
Home » Posts tagged 'JAPAN 2022' (Page 2)
Tag Archives: JAPAN 2022
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
NOVAWAX, NVX-CoV2373,
NOVAWAX
SARS-CoV-2 rS Nanoparticle Vaccine
MCDC OTA agreement number W15QKN-16-9-1002
Novavax COVID-19 vaccine, Coronavirus disease 19 infection
SARS-CoV-2 rS, TAK 019
Novavax, Inc. is an American vaccine development company headquartered in Gaithersburg, Maryland, with additional facilities in Rockville, Maryland and Uppsala, Sweden. As of 2020, it had an ongoing Phase III clinical trial in older adults for its candidate vaccine for seasonal influenza, NanoFlu and a candidate vaccine (NVX-CoV2373) for prevention of COVID-19.
NVX-CoV2373 is a SARS-CoV-2 rS vaccine candidate and was shown to have high immunogenicity in studies. The vaccine is created from the genetic sequence of COVID-19 and the antigen derived from the virus spike protein is generated using recombinant nanoparticle technology. The vaccine was developed and tested by Novavax. As of May 2020, the company is pursuing a Phase 1 clinical trial (NCT04368988) to test the vaccine.
History
Novavax was founded in 1987. It focused principally on experimental vaccine development, but did not achieve a successful launch up to 2021.[4]
In June 2013, Novavax acquired the Matrix-M adjuvant platform with the purchase of Swedish company Isconova AB and renamed its new subsidiary Novavax AB.[5]
In 2015, the company received an $89 million grant from the Bill & Melinda Gates Foundation to support the development of a vaccine against human respiratory syncytial virus for infants via maternal immunization.[6][7][8][9]
In March 2015 the company completed a Phase I trial for its Ebola vaccine candidate,[10] as well as a phase II study in adults for its RSV vaccine, which would become ResVax.[11] The ResVax trial was encouraging as it showed significant efficacy against RSV infection.[11]
2016 saw the company’s first phase III trial, the 12,000 adult Resolve trial,[11] for its respiratory syncytial virus vaccine, which would come to be known as ResVax, fail in September.[3] This triggered an eighty-five percent dive in the company’s stock price.[3] Phase II adult trial results also released in 2016 showed a stimulation of antigencity, but failure in efficacy.[11] Evaluation of these results suggested that an alternative dosing strategy might lead to success, leading to plans to run new phase II trials.[3] The company’s difficulties in 2016 led to a three part strategy for 2017: cost reduction through restructuring and the termination of 30% of their workforce; pouring more effort into getting ResVax to market; and beginning clinical trials on a Zika virus vaccine.[3]
Alongside the adult studies of ResVax, the vaccine was also in 2016 being tested against infant RSV infection through the route of maternal immunization.[11]
In 2019, late-stage clinical testing of ResVax, failed for a second time, which resulted in a major downturn in investor confidence and a seventy percent reduction in capital value for the firm.[12][13] As a secondary result, the company was forced to conduct a reverse stock split in order to maintain Nasdaq minimum qualification, meaning it was in risk of being delisted.[13]
The company positions NanoFlu for the unmet need for a more effective vaccine against influenza, particularly in the elderly who often experience serious and sometimes life-threatening complications. In January 2020, it was granted fast track status by the U.S. Food and Drug Administration (FDA) for NanoFlu.
External sponsorships
In 2018, Novavax received a US$89 million research grant from the Bill and Melinda Gates Foundation for development of vaccines for maternal immunization.[14]
In May 2020, Novavax received US$384 million from the Coalition for Epidemic Preparedness Innovations to fund early-stage evaluation in healthy adults of the company’s COVID-19 vaccine candidate NVX-CoV2373 and to develop resources in preparation for large-scale manufacturing, if the vaccine proves successful.[15] CEPI had already invested $4 million in March.[15]
Drugs in development
ResVax is a nanoparticle-based treatment using a recombinant F lipoprotein or saponin, “extracted from the Quillaja saponaria [or?] Molina bark together with cholesterol and phospholipid.”[16] It is aimed at stimulating resistance to respiratory syncytial virus infection, targeting both adult and infant populations.[11]
In January 2020, Novavax was given Fast Track status by the FDA to expedite the review process for NanoFlu, a candidate influenze vaccine undergoing a Phase III clinical trial scheduled for completion by mid-2020.[17]
COVID-19 vaccine candidate
See also: NVX-CoV2373 and COVID-19 vaccine
In January 2020, Novavax announced development of a vaccine candidate, named NVX-CoV2373, to establish immunity to SARS-CoV-2.[18] NVX-CoV2373 is a protein subunit vaccine that contains the spike protein of the SARS-CoV-2 virus.[19] Novavax’s work is in competition for vaccine development among dozens of other companies.
In January 2021, the company released phase 3 trials showing that it has 89% efficacy against Covid-19, and also provides strong immunity against new variants.[20] It has applied for emergency use in the US and UK but will be distributed in the UK first.Novavax COVID-19 Vaccine Demonstrates 89.3% Efficacy in UK Phase 3 TrialJan 28, 2021 at 4:05 PM ESTDownload PDF
First to Demonstrate Clinical Efficacy Against COVID-19 and Both UK and South Africa Variants
- Strong efficacy in Phase 3 UK trial with over 50% of cases attributable to the now-predominant UK variant and the remainder attributable to COVID-19 virus
- Clinical efficacy demonstrated in Phase 2b South Africa trial with over 90% of sequenced cases attributable to prevalent South Africa escape variant
- Company to host investor conference call today at 4:30pm ET
GAITHERSBURG, Md., Jan. 28, 2021 (GLOBE NEWSWIRE) — Novavax, Inc. (Nasdaq: NVAX), a biotechnology company developing next-generation vaccines for serious infectious diseases, today announced that NVX-CoV2373, its protein-based COVID-19 vaccine candidate, met the primary endpoint, with a vaccine efficacy of 89.3%, in its Phase 3 clinical trial conducted in the United Kingdom (UK). The study assessed efficacy during a period with high transmission and with a new UK variant strain of the virus emerging and circulating widely. It was conducted in partnership with the UK Government’s Vaccines Taskforce. Novavax also announced successful results of its Phase 2b study conducted in South Africa.
“With today’s results from our UK Phase 3 and South Africa Phase 2b clinical trials, we have now reported data on our COVID-19 vaccine from Phase 1, 2 and 3 trials involving over 20,000 participants. In addition, our PREVENT-19 US and Mexico clinical trial has randomized over 16,000 participants toward our enrollment goal of 30,000. NVX-CoV2373 is the first vaccine to demonstrate not only high clinical efficacy against COVID-19 but also significant clinical efficacy against both the rapidly emerging UK and South Africa variants,” said Stanley C. Erck, President and Chief Executive Officer, Novavax. “NVX-CoV2373 has the potential to play an important role in solving this global public health crisis. We look forward to continuing to work with our partners, collaborators, investigators and regulators around the world to make the vaccine available as quickly as possible.”
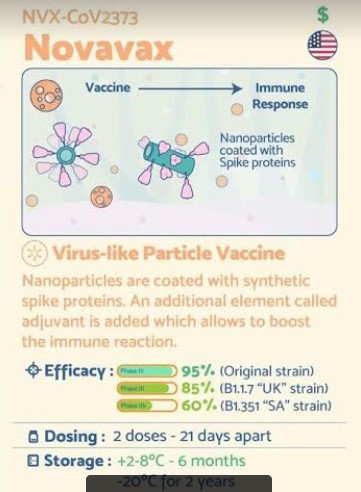
NVX-CoV2373 contains a full-length, prefusion spike protein made using Novavax’ recombinant nanoparticle technology and the company’s proprietary saponin-based Matrix-M™ adjuvant. The purified protein is encoded by the genetic sequence of the SARS-CoV-2 spike (S) protein and is produced in insect cells. It can neither cause COVID-19 nor can it replicate, is stable at 2°C to 8°C (refrigerated) and is shipped in a ready-to-use liquid formulation that permits distribution using existing vaccine supply chain channels.
UK Phase 3 Results: 89.3% Efficacy
The study enrolled more than 15,000 participants between 18-84 years of age, including 27% over the age of 65. The primary endpoint of the UK Phase 3 clinical trial is based on the first occurrence of PCR-confirmed symptomatic (mild, moderate or severe) COVID-19 with onset at least 7 days after the second study vaccination in serologically negative (to SARS-CoV-2) adult participants at baseline.
The first interim analysis is based on 62 cases, of which 56 cases of COVID-19 were observed in the placebo group versus 6 cases observed in the NVX-CoV2373 group, resulting in a point estimate of vaccine efficacy of 89.3% (95% CI: 75.2 – 95.4). Of the 62 cases, 61 were mild or moderate, and 1 was severe (in placebo group).
Preliminary analysis indicates that the UK variant strain that was increasingly prevalent was detected in over 50% of the PCR-confirmed symptomatic cases (32 UK variant, 24 non-variant, 6 unknown). Based on PCR performed on strains from 56 of the 62 cases, efficacy by strain was calculated to be 95.6% against the original COVID-19 strain and 85.6% against the UK variant strain [post hoc].
The interim analysis included a preliminary review of the safety database, which showed that severe, serious, and medically attended adverse events occurred at low levels and were balanced between vaccine and placebo groups.
“These are spectacular results, and we are very pleased to have helped Novavax with the development of this vaccine. The efficacy shown against the emerging variants is also extremely encouraging. This is an incredible achievement that will ensure we can protect individuals in the UK and the rest of the world from this virus,” said Clive Dix, Chair, UK Vaccine Taskforce.
Novavax expects to share further details of the UK trial results as additional data become available. Additional analysis on both trials is ongoing and will be shared via prepublication servers as well as submitted to a peer-reviewed journal for publication. The company initiated a rolling submission to the United Kingdom’s regulatory agency, the MHRA, in mid-January.
South Africa Results: Approximately 90% of COVID-19 cases attributed to South Africa escape variant
In the South Africa Phase 2b clinical trial, 60% efficacy (95% CI: 19.9 – 80.1) for the prevention of mild, moderate and severe COVID-19 disease was observed in the 94% of the study population that was HIV-negative. Twenty-nine cases were observed in the placebo group and 15 in the vaccine group. One severe case occurred in the placebo group and all other cases were mild or moderate. The clinical trial also achieved its primary efficacy endpoint in the overall trial population, including HIV-positive and HIV-negative subjects (efficacy of 49.4%; 95% CI: 6.1 – 72.8).
This study enrolled over 4,400 patients beginning in August 2020, with COVID-19 cases counted from September through mid-January. During this time, the triple mutant variant, which contains three critical mutations in the receptor binding domain (RBD) and multiple mutations outside the RBD, was widely circulating in South Africa. Preliminary sequencing data is available for 27 of 44 COVID-19 events; of these, 92.6% (25 out of 27 cases) were the South Africa escape variant.
Importantly in this trial, approximately 1/3 of the patients enrolled (but not included in the primary analyses described above) were seropositive, demonstrating prior COVID-19 infection at baseline. Based on temporal epidemiology data in the region, the pre-trial infections are thought to have been caused by the original COVID-19 strain (i.e., non-variant), while the subsequent infections during the study were largely variant virus. These data suggest that prior infection with COVID-19 may not completely protect against subsequent infection by the South Africa escape variant, however, vaccination with NVX-CoV2373 provided significant protection.
“The 60% reduced risk against COVID-19 illness in vaccinated individuals in South Africans underscores the value of this vaccine to prevent illness from the highly worrisome variant currently circulating in South Africa, and which is spreading globally. This is the first COVID-19 vaccine for which we now have objective evidence that it protects against the variant dominating in South Africa,” says Professor Shabir Maddi, Executive Director of the Vaccines and Infectious Diseases Analytics Research Unit (VIDA) at Wits, and principal investigator in the Novavax COVID-19 vaccine trial in South Africa. “I am encouraged to see that Novavax plans to immediately begin clinical development on a vaccine specifically targeted to the variant, which together with the current vaccine is likely to form the cornerstone of the fight against COVID-19.”
Novavax initiated development of new constructs against the emerging strains in early January and expects to select ideal candidates for a booster and/or combination bivalent vaccine for the new strains in the coming days. The company plans to initiate clinical testing of these new vaccines in the second quarter of this year.
“A primary benefit of our adjuvanted platform is that it uses a very small amount of antigen, enabling the rapid creation and large-scale production of combination vaccine candidates that could potentially address multiple circulating strains of COVID-19,” said Gregory M. Glenn, M.D., President of Research and Development, Novavax. “Combined with the safety profile that has been observed in our studies to-date with our COVID-19 vaccine, as well as prior studies in influenza, we are optimistic about our ability to rapidly adapt to evolving conditions.”
The Coalition for Epidemic Preparedness Innovations (CEPI) funded the manufacturing of doses of NVX-CoV2373 for this Phase 2b clinical trial, which was supported in part by a $15 million grant from the Bill & Melinda Gates Foundation.
Significant progress on PREVENT-19 Clinical Trial in US and Mexico
To date, PREVENT-19 has randomized over 16,000 participants and expects to complete our targeted enrollment of 30,000 patients in the first half of February. PREVENT-19 is being conducted with support from the U.S. government partnership formerly known as Operation Warp Speed, which includes the Department of Defense, the Biomedical Advanced Research and Development Authority (BARDA), part of the U.S. Department of Health and Human Services (HHS) Office of the Assistant Secretary for Preparedness and Response, and the National Institute of Allergy and Infectious Diseases (NIAID), part of the National Institutes of Health (NIH) at HHS. BARDA is also providing up to $1.75 billion under a Department of Defense agreement.
PREVENT-19 (the PRE-fusion protein subunit Vaccine Efficacy Novavax Trial | COVID-19) is a Phase 3, randomized, placebo-controlled, observer-blinded study in the US and Mexico to evaluate the efficacy, safety and immunogenicity of NVX-CoV2373 with Matrix-M in up to 30,000 subjects 18 years of age and older compared with placebo. The trial design has been harmonized to align with other Phase 3 trials conducted under the auspices of Operation Warp Speed, including the use of a single external independent Data and Safety Monitoring Board to evaluate safety and conduct an unblinded review when predetermined interim analysis events are reached.
The trial’s primary endpoint is the prevention of PCR-confirmed, symptomatic COVID-19. The key secondary endpoint is the prevention of PCR-confirmed, symptomatic moderate or severe COVID-19. Both endpoints will be assessed at least seven days after the second study vaccination in volunteers who have not been previously infected with SARS-CoV-2.
Conference Call
Novavax will host a conference call today at 4:30pm ET. The dial-in numbers for the conference call are (877) 212-6076 (Domestic) or (707) 287-9331 (International), passcode 7470222. A replay of the conference call will be available starting at 7:30 p.m. ET on January 28, 2021 until 7:30 p.m. ET on February 4, 2021. To access the replay by telephone, dial (855) 859-2056 (Domestic) or (404) 537-3406 (International) and use passcode 7470222.
A webcast of the conference call can also be accessed on the Novavax website at novavax.com/events. A replay of the webcast will be available on the Novavax website until April 28, 2021.
About NVX-CoV2373
NVX-CoV2373 is a protein-based vaccine candidate engineered from the genetic sequence of SARS-CoV-2, the virus that causes COVID-19 disease. NVX-CoV2373 was created using Novavax’ recombinant nanoparticle technology to generate antigen derived from the coronavirus spike (S) protein and is adjuvanted with Novavax’ patented saponin-based Matrix-M™ to enhance the immune response and stimulate high levels of neutralizing antibodies. NVX-CoV2373 contains purified protein antigen and can neither replicate, nor can it cause COVID-19. Over 37,000 participants have participated to date across four different clinical studies in five countries. NVX-CoV2373 is currently being evaluated in two pivotal Phase 3 trials: a trial in the U.K that completed enrollment in November and the PREVENT-19 trial in the U.S. and Mexico that began in December.
About Matrix-M™
Novavax’ patented saponin-based Matrix-M™ adjuvant has demonstrated a potent and well-tolerated effect by stimulating the entry of antigen presenting cells into the injection site and enhancing antigen presentation in local lymph nodes, boosting immune response.
About Novavax
Novavax, Inc. (Nasdaq: NVAX) is a biotechnology company that promotes improved health globally through the discovery, development and commercialization of innovative vaccines to prevent serious infectious diseases. The company’s proprietary recombinant technology platform combines the power and speed of genetic engineering to efficiently produce highly immunogenic nanoparticles designed to address urgent global health needs. Novavax is conducting late-stage clinical trials for NVX-CoV2373, its vaccine candidate against SARS-CoV-2, the virus that causes COVID-19. NanoFlu™, its quadrivalent influenza nanoparticle vaccine, met all primary objectives in its pivotal Phase 3 clinical trial in older adults and will be advanced for regulatory submission. Both vaccine candidates incorporate Novavax’ proprietary saponin-based Matrix-M™ adjuvant to enhance the immune response and stimulate high levels of neutralizing antibodies.
For more information, visit www.novavax.com and connect with us on Twitter and LinkedIn.
Candidate: NVX-CoV2373
Category: VAX
Type: Stable, prefusion protein made using Novavax’ proprietary nanoparticle technology, and incorporating its proprietary saponin-based Matrix-M™ adjuvant.
2021 Status: Novavax on March 11 announced final efficacy of 96.4% against mild, moderate and severe disease caused by the original COVID-19 strain in a pivotal Phase III trial in the U.K. of NVX–CoV2373. The study enrolled more than 15,000 participants between 18-84 years of age, including 27% over the age of 65.
The company also announced the complete analysis of its Phase IIb trial in South Africa, showing the vaccine had an efficacy of 55.4% among a cohort of HIV-negative trial participants, and an overall efficacy of 48.6% against predominantly variant strains of SARS-CoV-2 among 147 PCR-positive cases (51 cases in the vaccine group and 96 in the placebo group). Across both trials, NVX-CoV2373 demonstrated 100% protection against severe disease, including all hospitalization and death.
Philippines officials said March 10 that they secured 30 million doses of NVX-CoV2373 through an agreement with the Serum Institute of India, the second vaccine deal signed by the national government, according to Agence France-Presse. The first was with AstraZeneca for 2.6 million doses of its vaccine, developed with Oxford University.
The Novavax vaccine will be available from the third quarter, at a price that has yet to be finalized. The government hopes to secure 148 million doses this year from seven companies—enough for around 70% of its population.
In announcing fourth quarter and full-year 2020 results on March 1, Novavax said it could file for an emergency use authorization with the FDA in the second quarter of 2021. Novavax hopes it can use data from its Phase III U.K. clinical trial in its FDA submission, and expects the FDA to examine data in May, a month after they are reviewed by regulators in the U.K., President and CEO Stanley C. Erck said on CNBC. Should the FDA insist on waiting for U.S. data, the agency may push the review timeline by one or two months, he added.
The company also said that NVX-CoV2373 showed 95.6% efficacy against the original strain of COVID-19 and 85.6% against the UK variant strain, and re-stated an earlier finding that its vaccine met the Phase III trial’s primary endpoint met with an efficacy rate of 89.3%.
Novavax said February 26 that it signed an exclusive license agreement with Takeda Pharmaceutical for Takeda to develop, manufacture, and commercialize NVX-CoV2373 in Japan.
Novavax agreed to transfer the technology for manufacturing of the vaccine antigen and will supply its Matrix-M™ adjuvant to Takeda. Takeda anticipated the capacity to manufacture over 250 million doses of the COVID-19 vaccine per year. Takeda agreed in return to pay Novavax undisclosed payments tied to achieving development and commercial milestones, plus a portion of proceeds from the vaccine.
Takeda also disclosed that it dosed the first participants in a Phase II clinical trial to test the immunogenicity and safety of Novavax’ vaccine candidate in Japanese participants.
Novavax on February 18 announced a memorandum of understanding with Gavi, the Vaccine Alliance (Gavi), to provide 1.1 billion cumulative doses of NVX-CoV2373 for the COVAX Facility. Gavi leads the design and implementation of the COVAX Facility, created to supply vaccines globally, and has committed to working with Novavax to finalize an advance purchase agreement for vaccine supply and global distribution allocation via the COVAX Facility and its partners.
The doses will be manufactured and distributed globally by Novavax and Serum Institute of India (SII), the latter under an existing agreement between Gavi and SII.
Novavax and SK Bioscience said February 15 that they expanded their collaboration and license agreement, with SK finalizing an agreement to supply 40 million doses of NVX-CoV2373 to the government of South Korea beginning in 2021, for an undisclosed price. SK also obtained a license to manufacture and commercialize NVX-CoV2373 for sale to South Korea, as a result of which SK said it will add significant production capacity.
The agreement also calls on Novavax to facilitate technology transfer related to the manufacturing of its protein antigen, its Matrix M adjuvant, and support to SK Bioscience as needed to secure regulatory approval.
Rolling review begins—On February 4, Novavax announced it had begun a rolling review process for authorization of NVX-CoV2373 with several regulatory agencies worldwide, including the FDA, the European Medicines Agency, the U.K. Medicines and Healthcare products Regulatory Agency (MHRA), and Health Canada. The reviews will continue while the company completes its pivotal Phase III trials in the U.S. and U.K., and through initial authorization for emergency use granted under country-specific regulations, and through initial authorization for emergency use.
A day earlier, Novavax executed a binding Heads of Terms agreement with the government of Switzerland to supply 6 million doses of NVX-CoV2373, to the country. Novavax and Switzerland plan to negotiate a final agreement, with initial delivery of vaccine doses slated to ship following successful clinical development and regulatory review.
On January 28, Novavax electrified investors by announcing that its COVID-19 vaccine NVX-CoV2373 showed efficacy of 89.3% in the company’s first analysis of data from a Phase III trial in the U.K., where a variant strain (B.1.1.7) accounted for about half of all positive cases.
However, NVX-CoV2373 achieved only 60% efficacy in a Phase IIb trial in South Africa, where that country’s escape variant of the virus (B.1.351, also known as 20H/501Y.V2) was seen in 90% of cases, Novavax said.
Novavax said January 7 it executed an Advance Purchase Agreement with the Commonwealth of Australia for 51 million doses of NVX-CoV2373 for an undisclosed price, with an option to purchase an additional 10 million doses—finalizing an agreement in principle announced in November 2020. Novavax said it will work with Australia’s Therapeutics Goods Administration (TGA), to obtain approvals upon showing efficacy in clinical studies. The company aims to deliver initial doses by mid-2021.
2020 Status: Phase III trial launched—Novavax said December 28 that it launched the pivotal Phase III PREVENT-19 trial (NCT04611802) in the U.S. and Mexico to evaluate the efficacy, safety and immunogenicity of NVX-CoV2373. The randomized, placebo-controlled, observer-blinded study will assess the efficacy, safety and immunogenicity of NVX-CoV2373 in up to 30,000 participants 18 years of age and older compared with placebo. The trial’s primary endpoint is the prevention of PCR-confirmed, symptomatic COVID-19. The key secondary endpoint is the prevention of PCR-confirmed, symptomatic moderate or severe COVID-19. Both endpoints will be assessed at least seven days after the second study vaccination in volunteers who have not been previously infected with SARS-CoV-2.
Two thirds of the participants will be assigned to randomly receive two intramuscular injections of the vaccine, administered 21 days apart, while one third of the trial participants will receive placebo. Trial sites were selected in locations where transmission rates are currently high, to accelerate the accumulation of positive cases that could show efficacy. Participants will be followed for 24 months following the second injection
PREVENT-19 is being conducted with support from federal agencies involved in Operation Warp Speed, the Trump administration’s effort to promote development and distribution of COVID-19 vaccines and drugs. Those agencies include the Department of Defense (DoD), the NIH’s National Institute of Allergy and Infectious Diseases (NIAID), and the Biomedical Advanced Research and Development Authority (BARDA)—which has committed up to $1.6 billion to Novavax under a DoD agreement (identifier MCDC OTA agreement number W15QKN-16-9-1002).
Novavax is also conducting a pivotal Phase III study in the United Kingdom, a Phase IIb safety and efficacy study in South Africa, and an ongoing Phase I/II trial in the U.S. and Australia. Data from these trials are expected as soon as early first quarter 2021, though timing will depend on transmission rates in the regions, the company said.
Novavax said November 9 that the FDA granted its Fast Track designation for NVX-CoV2373. By the end of November, the company expected to finish enrollment in its Phase III U.K. trial, with interim data in that study expected as soon as early first quarter 2021.
Five days earlier, Novavax signed a non-binding Heads of Terms document with the Australian government to supply 40 million doses of NVX-CoV2373 to Australia starting as early as the first half of 2021, subject to the successful completion of Phase III clinical development and approval of the vaccine by Australia’s Therapeutic Goods Administration (TGA). The vaccine regimen is expected to require two doses per individual, administered 21 days apart.
Australia joins the U.S., the U.K., and Canada in signing direct supply agreements with Novavax. The company is supplying doses in Japan, South Korea, and India through partnerships. Australian clinical researchers led the global Phase I clinical trial in August, which involved 131 Australians across two trial sites (Melbourne and Brisbane). Also, approximately 690 Australians have participated in the Phase II arm of the clinical trial, which has been conducted across up to 40 sites in Australia and the U.S.
Novavax joined officials in its headquarters city of Gaithersburg, MD, on November 2 to announce expansion plans. The company plans to take 122,000 square feet of space at 700 Quince Orchard Road, and has committed to adding at least 400 local jobs, nearly doubling its current workforce of 450 worldwide. Most of the new jobs are expected to be added b March 2021.
Maryland’s Department of Commerce—which has prioritized assistance to life sciences companies—approved a $2 million conditional loan tied to job creation and capital investment. The state has also approved a $200,000 Partnership for Workforce Quality training grant, and the company is eligible for several tax credits, including the Job Creation Tax Credit and More Jobs for Marylanders.
Additionally, Montgomery County has approved a $500,000 grant tied to job creation and capital investment, while the City of Gaithersburg said it will approve a grant of up to $50,000 from its Economic Development Opportunity Fund. The city accelerated its planning approval process to accommodate Novavax’ timeline, given the company’s role in fighting COVID-19 and resulting assistance from Operation Warp Speed, the Trump administration’s effort to accelerate development of COVID-19 vaccines.
On October 27, Novavax said that it had enrolled 5,500 volunteers in the Phase III U.K. trial, which has been expanded from 10,000 to 15,000 volunteers. The increased enrollment “is likely to facilitate assessment of safety and efficacy in a shorter time period,” according to the company.
The trial, which is being conducted with the U.K. Government’s Vaccines Taskforce, was launched in September and is expected to be fully enrolled by the end of November, with interim data expected by early first quarter 2021, depending on the overall COVID-19 attack rate. Novavax has posted the protocol for the Phase III U.K. trial online. The protocol calls for unblinding of data once 152 participants have achieved mild, moderate or severe endpoints. Two interim analyses are planned upon occurrence of 66 and 110 endpoints.
Novavax also said it expects to launch a second Phase III trial designed to enroll up to 30,000 participants in the U.S. and Mexico by the end of November—a study funded through the U.S. government’s Operation Warp Speed program. The patient population will reflect proportional representation of diverse populations most vulnerable to COVID-19, across race/ethnicity, age, and co-morbidities.
The company cited progress toward large-scale manufacturing while acknowledging delays from original timeframe estimates. Novavax said it will use its contract manufacturing site at FUJIFILM Diosynth Biotechnologies’ Morrisville, NC facility to produce material for the U.S. trial.
On September 25, Novavax entered into a non-exclusive agreement with Endo International subsidiary Par Sterile Products to provide fill-finish manufacturing services at its plant in Rochester, MI, for NVX-CoV2373. Under the agreement, whose value was not disclosed, the Rochester facility has begun production of NVX-CoV2373 final drug product, with initial batches to be used in Novavax’ Phase III clinical trial in the U.S. Par Sterile will also fill-finish NVX-CoV2373 vaccine intended for commercial distribution in the U.S.
A day earlier, Novavax launched the U.K. trial. The randomized, placebo-controlled, observer-blinded study to evaluate the efficacy, safety and immunogenicity of NVX-CoV2373 with Matrix-M in up to 10,000 subjects 18-84 years of age, with and without “relevant” comorbidities, over the following four to six weeks, Novavax said. Half the participants will receive two intramuscular injections of vaccine comprising 5 µg of protein antigen with 50 µg Matrix‑M adjuvant, 21 days apart, while half of the trial participants will receive placebo. At least 25% of the study population will be over age 65.
The trial’s first primary endpoint is first occurrence of PCR-confirmed symptomatic COVID-19 with onset at least seven days after the second study vaccination in volunteers who have not been previously infected with SARS-CoV-2. The second primary endpoint is first occurrence of PCR-confirmed symptomatic moderate or severe COVID-19 with onset at least seven days after the second study vaccination in volunteers who have not been previously infected with SARS-CoV-2
“The data from this trial is expected to support regulatory submissions for licensure in the UK, EU and other countries,” stated Gregory M. Glenn, M.D., President, Research and Development at Novavax.
Maryland Gov. Larry Hogan joined state Secretary of Commerce Kelly M. Schulz and local officials in marking the launch of Phase III studies with a tour of the company’s facilities in Gaithersburg: “The coronavirus vaccine candidate that’s been developed by Novavax is one of the most promising in the country, if not the world.”
On August 31, Novavax reached an agreement in principle with the government of Canada to supply up to 76 million doses of NVX-CoV2373. The value was not disclosed. Novavax and Canada did say that they expect to finalize an advance purchase agreement under which Novavax will agree to supply doses of NVX-CoV2373 to Canada beginning as early as the second quarter of 2021.
The purchase arrangement will be subject to licensure of the NVX-CoV2373 by Health Canada, Novavax said. The vaccine is in multiple Phase II clinical trials: On August 24, Novavax said the first volunteers had been enrolled in the Phase II portion of its ongoing Phase I/II clinical trial (NCT04368988), designed to evaluate the immunogenicity and safety of two doses of of NVX-CoV2373 (5 and 25 µg) with and without 50 µg of Matrix‑M™ adjuvant in up to 1,500 volunteers ages 18-84.
The randomized, placebo-controlled, observer-blinded study is designed to assess two dose sizes (5 and 25 µg) of NVX-CoV2373, each with 50 µg of Matrix‑M. Unlike the Phase I portion, the Phase II portion will include older adults 60-84 years of age as approximately half of the trial’s population. Secondary objectives include preliminary evaluation of efficacy. The trial will be conducted at up to 40 sites in the U.S. and Australia, Novovax said.
NVX-CoV2373 is in a pair of Phase II trials launched in August—including a Phase IIb study in South Africa to assess efficacy, and a Phase II safety and immunogenicity study in the U.S. and Australia.
On August 14, the U.K. government agreed to purchase 60 million doses of NVX-CoV2373 from the company, and support its planned Phase III clinical trial in the U.K., through an agreement whose value was not disclosed. The doses are set to be manufactured as early as the first quarter of 2021.
The trial will be designed to evaluate the ability of NVX-CoV2373 to protect against symptomatic COVID-19 disease as well as evaluate antibody and T-cell responses. The randomized, double-blind, placebo-controlled efficacy study will enroll approximately 9,000 adults 18-85 years of age in the U.K., and is expected to start in the third quarter.
Novavax also said it will expand its collaboration with FUJIFILM Diosynth Biotechnologies (FDB), which will manufacture the antigen component of NVX-CoV2373 from its Billingham, Stockton-on-Tees site in the U.K., as well as at U.S. sites in Morrisville, NC, and College Station, TX. FDB’s U.K. sitevis expected to produce up to 180 million doses annually.
On August 13, Novavax said it signed a development and supply agreement for the antigen component of NVX-CoV2373 with Seoul-based SK bioscience, a vaccine business subsidiary of SK Group. The agreement calls for supply to global markets that include the COVAX Facility, co-led by Gavi, the Coalition for Epidemic Preparedness Innovations (CEPI) and the World Health Organization.
Novavax and SK signed a letter of intent with South Korea’s Ministry of Health and Welfare to work toward broad and equitable access to NVX-CoV2373 worldwide, as well as to make the vaccine available in South Korea. SK bioscience agreed to manufacture the vaccine antigen component for use in the final drug product globally during the pandemic, at its vaccine facility in Andong L-house, South Korea, beginning in August. The value of the agreement was not disclosed.
On August 7, Novavax licensed its COVID-19 vaccine technology to Takeda Pharmaceutical through a partnership by which Takeda will develop, manufacture, and commercialize NVX‑CoV2373 in Japan, using Matrix-M adjuvant to be supplied by Novavax. Takeda will also be responsible for regulatory submission to Japan’s Ministry of Health, Labour and Welfare (MHLW).
MHLW agreed to provide funding to Takeda—the amount was not disclosed in the companies’ announcement—for technology transfer, establishment of infrastructure, and scale-up of manufacturing. Takeda said it anticipated the capacity to manufacture over 250 million doses of NVX‑CoV2373 per year.
Five days earlier, Serum Institute of India agreed to license rights from Novavax to NVX‑CoV2373 for development and commercialization in India as well as low- and middle-income countries (LMIC), through an agreement whose value was not disclosed. Novavax retains rights to NVX-CoV2373 elsewhere in the world.
Novavax and Serum Institute of India agreed to partner on clinical development, co-formulation, filling and finishing and commercialization of NVX-CoV2373. Serum Institute will oversee regulatory submissions and marketing authorizations in regions covered by the collaboration. Novavax agreed to provide both vaccine antigen and Matrix‑M adjuvant, while the partners said they were in talks to have the Serum Institute manufacture vaccine antigen in India. Novavax and Seerum Institute plan to split the revenue from the sale of product, net of agreed costs.
A day earlier, Novavax announced positive results from the Phase I portion of its Phase I/II clinical trial (NCT04368988), designed to evaluate two doses of NVX-CoV2373 (5 and 25 µg) with and without Matrix‑M™ adjuvant in 131 healthy adults ages 18-59. NVX-CoV2373, adjuvanted with Matrix-M, elicited robust antibody responses numerically superior to human convalescent sera, according to data submitted for peer-review to a scientific journal.
All participants developed anti-spike IgG antibodies after a single dose of vaccine, Novavax said, many also developing wild-type virus neutralizing antibody responses. After the second dose, all participants developed wild-type virus neutralizing antibody responses. Both anti-spike IgG and viral neutralization responses compared favorably to responses from patients with clinically significant COVID‑19 disease, the company said—adding that IgG antibody response was highly correlated with neutralization titers, showing that a significant proportion of antibodies were functional.
For both dosages of NVX‑CoV2373 with adjuvant, the 5 µg dose performed “comparably” with the 25 µg dose, Novavax said. NVX‑CoV2373 also induced antigen-specific polyfunctional CD4+ T cell responses with a strong bias toward the Th1 phenotype (IFN-g, IL-2, and TNF-a).
Based on an interim analysis of Phase I safety and immunogenicity data, the trial was expanded to Phase II clinical trials in multiple countries, including the U.S. The trial—which began in Australia in May—is being funded by up-to $388 million in funding from the Coalition for Epidemic Preparedness Innovations (CEPI). If the Phase I/II trial is successful, CEPI said, it anticipates supporting further clinical development that would advance NVX-CoV2373 through to licensure.
On July 23, Novavax joined FDB to announce that FDB will manufacture bulk drug substance for NVX-CoV2373, under an agreement whose value was not disclosed. FDB’s site in Morrisville, NC has begun production of the first batch of NVX-CoV2373. Batches produced at FDB’s Morrisville site will be used in Novavax’s planned pivotal Phase III clinical trial, designed to assess NVX-CoV2373 in up to 30,000 participants, and set to start this fall.
The Phase III trial is among R&D efforts to be funded through the $1.6 billion awarded in July to Novavax through President Donald Trump’s “Operation Warp Speed” program toward late-stage clinical trials and large-scale manufacturing to produce 100 million doses of its COVID-19 vaccine by year’s end. Novavax said the funding will enable it to complete late-stage clinical studies aimed at evaluating the safety and efficacy of NVX-CoV2373.
In June, Novavax said biotech investor and executive David Mott was joining its board as an independent director, after recently acquiring nearly 65,000 shares of the company’s common stock. Also, Novavax was awarded a $60 million contract by the U.S. Department of Defense (DoD) for the manufacturing of NVX‑CoV2373. Through the Defense Health Program, the Joint Program Executive Office for Chemical, Biological, Radiological and Nuclear Defense Enabling Biotechnologies (JPEO-CBRND-EB) agreed to support production of several vaccine components to be manufactured in the U.S. Novavax plans to deliver this year for DoD 10 million doses of NVX‑CoV2373 that could be used in Phase II/III trials, or under an Emergency Use Authorization (EUA) if approved by the FDA.
Also in June, AGC Biologics said it will partner with Novavax on large-scale GMP production of Matrix-M– significantly increasing Novavax’ capacity to deliver doses in 2020 and 2021—through an agreement whose value was not disclosed. And Novavax joined The PolyPeptide Group to announce large-scale GMP production by the global CDMO of two unspecified key intermediate components used in the production of Matrix-M.
In May, Novavax acquired Praha Vaccines from the India-based Cyrus Poonawalla Group for $167 million cash, in a deal designed to ramp up Novavax’s manufacturing capacity for NVX-CoV2373. Praha Vaccines’ assets include a 150,000-square foot vaccine and biologics manufacturing facility and other support buildings in Bohumil, Czech Republic. Novavax said the Bohumil facility is expected to deliver an annual capacity of over 1 billion doses of antigen starting in 2021 for the COVID-19 vaccine.
The Bohumil facility is completing renovations that include the addition of Biosafety Level-3 (BSL-3) capabilities. The site’s approximately 150 employees with “significant experience” in vaccine manufacturing and support have joined Novavax, the company said.
On May 11, Novavax joined CEPI in announcing up to $384 million in additional funding for the company toward clinical development and large-scale manufacturing of NVX-CoV2373. CEPI agreed to fund preclinical as well as Phase I and Phase II studies of NVX-CoV2373. The funding multiplied CEPI’s initial $4 million investment in the vaccine candidate, made two months earlier. Novavax’s total $388 million in CEPI funding accounted for 87% of the total $446 million awarded by the Coalition toward COVID-19 vaccine R&D as of that date.
Novavax identified its COVID-19 vaccine candidate in April. The company said NVX-CoV2373 was shown to be highly immunogenic in animal models measuring spike protein-specific antibodies, antibodies that block the binding of the spike protein to the receptor, and wild-type virus neutralizing antibodies. High levels of spike protein-specific antibodies with ACE-2 human receptor binding domain blocking activity and SARS-CoV-2 wild-type virus neutralizing antibodies were also seen after a single immunization.
In March, Emergent Biosolutions disclosed it retained an option to allocate manufacturing capacity for an expanded COVID-19 program under an agreement with Novavax to provide “molecule-to-market” contract development and manufacturing (CDMO) services to produce Novavax’s NanoFlu™, its recombinant quadrivalent seasonal influenza vaccine candidate.
Earlier in March, Emergent announced similar services to support clinical development of Novavax’s COVID-19 vaccine candidate, saying March 10 it agreed to produce the vaccine candidate and had initiated work, anticipating the vaccine candidate will be used in a Phase I study within the next four months. In February, Novavax said it had produced and was assessing multiple nanoparticle vaccine candidates in animal models prior to identifying an optimal candidate for human testing.
References
- ^ “Company Overview of Novavax, Inc”. Bloomberg.com. Archived from the original on 24 February 2017. Retrieved 2 June2019.
- ^ https://www.globenewswire.com/news-release/2021/03/01/2184674/0/en/Novavax-Reports-Fourth-Quarter-and-Full-Year-2020-Financial-Results-and-Operational-Highlights.html
- ^ Jump up to:a b c d e Bell, Jacob (November 14, 2016). “Novavax aims to rebound with restructuring, more trials”. BioPharma Dive. Washington, D.C.: Industry Dive. Archived from the original on 2017-03-29. Retrieved 2017-03-28.
- ^ Thomas, Katie; Twohey, Megan (2020-07-16). “How a Struggling Company Won $1.6 Billion to Make a Coronavirus Vaccine”. The New York Times. ISSN 0362-4331. Retrieved 2021-01-29.
- ^ Taylor, Nick Paul (3 June 2013). “Novavax makes $30M bid for adjuvant business”. FiercePharma. Archived from the original on 14 September 2016. Retrieved 9 September 2016.
- ^ “Gaithersburg Biotech Receives Grant Worth up to $89 million”. Bizjournals.com. Archived from the original on 2017-04-01. Retrieved 2017-03-28.
- ^ “With promising RSV data in hand, Novavax wins $89M Gates grant for PhIII | FierceBiotech”. Fiercebiotech.com. Archivedfrom the original on 2017-04-14. Retrieved 2017-03-28.
- ^ “Novavax RSV vaccine found safe for pregnant women, fetus”. Reuters. 2016-09-29. Archived from the original on 2016-10-07. Retrieved 2017-03-28.
- ^ Herper, Matthew. “Gates Foundation Backs New Shot To Prevent Babies From Dying Of Pneumonia”. Forbes. Archived from the original on 2016-09-21. Retrieved 2017-03-28.
- ^ “Novavax’s Ebola vaccine shows promise in early-stage trial”. Reuters. 2017-07-21. Archived from the original on 2016-10-02. Retrieved 2017-03-28.
- ^ Jump up to:a b c d e f Adams, Ben (September 16, 2016). “Novavax craters after Phase III RSV F vaccine failure; seeks path forward”. FierceBiotech. Questex. Archived from the original on 18 August 2020. Retrieved 25 Jan 2020.
- ^ Shtrubel, Marty (December 12, 2019). “3 Biotech Stocks That Offer the Highest Upside on Wall Street”. Biotech. Nasdaq. Archived from the original on 2020-01-26. Retrieved 25 Jan 2020.
- ^ Jump up to:a b Budwell, George (January 20, 2020). “3 Top Biotech Picks for 2020”. Markets. Nasdaq. Novavax: A catalyst awaits. Archivedfrom the original on 2020-01-25. Retrieved 25 Jan 2020.
- ^ Mark Terry (February 16, 2018). “Why Novavax Could be a Millionaire-Maker Stock”. BioSpace. Archived from the original on 22 November 2020. Retrieved 6 March 2020.
- ^ Jump up to:a b Eric Sagonowsky (2020-05-11). “Novavax scores $384M deal, CEPI’s largest ever, to fund coronavirus vaccine work”. FiercePharma. Archived from the original on 2020-05-16. Retrieved 2020-05-12.
- ^ “Novavax addresses urgent global public health needs with innovative technology”. novavax.com. Archived from the original on 10 September 2020. Retrieved 30 August 2020.
- ^ Sara Gilgore (January 15, 2020). “Novavax earns key FDA status for its flu vaccine. Wall Street took it well”. Washington Business Journal. Archived from the original on 10 November 2020. Retrieved 6 March 2020.
- ^ Sara Gilgore (February 26, 2020). “Novavax is working to advance a potential coronavirus vaccine. So are competitors”. Washington Business Journal. Archived from the original on March 16, 2020. Retrieved March 6, 2020.
- ^ Nidhi Parekh (July 24, 2020). “Novavax: A SARS-CoV-2 Protein Factory to Beat COVID-19”. Archived from the original on November 22, 2020. Retrieved July 24, 2020.
- ^ “Covid-19: Novavax vaccine shows 89% efficacy in UK trials”. BBC news. Retrieved 1 February 2021.
Further reading
- “Novavax, Inc. Common Stock (NVAX) News Headlines”. Market Activity. Nasdaq. Retrieved 25 Jan 2020. Continuously updated listing of Nasdaq publications related to Novavax, newest items first.
External links
- Official website
- Business data for Novavax, Inc.:
General References
| Type | Public |
|---|---|
| Traded as | Nasdaq: NVAX Russell 2000 Component |
| Industry | Biotechnology |
| Founded | 1987; 34 years ago [1] |
| Headquarters | Gaithersburg, Maryland,United States |
| Area served | Worldwide |
| Key people | Stanley Erck (CEO) |
| Products | Vaccines |
| Revenue | |
| Number of employees | 500+[3] |
| Website | www.novavax.com |
The Novavax COVID-19 vaccine, codenamed NVX-CoV2373, and also called SARS-CoV-2 rS (recombinant spike) protein nanoparticle with Matrix-M1 adjuvant, is a COVID-19 vaccine candidate developed by Novavax and Coalition for Epidemic Preparedness Innovations (CEPI). It requires two doses[1] and is stable at 2 to 8 °C (36 to 46 °F) (refrigerated).[2]
Description
NVX-CoV2373 has been described as both a protein subunit vaccine[3][4][5] and a virus-like particle vaccine,[6][7] though the producers call it a “recombinant nanoparticle vaccine”.[8]
The vaccine is produced by creating an engineered baculovirus containing a gene for a modified SARS-CoV-2 spike protein. The baculovirus then infects a culture of Sf9 moth cells, which create the spike protein and display it on their cell membranes. The spike proteins are then harvested and assembled onto a synthetic lipid nanoparticle about 50 nanometers across, each displaying up to 14 spike proteins.[3][4][8]
The formulation includes a saponin-based adjuvant.[3][4][8]
Development
In January 2020, Novavax announced development of a vaccine candidate, codenamed NVX-CoV2373, to establish immunity to SARS-CoV-2.[9] Novavax’s work is in competition for vaccine development among dozens of other companies.[10]
In March 2020, Novavax announced a collaboration with Emergent BioSolutions for preclinical and early-stage human research on the vaccine candidate.[11] Under the partnership, Emergent BioSolutions will manufacture the vaccine at large scale at their Baltimore facility.[12] Trials have also taken place in the United Kingdom, and subject to regulatory approval, at least 60 million doses will be manufactured by Fujifilm Diosynth Biotechnologies in Billingham for purchase by the UK government.[13][14] They also signed an agreement with Serum Institute of India for mass scale production for developing and low-income countries.[15] It has also been reported, that the vaccine will be manufactured in Spain.[16] The first human safety studies of the candidate, codenamed NVX-CoV2373, started in May 2020 in Australia.[17][18]
In July, the company announced it might receive $1.6 billion from Operation Warp Speed to expedite development of its coronavirus vaccine candidate by 2021—if clinical trials show the vaccine to be effective.[19][20] A spokesperson for Novavax stated that the $1.6 billion was coming from a “collaboration” between the Department of Health and Human Services and Department of Defense,[19][20] where Gen. Gustave F. Perna has been selected as COO for Warp Speed. In late September, Novavax entered the final stages of testing its coronavirus vaccine in the UK. Another large trial was announced to start by October in the US.[21]
In December 2020, Novavax started the PREVENT-19 (NCT04611802) Phase III trial in the US and Mexico.[22][full citation needed][23]
On 28 January 2021, Novavax reported that preliminary results from the United Kingdom trial showed that its vaccine candidate was more than 89% effective.[24][2] However, interim results from a trial in South Africa showed a lower effectiveness rate against the 501.V2 variant of the virus, at around 50-60%.[1][25]
On 12 March 2021, they announced their vaccine candidate was 96.4% effective in preventing the original strain of COVID-19 and 86% effective against the U.K variant. It proved 55% effective against the South African variant in people without HIV/AIDS. It was also 100% effective at preventing severe illness.[citation needed]
Deployment
On 2 February 2021, the Canadian Prime Minister Justin Trudeau announced that Canada has signed a tentative agreement for Novavax to produce millions of doses of its COVID-19 vaccine in Montreal, Canada, once it’s approved for use by Health Canada, making it the first COVID-19 vaccine to be produced domestically.[26]
References
- ^ Jump up to:a b Wadman M, Jon C (28 January 2021). “Novavax vaccine delivers 89% efficacy against COVID-19 in UK—but is less potent in South Africa”. Science. doi:10.1126/science.abg8101.
- ^ Jump up to:a b “New Covid vaccine shows 89% efficacy in UK trials”. BBC News. 28 January 2021. Retrieved 28 January 2021.
- ^ Jump up to:a b c Wadman M (November 2020). “The long shot”. Science. 370 (6517): 649–653. Bibcode:2020Sci…370..649W. doi:10.1126/science.370.6517.649. PMID 33154120.
- ^ Jump up to:a b c Wadman M (28 December 2020). “Novavax launches pivotal U.S. trial of dark horse COVID-19 vaccine after manufacturing delays”. Science. doi:10.1126/science.abg3441.
- ^ Parekh N (24 July 2020). “Novavax: A SARS-CoV-2 Protein Factory to Beat COVID-19”. Archived from the original on 22 November 2020. Retrieved 24 July 2020.
- ^ Chung YH, Beiss V, Fiering SN, Steinmetz NF (October 2020). “COVID-19 Vaccine Frontrunners and Their Nanotechnology Design”. ACS Nano. 14 (10): 12522–12537. doi:10.1021/acsnano.0c07197. PMC 7553041. PMID 33034449.
- ^ Medhi R, Srinoi P, Ngo N, Tran HV, Lee TR (25 September 2020). “Nanoparticle-Based Strategies to Combat COVID-19”. ACS Applied Nano Materials. 3 (9): 8557–8580. doi:10.1021/acsanm.0c01978. PMC 7482545.
- ^ Jump up to:a b c “Urgent global health needs addressed by Novavax”. Novavax. Retrieved 30 January 2021.
- ^ Gilgore S (26 February 2020). “Novavax is working to advance a potential coronavirus vaccine. So are competitors”. Washington Business Journal. Archived from the original on 16 March 2020. Retrieved 6 March 2020.
- ^ “COVID-19 vaccine tracker (click on ‘Vaccines’ tab)”. Milken Institute. 11 May 2020. Archived from the original on 6 June 2020. Retrieved 12 May 2020. Lay summary.
- ^ Gilgore S (10 March 2020). “Novavax’s coronavirus vaccine program is getting some help from Emergent BioSolutions”. Washington Business Journal. Archived from the original on 9 April 2020. Retrieved 10 March 2020.
- ^ McCartney R. “Maryland plays an outsized role in worldwide hunt for a coronavirus vaccine”. Washington Post. Archived from the original on 7 May 2020. Retrieved 8 May 2020.
- ^ Boseley S, Davis N (28 January 2021). “Novavax Covid vaccine shown to be nearly 90% effective in UK trial”. The Guardian. Retrieved 29 January 2021.
- ^ Brown M (14 August 2020). “60m doses of new covid-19 vaccine could be made in Billingham – and be ready for mid-2021”. TeesideLive. Reach. Retrieved 29 January 2021.
- ^ “Novavax signs COVID-19 vaccine supply deal with India’s Serum Institute”. Reuters. 5 August 2020.
- ^ “Spain, again chosen to produce the vaccine to combat COVID-19”. This is the Real Spain. 18 September 2020.
- ^ Sagonowsky E (11 May 2020). “Novavax scores $384M deal, CEPI’s largest ever, to fund coronavirus vaccine work”. FiercePharma. Archived from the original on 16 May 2020. Retrieved 12 May 2020.
- ^ “Novavax starts clinical trial of its coronavirus vaccine candidate”. CNBC. 25 May 2020. Archived from the original on 26 May 2020. Retrieved 26 May 2020.
- ^ Jump up to:a b Thomas K (7 July 2020). “U.S. Will Pay $1.6 Billion to Novavax for Coronavirus Vaccine”. The New York Times. Archived from the original on 7 July 2020. Retrieved 7 July 2020.
- ^ Jump up to:a b Steenhuysen J (7 July 2020). “U.S. government awards Novavax $1.6 billion for coronavirus vaccine”. Reuters. Archived from the original on 14 September 2020. Retrieved 15 September 2020.
- ^ Thomas K, Zimmer C (24 September 2020). “Novavax Enters Final Stage of Coronavirus Vaccine Trials”. The New York Times. ISSN 0362-4331. Archived from the original on 28 September 2020. Retrieved 28 September 2020.
- ^ Clinical trial number NCT04611802 for “A Study Looking at the Efficacy, Immune Response, and Safety of a COVID-19 Vaccine in Adults at Risk for SARS-CoV-2” at ClinicalTrials.gov
- ^ “Phase 3 trial of Novavax investigational COVID-19 vaccine opens”. National Institutes of Health (NIH). 28 December 2020. Retrieved 28 December 2020.
- ^ Lovelace B (28 January 2020). “Novavax says Covid vaccine is more than 89% effective”. CNBC.
- ^ Facher L, Joseph A (28 January 2021). “Novavax says its Covid-19 vaccine is 90% effective in late-stage trial”. Stat. Retrieved 29 January 2021.
- ^ “Canada signs deal to produce Novavax COVID-19 vaccine at Montreal plant”. CP24. 2 February 2021. Retrieved 2 February2021.
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | Subunit |
| Clinical data | |
| Other names | NVX-CoV2373 |
| Routes of administration | Intramuscular |
| ATC code | None |
| Identifiers | |
| DrugBank | DB15810 |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| SARS-CoV-2 (virus)COVID-19 (disease) |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showImpact |
| COVID-19 Portal |
| vte |
////////////// Novavax, COVID-19, vaccine, CORONA VIRUS, NVX-CoV2373, SARS-CoV-2 rS, TAK 019
#Novavax, #COVID-19, #vaccine, #CORONA VIRUS, #NVX-CoV2373, #SARS-CoV-2 rS, #TAK 019
UPDATE
SARS-CoV-2 Spike glycoprotein vaccine antigen nvx-cov2373
SARS-CoV-2 rS;
Novavax Covid-19 vaccine (TN);
Nuvaxovid (TN)
SARS-CoV-2 rS;
組換えコロナウイルス (SARS-CoV-2) ワクチン;
コロナウイルス(SARS-CoV-2)スパイク糖タンパク質抗原nvx-cov2373ワクチン;
SARS-CoV-2 Spike glycoprotein vaccine antigen nvx-cov2373;
SARS-CoV-2 rS
APPROVED JAPAN Nuvaxovid, 2022/4/19
//////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
/////////SARS-CoV-2 Spike glycoprotein vaccine antigen nvx-cov2373, JAPAN 2022, APPROVALS 2022, VACCINE, COVID 19. CORONA VACCINE, SARS-CoV-2
Cabotegravir, GSK 744

Cabotegravir, GSK 744,
PMDA APPROVED 2022/5/31, JAPAN
(3S,11aR)-N-(2,4-Difluorobenzyl)-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide
3S, 1 1 aR)- N-[(2,4-difluorophenyl)methyl]-2,3,5,7, 1 1 , 1 1 a-hexahydro-6-hydroxy-3- methyl-5,7- dioxo-oxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxamide
OTHER ISOMER
(3R,11 aS)-N-[(2,4-Diflυorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7, 11, 11a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide
VIIV HEALTHCARE …INNOVATOR
-
GSK1265744, CAS 1051375-10-0, S-265744 LAP
-
C19-H17-F2-N3-O5
- 405.3553
- 744 LA
- GSK 1265744
- GSK 744
- GSK-1265744A
- GSK1265744
- GSK1265744A
- GSK744
- GSK744 LA
- GSK744 LAP
- S-265744
- S/GSK1265744
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Cabotegravir sodium | 3L12PT535M | 1051375-13-3 | AEZBWGMXBKPGFP-KIUAEZIZSA-M |
Cabotegravir, sold under the brand name Vocabria among others, is a antiretroviral medication used for the treatment of HIV/AIDS. It is available in the form of tablets and as an intramuscular injection, as well as in an injectable combination with rilpivirine under the brand name Cabenuva.[6][9]
It is an integrase inhibitor with a carbamoyl pyridone structure similar to that of dolutegravir.[10]
In December 2021, the U.S. Food and Drug Administration approved cabotegravir for pre-exposure prophylaxis (PrEP) in at-risk people under the brand name Apretude.[11]
GSK744 (also known as S/GSK1265744) is an investigational new drug under development for the treatment of HIV infection. It is anintegrase inhibitor, with a carbamoyl pyridone structure similar to dolutegravir. In investigational studies, the agent has been packaged into nanoparticles (GSK744LAP) conferring an exceptionally long half-life of 21–50 days following a single dose. In theory, this would make possible suppression of HIV with dosing as infrequently as once every three months.[1]
S-265744 LAP is in phase II clinical development at Shionogi-GlaxoSmithKline for the treatment of HIV infection. Phase III clinical trials had been ongoing for this indication; however, no recent development has been reported for this study.
Cabotegravir, or GSK1265744, is an HIV-1 integrase inhibitor that is prescribed with the non-nucleoside reverse transcriptase inhibitor, rilpivirine.4,6,7 Early research into cabotegravir showed it had lower oral bioavailability than dolutegravir,4 which resulted in the development of long acting monthly intramuscular injection formulation for cabotegravir.4,7
Cabotegravir was granted FDA approval on 21 January 2021 in combination with rilpivirine to treat HIV-1 infection in virologically suppressed individuals.8 While previously administered once monthly only, this combination product was granted FDA approval for dosing every two months on February 01, 2022 11 and without the need for an oral lead-in period prior.7
The human immunodeficiency virus (“HIV”) is the causative agent for acquired immunodeficiency syndrome (“AIDS”), a disease characterized by the destruction of the immune system, particularly of CD4+ T-cells, with attendant susceptibility to opportunistic infections, and its precursor Al DS-related complex (“ARC”), a syndrome characterized by symptoms such as persistent generalized lymphadenopathy, fever and weight loss. HIV is a retrovirus; the conversion of its RNA to DNA is accomplished through the action of the enzyme reverse transcriptase. Compounds that inhibit the function of reverse transcriptase inhibit replication of HIV in infected cells. Such compounds are useful in the prevention or treatment of HIV infection in humans.
A required step in HIV replication in human T-cells is the insertion by virally-encoded integrase of proviral DNA into the host cell genome. Integration is believed to be mediated by integrase in a process involving assembly of a stable nucleoprotein complex with viral DNA sequences, cleavage of two nucleotides from the 3′ termini of the linear proviral DNA and covalent joining of the recessed 3′ OH termini of the proviral DNA at a staggered cut made at the host target site. The repair synthesis of the resultant gap may be accomplished by cellular enzymes. There is continued need to find new therapeutic agents to treat human diseases. HIV integrase is an attractive target for the discovery of new therapeutics due to its important role in viral infections, particularly HIV infections. Integrase inhibitors are disclosed in WO2006/116724.
(3S, 1 1 aR)- N-[(2,4-difluorophenyl)methyl]-2,3,5,7, 1 1 , 1 1 a-hexahydro-6-hydroxy-3- methyl-5,7- dioxo-oxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxamide, a compound of formula (I), also referred to as compound (I), has proven antiviral activity against human immunodeficiency virus (HIV).

The present invention features pharmaceutical compositions comprising the active ingredient (3S, 1 1 aR)- N-[(2,4-difluorophenyl)methyl]-2,3,5,7, 1 1 , 1 1 a-hexahydro-6-hydroxy-3- methyl-5,7- dioxo-oxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxamide, or a pharmaceutically acceptable salt thereof, suitable for administration once monthly or longer.
Medical uses
Cabotegravir in combination with rilpivirine is indicated for the treatment of human immunodeficiency virus type-1 (HIV-1) in adults.[1][5] The combination injection is intended for maintenance treatment of adults who have undetectable HIV levels in the blood (viral load less than 50 copies/mL) with their current antiretroviral treatment, and when the virus has not developed resistance to non-nucleoside reverse transcriptase inhibitors (NNRTIs) and integrase strand transfer inhibitors.[5] The tablets are used to check whether a person tolerates the treatment before the injection therapy is started.[12][5]
The two medicines are the first antiretroviral drugs that come in a long-acting injectable formulation.[12]
Cabotegravir (Apretude) is indicated for use in at-risk people weighing at least 35 kilograms (77 lb) for pre-exposure prophylaxis (PrEP) to reduce the risk of sexually acquired HIV.[11]
Contraindications and interactions
Cabotegravir must not be combined with the drugs rifampicin, rifapentine, carbamazepine, oxcarbazepine, phenytoin or phenobarbital, which induce the enzyme UGT1A1.[5] These drugs significantly decrease cabotegravir concentrations in the body and thus may reduce its effectiveness.[9][5] Additionally, they induce the enzyme CYP3A4, which leads to reduced rilpivirine concentrations in the body.[5][13][14][15] Additionally, patients who are breastfeeding or plan to breastfeed should not take Cabotegravir because it is not known if it will pass within the breast milk.[16]
Adverse effects
The most common side effects of the injectable combination therapy with rilpivirine are reactions at the injection site (in up to 84% of patients) such as pain and swelling, as well as headache (up to 12%) and fever or feeling hot (in 10%). For the tablets, headache and a hot feeling were slightly less frequent. Less common side effects (under 10%) for both formulations are depressive disorders, insomnia, and rashes.[9]
Pharmacology
Mechanism of action
Cabotegravir is an integrase strand transfer inhibitor. This means it blocks the HIV’s enzyme integrase, thereby preventing its genome from being integrated into the human cells’ DNA.[9] As this is a necessary step for the virus to replicate, its further spread is hampered.[9]
Pharmacokinetics

Cabotegravir glucuronide, the main metabolite in human bile and urine[17]
When taken by mouth, cabotegravir reaches highest blood plasma levels after three hours. Taking the drug together with food slightly increases its concentrations in the blood, but this is not clinically relevant. After injection into the muscle, cabotegravir is slowly absorbed into the bloodstream, reaching its highest blood plasma levels after about seven days.[9]
Over 99% of the substance are bound to plasma proteins. The drug is inactivated in the body by glucuronidation, mainly by the enzyme UGT1A1, and to a much lesser extent by UGT1A9. More than 90% of the circulating substance are the unchanged cabotegravir, however. The biological half-life is 41 hours for the tablets and 5.6 to 11.5 weeks for the injection.[9]
Elimination has only been studied for oral administration: Most of the drug is eliminated via the faeces in unchanged form (47%). It is not known how much of this amount comes from the bile, and how much was not absorbed in the first place. (The bile actually contains the glucuronide, but this could be broken up again in the gut lumen to give the parent substance that is observed in the faeces.) To a lesser extent it is excreted via the urine (27%), almost exclusively as the glucuronide.[9]
Pharmacogenomics
UGT1A1 poor metabolizers have 1.3- to 1.5-fold increased cabotegravir concentrations in the body. This is not considered clinically significant.[9]
Chemistry
Cabotegravir is a white to off-white, crystalline powder that is practically insoluble in aqueous solutions under pH 9, and slightly soluble above pH 10. It is slightly acidic with a pKa of 7.7 for the enolic acid and 1.1 (calculated) for the carboxamide. The molecule has two asymmetric carbon atoms; only one of the four possible configurations is present in the medication.[18]
Formulation
In studies, the agent was packaged into nanoparticles (GSK744LAP) conferring a biological half-life of 21 to 50 days[citation needed] following a single dose. The marketed injection achieves its long half-life not via nanoparticles but with a suspension of the free cabotegravir acid. The tablets contain cabotegravir sodium salt.[18]
History
Cabotegravir was examined in the clinical trials HPTN 083 and HPTN 084.[19][20] In 2020, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Vocabria intended for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in combination with rilpivirine injection.[21] The EMA also recommended marketing authorization be given for rilpivirine and cabotegravir injections to be used together for the treatment of people with HIV-1 infection.[12] Cabotegravir was approved for medical use in the European Union in December 2020.[8]
Society and culture
Names
Cabotegravir is the United States Adopted Name (USAN)[22] and the international nonproprietary name (INN).[23]
Research
Pre-exposure prophylaxis
In 2020, results for some studies were released showing success in using injectable cabotegravir for long-acting pre-exposure prophylaxis (PrEP) with greater efficacy than the emtricitabine/tenofovir combination being widely used for PrEP at the time.[24][25]
The safety and efficacy of cabotegravir to reduce the risk of acquiring HIV were evaluated in two randomized, double-blind trials that compared cabotegravir to emtricitabine/tenofovir, a once daily oral medication for HIV PrEP.[11] Trial 1 included HIV-uninfected men and transgender women who have sex with men and have high-risk behavior for HIV infection.[11] Trial 2 included uninfected cisgender women at risk of acquiring HIV.[11]
In Trial 1, 4,566 cisgender men and transgender women who have sex with men received either cabotegravir or emtricitabine/tenofovir.[11] The trial measured the rate of HIV infections among trial participants taking daily cabotegravir followed by cabotegravir injections every two months compared to daily oral emtricitabine/tenofovir.[11] The trial showed participants who took cabotegravir had 69% less risk of getting infected with HIV when compared to participants who took emtricitabine/tenofovir.[11]
In Trial 2, 3,224 cisgender women received either cabotegravir or emtricitabine/tenofovir.[11] The trial measured the rate of HIV infections in participants who took oral cabotegravir and injections of cabotegravir compared to those who took emtricitabine/tenofovir orally.[11] The trial showed participants who took cabotegravir had 90% less risk of getting infected with HIV when compared to participants who took emtricitabine/tenofovir.[11]
In December 2021, the U.S. Food and Drug Administration (FDA) approved cabotegravir for pre-exposure prophylaxis.[11] The FDA granted the approval of Apretude to Viiv.[11]
Methods for the preparation of a compound of formula (I) are described in WO 2006/1 16764, WO2010/01 1814, WO2010/068262, and WO2010/068253
WO 2006116764
http://www.google.com/patents/WO2006116764A1?cl=en

[Chemical formula 68] is UNDESIRED ISOMER………..amcrasto@gmail.com

Example Z-1:
(3R,11 aS)-N-[(2,4-Diflυorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7, 11, 11a
-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide sodium salt.

(3R,11aS)-N-[(2,4-Diflυorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,
3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide. To a solution of 16a (409 mg, 0.87 mmol) in dichloroethane (20 mL) was added (2R)-2-amino-1-propanol (0,14 mL, 1.74 mmol) and 10 drops of glacial acetic acid.
The resultant solution was heated at reflux for 2 h. Upon cooling, Celite was added
to the mixture and the solvents removed in vacuo and the material was purified via
silica gel chromatography (2% CH3OH/CH2CI2 gradient elution) to give
(3R),11aS)-N-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6- [(phenylmethyl)oxy]-2,
3,5,7, 1 l , 11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazinc-8-carboxamide (396
mg, 92%) as a glass, JH NMR (CDCIo) δ 10.38 (m, 1 H), 8.42 (s, 1 H), 7,54-7,53 (m, 2
H), 7,37-7.24 (m, 4 H), 6.83-6,76 (m, 2 H), 5.40 (d, J = 10.0 Hz, 1 H), 5.22 (d, J = 10,0
Hz, 1 H), 5.16 (dd, J – 9,6, 6.0 Hz, 1 H), 4,62 (m, 2 H), 4.41 (m, 1 H), 4.33-4.30 (m, 2
H), 3.84 (dd, J= 12.0, 10.0 Hz, 1 H), 3.63 (dd, J= 8,4, 7.2 Hz, 1 H), 1.37 (d, J= 6.0 Hz,
3 H); ES+ MS: 496 (M+1).
b)
(3R, 11aS)-N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7, 11, 1la
-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8vcarboxamide sodium salt. To a
solution of
(37?, 11aS)-N-[(2,4-difluo]-ophenyl)methyl]-3-methyl-5,7-dioxo-6- [(phenylmethyl)oxy] -2,
3,5,7,11,11 a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (396
mg, 0.80 mmol) in methanol (30 mL) was added 10% Pd/C (25 mg). Hydrogen was
bubbled through the reaction mixture via a balloon for 2 h. The resultant mixture
was filtered through Celite with methanol and dichloromethanc. The filtrate was
concentrated in vacuo to give
(3R, l] aS)-N-f(2,4-difliιorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7, υ , 11a- hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide as a pink tinted
white solid (278 mg, 86%), 1H NMR (ODCU) δ 11.47 (m, 1 H), 10.29 (m, 1 H), 8,32 (s,
1 H), 7.36 (m, 1 H), 6.82 (m, 2 H), 5.31 (dd, J – 9.6, 3.6 Hz, 1 H), 4.65 (m, 2 H),
4,47-4,38 (m, 3 H), 3.93 (dd, J= 12.0, 10.0 Hz, 1 H), 3,75 (m, 1 H), 1.49 (d, J= 5.6 Hz,
3 H); BS1 MS: 406 (M+ 1). The above material (278 mg, 0,66 mmol) was taken up
m cthanol (10 mL) and treated with 1 Nsodium hydroxide (aq) (0.66 mL, 0.66 mmol).
The resulting suspension was stirred at room temperature for 30 min, Ether was
added and the liquids were collected to provide the sodium salt of the title compound
as a white powder (291 mg, 99%).‘ 1H NMR (OMSO- do) δ 30.68 (m, 1 H), 7,90 (s, 1 H),
7.35 (m, 1 H), 7.20 (m, 1 H), 7,01 (m, 1 H), 5,20 (m, 1 H), 4,58 (m, I H), 4.49 (m, 2 H),
4.22 (m, 2 H), 3 74 (dd, J= 11.2, 10.4 Hz, 1 H), 3.58 (m, 1 H), 1.25 (d, J=- 4.4 Hz, 3 H)
Example Z-9-
(3£ 11aΛ^N-[(2.4-D-fluoroDhonyl)methyl] -6-hvdroxy-3-methyl-5.7-dioxo-2,3,5.7, n , 11 a
-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazino-8-carboxamide sodium salt.

The title compound was made in two steps using a similar process to that described
in example Z-I. 16a (510 mg, 1.08 mmol) and (2«5)-2-amino-1-propanol (0.17 mL, 2,17 mmol) were reacted in 1,2-dichloroethane (20 mL) with acetic acid to give
(3S, 11aR)-i\A[(2,4-diflιιorophenyl)methyl]-3-methyl-5,7-d.ioxo-6-[(phenylmethyl)oxy]-2,
3,5,7,11,1la-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (500
mg, 93%). This material was hydrogenated in a second step as described in example
Z- I to give
3S, 11a R)-7N-[(2,4-Diiαuorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7, 11, 11a-
hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyraziine-8-carboxamide (386 mg, 94%) as a
tinted white solid. Η NMR (CDCL3) δ 11.46 (m, 1 H), 10.28 (m, 1 H), 8.32 (s, 1 H),
7.35 (m, 1 H), 6.80 (m, 2 H), 5.30 (dd, J = 10.0, 4.0 Hz, 1 H), 4.63 (m, 2 H), 4.48-4.37
(m, 3 H), 3.91 (dd, J = 12.0, 10.0 Hz, 1 H), 3.73 (m, 1 H), 1.48 (d, J – 6.0 Hz, 3 H);
ES 1 MS: 406 (M+ 1). This material (385 mg, 0.95 mmol) was treated with sodium
hydroxide (0,95 mL, 1.0 M, 0.95 mmol) m ethanol (15 mL) as described in example Z-1
to provide its corresponding sodrum sail (381 mg, 94%) as a white solid. 1H NMR
(DMSO- Λ) δ 10.66 (m, 1 PI), 7.93 (s, 1 H), 7.33 (m, 1 H), 7.20 (m, 1 H), 7.01 (m, 1 H),
5.19 (m, 1 H), 4.59 (m, 1 H), 4 48 (m, 2 H), 4.22 (m, 2 H), 3,75 (m, 1 H), 3.57 (m, 1 H),
1.24 (d, J= 5 6 Hz, 3 H).
WO 2010068253
http://www.google.com/patents/WO2010068253A1?cl=en
Example A
The starting material of Example A is compound 8, which is identical to formula (Ia). Thus, Example A depicts a process in providing an intermediate for the compound of formula 17 below which is isomeric to the compound ZZ-2 at page 237 of WO 2006/116764 to Brian Johns et al.

14


Example Aa After dissolution of mixture of 320 g of compound 8 (1.0 eq.) in 3.20 L of MeOH by heating, the solution was concentrated. To the residue, 1.66 L of MeCN, 5.72 mL of AcOH(0.1 eq.) and 82.6 g of (S)-2-Amino-propan-1-ol(1.1 eq.) were added and the mixture was heated to 70 °C, stirred at 70 0C for 4 h and concentrated. To the residue, 1.67 L of 2-propanol was added and the mixture was concentrated (twice). After cooling of the residue, filtration, washing with 500 mL of cold 2-propanol and drying provided 167 g of compound 14 (52% yield) as a crystal. 1H NMR(300 MHz1 CDCI3) δ 7.61-7.55 (m, 2H), 7.40-7.20 (m, 4H), 6.53 (d, J = 7.2, 1H), 5.46 (d, J = 10.5 Hz, 1H), 5.23 (d, J = 10.2 Hz, 1H), 5.20 (dd, J = 3.9, 9.6 Hz, 1H), 4.46- 4.34 (m, 1H)1 4.31 (dd, J = 6.6, 8.7 Hz, 1H)1 4.14 (dd, J = 3.9, 12.3 Hz1 1H)1 3.79 (dd, J = 9.9, 12.3 Hz1 1 H), 3.62 (dd, J = 6.9, 8.7 Hz1 1 H), 1.38 (d, J = 6.3 Hz1 3H).
Example Ab
To slurry of 156 g of compound 14 (1.0 eq.) in 780 ml_ of NMP was added 93.6 g of NBS(1.1 eq.) and the mixture was stirred at room temperature for 2.5 h. The reaction mixture was added to 3.12 L of H2O. Filtration, washing with 8.0 L of H2O and drying provided 163 g of compound 15 (84% yield) as a crystal.
1H NMR(300 MHz, DMSO-CT6) δ 8.37 (s, 1H), 7.55-7.50 (m, 2H), 7.42-7.25 (m, 3H), 5.34 (dd, J = 3.6, 9.9 Hz, 1H), 5.18 (d, J = 10.8 Hz, 1H), 5.03 (d, J = 10.5 Hz, 1H), 4.53 (dd, J = 3.6, 12.0 Hz, 1H)1 4.40-4.20 (m, 2H), 3.99 (dd, J = 9.9, 11.7 Hz1 1H), 3.64 (dd, J = 5.7, 8.1 Hz1 1 H)1 1.27 (d, J = 6.3 Hz1 3H).
Example Ac
Under carbon mono-oxide atmosphere, a mixture of 163 g of compound 15 (1.0 eq.), 163 mL of /-Pr2NEt(2.5 eq.), 68.4 ml_ of 2,4-difluorobenzylamine(1.5 eq.) and 22.5 g of Pd(PPh3)4(0.05 eq.) in 816 mL of DMSO was stirred at 90 0C for 7 h. After cooling, removal of precipitate, washing with 50 mL of DMSO and addition of 11.3 g of
Pd(PPh3)4(0.025 eq.), the reaction mixture was stirred at 90 0C for 2 h under carbon mono-oxide atmosphere again. After cooling, removal of precipitate and addition of 2.0 L of AcOEt and 2.0 L of H2O1 the organic layer was washed with 1.0 L of 1 N HCIaq. and 1.0 L of H2O (twice) and the aqueous layer was extracted with 1.0 L of AcOEt. The organic layers were combined and concentrated. Silica gel column chromatography of the residue provided 184 g of compound 16 (96% yield) as foam.
1H NMR(300 MHz, CDCI3) δ 10.38 (t, J = 6.3 Hz1 1H)1 8.39 (s, 1H)1 7.75-7.25 (m, 7H), 6.90-6.70 (m, 2H), 5.43 (d, J = 10.2 Hz, 1H), 5.24 (d, J = 10.2 Hz, 1H)1 5.19 (dd, J = 3.9, 9.9 Hz, 1H)1 4.63 (d, J = 6.0 Hz, 2H), 4.50-4.25 (m, 3H)1 3.86 (dd, J = 9.9, 12.3 Hz, 1H), , 3.66 (dd, J = 6.9, 8.4 Hz1 1 H), 1.39 (d, J = 6.0 Hz, 3H).
Example Ad
Under hydrogen atmosphere, a mixture of 184 g of compound 16 (1.0 eq.) and 36.8 g of 10%Pd-C in 3.31 L of THF and 0.37 L of MeOH was stirred for 3 h. After filtration of precipitate(Pd-C), washing with THF/MeOH(9/1 ) and addition of 36.8 g of 10% Pd-C, the mixture was stirred for 20 min under hydrogen atmosphere. After filtration of precipitate(Pd-C) and washing with THF/MeOH(9/1), the filtrate was concentrated. After 200 ml_ of AcOEt was added to the residue, filtration afforded crude solid of compound 17. The precipitates were combined and extracted with 4.0 L of CHCl3/MeOH(5/1). After concentration of the CHCI3ZMeOH solution and addition of 250 ml_ of AcOEt to the residue, filtration afforded crude solid of compound 17. The crude solids were combined and dissolved in 8.2 L of MeCN/H2O(9/1 ) by heating. After filtration, the filtrate was concentrated. To the residue, 1.5 L of EtOH was added and the mixture was concentrated (three times). After cooling of the residue, filtration and drying provided 132 g of compound 17 (88% yield) as a crystal. 1H NMR(300 MHz, DMSO-cfe) δ 11.47 (brs, 1H), 10.31 (t, J = 6.0 Hz, 1H), 8.46 (s, 1H), 7.40 (td, J = 8.6, 6.9 Hz, 1H), 7.24 (ddd, J = 2.6, 9.4, 10.6, 1H), 7.11-7.01 (m, 1H), 5.39 (dd, J = 4.1, 10.4 Hz, 1H), 4.89 (dd, J = 4.2, 12.3 Hz, 1H), 4.55 (d, J = 6.0 Hz, 2H), 4.40 (dd, J = 6.8, 8.6 Hz, 1H), 4.36-^.22 (m, 1H)1 4.00 (dd, J = 10.2, 12.3 Hz, 1H), 3.67 (dd, J = 6.7, 8.6 Hz, 1H), 1.34 (d, J = 6.3 Hz, 3H).
Example Ae
After dissolution of 16.0 g of compound 17 (1.0 eq.) in 2.56 L of EtOH and 0.64 L of H2O by heating, followed by filtration, 39 ml_ of 1N NaOHaq.(1.0 eq.) was added to the solution at 75 0C. The solution was gradually cooled to room temperature. Filtration, washing with 80 ml_ of EtOH and drying provided 13.5 g of compound 18 (80% yield) as a crystal.
1H NMR(300 MHz, DMSO-cfe) δ 10.73 (t, J = 6.0 Hz, 1H), 7.89 (s, 1H), 7.40-7.30 (m, 1H), 7.25-7.16 (m, 1H), 7.07-6.98 (m, 1H), 5.21 (dd, J = 3.8, 10.0 Hz, 1H), 4.58 (dd, J = 3.8, 12.1 Hz, 1H), 4.51 (d, J = 5.4 Hz, 2H), 4.3CM.20 (m, 2H), 3.75 (dd, J = 10.0, 12.1 Hz, 1H), 3.65-3.55 (m, 1H), 1.27 (d, J = 6.1 Hz, 3H).
………………
WO2010011814
http://www.google.st/patents/WO2010011814A1?cl=en&hl=pt-PT
Scheme 1

2a 2b
Scheme 2

Scheme 3

Scheme 4
phosphorylation


Scheme 5
Hydrogenolysis



The following examples are intended for illustratation only and are not intended to limit the scope of the invention in any way. Preparation 1 : (3S.11 af?VΛ/-r(2.4-DifluoroDhenvnmethyll-6-hvdroxy-3-methyl-5.7-dioxo- 2,3,5,7, 11 ,11 a-hexahydroM ,31oxazolor3,2-alpyridori ,2-c/1pyrazine-8-carboxamide sodium salt (compound 1 b, scheme 2).
I) MsCI, Et3N

2) DBU
P-1 P-2 P-3

a) Synthesis of 2-methyl-3-[(phenylmethvl)oxvl-4/-/-pvran-4-one (compound P-2). To a slurry of 2000 g of compound P-1(1.0 eq.) in 14.0 L of MeCN were added 2848 g of benzyl bromide(1.05 eq.) and 2630 g of K2CO3(1.2 eq.). The mixture was stirred at 80 0C for 5 h and cooled to 13°C. Precipitate was filtered and washed with 5.0 L of MeCN. The filtrate was concentrated and 3.0 L of THF was added to the residue. The THF solution was concentrated to give 3585 g of crude compound P-2 as oil. Without further purification, compound P-2 was used in the next step. 1H NMR(300 MHz, CDCI3) δ 7.60 (d, J = 5.7 Hz, 1 H), 7.4-7.3 (m, 5H), 6.37 (d, J = 5.7 Hz, 1 H), 5.17 (s, 2H), 2.09 (s, 3H).
b) Synthesis of 2-(2-hydroxy-2-phenylethyl)-3-[(phenylmethyl)oxy]-4H-pyran-4-one (compound P-3). To 904 g of the crude compound P-2 was added 5.88 L of THF and the solution was cooled to -60 0C. 5.00 L of 1.0 M of Lithium bis(trimethylsilylamide) in THF(1.25 eq.) was added dropwise for 2 h to the solution of compound 2 at -60 0C. Then, a solution of 509 g of benzaldehyde(1.2 eq.) in 800 ml. of THF was added at -60 0C and the reaction mixture was aged at -60 0C for 1 h. The THF solution was poured into a mixture of 1.21 L of conc.HCI, 8.14 L of ice water and 4.52 L of EtOAc at less than 2 0C.
The organic layer was washed with 2.71 L of brine (twice) and the aqueous layer was extracted with 3.98 L of EtOAc. The combined organic layers were concentrated. To the mixture, 1.63 L of toluene was added and concentrated (twice) to provide toluene slurry of compound P-3. Filtration, washing with 0.90 L of cold toluene and drying afforded 955 g of compound P-3 (74% yield from compound P-1 ) as a solid. 1H NMR(300 MHz, CDCI3) δ
7.62 (d, J = 5.7 Hz, 1 H), 7.5-7.2 (m, 10H), 6.38 (d, J = 5.7 Hz, 1 H), 5.16 (d, J = 11.4 Hz, 1 H), 5.09 (d, J = 11.4 Hz, 1 H), 4.95 (dd, J = 4.8, 9.0 Hz, 1 H), 3.01 (dd, J = 9.0, 14.1 Hz, 1 H), 2.84 (dd, J = 4.8, 14.1 Hz, 1 H).
c) Synthesis of 2-[(£)-2-phenylethenyl]-3-[(phenylmethyl)oxy]-4H-pyran-4-one (compound
P-4). To a solution of 882 g of compound P-3 (1.0 eq.) in 8.82 L of THF were added 416 g of Et3N(1.5 eq.) and 408 g of methanesulfonyl chloride(1.3 eq.) at less than 30 0C. After confirmation of disappearance of compound P-3, 440 ml. of NMP and 1167 g of DBU(2.8 eq.) were added to the reaction mixture at less than 30 0C and the reaction mixture was aged for 30 min. The mixture was neutralized with 1.76 L of 16% sulfuric acid and the organic layer was washed with 1.76 L of 2% Na2S03aq. After concentration of the organic layer, 4.41 L of toluene was added and the mixture was concentrated (tree times). After addition of 4.67 L of hexane, the mixture was cooled with ice bath. Filtration, washing with 1.77 L of hexane and drying provided 780 g of compound P-4 (94% yield) as a solid. 1H NMR(300 MHz, CDCI3) δ 7.69 (d, J = 5.7 Hz, 1 H), 7.50-7.25 (m, 10H), 7.22 (d, J = 16.2
Hz, 1 H), 7.03 (d, J = 16.2 Hz, 1 H), 6.41 (d, J = 5.7 Hz, 1 H), 5.27 (s, 2H). d) Synthesis of 4-oxo-3-[(phenylmethyl)oxy]-4H-pyran-2-carboxylic acid (compound P-5). To a mixture of 822 g of compound P-4 (1.0 eq.) and 1 1.2 g of RuCI3-nH2O(0.02 eq.) in 2.47 L of MeCN, 2.47 L of EtOAc and 2.47 L of H2O was added 2310 g of NalO4(4.0 eq.) at less than 25 0C. After aging for 1 h, 733 g of NaCIO2(S-O eq.) was added to the mixture at less than 25 0C. After aging for 1 h, precipitate was filtered and washed with 8.22 L of
EtOAc. To the filtrate, 1.64 L of 50% Na2S203aq, 822 ml. of H2O and 630 ml. of coc.HCI were added. The aqueous layer was extracted with 4.11 L of EtOAc and the organic layers were combined and concentrated. To the residue, 4 L of toluene was added and the mixture was concentrated and cooled with ice bath. Filtration, washing with 1 L of toluene and drying provided 372 g of compound P-5 (56% yield) as a solid. 1H NMR(300 MHz,
CDCI3) δ 7.78 (d, J = 5.7 Hz, 1 H), 7.54-7.46 (m, 2H), 7.40-7.26 (m, 3H), 6.48 (d, J = 5.7 Hz, 1 H), 5.6 (brs, 1 H), 5.31 (s, 2H).
e) Synthesis of 1-(2,3-dihydroxypropyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylic acid (compound P-6). A mixture of 509 g of compound P-5 (1.0 eq.) and
407 g of 3-amino-propane-1 ,2-diol(2.5 eq.) in 1.53 L of EtOH was stirred at 65 0C for 1 h and at 80 0C for 6 h. After addition of 18.8 g of 3-Amino-propane-1 ,2-diol(0.1 eq.) in 200 ml. of EtOH, the mixture was stirred at 80 0C for 1 h. After addition of 18.8 g of 3-amino- propane-1 ,2-diol (0.1 eq.) in 200 ml. of EtOH, the mixture was stirred at 80 0C for 30 min. After cooling and addition of 509 ml. of H2O, the mixture was concentrated. To the residue,
2.54 L of H2O and 2.54 L of AcOEt were added. After separation, the aqueous layer was washed with 1.02 L of EtOAc. To the aqueous layer, 2.03 L of 12% sulfuric acid was added at less than 12 0C to give crystal of compound P-6. Filtration, washing with 1.53 L of cold H2O and drying provided 576 g of compound P-6 (83% yield) as a solid. 1H NMR(300 MHz, DMSO-de) δ 7.67 (d, J = 7.5 Hz, 1 H), 7.5-7.2 (m, 5H), 6.40 (d, J = 7.5 Hz, 1 H), 5.07
(s, 2H), 4.2-4.0 (m, 1 H), 3.9-3.6 (m, 2H), 3.38 (dd, J = 4.2, 10.8 Hz, 1 H), 3.27 (dd, J = 6.0, 10.8 Hz, 1 H).
f) Synthesis of methyl 1-(2,3-dihydroxypropyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylate (compound P-7). To a slurry of 576 g of compound P-6 (1.0 eq.: 5.8% of H2O was contained) in 2.88 L of NMP were added 431 g of NaHCO3(3.0 eq.) and 160 ml. of methyl iodide(1.5 eq.) and the mixture was stirred at room temperature for 4 h. After cooling to 5 0C, 1.71 L of 2N HCI and 1.15 L of 20% NaClaq were added to the mixture at less than 10 0C to give crystal of compound 7. Filtration, washing with 1.73 L of H2O and drying provided 507 g of compound P-7 (89% yield) as a solid. 1H NMR(300 MHz, DMSO- cfe) δ 7.59 (d, J = 7.5 Hz, 1 H), 7.40-7.28 (m, 5H), 6.28 (d, J = 7.5 Hz, 1 H), 5.21 (d, J = 5.4 Hz, 1 H), 5.12 (d, J = 10.8 Hz, 1 H), 5.07 (d, J = 10.8 Hz, 1 H), 4.83 (t, J = 5.7 Hz, 1 H), 3.97 (dd, J = 2.4, 14.1 Hz, 1 H), 3.79 (s, 3H), 3.70 (dd, J = 9.0, 14.4 Hz, 1 H), 3.65-3.50 (m, 1 H), 3.40-3.28 (m, 1 H), 3.26-3.14 (m, 1 H).
g) Synthesis of methyl 1-(2,2-dihydroxyethyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylate (compound P-8). To a mixture of 507 g of compound P -7 (1.0 eq.) in
5.07 L of MeCN, 5.07 L of H2O and 9.13 g of AcOH(0.1 eq.) was added 390 g of NaIO4(1.2 eq.) and the mixture was stirred at room temperature for 2 h. After addition of 1.52 L of 10% Na2S2OsBq., the mixture was concentrated and cooled to 10 0C. Filtration, washing with H2O and drying provided 386 g of compound P-8 (80% yield) as a solid. 1H NMR(300 MHz, DMSO-d6) δ 7.62 (d, J = 7.5 Hz, 1 H), 7.42-7.30 (m, 5H), 6.33 (d, J = 6.0 Hz, 2H),
6.29 (d, J = 7.5 Hz, 1 H), 5.08 (s, 2H), 4.95-4.85 (m, 1 H), 3.80 (s, 3H), 3.74 (d, J = 5.1 Hz, 2H).
h) Synthesis of (3S, 11 aR)-3-methyl-6-[(phenylmethyl)oxy]-2,3, 1 1 ,1 1a- tetrahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-5,7-dione (compound P-9). After dissolution of mixture of 320 g of compound P-8 (1.0 eq.) in 3.20 L of MeOH by heating, the solution was concentrated. To the residue, 1.66 L of MeCN, 5.72 ml. of AcOH(0.1 eq.) and 82.6 g of (S)-2-Amino-propan-1-ol(1.1 eq.) were added and the mixture was heated to 70 0C, stirred at 70 0C for 4 h and concentrated. To the residue, 1.67 L of 2-propanol was added and the mixture was concentrated (twice). After cooling of the residue, filtration, washing with 500 ml. of cold 2-propanol and drying provided 167 g of compound P-9 (52% yield) as a solid. 1H NMR(300 MHz, CDCI3) δ 7.61-7.55 (m, 2H), 7.40-7.20 (m, 4H), 6.53 (d, J = 7.2, 1 H), 5.46 (d, J = 10.5 Hz, 1 H), 5.23 (d, J = 10.2 Hz, 1 H), 5.20 (dd, J = 3.9, 9.6 Hz, 1 H), 4.46-4.34 (m, 1 H), 4.31 (dd, J = 6.6, 8.7 Hz, 1 H), 4.14 (dd, J = 3.9, 12.3 Hz, 1 H), 3.79 (dd, J = 9.9, 12.3 Hz, 1 H), 3.62 (dd, J = 6.9, 8.7 Hz, 1 H), 1.38 (d, J = 6.3 Hz, 3H).
i) Synthesis of (3 S, 1 1 aR)-8-bromo-3-methyl-6-[(phenylmethyl)oxy]-2,3, 11 ,11a- tetrahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-5,7-dione (compound P-10). To slurry of 156 g of compound P-9 (1.0 eq.) in 780 ml. of NMP was added 93.6 g of NBS(1.1 eq.) and the mixture was stirred at room temperature for 2.5 h. The reaction mixture was added to 3.12 L of H2O. Filtration, washing with 8.0 L of H2O and drying provided 163 g of compound P-10 (84% yield) as a solid. 1H NMR(300 MHz, DMSO-d6) δ 8.37 (s, 1 H), 7.55- 7.50 (m, 2H), 7.42-7.25 (m, 3H), 5.34 (dd, J = 3.6, 9.9 Hz, 1 H), 5.18 (d, J = 10.8 Hz, 1 H), 5.03 (d, J = 10.5 Hz, 1 H), 4.53 (dd, J = 3.6, 12.0 Hz, 1 H), 4.40-4.20 (m, 2H), 3.99 (dd, J = 9.9, 1 1.7 Hz, 1 H), 3.64 (dd, J = 5.7, 8.1 Hz, 1 H), 1.27 (d, J = 6.3 Hz, 3H). j) Synthesis of (3S,1 1aS)-Λ/-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6- [(phenylmethyl)oxy]-2,3,5,7, 11 ,1 1 a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-8- carboxamide (compound P-11). Under carbon mono-oxide atmosphere, a mixture of 163 g of compound P-10 (1.0 eq.), 163 mL of /-Pr2NEt(2.5 eq.), 68.4 mL of 2,4- difluorobenzylamine(1.5 eq.) and 22.5 g of Pd(PPh3)4(0.05 eq.) in 816 mL of DMSO was stirred at 90 0C for 7 h. After cooling, removal of precipitate, washing with 50 mL of DMSO and addition of 1 1.3 g of Pd(PPh3)4(0.025 eq.), the reaction mixture was stirred at 90 0C for 2 h under carbon mono-oxide atmosphere again. After cooling, removal of precipitate and addition of 2.0 L of AcOEt and 2.0 L of H2O, the organic layer was washed with 1.0 L of 1 N HCIaq. and 1.0 L of H2O (twice) and the aqueous layer was extracted with 1.0 L of AcOEt.
The organic layers were combined and concentrated. Silica gel column chromatography of the residue provided 184 g of compound P-11 (96% yield) as foam. 1H NMR(300 MHz, CDCI3) δ 10.38 (t, J = 6.3 Hz, 1 H), 8.39 (s, 1 H), 7.75-7.25 (m, 7H), 6.90-6.70 (m, 2H), 5.43 (d, J = 10.2 Hz, 1 H), 5.24 (d, J = 10.2 Hz, 1 H), 5.19 (dd, J = 3.9, 9.9 Hz, 1 H), 4.63 (d, J = 6.0 Hz, 2H), 4.50-4.25 (m, 3H), 3.86 (dd, J = 9.9, 12.3 Hz, 1 H), 3.66 (dd, J = 6.9, 8.4 Hz,
1 H), 1.39 (d, J = 6.0 Hz, 3H).
k) Synthesis of (3S,1 1aR)-Λ/-[(2,4-difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo- 2,3,5,7, 11 ,11 a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-8-carboxamide (compound 1a). Under hydrogen atmosphere, a mixture of 184 g of compound P-11 (1.0 eq.) and 36.8 g of 10%Pd-C in 3.31 L of THF and 0.37 L of MeOH was stirred for 3 h. After filtration of precipitate(Pd-C), washing with THF/MeOH(9/1 ) and addition of 36.8 g of 10% Pd-C, the mixture was stirred for 20 min under hydrogen atmosphere. After filtration of precipitate(Pd-C) and washing with THF/MeOH(9/1 ), the filtrate was concentrated. After 200 mL of AcOEt was added to the residue, filtration afforded crude solid of compound 1 a.
The precipitates were combined and extracted with 4.0 L of CHCI3/Me0H(5/1 ). After concentration of the CHCI3/MeOH solution and addition of 250 mL of AcOEt to the residue, filtration afforded crude solid of compound 1a. The crude solids were combined and dissolved in 8.2 L of MeCN/H2O(9/1 ) by heating. After filtration, the filtrate was concentrated. To the residue, 1.5 L of EtOH was added and the mixture was concentrated
(three times). After cooling of the residue, filtration and drying provided 132 g of compound 1a (88% yield) as a solid. 1H NMR(300 MHz, DMSO-d6) δ 11.47 (brs, 1 H), 10.31 (t, J = 6.0 Hz, 1 H), 8.46 (s, 1 H), 7.40 (td, J = 8.6, 6.9 Hz, 1 H), 7.24 (ddd, J = 2.6, 9.4, 10.6, 1 H), 7.11- 7.01 (m, 1 H), 5.39 (dd, J = 4.1 , 10.4 Hz, 1 H), 4.89 (dd, J = 4.2, 12.3 Hz, 1 H), 4.55 (d, J = 6.0 Hz, 2H), 4.40 (dd, J = 6.8, 8.6 Hz, 1 H), 4.36-4.22 (m, 1 H), 4.00 (dd, J = 10.2, 12.3 Hz, 1 H), 3.67 (dd, J = 6.7, 8.6 Hz, 1 H), 1.34 (d, J = 6.3 Hz, 3H).
I) Synthesis of (3S,1 1aR)-Λ/-[(2,4-difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo- 2,3,5,7, 11 ,11 a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-8-carboxamide sodium salt (compound 1 b). After dissolution of 16.0 g of compound 1a (1.0 eq.) in 2.56 L of EtOH and 0.64 L of H2O by heating, followed by filtration, 39 ml. of 1 N NaOHaq.(1.0 eq.) was added to the solution at 75 0C. The solution was gradually cooled to room temperature. Filtration, washing with 80 ml. of EtOH and drying provided 13.5 g of compound 1b (80% yield) as a solid. 1H NMR(300 MHz, DMSO-d6) δ 10.73 (t, J = 6.0 Hz, 1 H), 7.89 (s, 1 H), 7.40-7.30 (m, 1 H), 7.25-7.16 (m, 1 H), 7.07-6.98 (m, 1 H), 5.21 (dd, J = 3.8, 10.0 Hz, 1 H), 4.58 (dd, J = 3.8, 12.1 Hz, 1 H), 4.51 (d, J = 5.4 Hz, 2H), 4.30-4.20 (m, 2H), 3.75 (dd, J = 10.0, 12.1 Hz, 1 H), 3.65-3.55 (m, 1 H), 1.27 (d, J = 6.1 Hz, 3H).
updates

An Efficient and Highly Diastereoselective Synthesis of GSK1265744, a Potent HIV Integrase Inhibitor
Abstract

Bictegravir and dolutegravir are two recently approved integrase inhibitors for the treatment of HIV. A third inhibitor, cabotegravir, is in Phase 3 development. As a continuation of a series of articles on synthetic routes to newly approved drugs, the current article reviews the patent and journal literature regarding synthetic routes and final forms of these drug





References
- ^ Jump up to:a b c “Vocabria (cabotegravir) film-coated tablets Product Information”. TGA eBS. 12 June 2021. Retrieved 12 June 2021.
- ^ Jump up to:a b “Vocabria”. Therapeutic Goods Administration (TGA). 26 February 2021. Retrieved 8 September 2021.
- ^ “Summary for ARTG Entry: 323721 VOCABRIA cabotegravir (as sodium) 30 mg film-coated tablet, bottle”. Therapeutic Goods Administration. The G overnment of Australia. 13 August 2021.
- ^ “Vocabria Product information”. Health Canada. 25 April 2012. Retrieved 22 January 2021.
- ^ Jump up to:a b c d e f g “Vocabria- cabotegravir sodium tablet, film coated”. DailyMed. Retrieved 12 June 2021.
- ^ Jump up to:a b “FDA Approves First Extended-Release, Injectable Drug Regimen for Adults Living with HIV”. U.S. Food and Drug Administration (FDA) (Press release). 21 January 2021. Retrieved 21 January 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Apretude- cabotegravir kit”. DailyMed. Retrieved 24 December 2021.
- ^ Jump up to:a b “Vocabria EPAR”. European Medicines Agency (EMA). Retrieved 22 January 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b c d e f g h i “Vocabria: EPAR – Product information” (PDF). European Medicines Agency. 5 January 2021.
- ^ Borrell B (March 2014). “Long-acting shot prevents infection with HIV analogue”. Nature News. doi:10.1038/nature.2014.14819. S2CID 184399045.
- ^ Jump up to:a b c d e f g h i j k l m “FDA Approves First Injectable Treatment for HIV Pre-Exposure Prevention”. U.S. Food and Drug Administration (FDA) (Press release). 20 December 2021. Retrieved 21 December 2021. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c “First long-acting injectable antiretroviral therapy for HIV recommended approval” (Press release). European Medicines Agency (EMA). 16 October 2020. Retrieved 16 October 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Cabenuva- cabotegravir and rilpivirine kit”. DailyMed. Retrieved 12 June 2021.
- ^ “Edurant- rilpivirine hydrochloride tablet, film coated”. DailyMed. Retrieved 12 June 2021.
- ^ “Rilpivirine Monograph for Professionals”. Drugs.com. 24 September 2020. Retrieved 12 June 2021.
- ^ “A Treatment Option | CABENUVA (cabotegravir; rilpivirine)”. http://www.cabenuva.com. Retrieved 16 April 2022.
- ^ Patel M, Eberl HC, Wolf A, Pierre E, Polli JW, Zamek-Gliszczynski MJ (August 2019). “Mechanistic Basis of Cabotegravir-Glucuronide Disposition in Humans”. The Journal of Pharmacology and Experimental Therapeutics. 370 (2): 269–277. doi:10.1124/jpet.119.258384. PMID 31175220. S2CID 182950312.
- ^ Jump up to:a b “Vocabria: EPAR – Public assessment report” (PDF). European Medicines Agency. 5 January 2021.
- ^ “HPTN083 — Prevention Now”. HIV Prevention Trials Network. Retrieved 2 December 2017.
- ^ “HPTN084 — Prevention Now”. HIV Prevention Trials Network. Retrieved 2 December 2017.
- ^ “Vocabria: Pending EC decision”. European Medicines Agency (EMA). 16 October 2020. Retrieved 16 October 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Adopted USANs” (PDF). American Medical Association. Retrieved 19 September 2014.
- ^ World Health Organization (2015). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 73”. WHO Drug Information. 29 (1): 70–1. hdl:10665/331088.
- ^ Ryan G (7 July 2020). “Injectable PrEP Is Even More Effective Than Daily Truvada”. Poz. Retrieved 9 November 2020.
- ^ Ryan G (9 November 2020). “For Women, Injectable Cabotegravir Is More Effective Than Truvada as PrEP”. Poz. Retrieved 9 November 2020.
External links
- “Cabotegravir”. Drug Information Portal. U.S. National Library of Medicine.
References
- PrEP GSK744 Integrase Administered Monthly Perhaps Quarterly Prevents HIV-Infection in Monkeys. 20th Conference on Retroviruses and Opportunistic Infections. Atlanta, GA March 3–6, 2013.
- http://www.natap.org/2013/CROI/croi_38.htm
SMILES [H][C@@]12CN3C=C(C(=O)NCC4=C(F)C=C(F)C=C4)C(=O)C(O)=C3C(=O)N1[C@@H](C)CO2
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2006116764A1 * | Apr 28, 2006 | Nov 2, 2006 | Shionogi & Co | Polycyclic carbamoylpyridone derivative having hiv integrase inhibitory activity |
| US6919351 * | Oct 9, 2001 | Jul 19, 2005 | Merck & Co., Inc. | Aza-and polyaza-naphthalenyl-carboxamides useful as HIV integrase inhibitors |
| WO2012018065A1 * | Aug 4, 2011 | Feb 9, 2012 | Shionogi & Co., Ltd. | Process for preparing compound having hiv integrase inhibitory activity |
| WO2012151361A1 | May 3, 2012 | Nov 8, 2012 | Concert Pharmaceuticals Inc. | Carbamoylpyridone derivatives |
| WO2013038407A1 * | Sep 2, 2012 | Mar 21, 2013 | Mapi Pharma Ltd. | Amorphous form of dolutegravir |
| US8552187 | Dec 9, 2009 | Oct 8, 2013 | Shionogi & Co., Ltd. | Processes and intermediates for carbamoylpyridone HIV integrase inhibitors |
| US8580967 | Jul 23, 2009 | Nov 12, 2013 | Shionogi & Co., Ltd. | Methyl 3-(benzyloxy)-1-(2,2-dihydroxyethyl)-4-oxo-1,4-dihydropyridine-2-carboxylate and processes for the preparation thereof |
| US8624023 | Dec 8, 2009 | Jan 7, 2014 | Shionogi & Co., Ltd. | Synthesis of carbamoylpyridone HIV integrase inhibitors and intermediates |
SYN
Synthetic Reference
Dolutegravir HELP . ADDED TO AID HELP
Dolutegravir HELP . ADDED TO AID HELP
Dolutegravir HELP . ADDED TO AID HELP
Dolutegravir HELP . ADDED TO AID HELP
Aoyama, Yasunori; Hakogi, Toshikazu; Fukui, Yuki; Yamada, Daisuke; Ooyama, Takao; Nishino, Yutaka; Shinomoto, Shoji; Nagai, Masahiko; Miyake, Naoki; Taoda, Yoshiyuki; Yoshida, Hiroshi; Yasukata, Tatsuro. Practical and Scalable Synthetic Method for Preparation of Dolutegravir Sodium: Improvement of a Synthetic Route for Large-Scale Synthesis. Organic Process Research & Development. Volume 23. Issue 4. Pages 558-564. Journal; Online Computer File. (2019).
Synthetic Reference 2
Wang, Xianheng; Chen, Song; Zhao, Changkuo; Long, Liangye; Wang, Yuhe. Preparation of Dolutegravir Intermediate Diastereomer. Journal of Heterocyclic Chemistry. Volume 56. Issue 7. Pages 2063-2067. Journal; Online Computer File. (2019).

Synthetic Reference 3
Ziegler, Robert E.; Desai, Bimbisar K.; Jee, Jo-Ann; Gupton, B. Frank; Roper, Thomas D.; Jamison, Timothy F. 7-Step Flow Synthesis of the HIV Integrase Inhibitor Dolutegravir. Angewandte Chemie, International Edition. Volume 57. Issue 24. Pages 7181-7185. Journal; Online Computer File. (2018).

SYN 4
Synthetic Reference
Rajan, Srinivasan Thirumalai; Eswaraiah, Sajja; Reddy, Ghojala Venkat; Reddy, Sagyam Rajeshwar; Markandeya, Bekkam; Rajesham, Boge. Novel crystalline polymorph of sodium (4R,12aS)-9-{[(2,4-difluorophenyl)methyl]carbamoyl}-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazin-7-olate and process for preparation thereof. Assignee MSN Research & Development Center, India. IN 201641037221. (2018).

Synthetic Reference 5
Sharma, Pramodkumar; Rao, Bhatraju Srinivasa; Deo, Keshav. A process for the preparation of Dolutegravir or its pharmaceutical acceptable salts thereof. Assignee Wockhardt Limited, India. IN 2015MU01007. (2016).

Synthetic Reference 6
Weaver, Jimmie Dean. Preparation of fluoroarenes via hydrogen bond directed photocatalytic hydrodefluorination of perfluoroarenes. Assignee The Board of Regents for Oklahoma State University, USA. WO 2018187336. (2018).

SYN 7
Synthetic Reference
Li, Xuguang; Chen, Shuiku; Zhu, Songlin; Zhang, Fangjie; Fang, Shuixia; Liu, Congjun; Zhu, Huifeng; Luo, Qi; Meng, Qingyue; Cui, Hao. Method for preparation of dolutegravir. Kaifeng Pharmaceutical (Group) Co., Ltd.; Henan Furen Pharmaceutical Technology Development Co., Ltd. CN 108299466. (2018).


SYN 8
Synthetic Reference
Vellanki, Sivaram Prasad; Nadella, Madumurthy; Bhalme, Mitali; Ramabhotla, Revathi Srinivas. Process for the preparation of dolutegravir, an integrase inhibitor for HIV-1 infection therapy. Assignee Mylan Laboratories Ltd., India. IN 2015CH00588. (2016).

SYN 9
Synthetic Reference
Sankareswaran, Srimurugan; Mannam, Madhavarao; Chakka, Veerababu; Mandapati, Srirami Reddy; Kumar, Pramod. Identification and Control of Critical Process Impurities: An Improved Process for the Preparation of Dolutegravir Sodium. Organic Process Research & Development. Volume 20. Issue 8. Pages 1461-1468. Journal; Online Computer File. (2016).
TOLVAPTAN

TOLVAPTAN
的合成
N-(4-{[(5R)-7-chloro-5-hydroxy-2,3,4,5-tetrahydro-1H-1-benzazepin-1-yl]carbonyl}-3-methylphenyl)-2-methylbenzamide
| Formula | C26H25ClN2O3 |
|---|---|
| Mol. mass | 448.941 g/mol |
150683-30-0 CAS NO
+ form 331947-66-1 Rform
OPC-41061
Otsuka…..innovator
UPDATE 2022
Tolvaptan sodium phosphate, Samtasu,
|
トルバプタンリン酸エステルナトリウム
|
| 2022/3/28 JAPAN PPROVED |
| Formula |
C26H24ClN2O6P. 2Na
|
|---|---|
| CAS |
|
| Mol weight |
572.8849
|
Tolvaptan sodium phosphate
disodium;[(5R)-7-chloro-1-[2-methyl-4-[(2-methylbenzoyl)amino]benzoyl]-2,3,4,5-tetrahydro-1-benzazepin-5-yl] phosphate
European Medicines Agency (EMA) Accepts Otsuka’s Marketing Authorisation Application (MAA) for Tolvaptan, an Investigational Compound for Autosomal Dominant Polycystic Kidney Disease (ADPKD)
•Tolvaptan was discovered by Otsuka in Japan and, if approved by the EMA, would become the first pharmaceutical therapy in Europe for patients with ADPKD
•ADPKD is an inherited genetic disease that causes cyst growth in the kidneys, which gradually impairs their functioning. There is no current pharmaceutical treatment option
•Otsuka’s development of tolvaptan as a treatment for ADPKD illustrates the company’s commitment to address significant patient needs for diseases that traditionally have not been a priority for the pharmaceutical industry
TOKYO–(BUSINESS WIRE)–Otsuka Pharmaceutical Co., Ltd. announced today that the European Medicines Agency (EMA) has accepted the submission of a marketing authorisation application (MAA) for the potential approval of tolvaptan for the treatment of autosomal dominant polycystic kidney disease (ADPKD). Phase III clinical trial results that form the basis of the regulatory filing were published in the New England Journal of Medicine.
http://www.pharmalive.com/ema-accepts-otsukas-maa-for-tolvaptan
Tolvaptan is a selective vasopressin V2-receptor antagonist with an affinity for the V2-receptor that is 1.8 times that of native arginine vasopressin (AVP).
Tolvaptan is (±)-4′-[(7-chloro-2,3,4,5-tetrahydro-5-hydroxy-1H-1-benzazepin-1-yl) carbonyl]-otolu-m-toluidide. The empirical formula is C26H25ClN2O3. Molecular weight is 448.94. The chemical structure is:
 |
SAMSCA tablets for oral use contain 15 mg or 30 mg of tolvaptan. Inactive ingredients include corn starch, hydroxypropyl cellulose, lactose monohydrate, low-substituted hydroxypropyl cellulose, magnesium stearate and microcrystalline cellulose and FD&C Blue No. 2 Aluminum Lake as colorant.
SEE NEW UPDATE AT END OF PAGE
Tolvaptan (INN), also known as OPC-41061, is a selective, competitive vasopressin receptor 2 antagonist used to treat hyponatremia (low blood sodium levels) associated withcongestive heart failure, cirrhosis, and the syndrome of inappropriate antidiuretic hormone(SIADH). Tolvaptan was approved by the U.S. Food and Drug Administration (FDA) on May 19, 2009, and is sold by Otsuka Pharmaceutical Co. under the trade name Samsca and in India is manufactured & sold by MSN laboratories Ltd. under the trade name Tolvat & Tolsama.
Tolvaptan is also in fast-track clinical trials[2] for polycystic kidney disease. In a 2004 trial, tolvaptan, when administered with traditional diuretics, was noted to increase excretion of excess fluids and improve blood sodium levels in patients with heart failure without producing side effects such as hypotension (low blood pressure) or hypokalemia(decreased blood levels of potassium) and without having an adverse effect on kidney function.[3] In a recently published trial (TEMPO 3:4 ClinicalTrials.gov number, NCT00428948) the study met its primary and secondary end points. Tolvaptan, when given at an average dose of 95 mg per day over a 3-year period, slowed the usual increase in kidney volume by 50% compared to placebo (2.80% per year versus 5.51% per year, respectively, p<0.001) and reduced the decline in kidney function when compared with that of placebo-treated patients by approximately 30% (reciprocal serum creatinine, -2.61 versus -3.81 (mg/mL)-1 per year, p <0.001)[4]
Tolvaptan was first approved by the U.S. Food and Drug Administration (FDA) on May 19, 2009, then approved by the European Medicines Agency (EMA) on August 3, 2009 and approved by Pharmaceuticals and Medical Devices Agency of Japan on Feb 4, 2013. It was developed and marketed as Samsca® by Otsuka in the US, DE and JP.
UPDATED
Tolvaptan is a selective vasopressin V2-receptor antagonist with an affinity for the V2-receptor that is 1.8 times that of native arginine vasopressin (AVP) and that is 29 times greater than for the V1a-receptor. When taken orally, 15 to 60 mg doses of tolvaptan antagonize the effect of vasopressin and cause an increase in urine water excretion that results in an increase in free water clearance (aquaresis), a decrease in urine osmolality, and a resulting increase in serum sodium concentrations. It is indicated for the treatment of clinically significant hypervolemic and euvolemic hyponatremia [serum sodium < 125 mEq/L or less marked hyponatremia that is symptomatic and has resisted correction with fluid restriction], including patients with heart failure, cirrhosis, and syndrome of inappropriate antidiuretic hormone (SIADH).
Samsca® is available as tablet for oral use, containing 7.5 mg/15 mg/30 mg of free Tolvaptan. The recommended starting dose is 15 mg once daily and it may be increased at intervals ≥ 24 hr to 30 mg once daily, and to a maximum of 60 mg once daily as needed to raise serum sodium.
| PATENT NUMBER | PEDIATRIC EXTENSION | APPROVED | EXPIRES (ESTIMATED) | |
|---|---|---|---|---|
| US5258510 | No | 1993-11-02 | 2010-11-02 |  |
| US5753677 | No | 1998-05-19 | 2020-05-19 | |
| US8501730 | No | 2013-08-06 | 2026-09-01 | |
| US5972882 | No | 1999-10-26 | 2018-12-14 | |
| US10905694 | No | 2021-02-02 | 2030-04-07 | |
Synthesis Reference
Bandi Parthasaradhi Reddy, “PROCESS FOR PREPARING TOLVAPTAN INTERMEDIATES.” U.S. Patent US20130190490, issued July 25, 2013.
US20130190490

Reference:1. US5258510A.

Reference:1. Bio. Med. Chemistry 2006, 14, 6165–6173.

Reference:1. Bio. Med. Chem. Lett. 2007, 17, 6455–6458.

Reference:1. CN102060769B.

Reference:1. Org. Lett. 2014, 16, 6041−6043.

Reference:1. Bio. Med. Chem. 1999, 7, 1743-1754.
2. WO2007026971A2.
SYN
SYN
Chemical synthesis:[5] 
Tolvaptan is chemically, N-[4-[(7-chloro-2,3,4,5-tetrahydro-5-hydroxy1H-1-benzazepin-1-yl)carbonyl]-3-methylphenyl]-2-methylbenzamide. Tolvaptan is represented by the following structure:
Tolvaptan, also known as OPC-41061, is a selective, competitive arginine vasopressin receptor 2 antagonist used to treat hyponatremia (low blood sodium levels) associated with congestive heart failure, cirrhosis, and the syndrome of inappropriate antidiuretic hormone (SIADH). Tolvaptan is sold by Otsuka Pharmaceutical Co. under the trade name Samsca.
Tolvaptan and its process for preparation were disclosed in U.S. Pat. No. 5,258,510.
Processes for the preparation of 7-chloro-2,3,4,5-tetrahydro-1H-1-benzazepin-5-one, 7-chloro-1-(2-methyl-4-nitrobenzoyl)-5-oxo-2,3,4,5-tetrahydro-1H-1-benzazepine and 7-chloro-1-[2-methyl-4-[(2-methylbenzoyl)amino]benzoyl]-5-oxo-2,3,4,5-tetrahydro-1H-1-benzazepine were reported in Bioorganic & medicinal chemistry 7 (1999), 1743-1754. According to the journal, 7-chloro-2,3,4,5-tetrahydro-1H-1-benzazepin-5-one can be prepared by reacting 7-chloro-4-ethoxycarbonyl-5-oxo-N-p-toluenesufonyl-2,3,4,5-tetrahydro-1H-1-benzazepine with acetic acid in the presence of hydrochloric acid and water to obtain 7-chloro-5-oxo-2,3,4,5-tetrahydro-1-p-toluenesulfonyl-1H-1-benzazepine, and then reacted with polyphospholic acid. According to the journal, 7-chloro-1-(2-methyl-4-nitrobenzoyl)-5-oxo-2,3,4,5-tetrahydro-1H-1-benzazepine can be prepared by reacting 7-chloro-5-oxo-2,3,4,5-tetrahydro-1H-1-benzazepine with 2-methyl-4-nitobenzoyl chloride in the presence of triethylamine.
According to the journal, 7-chloro-1-[2-methyl-4-[(2-methylbenzoyl)amino]benzoyl]-5-oxo-2,3,4,5-tetrahydro-1H-1-benzazepine can be prepared by reacting 1-(4-amino-2-methylbenzoyl)-7-chloro-5-oxo-2,3,4,5-tetrahydro-1H-1-benzazepine with 2-methylbenzoylchloride in the presence of triethylamine.
PCT publication no. WO 2007/026971 disclosed a process for the preparation oftolvaptan can be prepared by the reduction of 7-chloro-1-[2-methyl-4-(2-methylbenzoylamino)benzoyl]-2,3,4,5-tetrahydro-1H-1-benzazepin-5-one with sodium borohydride.
7-Chloro-2,3,4,5-tetrahydro-1H-1-benzazepin-5-one is a key intermediate for the preparation of tolvaptan.
Biooganic and Medicinal Chemistry I (2007) 6455-6458, Biooganic andMedicinal Chemistry 14 (2000) 2493-2495 reported in the literature of the intermediate 2 – carboxylic acid -5 – (2 – methyl-benzoylamino) toluene synthesis method,


5-Chloro-2-nitrobenzoic acid (I) was converted into methyl ester (II) using dimethyl sulfate and K2CO3 in acetone. The nitro group of (II) was then reduced with SnCl2 to afford aniline (III), which was protected as the p-toluenesulfonamide (IV) with tosyl chloride in pyridine. Alkylation of (IV) with ethyl 4-bromobutyrate (V) yielded diester (VI). Subsequent Dieckmann cyclization of (VI) in the presence of potassium tert-butoxide provided benzazepinone (VIIa-b) as a mixture of ethyl and methyl esters, which was decarboxylated to (VIII) by heating with HCl in AcOH. Deprotection of the tosyl group of (VIII) was carried out in hot polyphosphoric acid. The resulting benzazepinone (IX) was condensed with 2-methyl-4-nitrobenzoyl chloride (X) to give amide (XI). After reduction of the nitro group of (XI) to the corresponding aniline (XII), condensation with 2-methylbenzoyl chloride (XIII) provided diamide (XIV). Finally, ketone reduction in (XIV) by means of NaBH4 led to the target compound.
……………………………………….
PATENT

……………………………………………..
PATENT

Synthesis of Intermediate III: 1.
Example
2-methyl-4-nitrobenzoic acid (available from Alfa Aesar Tianjin Chemical Co., purity> 99%, 25g,
0.14mol) was added to a 250ml reaction flask, is reacted with thionyl chloride under reflux conditions for 3h, thionyl chloride was distilled off under reduced pressure to give 2-methyl-4-nitrobenzoyl chloride (26.Sg, light yellow oily liquid), without purification, was used directly in the next step.
Intermediate II (20g, 0.1moI) and 2_ methyl _4_ nitrobenzoylchloride (22.4g, 0.llmol) was added to a 250ml reaction flask. Dichloromethane (50ml), cooled to ice bath with stirring to dissolve O~5 ° C, was slowly added dropwise N- methylmorpholine (11.2g, 0.llmol), Bi dropwise with stirring while, at room temperature the reaction 4h. TLC [developing solvent: ethyl acetate – petroleum ether (I: I), hereinafter] is displayed after completion of the reaction, saturated aqueous sodium bicarbonate (20ml), stirred for lOmin, filtered, the filter cake with dichloromethane (15ml X 2 ) washing. The filtrate and washings were combined, washed with saturated sodium chloride solution (30ml X 3), dried over anhydrous sodium sulfate and filtered. The filtrate under reduced pressure to recover the solvent, the residue was recrystallized from anhydrous methanol to give a white powder 111 (27.5g, 75.2%), mp 154.8 ~155.6 ° C. Purity 97.9% (HPLC normalization method).
Synthesis of Intermediate IV:
Intermediate III (10g, 28mmol) was added to a 250ml reaction flask, concentrated hydrochloric acid (40ml) and ethanol (50ml), with stirring, was slowly added dropwise stannous chloride (20g, 88mmol) in ethanol (40ml) . Bi room temperature drops 5h. After TLC showed completion of the reaction, ethanol was distilled off under reduced pressure to about 70ml, the residue was -10 ° C -0 ° C allowed to stand overnight to cool. Filtered, and the filter cake was washed with water poured into water (40ml) in. Plus 20% sodium hydroxide solution (approximately 60ml) was adjusted to pH 9. Filtered, washed with ethanol and recrystallized to give a pale yellow powdered solid IV (6.3g, 68.7%), mp 190.4~191.1 ° C. Purity 97.2% (HPLC normalization method).
Synthesis of intermediate V:
Intermediate IV (5g, 15mmol) and triethylamine (2.3g, 23mmol) was added followed by IOOml reaction flask was added dichloromethane (30ml), stir until dissolved. Solution of o-methylbenzoyl chloride (2.8g, 18mmol), dropwise at room temperature completion of the reaction Ih0 TLC showed the reaction was complete was poured into ice-water (about 40ml) in, (20ml X 3) and extracted with dichloromethane, the combined organic phases, and saturated sodium chloride solution successively (25ml X 3), dried over anhydrous sodium sulfate and filtered with 5% hydrochloric acid (25ml X 3). The filtrate under reduced pressure to recover the solvent (about 50ml), dried over anhydrous methanol residue – petroleum ether (2: 1) and recrystallized to give white crystals of Intermediate V (6.2g, 90.9%), mp 121.1 ~123.6 ° C. Purity 98.6% (HPLC normalization method).
Synthesis of tolvaptan: Example 4
Intermediate V (5g, Ilmmol) IOOml added to the reaction flask, was added anhydrous methanol (25ml), stirred and then added portionwise sodium borohydride (0.65g, 17mmol) to the reaction mixture, addition was complete the reaction at room temperature lh. After TLC showed the reaction was complete, the methanol recovered under reduced pressure (approximately 20ml), the residue was added methylene chloride (25ml), (25mlX3) and washed with saturated sodium chloride solution. Anhydrous sodium sulfate and filtered, and the filtrate under reduced pressure to recover the solvent, the residue with absolute methanol – petroleum ether (2: 1) and recrystallized tolvaptan white crystals (4.85g, 96.6%), mp 220.1~221.5 ° C. Purity 99.2% (HPLC normalization method). ES1-HRMS (C26H25C1N203, m / z) found (calc): 447.1476 (447.1481) [MH] – “
…………..
PATENT
http://www.google.com/patents/WO2012046244A1?cl=en
Tolvaptan is chemically, N-[4-[(7-chloro-2,3,4,5-tetrahydro-5-hydroxylH-l- benzazepin- 1 -yl)carbonyl]-3-methylphenyl]-2-methylbenzamide. Tolvaptan is represented by the following structure:

Tolvaptan, also known as OPC-41061, is a selective, competitive arginine vasopressin receptor 2 antagonist used to treat hyponatremia (low blood sodium levels) associated with congestive heart failure, cirrhosis, and the syndrome of inappropriate antidiuretic hormone (SIADH). Tolvaptan is sold by Otsuka Pharmaceutical Co. under the trade name Samsca.
Tolvaptan and its process for preparation were disclosed in U.S. patent no. 5,258,510. Processes for the preparation of 7-chloro-2,3,4,5-tetrahydro-lH-l-benzazepin-5- one, 7-chloro-l-(2-methyl-4-nitrobenzoyl)-5-oxo-2,3,4,5-tetrahydro-lH-l-benzazepine and 7-chloro- 1 -[2-methyl-4-[(2-methylbenzoyl)amino]benzoyl]-5-oxo-2,3,4,5- tetrahydro-lH-l-benzazepine were reported in Bioorganic & medicinal chemistry 7 (1999), 1743-1754. According to the journal, 7-chloro-2,3,4,5-tetrahydro-lH-l- benzazepin-5-one can be prepared by reacting 7-chloro-4-ethoxycarbonyl-5-oxo-N-p- toluenesufonyl-2,3,4,5-tetrahydro-lH-l-benzazepine with acetic acid in the presence of hydrochloric acid and water to obtain 7-chloro-5-oxo-2,3,4,5-tetrahydro-l-p- toluenesulfonyl-lH-l-benzazepine, and then reacted with polyphospholic acid.
According to the journal, 7-chloro- 1 -(2 -methyl-4-nitrobenzoyl)-5-oxo-2,3,4,5- tetrahydro-lH-l-benzazepine can be prepared by reacting 7-chloro-5-oxo-2,3,4,5- tetrahydro-lH-l-benzazepine with 2-methyl-4-nitobenzoyl chloride in the presence of triethylamine.
According to the journal, 7-chloro- l-[2-methyl-4-[(2- methylbenzoyl)amino]benzoyl]-5-oxo-2,3,4,5-tetrahydro-lH-l-benzazepine can be prepared by reacting l-(4-amino-2-methylbenzoyl)-7-chloro-5-oxo-2,3,4,5-tetrahydro- lH-l-benzazepine with 2-methylbenzoylchloride in the presence of triethylamine.
PCT publication no. WO 2007/026971 disclosed a process for the preparation of tolvaptan can be prepared by the reduction of 7-chloro- l-[2-methyl-4-(2- methylbenzoylamino)benzoyl]-2,3,4,5-tetrahydro-lH-l-benzazepin-5-one with sodium borohydride.
7-Chloro-2,3,4,5-tetrahydro-lH-l-benzazepin-5-one is a key intermediate for the preparation of tolvaptan.
SYNTHESIS CONSTRUCTION


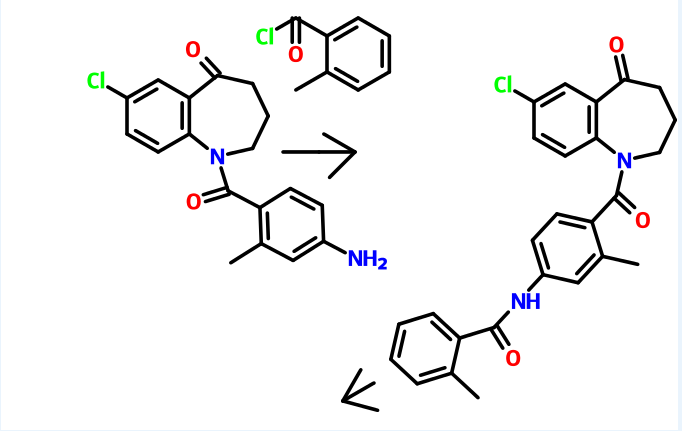

Reference example 1 :
Preparation of methyl 5-chloro-2-nitrobenzoate
Potassium carbonate (515 gm) was added to a solution of 5-chloro-2-nitro benzoic acid (500 gm) in acetone (2750 ml) at room temperature. Dimethyl sulphate (306.5 gm) was added to the reaction mixture slowly and heated to reflux for 30 minutes. The reaction mass was filtered and then concentrated to obtain a residual mass. The residual mass was poured to the ice water and extracted with methylene chloride. The solvent was distilled off under reduced pressure to obtain a residual solid of methyl 5- chloro-2-nitrobenzoate (534 gm). Reference example 2:
Preparation of methyl 2-amino-5-chlorobenzoate
A mixture of methyl 5-chloro-2-nitrobenzoate (534 gm) as obtained in reference example 1 and concentrated hydrochloric acid (2250 ml) was added to ethyl acetate (1120 ml). To the reaction mixture was added a solution of tin chloride (1680 gm) in ethyl acetate (2250 ml). The reaction mass was stirred for 16 hours at room temperature and then poured to the ice water. The pH of the reaction mass was adjusted to 8.0 to 9.0 with aqueous sodium hydroxide solution (2650 ml). The separated aqueous layer was extracted with ethyl acetate and then concentrated to obtain a residual solid of methyl 2- amino-5-chlorobenzoate (345 gm). Reference example 3:
Preparation of methyl 5-chIoro-2-(N-p-toluenesulfonyl)aminobenzoate
To a solution of methyl-2-amino-5-chloro benzoate (345 gm) as obtained in reference example 2 in pyridine (1725 ml) was added p-toluenesulfonyl chloride (425 gm). The reaction mixture was stirred for 2 hours at room temperature and poured to the ice water. The separated solid was filtered and dried to obtain 585 gm of methyl 5- chloro-2-(N-p-toluenesulfonyl)aminobenzoate.
Reference example 4:
Preparation of methyl 5-chloro-2-[N-(3-ethoxycarbonyI)propyI-N-p- toluenesulfonyl] aminobenzoate
Methyl 5-chloro-2-(N-p-toluenesulfonyl)aminobenzoate (585 gm) as obtained in reference example 3, ethyl-4-bromo butyrate (369.6 gm) and potassium carbonate (664 gm) in dimethylformamide (4400 ml) were added at room temperature. The contents were heated to 120°C and maintained for 2 hours. The reaction mass was poured into water and filtered. The solid obtained was dried to give 726 gm of methyl 5-chloro-2-[N- (3 -ethoxycarbonyl)propyl-N-p-toluenesulfonyl] aminobenzoate.
Reference example 5:
Preparation of 7-chloro-4-ethoxycarbonyI-5-oxo-N-p-toluenesufonyl-2,3,4,5- tetrahydro-lH-l-benzazepine
To a heated mixture of potassium tetrabutoxide (363 gm) in toluene (1000 ml) at 70°C was added portion wise methyl 5-chloro-2-[N-(3-ethoxycarbonyl)propyl-N-p- toluenesulfonyl]aminobenzoate (726 gm) as obtained in reference example 4. The contents were heated to reflux and maintained for 30 minutes. The reaction mass was then cooled to room temperature and then poured to the ice water. The layers were separated and the aqueous layer was extracted with toluene. The solvent was distilled off under reduced pressure to obtain a residual solid of 7-chloro-4-ethoxycarbonyl-5-oxo-N- p-toluenesufonyl-2,3,4,5-tetrahydro-lH-l-benzazepine (455 gm).
Example 1:
Preparation of 7-chIoro-5-oxo-2,3,4,5-tetrahydro-lH-l-benzazepine
7-Chloro-4-ethoxycarbonyl-5-oxo-N-p-toluenesufonyl-2,3,4,5-tetrahydro- 1 H- 1 – benzazepine (455 gm) as obtained in reference example 5 was added to aqueous sulfuric acid (80%, 2275 ml). The contents heated to 75°C and maintained for 2 hours. The reaction mass was then cooled to room temperature and then poured to the ice water. The pH of the reaction mass was adjusted to 7.5 to 8.0 with sodium hydroxide solution (2575 ml). The solid obtained was collected by filtration and dried to give 160 gm of 7- chloro-5-oxo-2,3 ,4,5-tetrahydro- 1 H- 1 -benzazepine.
Example 2:
Preparation of 7-chIoro-l-(2-methyl-4-nitrobenzoyl)-5-oxo-2,3,4,5-tetrahydro-lH-l- benzazepine
7-Chloro-5-oxo-2,3,4,5-tetrahydro-lH-l -benzazepine (160 gm) as obtained in example 1 was dissolved in methylene dichloride (480 ml) and then added aqueous sodium bicarbonate solution (20%, 68.75 gm). The reaction mixture was then cooled to 0 to 5°C and then added 2-methyl-4-nitrobenzoylchloride (180 gm) slowly. The pH of the reaction mass was adjusted to 7.0 to 8.0 with aqueous sodium bicarbonate solution (170 ml). The layers were separated and the aqueous layer was extracted with methylene chloride. The solvent was distilled off under reduced pressure to obtain a residual mass. To the residual mass was dissolved in isopropyl alcohol (7300 ml) and maintained for 2 hours at reflux temperature. The separated solid was filtered and dried to obtain 250 gm of 7-chloro-l-(2-methyl-4-nitrobenzoyl)-5-oxo-2,3,4,5-tetrahydro-lH-l-benzazepine. Example 3:
Preparation of l-(4-amino-2-methylbenzoyl)-7-chIoro-5-oxo-2,3,4,5-tetrahydro-lH- 1-benzazepine
7-Chloro- 1 -(2-methyl-4-nitrobenzoyl)-5-oxo-2,3 ,4,5-tetrahydro- 1 H- 1 – benzazepine (250 gm) as obtained in example 2 was dissolved in methanol (575 ml) and then added a solution of tin chloride (630 gm) in methanol (1130 ml). The reaction mixture was stirred for 16 hours at room temperature and then poured to the ice water. The pH of the reaction mass was adjusted to 8.0 to 9.0 with sodium hydroxide solution (1250 ml). The layers were separated and the aqueous layer was extracted with ethyl acetate. The solvent was distilled off under vacuum to obtain a residual solid of l-(4- amino-2-methylbenzoyl)-7-chloro-5-oxo-2,3,4,5-tetrahydro- 1 H- 1 -benzazepine (185 gm).
Example 4:
Preparation of 7-chloro-l-[2-methyl-4-[(2-methylbenzoyl)amino]benzoyl]-5-dxo- 2,3,4,5-tetrahydro-lH-l-benzazepine
1 -(4-Amino-2-methylbenzoyl)-7-chloro-5-oxo-2,3 ,4,5-tetrahydro- 1 H- 1 – benzazepine (185 gm) as obtained in example 3 was dissolved in methylene chloride (4000 ml) and then added sodium bicarbonate solution (10%, 47.3 gm). The reaction mass was cooled to 0 to 5°C and then added 2-methyl benzoyl chloride (95.7 gm) slowly. -The pH of the reaction mass was adjusted to 7.0 to 8.0 with aqueous sodium bicarbonate solution (120 ml). The separated aqueous layer was extracted with methylene chloride and then concentrated to obtain a residual solid of 7-chloro-l-[2- methyl-4-[(2-methylbenzoyl)amino]benzoyl]-5-oxo-2,3,4,5-tetrahydro- 1 H- 1 – benzazepine (185 gm). Example 5:
Preparation of tolvaptan
7-Chloro- 1 -[2-methyl-4-[(2-methylbenzoyl)amino]benzoyl]-5-oxo-2,3,4,5- tetrahydro-lH-1 -benzazepine (63 gm) as obtained in example 4 was dissolved in methanol (570 ml) and then added sodium borohydride (2.07 gm) at room temperature. The reaction mass was stirred for 1 hour and pH of the reaction mass was adjusted to 6.0 to 7.0 with hydrochloric acid solution (1%, 630 ml). The separated solid was filtered and dried to obtain 57 gm of tolvaptan.
……………….

Process used to prepare Tolvaptan involves condensing 7-chloro-1, 2, 3, 4-tetrahydro-benzo[b]azepin-5-one with 2-methyl, 4-nitro benzoyl chloride, followed by reduction using SnCl2/HCl catalyst resulting in amine which is then condensed with o-toluoyl chloride followed by reduction with sodium borohydride to give Tolvaptan
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
SYN 1
Synthetic Reference
Cordero-Vargas, Alejandro; Quiclet-Sire, Beatrice; Zard, Samir Z. A flexible approach for the preparation of substituted benzazepines: Application to the synthesis of tolvaptan. Bioorganic & Medicinal Chemistry. Volume 14. Issue 18. Pages 6165-6173. 2006.

SYN 2
Synthetic Reference
Torisawa, Yasuhiro; Abe, Kaoru; Muguruma, Yasuaki; Fujita, Shigekazu; Ogawa, Hidenori; Utsumi, Naoto; Miyake, Masahiro. Process for preparation of benzoylaminobenzoylbenzazepinones by reaction of benzazepinones with benzoylaminophenyl halides in the presence of carbonylating agents. Assignee Otsuka Pharmaceutical Co.,

SYN 3
Synthetic Reference
Zard, Samir; Cordero Vargas, Alejandro; Sire, Beatrice. Improved process for the preparation of benzazepines and their derivatives. Assignee Centre National de la Recherche Scientifique CNRS, Fr.; Ecole Polytechnique. FR 2867187. (2005).

SYN 4
Synthetic Reference
Gao, Junlong; Li, Peng; Liu, Kai; Guo, Dapeng. Method for preparing high-purity Tolvaptan intermediate. Assignee Jiangsu Hengrui Medicine Co., Ltd., Peop. Rep. China. CN 108503586. (2018).

SYN 5
Synthetic Reference
Han, Shin; Jeon, Seong Hyeon; Lee, Shin Yoon. Improved method for preparing synthetic intermediates for tolvaptan. Assignee Hexa Pharmatec Co., Ltd., S. Korea. JP 2018012690. (2018).

SYN 6
Synthetic Reference
Guo, Xinfu; Wang, Qiang; Liu, Zhaoguo; Wang, Zhipeng. Preparation method of tolvaptan. Assignee Tianjin Taipu Pharmaceutical Co., Ltd., Peop. Rep. China. CN 106883175. (2017).

SYN 7
Synthetic Reference
Lixin, Juanzi; Li, Jianzhi; Ma, Xilai; Chi, Wangzhou; Liu, Hai; Hu, Xuhua; Zheng, Xiaoli; Zhai, Zhijun; Li, Jianxun. Process for the preparation of tolvaptan. Assignee Shanghai Tianci International Pharmaceutical Co., Ltd., Peop. Rep. China. CN 105753735. (2016).

STR8
Synthetic Reference
Patel, Dhaval J.; Shah, Tejas C.; Singh, Manoj Kumar. A process for the preparation of tolvaptan. Assignee Cadila Healthcare Limited, India. IN 2012MU01559. (2014).

STR9
Synthetic Reference
Sethi, Madhuresh Kumar; Rawat, Vijendrasingh; Thirunavukarasu, Jayaprakash; Yerramala, Raja Krishna; Kumar, Anish. Improved process for the preparation of tolvaptan. Assignee Matrix Laboratories Ltd., India. IN 2011CH01303. (2013).

/////////////////////

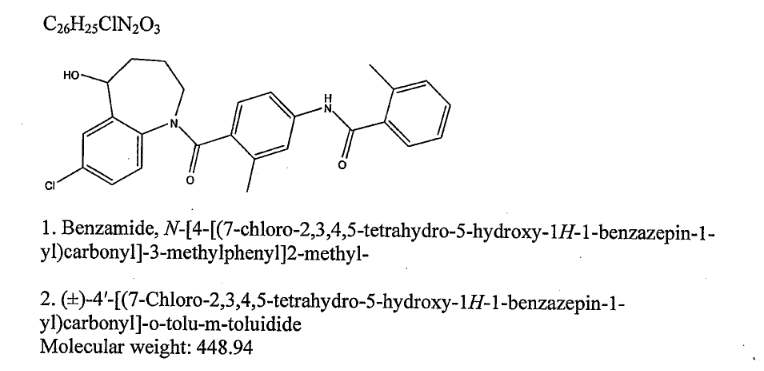

Title: Tolvaptan
CAS Registry Number: 150683-30-0
CAS Name: N-[4-[(7-Chloro-2,3,4,5-tetrahydro-5-hydroxy-1H-1-benzazepin-1-yl)carbonyl]-3-methylphenyl]-2-methylbenzamide
Additional Names: 7-chloro-5-hydroxy-1-[2-methyl-4-(2-methylbenzoylamino)benzoyl]-2,3,4,5-tetrahydro-1H-1-benzazepine
Manufacturers’ Codes: OPC-41061
Molecular Formula: C26H25ClN2O3
Molecular Weight: 448.94
Percent Composition: C 69.56%, H 5.61%, Cl 7.90%, N 6.24%, O 10.69%
Literature References: Nonpeptide arginine vasopressin V2 receptor antagonist. Prepn: H. Ogawa et al., WO 9105549; eidem, US 5258510 (1991, 1993 both to Otsuka); K. Kondo et al., Bioorg. Med. Chem. 7, 1743 (1999). Pharmacology: Y. Yamamura et al., J. Pharmacol. Exp. Ther. 287, 860 (1998). Clinical trial in heart failure: M. Gheorghiade et al., J. Am. Med. Assoc. 291, 1963 (2004).
Properties: Colorless prisms, mp 225.9°.
Melting point: mp 225.9°
Therap-Cat: In treatment of congestive heart failure.
Keywords: Vasopressin Receptor Antagonist.
- Shoaf S, Elizari M, Wang Z, et al. (2005). “Tolvaptan administration does not affect steady state amiodarone concentrations in patients with cardiac arrhythmias”. J Cardiovasc Pharmacol Ther 10 (3): 165–71. doi:10.1177/107424840501000304. PMID 16211205.
- Otsuka Maryland Research Institute, Inc.
- Gheorghiade M, Gattis W, O’Connor C, et al. (2004). “Effects of tolvaptan, a vasopressin antagonist, in patients hospitalized with worsening heart failure: a randomized controlled trial”. JAMA 291 (16): 1963–71. doi:10.1001/jama.291.16.1963. PMID 15113814.
- (2012) Tolvaptan in Patients with Autosomal Dominant Polycystic Kidney Disease
- Kondo, K.; Ogawa, H.; Yamashita, H.; Miyamoto, H.; Tanaka, M.; Nakaya, K.; Kitano, K.; Yamamura, Y.; Nakamura, S.; Onogawa, T.; et al.; Bioor. Med. Chem. 1999, 7, 1743.
- http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm350185.htm?source=govdelivery
- Gheorghiade M, Niazi I, Ouyang J et al. (2003). “Vasopressin V2-receptor blockade with tolvaptan in patients with chronic heart failure: results from a double-blind, randomized trial”. Circulation 107 (21): 2690–6. doi:10.1161/01.CIR.0000070422.41439.04.PMID 12742979.
G. R. Belum, V. R. Belum, S. K. Chaitanya Arudra, and B. S. N. Reddy, “The Jarisch-Herxheimer reaction: revisited,” Travel Medicine and Infectious Disease, vol. 11, no. 4, pp. 231–237, 2013.
H. D. Zmily, S. Daifallah, and J. K. Ghali, “Tolvaptan, hyponatremia, and heart failure,” International Journal of Nephrology and Renovascular Disease, vol. 4, pp. 57–71, 2011.
M. N. Ferguson, “Novel agents for the treatment of hyponatremia: a review of conivaptan and tolvaptan,” Cardiology in Review, vol. 18, no. 6, pp. 313–321, 2010.
H. Ogawa, H. Miyamoto, K. Kondo, et al., US5258510, 1993.
K. Kondo, H. Ogawa, H. Yamashita et al., “7-Chloro-5-hydroxy-1-[2-methyl-4-(2-methylbenzoylamino)benzoyl]-2,3,4,5- tetrahydro-1H-1-benzazepine (OPC-41061): a potent, orally active nonpeptide arginine vasopressin V2 receptor antagonist,” Bioorganic and Medicinal Chemistry, vol. 7, no. 8, pp. 1743–1754, 1999.
| WO2012046244A1 * | Aug 23, 2011 | Apr 12, 2012 | Hetero Research Foundation | Process for preparing tolvaptan intermediates |
| CN102060769A * | Dec 20, 2010 | May 18, 2011 | 天津药物研究院 | Preparation method of tolvaptan |
| CN102060769B | Dec 20, 2010 | Sep 18, 2013 | 天津药物研究院 | Preparation method of tolvaptan |
| US9024015 | Aug 23, 2011 | May 5, 2015 | Hetero Research Foundation | Process for preparing tolvaptan intermediates |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN101817783A | May 12, 2010 | Sep 1, 2010 | 天津泰普药品科技发展有限公司 | Method for preparing tolvaptan intermediate |
| WO2007026971A2 | Sep 1, 2006 | Mar 8, 2007 | Otsuka Pharma Co Ltd | Process for preparing benzazepine compounds or salts thereof |
| Reference | ||
|---|---|---|
| 1 | Cordero-Vargas, Alejandro | |
| 2 | Kondo, Kazumi et al.7-chloro-5-hydroxy-1-[2-methyl-4-(2-methylbenzoyl-amino)benzoyl]-2,3,4,5-tetrahydro-1H-1-benzazepine (OPC-41061): A potent, orally active nonpeptide arginine vasopressin V2 receptor antagonist.《Bioorganic & Medicinal Chemistry》.1999,1743-1757. | |
| 3 | Quiclet-Sire, Beatrice | |
| 4 | Torisawa, Yasuhiro et al.Aminocarbonylation route to tolvaptan.《Bioorganic & Medicinal Chemistry Letters》.2007,6455-6458. | |
| 5 | Zard, Samir Z.A flexible approach for the preparation of substituted benzazepines: Application to the synthesis of tolvaptan.《Bioorganic & Medicinal Chemistry》.2006,6165-6173. | |
///////////////
CC1=CC=CC=C1C(=O)NC2=CC(=C(C=C2)C(=O)N3CCCC(C4=C3C=CC(=C4)Cl)OP(=O)([O-])[O-])C.[Na+].[Na+]
////////////UPDATE 2022
-Tolvaptan_Structural_Formula_V1.svg) |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Samsca, Jinarc, Jynarque, others |
| Other names | OPC-41061 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a609033 |
| License data | |
| Pregnancy category |
|
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | Unknown (40% absorbed) |
| Protein binding | 99% |
| Metabolism | Liver (CYP3A4-mediated)[7] |
| Elimination half-life | 12 hours (terminal) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.219.212 |
| Chemical and physical data | |
| Formula | C26H25ClN2O3 |
| Molar mass | 448.95 g·mol−1 |
| 3D model (JSmol) | |
| |
|
Tolvaptan, sold under the brand name Samsca among others, is an aquaretic drug that functions as a selective, competitive vasopressin receptor 2 (V2) antagonist used to treat hyponatremia (low blood sodium levels) associated with congestive heart failure, cirrhosis, and the syndrome of inappropriate antidiuretic hormone (SIADH). Tolvaptan was approved by the U.S. Food and Drug Administration (FDA) on May 19, 2009, and is sold by Otsuka Pharmaceutical Co. under the trade name Samsca.[8] Tolvaptan, as Jynarque, was granted approval for medical use in the United States in April 2018.[9]
The U.S. Food and Drug Administration (FDA) granted tolvaptan a fast track designation for clinical trials investigating its use for the treatment of polycystic kidney disease.[10] The FDA granted Jynarque an orphan drug designation in April 2012, for the treatment of autosomal dominant polycystic kidney disease.[11]
Tolvaptan is available as a generic medication.[12]
Medical uses
Tolvaptan (Samsca) is indicated for the treatment of clinically significant hypervolemic and euvolemic hyponatremia.[13]
Tolvaptan (Jynarque) is indicated to slow kidney function decline in adults at risk of rapidly progressing autosomal dominant polycystic kidney disease (ADPKD).[14]
Side effects
The FDA has determined that tolvaptan should not be used for longer than 30 days and should not be used in patients with underlying liver disease because it can cause liver injury, potentially leading to liver failure.[15] When using to treat hyponatremia, it may cause too rapid correction of hyponatremia resulting in fatal osmotic demyelination syndrome.[16]
Pharmacology
Tolvaptan is a selective vasopressin V2 receptor antagonist.[13][14]
Chemistry
Tolvaptan is a racemate, a 1:1 mixture of the following two enantiomers:[17]
| Enantiomers of tolvaptan | |
|---|---|
-Tolvaptan_Structural_Formula_V1.svg) (R)-Tolvaptan CAS number: 331947-66-1 |
-Tolvaptan_Structural_Formula_V1.svg) (S)-Tolvaptan CAS number: 331947-44-5 |
References
- ^ “Samsca 15 mg tablets – Summary of Product Characteristics (SmPC)”. (emc). Retrieved 14 December 2020.
- ^ “Jinarc 15 mg tablets – Summary of Product Characteristics (SmPC)”. (emc). 21 April 2020. Retrieved 14 December 2020.
- ^ “Jynarque- tolvaptan kit Jynarque- tolvaptan tablet”. DailyMed. 31 March 2020. Retrieved 14 December 2020.
- ^ “Samsca- tolvaptan tablet”. DailyMed. 26 October 2020. Retrieved 14 December 2020.
- ^ “Samsca EPAR”. European Medicines Agency (EMA). Retrieved 14 December 2020.
- ^ “Jinarc EPAR”. European Medicines Agency (EMA). Retrieved 14 December 2020.
- ^ Shoaf S, Elizari M, Wang Z, et al. (2005). “Tolvaptan administration does not affect steady state amiodarone concentrations in patients with cardiac arrhythmias”. J Cardiovasc Pharmacol Ther. 10 (3): 165–71. doi:10.1177/107424840501000304. PMID 16211205. S2CID 39158242.
- ^ “Drug Approval Package: Samsca (Tolvaptan) Tablets NDA #022275”. U.S. Food and Drug Administration (FDA). 21 July 2009. Retrieved 15 August 2020. Lay summary (PDF).
{{cite web}}: Cite uses deprecated parameter|lay-url=(help) - ^ “Drug Approval Package: Jynarque (tolvaptan)”. U.S. Food and Drug Administration (FDA). 8 June 2018. Retrieved 15 August 2020.
- ^ “Otsuka Maryland Research Institute, Inc. Granted Fast Track Designation For Tolvaptan In PKD”. Medical News Today. Healthline Media UK Ltd. Retrieved 6 December 2018.
- ^ “Tolvaptan Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 6 April 2012. Retrieved 15 August 2020.
- ^ “Drugs@FDA: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 15 August 2020.
- ^ Jump up to:a b “Samsca- tolvaptan tablet”. DailyMed. 28 May 2019. Retrieved 15 August 2020.
- ^ Jump up to:a b “Jynarque- tolvaptan kit Jynarque- tolvaptan tablet”. DailyMed. 31 March 2020. Retrieved 15 August 2020.
- ^ “U.S. Food and Drug Administration.” Samsca (Tolvaptan): Drug Safety Communication. N.p., 30 Apr. 2013. Web. 1 June 2014. <http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm350185.htm>[dead link]
- ^ Goodman & Gilman’s the pharmacological basis of therapeutics. Brunton, Laurence L, Knollmann, Björn C, Hilal-Dandan, Randa (Thirteenth ed.). New York. 5 December 2017. ISBN 9781259584732. OCLC 994570810.
- ^ Rote Liste Service GmbH (Hrsg.): Rote Liste 2017 – Arzneimittelverzeichnis für Deutschland (einschließlich EU-Zulassungen und bestimmter Medizinprodukte). Rote Liste Service GmbH, Frankfurt/Main, 2017, Aufl. 57, ISBN 978-3-946057-10-9, S. 222.
Further reading
- Gheorghiade M, Niazi I, Ouyang J, et al. (2003). “Vasopressin V2-receptor blockade with tolvaptan in patients with chronic heart failure: results from a double-blind, randomized trial”. Circulation. 107 (21): 2690–6. doi:10.1161/01.CIR.0000070422.41439.04. PMID 12742979.
External links
- “Tolvaptan”. Drug Information Portal. U.S. National Library of Medicine.
Synthesis
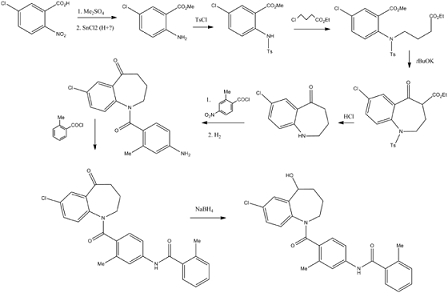
Fugure 1: Synthesis of Tolvaptan

NEW DRUG APPROVALS
HELP TO MAINTAIN THIS BLOG SUBSCRIPTION
$10.00
Title: Tolvaptan
CAS Registry Number: 150683-30-0
CAS Name: N-[4-[(7-Chloro-2,3,4,5-tetrahydro-5-hydroxy-1H-1-benzazepin-1-yl)carbonyl]-3-methylphenyl]-2-methylbenzamide
Additional Names: 7-chloro-5-hydroxy-1-[2-methyl-4-(2-methylbenzoylamino)benzoyl]-2,3,4,5-tetrahydro-1H-1-benzazepine
Manufacturers’ Codes: OPC-41061
Molecular Formula: C26H25ClN2O3
Molecular Weight: 448.94
Percent Composition: C 69.56%, H 5.61%, Cl 7.90%, N 6.24%, O 10.69%
Literature References: Nonpeptide arginine vasopressin V2 receptor antagonist. Prepn: H. Ogawa et al., WO 9105549; eidem, US 5258510 (1991, 1993 both to Otsuka); K. Kondo et al., Bioorg. Med. Chem. 7, 1743 (1999). Pharmacology: Y. Yamamura et al., J. Pharmacol. Exp. Ther. 287, 860 (1998). Clinical trial in heart failure: M. Gheorghiade et al., J. Am. Med. Assoc. 291, 1963 (2004).
Properties: Colorless prisms, mp 225.9°.
Melting point: mp 225.9°
Therap-Cat: In treatment of congestive heart failure.
Keywords: Vasopressin Receptor Antagonist.

{kind=link}
{kind=link}
{kind=link}
.svg){kind=link}
.svg){kind=link}
{kind=link}