
Home » Posts tagged 'INDIA 2021'
Tag Archives: INDIA 2021
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
BIAPENEM

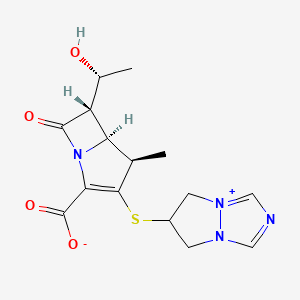
Biapenem
RPX7009
- Molecular FormulaC15H18N4O4S
- Average mass350.393 Da
Biapenern
CL 186-815LJ
C10,627LJ
C10627LJC 10627
omegacin
YR5U3L9ZH1
(4R,5S,6S)-3-((6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazol-4-ium-6-yl)thio)-6-((R)-1-hydroxyethyl)-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylate
[4R-[4a,5b,6b(R*)]]-6-[[2-Carboxy-6-(1-hydroxyethyl)-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-en-3-yl]thio]-6,7-dihydro-5 H-pyrazolo[1,2-a][1,2,4]triazol-4-ium inner salt
120410-24-4[RN]
5H-Pyrazolo[1,2-a][1,2,4]triazol-4-ium, 6-[[(4R,5S,6S)-2-carboxy-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-en-3-yl]thio]-6,7-dihydro-, inner salt [ACD/Index Name]
6-[[(4R,5S,6S)-2-Carboxy-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-en-3-yl]thio]-6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazol-4-ium inner salt
7074
(4R,5S,6S)-3-(6,7-Dihydro-5H-pyrazolo[1,2-a][1,2,4]triazol-4-ium-6-ylsulfanyl)-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylate
TL8000539UNII:YR5U3L9ZH1UNII-YR5U3L9ZH1биапенем
بيابينام比
阿培南
INDIA CDSCO APPROVED 25 SEPT 2021, BDR PHARMA, 
https://www.cdsco.gov.in/opencms/resources/UploadCDSCOWeb/2018/UploadCTApprovals/BDR.pdfhttps://medicaldialogues.in/news/industry/pharma/bdr-pharma-gets-dcgi-nod-for-generic-antibiotic-drug-biapenem-82384

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter a
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
Biapenem (INN) is a carbapenem antibiotic. It has in vitro activity against anaerobes.[1] 1-β-methyl-carbapenem antibiotic. Approved in Japan in 2001.
PATENT
EP 168707
EP 289801
JP 02088578
ZA 9100014
EP 533149
CN 1995040
IN 2006DE01555
CN 101121716
IN 2008CH00177
CN 101805359
CN 101851206
CN 101935321
CN 111875622
WO 2018074916
WO 2016059622
US 20150328323
WO 2015151081
WO 2015155753
WO 2015151078
US 20150284416
WO 2015151080
US 20150038726
WO 2014104488
IN 2013MU00181
WO 2014111957
CN 103570750
WO 2014097221
IN 2012CH01371
WO 2013150550
PAPERS
Journal of Organic Chemistry (1992), 57(15), 4243-9.
Heterocycles (1993), 36(8), 1729-34.
Journal of Antibiotics (1993), 46(12), 1866-82.
e-EROS Encyclopedia of Reagents for Organic Synthesis (2008), 1-3.
Bioorganic & medicinal chemistry letters (2009), 19(17), 5162-5.
IP.com Journal (2014), 14(12A), 1-3
IP.com Journal (2014), 14(10A), 1-2.
Bioorganic & medicinal chemistry (2013), 21(18), 5841-50.

NEW DRUG APPROVALS
one time
$10.00
PATENT
https://patents.google.com/patent/WO2014097221A1/esBiapenem is chemically known as 6-[[2(4R,5S,6S)-carboxy-6-[(lR)- hydroxy ethyl] -4-methyl-7-oxo- 1 -azabicyclo [3.2.0]hept-2-en-3 -yljthio] 6,7-dihydro-5H- pyrazolo[l,2-a][l,2,4]triazol-4-ium inner salt, and is represented by Formula 1. It is indicated for the treatment of bacterial infection and sepsis.

Formula 1U.S. Patent No. 4,866,171, in Example 6, discloses the purification of biapenem using chromatography and/or lyophilization techniques. This patent also describes a process for the conversion of amorphous biapenem into a crystalline form by dissolving the amorphous biapenem in water while heating, followed by cooling, then washing the obtained crystals with a 50% aqueous ethanol solution.U.S. Patent No. 5,241,073 describes a process for the purification of biapenem involving column chromatography and crystallization with ethanol.U.S. Patent No. 5,286,856 describes a process for the crystallization of biapenem from an aqueous solution, comprising maintaining the temperature of the aqueous solution from eutectic temperature (-10°C to -2°C) to a temperature lower than 0°C, followed by lyophilization.The Journal of Organic Chemistry, 63(23):8145-8149 (1998) describes the purification of biapenem involving resin chromatography.The present invention provides an alternate process for the purification of biapenem that avoids making use of tedious techniques like chromatography and lyophilization. At the same time, it results in a high yield and high purity of the final product. Advantageously, the crystalline biapenem of this invention can be directly isolated from the reaction mixture. Further, the process of the present invention involves fewer steps, is easily scalable, and industrially advantageous.EXAMPLESExample 1 : Purification of BiapenemBiapenem (12 g) was added into water (300 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.6 g) was added to the reaction mixture and stirred for 10 minutes to 15 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (36 mL). The filtrate obtained was passed through a 0.45 micron filter, and its pH was adjusted to 5.5 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (336 mL) was added to the reaction mixture at 5°C to 10°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (60 mL). The solid was dried under reduced pressure (720 mmHg) at 30°C to 35°C to obtain the title product as white crystals.Yield: 84%HPLC Purity: 99.87% Example 2: Purification of BiapenemBiapenem (18 g) was added into water (450 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.9 g) was added to the reaction mixture and stirred for 30 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (54 mL). The filtrate obtained was passed through a 0.45 micron filter and its pH was adjusted to 4.9 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (504 mL) was added to the reaction mixture at 10°C to 15°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (90 mL). The solid was dried under reduced pressure (720 mmHg) at 35°C to 40°C to obtain the title product as white crystals.Yield: 81.77%HPLC Purity: 99.80%
PATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2013150550
The present invention relates to an improved process for the preparation of carbapenem antibiotic; more particularly relates to the preparation of Ertapenem monosodium salt of formula (I) having purity greater than 98.5% and having pharmaceutically acceptable level of residual solvent and palladium content.
The US patents namely US 5,478,820 and US 5,856,321 disclose various processes for preparing Ertapenem and its sodium salt. Example 12 of US 5,478,820 discloses a process in which the Ertapenem was isolated using column purification followed by freeze-drying technique. According to Example-4 of this patent disodium salt of Ertapenem was prepared by dissolving crude product in water using NaHCO3, followed by purification using column chromatography and subsequent lyophilization.
US 6,504,027 provides a process for preparing Ertapenem in crystalline form which comprises deprotecting and extracting a polar organic solution containing a crude mono-protected Ertapenem of formula
wherein P represents protecting group and X represents charge balancing group like sodium
with C4.10 alcohol in the presence of ion-pairing reagent followed by adjusting the pH of the aqueous layer to 5.5 and crystallizing using methanol and 1-propanol to produce a crystalline compound; this patent process involves operations like
multiple extractions which is cumbersome in plant and said operation affects the overall yield.
US 7,145,002 provides a process for producing Ertapenem or its sodium salt and/or its solvate in crystalline form. This patent states (refer para 3, lines 31-41) that contact of Ertapenem sodium with water and alcoholic solvents results in the formation of crystalline solvates. The processes reported in examples- 1 & 2 provide crystalline Ertapenem monosodium which is isolated from a mixture of methanol, 1-propanol and water followed by washing with aqueous isopropyl alcohol which results in the formation of crystalline solvate of Ertapenem sodium. Applicant found the Ertapenem monosodium obtained according to this process contain higher amount of residual solvent and palladium content.
US 7,022,841 provide a process for reducing the levels of organic solvents in Ertapenem to pharmaceutically acceptable levels. This patent discloses (Refer para 1, lines 52-60) that Ertapenem sodium obtained from water/alcohol mixture according to US 7, 145,002 becomes amorphous when water content of the solid is reduced and further the organic solvent present in the solid is not readily removed. In view of this drawback, this patent provides a process wherein the water content of Ertapenem sodium is maintained between 13-25% during the washing and drying process. This patent further discloses that (Refer para 9, lines 6-14) the washing of Ertapenem sodium can be carried out using anhydrous solvents which results in the formation of amorphous solid, which is then dried using hydrated nitrogen by increasing the water content of the solid. Due to the hygroscopic and unstable nature of Ertapenem sodium when in contact with water, the above processes result in more degradation of Ertapenem. The patent further discloses in example 5 that the degradation of Ertapenem sodium is more when it takes more time for drying.
Further this patent requires repetitive washing and control of moisture content to get the desired results.
For isolation of Ertapenem sodium from the reaction mass, all the above discussed prior art patents utilize methanol and 1-propanol as crystallization solvent. The filtration of Ertapenem sodium formed by using these solvents or their mixture takes longer time duration and subsequent drying for the removal of residual solvent also takes several hours due to occlusion of solvent into Ertapenem sodium. During these operations the Ertapenem sodium degrades an results in the formation of many impurities such as several dimers, methanolysis impurity etc., and hence the reported processes is not suitable to manufacture Ertapenem sodium on commercial scale with purity greater than 98.5% and with pharmaceutically acceptable level of residual solvent content.
Methanolysis impurity Dimer-I
Dimer-II
Further the applicant found that Ertapenem monosodium isolated by following the process reported in prior art was having palladium content above the pharmaceutically acceptable level. Hence the process reported in prior art is not suitable on manufacturing scale where maintaining stringent technological condition is cumbersome and involves higher operating cost.
Thus all the reported processes suffer in terms of one or more of the following facts:
■ Filtration time of Ertapenem sodium takes several hours.
■ Drying time takes several hours due to occlusion of solvent and nature of the solid.
■ Stringent technological condition is required for maintenance of moisture content during washing & drying operation.
■ Palladium content is found to be higher (greater than 25 ppm) which is not acceptable for pharmaceutical products.
■ The isolated Ertapenem sodium is having higher amount of residual solvents.
■ The purity is reduced over to several hours of filtration & drying.
With our continued research for developing a process for the preparation of Ertapenem monosodium of formula (I) to overcome the above mentioned drawbacks, we surprisingly found that when esters of organic acid were used as solvents in place of 1-propanol, the solid obtained was easily filterable with less cycle time. Further the washing with hydrocarbon solvents containing 0-75% alcoholic solvent followed by drying results in Ertapenem having residual solvent content well below the pharmaceutically acceptable levels. The use of thiourea, thiosemicarbazide or their N-substituted derivatives in the presence of organic solvents during isolation brings down the palladium content to pharmaceutically acceptable level.
The Ertapenem or its sodium salt can be prepared according the processes provi
(I)
P’ and P” represent carboxylic protecting groups and X is H or Na
Scheme-1
The present invention is illustrated with the following examples, which should not be construed to limit the scope of the invention.
Example- I
Preparation of Ertapenem monosodium of formula (I)
Step-I:
To a stirred solution of p-nitrobenzyl (4R,5S,6S)-3-(diphenyloxy)phosphoryloxy-6-[(lR)-l-hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3,2,0]hept-2-ene-2-carboxylate (compound II) (100 g) and (2S,4S)-2-[[(3-carboxyphenyl) amino]carbonyl]-4-mercapto-l-(4-nitrobenzyl)pyrrolidinecarboxylate (compound III) (75 g) in N,N-dimethylformamide was added Ν,Ν-diisopropylethylamine at -30 to -40° C and stirred. The reaction mass, after completion of the reaction, was quenched with a mixture of phosphate buffer solution-ethyl acetate and the pH was adjusted to 5 – 6 with phosphoric acid. The organic layer was separated, washed with water and subjected to carbon treatment. To the organic layer containing the compound of formula (IV) (wherein P’ and P” refers to p-nitrobenzyl), a solution of sodium 2-ethylhexanoate (42 g in 500 mL methanol) was added and taken to next step as such. (If required the compound of formula (IV) is isolated either as sodium salt or as free acid by following the process reported in prior art and taken further)
Step-II:
To the Step-I organic layer containing the compound of formula (IV) (wherein P’ and P” refers to p-nitrobenzyl & X is Na), 3-(N-morpholino)propanesulfonic acid solution was added and subjected to hydrogenation using palladium on carbon at 8- 10° C with 9-10 kg hydrogen pressure. The reaction mass, after completion of reaction, was filtered to remove palladium on carbon. To the filtrate, thiourea (5 g) and tetrahydrofuran were added and stirred. The aqueous layer was separated and treated with carbon and neutral alumina at 10-15° C while degassing and filtered. The filtrate was added to methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with a mixture of cyclohexane: ethanol (200 ml) and dried under vacuum. Yield: 46 g; Purity by HPLC: 98.93%; Palladium content: 1.8 ppm by ICP MS
The HPLC purity of Ertapenem monosodium was checked using the following parameters
Column : Zorbax Eclipse plus C8, (50 mm x 4.6 mm), 1.8μ).
Mobile phase : Ammoniam acetate buffer: Acetonitile: water
Detector : UV at 250 nm
Flow rate : 0.5 mL/min
Run time : 45 min.
Example- II
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and subjected to hydrogenation using palladium on carbon at 8-10° C with 9-10 kg hydrogen pressure. The reaction mass, after completion of reaction, was filtered and the filtrate was treated with thiourea and 2-methyltetrahydrofuran and the layers separated. The aqueous layer was treated with carbon & neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with cyclohexane (200 ml) and
dried under vacuum. Yield: 44 g; Purity by HPLC: 98.84%; Palladium content: 0.93 ppm by ICP MS
The term ICP MS method refers to the inductively coupled plasma mass spectrometry. The following parameter was used to determine the content of palladium.
The carbapenem was digested in a closed vessel system in presence of reagents Nitric acid, Hydrogen peroxide and Hydrochloric acid by using Microwave reaction system with microwave radiation power 1200 Watts. The digested sample was introduced into inductively coupled plasma mass spectrometer by help of Peltier cooled spray chamber. The sample aerosol is getting atomized then ionized in the argon plasma. The ionized Palladium was estimated by using Quadrupole mass detector. The sample was quantified against NIST traceable reference standards at mass number ! 05.
Example- III
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction, was filtered and the filtrate was treated with thiourea and tetrahydrofuran and the layers separated. The aqueous layer was separated and treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with a mixture of toluene: ethanol (200 ml) and dried under vacuum. Yield: 42 g; Purity by HPLC: 99.03%
Example- IV
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction was filtered and the filtrate was treated with thiosemicarbazide and tetrahydrofuran and the layers separated. The aqueous layer was treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C followed by the addition of ethyl acetate and stirred. The solid obtained was filtered, washed with a mixture of cyclohexane: ethanol (200 ml) and dried under vacuum. Yield: 41 g; Purity by HPLC: 99.13%; Palladium content: 1.71 ppm by ICP MS
Example- V
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and subjected to hydrogenation using palladium on carbon at 8-10° C with 9-10 kg hydrogen pressure. The reaction mass, after completion of reaction, was filtered and the filtrate was treated with thiourea and 2-methyltetrahydrofuran and the layers separated. The aqueous layer was treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, a mixture of ethyl acetate containing 10% methyl acetate was added and stirred. The solid obtained was
filtered, washed with cyclohexane:ethanol and dried under vacuum. Yield: 40.5 g; Purity by HPLC: 98.77%; Palladium content: 1.43 ppm by ICP MS
Example-VI
(V ) (V I )
The diprotected Meropenem of formula (V) (where P and P’ were p-nitrobenzyl) was dissolved in tetrahydrofuran and 3-(N-morpholino)propanesulfonic acid buffer and hydrogenated using palladium on carbon at 9-10 kg hydrogen pressure. The mass was filtered and the filtrate was washed with ethyl acetate. The aqueous layer was treated with thiourea and 2-methyltetrahydrofuran. The aqueous layer was separated, treated with carbon and degassed. The carbon was filtered off and acetone was added to the filtrate to crystallize Meropenem trihydrate of formula (VI). The product was filtered and washed with aq. acetone and dried under vacuum to get Meropenem trihydrate. Purity: 99.8%; Pd content: 0.08 ppm
Reference example-I:
Preparation of Ertapenem monosodium of formula (I)
To Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction, was filtered. The filtrate was treated with thiourea and tetrahydrofuran and the layers separated. The aqueous layer was treated with carbon and neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5.5-5.7 using aqueous acetic acid. To the mass ethyl acetate was added and stirred. The solid obtained was filtered, washed with ethanol (5 * 100 ml) and dried under vacuum. Yield: 31 g; Purity by HPLC: 96.76%
Reference example-II:
Preparation of Ertapenem monosodium of formula (I)
To the Step-I reaction mass , as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction was filtered and the layers separated. The aqueous layer was treated with carbon and neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5.5-5.7 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with a mixture of cyclohexane: ethanol and dried under vacuum. Yield: 43 g; Purity by HPLC: 98.6%; Palladium content: 35.8 ppm by ICP MS.
Reference example-HI:
Preparation of Ertapenem monosodium of formula (I)
To the Step-I reaction mass as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction, was filtered and the layers separated. The aqueous layer was treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with 1-propanol at -5° C and the pH was adjusted to 5.5-5.7 using aqueous acetic acid. To the mass methanol and 1-propanol were added and stirred. The solid obtained was filtered, washed with ethanol and dried under nitrogen atmosphere in vacuum. Yield: 25 g; Purity by HPLC: 97 %.: palladium content: 38.2 ppm
The following tables illustrate the advantages of the present invention over prior art process:
Table-I: Comparison of present process with prior art process
The crystallization and washing method disclosed in US 7,022,841 was followed.
The above table indicates that the use of ethyl acetate as crystallization solvent results with improved yield and high purity with less filtration and drying time thereby increasing the productivity significantly on manufacturing scale. Further the use of thiourea or thiosemicarbazide as reagents in the present process results in the pharmaceutically acceptable level of palladium content.
Table-II: Comparison of solvents for washing Ertapenem monosodium
The above table indicates that the use of hydrocarbon solvents containing 0-75% of alcoholic solvent helps in washing to remove the residual solvent content in shorter duration and with single run wash. On the other hands the use of ethanol alone results in Ertapenem monosodium having less yield and purity requiring repetitive washing.
Table-IH: Effect of different reagent in reduction of palladium content
Reagent : thiourea, thiosemicarbazide or its N-substituted derivatives
Advantages of the process of the present invention:
> The use of ester of an organic acid for the crystallization of Ertapenem sodium results in fast filtration and reduced cycle time, thereby increasing the productivity.
> Washing of Ertapenem sodium with hydrocarbon solvent optionally containing alcohol results in improved physical nature of Ertapenem sodium resulting in reduced washing and drying time thereby avoid the degradation of Ertapenem and providing Ertapenem sodium with purity greater than 98.5% by HPLC.
Use of thiourea, thiosemicarbazide or their N-substituted derivatives in the process results in Ertapenem sodium having pharmaceutically acceptable level of palladium content.
PATENT
https://patents.google.com/patent/WO2002057266A1/enEXAMPLE

PNB = p-nitrobenzyl

Ia’A hydrogenator is charged with 63 g of 10% Pd on carbon catalyst (dry weight) in 1.8 L of water. The vessel is placed under hydrogen then vented and placed under nitrogen. Sodium hydroxide (68 g, 50%) is charged adjusting the pH to about 7.5 with carbon dioxide.The enol phosphate (170 g) and the thiol (86 g) are dissolved in 1.3‘L of N- ethylpyrrolidinone (NEP). The mixture is cooled to below -40°C and 1,1,3,3- tetramethylguanidine (109 g) is added. After 3 hours, the reaction mixture is quenched into the hydrogenator at below 15°C adjusting the pH to about 8 with carbon dioxide. The vessel is placed under hydrogen. When the reaction is complete, the hydrogen is vented and the reaction mixture is treated with activated carbon and filtered. The filtrate is extracted with iso-amyl alcohol containing diphenylphosphoric acid (240 g) and 50% NaOH (44 g). The resulting aqueous solution is further extracted with iso-amyl alcohol to give an aqueous solution containing at least 90 mg/mL of the product. Both extractions are performed using two CINC centrifugal separators set in series for countercurrent extraction. The pH is adjusted to 5.5 with acetic acid. The product is crystallized by adding equal volumes of methanol and 1- propanol at below -5°C and isolated by filtration. The solid is washed with a mixture of 2-propanol and water (85: 15 v/v) then dried to yield a compound of formula la’.While certain preferred embodiments of the invention have been described herein in detail, numerous alternative embodiments are contemplated as falling within the scope of the appended claims. Consequently the invention is not to be limited thereby.
Patent Citations
Publication numberPriority datePublication dateAssigneeTitleUS4866171A1987-04-111989-09-12Lederle (Japan), Ltd.(1R,5S,6S)-2-[(6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazolium-6-yl)]thio-6-[R-1-hydroxyethyl]-1-methyl-carbapenum-3-carboxylateUS5241073A1990-10-121993-08-31Lederle (Japan)Process for preparing (1R,5S,6S)-2-[(6,7-dihydro-5H-pyrazolo [1,2-a][1,2,4]triazolium-6-yl)]thio-6-[(R)-1-hydroxyethyl]-1-methyl-carbapenem-3-carboxylate and starting materials thereofUS5286856A1991-09-201994-02-15Takeda Chemical Industries, Ltd.Production of crystalline penemWO2002057266A1 *2001-01-162002-07-25Merck & Co., Inc.Improved process for carbapenem synthesisWO2009047604A1 *2007-10-082009-04-16Orchid Chemicals & Pharmaceuticals LimitedProcess for the preparation of carbapenem antibioticCN102268025A *2011-07-152011-12-07海南美兰史克制药有限公司一种比阿培南化合物及其制法
References
- ^ Aldridge KE, Morice N, Schiro DD (April 1994). “In vitro activity of biapenem (L-627), a new carbapenem, against anaerobes”. Antimicrob. Agents Chemother. 38 (4): 889–93. doi:10.1128/aac.38.4.889. PMC 284564. PMID 8031067.
External links
- (in Japanese) Omegacin
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | IV |
| ATC code | J01DH05 (WHO) |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 120410-24-4 |
| PubChem CID | 71339 |
| ChemSpider | 64442 |
| UNII | YR5U3L9ZH1 |
| ChEBI | CHEBI:3089 |
| ChEMBL | ChEMBL285347 |
| CompTox Dashboard (EPA) | DTXSID5046435 |
| Chemical and physical data | |
| Formula | C15H18N4O4S |
| Molar mass | 350.39 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
ClinicalTrials.gov
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT04552444 | Clinical Efficacy of Combination Therapy Based on High-dose Biapenem in CRKP Infections | Recruiting | 2020-09-17 | |
| NCT01772836 | Safety Study of Intravenous Biapenem (RPX2003) and RPX7009 Given Alone and in Combination | Phase 1 | Completed | 2013-07-11 |
| NCT01702649 | Safety, Tolerability, Pharmacokinetics of Intravenous RPX2003 (Biapenem) in Healthy Adult Subjects | Phase 1 | Completed | 2012-12-03 |
NIPH Clinical Trials Search of Japan
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| UMIN000017219 | Feasibility and efficacy of the de-escalation therapy by Biapenem for postoperative bacterial pneumonia. | None | Recruiting | 2015-04-22 |
| UMIN000003964 | Clinical evaluation of Biapenem 0.3g, three times daily dosing in eldery patients with pneumonia (moderate and severe infection) | Not applicable | Complete: follow-up complete | 2010-07-29 |
/////////BIAPENEM, TL8000539, UNII:YR5U3L9ZH1, UNII-YR5U3L9ZH1, биапенем, بيابينام ,比阿培南 , Biapenern, CL 186-815, CL 186815, L 627, LJC 10627, Omegacin, Antibacterial, Antibiotics, Lactams, Carbapenems, ind 2021, india 2021, approvals 2021
CC1C2C(C(=O)N2C(=C1SC3CN4C=NC=[N+]4C3)C(=O)[O-])C(C)O
https://clinicaltrials.gov/search/intervention=Biapenem

updated
Biapenem is chemically known as 6-[[2(4R,5S,6S)-carboxy-6-[(lR)-hydroxy ethyl] -4-methyl-7-oxo- 1 -azabicyclo [3.2.0]hept-2-en-3-yljthio] 6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazol-4-ium inner salt, and is represented by Formula 1. It is indicated for the treatment of bacterial infection and sepsis.
Formula 1
U.S. Patent No. 4,866,171, in Example 6, discloses the purification of biapenem using chromatography and/or lyophilization techniques. This patent also describes a process for the conversion of amorphous biapenem into a crystalline form by dissolving the amorphous biapenem in water while heating, followed by cooling, then washing the obtained crystals with a 50% aqueous ethanol solution.
U.S. Patent No. 5,241,073 describes a process for the purification of biapenem involving column chromatography and crystallization with ethanol.
U.S. Patent No. 5,286,856 describes a process for the crystallization of biapenem from an aqueous solution, comprising maintaining the temperature of the aqueous solution from eutectic temperature (-10°C to -2°C) to a temperature lower than 0°C, followed by lyophilization.
The Journal of Organic Chemistry, 63(23):8145-8149 (1998) describes the purification of biapenem involving resin chromatography.
The present invention provides an alternate process for the purification of biapenem that avoids making use of tedious techniques like chromatography and lyophilization. At the same time, it results in a high yield and high purity of the final product. Advantageously, the crystalline biapenem of this invention can be directly isolated from the reaction mixture. Further, the process of the present invention involves fewer steps, is easily scalable, and industrially advantageous.
EXAMPLES
Example 1 : Purification of Biapenem
Biapenem (12 g) was added into water (300 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.6 g) was added to the reaction mixture and stirred for 10 minutes to 15 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (36 mL). The filtrate obtained was passed through a 0.45 micron filter, and its pH was adjusted to 5.5 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (336 mL) was added to the reaction mixture at 5°C to 10°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (60 mL). The solid was dried under reduced pressure (720 mmHg) at 30°C to 35°C to obtain the title product as white crystals.
Yield: 84%
HPLC Purity: 99.87%
Example 2: Purification of Biapenem
Biapenem (18 g) was added into water (450 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.9 g) was added to the reaction mixture and stirred for 30 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (54 mL). The filtrate obtained was passed through a 0.45 micron filter and its pH was adjusted to 4.9 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (504 mL) was added to the reaction mixture at 10°C to 15°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (90 mL). The solid was dried under reduced pressure (720 mmHg) at 35°C to 40°C to obtain the title product as white crystals.
Yield: 81.77%
HPLC Purity: 99.80%
PATENT
Background of the Invention Biapenem is a synthetic broad-spectrum carbapenem antibiotic which suppresses bacterial growth by inhibiting the enzymes responsible for bacterial cell wall synthesis, and shows broad-spectrum antibacterial activity both against gram-positive bacteria and gram-negative bacteria. Biapenem is chemically known as (4R,5S,6S)-3-(6,7-dihydro-5H-pyrazolo[l,2-a][ 1,2,4] triazol-8-ium-6-ylsulfanyl)-6-( 1 -hydroxyethyl)-4-methyl-7-oxo-1 -azabicyclo [3.2.0]hept-2-ene-2-carboxylate and marketed in Japan as OMEGACIN®.Various methods are reported in the prior art for the preparation of Biapenem of formula (I) which includes the condensation of compound of formula (II) with compound of formula (III) and subsequent deprotection of the protecting group as shown in scheme-1. wherein R1 is hydrogen or hydroxy protecting group such as tert-butyl dimethyl silyl and the like, R2 is hydrogen or carboxyl protecting group such as p-nitrobenzyl, p-methoxy benzyl, allyl and the like, A is an activating group such as P(0)(OR)2, SO2R and the like wherein R is selected from substituted or unsubstituted C1-6 alkyl, aralkyl or aryl to form the compound of formula (II). The X” in compound of formula (III) is halogen selected from Br or CI.Biapenem was first disclosed in US 4,866,171 and the said patent also discloses a process for the preparation of the same. US 5,241,073 disclosed the method for the preparation of compound of formula (III) followed by condensation with compound of general formula (II) using base such as N-ethyldiisopropylamine and subsequent deprotection yields Biapenem which was isolated by column chromatography followed by crystallization from ethanol.EP 0289801 discloses a process for the preparation of crystalline Biapenem wherein Biapenem was dissolved in water and lyophilized to get amorphous compound. The amorphous compound was dissolved in water at 40° C followed by cooling to get crystalline product. This patent further provides the PXRD values of the crystalline Biapenem. The Biapenem obtained according to the process provided in this patent takes longer time for reconstitution and hence not suitable.US 5,286,856 and US 5,424,069 provide a process for the crystallization of Biapenem which utilizes freeze-drying technique and vial lyophillisation method respectively. These patents disclose (refer para 1, lines 10-33 of US’ 856) that the process provided in EP 0289801 results with Biapenem crystals which take relatively longer time for dissolution during use. To overcome the above issues, these patents utilize the freeze-drying and vial lyophillisation methods. The said methods involve freezing of the solution containing Biapenem followed by raising the temperature and repeating the cooling and heating process followed by lyophillisation to get the crystalline product. Lyophillisation and related process are capital intensive techniques and uneconomical in commercial scale operations.All the above said prior arts utilize either the lyophillisation technique or preparing the amorphous material and crystallizing it from water to get crystalline Biapenem.Biapenem is available as powder for injection which needs to be reconstituted with water or saline solution before injection. The process of preparing a solution having an appropriate concentration of an active ingredient for the administration is called “reconstitution”. The reconstitution time (RCT) plays a critical role in injectable powders. Short reconstitution time is preferable for both a member of medical center and patients. If the reconstitution time is too long, it will increase the preparation time thus making it difficult to administrate it to many patients at the same, which will eventually lower the competitiveness of the drug. The problem before the applicants is to find economic and robust process for the preparation of Biapenem with high purity and yield which should dissolve in water in less than 25 seconds (reconstitution time). With our continued intensive and diligent research for developing a process for the preparation of Biapenem having high purity and yield with reconstitution time of less than 25 seconds, we have identified an improved process which is commercially viable and eliminates the issues associated with reconstitution time. The process of this invention is simple and obviates the use of freeze crystallization. Further the present invention fulfils the need for a process for the manufacture of Biapenem which is convenient to operate in commercial scale
Objectives of the inventionThe main objective of the present invention is to provide a simple and commercially viable, industrially scalable process for the crystallization of Biapenem of formula (I) with high purity and good yield.Yet another objective of the present invention is to provide a simple and commercially suitable process for the preparation of Biapenem of formula (I) with reconstitution time less than 25 seconds. The reconstitution time is calculated by the time taken to dissolve 300 mg of Biapenem in 100 ml of water or saline solution.Summary of the inventionAccordingly the primary aspect of the present invention is to provide an improved process for the preparation of Biapenem of formula (I) the said process comprises;(i) obtaining a solution of Biapenem in water containing co-solvent; and(ii) adding anti-solvent in to the solution of step (i) or vice-versa to crystallize Biapenem followed by filtration. Detailed Description In an embodiment of the present invention, the co-solvent used in step (i) is selected from alcoholic solvents consisting of methanol, ethanol, isopropyl alcohol, n-propanol, n-butanol and iso-butanol or mixtures thereof; preferably methanol, ethanol and isopropyl alcohol; more preferably methanol.In another embodiment of the present invention the anti-solvent used in step (ii) is selected from acetone, methyl ethyl ketone, methyl isobutyl ketone, ethyl acetate, methyl acetate, butyl acetate, tetrahydrofuran or mixtures thereof; preferably acetone. In yet another embodiment of the present invention, the solution of Biapenem in step (i) can be obtained by (a) dissolving Biapenem in water followed by addition of co-solvent (b) dissolving Biapenem in water containing the co-solvent (c) the aqueous solution containing Biapenem can be obtained directly from the reaction mass followed by addition of co-solvent (d) the aqueous solution of Biapenem containing co-solvent can be obtained directly from the reaction mass. The said solutions, if necessary can be subjected to sterile filtration before the addition of anti-solvent. Thus the present invention provided a process for the preparation of sterile Biapenem having reconstitution time less than 25 seconds, more preferably less than 15 seconds.The prior art lyophillisation process for the preparation of Biapenem requires capital investment and high operating cost due to the involvement of repetitive heating and cooling process which is tedious technology in commercial scale operations. The reported prior art process for the crystallization of Biapenem of formula (I) from water results in the formation of crystalline powder which takes longer time for dissolution in water or saline solution (reconstitution time). Surprisingly, applicant found that the use of co-solvents during the crystallization of Biapenem results with Biapenem having reconstitution time of less than 25 seconds. This constitutes the novelty of the present invention.In this present invention the Biapenem of formula (I) is obtained as crystalline solid with purity above 99.0 % by HPLC with good stability and further can be easily filled in vials.
The following examples are provided by way of illustration only and should not be construed to limit the scope of the invention.
Crystallization of (4R,5S,6S)-3-(6,7-dihvdro-5H-pyrazolo[l,2-al 11,2,41 triazol-8-ium-6-vlsulfanvl)-6-(l-hydroxvethvl)-4-methvl-7-oxo-l-azabicyclo [3.2.01hept-2-ene-2-carboxvlate [Biapenem of formula (1)1:Example -1:To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. Activated carbon and EDTA were added to the clear solution and filtered through hi-flow bed, washed with water followed by filtration through micron filters in sterile area. To the filtrate, methanol (600 mL) was added followed by acetone under stirring. To the reaction mass, Biapenem seed material was added and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem.Yield: 85 g Purity by HPLC: 99.5% Reconstitution time (RCT): < 15 seconds
Example -2:To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. To the filtrate, isopropyl alcohol (500 ml) was added followed by acetone under stirring. The mass was cooled and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem.Yield: 83 g Purity by HPLC: 99.6% Reconstitution time: < 15 seconds
Example -3;To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. The solution was filtered through micron filters. To the filtrate, ethanol (600 ml) was added followed by acetone and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem.Yield: 84 g Purity by HPLC: 99.5% Reconstitution time : < 15 seconds
Example -4:To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. The solution was filtered through hi-flow bed, washed with water followed by filtration through micron filters. To the filtrate, methanol (450 ml) was added followed by acetone and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem. Yield: 87 g Purity by HPLC: 99.4% Reconstitution time (RCT): < 15 seconds
Reference example -1:Preparation of Biapenem (Non-Sterile)Step-I: Preparation of p-Nitrobenzyl (4R,5S,6S)-3-(6,7-dihydro-5H-pvrazolofl,2-al[l,2,41triazol-8-ium-6-vlsulfanvn-6-(l-hvdroxvethyl)-4-methvI-7-oxo-l-azabicvclo[3.2.01hept-2-ene-2-carboxylate [Compound of formula (IV)1To a mixture of acetonitrile and DMF, P-Nitrobenzyl (4R,5S,6S)-3-(dipheny loxy)phosphory loxy-6- [(1R)-1 -hydroxyethy 1] -4-methy 1-7-oxo-1 -azabicyclo[3,2,0]hept-2-ene-2-carboxylate (compound of formula II) and 6,7-dihydro-6-mercapto-5H-pyrazolo[l,2-a] [1,2,4] triazole chloride (compound of formula III) were added and cooled to 0-5° C. To this mixture, N-ethyldiisopropyl amine was added and stirred till the completion of the reaction, followed by the addition of dichloromethane to crystallize the p-Nitrobenzyl (4R,5S,6S)-3-(6,7-dihydro-5H-pyrazolo[l,2-a][l,2,4]triazol-8-ium-6-ylsulfanyl)-6-(l -hydroxyethyl)-4-methyl-7-oxo-1 -azabicyclo[3.2.0] hept-2-ene-2-carboxylate which was filtered and dried under nitrogen.
Step-II: Preparation of BiapenemTo a solution of MOPS buffer and THF, p-Nitrobenzyl (4R,5S,6S)-3-(6,7-dihydro-5H-pyrazolo[l,2-a][l,2,4]triazol-8-ium-6-ylsulfanyl)-6-(l-hydroxy ethyl)-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene-2-carboxylate (Compound of formula-IV) was added at pH 7-8 and cooled to 5-10° C. The mixture was hydrogenated using palladium on carbon as catalyst. The catalyst was filtered and the filtrate was treated with activated carbon and filtered. The filtrate was extracted with dichloromethane and the layers separated. The aqueous layer was degassed. To the aqueous layer, acetone was added to crystallize Biapenem at 20-25° C. The product was filtered, washed with aqueous acetone and dried under vacuum to get Biapenem (Non-Sterile).
Reference example -2: Crystallization of Biapenem
Example -1 was repeated without the addition of methanol.Yield: 84 g Purity by HPLC: 99.5%Reconstitution time : > 90 secondsThe reconstitution time is calculated by the time taken to dissolve 300 mg of Biapenem in 100 ml of water or saline solution.Table-1: Comparative Data:The comparative data provided in the table-1 clearly indicates that the addition of co-solvent during crystallization provides Biapenem with reconstitution time less than 25 seconds.
Benzonatate
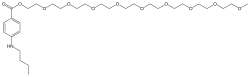


Benzonatate
- Molecular FormulaC30H53NO11
- Average mass603.742 Da
104-31-4[RN]2,5,8,11,14,17,20,23,26-Nonaoxaoctacosan-28-yl 4-(butylamino)benzoateбензонататبنزوناتات苯佐那酯ベンゾナテート;KM 652,5,8,11,14,17,20,23,26-nonaoxaoctacosan-28-yl 4-(butylamino)benzoate2-[2-[2-[2-[2-[2-[2-[2-(2-methoxyethoxy)ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethyl 4-(butylamino)benzoate
Benzonatate bulk and Benzonatate capsules 100mg, cdsco india 2021, 15.07.2021
For the treatment of refractory coughCAS Registry Number: 104-31-4CAS Name: 4-(Butylamino)benzoic acid 3,6,9,12,15,18,21,24,27-nonaoxaoctacos-1-yl esterAdditional Names: nonaethyleneglycol monomethyl ether p-n-butylaminobenzoate; p-butylaminobenzoic acid w-O-methylnonaethyleneglycol ester; benzononatineTrademarks: Exangit; Tessalon (Forest)Molecular Formula: C30H53NO11Molecular Weight: 603.74Percent Composition: C 59.68%, H 8.85%, N 2.32%, O 29.15%Literature References: Prepn: Matter, US2714608 (1955 to Ciba).Properties: Colorless to faintly yellow oil. Soluble in most organic solvents except aliphatic hydrocarbons.Therap-Cat: Antitussive.Keywords: Antitussive.
Synthesis Reference
Matter, M.; U.S. Patent 2,714,608; August 2, 1955; assigned to Ciba Pharmaceutical Products, Inc.
Synthesis Path
Substances Referenced in Synthesis Path
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 94-32-6 | C13H19NO2 | ethyl 4-butylaminobenzoate | Benzoic acid, 4-(butylamino)-, ethyl ester |
| 6048-68-6 | C19H40O10 | nonaethylene glycol monomethyl ether | 2,5,8,11,14,17,20,23,26-Nonaoxaoctacosan-28-ol |
Benzonatate, sold under the brand name Tessalon among others, is a medication used to try to help with the symptoms of cough and hiccups.[1][2] It is taken by mouth.[1] Use is not recommended in those under the age of ten.[3] Effects generally begin within 20 minutes and last up to eight hours.[1][4]
Side effects include sleepiness, dizziness, headache, upset stomach, skin rash, hallucinations, and allergic reactions.[1] Excessive doses may cause seizures, irregular heartbeat, and death.[3] Chewing or sucking on the capsule can lead to laryngospasm, bronchospasm, and circulatory collapse.[1] It is unclear if use in pregnancy or breastfeeding is safe.[5] It works by numbing stretch receptors in the lungs and suppressing the cough reflex in the brain.[1]
Benzonatate was approved for medical use in the United States in 1958.[1] It is available as a generic medication.[3] It is not available in many countries.[6] In 2018, it was the 113th most commonly prescribed medication in the United States, with more than 6 million prescriptions.[7][8]
Medical uses

100mg generic benzonatate capsules
Cough
Benzonatate is a prescription non-opioid alternative for the symptomatic relief of cough.[1][3] It has been shown to improve cough associated with a variety of respiratory conditions including asthma, bronchitis, pneumonia, tuberculosis, pneumothorax, opiate-resistant cough in lung cancer, and emphysema.[1][9][10]
Benzonatate also reduces the consistency and volume of sputum production associated with cough in those with chronic obstructive pulmonary disorder (COPD).[9]
Compared to codeine, benzonatate has been shown to be more effective in reducing the frequency of induced cough in experiments.[1]
Benzonatate does not treat the underlying cause of the cough.[11]
Hiccups
Benzonatate has been shown to have use in the suppression of hiccups.[2]
Intubation
Benzonatate acts as a local anesthetic and the liquid inside the capsule can be applied in the mouth to numb the oropharynx for awake intubation.[1] However, there can be life-threatening adverse effects when the medication is absorbed by the oral mucosa, including choking, hypersensitivity reactions, and circulatory collapse.[1]
Contraindications
Hypersensitivity to benzonatate or any related compounds is a contraindication to its administration.[4]
Side effects
Benzonatate is generally well-tolerated[vague][specify] if the liquid-capsule is swallowed intact.[1] Potential adverse effects to benzonatate include:
- Constipation, dizziness, fatigue, stuffy nose, nausea, headache are frequently reported.[12]
- Sedation, a feeling of numbness in the chest, sensation of burning in the eyes, a vague “chilly” sensation, itchiness, and rashes are also possible.[1][4]
- Ingestion of a small handful of capsules has caused seizures, cardiac arrhythmia, and death in adults.[13]
Hypersensitivity reactions
Benzonatate is structurally related to anesthetic medications of the para-aminobenzoic acid (PABA) class which includes procaine and tetracaine.[4][13] Procaine and tetracaine, previously used heavily in the fields of dentistry and anesthesiology, have fallen out of favor due to allergies associated with their metabolites.[13] Similarly, severe hypersensitivity reactions to benzonatate have been reported and include symptoms of laryngospasm, bronchospasm, and cardiovascular collapse.[4][14] These reactions are possibly associated with chewing, sucking, or crushing the capsule in the mouth.[4][13]
Improper use
Benzonatate should be swallowed whole.[4] Crushing or sucking on the liquid-filled capsule, or “softgel,” will cause release of benzonatate from the capsule and can produce a temporary local anesthesia of the oral mucosa.[4] Rapid development of numbness of the tongue and choking can occur.[4][13] In severe cases, excessive absorption can lead to laryngospasm, bronchospasm, seizures, and circulatory collapse.[4][13] This may be due to a hypersensitivity reaction to benzonatate or a systemic local anesthetic toxicity, both of which have similar symptoms.[13] There is a potential for these adverse effects to occur at a therapeutic dose, that is, a single capsule, if chewed or sucked on in the mouth.[13]
Psychiatric effects
Isolated cases of bizarre behavior, mental confusion, and visual hallucinations have been reported during concurrent use with other prescribed medications.[4] Central nervous system effects associated with other para-aminobenozic acid (PABA) derivative local anesthetics, for example procaine or tetracaine, could occur with benzonatate and should be considered.[1]
Children
Safety and efficacy in children below the age of 10 have not been established.[4] Accidental ingestion resulting in death has been reported in children below the age of 10.[4] Benzonatate may be attractive to children due to its appearance, a round-shaped liquid-filled gelatin capsule, which looks like candy.[14][15] Chewing or sucking of a single capsule can cause death of a small child.[4][15] Signs and symptoms can occur rapidly after ingestion (within 15–20 minutes) and include restlessness, tremors, convulsions, coma, and cardiac arrest.[15] Death has been reported within one hour of ingestion.[12][15]
Pregnancy and breast feeding
In the U.S., benzonatate is classified by the U.S. Food and Drug Administration (FDA) as pregnancy category C.[5] It is not known if benzonatate can cause fetal harm to a pregnant woman or if it can affect reproduction capacity.[4][5] Animal reproductive studies have not yet been conducted with benzonatate to evaluate its teratogenicity.[4] Benzonatate should only be given to a pregnant woman if it is clearly needed.[4][5]
It is not known whether benzonatate is excreted in human milk.[4][5] It is recommended to exercise caution when benzonatate is given to a nursing woman.[4][5]
Overdose
Benzonatate is chemically similar to other local anesthetics such as tetracaine and procaine, and shares their pharmacology and toxicology.[13]
Benzonatate overdose is characterized by symptoms of restlessness, tremors, seizures, abnormal heart rhythms (cardiac arrhythmia), cerebral edema, absent breathing (apnea), fast heart beat (tachycardia), and in severe cases, coma and death.[1][4][16][11] Symptoms develop rapidly, typically within 1 hour of ingestion.[4][11] Treatment focuses on removal of gastric contents and on managing symptoms of sedation, convulsions, apnea, and cardiac arrhythmia.[4]
Despite a long history of safe and appropriate usage, the safety margin of benzonatate is reportedly narrow.[13] Toxicity above the therapeutic dose is relatively low and ingestion of a small handful of pills can cause symptoms of overdose.[13][11] Children are at an increased risk for toxicity, which have occurred with administration of only one or two capsules.[15][16][11]
Due to increasing usage of benzonatate and rapid onset of symptoms, there are accumulating cases of benzonatate overdose deaths, especially in children.[11]
Pharmacology
Benzonatate is chemically similar to other local anesthetics such as tetracaine and procaine, and shares their pharmacology.[13]
Mechanism of action
Similar to other local anesthetics, benzonatate is a potent voltage-gated sodium channel inhibitor.[13] After absorption and circulation to the respiratory tract, benzonatate acts as a local anesthetic, decreasing the sensitivity of vagal afferent fibers and stretch receptors in the bronchi, alveoli, and pleura in the lower airway and lung.[1][2] This dampens their activity and reduces the cough reflex.[1][4] Benzonatate also has central antitussive activity on the cough center in central nervous system at the level of the medulla.[1][9] However, there is minimal inhibition of the respiratory center at a therapeutic dosage.[4]
Pharmacokinetics
The antitussive effect of benzonatate begins within 15 to 20 minutes after oral administration and typically lasts between 3 and 8 hours.[4][9]
Benzonatate is hydrolyzed by plasma butyrylcholinesterase (BChE) to the metabolite 4-(butylamino)benzoic acid (BABA) as well as polyethylene glycol monomethyl esters.[13] Like many other local anesthetic esters, the hydrolysis of the parent compound is rapid.[13] There are concerns that those with pseudocholinesterase deficiencies may have an increased sensitivity to benzonatate as this hydrolysis is impaired, leading to increased levels of circulating medication.[13]
Chemical structure
Benzonatate is a butylamine, structurally related to other polyglycol ester local anesthetics such as procaine and tetracaine.[13] The molecular weight of benzonatate is 603.7 g/mol.[4] However, the reference standard for benzonatate is a mixture of n-ethoxy compounds, differing in the abundance of 7-9 repeating units, with an average molecular weight of 612.23 g/mol.[13] There is also evidence that the compound is not uniform between manufacturers.[13]
Society and culture
Benzonatate was first made available in the U.S. in 1958 as a prescription medication for the treatment of cough in individuals over the age of 10.[15][16] There are a variety of prescription opioid-based cough relievers, such as hydrocodone and codeine, but have unwanted side effects and potential of abuse and diversion.[13] However, benzonatate is currently the only prescription non-opioid antitussive and its usage has been rapidly increasing.[13][11] The exact reasons of this increase are unclear.[11]
Economics
In the United States between 2004 and 2009, prescriptions increased 50% from 3.1 million to 4.7 million, the market share of benzonatate among antitussives increased from 6.3% to 13%, and the estimated number of children under the age of 10 years receiving benzonatate increased from 10,000 to 19,000.[13][11] Throughout this same period, greater than 90% of prescriptions were given to those 18 or older.[11] The majority of prescriptions were given by general, family, internal, and osteopathic physicians with pediatricians account for about 3% of prescribed benzonatate.[11]
In 2018, it was the 113th most commonly prescribed medication in the United States, with more than 6 million prescriptions.[7][8]
Brand names
Tessalon is a brand name version of benzonatate manufactured by Pfizer, Inc.[13][11] It is available as perles or capsules.[17] Zonatuss was a brand name manufactured by Atley Pharmaceuticals, Inc. and Vertical Pharmaceuticals, Inc.[18][19]
References
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s “Benzonatate Monograph for Professionals”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 23 March 2019.
- ^ Jump up to:a b c Becker, DE (2010). “Nausea, vomiting, and hiccups: a review of mechanisms and treatment”. Anesthesia Progress. 57 (4): 150–6, quiz 157. doi:10.2344/0003-3006-57.4.150. PMC 3006663. PMID 21174569.
- ^ Jump up to:a b c d “Drugs for cough”. The Medical Letter on Drugs and Therapeutics. 60 (1562): 206–208. 17 December 2018. PMID 30625123.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u v w x y z “Tessalon – benzonatate capsule”. DailyMed. 20 November 2019. Retrieved 21 April 2020.
- ^ Jump up to:a b c d e f “Benzonatate Use During Pregnancy”. Drugs.com. 10 October 2019. Retrieved 20 February 2020.
- ^ Walsh, T. Declan; Caraceni, Augusto T.; Fainsinger, Robin; Foley, Kathleen M.; Glare, Paul; Goh, Cynthia; Lloyd-Williams, Mari; Olarte, Juan Nunez; Radbruch, Lukas (2008). Palliative Medicine E-Book. Elsevier Health Sciences. p. 751. ISBN 9781437721942.
- ^ Jump up to:a b “The Top 300 of 2021”. ClinCalc. Retrieved 18 February2021.
- ^ Jump up to:a b “Benzonatate – Drug Usage Statistics”. ClinCalc. Retrieved 18 February 2021.
- ^ Jump up to:a b c d Homsi, J.; Walsh, D.; Nelson, K. A. (November 2001). “Important drugs for cough in advanced cancer”. Supportive Care in Cancer. 9 (8): 565–574. doi:10.1007/s005200100252. ISSN 0941-4355. PMID 11762966. S2CID 25881426.
- ^ Estfan, Bassam; LeGrand, Susan (November 2004). “Management of cough in advanced cancer”. The Journal of Supportive Oncology. 2 (6): 523–527. ISSN 1544-6794. PMID 16302303.
- ^ Jump up to:a b c d e f g h i j k l McLawhorn, Melinda W.; Goulding, Margie R.; Gill, Rajdeep K.; Michele, Theresa M. (January 2013). “Analysis of benzonatate overdoses among adults and children from 1969-2010 by the United States Food and Drug Administration”. Pharmacotherapy. 33 (1): 38–43. doi:10.1002/phar.1153. ISSN 1875-9114. PMID 23307543. S2CID 35165660.
- ^ Jump up to:a b “Benzonatate (Professional Patient Advice)”. Drugs.com. 4 March 2020. Retrieved 21 April 2020.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u v w Bishop-Freeman SC, Shonsey EM, Friederich LW, Beuhler MC, Winecker RE (June 2017). “Benzonatate Toxicity: Nothing to Cough At”. J Anal Toxicol. 41 (5): 461–463. doi:10.1093/jat/bkx021. PMID 28334901.
- ^ Jump up to:a b “Drugs for Cough”. The Medical Letter on Drugs and Therapeutics. 60 (1562): 206–208. 17 December 2018. PMID 30625123.
- ^ Jump up to:a b c d e f “FDA Drug Safety Communication: Death resulting from overdose after accidental ingestion of Tessalon (benzonatate) by children under 10 years of age”. U.S. Food and Drug Administration (FDA). 28 June 2019. Retrieved 22 April 2020.
- ^ Jump up to:a b c “In brief: benzonatate warning”. The Medical Letter on Drugs and Therapeutics. 53 (1357): 9. 7 February 2011. ISSN 1523-2859. PMID 21304443.
- ^ “Tessalon- benzonatate capsule”. DailyMed. 20 November 2019. Retrieved 25 April 2020.
- ^ “Zonatuss (Benzonatate Capsules USP, 150 mg)”. DailyMed. 2 June 2010. Retrieved 20 August 2020.
- ^ “Zonatuss (Benzonatate Capsules USP, 150 mg)”. DailyMed. 31 October 2016. Retrieved 20 August 2020.
External links
- “Benzonatate”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Tessalon, Zonatuss, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682640 |
| License data | US DailyMed: Benzonatate |
| Routes of administration | By mouth |
| ATC code | R05DB01 (WHO) |
| Legal status | |
| Legal status | US: ℞-only |
| Pharmacokinetic data | |
| Elimination half-life | 3-8 hours |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 32760-16-0 |
| PubChem CID | 7699 |
| IUPHAR/BPS | 7611 |
| DrugBank | DB00868 |
| ChemSpider | 7413 |
| UNII | 5P4DHS6ENR |
| KEGG | D00242 |
| ChEBI | CHEBI:3032 |
| ChEMBL | ChEMBL1374379 |
| CompTox Dashboard (EPA) | DTXSID9022655 |
| ECHA InfoCard | 100.002.904 |
| Chemical and physical data | |
| Formula | C30H53NO11 |
| Molar mass | 603.750 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
///////////Benzonatate, refractory cough , INDIA 2021, APPROVALS 2021, бензонатат , بنزوناتات , 苯佐那酯 , KM 65 , ベンゾナテート, ANTITUSSIVE, IND 2021
CCCCNC1=CC=C(C=C1)C(=O)OCCOCCOCCOCCOCCOCCOCCOCCOCCOC

NEW DRUG APPROVALS
ONE TIME
$10.00
ZyCoV-D

ZyCoV-D
CAS 2541524-47-2
DNA vaccine construct encoding a spike protein antigen of SARS-CoV-2 virus (Zydus-Cadila)
UPDATE. APPROVED IN INDIA AUG 2021
http://ctri.nic.in/Clinicaltrials/showallp.php?mid1=51254&EncHid=&userName=ZyCoV-D
bioRxiv (2021), 1-26.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7423510/
| ZyCoV-D | (CTRI/2020/07/026352, 2020, CTRI/2020/07/026352, 2020; Myupchar, 2020) | ZYDUS CADILA |
ZyCoV-D is a genetically engineered DNA plasmid based vaccine encoding for the membrane proteins of the virus. The clinical trials to study the immunogenicity, and safety of the vaccine, will administer three doses at an interval of 28 days in 1048 individuals.
Phase 1/2: CTRI/2020/07/026352
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | DNA |
| Clinical data | |
| Routes of administration |
Intradermal |
| ATC code | None |
| Identifiers | |
| DrugBank | DB15892 |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| SARS-CoV-2 (virus)COVID-19 (disease) |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showImpact |
| COVID-19 portal |
ZyCoV-D is a DNA plasmid based COVID-19 vaccine being developed by Cadila Healthcare with support from the Biotechnology Industry Research Assistance Council.
The ZYCOV-D vaccine candidate was developed by Cadila Healthcare Ltd. based in India1. The vaccine was developed using a DNA vaccine platform with a non-replicating and non-integrating plasmid carrying the gene of interest3. Once the plasmid DNA is introduced into host cells and the viral protein is translated, it elicits a strong immune response, stimulating the humoral and cellular components of the immune system3. The DNA vaccine platform offers minimal biosafety requirements, more improved vaccine stability, and lower cold chain requirements3. Phase I clinical trials of this vaccine candidate were completed in July 2020, with the company reporting successful dosing and tolerance1,2. As of August, 2020 the candidate is in Phase II clinical trials1.
NEW DRUG APPROVALS
ONE TIME
$10.00
Clinical research
Phase I and II trials
In February 2020, Cadila Healthcare decided to develop a DNA plasmid based COVID-19 vaccine at their Vaccine Technology Centre (VTC) in Ahmedabad.[1] The vaccine candidate was able to pass the pre-clinical trials on animal models successfully. A report of the study was made available via bioRxiv.[2] Thereafter, human trials for Phase I and II were approved by the regulator.[3]
The Phase II trials of the vaccine candidate were conducted in over 1,000 volunteers as part of the adaptive Phase I/II multi-centric, dose escalation, randomised, double-blind placebo controlled method.[4][5]
Phase III trials
In November 2020, the company announced it would test the vaccine candidate on 30,000 patients in Phase III trials.[6] The vaccine would be given out in three doses at five sites across four cities of India.[7] In January 2021, the Drugs Controller General of India (DCGI) granted permission to conduct the Phase III clinical trials for 28,216 Indian participants.[8][9]
In April 2021, the company reported that they expected to have initial data for the Phase III trials by May 2021.[10]
Production
On 23 April 2021, production of the ZyCoV-D vaccine was started, with a yearly capacity of 240 million doses. It is expected to get emergency use authorization in May or June.[11]
References
- ^ “Zydus Cadila launches a fast tracked programme to develop vaccine for the novel coronavirus, 2019-nCoV (COVID-19)”(PDF). http://www.zyduscadila.com. Cadila Healthcare.
- ^ Dey A, Rajanathan C, Chandra H, Pericherla HP, Kumar S, Choonia HS, et al. (26 January 2021). “Immunogenic Potential of DNA Vaccine candidate, ZyCoV-D against SARS-CoV-2 in Animal Models”. bioRxiv: 2021.01.26.428240. doi:10.1101/2021.01.26.428240. S2CID 231777527.
- ^ “A prospective, randomized, adaptive, phase I/II clinical study to evaluate the safety and immunogenicity of Novel Corona Virus −2019-nCov vaccine candidate of M/s Cadila Healthcare Limited by intradermal route in healthy subjects”. ctri.nic.in. Clinical Trials Registry India. 15 December 2020. CTRI/2020/07/026352. Archived from the original on 22 November 2020.
- ^ “Zydus Cadila’s ZyCov-D vaccine found to be ‘safe and immunogenic'”. @businessline. The Hindu. 24 December 2020.
- ^ Rawat K, Kumari P, Saha L (February 2021). “COVID-19 vaccine: A recent update in pipeline vaccines, their design and development strategies”. European Journal of Pharmacology. 892: 173751. doi:10.1016/j.ejphar.2020.173751. PMC 7685956. PMID 33245898.
- ^ Thacker T (7 November 2020). “Zydus Cadila to test ZyCoV-D on 30,000 patients in Phase-3 trials”. The Economic Times.
- ^ “Covid 19 vaccine in India: Zydus Cadila begins enrolment for Phase 3 trial of ZyCoV-D in 4 cities”. The Financial Express. 22 January 2021.
- ^ “DBT-BIRAC supported indigenously developed DNA Vaccine Candidate by Zydus Cadila, approved for Phase III clinical trials”. pib.gov.in. Press Information Bureau. 3 January 2021.
- ^ “Novel Corona Virus-2019-nCov vaccine by intradermal route in healthy subjects”. ctri.nic.in. Clinical Trials Registry – India. Retrieved 10 April 2021.
- ^ Das, Sohini (22 April 2021). “Cadila Healthcare testing two-shot regimen for ZyCoV-D, data likely by May”. Business Standard India.
- ^ Writer, Staff (24 April 2021). “Cadila Healthcare starts production of Covid vaccine candidate”. mint. Retrieved 27 April 2021.
Zydus Cadila Covid vaccine close to getting approved in India, says MD Sharvil Patel
In an exclusive interview with India Today TV, Managing Director of Zydus Cadila Dr Sharvil Patel said the company’s Covid vaccine candidate ZyCoV-D against the Covid-19 infection is very close to getting approved in India. They are likely to apply for emergency use authorisation this month.
Ahmedabad-based pharmaceutical company Zydus Cadila is likely to submit the application for emergency use authorisation of its Covid-19 vaccine candidate ‘ZyCoV-D’ in India this month. The company is confident that the vaccine will be approved in May itself. The company plants to produce one crore doses of its ‘painless’ Covid-19 vaccine per month.
If approved, ZyCoV-D will be the fourth vaccine to be used in India’s Covid-19 vaccination drive. Made in India, the company plans to ramp up the vaccine’s production to 3-4 crore doses per month and is already in talks with two other manufacturing companies for the same
Although the vaccine should ideally be stored between 2 and 8 degrees Celsius, it remains stable even at room temperature conditions at 25 degrees Celsius. It is easy to administer, the developers said, and will be administered via intradermal injection.
If approved for emergency use, ZyCoV-D could help India fill the vacuum of vaccine doses currently being experienced in the country’s immunisation drive.
Earlier in April, Zydus Cadila announced that its drug Virafin had received restricted emergency use approval from the Drug Controller General of India for the treatment of mild cases of Covid-19.
In an exclusive interview with India Today TV, Sharvil Patel sheds details on all aspects of the Covid-19 vaccine ZyCoV-D.
When asked the status of Covid vaccine candidate ZyCoV-D and when exactly Zydus Cadila would apply for emergency use authorisation in India, Dr Sharvil Patel said the vaccine was getting very close to getting approved in the country.
“I am very happy to say that India’s first indigenously developed DNA vaccine candidate against Covid, which is our ZyCoV-D, is getting very close to approval,” he said.
“We have almost completed all our recruitment for the clinical trials. We have, by far, recruited the largest number of patients for a Covid vaccine trial in India. The number of volunteers who have been vaccinated as a part of the trial is 28,000,” Sharvil Patel said.
Sharvil Patel also said that his company has also included children in the 12-17 age group for the vaccine trials.
He said, “The recruitment holds very important milestones in terms of cohorts because not only have we included the elderly and those with co-morbidities, but also children in the age group of 12 to 17 years.”
Sharvil Patel said as soon as the efficacy data is obtained, Sydus Cadila will file for emergency use authorisation. As soon as the approval is granted, Zydus Cadila will start production of Covid-19 vaccines from July, he said.
“We hope to see our efficacy data in the middle of May. As soon as we see strong efficacy which correlates to the vaccine’s strong immunogenicity in Phase 2, we will file for emergency use authorization. We hope to produce a good quantity of the vaccine from July onwards to make sure it is available to the people. That is the need of the hour right now,” Sharvil Patel said.
He said by May the company will be in a position to talk to the regulators about the restricted use of the Covid-19 vaccine. “The regulatory process is a rolling one. I believe the regulators look at the data in a short period of time,” Sharvil Patel said.
“We have submitted a lot of data already so that it will aid the regulators once we provide them with the efficacy results. We are, hence, expecting to get the approval in May itself,” Sharvil Patel said.
///////////ZyCoV-D, COVID 19, CORONA VIRUS, VACCINE, INDIA 2021, APPROVALS 2021, SARS-CoV-2
2-Deoxy-D-glucose

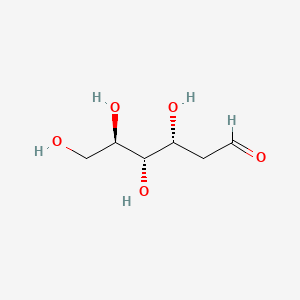
2-Deoxy-D-glucose
- Molecular FormulaC6H12O5
- Average mass164.156 Da
2-Deoxy-D-glucose
(4R,5S,6R)-6-(Hydroxymethyl)tetrahydro-2H-pyran-2,4,5-triol(4R,5S,6R)-6-(Hydroxyméthyl)tétrahydro-2H-pyran-2,4,5-triol
154-17-6[RN]
- 2-Deoxy-D-arabino-hexose
- 2 DG
- 2-Deoxy-D-glucose
- 2-Deoxy-D-mannose
- 2-Deoxyglucose
- 2-Desoxy-D-glucose
- Ba 2758
- D-Glucose, 2-deoxy-
- NSC 15193
2-Deoxy-D-arabino-hexopyranose2-deoxy-D-glucopyranose2-deoxyglucose
2-DGD-arabino-2-DesoxyhexoseD-arabino-Hexopyranose, 2-deoxy- [(4R,5S,6R)-6-(Hydroxymethyl)oxane-2,4,5-triol2-deoxyglucopyranose2-deoxymannopyranose2-dGlc
D-arabino-2-Deoxyhexoseglucitol, 2,5-anhydro-
2-Deoxy-D-glucose
CAS Registry Number: 154-17-6
CAS Name: 2-Deoxy-D-arabino-hexose
Additional Names: D-arabino-2-desoxyhexose; 2-deoxyglucose; 2-DGManufacturers’ Codes: Ba-2758Molecular Formula: C6H12O5Molecular Weight: 164.16Percent Composition: C 43.90%, H 7.37%, O 48.73%Literature References: Antimetabolite of glucose, q.v., with antiviral activity.
Synthesis: M. Bergmann et al.,Ber.55, 158 (1922); 56, 1052 (1923); J. C. Sowden, H. O. L. Fischer, J. Am. Chem. Soc.69, 1048 (1947); H. R. Bolliger, Helv. Chim. Acta34, 989 (1954); H. R. Bolliger, M. D. Schmid, ibid. 1597, 1671; H. R. Bolliger, “2-Deoxy-D-arabino-hexose (2-Deoxy-D-glucose)” in Methods in Carbohydrate Chemistryvol. I, R. L. Whistler, M. L. Wolfrom, Eds. (Academic Press, New York, 1962) pp 186-189.
Inhibition of influenza virus multiplication: E. D. Kilbourne, Nature183, 271 (1959).
Effects on herpes simplex virus: R. J. Courtney et al.,Virology52, 447 (1973). Mechanism of action studies: M. R. Steiner et al.,Biochem. Biophys. Res. Commun.61, 745 (1974); E. K. Ray et al.,Virology58, 118 (1978). Use in human genital herpes infections: H. A. Blough, R. L. Giuntoli, J. Am. Med. Assoc.241, 2798 (1979); L. Corey, K. K. Holmes, ibid.243, 29 (1980). Effect vs respiratory syncytial viral infections in calves: S. B. Mohanty et al.,Am. J. Vet. Res.42, 336 (1981).
Properties: Cryst from acetone or butanone, mp 142-144°. [a]D17.5 +38.3° (35 min) ®+45.9° (c = 0.52 in water); +22.8° (24 hrs) ® +80.8° (c = 0.57 in pyridine).
Melting point: mp 142-144°
Optical Rotation: [a]D17.5 +38.3° (35 min) ®+45.9° (c = 0.52 in water); +22.8° (24 hrs) ® +80.8° (c = 0.57 in pyridine) Derivative Type: a-Form
Properties: Cryst from isopropanol, mp 134-136°. [a]D26 +156° ® +103° (c = 0.9 in pyridine).Melting point: mp 134-136°Optical Rotation: [a]D26 +156° ® +103° (c = 0.9 in pyridine) Use: Exptlly as an antiviral agent.


Source Temperature: 210 °C Sample Temperature: 150 °C Direct, 75 eV
14.0 2.2
15.0 11.5
17.0 3.9
18.0 19.4
19.0 13.7
26.0 2.5
27.0 12.1
28.0 21.9
29.0 31.2
30.0 4.6
31.0 41.3
32.0 12.4
39.0 5.9
40.0 2.1
41.0 10.9
42.0 12.4
43.0 46.3
44.0 31.5
45.0 34.3
46.0 2.8
47.0 4.1
53.0 1.5
54.0 2.0
55.0 14.4
56.0 35.3
57.0 55.7
58.0 11.4
59.0 2.0
60.0 100.0
61.0 31.1
62.0 2.3
68.0 4.6
69.0 12.2
70.0 3.0
71.0 34.9
72.0 7.0
73.0 25.3
74.0 46.6
75.0 5.1
81.0 1.5
82.0 2.4
83.0 1.3
84.0 1.3
85.0 18.1
86.0 55.3
87.0 4.6
89.0 1.2
91.0 1.5
97.0 3.6
98.0 2.9
99.0 1.7
100.0 3.5
102.0 1.1
103.0 19.8
104.0 1.4
111.0 1.6
115.0 25.2
116.0 3.0
117.0 2.1
120.0 3.3
128.0 1.0
129.0 2.5
133.0 1.8
147.0 2.2
1H NMR DMSO D6


1H NMR D20

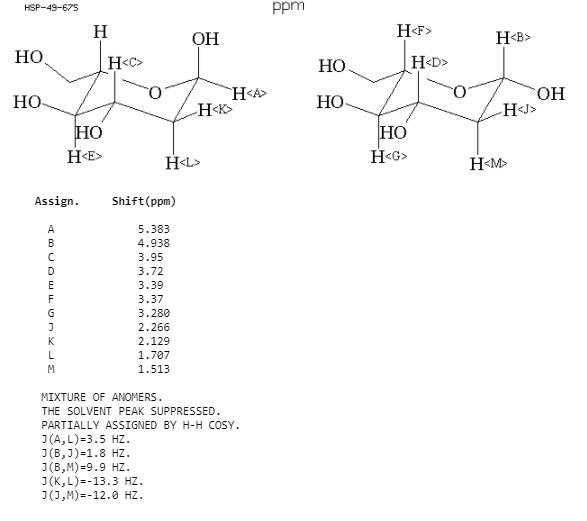
IR NUJOL MULL

IR KBR

PAPERCollection of Czechoslovak Chemical Communications (1955), 20, 42-5. http://cccc.uochb.cas.cz/20/1/0042/
Preparation of 2-deoxy-D-glucose
By: Stanek, Jaroslav; Schwarz, Vladimir
Triacetyl-D-glucal (I) adds (BzO)2IAg and (BzO)2BrAg, to give 1-benzoyl-3,4,6-triacetyl-2-deoxy-2-iodo-α-D-glucopyranose (II) and 1-benzoyl-3,4,6-triacetyl-2-deoxy-2-bromo-α-D-glucopyranose (III), resp. Both halogen derivs. give 2-deoxy-D-glucose (IV) by reduction. Adding a C6H6 soln. of 16.7 g. iodine into a suspension of 33.6 g. dry BzOAg in 200 ml. C6H6, treating the mixt. with a soln. of 20 g. I in 200 ml. C6H6, heating the mixt. 7 hrs. on the steam bath, removing the AgI, evapg. the solvent, and crystg. the residue from EtOH gave 20.8 g. (54.7%) II, m. 129-30°, [α]21D 21.7°. Analogous procedure with 13.4 g. BzOAg, 4.6 g. Br, and 8 g. I gave 3.9 g. (33%) III, m. 139-40°, [α]17D 33.5°. The same compd. (3 g.), m. 140°, [α]18D 33.6°, was obtained by adding 3.2 g. Br to a soln. of 5.44 g. I in 50 ml. CCl4, by refluxing the mixt. 2 hrs. with 6 g. BzOAg, filtering off the AgBr, and evapg. the solvent. Reducing 8 g. II or an equiv. III in 150 ml. MeOH with 60 g. Zn activated by 1 hr. immersion in a soln. of 60 g. CuSO4 in 1500 ml. H2O, removing Zn after 8 hrs., evapg. the MeOH, and sapong. the residue with Ba(OH)2 yielded 0.42 g. (20%) IV, m. 145°, [α]18D 46.1°.
Wavlen: 589.3 nm; Temp: 18 °C, +46.1 ° ORD
PATENT
https://patents.google.com/patent/WO2004058786A1/enThe present invention relates to a process for the synthesis of 2-deoxy-D-glucose. Background of the invention 2-deoxy-D-glucose is useful in control of respiratory infections and for application as an antiviral agent for treatment of human genital herpes.Prior art for preparation of 2-deoxy-D-glucose while operable, tend to be expensive and time consuming. Reference may be made to Bergmann, M., Schotte, H., Lechinsky, W., Ber, 55, 158 (1922) and Bergmann, M., Schotte, H., Lechinsky, W., Ber 56, 1052 (1923) which disclose the preparation of 2-deoxy-D-glucose in low yield by mineral acid catalyzed addition of water to D-glucal. Another method of producing 2-deoxy-D-glucose is from diethyldithioacetal derivative of D-glucose (Bolliger, H.R. Schmid, M.D., Helv. Chim. Ada 34, 989 (1951); Bolliger, H.R., Schmid, M.D., Helv. Chim. A a 34, 1597 (1951); Bolliger, H.R. Schmid, M.D., Helv. Chim. Ada 34, 1671 (1951) and from D-arabhiose by reaction with nitromethane followed by acetylation, reduction and hydrolysis (Sowden, J.C, Fisher, H.O.L., J. Am. Chem., 69, 1048 (1947). However these methods result in the formation of 2- deoxy-D-glucose in low yield and of inferior purity due to the formation of several byproducts and involve use of toxic reagents such as ethanethiol and nitromethane. As a result purification of 2-deoxy-D-glucose has to be done by recrystallisation which is tedious, time consuming and difficult.Accordingly it is important to develop a process for synthesis of 2-deoxy-D-glucose which obviates the drawbacks as detailed above and results in good yield and good purity. Objects of the inventionThe main object of the present invention is to provide a process for the synthesis of 2- deoxy-D-glucose resulting in good yield and with good purity.Another object of the invention is to provide an economical process for the synthesis of 2-deoxy-D-glucose. Summary of the inventionA process that would produce 2-deoxy-D-glucose economically and with desired purity, is a welcome contribution to the art. This invention fulfills this need efficiently.Accordingly the present invention relates to a process for the synthesis of 2-deoxy- D-glucose comprising haloalkoxylation of R-D-Glucal wherein R is selected from H and 3, 4, 6-tri-O-benzyl, to obtain alkyl 2-deoxy-2-halo-R-α/ -D-gluco/mannopyranoside, converting alkyl 2-deoxy-2-halo-R-α/β-D-gluco/mannopyranoside by reduction to alkyl 2- deoxy-α/β-D-glucopyranoside, hydrolysing alkyl 2-deoxy-α/β-D-glucopyranoside to 2- deoxy-D-glucose.In one embodiment of the invention, the alkyl 2-deoxy-α/β-D-glucopyranoside is obtained by (a) haloalkoxylating 3,4,6,-tri-O-benzyl-D-glucal to alkyl 2-deoxy-2-halo-3,4,6-tri-O- benzyl-α/β-D-gluco-/mannopyranoside; (b) subjecting alkyl 2-deoxy-2-halo-3,4,6-tri-O-benzyl-α/β-D-gluco/mannopyranoside to reductive dehalogenation and debenzylation to obtain alkyl 2-deoxy -α/β-D- glucopyranoside. In another embodiment of the invention, in step (a) haloalkoxylation of 3,4,6-tri-O- benzyl-D-glucal is carried out by reaction with a haloalkoxylating agent selected from a N- halosuccinimide and a N-haloacetamide, and alcohol.The reaction scheme for the reactions involved in the process of the invention are also given below:

in R’=CH3I R=C6H5CH2 H R=C6H5CH2, X=Br, R’=CH3 IV R=H V R=CH3, C2HSJ C6H5CH3, iPr, X=Br

Such overall synthesis may be depicted as follows where R=H, CH3, C2H5, (CH3)2CH, C6H5CH ; RX-CH3; X-CL, Br.Example 1 To a solution of 3,4,6-tri-O-benzyl-D-glucal (39 g, 0.09 mol) in dichloromethane (20ml) and methanol (100 ml) was added N-bromosuccinimide (18.7 g, 0.09 mil) during 10 min. at room temperature and stirred for 4 h. After completion of the reaction solvent was distilled off. The resultant residue extracted into carbon tetrachloride (2×100 ml) and organic phase concentrated to obtain methyl 2-bromo 2-deoxy-3,4,6-tri-O-benzyl-α/β-D-gluco- /mannopyranoside as a syrup. Quantity obtained 50 g. 1H NMR (200 MHz, CDC13) 3.40-4.00 (m, 7H, H-2,5,6,6′ and OCH3) 4.30-5.10 (m, 9H, H-1,3,4 and 3xPhCH2O), 7.10-7.60 (m, 15H, Ar-H). A solution of methyl 2-bromo-2-deoxy-3,4,6-tri-O-benzyl-α/β-D-gluco- /mannopyranoside (50 g) in methanol (300) was charged into one litre autoclave along with Raney nickel (10 ml) Et3N (135 ml) and subjected to hydrogenation at 120 psi pressure at 50°C for 8 h. After completion of the reaction the catalyst was filtered off and the residue washed with methanol (25 ml). The filtrate was concentrate to obtain methyl 2-deoxy-3,4,6- tri-O-benzyl-α/β-D-glucopyranoside as a syrup (37.9 g, 89%). 1H NMR (200 MHz, CDC13): δ 1.50-2.40 (m,2H,H-2,2′)5 3.32, 3.51 (2s, 3H, OCH3) 3.55-4.00 (m, 5H, H-3,4,5,6,6′), 4.30-5.00 (m, 7H, 3xPhCH2, H-l), 7.10-7.45 (m, 15H, Ar-H). The syrup of methyl 2-deoxy-3,4,6- tri-O-benzyl-α/β-D-glucopyranoside (37.9g) was dissolved in methanol (200 ml). 1 g of 5%Pd/C was added and hydrogenated at 150 psi pressure at room temperature. After 5 hours catalyst was filtered off and solvent evaporated. Quantity of the methyl 2-deoxy-α/β-D- glucopyranoside obtained 10.5 g (70%). [ ]D + 25.7° (c 1.0, MeOH), 1H NMR (200 MHz, D2O); δ 1.45-2.40 (m, 2H, H-2,2′) 3.20-4.80, (m 9H, H- 1,3,4,5,6,6′ – OCH3).Example 2 To a solution of D-glucal (64.6g, 0.44 mol) in methanol (325 ml) at 10°C was addedN-bromosuccinimide (78.7 g, 0.44 mol) during 40 min. maintaining the temperature between 10-15°C during the addition. The reaction mixture was stirred at room temperature. After 5 hours solvent was evaporated to obtain a residue which was refluxed in ethyl acetate (100 ml). Ethyl acetate layer was discarded to leave a residue of methyl 2-bromo-2-deoxy-α/β-D- gluco/mannopyranoside (105 g) as a syrup. [α]D + 36° (c 1.0, MeOH). 1H NMR (200 MHz, D2O): δ 3.47, 3.67 (2s, 3H, OCH3), 3.70-4.05 (m, 6h, H-23,4,5,6,6′), 4.48-5.13 (2s, 1H, H-l). The syrupy methyl 2-bromo-2-deoxy-α/β-D-gluco-/mannopyranoside was dissolved in methanol (400 ml), a slurry of 80 g Raney nickel (a 50% slurry in methanol), Et3N (30 ml) and hydrogenated in a Parr apparatus at 120 psi. After 8-9 hours, the reaction mixture was filtered through a Celite filter pad and washed with MeOH. The washings and filtrate were combined and triturated with hexane to separate and remove by filtration insoluble triethylamine hydrobromide and traces of succinimide. The filtrate was concentrated to a residue. The isolated yield of methyl 2-deoxy-α/β-D-glucopyranoside was 89%. Ethyl 2-bromo-2deoxy-α/β-D-gluco-/mannopyranoside: When solvent was ethanol instead of methanol the compound obtained was ethyl 2- bromo-2-deoxy-α/β-D-gluco-/mannopyranoside. 1HNMR (200 MHz, D2O): δ 1.10-1.32 (m, 3H, CH3), 2.80 (s, 4H, -CO(CH2)2CO-NH-), 3.40-4.10 (m, 8H, H-2,3,4,5,6,6′, CH2), 4.40, 5.20 (2s 1H, H-l α/β).Isopropyl 2-bromo-2-deoxy- /β-D-gluco-/mannopyranoside: When isopropanol instead of methanol was used as a solvent the compound obtained was isopropyl 2-bromo-2-deoxy-α/β-D-gluco/mannopyranoside. 1H NMR (200 MHz, D2O): δ 1.10-1.30 (m, 6H, 2xCH3) 2.80 (s, 4H, -CO(CH2)2CO-NH-), 3.60-4.60 (m 8H,H- 2,3,4,5,6,6′, CH2) 4.40, 5.30 (2s, 1H, H-l, α/β).Example 3 A mixture of D-glucal (64.6 g), methanol (400 ml), N-bromosuccinimide (79 g) were stirred at 15 C for 6 h. The reaction mixture was hydrogenated in a Parr apparatus in presence of 60 g of Raney nickel catalyst (a 50% slurry in methanol) and triethylamine (62 ml). After 8-9 h, the reaction mixture was filtered on a Celite filter pad. The Celite pad was washed with methanol. The washings and filtrate were combined, concentrated to a thick heavy syrup, dissolve in chloroform (500 ml), pyridine (400 ml) and acetic anhydride (251 ml) was added while stirring, maintaining the temperature between 5-10°C. After 12 hours, the reaction mixture was diluted with CHC13 (500 ml) transferred to a separating funnel and organic phase was washed with water. The organic phase was separated, dried (Na2SO4) and concentrated to obtain methyl 2-deoxy-3,4,6-tri-O-acetyl-2 deoxy-α/β-D-glucopyranoside as a syrup (163.43 g, 87%). [α]D + 65.0° (c 1.0, CHC13) 1H NMR (200 MHz, CDC13): δ 1.55-1.90 (m, 2H, H-2,2′), 2.01, 2.04,2.11, 2.15, (4s, 9H, 3xOCOCH3), 2.18,3.40 (2s, 3H, OCH3), 3.45-50 (m, 3H, H-5, 6,6′) 4.80-5.40 (m, 3H,H-1,3,4). The syrup was dissolved in methanol (600 ml) IN NaOMe in methanol (25ml) was added and left at room temperature. After 6-10 h, dry CO2 gas was passed into the reaction mixture, solvent was evaporated to obtain a syrupy residue. The residue was once again extracted into dry methanol and concentrated to obtain methyl 2-deoxy-α/β-D-glucopyranoside as syrup. Quantity obtained 81 g (92%).Example 4 A 500 ml round bottom flask equipped with magnetic stir bar was charged with a solution of D-glucal (32.3 g) in methanol (175 ml), cooled to 15°C, N-bromosucci-t imide (NBS) (39.4 g) was added and stirred for 6 hours at 15°C. The reaction mixture was concentrated to half the volume, cooled to 0°C and separated succinimide was removed by filtration. To the filtrate was added a slurry of 30 g Raney nickel (a 50% slurry in methanol) Et3N (32 ml) and hydrogenated in a Parr apparatus at 120 psi. After 7-8 hours, the reaction mixture was filtered through a Celite filter pad, and washed with MeOH. The washings and filtrate were combined and triturate with hexane to separate and remove by filtration insoluble triethylamine hydrobromide and succinimide. The filtrate was concentrated to a residue, dissolved in methanol and triturated with hexane to remove most of the triethylamine hydrobromide and succinimide. The filtrate was concentrated to obtain methyl 2-deoxy-α/β- D-glucopyranoside (85%).Example 5 To a stirred solution of methyl 3,4,6-tri-O-acetyl-2-deoxy-α/β-D-glucopyranoside (47 g) (from example 3) in acetic acid (40 ml) and acetic anhydride (110 ml) was added concentrated sulphuric acid (0.94 ml) at 0°. The reaction mixture was brought to room temperature and stirred. After 2 hours the reaction mixture was diluted with water (50 ml) and extracted into CH2C12 (3×150 ml). The organic phase was separated, washed with saturated NaHCO3 solution, H2O dried over Na2SO and concentrated to obtain 2-deoxy- 1,3,4,6-tetra-O-acetyl-α/β-D-glucopyranoside as a crystalline compound, mp. 115-118°C. Quantity obtained 44.5 g (86%). [α]D + 21.5° (c 1.0, CHC13). 1H NMR (200 MHz, CDC13): δ 1.50-2.45 (m, 14H, H-2,2′, 4xOCOCH3), 3.85-5.40, (m, 5H, H-3,4,5,6,6′), 5.75-6.20 (m, 1H, H-l,α/ β). To a heterogeneous mixture of l,3,4,6-tetra-O-acetyl-2-deoxy-α/β-D- glucopyranoside (10 g) in water (100 ml) was added acetyl chloride (10 ml) and heated to 80°C. After 6 hours the reaction mixture was cooled to room temperature, neutralised with saturated aq. Ba(OH)2, concentrated to half the volume and filtered on a Celite pad. Filtrate was concentrated on a rotary evaporator and dried over anhydrous P2O5 to obtain a residue which was dissolved in hot isopropyl alcohol and filtered on a pad of Celite to obtain a clear filtrate. The filtrate was concentrated to a residue, dissolved in hot isopropyl alcohol (50 ml), acetone (75 ml) and seeded with a few crystals of 2-deoxy-D-glucose. After 15-18 hours at 5°C crystalline title product was filtered. Quantity obtained 3.21 g (64.9%) m.p. 148-149°C.Example 6 A heterogeneous mixture of l,3,4,6-tetra-O-acetyl-2-deoxy-α/β-D-glucopyranoside (9 g) (from example 5), water (30 ml) and 11% aq. H2SO (0.3 ml) was stirred at 85°C for 7 h to obtain a homogenous solution. The reaction mixture was cooled, neutralised with aq. Ba(OH)2 solution and filtered. The filtrate obtained was concentrated to half the volume and solids separated were filtered. To the filtrate was added activated carbon (1 g) and filtered. The filtrate was concentrated on a rotary evaporator and dried over P2O5 to obtain 2-deoxy- D-glucose that was crystallized from methyl alcohol (27 ml) and acetone (54 ml). Quantity obtained 2.4 g. mp. 146-149°C.Example 7A heterogeneous mixture of l,3,4,6-tetra-O-acetyl-2-deoxy-α/β-D-glucopyranoside(25g) (from example 5), H2O (250 ml), toluene (250 ml) and glacial acetic acid (1.25 ml) was heated to reflux for 10-12 hours, while it was connected to a Dean- Stark azeotropic distillation apparatus. An azeotropic mixture of acetic acid, toluene was collected to remove acetic acid and every one hour fresh toluene (50 ml) was introduced. After completion of the reaction, toluene was removed by distillation from the reaction mixture to obtain a residue that was dissolved in methanol, treated with charcoal and filtered. The filtrate was separated, concentrated to a residue and crystallized from isopropyl alcohol and acetone to obtain 2- deoxy-D-glucose (7.33 g, 59%). mp. 148-151°C.Example 8 A heterogeneous mixture of l,3,4,5-tetra-O-acetyl-2-deoxy-α/β-D-glucopyranoside (lOg) (from example 5), H2O (200 ml) cone. HC1 (0.3 ml) and glacial acetic acid (0.5 ml) was heated to 85°C. After 6 hours the reaction mixture was cooled to room temperature, neutralized with aq. Ba(OH)2 and filtered on a pad of Celite. Filtrate was separated, treated with charcoal and filtered. The filtrate was concentrated to a residue and crystallized from MeOH, acetone to obtain the product. Quantity obtained 2.75 g. mp. 147-148°C.Example 9 A heterogeneous mixture of l,3,4,5-tetra-O-acetyl-2-deoxy-α/β-D-glucopyranoside(lOg) (from example 3) water (100 ml) and cone. HCI (0.5ml) was heated to 80°C. After 2-5 hours the reaction mixture was cooled to room temperature, neutralized with aq. Ba(OH)2 and filtered on a pad of Celite. The filtrate was concentrated to a residue, dissolved in ethanol, treated with charcoal and filtered. The filtrate was concentrated to a solid residue andcrystallized from methanol-acetone to obtain the title product. Quantity obtained 3.15g mp. 148-151°C.Example 10A solution of methyl 2-deoxy-α/β-D-glucopyranoside (30g) (from example 2) water(15 ml) and cone. HCI (1.5 ml) was heated to 80-85°C. After 3-5 hours the reaction mixture was cooled to room temperature, neutralized with aq. Ba(OH)2 and filtered to remove insoluble salts. The filtrate was concentrated to a residue, crystallized from MeOH, acetone and hexane to obtain 2-deoxy-D-glucose (11.77 g) mp. 149-151°C.Example 11A solution of methyl 2-deoxy-α/β-D-glucopyranoside (30g) (from example 2) water (195 ml) and cone. H2SO (5.9 ml) was heated to 80°C. After 2-3 hours the reaction mixture was cooled, neutralized with aq. Ba(OH)2 and filtered. The filtrate was separated, treated with charcoal and filtrate. The Filtrate was concentrated to a residue and crystallized from isopropyl alcohol to obtain the title product. Quantity obtained 5.2 g. mp. 152-154°C.Example 12 A mixture of methyl 2-deoxy-α/β-D-glucopyranoside (24g) (from example 2) water(125 ml) and IR 120 H+ resin (7.5 ml) was heated to 90-95°C for 2h. The reaction mixture was cooled to room temperature, filtered and the resin was washed with water (20 ml). The filtrate was concentrated to residue and crystallized from ethanol to obtain 2-deoxy-D- glucose (8.8 g), mp. 150-152°C. The main advantages of the present invention are:-1). It does not involve the use of toxic mercaptans like ethane thiol. 2). This process does not involve reaction of D-glucal with mineral acid, thereby avoiding the formation of Ferrier by-products.
2-Deoxy-d-glucose is a glucose molecule which has the 2-hydroxyl group replaced by hydrogen, so that it cannot undergo further glycolysis. As such; it acts to competitively inhibit the production of glucose-6-phosphate from glucose at the phosphoglucoisomerase level (step 2 of glycolysis).[2] In most cells, glucose hexokinase phosphorylates 2-deoxyglucose, trapping the product 2-deoxyglucose-6-phosphate intracellularly (with exception of liver and kidney)[; thus, labelled forms of 2-deoxyglucose serve as a good marker for tissue glucose uptake and hexokinase activity. Many cancers have elevated glucose uptake and hexokinase levels. 2-Deoxyglucose labeled with tritium or carbon-14 has been a popular ligand for laboratory research in animal models, where distribution is assessed by tissue-slicing followed by autoradiography, sometimes in tandem with either conventional or electron microscopy.
2-DG is uptaken by the glucose transporters of the cell. Therefore, cells with higher glucose uptake, for example tumor cells, have also a higher uptake of 2-DG. Since 2-DG hampers cell growth, its use as a tumor therapeutic has been suggested, and in fact, 2-DG is in clinical trials. [3] A recent clinical trial showed 2-DG can be tolerated at a dose of 63 mg/kg/day, however the observed cardiac side-effects (prolongation of the Q-T interval) at this dose and the fact that a majority of patients’ (66%) cancer progressed casts doubt on the feasibility of this reagent for further clinical use.[4] However, it is not completely clear how 2-DG inhibits cell growth. The fact that glycolysis is inhibited by 2-DG, seems not to be sufficient to explain why 2-DG treated cells stop growing.[5] Because of its structural similarity to mannose, 2DG has the potential to inhibit N-glycosylation in mammalian cells and other systems, and as such induces ER stress and the Unfolded Protein Response (UPR) pathway.[6][7][8]
Clinicians have noted that 2-DG is metabolised in the pentose phosphate pathway in red blood cells at least, although the significance of this for other cell types and for cancer treatment in general is unclear.
Work on the ketogenic diet as a treatment for epilepsy have investigated the role of glycolysis in the disease. 2-Deoxyglucose has been proposed by Garriga-Canut et al. as a mimic for the ketogenic diet, and shows great promise as a new anti-epileptic drug.[9][10] The authors suggest that 2-DG works, in part, by increasing the expression of Brain-derived neurotrophic factor (BDNF), Nerve growth factor (NGF), Arc (protein) (ARC), and Basic fibroblast growth factor (FGF2).[11] Such uses are complicated by the fact that 2-deoxyglucose does have some toxicity.
A study found that by combining the sugar 2-deoxy-D-glucose (2-DG) with fenofibrate, a compound that has been safely used in humans for more than 40 years to lower cholesterol and triglycerides, an entire tumor could effectively be targeted without the use of toxic chemotherapy.[12][13]
2-DG has been used as a targeted optical imaging agent for fluorescent in vivo imaging.[14][15] In clinical medical imaging (PET scanning), fluorodeoxyglucose is used, where one of the 2-hydrogens of 2-deoxy-D-glucose is replaced with the positron-emitting isotope fluorine-18, which emits paired gamma rays, allowing distribution of the tracer to be imaged by external gamma camera(s). This is increasingly done in tandem with a CT function which is part of the same PET/CT machine, to allow better localization of small-volume tissue glucose-uptake differences.
Resistance to 2-DG has been reported in HeLa cells [16] and in yeast;[17][8] in the latter, it involves the detoxification of a metabolite derived from 2-DG (2DG-6-phosphate) by a phosphatase. Despite the existence of such a phosphatase in human (named HDHD1A) However it is unclear whether it contributes to the resistance of human cells to 2DG or affects FDG-based imaging.
SYN
Indian Pat. Appl., 2004DE02075,

SYN
CN 106496288,
STARTING MATERIAL CAS 69515-91-9
C14 H20 O9, 332.30
D-arabino-Hexopyranose, 2-deoxy-, 1,3,4,6-tetraacetate
SYN
Bioorganic & Medicinal Chemistry Letters, 22(10), 3540-3543; 2012
https://www.sciencedirect.com/science/article/abs/pii/S0960894X12004258
PATENT
https://patents.google.com/patent/US6933382B2/en2-deoxy-D-glucose is useful in control of respiratory infections and for application as an antiviral agent for treatment of human genital herpes.Prior art for preparation of 2-deoxy-D-glucose while operable, tend to be expensive and time consuming. Reference may be made to Bergmann M., Schotte, H., Lechinsky, W., Ber, 55, 158 (1922) and Bergmann, M., Schotte, H., Lechinsky, W., Ber 56, 1052 (1923) which disclose the preparation of 2-deoxy-D-glucose in low yield by mineral acid catalyzed addition of water to D-glucal. Another method of producing 2-deoxy-D-glucose is from diethyldithioacetal derivative of D-glucose (Bolliger, H. R. Schmid, M. D., Helv. Chim. Acta 34, 989 (1951); Bolliger, H. R., Schmid, M. D., Helv, Chim. Acta 34, 1597 (1951); Bolliger, H. R Schmid, M. D., Helv. Chim. Acta 34, 1671 (1951) and from D-arabinose by reaction with nitromethane followed by acetylation, reduction and hydrolysis (Sowden, J. C., Fisher, H. O. L., J. Am. Chem., 69, 1048 (1947). However these methods result in the formation of 2-deoxy-D-glucose in low yield and of inferior purity due to the formation of several by-products and involve use of toxic reagents such as ethanethiol and nitromethane. As a result purification of 2-deoxy-D-glucose has to be done by recrystallisation which is tedious, time consuming and difficult.

EXAMPLE 1To a solution of 3,4,6-tri-O-benzyl-D-glucal (39 g, 0.09 mmol) in dichloromethane (20 ml) and methanol (100 ml) was added N-bromosuccinimide (18.7 g, 0.09 mil) during 10 min. at room temperature and stirred for 4 h. After completion of the reaction solvent was distilled off. The resultant residue extracted into carbon tetrachloride (2×100 ml) and organic phase concentrated to obtain methyl 2-bromo 2-deoxy-3,4,6-tri-O-benzyl-α/β-D-gluco-/mannopyranoside as a syrup. Quantity obtained 50 g. 1H NMR (200 MHz, CDCl3) 3.40-4.00 (m, 7H, H-2,5,6,6′ and OCH3) 4.30-5.10 (m, 9H, H-1,3,4 and 3×PhCH2O), 7.10-7.60 (m 15H, Ar—H). A solution of methyl 2-bromo-2-deoxy-3,4,6-tri-O-benzyl/α/β-D-gluco-/mannopyranoside (50 g) in methanol (300) was charged into one liter autoclave along with Raney nickel (10 ml) Et3N (135 ml) and subjected to hydrogenation at 120 psi pressure at 50° C. for 8 h. After completion of the reaction the catalyst was filtered off and the residue washed with methanol (25 ml). The filtrate was concentrate to obtain methyl 2-deoxy-3,4,6-tri-O-benzyl-α/β-D-glucopyranoside as a syrup (37.9 g, 89%). 1H NMR (200 MHz CDCl3): δ 1.50-2.40 (m,2H,H-2,2′), 3.32, 3.51 (2s, 3H, OCH3) 3.55-4.00 (m, 5, H-3,4,5,6,6′) 4.30-5.00 (M 7H, 3×PhCH2, H-1), 7.10-7.45 (m, 15H, Ar—H). The syrup of methyl 2-deoxy-3,4, 6-tri-O-benzyl-α/β-D-glucopyranoside (37.9 g) was dissolved in methanol (200 ml). 1 g of 5% Pd/C was added and hydrogenated at 150 psi pressure at room temperature. After 5 hours catalyst was filtered off and solvent evaporated. Quantity of the methyl 2-deoxy-α/β-D-glucopyranoside obtained 10.5 g (70%). [α]D+25.7° (c 1.0, MeOH), 1H NMR (200 MHz, D2O); δ 1.45-2.40 (m, 2H, H-2,2′) 3.20-4.80, (m 9H, H-1,3,4,5,6,6′—OCH3).EXAMPLE 2To a solution of D-glucal (64.6 g, 0.44 mmol) in methanol (325 ml) at 10° C. was added N-bromosuccinimide (78.7 g, 0.44 mol) during 40 min. maintaining the temperature between 10-15° C. during the addition. The reaction mixture was stirred at room temperature. After 5 hours solvent was evaporated to obtain a residue which was refluxed in ethyl acetate (100 ml). Ethyl acetate layer was discarded to leave a residue of methyl 2-bromo-2-deoxy-α/β-D-gluco/mannopyranoside (105 g) as a syrup. [α]D+36° (c 1.0, MeOH). 1H NMR (200 MHz, D2O): δ 3.47, 3.67 (2s, 3H, OCH3), 3.70-4.05 (m, 6h, H-2,3,4,5,6,6′), 4.48-5.13 (28, 1H, 1H, H-1). The syrupy methyl 2-bromo-2-deoxy-α/β-D-gluco-/mannopyranoside was dissolved in methanol (400 ml), a slurry of 80 g Raney nickel (a 50% slurry in methanol), Et3N (30 ml) and hydrogenated in a Parr apparatus at 120 psi. After 8-9 hours, the reaction mixture was filtered through a Celite filter pad and washed with MeOH. The washings and filtrate were combined and triturated with hexane to separate and remove by filtration insoluble triethylamine hydrobromide and traces of succinimide. The filtrate was concentrated to a residue. The isolated yield of methyl 2-deoxy-α/β-D-glucopyranoside was 89%.Ethyl 2-bromo-2deoxy-α/β-D-gluco-/mannopyranoside:When solvent was ethanol instead of methanol the compound obtained was ethyl 2-bromo-2deoxy-α/β-D-gluco-/mannopyranoside. 1H NMR (200 MHz, D2O): δ 1.10-1.32 (m, 3H, CH3), 2.80 (s, 4H, —CO(CH2)2CO—NH—), 3.40-4.10 (m, 8H, H-2,3,4,5,6,6′, CH2), 4.40, 5.20 (2s 1H, H-1, α/β).Isopropyl 2-bromo-2-deoxy-α/β-D-gluco-/mannopyranoside:When isopropanol instead of methanol was used as a solvent the compound obtained was isopropyl 2-bromo-2-deoxy-α/β-D-gluco/mannopyranoside, 1H NMR (200 MHz, D2O): δ 1.10-1.30 (m, 6H, 2×CH3) 2.80 (s, 4H, —CO(CH2)2CO—NH—), 3.60-4.60 (m 8H,H-2,3,4,5,6,6′, CH2) 4.40, 5,30 (2s, 1H, H-1, α/β.EXAMPLE 3A mixture of D-glucal (64.6 g), methanol (400 ml), N-bromosuccinimide (79 g) were stirred at 15° C. for 6 h. The reaction mixture was hydrogenated in a Parr apparatus in presence of 60 g of Raney nickel catalyst (a 50% slurry in methanol) and triethylamine (62 ml). After 8-9 h, the reaction mixture was filtered on a Celite filter pad. The Celite pad was washed with methanol. The washings and filtrate were combined, concentrated to a thick heavy syrup, dissolve in chloroform (500 ml), pyridine (400 ml) and acetic anhydride (251 ml) was added while stirring, maintaining the temperature between 5-10° C. After 12 hours, the reaction mixture was diluted with CHCl3 (500 ml) transferred to a separating funnel and organic phase was washed with water. The organic phase was separated, dried (Na2SO4) and concentrated to obtain methyl 2-deoxy-3,4,6-tri-O-acetyl-2 deoxy-α/β-D-glucopyranoside as a syrup (163.43 g, 87%). [α]D+65.0° (c 1.0, CHCl3) 1H NMR (200 MHz, CDCl3): δ 1.55-1.90 (m, 2H, H-22′), 2.01, 2.04, 2.11, 2.15, (4s, 9H, 3×OCOCH3), 2.18, 3.40 (2s, 3H, OCH3), 3.45-50 (m, 3H, H-5, 6,6′) 4.80-5.40 (m, 3H,H-1,3,4). The syrup was dissolved in methanol (600 ml) 1N NaOMe in methanol (25 ml) was added and left at room temperature. After 6-10 h, dry CO2 gas was passed into the reaction mixture, solvent was evaporated to obtain a syrupy residue. The residue was once again extracted into dry methanol and concentrated to obtain methyl 2-deoxy-α/β-D-glucopyranoside as syrup. Quantity obtained 81 g (92%).EXAMPLE 4A 500 ml round bottom flask equipped with magnetic stir bar was charged with a solution of D-glucal (323 g) in methanol (175 ml), cooled to 15° C., N-bromosuccinimide (NIBS) (39.4 g) was added and stirred or 6 hours at 15° C., The reaction mixture was concentrated to half the volume, cooled to 0° C. and separated succinimide, was removed by filtration. To the filtrate was added a slurry of 30 g Raney nickel (a 50% slurry in Methanol) Et3N (32 ml) and hydrogenated in a Parr apparatus at 120 psi. After 7-8 hours, the reaction mixture was filtered through a Celite filter pad, and washed with MeOH. The washings and filtrate were combined and triturate with hexane to separate and remove by filtration insoluble triethylamine hydrobromide and succinimide. The filtrate was concentrated to a residue, dissolved in methanol and triturated with hexane to remove most of the triethylamine hydrobromide and succinimide. The filtrate was concentrated to obtain methyl 2-deoxy-α/β-D-glucopyranoside (85%).EXAMPLE 5To a stirred solution of methyl 3,4,6-tri-O-acetyl-2-deoxy-α/β-D-glucopyranoside (47 g) (from example 3) in acetic acid (40 ml) and acetic anhydride (110 ml) was added concentrated sulphuric acid (0.94 ml) at 0°. The reaction mixture was brought to room temperature and stirred. After 2 hours the reaction mixture was diluted with water (50 ml) and extracted into CH2Cl2 (3×150 ml). The organic phase was separated, washed with saturated NaHCO3 solution H2O dried over Na2SO4 and concentrated to obtain 2-deoxy-1,3,4,6-tetra-O-acetyl-α/β-D-glucopyranoside as a crystalline compound. mp. 115-118° C. Quantity obtained 44.5 g (86%). [α]D+21.5° (c 1.0, CHCl3). 1H NMR (200 MHz, CDCl3): δ 1.50-2.45 (m, 14H, H-2,2′, 4×OCOCH3), 3.85-5.40, (m, 5H, H-3,4,5,6,6′), 5.75-6.20 (m, 1H, H-1, α/β). To a heterogeneous mixture of 1,3,4,6-tetra-O-acetyl-2-deoxy-α/β-D-glucopyranoside (10 g) in water (100 ml) was added acetyl chloride (10 ml) and heated to 80° C. After 6 hours the reaction mixture was cooled to room temperature, neutralised with saturated aq. Ba(OH)2, concentrated to half the volume and filtered on a Celite pad, Filtrate was concentrated on a rotary evaporator and dried over anhydrous P2O5 to obtain a residue which was dissolved in hot isopropyl alcohol and filtered on a pad of Celite to obtain a clear filtrate. The filtrate was concentrated to a residue, dissolved in hot isopropyl alcohol (50 ml), acetone (75 ml) and seeded with a few crystals of 2-deoxy-D-glucose. After 15-18 hours at 5° C. crystalline title product was filtered. Quantity obtained 3.21 g (64.9%) m.p. 148-149° C.EXAMPLE 6A heterogeneous mixture of 1,3,4,6-tetra-O-acetyl-2-deoxy-α/β-D-glucopyranoside (9 g) (from example 5), water (30 ml) and 11% aq. H2SO4 (0.3 ml) was stirred at 85° C. for 7 h to obtain a homogenous solution. The reaction mixture was cooled, neutralised with aq. Ba(OH)2 solution and filtered. The filtrate obtained was concentrated to half the volume and solids separated were filtered. To the filtrate was added activated carbon (1 g) and filtered. The filtrate was concentrated on a rotary evaporator and dried over P2O5 to obtain 2-deoxy-D-glucose that was crystallized from methyl alcohol (27 ml) and acetone (54 ml). Quantity obtained 2.4 g. mp. 146-149° C.,EXAMPLE 7A heterogeneous mixture of 1,3,4,tetra-O-acetyl-2-deoxy-α/β-D-glucopyranoside (25 g) (from example 5), H2O (250 ml), toluene (250 ml) and glacial acetic acid (1.25 ml) was heated to reflux for 10-12 hours, while it was connected to a Dean-Stark azeotropic distillation apparatus. An azeotropic mixture of acetic acid, toluene was collected to remove acetic acid and every one hour fresh toluene (50 ml) was introduced. After completion of the reaction, toluene was removed by distillation from the reaction mixture to obtain a residue that was dissolved in methanol, treated with charcoal and filtered. Be filtrate was separated, concentrated to a residue and crystallized from isopropyl alcohol and acetone to obtain 2-deoxy-D-glucose (7.33 g, 59%). mp. 148-151° C.EXAMPLE 8A heterogeneous mixture of 1,3,4,5-tetra-O-acetyl-2-deoxy-α/β-D-glucopyranoside (10 g) (tom example 5), H2O (200 ml) conc. HCl (0.3 ml) and glacial acetic acid (0.5 ml) was heated to 85° C. After 6 hours the reaction mixture was cooled to room temperature, neutralized with aq. Ba(OH)2 and filtered on a pad of Celite. Filtrate was separated, treated with charcoal and filtered. The filtrate was concentrated to a residue and crystallized from MeOH, acetone to obtain the product. Quantity obtained 275 g. mp. 147-148° C.EXAMPLE 9A heterogeneous mixture of 1,3,4,5-tetra-O-acetyl-2-deoxy-α/β-D-glucopyranoside (10 g) (from example 3) water (100 ml) and conc. HCl (0.5 ml) was heated to 80° C. After 2-5 hours the reaction mixture was cooled to room temperature, neutralized with aq. Ba(OH)2 and filtered on a pad of Celite. The filtrate was concentrated to a residue, dissolved in ethanol, treated with charcoal and filtered. The filtrate was concentrated to a solid residue and crystallized from methanol-acetone to obtain the title product. Quantity obtained 3.15 g mp. 148-151° C.,EXAMPLE 10A solution of methyl 2-deoxy-α/β-D-glucopyranoside (30 g) (from example 2) water (15 ml) and conc. HCl (1.5 ml) was heated to 80-85° C. After 3-5 hours the reaction mixture was cooled to room temperature, neutralize with aq. Ba(OH)2 and filtered to remove insoluble salts. The filtrate was concentrated to a residue, crystallized from MeOH, acetone and hexane to obtain 2-deoxy-D-glucose (11.77 g) mp. 149-151° C.EXAMPLE 11A solution of methyl 2-deoxy-α/β-D-glucopyranoside (30 g) (form example 2) water (195 ml) and conc. H2SO4 (5.9 ml) was heated to 80° C. After 2-3 hours the reaction mixture was cooled, neutralized with aq. Ba(OH)2 and filtered. The filtrate was separated, treated with charcoal and filtrate. The Filtrate was concentrated to a residue and crystallized from isopropyl alcohol to obtain the title product. Quantity obtained 5.2 g. mp. 152-154° C.EXAMPLE 12A mixture of methyl 2-deoxy-α/β-D-glucopyranoside (24 g) (from example 2) water (125 ml) and IR 120H+resin (7.5 ml) was heated to 90-95° C. for 2 h. The reaction mixture was cooled to room temperature, filtered and the resin was washed with water (20 ml). The filtrate was concentrated to residue and crystallized from ethanol to obtain 2-deoxy-D-glucose (8.8 g), mp. 150-152° C.CLIP
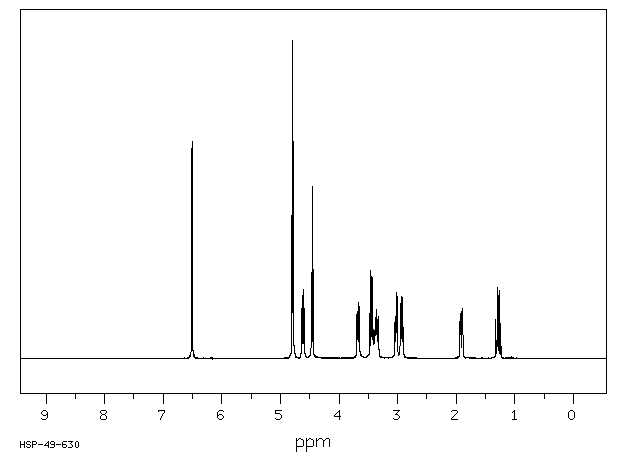
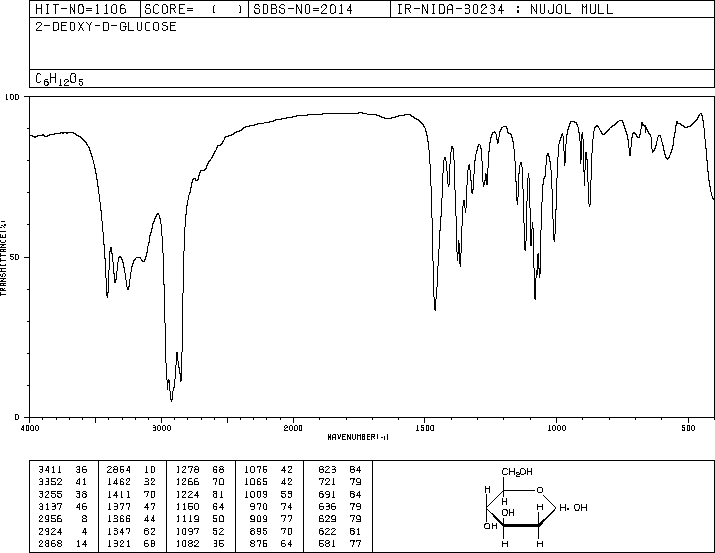
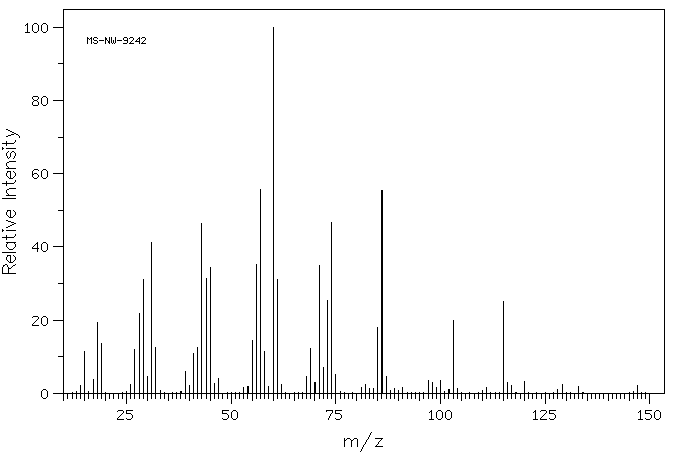
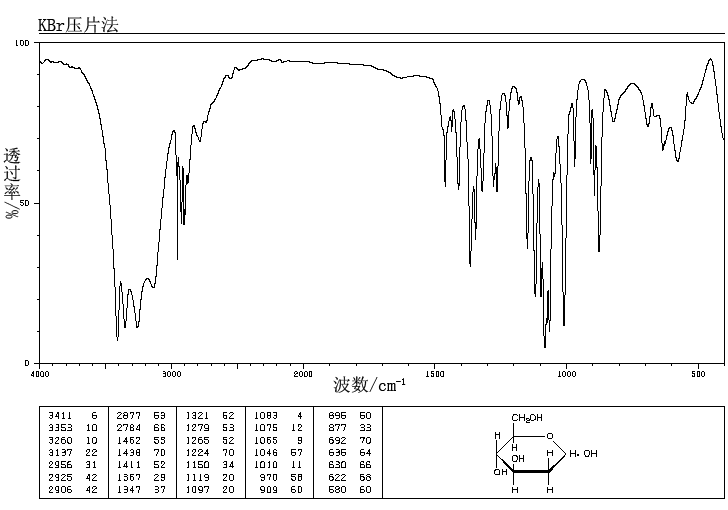
References
- ^ Merck Index, 11th Edition, 2886.
- ^ Wick, AN; Drury, DR; Nakada, HI; Wolfe, JB (1957). “Localization of the primary metabolic block produced by 2-deoxyglucose”(PDF). J Biol Chem. 224 (2): 963–969. doi:10.1016/S0021-9258(18)64988-9. PMID 13405925.
- ^ Pelicano, H; Martin, DS; Xu, RH; Huang, P (2006). “Glycolysis inhibition for anticancer treatment”. Oncogene. 25 (34): 4633–4646. doi:10.1038/sj.onc.1209597. PMID 16892078.
- ^ Raez, LE; Papadopoulos, K; Ricart, AD; Chiorean, EG; Dipaola, RS; Stein, MN; Rocha Lima, CM; Schlesselman, JJ; Tolba, K; Langmuir, VK; Kroll, S; Jung, DT; Kurtoglu, M; Rosenblatt, J; Lampidis, TJ (2013). “A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors”. Cancer Chemother. Pharmacol. 71 (2): 523–30. doi:10.1007/s00280-012-2045-1. PMID 23228990. S2CID 2990078.
- ^ Ralser, M.; Wamelink, M. M.; Struys, E. A.; Joppich, C.; Krobitsch, S.; Jakobs, C.; Lehrach, H. (2008). “A catabolic block does not sufficiently explain how 2-deoxy-D-glucose inhibits cell growth”. Proceedings of the National Academy of Sciences. 105 (46): 17807–17811. Bibcode:2008PNAS..10517807R. doi:10.1073/pnas.0803090105. PMC 2584745. PMID 19004802.
- ^ Kurtoglu, M.; Gao, N.; Shang, J.; Maher, J. C.; Lehrman, M. A.; Wangpaichitr, M.; Savaraj, N.; Lane, A. N.; Lampidis, T. J. (2007-11-07). “Under normoxia, 2-deoxy-D-glucose elicits cell death in select tumor types not by inhibition of glycolysis but by interfering with N-linked glycosylation”. Molecular Cancer Therapeutics. 6 (11): 3049–3058. doi:10.1158/1535-7163.mct-07-0310. ISSN 1535-7163. PMID 18025288.
- ^ Xi, Haibin; Kurtoglu, Metin; Liu, Huaping; Wangpaichitr, Medhi; You, Min; Liu, Xiongfei; Savaraj, Niramol; Lampidis, Theodore J. (2010-07-01). “2-Deoxy-d-glucose activates autophagy via endoplasmic reticulum stress rather than ATP depletion”. Cancer Chemotherapy and Pharmacology. 67 (4): 899–910. doi:10.1007/s00280-010-1391-0. ISSN 0344-5704. PMC 3093301. PMID 20593179.
- ^ Jump up to:a b Defenouillère, Quentin; Verraes, Agathe; Laussel, Clotilde; Friedrich, Anne; Schacherer, Joseph; Léon, Sébastien (2019-09-03). “The induction of HAD-like phosphatases by multiple signaling pathways confers resistance to the metabolic inhibitor 2-deoxyglucose”. Science Signaling. 12 (597): eaaw8000. doi:10.1126/scisignal.aaw8000. ISSN 1945-0877. PMID 31481524. S2CID 201829818.
- ^ Garriga-Canut, Mireia; Schoenike, Barry; Qazi, Romena; Bergendahl, Karen; Daley, Timothy J.; Pfender, Rebecca M.; Morrison, John F.; Ockuly, Jeffrey; Stafstrom, Carl; Sutula, Thomas; Roopra, Avtar (2006). “2-Deoxy-D-glucose reduces epilepsy progression by NRSF-CTBP–dependent metabolic regulation of chromatin structure”. Nature Neuroscience. 9 (11): 1382–1387. doi:10.1038/nn1791. PMID 17041593. S2CID 10175791.
- ^ Garriga-Canut, M.; Schoenike, B.; Qazi, R.; Bergendahl, K.; Daley, T. J.; Pfender, R. M.; Morrison, J. F.; Ockuly, J.; Stafstrom, C.; Sutula, T.; Roopra, A. (2006). “2-Deoxy-D-glucose reduces epilepsy progression by NRSF-CtBP–dependent metabolic regulation of chromatin structure”. Nature Neuroscience. 9 (11): 1382–1387. doi:10.1038/nn1791. PMID 17041593. S2CID 10175791.
- ^ Jia Yao, Shuhua Chen, Zisu Mao, Enrique Cadenas, Roberta Diaz Brinton “2-Deoxy-D-Glucose Treatment Induces Ketogenesis, Sustains Mitochondrial Function, and Reduces Pathology in Female Mouse Model of Alzheimer’s Disease”, PLOS ONE
- ^ Researchers develop novel, non-toxic approach to treating variety of cancers. ScienceDaily
- ^ Liu, Huaping; Kurtoglu, Metin; León-Annicchiarico, Clara Lucia; Munoz-Pinedo, Cristina; Barredo, Julio; Leclerc, Guy; Merchan, Jaime; Liu, Xiongfei; Lampidis, Theodore J. (2016). “Combining 2-deoxy-D-glucose with fenofibrate leads to tumor cell death mediated by simultaneous induction of energy and ER stress”. Oncotarget. 7 (24): 36461–36473. doi:10.18632/oncotarget.9263. PMC 5095013. PMID 27183907.
- ^ Kovar, Joy L.; Volcheck, William; Sevick-Muraca, Eva; Simpson, Melanie A.; Olive, D. Michael (2009). “Characterization and performance of a near-infrared 2-deoxyglucose optical imaging agent for mouse cancer models”. Analytical Biochemistry. 384(2): 254–262. doi:10.1016/j.ab.2008.09.050. PMC 2720560. PMID 18938129.
- ^ Cheng, Z., Levi, J., Xiong, Z., Gheysens, O., Keren, S., Chen, X., and Gambhir, S., Bioconjugate Chemistry, 17(3), (2006), 662-669
- ^ Barban, Stanley (December 1962). “Induced resistance to 2-deoxy-d-glucose in cell cultures”. Biochimica et Biophysica Acta. 65(2): 376–377. doi:10.1016/0006-3002(62)91065-x. ISSN 0006-3002. PMID 13966473.
- ^ Sanz, Pascual; Randez-Gil, Francisca; Prieto, José Antonio (September 1994). “Molecular characterization of a gene that confers 2-deoxyglucose resistance in yeast”. Yeast. 10 (9): 1195–1202. doi:10.1002/yea.320100907. ISSN 0749-503X. PMID 7754708. S2CID 9497505.
The Drugs Controller General of India (DCGI) has given permission for the emergency use of drug 2-deoxy-D-glucose (2-DG) as an adjunct therapy in moderate to severe Covid-19 cases, said Defence Research and Development Organisation on Saturday.
“Being a generic molecule and analogue of glucose, it can be easily produced and made available in plenty,” said the DRDO in a statement.
An adjunct therapy refers to an alternative treatment that is used together with the primary treatment. Its purpose is to assist the primary treatment.
“The drug has been developed by DRDO lab Institute of Nuclear Medicine and Allied Sciences in collaboration with Dr Reddy’s Laboratories. Clinical trial have shown that this molecule helps in faster recovery of hospitalized patients and reduces supplemental oxygen dependence,” the statement read.
According to DRDO, the patients treated with 2-DG showed faster symptomatic cure than Standard of Care (SoC) on various endpoints in the efficacy trends.
“A significantly favourable trend (2.5 days difference) was seen in terms of the median time to achieving normalization of specific vital signs parameters when compared to SOC,” it said.
The drug comes in powder form in sachets, which is taken orally by dissolving it in water.
“It accumulates in the virus-infected cells and prevents virus growth by stopping viral synthesis and energy production,” said the DRDO.
In April 2020, during the first wave of the Covid-19 pandemic, INMAS-DRDO scientists conducted laboratory experiments of 2-DG with the help of the Centre for Cellular and Molecular Biology (CCMB), Hyderabad.
They found that this molecule works effectively against the SARS-CoV-2 virus and inhibits viral growth.
Based on the results, the DCGI had in May 2020 permitted Phase-II clinical trial of 2-DG in Covid-19 patients.
In Phase-II trials (including dose-ranging) conducted from May to October 2020, the drug was found to be safe and showed significant improvement in the patients’ recovery.
“Phase IIa was conducted in 6 hospitals and Phase IIb (dose-ranging) clinical trial was conducted at 11 hospitals all over the country. Phase-II trial was conducted on 110 patients,” said the DRDO.
NEW DRUG APPROVALS
ONE TIME
$10.00
| Names | |
|---|---|
| IUPAC name(4R,5S,6R)-6-(hydroxymethyl)oxane-2,4,5-triol | |
| Other names2-Deoxyglucose 2-Deoxy-d-mannose 2-Deoxy-d-arabino-hexose 2-DG | |
| Identifiers | |
| CAS Number | 154-17-6 |
| 3D model (JSmol) | Interactive image |
| ChEMBL | ChEMBL2074932 |
| ChemSpider | 388402 |
| EC Number | 205-823-0 |
| IUPHAR/BPS | 4643 |
| PubChem CID | 108223 |
| UNII | 9G2MP84A8W |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C6H12O5 |
| Molar mass | 164.16 g/mol |
| Melting point | 142 to 144 °C (288 to 291 °F; 415 to 417 K) |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). |
////////////2-Deoxy-D-glucose, 2 dg, 2-dg, 2 DEOXY D GLUCOSE, COVID 19, CORONA VIRUS, INDIA 2021, DCGI, DRDO, DR REDDYS
C(C=O)C(C(C(CO)O)O)O
Pegylated Interferon alpha-2b, (PegIFN), Virafin
DB00022 sequence CDLPQTHSLGSRRTLMLLAQMRRISLFSCLKDRHDFGFPQEEFGNQFQKAETIPVLHEMI QQIFNLFSTKDSSAAWDETLLDKFYTELYQQLNDLEACVIQGVGVTETPLMKEDSILAVR KYFQRITLYLKEKKYSPCAWEVVRAEIMRSFSLSTNLQESLRSKE
CDLPQTHSLG SRRTLMLLAQ MRRISLFSCL KDRHDFGFPQ EEFGNQFQKA ETIPVLHEMI
QQIFNLFSTK DSSAAWDETL LDKFYTELYQ QLNDLEACVI QGVGVTETPL MKEDSILAVR
KYFQRITLYL KEKKYSPCAW EVVRAEIMRS FSLSTNLQES LRSKE

Chemical structure of peginterferon α-2a and α-2b. Abbreviations: PeG-IFN, peginterferon; IFN, interferon; Lys, lysine; His, histidine; Cys, cysteine; Ser, serine.
Pegylated Interferon alpha-2b
(PegIFN), Virafin

| Formula | C860H1353N229O255S9 |
|---|---|
| CAS | 99210-65-8, 98530-12-2, 215647-85-1 |
| Mol weight | 19268.9111 |
- Interferon α2b, pegylated
- PegIFN a-2b
- PegIFN a-2b (biologics)
- PegIFN α-2b
- PegIntron
- Pegaferon
- PegiHep
- Peginterferon alfa-2b
- Peginterferon α-2b
- Pegylated interferon alfa-2b
- Pegylated interferon α-2b
- Pegylated interferons, PegIFN a-2b
- Proteinaceous biopharmaceuticals, PegIFN a-2b
- Sch 54031
- Sylatron
- ViraferonPeg
Active Moieties
| NAME | KIND | UNII | CAS | INCHI KEY |
|---|---|---|---|---|
| Interferon alfa-2b | unknown | 43K1W2T1M6 | 98530-12-2 | Not applicable |
| Clinical data | |
|---|---|
| Trade names | PegIntron, Sylatron, ViraferonPeg, others |
| AHFS/Drugs.com | Professional Drug Facts |
| MedlinePlus | a605030 |
| License data | EU EMA: by INN |
| Routes of administration | Subcutaneous injection |
| ATC code | L03AB10 (WHO) |
| Legal status | |
| Legal status | US: ℞-only [1][2]EU: Rx-only |
| Pharmacokinetic data | |
| Elimination half-life | 22–60 hrs |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 215647-85-1 |
| IUPHAR/BPS | 7462 |
| DrugBank | DB00022 |
| ChemSpider | none |
| UNII | G8RGG88B68 |
| KEGG | D02745 |
| ChEMBL | ChEMBL1201561 |
| ECHA InfoCard | 100.208.164 |
| Chemical and physical data | |
| Formula | C860H1353N229O255S9 |
| Molar mass | 19269.17 g·mol−1 |
NEW DRUG APPROVALS
ONE TIME
$10.00
New Delhi: ,,,,,,https://www.ndtv.com/india-news/zydus-virafin-gets-emergency-use-approval-for-treating-moderate-covid-19-cases-2420358
Zydus Cadila received emergency use approval from the Drugs Controller General of India (DGCI) on Friday for the use of “Virafin”, Pegylated Interferon alpha-2b (PegIFN) in treating moderate COVID-19 infection in adults.
A single-dose subcutaneous regimen of the antiviral Virafin will make the treatment more convenient for the patients. When administered early on during COVID-19, Virafin will help patients recover faster and avoid much of the complications, the company said.
In a release, Cadila Health highlighted that “the drug has also shown efficacy against other viral infections.”
Speaking on the development, Dr Sharvil Patel, Managing Director, Cadila Healthcare Limited said, “The fact that we are able to offer a therapy which significantly reduces the viral load when given early on can help in better disease management. It comes at a much-needed time for patients and we will continue to provide them access to critical therapies in this battle against COVID-19.”
In its Phase III clinical trials, the therapy had shown better clinical improvement in the patients suffering from COVID-19. During the trials, a higher proportion of patients administered with PegIFN arm were RT-PCR negative by day 7. The drug ensures faster viral clearance and has several add-on advantages compared to other anti-viral agents, the release further reads.
The development and the nod from DGCI come at a time when India is combating the second wave of coronavirus.
The central government in one of its major announcements decided to administer COVID-19 vaccines to all age above 18 years.
India recorded 3,32,730 new COVID-19 cases in the last 24 hours, the highest single-day spike since the pandemic broke out last year. India has crossed the mark of 3 lakh COVID-19 cases for two consecutive days now. This has taken the cumulative count of the COVID infection in the country to 1,62,63,695.
2CommentsThe country has recorded 2,263 new deaths due to COVID-19 in the last 24 hours. As many as 1,86,920 people have succumbed to the viral infection in India so far. There are 24,28,616 active COVID-19 cases in the country now.
PATENT
https://patents.google.com/patent/EP1562634B1/en
- Interferon alpha-2a plays an important role for the treatment of chronic hepatitis C, but it is limited in its efficacy by the short in vivo half-life. To improve the half-life and efficacy, interferon alpha-2a was conjugated with a polyethylene glycol moiety. Pegylation changes physicochemical and biological properties of the protein. One effect is the decrease of the proteolytic degradation and the renal clearance. This increases the half-life of the pegylated protein in blood. Another effect is the altered distribution in the body, depending on the size of the PEG moiety of the protein. Interferon alpha 2a pegylated with a large polyethylene glycol moiety (PEG moiety) such as a 40 kDa branched polyethylene moietywherein R and R’ are independently lower alkyl; n and n’ are integers having a sum of from 600 to 1500; and the average molecular weight of the polyethylene glycol units in said conjugate is from about 26,000 daltons to about 66,000 daltons;
has an improved biological activity and exhibits sustained adsorption and reduced renal clearance, resulting in a strong antiviral pressure throughout a once-weekly dosing schedule, see Perry M. C., et al. Drugs, 2001,15,2263-2288 and Lamb M. W., et al. The Annals of Pharmacotherapy, 2002, 36, 933-938. - [0003]See also Monkarsh et al. Analytical Biochemistry, 1997, 247, 434- 440 (Positional Isomers of Mono-pegylated Interferon α-2a) and Bailon et al. Bioconjugate Chemistry, 2001, 12, 195-202 (Rational Design of a Potent, Long-Lasting Form of interferon).
- [0004]The method for the pegylation of interferon alpha-2a is described in EP A 809 996. Since this pegylation is performed by reaction of PEG2-NHS of formulawith primary amino groups on for example lysine or to the N-terminus of the interferon alpha.one or more PEG moieties may be attached and form a mixture of unpegylated, mono- and multiple-pegylated interferon. Monopegylated interferon alpha can be isolated from the mixture by methods known in the art. Furthermore, since interferon alpha-2a molecule exhibits 12 sites for pegylation (11 lysines and the N-terminus) it is a mixture of positional isomers. From these possible twelve isomers, nine were isolated and characterized, each of these being conjugated to the branched polyethylene glycol chain at a specific lysine, namely,
at Lys(31) to form interferon alpha 2a pegylated at Lys(31) [referred to as PEG-Lys(31)],
at Lys(49) to form interferon.alpha 2a pegylated at Lys(49) [referred to as PEG-Lys(49)],
at Lys(70) to form interferon alpha 2a pegylated at Lys(70) [referred to as PEG-Lys(70)],
at Lys(83) to form interferon alpha 2a pegylated at Lys(83) [referred to as PEG-Lys(83)],
at Lys(112) to form interferon alpha 2a pegylated at Lys(112) [referred to as PEG-Lys(112)],
at Lys(121) to form interferon alpha 2a pegylated at Lys(121) [referred to as PEG-Lys(121)],
at Lys(131) to form interferon alpha 2a pegylated at Lys(131) [referred to as PEG-Lys(131)],
at Lys(134) to form interferon alpha 2a pegylated at Lys(134) [referred to as PEG-Lys(134)],
at Lys(164) to form interferon alpha 2a pegylated at Lys(164) [referred to as PEG-Lys(164)]. - [0005]It has been found that PEG-Lys(31) and PEG-Lys(134) have higher activities in an antiviral assay than the mixture, the activity of PEG-Lys(164) was equal to the mixture, whereas the activities of PEG-Lys(49), PEG-Lys(70), PEG-Lys(83), PEG-Lys(112), PEG-Lys(121) and PEG-Lys(131) were lower.
- The following examples will further illustrate the invention
Example 1A Separation of the positional isomers
- [0035]A two-step isolation and purification scheme was used to prepare the monopegylated isoforms of PEG-interferon alpha 2a.
- a) The first step was a separation of the positional isomers on a preparative low pressure liquid chromatography column with a weak-cation exchange matrix (TOSOH-BIOSEP, Toyopearl CM-650S, e.g. Resin Batch no. 82A the diameter of the column being 16 mm, the length 120 cm). A linear pH-gradient of increasing sodium acetate concentration (25 mM, pH 4.0 up 75 mM to pH 7.8) was applied at a flow rate of 0.7 mL/min. Detection was at 280 nm. With this chromatographic step species 1, 2, 5,6 and a mixture of 3, 4, 4a, 7 and 8 could be collected, see Table 1.
- b) The fractions were further separated and purified in the second preparation step. A preparative column with the same matrix as the analytical strong-cation exchange column (Resin Batch no. 82A having a ion exchange capacity of 123 mEq/ml) as described above but larger dimensions (30 mm i.d. and 70 mm length), further a higher flow rate and an extended run time was used. As for the analytical method the column was pre-equilibrated with 3.4 mM sodium acetate, 10% ethanol and 1% diethylene glycol, adjusted to pH 4.4 (buffer A). After loading the PEG-IFN samples, the column was washed with buffer A, followed by an ascending linear gradient to 10 mM dibasic potassium phosphate, 10% ethanol and 1% diethylene glycol, adjusted to pH 6.6 (buffer B). The flow rate was 1.0 mL/min and the detection at 218 nm.
- [0036]The protein concentration of the PEG-IFN alpha 2a isomer was determined by spectrophotometry, based on the 280 nm absorption of the.protein moiety of the PEG-IFN alpha 2a.
- [0037]An analytical elution profile of 180 µg of PEG-IFN alpha 2a is shown in Figure 1. The result of this method is a separation into 8 peaks, 2 peaks with baseline separation and 6 with partial separation. The decrease of the baseline absorption towards the end of the chromatogram suggests that there were no other monopegylated species of IFN alpha 2a eluting at higher retention time.
- [0038]In addition, looking carefully at the IEC-chromatogram a further peak close to the detection limit is visible between peaks 2 and 3 indicating the presence of additional positional isomers that should also contribute to the specific activity of the PEG-IFN alpha 2a mixture. Additional species were expected as the interferon alpha-2a molecule exhibits 12 sites for pegylation (11 lysines and the N-terminus). However, given the low abundance of the these species, they were not isolated and characterised.
- [0039]Isomer samples derived from IEC optimisation runs were investigated directly after the isolation (t = 0) and after 2 of weeks of storage at 5°C (data not shown). No significant differences were observed for the protein derived from IEC-peaks with regard to the protein content as determined by spectrometric methods; nor were any changes to be detected in the monopegylation site, the content of oligo-PEG-IFN alpha 2a, the amount of aggregates and the bioassay activity. Taking into account the relative abundance of the individual isomers – as determined by the IEC method – as well as the specific activities – as determined in the anti-viral assay – almost the total specific bioactivity of the PEG-IFN alpha 2a mixture used for their isolation is recovered (approximately 93%).
- [0040]The analytical IE-HPLC was used to check the purity of the individual isomers with respect to contamination with other positional isomers in the IEC fractions. The peaks 2, 3, 4, 4a, 5 and 7 had more than 98%, the peaks 1 and 8 had 93% and peak 6 had 88 % purity. Table 1:PEG-peptides identified by comparison of the Lys-C digest spectra of the isomers and the reference standard.Identified PEG Sites in the separated PEG-IFN SpeciesPeakmissing peaks in peptide mapPEG-IFNPEG siteMr (DA)SequencePeak 1K31A,E24-49Peak 2K134I, I’134-164Peak 3K131C122-131aPeak 4K121B, C113-131Peak 4aK164b134-164a,bPeak 5K70D, F50-83Peak 6K83D, H71-112Peak 7K49E, F32-70Peak 8K112B, H84-121a132-133 too small to detect.a,b RP-HPLC.
- [0041]The fractions were characterised by the methods described in examples 2 to 6.
Example 1B Analytical separation of positional isomers of mono-pegylated interferon alpha 2a
- [0042]HPLC Equipment:HP1100Column:SP-NPR, TosoH Bioscience, Particle size: 2.5µm, nonporous, Order#: 13076Injection:5-10 µg monopegylated IFNmobile Phase:Buffer A: 10% v/vEthanol 1% v/vDiethylenglycol 2.3 mMNa-Acetat 5.2 mMAcetic acid, in purified water, no pH adjustment Buffer B: 10% v/vEthanol 1% v/vDiethylenglycol 16.4 mMKH2PO4 4.4 mMK2HPO4, in purified water, no pH adjustmentGradient:0 Min40 %B 2 Min40 %B 2.1 Min48 %B 25 Min68 %B 27 Min75 %B 30 Min75 %B 34 Min40 %B 40 Min40 %BFlow:1.0 ml/min Column Temperature:25°C Detection:218 nm a typical Chromatogram is given in Figure 8.
Example 2 Analysis of the fractions by mass spectrometry peptide mapping
- [0043]Mass spectra were recorded on a MALDI-TOF MS instrument (PerSeptive Biosystems Voyager-DE STR with delayed extraction). Each IEC fraction (Ion Exchange Chromatography) was desalted by dialysis, reduced with 0.02 M 1,4-dithio-DL-threitol (DTT) and alkylated with 0.2 M 4-vinyl pyridine. Then the proteins were digested with endoproteinase Lys-C (Wako Biochemicals) in 0.25 M Tris (tris(hydroxymethyl)-aminoethane) at pH 8.5 with an approximate enzyme to protein ratio of 1:30. The reaction was carried out over night at 37 °C.
- [0044]A solution of 20 mg/ml α-cyano-4-hydroxycinnamic acid and 12 mg/ml nitrocellulose in acetone/isopropanol 40/60 (v/v) was used as matrix (thick-layer application). First, 0.5 µL of matrix was placed on the target and allowed to dry. Then, 1.0 µL of sample was added. The spectra were obtained in linear positive ionisation mode with an accelerating voltage of 20.000 V and a grid voltage of 95 %. At least 190 laser shots covering the complete spot were accumulated for each spectrum. Des-Arg1-bradykinin and bovine insulin were used for internal calibration.
Example 3 high-performance liquid chromatography (RP-HPLC) Peptide Mapping
- [0045]The peptides were characterized by reverse-phase high-performance liquid chromatography (RP-HPLC) Peptide Mapping. The IEC fractions were reduced, alkylated and digested with endoproteinase Lys-C as described for the MALDI-TOF MS peptide mapping. The analysis of the digested isomers was carried out on a Waters Alliance HPLC system with a Vydac RP-C18 analytical column (5 µm, 2.1 × 250 mm) and a precolumn with the same packing material. Elution was performed with an acetonitrile gradient from 1 % to 95 % for 105 min in water with a flow rate of 0.2 mL/min. Both solvents contained 0.1 % (v/v) TFA. 100 µL of each digested sample were injected and monitored at 215 nm.
Example 4 MALDI-TOF spectra of undigested protein
- [0046]An 18 mg/ml solution of trans-3-indoleacrylic acid in acetonitrile/0.1 % trifluoroacetic acid 70/30 (v/v) was premixed with the same volume of sample solution. Then 1.0 µL of the mixture was applied to the target surface. Typically 150 – 200 laser shots were averaged in linear positive ionisation mode. The accelerating voltage was set to 25.000 V and the grid voltage to 90 %. Bovine albumin M+ and M2+ were used for external calibration.
Example 5 SE-HPLC (size exclusion HPLC)
- [0047]SE-HPLC was performed with a Waters Alliance 2690 HPLC system equipped with a TosoHaas TSK gel G 4000 SWXL column (7.8 × 300 mm). Proteins were eluted using a mobile phase containing 0.02 M NaH2PO4, 0.15 M NaCl, 1% (v/v) diethylene glycol and 10 % (v/v) ethanol (pH 6.8) at a flow rate of 0.4 mL/min and detected at 210 nm. The injection amounts were 20 µg of each isomers.
- [0048]Size Exclusion HPLC and SDS-PAGE were used to determine the amount of oligo-PEG-IFN alpha 2a forms and aggregates in the different IEC fractions. The reference material contains 2.3 % aggregates and 2.2 % oligomers (Figure 4).
- [0049]Peaks 1, 4, 4a, 5, 6 and 8 contain < 0.7 % of the oligopegylated IFN alpha 2a forms, whereas in,peaks 2, 3, and 7 the percentage of the oligopegylated IFN alpha 2a forms are under the detection limit (< 0.2 %). In the case of the aggregates a different trend could be seen. In all peaks the amount of aggregates is below 0.9 %.
Example 6 SDS-PAGE
- [0050]SDS-PAGE was carried out both under non-reducing and under reducing conditions using Tris-Glycine gels of 16 % (1.5 mm, 10 well). Novex Mark 12 molecular weight markers with a mass range from 2.5 to 200 kDa were used for calibration, bovine serum albumin (BSA) was used as sensitivity standard (2 ng). Approximately 1 µg of all the samples and 0.5 µg of standard were applied to the gel. The running conditions were 125 V and 6 W for 120 min. The proteins were fixed and stained using the silver staining kit SilverXpress from Novex.
- [0051]The gels that were recorded under non-reducing conditions for the IEC fractions 1- 8 (Figure 2) show a pattern that is comparable to that of the PEG-IFN alpha 2a reference standard.
- [0052]Under reducing conditions, the gels show an increase in intensity of the minor bands at about 90 kDa as compared to the standard. Between 6 and 10 kDa protein fragments appear for peaks 6, 7 and 8 (Figure 3). Both bands together correspond to approximately 1 % of clipped material. In the lanes of isomer 1, 5, 6, 7, 8 additional bands with more than 100 kDa can be seen which are also present in the standard. These can be assigned to oligomers. Thus SDS-PAGE confirms the results of the SE-HPLC analysis.
- [0053]Overall, RP-HPLC and SDS-PAGE experiments indicate that the purity of the IEC fractions can be considered comparable to the PEG-IFN alpha 2a reference standard.
- [0054]The structure of the PEG-IFN alpha 2a species derived from the 9 IEC-fractions were identified based on the results of the methods described above using the strategy mentioned above.
Example 7 The antiviral activity (AVA)
- [0055]The antiviral activity was estimated by its protective effect on Madin-Darby bovine kidney (MDBK) cells against the infection by vesticular stomatitis virus (VSV) and compared with a PEG-IFN alpha 2a standard. Samples and reference standard were diluted in Eagle’s Minimum Essential Medium (MEM) containing 10 % fetal bovine serum to a final concentration of 10 ng/mL (assay starting concentration). Each sample was assayed in quadruplicate.
- [0056]The antiviral protection of Madin-Darby bovine kidney cells (MDBK) with vesicular stomatitis virus was tested according to the method described in Virol. 1981, 37, 755-758. All isomers induced an activity in the anti-viral assay as presented in Table 2. The activities range between 1061 and 339 U/µg, indicating that the difference in specific activities of the protein in the positional isomers is significant. The know-how and the results generated so far will allow the initiation of further investigations to establish this structure-function relationship between the positional isomers and the IFN alpha receptors. Table 2:In Vitro Antiviral Activities of PEG-IFN alpha 2a and individual PEG-IFN alpha 2a isomers. The Antiviral activity was determined in MDBK cells infected with vesicular stomatitis virus. The results present the averages of three assays performed independently.Antiviral Assay of PEG-IFNPeakU/µgPEG-IFN1061 ± 50Peak 11818 ± 127Peak 21358 ± 46Peak 3761197Peak 4339 ± 33Peak 4a966 ± 107Peak 5600 ± 27Peak 6463 ± 25Peak7513 ± 20Peak 8468 ± 23
- [0057]The results are further illustrated by the following figures
- Figure 1: Analytical IEC-HPLC of 180µg of PEG-IFN alpha 2a. An analytical strong-cation exchange column was used to check the purity of the separated positional isomers from each purification step (TOSOH-BIOSEP, SP-SPW,10 µm particle size, 7.5 mm diameter, 7.5 cm length).
- Figure 2: A/B: SDS-PAGE analysis with Tris-glycine (16%), the samples were electrophoresed under non-reduced conditions. The gels were stained for protein with Silver Stain. Lanes: M, molecular weight marker proteins/ 2, Peak 1/ 3, Peak 2/ 4, Peak 3/ 5, Peak 4/ 6, Peak 4a/ 7, Peak 5/ 8, Peak 6/ 9, Peak 7/10, Peak 8/ 11, Ix PEG-IFN standard/ 12, 1.5x PEG-IFN standard/ C1, IFN standard.
- Figure 3: A/B: SDS-PAGE analysis with Tris-glycine (16%), the samples were electrophoresed under reduced conditions. The gels were stained for protein with Silver Stain. Lanes: M, molecular weight marker proteins/ 2, Peak 1/ 3, Peak 2/ 4, Peak 3/ 5, Peak 4/ 6, Peak 4a/ 7, Peak 5/ 8, Peak 6/ 9, Peak 7/ 10, Peak 8/ 11, 1x PEG-IFN standard/ 12, 1.5x PEG-IFN standard/ C1, IFN standard.
- Figure 4: Size Exclusion (SE-) HPLC was used to determine the amount of oligo PEG-IFN forms and aggregates in the different IEC fractions. SE-HPLC was performed with a TosoHaas TSK gel G 4000 SWXL column (7.8 × 300 mm).
- Figure 5: MALDI-TOF spectrometry was used to determine the molecular weight of each isomer in order to ensure that the PEG-IFN molecules were still intact after IEC chromatography and to confirm the monopegylation.
- Figure 6: MALDI-TOF Lys-C peptide maps of the PEG-IFN reference standard and the peaks 1, 2, 3, 4, 4a, 5, 6, 7, 8. Missing peaks compared to the standard are indicated by arrows.
- Figure 7: RP-HPLC chromatograms of the Lys-C digests of the PEG-IFN reference and peak 4a
- Figure 8: Analytical HPLC of 5-10µg of PEG-IFN alpha 2a mixture of positional isomers on a column charged with SP-NPR, TosoH Bioscience, Particle size: 2.5µm, nonporous as described in Example 1B..
- Figure 9: Ribbon structure of interferon alpha-2a showing the pegylation sites. This is the high resolution structure of human interferon alpha-2a determined with NMR spectroscopy see J. Mol. Biol. 1997, 274, 661-675. The pegylation sites of pegylated interferon alpha-2a are coloured red and labelled with residue type and residue number.
Pegylated interferon alfa-2b, sold under the brand name PegIntron among others, is a medication used to treat hepatitis C and melanoma.[3] For hepatitis C it is typically used with ribavirin and cure rates are between 33 and 82%.[3][4] For melanoma it is used in addition to surgery.[3] It is given by injection under the skin.[3]
Side effects are common.[5] They may include headache, feeling tired, mood changes, trouble sleeping, hair loss, nausea, pain at the site of injection, and fever.[3] Severe side effects may include psychosis, liver problems, blood clots, infections, or an irregular heartbeat.[3] Use with ribavirin is not recommended during pregnancy.[3] Pegylated interferon alfa-2b is in the alpha interferon family of medications.[3] It is pegylated to protect the molecule from breakdown.[5]
Pegylated interferon alfa-2b was approved for medical use in the United States in 2001.[3] It is on the World Health Organization’s List of Essential Medicines.[6]
Peginterferon alfa-2b is a form of recombinant interferon used as part of combination therapy to treat chronic Hepatitis C, an infectious liver disease caused by infection with Hepatitis C Virus (HCV). HCV is a single-stranded RNA virus that is categorized into nine distinct genotypes, with genotype 1 being the most common in the United States, and affecting 72% of all chronic HCV patients 3. Treatment options for chronic Hepatitis C have advanced significantly since 2011, with the development of Direct Acting Antivirals (DAAs) resulting in less use of Peginterferon alfa-2b. Peginterferon alfa-2b is derived from the alfa-2b moeity of recombinant human interferon and acts by binding to human type 1 interferon receptors. Activation and dimerization of this receptor induces the body’s innate antiviral response by activating the janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway. Use of Peginterferon alfa-2b is associated with a wide range of severe adverse effects including the aggravation and development of endocrine and autoimmune disorders, retinopathies, cardiovascular and neuropsychiatric complications, and increased risk of hepatic decompensation in patients with cirrhosis. The use of Peginterferon alfa-2b has largely declined since newer interferon-free antiviral therapies have been developed.
In a joint recommendation published in 2016, the American Association for the Study of Liver Diseases (AASLD) and the Infectious Diseases Society of America (IDSA) no longer recommend Peginterferon alfa-2b for the treatment of Hepatitis C 2. Peginterferon alfa-2b was used alongside Ribavirin(https://go.drugbank.com/drugs/DB00811) with the intent to cure, or achieve a sustained virologic response (SVR), after 48 weeks of therapy. SVR and eradication of HCV infection is associated with significant long-term health benefits including reduced liver-related damage, improved quality of life, reduced incidence of Hepatocellular Carcinoma, and reduced all-cause mortality 1.
Peginterferon alfa-2b is available as a variable dose injectable product (tradename Pegintron) used for the treatment of chronic Hepatitis C. Approved in 2001 by the FDA, Pegintron is indicated for the treatment of HCV with Ribavirin or other antiviral drugs Label. When combined together, Peginterferon alfa-2b and Ribavirin have been shown to achieve a SVR between 41% for genotype 1 and 75% for genotypes 2-6 after 48 weeks of treatment.
Medical uses
It is used to treat hepatitis C and melanoma. For hepatitis C it is typically used with ribavirin. For melanoma it is used in addition to surgery.[3]
For hepatitis C it may also be used with boceprevir, telaprevir, simeprevir, or sofosbuvir.[5]
In India, in 2021, DGCI approved emergency use of Zydus Cadila‘s Virafin in treating moderate COVID-19 infection.[7]
Host genetic factors
For genotype 1 hepatitis C treated with pegylated interferon-alfa-2a or pegylated interferon-alfa-2b combined with ribavirin, it has been shown that genetic polymorphisms near the human IL28B gene, encoding interferon lambda 3, are associated with significant differences in response to the treatment. This finding, originally reported in Nature,[8] showed that genotype 1 hepatitis C patients carrying certain genetic variant alleles near the IL28B gene are more likely to achieve sustained virological response after the treatment than others. A later report from Nature[9] demonstrated that the same genetic variants are also associated with the natural clearance of the genotype 1 hepatitis C virus.
Side effects
Common side effects include headache, feeling tired, mood changes, trouble sleeping, hair loss, nausea, pain at the site of injection, and fever. Severe side effects may include psychosis, liver problems, blood clots, infections, or an irregular heartbeat.[3] Use with ribavirin is not recommended during pregnancy.[3]
Mechanism of action
One of the major mechanisms of PEG-interferon alpha-2b utilizes the JAK-STAT signaling pathway. The basic mechanism works such that PEG-interferon alpha-2b will bind to its receptor, interferon-alpha receptor 1 and 2 (IFNAR1/2). Upon ligand binding the Tyk2 protein associated with IFNAR1 is phosphorylated which in turn phosphorylates Jak1 associated with IFNAR2. This kinase continues its signal transduction by phosphorylation of signal transducer and activator of transcription (STAT) 1 and 2 via Jak 1 and Tyk2 respectively. The phosphorylated STATs then dissociate from the receptor heterodimer and form an interferon transcription factor with p48 and IRF9 to form the interferon stimulate transcription factor-3 (ISGF3). This transcription factor then translocates to the nucleus where it will transcribe several genes involved in cell cycle control, cell differentiation, apoptosis, and immune response.[10][11]
PEG-interferon alpha-2b acts as a multifunctional immunoregulatory cytokine by transcribing several genes, including interleukin 4 (IL4). This cytokine is responsible for inducing T helper cells to become type 2 helper T cells. This ultimately results in the stimulation of B cells to proliferate and increase their antibody production. This ultimately allows for an immune response, as the B cells will help to signal the immune system that a foreign antigen is present.[12]
Another major mechanism of type I interferon alpha (IFNα) is to stimulate apoptosis in malignant cell lines. Previous studies have shown that IFNα can cause cell cycle arrest in U266, Daudi, and Rhek-1 cell lines.[13]
A follow-up study researched to determine if the caspases were involved in the apoptosis seen in the previous study as well as to determine the role of mitochondrial cytochrome c release. The study confirmed that there was cleavage of caspase-3, -8, and -9. All three of these cysteine proteases play an important role in the initiation and activation of the apoptotic cascade. Furthermore, it was shown that IFNα induced a loss in the mitochondrial membrane potential which resulted in the release of cytochrome c from the mitochondria. Follow-up research is currently being conducted to determine the upstream activators of the apoptotic pathway that are induced by IFNα.[14]
History
It was developed by Schering-Plough. Merck studied it for melanoma under the brand name Sylatron. It was approved for this use in April 2011.
References
- ^ “PegIntron- peginterferon alfa-2b injection, powder, lyophilized, for solution PegIntron- peginterferon alfa-2b kit”. DailyMed. Retrieved 28 September 2020.
- ^ “Sylatron- peginterferon alfa-2b kit”. DailyMed. 28 August 2019. Retrieved 28 September 2020.
- ^ Jump up to:a b c d e f g h i j k l “Peginterferon Alfa-2b (Professional Patient Advice) – Drugs.com”. http://www.drugs.com. Archived from the original on 16 January 2017. Retrieved 12 January 2017.
- ^ “ViraferonPeg Pen 50, 80, 100, 120 or 150 micrograms powder and solvent for solution for injection in pre-filled pen CLEAR CLICK – Summary of Product Characteristics (SPC) – (eMC)”. http://www.medicines.org.uk. Archived from the original on 13 January 2017. Retrieved 12 January 2017.
- ^ Jump up to:a b c “Peginterferon alfa-2b (PegIntron)”. Hepatitis C Online. Archived from the original on 23 December 2016. Retrieved 12 January 2017.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ https://www.aninews.in/news/national/general-news/dgci-approves-emergency-use-of-zyduss-virafin-in-treating-moderate-covid-19-infection20210423163622/
- ^ Ge D, Fellay J, Thompson AJ, et al. (2009). “Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance”. Nature. 461 (7262): 399–401. Bibcode:2009Natur.461..399G. doi:10.1038/nature08309. PMID 19684573. S2CID 1707096.
- ^ Thomas DL, Thio CL, Martin MP, et al. (2009). “Genetic variation in IL28B and spontaneous clearance of hepatitis C virus”. Nature. 461 (7265): 798–801. Bibcode:2009Natur.461..798T. doi:10.1038/nature08463. PMC 3172006. PMID 19759533.
- ^ Ward AC, Touw I, Yoshimura A (January 2000). “The JAK-STAT pathway in normal and perturbed hematopoiesis”. Blood. 95 (1): 19–29. doi:10.1182/blood.V95.1.19. PMID 10607680. Archived from the original on 2014-04-26.
- ^ PATHWAYS :: IFN alpha[permanent dead link]
- ^ Thomas H, Foster G, Platis D (February 2004). “Corrigendum toMechanisms of action of interferon and nucleoside analogues J Hepatol 39 (2003) S93–8″. J Hepatol. 40 (2): 364. doi:10.1016/j.jhep.2003.12.003.
- ^ Sangfelt O, Erickson S, Castro J, Heiden T, Einhorn S, Grandér D (March 1997). “Induction of apoptosis and inhibition of cell growth are independent responses to interferon-alpha in hematopoietic cell lines”. Cell Growth Differ. 8 (3): 343–52. PMID 9056677. Archived from the original on 2014-04-26.
- ^ Thyrell L, Erickson S, Zhivotovsky B, et al. (February 2002). “Mechanisms of Interferon-alpha induced apoptosis in malignant cells”. Oncogene. 21 (8): 1251–62. doi:10.1038/sj.onc.1205179. PMID 11850845.
External links
- Peginterferon alfa-2b in the U.S. National Library of Medicine’s Drug Information Portal
- Medicines patent loophole ‘found’ at the BBC, 2007
- PEG-Intron (Peginterferon Alfa-2B) — Platelet Count Decreased from DrugLib.com
///////////Pegylated Interferon alpha-2b, PegIFN, Virafin, COVID 19, CORONA VIRUS, INDIA 2021, APPROVALS 2021
COVAXIN, BBV 152


COVAXIN
CAS 2501889-19-4
- Whole-Virion Inactivated SARS-CoV-2 Vaccine
- UNII76JZE5DSN6
- BBV 152
- A whole virion inactivated COVID-19 vaccine candidate derived from SARS-CoV-2 strain NIV-2020-770
REF
medRxiv (2020), 1-21.
bioRxiv (2020), 1-32.
BBV152 (also known as Covaxin) is an inactivated virus-based COVID-19 vaccine being developed by Bharat Biotech in collaboration with the Indian Council of Medical Research.
BBV152 is a vaccine candidate created by the Indian Council of Medical Research (ICMR). The candidate, a whole virion inactivated SARS-CoV-2 vaccine, was developed from a well-known SARS-CoV-2 strain and a vero cell platform (CCL-81) with adjuncts of either aluminum hydroxide gel (Algel) or a novel TLR7/8 agonist adsorbed gel. The components of the vaccine include BBV152A, BBV152B, and BBV152C. Animal studies in mice, rats, and rabbits reported BBV152 immunogenicity at two separate antigen concentrations with both types of adjuvants. The formulation with the TLR7/8 adjuvant specifically induced significant Th1 biased antibody responses and increased SARS-CoV-2 lymphocyte responses. Thus, as of July 2020, BBV152 is in Phase 1/2 clinical trials assessing safety and immunogenicity in humans (NCT04471519).
Clinical research
Phase I and II trials
In May 2020, Indian Council of Medical Research’s (ICMR‘s) National Institute of Virology approved and provided the virus strains for developing a fully indigenous COVID-19 vaccine.[1][2] In June 2020, the company got permission to conduct Phase I and Phase II human trials of a developmental COVID-19 vaccine named Covaxin, from the Drugs Controller General of India (DCGI), Government of India.[3] A total of 12 sites were selected by the Indian Council for Medical Research for Phase I and II randomised, double-blind and placebo-controlled clinical trials of vaccine candidate.[4][5][6]
In December 2020, the company announced the report for Phase I trials and presented the results through medRxiv preprint;[7][8] the report was later published in the The Lancet.[9]
On March 8, 2021, Phase II results were published in The Lancet. The study showed that Phase II trials had a higher immune response and induced T-cell response due to the difference in dosing regime from Phase I. The doses in Phase II were given at 4 weeks interval as opposed to 2 weeks in Phase I. Neutralization response of the vaccine were found significantly higher in Phase II.[10]
Phase III trials[edit]
In November 2020, Covaxin received the approval to conduct Phase III human trials[11] after completion of Phase I and II.[12] The trial involves a randomised, double-blinded, placebo-controlled study among volunteers of age group 18 and above and started on 25 November.[13] The Phase III trials involved around 26,000 volunteers from across India.[14] The phase III trials covered a total of 22 sites consisting several states in the country, including Delhi, Karnataka and West Bengal.[15] Refusal rate for Phase III trials was much higher than that for Phase I and Phase II. As a result only 13,000 volunteers had been recruited by 22 December with the number increasing to 23,000 by 5 January. [16][17]
As on March 2021, the stated interim efficacy rate for phase III trial is 81%.[18][10]
B.1.1.7 (United Kingdom) variant
In December 2020, a new SARS‑CoV‑2 variant, B.1.1.7, was identified in the UK.[19] A study on this variant was carried and preliminary results presented in biorxiv have shown Covaxin to be effective in neutralizing this strain.[20]
Manufacturing
The vaccine candidate is produced with Bharat Biotech’s in-house vero cell manufacturing platform[21] that has the capacity to deliver about 300 million doses.[22] The company is in the process of setting up a second plant at its Genome Valley facility in Hyderabad to make Covaxin. The firm is in talks with other state governments like Odisha[23] for another site in the country to make the vaccine. Beside this, they are also exploring global tie-ups for Covaxin manufacturing.[24]
In December 2020, Ocugen Inc entered a partnership with Bharat Biotech to co-develop Covaxin for the U.S. market.[25][26] In January 2021, Precisa Med entered an agreement with Bharat Biotech to supply Covaxin in Brazil[27]
Emergency use authorisation
See also: COVID-19 vaccine § Trial and authorization status
Bharat Biotech has applied to the Drugs Controller General of India (DCGI), Government of India seeking an emergency use authorisation (EUA).[31] It was the third firm after Serum Institute of India and Pfizer to apply for emergency use approval.[32]
On 2 January 2021, the Central Drugs Standard Control Organisation (CDSCO) recommended permission for EUA,[33] which was granted on 3 January.[34] The emergency approval was given before Phase III trial data was published. This was criticized in some sections of the media.[35][36]
The vaccine was also approved for Emergency Use in Iran and Zimbabwe.[30][29]
References
- ^ “ICMR teams up with Bharat Biotech to develop Covid-19 vaccine”. Livemint. 9 May 2020.
- ^ Chakrabarti A (10 May 2020). “India to develop ‘fully indigenous’ Covid vaccine as ICMR partners with Bharat Biotech”. ThePrint.
- ^ “India’s First COVID-19 Vaccine Candidate Approved for Human Trials”. The New York Times. 29 June 2020.
- ^ “Human clinical trials of potential Covid-19 vaccine ‘COVAXIN’ started at AIIMS”. DD News. Prasar Bharati, Ministry of I & B, Government of India. 25 July 2020.
- ^ Press, Associated (25 July 2020). “Asia Today: Amid new surge, India tests potential vaccine”. Washington Post. Retrieved 17 December 2020.
- ^ “Delhi: 30-year-old is first to get dose of trial drug Covaxin”. The Indian Express. 25 July 2020.
- ^ Perappadan, Bindu Shajan (16 December 2020). “Coronavirus | Covaxin phase-1 trial results show promising results”. The Hindu. Retrieved 17 December 2020.
- ^ Sabarwal, Harshit (16 December 2020). “Covaxin’s phase 1 trial result shows robust immune response, mild adverse events”. Hindustan Times. Retrieved 17 December 2020.
- ^ Ella, Raches; Vadrevu, Krishna Mohan; Jogdand, Harsh; Prasad, Sai; Reddy, Siddharth; Sarangi, Vamshi; Ganneru, Brunda; Sapkal, Gajanan; Yadav, Pragya; Abraham, Priya; Panda, Samiran; Gupta, Nivedita; Reddy, Prabhakar; Verma, Savita; Rai, Sanjay Kumar; Singh, Chandramani; Redkar, Sagar Vivek; Gillurkar, Chandra Sekhar; Kushwaha, Jitendra Singh; Mohapatra, Satyajit; Rao, Venkat; Guleria, Randeep; Ella, Krishna; Bhargava, Balram (21 January 2021). “Safety and immunogenicity of an inactivated SARS-CoV-2 vaccine, BBV152: a double-blind, randomised, phase 1 trial”. The Lancet Infectious Diseases. doi:10.1016/S1473-3099(20)30942-7. PMC 7825810. PMID 33485468.
- ^ Jump up to:a b Ella, Raches; Reddy, Siddhart; Jogdand, Harsh; Sarangi, Vamsi; Ganneru, Brunda; Prasad, Sai; Das, Dipankar; Dugyala, Raju; Praturi, Usha; Sakpal, Gajanan; Yadav, Pragya; Reddy, Prabhakar; Verma, Savita; Singh, Chandramani; Redkar, Sagar Vivek; Singh, Chandramani; Gillurkar, Chandra Sekhar; Kushwaha, Jitendra Singh; Mohapatra, Satyajit; Mohapatra, Satyajit; Bhate, Amit; Rai, Sanjay; Panda, Samiran; Abraham, Priya; Gupta, Nivedita; Ella, Krishna; Bhargav, Balram; Vadrevu, Krishna Mohan (8 March 2021). “Safety and immunogenicity of an inactivated SARS-CoV-2 vaccine, BBV152: interim results from a double-blind, randomised, multicentre, phase 2 trial, and 3-month follow-up of a double-blind, randomised phase 1 trial”. The Lancet Infectious Diseases. doi:10.1016/S1473-3099(21)00070-0.
- ^ “Coronavirus | Covaxin Phase III trial from November”. The Hindu. 23 October 2020.
- ^ Ganneru B, Jogdand H, Daram VK, Molugu NR, Prasad SD, Kannappa SV, et al. (9 September 2020). “Evaluation of Safety and Immunogenicity of an Adjuvanted, TH-1 Skewed, Whole Virion InactivatedSARS-CoV-2 Vaccine – BBV152”. doi:10.1101/2020.09.09.285445. S2CID 221635203.
- ^ “An Efficacy and Safety Clinical Trial of an Investigational COVID-19 Vaccine (BBV152) in Adult Volunteers”. clinicaltrials.gov(Registry). United States National Library of Medicine. NCT04641481. Retrieved 26 November 2020.
- ^ “Bharat Biotech begins Covaxin Phase III trials”. The Indian Express. 18 November 2020.
- ^ Sen M (2 December 2020). “List of states that have started phase 3 trials of India’s first Covid vaccine”. mint.
- ^ “70%-80% Drop In Participation For Phase 3 Trials Of Covaxin: Official”. NDTV. 17 December 2020.
- ^ “Bharat Biotech’s Covaxin given conditional nod based on incomplete Phase 3 trial results data”. The Print. 3 January 2021.
- ^ Kumar, N. Ravi (3 March 2021). “Bharat Biotech says COVID-19 vaccine Covaxin shows 81% efficacy in Phase 3 clinical trials”. The Hindu.
- ^ “Inside the B.1.1.7 Coronavirus Variant”. The New York Times. 18 January 2021. Retrieved 29 January 2021.
- ^ Sapkal, Gajanan N.; Yadav, Pragya D.; Ella, Raches; Deshpande, Gururaj R.; Sahay, Rima R.; Gupta, Nivedita; Mohan, V. Krishna; Abraham, Priya; Panda, Samiran; Bhargava, Balram (27 January 2021). “Neutralization of UK-variant VUI-202012/01 with COVAXIN vaccinated human serum”. bioRxiv: 2021.01.26.426986. doi:10.1101/2021.01.26.426986. S2CID 231777157.
- ^ Hoeksema F, Karpilow J, Luitjens A, Lagerwerf F, Havenga M, Groothuizen M, et al. (April 2018). “Enhancing viral vaccine production using engineered knockout vero cell lines – A second look”. Vaccine. 36 (16): 2093–2103. doi:10.1016/j.vaccine.2018.03.010. PMC 5890396. PMID 29555218.
- ^ “Coronavirus vaccine update: Bharat Biotech’s Covaxin launch likely in Q2 of 2021, no word on pricing yet”. http://www.businesstoday.in. India Today Group. Retrieved 13 December2020.
- ^ “Odisha fast tracks coronavirus vaccine manufacturing unit”. The New Indian Express. 7 November 2020.
- ^ Raghavan P (24 September 2020). “Bharat Biotech exploring global tie-ups for Covaxin manufacturing”. The Indian Express.
- ^ Reuters Staff (22 December 2020). “Ocugen to co-develop Bharat Biotech’s COVID-19 vaccine candidate for U.S.” Reuters. Retrieved 5 January 2021.
- ^ “Bharat Biotech, Ocugen to co-develop Covaxin for US market”. The Economic Times. Retrieved 5 January 2021.
- ^ “Bharat Biotech inks pact with Precisa Med to supply Covaxin to Brazil”. mint. 12 January 2021.
- ^ Schmall E, Yasir S (3 January 2021). “India Approves Oxford-AstraZeneca Covid-19 Vaccine and 1 Other”. The New York Times. Retrieved 3 January 2021.
- ^ Jump up to:a b “Iran issues permit for emergency use for three other COVID-19 vaccines: Official”. IRNA English. 17 February 2021.
- ^ Jump up to:a b Manral, Karan (4 March 2021). “Zimbabwe approves Covaxin, first in Africa to okay India-made Covid-19 vaccine”. Hindustan Times. Retrieved 6 March 2021.
- ^ Ghosh N (7 December 2020). “Bharat Biotech seeks emergency use authorization for Covid-19 vaccine”. Hindustan Times.
- ^ “Coronavirus | After SII, Bharat Biotech seeks DCGI approval for Covaxin”. The Hindu. 7 December 2020.
- ^ “Expert panel recommends granting approval for restricted emergency use of Bharat Biotech’s Covaxin”. The Indian Express. 2 January 2021.
- ^ “Coronavirus: India approves vaccines from Bharat Biotech and Oxford/AstraZeneca”. BBC News. 3 January 2021. Retrieved 3 January 2021.
- ^ “Disputes Mount, but Heedless Govt Intent on Rolling Vaccine Candidates Out”. The Wire. 12 January 2021.
- ^ “AIPSN urges govt to reconsider emergency approval for Covaxin till Phase 3 data is published – Health News , Firstpost”. Firstpost. 8 January 2021.
External links
| Scholia has a profile for Covaxin / BBV152 (Q98703813). |
COVAXIN®, India‘s indigenous COVID-19 vaccine by Bharat Biotech is developed in collaboration with the Indian Council of Medical Research (ICMR) – National Institute of Virology (NIV).
The indigenous, inactivated vaccine is developed and manufactured in Bharat Biotech’s BSL-3 (Bio-Safety Level 3) high containment facility.
The vaccine is developed using Whole-Virion Inactivated Vero Cell derived platform technology. Inactivated vaccines do not replicate and are therefore unlikely to revert and cause pathological effects. They contain dead virus, incapable of infecting people but still able to instruct the immune system to mount a defensive reaction against an infection.
Why develop Inactivated Vaccine? Conventionally, inactivated vaccines have been around for decades. Numerous vaccines for diseases such as Seasonal Influenza, Polio, Pertussis, Rabies, and Japanese Encephalitis use the same technology to develop inactivated vaccines with a safe track record of >300 million doses of supplies to date. It is the well-established, and time-tested platform in the world of vaccine technology.
Key Attributes:
- COVAXIN® is included along with immune-potentiators, also known as vaccine adjuvants, which are added to the vaccine to increase and boost its immunogenicity.
- It is a 2-dose vaccination regimen given 28 days apart.
- It is a vaccine with no sub-zero storage, no reconstitution requirement, and ready to use liquid presentation in multi-dose vials, stable at 2-8oC.
- Pre-clinical studies: Demonstrated strong immunogenicity and protective efficacy in animal challenge studies conducted in hamsters & non-human primates. For more information about our animal study, please visit our blog page on Non-Human Primates.
- The vaccine received DCGI approval for Phase I & II Human Clinical Trials in July, 2020.
- A total of 375 subjects have been enrolled in the Phase 1 study and generated excellent safety data without any reactogenicity. Vaccine-induced neutralizing antibody titers were observed with two divergent SARS-CoV-2 strains. Percentage of all the side-effects combined was only 15% in vaccine recipients. For further information, visit our blog page on phase 1 study.
- In Phase 2 study, 380 participants of 12-65 years were enrolled. COVAXIN® led to tolerable safety outcomes and enhanced humoral and cell-mediated immune responses. Know more about our phase 2 study.

- A total of 25,800 subjects have been enrolled and randomized in a 1:1 ratio to receive the vaccine and control in a Event-Driven, randomized, double-blind, placebo-controlled, multicentre phase 3 study.
The purpose of this study is to evaluate the efficacy, safety, and immunogenicity of COVAXIN® in volunteers aged ≥18 years.
Of the 25,800 participants, >2400 volunteers were above 60 years of age and >4500 with comorbid conditions.
COVAXIN® demonstrated 81% interim efficacy in preventing COVID-19 in those without prior infection after the second dose.
COVAXIN® effective against UK variant strain:
Analysis from the National Institute of Virology indicates that vaccine-induced antibodies can neutralize the UK variant strains and other heterologous strains.
Global Acceptance of COVAXIN®:
Bharat biotech has been approached by several countries across the world for the procurement of COVAXIN®.
- Clinical trials in other countries to commence soon.
- Supplies from government to government in the following countries to take place: Mongolia, Myanmar, Sri Lanka, Philippines, Bahrain, Oman, Maldives and Mauritius.
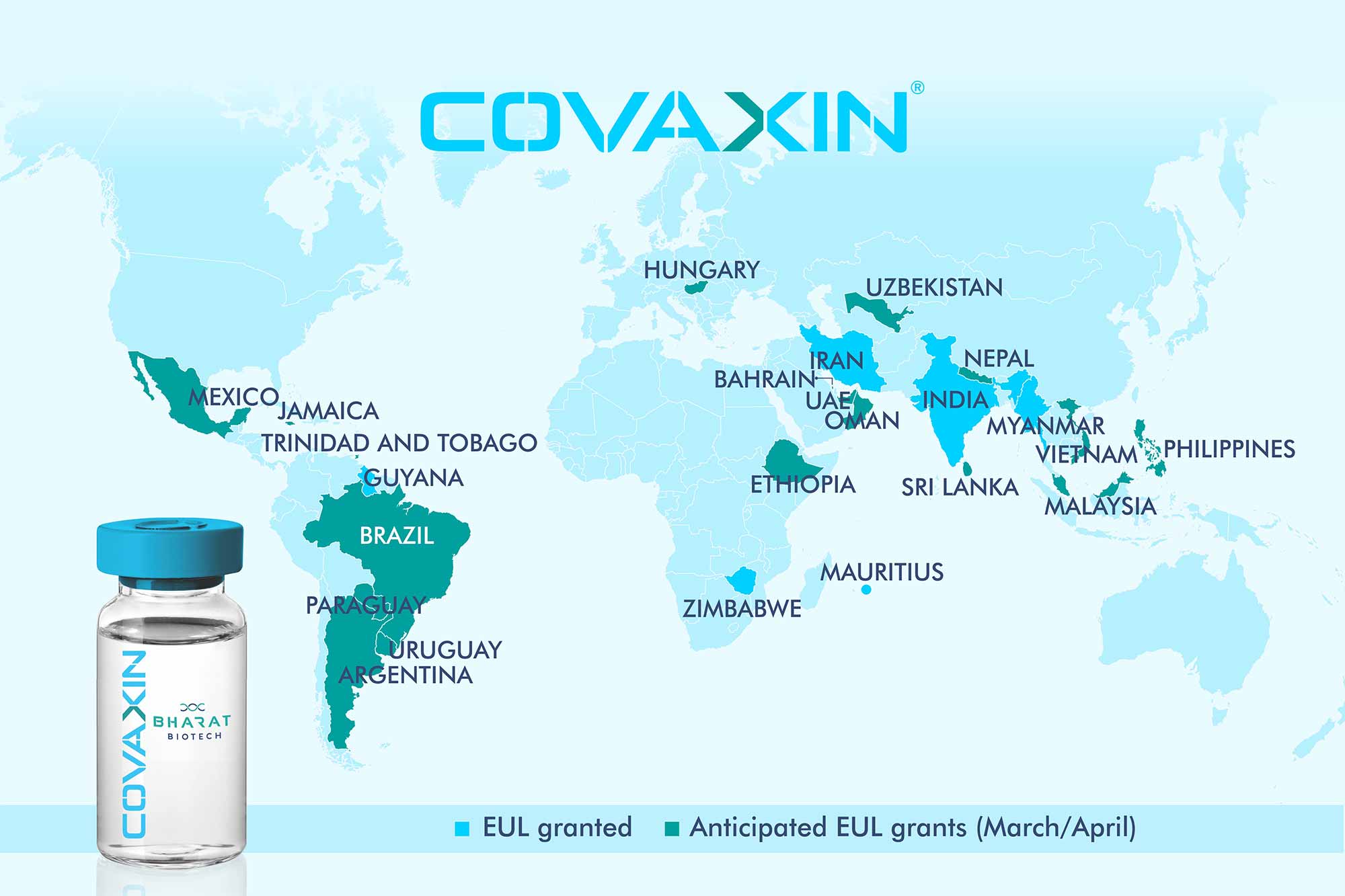
| A person holding a vial of the Covaxin vaccine | |
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | Inactivated |
| Clinical data | |
| Trade names | Covaxin |
| Routes of administration | Intramuscular |
| ATC code | None |
| Legal status | |
| Legal status | EUA : IND, IRN, ZBW |
| Identifiers | |
| DrugBank | DB15847 |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| SARS-CoV-2 (virus)COVID-19 (disease) |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showImpact |
| COVID-19 Portal |
| vte |
////////COVAXIN, BBV152, BBV 152, INDIA 2021, APPROVALS 2021, COVID 19, CORONA VIRUS, bharat biotech
#COVAXIN, #BBV152, #BBV 152, #INDIA 2021, #APPROVALS 2021, #COVID 19, #CORONA VIRUS, #bharat biotech

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
.svg){kind=link}