alcohol (2x200mL, 1x100mL), ethyl acetate (500mL) was added to the resultant residue, stirred for 30-60minutes and cooled to 10-15°C. The solution was filtered and filtrate was concentrated to get title compound IV (yield 56 g).
Example-8: Preparation of Hexahydrofuro [2, 3-b] furan-3-yl acetate (III).
Hexahydrofuro [2, 3-b] furan-3-ol (IV, 60g) was dissolved in dichloromethane (300mL) and cooled to 0-5°C. To the cooled solution triethylamine (58.2 g), N, N-dimethylaminopyridine (1.12g) was added, acetic anhydride (56.5g) was added for 30-60 minutes at the same temperature, the mass temperature was raised to 25-35°C and stirred for 2-4hours. After completion of reaction the mass was cooled to 10-20°C, water (120mL) was added, stirred for 30minutes, separated the organic layer, washed with 10% sodium chloride solution (120mL) and distilled out dichloromethane to get title compound (yield 72g). Further, the product was purified by fractional distillation to get pure Hexahydrofuro [2, 3-b] furan-3-yl acetate III (yield 54g).
1 H NMR : 1.9-2.09(m, 2H), 2.10(s, 3H), 3.0-3.1 (m, 1 H), 3.86-4.03(m, 2H), 3.73(dd, 1 H), 4.10(dd, 1 H), 5.19(m, 1 H), 5.72 (d, 1 H)
Example-9: Preparation of (3R, 3aS, 6aR)-Hexahydrofuro [2, 3-b] furan-3-yl acetate (II). To the buffer solution (104.3g of sodium dihydrogen orthophosphate dissolved in 530mL of water & pH adjusted to 6.0-6.5 with saturated sodium bicarbonate solution(68g in 680 mL water) solution) hexahydrofuro [2, 3-b] furan-3-yl acetate (111,115g) and CAL-B (17.25g) was added at 25-35°C, heated to 38-45°C and stirred for 24 hours. CAL-B (17.25g) was added stirred for 16 hours, again CAL- B (11.5g) was added at 38-45°C and stirred for 16 hours (pH should maintain 6.0-6.5). The reaction mass was cooled to 20-30°C, methylenedichloride (1 150mL) was added to the mass and stirred for 30 minutes. The reaction mass was filtered through hyflowbed then separated the organic layer and washed with 10%sodiumchloride solution (575mL). Organic layer was distilled completely under vacuum to get title compound II (yield 40. Og). Example-10: Preparation of (3R, 3aS, 6aR)-Hexahydrofuro [2, 3-b] furan-3-ol (I).
(3R, 3aS, 6aR)-Hexahydrofuro [2, 3-b] furan-3-yl acetate (II, 14.0g) was dissolved in methanol (42mL). Potassium carbonate (0.34g) was added and stirred at 25-35°C for 6-8hours. Methanol was distilled out completely under vacuum, to the distillate methylenedichloride (28mL) was added, stirred the mass for 30 minutes and again distilled the solvent to get residue. Dissolved the residue in dichloromethane (56mL), the resultant solution was treated with carbon and the solvent was completely distilled out get title compound I (yield 10.5g). Example-11 : Preparation of (3R, 3aS, 6aR)-Hexahydrofuro [2, 3-b]-furan-3-yl-4-nitrophenyl carbonate (2).
To the solution of (3R, 3aS, 6aR)-Hexahydrofuro [2, 3-b] furan-3-ol (l,100g) and Bis-nitrophenyl carbonate (257.2g) in methylene dichloride (1200mL), triethylamine solution (132 g in 300 mL of methylene dichloride) was added slowly at 20-30°C for 2-3hours. Maintained the reaction at the same temperature for 8-10hours, after completion of reaction water (500mL) was added for 30- 60minut.es and settled the reaction mass then separated the organic layer. Organic layer was washed with 10% acetic acid (100mL) and 10% sodium chloride solution (500mL), distilled the organic layer and co distilled with ethyl acetate (100mL). Ethyl acetate (300mL) was added to the distillate and heated to 50-55°C for 30-45minut.es to get clear solution, the solution was cooled to 5-10°C and maintained at the same temperature for 60 minutes. The obtained solid was filtered, washed with ethanol (100mL) and dried the wet material at 40-45°C for 10-14 hours to get title compound 2 (yield 160g). Example-12: Preparation of dimethylformamide solvate of Darunavir.
To a mixture of 4-amino-N-(2r,3S)(3-amino-2-hydroxy-4-phenylbutyl)-N-lsobutyl- benzenesulfonamide (1 ,25g) and N-methyl-2-pyrrolidinone (NMPO, 50mL), a solution of (3R,3aS,6aR)-Hexahydrofuro[2,3-b]-furan-3-yl-4-nitrophenyl carbonate (2, 8.85g) and N-methyl- 2-pyrrolidinone (75mL) was added at -5 to 0°C for 2 to 3 hours under nitrogen atmosphere. The mass temperature was slowly raised to 25 to 30°C and stirred for 6 to 8 hours. The reaction mass was quenched in to the solution of methylene chloride (125mL) and water (250mL) at 25-35°C for 30 to 45 minutes. Separated the organic layer followed by washed with 10% sodium carbonate solution (150mL), 10% sodium chloride solution (150mL) and with water (6x150mL). Organic layer was dried over sodium sulphate and distill off the solvent under vacuum at below 50°C to obtain darunavir as a residue. To the residue Ν,Ν-dimethyl formamide (50mL) was added and cooled to 0 to -5°C, water (25mL) was added to the solution and maintained for 12hours at 0 to -5 °C, the obtained solid was filtered and washed with pre-cooled mixture of N,N-dimethyl formamide & water (25mL+25mL) to get dimethylformamide solvate of darunavir.
Example-13: Preparation of non-solvated crystalline Darunavir.
To a mixture of 4-amino-N-(2r,3S)(3-amino-2-hydroxy-4-phenylbutyl)-N-lsobutyl- benzenesulfonamide (1, 25g) and N-methyl-2-pyrrolidinone (NMPO, 50mL), a solution of (3R,3aS,6aR)-Hexahydrofuro[2,3-b]-furan-3-yl-4-nitrophenyl carbonate (2, 18.85g) and N- methyl-2-pyrrolidinone (75mL) was added at -5 – 0°C for 2 to 3 hours under nitrogen atmosphere. The mass temperature was slowly raised to 25 – 30°C and stirred for 6 to 8 hours. The reaction mass was quenched in to the solution of methylene chloride (250mL) and water (250mL) at 25- 35°C for 30 – 45 minutes. Separated the organic layer followed by washed with 10% potassium carbonate solution (5x125mL), water (5x125mL), 20% sodium chloride solution (25mL), finally washed with 20% citric acid solution (125mL). The organic layer was treated with carbon and distilled off the solvent under vacuum at below 50°C to obtain darunavir as a residue. To the residue ethylacetate (250mL) was added and cooled to 0 to -5°C, to the cooled solution hexane (225mL) was added and maintained for 12hours at 0 to -5 °C, the obtained solid was filtered, washed with pre-cooled mixture of ethylacetate and hexane (25mL+25mL) and dried the compound to get non-solvated crystalline darunavir(yield 25g).
Example -14: Preparation of Amorphous Darunavir.
Darunavir (200g) as obtained in above example was dissolved in methylene dichloride (10L) and washed with water (3×1000 mL). Organic layer was taken into agitated thin film dryer (ATFD) feed tank. Applied initial temperature about 36 – 40°C and high vacuum (580mm/Hg) to the vessel. Slowly feed the solution to the Vessel (feed rate 5L r) over 1hour finally given the methylene chloride (3L) flushing. The material is collected in the material collecter. Dried at 58 -62°C for 40 hours to get amorphous darunavir (yield 160g).
………………..
http://www.google.com/patents/WO2011048604A2?cl=en

Preparation of Durumvir ethanolate
A solution of (3R,3aS,6a ?)-hexahydrofuro[2,3-D]furan-3-yl 4-nitrophenyl carbonate (5b, 75.4 g) in A -methyl-2-pyrrolidinone (300 mL) was added to a pre-cooled (-2 ± 2°C) solution of the compound of formula 4 (100 g) in W-methyl-2-pyrrolidinone (200 mL) at -4 to 0°C over a period of 2 h. The temperature of the reaction mass was slowly raised to 25 – 30°C and maintained for 8 h. After completion of the reaction (TLC monitoring), ethyl acetate (1000 mL) and purified water (500 mL) were added to the reaction mass. The layers were separated; organic layer was washed with sodium carbonate solution (2 X 500 mL) followed by sodium chloride solution. The organic layer was concentrated; ethanol (300 mL) was added, heated to 45 – 50°C, maintained for 1 h, filtered and washed with ethanol. The wet compound was taken into a mixture of ethyl acetate- ethanol (7:93, 600 mL), heated to reflux, charcoal was added and filtered. The resultant filtrate was cooled to 0 – 5°C, filtered the separated solid and washed with ethanol. The wet compound was dried at 45°C to obtain the in 124.3 g (yield-82.5%). The obtained Darunavir ethanolate had purity of 99.79% on area by HPLC and contained 0.08% on area by HPLC of the difuranyl impurity. Preparation of Amorphous Darunavir
Example – 4
A solution of Darunavir ethanolate (200 g) in dichloromethane (10 L) was taken into ATFD Feed tank. The solvent was evaporated by fed the solution slowly to the ATFD Vessel (feed rate 5 L /h) at 36 – 40°C and high vacuum (580 mm/Hg) over 2 h and then flushed with dichloromethane (3 L). The material is collected in the material collector in 160g with the HPLC Purity of 99.60% and particle size D50 of approximately 50 micrometers and Dgo of approximately 100 to 180 micrometers. Example-5
Darunavir Ethanolate (200 gm) was dissolved in Methylene chloride (1000 ml) and solvent was evaporated by applying vacuum followed by isolation of amorphous Darunavir as a solid as such or by charging n-Heptane or Isopropyl ether. Example – 6
Darunavir Ethanolate (10 g) was dissolved in ethyl acetate (50 mL). The solution was heated to 40 – 45°C and maintained for 30 min. Ethyl acetate was distilled off under vacuum completely to get residue in the form of semisolid. n-Heptane (50 mL) was added to the residue and stirred for 30 min. at ambient temperature. The separated solid was filtered, washed the wet cake with n-heptane (5 mL) and dried at 40 – 45°C under vacuum to get 8.0 g of amorphous Darunavir.
Example – 7
Darunavir Ethanolate (10 g) was placed into a dry round bottom flask and heated to 110 – 120°C to melt and maintained under vacuum for 4 h. The reaction mass was slowly cooled to 25 – 35°C. The obtained glass type crystal was broken into powder to afford 8.5 g of amorphous Darunavir.
Example – 8
Darunavir Ethanolate (5.0 g) was suspended into glycerol (25 g), heated to 110 – 120°C under vacuum and maintained for 30min. Water (50 mL) was added to the cooled reaction mass at 25 – 35°C under stirring and the obtained suspension was stirred for 30 min at 25 – 35°C. The separated solid was filtered and dried at 40 – 45°C under vacuum to yield 3.5 g of amorphous Darunavir. Example – 9
Carbonic acid [(1 R,2S)-1-{((4-amino-benzenesulfonyl)-isobutyl-amino)-methyl}-2- ((3/?,3aS,6aR)-hexahydro-furo[2,3-ft]furan-3-yloxycarbonylamino)-3-phenyl-propylester (3R,3aS,6a ?)-hexahydro-furo[2,3- )]furan-3-yl ester (difuranyl impurity, 1).
The difuranyl impurity (1) isolated from the mother liquor by preparative HPLC using a mixture of formic acid-water (1 :99) as eluent. The 1H-NMR, 13C-NMR and mass spectral data complies with proposed structure.

1H-NMR (DMSO-cfe, 300 MHz, ppm) – δ 0.79 (d, J=6.6 Hz, 6H, 15 & 15′), 1.14-1.20 (m, 1 H, 20Ha), 1.34-1.42 (m, 1 H, 20Hb), 1.75-1.85 (m, 2H, 20’Ha & 14), 1.94-2.01(m, 1 H, 20’Hb), 2.54-2.64 (m, 2H, 8Ha & 13Ha), 2.74-2.89 (m, 3H, 8Hb, 13Hb & 19), 3.00-3.11 (m, 2H, 5Ha & 19′), 3.34-3.39 (m, 1H, 5Hb), 3.54-2.63 (m, 3H, 21 Ha & 17Ha), 3.65-3.74 (m, 3H, 21’Ha, 21 Hb &17Hb), 3.81-3.89 (m, 2H, 21’Hb & 17’Ha), 3.94-4.04 (m, 2H, 7 & 17’Hb), 4.81-4.88 (m, 1 H, 6), 4.92-4.96 (m, 1 H, 18′), 5.03-5.10 (m, 1 H, 18), 5.11 (d, J=5.4 Hz, 1 H, 22′), 5.61 (d, J=5.1 Hz, 1 H, 22), 6.03 (brs, 2H, NH2, D20 exchangeable), 6.63 (d, J=8.7 Hz, 2H, 2 & 2″), 7.15-7.28 (m, 5H, 10H, 10Ή, 11 H, 11′ & 12), 7.40 (d, J=8.7 Hz, 2H, 3 & 3′), 7.55 (d, J=9.3 Hz, 1 H, NH, D20 exchangeable).
“H-NMR (DMSO-d6, 75 MHz, ppm)- δ 19.56 & 19.81 (15C & 15’C), 25.42 (20 ), 25.47 (20C), 26.28 (14C), 35.14 (8C), 44.45(19’C), 45.01 (19C), 49.21 (5C), 53.39 (7C), 57.55 (13C), 68.70 (21 ‘C), 68.74 (21C), 69.95 (17’C), 70.20(17C), 72.65 (6C), 76.27 (18C), 79.59 (18’C), 108.70 (22’C), 108.75 (22C), 112.69 (2C), 122.56 (4C), 126.12 (12C), 128.04 (11 C & 11’C), 129.03 (10C & 10’C), 129.08 (3C), 138.03 (9C), 152.99 (1C), 153.55 (16’C), 155.32 (16C).
DIP MS: m/z (%) 1108 [M+Hf, 1131 [M+Naf
……………
http://www.google.com/patents/US20130244297

According to the present invention Darunavir having the below impurity not more than 0.1, preferably 0.05%.

………….
DARUNAVIR

Zosuquidar

LY335979, RS-33295-198 (Zosuquidar)
Roche Palo Alto (Originator)
LY335979 (Zosuquidar) is a selective Pgp (P-glycoprotein) inhibitor with a Ki of 59 nM. LY335979 significantly enhanced the survival of mice implanted with Pgp-expressing murine leukemia (P388/ADR) when administered in combination with either daunorubicin, doxorubicin or etoposide.
LY335979 (Zosuquidar)
M.Wt: 636.99
Formula: C32H31F2N3O2.3HCl
Name: Zosuquidar trihydrochloride
Elemental Analysis: C, 60.34; H, 5.38; Cl, 16.70; F, 5.97; N, 6.60; O, 5.02
CAS : 167465-36-3
167354-41-8 (free base)
Roche Bioscience (Originator), Eli Lilly and Company (Licensee).
US5654304, WO1994024107A1, WO2000075121, US6570016
Drug Des Discov 1992, 9(1): 69, Bioorg Med Chem Lett 1995, 5(21): 2473, Drugs Fut 2003, 28(2): 125
Zosuquidar is currently under development. It is now in “Phase 3” of clinical tests in the United States. Its action mechanism consists of the inhibition of P-glycoproteins; other drugs with this mechanism include tariquidar and laniquidar. P-glycoproteins are proteins which convert the energy derived from the hydrolysis of ATP to structural changes in protein molecules, in order to perform coupling, thus discharging medicine from cells. If P-glycoprotein coded with the MDR1 gene manifests itself in cancer cells, it discharges much of the antineoplastic drugs from the cells, making cancer cells medicine tolerant, and rendering antineoplastic drugs ineffective. This protein also manifests itself in normal organs not affected by the cancer (such as the liver, small intestine, and skin cells in blood vessels of the brain), and participates in the transportation of medicine. The compound Zosuquidar inhibits this P-glycoprotein, causing the cancer cells to lose their medicine tolerance, and making antineoplastic drugs effective
Clinicial trials: Clinical report published in 2010 showed that zosuquidar did not improve outcome in older acute myeloid leukemia, in part, because of the presence P-gp independent mechanisms of resistance. (Blood. 2010 Nov 18;116(20):4077-85.)
Zosuquidar is a potent P-glycoprotein inhibitor, which binds with high affinity to P-glycoprotein and inhibits P-glycoprotein-mediated multidrug resistance (MDR). P-glycoprotein, encoded by the MDR-1 gene, is a member of the ATP-binding cassette superfamily of transmembrane transporters and prevents the intracellular accumulation of many natural product-derived cytotoxic agents
Zosuquidar
U.S. Patent No. 5,112,817 to Fukazawa et al. discloses certain quinoline derivatives useful as anticancer drug potentiators for the treatment of multidrug resistance. One of the initially promising active agents there-disclosed is MS-073, which has the following structure:

MS-073
U.S. Pat. Nos. 5,643,909 and 5,654,304 disclose a series of 10,11- methanobenzosuberane derivatives useful in enhancing the efficacy of existing cancer chemotherapeutics and for treating multidrug resistance. One such derivative having good activity, oral bioavailability, and stability, is zosuquidar, a compound of formula (2R)-anti-5-
3 – [4-( 10, 11 -difluoromethanodibenzosuber-5-yl)piperazin- 1 -yl]-2-hydroxypropoxy) quinoline.

Zosuquidar
Given the limitations of previous generations of MDR modulators, three preclinical critical success factors were identified and met for zosuquidar: 1) it is a potent inhibitor of P-glycoprotein; 2) it is selective for P-glycoprotein; and 3) no pharmacokinetic interaction with co-administered chemotherapy is observed.
Zosuquidar is extremely potent in vitro (Kj = 59 nM) and is among the most active modulators of P-gp-associated resistance described to date. Zosuquidar has also demonstrated good in vivo activity in preclinical animal studies. In addition, the compound does not appear to be a substrate for P-gp efflux, resulting in a relatively long duration of reversal activity in resistant cells even after the modulator has been withdrawn.
Another significant attribute of zosuquidar as an MDR modulator is the minimal pharmacokinetic (PK) interactions with several oncolytics tested in preclinical models. Such minimal PK interaction permits normal doses of oncolytics to be administered and also a more straightforward interpretation of the clinical results.
Zosuquidar is generally administered in the form of the trihydrochloride salt. Conventional zosuquidar trihydrochloride formulations include those containing zosuquidar (50 mg as free base), glycine (15 mg), and mannitol (200 mg) dissolved in enough water for injection, to yield a free base concentration of 5 mg/mL. The formulation is filled into vials and lyophilized to give a vial containing 50 mg of free base. For such formulations, a 30 mL vial size is necessary to contain 50 mg of thezosuquidar formulation. For a typical >200 mg dose of zosuquidar, multiple 50 mg vials are needed to contain the formulation, greatly increasing manufacturing costs and reducing convenience for the end user {e.g., a pharmacist). Modified Cyclodextrins
Cyclodextrins are cyclic oligomers of glucose; these compounds form inclusion complexes with any drug whose molecule can fit into the lipophile-seeking cavities of the cyclodextrin molecule. See U.S. Pat. No. 4,727,064 for a description of various cyclodextrin derivatives. Cyclodextrins of preferred embodiments can include α-, β-, and χ-cyclodextrins. The α-cyclodextrins include six glucopyranose units, the β- cyclodextrins include seven glucopyranose units, and the χ-cyclodextrins include eight glucopyranose units. The β -cyclodextrins are generally preferred as having a suitable cavity size for zosuquidar. Cyclodextrin can be in any suitable form, including amorphous and crystalline forms, with the amorphous form generally preferred. Cyclodextrins suitable for use in the formulations of preferred embodiments include the hydroxypropyl, hydroxyethyl, glucosyl, maltosyl, and maltotrosyl derivatives of β- cyclodextrin, carboxyamidomethyl-β-cyclodextrin, carboxymethyl-β-cyclodextrin, and diethylamino-β-cyclodextrin.
Pharmaceutical complexes including various cyclodextrins and cyclodextrin derivatives are disclosed in the following United States patents: U.S. Pat. No. 4,024,223; U.S. Pat. No. 4,228,160; U.S. Pat. No. 4,232,009; U.S. Pat. No. 4,351,846; U.S. Pat. No. 4,352,793; U.S. Pat. No. 4,383,992; U.S. Pat. No. 4,407,795; U.S. Pat. No. 4,424,209; U.S. Pat. No. 4,425,336; U.S. Pat. No. 4,438,106; U.S. Pat. No. 4,474,881; U.S. Pat. No. 4,478,995; U.S. Pat. No. 4,479,944; U.S. Pat. No. 4,479,966; U.S. Pat. No. 4,497,803; U.S. Pat. No. 4,499,085; U.S. Pat. No. 4,524,068; U.S. Pat. No. 4,555,504; U.S. Pat. No. 4,565,807; U.S. Pat. No. 4,575,548; U.S. Pat. No. 4,598,070; U.S. Pat. No. 4,603,123; U.S. Pat. No. 4,608,366; U.S. Pat. No. 4,659,696; U.S. Pat. No. 4,623,641; U.S. Pat No. 4,663,316; U.S. Pat. No. 4,675,395; U.S. Pat. No. 4,728,509; U.S. Pat. No. 4,728,510; and U.S. Pat. No. 4,751,095.
Chemically modified and substituted α-, β-, and χ-cyclodextrins are generally preferred over unmodified α-, β-, and χ-cyclodextrins due to improved toxicity and solubility properties. The degree of substitution of the hydroxy 1 groups of the glucopyranose units of the cyclodextrin ring can affect solubility. In general, a higher average degree of substitution of substituent groups in the cyclodextrin molecule yields a cyclodextrin of higher solubility.
Examples for Pgp inhibitors are cyclosporine A, valpodar, elacridar, tariquidar, zosuquidar, laniquidar, biricodar, S-9788, MS-209, BIBW-22 (BIBW-22-BS) , toremifene, verapamil, dexverapamil , quinine, quinidine, trans- flupentixol, chinchonine and others (J. Roberts, C. Jarry (2003) : J. Med. Chem. 46, 4805 – 4817) . The list of inhibitors of P-glycoprotein is increasing (e.g. Wang et al . (2002) : Bioorg. Med. Chem. Lett. 12, 571 – 574) .
Figure 2: Structures of BIBW-22, MS-209 and S-9788
|
7-12-2000
|
10,11-methanodibenzosuberane derivatives
|
|
|
10-17-2007
|
Salt and crystalline forms of (2R)-anti-5-{3-[4-(10,11-difluoromethanodibenzosuber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinoline
|
|
|
9-2-2009
|
Salt and crystalline forms of (2R)-anti-5-{3-[4-(10,11-difluoromethanodibenzosuber-5-YL)piperazin-1-YL]-2-hydroxypropoxy}quinoline
|
……………………

U.S. Pat. Nos. 5,643,909 and 5,654,304, incorporated herein by reference, disclose a series of 10,11-methanobenzosuberane derivatives useful in enhancing the efficacy of existing cancer chemotherapeutics and for treating multidrug resistance. (2R)-anti-5-{3-[4-(10,11-difluoromethanodibenzosuber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinoline trihydrochloride disclosed therein, is currently under development as a pharmaceutical agent.
U.S. pat. No. 5,654,304 (‘304), incorporated by reference herein, discloses a series of 10,11-(optionally substituted)methanodibenzosuberane derivatives useful in enhancing, the efficacy of existing cancer chemotherapeutics and for treating multidrug resistance. (2R)-anti-5-{3-[4-(10,11-Difluoromethanodibenzosuber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinolone trihydrochloride is disclosed in ‘304 and is currently under development as a pharmaceutical agent. WO00/75121 discloses Form I, a crystalline form of (2R)-anti-5-{3-[4-(10,11-difluoromethanodibenzosuber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinolone trihydrochloride.
The art disclosed in U.S. Pat. No. 5,776,939, and U.S. Pat. No. 5,643,909 both incorporated herein by reference, and PCT Patent Applications (Publication numbers WO 94/24107 and 98/22112) teach the use of 1-formylpiperazine to introduce the piperazine group of the compound of formula II

Compound II is a mixture of syn isomer (III)

and anti isomer (IV)

The process as disclosed in U.S. Pat. Nos. 5,643,909 and 5,654,304 (represented by scheme A, below) involves (a) chromatographic separation(s) of the formyl piperazine compound; and (b) deformylation of the formyl piperazine compound to provide compound IV.https://www.google.co.in/patents/US6570016?cl=en
The process of the present invention uses piperazine to react with the (1aα,6α,10bα)-6-halo-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cycloheptene compound or derivative, instead of formylpiperazine.
The process of the present invention is advantageous because piperazine is readily available in commercial quantities whereas 1-formylpiperazine, which was utilized in the process disclosed in U.S. Pat. No. 5,643,909 is often not readily available in commercial quantities. Additionally piperazine enjoys a significant cost advantage over 1-formylpiperazine.
The use of piperazine instead of 1-formylpiperazine is a significant advancement over the prior art because it obviates the need to deformylate or hydrolyze off the formyl group (step 6, scheme A), thereby providing fewer operational steps. U.S. Pat. No. 5,643,909 teaches the separation of the 1-formylpiperazine compounds by chromatography or repeated crystallization. The present invention obviates the need for chromatographic separations of the formylpiperazine diastereomeric addition compounds (see step 4, scheme A)
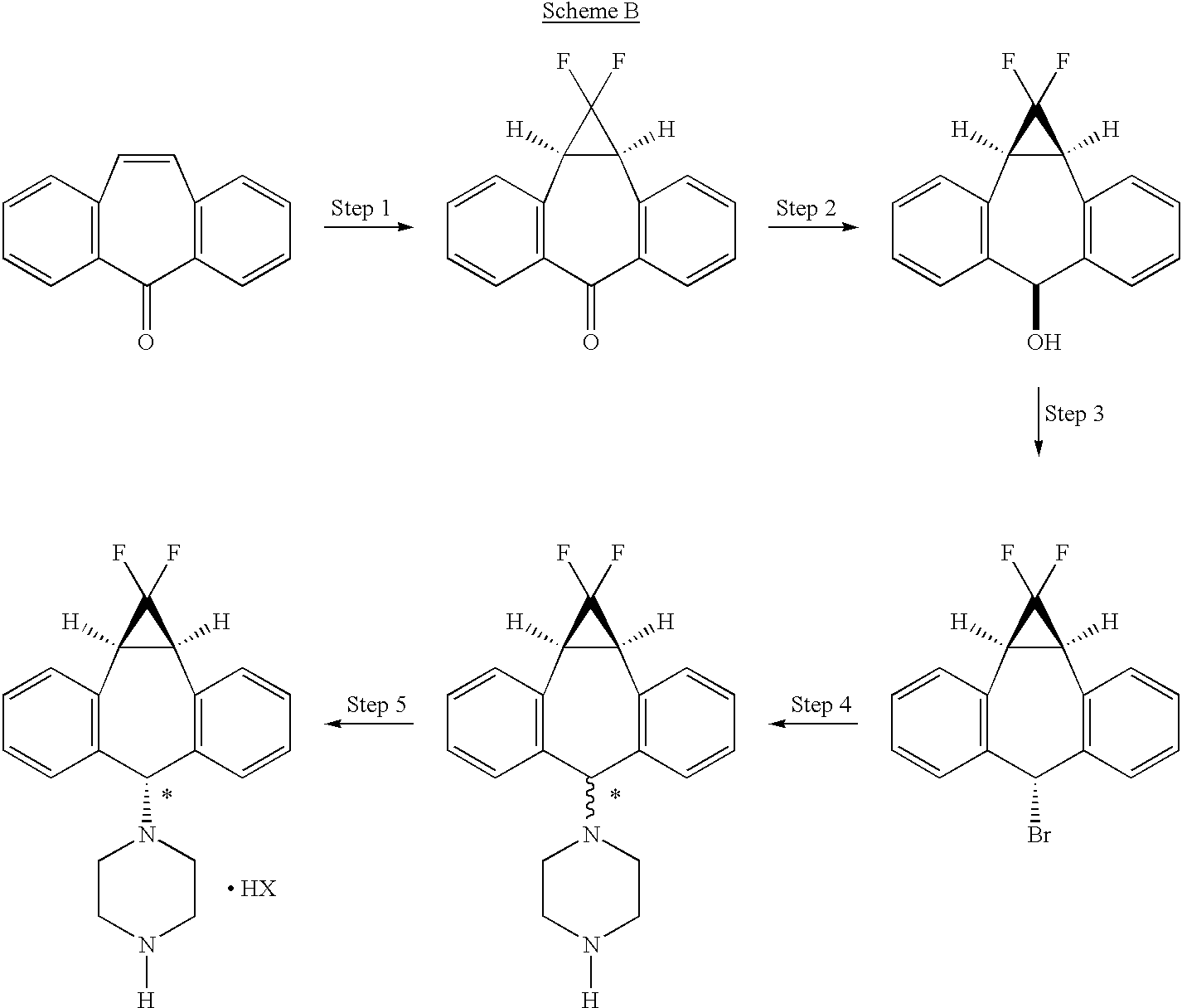
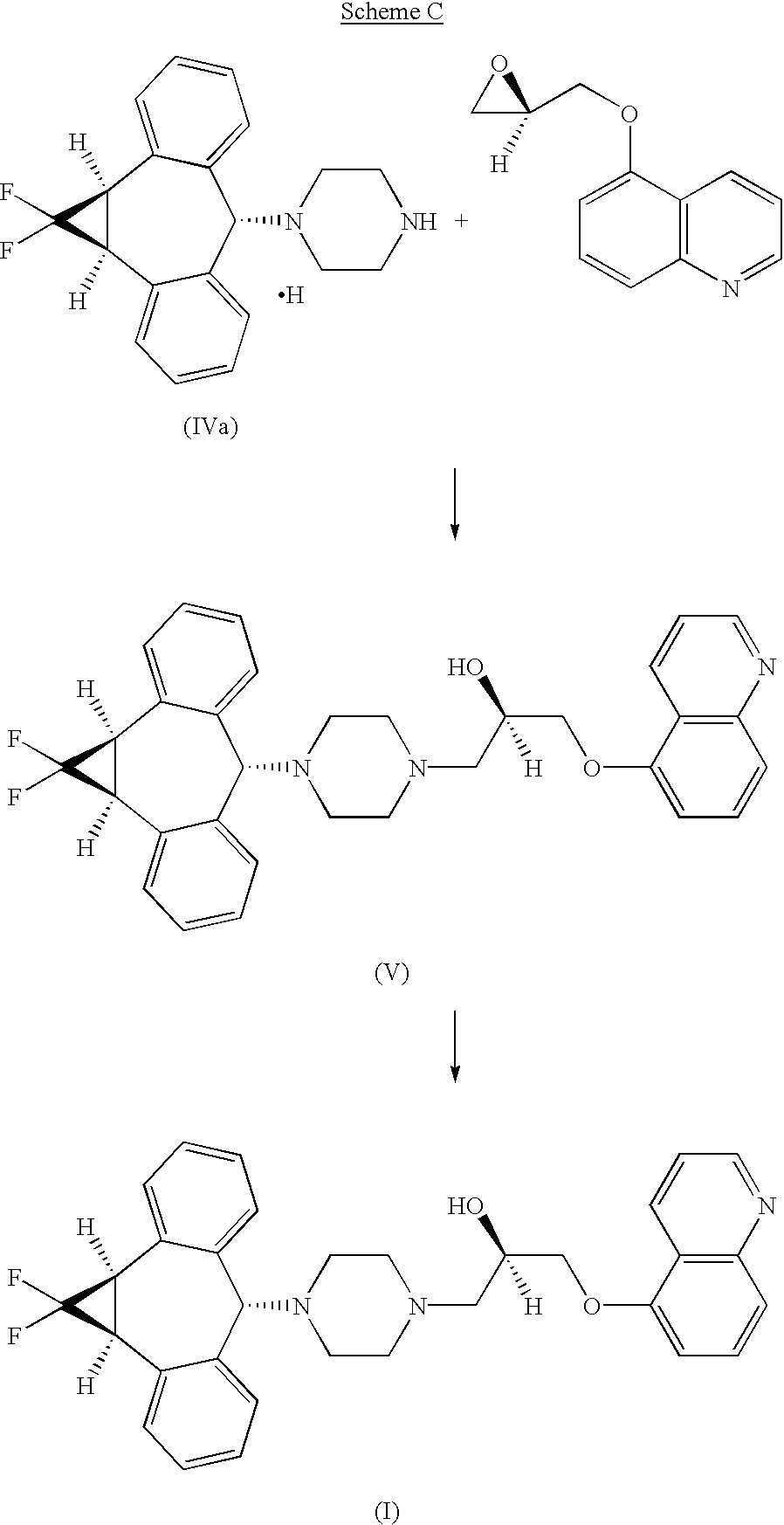
EXAMPLES
The following examples and preparations are illustrative only and are not intended to limit the scope of the invention in any way.
Preparation 1 R-1-(5-Quinolinyloxy)-2,3-epoxypropane
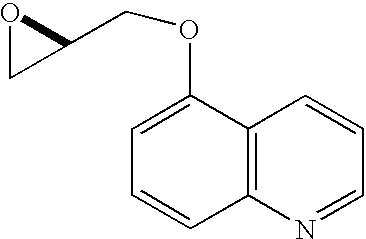
A mixture of 5-hydroxyquinoline (5.60 g, 38.6 mmol), R-glycidyl nosylate (10.0 g, 38.6 mmol), powdered potassium carbonate (11.7 g, 84.9 mmol), and N,N-dimethylformamide (100 mL) was stirred at ambient temperature until HPLC analysis (40% acetonitrile/60% of a 0.5% aqueous ammonium acetate solution, 1 mL/min, wavelength=230 nm, Zorbax RX-C8 25 cm×4.6 mm column) indicated complete disappearance of glycidyl nosylate (approximately 6 hours). The reaction mixture was filtered through paper and the filter cake was washed with 200 mL of a 3:1 mixture of MTBE and methylene chloride. The filtrate was washed with 200 mL of water and the aqueous layer was extracted four times with 100 mL of 3:1 MTBE/methylene chloride. The combined organic layers were dried over 30 grams of magnesium sulfate and the dried solution was then stirred with 50 grams of basic alumina for 30 minutes. The alumina was removed by filtration and the filter cake was washed with 200 mL of 3:1 MTBE/methylene chloride. The filtrate was concentrated to a volume of 100 mL, 300 mL of MTBE were added, and the solution was again concentrated to 80 mL. After heating to 50° C., the solution was treated with 160 mL of heptane dropwise over 15 minutes, allowed to cool to 40° C., and seeded, causing the formation of a crystalline precipitate. The mixture was stirred for two hours at ambient temperature and then at 0-5° C. for an additional 2 hours. The crystals were filtered, washed with cold heptane, and dried to provide 5.68 g (73.2%) of (2R)-1-(5-quinolinyloxy)-2,3-epoxypropane as white needles.
mp 79-81° C.;
[α]25 D−36.4° (c 2.1, EtOH);
1H NMR (500 MHz, CDCl3)δ 2.83 (dd, J=4.8, 2.7 Hz, 1H), 2.97 (m, 1H), 3.48 (m, 1H), 4.10 (dd, J=11.0, 6.0 Hz, 1H), 4.43 (dd, J=11.0, 2.7 Hz, 1H), 6.85 (d, J=7.8 Hz, 1H), 7.38 (dd, J=8.5 Hz, 4.1 Hz, 1H), 7.59 (m, 1H), 7.71 (d, J=8.5 Hz, 1H), 8.61 (m, 1H), 8.90 (m, 1H).
Example 1 (2R)-Anti-1-[4-(10,11-difluoromethano-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)-piperazin-1-yl]-3-qunolin-5-yloxy)-propan-2-ol Trihydrochloride

Preparation of the above compound is exemplified in the following preparative steps.
Step 1 1,1-Difluoro-1a,10b-dihydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6 (1H)-one

A solution of sodium chlorodifluoroacetate (350 g) in diglyme (1400 mL) was added dropwise over 4 to 8 hours, preferably over 6 hours, to a solution of 5H-dibenzo[a,d]cyclo-hepten-5-one (25 g) in diglyme (500 mL), with stirring, and under nitrogen, maintaining the reaction temperature at 160°-165° C. The cooled reaction mixture was poured into water (1.8 L) and extracted with ether (1.8 L). The organic phase was washed with water, dried over sodium sulfate (Na2SO4), and evaporated. The residue was recrystallized from ethanol, then from acetone/hexane to give 14 g of 1,1-difluoro-1a,10b-dihydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6(1H)-one.
mp 149.6° C.
Flash chromatography of the combined mother liquors on silica gel, eluting with 20% acetone/hexane, gave an additional 6.5 g of the target compound.
Step 2 (1aα,6β,10bα)-1,1-Difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cyclohepten-6-ol

A solution of 1,1-difluoro-1a,10b-dihydro-dibenzo[a,e]cyclopropa[c]cyclohepten-6(1H)-one (20.4 g) in tetrahydrofuran/methanol (1:2, 900 mL) was cooled in an ice bath. Sodium borohydride (12 g) was added in portions. The cooling bath was removed and the reaction mixture was stirred at ambient temperature for 2 hours, then poured into water. The product was filtered off, washed with water, and dried to give 20 g of (1aα,6β,10bα)-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cyclohepten-6-ol (ii).
mp 230.1°-230.6° C.
Step 2A Combined Steps 1 and 2 Procedure (1aα,6β,10bα)-1,1-Difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cyclohepten-6-ol

To a solution of 103.1 g (0.500 mol) of 5H-dibenzo[a,d]cyclohepten-5-one (2) in 515 mL of triethylene glycol dimethyl ether heated to between 180° C. and 210° C. was added over 7 hours, 293.3 g (2.15 mol) of chlorodifluoroacetic acid lithium salt (as a 53% by weight solution in ethylene glycol dimethyl ether). The ethylene glycol dimethyl ether was allowed to distill from the reaction as the salt addition proceeded. The GC analysis of an aliquot indicated that all of the 5H-dibenzo[a,d]cyclohepten-5-one had been consumed. The reaction was cooled to ambient temperature and then combined with 400 mL of ethyl acetate and 75 g of diatomaceous earth. The solids were removed by filtration and washed with 300 mL of ethyl acetate. The washes and filtrate were combined and the ethyl acetate was removed by concentration under vacuum leaving 635 g of dark liquid. The dark liquid was cooled to 18° C. and to this was added, over 15 minutes, 6.62 g (0.175 mol) of sodium borohydride (as a 12% by wt solution in 14 M NaOH). After stirring for 2 h the reaction was quenched by careful addition of 900 mL of a 1:3.5:4.5 solution of conc. HCl-methanol-water. The suspension was stirred for 30 min and the crude product was collected by filtration, washed with 600 mL of 1:1 methanol-water and dried to 126.4 g of dark brown solid. The crude product was slurried in 600 mL of methylene chloride, filtered, washed twice with 150 mL portions of methylene chloride, and dried to 91.6 g (71%) of (1aα,6β,10bα)-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-ol. Gas Chromatography (GC) Conditions; Column: JW Scientific DB-1, Initial Temperature 150° C. for 5 min, 10° C./min ramp, Final temp 250° C. for 5 min. tR: intermediate, 11.5 min; reaction product (alcohol), 11.9 min; starting material, 12.3 minutes.
Step 3 Preparation of (1aα,6α,10b)-6-bromo-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa-[c]cycloheptene

A slurry of (1aα,6β,10bα)-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-ol (3.0 g, 11.6 mmol, 1.0 equiv) in heptane (24 mL) was treated with 48% HBr (1.58 mL, 14.0 mmol, 1.2 equiv) and the reaction was heated at reflux with vigorous stirring for 2.5 hr. Solvent was then removed by atmospheric distillation (bp 95-98° C.) until approximately 9 mL of distillate was collected. The reaction was cooled and treated with EtOAc (15 mL), Na2SO4 and activated charcoal. The mixture was stirred at RT for 15 min and filtered through hyflo. The filter cake was washed with 50:50 EtOAc:heptane and the filtrate was concentrated in vacuo to provide the title product as a crystalline solid.
mp 119° C. (3.46 g corr., 93%);
1H NMR (500 MHz CDCl3) δ 7.20-7.41 (8H, m), 5.81 (1H, s), 3.41 (2H, d, J 12.5 Hz);
13CNMR (126 MHz CDCl3) δ 141.3, 141.2, 133.5, 130.1, 129.8, 128.3, 128.2, 112.9, 110.6, 110.5, 108.3, 53.6, 30.2, 30.1, 30.0.
Anal. Calcd. For C16H11BrF2: C, 59.84; H, 3.45. Found: C, 60.13; H, 3.50.
Step 3A Preparation of (1aα,6α,10bα)-6-Bromo-1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cycloheptene

To a stirred suspension of (1aα,6β,10bα)-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-ol, (18.4 g, 71.2 mmol) in 151 mL of methylene chloride which had been cooled to 10-17° C. was added phosphorous tribromide (9.6 g, 35.6 mmol) dropwise over 15 minutes. The cooling bath was removed and the reaction was stirred for 2 hours at ambient temperature. Analysis by gas chromatography indicated complete consumption of starting material. Cold water (92 mL) and activated carbon (1.84 g) were added and the resulting mixture was stirred for 30 minutes. The activated carbon was removed by filtration through Hyflo brand filter aid and the two phases were separated. The organic phase was washed with water (184 mL×2), brine (184 ml), dried over magnesium sulfate and concentrated to dryness under vacuum, affording 21.7 g (94.8%) of (1aα,6α,10bα)-6-bromo-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cycloheptene.
1H NMR (CDCl3, 300 MHz) δ 3.36 (s, 1H), 3.40 (s, 1H), 5.77 (s, 1H), 7.16-7.38 (m, 8H).
Steps 4 and 5 (1aα,6α,10bα)-1-(1,1-Difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cyclohepten-6-yl)-piperazine, Hydrobromide Salt

To a solution of 237.5 g (0.739 mol) of (1aα,6α,10bα)-6-bromo-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]-cyclopropa[c]cycloheptene in 3.56 L of acetonitrile was added 207.7 g (2.41 mol) of piperazine and the mixture was heated to reflux for 2 hours, at which time analysis by gas chromatography showed complete consumption of (1aα,6α,10bα)-6-bromo-1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cycloheptene (iii) and formation of a mixture of syn and anti piperazine compounds (III and IV) in an anti-syn ratio of 55:45. The reaction was cooled to about 7° C. and stirred for 30 minutes at that temperature. The reaction mixture was filtered to remove the precipitated syn-isomer (III) and the filter cake was washed with 250 mL of acetonitrile. The combined filtrate and wash were concentrated under vacuum to 262.4 grams of a foam which was dissolved in 450 mL of acetonitrile with heating. The solution was cooled to about 12° C. in an ice bath and stirred for 1 hour at that temperature. The precipitated syn-piperazine compound of formula (III) was filtered and washed with 125 ml of acetonitrile. The combined filtrate and wash were concentrated under vacuum to 194.1 g and dissolved in 1.19 L of ethyl acetate. The organic solution was washed sequentially with 500 mL portions of 1N sodium hydroxide, water, and saturated sodium chloride. The ethyl acetate solution was dried over sodium sulfate and concentrated to give 137.0 grams of residue which was dissolved in 1.37 L of methylene chloride and seeded with (1aα,6α,10bα)-1-(1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-yl)-piperazine, hydrobromide salt, followed by the addition of 70.8 grams of 48% aqueous hydrobromic acid. The mixture was stirred for about 45 minutes, causing the anti-isomer to crystallize as its hydrobromide salt. The crystals were filtered, washed with methylene chloride, and dried to provide purified hydrobromide salt of compound (IVa), shown by HPLC to have an anti-syn ratio of 99.3:0.7. Treatment of the isolated hydrobromide salt of compound (IVa) with aqueous sodium hydroxide, extraction into methylene chloride, separation of the aqueous layer and concentration to dryness gave 80.1 grams (33.2% yield based on starting material) of (1aα,6α,10bα)-1-(1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-yl)-piperazine as the free base. Acidification of a solution of the free base in 800 mL of methylene chloride by addition of 41.2 g of 48% hydrobromic acid as described above afforded 96.4 g of pure hydrobromide salt (title compound) with an anti-syn ratio of 99.8:0.2 (HPLC), mp 282-284° C. 1H NMR (DMSO-d6) δ 2.41 (m, 4H), 3.11 (m, 4H), 3.48 (d, J=12.4 Hz, 2H), 4.13 (s, 1H), 7.2 (m, 8H), 8.65 (bs, 2H). 13C NMR (DMSO-d6) δ 28.0, 42.9, 48.0, 75.1, 108.5, 112.9, 117.3, 127.5, 128.0, 128.6, 129.6, 132.4, 141.3. IR: (KBr) 3019, 2481, 1587, 1497, 1298 cm−1. Anal. Calcd for C20H21BrF2N2: C, 58.98; H, 5.20; N, 6.88. Found: C, 58.75; H, 5.29; N, 7.05.
Step 6 Preparation of (2R)-Anti-1-[4-(10,11-difluoromethano-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)-piperazin-1-yl]-3-quinolin-5-yloxy)propan-2-ol Trihydrochloride
A suspension of (1aα,6α,10bα)-1-(1,1-difluoro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]-cyclohepten-6-yl)-piperazine, hydrochloride compound of formula IVa (5.41 g, 14.9 mmol) and powdered sodium carbonate (3.16 g, 29.8 mmol) in 54 mL of 3A ethanol was stirred at ambient temperature for 1 hour. R-1-(5-quinolinyloxy)-2,3-epoxypropane (3.00 g, 14.9 mmol) was added in one portion and the reaction mixture was heated to 65° C. for 19 hours. HPLC analysis (Gradient system with solvent A (acetonitrile) and solvent B (0.02M sodium monophosphate buffer containing 0.1% triethylamine adjusted to pH 3.5 with phosphoric acid) as follows: 0-12 min, 30% solvent A/70% solvent B; 12-30 min, linear gradient from 30% to 55% solvent A/70% to 45% solvent B; 30-35 min, 55% solvent A/45% solvent B, 1 mL/min, 1=240 nm, Synchropak SCD-100 25 cm×4.6 mm column) indicated the total consumption of the piperazinyl compound of formula (IV). The mixture was allowed to cool to room temperature, filtered through a plug of silica gel, and eluted with an additional 90 mL of ethanol. The eluent was concentrated to a volume of approximately 60 mL and heated to 65° C. with stirring. A solution of HCl in ethanol (16.1 g at 0.135 g/g of solution, 59.6 mmol) was added dropwise over 10 minutes and the resultant product solution was seeded, causing the trihydrochloride salt to precipitate. The mixture was allowed to cool to ambient temperature and stirred slowly (less than 100 RPM) for 2 hours. The precipitate was filtered, washed with ethanol, and dried in vacuo at 50° C. to give the crude trihydrochloride salt which was further purified by recrystallization from methanol/ethyl acetate to provide 7.45 g (78.4%) of (2R)-anti-1-[4-(10,11-difluoromethano-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)-piperazin-1-yl]-3-quinolin-5-yloxy)-propan-2-ol trihydrochloride.
Step 6a
The syn isomer compound of formula (III) isolated as described supra (combined steps 4 and 5), can be utilized to produce the corresponding syn-5-{3-[4-(10,11-difluoromethano-dibenzosuber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinoline trihydrochloride (XII) essentially as shown below for the free base of the anti isomer (IVa)in step 6.
https://www.google.co.in/patents/US6570016?cl=en
………………………………………
http://www.google.it/patents/WO1994024107A1?cl=en
REACTION SCHEME 1

FormuIa 1
Formula 1

Formula 2 Formula 2

Formula 3
Formula 3

Formula 4

Formula I
……………………………………….
http://www.google.com/patents/WO2000075121A3

1HNMR (500 MHz DMSO-d6) δ9.41 (2H, br. s), 7.17-7.31 (8H, m), 4.17 (1H, s), 3.52 (2H, d, J=12.4 Hz), 3.11 (4H, br. s), 2.48-2.51 (4H, m)
13CNMR (126 MHz DMSO-d6) δ142.3, 133.4, 130.5, 129.6, 129.0, 128.4, 115.9, 113.6, 111.3, 76.2, 49.0, 43.6, 29.2, 29.1, 29.0; FD MS: m/e 326 (M+).
Anal. Calcd. For C20H21ClF2N2: C, 66.20; H, 5.83; N, 7.72.
Found: C, 66.08; H, 5.90; N, 7.72.
…………………………………………..
http://www.google.com/patents/US6570016?cl=fr


(2R)-Anti-1-[4-(10,11-difluoromethano-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5-yl)-piperazin-1-yl]-3-qunolin-5-yloxy)-propan-2-ol Trihydrochloride
……………….
Chemical Shift Data and Peak Assignments for the Crystal Forms.

Form II has a solid-state 13C NMR spectrum comprised of isotropic peaks at the following chemical shifts: 29.9, 50.1, 55.3, 62.0, 66.5, 72.0, 75.8, 104.8, 107.5, 108.2, 109.1, 110.2, 112.0, 118.4, 119.5, 120.1, 123.1, 128.7, 131.1, 133.0, 134.8, 136.4, 136.9, 139.9, 140.0, 142.3, 144.5, 146.6, 149.0, 144.2, 153.0 and 153.6 ppm.
Form III has a solid-state 13C NMR spectrum comprised of isotropic peaks at the following chemical shifts: 30.3, 50.4, 59.1, 63.2, 72.8, 77.2, 109.1, 110.2, 112.2, 112.8, 118.7, 119.5, 119.9, 121.0, 122.2, 123.0, 128.9, 130.6, 132.7, 134.0, 136.4, 140.0, 141.0, 141.8, 142.5, 143.3, 146.1, 153.1, 153.8 and 154.7 ppm.
Sprout Pharmaceuticals Appeals FDA Decision on NDA for Flibanserin to Treat Hypoactive Sexual Desire Disorder in Premenopausal Women

Flibanserin, girosa
167933-07-5 cas no
147359-76-0 (monoHCl)
- Bimt 17
- BIMT 17 BS
- Bimt-17
- Flibanserin
- Girosa
- UNII-37JK4STR6Z
December 11, 2013 – Sprout Pharmaceuticals today announced that it has received and appealed the Food and Drug Administration’s (FDA) Complete Response Letter (CRL) for flibanserin through the Formal Dispute Resolution process.
Flibanserin is an investigational, once-daily treatment for Hypoactive Sexual Desire Disorder, or HSDD, in premenopausal women. HSDD is the most commonly reported form of female sexual dysfunction
read all here
A new drug being developed by Boehringer Ingelheim could give a boost to the sex drive of women with low libido. The drug, known as flibanserin, has been shown in clinical trials to increase their sexual desire when taken once a day at bedtime.
The results from four pivotal Phase III clinical trials on women with hypoactive sexual desire disorder (HSDD) were presented this week at the European Society for Sexual Medicine’s congress in Lyon, France. The trials showed that participants taking flibanserin had a significant improvement in their sexual desire compared to those given a placebo. They also experienced less of the distress associated with sexual dysfunction.
The drug was initially being investigated as a treatment for depression, and acts on the serotonin receptors in the brain – it is both a 5-HT1A receptor agonist and a 5-HT2A receptor antagonist. It is also a partial agonist at the dopamine D4 receptor.

Neurotransmitters such as serotonin are believed to be involved in sexual function, and antidepressants are commonly associated with a loss of libido, so this was an obvious side-effect to look out for during clinical trials in depression. But far from suppressing the libido in women, it appeared to have the opposite effect, so trials in women with HSDD were initiated.
Hormone replacement can improve the libido of women who have had their ovaries removed, but there is no available drug to treat those who have not. There have been accusations that pharma companies invent new diseases like HSDD in order to sell more medicines, but according to Kathleen Segraves, an assistant professor at Case Western Reserve University in the US who has worked in the field of sexual functioning for many years, this is not the case here. HSDD is a very real disorder, she says, and the potential for a treatment for these women is very exciting.
Flibanserin (code name BIMT-17; proposed trade name Girosa) is a drug that was investigated by Boehringer Ingelheim as a novel, non-hormonal treatment for pre-menopausal women with Hypoactive Sexual Desire Disorder (HSDD).[1][2] Development was terminated in October 2010 following a negative report by the U.S. Food and Drug Administration.[3]
HSDD is the most commonly reported female sexual complaint and characterized by a decrease in sexual desire that causes marked personal distress and/or personal difficulties. According to prevalence studies about 1 in 10 women reported low sexual desire with associated distress, which may be HSDD.[4] The neurobiological pathway of female sexual desire involves interactions among multiple neurotransmitters, sex hormones and various psychosocial factors. Sexual desire is modulated in distinct brain areas by a balance between inhibitory and excitatory neurotransmitters, serotonin acting as an inhibitor while dopamine and norepinephrine act as a stimulator of sexual desire.[5][6]Flibanserin is a 5-HT1A receptor agonist and 5-HT2A receptor antagonist that had initially been investigated as an antidepressant. Preclinical evidence suggested that flibanserin targets these receptors preferentially in selective brain areas and helps to restore a balance between these inhibitory and excitatory effects.[6] HSDD has been recognized as a distinct sexual function disorder for more than 30 years.
The proposed mechanism of action refers back to the Kinsey dual control model. Several sex steroids, neurotransmitters, and hormones have important excitatory or inhibitory effects on the sexual response. Among the neurotransmitters, the excitatory activity is driven by dopamine and norepinephrine, while the inhibitory activity is driven by serotonin. The balance between these systems is relevant for a healthy sexual response. By modulating these neurotransmitters in selective brain areas, flibanserin, a 5-HT1A receptoragonist and 5-HT2A receptor antagonist, is likely to restore the balance between these neurotransmitter systems.[6]
Several large pivotal Phase III studies with Flibanserin were conducted in the USA, Canada and Europe. They involved more than 5,000 pre-menopausal women with generalized acquired Hypoactive Sexual Desire Disorder (HSDD). The results of the Phase III North American Trials demonstrated that
Although the two North American trials that used the flibanserin 100 mg qhs dose showed a statistically significant difference between flibanserin and placebo for the endpoint of [satisfying sexual events], they both failed to demonstrate a statistically significant improvement on the co-primary endpoint of sexual desire. Therefore, neither study met the agreed-upon criteria for success in establishing the efficacy of flibanserin for the treatment of [Hypoactive Sexual Desire Disorder].
These data were first presented on November 16, 2009 at the congress of the European Society for Sexual Medicine in Lyon, France. The women receiving Flibanserin reported that the average number of times they had “satisfying sexual events” rose from 2.8 to 4.5 times a month. However, women receiving placebo reported also an increase of “satisfying sexual events” from 2.7 to 3.7 times a month.
Evaluation of the overall improvement of their condition and whether the benefit was meaningful to the women, showed a significantly higher rate of a meaningful benefit in the flibanserin-treated patient group versus the placebo group.The onset of the Flibanserin effect was seen from the first timepoint measured after 4 weeks of treatment and maintained throughout the treatment period.
The overall incidence of adverse events among women taking flibanserin was low, the majority of adverse events being mild to moderate and resolved during the treatment. The most commonly reported adverse events included dizziness, nausea, fatigue, somnolence and insomnia.

On June 18, 2010, a federal advisory panel to the U.S. Food and Drug Administration (FDA) unanimously voted against recommending approval of Flibanserin.
Earlier in the week, a FDA staff report also recommended non-approval of the drug. While the FDA still might approve Flibanserin, in the past, negative panel votes tended to cause the FDA not to approve.
On October 8, 2010, Boehringer Ingelheim announced that it would discontinue its development of flibanserin in light of the FDA advisory panel’s recommendation.
On June 27, 2013, Sprout Pharmaceuticals confirmed they had resubmitted flibanserin for FDA approval.
Flibanserin, chemically 1 -[2-(4-(3-trifluoromethylphenyl)piperazin-1 – yl)ethyl]-2,3-dihydro-1 H-benzimidazole-2-one was disclosed in form of its hydrochloride in European Patent No. 526,434 (‘434) and has the following chemical structure:

Process for preparation of flibanserin were disclosed in European Patent No. ‘434, U.S. Application Publication No. 2007/0032655 and Drugs of the future 1998, 23(1): 9-16.
According to European Patent No. ‘434 flibanserin is prepared by condensing 1-(2-chloroethyl)-2,3-dihydro-1 H-benzimidazol-one with m- trifluoromethyl phenyl piperazine. According to U.S. Application Publication No. 2007/0032655 flibanserin is prepared by condensing 1-[(3-trifluoromethyl)phenyl]-4-(2- chloroethyl)piperazine with 1 -(2-propenyl)-1 ,3-dihydro-benzimidazol-2H-one.
According to Drugs of the future 1998, 23(1): 9-16 flibanserin is prepared by reacting 1-(2-chloroethyl)-2,3-dihydro-1 H-benzimidazol-one with m- trifluoromethylphenylpiperazine.
…………………
1-[2-(4-(3-trifluoromethyl-phenyl)piperazin-1-yl)ethyl]-2,3-dihydro-1H-benzimidazol-2-one
Compound 3
- Hydrochloride salt (isopropanol) M.p. 230-231°C
Analysis
-

¹H NMR (DMSO-d₆/CDCL₃ 5:2) 11.09 (b, 1H), 11.04 (s, 1H), 7.5-6.9 (8H), 4.36 (t, 2H), 4.1-3.1 (10H)

…………………………………

The compound 1-[2-(4-(3-trifluoromethyl-phenyl)piperazin-1-yl)ethyl]-2,3-dihydro-1 H- benzimidazol-2-one (flibanserin) is disclosed in form of its hydrochlorid in European Patent Application EP-A-526434 and has the following chemical structure:

Flibanserin shows affinity for the 5-HTιA and 5-HT2-receptor. It is therefore a promising therapeutic agent for the treatment of a variety of diseases, for instance depression, schizophrenia, Parkinson, anxiety, sleep disturbances, sexual and mental disorders and age associated memory impairment.

EXAMPLE……… EP1518858A1
375 kg of 1-[(3-trifluoromethyl)phenyl]-4-(2-cloroethyl)piperazin are charged in a reactor with 2500 kg of water and 200 kg of aqueous Sodium Hydroxide 45%. Under stirring 169.2 kg of 1-(2-propenyl)-1,3-dihydro-benzimidazol-2H-one, 780 kg of isopropanol, 2000 kg of water and 220 kg of aqueous Sodium Hydroxide 45% are added. The reaction mixture is heated to 75-85° C. and 160 kg of concentrated hydrochloric acid and 200 kg of water are added.
The reaction mixture is stirred at constant temperature for about 45 minutes. After distillation of a mixture of water and Isopropanol (about 3000 kg) the remaining residue is cooled to about 65-75° C. and the pH is adjusted to 6.5-7.5 by addition of 125 kg of aqueous Sodium Hydroxide 45%. After cooling to a temperature of 45-50° C., the pH value is adjusted to 8-9 by addition of about 4 kg of aqueous Sodium Hydroxide 45%. Subsequently the mixture is cooled to 30-35° C. and centrifuged. The residue thus obtained is washed with 340 l of water and 126 l of isopropanol and then with water until chlorides elimination.
The wet product is dried under vacuum at a temperature of about 45-55° C. which leads to 358 kg of crude flibanserin polymorph A. The crude product thus obtained is loaded in a reactor with 1750 kg of Acetone and the resulting mixture is heated under stirring until reflux. The obtained solution is filtered and the filtrate is concentrated by distillation. The temperature is maintained for about 1 hour 0-5° C., then the precipitate solid is isolated by filtration and dried at 55° C. for at least 12 hours.
The final yield is 280 kg of pure flibanserin polymorph A.
………………………….
Flibanserin may be prepared by reacting 1-(phenylvinyl)-2,3-dihydro-1H-benzimidazol-2-one (I) with 1,2-dichloroethane (II) in the presence of NaH in warm dimethylformamide. The resulting 1-(2-chloroethyl)-2,3-dihydro-1H-benzimidazol-one (III) is in turn coupled with commercially available m-trifluoromethylphenylpiperazine hydrochloride (IV) in the presence of sodium carbonate and catalytic potassium iodide in refluxing ethanol. The crude flibanserin hydrochloride (V) is then dissolved in aqueous ethanol and the pure base is precipitated upon addition of sodium hydroxide.
PICK UP INTERMEDIATES FROM CHEM24H.COM
1-(1-phenylvinyl)-1,3-dihydro-2H-benzimidazol-2-one (I)
1,2-dichloroethane (II)
1-(2-chloroethyl)-1,3-dihydro-2H-benzimidazol-2-one (III)
1-[3-(trifluoromethyl)phenyl]piperazine; N-[3-(trifluoromethyl)phenyl]piperazine (IV)
1-(2-[4-[3-(trifluoromethyl)phenyl]piperazino]ethyl)-1,3-dihydro-2H-benzimidazol-2-one (V)
………………………..


wherein R2 is as defined in formula X; with a compound of formula Xl:

According to another aspect of the present invention there is provided a novel compound or a salt thereof selected from the compounds of formula I, IV and VII:


Wherein R is hydrogen or an amino protecting group.
Preferable the amino protecting groups are selected from butyl, 1 ,1- diphenylmethyl, methoxymethyl, benzyloxymethyl, trichloroethoxymethyl, pyrrolidinomethyl, cyanomethyl, pivaloyloxymethyl, allyl, 2-propenyl, t- butyldimethylsilyl, methoxy, thiomethyl, phenylvinyl, 4-methoxyphenyl, benzyl, A- methoxybenzyl, 2,4-dimethoxybenzyl, 2-nitrobenzyl, t-butoxycarbonyl, benzyloxycarbonyl, phenoxycarbonyl, 4-chlorophenoxycarbonyl, A- nitrophenoxycarbonyl, methoxycarbonyl and ethoxycarbonyl. Still more preferable protecting groups are selected from t- butoxycarbonyl, ethoxycarbonyl, methoxycarbonyl, benzyloxycarbonyl, phenoxycarbonyl, phenylvinyl and 2-propenyl.
R1 is independently selected from chlorine, bromine, iodine, methanesulphonate, trifluoromethanesulphonate, paratoluenesulphonate or benzenesulphonate. Preferable R1 is independently selected from chlorine, bromine or iodine and more preferable R1 is chlorine.
Wherein R2 is hydrogen or an amino protecting group.
The amino protecting group may be any of the groups commonly used to protect the amino function such as alkyl, substituted alkyl, hetero substituted alkyl, substituted or unsubstituted unsaturated alkyl, alkyl substituted hetero atoms, substituted or unsubstituted phenyl, substituted or unsubstituted benzyl, alkyoxy carbonyl groups and aryloxy carbonyl groups.
Preferable the amino protecting groups are selected from butyl, 1 ,1 – diphenylmethyl, methoxymethyl, benzyloxymethyl, trichloroethoxymethyl, pyrrolidinomethyl, cyanomethyl, pivaloyloxymethyl, allyl, 2-propenyl, t- butyldimethylsilyl, methoxy, thiomethyl, phenylvinyl, 4-methoxyphenyl, benzyl, A- methoxybenzyl, 2,4-dimethoxybenzyl, 2-nitrobenzyl, t-butoxycarbonyl, benzyloxycarbonyl, phenoxycarbonyl, 4-chlorophenoxycarbonyl, A- nitrophenoxycarbonyl, methoxycarbonyl and ethoxycarbonyl. Still more preferable protecting groups are selected from t- butoxycarbonyl, ethoxycarbonyl, methoxycarbonyl, benzyloxycarbonyl, phenoxycarbonyl, phenylvinyl and 2-propenyl. The following examples are given for the purpose of illustrating the present invention and should not be considered as limitations on the scope and spirit of the invention.
EXAMPLES Example 1
A mixture of sodium hydroxide (47 gm) and i-(α-methylvinyl) benzimidazol-2-one (100 gm) in dimethylformamide (400 ml) was .stirred for 1 hour at room temperature. Dibromoethane (217 gm) was slowly added to the mixture and stirred at 1 hour 30 minutes. The resulting solution after addition water (500 ml) was extracted with ethyl acetate. The combined ethyl acetate extract washed with water. After drying the solvent was removed under vacuum to yield 132 gm of 1 ,3-dihydro-1-(2-bromoethyl)-3-isopropenyl-2H-benzimidazol- 2-one as a yellow oily liquid.
Example 2 A mixture of 1 ,3-dihydro-1-(2-bromoethyl)-3-isopropenyl-2H- benzimidazol-2-one (100 gm), diethanolamine (175 ml), sodium carbonate (40 gm) and potassium iodide (10 gm) was heated to 90 to 95 deg C and stirred for 2 hours. The reaction mass was cooled to room temperature and added water (500 ml). The resulting mixture extracted into ethyl acetate and the organic layer washed with water. After drying the solvent was removed under vacuum to yield 105 gm of 1 ,3-dihydro-1-[2-[N-bis-(2-hydroxyethyl)amino]ethyl]-3-isopropenyl- 2H-benzimidazol-2-one as a thick yellow oily liquid.
Example 3
To the mixture of 1 ,3-dihydro-1-[2-[N-bis-(2-hydroxyethyl)amino]ethyl]-3- isopropenyl-2H-benzimidazol-2-one (100 gm) obtained as in example 2 and chloroform (300 ml), thionyl chloride (95 ml) was slowly added. The mixture was heated to reflux and stirred for 2 hours. The excess thionyl chloride and chloroform was distilled off to yield 98 gm of 1 ,3-dihydro-1-[2-[N-[bis-(2- chloroethyl)amino]ethyl]-3-isopropenyl-2H-benzimidazol-2-one as a brown coloured sticky residue.
Example 4
1 ,3-dihydro-1-[2-[N-[bis-(2-chloroethyl)amino]ethyl]-3-isopropenyl-2H- benzimidazol-2-one (98 gm) obtained as in example 3 was added to water (500 ml) and concentrated hydrochloric acid (200 ml) mixture. The mixture was heated to 60 to 65 deg C and stirred for 1 hour. The contents of the flask cooled to room temperature and pH of the solution adjusted to 9 – 10 with 10% sodium hydroxide solution. The resulting solution extracted with ethyl acetate and washed the organic layer with water. Evaporate the solvent under reduced pressure to yield 82 gm of 1 ,3-dihydro-1-[2-[N-bis-(2-chloroethyl)amino]ethyl]- 2H-benzimidazol-2-one as a dark brown coloured oily liquid
Example 5
A mixture of 1 ,3-dihydro-1-[2-[N-bis-(2-chloroethyl)amino]ethyl]-1,2-H- benzimidazol-2-one (82 gm) obtained as in example 4, xylene (300 ml) and m- trifluoromethyl aniline (58 gm) was refluxed for 64 hours. The reaction mass was cooled to room temperature and filtered to obtain 1-[2-(4-(3- thfluoromethylphenyl)piperazin-1-yl)ethyl]-2,3-dihydro-1 H-benzimidazole-2-one hydrochloride (Flibanserin hydrochloride) as a light brown coloured solid.
The crude flibanserin hydrochloride was purified in isopropyl alcohol to give 85 gm of pure flibanserin hydrochloride as off white solid.
Example 6
Piperazine (12 gm), toluene(60 ml) and tetra butyl ammonium bromide (1 gm) mixture was heated to 60 deg C, added 1 ,3-dihydro-1-(2-bromoethyl)-3- isopropenyl-2H-benzimidazol-2-one (10 gm) and stirred for 4 hours at 90 to 95 deg C. The mixture was cooled to 60 deg C and added water (50 ml). The separated toluene layer distilled under vacuum to give 8.5 gm of 1 ,3-dihydro-1- (2-piperazinyl)ethyl-3-isopropenyl-2H-benzimidazol-2-one as a white solid.
Example 7
To the mixture of concentrated hydrochloric acid (20 ml) and water (100 ml) was added 1 ,3-dihydro-1-(2-piperazinylethyl)-3-isopropenyl-2H- benzimidazol-2-one (10 gm) obtained as in example 6 and heated to 60 to 65 deg C 1 hour. The mixture was cooled to room temperature and pH of the solution was adjusted to 9 – 10 with 10% sodium hydroxide solution, extracted with ethyl acetate and the organic layer was washed with water. After drying the solvent was removed under vacuum to yield 8.5 gm of 1 ,3-dihydro-1-(2- piperazinyl ethyl)-2H-benzimidazol-2-one as a white solid.
Example 8
3-trifluoromethylaniline (40 gm) and hydrobromic acid (85 ml; 48- 50%w/w) mixture was cooled to 0 to 5 deg C. To this mixture added sodium nitrite solution (18.5 gm in 25 ml of water) at 5 to 10 deg C and copper powder (1 gm). The temperature was slowly raised to 50 to 55 deg C and stirred for 30 minutes. Added water (200 ml) to reaction mass and applied steam distillation, collected m-trifluoromethylbromobenzene as oily liquid. The oily liquid washed with sulfuric acid for two times (2 X 10 ml) followed by washed with water (2 X 20 ml) and dried the liquid with sodium sulphate to give 22 gm of m- trifluoromethylbromobenzene.
Example 9
To a mixture of 1 ,3-dihydro-1-(2-piperazinyl ethyl)-2H-benzimidazol-2- one (10 gm) obtained as in example 7, m-trifluoromethylbromobenzene (9 gm) obtained as in example 8, sodium tert-butoxide (5.5 gm), palladium acetate (4.5 mg) and xylene (80 ml) was added tri-tert.-butylphosphine (0.2 ml). The mixture was heated to 120 deg C and stirred for 3 hours. The reaction mass was cooled, added water (100 ml) and extracted with ethyl acetate and the organic layer was washed with water. After drying the solvent was removed under vacuum to yield
10 gm of 1-[2-(4-(3-trifluoromethylphenyl)piperazin-1-yl)ethyl]-2,3-dihydro-1 H- benzimidazole-2-one (Flibanserin).
Example 10
To a mixture of 1 ,3-dihydro-1-[2-[N-bis-(2-hydroxyethyl)amino]ethyl]-3- isopropenyl-2H-benzimidazol-2-one (100 gm) obtained as in example 3, cyclohexane (400 ml) and sodium carbonate (35 gm) was added benzene sulfonyl chloride (116 gm) at room temperature. The mixture was heated to 80 to
85 deg C and stirred for 8 hours . The contents were cooled to room temperature and added water (500 ml). Distilled the organic layer to give 182 gm of 1 ,3-dihydro-1-[2-[N-[bis-(2-benzenesulfonyloxy)- ethyl]amino]ethyl]-3- isopropenyl- 2H-benzimidazol-2-one.
Example 11
1 ,3-dihydro-1 -[2-[N-[bis-(2-benzenesulfonyloxy)- ethyl]amino]ethyl]-3- isopropenyl- 2H-benzitηidazol-2-one (100 gm) obtained as in example 10, dimethylformamide (500 ml) and sodium corbonate (18 gm) was mixed and heated to 70 deg C. To the mixture was added m-trifluoromethyl aniline (27 gm) and heated to 80 to 85 deg C, stirred for 5 hours. The reaction mass was cooled and added water (2000 ml), filtered the solid to yield 1 ,3-dihydro-1-[2-[4-(3- trifluoromethylphenyl)piperazinyl]ethyl]-3-isopropenyl-2H benzimidazol-2-one. Example 12
1 ,3-dihydro-1-[2-[N-[bis-(2-benzenesulfonyloxy)- ethyl]amino]ethyl]-3- isopropenyl- 2H-benzimidazol-2-one (100 gm) obtained as in example 11 added to the mixture of water (500 ml) and concentrated hydrochloric acid (200 ml), heated to 65 deg C and stirred for 1 hour. The reaction mass was cooled to room temperature and pH adjusted to 10 to 10-5 with 10% sodium hydroxide solution. The resulting mixture was extracted with ethyl acetate and the organic
■ layer was washed with water. After drying the solvent was removed under vacuum to yield 87 gm of 1-[2-(4-(3-trifluoromethylphenyl)piperazin-1-yl)ethyl]- 2,3-dihydro-1 H-benzimidazole -2-one (Flibanserin).
…………………..
Paper
Journal of Pharmaceutical and Biomedical Analysis, v.57, 2012 Jan 5, p.104(5)
Isolation and structural elucidation of flibanserin as an adulterant in a health supplement used for female sexual performance enhancement
Low, Min-Yong et al
http://www.sciencedirect.com/science/article/pii/S0731708511004833

This proposed formula and structure was further confirmed by 1H and 13C NMR data which indicated the presence of 20 carbon atoms and 21 protons.
1H NMR
13C NMR
1D and 2DNMR data were used to assign the protons and carbon atoms.
In the1H NMR spectrum , a sharp singlet at 10.00 ppm integrating for one
proton is a typical proton attached to nitrogen. HMBC correlated this proton to C-2, C-4, and C-9 suggesting that it was H-3.
Complex signals were observedbetween 7.00 to 7.31 ppm, integrating for eight protons. A triplet at 7.31 ppm,integrating for a proton has a coupling constant of 8.0 Hz. HMBC correlated thisproton with C-16, C-19, and C-21 suggesting that it was H-20.
A double-doubletsplitting pattern at chemical shift 7.11 ppm, integrating for a proton, has couplingconstants of 6.3 Hz and 1.6 Hz.
HMBC correlated this proton to C-6, C-7, and C-9 showing that it was H-8. Overlapped signals were observed from 7.04 ppm to7.10 ppm, integrating for five protons. A double-doublet splitting pattern at 7.01ppm with coupling constant 8.0 Hz and 2.0 Hz, integrating for a proton was
observed.
HMBC correlated this proton to C-17 suggesting that it was either H-19or H-21. Four triplet signals were also observed from 2.73 ppm to 4.08 ppm,integrating for a total of twelve protons.
Two of these triplet signals at 2.74 ppmand 3.22 ppm integrated for four protons each, suggesting overlapping signals ofmethylene protons. This was further confirmed by 13C and DEPT NMR.
13C and DEPT NMR data showed the signals of four methylene, eight methineand six quaternary carbon atoms. The DEPT signals at 53.1 ppm and 48.6 ppmhave intensities which were double of those from the rest of the methylene carbonsignals, suggesting two methylene carbon atoms each contributing to the signal at 53.1 ppm and 48.6 ppm.
DEPT
HMQC results further indicated that these two methylene carbon signals at 53.1 ppm and 48.6 ppm were correlated to the protons signal at 2.73 ppm and 4.08 ppm respectively, which corresponded to four protons each. The finding confirmed overlapping methylene carbon signals (at 53.1 ppm and 48.6 ppm) and methylene proton signals (at 2.73 ppm and 4.08 ppm). Hence, the unknown compound has six methylene carbon atoms with a total of twelve methylene protons.
The chemical shifts of the twelve methylene protons suggested that they were attached to relatively electronegative atoms. It was speculated that the six methylene groups were attached to the nitrogen atoms and the electron withdrawing effect of these electronegative nitrogen atoms resulted in the deshielding of the protons. HMBC and COSY correlations were used to assign the rest of the protons
The 13C NMR data showed that there were two quaternary carbon at
155.6 ppm and 151.3 ppm. The carbon with chemical shift 155.6 ppm was C-2. Inthe structure of imidazolone, carbonyl carbon C-2 was attached to two nitrogenatoms which helped to withdraw electrons from oxygen to C-2. Hence, C-2 wasless deshielded as compared to a normal carbonyl carbon which has chemical shiftabove 170 ppm.
Eight methine carbons and two quaternary carbons with chemicalshifts above 108 ppm suggested the presence of two aromatic rings. Thequaternary carbon with chemical shift 125.4 ppm was C-22 which was attached tothree fluorine atoms. Due to the strong electron withdrawing effect of the fluorineatoms, C-22 was highly deshielded and had a high chemical shift.
The IR spectrum of the isolated compound showed absorption bands of amide (νC=O 1685 cm-1, νN-H (stretch) 3180 cm-1, νN-H (bending) 1610 cm-1), alkyl fluoride (νC-F1077 cm-1, 1112 cm-1, 1158 cm-1), aromatic ring (ν Ar-H 3028 cm-1, 3078 cm-1 andνC=C 1401 cm-1, 1446 cm-1, 1453 cm-1, 1468 cm-1, 1487 cm-1) and alkane (νC-H2891 cm-1, 2930 cm-1 2948 cm-).
COSY
……………………………….
US5576318, 1996
1 H NMR (DMSO-d6 /CDCL3 5:2) 11.09 (b, 1H), 11.04 (s, 1H), 7.5-6.9 (SH), 4.36 (t, 2H), 4.1-3.1 (10 H)
,,,,,,,,,,,,,,,,,,
- Borsini F, Evans K, Jason K, Rohde F, Alexander B, Pollentier S (summer 2002). “Pharmacology of flibanserin”. CNS Drug Rev. 8 (2): 117–142. doi:10.1111/j.1527-3458.2002.tb00219.x. PMID 12177684.
- Jolly E, Clayton A, Thorp J, Lewis-D’Agostino D, Wunderlich G, Lesko L (April 2008). “Design of Phase III pivotal trials of flibanserin in female Hypoactive Sexual Desire Disorder (HSDD)”. Sexologies 17 (Suppl 1): S133–4. doi:10.1016/S1158-1360(08)72886-X.
- Spiegel online: Pharmakonzern stoppt Lustpille für die Frau, 8 October 2010 (in German)
- Nygaard I (November 2008). “Sexual dysfunction prevalence rates: marketing or real?”. Obstet Gynecol 112 (5): 968–9.doi:10.1097/01.AOG.0000335775.68187.b2. PMID 18978094.
- Clayton AH (July 2010). “The pathophysiology of hypoactive sexual desire disorder in women”. Int J Gynaecol Obstet 110 (1): 7–11.doi:10.1016/j.ijgo.2010.02.014. PMID 20434725.
- Pfaus JG (June 2009). “Pathways of sexual desire”. J Sex Med 6 (6): 1506–33. doi:10.1111/j.1743-6109.2009.01309.x.PMID 19453889.
- Yves Aubert, Thesis, Leiden University. (Dec 11, 2012) Sex, aggression and pair-bond: a study on the serotonergic regulation of female sexual function in the marmoset monkey
- Viagra for women?
- Marazziti D, Palego L, Giromella A, et al. (June 2002). “Region-dependent effects of flibanserin and buspirone on adenylyl cyclase activity in the human brain”. Int. J. Neuropsychopharmacol. 5 (2): 131–40. doi:10.1017/S1461145702002869.PMID 12135537.
- Podhorna J, Brown RE (June 2000). “Flibanserin has anxiolytic effects without locomotor side effects in the infant rat ultrasonic vocalization model of anxiety”. Br J Pharmacol 130 (4): 739–746. doi:10.1038/sj.bjp.0703364. PMC 1572126.PMID 10864879.
- Brambilla A, Baschirotto A, Grippa N, Borsini F (December 1999). “Effect of flibanserin (BIMT 17), fluoxetine, 8-OH-DPAT and buspirone on serotonin synthesis in rat brain”. Eur Neuropsychopharmacol 10 (1): 63–7. doi:10.1016/S0924-977X(99)00056-5.PMID 10647099.
| EP0200322A1 * | Mar 18, 1986 | Nov 5, 1986 | H. Lundbeck A/S | Heterocyclic compounds |
| BE904945A1 * | Title not available | |||
| GB2023594A * | Title not available | |||
| US3472854 * | May 29, 1967 | Oct 14, 1969 | Sterling Drug Inc | 1-((benzimidazolyl)-lower-alkyl)-4-substituted-piperazines |
| US4954503 * | Sep 11, 1989 | Sep 4, 1990 | Hoechst-Roussel Pharmaceuticals, Inc. | 3-(1-substituted-4-piperazinyl)-1H-indazoles |
