
Home » Posts tagged 'Antineoplastic'
Tag Archives: Antineoplastic
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Ranosidenib
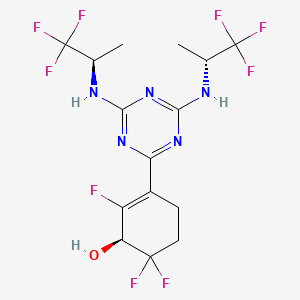
Ranosidenib
CAS 2301974-60-5
MF C15H16F9N5O MW 453.31 g/mol
(1S)-3-[4,6-bis[[(2R)-1,1,1-trifluoropropan-2-yl]amino]-1,3,5-triazin-2-yl]-2,6,6-trifluorocyclohex-2-en-1-ol
(1S)-3-(4,6-bis{[(2R)-1,1,1-trifluoropropan-2-yl]amino}-1,3,5-triazin-2-yl)-2,6,6-trifluorocyclohex-2-en-1-ol
isocitrate dehydrogenase (IDH) inhibitor, antineoplastic, [14C] HMPL-306, HMPL 306, PC64OXS2C2
- OriginatorHutchison MediPharma
- DeveloperHUTCHMED
- ClassAntineoplastics; Small molecules
- Mechanism of ActionIsocitrate dehydrogenase 1 inhibitors; Isocitrate dehydrogenase 2 inhibitors
- Phase IIIAcute myeloid leukaemia
- No development reportedHaematological malignancies; Solid tumours
- 28 Sep 2025No recent reports of development identified for phase-I development in Haematological-malignancies(Late-stage disease, Second-line therapy or greater) in Spain (PO)
- 28 Sep 2025No recent reports of development identified for phase-I development in Haematological-malignancies(Late-stage disease, Second-line therapy or greater) in USA (PO)
- 19 Sep 2025No development reported – Phase-I for Solid tumours (Late-stage disease, Metastatic disease, Second-line therapy or greater) in USA (PO)
Ranosidenib is a small molecule drug. Ranosidenib is under investigation in clinical trial NCT06387069 (A Study to Evaluate HMPL-306 in Patients With IDH1- and IDH2-mutated Acute Myeloid Leukemia). Ranosidenib has a monoisotopic molecular weight of 453.12 Da.
Ranosidenib is an orally bioavailable inhibitor of mutated forms of both isocitrate dehydrogenase type 1 (IDH1, IDH1 [NADP+] soluble) in the cytoplasm and type 2 (IDH2, isocitrate dehydrogenase [NADP+], mitochondrial) in the mitochondria, with potential antineoplastic activity. Upon administration, ranosidenib specifically targets and inhibits mutant forms of IDH1 and IDH2, thereby inhibiting the formation of the oncometabolite 2-hydroxyglutarate (2HG) from alpha-ketoglutarate (a-KG). This prevents 2HG-mediated signaling and leads to both an induction of cellular differentiation and an inhibition of cellular proliferation in tumor cells expressing IDH mutations. IDH1 and 2, metabolic enzymes that catalyze the conversion of isocitrate into a-KG, play key roles in energy production and are mutated in a variety of cancer cell types. Mutant forms of IDH1 and 2 catalyze the formation of 2HG and drive cancer growth by blocking cellular differentiation and inducing cellular proliferation.
A Study of HMPL-306 in Advanced Hematological Malignancies With mIDHCTID: NCT04764474Phase: Phase 1Status: TerminatedDate: 2026-01-29
A Study of HMPL-306 in Advanced Solid Tumors With IDH MutationsCTID: NCT04762602Phase: Phase 1Status: TerminatedDate: 2025-09-16
A Study to Evaluate HMPL-306 in Patients With IDH1or IDH2-mutated Acute Myeloid LeukemiaCTID: NCT06387069Phase: Phase 3Status: RecruitingDate: 2025-08-14
Phase I Study of HMPL-306 for the Treatment of Gliomas With IDH1 and/or IDH2 MutationsCTID: NCT07025018Phase: Phase 1Status: RecruitingDate: 2025-08-01
A Study of HMPL-306 in Patients With IDH1 and/or IDH2 Mutation of Relapsed/Refractory Myeloid Leukemia/NeoplasmsCTID: NCT04272957Phase: Phase 1Status: Unknown statusDate: 2020-06-16
SYN
https://pubs.acs.org/doi/10.1021/acsmedchemlett.4c00625
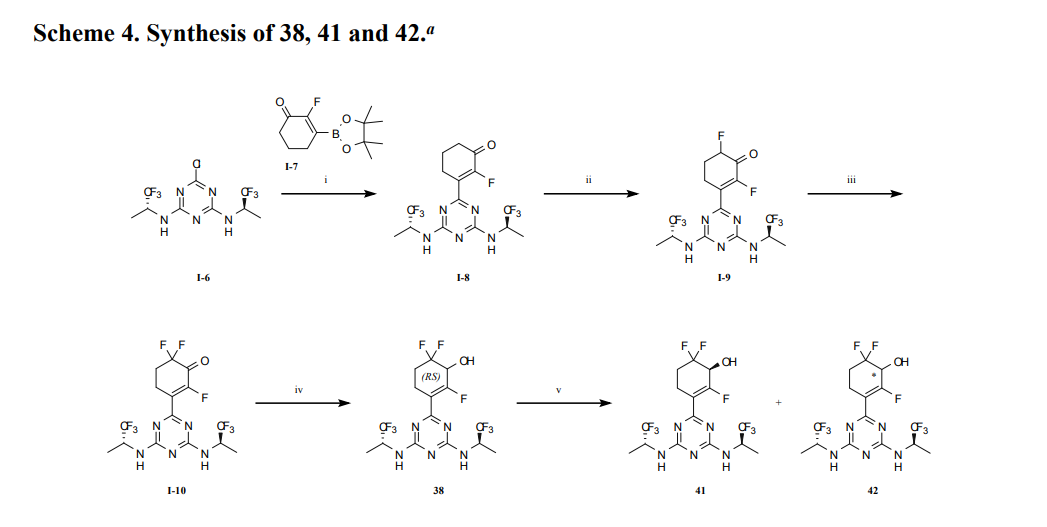
aReagents and conditions: (i) Na2PdCl4, DTBPPS, K2CO3, MeCN, H2O, 60 ℃; (ii) TBSOTf, Et3N,
DCM, 0~5 ℃; Selectfluor®, MeCN, 0~5 ℃; (iii) TBSOTf, Et3N, DCM, 0~5 ℃; Selectfluor®,
MeCN, 0~5 ℃; (iv) NaBH4, CeCl3·7H2O, EtOH, 0 ℃; (v) SFC separation.
Pat
Cycloolefin substituted heteroaromatic compounds and their use
Publication Number: US-2021363115-A2
Priority Date: 2017-09-07
PAT
Intermediate I-3
6-Chloro-N2,N4-bis((R)-1,1,1-trifluoropropan-2-yl)-1,3,5-triazine-2,4-diamine
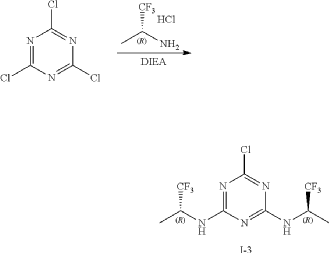
At 0° C., to a flask were added 1,4-dioxane (50 mL), 2,4,6-trichloro-1,3,5-triazine (1.84 g, 10 mmo), (R)-1,1,1-trifluoropropan-2-amine hydrochloride (2.99 g, 20 mmol) and DIEA (5.17 g, 40 mmol). The reaction was heated to 60° C. and stirred for 4 hours. After the reaction was completed, the mixture was condensed and purified by flash column chromatography (eluting with gradient water/MeOH=100:0-0:100) to give Intermediate I-3 as yellow solid (2.50 g, yield: 74%). MS (m/z): 338.0 [M+H] +
Compounds 197 and 198
3-(4,6-Bis(((R)-1,1,1-trifluoropropan-2-yl)amino)-1,3,5-triazin-2-yl)-2,6,6-trifluorocyclohex-2-en-1-ol, optically pure diastereoisomers
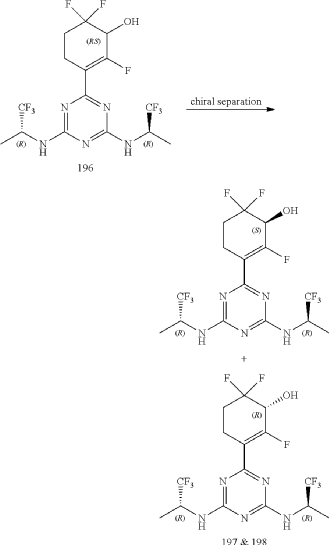
| The Compound 196 was resolved by chiral HPLC to provide a pair of optically pure diastereoisomers, Compounds 197 and 198 (Chiral HPLC conditions: Preparation instrument: Shimadzu LC-10AD vp; Column: Daicel AD-H(250 mm*30 mm, 5 um); mobile phase: n-heptane/isopropanol=90/10; flow rate: 40 mL/min; column temperature: 40° C.). The first eluent (RT=4.203 min) was concentrated and purified by flash column chromatography (eluting with gradient PE/EA=100:0-0:100) to give a compound named as Compound 197, de %=99.27%, MS (m/z): 454.1 [M+1] +. The second eluent (RT=5.906 min) was concentrated and purified by flash column chromatography (eluting with gradient PE/EA=100:0-0:100) to give a compound named as Compound 198, de %=97.82%, MS (m/z): 454.2 [M+1] +. |
| Compound 197: 1H NMR (400 MHz, CD 3OD): δ 5.00-4.86 (m, 2H), 4.36-4.17 (m, 1H), 2.80-2.65 (m, 1H), 2.58-2.42 (m, 1H), 2.25-2.05 (m, 2H), 1.37-1.31 (m, 6H). |
| Compound 198: 1H NMR (400 MHz, CD 3OD): δ 5.00-4.86 (m, 2H), 4.36-4.17 (m, 1H), 2.80-2.65 (m, 1H), 2.58-2.42 (m, 1H), 2.25-2.05 (m, 2H), 1.37-1.31 (m, 6H). |



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

/////////ranosidenib, isocitrate dehydrogenase (IDH) inhibitor, antineoplastic, [14C] HMPL-306, HMPL 306, PC64OXS2C2
Lead (212Pb) bamzireotide navoxetan
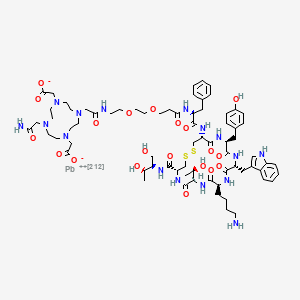
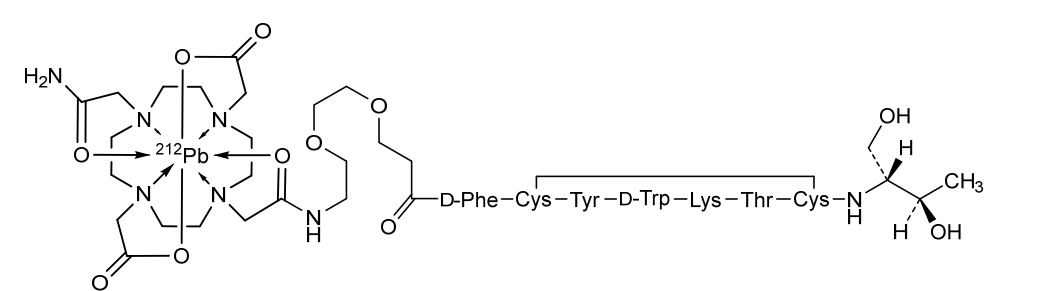
Lead (212Pb) bamzireotide navoxetan
CAS 2941523-47-1
MF C72H104N16O20212PbS2 MW1789.8 g/mol
2-[4-[2-[2-[2-[3-[[(2R)-1-[[(4R,7S,10S,13R,16S,19R)-10-(4-aminobutyl)-4-[[(2R,3R)-1,3-dihydroxybutan-2-yl]carbamoyl]-7-[(1R)-1-hydroxyethyl]-16-[(4-hydroxyphenyl)methyl]-13-(1H-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-1,2-dithia-5,8,11,14,17-pentazacycloicos-19-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-oxopropoxy]ethoxy]ethylamino]-2-oxoethyl]-10-(2-amino-2-oxoethyl)-7-(carboxylatomethyl)-1,4,7,10-tetrazacyclododec-1-yl]acetate;lead-212(2+)

ANTINEOPLASTIC, D2A42X3LCS
SYN
US 11037690 B2
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
///////////lead (212Pb) bamzireotide navoxetan, ANTINEOPLASTIC, D2A42X3LCS
Pebezertinib
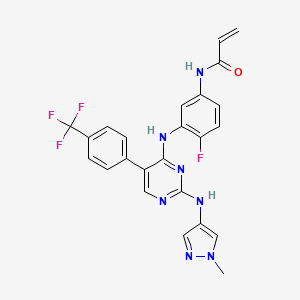
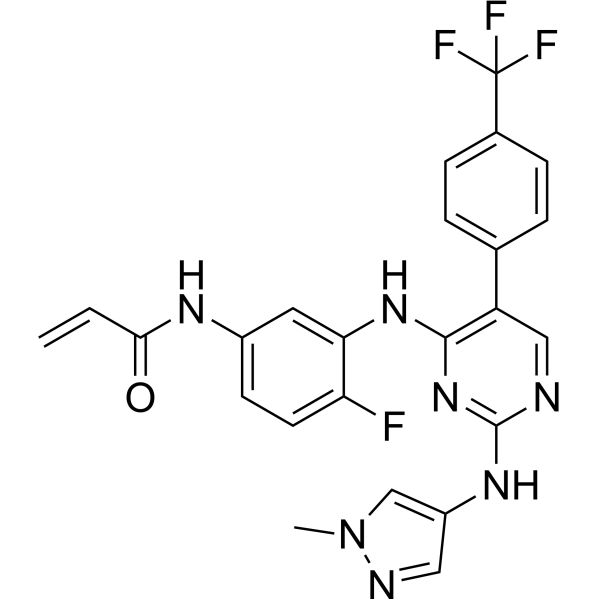
Pebezertinib
CAS 2769954-39-2
MF C24H19F4N7O MW 497.4 g/mol
N-[4-fluoro-3-[[2-[(1-methylpyrazol-4-yl)amino]-5-[4-(trifluoromethyl)phenyl]pyrimidin-4-yl]amino]phenyl]prop-2-enamide
N-[4-fluoro-3-({2-[(1-methyl-1H-pyrazol-4-yl)amino]-5-[4-(trifluoromethyl)phenyl]pyrimidin-4-yl}amino)phenyl]prop-2-enamide
epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor, antineoplastic, BLU 451, BLU 203139, G8G5AU5GLJ, LNG 451
Pebezertinib is a small molecule drug. The usage of the INN stem ‘-ertinib’ in the name indicates that Pebezertinib is a epidermal growth factor receptor (EGFR) inhibitor. Pebezertinib is under investigation in clinical trial NCT05241873 ((Concerto) Study of BLU-451 in Advanced Cancers With EGFR Exon 20 Insertion Mutations). Pebezertinib has a monoisotopic molecular weight of 497.16 Da.
Pebezertinib is an orally bioavailable, central nervous system (CNS) penetrating, mutant-selective covalent inhibitor of epidermal growth factor receptor (EGFR) exon 20 insertion (Ex20ins) activating mutations, with potential antineoplastic activity. Upon oral administration, pebezertinib selectively targets, irreversibly binds to and inhibits the activity of EGFR Ex20ins and some other oncogenic point mutations. This prevents EGFR Ex20ins-mediated signaling. This may induce cell death and inhibit tumor growth in EGFR Ex20ins-overexpressing tumor cells. EGFR, a receptor tyrosine kinase mutated in many tumors, plays a key role in tumor cell proliferation and tumor vascularization. Pebezertinib is able to penetrate the blood-brain-barrier (BBB) and may therefore exert its activity against EGFR Ex20ins-driven CNS primary tumors and CNS metastases. Pebezertinib does not inhibit the activity of wild-type (WT) EGFR. EGFR Ex20ins are oncogenic driver mutations that constitutively upregulate kinase activity.
(Concerto) Study of BLU-451 in Advanced Cancers With EGFR Exon 20 Insertion Mutations
CTID: NCT05241873
Phase: Phase 1
Status: Terminated
Date: 2025-02-10
Conditions: Lung Neoplasm Malignant; Carcinoma, Non-Small-Cell Lung; Respiratory Tract Neoplasms; Neoplasms; Neoplasms by Site; Lung Diseases; Respiratory Tract Disease; Carcinoma, Bronchogenic; Bronchial Neoplasms; Adenocarcinoma; Carcinoma; Neoplasms by Histologic Type; EGFR Exon 20 Mutation; EGFR Exon 20 Insertion Mutation; EGFR Activating Mutation; Antineoplastic Agents; Metastatic Lung Cancer; Brain Metastases; EGFR-mutated NSCLC; EGFR Atypical Mutations, Including G719X and L861Q
Interventions: Pemetrexed
Linked Compound CID: 426756; 135410875; 10339178; 163280903
SYN
Scheme 21: Synthesis of N-(4-fluoro-3-((2-((1-methyl-1H-pyrazol-4-yl)amino)-5-(4-(trifluoromethyl)phenyl)pyrimidin-4-yl)amino)phenyl)acrylamide (Compound 37):
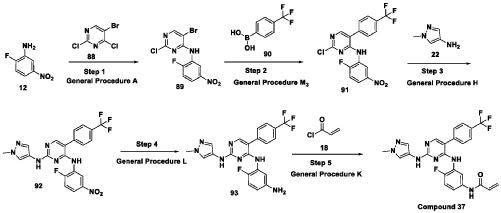
Step 1: Synthesis of 5-bromo-2-chloro-N-(2-fluoro-5-nitrophenyl)pyrimidin-4-amine (89):
[0286] Title compound was prepared in a manner substantially similar to procedure mentioned in General Procedure A. The crude was purified by combiflash eluted with 40% ethyl acetate in hexane to get (89) as pale yellow solid (1.3 g, Yield: 44.24 %). MS: [M+H]+ 346.97.
Step 2: Synthesis of 2-chloro-N-(2-fluoro-5-nitrophenyl)-5-(4-(trifluoromethyl)phenyl)pyrimidin-4-amine (91):
[0287] Title compound was prepared in a manner substantially similar to procedure mentioned in General Procedure M3. The crude was purified by combiflash eluted with 35% ethyl acetate in hexane to get desired product (91) as light yellow solid (700 mg; Yield: 50.12%). MS:
[M+H]+ 413.10
Step 3: Synthesis of N4-(2-fluoro-5-nitrophenyl)-N2-(1-methyl-1H-pyrazol-4-yl)-5-(4-(trifluoromethyl)phenyl)pyrimidine-2,4-diamine (92):
[0288] Title compound was prepared in a manner substantially similar to procedure mentioned in General Procedure H. The crude was purified by combiflash eluted with 1% methanol in dichloromethane to get desired product (92) as pale yellow solid (500 mg; Yield: 70.24%). MS:
[M+H]+ 474.09
Step 4: Synthesis of N4-(5-amino-2-fluorophenyl)-N2-(1-methyl-1H-pyrazol-4-yl)-5-(4-(trifluoromethyl)phenyl)pyrimidine-2,4-diamine (93):
[0289] Title compound was prepared in a manner substantially similar to procedure mentioned in General Procedure L to get (93) as semi solid (350 mg; Yield: 74.78%). MS: [M+H]+ 444.11
Step 5: Synthesis of N-(4-fluoro-3-((2-((1-methyl-1H-pyrazol-4-yl)amino)-5-(4-(trifluoromethyl)phenyl)pyrimidin-4-yl)amino)phenyl)acrylamide (Compound 37):
[0290] Title compound was prepared in a manner substantially similar to procedure mentioned in General Procedure K. The crude was purified by Prep HPLC to get Compound 37 as off white solid (30 mg, Yield: 13.33%).1H NMR (400 MHz, DMSO-d6): δ 10.21 (bs, 1H), 9.24 (bs, 1H), 8.53 (bs, 1H), 7.99 (s, 1H), 7.71-7.81 (m, 5H), 7.57 (s, 1H), 7.08-7.16 (m, 3H), 6.37-6.44 (m, 1H), 6.21-6.26 (m, 1H), 5.74 (d, J = 8.4 Hz, 1H), 3.54 (s, 3H). LCMS: [M+H]+ 498.35.
SYN
International Patent Application No. PCT/US2021/057472, the entire teachings of which are incorporated herein by reference, discloses selective inhibitors of EGFR, including exon 20 mutant proteins, which can be used to treat various cancers. The structure of one of the inhibitors disclosed in PCT Patent Application No. PCT/US2021/057472, referred to
herein as “Compound (I)” is shown below:
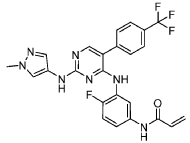
Example 1 : Preparation of Compound (I)
Synthesis of N-(4-fluoro-3-((2-((l-methyl-lH-pyrazol-4-yl)amino)-5-(4-(trifluoro methyl)phenyl)pyrimidin-4-yl)amino)phenyl)acrylamide (Compound I):
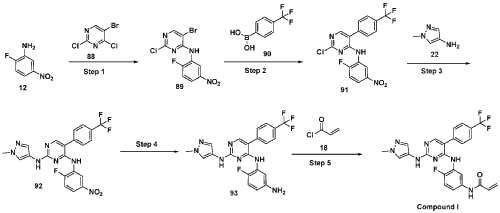
Step 1 : Synthesis of 5-bromo-2-chloro-N-(2-fluoro-5-nitrophenyl)pyrimidin-4-amine (89):
To an ice cold solution of 2-fluoro-5-nitroaniline (12) (1.0 eq) in tetrahydrofuran was added sodium hydride (60% dispersion in mineral oil, 3.0 eq) portion-wise. The resulting reaction mixture was stirred at room temperature for 30 minutes and followed by the addition of 2, 4-di chi oro-5 -bromopyrimidine (88) (1.0 eq). The resulting reaction mixture was heated at 60 °C for 16 hours. After completion (TLC monitoring), quenched with ice, extracted with ethyl acetate (3 times). The combined organic layers were washed with water, brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude was purified by combiflash eluted with 40% ethyl acetate in hexane to get (89) as pale yellow solid (1.3 g, Yield: 44.24 %). MS: [M+H]+ 346.97.
Step 2: Synthesis of 2-chloro-N-(2-fluoro-5-nitrophenyl)-5-(4-(trifluoromethyl)phenyl) pyrimidin-4-amine (91):
To a solution of halo derivative (89) (1.0 eq) and respective boronate acid/ester derivative (90) (1.1 eq) in A A i methyl form am ide: water (4: 1) was added sodium carbonate or sodium bicarbonate (2.0 eq). The resulting reaction mixture was degassed under argon atmosphere for 15 minutes, followed by addition of tetrakis(triphenylphosphine)palladium(0) (0.1 eq). The resulting reaction mixture was heated at 90 °C for 16 hours. After completion of reaction (TLC monitoring), the reaction mixture was cooled to room temperature, water was added and extracted with ethyl acetate (3 times). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude was purified by combiflash eluted with 35% ethyl acetate in hexane to get desired product (91) as light yellow solid (700 mg; Yield: 50.12%). MS: [M+H]+413.10.
Step 3 : Synthesis of N4-(2-fluoro-5-nitrophenyl)-N2-(l-methyl-lH-pyrazol-4-yl)-5-(4-(trifluoromethyl)phenyl)pyrimidine-2,4-diamine (92):
To an ice-cold solution of chloro compound (91) (1.0 eq) in isopropanol was added amine (22) (1.2 eq) and trifluoroacetic acid (2.0 eq). The reaction mixture was heated at 110 °C for 16 hours. After completion of the reaction (TLC monitoring), the reaction mixture was concentrated under reduced pressure, added saturated solution of sodium bicarbonate and extracted with dichloromethane (3 times). The combined organic layers were washed with brine solution, dried over anhydrous sodium sulfate and evaporated under reduced pressure. The crude was purified by combiflash eluted with 1% methanol in di chloromethane to get desired product (92) as pale yellow solid (500 mg; Yield: 70.24%). MS: [M+H]+ 474.09.
Step 4: Synthesis of N4-(5-amino-2-fluorophenyl)-N2-(l-methyl-lH-pyrazol-4-yl)-5-(4-(trifluoromethyl)phenyl)pyrimidine-2,4-diamine (93):
To an ice cold solution of nitro derivative (92) (1.0 eq) in methanol: tetrahydrofuran: water (2:2: 1) were added zinc-dust or iron powder (5 eq) and ammonium chloride (5 eq). The resultant reaction mixture was stirred at room temperature for 2 hours. After completion of reaction (TLC monitoring), reaction mixture passed through celite bed washed with 5% methanol in dichloromethane. The filtrate was washed with water, brine, dried over anhydrous sodium sulfate, filtered and concentrated to dryness to get the desired product (93) as semi solid (350 mg; Yield: 74.78%). MS: [M+H]+ 444.11.
Step 5 : Synthesis of N-(4-fluoro-3-((2-((l-methyl-lH-pyrazol-4-yl)amino)-5-(4-(trifluoromethyl)phenyl)pyrimidin-4-yl)amino)phenyl)acrylamide (Compound I):
To a solution of amino compound (93) (1.0 eq) in dichloromethane: tetrahydrofuran (1 :1) was cooled to -40 °C followed by triethylamine (3-5 eq) and acryloyl chloride (1.0 eq) were added. The mixture was stirred at the same temperature for 2 hours. After completion of reaction (monitored by TLC), added water and extracted with dichloromethane (3 times). The combined organic layers washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crudes were purified by Prep-HPLC purification to to obtain Compound I as off white solid (30 mg, Yield: 13.33%). ‘H NMR (400 MHz, DMSO-de): 8 10.21 (bs, 1H), 9.24 (bs, 1H), 8.53 (bs, 1H), 7.99 (s, 1H), 7.71-7.81 (m, 5H), 7.57 (s, 1H), 7.08-7.16 (m, 3H), 6.37-6.44 (m, 1H), 6.21-6.26 (m, 1H), 5.74 (d, J= 8.4 Hz, 1H), 3.54 (s, 3H). LCMS: [M+H]+ 498.35.
PAT
- Pyrimidine compounds, compositions, and medicinal applications thereofPublication Number: WO-2022094354-A1Priority Date: 2020-10-30Linked Compounds: 1,056Linked Substances: 1,365
- Pyrimidine compounds, compositions and pharmaceutical uses thereofPublication Number: CN-116685583-APriority Date: 2020-10-30Linked Compounds: 921Linked Substances: 1,108
- Pyrimidine compounds, compositions, and medicinal applications thereofPublication Number: TW-202233603-APriority Date: 2020-10-30Linked Compounds: 531Linked Substances: 575
- Pyrimidine compounds, compositions, and their medicinal applicationsPublication Number: KR-20230116795-APriority Date: 2020-10-30Linked Compounds: 699Linked Substances: 744
- Egfr inhibitors for treatment of cancerPublication Number: WO-2024097270-A1Patent Family: TW-202432143-A; WO-2024097270-A1Priority Date: 2022-11-01Inventor(s): ANKROM WENDY; MAR BRENTON; PANDEY ANJALI; PEARSON PAUL; ZALUTSKAYA ALENAAssignee(s): BLUEPRINT MEDICINES CORPClassification: A61K31/506; A61K31/519; A61K31/555; A61P35/00; A61P35/04Abstract: The present disclosure provides improved methods of treating non-small cell lung cancer characterized by EGFR mutation using Compound (I): or a pharmaceutically acceptable salt thereof.Linked Compounds: 27Linked Substances: 28
- Salt and crystal forms of an epidermal growth factor receptor inhibitorPublication Number: US-2025282761-A1Patent Family: AU-2023265064-A1; CN-119923392-A; EP-4519254-A1; IL-316663-A; JP-2025517634-A; KR-20250012078-A; MX-2024013485-A; TW-202409016-A; US-2025282761-A1; WO-2023215431-A1Priority Date: 2022-05-04Inventor(s): GRUFF ERIC; Kuang Shanming; PANDEY ANJALI; SHAH HARSH; XIE TIANAssignee(s): BLUEPRINT MEDICINES CORPClassification: A61K31/506; C07D403/12Abstract: Various salt forms and free base solid forms of Compound (I) represented by the following formula are disclosed. Pharmaceutical compositions comprising the same, methods of treating a disease associated with an epidermal growth factor receptor (EGFR) family kinase using the same, and methods for making the salt forms of Compound (I) and crystalline forms thereof are also disclosed.Linked Compounds: 11Linked Substances: 13
- Salt and crystal forms of an epidermal growth factor receptor inhibitorPublication Number: WO-2023215431-A1Priority Date: 2022-05-04Linked Compounds: 18Linked Substances: 22
- Salt and crystal forms of an epidermal growth factor receptor inhibitorPublication Number: EP-4519254-A1Priority Date: 2022-05-04Linked Compounds: 14Linked Substances: 16
- Pyrimidine compounds, compositions, and medicinal applications thereofPublication Number: EP-4237418-A1Priority Date: 2020-10-30Linked Compounds: 821Linked Substances: 918
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
[1].
Zhou Y, et al., Anti-PD-1/L1 antibody plus anti-VEGF antibody vs. more VEGFR-targeted TKI as first-line therapy for unresectable hepatocellular carcinoma: a network meta-analysis. Explor Target Antitumor Ther. 2024;5(3):568-580. [Content Brief]
//////////pebezertinib, antineoplastic, BLU 451, BLU 203139, G8G5AU5GLJ, LNG 451
Orenasitecan
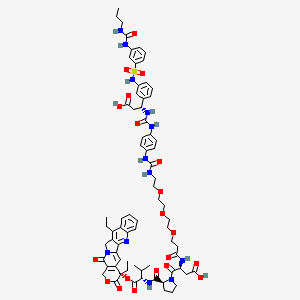
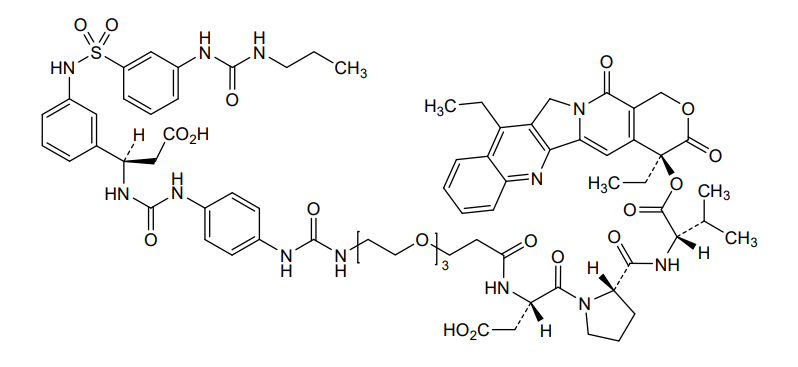

Orenasitecan
CAS 2418533-89-6
MF C72H86N12O20S MW1471.59
(3S)-3-[3-[2-[2-[2-[[4-[[(1R)-2-carboxy-1-[3-[[3-(propylcarbamoylamino)phenyl]sulfonylamino]phenyl]ethyl]carbamoylamino]phenyl]carbamoylamino]ethoxy]ethoxy]ethoxy]propanoylamino]-4-[(2S)-2-[[(2S)-1-[[(19S)-10,19-diethyl-14,18-dioxo-17-oxa-3,13-diazapentacyclo[11.8.0.02,11.04,9.015,20]henicosa-1(21),2,4,6,8,10,15(20)-heptaen-19-yl]oxy]-3-methyl-1-oxobutan-2-yl]carbamoyl]pyrrolidin-1-yl]-4-oxobutanoic acid
(4S)-4,11-diethyl-3,14-dioxo-3,4,12,14-tetrahydro-1Hpyrano[3′,4′:6,7]indolizino[1,2-b]quinolin-4-yl N-{1-[4-({[(1R)-2-
carboxy-1-(3-{3-[(propylcarbamoyl)amino]benzene-1-sulfonamido}phenyl)ethyl]carbamoyl}amino)anilino]-1-oxo-5,8,11-
trioxa-2-azatetradecan-14-oyl}-L-α-aspartyl-L-prolyl-L-valinate
antineoplastic, L3KV5NR5PG
Orenasitecan is a small molecule drug. The usage of the INN stem ‘-tecan’ in the name indicates that Orenasitecan is a antineoplastic, topoisomerase I inhibitor. Orenasitecan has a monoisotopic molecular weight of 1470.58 Da.
ORENASITECAN is a small molecule drug with a maximum clinical trial phase of II and has 1 investigational indication.
PAT
Cytostatic conjugates with integrin ligands
Publication Number: US-2021386864-A1
Priority Date: 2018-11-05
https://patents.google.com/patent/US20210386864A1
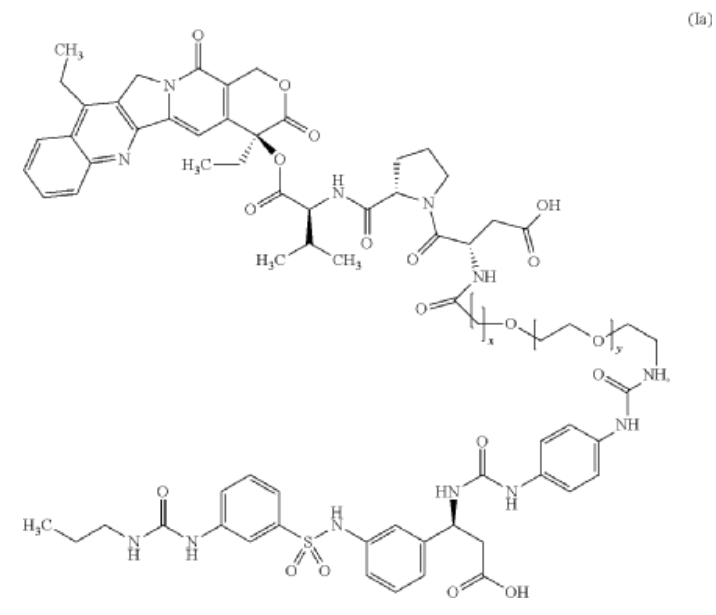
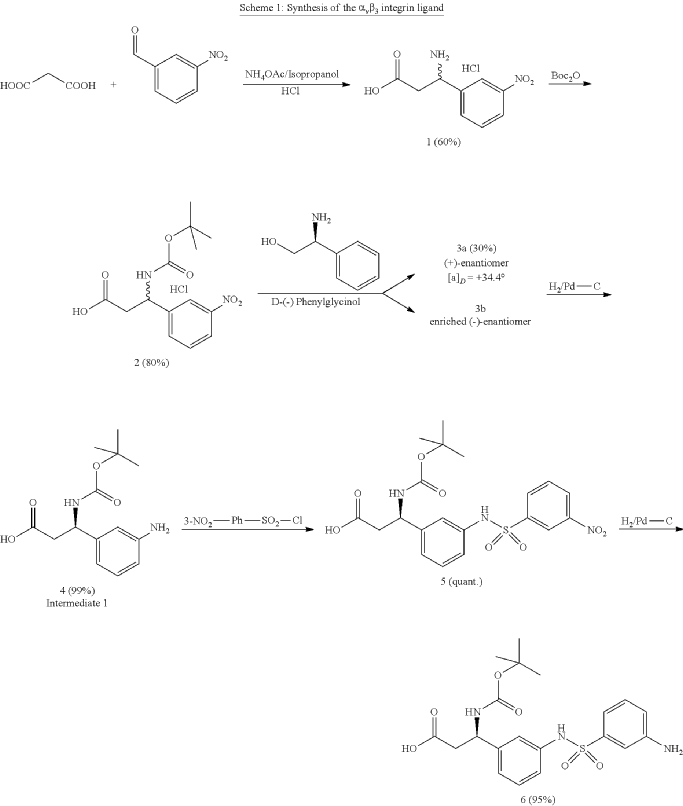
Separation of enantiomers can also be accomplished on different steps via chromatography using chiral columns
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
/////////orenasitecan, antineoplastic, L3KV5NR5PG
Olomorasib
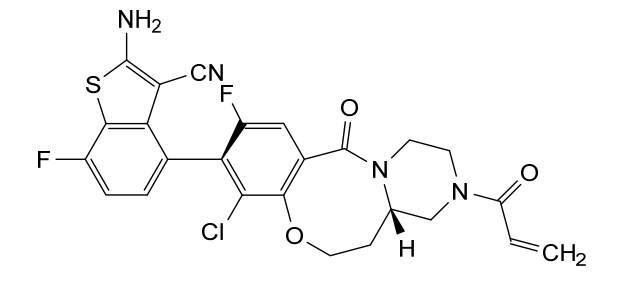
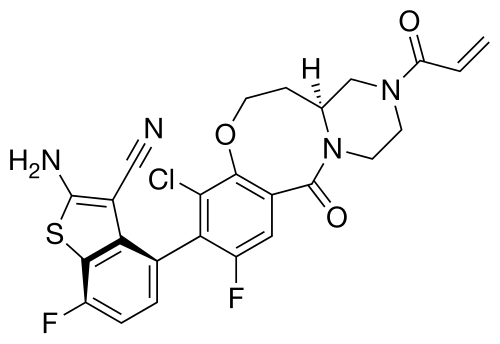
Olomorasib
CAS 2771246-13-8
MF C25H19ClF2N4O3S MW528.96
4-[(13aS)-10-chloro-8-fluoro-6-oxo-2-prop-2-enoyl-1,3,4,12,13,13a-hexahydropyrazino[2,1-d][1,5]benzoxazocin-9-yl]-2-amino-7-fluoro-1-benzothiophene-3-carbonitrile
(4M)-2-amino-4-[(4aS)-8-chloro-10-fluoro-12-oxo-3-(prop-2-enoyl)-2,3,4,4a,5,6-hexahydro-1H,12H-pyrazino[2,1-d][1,5]benzoxazocin-9-yl]-7-fluoro-1-benzothiophene-3-carbonitrile
Kirsten rat sarcoma viral oncogene homolog (KRAS) inhibitor, antineoplastic, LY3537982, LY 3537982, KRAS-G12C-II, LY-3537982, C2VJ83PSN7,
Olomorasib (LY3537982) is an investigational, oral, second-generation KRAS G12C inhibitor designed to treat advanced solid tumors, particularly non-small cell lung cancer (NSCLC). Developed by Eli Lilly and Company, it shows promising antitumor activity and a manageable safety profile, often combined with pembrolizumab (Keytruda). 
Key details about olomorasib include:
- Mechanism & Target: It targets the KRAS G12C mutation, a common driver in lung and colorectal cancers.
- Clinical Status: It is undergoing Phase 1/2 (LOXO-RAS-20001) and Phase 3 (SUNRAY-01) clinical trials.
- Breakthrough Therapy: The FDA granted Breakthrough Therapy designation for first-line treatment of advanced NSCLC (PD-L1
50%) in September 2025.
- Combination Efficacy: When combined with pembrolizumab, it showed an objective response rate of 73.9% in first-line patients, with higher efficacy in those with high PD-L1 expression.
- Safety Profile: Common adverse events include diarrhea, elevated liver enzymes (ALT/AST), and rash, which were generally manageable.
Eli Lilly and Company +4
Olomorasib is designed to be more potent with potentially better tolerability than earlier KRAS G12C inhibitors, aiming to improve outcomes in first-line settings.
- OriginatorEli Lilly and Company
- ClassAntineoplastics; Small molecules
- Mechanism of ActionKRAS protein inhibitors
- Phase IIINon-small cell lung cancer
- Phase ISolid tumours
- 05 Jan 2026Eli Lilly and Company completes a phase-I trial (In volunteers) in Japan (PO, Capsule) (NCT07124013)
- 22 Dec 2025Phase-I/II clinical trials in Non-small cell lung cancer (Metastatic disease, Second-line therapy or greater, Combination therapy) in USA, Canada, China, South Korea (PO) (NCT07227025)
- 12 Nov 2025Janssen Research & Development plans a phase I/II (KaRAnaSa) trial for Non-small cell lung cancer (Combination Therapy, Metastatic disease, Second-line therapy or greater) in December 2025 (NCT07227025)
Olomorasib is an orally available inhibitor of the oncogenic KRAS substitution mutation, G12C, with potential antineoplastic activity. Upon oral administration, olomorasib selectively targets the KRAS G12C mutant and inhibits KRAS G12C mutant-dependent signaling. KRAS, a member of the RAS family of oncogenes, serves an important role in cell signaling, division and differentiation. Mutations of KRAS may induce constitutive signal transduction leading to tumor cell growth, proliferation, invasion, and metastasis.
Olomorasib (LY3537982) is an experimental anticancer drug which acts as an inhibitor of the G12C mutant form of Kirsten rat sarcoma virus (KRAS), an oncogene commonly present in several forms of cancer. It is in early stage clinical trials against lung and colorectal cancers and advanced solid tumors.[1][2][3][4][5]
PAPER
ACS Omega. 2025 Jul 4;10(27):29637-29646. [Abstract]
PATENT
•Patent. US20240307395A1.
PAPER
https://www.nature.com/articles/s41598-025-07532-2
SYN
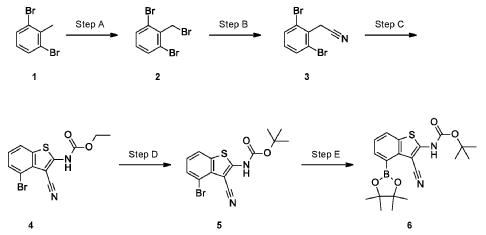
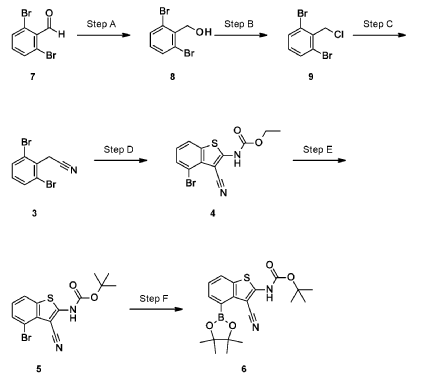

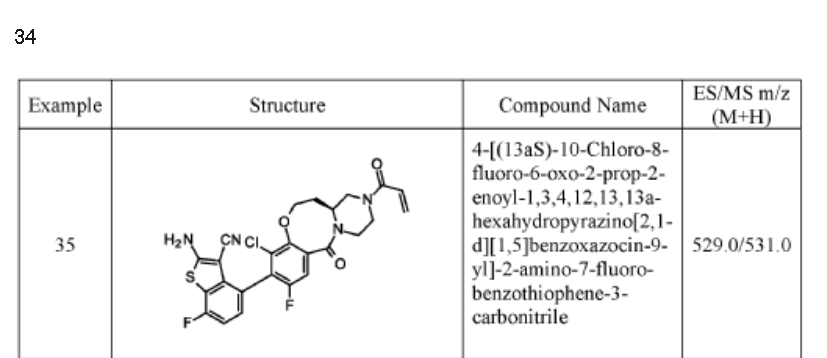
Example 34
4-[( 13 aS)- 10-Chloro-8-fluoro-6-oxo-2-prop-2-enoyl- 1,3,4,12, 13,13 a- hexahydropyrazino[2,ld][1,5]benzoxazocin-9-yl]-2-amino-benzothiophene-3- carbonitrile
A suspension of 4-[(13aS)-10-chloro-8-fluoro-6-oxo-2,3,4,12,13,13a-hexahydro-lH-pyrazino[2,ld][l,5]benzoxazocin-9-yl]-2-amino-benzothiophene-3-carbonitrile (1.58 g, 3.46 mmol) in EtOAc (35 mL), THF (15 mL) and water (40 mL) is charged with potassium carbonate (1.90 g, 13.7 mmol). The mixture is stirred rapidly and cooled to 0 °C. Acryloyl chloride in DCM (13.0 mL, 3.25 mmol, 0.25M) is added dropwise through a dropping funnel. After 10 minutes of stirring in an ice bath, the mixture is diluted with EtOAc and poured into a separatory funnel. The layers are separated and the aqueous layer is again extracted with EtOAc. The combined organic extracts are washed with saturated aqueous sodium chloride solution, dried over magnesium sulfate, filtered, and concentrated in vacuo. The residue is purified by silica gel flash column chromatography, eluting first with 0-100% (10% MeOH in DCM) / DCM, and second with 0-100% [10% (7N NH 3 in MeOH) in DCM] / DCM to give the desired product as fluffy solid. The solid is sonicated in ether for 30 minutes, filtered, and dried in high vacuum to give the title compound (1.60 g, 91%). ES/MS m/z ( 35 C1/ 37 C1) 511.0/513.0 [M+H] + .
Table 22: Compounds synthesized in a manner essentially analogous to that of Example
PAT
- Methods of Delaying, Preventing, and Treating Acquired Resistance to RAS InhibitorsPublication Number: KR-20230042600-APriority Date: 2020-06-18
- Methods for delaying, preventing, and treating acquired resistance to ras inhibitorsPublication Number: EP-4168002-A1Priority Date: 2020-06-18
- KRAS Gl2C INHIBITORSPublication Number: US-2023339968-A1Priority Date: 2019-12-11
- Kras g12c inhibitorsPublication Number: US-2021179633-A1Priority Date: 2019-12-11
- KRas G12C inhibitorsPublication Number: US-11731984-B2Priority Date: 2019-12-11Grant Date: 2023-08-22
- Combination of antibody-drug conjugate and rasg12c inhibitorPublication Number: WO-2023126822-A1Priority Date: 2021-12-28
- Conjugates comprising covalent binders for targeting intracellular kras g12c proteinsPublication Number: US-2024252694-A1Priority Date: 2021-08-06
- Conjugates containing covalent binders to target intracellular KRAS G12C proteinPublication Number: KR-20240099134-APriority Date: 2021-08-06
- Checkpoint kinase 1 (chk1) inhibitors and uses thereofPublication Number: CA-3219348-A1Priority Date: 2021-05-27
- Methods for inhibiting rasPublication Number: AU-2022281343-A1Priority Date: 2021-05-25
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Peng SB, Si C, Zhang Y, Van Horn RD, Lin X, Gong X, et al. (July 2021). “Preclinical characterization of LY3537982, a novel, highly selective and potent KRAS-G12C inhibitor”. Cancer Research. 81 (13_Supplement): 1259. doi:10.1158/1538-7445.AM2021-1259.
- Miyashita H, Hong DS (2024). “Combining EGFR and KRAS G12C Inhibitors for KRAS G12C Mutated Advanced Colorectal Cancer”. Journal of Cancer Immunology. 6 (2): 62–69. doi:10.33696/cancerimmunol.6.086. PMC 11340593. PMID 39175850.
- Hollebecque A, Kuboki Y, Murciano-Goroff YR, Yaeger R, Cassier PA, Heist RS, et al. (2024). “Efficacy and safety of LY3537982, a potent and highly selective KRAS G12C inhibitor in KRAS G12C-mutant GI cancers: Results from a phase 1 study”. Journal of Clinical Oncology. 42 (3_suppl): 94. doi:10.1200/JCO.2024.42.3_suppl.94.
- Burns TF, Dragnev KH, Fujiwara Y, Murciano-Goroff YR, Lee DH, Hollebecque A, et al. (2024). “Efficacy and safety of olomorasib (LY3537982), a second-generation KRAS G12C inhibitor (G12Ci), in combination with pembrolizumab in patients with KRAS G12C-mutant advanced NSCLC”. Journal of Clinical Oncology. 42 (16_suppl): 8510. doi:10.1200/JCO.2024.42.16_suppl.8510.
- Heist RS, Koyama T, Murciano-Goroff YR, Hollebecque A, Cassier PA, Han J, et al. (2024). “Pan-tumor activity of olomorasib (LY3537982), a second-generation KRAS G12C inhibitor (G12Ci), in patients with KRAS G12C-mutant advanced solid tumors”. Journal of Clinical Oncology. 42 (16_suppl): 3007. doi:10.1200/JCO.2024.42.16_suppl.3007.
| Clinical data | |
|---|---|
| Other names | LY3537982 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2649788-46-3 |
| PubChem CID | 156472638 |
| ChemSpider | 115009373 |
| UNII | C2VJ83PSN7 |
| KEGG | D12853 |
| Chemical and physical data | |
| Formula | C25H19ClF2N4O3S |
| Molar mass | 528.96 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
///////olomorasib, Kirsten rat sarcoma viral oncogene homolog (KRAS) inhibitor, antineoplastic, LY3537982, LY 3537982, KRAS-G12C-II, LY-3537982, C2VJ83PSN7,
Naxtarubicin, Annamycin
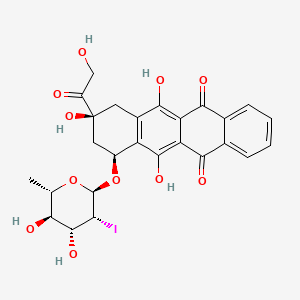
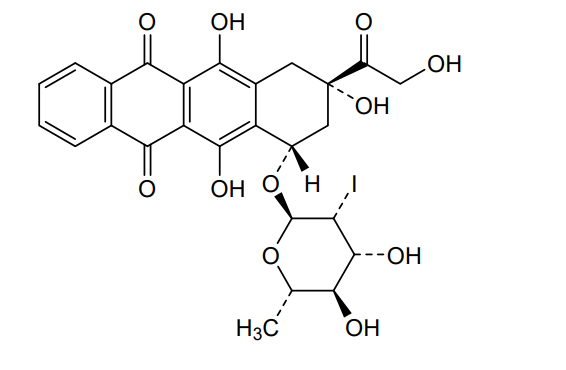
Naxtarubicin, Annamycin
CAS 92689-49-1
MF C26H25IO11 MW 640.4 g/mol
2′-Iodo-3′-hydroxy-4′-epi-4-demethoxydoxorubicin
(7S,9S)-7-[(2R,3R,4R,5R,6S)-4,5-dihydroxy-3-iodo-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-8,10-dihydro-7H-tetracene-5,12-dione
(7S,9S)-7-[(2,6-dideoxy-2-iodo-α-L-mannopyranosyl)oxy]-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-7,8,9,10-
tetrahydrotetracene-5,12-dione
DNA topoisomerase II inhibitor, antineoplastic, Annamycin, Annamycin-LF, Annamycin-liposomal, L-ANNA, L-annamycin, Liposomal annamycin, S-ANNA, SNU299M83Q
- OriginatorUniversity of Texas M. D. Anderson Cancer Center
- DeveloperAronex Pharmaceuticals; Callisto Pharmaceuticals; Moleculin Biotech; University of Texas M. D. Anderson Cancer Center
- ClassAnthracyclines; Antineoplastics; Cytostatic antibiotics; Small molecules
- Mechanism of ActionType II DNA topoisomerase inhibitors
- Orphan Drug StatusYes – Soft tissue sarcoma; Precursor cell lymphoblastic leukaemia-lymphoma; Acute myeloid leukaemia
- Phase II/IIIAcute myeloid leukaemia
- Phase IIOvarian cancer
- Phase I/IISoft tissue sarcoma
- PreclinicalColorectal cancer; Liver cancer; Pancreatic cancer; Solid tumours
- DiscontinuedChronic myeloid leukaemia; Precursor cell lymphoblastic leukaemia-lymphoma; Triple negative breast cancer
- 30 Oct 2025Moleculin biotech plans future regulatory filings based on data from phase III MIRACLE trial
- 29 Oct 2025Moleculin Biotech has patent protection for Naxtarubicin in Australia
- 23 Oct 2025Moleculin Biotech plans to submit an IND application to the US FDA for Pancreatic cancer
Naxtarubicin is a lipophilic, anthracycline antineoplastic antibiotic.Naxtarubicin intercalates into DNA and inhibits topoisomerase II, thereby inhibiting DNA replication and repair as well as inhibiting RNA and protein synthesis. This agent appears to not be a substrate for the p-glycoprotein associated multidrug-resistance (MDR) transporter; therefore, overcoming the resistance pattern seen with other anthracycline compounds.
Annamycin is an anthracycline antibiotic being investigated for the treatment of cancer.
SYN
US 4537882
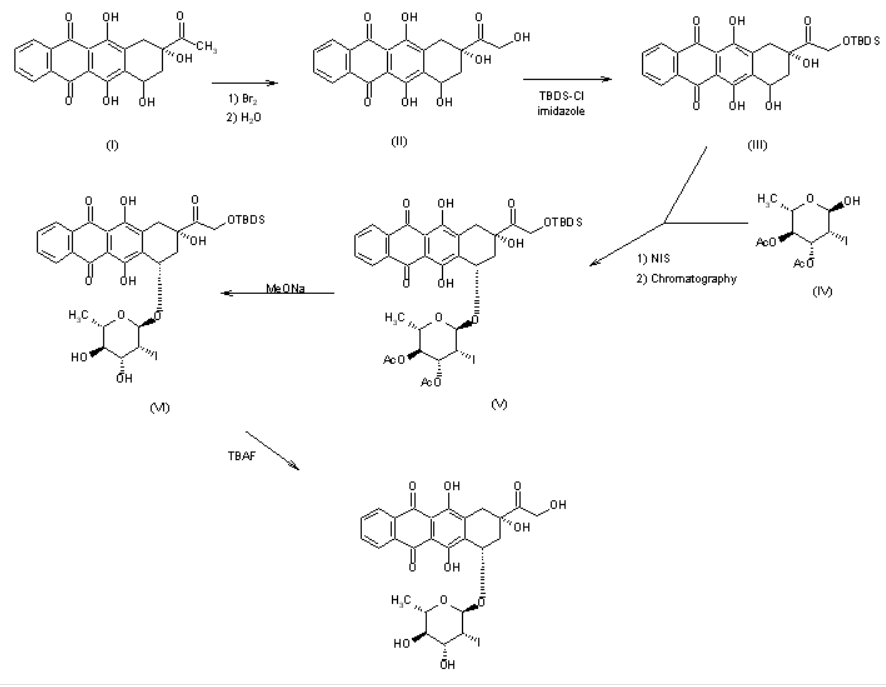
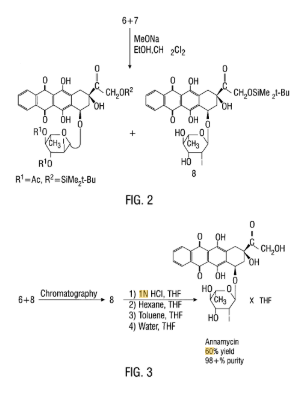
The reaction of racemic 4-demethoxydaunomycinone (I) with Br2 followed by hydrolysis in basic medium gives 4-demethoxyadriamycinone (II), which is treated with tert-butyldimethylsilyl chloride and imidazole in DMF to yield the monoprotected compound (III). The condensation of (III) with 3,4-di-O-acetyl-2,6-dideoxy-2-iodo-alpha-L-mannopyranose (IV) by means of N-iodosuccinimide (NIS), followed by chromatographic separation of the diastereomers affords (7S,9S)-14-O-(tert-butyldimethylsilyl)-4-demethoxy-7-O-(3,4-di-O-acetyl-2,6-dideoxy-2-iodo-alpha-L-mannopyranosyl)adriamycinone (V). The hydrolysis of (V) with sodium methoxide in methanol gives the silylated compound (VI), which is finally desilylated with tetrabutylammonium fluoride (TBAF) in dichloromethane/THF/pyridine.
SYN
https://patents.google.com/patent/US5977327A/en

EXAMPLE VIIIPURIFICATION OF ANNAMYCIN
Crude product was purified further by triple precipitation from THF. To accomplish this, approximately 87 mL of THF was used to redissolve each gram of Annamycin product and an equal volume of one of the following solvents was added to precipitate the Annamycin in each successive precipitation step. In the preferred method, the first precipitation was accomplished by adding an equal volume of a 7:3 mixture of hexane\diethylether, the second precipitation was accomplished by the addition of an equal volume of hexane, and the third precipitation was by addition of an equal volume of water and evaporation of half of the THF. Product obtained in this way (9.0146 g; 59%) was a complex containing 3 molecules of Annamycin per 2 molecules of THF and its purity by HPLC analysis was better than 98%. HPLC analysis was on an analytical C-18 reverse phase column with increasing concentrations of methanol/acetonitrile in water. The purity was determined by measuring the area of the absorbance peaks. 1 H NMR (DMSO-d6) d 1.20 (d, 3H, J6′, 5′ =6.2 Hz, H-6′), 1.75 (m, 2.7H, Ha from THF), 2.10 (dd, 1H, J8a,7 =5.6 Hz, J8a,8e =14.5 Hz, H-8a), 2.18 (dd, 1H, J8e,8a =14.8 Hz, J8e,7 =2.9 Hz, H-8e), 250 (DMSO peak), 2.75 (dd, 1H, J3′,2′ =3.9 Hz, J3′,4′ =8.8 Hz, H-3′), 2.95 (d, 1H, J10a,10e =18.4 Hz, H-10a), 3.00 (d, 1H, J10e,10a =18.4 Hz, H-10e), 3.20 (t, 1H, SJ=18.1 Hz, H-4′), 3.59 (m, 2.7H, Hb from THF), 3.95 (m, 1H, H-5′), 4.30 (d, 1H, J2′,3′ =4.0 Hz, H-2′), 4.55 (s, 2H, H-14), 4.89 (t, 1H, exchangeable, OH), 4.92 (m, 1H, H-7), 5.18 (d, 1H, exchangeable, OH), 5.38 (d, 1H, exchangeable, OH), 5.49 (s, 1H, H-1′), 5.50 (d, 1H, exchangeable, OH), 7.9, 8.4 (2m, 4H,H-1,2,3,4); 13 C NMR (DMSO-d6) d 17.0(s, 1C, C-6′), 24.5 (s, 1C, THFb), 31.7 (s, 1C, C-2′), 31.9 (s, 1C, C-10), 36.4 (s, 1C, C-8), 63.0 (s, 1C, C-3′), 66.4 (s, 1C, C-5′), 67.4 (s, 1C, THFa), 69.4, 13 C-NMR (DMSO-d6) δ 17.9 (s, 1C, C-6′), 25.1 (s, 1C, THFb), 40.6, 36.6, 32.1 (3s, 3C, C-2′,8,10), 63.6 (s, 1C, C-14), 67.0, 67.5, 70.4, 69.7 (4s, 4C, C-7, 5′, 3′, THFa), 74.2, 74.7 (2s, 2C, C-9, 4′), 104.5 (s, 1C, C-1′), 110.1, 110.8 (2s, 1C, C-11a, 5a), 126.6, 132.6, 132.8, 134.4, 135.1, 135.0, 136.0 (7s, 8C, C-2, 3, 1, 4, 4a, 12a, 10a), 136.0 (s, 1C, C-6a), 155.1, 156.4 (2s, 2C, C-6, 11), 186.2, 186.3 (2s, 2C, C-5, 12), 214 (s, 1C, C-13).
SYN
SYN
SYN
EXAMPLE I. SYNTHESIS OF (+)-4-DEMETHOXY-14-O-TERT-BUTYL
DIMETHYLSILYL-7-O-(2,6-DIDEOXY-2-IODO-α-L-MANNOPYRANOSYL)ADRIAMYCINONE (8)
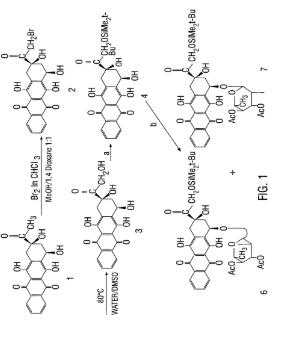
To a solution consisting of a mixture of compounds (6) and (7), shown in Figure 1 , (1.8530g, 2.21mmol) in CH2CI2 (48 mL) and EtOH (16 mL), a IN MeONa solution in MeOH (1.6 mL) was added at room temperature with stirring. Next 1.6 mL of a IN MeONa solution in MeOH (1.6 mL) was added after 50 min. After 1.5 hr. the reaction was checked by TLC developed with CCl4/MeOH (96:4), and the reaction mixture was diluted with dichloromethane (300 mL) and 0.05N HCL (100 mL) was added. The resulting mixture was shaken in a separatory funnel and, after separation, the organic layer was washed with water (2 x 50 mL), dried over Na2SO4, filtered and evaporated. The residue left after evaporation was precipitated from 4 mL of CH2CI2 by addition of 35 mL of hexane. The precipitate was filtered, washed with hexane (40 mL) and then dried in vacuo (1 lmbar) at ambient temperature for 30 min. to give crude product (8) (1.3618g, 82%). The crude product was then filtered through silica with a solution of 95:5 toluene/acetone and precipitated from CH2CI2 by addition of hexane. Product was then dried in vacuo (1 lmbar) at ambient temperature for 30 minutes to give pure compound (8) (1.358g; 55%): ^H NMR d 0.15 (s, 6H, Me2Si), 0.95 (s, 9H, CMe3), 1.40 (d, 3H, J6′,5’=6.2Hz, H-6′), 2.18 (dd, 1H, J8a,7=4.4Hz, J8a,8e=l 5.0Hz, H-8a), 2.35 (d, 1H, J8e,8a=14.9Hz, H-8e), 2.85 (dd, 1H, J3′,2’*=4-0Hz, J3′54’=8.9Hz, H-3′), 3.02 (d, 1H, Jl0a,10e=19.0Hz, H-lOa), 3.24 (d, 1H, Jl0e,10a=l 9.0Hz, H-lOe), 3.58 (t, 1H, SJ=18.2Hz, H-4′), 3.94 (m, 1H, H-5′), 4.18 (s, 1H, 9OH), 4.54 (d, 1H, J2′,3’=3.9Hz, H-2′) 4.84, 4.90 (2d, 2H, H-14), 5.22 (bs, 1H, H-7), 5.75 (s, 1H, H-l’), 7.9, 8.4 (2m, 4H, H-1,2,3,4).
EXAMPLE II. DESILYLATION IN THF/HCl
To a solution of compound (8), (16.5928g, 21.99mmol) in THF (415 mL), IN HC1 (415 mL) was added. After 25 minutes the progress of the reaction was checked by TLC developed in toluene/acetone (6:4 or 5:1) and half of the THF was evaporated in vacuo at 20°C (35mbar). The precipitate was filtered off and washed with water until the pH reached neutral (14 x 40 mL), then washed with ether (Et2θ, 5 x 32 mL) and subsequently with water (3 x 40 mL). The crude product was pre-dried on a Buchner funnel and then dried in vacuo (0.08mbar) at room temperature for 38 hrs.
EXAMPLE III. DESILYLATION IN METHANOL/HC1
To a solution/suspension of compound (8) (1.0064 g, 1.33 mmol) in methanol (45 mL), IN HC1 (10 mL) was added. The progress of the reaction was monitored by TLC developed in toluene/acetone, 6:4 and chloroform methanol, 94:6. After 45 min. 5 mL of IN HC1 solution was added to the reaction mixture. After 1 hr. 15 min. the product of the reaction was precipitated by addition of 30 mL water and filtered off. Product was washed with water until neutral pH (4 x 10 mL), diethylether (3 x 10 mL) and again with water (2 x 10 mL). Crude product was pre-dried on Buchner funnel and then dried in vacuo (0.1 mbar) at room temperature for 24 hrs. to give 0.6722 g (79% yield) of deep red powder.
EXAMPLE IV. DESILYLATION IN METHANOL/H2SO4
To a solution suspension of compound (8) (1.0065 g, 1.33 mmol) in methanol (45 mL), 10 mL of IN H2SO4 was added. The progress of the reaction was monitored by TLC developed in toluene/acetone, 6:4 and chloroform/methanol, 94:6. After 15 min. the product of the reaction was precipitated by adding 35 mL of water and filtered off. Product was washed with water until neutral pH (4 x 10 mL), diethylethe (3 x 10 mL) and again with water (2 x 10 mL). Crude product was pre-dried on Buchner funnel and then dried in vacuo (0.1 mbar) at room temperature for 24 hrs. to give 0.6318 g (74% yield) of deep red powder. EXAMPLE V. DESILYLATION IN ACETONE/H2SO4
To a solution of compound (8) (0.7592 g, 1.01 mmol) in acetone (30 mL) 3.5 mL IN H2SO4 was added. The progress of the reaction was monitored by TLC developed in toluene/acetone, 6:4 and chloroform/methanol, 94:6. After 1 hr. the product of the reaction was precipitated by addition of 35 mL water and filtered off. The product was washed with water until neutral pH (4 x 10 mL), dicthyleher (3 x 10 mL) and again with water (2 x 10 mL). Crude product was pre-dried on Buchner funnel and then dried in vacuo (0.1 mbar) at room temperature for 48 hrs. to give 0.4994 g (77% yield) of deep red powder.
EXAMPLE VI. DESILYLATION IN DMSO/HC1
To a solution of compound (8) (0.7815 g, 1.04 mmol) in DMSO (30 mL) 7.5 mL of IN HC1 was added. Progress of the reaction was monitored by TLC developed in toluene/acetone, 6:4 and chloroform/methanol, 94:6. After 1 hr. 20 min. the product of the reaction was precipitated by addition of water (37 mL) and filtered off. The product was washed with water until neutral pH (4 x 10 mL), dietheylether (3 x 10 mL) and again with water (2 x 10 mL). Crude product was pre-dried on Buchner funnel and then dried in vacuo (0.1 mbar) at room temperature for 48 hrs. to give 0.5165 g (78% yield) of deep red powder. EXAMPLE VII. DESILYLATION IN DMSO/H2SO
To a solution of compound (8) (0.7613 g, 1.01 mmol) in DMSO (5 mL) and ethanol
(10 mL) 1 mL of IN H2SO4 was added. Progress of the reaction was monitored by TLC developed in toluene/acetone, 6:4 and chloroform/methanol, 94:6. After 1 hr. 10 min. product of the reaction was precipitated by addition of water (15 mL) and filtered off. Product was washed with water until neutral pH (4 x 10 mL), diethylether (3 x 10 mL) and again with water (2 x 10 mL). Crude product was pre-dried on Buchner funnel and then dried in vacuo (0.1 mbar) at room temperature for 48 hrs. to give 0.5338 g (83% yield) of deep red powder. EXAMPLE VIII. PURIFICATION OF ANNAMYCIN
Crude product was purified further by triple precipitation from THF. To accomplish this, approximately 87 mL of THF was used to redissolve each gram of Annamycin product and an equal volume of one of the following solvents was added to precipitate the .Annamycin in each successive precipitation step. In the preferred method, the first precipitation was accomplished by adding an equal volume of a 7:3 mixture of hexane\diethylether, the second precipitation was accomplished by the addition of an equal volume of hexane, and the third precipitation was by addition of an equal volume of water and evaporation of half of the THF. Product obtained in this way (9.0146g; 59%) was a complex containing 3 molecules of .Annamycin per 2 molecules of THF and its purity by HPLC analysis was better than 98%. HPLC analysis was on an analytical C-18 reverse phase column with increasing concentrations of methanol/acetonitrile in water. The purity was determined by measuring the area of the absorbance peaks. H NMR (DMSO-d6) d 1.20 (d, 3H, J6′,5′-=6.2Hz, H-6′), 1.75 (m, 2.7H, Ha from THF), 2.10 (dd, IH, J8a,7=5.6Hz, J8a,8e=14.5Hz, H-8a), 2.18 (dd, IH, J8e,8a=14.8Hz, J8e,7=2.9Hz, H-8e), 2.50 (DMSO peak), 2.75 (dd, IH, J3′,2’=3.9Hz, J3′,4’=8.8Hz, H-3′), 2.95 (d, IH, Jl0a,10e=18.4Hz, H-10a), 3.00 (d, IH, Jl0e,10a=18.4Hz, H-lOe), 3.20 (t, IH, SJ=18.1Hz, H-4′), 3.59 (m, 2.7H, Hb from THF), 3.95 (m, IH, H-5′), 4.30 (d, IH, J2′,3’=4.0Hz, H-2′), 4.55 (s, 2H, H-14), 4.89 (t, IH, exchangeable, OH), 4.92 (m, IH, H-7), 5.18 (d, IH, exchangeable, OH), 5.38 (d, IH, exchangeable, OH), 5.49 (s, IH, H-l’), 5.50 (d, IH, exchangeable, OH), 7.9, 8.4 (2m, 4H,H- 1,2,3,4); 13C NMR (DMSO-d6) d 17.0(s, IC, C-6′), 24.5 (s, IC, THFb), 31.7 (s, IC, C-2′), 31.9 (s, IC, C-10), 36.4 (s, IC, C-8), 63.0 (s, IC, C-3′), 66.4 (s, IC, C-5′), 67.4 (s, IC, THFa), 69.4, ,3C-NMR (DMSO-d6) δ 17.9 (s, IC, C-6′), 25.1 (s, IC, THFb), 40.6, 36.6, 32.1 (3s, 3C, C-2′, 8, 10), 63.6 (s, IC, C-14), 67.0, 67.5, 70.4, 69.7 (4s, 4C, C-7, 5′, 3′, THFa), 74.2, 74.7 (2s, 2C, C-9, 4′), 104.5 (s, IC, C-l’), 110.1, 110.8 (2s, IC, C-lla, 5a), 126.6, 132.6, 132.8, 134.4, 135.1, 135.0, 136.0 (7s, 8C, C-2, 3, 1, 4, 4a, 12a, 10a), 136.0 (s, IC, C-6a), 155.1, 156.4 (2s, 2C, C-6, 11), 186.2, 186.3 (2s, 2C, C-5, 12), 214 (s, IC, C-13).
PAT
- Co-solvent compositions and methods for improved delivery of dantrolene therapeutic agentsPublication Number: US-2011160261-A2
- HER2 mutation inhibitorsPublication Number: US-12447153-B2Grant Date: 2025-10-21
- Submicron liposome suspensions obtained from preliposome lyophilizatesPublication Number: US-7238366-B1Priority Date: 1995-06-06Grant Date: 2007-07-03
- Submicron liposome suspension obtained from freeze-dried preliposomePublication Number: JP-H11507369-APriority Date: 1995-06-06
- Submicron liposome suspensions obtained from preliposome lyophilizatesPublication Number: EP-1800665-A3Priority Date: 1995-06-06
- Submicron liposome suspensions obtained from preliposome lyophilizatesPublication Number: EP-0831781-A1Priority Date: 1995-06-06
- Process for preparing aminoglycsidic antibioticsPublication Number: CS-202570-B2Priority Date: 1976-04-14
- Methods and systems for assessing biological materials using optical and spectroscopic detection techniquesPublication Number: US-2004052730-A1Priority Date: 1995-10-04
- Submicron liposome suspensions obtained from preliposome lyophilizatesPublication Number: EP-1800665-A2Priority Date: 1995-06-06
- Submicron liposome suspensions obtained from preliposome lyophilizatesPublication Number: US-5902604-APriority Date: 1995-06-06Grant Date: 1999-05-11
- Submicron liposome suspensions obtained from preliposome lyophilizatesPublication Number: WO-9639121-A1Priority Date: 1995-06-06
- Submicron liposome suspensions obtained from preliposome lyophilizatesPublication Number: CA-2221341-A1Priority Date: 1995-06-06
- Compounds and methods for the selective treatment of cancer and bacterial infectionsPublication Number: WO-9739007-A1Priority Date: 1996-04-12
- Compounds and methods for the selective treatment of cancer and bacterial infectionsPublication Number: US-6218519-B1Priority Date: 1996-04-12Grant Date: 2001-04-17
- Methods and systems for assessing biological materials using optical and spectroscopic detection techniquesPublication Number: US-6573063-B2Priority Date: 1995-10-04Grant Date: 2003-06-03
- Methods and systems for assessing biological materials using optical and spectroscopic detection techniquesPublication Number: US-6319682-B1Priority Date: 1995-10-04Grant Date: 2001-11-20
- Methods and systems for assessing biological materials using optical and spectroscopic detection techniquesPublication Number: US-2002055092-A1Priority Date: 1995-10-04

AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
Further reading
- Priebe W (1995). “Mechanism of action-governed design of anthracycline antibiotics: a “turn-off/turn-on” approach”. Current Pharmaceutical Design. 1 (1): 51–68. doi:10.2174/1381612801666220524190711. S2CID 90406009.
- Trevino AV, Woynarowska BA, Herman TS, Priebe W, Woynarowski JM (November 2004). “Enhanced topoisomerase II targeting by annamycin and related 4-demethoxy anthracycline analogues”. Mol Cancer Ther. 3 (11): 1403–10. doi:10.1158/1535-7163.1403.3.11. PMID 15542779.
External links
| Clinical data | |
|---|---|
| ATC code | none |
| Identifiers | |
| IUPAC name | |
| CAS Number | 92689-49-1 |
| PubChem CID | 115212 |
| ChemSpider | 103088 |
| UNII | SNU299M83Q |
| KEGG | D12844 |
| CompTox Dashboard (EPA) | DTXSID901027238 |
| ECHA InfoCard | 100.235.298 |
| Chemical and physical data | |
| Formula | C26H25IO11 |
| Molar mass | 640.379 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////////naxtarubicin, DNA topoisomerase II inhibitor, antineoplastic, Annamycin, Annamycin-LF, Annamycin-liposomal, L-ANNA, L-annamycin, Liposomal annamycin, S-ANNA, SNU299M83Q
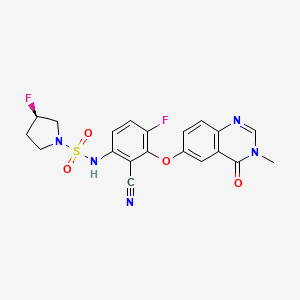
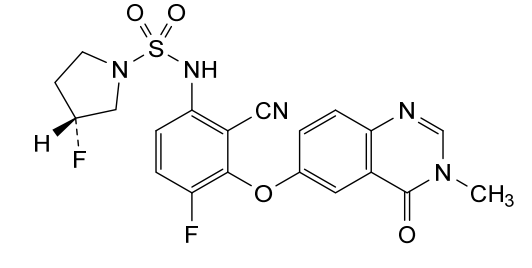
Mosperafenib
Mosperafenib
CAS 2649372-20-1
MF C20H17F2N5O4S MW 461.4 g/mol
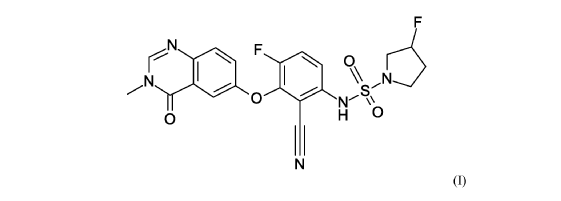
- (3R)-N-{2-cyano-4-fluoro-3-[(3-methyl-4-oxo-3,4-dihydroquinazolin-6-yl)oxy]phenyl}-3-fluoropyrrolidine-1-sulfonamide
- (3R)-N-(2-cyano-4-fluoro-3-((3-methyl-4-oxo-3,4-dihydroquinazolin-6-yl)oxy)phenyl)-3-fluoropyrrolidine-1-sulfonamide
(3R)-N-{2-cyano-4-fluoro-3-[(3-methyl-4-oxo-3,4-dihydroquinazolin-6-yl)oxy]phenyl}-3-fluoropyrrolidine-1-sulfonamide
B-Raf (BRAF) inhibitor, antineoplastic, RG6344, RO7276389, RG 6344, RO 7276389, 881-730-4, B-Raf IN 2
Mosperafenib is a small molecule drug. The usage of the INN stem ‘-rafenib’ in the name indicates that Mosperafenib is a Raf (rapidly accelerated fibrosarcoma) kinase inhibitor. Mosperafenib has a monoisotopic molecular weight of 461.1 Da.
Mosperafenib (RG6344, RO7276389) is an investigational, oral, “paradox-breaker” BRAF inhibitor developed by Roche for treating BRAF-mutated cancers, particularly BRAF V600E-mutant metastatic colorectal cancer. It acts as a potent, selective inhibitor that avoids MAPK pathway overactivation in non-V600E contexts, showing superior preclinical activity and brain penetration compared to existing inhibitors like encorafenib.
Key Aspects of Mosperafenib:
- Mechanism: As a “paradox-breaker” BRAF inhibitor, it avoids the paradoxical MAPK pathway activation seen with earlier inhibitors. It inhibits BRAF mutants (
) and is effective in RAF dimer-mediated resistant models.
- Clinical Development: Currently in Phase I clinical trials for BRAF V600E-mutant colorectal cancer.
- Preclinical Performance: In studies, it demonstrated higher antitumor activity than encorafenib/cetuximab combinations, even in BRAFi-naïve models.
- Combination Potential: It is being evaluated in combination with cetuximab and FOLFOX.
- Targeting: It targets BRAF V600E/K/A/D mutations.
- OriginatorRoche
- ClassAntineoplastics; Fluorinated hydrocarbons; Fluorobenzenes; Nitriles; Phenyl ethers; Pyridones; Pyrrolidines
- Mechanism of ActionProto-oncogene protein b-raf inhibitors
- Phase IMalignant melanoma; Solid tumours
- 18 Sep 2025Chemical structure information added.
- 30 May 2025Efficacy, pharmacokinetics and adverse events data from a phase I trial in Solid tumors presented at the 61st Annual Meeting of the American Society of Clinical Oncology (ASCO-2025)
- 25 Apr 2025Efficacy, pharmacokinetics and adverse events data from a phase I trial in Solid tumors presented at the 116th Annual Meeting of the American Association for Cancer Research (AACR-2025)
SYN
SYN
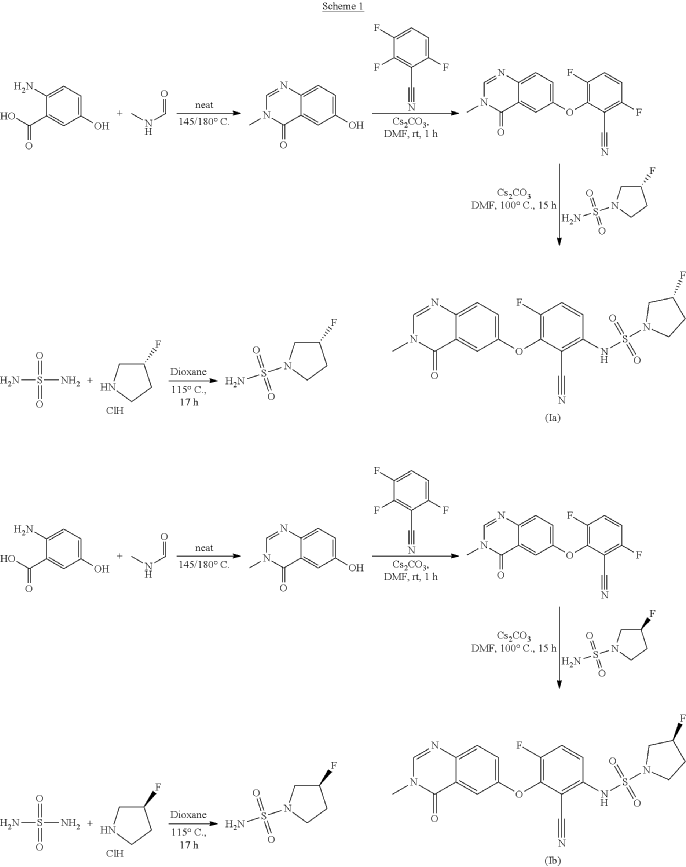
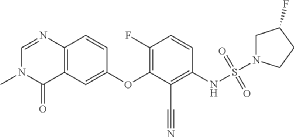
3R)-N-[2-cyano-4-fluoro-3-(3-methyl-4-oxo-quinazolin-6-yl)oxy-phenyl]-3-fluoro-pyrrolidine-1-sulfonamide (Example 1)
R)-3-Fluoropyrrolidine-1-sulfonamide (1.26 g, 7.51 mmol, Eq: 2.1) and cesium carbonate (2.56 g, 7.87 mmol, Eq: 2.2) were suspended in dry DMF (10.2 ml) under an argon atmosphere. The reaction was stirred at 50° C. for 30 min. The reaction mixture was cooled to rt and a solution of 3,6-difluoro-2-((3-methyl-4-oxo-3,4-dihydroquinazolin-6-yl)oxy)benzonitrile (1.12 g, 3.58 mmol, Eq: 1.0) in DMF (25.5 ml) was added. The reaction mixture was stirred at 100° C. for 15 h, then concentrated in vacuo. The residue was taken up in sat. aq. NH 4Cl (100 mL) and EtOAc (100 mL). The phases were separated, and the aqueous layer was extracted further with 2×100 mL EtOAc. The combined organic layers were washed with water (200 mL) and brine (200 mL), dried (Na 2SO 4), filtered and concentrated in vacuo. The water layer was back-extracted with EtOAc (3×100 mL). The combined organic extracts were washed with brine (200 mL), dried (Na 2SO 4), filtered and concentrated in vacuo. The residue was diluted with DCM and MeOH, and concentrated onto silica. Purification by flash chromatography (120 g, 0.5-2% MeOH/DCM) gave an off-white solid which was triturated with 1:1 heptane/DCM (20 mL) with sonication, then dried in vacuo to give the title compound as a colourless solid (1.087 g, 66% yield). MS (ESI) m/z: 426.2 [M+H] +. Chiral SFC: RT=4.594 min [Chiralpak IC column, 4.6×250 mm, 5 μm particle size (Daicel); gradient of 20-40% MeOH containing 0.2% NHEt 2 over 8 min; flow: 2.5 mL/min; 140 bar backpressure].
SYN
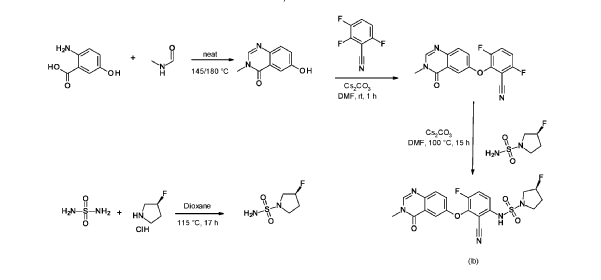
Refences compounds AR-25, AR-30 and AR-31 were prepared according to the synthesis disclosed in WO2012/118492 in example 25, example 30 and example 31 respectively.
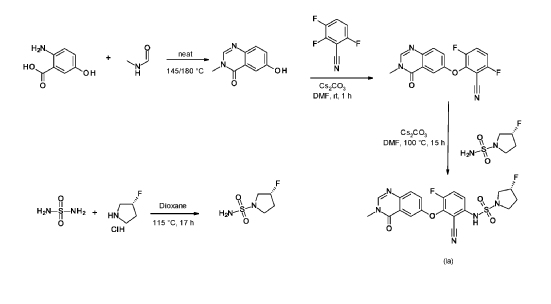
6-hydroxy-3-methyl-quinazolin-4-one
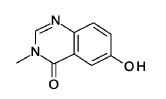
2-Amino-5-hydroxybenzoic acid (10 g, 65.3 mmol, Eq: 1.0) and A-methylformamide (30 g, 29.9 mL, 503 mmol, Eq: 7.7) were heated at 145 °C for 21 h 45 min, then cooled to rt. The reaction mixture was diluted with 50 mL H2O and stirred at rt for 20 min. The resulting precipitate was collected by filtration. The light brown solid was washed 3 × with 20 mL water. The solid was taken up in toluene and evaporated to dryness (3 ×). The solid was dried in vacuo at 40 °C overnight under high vacuum to give the title compound as a light brown solid (10.3 g, 89% yield). MS (ESI) mlz: 177.1 [M+H]+.
3.6-difluoro-2-(3-methyl-4-oxo-quinazolin-6-yl)oxy-benzonitrile
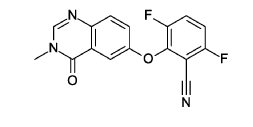
Cesium carbonate (3.22 g, 9.79 mmol, Eq: 1.15) was added at rt to a solution of 6-hydroxy-3-methylquinazolin-4-one (1500 mg, 8.51 mmol, Eq: 1.0) in N,N-dimethylformamide (35 mL). The mixture was stirred for 30 min at rt then 2,3,6-trifluorobenzonitrile (1.47 g, 1.08 ml, 9.37 mmol, Eq: 1.1) was added. After 1 h, the reaction was cooled on ice and diluted with water (120 mL). The resultant solid was collected by filtration, washed with iced water (100 mL) and heptane (100 mL) and suction-dried. The solid was taken up in toluene and evaporated to dryness (3 ×) then dried overnight in vacuo to give the title compound as a light brown solid (2.58 g, 97% yield). MS (ESI) m/z: 314.1 [M+H]+.
(3R)-3 -fluoropyrrolidine- 1 -sulfonamide
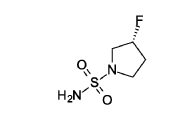
(R)-3 -Fluoropyrrolidine hydrochloride (1.8 g, 14.3 mmol, Eq: 1.2) was added to a solution of sulfuric diamide (1.148 g, 11.9 mmol, Eq: 1.0) and triethylamine (2.42 g, 3.33 mL, 23.9 mmol, Eq: 2) in dioxane (10 mL). The reaction was stirred in a sealed tube at 115 °C for 15.5 h then cooled to rt and concentrated in vacuo. The residue was diluted with DCM, evaporated with silica gel to dryness and transferred to a column. Purification by flash chromatography (40 g silica, 80% EtOAc) gave the title compound as a white crystalline solid (1.82 g, 91% yield). MS (ESI) m/z: 169.1 [M+H]+.
(3S)-3 -fluoropyrrolidine- 1 -sulfonamide
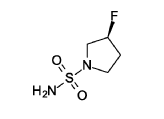
Triethylamine (304 mg, 419 μl, 3.01 mmol, Eq: 2.0) was added to a suspension of sulfuric diamide (146 mg, 1.5 mmol, Eq: 1.0) and (S)-3 -fluoropyrrolidine hydrochloride (234 mg, 1.8 mmol, Eq: 1.2) in dioxane (1.3 ml). The reaction was stirred in a sealed tube at 115°C for 16 h 35 min, then concentrated in vacuo. The residue was diluted with MeOH and evaporated with silica gel to dryness and transferred to a column. Purification by flash chromatography (40 g silica, 0-8% MeOH/DCM) gave the title compound as a light yellow solid (193 mg, 75% yield). MS (ESI) m/z: 169.1 [M+H]+.
(3R)-N-[2-cyano-4-fluoro-3-(3-methyl-4-oxo-quinazolin-6-yl)oxy-phenyl]-3-fluoro-pyrrolidine-1 -sulfonamide (Example 1)
(R)-3-Fluoropyrrolidine-1-sulfonamide (1.26 g, 7.51 mmol, Eq: 2.1) and cesium carbonate (2.56 g, 7.87 mmol, Eq: 2.2) were suspended in dry DMF (10.2 ml) under an argon atmosphere. The reaction was stirred at 50 °C for 30 min. The reaction mixture was cooled to rt and a solution of 3,6-difluoro-2-((3-methyl-4-oxo-3,4-dihydroquinazolin-6-yl)oxy)benzonitrile (1.12 g, 3.58 mmol, Eq: 1.0) in DMF (25.5 ml) was added. The reaction mixture was stirred at 100 °C for 15 h, then concentrated in vacuo. The residue was taken up in sat. aq. NH4Cl (100 mL) and EtOAc (100 mL). The phases were separated, and the aqueous layer was extracted further with 2 x 100 mL EtOAc. The combined organic layers were washed with water (200 mL) and brine (200 mL), dried (Na2SO4), filtered and concentrated in vacuo. The water layer was back-extracted with EtOAc (3 x 100 mL). The combined organic extracts were washed with brine (200 mL), dried (Na2SO4), filtered and concentrated in vacuo. The residue was diluted with DCM and MeOH, and concentrated onto silica. Purification by flash chromatography (120 g, 0.5-2% MeOH/DCM) gave an off-white solid which was triturated with 1 : 1 heptane/DCM (20 mL) with sonication, then dried in vacuo to give the title compound as a colourless solid (1.087 g, 66% yield). MS (ESI) mlz: 426.2 [M+H]+. Chiral SFC: RT = 4.594 min [Chiralpak IC column, 4.6 x 250 mm, 5μm particle size (Daicel); gradient of 20 – 40% MeOH containing 0.2% NHEt2 over 8 min; flow: 2.5 mL/min; 140 bar backpressure],
(3S)-N-[2-cyano-4-fluoro-3-(3-methyl-4-oxo-quinazolin-6-yl)oxy-phenyl]-3-fluoro-pyrrolidine-1 -sulfonamide (Example 2)
(S)-3-Fluoropyrrolidine-1-sulfonamide (181 mg, 1.08 mmol, Eq: 2.1) was dissolved in DMF (1.6 ml). At rt cesium carbonate (368 mg, 1.13 mmol, Eq: 2.2) was added and the reaction mixture was stirred at 50 °C for 30 min. The reaction mixture was cooled to rt and a solution of 3,6-difluoro-2-((3-methyl-4-oxo-3,4-dihydroquinazolin-6-yl)oxy)benzonitrile (160.8 mg, 513 μmol, Eq: 1.0) in DMF (4 ml) was added. The reaction mixture was stirred at 105 °C for 2 h 50 min then concentrated in vacuo. The residue was taken up in DCM and washed with sat. aq. NH4Cl. The aq. layer was back-extracted twice with DCM. The combined organic layers were dried over Na2SO4, filtrated and evaporated. The residue (brown oil) was diluted with DCM and transferred to a column. Purification by flash chromatography (80 g, 0-100% EtOAc in DCM) gave a solid which was further purified by SFC to give the title compound as a light yellow solid (119 mg, 50% yield). MS (ESI) m/z: 426.2 [M+H]+. Chiral SFC: RT = 4.411 min [Chiralpak IC column, 4.6 x 250 mm, 5μm particle size (Daicel); gradient of 20 – 40% MeOH containing 0.2% NHEt2 over 8 min; flow: 2.5 mL/min; 140 bar backpressure].
PAT
New methylquinazolinone derivatives
Publication Number: AU-2020403443-A1
Priority Date: 2019-12-10
- Methylquinazolinone derivativesPublication Number: US-2024174621-A1Priority Date: 2019-12-10
- New methylquinazolinone derivativesPublication Number: EP-4073065-B1Priority Date: 2019-12-10Grant Date: 2025-02-19
- Methylquinazolinone derivativesPublication Number: US-2022298119-A1Priority Date: 2019-12-10
- Novel BRAF inhibitors as anomalous breakersPublication Number: CN-114746405-BPriority Date: 2019-12-10Grant Date: 2024-03-26
- New BRAF inhibitors as paradox breakersPublication Number: AU-2020403082-A1Priority Date: 2019-12-10
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
////////////////mosperafenib, B-Raf (BRAF) inhibitor, antineoplastic, RG6344, RO7276389, RG 6344, RO 7276389, 881-730-4, B-Raf IN 2
Mocaciclib
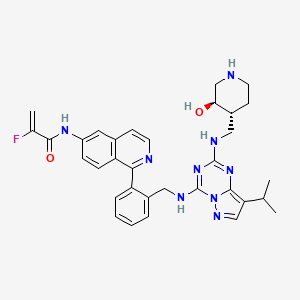
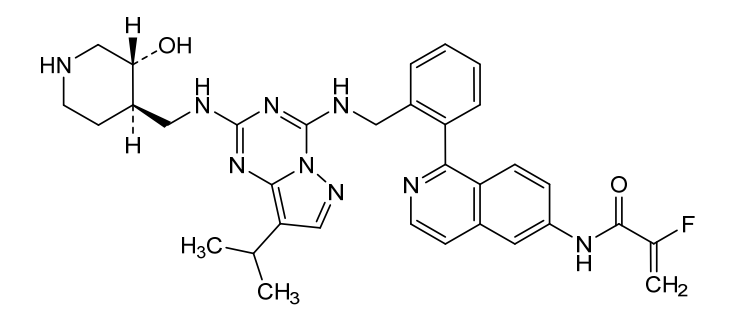
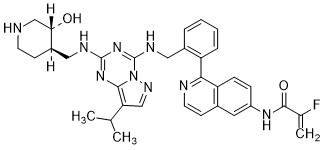
Mocaciclib
CAS 2766124-39-2
MF C33H36FN9O2 MW609.71
- 2-fluoro-N-[1-[2-[[[2-[[(3R,4R)-3-hydroxypiperidin-4-yl]methylamino]-8-propan-2-ylpyrazolo[1,5-a][1,3,5]triazin-4-yl]amino]methyl]phenyl]isoquinolin-6-yl]prop-2-enamide
- 2-Fluoro-N-[1-[2-[[[2-[[[(3R,4R)-3-hydroxy-4-piperidinyl]methyl]amino]-8-(1-methylethyl)pyrazolo[1,5-a]-1,3,5-triazin-4-yl]amino]methyl]phenyl]-6-isoquinolinyl]-2-propenamide
- 2-fluoro-N-[1-[2-[[[2-[[(3R,4R)-3-hydroxypiperidin-4-yl]methylamino]-8-propan-2-ylpyrazolo[1,5-a][1,3,5]triazin-4-yl]amino]methyl]phenyl]isoquinolin-6-yl]prop-2-enamide

cyclin-dependent kinase (CDK) inhibitor, antineoplastic, Q 901, CDK7-IN-21,
- OriginatorThe Lead Discovery Center; The Max Planck Institute of Biochemistry
- DeveloperQurient Co
- ClassAntineoplastics; Small molecules
- Mechanism of ActionCyclin-dependent kinase-activating kinase inhibitors
- Phase I/IISolid tumours
- 31 May 2024Preliminary efficacy, pharmacodynamics, pharmacokinetics and adverse events data from a phase I/II trial in Solid tumours presented at the 60th Annual Meeting of the American Society of Clinical Oncology (ASCO-2024)
- 21 May 2024Qurient Therapeutics enters into an Cooperative Research and Development Agreement (CRADA) with the US National Cancer Institute (NCI) for phase I/II trial in Small cell lung cancer (SCLC) and Solid tumours
- 21 May 2024Qurient Therapeutics plans phase I/II trial in Small cell lung cancer (SCLC) and Solid tumours
Mocaciclib (Q-901) is an orally bioavailable, selective cyclin-dependent kinase (CDK) inhibitor with potent activity against CDK2, CDK4, and CDK6. Preclinical data show that Mocaciclib inhibits CDK2/cyclin E with an IC₅₀ of 1.1 nM, CDK4/cyclin D1 with an IC₅₀ of 2.5 nM, and CDK6/cyclin D3 with an IC₅₀ of 4.1 nM, demonstrating high potency in enzymatic assays. In cancer cell lines, Mocaciclib suppresses retinoblastoma protein (Rb) phosphorylation, leading to G1 cell cycle arrest and growth inhibition in Rb-positive tumor models. It has shown antiproliferative effects in various preclinical models, including breast and lung cancers.
Mocaciclib is a selective inhibitor of cyclin-dependent kinase 7 (CDK7), with potential antineoplastic activity. Upon administration, mocaciclib selectively targets, covalently binds to and inhibits the activity of CDK7, thereby inhibiting CDK7-mediated signaling. The inhibition of CDK7 prevents phosphorylation of the carboxy-terminal domain (CTD) of RNA polymerase II, thereby preventing transcription of important cancer-promoting genes. It prevents phosphorylation of the cell cycle kinases CDK1, 2, 4, and 6, thereby disrupting uncontrolled cell cycle progression. Altogether, this may induce apoptosis, cause cell cycle arrest, inhibit DNA damage repair and inhibit tumor cell proliferation in certain cancers that are dependent on CDK7-mediated transcriptional regulation and signaling. CDK7, a serine/threonine kinase, plays a role in controlling cell cycle progression and transcriptional regulation, and promotes the expression of key oncogenes through the phosphorylation of RNA polymerase II. It is overexpressed in multiple cancers.
SYN
SYN
This is compound 64, as disclosed in WO2O19/197546.
PAT
- Compounds having cyclin-dependent kinase(cdk)-inhibitory functionPublication Number: WO-2022117504-A1Priority Date: 2020-12-02
- Substituted pyrazolo[1,5-a]pyrimidines and pyrazolo[1,5-a][1,3,5]triazines as CDK inhibitorsPublication Number: US-11858937-B2Priority Date: 2018-04-11Grant Date: 2024-01-02
- Pharmaceutically active pyrazolo-triazine and/or pyrazolo-pyrimidine derivativesPublication Number: US-2021139483-A1Priority Date: 2018-04-11
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
//////////mocaciclib, cyclin-dependent kinase (CDK) inhibitor, antineoplastic, Q 901, CDK7-IN-21,
Mobinitinib
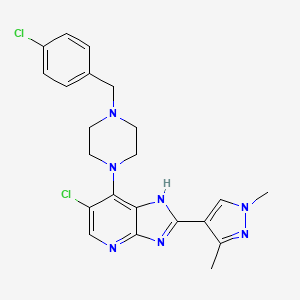
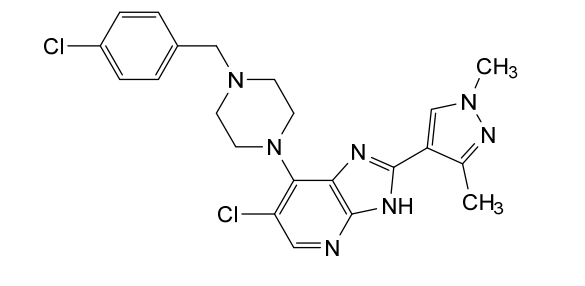
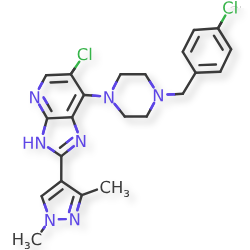
Mobinitinib
CAS1402709-93-6
MF C22H23Cl2N7 MW456.37
6-chloro-7-{4-[(4-chlorophenyl)methyl]piperazin-1-yl}-2-(1,3-dimethyl-1Hpyrazol-4-yl)-3H-imidazo[4,5-b]pyridine
6-chloro-7-{4-[(4-chlorophenyl)methyl]piperazin-1-yl}-2-(1,3-dimethyl-1Hpyrazol-4-yl)-3H-imidazo[4,5-b]pyridine
dual FMS-like tyrosine kinase-3 (FLT3)-Aurora kinase inhibitor, antineoplastic, CCT241736, CCT 241736, ZE94SP78UG, EP0042, EP 0042
Mobinitinib (CCT241736) is an investigational, orally bioavailable, small-molecule, dual inhibitor targeting Aurora kinase and FLT3 (including ITD and D835Y mutations). It shows potent antineoplastic activity in preclinical models, including acute myeloid leukemia (AML), by inhibiting tumor cell proliferation and disrupting mitotic spindle assembly. It is a distinct compound from similarly named drugs like Momelotinib or Binimetinib.
Key Details About Mobinitinib (CCT241736)
- Mechanism of Action: Acts as a dual inhibitor of Aurora kinases (A and B) and FMS-related tyrosine kinase 3 (FLT3). By inhibiting these kinases, it interferes with mitotic spindle assembly and chromosome segregation, leading to cell cycle arrest.
- Target Indications: Primarily studied for its potential to treat hematological malignancies and solid tumors that overexpress FLT3 or Aurora kinases. Preclinical studies show effectiveness in FLT3-ITD positive AML cell lines (e.g., MOLM-13, MV4-11).
- Preclinical Activity: Demonstrates strong anti-proliferative activity with
values in the sub-micromolar range (e.g., 0.1–0.3
M) in certain human tumor cell lines. It has shown significant tumor growth inhibition in mouse xenograft models at doses of 50-100 mg/kg.
- Chemical Properties: It is a 1H-imidazo[5-b]pyridine derivative.
It is important to distinguish mobinitinib (CCT241736) from momelotinib, a JAK1/JAK2 inhibitor approved for myelofibrosis, and binimetinib, a MEK inhibitor for melanoma.
Mobinitinib is an orally bioavailable inhibitor of both the serine/threonine protein kinase Aurora kinase and FMS-related tyrosine kinase 3 (FLT3; STK1; CD135; FLK2), with potential antineoplastic activity. Upon oral administration, mobinitinib specifically binds to and inhibits Aurora kinase and FLT3, which interferes with the activation of Aurora kinase- and FLT3-mediated signal transduction pathways. This may result in the disruption of the assembly of the mitotic spindle apparatus, the disruption of chromosome segregation and the inhibition of cell proliferation in tumor cells that overexpress Aurora kinase and/or FLT3. Aurora kinase plays essential roles in mitotic checkpoint control during mitosis. Aurora kinase and FLT3 are overexpressed in a variety of cancers and play key roles in tumor cell proliferation.
MOBINITINIB is a small molecule drug with a maximum clinical trial phase of II and has 1 investigational indication.
Study to Evaluate the Safety and Tolerability of EP0042
CTID: NCT04581512
Phase: Phase 1/Phase 2
Status: Recruiting
Date: 2025-10-14
SYN
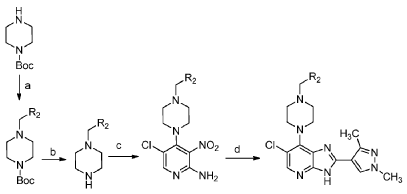
5-Chloro-4-(4-(4-chlorobenzyl)piperazin-1-yl)-3-nitropyridin-2-amine
[00119] To a mixture of 2-amino-4,5-dichloro-3-nitropyridine (0.152 g, 0.73 mmol) and isopropanol (22 mL) was added 1 -(4-chlorobenzyl)piperazine (0.165 g, 0.78 mmol) followed by diisopropylethylamine (0.17 mL, 0.97 mmol). The reaction mixture was heated at 45 °C for 18 h, then allowed to cool to room temperature, and diluted with isopropanol (5 mL). The precipitate was collected by filtration, washed with isopropanol and diethyl ether. The title compound was thus obtained as a yellow solid (0.215 g, 77%); 1H-NMR (500 MHz, DMSO-d6) 2.48 (br s, obscured by DMSO peak, 4H, piperazine C-H), 3.06 (br t, J = 4.3 Hz, 4H, piperazine C-H), 3.52 (s, 2H, NCH2C6H4Cl), 6.95 (s, 2H, NH2), 7.35 (d, J = 8.5 Hz, 2H) and 7.38 (d, J = 8.5 Hz, 2H) (3,5-ArH and 2,6- ArH), 8.06 (s, 1 H, 6-H); LC – MS (ESI, m/z): Rt = 1 .70 min – 382, 384, 386 [(M+H)+, Cl2 isotopic pattern].
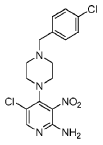
6-Chloro-7-(4-(4-chlorobenzyl)piperazin-1-yl)-2-(1,3-dimethyl-1H-pyrazol-4-yl)-3H-imidazo[4,5-b]pyridine
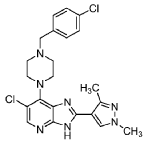
[00120] To a mixture of 5-chloro-4-(4-(4-chlorobenzyl)piperazin-1 -yl)-3-nitropyridin-2-amine (0.076 g, 0.20 mmol) and EtOH (4.0 ml.) was added 1 ,3-dimethyl-1 H-pyrazole-4-carbaldehyde (0.027 g, 0.22 mmol) followed by a freshly prepared aqueous solution of Na2S2O4 (1 M; 0.85 mL, 0.85 mmol). The reaction mixture was stirred at 80 °C for 24 h, it was then allowed to cool to room temperature, concentrated in vacuo, and the residue was absorbed on silica gel and placed on a 10 g isolute silica column. Elution with ethyl acetate / dichloromethane (v/v; 1 :1 ), and then 4% methanol in ethyl acetate / dichloromethane (v/v; 1 :1 ) afforded the title compound as a white solid after trituration with diethyl ether (0.023 g, 25%).
[00121 ] 1 H-NMR (500 MHz, DMSO-d6) 2.51 (s, obscured by solvent peak, pyrazole 3-CH3), 2.57 (br s, 4H, piperazine C-H), 3.54 (s, 2H, N-CH2C6H4Cl), 3.68 (br s, 4H, piperazine C-H), 3.84 (s, 3H, pyrazole N-Me), 7.37 (d, J = 8.5 Hz, 2H) and 7.40 (d, J = 8.5 Hz, 2H) (C6H4Cl), 8.02 (s, 1 H), and 8.18 (s, 1 H) (pyrazole 5-H, and imidazo[4,5-b]pyridine 5-H), 12.95 (br s, 1 H, imidazo[4,5-b]pyridine N-H); LC – MS (ESI, m/z): Rt = 1.97 min – 456, 458, 460 [(M+H)+, Cl2 isotopic pattern].
[00122] HRMS: Found: 456.1457, calculated for C22H24Cl2N7 (M+H)+: 456.1465.
LIT
- FLT3 Inhibitors in Acute Myeloid Leukemia: Challenges and Recent Developments in Overcoming ResistancePublication Name: Journal of Medicinal ChemistryPublication Date: 2021-03-10PMID: 33719439DOI: 10.1021/acs.jmedchem.0c01851
- Quizartinib-resistant FLT3-ITD acute myeloid leukemia cells are sensitive to the FLT3-Aurora kinase inhibitor CCT241736Publication Name: Blood AdvancesPublication Date: 2020-04-13PMCID: PMC7160287PMID: 32282883DOI: 10.1182/bloodadvances.2019000986
- Metabolism of the dual FLT-3/Aurora kinase inhibitor CCT241736 in preclinical and human in vitro models: Implication for the choice of toxicology speciesPublication Name: European journal of pharmaceutical sciences : official journal of the European Federation for Pharmaceutical SciencesPublication Date: 2019-11-01PMCID: PMC6892276PMID: 30953752DOI: 10.1016/j.ejps.2019.04.004
- Optimization of Imidazo[4,5-b]pyridine-Based Kinase Inhibitors: Identification of a Dual FLT3/Aurora Kinase Inhibitor as an Orally Bioavailable Preclinical Development Candidate for the Treatment of Acute Myeloid LeukemiaPublication Name: Journal of Medicinal ChemistryPublication Date: 2012-10-08PMCID: PMC3483018PMID: 23043539DOI: 10.1021/jm300952s
PAT
- Pharmaceutically active compoundsPublication Number: WO-2013190319-A1Priority Date: 2012-06-21
- Compound and, pharmaceutical compositionPublication Number: BR-112014032142-B1Priority Date: 2012-06-21
- Pharmaceutically active compoundsPublication Number: US-9447092-B2Priority Date: 2012-06-21Grant Date: 2016-09-20
- Pharmaceutically active compoundsPublication Number: CA-2876357-A1Priority Date: 2012-06-21
- Pharmaceutically active compoundsPublication Number: US-2015266868-A1Priority Date: 2012-06-21
- PHARMACEUTALLY ACTIVE COMPOUNDSPublication Number: RU-2015101702-APriority Date: 2012-06-21
- Pharmaceutically active compoundsPublication Number: SI-2864328-T1Priority Date: 2012-06-21
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
////////////////mobinitinib, antineoplastic, CCT241736, CCT 241736, ZE94SP78UG, EP0042, EP 0042
Mevrometostat
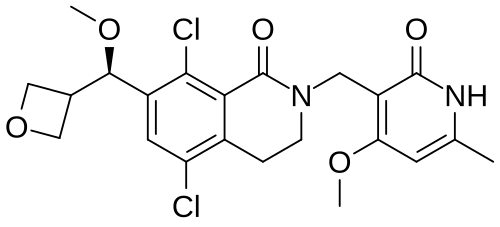
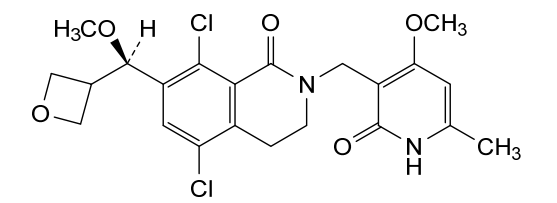
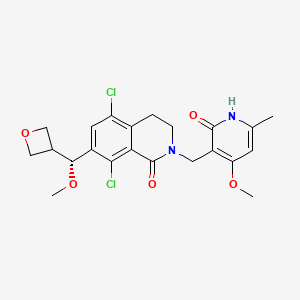
Mevrometostat
CAS 1844849-10-0
MF C22H24Cl2N2O5 MW467.3 g/mol
5,8-dichloro-2-[(4-methoxy-6-methyl-2-oxo-1H-pyridin-3-yl)methyl]-7-[(R)-methoxy(oxetan-3-yl)methyl]-3,4-dihydroisoquinolin-1-one
5,8-dichloro-7-[(R)-methoxy(oxetan-3-yl)methyl]-2-[(4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl]-3,4-
dihydroisoquinolin-1(2H)-one
enhancer of zeste homolog 2 (EZH2) inhibitor, antineoplastic, PF-06821497, PF 06821497, S4L4MM20B6
Mevrometostat (development code PF-06821497) is an investigational anticancer drug that functions as a potent and selective inhibitor of enhancer of zeste homolog 2 (EZH2).[1][2] Currently under development by Pfizer, mevrometostat is being investigated primarily for the treatment of metastatic castration-resistant prostate cancer (mCRPC) in combination with enzalutamide.
PF-06821497 is under investigation in clinical trial NCT03460977 (PF-06821497 Treatment Of Relapsed/Refractory SCLC, Castration Resistant Prostate Cancer, and Follicular Lymphoma).
Mevrometostat is an orally available selective inhibitor of the histone lysine methyltransferase (HMT) enhancer of zeste homolog 2 (EZH2), with potential antineoplastic activity. Upon oral administration, mevrometostat selectively targets, binds to and inhibits the activity of EZH2. Inhibition of EZH2 specifically prevents the methylation of histone H3 on lysine 27 (H3K27). This decrease in histone methylation alters gene expression patterns associated with cancer pathways and results in decreased proliferation of EZH2-expressing cancer cells. EZH2, an HMT class enzyme and the catalytic subunit of the polycomb repressive complex 2 (PRC2), is overexpressed or mutated in a variety of cancer cells and plays a key role in tumor cell proliferation; its expression is correlated with tumor initiation, progression, stem cell self-renewal, migration and angiogenesis.
MEVROMETOSTAT is a small molecule drug with a maximum clinical trial phase of II and has 1 investigational indication.
Synthesis
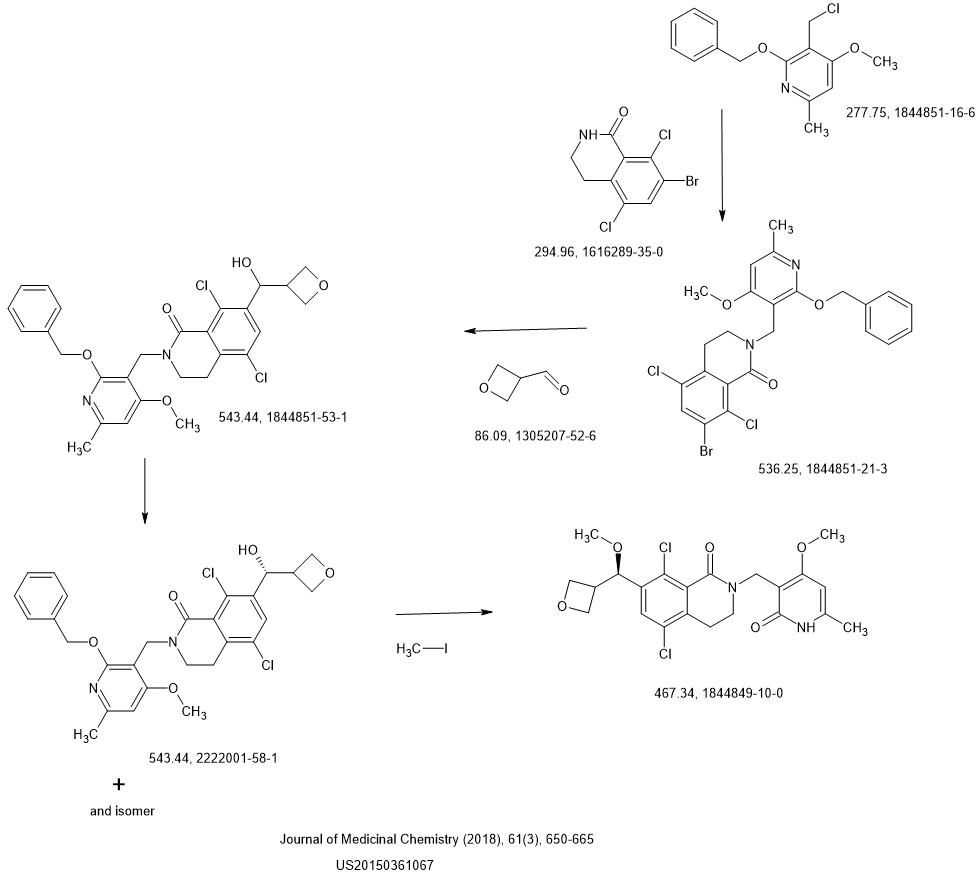
LAST STEP CONDITIONS
METHYL IODIDE REAGENT, Tetrahydrofuran , Potassium tert-butoxide
NEXT Hydrogen, Platinum dioxide,
SYN
Optimization of Orally Bioavailable Enhancer of Zeste Homolog 2 (EZH2) Inhibitors Using Ligand and Property-Based Design Strategies: Identification of Development Candidate (R)-5,8-Dichloro-7-(methoxy(oxetan-3-yl)methyl)-2-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (PF-06821497)Publication Name: Journal of Medicinal ChemistryPublication Date: 2017-12-27PMID: 29211475DOI: 10.1021/acs.jmedchem.7b01375
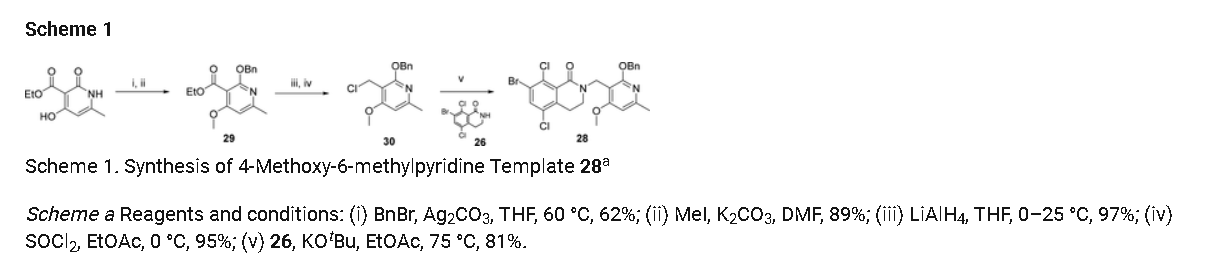
5,8-dichloro-2-[(4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl]-7-[(R)-
methoxy(oxetan-3-yl)methyl]-3,4-dihydroisoquinolin-1(2H)-one (23a) and 5,8-dichloro-2-[(4-
methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl]-7-[(S)-methoxy(oxetan-3-yl)methyl]-
3,4-dihydroisoquinolin-1(2H)-one (23b)
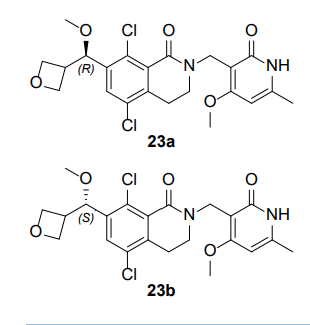
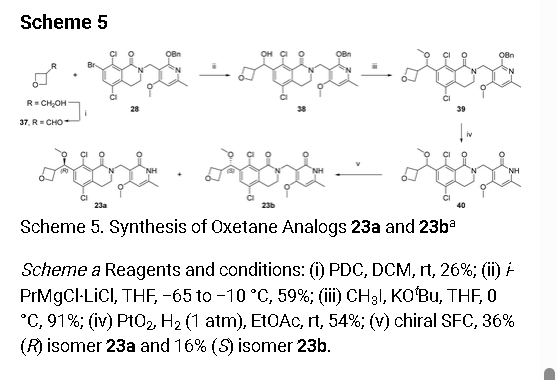
Multiple batches of (±)-5,8-dichloro-2-[(4-methoxy-6-
methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl]-7-[methoxy-
(oxetan-3-yl)methyl]-3,4-dihydroisoquinolin-1(2H)-one 40
were combined (140 mg total), and the enantiomers separated
by preparative chiral SFC [Column: (R,R)Whelk O1
250mm*30mm,5µ; mobile phase: EtOH; wavelength: 220
nm] to give, after lyophilization, 23a (50.3 mg, 36%) as a
white solid, and 23b (22.8 mg, 16%) as a white solid. A
small-molecule X-Ray crystal structure of 23a showed it to
have absolute (R) stereochemistry. A small-molecule X-Ray crystal structure of 23b confirmed
the expected absolute (S) stereochemistry.
5,8-dichloro-2-[(4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl]-7-[(R)-
methoxy(oxetan-3-yl)methyl]-3,4-dihydroisoquinolin-1(2H)-one (23a). After chiral SFC and
lyophilization, 23a (50.3 mg, 36%) was obtained as a white solid. LCMS m/z 489 [M+Na]+; 1H
NMR (400 MHz, CDCl3) δ 12.34 (br s, 1H), 7.49 (s, 1H), 5.93 (s, 1H), 5.05 (d, J=6.0 Hz, 1H),
4.78-4.61 (m, 6H), 3.88 (s, 3H), 3.50-3.48 (m, 2H), 3.38-3.37 (m, 1H), 3.31 (s, 3H), 2.94 (t,
J=6.2 Hz, 2H), 2.35 (s, 3H). [α]D
22 +67.7° (c 0.1, MeOH); Chiral analysis: 100% ee; retention
time 9.85 min; column (R,R)Whelk O1, 250×4.6mm I.D., 5µ; mobile phase 50% ethanol (0.05%
DEA) in CO2; wavelength 220 nm. A crystalline sample of 23a was obtained by dissolving the
lyophilized powder in hot isopropanol in a 1 dram vial, then letting the vial stand in a capped
TLC chamber containing a layer of hexanes in the bottom, which allowed slow diffusion of hexanes into isopropanol. After two days, crystals (square plates) were collected. A smallmolecule X-Ray crystal structure of 23a showed it to have absolute (R) stereochemistry.
Crystallographic data are available in the Supporting Information.
syn
Pfizer Inc.
United States, US20150361067
REF
- Computational exploration in search for novel natural product-derived EZH2 inhibitors for advancing anti-cancer therapyPublication Name: Molecular DiversityPublication Date: 2025-02-19PMID: 39969739DOI: 10.1007/s11030-025-11128-3
- TTD: Therapeutic Target Database describing target druggability informationPublication Name: Nucleic Acids ResearchPublication Date: 2023-09-15PMCID: PMC10767903PMID: 37713619DOI: 10.1093/nar/gkad751
- BLM overexpression as a predictive biomarker for CHK1 inhibitor response in PARP inhibitor–resistant BRCA -mutant ovarian cancerPublication Name: Science Translational MedicinePublication Date: 2023-06-21PMCID: PMC10758289PMID: 37343085DOI: 10.1126/scitranslmed.add7872
- Structural modification aimed for improving solubility of lead compounds in early phase drug discoveryPublication Name: Bioorganic & Medicinal ChemistryPublication Date: 2022-02-15PMID: 35033884DOI: 10.1016/j.bmc.2022.116614
- High-Throughput Screening to Identify Inhibitors of the Type I Interferon–Major Histocompatibility Complex Class I Pathway in Skeletal MusclePublication Name: ACS Chemical BiologyPublication Date: 2020-05-27PMCID: PMC7859889PMID: 32459468DOI: 10.1021/acschembio.0c00343
- Translational Pharmacokinetic-Pharmacodynamic Modeling for an Orally Available Novel Inhibitor of Epigenetic Regulator Enhancer of Zeste Homolog 2Publication Name: The Journal of Pharmacology and Experimental TherapeuticsPublication Date: 2020-05PMID: 32094296DOI: 10.1124/jpet.119.263491
- Optimization of Orally Bioavailable Enhancer of Zeste Homolog 2 (EZH2) Inhibitors Using Ligand and Property-Based Design Strategies: Identification of Development Candidate (R)-5,8-Dichloro-7-(methoxy(oxetan-3-yl)methyl)-2-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (PF-06821497)Publication Name: Journal of Medicinal ChemistryPublication Date: 2017-12-27PMID: 29211475DOI: 10.1021/acs.jmedchem.7b01375
- Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylationPublication Name: BloodPublication Date: 2011-02-24PMCID: PMC3062411PMID: 21190999DOI: 10.1182/blood-2010-11-321208
PAT
- Novel 3-amino-3,4,6-trideoxyglycals, their production methods and anthracyclines obtained from these glycalsPublication Number: JP-S6110572-APriority Date: 1984-06-15
- CARBON HYDROGENATION METHOD WITH INCREASED RETENTION OF SOLIDS IN FLUID BED REACTORSPublication Number: DE-3245494-A1Priority Date: 1981-12-21
- Group extraction of organic compounds present in liquid samplesPublication Number: US-3966410-APriority Date: 1972-07-24Grant Date: 1976-06-29
- PRMT5 inhibitors and uses thereofPublication Number: US-12448388-B2Grant Date: 2025-10-21
- KRAS G12D modulating compoundsPublication Number: US-12448400-B2Grant Date: 2025-10-21
- Fungicidal, acaricidal and insecticidal composition comprising imidazole derivatives as active ingredient and process for producing the active ingredientsPublication Number: HU-206245-BPriority Date: 1987-03-13
- Fluorine derivatives of vitamin d _and process for producing the samePublication Number: CA-1297869-CPriority Date: 1986-10-20Grant Date: 1992-03-24
- Fluorine derivatives of vitamin d3 and process for producing the samePublication Number: EP-0264880-B1Priority Date: 1986-10-20Grant Date: 1991-03-13
- Fluorine derivatives of vitamin D3 and process for producing the samePublication Number: EP-0264880-A1Priority Date: 1986-10-20
- Process for production of new derivatives of imidasolil-alkyl-guanidin and medical preparatives containing these substancesPublication Number: HU-198024-BPriority Date: 1985-04-02
WO-0003990-A1 WO-0059513-A1 WO-0127114-A1 WO-02100354-A2 WO-2004029040-A1 WO-2004033420-A1 WO-2004078712-A2 WO-2004092118-A2 WO-2005000869-A1 WO-2005040109-A1 WO-2005082368-A1 WO-2006083424-A2 WO-2007022638-A1 WO-2007030089-A1 WO-2007058503-A1 WO-2008116833-A1 WO-2009005690-A2 WO-2009142969-A1 WO-2010015656-A2 WO-2010123599-A9 WO-2011102800-A1 WO-2011115818-A1 WO-2011139702-A2 WO-2012033149-A1 WO-2012037372-A2 WO-2012069856-A1 WO-2012097013-A1 WO-2012112969-A1 WO-2012129673-A1 WO-2012130912-A1 WO-2013096679-A1 WO-2013096680-A1 WO-2013102242-A1 WO-2013142525-A1 WO-2013177983-A1 WO-2014037498-A2 WO-2014057415-A2 WO-2015193765-A1 WO-2020190754-A1 WO-2021163064-A9 WO-2022053990-A1 WO-2022058405-A2 WO-2022087149-A2 WO-2022087230-A1 WO-2022164789-A1 WO-2022164789-A9 WO-2022212746-A1 WO-2022212748-A1 WO-2022221304-A1 WO-2022232435-A1 WO-2022261243-A1 WO-2022271650-A1 WO-2022271659-A1 WO-2022271677-A1 WO-2022271684-A1 WO-2023015164-A1 WO-2023041674-A1 WO-2023055885-A2 WO-2023061446-A1 WO-2023076554-A9 WO-2023076983-A1 WO-2023077030-A1 WO-2023100131-A1 WO-2023108563-A1 WO-2023111810-A1 WO-2023114460-A1 WO-2023122581-A2 WO-2023122615-A1 WO-2023147418-A1 WO-2023166418-A2 WO-2023166420-A1 WO-2023178181-A1 WO-2023178354-A1 WO-2023183817-A1 WO-2023196784-A1 WO-2023205719-A1 WO-2023205850-A1 WO-2023212504-A1 WO-2023218320-A1 WO-2023242769-A1 WO-2023249714-A1 WO-2024003773-A1 WO-2024006929-A1 WO-2024009191-A1 WO-2024015566-A1 WO-2024019995-A1 WO-2024020534-A2 WO-2024033513-A1 WO-2024035688-A1 WO-2024064668-A1 WO-2024074977-A1 WO-2024086094-A1 WO-2024105563-A1 WO-2024187153-A1 WO-2024189607-A1 WO-2024192373-A1 WO-2024193570-A1 WO-2024193570-A9 WO-2024209339-A1 WO-2024213979-A1 WO-2024215754-A1 WO-2024218686-A1 WO-2024220917-A1 WO-2024246965-A1 WO-2025006704-A1 WO-2025006720-A1 WO-2025024663-A1 WO-2025024811-A1 WO-2025031307-A1 WO-2025038726-A2 WO-2025049966-A2 WO-2025049966-A9 WO-2025054347-A1 WO-2025054530-A1 WO-2025059027-A1 WO-2025085536-A1 WO-2025094035-A1 WO-2025096589-A1 WO-2025121805-A1 WO-2025121807-A1 WO-2025137640-A1 WO-2025151706-A1 WO-2025245003-A1 WO-9012007-A1 WO-9522521-A1 WO-9954305-A1
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
Mechanism of action
Mevrometostat is a small molecule inhibitor that targets EZH2, the catalytic subunit of polycomb repressive complex 2 (PRC2).[1][3] EZH2 plays a crucial role in epigenetic regulation by modifying gene expression patterns that control cellular fate decisions, including differentiation and self-renewal.[1]
In prostate cancer, EZH2 dysregulation contributes to treatment resistance through multiple pathways, including:
- Silencing of tumor suppressor genes
- Activation of androgen receptor transcription factors
- Promotion of neuroendocrine transdifferentiation[4]
Mevrometostat demonstrates dose-dependent EZH2 inhibition, leading to reactivation of tumor suppressor genes while suppressing genes involved in tumor progression.[5]
Clinical development
Phase I/II trials
The primary clinical evaluation of mevrometostat is being conducted through a phase 1/2 dose-expansion study (NCT03460977) investigating the combination of mevrometostat with enzalutamide and androgen deprivation therapy in patients with mCRPC.[6]
The dose-expansion portion of this study enrolled patients with mCRPC who had previously received abiraterone, with evidence of disease progression per modified Prostate Cancer Working Group 3 criteria.[2]
Key efficacy results
In the randomized dose-expansion study, the combination of mevrometostat (1,250 mg twice daily on an empty stomach) plus enzalutamide demonstrated:
- 49% relative reduction in the rate of progression or death
- Approximately 8-month improvement in median radiographic progression-free survival (rPFS)
- Hazard ratio of 0.51 (90% CI: 0.28–0.95)[7]
The median radiographic progression-free survival was 14.3 months with the combination therapy compared to 6.2 months with enzalutamide alone.[8]
Phase III trials
Based on promising phase I/II results, Pfizer has initiated multiple phase 3 clinical trials:
MEVPRO-1 study
The MEVPRO-1 study (NCT06551324) is a randomized phase 3 trial evaluating mevrometostat in combination with enzalutamide versus physician’s choice of therapy in patients with mCRPC previously treated with abiraterone acetate.[9][10]
- Study design: Randomized 1:1 to receive mevrometostat (875 mg twice daily with food) plus enzalutamide (160 mg daily) versus physician’s choice of enzalutamide or docetaxel
- Target enrollment: Approximately 600 patients
- Primary endpoint: Blinded independent central review-assessed rPFS per RECIST 1.1 and PCWG3 criteria
- Key secondary endpoint: Overall survival
MEVPRO-2 study
The MEVPRO-2 study (NCT06629779) is evaluating mevrometostat plus enzalutamide in androgen receptor pathway inhibitor (ARPI)-naïve patients with mCRPC.[11][12]
Additional development
Pfizer has also initiated phase 3 trials evaluating mevrometostat plus enzalutamide in first-line metastatic castration-sensitive prostate cancer.[8][13]
Safety profile
The most common adverse events considered related to mevrometostat treatment include:
Dose optimization studies found that mevrometostat 875 mg twice daily with food showed similar efficacy and better safety compared to the 1,250 mg dose on an empty stomach.[15]
Pharmacokinetics
Based on safety and pharmacokinetic findings from phase 1 trials, mevrometostat 875 mg twice daily with food was selected as the recommended dose for phase 3 clinical development in combination with enzalutamide.[16]
Regulatory status
As of 2025, mevrometostat remains an investigational agent under clinical development by Pfizer. The drug has not received regulatory approval from the Food and Drug Administration (FDA), European Medicines Agency (EMA), or other regulatory authorities.
See also
- Polycomb repressive complex 2
- Tazemetostat (approved EZH2 inhibitor)
- Prostate cancer
- Enzalutamide
References
- “Mevrometostat (PF-06821497)”. Pfizer Oncology Development. Retrieved 11 September 2025.
- Schweizer MT, Calvo M, Moreno V, Mellado B, Castellano D, Spira AI, et al. (2025). “Mevrometostat (PF-06821497), an enhancer of zeste homolog 2 (EZH2) inhibitor, in combination with enzalutamide in patients with metastatic castration-resistant prostate cancer (mCRPC): A randomized dose-expansion study”. Journal of Clinical Oncology. 43 (5_suppl) LBA138. doi:10.1200/JCO.2025.43.5_suppl.LBA138.
- Schweizer MT, Penkov K, Choudhury AD, Calvo E, Frank RC, Liu L, et al. (2024). “Phase 1 trial of mevrometostat (PF-06821497), a potent and selective inhibitor of enhancer of zeste homolog 2 (EZH2), in castration-resistant prostate cancer (CRPC)”. Journal of Clinical Oncology. 42 (16_suppl): 5061. doi:10.1200/JCO.2024.42.16_suppl.5061.
- “SUO 2024: Mevrometostat (PF-06821497) in Combination with Enzalutamide in Patients with Metastatic Castration-Resistant Prostate Cancer Previously Treated with Abiraterone Acetate”. UroToday. Retrieved 11 September 2025.
- “Mevrometostat and enzalutamide in mCRPC: gene expression and EZH2 modulation”. VJ Oncology. 17 February 2025. Retrieved 11 September 2025.
- Pfizer (4 September 2025). A PHASE I DOSE ESCALATION AND EXPANDED COHORT STUDY OF PF 06821497 (MEVROMETOSTAT) IN THE TREATMENT OF ADULT PATIENTS WITH RELAPSED/REFRACTORY SMALL CELL LUNG CANCER (SCLC), CASTRATION RESISTANT PROSTATE CANCER (CRPC) AND FOLLICULAR LYMPHOMA (FL) (Report). clinicaltrials.gov.
- “ASCO GU 2025: Mevrometostat (PF-06821497), an EZH2 Inhibitor, in Combination with Enzalutamide in Patients with mCRPC”. UroToday. Retrieved 11 September 2025.
- “Mevrometostat/enzalutamide combo shows rPFS benefit in mCRPC”. Urology Times. 21 February 2025. Retrieved 11 September 2025.
- Agarwal N, Schweizer MT, Castro E, Azad A, George DJ, Chakrabarti J, et al. (2025). “Mevrometostat (PF-06821497) in combination with enzalutamide in patients with metastatic castration-resistant prostate cancer previously treated with abiraterone acetate: The phase 3, randomized MEVPRO-1 study”. Journal of Clinical Oncology. 43 (5_suppl) TPS288. doi:10.1200/JCO.2025.43.5_suppl.TPS288.
- Pfizer (4 September 2025). A PHASE 3, RANDOMIZED, OPEN-LABEL STUDY OF PF-06821497 (MEVROMETOSTAT) IN COMBINATION WITH ENZALUTAMIDE COMPARED WITH ENZALUTAMIDE OR DOCETAXEL IN PARTICIPANTS WITH METASTATIC CASTRATION RESISTANT PROSTATE CANCER PREVIOUSLY TREATED WITH ABIRATERONE ACETATE (MEVPRO-1) (Report). clinicaltrials.gov.
- “ASCO GU 2025: Mevrometostat (PF-06821497) in Combination With Enzalutamide for ARPI-Naïve Patients With mCRPC: The Phase 3, Randomized MEVPRO-2 Trial”. UroToday. Retrieved 11 September 2025.
- Pfizer (4 September 2025). A PHASE 3, RANDOMIZED, DOUBLE BLIND, PLACEBO CONTROLLED STUDY OF PF-06821497 (MEVROMETOSTAT) WITH ENZALUTAMIDE IN METASTATIC CASTRATION RESISTANT PROSTATE CANCER (MEVPRO-2) (Report). clinicaltrials.gov.
- Pfizer (4 September 2025). A Phase 3, Randomized, Double-Blind, Placebo-Controlled Study of Mevrometostat (PF-06821497) With Enzalutamide in Metastatic Castration-Sensitive Prostate Cancer (MEVPRO-3) (Report). clinicaltrials.gov.
- “ASCO 2025: Mevrometostat in Combination with Enzalutamide in Patients with mCRPC Previously Treated with Abiraterone Acetate”. UroToday. Retrieved 11 September 2025.
- “Mevrometostat Plus Enzalutamide Improves rPFS vs Enzalutamide in Metastatic CRPC”. OncLive. 21 February 2025. Retrieved 11 September 2025.
- “ASCO 2025: Safety and Pharmacokinetics of Mevrometostat in Combination with Enzalutamide in Patients with mCRPC”. UroToday. Retrieved 11 September 2025.
| Clinical data | |
|---|---|
| Other names | PF-06821497 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1844849-10-0 |
| PubChem CID | 118572065 |
| IUPHAR/BPS | 10516 |
| DrugBank | DB14799 |
| ChemSpider | 65321668 |
| UNII | S4L4MM20B6 |
| KEGG | D12845 |
| ChEMBL | ChEMBL4080228 |
| PDB ligand | CJD (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C22H24Cl2N2O5 |
| Molar mass | 467.34 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////mevrometostat, enhancer of zeste homolog 2 (EZH2) inhibitor, antineoplastic, PF-06821497, PF 06821497, S4L4MM20B6
#mevrometostat, #enhancer of zeste homolog 2 (EZH2) inhibitor, #antineoplastic, #PF-06821497, #PF 06821497, #S4L4MM20B6

{kind=link}
{kind=link}
{kind=link}