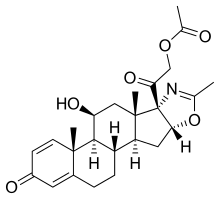
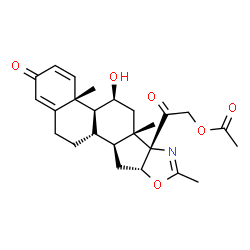
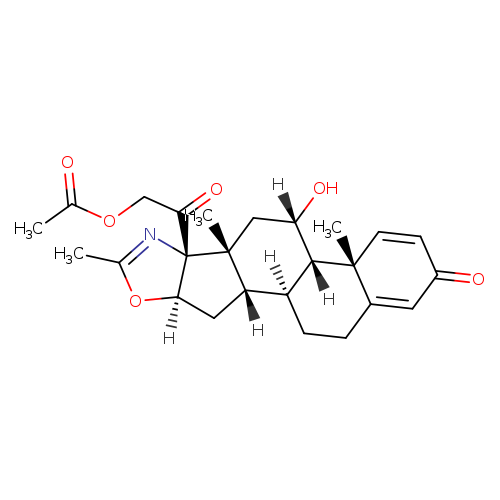
Deflazacort
- Molecular Formula C25H31NO6
- Average mass 441.517 Da
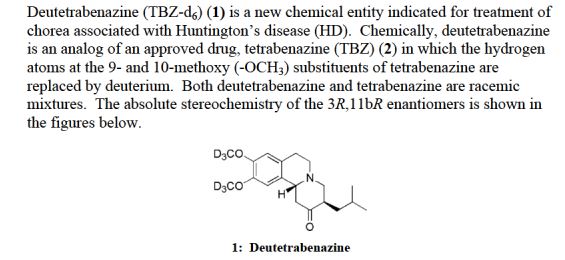
(11b,16b)-21-(Acetyloxy)-11-hydroxy-2′-methyl-5’H-pregna-1,4-dieno[17,16-d]oxazole-3,20-dione
11b,21-Dihydroxy-2′-methyl-5’bH-pregna-1,4-dieno[17,16-d]oxazole-3,20-dione 21-acetate
2-[(4aR,4bS,5S,6aS,6bS,9aR,10aS,10bS)-5-Hydroxy-4a,6a,8-trimethyl-2-oxo-2,4a,4b,5,6,6a,9a,10,10a,10b,11,12-dodecahydro-6bH-naphtho[2′,1′:4,5]indeno[1,2-d][1,3]oxazol-6b-yl]-2-oxoethyl acetate
- 5’βH-Pregna-1,4-dieno[17,16-d]oxazole-3,20-dione, 11β,21-dihydroxy-2′-methyl-, 21-acetate (8CI)
- (11β,16β)-21-(Acetyloxy)-11-hydroxy-2′-methyl-5’H-pregna-1,4-dieno[17,16-d]oxazole-3,20-dione
- 2H-Naphth[2′,1′:4,5]indeno[1,2-d]oxazole, 5’H-pregna-1,4-dieno[17,16-d]oxazole-3,20-dione deriv.
- Azacort
- Azacortinol
- Calcort
| Optical Rotatory Power |
+62.3 ° |
Conc: 0.5 g/100mL; Solv: chloroform (67-66-3); Wavlength: 589.3 nm |
…………..REF, “Drugs – Synonyms and Properties” data were obtained from Ashgate Publishing Co. (US)Hoechst Marion Roussel (now Aventis Pharma) has developed and launched Deflazacort (Dezacor; Flantadin; Lantadin; Calcort) a systemic corticosteroid developed for the treatment of a variety of inflammatory conditions .
In March 1990, the drug was approved in Spain, and by January 2013, the drug had been launched by FAES Farma . By the end of 1999, the product had been launched in Germany, Italy, Belgium, Switzerland and South Korea
Deflazacort is a corticosteroid first launched in 1985 by Guidotti in Europe for the oral treatment of allergic asthma, rheumatoid arthritis, arthritis, and skin allergy.
In 2017, an oral formulation developed at Marathon Pharmaceuticals was approved by the FDA for the treatment of Duchenne’s muscular dystrophy in patients 5 years of age and older.
Deflazacort (trade name Emflaza or Calcort among others) is a glucocorticoid used as an anti-inflammatory and immunosuppressant.
In 2013, orphan drug designation in the U.S. was assigned to the compound for the treatment of Duchenne’s muscular dystrophy. In 2015, additional orphan drug designation in the U.S. was assigned for the treatment of pediatric juvenile idiopathic arthritis (JIA) excluding systemic JIA.
Also in 2015, deflazacort was granted fast track and rare pediatric disease designations in the U.S. for the treatment of Duchenne’s muscular dystrophy.
| Deflazacort is a glucocorticoid used as an anti-inflammatory and immunosuppressant. It was approved in February, 2017 by the FDA for use in treatment of Duchenne muscular dystrophy (trade name Emflaza). |
- Aventis Pharma (Originator), Lepetit (Originator), Guidotti (Licensee), Shire Laboratories (Licensee)
February 9, 2017 FDA approved
The U.S. Food and Drug Administration today approved Emflaza (deflazacort) tablets and oral suspension to treat patients age 5 years and older with Duchenne muscular dystrophy (DMD), a rare genetic disorder that causes progressive muscle deterioration and weakness. Emflaza is a corticosteroid that works by decreasing inflammation and reducing the activity of the immune system.
Corticosteroids are commonly used to treat DMD across the world. This is the first FDA approval of any corticosteroid to treat DMD and the first approval of deflazacort for any use in the United States.

“This is the first treatment approved for a wide range of patients with Duchenne muscular dystrophy,” said Billy Dunn, M.D., director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “We hope that this treatment option will benefit many patients with DMD.”
DMD is the most common type of muscular dystrophy. DMD is caused by an absence of dystrophin, a protein that helps keep muscle cells intact. The first symptoms are usually seen between 3 and 5 years of age and worsen over time. The disease often occurs in people without a known family history of the condition and primarily affects boys, but in rare cases it can affect girls. DMD occurs in about one of every 3,600 male infants worldwide.
People with DMD progressively lose the ability to perform activities independently and often require use of a wheelchair by their early teens. As the disease progresses, life-threatening heart and respiratory conditions can occur. Patients typically succumb to the disease in their 20s or 30s; however, disease severity and life expectancy vary.
The effectiveness of deflazacort was shown in a clinical study of 196 male patients who were 5 to 15 years old at the beginning of the trial with documented mutation of the dystrophin gene and onset of weakness before age 5. At week 12, patients taking deflazacort had improvements in a clinical assessment of muscle strength across a number of muscles compared to those taking a placebo. An overall stability in average muscle strength was maintained through the end of study at week 52 in the deflazacort-treated patients. In another trial with 29 male patients that lasted 104 weeks, deflazacort demonstrated a numerical advantage over placebo on an assessment of average muscle strength. In addition, although not statistically controlled for multiple comparisons, patients on deflazacort appeared to lose the ability to walk later than those treated with placebo.
The side effects caused by Emflaza are similar to those experienced with other corticosteroids. The most common side effects include facial puffiness (Cushingoid appearance), weight gain, increased appetite, upper respiratory tract infection, cough, extraordinary daytime urinary frequency (pollakiuria), unwanted hair growth (hirsutism) and excessive fat around the stomach (central obesity).
Other side effects that are less common include problems with endocrine function, increased susceptibility to infection, elevation in blood pressure, risk of gastrointestinal perforation, serious skin rashes, behavioral and mood changes, decrease in the density of the bones and vision problems such as cataracts. Patients receiving immunosuppressive doses of corticosteroids should not be given live or live attenuated vaccines.
The FDA granted this application fast track designation and priority review. The drug also received orphan drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The sponsor is receiving a rare pediatric disease priority review voucher under a program intended to encourage development of new drugs and biologics for the prevention and treatment of rare pediatric diseases. A voucher can be redeemed by a sponsor at a later date to receive priority review of a subsequent marketing application for a different product. This is the ninth rare pediatric disease priority review voucher issued by the FDA since the program began.
Emflaza is marketed by Marathon Pharmaceuticals of Northbrook, Illinois.
Medical uses
The manufacturer lists the following uses for deflazacort:[1]
In the United States, deflazacort is only FDA-approved for the treatment of Duchenne muscular dystrophy in people over the age of 5.




Adverse effects
Deflazacort carries the risks common to all corticosteroids, including immune suppression, decreased bone density, and endocrine insufficiency. In clinical trials, the most common side effects (>10% above placebo) were Cushing’s-like appearance, weight gain, and increased appetite.[2]
Pharmacology
Mechanism of action
Deflazacort is an inactive prodrug which is metabolized rapidly to the active drug 21-desacetyldeflazacort.[3]
Relative potency
Deflazacort’s potency is around 70–90% that of prednisone.[4] A 2017 review found its activity of 7.5 mg of deflazacort is approximately equivalent to 25 mg cortisone, 20 mg hydrocortisone, 5 mg of prednisolone or prednisone, 4 mg of methylprednisolone or triamcinolone, or 0.75 mg of betamethasone or dexamethasone. The review noted that the drug has a high therapeutic index, being used at initial oral doses ranging from 6 to 90 mg, and probably requires a 50% higher dose to induce the same demineralizing effect as prednisolone. Thus it has “a smaller impact on calcium metabolism than any other synthetic corticosteroid, and therefore shows a lower risk of growth rate retardation in children and of osteoporosis” in the elderly, and comparatively small effects on carbohydrate metabolism, sodium retention, and hypokalemia.[5]
History
In January 2015, the FDA granted fast track status to Marathon Pharmaceuticals to pursue approval of deflazacort as a potential treatment for Duchenne muscular dystrophy, a rare, “progressive and fatal disease” that affects boys.[6] Although deflazacort was approved by the FDA for use in treatment of Duchenne muscular dystrophy on February 9, 2017,[7][8] Marathon CEO announced on February 13, 2017 that the launch of deflazacort (Emflaza) would be delayed amidst controversy over the steep price Marathon was asking for the drug – $89,000-a-year. In Canada the same drug can be purchased for around $1 per tablet.[9] Marathon has said that Emflaza is estimated to cost $89,000/year which is “roughly 70 times” more than it would cost overseas.[10] Deflazacort is sold in the United Kingdom under the trade name Calcort;[4] in Brazil as Cortax, Decortil, and Deflanil; in India as Moaid, Zenflav, Defolet, DFZ, Decotaz, and DefZot; in Bangladesh as Xalcort; in Panama as Zamen; Spain as Zamene; and in Honduras as Flezacor.[11]
SYNTHESIS

Worlddrugtracker drew this
1 Protection of the keto groups in pregna-1,4-diene derivative with NH2NHCOOMe using HCOOH, yields the corresponding methyl ester.
2 Cleavage of epoxide with NH3 in DMAc/DMF gives amino-alcohol,
3 which on esterification with acetic anhydride in the presence of AcOH furnishes acetate.
4 Cyclization of amine using NaOH, Na2CO3 or K2CO3 produces oxazoline derivative ,
5 which is finally deprotected with HCl to afford Deflazacort
SYNTHESIS FROM CHEMDRUG

The cyclization of 17alpha-azido-3beta,16alpha-acetoxy-5alpha-pregnane-11,20-dione (I) by hydrogenation with H2 over Pt in methanol, followed by a treatment with 10% HCl gives 3beta-hydroxy-5alpha-pregnane-11,20-dione-[17alpha,16alpha-d]-2′-methyloxazoline (II), which is converted into the semicarbazone (III) by treatment with semicarbazide hydrochloride (A) and pyridine in refluxing methanol. The reduction of one ketonic group of (III) with NaBH4 in refluxing ethanol yields the dihydroxy-semicarbazone (IV), which is hydrolyzed with 10% HCl in refluxing methanol to afford the ketodiol (V). The oxidation of (V) with cyclohexanone and aluminum isopropoxide in refluxing toluene gives 11beta-hydroxy-5alpha-pregnane-3,20-dione-[17alpha,16alpha-d]-2′-methyloxazoline (VI). The dehydrogenation of (VI) by treatment with Br2 in dioxane-acetic acid, followed by treatment with Li2CO3 in DMF at 140 C yields the corresponding 1,4-diene derivative (VII). Finally, the reaction of (VII) with I2 by means of azobisisobutyronitrile in CH2Cl2 affords the corresponding 21-iodo compound, which is then acetylated with triethylammonium acetate in refluxing acetone.

The monoacetylation of (V) with acetic anhydride and pyridine at 100 C gives the 3-acetoxy-11-hydroxy compound (IX), which is dehydrated by treatment with methanesulfonyl chloride and then with sodium acetate yielding 3beta-acetoxy-5alpha-pregn-9(11)-ene-20-one-[17alpha,16alpha-d]-2′-methyloxazoline (X). The hydrolysis of (X) with KOH in refluxing methanol affords the corresponding hydroxy compound (XI), which is acetoxylated by treatment with I2 and AZBN as before giving the iodo derivative (XII), and then with triethylammonium acetate also as before, yielding 3beta-hydroxy-21-acetoxy-5alpha-pregn-9(11)-ene-20-one-[17alpha,16alpha-d]-2′-methyloxazoline (XIII). The oxidation of (XIII) with CrO3 in acetone yields the 3,20-diketone (XIV), which by treatment with Br2 and Li2CO3 as before is dehydrogenated affording the 1,4,9(11)-pregnatriene (XV). Finally, the reaction of (XV) with N-bromoacetamide in THF yields 9alpha-bromo-11beta-hydroxy-21-acetoxy-5alpha-pregna-1,4-dieno-3,20-dione-[17alpha,16alpha-d]-2′-methyloxazoline (XVI), which is then debrominated by reaction with chromous acetate and butanethiol in DMSO.
PAPER
Journal of Medicinal Chemistry (1967), 10(5), 799-802
Steroids Possessing Nitrogen Atoms. III. Synthesis of New Highly Active Corticoids. [17α,16α,-d]Oxazolino Steroids
J. Med. Chem., 1967, 10 (5), pp 799–802
DOI: 10.1021/jm00317a009
PATENT
CN 105622713
PATENT CN 106008660
MACHINE TRANSLATED FROM CHINESE may seem funny



Description of the drawings
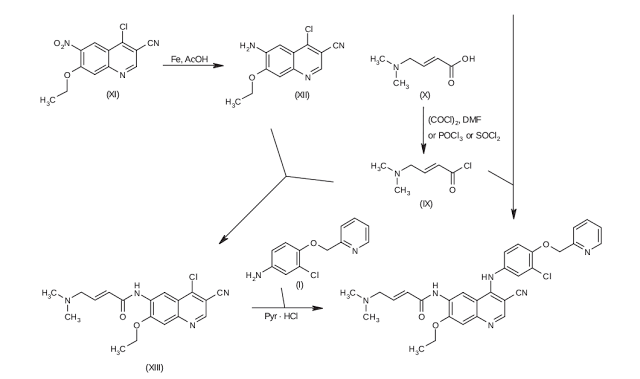
[0007] Figure 1 is a map of the traditional method of the combination process;
Figure 2 is a two-step method of the present invention.
detailed description
[0008] In order to more easily illustrate the gist and spirit of the present invention, the following examples illustrate:
Example 1
A: Preparation of hydroxylamine
In a 100 ml three-necked flask, 20 g of 16 (17) a-epoxy prednisolone, 30 ml of DMF, 300 ml of chloroform was added and incubated at 30-35 ° C with 8 g of ammonia gas at 1-2 atmospheres Reaction 16 ~ 20 hours, TLC detection reaction end point, after the reaction, the vacuum exhaust ammonia gas, add 3x100ml saturated brine washing 3 times, plus 10ml pure water washing times, then, under reduced pressure to chloroform to dry, add 200ml Ethyl acetate, Ig activated carbon, stirring reflux 60-90 minutes, cooling to 50-55 degrees, hot filter, l-2ml ethyl acetate washing carbon, combined filtrate and lotion, and then below 500C concentrated under pressure 95 % Of ethyl acetate, the system cooled to -5-0 ° C, stirring crystallization 2 ~ 3 hours, filter, 0.5-lml ethyl acetate washing, lotion and filtrate combined sets of approved; filter cake below 70 ° C Drying, get hydroxylamine 18.2g, HPLC content of 99.2%, weight loss of 91%.
[0009] B: Preparation of terracavir
Add 10 g of hydroxylamine, 150 ml of glacial acetic acid and 150 ml of acetic anhydride in a 100 ml three-necked flask. Add 5 g of concentrated sulfuric acid under stirring at room temperature. The reaction was carried out at 30-35 ° C for 12-16 hours. TLC confirmed the end of the reaction. Add 500ml of pure water, and adjust the pH of 7.5.5 with liquid alkali, cool to 10 ~ 15 ° C, stirring crystallization 2-3 hours, filtration, washing to neutral, combined filtrate and lotion, pretreated into Waste water treatment tank, filter cake below 70 V drying, Texaco can be special crude 112.5g, HPLC content of 98.2%, the yield of 112.5% ο the above terracotta crude dissolved in 800ml of alcohol, add 5g activated carbon, Decolorization 1-1.5 hours, hot filter, 10ml alcohol detergent cake, lotion and filtrate combined, atmospheric pressure recovery of about 90% of the alcohol, and then cooled to -5-0 ° C, frozen crystal 2-3 hours, Filtration, filter cake with 4-5ml alcohol washing, 70 ° C below drying, digoxin special product 89.2g, melting point 255.5-256.0 degrees, HPLC content of 99.7%, yield 89.2%. The mother liquor is recycled with solvent and crude.
[0010] Example II
A: Preparation of hydroxylamine
In a 100 ml three-necked flask, 20 g of 16 (17) a-epoxy prednisolone, 120 ml of toluene was added and incubated at 30-35 ° C with 8 g of ammonia and 16 to 20 at atmospheric pressure The reaction was carried out in the presence of 3 x 50 ml of saturated brine and 50 ml of pure water was added. Then, the toluene was dried under reduced pressure to dryness, and 200 ml of ethyl acetate, Ig activated carbon was added, and the mixture was stirred. Reflux 60-90 minutes, cool to 50-55 ° C, hot filter, l2ml ethyl acetate wash carbon, combined filtrate and lotion, and then below 500C under reduced pressure 95% ethyl acetate, the system cooling To 5-0C, stirring crystallization 2 ~ 3 hours, filter, 0.5-lml ethyl acetate washing, lotion and filtrate combined sets of the next batch; filter cake 70 ° C below drying, hydroxylamine 18.0g, HPLC content 99.1%, 90% by weight.
[0011] B: Preparation of terracavir
Add 10 g of hydroxylamine, 500 ml of chloroform and 150 ml of acetic anhydride in a 100 ml three-necked flask, add 5 g of p-toluenesulfonic acid under stirring at room temperature, and incubate at 30-35 ° C for 12-16 hours. TLC confirms the reaction end, After the addition of 500ml of pure water, and with the liquid alkali pH 7.55, down to 10 ~ 15 ° C, stirring 0.5_1 hours, separate the water layer, washed to neutral, combined with water and lotion, pretreated into Waste water treatment tank, organic layer under reduced pressure concentrated chloroform to near dry, adding 200ml hexane, reflux 0.5-1 hours, slowly cooling to -5 ~ O0C, stirring crystallization 2-3 hours, filter, filter cake with 4-5ml Alcohol washing, the filtrate and lotion combined apply to the next batch, the filter cake below 70 ° C drying, Texaco can crude 110.5g, HPLC content of 98.4%, the yield of 110.5%. The above-mentioned diltiazem crude product dissolved in 800ml alcohol, add 5g activated carbon, temperature reflux bleaching 1-1.5 hours, hot filter, 10ml alcohol washing cake, lotion and filtrate combined, atmospheric pressure recovery of about 90% of the alcohol And then cooled to -500C, frozen crystallization for 2-3 hours, filtration, filter cake with 4-5ml alcohol washing, 70 ° C the following drying, digester can special products 88.6g, melting point 255.0-256.0 degrees, HPLC content of 99.5%, the yield of 88.6%. The mother liquor is recycled with solvent and crude.
[0012] Example 3
A: Preparation of hydroxylamine
Add 20 g of 16 (17) a-epoxy prednisolone to 120 ml of ethanol in a 100 ml three-necked flask and incubate at 30-35 ° C with stirring to give Sg ammonia at 16 to 20 hours , TLC test reaction end point, after the reaction, vacuum exhaust ammonia gas, concentrated ethanol to the near dry, cooling, adding 300ml chloroform, stirring dissolved residue, and then add 3x100ml saturated brine washing, plus 10ml pure water washing, washing And then concentrated to reduce the chloroform to dry, add 200ml of ethyl acetate, Ig activated carbon, stirring reflux 60-90 minutes, cooling to 50-55 ° C, hot filter, l2ml ethyl acetate washing carbon, combined filtrate and lotion And then concentrated below 50 ° C to 95% ethyl acetate under reduced pressure. The system was cooled to -5-0 0C, stirred for 2 to 3 hours, filtered, 0.5-l of ethyl acetate, washed and filtrate The filter cake was dried at 70 ° C, 18.6 g of hydroxylamine, 99.5% of HPLC, and 93% by weight.
[0013] B: Preparation of terracavir
In a 100ml three-necked flask, add 10g of hydroxylamine, 500ml toluene, 150ml acetic anhydride, stirring at room temperature by adding 5g concentrated sulfuric acid, insulation at 30-35 degrees stirring reaction 12-16 hours, TLC confirmed the end of the reaction, after the reaction, Add 500ml of pure water, and liquid pH adjustment pH 7.5, cooling to 1 ~ 15 ° C, stirring 0.5-1 hours, the water layer, washed to neutral, combined with water and lotion, pretreated into the wastewater The cells were dried and the organic layer was concentrated to dryness under reduced pressure. 200 ml of hexane was added and refluxed
0.5-1 hours, slowly cool to -5 ~ O0C, stirring crystallization 2-3 hours, filtration, filter cake with 4-5ml hexane, the filtrate and lotion combined apply to the next batch, filter cake below 70 ° C Drying, digoxin crude 112.5g, HPLC content of 97.4%, the yield of 112,5% ο will be the above terracotta crude dissolved in 800ml of alcohol, add 5g activated carbon, heating reflux bleaching 1-1.5 hours, while Hot filter, 10ml alcohol detergent cake, lotion and filtrate combined, atmospheric pressure recovery of about 90% of the alcohol, and then cooled to -500C, frozen crystallization for 2-3 hours, filter, filter cake with 4-5ml alcohol Washing, 70 ° C below the dry, Diges can special products 86.2g, melting point 255.5-256.0 degrees, HPLC content of 99.8%, the yield of 86.2%. The mother liquor is recycled with solvent and crude.
PATENT
https://www.google.com/patents/CN101418032A?cl=en


Example 1
21- bromo -ll (3- hydroxy – pregna–l, 4- diene -3, 20-dione [170, 16o-d] -2′- methyl-oxazoline (4) Preparation:
A dry fitted with a thermometer, a reflux condenser, magnetically stirred flask was added 250mL three compound (2) (19.17 g; Fw: 383.48; 50 mmol), N- bromosuccinimide (9.79 g; Fw: 178.00; 55 mmol), 150 ml of ether; then ammonium acetate (0.39 g; Fw: 77.08; 0.005 mmol) added to the system. System continues to stir at 20 ° C 0.5 h, the reaction is complete. After completion of the reaction was filtered to remove the white precipitate cake was washed with 50 mL of dichloromethane, and the combined organic Xiangde pale yellow clear liquid, the solvent was evaporated under reduced pressure to give a pale yellow solid 21.27 g, yield: 92%, HPLC content of greater than 95%.
Example 2
21- bromo -lip- hydroxy – pregna–l, 4- diene -3, 20-dione [17 “16o-d] -2′- methyl-oxazoline (4) Preparation:
A dry fitted with a thermometer, a reflux condenser, magnetically stirred flask were added sequentially 250mL three compound (2) (19.17 g; Fw: 383.48; 50 mmol), N- bromosuccinimide (9.79 g; Fw : 178.00; 55 mmol), 150 ml of toluene; then ammonium acetate (0.39 g; Fw: 77.08; 0.005 mmol) added to the system. System continues to stir at 110 ° C 5 h, the reaction is complete. After completion of the reaction was cooled to room temperature, the white precipitate was removed by filtration cake was washed with 50 mL of dichloromethane, and the combined organic Xiangde pale yellow clear liquid, concentrated under reduced pressure to remove the solvent to give a pale yellow solid 19.65 g, yield: 85%, HPLC content greater than 95%.
Example 3
21 Jie bromo -11 – hydroxy – pregna-1,4-diene -3, 20-dione [17a, 16o-d] -2′- methyl-oxazoline (4) Preparation:
A dry fitted with a thermometer, a reflux condenser, magnetically stirred flask were added sequentially 250mL three compound (2) (19.17 g; Fw: 383.48; 50 mmol), 1,3- dibromo-5,5-dimethyl- Hein (35.74 g; Fw: 285.94; 125 mmol), 150 ml of ether; then ammonium acetate (0.39 g; Fw: 77.08; 0.005 mmol) added to the system. System Stirring was continued at reflux for 3 h, the reaction was completed. After completion of the reaction a white precipitate was removed by filtration and the cake was washed with 50 mL of diethyl ether, and the combined organic Xiangde pale yellow clear liquid, concentrated under reduced pressure to remove the solvent to give a pale yellow solid 16.18 g, yield: 70%, HPLC content greater than 92%.
Example 4
21- bromo -11 Jie – hydroxy – pregna-1,4-diene -3, 20- dione [17c, 16o-d] -2′- methyl-oxazoline (4) Preparation:
A dry fitted with a thermometer, a reflux condenser, magnetically stirred flask were added sequentially 250mL three compound (2) (19.17 g; Fw: 383.48; 50 mmol), 1,3- dibromo-5,5-dimethyl- Hein (35.74 g; Fw: 285.94; 125 mmol), 150 ml dichloromethane; followed by ammonium acetate (0.039 g; Fw: 77.08; 0.0005 mmol) added to the system. System Stirring was continued at reflux for 24 h, the reaction was completed. After completion of the reaction a white precipitate was removed by filtration and the cake was washed with 50 mL of diethyl ether, and the combined organic Xiangde pale yellow clear liquid, concentrated under reduced pressure to remove the solvent to give a pale yellow solid 16.41 g, yield: 71%, HPLC content of greater than 92. / 0.
Example 5
Deflazacort Preparation:
In a nitrogen-filled dry fitted with a thermometer, magnetic stirring and a reflux condenser 100 mL three-necked flask was charged with Compound (4) (11.56 g; Fw: 462.38; 25 mmol), followed by addition of sodium acetate (8.20g; Fw: 82.03; lOOmmol), 50 mL methanol was added to the system.
Then tetrabutylammonium bromide (O. 81g; Fw: 322.38; 2.5 mmol). Warmed to 50 ° C with stirring
48 h. Until after the completion of the reaction was cooled to room temperature. After completion of the reaction, temperature of the system was cooled to room temperature, the system was supplemented with chloroform 50mL, filtered, and the filter cake was washed with small amount of chloroform and then to confirm that no product was dissolved, and the combined organic phases, the organic phase washed with 10% aqueous sodium carbonate paint 3 times, saturated sodium chloride once. The organic phase was dried over anhydrous sodium sulfate, the inorganic salt was removed to give a pale yellow liquid, was concentrated to dryness, purified ethyl acetate to give the product 9.93g, yield 90%, HPLC content> 990/0.
Example 6
Deflazacort Preparation –
In a nitrogen-filled dry fitted with a thermometer, magnetic stirring and a reflux condenser 100 mL three-necked flask was charged with Compound (4) (11.56 g; Fw: 462.38; 25 mmol), followed by addition of anhydrous potassium acetate (3.68g; Fw: 98.14; 37.5 mmol), 50 mL acetone was added to the system. Followed by tetrabutylammonium iodide (0.10g; Fw: 369.37; 0.25 mmol). Heated to reflux with stirring 2h. Until after the completion of the reaction was cooled to room temperature. After completion of the reaction, temperature of the system was cooled to room temperature, the system was supplemented with chloroform 50mL, filtered, and the filter cake was washed with small amount of chloroform and then to confirm that no product was dissolved, and the combined organic phases, the organic phase was washed 3 times with 10% aqueous sodium carbonate , washed once with saturated sodium chloride. The organic phase was dried over anhydrous sodium sulfate, the inorganic salt was removed to give a pale yellow liquid, was concentrated to dryness, ethyl acetate was purified to give the product 10.93 g, yield 99%, HPLC content> 99%.
Example 7
Deflazacort Preparation:
In a nitrogen-filled dry fitted with a thermometer, magnetic stirring and a reflux condenser 100 mL three-necked flask was charged with Compound (4) (11.56 g; Fw: 462.38; 25 mmol), followed by addition of anhydrous potassium acetate (3.68g; Fw: 98.14; 37.5 mmol), 50 mL acetonitrile was added to the system. Followed by tetrabutylammonium iodide (0.10g; Fw: 369.37; 0.25 mmol). Heated to reflux with stirring 2h. Until after the completion of the reaction was cooled to room temperature. After completion of the reaction, temperature of the system was cooled to room temperature, the system was supplemented with chloroform 50mL, filtered, and the filter cake was washed with small amount of chloroform and then to confirm that no product was dissolved, and the combined organic phases, the organic phase was washed 3 times with 10% aqueous sodium carbonate , washed once with saturated sodium chloride. The organic phase was dried over anhydrous sodium sulfate, the inorganic salt was removed to give a pale yellow liquid, was concentrated to dryness, ethyl acetate was purified to give the product 10.93 g, yield 99%, HPLC content> 99%.
Example 8
Deflazacort Preparation:
In a nitrogen-filled dry fitted with a thermometer, magnetic stirring and a reflux condenser 100 mL three-necked flask was charged with Compound (4) (11.56 g; Fw: 462.38; 25 mmol), followed by addition of anhydrous potassium acetate (2.45g; Fw: 98.14; 25 mmol), the N, N- dimethylformamide, 50 mL added to the system. Followed by tetrabutylammonium iodide (O.IO g; Fw: 369.37; 0.25 mmol). Warmed to 120. C stirring 2h. Until after the completion of the reaction was cooled to room temperature. After completion of the reaction, temperature of the system was cooled to room temperature, the system was supplemented with chloroform 50mL, filtered, and the filter cake was washed with small amount of chloroform and then to confirm that no product was dissolved, and the combined organic phases, the organic phase was washed 3 times with 10% aqueous sodium carbonate , washed once with saturated sodium chloride. The organic phase was dried over anhydrous sodium sulfate, the inorganic salt was removed to give a pale yellow liquid, was concentrated to dryness, ethyl acetate was purified to give the product 10.93 g, yield 99%, HPLC content> 99o / q.
PATENT
https://www.google.com/patents/WO1997021722A1?cl=zh
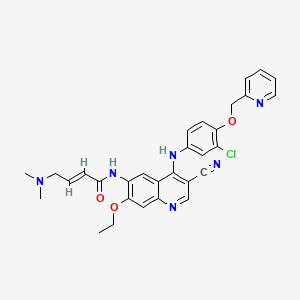
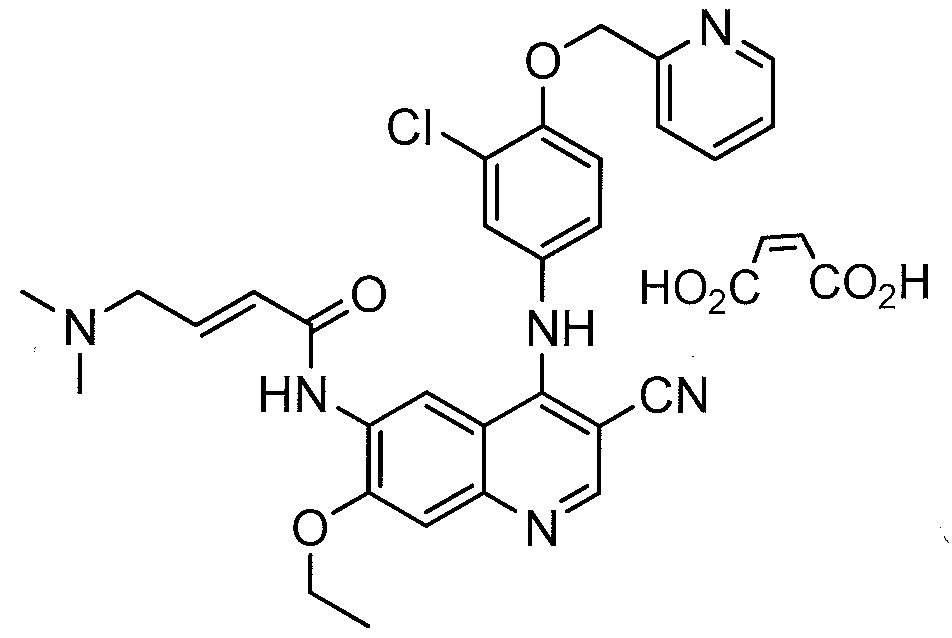
compound (llβ,16β)-21-(acetyloxy)-11- hydroxy-2 ‘ -methyl-5 ‘H-pregna-1, -dieno[17 , 16-d Joxazole- 3,20-dione, also known, and hereinafter referred to, with the INN (International Nonproprietary Name) deflazacort. Deflazacort is represented by the following formula I
Deflazacort is employed in therapy aince some years as a calcium-sparing corticoid agent. This compound belongs to the more general class of pregneno-oxazolines, for which anti-inflammatory, glucocorticoid and hormone-like pharmacological activities are reported. Examples of compounds of the above class, comprising deflazacort, are disclosed in US 3413286, where deflazacort is referred to as llβ-21-dihydroxy-2 ‘ -methyl-5 ‘ βH-pregna-1,4-dieno.17 , 16- d]oxazole-3,20-dione 21-acetate.
According to the process disclosed by US 3413286, deflazacort is obtained from 5-pregnane-3β-ol-ll , 20- dione-2 ‘-methyloxazoline by 2 , -dibromination with Br2– dioxane, heating the product in the presence of LiBr- iC03 for obtaining the 1,4-diene, and converting this latter into the 21-iodo and then into the desired 21- acetyloxy compound. By hydrolysis of deflazacort, the llβ-21-dihydroxy-2 ‘ -methyl-5 ‘βH-pregna-1, -dieno[ 17 , 16- d-]oxazoline-3, 20-dione of formula II is obtained:
The compound of formula II is preferably obtained according to a fermentation process disclosed in
EP-B-322630; in said patent, the compound of formula II is referred to as llβ-21-dihydroxy-2 ‘-methyl-5 ‘ βH- pregna-1,4-dieno[17,16-d-]oxazoline-3,20-dione.
The present invention provides a new advantageous single-step process for obtaining deflazacort, by acetylation of the compound of formula II.
CLIP

tructure of deflazacort and its forced degradation product (A), chromatogram plot of standard deflazacort (B), contour plot of deflazacort (C). Deflazacort was found to be a stable drug under stress condition such as thermal, neutral and oxidative condition. However, the forceddegradation study on deflazacort showed that the drug degraded under alkaline, acid and photolytic conditions.

Mass fragmentation pathway for degradant product of deflazacort.
PATENT
CN 103059096

Example 1: Protective reaction To the reaction flask was added 20 g of 1,4-diene-11? -hydroxy-16,17-epoxy_3,20-dione pregnone (Formula I) 20% of the aqueous solution of glacial acetic acid 300g, stirring 5 minutes, temperature 10 ° C ~ 15 ° C, adding ethyl carbazate 14g, temperature control 30 ° C reaction 6 hours; TLC detection reaction is complete, cooling to 0 ° C ~ 5 ° C for 2 hours, until dry, washed to neutral; 60 ° C vacuum dry to dry creatures 20. 5g; on P, oxazoline ring reaction The above protective products into the reaction bottle, add 41ml Of the DMAC dissolved, temperature 25 ~ 30 ° C, access to ammonia, to keep the reaction bottle micro-positive pressure, the reaction of 32 hours, atmospheric pressure exhaust ammonia and then decompression pumping ammonia for 30 minutes; 5 ° C, temperature 5 ~ 0 ° C by adding 5ml glacial acetic acid, then add 21ml acetic anhydride, heated to 35 ° C reaction 4 hours, the sample to confirm the reaction completely; slowly add 5% sodium hydroxide solution 610ml and heated to 60 ~ 70 ° C reaction 2 hours; point plate to confirm the end of the reaction, cooling to 50 ° C, half an hour by adding refined concentrated hydrochloric acid 40ml, insulation 50 ~ 55 ° C reaction 10 hours; to the end of the reaction temperature to room temperature, chloroform Extraction, drying and filtration, concentration of at least a small amount of solvent, ethyl acetate entrained twice, leaving a small amount of solvent, frozen crystallization filter high purity [17a, 16a-d] terfu Kete intermediate. Example 2: Protective reaction 20 g of 1,4-diene-l1-la-hydroxy-16,17-epoxy_3,20_dione progestin (Formula I) was added to the reaction flask and 15% Formic acid solution 300g, stirring for 5 minutes, temperature 10 ~ 15 ° C, adding methyl carbazate 12g, temperature control 30 ° C reaction 5 hours to test the end of the reaction, cooling to O ~ 5 ° C stirring 2 hours crystallization, Suction to dry, washed to neutral; 60 ° C vacuum drying to dry protection of 20g; on P, oxazoline ring reaction The protection of the reaction into the reaction flask, add 30ml of DMF dissolved, temperature control 25 ~ 30 ° C, access to ammonia, keep the reaction bottle in the micro-positive pressure, reaction 30 hours, atmospheric pressure exhaust ammonia and then decompression pumping ammonia for 30 minutes, ice water cooled to 5 ° C, temperature 5 ~ 10 ° C add 5ml of glacial acetic acid, then add 20ml acetic anhydride, heated to 30 ° C reaction for 5 hours to confirm the reaction is complete; slowly add 20% sodium carbonate aqueous solution 500ml and heated to 60 ~ 70 ° C reaction 4 hours, the point plate to confirm the reaction The temperature of 55 ~ 60 ° C for 10 hours; to be the end of the reaction temperature to room temperature, chloroform extraction, drying and filtration, concentration of a small amount of solvent, acetic acid isopropyl The ester was entrained twice, leaving a small amount of solvent, frozen and crystallized to obtain high purity [17a, 16a-d] oxazoline residues. [0024] Example 3: Protective reaction 20 g of I, 4-diene-16,17-epoxy-3,11,20-triketone pregnone (Formula I) was added to the reaction flask and 20% Formic acid solution 300g, stirring for 5 minutes, temperature 10 ~ 15 ° C, adding hydrazine carbamate 15g, temperature control 30 ° C reaction 5 hours to test the end of the reaction, cooling to O ~ 5 ° C stirring 2 hours crystallization, To the dry, washed to neutral; 60 ° C vacuum drying to dry protection of 22g; on P, oxazoline ring reaction of the protection of the reaction into the bottle, add 30ml of DMAC dissolved temperature control 35 ~ 40 ° C, access to ammonia, keep the reaction bottle in the micro-positive pressure, reaction 40 hours, atmospheric pressure exhaust ammonia and then decompression pumping ammonia for 30 minutes, ice water cooling to 5 ° C, temperature 5 ~ 10 ° C add 5ml of glacial acetic acid, then add 20ml acetic anhydride, heated to 40 ° C reaction 5 hours to confirm the reaction is complete; slowly add 20% potassium carbonate aqueous solution 500ml and heated to 60 ~ 70 ° C reaction 7 hours, the point plate to confirm the reaction The temperature of the reaction to the end of the temperature to room temperature, chloroform extraction, drying filter, concentrated to a small amount of solvent, acetic acid isopropyl The ester was entrained twice, leaving a small amount of solvent, frozen and crystallized to obtain high purity [17a, 16a-d] oxazoline residues.
PATENT
CN 102936274

xample 1
[0028] A 30 g 16, 17 α- epoxy – pregn -20- substituting methyl hydrazine -3-acetyl-1,4-diene, 11- dione (a) and 150 mL of chloroform and 15 mLDMF mixed, pressure reactor, stirring ammonia gas to the reactor pressure to 0.15 MPa (during ventilation control the reaction temperature at 10-15 ° C), 30 ° C heat reaction, TLC track the progress of the reaction. Completion of the reaction, the material was transferred to a glass reaction flask, the temperature of the material to be reduced to below 10 ° C, add acetic acid adjusted to pH 5 to 6, the solvent was removed under reduced pressure; reaction flask was added 30 mL of acetic acid, 30 g of acetic anhydride, The reaction temperature was controlled at 30 ° C, the reaction 6 hours, the reaction mixture was poured into cold 500 mL10% sodium hydroxide solution, stirred for 1 hour, filtration to give product 30.6 g, 102% mass yield, product by HPLC , a purity of 95.2%.
[0029] Example 2
[0030] A 30 g 16, 17 α- epoxy – pregn -20- substituting methyl hydrazine -3-acetyl-1,4-diene, 11- dione (a) and 150 mL of chloroform and 30 mL of pyridine were mixed, added pressure reactor, stirring ammonia gas to the reactor pressure to 0. 15 MPa (during ventilation control the reaction temperature at 10~15 ° C), 15 ° C heat reaction, TLC track the progress of the reaction. Completion of the reaction, the material was transferred to a glass reaction flask, the temperature of the material to be reduced to below 10 ° C, add acetic acid adjusted to pH 5 to 6, the solvent was removed under reduced pressure; reaction flask was added 30 mL of acetic acid, 30 g of acetic anhydride, The reaction temperature was controlled at 30 ° C, the reaction 6 hours, the reaction mixture was poured into cold 500 mL10% sodium hydroxide solution, stirred for 1 hour, filtration to give product 28.6 g, yield 95% by mass, product by HPLC , a purity of 94.8%.
[0031] Example 3
[0032] A 30 g 16, 17 α- epoxy – pregn -20- substituting methyl hydrazine -3-acetyl-1,4-diene, 11- dione (a) and 150 mL of chloroform and 30 mLDMF mixed, pressure reactor, stirring ammonia gas to the reactor pressure to 0.15 MPa (during ventilation control the reaction temperature at 10~15 ° C), 40 ° C heat reaction, TLC track the progress of the reaction.Completion of the reaction, the material was transferred to a glass reaction flask, the temperature of the material to be reduced to below 10 ° C, add acetic acid adjusted to pH 5 to 6, the solvent was removed under reduced pressure; reaction flask was added 30 mL of acetic acid, 30 g of acetic anhydride, The reaction temperature was controlled at 30 ° C, the reaction for 6 hours. The reaction mixture was poured into cold 500 mL10% sodium hydroxide solution, stirred for 1 hour, filtration to give the product 31.2 g, yield 104% quality products by HPLC , a purity of 95.4%.
[0033] Example 4
[0034] A 30 g 16, 17 α- epoxy – pregn -20- substituting methyl hydrazine -3-acetyl-1,4-diene, 11- dione (a) and 150 mL of chloroform and 30 mLDMF mixed, pressure reactor, stirring ammonia gas to the reactor pressure to 0.5 MPa (during ventilation control the reaction temperature at 10~15 ° C), 40 ° C heat reaction, TLC track the progress of the reaction. Completion of the reaction, the material was transferred to a glass reaction flask, the temperature of the material to be reduced to below 10 ° C, add acetic acid adjusted to pH 5 to 6, the solvent was removed under reduced pressure; reaction flask was added 30 mL of acetic acid, 30 g of acetic anhydride, The reaction temperature was controlled at 30 ° C, the reaction 6 hours, the reaction mixture was poured into cold 500 mL10% sodium hydroxide solution, stirred for 1 hour, filtration to give the product 31. I g, 102% mass yield, product by by HPLC, the purity was 95.2%.
[0035] Example 5
[0036] A 30 g 16, 17 α- epoxy – pregn -20- substituting methyl hydrazine -3-acetyl-1,4-diene, 11- dione (a) and 150 mL of chloroform and 30 mLDMF mixed, pressure reactor, stirring ammonia gas to the reactor pressure to 0.15 MPa (during ventilation control the reaction temperature at 10~15 ° C), 40 ° C heat reaction, TLC track the progress of the reaction. Completion of the reaction, the material was transferred to a glass reaction flask, the temperature of the material to be reduced to below 10 ° C, add acetic acid adjusted to pH 5 to 6, the solvent was removed under reduced pressure; reaction flask was added 60 mL of acetic acid, 15 g of acetic anhydride, The reaction temperature was controlled at 30 ° C, the reaction 6 hours, the reaction mixture was poured into cold 500 mL10% sodium hydroxide solution, stirred for 1 hour, filtration to give the product 29. 5 g, yield 98% by mass, the product of by HPLC, purity of 95%.
[0037] Example 6
[0038] A 30 g 16, 17 α- epoxy – pregn -20- substituting methyl hydrazine -3-acetyl-1,4-diene, 11- dione (a) and 150 mL of chloroform and 30 mLDMF mixed, pressure reactor, stirring ammonia gas to the reactor pressure to 0.15 MPa (during ventilation control the reaction temperature at 10~15 ° C), 40 ° C heat reaction, TLC track the progress of the reaction. The reaction was complete, the material was transferred to a glass reaction flask until the material temperature drops below 10 ° C, plus acetic acid to adjust the pH to 5 to 6, the solvent was removed under reduced pressure; the reaction flask was added 30 mL of acetic acid, 30 g of maleic dianhydride, the reaction temperature was controlled at 30 ° C, the reaction 6 hours, the reaction mixture was poured into cold 500 mL10% sodium hydroxide solution, stirred for 1 hour, filtration to give the product 30 g, 100% mass yield, product by HPLC purity of 95.2%.
[0039] Example 7
[0040] A 30 g 16, 17 α- epoxy – pregn -20- substituting methyl hydrazine -3-acetyl-1,4-diene, 11- dione (a) and 150 mL of chloroform and 30 mLDMF mixed, pressure reactor, stirring ammonia gas to the reactor pressure to 0.15 MPa (during ventilation control the reaction temperature at 10~15 ° C), 40 ° C heat reaction, TLC track the progress of the reaction. Completion of the reaction, the material was transferred to a glass reaction flask, the temperature of the material to be reduced to below 10 ° C, add acetic acid adjusted to pH 5 to 6, the solvent was removed under reduced pressure; reaction flask was added 30 mL of acetic acid, 30 g of propionic anhydride, The reaction temperature was controlled at 30 ° C, the reaction for 6 hours. The reaction mixture was poured into cold 500 mL10% sodium hydroxide solution, stirred for 1 hour, filtration to give the product 27.6 g, 92% yield of quality products by HPLC , a purity of 93.5%.
[0041] Example 8
[0042] A 30 g 16, 17 α- epoxy – pregn -20- substituting methyl hydrazine -3-acetyl-1,4-diene, 11- dione (a) and 150 mL of chloroform and 30 mLDMF mixed, pressure reactor, stirring ammonia gas to the reactor pressure to 0.15 MPa (during ventilation control the reaction temperature at 10~15 ° C), 40 ° C heat reaction, TLC track the progress of the reaction. Completion of the reaction, the material was transferred to a glass reaction flask, the temperature of the material to be reduced to below 10 ° C, add acetic acid adjusted to pH 5 to 6, the solvent was removed under reduced pressure; reaction flask was added 30 mL of acetic acid, 30 g of acetic anhydride, The reaction temperature is controlled at 50 ° C, the reaction for 6 hours. The reaction mixture was poured into cold 500 mL10% sodium hydroxide solution, stirred for 1 hour, filtration to give the product 29.8 g, 99% yield of quality products by HPLC , a purity of 94.8%.
References
- Jump up^ “Refla: deflazacort” (PDF).
- Jump up^http://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208684s000,208685s000lbl.pdf
- Jump up^ Möllmann, H; Hochhaus, G; Rohatagi, S; Barth, J; Derendorf, H (1995). “Pharmacokinetic/pharmacodynamic evaluation of deflazacort in comparison to methylprednisolone and prednisolone”. Pharmaceutical Research. 12 (7): 1096–100. PMID 7494809.
- ^ Jump up to:a b “Calcort”. electronic Medicines Compendium. June 11, 2008. Retrieved on October 28, 2008.
- Jump up^ Luca Parente (2017). “Deflazacort: therapeutic index, relative potency and equivalent doses versus other corticosteroids”. BMC Pharmacol Toxicol. doi:10.1186/s40360-016-0111-8.
- Jump up^ Ellen Jean Hirst (January 19, 2015), Duchenne muscular dystrophy drug could get OK for U.S. sales in 2016, The Chicago Tribune, retrieved February 13, 2017,
has been shown to prolong lives … a progressive and fatal disease that has no drug treatment available in the US
- Jump up^ “FDA approves drug to treat Duchenne muscular dystrophy”. http://www.fda.gov. 2017-02-09. Retrieved 2017-02-10.
- Jump up^ “Marathon Pharmaceuticals to Charge $89,000 for Muscular Dystrophy Drug”. http://www.wsj.com. 2017-02-10. Retrieved 2017-02-10.
- Jump up^ Clifton Sy Mukherjee (February 10, 2017). “Brainstorm Health Daily”. Retrieved February 13, 2017.
- Jump up^ Joseph Walker and Susan Pulliam (February 13, 2017), Marathon Pharmaceuticals to Charge $89,000 for Muscular Dystrophy Drug After 70-Fold Increase, The Wall Street Journal, retrieved February 13, 2017,
FDA-approved deflazacort treats rare type of disease affecting boys
- Jump up^ “Substâncias: DEFLAZACORT” (in Portuguese). Centralx. 2008. Retrieved on October 28, 2008.
| CN102746358A * |
Apr 22, 2011 |
Oct 24, 2012 |
天津金耀集团有限公司 |
Novel technology for synthesis of pregnane 21-bit bromide |
| CN102746358B * |
Apr 22, 2011 |
Feb 10, 2016 |
天津金耀集团有限公司 |
一种合成孕甾21位溴化物的工艺 |
| CN102936274A * |
Nov 12, 2012 |
Feb 20, 2013 |
浙江仙居君业药业有限公司 |
Preparation method for [17alpha, 16alpha-d] methyl oxazoline |
| CN102936274B * |
Nov 12, 2012 |
Apr 1, 2015 |
江西君业生物制药有限公司 |
Preparation method for [17alpha, 16alpha-d] methyl oxazoline |
///////FDA 2017, Emflaza, Calcort, Deflazacort, orphan drug designation, FAST TRACK
|
[H][C@@]12C[C@@]3([H])[C@]4([H])CCC5=CC(=O)C=C[C@]5(C)[C@@]4([H])[C@@]([H])(O)C[C@]3(C)[C@@]1(N=C(C)O2)C(=O)COC(C)=O
|

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....









































































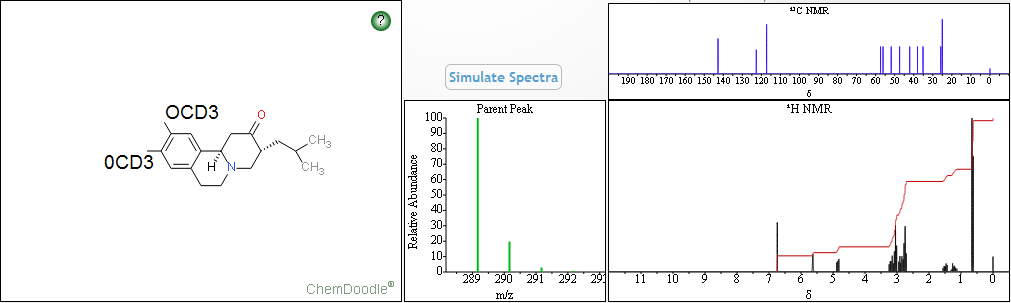
-9,10-di((2H3)methoxy)-3-(2-methylpropyl)-1,3,4,6,7,11b-hexahydro-2H-benzo(a)quinolizin-2-one")















































 LIONEL MY SON
LIONEL MY SON



























































{kind=link}