
New Website goflow.at online!

Professor at University of Graz
Institute of Chemistry
Univ.-Prof. Mag. Dr.rer.nat.
+43 316 380-5352
+43 (0) 316 380 – 9840
Oliver.kappe (at) uni-graz.at
Research in the Kappe lab focuses on flow chemistry, microreactor technology, process intensification and the continuous generation of active pharmaceutical ingredients (APIs). Check out our new webiste at: goflow.at
Recent Hot Papers from the Kappe Lab
Web of Science Highly Cited and Hot Article
Continuous Flow Technology – A Tool for the Manufacturing of Active Pharmaceutical Ingredients
B. Gutmann, D. Cantillo, C. O. Kappe, Angew. Chem. Int. Ed. 2015 , 54, 6688-6729.
DOI: 10.1002/anie.201409318
Chemistry – A European Journal Hot Paper
Continuous Flow Homolytic Aromatic Substitution with Electrophilic Radicals – A Fast and Scalable Protocol for Trifluoromethylation
J. L. Monteiro, P. F. Carneiro, P. Elsner, D. Roberge, P. G. M. Wuts, K. Kurjan, B. Gutmann, C. O. Kappe,
Chem. Eur. J. 2017 , 23, in press.
DOI: 10.1002/chem.201604579
Journal of Organic Chemistry Featured Article
A Lab-Scale Membrane Reactor for the Generation of Anhydrous Diazomethane
D. Dallinger, V. D. Pinho, B. Gutmann, C. O. Kappe, J. Org. Chem. 2016 , 81, 5814-5823.
DOI: 10.1021/acs.joc.6b01190
Active Pharmaceutical Ingredients (APIs) in Flow
Continuous flow processes form the basis of the petrochemical and bulk chemicals industry where strong competition, stringent environmental and safety regulations, and low profit margins drive the need for highly performing, cost effective, safe and atom efficient chemical operations. In contrast to the commodity chemical industry, however, the fine chemical industry primarily relies on its existing infrastructure of multipurpose batch or semi-batch reactors. Fine chemicals, such as drug substances and active pharmaceutical ingredients (APIs), are generally considerably more complex than commodity chemicals and usually require numerous, widely diverse reaction steps for their synthesis (typically 6 to 10 synthetic steps), and multiple rounds of quenching, separation and purification. These requirements, together with the comparatively low production volumes and often short life time of many of these materials, make versatile and reconfigurable multipurpose batch reactors the technology of choice for their preparation. However, the advantages of continuous flow processing are increasingly being appreciated also by the pharmaceutical industry and, thus, a growing number of scientists, from research chemists in academia to process chemists and chemical engineers in pharmaceutical companies, are now starting to employ continuous flow technologies on a more routine basis. Together with our industrial partners, the Kappe laboratories are involved in numerous flow API synthesis projects.

Key Publications
Review: Continuous-Flow Technology—A Tool for the Safe Manufacturing of Active Pharmaceutical Ingredients
B. Gutmann, D. Cantillo, C. O. Kappe, Angew. Chem. Int. Ed. 2015, 54, 6688-6729. DOI: 10.1002/anie.201409318 (Web of Science “Highly Cited Paper”).
Towards the Synthesis of Noroxymorphone via Aerobic Palladium-Catalyzed Continuous Flow N-Demethylation Strategies. B. Gutmann, P. Elsner, D. P. Cox, U. Weigl, D. M. Roberge, C. O. Kappe, ACS Sust. Chem. Eng. 2016, 4, in press. DOI: 10.1021/acssuschemeng.6b01371
Batch and Continuous Flow Aerobic Oxidation of 14-Hydroxy Opioids to 1,3-Oxazolidines – A Concise Synthesis of Noroxymorphone
B. Gutmann, U. Weigl, D. P. Cox, C. O. Kappe, Chem. Eur. J. 2016, 22, 10393–10398. DOI:10.1002/chem.201601902 (selected as ”Hot Paper” by the Editors).
Selective Olefin Reduction in Thebaine Using Hydrazine Hydrate and O2 under Intensified Continuous Flow Conditions
B. Pieber, D. P. Cox, C. O. Kappe, Org. Process Res. Develop. 2016, 20, 376−385. DOI: 10.1021/acs.oprd.5b00370
Process Intensified Flow Synthesis of 1H-4-Substituted Imidazoles: Toward the Continuous Production of Daclatasvir
P. F. Carneiro, B. Gutmann, R. O. M. A. de Souza, C. O. Kappe, ACS Sust. Chem. Eng. 2015, 3, 3445−3453. DOI: 10.1021/acssuschemeng.5b01191
Continuous Flow Reduction of Artemisinic Acid Utilizing Multi-Injection Strategies – Closing the Gap Towards a Fully Continuous Synthesis of Antimalarial Drugs
B. Pieber, T. Glasnov, C. O. Kappe, Chem. Eur. J. 2015, 21, 4368-4376. DOI: 10.1002/chem.201406439 (selected as “Hot Paper“ by the Editors, covered by Chemical & Engineering News).
Development of a Continuous Flow Sulfoxide Imidation Protocol Using Azide Sources under Superacidic Conditions
B. Gutmann, P. Elsner, A. O’Kearney-McMullan, W. Goundry, D. M. Roberge, C. O. Kappe, Org. Process Res. Develop. 2015, 19, 1062-1067. DOI: 10.1021/acs.oprd.5b00217
Continuous Flow Synthesis of alpha-Haloketones – Essential Building Blocks of Antiretroviral Agents
V. D. Pinho, B. Gutmann, L. S. M. Miranda, R. O. M. A. de Souza, C. O. Kappe, J. Org. Chem. 2014, 79, 1555-1562. DOI: 10.1021/jo402849z (selected as “Featured Article” by the Editors).
Combined Batch and Continuous Flow Procedure to the Chemo-Enzymatic Synthesis of Biaryl Moiety of Odanacatib.
R. de Oliveira Lopes, A. S. de Miranda, B. Reichart, T. Glasnov, C. O. Kappe, R. C. Simon, W. Kroutil, L. S. M. Miranda, I. C. R.Leal, R. O. M. A. de Souza, J. Mol. Catal. B. 2014, 104, 101-107. DOI: 10.1016/j.molcatb.2014.03.017
On the Fischer Indole Synthesis of 7-Ethyltryptophol- Mechanistic and Process Intensification Studies under Continuous Flow Conditions.
B. Gutmann, M. Gottsponer, P. Elsner, D. Cantillo, D. M. Roberge, C. O. Kappe, Org. Process Res. Develop. 2013, 17, 294-302. DOI: 10.1021/op300363s
A Three Step Continuous Flow Synthesis of the Biaryl Unit of the HIV Protease Inhibitor Atazanavir.
L. Dalla-Vechia, B. Reichart, T. N. Glasnov, L. S. M. Miranda, C. O. Kappe, R. O. M. A. de Souza, Org. Biomol. Chem. 2013, 11, 6806-6813. DOI: 10.1039/c3ob41464g
A Scalable Two-Step Continuous Flow Synthesis of Nabumetone and Related 4-Aryl-2-butanones.
M. Viviano, T. N. Glasnov, B. Reichart, G. Tekautz, C. O. Kappe, Org. Process Res. Develop. 2011, 15, 858-870. DOI: 10.1021/op2001047


DR SANJAY BAJAJ,,,,,,,,,,,,,,,,,,,DR ANTHONY CRASTO…………PROF OLIVER KAPPE FLOW CHEM CONFERENCE , MUMBAI, 22 JAN 2015……SELECTBIO
////////////New Website, goflow.at, online, C. Oliver Kappe, University of Graz, flow chemistry

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....












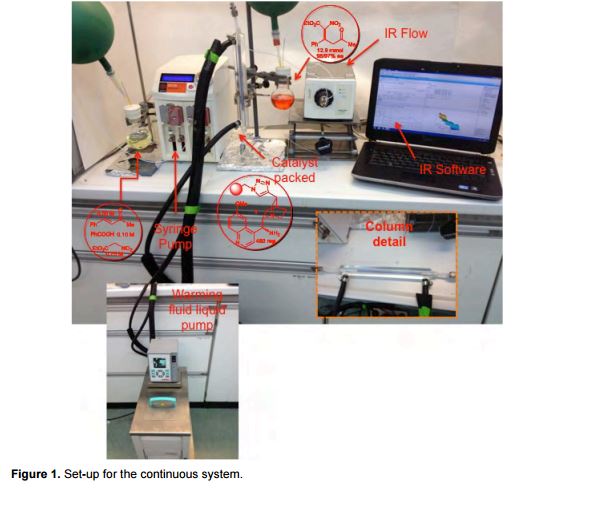













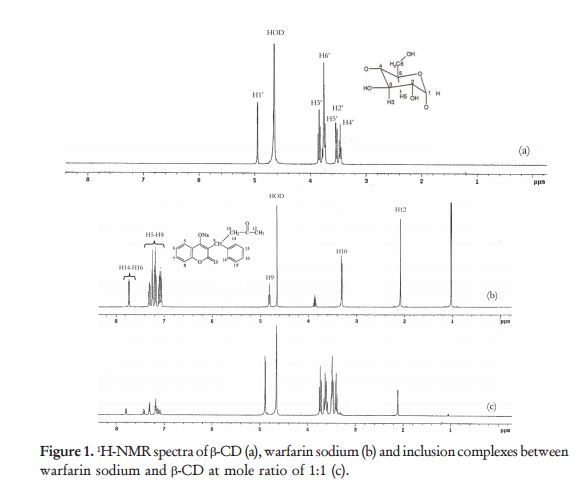
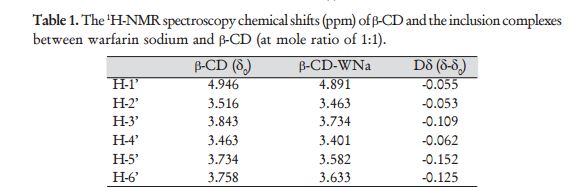











 Reaction of 4-hydroxycoumarin with benzylacetone under
Reaction of 4-hydroxycoumarin with benzylacetone under


 Vitamin K is a cofactor in the synthesis of blood clotting factors II, VII, IX and X*, this step occurs in the liver and involves the gammacarboxylation of the first 10 glutamic acid residues in the amino-terminal region of the prothrombin clotting factor to generategamma-carboxyglutamate. The gamma-carboxyglutamatee amino acid groups can chelate Ca2+ better than ten replaced glutamate residues, thus providing binding sites for four Vitamin Ks onto the phospholipid membrane during coagulation. The clotting occurs via a cascade*, a kind of biochemical chain reaction. {See
Vitamin K is a cofactor in the synthesis of blood clotting factors II, VII, IX and X*, this step occurs in the liver and involves the gammacarboxylation of the first 10 glutamic acid residues in the amino-terminal region of the prothrombin clotting factor to generategamma-carboxyglutamate. The gamma-carboxyglutamatee amino acid groups can chelate Ca2+ better than ten replaced glutamate residues, thus providing binding sites for four Vitamin Ks onto the phospholipid membrane during coagulation. The clotting occurs via a cascade*, a kind of biochemical chain reaction. {See  To work, the Vitamin K must be reduced to its quinol or hydroquinone form. This is achieved with Vitamin K Oxide reductase, which is the step inhibited by S-warfarin, being some three times more potent than R-warfarin. S-warfarin is metabolized primarily by the CYP2C9 enzyme of the cytochrome P450 system. The R-warfarin is metabolized by the two cytochrome P450 enzymes, CP1A4Y and CYP3A4. Warfarin is very soluble in water, and is absorbed into the blood stream within 90 minutes of taking the pills.
To work, the Vitamin K must be reduced to its quinol or hydroquinone form. This is achieved with Vitamin K Oxide reductase, which is the step inhibited by S-warfarin, being some three times more potent than R-warfarin. S-warfarin is metabolized primarily by the CYP2C9 enzyme of the cytochrome P450 system. The R-warfarin is metabolized by the two cytochrome P450 enzymes, CP1A4Y and CYP3A4. Warfarin is very soluble in water, and is absorbed into the blood stream within 90 minutes of taking the pills.





















































 Food in mumbai
Food in mumbai mumbai skyline
mumbai skyline










































