
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....





















 Wherein R is -OH or -CI
Wherein R is -OH or -CI


- Betanis
- Myrbetriq
- UNII-MVR3JL3B2V
- YM 178
- YM178
Мирабегронميرابيغرون 米拉贝隆
Mirabegron (YM-178) is the first β3-adrenoceptor agonist that is clinically effective for overactive bladder. Mirabegron (0.3 and 1 mg/kg) inhibits mechanosensitive single-unit afferent activities (SAAs) of Aδ fibers in response to bladder filling. Mirabegron activates the β3 adrenergic receptor in the detrusor muscle in the bladder, which leads to muscle relaxation and an increase in bladder capacity. Mirabegron (YM-178) acts partly as an irreversible or quasi-irreversible metabolism-dependent inhibitor of CYP2D6. Mirabegron at a dose of 3 mg/kg i.v. decreased the frequency of rhythmic bladder contraction induced by intravesical filling with saline without suppressing its amplitude in anesthetized rats. Mirabegron decreases primary bladder afferent activity and bladder microcontractions in rats. Mirabegron (YM-178) also reduced non-micturition bladder contractions in an awake rat model of bladder outlet obstruction.
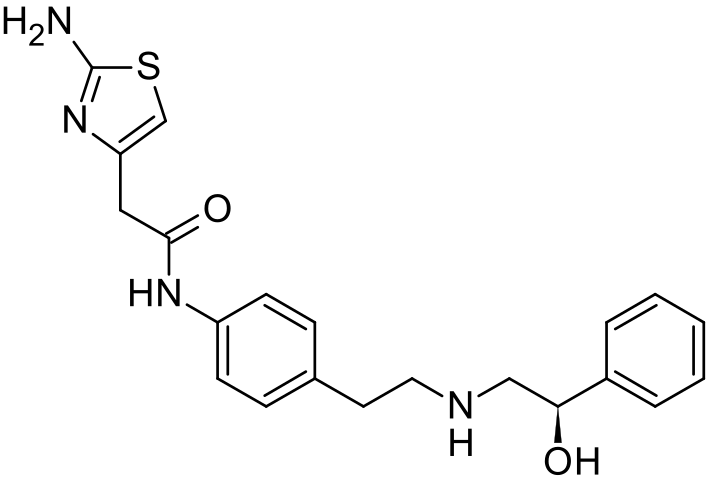
Mirabegron is a white crystalline powder, not hygroscopic and freely soluble in dimethyl sulfoxide, soluble in methanol and soluble in water between neutral to acidic pH. The chemical name is 2-(2- Amino-1,3-thiazol-4-yl)-N-[4-(2-{[(2R)-2-hydroxy-2- phenylethyl]amino}ethyl)phenyl]acetamide., Mirabegron exhibits stereoisomerism due to the presence of one chiral centre. The R enantiomer has been used in the manufacture of the finished product. The enantiomeric purity is controlled routinely by chiral HPLC-UV. Polymorphism has been observed for the active substance. The polymorphic form α is routinely and consistently produced by the synthetic process and it is used in the manufacture of the finished product…….http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002388/WC500137308.pdf
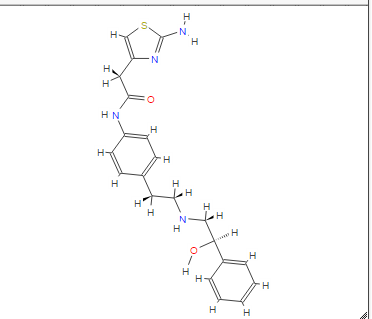
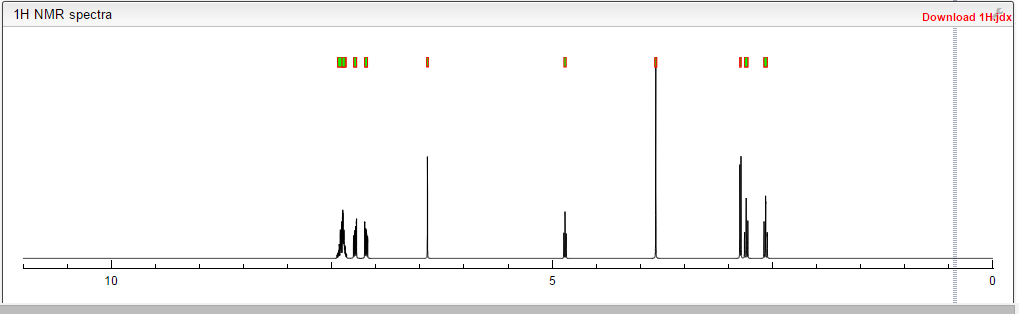
COSY PREDICT
 CN 103896872
CN 103896872http://www.google.com/patents/CN103896872A?cl=en


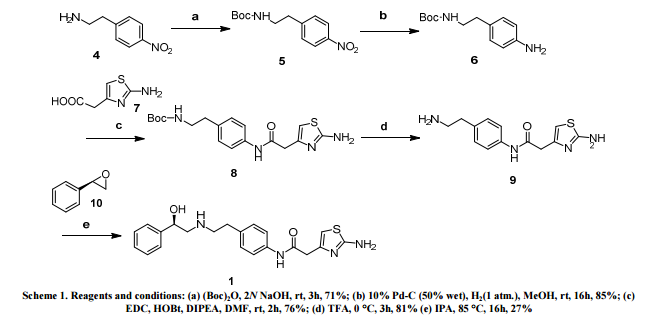
Third, Mira Veron synthesis:
reaction:

in 500mL three-necked flask, 2- (2-aminothiazol-4-yl) acetic acid 17.42g (0.086mol), N, N- dimethylformamide 180mL, then added H0BT15.12g (0.104 mol), was added (R) _2 _ ((4- aminophenyl) amino) phenyl-ethan-l-ol -1_ 20g (0.078mol), was added triethylamine 13.04g (0.13mol), was added portionwise EDCI21. 46g (0.104mol), under magnetic stirring, room temperature for 5h, TLC until the reaction was complete tracking.
After treatment: After the completion of the reaction, the reaction solution was poured into 900mL saturated saline water, and then extracted with 400mL of dichloromethane each time, and extracted three times, each time the organic phase is then washed with 200mL of saturated aqueous sodium carbonate solution, washed three times, each time with distilled water and then 200mL of water, washed three times, the organic phase was dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give a white solid in methylene chloride was distilled off Mira Veron crude, the crude product was recrystallized from methanol solution, wherein the methanol solution of methanol and water, the volume ratio of 10: 4, and recrystallized to give 25.08g, yield 81.0%.
The present embodiment Mira Veron synthesized for testing and structural identification:
mp138 ~ 140 ° C (137 ~ 139 ° C)
[α] 20-18. ~ -22. (CH3OH)
chemical purity HPLC: 99.96%
Optical purity: 97.55ee%
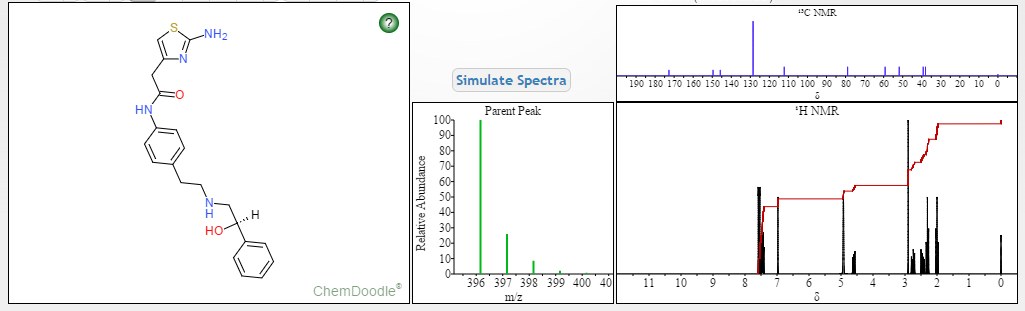
HRMS (ES1-MS, m / z) calcd: for C21H25N4O2S [M + H] + 397.16.Found:. 397.16
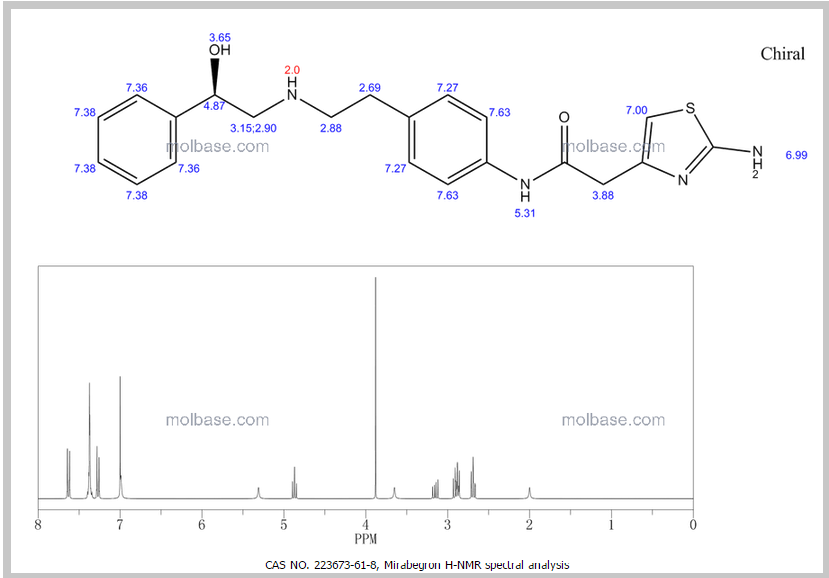
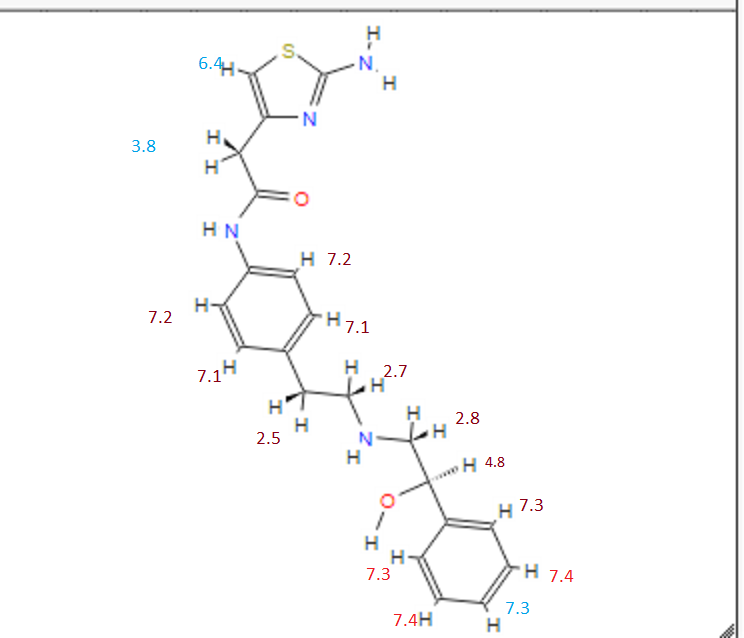
1H Mffi (400MHz, DMS0) Sl0.00 (s, lH), 7.50 ( d, J = 8.5Hz, 2H), 7.30 (dd, J = 9.5,5.1Hz, 4H), 7.23 (dd, J = 6.0, 2.7Hz, 1H), 7.12 (d, J = 8.5Hz, 2H), 6.90 (s, 2H), 6.30 (s, 1H), 5.24 (s, 1H), 4.60 (s, 1H), 3.45 (s, 2H), 2.74 (dd, J = 9.8, 3.5Hz, 2H), 2.64 (m, 4H).
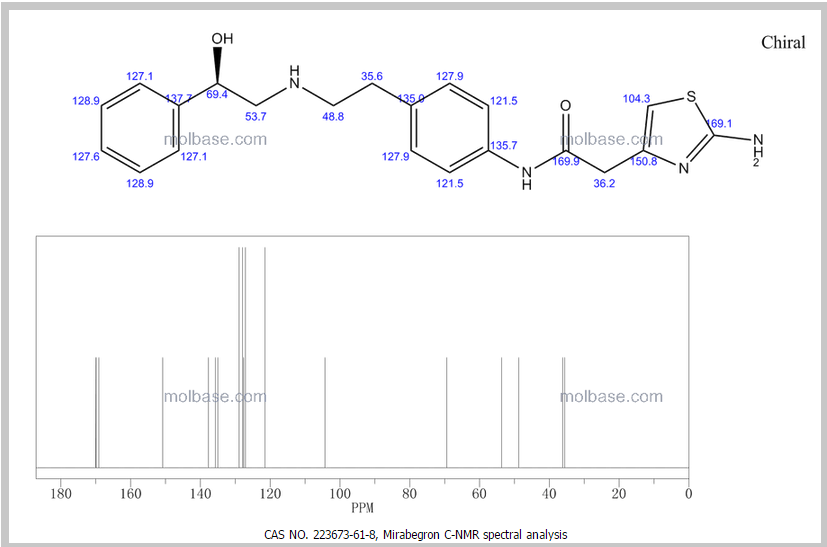
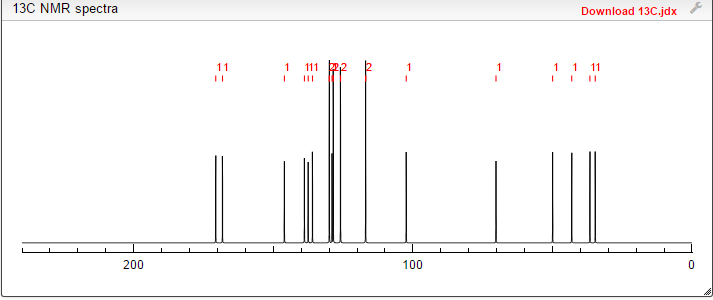
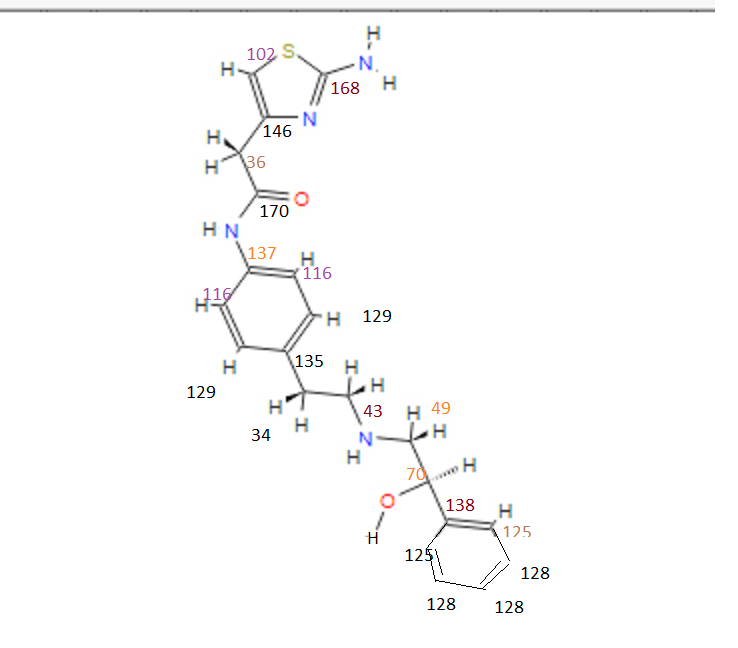
13C NMR (101MHz, DMSO) δ 168.69 (s), 168.26 (s), 146.35 (s), 145.03 (s), 137.66 (s), 135.51 (s), 129.24 (s ), 128.38 (s), 127.22 (s), 126.33 (s), 119.46 (s), 103.03 (s), 71.88 (s), 57.94 (s), 51.20 (s), 40.40 (s), 40.20 (s ), 39.99 (s), 39.78 (s), 39.57 (s), 35.77 (s)
1H NMR FIG2…SEE…….http://orgspectroscopyint.blogspot.in/2015/08/mirabegron.html

13C NMR FIG3

………….
CN 103193730
http://www.google.com/patents/CN103193730A?cl=en

By and O ° C under nitrogen protection temperature conditions, 7.3g (R) -2- amino _1_ benzeneethanol added 250mL three-necked flask, the stirring was dissolved in 50mL of dichloromethane Mira Veron Intermediate C was added dropwise to the reaction solution to form three-necked flask. Stirred for I hour under nitrogen, with stirring 4.12g of sodium borohydride was added to the reaction mixture. The reaction mixture was stirred (under TC 3 hours to TLC the reaction was complete. The reaction is complete the reaction mixture was added dropwise a saturated aqueous ammonium chloride solution IOmL quenched reaction was washed twice with 40mL of water, the organic phase was separated. The The organic phase at the conditions at 0 ° C was added concentrated sulfuric acid was stirred IOmL until TLC after 0.5 hours the reaction was complete, then was added 20mL of 20% aqueous sodium hydroxide solution to complete the reaction of the organic phase was adjusted to pH 10 and stirred for 15 minutes minutes solution. The organic phase first with 50mL saturated brine I times with IOg anhydrous sodium sulfate and concentrated to give crude product was recrystallized from methanol and water to give 18.7g of the final product Mira Veron purity of 99.33%, chiral purity of 99.01%, a yield of 88.12%.
Mira Veron use randomly selected samples prepared by the synthesis method of the present invention is detected by liquid chromatography.
Test conditions: Instrument: Agilent 1100 HPLC;
Column: Luna C18, 4.6mmX 250mm, 5 μ m;
Column temperature: 25 ° C;
flow rate: 1.0mL / min;
The detection wavelength: 2IOnm;
Injection volume: 5ul;
Mobile phase A: acetonitrile;
Mobile phase B: 0.1% phosphoric acid aqueous solution;
Running time: 40min.
FIG liquid chromatography after detection of the sample shown in Figure 1; results are shown in Table I.
Table 1: The Mira Veron chromatographic analysis sample preparation method of the present invention

……….
http://www.google.co.in/patents/EP1440969A1?cl=en

- Example 4 (Production of the α-form crystal from wet cake of the β-form crystal) :
-
The same procedures as in Example 2 were followed to obtain 23.42 kg of a wet cake of the β-form crystal of (R)-2-(2-aminothiazol-4-yl)-4′-[2-[(2-hydroxy-2-phenylethyl)amino]ethyl]acetanilide from 6.66 kg of (R)-2-[[2-(4-aminophenyl)ethyl]amino]-1-phenylethanol monohydrochloride. This cake was added with and dissolved in 92 L of water and 76 L of ethanol by heating at about 80°C, and the solution was cooled at a rate of about 10°C per hour, to which was then added 8.4 g of the α-form crystal at 55°C. Thereafter, the mixture was cooled to 20°C. A crystal was filtered and dried to obtain 6.56 kg of the α-form crystal of (R)-2-(2-aminothiazol-4-yl)-4′-[2-[(2-hydroxy-2-phenylethyl)amino]ethyl]acetanilide.
-
Powder X-ray diffraction diagram and thermal analysis diagram of the α-form crystal are shown in Fig. 4 and Fig. 5, respectively.
1H-NMR (DMSO-d 6, 500 MHz) δ (ppm) = 1.60 (1H, s), 2.59 to 2.66 (4H, m), 2.68 to 2.80 (2H, m), 3.45 (2H, s), 4.59 (1H, br), 5.21 (1H, br), 6.30 (1H, s), 6.89 (2H, s), 7.11 (2H, d, J = 8.5 Hz), 7.19 to 7.23 (1H, m), 7.27 to 7.33 (4H, m), 7.49 (2H, d, J = 8.5 Hz), 9.99 (1H,s). FAB-MS m/z: 397 (M+H)+.
References
- “mirabegron (Rx) – Myrbetriq”. Medscape Reference. WebMD. Retrieved 17 November 2013.
- Gras, J (2012). “Mirabegron for the treatment of overactive bladder”. Drugs of today (Barcelona, Spain : 1998) 48 (1): 25–32. doi:10.1358/dot.2012.48.1.1738056. PMID 22384458.
- Sacco, E; Bientinesi, R et al. (Apr 2014). “Discovery history and clinical development of mirabegron for the treatment of overactive bladder and urinary incontinence”. Expert Opin Drug Discov9 (4): 433–48. doi:10.1517/17460441.2014.892923. PMID 2455903.
- “New Drug Approvals 2012 – Pt. XIV – Mirabegron (MyrbetriqTM)”. ChEMBL. 5 July 2012. Retrieved 28 September 2012.
- “MYRBETRIQ (mirabegron) tablet, film coated, extended release [Astellas Pharma US, Inc.]“. DailyMed. Astellas Pharma US, Inc. September 2012. Retrieved 17 November 2013.
- “Betmiga 25mg & 50mg prolonged-release tablets”. electronic Medicines Compendium. Astellas Pharma Ltd. 22 February 2013. Retrieved 17 November 2013.
- Cypess, Aaron; Weiner, Lauren; Roberts-Toler, Carla; Elía, Elisa; Kessler, Skyler; Kahn, Peter; English, Jeffrey; Chatman, Kelly; Trauger, Sunia; Doria, Alessandro; Kolodny, Gerald (6 January 2015). “Activation of Human Brown Adipose Tissue by a β3-Adrenergic Receptor Agonist”. Cell Metabolism 21 (1): 33–38. doi:10.1016/j.cmet.2014.12.009. PMID 25565203. Retrieved 26 January 2015.
External links
- Sacco, E.; Bientinesi, R. (2012). “Mirabegron: A review of recent data and its prospects in the management of overactive bladder”. Therapeutic Advances in Urology 4 (6): 315–24. doi:10.1177/1756287212457114. PMC 3491758. PMID 23205058.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
2-(2-Amino-1,3-thiazol-4-yl)-N-[4-(2-{[(2R)-2-hydroxy-2-phenylethyl]amino}ethyl)phenyl]acetamide
|
|
| Clinical data | |
| Trade names | Myrbetriq (US), Betanis (Japan), Betmiga (EU) |
| Licence data | EMA:Link, US FDA:link |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 29-35%[1] |
| Protein binding | 71%[1] |
| Metabolism | Hepatic via (direct) glucuronidation, amide hydrolysis, and minimal oxidative metabolism in vivo byCYP2D6 and CYP3A4. Some involvement of butylcholinesterase[1] |
| Biological half-life | 50 hours[1] |
| Excretion | Urine (55%), faeces (34%)[1] |
| Identifiers | |
| CAS Registry Number | 223673-61-8 |
| ATC code | G04BD12 |
| PubChem | CID: 9865528 |
| ChemSpider | 8041219 |
| Synonyms | YM-178 |
| Chemical data | |
| Formula | C21H24N4O2S |
| Molecular mass | 396.506 g/mol |
| Patent | Submitted | Granted |
|---|---|---|
| Alpha-form or beta-form crystal of acetanilide derivative [US7342117] | 2005-01-06 | 2008-03-11 |
| Pharmaceutical composition for treating stress incontinence and/or mixed incontinence [US2006004105] | 2006-01-05 | |
| Pharmaceutical composition comprising a beta-3-adrenoceptor agonist and a serotonin and/or norepinephrine reuptake inhibitor Pharmaceutical composition comprising a beta-3-adrenoceptor agonist and a serotonin and/or norepinephrine reuptake inhibitor [US2009012161] | 2005-11-24 | |
| Pharmaceutical composition consisting of a beta-3-adrenoceptor agonist and alpha-agonist [US2005154041] | 2005-07-14 | |
| Pharmaceutical composition consisting of a beta-3-adrenoceptor agonist and an active substance which influences prostaglandin metabolism [US2005119239] | 2005-06-02 | |
| Pharmaceutical Composition For Treating Stress Incontinence And/Or Mixed Incontinence [US2007129435] | 2007-06-07 | |
| Remedy for overactive bladder comprising acetic acid anilide derivative as the active ingredient [US7750029] | 2006-06-01 | 2010-07-06 |
| [alpha]-form or [beta]-form crystal of acetanilide derivative [US7982049] | 2008-09-04 | 2011-07-19 |
| BETA ADRENERGIC RECEPTOR AGONISTS FOR THE TREATMENT OF B-CELL PROLIFERATIVE DISORDERS [US2010009934] | 2010-01-14 | |
| PHARMACEUTICAL COMPOSITION FOR IMPROVING LOWER URINARY TRACT SYMPTOMS [US2010261770] | 2010-10-14 |
|
11 to 16 of 16
|
||
|---|---|---|
| Patent | Submitted | Granted |
| PHARMACEUTICAL COMPOSITION FOR MODIFIED RELEASE [US2010144807] | 2010-06-10 | |
| BENZYLAMINE DERIVATIVE OR PHARMACEUTICALLY ACCEPTABLE ACID ADDITION SALT THEREOF, AND USE THEREOF FOR MEDICAL PURPOSES [US8148427] | 2010-04-22 | 2012-04-03 |
| Pharmaceutical composition containing a beta-3-adrenoceptor agonist and an alpha antagonist and/or a 5-alpha reductase inhibitor [US2005101607] | 2005-05-12 | |
| REMEDY FOR OVERACTIVE BLADDER COMPRISING ACETIC ACID ANILIDE DERIVATIVE AS THE ACTIVE INGREDIENT [US2009093529] | 2009-04-09 | |
| PHARMACEUTICAL COMPOSITION FOR TREATING OVERACTIVE BLADDER [US2010240697] | 2010-09-23 | |
| Pharmaceutical composition comprising beta-3-adrenoceptor-agonists and antimuscarinic agents [US2005261328] | 2005-11-24 | |
| US Patent No | Patent Expiry | patent use |
|---|---|---|
| 6346532 | Oct 15, 2018 | |
| 6562375 | Aug 1, 2020 | |
| 6699503 | Sep 10, 2013 | |
| 7342117 | Nov 4, 2023 | |
| 7750029 | Dec 18, 2023 | U-913 |
| 7982049 | Nov 4, 2023 |
| Exclusivity Code | Exclusivity Date |
|---|---|
| NCE | Jun 28, 2017 |
U-913……….TREATMENT OF OVERACTIVE BLADDER WITH SYMPTOMS OF URGE URINARY INCONTINENCE, URGENCY, AND FREQUENCY
//////Mirabegron, Overactive bladder, FDA 2012, ASTELLAS PHARMA, YM-178, Myrbetriq, Betmiga
Updates…….

Overactive bladder (OAB) is characterized by symptoms of urinary urgency, with or without urgency incontinence, usually with increased daytime frequency and nocturia.(1-3) Current guidelines recommend oral antimuscarinics drugs as the first-line pharmacologic therapy in the management of OAB despite the companion adverse effects.(4, 5) Mirabegron is an orally active β3 adrenoceptor agonist approved by the FDA for treatment of OAB in 2012, which is an important step toward the better treatment options for the management of OAB.(6)
(R)-Styrene oxide 1 and 4-nitrophenethylamine 2 were exploited as starting materials in the first synthesis of mirabegron (Scheme 1). Heating 1 and 2 in i-propanol afforded amino alcohol 3, and then the amino group was protected by di-tert-butyl dicarbonate (Boc2O), followed by a condensation with 2-aminothiazol-4-acetic acid. Deprotection of the condensation product 7 finally afforded mirabegron.(7-10) Although reactions in the whole process were all conventional reactions, optically pure 1 was not industrially available, which restricted its application in industry.
(R)-Mandelic 8 and 4-nitrophenethylamine hydrochloride 9 were exploited as starting materials in an alternate route (Scheme 2). Condensation of 8 and 9 in the presence of 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide hydrochloride (EDCI), 1-hydroxybenzotriazole (HOBt), and triethylamine in N,N-dimethylformamide (DMF) furnished the corresponding amide 10, which was further reduced in the presence of borane-tetrahydrofuran complex in a mixed solution of 1,3-dimethyl-2-imidazolidinone (DMI) and tetrahydrofuran (THF), affording amine 11. The nitro group of 11 was then reduced by hydrogenation affording aniline 12 which was further amidated by an aqueous EDCI coupling affording mirabegron. This route was rather concise with only four steps, in which the sole stereogenic center was introduced via a bulk starting material 8.(11-13) However, usage of the costly EDCI twice, especially in the first step, led to a high cost and more impurities.

mail: chm_zhenggx@ujn.edu.cn, chm_zhenggx@ujn.edu.cn
-
1 Abrams, P.; Cardozo, L.; Fall, M.; Griffiths, D.; Rosier, P.; Ulmsten, U.; Van Kerrebroeck, P.; Victor, A.; Wein,A. UROLOGY 2003, 61, 37, DOI: 10.1016/S0090-4295(02)02243-4
-
2.Abrams, P.; Chapple, C.; Khoury, S.; Roehrborn, C.; de la Rosette, J. J. Urol. 2009, 181, 1779, DOI: 10.1016/j.juro.2008.11.127
-
3.Jaiprakash, H.; Benglorkar, G. M. RJPBCS 2014, 5 ( 3) 213
-
4.Lucas, M. G.; Ruud, J. L.; Bosch, R. J. L.; Burkhard, F. C.; Cruz, F.; Madden, T. B.; Nambiar, A. K.; Neisius,A.; de Ridder, D. J. M. K.; Tubaro, A.; Turner, W.; Pickard, R. Eur. Urol. 2012, 62, 1130, DOI: 10.1016/j.eururo.2012.08.047
-
5.Gormley, E. A.; Lightner, D. J.; Burgio, K. L.; Chai, T. C.; Clemens, J. Q.; Culkin, D. J.; Das, A. K.; Foster, H. E.; Scarpero, H. M.; Tessier, C. D.; Vasavada, S. P. J. Urol. 2012, 188, 2455, DOI: 10.1016/j.juro.2012.09.079
-
6.Sacco, E.; Bientinesi, R. World J. Obstet Gynecol 2013, 2 ( 4) 65, DOI: 10.5317/wjog.v2.i4.65
-
7.Maruyama, T.; Suzuki, T.; Onda, K.; Hayakawa, M.; Moritomo, H.; Kimizuka, T.; Matsui, T. US6346532,2002.
-
8.Kawazoe, S.; Sakamoto, K.; Awamura, Y.; Maruyama, T.; Suzuki, T.; Onda, K.; Takasu, T. EP144096A1,2004.
-
9.Takasu, T.; Sato, S.; Ukai, M.; Maruyama, T. EP1559427A1, 2005.
-
10Zhang, H.; Li, Y.; Chen, S.; Shen, M.; Wang, X. CN103896872A, 2014
//////////
