
Home » Posts tagged 'Chronic Obstructive Pulmonary Disease'
Tag Archives: Chronic Obstructive Pulmonary Disease
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
ICENTICAFTOR
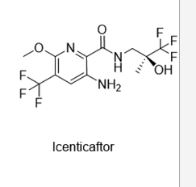
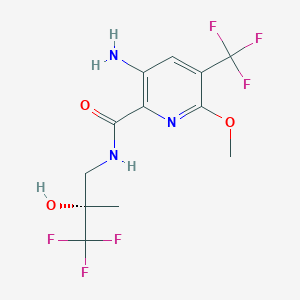
QBW 251, ICENTICAFTOR
- Molecular FormulaC12H13F6N3O3
- Average mass361.240 Da
Icenticaftor (development code QBW251) is a drug candidate for the treatment of chronic obstructive pulmonary disease (COPD)[1][2] and cystic fibrosis.[3][4] The drug is being developed by Novartis.[5]
Like ivacaftor (which is marketed as Kalydeco), icenticaftor functions by acting as a stimulator of the protein cystic fibrosis transmembrane conductance regulator (CFTR).[5]
Icenticaftor (QBW251) is an orally active CFTR channel potentiator, with EC50s of 79 nM and 497 nM for F508del and G551D CFTR, respectively. Icenticaftor can be used for chronic obstructive pulmonary disease (COPD) and cystic fibrosis research.
Cystic fibrosis (CF) is the most prevalent life-threatening Mendelian disorder in Caucasian populations. CF arises from mutations of the gene for the cystic fibrosis transmembrane conductance regulator (CFTR) protein. The CFTR ion channel orchestrates gating of chloride and bicarbonate ions across epithelial cell membranes in various tissues, including the lung, pancreas, intestine, reproductive tract, and sweat glands. While CF is a systemic disorder, the primary mortality derives from reduced CFTR activity in the airways. Subsequent acidification3 and dehydration leads to accumulation of a viscous mucus layer, occluding the airways and trapping bacteria, leading to infections, reduced lung function, and ultimately, respiratory failure. The most common CFTR mutation, F508del (Class II, found in 90% of CF patients), impairs folding of the CFTR protein (a Class II trafficking defect), resulting in a reduced amount of channel present at the plasma membrane. With the G551D mutation (class III), theamount of protein at the membrane is unaffected, but its open probability (Po) is reduced, also resulting in a reduced channel gating. Thus, to address the underlying causes of CF, two distinct CFTR modulators are required: correctors to increase CFTR levels at the plasma membrane and potentiators to enable effective opening of the channel
Chronic obstructive pulmonary disease (COPD) is anticipated to shortly become the third leading cause of death globally. COPD is characterized by persistent airflow obstruction with cigarette smoke exposure recognized as the primary risk factor. Airflow limitation is associated with all COPD patients; however, the disease is heterogeneous, with variable phenotypes ranging from chronic bronchitis (CB) to emphysema. Small airway disease exhibits increased numbers of goblet cells and mucus plugging with associated smooth muscle hyperplasia, airway fibrosis, and increased inflammation. Excess mucus secretion is believed to play an important role in COPD pathogenesis and is associated with progression of the disease.
Cystic fibrosis (CF) is a fatal genetic disease caused by mutations in the gene encoding the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR), a protein kinase A activated epithelial anion channel involved in salt and fluid transport in multiple organs, including the lung. Most CF mutations either reduce the number of CFTR channels at the cell surface (e.g. synthesis or processing mutations) or impair channel function (e.g. gating or conductance mutations) or both.
PCT publication No. WO 2011/113894 describes compounds which restore or enhance the function of mutant and/or wild type CFTR for the treatment of cystic fibrosis, primary ciliary dyskinesia, chronic bronchitis, chronic obstructive pulmonary disease, asthma and other CFTR related diseases. The compounds described therein include (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide (Example 5 of WO 2011/113894).
The synthesis described in WO 2011/113894 to make (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide is long, uses expensive starting materials and toxic reagents. Schemes 1 and 2 outline a synthesis from WO 2011/113894 used to make(S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide.
In Scheme 1, the intermediate ethyl 3-amino-5-(trifluoromethyl)picolinate (B4) is made via a Buchwald-Hartwig coupling reaction which requires the use of an expensive starting material (B1) and an expensive palladium catalyst which has to be controlled in the final product. Also, the conversion of B4 to B5 requires the use of NBS, a mutagenic reagent which has to be controlled in the API.
Moreover, the conversion of B5 to B8 is accomplished through the addition of 2,5-hexanedione, a well-known neurotoxin, as shown in Scheme 2. Transformation of the pyrrole in B8 to the amine B9 uses hydroxylamine which is a mutagenic and thermally unstable compound that is dangerous to use in large quantities. The overall process described in WO 2011/113894 requires many protecting group manipulations that lead to a low atom economy and afford a lot of waste. Thus there is a need for an improved synthetic process for making (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide.
PATENT
WO 2018116139,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018116139&_cid=P21-L7D5PQ-39961-1
xample 1: 3-Bromo-6-methoxy-5-(trifluoromethyl)picolinic acid
5-bromo-2-methoxy-3-(trifluoromethyl)pyridine (III) (1.4 kg, 5.47 mol), tetramethyl ethylene diamine (TMEDA) (1.75 kg, 15 mol) and tetrahydrofuran (THF) (10kg) were charged to a dry and inert reactor. At -25°C a solution of 2,2,6,6-tetramethyl-piperidinylmagnesium chloride lithiumchloride complex, 1 M in THF/toluene (TMPMgCl.LiCl)(14.5 kg, 15 mol) was slowly added. After stirring the reaction mixture for 30 min., CO2 gas was carefully bubbled into the reactor so that the temperature of the exothermic reaction did not exceed -20°C. The reaction mixture was then quenched onto a mixture of t-butyl methyl ether (TBME) and 5% aq. H2SO4 (50 kg). The biphasic mixture was separated and the organic phase was extracted with 2M NaOH solution. The aqueous phase was acidified to pH 1-2 with 5% aq. H2SO4 and extracted with TBME. After a distillative solvent change to cyclohexane the product was crystallized from cyclohexane to yield 1.1 kg 3-bromo-6-methoxy-5-(trifluoromethyl)picolinic acid (65% yield).
1H NMR (400 MHz, CDCl3): δ ppm 8.24 (d,J = 0.7Hz, 1 H), 4.12 (s, 3H)
13C NMR (101 MHz, DMSO-d6): δ ppm 54.84, 106.37, 114 (m), 117.6/120.3/123.0/125.7 (m), 141.74, 152.43, 158.63, 165.63
HRMS: [M-H]- expected C8H4BrF3NO3, 297.9405; found C8H4BrF3NO3, 297.9337
Example 2: Methyl 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate
5-bromo-2-methoxy-3-(trifluoromethyl)pyridine (III) (5.0 g, 19.53 mmol) was added to a 100 ml reactor followed by toluene (20 ml) and dimethylcarbonate (17.59 g, 195.30 mmol). To the stirred solution at 20 °C was slowly added 2,2,6, 6-tetramethyl-piperidinylmagnesium chloride lithium chloride complex as a 1 M solution in THF/toluene (27.34 ml, 27.34 mmol) within 45 minutes. A sample was taken and diluted in acetic acid for HPLC analysis in order to confirm full conversion of II to the methylester. Within the same vessel 5% aq. H2SO4 (36 ml) was slowly added to the reaction mixture until a pH below 2 was obtained (caution, exothermic). The biphasic mixture was separated and the lower aqueous phase back-extracted with toluene (10 ml).
In order to isolate the methylester the organic phases were combined and concentrated by rotary evaporation to yield a residue which was chromatographed on reverse-phase silica to yield the final product: methyl 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate as a yellow solid, 5.3 g, 86 % yield. The solid was optionally recrystallized from methanol and water to further increase purity.
1H NMR (400MHz, CDCl3): δ ppm 8.08 (br s, 1 H), 4.07 (s, 3H), 4.02 (s, 3H)
13C NMR (CDCl3): δ ppm 164.76, 159.22, 149.90, 141.49, 122.83, 120.12, 116.12, 108.05, 54.93, 53.09
HRMS: MH+ expected C9H8BrF3NO3, 313.9561 ; found C9H8BrF3NO3, 313.9634
HPLC Conditions:
HPLC: Column : Agilent Zorbax SB-C18 (150 mm x 3.0 mm, particle size 3.5 urn)
Eluent A : Water / TFA = 1000/1 (v/v)
Eluent B: Acetonitrile / TFA = 1000/1 (v/v)
Wavelength : 230 nm
Flow-rate : 0.8 ml/min
Gradient: eluent B: 45% to 90% over 9 mins
Retention time 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate: 5.80 min
Alternative synthesis for 3-bromo-6-methoxy-5-(trifluoromethyl)picolinic acid:
Isolation of Example 1
In order to proceed to Example 1 without the isolation of VII, the work-up continues from the combined toluene phases post-H2SO4 quench as follows:
To the combined organic phases was slowly added 50% aq. sodium hydroxide (30 ml) until a pH of above 10 was obtained. The reaction mixture was heated to 35 °C and after 15 mins addition of water (30 ml) followed by 30 mins further stirring preceded sample-taking to ensure full hydrolysis of the methylester to Example 1 by HPLC. Water was added (130 ml), followed by TBME (60 ml) and the phases separated. To the aqueous phase was cautiously added concentrated H2SO4 (30 g) until a pH of below 2.5 was obtained (caution, exothermic and release of CO2 causes foaming). TBME (100 ml) was added and the phases separated. The organic phase contained the C2, and could be evaporated to dryness by rotary evaporation to confirm the yield, 5.4 g C2, 92 % yield.
1H NMR (400 MHz,CDCl3): δ ppm 8.24 (d,J=0.7Hz, 1 H), 4.12 (s, 3H)
13C NMR (101 MHz,DMSO-d6): δ ppm 54.84, 106.37, 114 (m), 117.6/120.3/123.0/125.7 (m), 141.74, 152.43, 158.63, 165.63
HRMS: M-H- expected C8H4BrF3NO3, 297.9405; found C8H4BrF3NO3, 297.9333
For HPLC method details see above. Retention time C2: 2.94 min
Alternative synthesis for ethyl 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate:
5-bromo-2-methoxy-3-(trifluoromethyl)pyridine (III) (0.5 g, 1.95 mmol) was added to a reactor followed by THF (2 ml) and the solution cooled to 0 °C. To the mixture was added 2,2,6,6-tetramethyl-piperidinylmagnesium chloride lithium chloride complex as a 1 M solution in THF/toluene (4.88 ml, 3.91 mmol), and the mixture was left to stir for 15 minutes at 0 °C. An aliquot of the solution (50 ul) was then added to a reactor containing diethylcarbonate (20 ul, 19.5 mmol). A second aliquot (50 ul) was taken of the metallated II and added to a reactor containing ethyl chloroformate (14 ul, 19.5 mmol). After 2 minutes both reactors were quenched with a 1 :1 mixture of acetonitrile/HCl (1 M). The reaction with diethylcarbonate gave 56 A% of ethyl 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate and the reaction with ethyl chloroformate gave 68 A% of ethyl 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate product according to the HPLC method described above.
Example 3: Synthesis of (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide
Step 1: 3-bromo-6-methoxy-5-(trifluoromethyl)picolinic acid (1.3 kg, 4.33 mol) and
copper(II)sulfate pentahydrate (0.108 kg, 0.433 mol) were charged into an inert autoclave
followed by aqueous ammonia 25% (12 kg). The mixture was stirred and heated up to 100 °C, whereby a pressure of 7 bar resulted. The solution was stirred for 2 hr and then cooled down to
5 °C. Sulfuric acid (8 M) was dosed upon cooling, so that a temperature range of 5 °C to 30 °C was held until a pH of about 5 was reached. Isopropylacetate was added and the pH was
further adjusted to 1-2. The phases were separated and the organic phase was azeotropically dried by partial distillation. n-Heptane was added and the mixture stirred for 15 hr at 20 °C
during which the product crystallized out. After filtration and drying 3-amino-6-methoxy-5-(trifluoromethyl)picolinic acid was obtained as a yellow solid (0.92 kg, 90%).
1H NMR (400 MHz, DMSO-d6): δ ppm 7.70 (s, 1 H), 3.89 (s, 3H)
13C NMR (101 MHz, DMSO-d6): δ ppm 53.59, 116.76 m, 123.27, 126.36-117.40 m, 128.04, 142.56, 148.65, 167.62
Step 2: 3-amino-6-methoxy-5-(trifluoromethyl) picolinic acid (20 g, 84.7 mmol) and HATU (38.6 g, 101.6 mmol) were charged to a reactor followed by a solution of (S)-3-amino-1 ,1 ,1-trifluoro-2- methylpropan-2-ol in isopropylacetate (7 %, 188 g, 93 mmol). The solution was stirred at room temperature, diisopropyl ethyl amine (21.9 g, 169 mmol) was added and stirring was continued for at least 16h at 25 °C. Water (250 ml) was then added dropwise within 15 min. keeping the temperature below 25 °C. The water phase was separated and the organic phase was extracted with 5% aqueous HCl , 5% potassium carbonate solution, and water. The organic layer was concentrated to about 60% solution. At 50 °C n-heptane (41 g) was added and the solution was cooled by a linear ramp to 5 °C while adding more n-heptane (131 g). The precipitate was filtered off and dried at 50 °C resulting in a yellow to beige product (S)-3-amino-6-methoxy-N- (3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide (21.1 g, 69 % yield).
1H NMR (400 MHz, DMSO-d6): δ ppm 8.30 (m,1 H), 7.68 (s,1 H), 6.69 (s,2H), 6.29(s,1 H), 3.93(s,3H), 3.7-3.4(m,2H), 1.26(s,3H)
13C NMR (101 MHz, DMSO-d6): δ ppm 18.92, 42.15, 53.52, 72.40, 115.5-116.5 m, 118-126 m, 122-130.7 m, 124.82, 128.3 m, 140.95, 148.49, 166.27
Example 4: Telescoped process for the synthesis of the HCl salt of 3-amino-6-methoxy- 5-(trifluoromethyl)picolinic acid (V)
1 Equivalent* of (III) and 6 equivalents of dimethyl carbonate (DMC) were dissolved in 3.5 parts** of toluene at room temperature. To this solution 1.5 equivalent of TMPMgCl.LiCl solution in THF was added at 15-25°C within ca. 1 h. Tert butyl methyl ether (MTBE, 5.9 parts) was added and the mixture was quenched in 7.3 parts of 10% sulfuric acid at 25-40°C. The water phase was discarded and to the organic phase 6.2 parts of 30% sodium hydroxide solution were added. The mixture was stirred well at 40°C for 1-2h. After the successful conversion of (VIII) to (IV), 2.5 parts of water were added to dissolve the partially precipitated sodium carbonate. The water phase was discarded and the organic phase was cooled to 20°C and extracted with 4.8 parts of 25% aqueous ammonia. The aqueous phase was transferred in an autoclave and 0.0979 parts (10mol%) of copper sulfate pentahydrate were added. The autoclave was well inertized by a pressure method and heated up to 100°C, while the pressure raises up to ca. 8 bar absolute pressure. After the successful conversion of (IV) to (V), the green solution was added to a mixture of 3.7 parts of MTBE and 6.8 parts of 50% sulfuric acid resulting in a biphasic solution of pH 1-2. The water phase was separated and the organic phase washed two times with 2.5 parts of water each. The organic phase was dried by distillation at JT 50°C/400mbar while 3.7 parts of MTBE were added/replaced. To the dried organic solution 0.41 parts of HCl gas was dosed at 0-5°C under or over solvent level. The suspension was stirred for ca.1 h, then filtered off and washed with 48 parts of TBME. The product was dried at 40°C/20 mbar for ca. 12h. (yield from (III): 72%, slightly beige solid).
*equivalents are based on the molar amount of the starting material (III) = 1 equivalent
**parts = weight/weight (III)
1H NMR (400 MHz, DMSO-d6): δ ppm 7.70 (s, 1 H), 3.89 (s, 3H)
13C NMR (101 MHz, DMSO-d6): δ ppm 53.59, 116.76 m, 123.27, 126.36-117.40 m, 128.04, 142.56, 148.65, 167.62
Example 5: Alternative synthesis of (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide
Step 1 : (VIII) (1.0 g), (S)-3-amino-1 ,1 ,1-trifluoro-2-methylpropan-2-ol as mandellic acid salt (1.128 g, 1.2 eq.) and 2,3,4,6, 7, 8-hexahydro-1H-pyrimido[1,2-a]pyrimidine (TBD, 0.588 g, 1.3 eq.) were added to a pre-dried flask as solids. To this was added the anhydrous THF (10 ml) and the cloudy solution heated to 55 °C. Sampling and analytical determination of purity at 2.5 hrs confirmed 88 A% product upon which water (10 ml) was added and the phases separated. The organic phase was distilled to a concentrated mixture upon which toluene (20 ml) was added. The organic layer was extracted with 10% aq. citric acid (10 ml) followed by three consecutive extractions with 1 M aq. NaOH. The organic phase was then dried with magnesium sulfate and evaporated to dryness to give 1.196 g of (S)-3-bromo-6-methoxy-N-(3,3,3-trifluoro- 2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide (IX) as a white solid (95 A%, 88% yield).
1H NMR, CDCl3: δ ppm 8.08 (s, 1 H), 7.83 (br s, 1 H), 3.99 (s, 3H), 3.78-3.60 (m, 2H), 3.51 (br s, 1 H), 1.36 (s, 3H)
19F NMR, CDCl3: δ ppm -64.28, -81.44
13C DEPT135, CDCl3: δ ppm 144.20 (CH), 54.70 (CH3), 44.26 (CH2), 19.71 (CH3)
HRMS: MH+ expected C12H12BrF6N2O3, 424.9857; found C12H12BrF6N2O3, 424.9931
HPLC (method described above): retention time = 4.94 min
Step 2: IX (79 mg, 0.186 mmol) was combined with copper(II)sulfate pentahydrate (4.6 mg, 0.019 mmol), methanol (0.6 ml) and 23% aqueous ammonium hydroxide solution (559 ul) within a glass microwave vial. The headspace was inertized with nitrogen, then the vial sealed and placed in the microwave unit for heating to 105 °C for 7.5 hrs. Isopropylacetate (5 ml) was added to the deep green reaction mixture and a solvent-switch brought about by rotary evaporation. To the mixture now in water and isopropyl acetate was added 8M H2SO4 (5 ml), the phases mixed and then left to separate. The aqueous phase was further extracted with isopropylacetate and the combined organic phases washed with aq. NaCl (5 ml). The organic phase was dried over MgSO4 and evaporated to yield of a yellow residue, 66 mg.
A portion of the residue (16 mg) was re-dissolved in heptane / ethyl acetate and submitted for combiflash purification (n-heptane / ethyl acetate gradient, elution at 20% ethyl acetate) providing (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide (VII) as a residue on evaporation in 91 A% purity containing trace residual solvents (17 mg, corrected to 13 mg by 1H NMR, 80 % yield back-calculated).
1H NMR, CDCl3: δ ppm 8.11 (br s, 1 H), 7.37 (s, 1 H), 3.97 (s, 3H), 3.76-3.72 (d, 2H, J=6.3Hz), 1.42 (s, 3H)
13C NMR, CDCl3: δ ppm 168.86, 150.55, 140.21 , 128.63, 127.26, 125.35, 124.42, 123.39, 120.68, 118.60, 74.16, 53.73, 44.39, 19.55
ESI-MS: expected mass 361.2. ELS detector, 100 A%, MH+ 362.1 , M- 360.1
HPLC (method described above): retention time = 4.39 min
PATENT
US20200383960
https://patentscope.wipo.int/search/en/detail.jsf?docId=US312969607&_cid=P21-L7D5H8-38258-1
Examples 4, 5 and 6: 3-Amino-6-methoxy-5-trifluoromethyl-pyridine-2-carboxylic acid (3,3,3-trifluoro-2-hydroxy-2-methyl-propyl)-amide and its enantiomers
was prepared according to the following procedure:
Examples 5 and 6 are Entantiomers
SYN
J. Med. Chem. 2021, 64, 11, 7241–7260
Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) ion channel are established as the primary causative factor in the devastating lung disease cystic fibrosis (CF). More recently, cigarette smoke exposure has been shown to be associated with dysfunctional airway epithelial ion transport, suggesting a role for CFTR in the pathogenesis of chronic obstructive pulmonary disease (COPD). Here, the identification and characterization of a high throughput screening hit 6 as a potentiator of mutant human F508del and wild-type CFTR channels is reported. The design, synthesis, and biological evaluation of compounds 7–33 to establish structure–activity relationships of the scaffold are described, leading to the identification of clinical development compound icenticaftor (QBW251) 33, which has subsequently progressed to deliver two positive clinical proofs of concept in patients with CF and COPD and is now being further developed as a novel therapeutic approach for COPD patients.


a Reagents and conditions: (i) aq NaOH, THF, RT, 97%; (ii) aq Me2NH or MeNH2, THF, RT, 56−92%; (iii) 41, HATU, Et3N, NMP, RT, 52− 78%; (iv) NH2OH·HCl, Et3N, EtOH−water, reflux, then chiral HPLC, 34−36%; (v) aq NaOH, MeOH, 60°C, 97%; (vi) cat H2SO4, MeOH, reflux, 75%; (vii) TMSCl, KI, MeCN, reflux, 54%; (viii) EtOH, DEAD, Ph3P, dioxane, RT, 61%; (ix) aq NaOH, THF, reflux, 26%; (x) (S)-41, HATU, DIPEA, DMF, RT, 89%; (xi) NH2OH·HCl, Et3N, EtOH−water, reflux, 37−53%; (xii) (S)-41, HATU, DIPEA, NMP, RT, 59%.
(S)-3-Amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2- methylpropyl)-5-(trifluoromethyl)picolinamide
(S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2- methylpropyl)-5-(trifluoromethyl)picolinamide 33 as a white solid (33.6 g, 59%). LRMS C12H13F6N3O3 requires M+ 361.08, found [MH]+ 362.2. Elemental analysis requires C, 39.90%; H, 3.63%; N, 11.63% found C, 40.22 ± 0.06%; H, 3.68 ± 0.11%; N, 11.76 ± 0.04%. 1 H NMR (DMSO-d6) 1.26 (3H s), 3.46 (1H dd J = 13.3, 5.6), 3.66 (1H dd J = 13.7, 7.3), 3.92 (3H s), 6.29 (1H s), 6.69 (2H br s), 7.68 (1H s), 8.30 (1H t J = 6.4). 13C NMR (DMSO-d6) 18.95 (q), 42.19 (t), 53.56 (q), 72.27 (s JF = 26.8), 116.07 (s JF = 32.3), 122.40 (s JF = 272.1), 124.85 (s), 126.43 (s JF = 287.1), 128.29 (d JF = 5.2), 141.0 (s), 148.51 (s), 166.3 (s). 19F NMR (DMSO-d6) −62.71 (s), −80.46 (s).
/////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Clinical data | |
|---|---|
| Other names | QBW251 |
| ATC code |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| Chemical and physical data | |
| Formula | C12H13F6N3O3 |
| Molar mass | 361.244 g·mol−1 |
| 3D model (JSmol) | |
References
- ^ Rowe SM, Jones I, Dransfield MT, Haque N, Gleason S, Hayes KA, et al. (2020). “Efficacy and Safety of the CFTR Potentiator Icenticaftor (QBW251) in COPD: Results from a Phase 2 Randomized Trial”. International Journal of Chronic Obstructive Pulmonary Disease. 15: 2399–2409. doi:10.2147/COPD.S257474. PMC 7547289. PMID 33116455.
- ^ Grand DL, Gosling M, Baettig U, Bahra P, Bala K, Brocklehurst C, et al. (June 2021). “Discovery of Icenticaftor (QBW251), a Cystic Fibrosis Transmembrane Conductance Regulator Potentiator with Clinical Efficacy in Cystic Fibrosis and Chronic Obstructive Pulmonary Disease”. Journal of Medicinal Chemistry. 64 (11): 7241–7260. doi:10.1021/acs.jmedchem.1c00343. ISSN 0022-2623. PMID 34028270.
- ^ Kazani S, Rowlands DJ, Bottoli I, Milojevic J, Alcantara J, Jones I, et al. (March 2021). “Safety and efficacy of the cystic fibrosis transmembrane conductance regulator potentiator icenticaftor (QBW251)”. Journal of Cystic Fibrosis. 20 (2): 250–256. doi:10.1016/j.jcf.2020.11.002. PMID 33293212.
- ^ Ray F (December 9, 2020). “Icenticaftor Effective in CF Patients With Certain Mutations, Phase 1/2 Trial Shows”. cysticfibrosisnewstoday.com. BioNews Services.
- ^ Jump up to:a b “Icenticaftor – Novartis”. Adis Insight. Springer Nature Switzerland AG.
////////////QBW 251, ICENTICAFTOR, NOVARTIS, chronic obstructive pulmonary disease, COPD, cystic fibrosis,
C[C@](CNC(=O)C1=C(C=C(C(=N1)OC)C(F)(F)F)N)(C(F)(F)F)O
PDE4 Inhibitor, SB-207499, Cilomilast……….REVISTED

Cilomilast (Ariflo, SB-207,499)
cas 153259-65-5
cis-{-4-cyano-4-[3- (trans-3-hydroxycyclopentyloxy)-4-methoxyphenyl]cyclohexane-l -carboxylic acid}
cis-4-Cyano-4-[3-(cyclopentyloxy)-4-(methoxyphenyl)]-r-1-cyclohexanecarboxylic acid
C20-H25-N-O4, 343.4205
GSK….INNOVATOR
- Ariflo
- Cilomilast
- SB 207499
- SB207499
- UNII-8ATB1C1R6X
A selective phosphodiesterase-4 inhibitor for treatment of patients with chronic obstructive pulmonary disease.
CLINICAL https://clinicaltrials.gov/search/intervention=Cilomilast
Cilomilast (Ariflo, SB-207,499) is a drug which was developed for the treatment of respiratory disorders such as asthma and Chronic Obstructive Pulmonary Disease (COPD). It is orally active and acts as a selective Phosphodiesterase-4 inhibitor.[1]
SB-207499 is a potent second-generation inhibitor of PDE4 (phosphodiesterase-4) with decreased side effects versus those of the well-known first-generation inhibitor, (R)-rolipram. SB-207499 is in clinical development both for asthma and chronic obstructive pulmonary disease (COPD)……..J. Med. Chem. 1998, 41, 821
Cilomilast (Ariflo™, SB 207499) is an orally active, second-generation phosphodiesterase (PDE) 4 inhibitor that is being developed by GlaxoSmithkline for the treatment of chronic obstructive pulmonary disease (COPD). The results of Phase I and Phase II studies have demonstrated that cilomilast significantly improves lung function and quality of life to a clinically meaningful extent, which has led to a comprehensive Phase III programme of research evaluating efficacy, safety and mechanism of action. However, the results of those Phase III studies are unremarkable and disappointing, raising doubt over the future of cilomilast as a novel therapy for COPD. This review summarizes data obtained from the Phase III clinical development programme, highlights some of the potential concerns both specific to cilomilast and to PDE4 inhibitors in general and assesses the likelihood that cilomilast will reach the market.
Cilomilast is GlaxoSmithKline’s selective phosphodiesterase type 4 (PDE4) inhibitor. The drug candidate had been preregistered in the U.S. for the maintenance of lung function in patients with chronic obstructive pulmonary disease (COPD) who are poorly responsive to albuterol. GlaxoSmithKline received an approval letter from the FDA in October 2003, however, in 2007, the company discontinued development of the compound. In 2008, the product was licensed to Alcon by GlaxoSmithKline for the treatment of eye disorders.

Phosphodiesterase (PDE) inhibitors, such as theophylline, have been used to treat Chronic Obstructive Pulmonary Disease (COPD) for centuries; however, the clinical benefits of these agents have never been shown to out-weigh the risks of their numerous adverse effects. Four clinical trials were identified evaluating the efficacy of cilomilast, the usual randomized, double-blind, and placebo-controlled protocols were used. It showed reasonable efficacy for treating COPD, but side effects were problematic and it is unclear whether cilomalast will be marketed, or merely used in the development of newer drugs.[2][3]
Cilomilast is a second-generation PDE4 inhibitor with antiinflammatory effects that target bronchoconstriction, mucus hypersecretion, and airway remodeling associated with COPD.

| 4-cyano-4-(3-cyclopentyloxy-4-methoxyphenyl)cyclohexane-1-carboxylic acid | |
| Clinical data | |
|---|---|
| Legal status |
?
|
| Identifiers | |
| CAS number | 153259-65-5 |
| ATC code | None |
| PubChem | CID 151170 |
| ChemSpider | 18826005 |
| UNII | 8ATB1C1R6X |
| Chemical data | |
| Formula | C20H25NO4 |
| Mol. mass | 343.417 g/mol |
Synthesis

Christensen, Siegfried B.; Guider, Aimee; Forster, Cornelia J.; Gleason, John G.; Bender, Paul E.; Karpinski, Joseph M.; Dewolf,, Walter E.; Barnette, Mary S. et al. (1998). “1,4-Cyclohexanecarboxylates: Potent and Selective Inhibitors of Phosophodiesterase 4 for the Treatment of Asthma”. Journal of Medicinal Chemistry 41 (6): 821–35. doi:10.1021/jm970090r. PMID 9526558.

The reaction of 3-cyclopentyloxy-4-methoxybenzaldehyde (I) with LiBr, trimethylsilyl chloride (TMS-Cl) and 1,1,3,3-tetramethyldisiloxane in acetonitrile gives the corresponding benzyl bromide (II), which by reaction with NaCN in DMF affords 2-(3-cyclopentyloxy-4-methoxyphenyl)acetonitrile (III).
The condensation of (III) with methyl acrylate (IV) by means of Triton B in refluxing acetonitrile yields the 4-cyanopimelate (V), which is cyclized by means of NaH in refluxing DME, giving the 2-oxocyclohexanecarboxylic ester (VI). The decarboxylation of (VI) by means of NaCl in DMSO/water at 150 C yields the cyclohexanone (VII), which is condensed with 2-(trimethylsilyl)-1,3-dithiane (VIII) by means of BuLi in THF, affording the cyclohexylidene-dithiane (IX).
The methanolysis of (IX) catalyzed by HgCl2 and HClO4 in refluxing methanol gives a mixture of the cis- and trans-4-cyano-4-(3-cyclopentyloxy-4-methoxyphenyl)cyclohexanecarboxylic acid methyl ester which is submitted to flash chromatography to obtain the cis-isomer (XII). Finally, this compound is hydrolyzed with KOH in methanol/THF/water.


The synthesis of SB-207499 is described. Investigation and development of new strategies for the homologation of ketone, 4-cyano-4-[3-(cyclopentyloxy)-4-(methoxyphenyl)]-cyclohexan-1-one 2 are described which produce SB-207499. Our ultimate route of synthesis to SB-207499 is robust and operationally simple and produces the final drug substance in good yield and purity.
cis-4-Cyano-4-[3-(cyclopentyloxy)-4-(methoxyphenyl)]-r-1-cyclohexanecarboxylic acid (1a):
mp 148−150 °C; IR (KBr pellet) cm–1 3300−2400, 2231, 1707, 1694;
1H (400 MHz, CDCl3) δ 11.75 (1Η, br s), 7.02 (1H, d, J = 2.3 Hz), 6.98 (1H, dd, J = 2.3, 8.4 Hz), 6.87 (1H, d, J = 8.4 Hz), 4.82 (1H, m), 3.86 (3H, s), 2.43 (1H, tt, J = 3.7, 12.2 Hz), 2.29 (2H, br d, J = 15.6 Hz), 2.25 (2H, br d, J = 16.4 Hz), 2.05 (2H, m), 1.94 (4H, m), 1.86 (2H, m), 1.82 (2H, m), 1.64 (2H, m); 13C (100 MHz, CDCl3) δ 180.5, 149.8, 147.8, 132.8, 122.2, 117.3, 112.9, 111.9, 80.7, 56.1, 43.0, 41.7, 36.4, 32.8, 25.9, 24.0.
………………………………………..
http://www.google.com/patents/WO1995024381A1?cl=en
cis-{-4-cyano-4-[3- (trans-3-hydroxycyclopentyloxy)-4-methoxyphenyl]cyclohexane-l -carboxylic acid} or the corresponding compounds as defined by Formula I. The preparation of any remaining compounds of the Formula (I) not described therein may be prepared by the analogous processes disclosed herein which comprise:

Example 1
Preparation of cis-r4-cvano-4-(3-cyclopentyloxy-4-methoxyphenyl)cvclohexane- 1 – carboxylic acid]
1 fa (3-Cyclopentyloxy-4-methoxyphenv acetonitrile
To a solution of 3-cyclopentyloxy-4-methoxybenzaldehyde (20 g, 90.8 mmol) in acetonitrile (100 mL) was added lithium bromide (15 g, 173 mmol) followed by the dropwise addition of trimethylsilylchloride (17.4 mL, 137 mmol). After 15 min, the reaction mixture was cooled to 0° C, 1,1,3,3-tetramethyldisiloxane (26.7 mL, 151 mmol) was added dropwise and the resulting mixture was allowed to warm to room temperature. After stirring for 3 h, the mixture was separated into two layers. The lower layer was removed, diluted with methylene chloride and filtered through Celite®. The filtrate was concentrated under reduced pressure, dissolved in methylene chloride and refiltered. The solvent was removed in vacuo to provide a light tan oil. To a solution of this crude a- bromo-3-cyclopentyloxy-4-methoxy toluene in dimethylformamide (160 mL) under an argon atmosphere was added sodium cyanide (10.1 g, 206 mmol) and the resulting mixture was stirred at room temperature for 18 h, then poured into cold water (600 mL) and extracted three times with ether. The organic extract was washed three times with water, once with brine and was dried (K2CO3). The solvent was removed in vacuo and the residue was purified by flash chromatography (silica gel, 10% ethyl acetate/hexanes) to provide an off-white solid ( m.p. 32-34g C); an additional quantity of slightly impure material also was isolated. Kb Dimethyl 4-cvano-4-(‘3-cvclopentyloxy-4-methoxyphenv pimelate
To a solution of (3-cyclopentyloxy-4-methoxyphenyl)acetonitrile (7 g, 30.3 mmol) in acetonitrile (200 mL) under an argon atmosphere was added a 40% solution of Triton-B in methanol (1.4 mL, 3.03 mmol) and the mixture was heated to reflux. Methyl acrylate (27 mL, 303 mmol) was added carefully, the reaction mixture was maintained at reflux for 5 h and then cooled. The mixture was diluted with ether, was washed once with IN hydrochloric acid and once with brine, was dried (MgSO4) and the solvent was removed in vacuo. The solid residue was triturated with 5% ethanol/hexane to provide a white solid (m.p. 81-82° C); an additional quantity was also obtained from the filtrate. Anal. (C22H29NO6) calcd: C 65.49, H 7.25, N 3.47. found: C 65.47, H 7.11, N 3.49. 1. c) 2-Caf bomethoxy-4-cvano-4-(3-cyclopentyloxy-4-methoxyphen vDcvclohexan- 1 -one To a suspension of sodium methoxide (350 mL, 1.55 mol, 25% w/w in methanol) in toluene (2.45 L) heated to 80° C under a nitrogen atmosphere was added a solution of dimethyl 4-cyano-4-(3-cyclopentyloxy-4-methoxyphenyl)pimelate (350.0 g, 0.87 mol) in toluene (1.05 L) over 10 min. The reaction was heated to 85° C by distilling away 250 mL of solvent and was vigorously stirred under nitrogen for 2 hours. The reaction was cooled to 50° C and was quenched with 3N (aq) HC1 (700 mL, 2.1 mol). The organic layer was isolated, was washed once with deionized water (700 mL) and once with brine (700 mL). The organic layer was concentrated via low vacuum distillation to afford crude 2- carbomethoxy-4-cyano-4-(3-cyclopentyloxy-4-methoxyphenyl)cyclohexane- 1 -one in toluene. This was dissolved in 4.2 L of dimethyl sulfoxide and used in the next step. 1 (d) 4-Cvano-4-f3-cyclopentyloxy-4-methoxyphenyl cvclohexan- 1-one
To a suspension of sodium chloride (315 g, 5.39 mol) and deionized water ( 315 mL) was added the dimethyl sulfoxide (4.2 L) solution of 2-carbomethoxy-4-cyano-4-(3- cyclopentyloxy-4-methoxyphenyl)cyclohexane-l-one ( 323 g, 0.87 mol) and the resulting suspension was heated to 155° C for 1.75 h. The reaction was cooled to 40° C, was quenched into 8 L of iced water (22 C) and was extracted with ethyl acetate (3.5 L). The aqueous layer was isolated and re-extracted with 2.5 L of ethyl acetate. The combined organic extract (6 L) was washed two times with deionized water (2 x 1 L) and once with brine (1 L). The organic layer was isolated and concentrated in vacuo to afford a residue. This residue was dissolved in refluxing isopropanol (500 mL), was cooled to 0° C and held at this temperature for 1 hour. The crystals were isolated by filtration, were washed with 250 mL of isopropanol (0° C), and were dried in a vacuum oven (45° C at 20 inches) to produce 4-cyano-4-(3-cyclopentyloxy-4-methoxyphenyl)cyclohexan-l -one . m.p. 111-112° C; Anal. (C19H23NO ) calcd: C 72.82, H 7.40, N 4.47; found: C 72.72, H 7.39, N 4.48. 1 (e) 2-r4-Cyano-4-G-cyclopentyloxy-4-methoxyphenyl)cvclohexylidenel- 1.3-dithiane To a solution of 2-trimethylsilyl-l,3-dithiane (9.25 mL, 48.7 mmol) in dry tetrahydrofuran (80 mL) at 0° C under an argon atmosphere was added rapidly n- butyllithium (2.5M in hexanes, 19.2 mL, 48 mmol). After 10 min, the mixture was cooled to -78° C and a solution of 4-cyano-4-(3-cyclopentyloxy-4-methoxyphenyl)cyclohexan-l- one (7.53 g, 23 mmol) in tetrahydrofuran (40 mL) was added. After 10 min, aqueous sodium chloride was added, the mixture was allowed to warm to room temperature and was diluted with water. This mixture was combined with the product of three substantially similar reactions conducted on ketone (3.04, 6.01 and 6.1 g, 48.3 mmol total), the combined mixture was extracted three times with methylene chloride, the extract was dried (MgSO4) and evaporated. Purification by flash chromatography (silica gel, 10% ethyl acetate/hexanes) provided a white solid, m.p. 115-116° C. \(f) cis-r4-Cyano-4-(3-cyclopentyloxy-4-methoxyphenyl‘)cyclohexane- 1 -carboxylic acidl
To a suspension of 2-[4-cyano-4-(3-cyclopentyloxy-4-methoxyphenyl)cyclo- hexylidene]-l,3-dithiane ( 140.0 g, 0.34 mol) in acetonitrile (500 mL) and deioinized water (140 mL) under nitrogen was added trifluoroacetic acid (136 g, 1.19 mol). The suspension was heated to 652 C for 1.25 h followed by the addition of 20% sodium hydroxide (420 g, 2.1 mol). The solution was heated at 70 to 75° C for an additional 1.25 h, was cooled to 45° C, deionized water (420 mL)was added followed by 3N (aq) HC1 (392 mL, 1.18 mol). The suspension was cooled to 5° C and held for 1 h. The suspension was filtered, was washed with cold (5e C) deionized water ( 200 mL), and was dried in a vacuum oven (40°C at 20 inches) to obtain crude cis-[4-cyano-4-(3-cyclopentyloxy-4- methoxyphenyl)cyclohexane-l -carboxylic acid]. This material was assayed at 98.5% and was found to a 98.8:1.2 mixture of cis-to-trans isomers, which was contaminated with 0.1% of residual 1,3-propanedithiol. This material was purified via an oxidative workup as follows.
To a hot solution (65° C) of crude cis-[4-cyano-4-(3-cyclopentyloxy-4- methoxyphenyl)cyclohexane-l -carboxylic acid] (85 g, 0.247 mol) in acetonitrile (425 mL) was added 1M sodium hydroxide ( 425 mL, 0.425 mol). To the solution (60° C) was added 4.25 g of calcium hypochlorite and the suspension was vigorously stirred for 2 h. The reaction was concentrated by distilling out 320 mL of solvent, followed by the addition of ethyl acetate ( 425 mL). The reaction was again concentrated by distilling out 445 mL of solvent, was cooled to 55° C followed by the addition of ethyl acetate (1.0 L) and 6N (aq.) HC1 (100 mL). The organic layer was isolated, was washed three times with deionized water (3 x 300 mL), was filtered and was concentrated by distilling out 530 mL of solvent. To the solution was added ethyl acetate (635 mL) with continued distillation to remove 750 mL of solvent. The solution was cooled to 65° C followed by the addition of hexane ( 340 mL). The suspension was cooled to 5° C, held at this temperature for 1 hour, was filtered and was washed with cold (5° C) 10% ethyl acetate/ hexane ( 200 mL). The solid was collected and was dried in a vacuum oven (40° C at 20 inches) to obtain cis- [4- cyano-4- (3-cyclopentyloxy-4-methoxyphenyl)cyclohexane- 1 -carboxylic acid] . This material was found to contain no trans isomer. Anal.(C2θH25-Nθ4) calcd: C 69.95, H 7.34, N 4.08; found: C 69.90, H 7.35, N 4.02. Example 2
Preparation of cis-f 4-cvano-4-r3-(trans-3-hydroxycyclopentyloxy)-4-methoxyphenyll- cyclohexane-1 -carboxylic acid)
2(a’) cis-F4-Cyano-4-(3-hvdroxy-4-methoxyphenvDcyclohexane- 1 -carboxylic acid]
To a solution of boron tribromide in dichlorormethane (0.1M, 335 mL, 33.5 mmol) under an argon atmosphere at -78° C was slowly added a solution of cis-[4-cyano-4-(3- cyclopentyloxy-4-methoxyphenyl)cyclohexane-l -carboxylic acid] (4.03 g, 11.7 mmol) in dichloromethane (180 mL). The mixture was stirred for 5 min, 15% sodium methoxide in methanol was added to pH 8-9 and the reaction was warmed to RT. Water (lOOmL) was added and the mixture was acidified with 3N aqueous hydrochloric acid to pH 1-2. The organic layer was separated, was dried (MgSO4/Na2SO4), was filtered and was evaporated. The residue was twice dissolved in chloroform and the solution was evaporated to yield a white solid. -1H NMR(400 MHz, CDCI3) δ 7.01 (d, J=2.4 Hz, 1H), 6.96 (d of d, J=2.4, 8.5 Hz, 1H), 3.89 (s, 3H), 2.31 (m, 1H), 2.21 (br t, J=13.6 Hz, 4H), 1.98 (m,2H), 1.77 (m, 2H); mp 190-193° C. Kb) Methyl cis- r-4-cvano-4-(3-hvdroxy-4-methoxyphenyl‘)cvclohexane-l-carboxylatel -Toluenesulfonic acid monohydrate (0.015 g, 0.08 mmol) was added to a solution of the compound of Example 2(a) (0.70 g, 2.54 mmol) in dry methanol (20 mL) under an argon atmosphere and the reaction was stirred for 6 h at 45-509 C. The reaction was cooled to RT and was stirred for an additional 16 h. The solution was evaporated and the residue was purified by flash chromatography (silica gel, 50% hexane/ethyl acetate) to yield the tide compound as a white solid. -1H NMR(400 MHz, CDC13) δ 7.01 (m, 2H), 6.85 (d, J=9.1 Hz, IH), 3.90 (s, 3H), 3.72 (s, 3H), 2.35 (t of t, J=3.6, 12.2 Hz, IH), 2.14-2.25 (m, 4H), 2.00 (app q, J=13.4 Hz, IH), 1.99 (app q, J=13.4 Hz, IH), 1.77 (app t, J=13.4 Hz, IH), 1.76 (app t, J=13.4 Hz, IH); mp 106-107° C.
2(c) Methyl cis- f -4-cvano-4-r3-(trans-3-hydroxycvclopentyloxy )-4-methoxyphenyl – cvclohexane- 1 -carboxylate 1
The compound of Example 2(b) (0.69 g, 2.37 mmol) was dissolved in tetrahydrofuran (20 mL) under an argon atmosphere and was treated with triphenylphosphine (1.24 g, 4.74 mmol) and cis-l,3-cyclopentanediol (0.49 g, 4.74 mmol). Diethyl azodicarboxylate (0.83 g, 4.74 mmol) was added and the mixture was stirred at RT for 16 h. The solution was evaporated, the residue was diluted with ether and the white solid was removed by filtration. The filtrate was concentrated and the residue was purified by flash chromatography (silica gel, 50% hexane/ethyl acetate) to yield a mixture of the title compound and triphenylphosphine oxide. The mixture was diluted with ether and the white solid triphenylphosphine oxide was removed by filtration. Evaporation of the filtrate yielded the title compound as a sticky, colorless semi-solid. 1H NMR(400 MHz, CDCI3) δ 7.07 (d, J=2.4 Hz, IH), 7.02 (d of d, J=2.4, 8.8 Hz, IH), 6.87 (d, J=8.8 Hz, IH), 4.99 (m, IH), 4.37 (m, IH), 3.85 (s, 3H), 3.74 (s, 3H), 3.16 (d, J=9.1 Hz, IH), 2.39 (m, IH), 1.88-2.25 (m, 12H), 1.80 (br t, J=13.5 Hz, 2H).
2(d) cis-f-4-cyano-4-r3-(trans-3-hydroxycyclopentyloxy )-4- methoxyphenyllcyclohexane-1 -carboxylic acid )
The compound of Example 2(c) (0.10 g, 0.27 mmol) was dissolved in 5:5:2 tetrahydrofuran methanol/water (5 mL), sodium hydroxide (0.035 g, 0.88 mmol) was added and the mixture was stirred at RT for 3 h. The solvent was evaporated, the residue was partitioned between 5% aqueous NaOH and dichloromethane and the layers were separated. The aqueous layer was acidified to pH 3 with 3N aqueous hydrochloric acid and was extracted three times with 5% methanol in chloroform. The organic extracts were combined, were dried (MgSO4), filtered and evaporated. The residue was purified by flash chromatography (silica gel, 90:10:1 chloroform/methanol water) to yield a solid which was slurried in ether, was collected by filtration and was dried in vacuo to afford the title compound. MS(d/NH3) m e 377 [M + NH ]+; 1H NMR(400 MHz, CDCI3) δ 7.08 (br s, IH), 7.03 (br d, J=8.5Hz, IH), 6.88 (d, J=8.5 Hz, IH), 4.98 (m, IH), 4.38 (m, IH), 3.84 (s, IH), 2.41 (m, IH), 1.77-2.29 (m, 16H); Anal. (C2oH25NO5-»0.9 H2O) calcd: C, 63.95; H,7.19; N,3.73. found: C, 64.06; H, 6.88; N, 3.77; mp 161-163° C.
Example 3 Preparation of cis- f 4-cvano-4-r3-(cis-3-hvdroxycvclopentyloxy)-4-methoxyphenyll- cyclohexane-1 -carboxylic acid) 3(a) Methyl cis-(-4-cvano-4-r3-(cis-3-formyloxycvclopentyloxy)-4-methoxyphenyll- cvclohexane- 1 -carboxylate ) The compound of Example 2(c) (0.68 g, 1.83 mmol) was dissolved in tetrahyrofuran (20 mL) under an argon atmosphere and was treated with triphenylphosphine ( 0.96 g, 3.66 mmol) and formic acid (0.17 g, 3.66 mmol). Diethyl azodicarboxylate (0.64 g, 3.66 mmol) was added and d e mixture was stirred at RT for 16 h. The solution was evaporated, ether was added and the white solid was removed by filtration. The filtrate was concentrated and die residue was purified by flash chromatography (silica gel, 65% hexane/ethyl acetate) to yield the title compound as a clear colorless oil. **-H NMR(400 MHz, CDC13) δ 8.02 (s,lH), 7.0 (d of d, J=2.4, 8.2 Hz, IH), 6.99 (d, J=2.4 Hz, 1 H), 6.87 (d, J=8.2 Hz, IH), 5.48 (m, IH), 4.95 (m, IH), 3.84 (s, 3H), 3.72 (s, 3H), 2.31-2.40 (m, 2H), 2.13-2.28 (m, 7H), 1.96-2.06 (m, 3H), 1.74-1.87 (m, 3H).
3(h) cis- ( -4-cvano-4-r3-(cis-3-hvdroxvcvclθDentvloxy)-4-methoχyphenyllcvclohexane- 1 -carboxylic acid)
The compound of Example 3(a) (0.52 g, 1.31 mmol) was dissolved in 5:5:2 tetrahydrofuran/methanol/water (20mL), sodium hydroxide (0.32 g, 8.0 mmol) was added and die mixture was stirred at RT for 2.5 h. The solvent was evaporated and the aqueous residue was acidified to pH 1-2 with 3N aqueous hydrochloric acid. The white solid product was collected, was washed with water and was dried in vacuo to afford the title compound as a white solid. MS(CI/NH3) m/e 377 [M + NH3]+;
IH NMR(250 MHz, CDCI3) δ 6.98 (m, 2H), 6.86 (d, J=8.2 Hz, IH), 4.97 (m, IH), 4.59 (m, IH), 3.85 (s, 3H), 1.64-2.47 (m, 17H);
mp 143-145° C.
References
- http://www.medscape.com/viewarticle/549357
- Torphy TJ, Barnette MS, Underwood DC, Griswold DE, Christensen SB, Murdoch RD, Nieman RB, Compton CH. Ariflo (SB 207499), a second generation phosphodiesterase 4 inhibitor for the treatment of asthma and COPD: from concept to clinic. Pulmonary Pharmacology and Therapeutics. 1999;12(2):131-5. PMID 10373396
- Ochiai H, Ohtani T, Ishida A, Kusumi K, Kato M, Kohno H, Kishikawa K, Obata T, Nakai H, Toda M. Highly potent PDE4 inhibitors with therapeutic potential. Bioorganic and Medicinal Chemistry Letters. 2004 Jan 5;14(1):207-10. PMID 14684329
| WO1993019747A1 * | Mar 5, 1993 | Oct 14, 1993 | Siegfried B Christensen Iv | Compounds useful for treating allergic and inflammatory diseases |
| WO1993019748A1 * | Mar 5, 1993 | Oct 14, 1993 | Paul Elliot Bender | Compounds useful for treating inflammatory diseases and for inhibiting production of tumor necrosis factor |
| WO1993019750A1 * | Mar 12, 1993 | Oct 14, 1993 | Paul Elliot Bender | Compounds useful for treating allergic or inflammatory diseases |
| US4795757 * | Nov 20, 1986 | Jan 3, 1989 | Rorer Pharmaceutical Corporation | Bisarylamines |
| US5096906 * | Dec 5, 1990 | Mar 17, 1992 | University Of Virginia Alumni Patents Foundation | Method of inhibiting the activity of leukocyte derived cytokines |
| WO1993019720A2 * | Mar 12, 1993 | Oct 14, 1993 | Paul Elliot Bender | Compounds |
FDA Approves Spiriva Respimat (tiotropium) for the Maintenance Treatment of COPD

Ridgefield, Conn., September 25, 2014 – Boehringer Ingelheim Pharmaceuticals, Inc. announced today that the U.S. Food and Drug Administration (FDA) approved Spiriva Respimat (tiotropium bromide) inhalation spray for the long-term, once-daily maintenance treatment of bronchospasm associated with chronic obstructive pulmonary disease (COPD), including chronic bronchitis and emphysema and to reduce exacerbations in COPD patients. Boehringer Ingelheim anticipates Spiriva Respimat to be available in January 2015.

Spiriva Respimat provides a pre-measured amount of medicine in a slow-moving mist that helps patients inhale the medicine. Spiriva Respimat was developed to actively deliver medication in a way that does not depend of how fast air is breathed in from the inhaler.
READ AT




MAKE IN INDIA
FDA Approves Striverdi Respimat, Olodaterol to Treat Chronic Obstructive Pulmonary Disease

FDA Approves Striverdi Respimat to Treat Chronic Obstructive Pulmonary Disease
July 31, 2014 — Today, the U.S. Food and Drug Administration approved
Striverdi Respimat (olodaterol) inhalation spray to treat patients with chronic
obstructive pulmonary disease (COPD), including chronic bronchitis and/or emphysema
that are experiencing airflow obstruction. Striverdi Respimat can be used once daily
over a long period of time.
read at
http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm407465.htm
See my old post cut paste here
BI launches COPD drug Striverdi, olodaterol in UK and Ireland
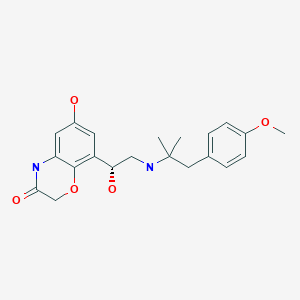
Olodaterol
オロダテロール
BI-1744
BI-1744-CL (hydrochloride) marketed as drug
Boehringer Ingelheim Pharma innovator
synthesis…..http://wendang.baidu.com/view/d4f95541e518964bcf847c22.html
Olodaterol (trade name Striverdi) is a long acting beta-adrenoceptor agonist used as an inhalation for treating patients with chronic obstructive pulmonary disease (COPD), manufactured by Boehringer-Ingelheim.[1]
see……….https://www.thieme-connect.de/DOI/DOI?10.1055/s-0029-1219649 ……… synfacts
Olodaterol is a potent agonist of the human β2-adrenoceptor with a high β1/β2 selectivity. Its crystalline hydrochloride salt is suitable for inhalation and is currently undergoing clinical trials in man for the treatment of asthma. Olodaterol has a duration of action that exceeds 24 hours in two preclinical animal models of bronchoprotection and it has a better safety margin compared with formoterol.
Olodaterol hydrochloride [USAN]
Bi 1744 cl
Bi-1744-cl
Olodaterol hydrochloride
Olodaterol hydrochloride [usan]
UNII-65R445W3V9
868049-49-4 [RN] FREE FORM
CAS 869477-96-3 HCL SALT
R ENANTIOMER
2H-1,4-Benzoxazin-3(4H)-one, 6-hydroxy-8-((1R)-1-hydroxy-2-((2-(4-methoxyphenyl)- 1,1-dimethylethyl)amino)ethyl)-, hydrochloride (1:1)
2H-1,4-benzoxazin-3(4H)-one, 6-hydroxy-8-((1R)-1-hydroxy-2-((2-(4-methoxyphenyl)- 1,1-dimethylethyl)amino)ethyl)-, hydrochloride (1:1)
6-Hydroxy-8-((1R)-1-hydroxy-2-((2-(4-methoxyphenyl)-1,1-dimethylethyl)amino)ethyl)- 2H-1,4-benzoxazin-3(4H)-one hydrochloride
clinical trialshttp://clinicaltrials.gov/search/intervention=Olodaterol+OR+BI+1744
Boehringer Ingelheim has launched a new chronic obstructive pulmonary disease drug, Striverdi in the UK and Ireland.
Striverdi (olodaterol) is the second molecule to be licenced for delivery via the company’s Respimat Soft Mist inhaler, following the COPD blockbuster Spiriva (tiotropium). The drug was approved in Europe in November based on results from a Phase III programme that included more than 3,000 patients with moderate to very severe disease.http://www.pharmatimes.com/Article/14-07-01/BI_launches_COPD_drug_Striverdi_in_UK_and_Ireland.aspx
Olodaterol hydrochloride is a drug candidate originated by Boehringer Ingelheim. The product, delivered once-daily by the Respimat Soft Mist Inhaler, was first launched in Denmark and the Netherlands in March 2014 for the use as maintenance treatment of chronic obstructive pulmonary disease (COPD), including chronic bronchitis and/or emphysema. In 2013, approval was obtained in Russia and Canada for the same indication, and in the U.S, the product was recommended for approval. Phase III clinical trials for the treatment of COPD are ongoing in Japan.
| Systematic (IUPAC) name | |
|---|---|
| 6-hydroxy-8-{(1R)-1-hydroxy-2-{[1-(4-methoxyphenyl)-2-methylpropan-2-yl]amino}ethyl}-4H-1,4-benzoxazin-3-one | |
| Clinical data | |
| Trade names | Striverdi |
| AHFS/Drugs.com | UK Drug Information |
| Pregnancy cat. | No experience |
| Legal status | POM (UK) |
| Routes | Inhalation |
| Identifiers | |
| CAS number | 868049-49-4; 869477-96-3 (hydrochloride) |
| ATC code | R03AC19 |
| PubChem | CID 11504295 |
| ChemSpider | 9679097 |
| UNII | VD2YSN1AFD |
| ChEMBL | CHEMBL605846 |
| Synonyms | BI 1744 CL |
| Chemical data | |
| Formula | C21H26N2O5 free form C21 H26 N2 O5 . Cl H; of hcl salt |
| Mol. mass | 386.44 g/mol free form; 422.902 as hyd salt |
Medical uses
Olodaterol is a once-daily maintenance bronchodilator treatment of airflow obstruction in patients with COPD including chronic bronchitis and/or emphysema, and is administered in an inhaler called Respimat Soft Mist Inhaler.[2][3][4][5][6][7]
As of December 2013, olodaterol is not approved for the treatment of asthma. Olodaterol monotherapy was previously evaluated in four Phase 2 studies in asthma patients. However, currently there are no Phase 3 studies planned for olodaterol monotherapy in patients with asthma.
In late January 2013, Olodaterol CAS# 868049-49-4 was the focus of an FDA committee reviewing data for the drug’s approval as a once-daily maintenance bronchodilator to treat chronic obstructive pulmonary disease (COPD), as well as chronic bronchitis and emphysema. The FDA Pulmonary-Allergy Drugs Advisory Committee recommended that the clinical data from the Boehringer Ingelheim Phase III studies be included in their NDA.
Also known as the trade name Striverdi Respimat, Olodaterol is efficacious as a long-acting beta-agonist, which patients self-administer via an easy to use metered dose inhaler. While early statistics from clinical trials of Olodaterol were encouraging, a new set of data was released earlier this week, which only further solidified the effectual and tolerable benefits of this COPD drug.
On September 10, 2013 results from two Phase 3 studies of Olodaterol revealed additional positive results from this formidable COPD treatment. The conclusion from these two 48 week studies, which included over 3,000 patients, showed sizable and significant improvements in the lung function of patients who were dosed with Olodaterol. Patients in the aforementioned studies were administered either a once a day dosage of Olodaterol via the appropriate metered-dose inhaler or “usual care”. The “usual care” included a variety of treatment options, such as inhaled corticosteroids (not Olodaterol), short and long acting anticholinergics, xanthines and beta agonists, which were short acting. The clinical trial participants who were dosed with Olodaterol displayed a rapid onset of action from this drug, oftentimes within the first five minutes after taking this medication. Additionally, patients dispensed the Olodaterol inhaler were successfully able to maintain optimum lung function for longer than a full 24 hour period. The participants who were given Olodaterol experienced such an obvious clinical improvement in their COPD symptoms, and it quickly became apparent that the “usual care” protocol was lacking in efficacy and reliability.
A staggering 24 million patients in the United States suffer from chronic obstructive pulmonary disease, and this patient population is in need of an effectual, safe and tolerable solution. Olodaterol is shaping up to be that much needed solution. Not only have the results from studies of Olodaterol been encouraging, the studies themselves have actually been forward thinking and wellness centered. Boehringer Ingelheim is the first company to included studies to evaluate exercise tolerance in patients with COPD, and compare the data to those patients who were dosed with Olodaterol. By including exercise tolerance as an important benchmark in pertinent data for Olodaterol, Boehringer Ingelheim has created a standard for COPD treatment expectations. The impaired lung function for patients with COPD contributes greatly to their inability to exercise and stay healthy. Patients who find treatments and management techniques to combat the lung hyperinflation that develops during exercise have a distinct advantage to attaining overall good health.
– See more at: http://www.lgmpharma.com/blog/olodaterol-offers-encouraging-results-patients-copd/#sthash.DOjcrGxc.dpuf
Data has demonstrated that Striverdi, a once-daily long-acting beta2 agonist, significantly improved lung function versus placebo and is comparable to improvements shown with the older LABA formoterol. The NHS price for the drug is £26.35 for a 30-day supply.
Boehringer cited Richard Russell at Wexham Park Hospital as saying that the licensing of Stirverdi will be welcomed by clinicians as it provides another option. He added that the trial results showing improvements in lung function “are particularly impressive considering the study design, which allowed participants to continue their usual treatment regimen. This reflects more closely the real-world patient population”.
Significantly, the company is also developing olodaterol in combination with Spiriva, a long-acting muscarinic antagonist. LAMA/LABA combinations provide the convenience of delivering the two major bronchodilator classes.
 Olodaterol is a novel, long-acting beta2-adrenergic agonist (LABA) that exerts its pharmacological effect by binding and activating beta2-adrenergic receptors located primarily in the lungs. Beta2-adrenergic receptors are membrane-bound receptors that are normally activated by endogenous epinephrine whose signalling, via a downstream L-type calcium channel interaction, mediates smooth muscle relaxation and bronchodilation. Activation of the receptor stimulates an associated G protein which then activates adenylate cyclase, catalyzing the formation of cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA). Elevation of these two molecules induces bronchodilation by relaxation of airway smooth muscles. It is by this mechanism that olodaterol is used for the treatment of chronic obstructive pulmonary disease (COPD) and the progressive airflow obstruction that is characteristic of it. Treatment with bronchodilators helps to mitigate associated symptoms such as shortness of breath, cough, and sputum production. Single doses of olodaterol have been shown to improve forced expiratory volume in 1 sec (FEV1) for 24 h in patients with COPD, allowing once daily dosing. A once-a-day treatment with a LABA has several advantages over short-acting bronchodilators and twice-daily LABAs including improved convenience and compliance and improved airflow over a 24-hour period. Despite similarities in symptoms, olodaterol is not indicated for the treatment of acute exacerbations of COPD or for the treatment of asthma.
Olodaterol is a novel, long-acting beta2-adrenergic agonist (LABA) that exerts its pharmacological effect by binding and activating beta2-adrenergic receptors located primarily in the lungs. Beta2-adrenergic receptors are membrane-bound receptors that are normally activated by endogenous epinephrine whose signalling, via a downstream L-type calcium channel interaction, mediates smooth muscle relaxation and bronchodilation. Activation of the receptor stimulates an associated G protein which then activates adenylate cyclase, catalyzing the formation of cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA). Elevation of these two molecules induces bronchodilation by relaxation of airway smooth muscles. It is by this mechanism that olodaterol is used for the treatment of chronic obstructive pulmonary disease (COPD) and the progressive airflow obstruction that is characteristic of it. Treatment with bronchodilators helps to mitigate associated symptoms such as shortness of breath, cough, and sputum production. Single doses of olodaterol have been shown to improve forced expiratory volume in 1 sec (FEV1) for 24 h in patients with COPD, allowing once daily dosing. A once-a-day treatment with a LABA has several advantages over short-acting bronchodilators and twice-daily LABAs including improved convenience and compliance and improved airflow over a 24-hour period. Despite similarities in symptoms, olodaterol is not indicated for the treatment of acute exacerbations of COPD or for the treatment of asthma.Adverse effects
Adverse effects generally were rare and mild in clinical studies. Most common, but still affecting no more than 1% of patients, were nasopharyngitis (running nose), dizziness and rash. To judge from the drug’s mechanism of action and from experiences with related drugs, hypertension (high blood pressure), tachycardia (fast heartbeat), hypokalaemia (low blood levels of potassium), shaking, etc., might occur in some patients, but these effects have rarely, if at all, been observed in studies.[1]
Interactions
Based on theoretical considerations, co-application of other beta-adrenoceptor agonists, potassium lowering drugs (e. g. corticoids, many diuretics, and theophylline), tricyclic antidepressants, and monoamine oxidase inhibitors could increase the likelihood of adverse effects to occur. Beta blockers, a group of drugs for the treatment of hypertension (high blood pressure) and various conditions of the heart, could reduce the efficacy of olodaterol.[1] Clinical data on the relevance of such interactions are very limited.
Pharmacology
Mechanism of action
Like all beta-adrenoceptor agonists, olodaterol mimics the effect of epinephrine at beta-2 receptors (β₂-receptors) in the lung, which causes the bronchi to relax and reduces their resistance to airflow.[3]
Olodaterol is a nearly full β₂-agonist, having 88% intrinsic activity compared to the gold standard isoprenaline. Its half maximal effective concentration (EC50) is 0.1 nM. It has a higher in vitro selectivity for β₂-receptors than the related drugs formoterol and salmeterol: 241-fold versus β₁- and 2299-fold versus β₃-receptors.[2] The high β₂/β₁ selectivity may account for the apparent lack of tachycardia in clinical trials, which is mediated by β₁-receptors on the heart.
Pharmacokinetics
Once bound to a β₂-receptor, an olodaterol molecule stays there for hours – its dissociation half-life is 17.8 hours –, which allows for once-a-day application of the drug[3] like with indacaterol. Other related compounds generally have a shorter duration of action and have to be applied twice daily (e.g. formoterol, salmeterol). Still others (e. g. salbutamol, fenoterol) have to be applied three or four times a day for continuous action, which can also be an advantage for patients who need to apply β₂-agonists only occasionally, for example in an asthma attack.[8]
History
On 29 January 2013 the U.S. Food and Drug Administration (FDA) Pulmonary-Allergy Drugs Advisory Committee (PADAC) recommended that the clinical data included in the new drug application (NDA) for olodaterol provide substantial evidence of safety and efficacy to support the approval of olodaterol as a once-daily maintenance bronchodilator treatment for airflow obstruction in patients with COPD.[9]
On 18 October 2013 approval of olodaterol in the first three European countries – the United Kingdom, Denmark and Iceland – was announced by the manufacturer.[10]

Figure Chemical structures of salmeterol, formoterol, inda- caterol, and emerging once-daily long-acting β2-agonists
CLIP
Synthetic approaches to the 2013 new drugs – ScienceDirect

Olodaterol hydrochloride was approved for long-term, once-daily maintenance treatment of chronic
obstructive pulmonary disease (COPD) in 2013 in the following countries: Canada, Russia, United
Kingdom, Denmark, and Iceland.142, 143 The drug has been recommended by a federal advisory panel for
approval by the FDA.142, 143 Developed and marketed by Boehringer Ingelheim, olodaterol is a longacting
β2-adrenergic receptor agonist with high selectivity over the β1- and β3-receptors (219- and 1622-fold, respectively).144 Upon binding to and activating the β2-adrenergic receptor in the airway, olodaterol
stimulates adenyl cyclase to synthesize cAMP, leading to the relaxation of smooth muscle cells in the
airway. Administered by inhalation using the Respimat®
Soft Mist inhaler, it delivers significant
bronchodilator effects within five minutes of the first dose and provides sustained improvement in
forced expiratory volume (FEV1) for over 24 hours.143 While several routes have been reported in the
patent and published literature,144-146 the manufacturing route for olodaterol hydrochloride disclosed in
2011 is summarized in Scheme 19 below.147
Commercial 2’,5’-dihydroxyacetophenone (122) was treated with one equivalent of benzyl bromide
and potassium carbonate in methylisobutylketone (MIBK) to give the 5’-monobenzylated product in
76% yield. Subsequent nitration occurred at the 4’-position to provide nitrophenol 123 in 87% yield.
Reduction of the nitro group followed by subjection to chloroacetyl chloride resulted in the construction
of benzoxazine 124 in 82% yield. Next, monobromination through the use of tetrabutylammonium
tribromide occurred at the acetophenone carbon to provide bromoketone 125, and this was followed by
asymmetric reduction of the ketone employing (−)-DIP chloride to afford an intermediate bromohydrin,
which underwent conversion to the corresponding epoxide 126 in situ upon treatment with aqueous
NaOH. This epoxide was efficiently formed in 85% yield and 98.3% enantiomeric excess. Epoxide
126 underwent ring-opening upon subjection to amine 127 to provide amino-alcohol 128 in in 84-90%
yield and 89.5-99.5% enantiomeric purity following salt formation with HCl. Tertiary amine 127 was
itself prepared in three steps by reaction of ketone 129 with methylmagnesium chloride, Ritter reaction
of the tertiary alcohol with acetonitrile, and hydrolysis of the resultant acetamide with ethanolic
potassium hydroxide. Hydrogenative removal of the benzyl ether within 128 followed by
recrystallization with methanolic isopropanol furnished olodaterol hydrochloride (XVI) in 63-70%
yield. Overall, the synthesis of olodaterol hydrochloride required 10 total steps (7 linear) from
commercially available acetophenone 122.
142. Gibb, A.; Yang, L. P. H. Drugs 2013, 73, 1841.
143. http://www.boehringeringelheim.com/news/news_releases/press_releases/2013/18_october_2013_olodaterol.html.
144. Bouyssou, T.; Hoenke, C.; Rudolf, K.; Lustenberger, P.; Pestel, S.; Sieger, P.; Lotz, R.; Heine,
C.; Buettner, F. H.; Schnapp, A.; Konetzki, I. Bioorg. Med. Chem. Lett. 2010, 20, 1410.
145. Trunk, M. J. F.; Schiewe, J. US Patent 20050255050A1, 2005.
146. Lustenberger, P.; Konetzki, I.; Sieger, P. US Patent 20090137578A1, 2009.
147. Krueger, T.; Ries, U.; Schnaubelt, J.; Rall, W.; Leuter, Z. A.; Duran, A.; Soyka, R. US Patent
20110124859A1, 2011.
PATENT
WO 2004045618 or
http://www.google.com/patents/EP1562603B1?cl=en
Example

a)
To a solution of 3.6 g 1,1-dimethyl-2-(4-methoxyphenyl)-ethylamine in 100 mL of ethanol at 70 ° C. 7.5 g of (6-benzyloxy-4H-benzo [1,4] oxazin-3-one )-glyoxal added and allowed to stir for 15 minutes. Then within 30 minutes at 10 to 20 ° C. 1 g of sodium borohydride added. It is stirred for one hour, with 10 mL of acetone and stirred for another 30 minutes. The reaction mixture is diluted with 150 mL ethyl acetate, washed with water, dried with sodium sulfate and concentrated. The residue is dissolved in 50 mL of methanol and 100 mL ethyl acetate and acidified with conc. Hydrochloric acid. After addition of 100 mL of diethyl ether, the product precipitates. The crystals are filtered, washed and recrystallized from 50 mL of ethanol. Yield: 7 g (68%; hydrochloride), mp = 232-234 ° C.
b)
6.8 g of the above obtained benzyl compound in 125 mL of methanol with the addition of 1 g of palladium on carbon (5%) was hydrogenated at room temperature and normal pressure. The catalyst is filtered and the filtrate was freed from solvent. Recrystallization of the residue in 50 mL of acetone and a little water, a solid is obtained, which is filtered and washed.
Yield: 5.0 g (89%; hydrochloride), mp = 155-160 ° C.
The (R) – and (S)-enantiomers of Example 3 can be obtained from the racemate, for example, by chiral HPLC (for example, column: Chirobiotic T, 250 x 1.22 mm from the company Astec). As the mobile phase, methanol with 0.05% triethylamine and 0.05% acetic acid. Silica gel with a grain size of 5 microns, to which is covalently bound the glycoprotein teicoplanin can reach as column material used. Retention time (R enantiomer) = 40.1 min, retention time (S-enantiomer) = 45.9 min. The two enantiomers can be obtained by this method in the form of free bases. According to the invention of paramount importance is the R enantiomer of Example 3
PATENT
WO 2005111005
http://www.google.fm/patents/WO2005111005A1?cl=en
Scheme 1.




Scheme 1:
Example 1 6-Hydroxy-8-{(1-hydroxy-2-r2-(4-methoxy-phenyl) – 1, 1-dimethyl-ethylamino]-ethyl)-4H-benzor 41oxazin-3-one – Hvdrochlorid

a) l-(5-benzyloxy-2-hydroxy-3-nitro-phenyl)-ethanone
To a solution of 81.5 g (0.34 mol) l-(5-benzyloxy-2-hydroxy-phenyl)-ethanone in 700 ml of acetic acid are added dropwise under cooling with ice bath, 18 mL of fuming nitric acid, the temperature does not exceed 20 ° C. increases. The reaction mixture is stirred for two hours at room temperature, poured onto ice water and filtered. The product is recrystallized from isopropanol, filtered off and washed with isopropanol and diisopropyl ether. Yield: 69.6 g (72%), mass spectroscopy [M + H] + = 288
b) l-(3-Amino-5-benzyloxy-2-hydroxy-phenyl)-ethanone
69.5 g (242 mmol) of l-(5-benzyloxy-2-hydroxy-3-nitro-phenyl)-ethanone are dissolved in 1.4 L of methanol and in the presence of 14 g of rhodium on carbon (10%) as catalyst at 3 bar room temperature and hydrogenated. Then the catalyst is filtered off and the filtrate concentrated. The residue is reacted further without additional purification. Yield: 60.0 g (96%), R f value = 0.45 (silica gel, dichloromethane).
c) 8-acetyl-6-benzyloxy-4H-benzoπ .4] oxazin-3-one
To 60.0 g (233 mmol) of l-(3-Amino-5-benzyloxy-2-hydroxy-phenyl)-ethanone and 70.0 g (506 mmol) of potassium carbonate while cooling with ice bath, 21.0 ml (258 mmol) of chloroacetyl chloride added dropwise. Then stirred overnight at room temperature and then for 6 hours under reflux. The hot reaction mixture is filtered and then concentrated to about 400 mL and treated with ice water. The precipitate is filtered off, dried and purified by chromatography on a short silica gel column (dichloromethane: methanol = 99:1). The product-containing fractions are concentrated, suspended in isopropanol, diisopropyl ether, and extracted with
Diisopropyl ether. Yield: 34.6 g (50%), mass spectroscopy [M + H] + = 298
d) 6-Benzyloxy-8-(2-chloro-acetyl)-4H-benzoFl, 4] oxazin-3-one 13.8 g (46.0 mmol) of 8-benzyloxy-6-Acetyl-4H-benzo [l, 4] oxazin -3-one and 35.3 g (101.5 mmol) of benzyltrimethylammonium dichloriodat are stirred in 250 mL dichloroethane, 84 mL glacial acetic acid and 14 mL water for 5 hours at 65 ° C. After cooling to room temperature, treated with 5% aqueous sodium hydrogen sulfite solution and stirred for 30 minutes. The precipitated solid is filtered off, washed with water and diethyl ether and dried. Yield: 13.2 g (86%), mass spectroscopy [M + H] + = 330/32.
e) 6-Benzyloxy-8-((R-2-chloro-l-hydroxy-ethyl)-4H-benzori ,41-oxazin-3-one The procedure is analogous to a procedure described in the literature (Org. Lett ., 2002, 4, 4373-4376).
To 13:15 g (39.6 mmol) of 6-benzyloxy-8-(2-chloro-acetyl)-4H-benzo [l, 4] oxazin-3-one and 25.5 mg (0:04 mmol) Cρ * RhCl [(S, S) -TsDPEN] (Cp * = pentamethylcyclopentadienyl and TsDPEN = (lS, 2S)-Np-toluenesulfonyl-l ,2-diphenylethylenediamine) in 40 mL of dimethylformamide at -15 ° C and 8 mL of a mixture of formic acid and triethylamine (molar ratio = 5: 2) dropwise. It is allowed for 5 hours at this temperature, stirring, then 25 mg of catalyst and stirred overnight at -15 ° C. The reaction mixture is mixed with ice water and filtered. The filter residue is dissolved in dichloromethane, dried with sodium sulfate and the solvent evaporated. The residue is recrystallized gel (dichloromethane / methanol gradient) and the product in diethyl ether / diisopropyl ether. Yield: 10.08 g (76%), R f value = 00:28 (on silica gel, dichloromethane ethanol = 50:1).
f) 6-Benzyloxy-8-(R-oxiranyl-4H-benzo [“L4] oxazin-3-one 6.10 g (30.1 mmol) of 6-benzyloxy-8-((R)-2-chloro-l-hydroxy- ethyl)-4H-benzo [l, 4] oxazin-3-one are dissolved in 200 mL of dimethylformamide. added to the solution at 0 ° C with 40 mL of a 2 molar sodium hydroxide solution and stirred at this temperature for 4 hours. the reaction mixture is poured onto ice water, stirred for 15 minutes, and then filtered The solid is washed with water and dried to give 8.60 g (96%), mass spectroscopy [M + H] + = 298..
g) 6-Benyloxy-8-{(R-l-hydroxy-2-r2-(4-methoxy-phenyl)-dimethyl-ll-ethvIaminol-ethyl)-4H-benzo-3-Tl A1oxazin
5.25 g (17.7 mmol) of 6-benzyloxy-8-(R)-oxiranyl-4H-benzo [l, 4] oxazin-3-one and 6.30 g (35.1 mmol) of 2 – (4-methoxy-phenyl 1, 1 – dimethyl-ethyl to be with 21 mL
Of isopropanol and stirred at 135 ° C for 30 minutes under microwave irradiation in a sealed reaction vessel. The solvent is distilled off and the residue chromatographed (alumina, ethyl acetate / methanol gradient). The product thus obtained is purified by recrystallization from a mixture further Diethylether/Diisopropylether-. Yield: 5:33 g (63%), mass spectroscopy [M + H] + = 477 h) 6-Hydroxy-8-{(R)-l-hydroxy-2-[2 – (4-methoxy-phenyl)-l, l-dimethyl-ethylamino] – ethyl}-4H-benzo [1, 4, 1 oxazin-3-one hydrochloride
A suspension of 5:33 g (11.2 mmol) of 6-Benyloxy-8-{(R)-l-hydroxy-2-[2 – (4-methoxy-phenyl)-l, l-dimethyl-ethylamino]-ethyl}-4H -benzo [l, 4] oxazin-3-one in 120 mL of methanol with 0.8 g of palladium on carbon (10%), heated to 50 ° C and hydrogenated at 3 bar hydrogen pressure. Then the catalyst is filtered off and the filtrate concentrated. The residue is dissolved in 20 mL of isopropanol, and 2.5 mL of 5 molar hydrochloric acid in isopropanol. The product is precipitated with 200 mL of diethyl ether, filtered off and dried. Yield: 4.50 g (95%, hydrochloride), mass spectroscopy [M + H] + = 387
PATENT
WO 2007020227
http://www.google.com.ar/patents/WO2007020227A1?cl=en
PATENT
WO 2008090193
or
http://www.google.com/patents/EP2125759B1?cl=en
PAPER
Discovery of olodaterol, a novel inhaled beta(2)-adrenoceptor agonist with a 24h bronchodilatory efficacy
Bioorg Med Chem Lett 2010, 20(4): 1410
http://www.sciencedirect.com/science/article/pii/S0960894X09018101
The discovery of the β2-adrenoceptor agonist (R)-4p designated olodaterol is described. The preclinical profile of the compound suggests a bronchoprotective effect over 24 h in humans.

CLIP
Australia
http://www.tga.gov.au/pdf/auspar/auspar-olodaterol-140327-pi.pdf
CLIP
DUTCH
FDA
Click to access 203108Orig1s000ChemR.pdf
NDA 203108
Striverdi® Respimat® (olodaterol) Inhalation Spray
Boehringer Ingelheim Pharmaceuticals, Inc.
References
- Striverdi UK Drug Information
- Bouyssou, T.; Casarosa, P.; Naline, E.; Pestel, S.; Konetzki, I.; Devillier, P.; Schnapp, A. (2010). “Pharmacological Characterization of Olodaterol, a Novel Inhaled 2-Adrenoceptor Agonist Exerting a 24-Hour-Long Duration of Action in Preclinical Models”. Journal of Pharmacology and Experimental Therapeutics334 (1): 53–62. doi:10.1124/jpet.110.167007. PMID20371707.
- Casarosa, P.; Kollak, I.; Kiechle, T.; Ostermann, A.; Schnapp, A.; Kiesling, R.; Pieper, M.; Sieger, P.; Gantner, F. (2011). “Functional and Biochemical Rationales for the 24-Hour-Long Duration of Action of Olodaterol”. Journal of Pharmacology and Experimental Therapeutics337 (3): 600–609. doi:10.1124/jpet.111.179259. PMID21357659.
- Bouyssou, T.; Hoenke, C.; Rudolf, K.; Lustenberger, P.; Pestel, S.; Sieger, P.; Lotz, R.; Heine, C.; Büttner, F. H.; Schnapp, A.; Konetzki, I. (2010). “Discovery of olodaterol, a novel inhaled β2-adrenoceptor agonist with a 24h bronchodilatory efficacy”. Bioorganic & Medicinal Chemistry Letters20 (4): 1410–1414. doi:10.1016/j.bmcl.2009.12.087. PMID20096576.
- Joos G, Aumann JL, Coeck C, et al. ATS 2012 Abstract: Comparison of 24-Hour FEV1 Profile for Once-Daily versus Twice-Daily Treatment with Olodaterol, A Novel Long-Acting ß2-Agonist, in Patients with COPD[dead link]
- Van Noord, J. A.; Smeets, J. J.; Drenth, B. M.; Rascher, J.; Pivovarova, A.; Hamilton, A. L.; Cornelissen, P. J. G. (2011). “24-hour Bronchodilation following a single dose of the novel β2-agonist olodaterol in COPD”. Pulmonary Pharmacology & Therapeutics24 (6): 666–672. doi:10.1016/j.pupt.2011.07.006. PMID21839850.
- van Noord JA, Korducki L, Hamilton AL and Koker P. Four Weeks Once Daily Treatment with BI 1744 CL, a Novel Long-Acting ß2-Agonist, is Effective in COPD Patients. Am. J. Respir. Crit. Care Med. 2009; 179: A6183[dead link]
- Haberfeld, H, ed. (2009). Austria-Codex (in German) (2009/2010 ed.). Vienna: Österreichischer Apothekerverlag. ISBN3-85200-196-X.
- Hollis A (31 January 2013). “Panel Overwhelmingly Supports Boehringer COPD Drug Striverdi”. FDA News/Drug Industry Daily.
- “New once-daily Striverdi (olodaterol) Respimat gains approval in first EU countries”. Boehringer-Ingelheim. 18 October 2013.
External links
The active moiety olodaterol is a selective beta2-adrenergic bronchodilator. The drug substance, olodaterol hydrochloride, is chemically described as 2H-1,4- Benzoxazin-3H(4H)-one, 6-hydroxy-8-[(1R)-1-hydroxy-2-[[2-(4-methoxyphenyl)-1,1-dimethylethyl]-amino]ethyl]-, monohydrochloride. Olodaterol hydrochloride is a white to off-white powder that is sparingly-slightly soluble in water and slightly soluble in ethanol. The molecular weight is 422.9 g/mole (salt): 386.5 g/mole (base), and the molecular formula is C21H26N2O5 x HCl as a hydrochloride. The conversion factor from salt to free base is 1.094.
The structural formula is:
 |
The drug product, STRIVERDI RESPIMAT, is composed of a sterile, aqueous solution of olodaterol hydrochloride filled into a 4.5 mL plastic container crimped into an aluminum cylinder (STRIVERDI RESPIMAT cartridge) for use with the STRIVERDI RESPIMAT inhaler.
Excipients include water for injection, benzalkonium chloride, edetate disodium, and anhydrous citric acid. The STRIVERDI RESPIMAT cartridge is only intended for use with the STRIVERDI RESPIMAT inhaler. The STRIVERDI RESPIMAT inhaler is a hand held, pocket sized oral inhalation device that uses mechanical energy to generate a slow-moving aerosol cloud of medication from a metered volume of the drug solution. The STRIVERDI RESPIMAT inhaler has a yellow-colored cap.
When used with the STRIVERDI RESPIMAT inhaler, each cartridge containing a minimum of 4 grams of a sterile aqueous solution delivers the labeled number of metered actuations after preparation for use. Each dose (1 dose equals 2 actuations) from the STRIVERDI RESPIMAT inhaler delivers 5 mcg olodaterol in 22.1 mcL of solution from the mouthpiece. As with all inhaled drugs, the actual amount of drug delivered to the lung may depend on patient factors, such as the coordination between the actuation of the inhaler and inspiration through the delivery system. The duration of inspiration should be at least as long as the spray duration (1.5 seconds).
| WO2002030928A1 | 28 Sep 2001 | 11 Apr 2003 | Boehringer Ingelheim Pharma | Crystalline monohydrate, method for producing the same and the use thereof in the production of a medicament |
| WO2003000265A1 | 8 Jun 2002 | 3 Jan 2003 | Boehringer Ingelheim Pharma | Crystalline anticholinergic, method for its production, and use thereof in the production of a drug |
| WO2004045618A2 * | 11 Nov 2003 | 3 Jun 2004 | Boehringer Ingelheim Pharma | Novel medicaments for the treatment of chronic obstructive pulmonary diseases |
| EP0073505A1 * | 28 Aug 1982 | 9 Mar 1983 | Boehringer Ingelheim Kg | Benzo-heterocycles |
| EP0321864A2 * | 15 Dec 1988 | 28 Jun 1989 | Boehringer Ingelheim Kg | Ammonium compounds, their preparation and use |
| US4460581 | 12 Oct 1982 | 17 Jul 1984 | Boehringer Ingelheim Kg | Antispasmodic agents, antiallergens |
| US4656168 * | 13 Oct 1983 | 7 Apr 1987 | Merck & Co., Inc. | Vision defects; adrenergic blocking and hypotensive agents |
Organic spectroscopy should be brushed up and you get confidence
read my blog
ORGANIC SPECTROSCOPY INTERNATIONAL is the blog
Organic chemists from Industry and academics to interact on Spectroscopy techniques for Organic compounds ie NMR, MASS, IR, UV Etc. email me ……….. amcrasto@gmail.com
http://orgspectroscopyint.blogspot.in/ is the link
 amcrasto@gmail.com
amcrasto@gmail.com
