
07/28/2016 07:53 AM EDT
The U.S. Food and Drug Administration approved Adlyxin (lixisenatide), a once-daily injection to improve glycemic control (blood sugar levels), along with diet and exercise, in adults with type 2 diabetes.
The U.S. Food and Drug Administration approved Adlyxin (lixisenatide), a once-daily injection to improve glycemic control (blood sugar levels), along with diet and exercise, in adults with type 2 diabetes.
“The FDA continues to support the development of new drug therapies for diabetes management,” said Mary Thanh Hai Parks, M.D., deputy director, Office of Drug Evaluation II in the FDA’s Center for Drug Evaluation and Research. “Adlyxin will add to the available treatment options to control blood sugar levels for those with type 2.”
Type 2 diabetes affects more than 29 million people and accounts for more than 90 percent of diabetes cases diagnosed in the United States. Over time, high blood sugar levels can increase the risk for serious complications, including heart disease, blindness and nerve and kidney damage.
Adlyxin is a glucagon-like peptide-1 (GLP-1) receptor agonist, a hormone that helps normalize blood sugar levels. The drug’s safety and effectiveness were evaluated in 10 clinical trials that enrolled 5,400 patients with type 2 diabetes. In these trials, Adlyxin was evaluated both as a standalone therapy and in combination with other FDA-approved diabetic medications, including metformin, sulfonylureas, pioglitazone and basal insulin. Use of Adlyxin improved hemoglobin A1c levels (a measure of blood sugar levels) in these trials.
In addition, more than 6,000 patients with type 2 diabetes at risk for atherosclerotic cardiovascular disease were treated with either Adlyxin or a placebo in a cardiovascular outcomes trial. Use of Adlyxin did not increase the risk of cardiovascular adverse events in these patients.
Adlyxin should not be used to treat people with type 1 diabetes or patients with increased ketones in their blood or urine (diabetic ketoacidosis).
The most common side effects associated with Adlyxin are nausea, vomiting, headache, diarrhea and dizziness. Hypoglycemia in patients treated with both Adlyxin and other antidiabetic drugs such as sulfonylurea and/or basal insulin is another common side effect. In addition, severe hypersensitivity reactions, including anaphylaxis, were reported in clinical trials of Adlyxin.
The FDA is requiring the following post-marketing studies for Adlyxin:
- Clinical studies to evaluate dosing, efficacy and safety in pediatric patients.
- A study evaluating the immunogenicity of lixisenatide.
Adlyxin is manufactured by Sanofi-Aventis U.S. LLC, of Bridgewater, New Jersey.
END……………….

| lixisenatide;Lixisenatide|Lixisenatide Acetate;Lixisenatide Acetate |
| CAS: |
320367-13-3 |
| MF: |
C215H347N61O65S |
| MW: |
4858.53 |
C215 H347 N61 O65 S
L-Lysinamide, L-histidylglycyl-L-α-glutamylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-α-aspartyl-L-leucyl-L-seryl-L-lysyl-L-glutaminyl-L-methionyl-L-α-glutamyl-L-α-glutamyl-L-α-glutamyl-L-alanyl-L-valyl-L-arginyl-L-leucyl-L-phenylalanyl-L-isoleucyl-L-α-glutamyl-L-tryptophyl-L-leucyl-L-lysyl-L-asparaginylglycylglycyl-L-prolyl-L-seryl-L-serylglycyl-L-alanyl-L-prolyl-L-prolyl-L-seryl-L-lysyl-L-lysyl-L-lysyl-L-lysyl-L-lysyl-
L-Histidylglycyl-L-α-glutamylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-α-aspartyl-L-leucyl-L-seryl-L-lysyl-L-glutaminyl-L-methionyl-L-α-glutamyl-L-α-glutamyl-L-α-glutamyl-L-alanyl-L-valyl-L-arginyl-L-leucyl-L-phenylalanyl-L-isoleucyl-L-α-glutamyl-L-tryptophyl-L-leucyl-L-lysyl-L-asparaginylglycylglycyl-L-prolyl-L-seryl-L-serylglycyl-L-alanyl-L-prolyl-L-prolyl-L-seryl-L-lysyl-L-lysyl-L-lysyl-L-lysyl-L-lysyl-L-lysinamide

Lixisenatide

Lixisenatide
IUPAC Condensed
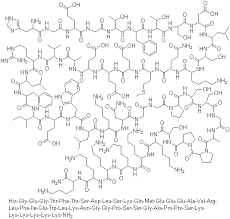
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Ser-Lys-Lys-Lys-Lys-Lys-Lys-NH2
from PubChem
LINUCS
[][L-Lys-NH2]{[(1+2)][L-Lys]{[(1+2)][L-Lys]{[(1+2)][L-Lys]{[(1+2)][L-Lys]{[(1+2)][L-Lys]{[(1+2)][L-Ser]{[(1+2)][L-Pro]{[(1+2)][L-Pro]{[(1+2)][L-Ala]{[(1+2)][Gly]{[(1+2)][L-Ser]{[(1+2)][L-Ser]{[(1+2)][L-Pro]{[(1+2)][Gly]{[(1+2)][Gly]{[(1+2)][L-Asn]{[(1+2)][L-Lys]{[(1+2)][L-Leu]{[(1+2)][L-Trp]{[(1+2)][L-Glu]{[(1+2)][L-Ile]{[(1+2)][L-Phe]{[(1+2)][L-Leu]{[(1+2)][L-Arg]{[(1+2)][L-Val]{[(1+2)][L-Ala]{[(1+2)][L-Glu]{[(1+2)][L-Glu]{[(1+2)][L-Glu]{[(1+2)][L-Met]{[(1+2)][L-Gln]{[(1+2)][L-Lys]{[(1+2)][L-Ser]{[(1+2)][L-Leu]{[(1+2)][L-Asp]{[(1+2)][L-Ser]{[(1+2)][L-Thr]{[(1+2)][L-Phe]{[(1+2)][L-Thr]{[(1+2)][Gly]{[(1+2)][L-Glu]{[(1+2)][Gly]{[(1+2)][L-His]{}}}}}}}}}}}}}}}}}}}}}}}}}}}}}}}}}}}}}}}}}}}}
from PubChem
Sequence
HGEGTFTSDLSKQMEEEAVRLFIEWLKNGGPSSGAPPSKKKKKK
from PubChem
PLN
H-HGEGTFTSDLSKQMEEEAVRLFIEWLKNGGPSSGAPPSKKKKKK-[NH2]
from PubChem
HELM
PEPTIDE1{H.G.E.G.T.F.T.S.D.L.S.K.Q.M.E.E.E.A.V.R.L.F.I.E.W.L.K.N.G.G.P.S.S.G.A.P.P.S.K.K.K.K.K.K.[am]}$$$$

Sanofi (formerly sanofi-aventis, formerly Aventis), under license from Zealand Pharma, has developed and launched lixisenatide
Lixisenatide (trade name Lyxumia) is a once-daily injectable GLP-1 receptor agonist for the treatment of diabetes, discovered by Zealand Pharma A/S of Denmark and licensed and developed by Sanofi.[1] Lixisenatide was accepted for review by the US FDA on February 19, 2013, and approved by the European Commission on February 1, 2013.[2] On September 12, 2013, Sanofi delayed the approval process in the US, citing internal data from a cardiovascular risk study. The drug will likely be resubmitted for approval in 2015.
Lixisenatide is a once-daily injectable GLP-1 receptor agonist discovered by Zealand Pharma A/S of Denmark and licensed and developed by Sanofi. As of September 2010 it is in clinical trials for diabetes. Lixisenatide was accepted for review by the US FDA on February 19, 2013, and approved by the European Commission on February 1, 2013. The drug will likely be resubmitted for approval in 2015.
Mechanism of action
GLP-1 is a naturally-occurring peptide that is released within minutes of eating a meal. It is known to suppress glucagon secretion from pancreatic alpha cells and stimulate insulin secretion by pancreatic beta cells. GLP-1 receptor agonists are used as an add-on treatment for type 2 diabetes and their use is endorsed by the European Association for the Study of Diabetes, the American Diabetes Association, the American Association of Clinical Endocrinologists and the American College of Endocrinology.
Physical and chemical properties
Lixisenatixe has been described as “des-38-proline-exendin-4 (Heloderma suspectum)-(1–39)-peptidylpenta-L-lysyl-L-lysinamide”, meaning it is derived from the first 39 amino acids in the sequence of the peptide exendin-4, found in the Gila monster (Heloderma suspectum), omitting proline at position 38 and adding six lysine residues. Its complete sequence is:[3]
- H–His–Gly–Glu–Gly–Thr–Phe–Thr–Ser–Asp–Leu–Ser–Lys–Gln–Met–Glu–Glu–Glu–Ala–Val–Arg–Leu–Phe–Ile–Glu–Trp–Leu–Lys–Asn–Gly–Gly–Pro–Ser–Ser–Gly–Ala–Pro–Pro–Ser–Lys–Lys–Lys–Lys–Lys–Lys–NH2
PATENT
US 20110313131
http://www.google.co.in/patents/US20110313131
PATENT
CN 105713082
The title method comprises the steps of: (1) coupling Fmoc-Lys(Boc)-OH and resin to obtain Fmoc-Lys(Boc)-resin, (2) protecting amino acid with Fmoc, conducting solid-phase synthesis to obtain lixisenatide wholly protected 20-44-peptide resin, (3) conducting solid-phase synthesis to obtain wholly protected 15-19-peptide resin, (4) coupling the wholly protected 20-44-peptide resin and wholly protected 15-19-peptide resin, (5) coupling other amino acids till solid-phase synthesis finishes, (6) cracking lixisenatide peptide resin to obtain crude peptide, and (7) purifying through RP-HPLC. The method improves crude peptide purity and purifn. yield.
PATENT
MACHINE TRANSLATION FROM CHINESE, PL BEAR WITH SOME IREGULARITES IN GRAMMAR
利西拉, the English name: Lixisenatide, is a polypeptide containing 44 amino acids, the structural formula is as follows: peptide sequence as follows:
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Al a-Val-Arg-Leu-Phe-IIe-Glu -Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pr O-Ser-Lys-Lys-Lys-Lys-Lys-Lys-NH 2 Li Xila to (Lixisenatide ) by Sanofi-Aventis developed once a day subcutaneously with glucagon-like peptide -I (GLP-I) receptor agonists, for the treatment of type II diabetes, on February 1, 2013 Sanofi Lee Division -Aventis of exenatide is approved EMEA, for the adjuvant treatment of poorly stable dose of basal insulin (or metformin) in the treatment of type II diabetes to improve HbAlc and postprandial blood glucose levels.
CN201210030151. 2 used in a pure solid phase sequential coupling method synthetic peptides. The method amino resin as the carrier, using conventional coupling sequence, the final cut to give Li Xila.
US6528486 patent for the compound, synthetic methods mentioned it to phase condensation method Fmoc / tBu strategy.
The [0005] W02005058954 synthesis method including the gradual condensation process Fmoc / tBu strategy, Boc strategy of gradual condensation methods and genetic engineering.
The W02001004156 synthesis method for the gradual condensation process Fmoc / tBu strategy.
Since Li Xila abroad mostly used to synthesize Fmoc solid phase synthesis method, a gradual shrinking gradually synthesis step more, resulting in more types of product impurities, US 20130284912 Special Report polypeptide impurity: Di-Ser33- Leisy pull and Di-Ala35- Li Xila come, Di-Ser 33- Li Xila come and Di-Ala35- Li Xila to atmosphere amino acid sequence as follows: Di-Ser33- Li Xila to the amino acid sequence: H-His -Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Al a-Val-Arg-Leu-Phe-IIe-Glu-Trp- Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Ser-Gly-Ala-Pr 〇-Pr〇-Ser-Lys_Lys_Lys_Lys_Lys_LyS-NH2 Di-Ala35- Li Xila to the amino acid sequence: H-His-Gly- Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Al a-Val-Arg-Leu-Phe-IIe-Glu-Trp-Leu-Lys -Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Ala-Pr 〇-Pr〇-Ser-Lys_Lys_Lys_Lys_Lys_LyS-NH2 toxicity of these impurities are impurities larger, and very difficult to separate from the main peak , the presence of the impurities seriously affect 利西拉 to content and the use of safety. Hence the need to find an effective way to remove it and to reach the high standard level of 0.1% or less. The present inventors have found that this impurity is difficult to remove by means of the prior art, although there are ways to remove part of, but removal is not ideal, it is difficult to achieve high quality standards is likely to cause 利西拉 level while reducing their yield.
In summary, the existing Li Xila to the solid phase synthesis, low yield of the synthesis, impurities, in particular, are not well controlled impurity Di-Ser 33- Li Xila come and Di-Ala35 – Li Xila to, does not apply to industrial production
Example i ^ a: Preparation 利西拉 to fine peptide acetate Weigh 利西拉 above 44. 70g to 45L crude peptide was dissolved in water, purified by C18 column, the first purification conditions: mobile phase: A phase: 0 I% TFA; B phase: acetonitrile; gradient program was: 15% B, 60 minutes to 60% B; detection wavelength 220 nm; peak fraction collection purposes. The second purification conditions: mobile phase was: A phase: 0 3% HAC; B phase: acetonitrile; gradient program was: 10% B, 60 minutes to 60% B; detection wavelength 220 nm; peak fraction collection purposes. Desalting conditions: Mobile phase: A phase: an aqueous solution of 20 mmol / L ammonium acetate: acetonitrile = 95: 5; B phase: water: acetonitrile = 95: 5; C phase: 0.03% aqueous solution of acetic acid: acetonitrile = 95 : 5; D phase: 0.03% aqueous solution of acetic acid: acetonitrile = 50: 50; gradient program: mobile phase A isocratic for 15 minutes, convert isocratic mobile phase B for 10 minutes, is converted into the flow Phase C isocratic 10 minutes, converted into a mobile phase D isocratic 25 minutes; detection wavelength 220 nm; peak fraction collection purposes; rotary evaporation concentrated and lyophilized to give Li Xila acetate fine peptide 22. 65g which HPLC spectrum shown in Figure 5, HPLC purity of 99.75% (area normalization method), Di-Ser33- Li Xila come to 0.03% (area normalization method), Di-Ala35- Li Xila to the content of 0.05% (area normalization method). Purification total yield of 51%, total yield 41%. Its mass spectrum as shown in Figure 6, [M + H] + = 4858. 691, 利西拉 precise molecular weight to the theoretical: 4857.53, the sample mass is consistent with the theoretical molecular weight.
PATENT
CN 103709243
MACHINE TRANSLATION FROM CHINESE, PL BEAR WITH SOME IREGULARITES IN GRAMMAR
Example 2: Preparation 利西拉 to crude peptide
利西拉 [0116] Example 24 was prepared to be placed 125.4g peptide resin cleavage reaction to 10ml / g resin ratio added lysis reagent (TFA: thioanisole: EDT: TIS: water = 86: 5 : 5: 3: 1 (V / V)), stirred at room temperature 2.5h. The reaction was purified by frit funnel filtration, the filtrate was collected, the resin was washed 3 times and then a small amount of TFA, the combined filtrates concentrated under reduced pressure. Frozen precipitation in anhydrous ether was added, washed three times with anhydrous diethyl ether, and dried in vacuo to give a white solid powder, i.e. Li Xila to crude peptide 47.lg, by weight of the crude peptide yield 97.2%, HPLC purity 63.8% 0
利西拉 to crude peptide preparation: 27 patients [0117] Example
利西拉 [0118] The Example 25 was prepared to be placed 123.7g peptide resin cleavage reaction to 10ml / g resin ratio added lysis reagent (TFA: thioanisole: EDT: TIS: water = 86: 5 : 5: 3: 1 (V / V)), stirred at room temperature 2.5h. The reaction was purified by frit funnel filtration, the filtrate was collected, the resin was washed 3 times and then a small amount of TFA, the combined filtrates concentrated under reduced pressure. Frozen precipitation in anhydrous ether was added, washed three times with anhydrous diethyl ether, and dried in vacuo to give a white solid powder, i.e. Li Xila to crude peptide 46.9g, yield the crude peptide by weight 96.5%, HPLC purity 64.2% 0
28 Example 2: Preparation 利西拉 to fine peptide acetate
Example weighed 26 to 27 after 利西拉 to any 30.0g crude peptide was dissolved in 3000ml of water using Waters2545RP-HPLC system, wavelength 230nm, 50 X 250mm column of reverse phase C18 column, 0.2% TFA conventional / acetonitrile mobile phase were fractionated peaks of fractions, refined peptide purity greater than 98.5%. The fine peptide solution using Waters2545RP-HPLC system, 50 X 250mm column was C18 reverse phase column, 0.1% acetic acid / acetonitrile mobile phase transfer salt, the purpose of peak fractions were collected, concentrated by rotary evaporation and lyophilized to give Li Xila acetate fine salt peptide> 9.0g, RP-HPLC purity ≥98.5%. Purification Yield ≥30%, total yield ≥29.0%.
PATENT
CN 102875663
MACHINE TRANSLATION FROM CHINESE, PL BEAR WITH SOME IREGULARITES IN GRAMMAR
http://www.google.at/patents/CN102875663B?cl=en
Example 9
[0239] The crude peptide Li Xila to 4000g (including Li Xila to 1139g) was dissolved with purified water 100L, collected by filtration and the filtrate set aside.
[0240] purification chromatographic conditions:
[0241] HPLC Model: Novasep LC450
Column: 450X250mm, built-phenyl silane bonded silica gel as stationary phase filler, the filler particle size of 10 μ m0
flow rate: 5000ml / min.
The detection wavelength: 280nm.
Mobile phase A phase: 10% 30mM D- 30mM sodium tartrate and disodium hydrogenphosphate in methanol / 90% aqueous (v / v), adjusted to pH 2.5 with phosphoric acid.
[0246] Mobile phase A phase preparation process: Weigh 1280g 2070g D- sodium tartrate and disodium hydrogenphosphate, after an appropriate amount of purified water was dissolved through 0.45 μ m membrane filter, the filtrate collected all 300L tank, added 30L chromatographically pure After methanol was added to the 300L scale purification of water, adjusted to pH 2.5 with phosphoric acid. Repeat preparation run.
[0247] The mobile phase B phase: HPLC grade acetonitrile.
[0249] sample volume: 250.0g (6250ml).
[0250] Purification: column equilibration the sample so that after 5 minutes, run a gradient purification, monitoring and staging purposes peak fractions were collected. The collected fractions (chromatographic conditions purity testing to the same conditions as above 利西拉 determination to area normalization method measured) purity test, the purity of greater than or equal to 98% of the fractions after removing most of the acetonitrile in turn salt; purity of 70% or more less than 98% of the fraction recovered after removal of most of the acetonitrile and the purification procedure is repeated, again collected purity greater than or equal to 98% of the fraction after removal of most of the acetonitrile are also used to turn salt; purity of less than 70 % of fractions by waste disposal.
[0251] points and 16 injections, repeat the above operation.
[0252] turn salt chromatographic conditions:
[0253] HPLC Model: Novasep LC450
[0254] Column: 450 X 250mm, built-C8 reversed-phase chromatography packing, the particle size of the filler is 10 μ m.
[0255] flow rate: 5000ml / min.
[0256] The detection wavelength: 280nm.
[0257] Mobile phase A phase: 0.2% acetic acid (v / v) solution.
[0258] The mobile phase B phase: HPLC grade acetonitrile.
[0259] gradient
[0260] sample volume: 2500ml.
[0261] Purification: The column equilibration the sample for 5 minutes, run a gradient purification, monitoring and collecting the target peak fractions. The purpose of the peak fractions were concentrated by rotary evaporation under reduced pressure to 9000ml after lyophilization.
[0262] After the freeze-dried to give a white powder refined peptide 704g. Purity of 98.39%, the impurity content of less than 0.5%. Purification yield 61.8% (in crude Li Xila to content), total yield of 17.6%.
PATENT
CN 102558338
MACHINE TRANSLATION FROM CHINESE, PL BEAR WITH SOME IREGULARITES IN GRAMMAR
Preparation of Fmoc-Lys (Boc) -Lys (Boc) -Lys (Boc) -Lys (Boc) -Rink Amide-MBHAResin:
[0096] To the resulting Fmoc-Lys (Boc) -Lys (Boc) -Lys (Boc) -RinkAmide-MBHAResin mouth of a 20% strength piperidine / DMF solution for 10 minutes, the reaction was drained, washed with DMF Resin 6 (50ml * 6). Weigh Fmoc-Lys (Boc) -〇H3.52g, H0Bt1.01g, HBTU2.84g, TMP1.98ml, DMF50ml added to dissolve slowly with stirring under ice-cooling for 3 minutes, at room temperature for 2 hours, the reaction Ninhydrin detection method completed, pumping off the reaction solution, DMF the resin was washed twice (50mlX2), DCM the resin was washed twice (50mlX2), to give Fmoc-Lys (B oc) -Lys (Boc) -Lys (Boc) -Lys (Boc) -RinkAmide-MBHAResin. As used in the above operation Fmoc-Lys (Boc) -OH: HOBt: HBTU: TMP ratio is 1: 1: 1: 2, wherein Fmoc-Lys (Boc) -OH is the number of moles of Fmoc-RinkAmide-MBHAResin number of moles 3 times.
[0097] Li Xila fully protected side chain was prepared to -Rink Amide-MBHA Resin:
[0098] To the resulting Fmoc-Lys (Boc) -Lys (Boc) -Lys (Boc) -Lys (Boc) -RinkAmide-MBHA Resin added 20% piperidine / DMF solution for 10 minutes, drained reaction solution, washed 6 times with DMF. Weigh Jie 111〇 (3-1 ^ 8 billion (3) -0 13.528, 1 (»Shu 1.018,01 (:!! 1.391111 added 50,111,101 ^ dissolve slowly stirring for 3 minutes in an ice bath, poured into the solid phase resin is mixed with the reaction column, at room temperature for 2 hours, the reaction Ninhydrin detection method is completed, the reaction solution was deprived, DMF the resin was washed twice (50ml X 2), DCM the resin was washed twice (50ml X 2), to give Fmoc-Lys ( Boc) -Lys (Boc) -Lys (Boc) -Lys (Boc) -Lys (Boc) -Rink Amide-MBHAResin above operation used by the Fmoc-Lys (Boc) -〇H:. HOBt: DIC ratio is 1: 1: L2, which Fmoc-Lys (Boc) is three times the number of moles -〇H Fmoc-Rink Amide-MBHA Resin moles of repeat after the coupling step, followed by the completion of the 39 lysine to first. connecting protected amino acids histidine, followed by addition of 20% piperidine / DMF solution for 10 minutes, the reaction was drained, DMF the resin was washed six times (50ml X 6), DCM the resin was washed six times (50ml X 6 ), MeOH contraction of the resin three times with MeOH 50ml, each contraction 5min. After the resin was dried in vacuo to give a full side-chain protected peptide resin to the Li Xila 27. 5g, weight resin 17. 5g.
[0099] Li Xila to crude peptide preparation:
[0100] Weigh side chains fully protected Li Xila to -Rink Amide-MBHA Resin 27. 5 grams, into a round bottom flask.Configuration 275 ml lysis buffer, wherein trifluoroacetic acid: thioanisole: ethanedithiol: anisole, phenol = 93: 4: 1: 1.5: 2 (volume ratio). Lysate in the refrigerator after the pre-freeze 1 hour before Sheng Youli put to Silas to -Rink Amide-MBHA Resin round bottom flask, stirred at room temperature for 2 hours. The reaction mixture was filtered, the resin was washed with 20ml TFA and the combined filtrate.
[0101] The volume of the filtrate was slowly poured into 2,750 ml of diethyl ether frozen (frozen advance ether), a white precipitate appears, at 3000 rpm / centrifuged 5 minutes, the resulting solid was washed twice with ether, then the solid was dried under vacuum to give Li Xila trifluoroacetate crude peptide to 15. 3g.
[0102] Li Xila to large scale production of fine peptide:
[0103] Sample Preparation: The crude peptide was dissolved in water, the sample was completely dissolved by membrane filtration, the filtrate was collected for use.
[0104] Purification conditions: Column: octadecyl silane bonded silica gel as stationary phase column, the column diameter and length: 300_X250mm. Mobile phase: A phase: 35mm〇l / L phosphoric acid solution adjusted with triethylamine to pH 6. 7; B phase: acetonitrile, flow rate: 2200ml / min, Gradient: B%: 12% ~32%, detection wavelength: 280nm . The injection volume was 75g. Purification process: the column with 50% acetonitrile rinse clean after balance sample, sample amount is 75g. Linear gradient 120min, the purpose of collecting peaks will be collected 利西拉 solution was concentrated by rotary evaporation under reduced pressure to about 80mg / ml and reserve the water temperature exceeds 40 ° C without conditions.
[0105] turn salt: turn salt conditions: Column: octadecyl silane bonded silica gel as stationary phase column, the column diameter and length: 300mmX250mm. Mobile phase: A phase: mass concentration of 0.2% aqueous acetic acid; B phase: HPLC grade acetonitrile, flow rate: 2200ml / min, detection wavelength: 280nm. Gradient: B%: 6% ~36%. The injection volume was 48-60g. Salt transfer process: the column with 50% acetonitrile rinse clean after the sample, the sample volume is 1600ml sample solution. Linear gradient 90min, the purpose of collecting peaks collected Li Xila to solutions were concentrated by rotary evaporation to about 80ml / g after go to the appropriate size vials, then freeze-dried to obtain the purity of greater than 99.5% The Li Xila come.
Old post
https://newdrugapprovals.org/2013/09/13/sanofi-to-withdraw-the-lixisenatide-new-drug-application-nda-in-the-u-s-the-company-plans-to-resubmit-the-nda-in-2015-after-completion-of-the-elixa-cv-study/
lixisenatide
Sanofi Provides Update on Lixisenatide New Drug Application in U.S.
Paris, France – September 12, 2013 – Sanofi (EURONEXT: SAN and NYSE: SNY) announced today its decision to withdraw the lixisenatide New Drug Application (NDA) in the U.S., which included early interim results from the ongoing ELIXA cardiovascular (CV) outcomes study. The company plans to resubmit the NDA in 2015, after completion of the ELIXA CV study.
The decision to withdraw the lixisenatide application follows discussions with the U.S. Food and Drug Administration (FDA) regarding its proposed process for the review of interim data. Sanofi believes that potential public disclosure of early interim data, even with safeguards, could potentially compromise the integrity of the ongoing ELIXA study. Sanofi’s decision is not related to safety issues or deficiencies in the NDA………………………read all at
http://www.pharmalive.com/sanofi-pulls-diabetes-drug-nda
EU

| US20070037807 * |
29 Oct 2004 |
15 Feb 2007 |
Satoru Oi |
Pyridine compounds as inhibitors of dipeptidyl peptidase IV |
| US20070191436 * |
12 Sep 2006 |
16 Aug 2007 |
Valerie Niddam-Hildesheim |
Diastereomeric purification of rosuvastatin |
| EP0708179A2 * |
13 Oct 1995 |
24 Apr 1996 |
Eli Lilly And Company |
Glucagon-like insulinotropic peptide analogs, compositions, and methods of use |
| Citing Patent |
Filing date |
Publication date |
Applicant |
Title |
| CN102584982A * |
10 Feb 2012 |
18 Jul 2012 |
深圳翰宇药业股份有限公司 |
Method for purifying solid-phase synthetic coarse liraglutide |
| WO2013117135A1 * |
29 Jan 2013 |
15 Aug 2013 |
Hybio Pharmaceutical Co., Ltd. |
Method for purifying solid-phase synthetic crude liraglutide |
| WO2014077802A1 * |
13 Nov 2012 |
22 May 2014 |
Ipsen Pharma S.A.S. |
Purification method of a glp-1 analogue |
| WO2014118797A1 |
1 Jul 2013 |
7 Aug 2014 |
Neuland Health Sciences Private Limited |
Purification of organic compounds using surrogate stationary phases on reversed phase columns |
| CN1839155A |
18. Aug. 2004 |
27. Sept. 2006 |
诺沃挪第克公司 |
Purification of glucagon-like peptides |
| WO2006041945A2 |
4. Okt. 2005 |
20. Apr. 2006 |
Novetide, Ltd. |
A counterion exchange process for peptides |
References
///////FDA 2016, SANOFI, FDA, approves , Adlyxin, lixisenatide, type 2 diabetes, Sanofi-Aventis U.S. LLC, Bridgewater, New Jersey, Lyxumia, 利西拉, PEPTIDE,
CCC(C)C(C(=O)NC(CCC(=O)O)C(=O)NC(Cc1c[nH]c2c1cccc2)C(=O)NC(CC(C)C)C(=O)NC(CCCCN)C(=O)NC(CC(=O)N)C(=O)NCC(=O)NCC(=O)N3CCCC3C(=O)NC(CO)C(=O)NC(CO)C(=O)NCC(=O)NC(C)C(=O)N4CCCC4C(=O)N5CCCC5C(=O)NC(CO)C(=O)NC(CCCCN)C(=O)NC(CCCCN)C(=O)NC(CCCCN)C(=O)NC(CCCCN)C(=O)NC(CCCCN)C(=O)NC(CCCCN)C(=O)N)NC(=O)C(Cc6ccccc6)NC(=O)C(CC(C)C)NC(=O)C(CCCNC(=N)N)NC(=O)C(C(C)C)NC(=O)C(C)NC(=O)C(CCC(=O)O)NC(=O)C(CCC(=O)O)NC(=O)C(CCC(=O)O)NC(=O)C(CCSC)NC(=O)C(CCC(=O)N)NC(=O)C(CCCCN)NC(=O)C(CO)NC(=O)C(CC(C)C)NC(=O)C(CC(=O)O)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C(Cc7ccccc7)NC(=O)C(C(C)O)NC(=O)CNC(=O)C(CCC(=O)O)NC(=O)CNC(=O)C(Cc8cnc[nH]8)N
AND
CCC(C)C(C(=O)NC(CCC(=O)O)C(=O)NC(CC1=CNC2=CC=CC=C21)C(=O)NC(CC(C)C)C(=O)NC(CCCCN)C(=O)NC(CC(=O)N)C(=O)NCC(=O)NCC(=O)N3CCCC3C(=O)NC(CO)C(=O)NC(CO)C(=O)NCC(=O)NC(C)C(=O)N4CCCC4C(=O)N5CCCC5C(=O)NC(CO)C(=O)NC(CCCCN)C(=O)NC(CCCCN)C(=O)NC(CCCCN)C(=O)NC(CCCCN)C(=O)NC(CCCCN)C(=O)NC(CCCCN)C(=O)N)NC(=O)C(CC6=CC=CC=C6)NC(=O)C(CC(C)C)NC(=O)C(CCCNC(=N)N)NC(=O)C(C(C)C)NC(=O)C(C)NC(=O)C(CCC(=O)O)NC(=O)C(CCC(=O)O)NC(=O)C(CCC(=O)O)NC(=O)C(CCSC)NC(=O)C(CCC(=O)N)NC(=O)C(CCCCN)NC(=O)C(CO)NC(=O)C(CC(C)C)NC(=O)C(CC(=O)O)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C(CC7=CC=CC=C7)NC(=O)C(C(C)O)NC(=O)CNC(=O)C(CCC(=O)O)NC(=O)CNC(=O)C(CC8=CN=CN8)N

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....



































 LESINURAD
LESINURAD

















