
Home » Posts tagged 'JAK inhibitor'
Tag Archives: JAK inhibitor
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
TOFACITINIB 的合成, トファシチニブ, Тофацитиниб, توفاسيتين يب SPECTRAL VISIT
Tofacitinib Citrate, 的合成
托法替布, トファシチニブクエン酸塩, Тофацитиниба Цитрат
3-{(3R,4R)-4-methyl-3-[methyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3-oxo-propionitrile citrate salt
CAS : 540737-29-9
ROTATION +
Tofacitinib; Tasocitinib;
477600-75-2 base ; CP-690550;
3-((3R,4R)-4-methyl-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)-3-oxopropanenitrile;
3-{(3R,4R)-4-methyl-3-rmethyl-(7H-pyrrolor2,3-dlpyrimidin-4-yl)-amino1- piperidin-1-yl}-3-oxo-propionitrile mono citrate salt
CP 690550 Tofacitinib; CP-690550; CP-690550-10; Xeljanz; Jakvinus; Tofacitinib citrate
Trademarks: Xeljanz; Jakvinus
MF: C16H20N6O
CAS : 477600-75-2 BASE ; 540737-29-9(citrate) 3-[(3R,4R)-4-methyl-3-[methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]piperidin-1-yl]-3-oxopropanenitrile
Molecular Weight: 312.369
SMILES: C[C@@H]1CCN(C[C@@H]1N(C)C2=NC=NC3=C2C=CN3)C(=O)CC#N
Activity: Treatment of Rheumatoid Arthritis; RA Treatment, JAK Inhibitor; Protein Kinase Inhibitor; JAK3 Inhibitor; Janus Kinase 3 Inhibitor; JAK-STAT Signaling Pathway; JAK1 Kinase Inhibitor; Selective Immunosuppressants
Status: Launched 2012
Originator: Pfizer 
Pfizer Inc’s oral JAK inhibitor tofacitinib was approved on November 6, 2012 by US FDA for the treatment of rheumatoid arthritis.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
Tofacitinib (trade names Xeljanz and Jakvinus, formerly tasocitinib,[1] CP-690550[2]) is a drug of the janus kinase (JAK) inhibitor class, discovered and developed by Pfizer. It is currently approved for the treatment of rheumatoid arthritis (RA) in the United States,Russia, Japan and many other countries, is being studied for treatment of psoriasis, inflammatory bowel disease, and other immunological diseases, as well as for the prevention of organ transplant rejection. 

An Improved and Efficient Process for the Preparation of Tofacitinib Citrate
Yogesh S. Patil, Nilesh L. Bonde, Ankush S. Kekan, Dhananjay G. Sathe*1, and Arijit Das
Publication Date (Web): November 17, 2014 (Article)
DOI: 10.1021/op500274j
MS m/z 313 (M+ + 1);
mp 201–202 °C;
|
1H NMR (CDCl3) δ 8.34 (s, 1H), δ 7.38 (d, 1H, J = 2.4 Hz), δ 6.93 (d, 1H, J = 2.4 Hz), δ 4.97 (m, 1H), δ 3.93–4.03 (m, 4H), δ 3.66 (m, 1H), δ 3.50 (m, 4H), δ 2.91 (d, 2H, J = 15.6 Hz), δ 2.80 (t, 2H, J = 12.8 Hz), δ 2.55 (m, 1H), δ 1.99 (m, 1H), δ 1.77 (m, 1H), δ 1.13–1.18 (m, 3H).
|



TEAMWORK
Part of the Pfizer group responsible for Xeljanz: Front row, from left: Sally Gut Ruggeri, Chakrapani Subramanyam, Eileen Elliott Mueller, and Frank Busch. Second row, from left: Matthew Brown, Mark Flanagan, and Robert Dugger. Back row, from left: Elizabeth Kudlacz and Douglas Ball.
Credit: Pfizer
Mark Flanagan, who was on the team at Pfizer that discovered Xeljanz, (tofacitinib citrate), an oral treatment for rheumatoid arthritis, remembers testing the drug in a rat model and seeing the drug decrease the level of inflammation in the rats’ footpads. “What we look for is physical measurements of the size of the joint. In the control animals, there was quite a bit of inflammation in the joints, whereas animals treated with different doses of the drug showed a dose-dependent decrease in the size of the joint. “Tofacitinib showed robust efficacy in the first such study run. I can remember the excitement that this data generated on the team,” he says.
Tofacitinib, chemically known as (3R,4R)-4-methyl-3-(methyl-7H-pyrrolo [2,3- d]pyrimidin-4-ylamino)-B-oxo-l -piperidinepi panenitrile, is represented Formula I. Tofacitinib citrate, a janus kinase inhibitor, is approved as XELJANZ® tablets for treatment .of rheumatoid arthritis.

Various intermediates and processes for preparation of tofacitinib are disclosed in patents like US7301 023 and US8232394.

Formula I or isomers or a mixture of isomers thereof by following any method provided in the prior art, for example, by following Example 14 of U.S. Patent No. RE41,783 or by following Example 6 of U.S. Patent No. 7,301,023. Tofacitinib of Formula I or isomers of tofacitinib or a mixture of isomers thereof may be converted into a salt by following any method provided in the prior art, for example, by following Example 1 of U.S. Patent No. 6,965,027 or by following Example 1 or Example 8 of PCT Publication No. WO 2012/135338. The potential significance of JAK3 inhibition was first discovered in the laboratory of John O’Shea, an immunologist at the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health (NIH).[5] In 1994, Pfizer was approached by the NIH to form a public-private partnership in order to evaluate and bring to market experimental compounds based on this research.[5] Pfizer initially declined the partnership but agreed in 1996, after the elimination of an NIH policy dictating that the market price of a product resulting from such a partnership would need to be commensurate with the investment of public taxpayer revenue and the “health and safety needs of the public.”[5] The drug discovery, preclinical development, and clinical development of tofacitinib took place exclusively at Pfizer.[6] In November 2012, the U.S. Food and Drug Administration (FDA) approved tofacitinib for treatment of rheumatoid arthritis. Once on the market, rheumatologists complained that the $2,055 a month wholesale price was too expensive, though the price is 7% less than related treatments.[6] A 2014 study showed that tofacitinib treatment was able to convert white fat tissues into more metabolically active brown fat, suggesting it may have potential applications in the treatment of obesity.[7] It is an inhibitor of the enzyme janus kinase 1 (JAK1) and janus kinase 3 (JAK 3) , which means that it interferes with the JAK-STAT signaling pathway, which transmits extracellular information into the cell nucleus, influencing DNA transcription.[3] Recently it has been shown in a murine model of established arthritis that tofacitinib rapidly improved disease by inhibiting the production of inflammatory mediators and suppressing STAT1-dependent genes in joint tissue. This efficacy in this disease model correlated with the inhibition of both JAK1 and 3 signaling pathways, suggesting that tofacitinib may exert therapeutic benefit via pathways that are not exclusive to inhibition of JAK3.[4]
Preparation of 3-{(3R,4R)-4-methyl-3-[methyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3-oxo-propionitrile citrate salt (Tofacitinib citrate, Xeljanz, CP-690550-10)
To a round-bottomed flask fitted with a temperature probe, condenser, nitrogen source, and heating mantle, methyl-[(3R,4R)-4-methyl-piperidin-3-yl]-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amine (5.0 g, 20.4 mmol) was added followed by 1-butanol (15 mL), ethyl cyanoacetate (4.6 g, 40.8 mmol), and DBU (1.6 g, 10.2 mmol). The resulting amber solution was stirred at 40 °C for 20 h. Upon reaction completion, citric acid monohydrate (8.57 g, 40.8 mmol) was added followed by water (7.5 mL) and 1-butanol (39.5 mL). The mixture was heated to 81 °C and held at that temperature for 30 min. The mixture was then cooled slowly to 22 ºC and stirred for 2 h. The slurry was filtered and washed with 1-butanol (20 mL). The filter cake was dried in a vacuum oven at 80 °C to afford 9.6 g (93%) of tofacitinib citrate as an off-white solid.
1H NMR (500 MHz, d6-DMSO): δ 8.14 (s, 1H), 7.11 (d, J=3.6 Hz, 1H), 6.57 (d, J=3.6 Hz, 1H), 4.96 (q, J=6.0 Hz, 1H), 4.00-3.90 (m, 2H), 3.80 (m, 2H), 3.51 (m, 1H), 3.32 (s, 3H), 2.80 (Abq, J=15.6 Hz, 2H), 2.71 (Abq, J=15.6 Hz, 2H), 2.52-2.50 (m, 1H), 2.45-2.41 (m, 1H), 1.81 (m, 1H), 1.69-1.65 (m, 1H), 1.04 (d, J=6.9 Hz, 3H).
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
PAPER
3-((3R,4R)-4-Methyl-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)-3-oxopropanenitrile (1) Monocitrate
J. Med. Chem., 2010, 53 (24), pp 8468–8484
DOI: 10.1021/jm1004286
1monocitrate as a white crystalline solid (mp = 201 dec).
LRMS: m/z 313.2 (MH+).
1H NMR (400 MHz) (D2O) δ HOD: 0.92 (2 H, d, J = 7.2 Hz), 0.96 (1 H, d, J = 7.6 Hz), 1.66 (1 H, m), 1.80 (1 H, m), 2.37 (1 H, m), 2.58 (2 H, 1/2 ABq, J = 15.4 Hz), 2.70 (2 H, 1/2 ABq, J = 15.4 Hz), 3.23 (2 H, s), 3.25 (1 H, s), 3.33 (1 H, m), 3.46 (1 H, m), 3.81 (4 H, m), 4.55 (1 H, m), 6.65 (1 H, d, J = 3.2 Hz), 7.20 (1 H, t, J = 3.2 Hz), 8.09 (1 H, m).
Anal. Calcd for C22H28N6O8: C, 52.38; H, 5.59; N, 16.66. Found: C, 52.32; H, 5.83; N, 16.30. For additional characterization of the monocitrate salt of 1 see WO 03/048162.
NMR PREDICT


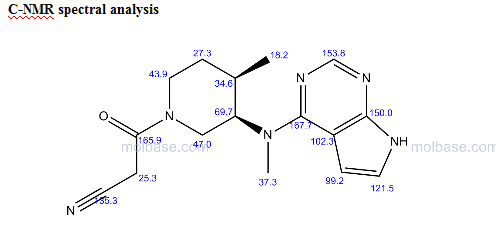

References:
Weiling Cai, James L. Colony,Heather Frost, James P. Hudspeth, Peter M. Kendall, Ashwin M. Krishnan,Teresa Makowski, Duane J. Mazur, James Phillips, David H. Brown Ripin, Sally Gut Ruggeri, Jay F. Stearns, and Timothy D. White; Investigation of Practical Routes for the Kilogram-Scale Production of cis-3-Methylamino-4-methylpiperidines; Organic Process Research & Development 2005, 9, 51−56
Ripin, D. H.B.; 3-amino-piperidine derivatives and methods of manufacture, US patent application publication, US 2004/0102627 A1
Ruggeri, Sally, Gut;Hawkins, Joel, Michael; Makowski, Teresa, Margaret; Rutherford, Jennifer, Lea; Urban,Frank,John;Pyrrolo[2,3-d]pyrimidine derivatives: their intermediates and synthesis, PCT pub. No. WO 2007/012953 A 2, US20120259115 A1, United States Patent US8232393. Patent Issue Date: July 31, 2012
Kristin E. Price, Claude Larrive´e-Aboussafy, Brett M. Lillie, Robert W. McLaughlin, Jason Mustakis, Kevin W. Hettenbach, Joel M. Hawkins, and Rajappa Vaidyanathan; Mild and Efficient DBU-Catalyzed Amidation of Cyanoacetates, Organic Letters, 2009, vol.11, No.9, 2003-2006
MORE NMR PREDICT

Tofacitinib 
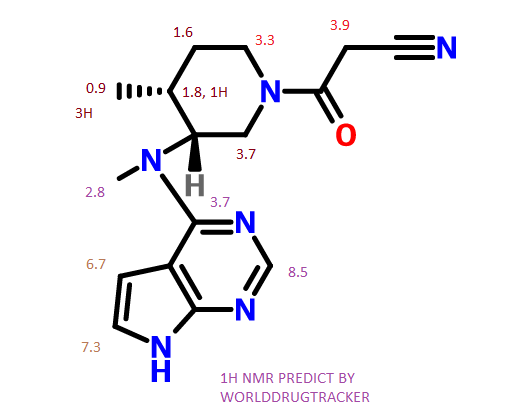
13C NMR PREDICT 

SEE…….https://newdrugapprovals.org/2015/07/24/tofacitinib-%E7%9A%84%E5%90%88%E6%88%90-spectral-visit/
COSY PREDICT 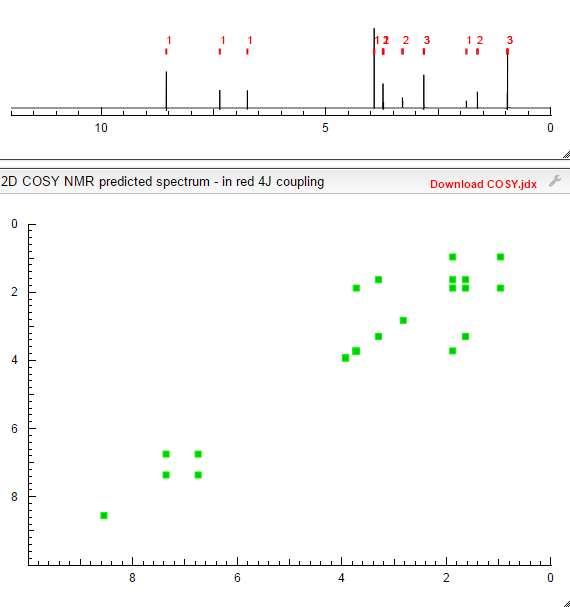 सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
SEE………http://orgspectroscopyint.blogspot.in/2014/12/tofacitinib-citrate.html
NMR PICTURE FROM THE NET

PAPER
Volume 54, Issue 37, 11 September 2013, Pages 5096–5098

Asymmetric total synthesis of Tofacitinib
- a Laboratory of Asymmetric Synthesis, Chemistry Institute of Natural Resources, University of Talca, P.O. Box 747, Talca, Chile
- b Laboratory of Natural Products, Department of Chemistry, University of Antofagasta, P.O. Box 170, Antofagasta, Chile
http://dx.doi.org/10.1016/j.tetlet.2013.07.042
Abstract
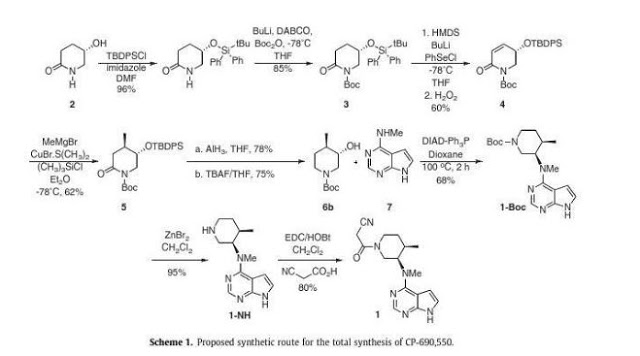
A novel stereoselective synthesis of Tofacitinib (CP-690,550), a Janus tyrosine kinase (JAK3) specific inhibitor, has been achieved starting from (5S)-5-hydroxypiperidin-2-one in 10 steps from 2 with a 9.5% overall yield. The potentiality of this synthetic route is the obtention of tert-butyl-(3S,4R)-3-hydroxy-4-methylpiperidine-1-carboxylate (6b) as a new chiral precursor involved in the synthesis of CP690,550, in a three-step reaction, without epimerizations, rather than the 5 or more steps used in described reactions to achieve this compound from analogues of 6b.
Graphical abstract

…………………. Tofacitinib synthesis: US2001053782A1

Tofacitinib synthesis: WO2002096909A1

Tofacitinib synthesis: Org Process Res Dev 2014, 18(12), 1714-1720 (also from a chinese publication, same procedure just slight changes in reagents/conditions)
References:
1. Blumenkopf, T. A.; et. al. Pyrrolo[2,3-d]pyrimidine compounds. US2001053782A1
2. Flanagan, M. E.; et. al. Optical resolution of (1-benzyl-4-methylpiperidin-3-yl) -methylamine and the use thereof for the preparation of pyrrolo 2,3-pyrimidine derivatives as protein kinases inhibitors. WO2002096909A1
3. Das, A.; et. al. An Improved and Efficient Process for the Preparation of Tofacitinib Citrate. Org Process Res Dev2014, 18(12), 1714-1720.
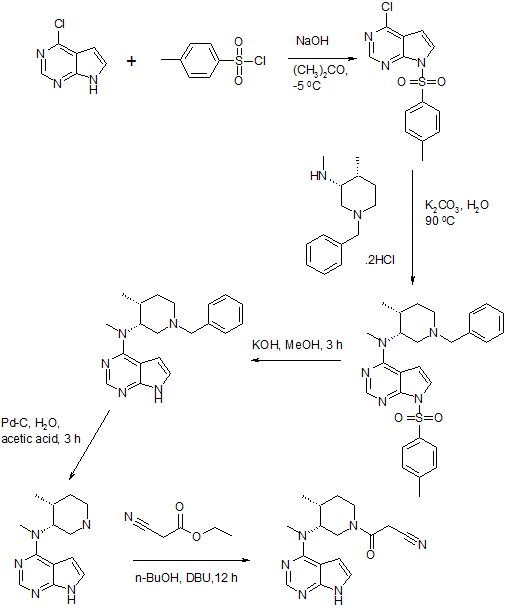
Synthesis of Tofacitinib Citrate

PATENT https://www.google.co.in/patents/WO2003048162A1?cl=en The crystalline form of the compound of this invention 3-{4-methyl-3-[methyl- (7H-pyrrolot2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3-oxo-propionitrile mono citrate salt is prepared as described below. Scheme 1


Scheme 2




Example 1 3-{(3R,4R)-4-methyl-3-rmethyl-(7H-pyrrolor2,3-dlpyrimidin-4-yl)-amino1- piperidin-1-yl}-3-oxo-propionitrile mono citrate salt Ethanol (13 liters), (3R, 4R)-methyl-(4-methyl-piperidin-3-yl)-(7H-pyrrolo[2,3- d]pyrimidin-4-yl)-amine (1.3 kg), cyano-acetic acid 2,5-dioxo-pyrrolidin-1-yl ester (1.5 kg), and triethylamine (1.5 liters) were combined and stirred at ambient temperature. Upon reaction completion (determined by High Pressure Liquid Chromotography (HPLC) analysis, approximately 30 minutes), the solution was filtered, concentrated and azeotroped with 15 liters of methylene chloride. The reaction mixture was washed sequentially with 12 liters of 0.5 N sodium hydroxide solution, 12 liters of brine and 12 liters of water. The organic layer was concentrated and azeotroped with 3 liters of acetone (final pot temperature was 42°C). The resulting solution was cooled to 20°C to 25°C followed by addition of 10 liters of acetone. This solution was filtered and then aqueous citric acid (0.8 kg in 4 liters of water) added via in-line filter. The reaction mixture was allowed to granulate. The slurry was cooled before collecting the solids by filtration. The solids were dried to yield 1.9 kg (71 %) (3R, 4R)- 3-{4-Methyl-3-[methyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3-oxo- propionitrile mono citrate. This material was then combined with 15 liters of a 1:1 ratio of ethanol/water and the slurry was agitated overnight. The solids were filtered and dried to afford 1.7 kg (63% from (3R, 4R)-methyl-(4-methyl-piperidin-3-yl)-(7H- pyrrolo[2,3-d]pyrimidin-4-yl)-amine) of the title compound as a white crystalline solid. 1H NMR (400 MH2)(D20) δ HOD: 0.92 (2H, d, J = 7.2 Hz), 0.96 (1H, d, J = 7.6 Hz), 1.66 (1H, m), 1.80 (1H, m), 2.37 (1H, m), 2.58 (2H, 1/2 ABq, J = 15.4 Hz), 2.70 (2H, 3 ABq, J = 154 Hz), 3.23 (2H, s), 3.25 (1H, s), 3.33 (1H, m), 3.46 (1H, m), 3.81 (4H, m), 4.55 (1 H, m), 6.65 (1 H, d, J = 3.2 Hz), 7.20 (1 H, t, J = 3.2 Hz), 8.09 (1 H, m).
Patent
http://www.google.co.in/patents/EP1913000A2?cl=en Example 10 Preparation of methyl-[(3R, 4R)-4-methyl-piperidin-3-yl]-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amine:
KEY INTERMEDIATE
To a clean, dry, nitrogen-purged 2 L hydrogenation reactor were charged 20 wt% Pd(OH)2/C (24.0 g, 50% water wet), water (160 ml), isopropanol (640 ml), (1-benzyl-4-methyl-piperidin-3-yI)-methyi- (7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amine (160.0 g, 0.48 mol), and acetic acid (28.65 g, 0.48 mol). The reactor was purged with three times at 50 psi with nitrogen and three times at 50 psi with hydrogen. Once purging was complete, the reactor was heated to 45-55°C and pressurized to 50 psi with hydrogen through a continuous feed. The hydrogen uptake was monitored until no hydrogen was consumed for 1 hour. The reactor was cooled to 20-300C and purged three times at 50 psi with nitrogen. The reaction mixture was filtered through wet Celite and the filtrate was sent to a clean, dry, nitrogen-purged vessel. A solution of sodium hydroxide (39.33 g) in water (290 ml) was charged and the mixture was stirred for a minimum of 1 hour then heated to 75-900C. The isopropanol was removed by distillation. The reaction mixture was cooled to 20-30°C and 2-methyltetrahydrofuran (1.6 L) was added. The aqueous layer was drained off and the 2-methyltetrahydrofuran was displaced with toluene (1.6 L). The distillation was continued until the final volume was 800 ml. The slurry was cooled to 20-30°C and held for a minimum of 7 hours. The resulting solids were isolated by filtration and washed with toluene (480 ml). After drying under vacuum between 40-50DC for a minimum of 24 hours with a slight nitrogen bleed 102.3 g (87.3%) of the title compound were isolated. Mp 158.6-159.8°C. 1H NMR (400 MHz, CDCI3): δ 11.38 (bs, 1H), 8.30 (s, 1H), 7.05 (d, J=3.5 Hz, 1H), 6.54 (d, J=3.5 Hz, 1H), 4.89-4.87 (m, 1H), 3.39 (s, 3H), 3.27 (dd, J=12.0, 9.3 Hz, 1 H), 3.04 (dd, J=12.0, 3.9 Hz, 1H), 2.94 (td, J=12.6, 3.1 Hz, 1H0, 2.84 (dt, J=12.6, 4.3 Hz, 1H), 2.51-2.48 (m, 1H), 2.12 (bs, 2H), 1.89 (ddt, J=13.7, 10.6, 4 Hz, 1 H), 1.62 (dq, J=13.7, 4Hz, 1 H), 1.07 (d, J=7.3 Hz, 3H). 13C NMR (400 MHz, CDCI3): δ 157.9, 152.0, 151.0, 120.0, 103.0, 102.5, 56.3, 46.2, 42.4, 34.7, 33.4, 32.4, 14.3.  KEY INT
KEY INT
Example 11 Preparation of 3-{(3R, 4R)-4-methyl-3-[methyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3- oxo-propionitrile….TOFACITINIB BASE
To a clean, dry, nitrogen-purged 1.0 L reactor were charged methyl-(4-methyl-piperidin-3-yI)-(7H- pyrroIo[2,3-d]pyrimidin-4-yl)-amine (32.0 g, 0.130 mol), toluene (160 ml), ethyl cyanoacetate (88.53 g, 0.783 mol) and triethyl amine (26.4 g, 0.261 mol). The reaction was heated to 1000C and held for 24 hours. The reaction was washed with water (160 ml). The organic layer concentrated to a volume of 10 ml and water (20 ml) was added. The residual toluene was removed by distillation and the mixture was cooled to room temperature. Acetone (224 ml) was added followed by citric acid (27.57 g, 0.144 mol) in water (76 ml). The resulting slurry was stirred for 7 hours. The solids were isolate by filtration, washed with acetone (96 ml), and dried under vacuum to afford 42.85 g (65.3%) of the title compound. Example 13 Preparation of 3-{(3R, 4R)~4-methyl-3-[methyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3-oxo- propionitrile citrate salt:…………..TOFACITINIB CITRATE To a clean, dry, nitrogen-purged 500 ml reactor were charged methyl-(4-methyl-piperidin-3-yl)-(7H- pyrrolo[2,3-d]pyrimidin-4-yl)-amine (25.0 g, 0.102 mol) and methylene chloride (250 ml). The mixture was stirred at room temperature for a minimum of 2.5 hours. To a clean, dry, nitrogen-purged 1 L reactor were charged cyanoacetic acid (18.2 g, 0.214 mol), methylene chloride (375 ml), and triethyl amine (30.1 ml, 0.214 mol). The mixture was cooled to -15.0— 5.00C over one hour and trimethylacetyl chloride (25.6 ml, 0.204 mol) was added at a rate to maintain the temperature below O0C. The reaction was held for a minimum of 2.5 hours, then the solution of the amine was added at a rate that maintained the temperature below O0C. After stirring for 1 hour, the mixture was warmed to room temperature and 1 M sodium hydroxide (125 ml) was added. The organic layer was washed with water (125 ml) The methylene chloride solution.was displaced with acetone until a volume of 500 ml and a temperature of 55-650C had been achieved. Water (75 ml) was charged to the mixture while maintaining the temperature at 55-65°C. A solution of citric acid (20.76 g, 0.107 mol) in water (25.0) was charged and the mixture was cooled to room temperature. The reactor was stirred for a minimum of 5 hours and then the resulting solids were isolated by filtration and washed with acetone (2×75 ml), which was sent to the filter. The salt was charged into a clean, dry, nitrogen-purged 1L reactor with 2B ethanol (190 ml) and water (190 ml). The slurry was heated to 75-850C for a minimum of 4 hours. The mixture was cooled to 20-300C and stirred for an additional 4 hours. The solids were isolated by filtration and washed with 2B ethanol (190 ml). After drying in a vacuum oven at 500C with a slight nitrogen bleed, 34.6 g (67.3%) of the title compound were isolated. 1H NMR (500 MHz, CZ6-DMSO): δ 8.14 (s, 1 H), 7.11 (d, J=3.6 Hz, 1 H), 6.57 (d, J=3.6 Hz, 1 H), 4.96 (q, J=6.0 Hz, 1 H), 4.00-3.90 (m, 2H), 3.80 (m, 2H), 3.51 (m, 1 H), 3.32 (s, 3H), 2.80 (Abq, J=15.6 Hz, 2H), 2.71 (Abq, J=15.6 Hz, 2H), 2.52-2.50 (m, 1 H), 2.45-2.41 (m, 1 H), 1.81 (m, 1 H), 1.69-1.65 (m, 1 H), 1.04 (d, J=6.9 Hz, 3H)
PAPER
Org. Lett., 2009, 11 (9), pp 2003–2006
DOI: 10.1021/ol900435t
http://pubs.acs.org/doi/full/10.1021/ol900435t 
PATENT
http://www.omicsonline.org/open-access/advances-in-the-inhibitors-of-janus-kinase-2161-0444.1000540.php?aid=29799  ……………..
……………..

 सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
Clinical trials
Rheumatoid arthritis
Phase II clinical trials tested the drug in rheumatoid arthritis patients that had not responded to DMARD therapy. In a tofacitinib monotherapy study, the ACR score improved by at least 20% (ACR-20) in 67% of patients versus 25% who received placebo; and a study that combined the drug with methotrexate achieved ACR-20 in 59% of patients versus 35% who received methotrexate alone. In a psoriasis study, the PASI score improved by at least 75% in between 25 and 67% of patients, depending on the dose, versus 2% in the placebo group.[8] The most important side effects in Phase II studies were increased blood cholesterol levels (12 to 25 mg/dl LDL and 8 to 10 mg/dl HDL at medium dosage levels) andneutropenia.[8] Phase III trials testing the drug in rheumatoid arthritis started in 2007 and are scheduled to run until January 2015.[9] In April 2011, four patients died after beginning clinical trials with tofacitinib. According to Pfizer, only one of the four deaths was related to tofacitinib.[10] By April 2011, three phase III trials for RA had reported positive results.[11] In November 2012, the U.S. FDA approved tofacitinib “to treat adults with moderately to severely active rheumatoid arthritis who have had an inadequate response to, or who are intolerant of, methotrexate.”[12]
Psoriasis
As of April 2011 a phase III trial for psoriasis is under way.[11]
Alopecia
In June 2014, scientists at Yale successfully treated a male patient afflicted with alopecia universalis. The patient was able to grow a full head of hair, eyebrows, eyelashes, facial, armpit, genitalia and other hair. No side effects were reported in the study.[13]
Ulcerative colitis
The OCTAVE study of Tofacitinib in Ulcerative Colitis started in 2012. It is currently enrolling patients, though the NIH trials page states that they expect the trial to close in June 2015.[14]
Vitiligo
In a June 2015 study, a 53-year-old woman with vitiligo showed noticeable improvement after taking tofacitinib for five months.[15]
Development of Safe, Robust, Environmentally Responsible Processes for New Chemical Entities
– Dr. V. Rajappa, Director & Head-Process R&D, Bristol-Myers Squibb, India
A PRESENTATION
 The presentation will load below
The presentation will load below
![]()
Scroll with mouse to view 76 pages


- Herper, Matthew (2 March 2011). “Why Pfizer’s Biggest Experimental Drug Got A Name Change”. Forbes. Retrieved 3 March 2011.
- Kremer, J. M.; Bloom, B. J.; Breedveld, F. C.; Coombs, J. H.; Fletcher, M. P.; Gruben, D.; Krishnaswami, S.; Burgos-Vargas, R. N.; Wilkinson, B.; Zerbini, C. A. F.; Zwillich, S. H. (2009). “The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis: Results of a double-blind, placebo-controlled phase IIa trial of three dosage levels of CP-690,550 versus placebo”. Arthritis & Rheumatism 60 (7): 1895–1905. doi:10.1002/art.24567. PMID 19565475.
- “Tasocitinib”. Drugs in R&D 10 (4): 271–284. 2010. doi:10.2165/11588080-000000000-00000. PMC 3585773. PMID 21171673.
- Ghoreschi, K.; Jesson, M. I.; Li, X.; Lee, J. L.; Ghosh, S.; Alsup, J. W.; Warner, J. D.; Tanaka, M.; Steward-Tharp, S. M.; Gadina, M.; Thomas, C. J.; Minnerly, J. C.; Storer, C. E.; Labranche, T. P.; Radi, Z. A.; Dowty, M. E.; Head, R. D.; Meyer, D. M.; Kishore, N.; O’Shea, J. J. (2011). “Modulation of Innate and Adaptive Immune Responses by Tofacitinib (CP-690,550)”. J Immunol. 186 (7): 4234–4243. doi:10.4049/jimmunol.1003668. PMC 3108067. PMID 21383241.
- ^ Jump up to:a b c “Seeking Profit for Taxpayers in Potential of New Drug”, Jonathan Weisman, New York Times, March 18, 2013
- Ken Garber (9 January 2013). “Pfizer’s first-in-class JAK inhibitor pricey for rheumatoid arthritis market”. Nature Biotechnology 31 (1): 3–4. doi:10.1038/nbt0113-3. PMID 23302910.
- Jump up^ Moisan A, et al. White-to-brown metabolic conversion of human adipocytes by JAK inhibition. Nature Cell Biology, 8 December 2014. DOI 10.1038/ncb3075
- “EULAR: JAK Inhibitor Effective in RA But Safety Worries Remain”. MedPage Today. June 2009. Retrieved 9 February 2011.
- Clinical trial number NCT00413699 for “Long-Term Effectiveness And Safety Of CP-690,550 For The Treatment Of Rheumatoid Arthritis” at ClinicalTrials.gov
- Matthew Herper. “Pfizer’s Key Drug Walks A Tightrope”. Forbes.
- “Two Phase III Studies Confirm Benefits of Pfizer’s Tofacitinib Against Active RA”. 28 Apr 2011.
- “FDA approves Xeljanz for rheumatoid arthritis”. 6 Nov 2012.
- “Hairless man grows full head of hair in yale arthritis drug trial”. 19 Jun 2014.
- https://clinicaltrials.gov/ct2/show/NCT01465763?term=A3921094&rank=1
- “This Drug Brought Pigment Back for Woman with Vitiligo”. TIME. June 27, 2015. Retrieved June 29, 2015.
- Nordqvist, Christian (27 April 2013). “Pfizer’s Arthritis Drug Xeljanz (tofacitinib) Receives A Negative Opinion In Europe”. Medical News Today. Retrieved 2 August 2013.
- “”XALEJANZ PRESCRIBING INFORMATION @ Labeling.Pfizer.com””.
SEE………http://orgspectroscopyint.blogspot.in/2014/12/tofacitinib-citrate.html
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
3-[(3R,4R)-4-methyl-3-[methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]piperidin-1-yl]-3-oxopropanenitrile
|
|
| Clinical data | |
| Trade names | Xeljanz, Jakvinus |
| AHFS/Drugs.com | entry |
| Licence data | US FDA:link |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration | Oral |
| Pharmacokinetic data | |
| Bioavailability | 74% |
| Protein binding | 40% |
| Metabolism | Hepatic (via CYP3A4 andCYP2C19) |
| Biological half-life | 3 hours |
| Excretion | Urine |
| Identifiers | |
| CAS Registry Number | 477600-75-2 |
| ATC code | L04AA29 |
| PubChem | CID: 9926791 |
| IUPHAR/BPS | 5677 |
| DrugBank | DB08183 |
| ChemSpider | 8102425 |
| UNII | 87LA6FU830 |
| ChEBI | CHEBI:71200 |
| ChEMBL | CHEMBL221959 |
| Synonyms | CP-690550 |
| Chemical data | |
| Formula | C16H20N6O |
| Molecular mass | 312.369 g/mol |
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.

सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
 LIONEL MY SON
LIONEL MY SONHe was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
//////
Decernotinib … JAK inhibitor for the treatment of autoimmune and inflammatory diseases, including rheumatoid arthritis.

Decernotinib
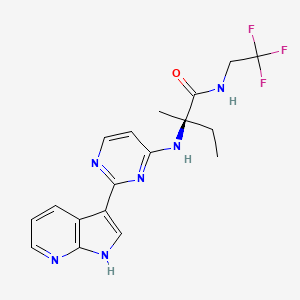
Decernotinib
N2-[2-(1H-Pyrrolo[2,3-b]pyridin-3-yl)pyrimidin-4-yl]-N-(2,2,2-trifluoroethyl)-D-isovalinamide
(R)-2-(2-(lH-pyrrolo[2,3-b]pyridin-3-yl)pyrimidin-4-ylamino)-2-methyl-N-(2,2,2- trifluoroethyl)butanamide
Vertex Pharmaceuticals Inc
UNII-MZK2GP0RHK, VX-509, VRT-831509, cas 944842-54-0
Molecular Formula: C18H19F3N6O
Molecular Weight: 392.37827
In phase 3 for the treatment of autoimmune and inflammatory diseases, including rheumatoid arthritis.
 DECERNOTINIB
DECERNOTINIB
The Janus kinases (JAK) are a family of tyrosine kinases consisting of JAK1, JAK2, JAK3, and TYK2. The JAKs play a critical role in cytokine signaling. The down-stream substrates of the JAK family of kinases include the signal transducer and activator of transcription (STAT) proteins. JAK/STAT signaling has been implicated in the mediation of many abnormal immune responses such as psoriasis. Moreover, JAK kinases represent an established therapeutic target for this disease.
For example, JAK kinases are an established therapeutic target for treating psoriasis. Stump K. L., et al., Arthritis Res. Ther. (201 1) 13:R68; Fridman J.S., et al., J Immunol. (2010) 184:5298-5307; West K., Curr. Op. Investig. Drugs (2009) 10:491-504; Kremer J. M. et al., Arthritis Rheumatism (2009) 60(7):1895- 1905; Xiong, W. et al., Ther Adv Musculoskelet Dis. (201 1) 3(5): 255-266; Panes, J. et al. 19th Ann. Eur. Gastroenterology Week (Oct 22-26, 2011) Stockholm, SE, PI 456; and Drugs in R & D “Tofacitinib” (2010) 10(4):271-84.
Compounds described as kinase inhibitors, particularly the JAK family kinases, are disclosed in WO 2005/095400 and WO 2007/084557. Also disclosed in these publications are processes and intermediates for preparing these compounds
Decernotinib ( VX-509 ) is an oral selective JAK3 inhibitor being evaluated for the treatment of rheumatoid arthritis ( RA ). This was a 24-week, randomized, placebo-controlled, double-blind, phase 2 study of four dosing regimens of Decernotinib, administered to patients with RA with inadequate response to Methotrexate ( MTX ).
The aim of the study was to assess the efficacy and safety of four dosing regimens of VX-509 administered to patients with rheumatoid arthritis on stable background Methotrexate therapy.
Patients with active rheumatoid arthritis ( C-reactive protein [ CRP ] greater than ULN, greater than or equal to 6 swollen joints [ of 66 ], and greater than or equal to 6 tender joints [ of 68 ] ) taking stable doses of MTX were randomized 1:1:1:1:1 to receive placebo or one of four dosing regimens of Decernotinib ( 100 mg QD, 150 mg QD, 200 mg QD, or 100 mg BID ) for a duration of 24 weeks.
The primary efficacy endpoints at week 12 were met and have previously been reported; 24-week efficacy and safety results are now reported.
A total of 358 patients were randomized and received greater than or equal to 1 dose of study drug; 81% of patients were female, with a mean age of 53 years.
At baseline, the mean tender joint count was 23.8, the mean swollen joint count was 16.1, and the average disease duration was 7.3 years.
After 24 weeks of treatment the proportion of patients achieving ACR20, ACR50, ACR70, DAS28 ( CRP ) less than 2.6 and DAS28 ( ESR ) less than 2.6 and the decrease from baseline in DAS28 ( CRP ) were statistically significantly greater in each of the Decernotinib dose groups than in the placebo group.
Over 24 weeks, the percentage of patients with any adverse event was higher in the Decernotinib group ( all Decernotinib dose groups combined ) ( 59.9% ) relative to placebo ( 42.3% ) and led to study discontinuation in 9.1% and 8.5% of patients in the Decernotinib and placebo groups, respectively.
The most common adverse reactions in the Decernotinib group were headache ( 8.7% ), hypercholesterolemia ( 5.2% ), and diarrhea ( 4.5% ).
Serious adverse reactions occurred in similar proportions of patients receiving Decernotinib ( 7.3% ) or placebo ( 5.6% ), but there were more serious infections in the Decernotinib group ( 3.5% ) compared with placebo ( 1.4% ).
Through 24 weeks there were two serious adverse effects that resulted in death; one was cardiac failure in the Decernotinib 100 mg BID group ( previously reported ) and one was pancytopenia in a patient with pneumonia in the Decernotinib 200 mg QD group.
Elevations in transaminase levels and decreases in median neutrophil and lymphocyte counts were observed in the Decernotinib groups and were generally mild.
Safety profiles were comparable across groups receiving Decernotinib.
In conclusion, all tested doses of Decernotinib significantly improved signs and symptoms of rheumatoid arthritis versus placebo when administered in combination with stable background Methotrexate therapy for 24 weeks.
Decernotinib was associated with small increases in adverse reactions rates, serious infections, and mostly minor laboratory abnormalities. ( Xagena )
Source: EULAR Meeting – van Vollenhoven R et al, Ann Rheum Dis 2014;73(Suppl2)
see
WO 2007084557
http://www.google.com/patents/WO2007084557A2?cl=en
………………………………………
WO 2013006634
http://www.google.com/patents/WO2013006634A2?cl=en

Formula I is:
The present invention provides a process for preparing (R)-2-(2-(lH-pyrrolo[2,3- b]pyridin-3-yl)pyrimidin-4-ylamino)-2-methyl-N-(2,2,2-trifluoroethyl)butanamide of Formula la:

la
comprising the steps of:
ivb) reacting lH-pyrrolo[2,3-b]pyridine (5a) with p-toluenesulfonyl chloride in the presence of an organic solvent to generate l-tosyl-lH-pyrrolo[2,3-b]pyridine (9a)

5a 9a
vb) reacting l-tosyl-lH-pyrrolo[2,3-b]pyridine (9a) in an organic solvent with N-bromosuccinimide to generate 3-bromo-l-tosyl-lH-pyrrolo[2,3-b]pyridine (7a)

vi) reacting 3-bromo-l-tosyl-lH-pyrrolo[2,3-b]pyridine (7a) with triisopropyl borate in the presence of a strong lithium base in an organic solvent to generate
l-tosyl-lH-pyrrolo[2,3-b]pyridin-3-ylboronic acid (8a) 0H

8a
vii) esterifying l-tosyl-lH-pyrrolo[2,3-b]pyridin-3-ylboronic acid (8a) with pinacolate alcohol in an organic solvent to generate
3 -(4,4,5 ,5 -tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 -tosyl- 1 H-pyrrolo[2,3 -bjpyridine (la) :

viiib) reacting 2,4-dichloropyrimidine (11a) with a hydrochloride salt of D-isovaline (15a) under coupling condition to generate a compound of Formula 2a

11a 2a
ixb) reacting the compound of Formula 2a with HC1 to generate the hydrochloride salt of the compound of Formula 2a;
i) reacting the compound of Formula la with the compound of Formula 2a with in the presence of water, an organic solvent, an inorganic base, and a transition metal catalyst to generate a compound of Formula 3a,

ii) deprotecting the compound of Formula 3a under basic conditions to generate a compound of Formula 4a

4a ; and iii) reacting the compound of Formula 4a with 2,2,2-trifluoroethylamine in the presence of a coupling agent and an organic solvent to generate the compound of Formula la.


– l13C4, 15N2]


……………………………………………………………….
WO 2013070606
http://www.google.com/patents/WO2013070606A1?cl=en
………………………………………………….
patent WO2014074471
WO2014074471 claiming use of heterocyclic compound (preferably decernotinib) for treating psoriasis. Vertex is developing decernotinib, an oral JAK 3 inhibitor, for the treatment of autoimmune and inflammatory diseases, including rheumatoid arthritis. As of July 2014, the drug is Phase 3 trials.
http://www.google.com/patents/WO2014074471A1?cl=en
Table 1:
COMPD 1 IS DECERNOTINIB


Example 1: Analytical Methods Used
[0260] (A) HPLC on C18 column. Mobile phase was acetonitrile/water/TFA (60:40:0.1). Flow rate was 1.0 mL/min. Detection at wavelength of 230 nm. Run time was 25-26 minutes.
[0261] (B) HPLC on C18 column. Mobile phase was acetonitrile/water/TFA (90: 10:0.1). Flow rate was 1.0 mL/min. Detection at wavelength of 230 nm.
[0262] (C) HPLC on a Waters XBridge Phenyl column, 4.6 x 150 mm, 3.5 μπι. Mobile phase A was water/1 M ammonium formate, pH 4.0 (99: 1). Mobile phase B was
acetonitrile/water/ 1M ammonium formate, pH 4.0 (90:9:1). Gradient 5 % to 90 % B in 15 minutes. Total run time 22 minutes. Flow rate 1.5 mL/min. Detection at UV, 245 nm.
T = 25 °C.
[0263] (D) HPLC on a Waters XBridge Phenyl column, 4.6 x 150 mm, 3.5 μπι. Mobile phase A was water/1 M ammonium formate, pH 4.0 (99: 1). Mobile phase B was
acetonitrile/water/ 1M ammonium formate, pH 4.0 (90:9: 1). Gradient 15% to 90 % B in 15 minutes. Total run time 22 minutes. Flow rate 1.5 mL/min. Detection at UV, 220 nm.
T = 35 °C.
[0264] Example 2: Preparation of Compounds of Formula I [0265] General Synthetic Scheme

[0266] The Boc-protected amino acid starting material (1) undergoes amidation in the presence of an activating agent, a coupling reagent, and the acid salt of the amine HNR7R17 to generate the Boc-protected amide intermediate (2). The amide intermediate (2) is
deprotected under acidic conditions and reacted with the halogenated heteroaryl (3) to generate the aminoheteroaryl intermediate (4). Boronated azaindole (5) is coupled with the aminoheteroaryl intermediate (4) under cross-coupling condition to generate the compound of Formula I.
………………………………………………………………………….
Patent
http://www.google.com/patents/US8163917
| 346 | M+H393.20 | RT 1.60 | (DMSO-d6, 300 MHz) 11.95 (bs, 1H), 8.7 (d, |
| 1H), 8.25 (m, 2H), 8.12 (d, 1H), 8.02 (d, 1H), | |||
| 7.28 (s, 1H), 7.13 (dd, 1H), 6.38 (bd, 1H), 3.75 | |||
| (m, 2H), 2.06 (m, 1H), 1.83 (m, 1H), 1.46 (s, | |||
| 3H), 0.8 (t, 3H); |
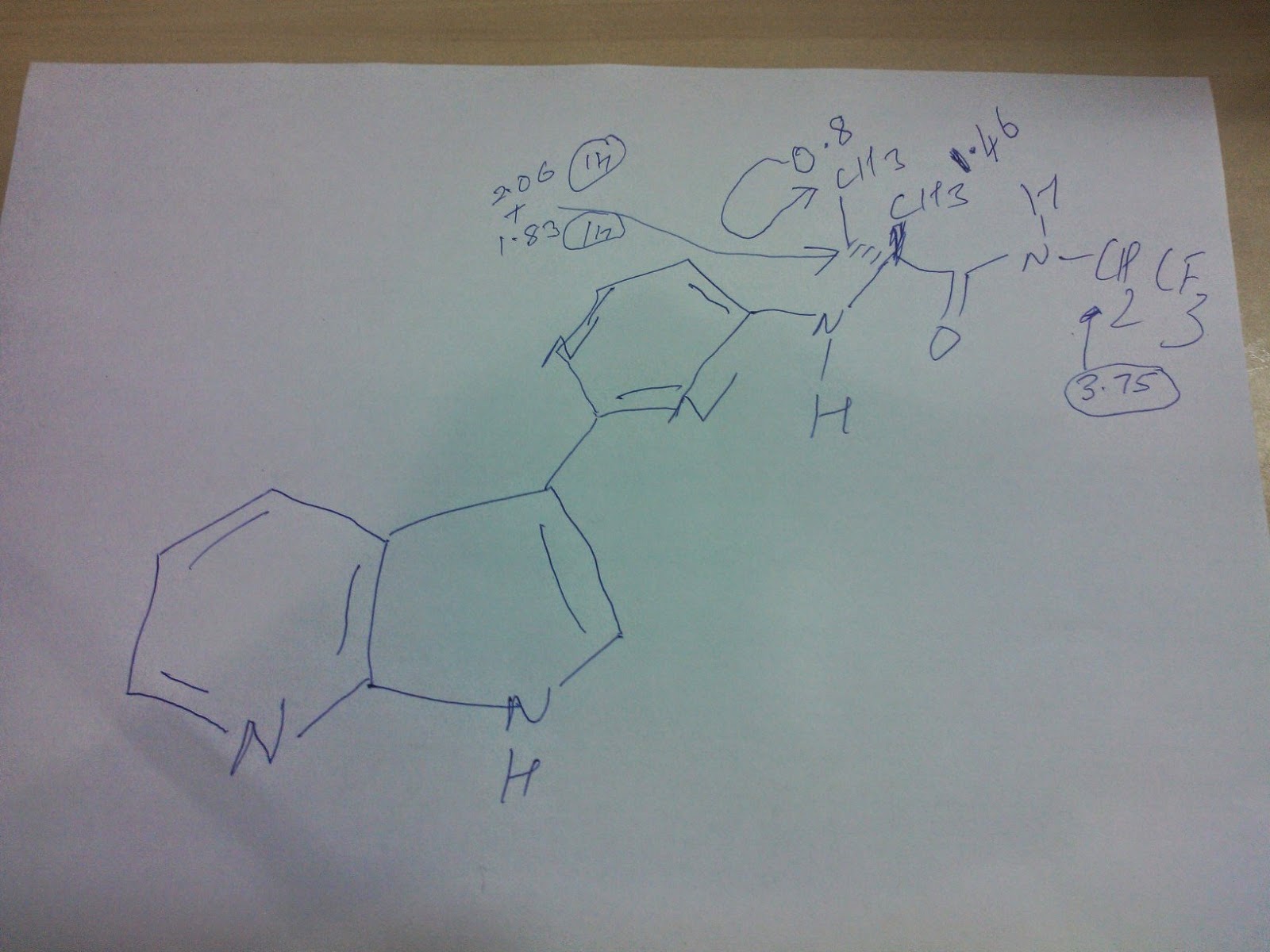
| 346 |
|
|
Example 1 Preparation of Compounds of the Invention
General Synthetic Scheme

Step 1
To a stirred solution of Boc-valine (1; R1 is Me; 3.8 g, 0.02 mol), EDC (4.63 g, 0.024 mol), HOBt (4.0 g, 0.026 mol), DIEA (10.5 mL, 0.06 mol) in 100 mL of DCM is added trifluoroethylamine HCl (2.92 g, 0.022 mol). The reaction mixture is stirred for 16 h. It is concentrated to dryness and redissolved in EtOAc, washed successively with 0.5N HCl, saturated aqueous solution of NaHCO3 and brine. The organic layer is dried (Na2SO4) and concentrated in vacuo to give 5.4 g (98%) of 2 as a white solid.
Step 2
Compound 2 (5.32 g, 0.0197 mol) is deprotected with a 1:1 mixture of DCM/TFA at rt for 45 min. Concentration to dryness gives the intermediate amine that is used directly for the next step. A mixture of 5-fluoro-2,4-dichloropyrimidine (3; R is F; 3.28 g, 0.0197 mol), the crude amine TFA salt (5.25 g, 0.0197 mol) and DIEA (10.27 mL, 0.059 mol) are stirred in isopropanol at rt for 16 h. The reaction mixture is concentrated in vacuo and redissolved in EtOAc, washed successively with 0.5N HCl, saturated aqueous solution of NaHCO3 and brine. The organic layer is dried (Na2SO4) and concentrated in vacuo to give a crude oil that is subjected to chromatography (50% EtOAc/50% hexanes) to yield the desired compound 4.
Step 3
A mixture of 5 (30 mg, 0.075 mmol; prepared according to WO 2005/095400), 4 (23 mg, 0.075 mmol), Pd (Ph3P)4 (9 mg, 0.0078 mmol) and sodium carbonate 2M (115 uL, 0.23 mmol) in 1 mL of DME is microwaved at 150° C. for 10 minutes. The reaction mixture is filtered through a short pad of silica gel with 30% EtOAc-70% hexanes as eluent to provide, after concentration to dryness, the crude intermediate that is used directly for the next step.
The crude intermediate is dissolved in 1 mL of dry methanol and 200 uL of sodium methoxide in methanol 25% was added. The reaction mixture is stirred at 60° C. for 1 h and quenched with 6N HCl (154 uL). The mixture is dried under a flow of nitrogen and purified by reverse phase HPLC (10-60 MeCN/water w/0.5% TFA) to provide the desired material of formula 6a.
Compounds of formulae 6b and 6c may be prepared in an analogous manner using the appropriate starting reagents. For instance, a compound of formula 6b may generally be made by substituting Cert-butyl 2-(2,2,2-trifluoroethylcarbamoyl)pyrrolidine-1-carboxylate for compound 1, while a compound of formula 6c may generally be made by substituting tert-butyl 2-(2,2,2-trifluoroethylcarbamoyl)propan-2-ylcarbamate for compound 1.
Example 2 Analytical Results
Tables 4, 5 and 6 below depicts exemplary 1H-NMR data (NMR) and liquid chromatographic mass spectral data, reported as mass plus proton (M+H), as determined by electrospray, and retention time (RT) for certain compounds of the present invention, wherein compound numbers in Tables 4, 5 and 6 correspond to the compounds depicted in Tables 1, 2 and 3, respectively (empty cells indicate that the test was not performed):
PATENTS
|
4-25-2012
|
Azaindoles Useful as Inhibitors of Janus Kinases
|
|
|
8-4-2010
|
Azaindoles useful as inhibitors of janus kinases
|
new patent
WO-2014110259
| US8450489 * | Mar 1, 2012 | May 28, 2013 | Vertex Pharmaceuticals Incorporated | Azaindoles useful as inhibitors of janus kinases |
| US8530489 * | May 22, 2012 | Sep 10, 2013 | Vertex Pharmaceuticals Incorporated | 5-cyano-4-(pyrrolo [2,3B] pyridine-3-yl)-pyrimidine derivatives useful as protein kinase inhibitors |
| US8686143 * | Oct 25, 2011 | Apr 1, 2014 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of Janus kinases |
| US20120157429 * | Oct 25, 2011 | Jun 21, 2012 | Wannamaker Marion W | Compounds useful as inhibitors of janus kinases |
| US20120165307 * | Mar 1, 2012 | Jun 28, 2012 | Vertex Pharmaceuticals Incorporated | Azaindoles useful as inhibitors of janus kinases |
| US20120309963 * | May 22, 2012 | Dec 6, 2012 | Vertex Pharmaceuticals Incorporated | 5-cyano-4- (pyrrolo [2,3b] pyridine-3-yl) -pyrimidine derivatives useful as protein kinase inhibitors |
| US20130237516 * | Apr 25, 2013 | Sep 12, 2013 | Vertex Pharmaceuticals Incorporated | Azaindoles useful as inhibitors of janus kinases |
| WO2013173506A2 | May 15, 2013 | Nov 21, 2013 | Rigel Pharmaceuticals, Inc. | Method of treating muscular degradation |
| WO2005095400A1 | Mar 30, 2005 | Oct 13, 2005 | Vertex Pharma | Azaindoles useful as inhibitors of jak and other protein kinases |
| WO2007084557A2 | Jan 17, 2007 | Jul 26, 2007 | Vertex Pharma | Azaindoles useful as inhibitors of janus kinases |
| WO2013070606A1 * | Nov 6, 2012 | May 16, 2013 | Vertex Pharmaceuticals Incorporated | Methods for treating inflammatory diseases and pharmaceutical combinations useful therefor |
Big boost for Incyte as Jakafi shines in PhII


ruxolitinib
Top-line results from a Phase II trial showed that its JAK inhibitor Jakafi (ruxolitinib), in combination with Roche’s Xeloda (capecitabine), improved survival in some patients with recurrent or treatment refractory advanced pancreatic cancer
http://www.pharmatimes.com/Article/13-08-
22/Big_boost_for_Incyte_as_Jakafi_shines_in_PhII.aspx
Ruxolitinib (trade names Jakafi and Jakavi, by Incyte Pharmaceuticals and Novartis) is a drug for the treatment of intermediate or high-risk myelofibrosis, a type of bone marrow cancer.It is also being investigated for the treatment of other types of cancer (such as lymphomas and pancreatic cancer), for polycythemia vera, and for plaque psoriasis.
The phase III Controlled Myelofibrosis Study with Oral JAK Inhibitor-I (COMFORT-I) and COMFORT-II trials showed significant benefits by reducing spleen size, relieving debilitating symptoms, and improving overall survival.
Mechanism of action
Ruxolitinib is a Janus kinase inhibitor with selectivity for subtypes 1 and 2 of this enzyme.
Side effects
Immunologic side effects have included herpes zoster (1.9%) and case reports of opportunistic infections.[10] Metabolic side effects have included weight gain (7.1%). Laboratory abnormalities have included alanine transaminase (ALT) abnormalities (25.2%), aspartate transaminase (AST) abnormalities (17.4%), and elevated cholesterol levels (16.8%).
Legal status
In November 2011, ruxolitinib was approved by the USFDA for the treatment of intermediate or high-risk myelofibrosis based on results of the COMFORT-I and COMFORT-II Trials.
Some analysts believe this to be a potential blockbuster drug.[3] As of the end of March 2012, and according to an Incyte spokesman, approximately 1000 physicians had prescribed the drug in the United States, out of a total 6500 hematologists and oncologists nationwide.

The US Food and Drug Administration had approved Incyte’s Jakafi (ruxolitinib) to treat patients with the bone marrow disease myelofibrosis (MF). Jakafi is the first and only drug granted license specifically for the treatment of the rare blood cancer.
Jakafi approved by FDA to treat rare bone marrow disease
Posted By Edward Su On November 17th, 2011
MF is a rare, potentially life-threatening blood cancer with limited treatment methods. Patients with the bone marrow disoder, characterized by bone marrow failure, enlarged spleen (splenomegaly), suffer from the symptoms of fatigue, night sweats and pruritus, poor quality of life, weight loss and shortened survival. The US drug firm Incyte estimates the disease affects about 16,000-18,500 people in the USA. Currently, the disease is treated with chemotherapy or bone marrow transplant.
Incyte’s Jakafi, the first drug to reach market from the Wilmington-based drug company, was approved by the FDA as a twice-a-day pill for the treatment of patients with intermediate or high-risk myelofibrosis (MF), including primary MF, post-polycythemia vera MF and post-essential thrombocythemia MF. The US regulators reviewed Jakafi under its priority review program for important new therapies.
The approval of Jakafi was based on the results from two clinical studies involved 528 patients with the disease. Patients in the Jakafi treatment arm experienced a significant reduction in the size of their spleen as well as a 50 percent decrease in symptoms, including pain, discomfort and night sweats.
Jakafi, generically known as ruxolitinib, works by blocking JAK1 and JAK2 enzymes associated with the disease. The company has co-developed the drug with Novartis as part of their collaboration signed in 2009. The Swiss drug firm has the rights to market Jakafi in other countries.
“The availability of Jakafi is a significant medical advancement for people living with myelofibrosis, a debilitating disease,” said Paul A. Friedman, M.D., President and Chief Executive Officer of Incyte. “This milestone marks a tremendous achievement for Incyte because a scientific discovery from our research laboratories has become the first JAK inhibitor to reach the market and provide a clinical benefit to patients.”
Richard Pazdur, director of the Office of Hematology and Oncology Drug Products in the FDA’s Center for Drug Evaluation and Research, said that Jakafi “represents another example of an increasing trend in oncology where a detailed scientific understanding of the mechanisms of a disease allows a drug to be directed toward specific molecular pathways”.
Incyte says Jakafi will be available next week, and the drug will cost $7,000 per month, or $84,000 for a year’s supply for insured patients. The company plans to provide Jakafi free to uninsured patients and will offer co-pay assistance to patients with financial need.
(JAK1, JAK2) inhibitor, developed by the Incyte Corporation, trade name Jakafi.
Ruxolitinib synthetic route as shown below. 4 – bromo-pyrazole ( 1 ) with ethyl vinyl ether ( 2 ) to protect, and then with a Grignard reagent to a halogen – exchanged with isopropyl magnesiumpinacol ester ( 3 ) quenching to obtain 4 . Compound 5 is obtained consisting of hydrogen is protected 6 , and then with a boronic acid ester 4 Suzuki coupling occurs under acidic conditions after removal of the protecting group pyrazolyl 7 , 7 and α, β-unsaturated aldehyde 8 chiral catalyst 9 of under the catalysis of asymmetric Michael addition to give ( R ) -10 (90% EE). ( R) -10 , after reaction with ammonia to obtain an imine oxidation with iodine nitrile 11 , respectively, with different conditions for the final removal of the protecting group to afford Ruxolitinib.
