
Home » Posts tagged 'Advinus'
Tag Archives: Advinus
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
PNQ 103 from Advinus for the potential treatment of COPD,; sickle cell disease (SCD)

Formula I and Formula II
OR

PNQ 103
STRUCTURE COMING…………
for the potential treatment of COPD & sickle cell disease (SCD)
Adenosine A2b receptor antagonist
Advinus Therapeutics Ltd
KEEP WATCHING THIS POST……….
PNQ-103 is a proprietary A2B Adenosine receptor (A2BAdoR antagonist), currently in the pre-clinical development stage for the potential treatment of COPD & sickle cell disease (SCD). Advinus is looking for partnering/co-development opportunities.
A2BAdenosine Receptor (A2BAdoR) Antagonist PNQ-103 for COPD and SCD
COPD
Chronic Obstructive Pulmonary Disease (COPD) is a disease that damages lung tissue or restricts airflow through the bronchioles and bronchi, and commonly leads to chronic bronchitis and emphysema. COPD, along with asthma, forms the third leading cause of death in both developed and developing countries and an annual direct and indirect cost of healthcare of more than $50 billion in the US alone. Current therapies suffer from lack of long term efficacy, patient compliance and a narrow therapeutic index.
Adenosine is a powerful bronchoconstrictor and pro-inflammatory agent in COPD and asthma. Adenosine regulates tissue function by activating its receptors: A1AdoR and A2AAdoR are high affinity receptors and A2BAdoR and A3AdoR are low affinity receptors. During pathological conditions in lung, local adenosine concentrations rise to high levels and activate A2BAdoR. A2BAdoR agonized by adenosine induces both bronchoconstriction and pro-inflammatory effects in lung by acting on multiple cell types that lead to airway hyperreactivity and chronic inflammation. Therefore, A2BAdoR antagonists are expected to be beneficial in COPD and asthma.
PNQ-103 is a proprietary A2BAdoR antagonist, currently in the pre-clinical development stage for the potential treatment of COPD. It is a potent, selective, orally bio-available agent with low clearance and small volume of distribution. PNQ-103 is efficacious in standard rodent asthma and lung fibrosis models. PNQ-103 was found to be safe in exploratory safety studies including a Drug Matrix Screen, mini-AMES test, and a test for cardiovascular liability in dog telemetry as well as a 30- day repeat dose study in rats.
SCD
Sickle Cell Disease (SCD) affects millions of people worldwide. It is caused by an autosomal mutation in the hemoglobin gene (substitution of amino-acid valine [Hb A] for glutamic acid [Hb S]. Hb S in low O2 condition polymerizes, leading to distortion of the cell membrane of red blood cells (RBC) into an elongated sickle shape. Sickled RBCs accumulate in capillaries causing occlusions, impair circulation and cause tissue damage and severe disabilities. Unfortunately, there is no targeted therapy for SCD.
Adenosine levels are elevated in SCD patients. Activation of the A2BAdoR by adenosine increases 2,3-DPG levels in RBCs, which reduces Hb S affinity to O2 and promotes its polymerization leading to RBC sickling. A recent study published in Nature Medicine (2011; 17:79-86) demonstrated potential utility of an A2BAdoR antagonist for the treatment of SCD, through selective inhibition of 2,3-DPG production in RBCs. Therefore, PNQ-103, a selective A2BAdoR antagonist, is expected to be useful for the treatment of SCD. In support, ex vivo PoC (selective inhibition of 2,3-DPG production) has been established for PNQ-103 in RBCs from normal and SCD patients.
EXAMPLES………
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012035548
Example 1: Phosphoric acid mono-{2-cyano-6-oxo-l-propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyraz -4-yl]-l,6-dihydr»-purin-7-ylmethyl} ester

Step I: Synthesis of l-(3-Trifiuoroirethyl-ben:ijl)-lH-pyrazole-4-carboxylic acid (6-amino-2,4-dioxo-3-propyl-l,2,354-tetrahydro-pyrimidin-5-yl)-amide
A mixture of 5,6-diamino-3-propyl-l H-pyrimidine-2,4-dione (4.25 g, 0.023 mol), l-(3-Trifluoromethyl-benzyl)-lH-pyrazole-4-carboxylic acid (6.23 g, 0.023 mol), prepared by conventional methods starting from pyrazole-4-carboxylic ester, in methanol (50 ml) were cooled to 0 °C and added EDCI.HC1 (8.82 g, 0.046 mol). The reaction mixture was stirred at 25 °C for 6 h and the organic volatiles were evaporated. To this residue water (50 ml) was added and the precipitate was filtered off, and washed with cold water (50 ml) to obtain l-(3-Trifluoromethyl-benzyl)- 1 H-pyrazole-4-carboxylic acid (6-amino-2,4-dioxo-3-propyl-l,2,3,4-tetrahydro-pyrimidin-5-yl)-amide (7.2 g, 72 %) as a pale yellow solid.
‘HNMR(400MHz, DMSO d6): δ 0.82 (t, J=7.6Hz, 3H); 1.46-1.51 (m, 2H); 3.64 (t, J=7.2Hz, 2H); 5.49 (s, 2H); 6.01 (s, 2H); 7.55-7.63 (m, 2H); 7.68-7.72 (m, 2H); 7.99 (s, 1H); 8.37 (s, 1H); 8.55 (s, 1H); 10.42 (s, 1H).
Step II: Preparation of l-Propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyrazoI-4-yl]-3,7-dihydro-purine-2,6-dione
A mixture of l-(3-Trifluoromethyl-benzyl)-lH-pyrazole-4-carboxylic acid (6-amino-2,4-dioxo-3-propyl-l,2,3,4-tetrahydro-pyrimidin-5-yl)-amide (30 g, 0.068 mol), P205(34.0g, 0.240.8 mol) and DMF (300ml) were heated at 100 °C for 30 minutes. The reaction mixture was cooled to 20-25 °C. The reaction mixture was slowly poured into water (1.5 L) with vigorous stirring. Solid material separated was filtered off, and washed with water (200ml) to obtain 1 -Propyl-8-[l -(3-trifluoromethyl-benzyl)-l H-pyrazol-4-yl]-3,7-dihydro-purine-2,6-dione (25 g, 88 %) as a pale yellow solid.
‘HNMR(400MHz, DMSO d6): δ 0.87 (t, J=7.2Hz, 3H); 1.53-1.60 (m, 2H); 3.98 (t, J=7.2Hz, 2H); 5.53 (s, 2H); 7.57-7.64 (m, 2H); 7.69-7.71 (m, 2H); 8.08 (s, 1H); 8.47 (s, 1H); 1 1.83 (s, 1H); 13.39 (s, 1H)
Step III: Preparation of 2-ChIoro-l-propyI-8-[l-(3-trifluoromethyI-benzyl)-lH-pyrazol-4-yl]-l,7-dihydro-purin-6-one
A mixture of l-Propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyrazol-4-yl]-3,7-dihydro-purine-2,6-dione (7.2 g, 0.017 mol), NH4C1 (4.54 g, 0.085 mol) and POCl3 (220 ml) were heated at 120-125 °C for 72 h. Reaction mixture was cooled to 20-25 °C. It was then concentrated under vacuum and quenched with cold water slowly and solid material was separated. It was filtered off and washed with water. The solid material was dried under vacuum. The crude product was purified by column chromatography using silica gel (230-400 mesh) and 0.5 to 4 % methanol in chloroform as an eluent to obtain 2-Chloro-l-propyl-8-[l-(3-trifluoromethyl-benzyl)- lH-pyrazol-4-yl]-l,7-dihydro-purin-6-one (4.2 g, 58 %) as a pale yellow solid.
‘HNMR(400MHz, CD3OD): 6 1.02 (t, J=7.2Hz, 3H); 1.78-1.84 (m, 2H); 4.29 (t, J=7.6Hz,
2H); 5.52 (s, 2H); 7.56-7.57 (m, 2H); 7.63 (m, 2H); 8.12 (s, 1H); 8.35 (s, 1 H)
Step IV: Preparation of 6-Oxo-l-propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyrazol-4-yl]-6,7-dihydro-lH-purine-2-carbonitrile
A mixture of 2-Chloro-l-propyl-8-[l-(3-trifluoromethyl-benzyl)-l H-pyrazol-4-yl]-l ,7-dihydro-purin-6-one (O. lg, 0.23 mmol), NaCN (0.016 g, 0.35 mmol), Nal (0.069g, 0.46 mmol) and DMF (2 ml) were stirred for 48 h at 65-70 °C. Reaction mixture was cooled to 20-25 °C and water was added. Solid material was separated. It was filtered off and washed with water. The product was dried under vacuum to obtain 6-Oxo-l-propyl-8-[l-(3-
trifluoromethyl-benzyl)-lH-pyrazol-4-yl]-6,7-dihydro-lH-puriiAe-2-carbonitrile (0.075 g, 77 %) as an off white solid.
‘HNMR(400MHz, DMSO d6): δ 0.97 (t, J=7.6Hz, 3H); 1.71-1.77 (m, 2H); 4.12 (t, J=7.6Hz, 2H); 5.51 (s, 2H); 7.57-7.67 (m, 4H); 8.14 (s, 1H); 8.55 (s, 1H); 14.01 (bs, 1H)
Preparation of hosphoric acid di-tert-butyl ester chloromethyl ester:

Step I: Phosphoric acid di-tert-butyl ester
A mixture of di-tert-butylphosphite (5 g, 0.026 mol), NaHC03 (3.71 g, 0.044 mol) and water (50 ml) were taken and cooled to 0-(-5 , °C. KMn04 (6.18 g, 0.039 mol) was added to the reaction mixture in portion wise over ¾ period of 30 minutes at that temperature. The reaction mixture was allowed to warm to 20-25 °C ana stirred for 1.5 hours at that temperature. To this reaction mixture activated charcoal (25 g) was added and stirred at 55-60 °C for 1 hour. The reaction mixture was cooled to room temperature and filtered off and washed with water (200 ml). The filtrate was concentrated to half of its volume and cooled to 0 °C. It was then acidified with con. HC1 (pH~l-2) to obtain solid. The solid material was filtered off, washed with ice cold water and dried under vacuum to obtain Phosphoric acid di-tert-butyl ester as white solid (3.44 g, 63 %).
Step II. Phosphoric acid di-tert-butyl ester chloromethyl ester
A mixture of Phosphoric acid di-tert-butyl ester (1 g, 0.0048 mol), NaHC03 (0.806 g, 0.0096 mol), tetra butyl ammonium hydrogen sulphate (0.163 g, 0.00048 mol), water (40 ml) and DCM (25 ml) were taken. The mixture was cooled to 0 °C and stirred at that temperature for 20 minutes. Chloromethyl chlorosulphatc (0.943g, 0.0057 mol) in DCM (15 ml) was added to it at 0 °C. The reaction mixture allc ed to warm to room temperature and stirred for 18 hours. The organic layer was separated and aqueous layer was extracted with DCM (30 ml). The organic layer was washed with brine (60 ml) solution and dried over Na2SC>4. The organic layer was evaporated to obtain Phosphoric acid di-tert-butyl ester chloromethyl ester as colorless oil (0.79 g, 64%).

Step I: Phosphoric acid di-tert-butyl ester 2-cyano-6-oxo-l-propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyrazol-4-yl]-l,6-dihydro-purin-7-ylmethyl ester
A mixture of 6-Oxo-l-propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyrazol-4-yl]-6,7-dihydro-lH-purine-2-carbonitrile (0.5 g, 0.0012mol), K2C03 (0.485 g, 0.0036 mol ) and acetone ( 10 ml) were taken and stirred for 20 minutes at room temperature. Nal (0.702 g, 0.0047 mol) was added and then Phosphoric acid di-ten-butyl ester chloromethyl ester (0.619 g, 0.0024 mol in 2 ml acetone) was added to the reaction mixture drop wise. The reaction mixture was heated at 45 °C for 16 h. The reaction mixture was filtered through celite and washed with acetone. The organic layer was concentrated and the residue was taken in ethyl acetate (30 ml) and saturated NaHC03 solution (20 ml). The organic layer was separated and washed with saturated sodium thiosulphate solution (20 ml). The organic layer was washed with 0.5 N HC1 solution (20 ml) and brine solution (20 ml). The organic layer was dried over sodium sulphate and evaporated to obtain brown colored mass. The crude product, which is a mixture of N7 and N9 isomers was purified by column chromatography (230-400 mesh silica gel and it was first treated with 5% triethyl amine in hexane) using 5-20 % acetone in hexane (with 0.5 to 1% triethyl amine) as an eluent to obtain N7 isomer (0.34g, 45 % ) and N9 isomer ( 0.1 lg, 14 % )
Phosphoric acid di-tert-butyl ester 2-cyano-6-oxo-l-propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyrazol-4-yl]-l,6-dihydro-purin-7-ylmethyl ester (N7-isomer).
Ή NMR (400MHz, DMSO d6):6 0.95 (t J=8Hz, 3H); 125 (s, 18 H); 1.75-1.80 (m, 2H); 4.18 (t, J=7.2Hz, 2H); 5.58 (s, 2H); 6.34 (d, ![]()
2H); 7.61-7.63 (m, 2H); 7.70-7.73 (m, 2H); 8.19 (s, 1H); 8.75 (s, 1H)
Phosphoric acid di-tert-butyl ester 2-cyano-8-[l-(3-trifluoromethyI-benzyl)-lH-pyrazol-4-yl]-6-oxo-l-propyl-l,6-dihydro-purin-9-ylmethyl ester (N9-isomer)
Ή NMR (400MHz, DMSO d6): δ 0.94 (t, J=8Hz, 3H); 125 (s, 18 H); 1.74-1.78 (m, 2H); 4.21 (t, J=7.2Hz, 2H); 5.59 (s, 2H); 6.05 (d, J=10.8Hz, 2H); 7.62-7.63 (m, 2H); 7.69-7.71 (m, 2H); 8.16 (s, 1H); 8.71 (s, 1H)
Step II: Phosphoric acid mono-{2-cyano-6-oxo-l-propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyrazol-4-yl]-l,6-dihydro-purin-7-ylmethyl} ester (N7-isomer).
The above product, N7 isomer (0.34 g, 0.52 mmol) was dissolved in DCM (20 ml) and TFA (0.29 ml, 4.2 mmol) was added to it. The reaction mixture was stirred at room temperature for 7 hours. The organic volatiles were evaporated and the residue was stirred with pentane: diethyl ether (3:1, 10 ml) and the solid material obtained was filtered off and washed with 10 % diethyl ether in pentane (10 ml) to obtain Phosphoric acid mono- {2-cyano-6-oxo-l -propyls’ [ 1 -(3 -trifluoromethyl-benzyl)- 1 H-pyrazol-4-yl]- 1 ,6-dihydro-purin-7-ylmethyl } ester (0.239g, 85 %) as an off white solid.
(400MHz, DMSO d6): δ 0.96 (t, J=7.6Hz, 3H); 1.75-1.81 (m, 2H); 4.16 (t, J=7.2Hz, 2H); 5.58 (s, 2H); 6.23 (d, J=6Hz, 2H); 7.61-7.63 (m, 2H); 7.69-7.75 (m, 2H); 8.22 (s, 1 H); 8.80 (s, 1H); (M+1): 538.2
Phosphoric acid mono-{2-cyano-6-oxo-l-propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyrazol-4-yl]-l,6-dihydro-purin-9-ylmethyl} ester (N9-isomer, 28%)
(400MHz, DMSO d6): δ 0.93 (t, J=7.6Hz, 3H); 1.72-1.80 (m, 2H); 4.16 (t, J=7.2Hz, 2H); 5.54 (s, 2H); 5.95 (d, J=6Hz, 2H); 7.59-7.60 (m, 2H); 7.67-7.73 (m, 2H); 8.17 (s, 1H); 8.72 (s, 1H).
Step III: Phosphoric acid mon -{2-cyano-6-oxo-l-propyl-8-[l-(3-trifluoromethyl-benzyI)-lH-pyrazol-4-yl]-l,6-dihydro-purin-7-yimethyl} ester di sodium salt
The above product (0.239g, 0.44 mmol) and water (25 ml) were taken. To the suspension formed, NaHC03 solution (0.1 12g, 1.3 mmol in 20 ml water) was added. The reaction mixture was stirred at room temperature for 1.5 h and the solid material obtained was filtered off. The clear solution was passed through reverse phase column chromatography (LCMS). The fraction obtained was evaporated. It was lyophilized to obtain pure Phosphoric acid mono-{2-cyano-6-oxo- 1 -propyl-8-[ 1 -(3 -trifluoromethyl-benzyl)- 1 H-pyrazol-4-yl]- 1 ,6-dihydro-purin-7-ylmethyl} ester di sodium salt (0.208g; 80%) as an off white solid.
Ή NMR: (400MHz, D20): δ 0.97 (t, J=7.6Hz, 3H); 1.80-1.86 (m, 2H); 4.28 (t, J=7.6Hz, 2H); 5.53 (s, 2H); 6.04 (d, J=3.2Hz, 2H); 7.52-7.53 (m, 2H); 7.62-7.64 (m, 2H); 8.22 (s, 1H); 8.74 (s, 1H)
31P NMR: (400MHz, D20): δ 0.447
EXAMPLES…………..
Patent
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2009118759
Example Al: 1, 3-Dipropyl-8-[l-(3-p-tolyl-prop-2ynyl)-lH-pyrazol-4-yI]-3, 7-dihydro-purine-2, 6-dione

Step I: l-(3-p-ToIyl-prop-2-ynyl)-lH-pyrazole-4-carboxylic acid ethyl ester
A mixture of l-prop-2-ynyl-lH-pyrazole-4-carboxylic acid ethyl ester obtained as given in example Bl (0.20Og, l.lmmol), 4-iodo toluene (0.254g, 1.1 mol), copper iodide (0.021g, O.l lmmol), dichlorobis (triphenylphosphine)-palladium (II) (39mg, O.Oόmmol), triethylamine (2ml), DMF (2ml) was degassed for lOmin. and stirred for 20hrs at 25-25 0C. Reaction mixture was diluted with water (10ml) and extracted with
• ethyl acetate. Organic layer was washed with brine solution and dried over Na2SO4.
The solvent was evaporated and crude product was purified by column chromatography
(Ethyl acetate: hexane-12:78) to obtain pure l-(3-p-tolyl-prop-2-ynyl)-lH-pyrazole-4- carboxylic acid ethyl ester compound (0.226g, 75%). 1HNMR^OOMHZ, CDCl3): δ 1.35 (t, J=6.8Hz, 3H); 2.37 (s, 3H); 4.31 (q, J=6.8Hz, 2H); 5.18 (s, 2H); 7.16 (d, J=7.6Hz, 2H); 7.38 (d, J=8Hz, 2H); 7.95 (s, IH); 8.21 (s, IH)
Step II: l-(3-p-Tolyl-prop-2-ynyl)-lH-pyrazole-4-carboxy!ic acid l-(3-p-Tolyl-prop-2-ynyl)-lH-pyrazole-4-carboxylic acid ethyl ester (0.226g, 0.84 mmol) was dissolved in a mixture of solvents THF: methanol: water (3:1:1, 10ml) and LiOH (0.07 Ig, 1.7mol) was added to the reaction mixture with stirring. The reaction mixture was then stirred at 20-25 0C for 2 hours. Solvents were evaporated and the residue was diluted with water (0.5 ml) and acidified with dil. HCl, filtered and dried to obtain off white precipitate, l-(3-p-Tolyl-prop-2-ynyl)-lH-pyrazole-4-carboxylic acid (0.182g, 90%).
1HNMR^OOMHZ, CDCl3): δ 2.37 (s, 3H); 5.2 (s, 2H); 7.16 (d, J=7.6Hz, 2H); 7.38 (d, J=8Hz, 2H); 8.01 (s, IH); 8.29 (s, IH) Step III: 1, 3-Dipropyl-8-[l-(3-p-tolyl-prop-2ynyl)-lH-pyrazol-4-yI]-3, 7-dihydro-‘ purine-2, 6-dione
A mixture of 5,6-diamino-l,3-dipropyl-lH-pyrimidine-2,4-dione (0.075g, 0.33 mmol), l-(3-p-tolyl-prop-2-ynyl)-lH-pyrazole-4-carboxylic acid (0.080gm, 0.33mmol), methanol (5ml), EDCI (0.089g, 0.46mmol) were taken and stirred for 12 hours at 20-25 0C. The reaction mixture was concentrated to obtain intermediate l-(3-p-tolyl-prop-2-ynyl)-lH-pyrazole-4-carboxylic acid (6-amino-2, 4-dioxo-l, 3-dipropyl)-l, 2, 3, 4-tetrahydro-pyrimidine-5yl) amide (50mg, 34%) which was dissolved in hexamethyldisilazane (HMDS). To this reaction mixture ammonium sulphate (0.01 Og) was added. The reaction mixture was refluxed at 140 0C for 18hrs. The organic volatiles were evaporated and the residue was treated with crushed ice, the precipitate formed was filtered off. The product was then purified by column chromatography (l%MeOH in CHCl3) to obtain 1, 3-dipropyl-8~[l-(3-p-tolyl-prop-2ynyl)-lH-pyrazol-4-yl]-3, 7-dihydro-purine-2, 6-dione (0.035g, 92%). ‘HNMR(400MHz, DMSO d6): δ 0.76-0.87 (m, 6H); 1.51-1.57 (m, 2H); 1.68-1.74 (m, 2H); 2.29 (s, 3H); 3.82 (t, J=7.2Hz, 2H); 3.95 (t, J=7.2Hz, 2H); 5.36 (s, 2H); 7.18 (d, J=8Hz, 2H); 7.35 (d. J=8Hz, 2H); 8.08 (s, IH); 8.49 (s, IH); 13.9 (bs,lH)
Happy new year wishes 2016


/////////
PNQ 201 from Advinus for for potential treatment of IBD.
formula I

PNQ 201
STRUCTURE COMING……
Adenosine A2b receptor antagonist
Advinus Therapeutics Ltd
KEEP WATCHING THIS POST……………
PNQ-201 is a proprietary orally active A2B Adenosine receptor (A2BAdoR) antagonist, currently in pre-clinical development for potential treatment of IBD. Advinus is looking for partnering/co-development opportunities.
A2BAdenosine Receptor (A2BAdoR) Antagonist PNQ-201 for IBD
Inflammatory Bowel Disease (IBD), which includes ulcerative colitis (UC) and Crohn’s disease (CD), is a multifactorial disease of an etiology not fully understood. It includes chronic inflammation of the gut, characterized by dysfunction of mucosal immunity. Current oral therapies are ineffective, non-specific, and have significant adverse effects. As such, there is a large unmet medical need for the development of new and specific therapies for IBD.
Adenosine is a stimulator of pro-inflammatory effects in the gastro-intestinal tract. Adenosine regulates tissue function by activating its receptors: A1AdoR and A2AAdoR are high affinity receptors and A2BAdoR and A3AdoR are low affinity receptors. A2BAdoR is highly expressed in cecum and colon, with expression increased even further in epithelial cells in human and murine colitis. A2BAdoR, agonized by adenosine induces cytokine secretion at the mucosal surface, inflammatory cell infiltration into intestinal wall, focal crypt damage and ulceration. Therefore, A2BAdoR antagonists are expected to be beneficial in IBD patients.
PNQ-201 is a proprietary orally active A2BAdoR antagonist, currently in pre-clinical development for the potential treatment of IBD. PNQ-201 is a potent and selective A2B antagonist. It is selected for development on the basis of poor systemic bioavailability and high exposure in colon/cecum. Negligible systemic bioavailability and maximum exposure at the sites of action in the lower gastrointestinal tract is expected to offer maximum therapeutic benefits while minimizing potential side effects. PNQ-201 has shown a robust efficacy profile in standard models of IBD, namely, the mouse DSS-induced colitis model and the rat TNBS-induced colitis model. PNQ-201 was found to be safe in exploratory safety studies including a Drug Matrix Screen, mini-AMES test, and a 14- day repeat dose toxicology study in rats.
PATENT
Example 1 : 8-(l-Benzyl~lH-pyrazol-4-yl)-l-propyl-l,4,5,7-tetrahydro-purin-6-one

Step 1: l-Benzyl-lH-pyrazole-4-carboxylic acid (6-amino-2,4-dioxo-3-propyl-l,2,3,4-tetrahydro-pyrimidin-5-yl)-amide
A mixture of 5,6-diamino-3-propyl-lH-pyrimidine-2,4-dione (1.6g, 8.55mmol), 1-benzyl-lH-pyrazole-4-carboxylic acid (1.75g, 8.65mmol) in methanol (10ml) were cooled to 0 0C and added EDCLHCl (2.32g, 12.11mmol). The reaction mixture was stirred at 25 0C for 20 hours and the solvents were removed under reduced pressure. To this residue water (10ml) was added and the precipitate was filtered off, and was washed sequentially with cold water (20ml) and DCM (25ml) to obtain l-Benzyl-lH-pyrazole-4-carboxylic acid (6-amino-2,4-dioxo-3 -propyl- 1 ,2,3,4-tetrahydro-pyrimidin-5-yl)-amide (1.5 g, 47 %) as a pale yellow solid.
1HNMR^OOMHZ5 DMSO d6): δ 0.82 (t, J=7.6Hzs 3H); 1.46-1.51 (m, 2H); 3.64 (t, J=7.2Hz, 2H);^5.36 (s, 2H); 6.01 (s, 2H); 7.26-7.38 (m, 5H); 7.96 (s, IH); 8.31 (s, IH); 8.54 (s, IH); 10.43 (s, IH).
Step 2 : 8-(l-Benzyl-lH-pyrazol-4-yl)-2-chloro-l-propyH,7-dihydro-purin-6-one A mixture of l-benzyl-lH-pyrazole-4-carboxylicacid(6-amino-2,4-dioxo-3-propyl-l,2,3,4-tetrahydro-ρyrimidin-5-yl)-amide (0.5g, 13.5mmol)s POCl3 (10ml) and DMF (0.1ml) were heated at 125-130 0C for 20 hours. Reaction mixture was cooled to 20-25 0C. It was then concentrated under vacuum. The residue was triturated with diethyl ether, dried. The crude product was purified by column chromatography using silica gel (100-200 mesh) and 2 to 4 % methanol in DCM as an eluent to obtain 8-( 1 -Benzyl- 1 H-pyrazol-4-yl)-2-chloro-l -propyl- l,7-dihydro-purin-6-one (0.04g, 8%) as a pale brown solid.
1HNMR^OOMHZ5 DMSO d6): δ 0.93 (t, J=7.6Hz, 3H); 1.67-1.73 (m, 2H); 4.15 (t, J=7.6Hz, 2H); 5.42 (s, 2H); 7.29-7.39 (m, 5H); 8.14 (s, IH); 8.49 (s, IH); 13.68 (bs, IH). Step 3: 8-(l-Benzyl-lH-pyrazol-4-yl)-l-propyl-l,7-dihydro-purin-6-one
A mixture of 8-(l -benzyl- lH-pyrazol-4-yl)-2-chloro-l -propyl- l,7-dihydro-purin-6-one (0.035 g, 0.094 mmol), Pd\C (10%) (0.025g), in ethanol (20ml) were stirred under hydrogen atmosphere for 20 hours. Reaction mixture was filtered through celite bed washed with methanol (20ml), and the solvents were removed under vacuum. The crude product was purified by column chromatography using silica gel (100-200 mesh) and 2 to 4 % methanol in DCM as an eluent to obtain 8-(l-Benzyl-lH-pyrazol-4-yl)-l-propyl-l,7-dihydro-purin-6-one (0.012g, 39%) as off white solid.
1HNMR^OOMHZ, DMSO d6): δ 0.89 (t, J=7.2Hz, 3H); 1.66-1.72 (m, 2H); 3.94 (t, J=7.6Hz, 2H); 5.41 (s, 2H); 7.302-7.38 (m, 5H); 8.03 (s, IH); 8.16 (s, IH); 8.34 (s, IH).
PATENT
WO 2011055391
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011055391
Preparation 1: 2-chloro-8-cyclopentyl-l-propyI-l, 7-dihydro-purin-6-one:

Step 1: Cyclopentane carboxylic acid (6-amino-2,4-dioxo-3-propyl-l,2,3,4-tetra hydro-pyriniidin-S-y -amide
To a solution of 5, 6-diamino-3-propyI-lH-pyrimidine-2, 4-dione (0.6 g, 2.72 mmol) in methanol (50 ml) was added cyclopentane carboxylic acid (0.310 g, 2.72 mmol). The reaction mixture was cooled to 0°C and then l-ethyl-3(3′-dimethylaminopropyl) carbodiimide hydrochloride (EDCI.HC1) (0.78 g, 4.1 mmol) was added. The resulting reaction mixture was stirred at room temperature overnight. The reaction mixture was concentrated under reduced pressure. The residue was dissolved in water. The solid was filtered and washed thoroughly with water followed by diethyl ether. The product obtained was dried under high vacuum. The crude product (0.40 g) was used for the next step without further purification.
Step 2: Preparation of 8-cyclopentyI-2-chloro-l-propyl-l, 7-dihydro-purin-6-one
To a suspension of cyclopentanecarboxylic acid (6-amino-2,4-dioxo-3-propyl- 1,2,3,4-tetrahydro-pyrimidin-5-yl)-amide (0.40 g, crude) obtained from step 1 in phosphorus oxychloride (25 ml) was added phosphorus pentachloride (0.10 g) and the resulting reaction mixture was refluxed overnight. Phosphorus oxychloride was evaporated under reduced pressure. The residue was slowly quenched with water. Ethyl acetate was added and the organic layer was separated and washed thoroughly with water followed by brine. The ethyl acetate layer was dried over anhydrous sodium sulphate and concentrated under vacuum. The crude product was purified by preparative TLC using dichloromethane, methanol (9:1) as the solvent system to give 0.075 g (19% over two steps) of the product as a white solid.
•H MR (400 MHz, DMSO d6): δ 0.9 (t, J = 8 Hz, 3H), 1.59-1.82 (m, 8H), 1.99 (m, 2H), 3.15 (t, J = 8 Hz, 1H), 4.12 (t, J = 8 Hz, 2H).
Preparations 2 to 7 were prepared following the experimental procedure as given for Preparation 1.
Preparation 2: 2-Chloro-8-cyclohexyl- 1 -propyl- 1 ,7-dihydro-purin-6-one,
Preparation 3: 2-Chloro-8-cyclopropyl-l -propyl- 1 ,7-dihydro-purin-6-one,
Preparation^ 2-Chloro-8-(hexahydro-2,5-methano-pentalen-3a-yl)-l -propyl- 1,7- dihydro-purin-6-one,
Preparation 5: 8-Bicyclo-[2.2.1]-hept-2-yl-2-chloro-l -propyl- 1, 7-dihydro-purin-6- one,
Preparation 6: 8-Adamantan-2-yl-2-chloro-l -propyl- 1, 7-dihydro-purin-6-one, Preparation7:3-[4-(2-Chloro-6-ox0-l-propyl-6,7-dihydro-lH-purin-8-yl)- bicyclo[2.2.2]oct-l-yl]-propionic acid.
Example 1: 8-Cyclopentyl-2-(3, 4-difluoro-phenoxy)-l-propyl-l, 7-dihydro-purin- 6-one:

To a solution of 8-cyclopentyl-2-chloro- 1 -propyl- l,7-dihydro-purin-6-one (0.06 g, 0.21 mmol) in N-methyl-2-pyrrolidone (0.2 ml) was added K2CO3 (0.044g, 0.32 mmol) followed by 3, 4-difluoro phenol and the reaction mixture was heated at 130 °C overnight. The reaction mixture was diluted with ethyl acetate and water. The layers were separated and ethyl acetate layer was washed with water. The ethyl acetate layer was dried over anhydrous sodium sulphate and concentrated under vacuum. The crude product was purified by preparative TLC using 3% methanol in DCM to give the product (0.015 g, 19 %) as a white solid.
‘HNMR (400 MHz, DMSO d6): δ 0.94 (t, J = 8 Hz, 3H), 1.59-1.74 (m, 6H), 1.94 (br.s, 2H), 3.12 (m, 2H), 4.09 (br. s, 2H), 7.21 (d, J = 8 Hz, 1H), 7.53-7.65 (m, 2H), 12.74 (br.s, 1H).
PNQ 370 useful in treating Parkinson’s disease from ADVINUS
2016

PNQ 370
Advinus Therapeutics Ltd
Adenosine A2a receptor antagonist
for treating disease or disorder susceptible to improvement by antagonism of A2A receptor.
Advinus Therapeutics is investigating PNQ-370, presumed to be lead from a series of small molecule therapeutics including PD-2 and PD-3, as adenosine A2a receptor antagonist, for the potential treatment of Parkinson’s disease . In November 2012, this drug was in preclinical development .
![]()
KEEP WATCHING THIS POST AS I ARRIVE AT THE STRUCTURE…………..








ONE OF THE ABOVE OR SIMILAR
INTRODUCTION
The effects of adenosine are mediated through at least four specific cell membrane receptors so far identified and classified as Ai, A2A, A2B and A3 belonging to G protein-coupled receptor family. The Ai and A3 receptors down-regulate cellular cAMP levels through their coupling to G protein, which inhibit adenylate cyclase. In contrast, A2A and A2B receptors couple to G protein that activate adenylate cyclase and increase intracellular levels of cAMP. Through these receptors, adenosine regulates the wide range of physiological functions.
Advances in understanding the role of adenosine and its receptors in physiology and pathophysiology, as well as new developments in medicinal chemistry of these receptors have identified potential therapeutic areas for drug development. With the combination of pharmacological data, using selective ligands and genetically modified mice, important progress has been made toward an understanding of the role of ARs in a variety of diseases, such as inflammatory conditions, sepsis, heart attack, ischemia-reperfusion injury, vascular injury, spinal cord injury, chronic obstructive pulmonary disease (COPD), asthma, diabetes, obesity, inflammatory bowel disease, retinopathy, and Parkinson’s Disease (PD).
Happy new year wishes 2016
Movement disorder constitutes a serious health problem, especially among the elderly. These movement disorders can often be the result of brain lesions. Disorders involving the basal ganglia which result in movement disorders include Parkinson’s disease, Huntington’s chorea and Wilson’s disease. Tremor, rigidity, akinesia and postural changes are four classic symptoms of Parkinson’s disease, it is also associated with depression, dementia and overall cognitive decline. Parkinson’s disease has a prevalence of 1 per 1000 of the total population and increases to 1 per 100 for those aged over 60 years. Degeneration of dopaminergic neurons in the substantia nigra and the subsequent reductions in the interstitial concentrations of dopamine in the striatum are critical to the development of Parkinson’s disease. About 80% of cells from the substantia nigra can be destroyed before the clinical symptoms of Parkinson’s disease become apparent
PD is a progressive, incurable disorder with no definite preventive treatment, although drugs are available to alleviate the symptoms and/or slow down the progress of the disease. Current therapy is based on dopamine replacement therapy, the most common drug treatments being dopaminomimetic agents, including L-DOPA, a dopamine precursor, as well as direct or indirect dopamine receptor agonists. L-DOPA is the mainstay in the treatment of PD, but because of tolerance problems and a wide range of adverse reactions, including involuntary movements and vomiting, a strong demand for new therapies exists. Among the various strategies, A2A AR blockers are considered a potential approach to treatment of the disease. Within the brain A2A ARs are richly expressed in the striatum, nucleus accumbens, and olfactory tubercle. A coexpression of A2A with D2 dopamine receptors has been reported in the GABAergic striatopallidal neurons where adenosine and dopamine agonists exert antagonistic effects in the regulation of locomotor activity. Activation of A2A ARs in striatopallidal neurons decreases the affinity of D2 receptors for dopamine, antagonizing the effects of D2 receptors.
The negative interaction between A2A and D2 receptors is at the basis of the use of A2A antagonists as a novel therapeutic approach in the treatment of PD. (Pharmacol. Ther. 2005, 105, 267). The recent discovery that the A2A can form functional heteromeric receptor complexes with other Gprote in-coupled receptors such as D2 and the mGlu5 receptors has also suggested new opportunities for the potential of A2A antagonists in PD. (J. Mol. Neurosci. 2005, 26, 209).
A2A knockout (KO) mice transient focal ischemia caused less neuronal damage in comparison to their wild-type (WT) littermates (J. Neurosci. 1999, 19, 9192.). Therefore, it seems that tonic activation of A2A ARs may be responsible for dangerous signal during injury, in contrast to the neuroprotective effects induced by endogenous Al activation. Recently, selective inactivation or reconstitution of A2A ARs in bone-marrow cells revealed their contribution to the development of ischemic brain injury (J.F. Nat. Med. 2004, 10, 1081) Blockade of A2A ARs has recently been implicated in the treatment of movement disorders such as Parkinson’s disease (Trends Pharmacol. Sci. 1997, 18, 338-344) and in the treatment of cerebral ischaemia (Life Sci. 1994, 55, 61-65).
The potential utility of A2A AR antagonists in the treatment of Parkinson’s disease has been reviewed (CNS drugs, 1998, 10, 31 1-320). One advantage of A2A AR antagonist therapy is that the underlying neurodegenerative disorder may also be treated ((Ann. N. Y. Acad. Sci. 1997, 825 (Neuroprotective Agents), 3048). In particular, blockade of A2A AR function confers neuroprotection against MPTP-induced neurotoxicity in mice (Neurosci. 2001, 21, RC143).
Alzheimer’s disease (AD) is a neurodegenerative disorder of the central nervous system manifested by cognitive and memory deterioration, a variety of neuropsychiatric symptoms, behavioral disturbances, and progressive impairment of daily life activities. Recent research suggests that adenosine receptors play important roles in the modulation of cognitive function. Epidemiological studies have found an association between coffee (a nonselective adenosine receptor antagonist) consumption and improved cognitive function in AD patients and in the elderly. Long-term administration of caffeine in transgenic animal models showed a reduced amyloid burden in brain with better cognitive performance.

Antagonists of adenosine A2A receptors mimic these beneficial effects of caffeine on cognitive function. Neuronal cell cultures with amyloid beta in the presence of an A2A receptor antagonist completely prevented amyloid beta-induced neurotoxicity. These findings suggest that the adenosinergic system constitutes a new therapeutic target for AD, and caffeine and A2A receptor antagonists may have promise to manage cognitive dysfunction in AD (Curr Neuropharmacol. 2009 September; 7(3): 207-216).
High expression of A2A ARs has been found in platelets, leukocytes, vascular smooth muscle, and endothelial cells with important implications in the regulation of inflammatory responses. It is now well established that stimulation of the A2A AR in immune cells induces anti-inflammatory effects, mostly due to its ability to increase cAMP levels, which has strong immunosuppressive effects (Trends Immunol. 2005, 26, 299). Stimulation of A2A ARs inhibits neutrophil adherence to the endothelium, degranulation of activated neutrophils and monocytes, plus superoxide anion generation. A2A ARs have been recently defined as sensors and terminators of proinflammatory activities. The strongest evidence for the key role of A2A in inflammation is derived by the elegant study using mice deficient in A2A ARs (Nature 2001, 414, 916).

In this model the lack of A2A subtype leads to increased tissue inflammation and damage, thus suggesting a negative and nonredundant regulatory role for the A2A AR. This model permits one to appreciate that adenosinergic regulation of immune cells is fundamental in normal physiological control of inflammation in vivo in spite of the fact that other Gs-protein-coupled receptors and cAMP elevating ligands are present, such as cathecolamines, prostaglandins, dopamine, and histamine (Trends Immunol. 2005, 26, 299). Interestingly, the A2A AR has been demonstrated to be involved in promotion of wound healing and angiogenesis in healing wounds (Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R283).
Moreover, it plays an active role in the pathogenesis of dermal fibrosis, suggesting a role for antagonists as novel therapeutic approach in the treatment and prevention of dermal fibrosis in diseases such as scleroderma (Arthritis Rheum. 2006, 54, 2632) as well as hepatic fibrosis (Br. J. Pharmacol. 2006 Aug; 148(8): 1 144-55). Studies also suggest that A2A receptor antagonists may be beneficial for social memory impairment and hypertension (Behav Brain Res. 2005 Apr 30;159(2):197-205), sepsis (J Immunol. 2006 May 1 ; 176(9): 5616-26), spinal cord injury and neuroprotection (J Neuroinflammation. 201 1 Apr 12;8:31), retinopathy (IVOS, Jan. 2000, vol. 41 (1), 230-243, depression (Neurology. 2003 Dec 9;61(1 1 Suppl 6):S82-7), narcolepsy and other sleep related disorders (Prog Neurobiol. 2007 Dec;83(5):332-47), attention-deficit hyperactivity disorder (ADHD) (Behav Pharmacol. 2009 Mar;20(2): 134-45; Clinical Genetics (2000), 58(1), 31-40 and references therein),

Dr Rashmi Barbhaiya, CEO & Managing Director

… Dr Rashmi Barbhaiya, CEO & Managing Director and Dr Kasim Mookthiar, Chief Scientific Officer and SVP, Drug Discovery, Advinus Therapeutics …
Antagonists of the A2A receptor are potentially useful therapies for the treatment of addiction. Major drugs of abuse (opiates, cocaine, ethanol, and the like) either directly or indirectly modulate dopamine signaling in neurons particularly those found in the nucleus accumbens, which contain high levels OfA2A adenosine receptors. Dependence has been shown to be augmented by the adenosine signaling pathway, and it has been shown that administration of an A2A receptor antagonist redues the craving for addictive substances (“The Critical Role of Adenosine A2A Receptors and Gi βγ Subunits in Alcoholism and Addiction: From Cell Biology to Behavior”, by Ivan Diamond and Lina Yao, (The Cell Biology of Addiction, 2006, pp 291-316) and “Adaptations in Adenosine Signaling in Drug Dependence: Therapeutic Implications”, by Stephen P. Hack and Macdonald J. Christie, Critical Review in Neurobiology, Vol. 15, 235-274 (2003)). See also Alcoholism: Clinical and Experimental Research (2007), 31(8), 1302-1307.
A2A receptors may be beneficial for the treatment or prevention of disorders such as a movement disorder, for example, Parkinson’s disease or progressive supernuclear palsy, Restless leg syndrome, nocturnal myoclonus, cerebral ischaemia, Huntington’s disease, multiple system atrophy, corticobasal degeneration, Wilson’s disease or other disorders of basal ganglia which results in dyskinesias, post traumatic stress disorder. See for example WO200013682, WO200012409, WO2009156737, WO20091 1442, WO2008121748, WO2001092264, WO2007038284, WO2008002596, WO20091 1 1449, WO20091 1 1442, WO2008121748, WO2009156737, WO2003022283, WO2005044245, WO2008077557, WO20091 1 1449, WO2009705138, WO20091 1 1442, WO2007035542, WO20080870661, WO2008070529, WO20051 16026, WO2009055548, WO2007133983, WO2010045006, WO2010045015, WO2010045008 WO2009015236.
![]()

centre: Mr Ratan Tata, Chairman, Tata Sons, flanked by Dr Rashmi Barbhaiya (left), Managing Director and CEO, Advinus, and Mr R. Gopalakrishnan, …
ONE EXAMPLE………..
| Molecular Formula: | C26H31N9O4 |
|---|---|
| Molecular Weight: | 533.58224 g/mol |
 A1
A15-amino-8-(furan-2-yl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin-1-yl]ethyl]-1-methyl-[1,2,4]triazolo[5,1-f]purin-2-one
Example Al :
5-amino-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin- 1 -yl]ethyl]- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -f]purin-2-one
5-Amino-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin-l-
5-amino-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin- 1 -yl]ethyl]- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -f]purin-2-one
Step-1 : 2-[(2,5-Diamino-6-chloro-pyrimidin-4-yI)amino]ethanol
A mixture of 4,6-dichloropyrimidine-2,5-diamine (28g, 156mmol), ethanolamine (18ml, 312mmol) and ethanol (250ml) were heated at 100-1 10 °C for 16 hours. The mixture was cooled and solvent was removed. To the residue methanol (100ml) was added and stirred for 20 minutes. The solid was filtered off to obtain 2-[(2,5-diamino-6-chloro-pyrimidin-4-yl)amino]ethanol (22.0g, 70%).
‘H MR(400MHz, DMSO d6): δ 3.36-3.40 (m, 2H); 3.50-3.54 (m, 2H); 3.88 (bs, 2H); 4.74 (t, J=5.6Hz, 1H); 5.63 (bs, 2H); 6.51 (t, J=5.6Hz, 1H)
Step-2: 2-Amino-6-chloro-9-(2-hydroxyethyl)-7H-purin-8-one
A mixture of 2-[(2,5-diamino-6-chloro-pyrimidin-4-yl)amino]ethanol obtained in step 1 (l O.Og, 49.26mmol) in acetonitrile (400ml) were cooled to 0 °C. To this reaction mixture K2C03 (20.39gm, 147.7mmol) and 4-nitrophenyl chloroformate (19.8g, 98.52mmol)was added and stirred at 25-27 °C for 24 hours. This reaction mixture was filtered and washed with acetonitrile (300ml) and diethyl ether (300ml) respectively. Solid obtained was dried to obtain crude 2-amino-6-chloro-9-(2-hydroxyethyl)-7H-purin-8-one as a yellow solid. Small amount of crude material was purified by column chromatography to obtain pure product. ‘HNMR(400MHz, DMSO d6): δ 3.61-3.66 (m, 2H); 3.72-3.75 (m, 2H); 4.85 (t, J=6Hz, 1H); 6.60 (s, 2H); 1 1.21 (s, 1 H)
Step-3: 2-Amino-6-chloro-9-(2-hydroxyethyl)-7-methyl-purin-8-one
A mixture of 2-amino-6-chloro-9-(2-hydroxyethyl)-7H-purin-8-one obtained in step 2 (13g, 56.7mmol) , K2C03 (1 1.5g, 84mmol), methyl iodide (12g, 85.15mmol) and DMF (130ml) were stirred at 25-30 °C for 16 hours. The reaction mixture was concentrated and purified by column chromatography using 60-120 silica gel and 4% methanol in DCM as an eluent to obtain 2-amino-6-chloro-9-(2-hydroxyethyl)-7-methyl-purin-8-one (8g, 58%) as an off white solid.
‘HNMR(400MHz, DMSO d6): δ 3.42 (s, 3H); 3.65 (t, J=5.6Hz, 2H); 3.78 (t, J=5.6Hz, 2H); 4.85 (t, J=5.6Hz, 1H); 6.69 (bs, 2H).
Step-4: 2-Amino-6-hydrazino-9-(2-hydroxyethyl)-7-methyI-purin-8-one
A mixture of 2-amino-6-chloro-9-(2-hydroxyethyl)-7-methyl-purin-8-one obtained in step 3 (8g, 32.9mmol) , Hydrazine hydrate (16ml ,32.9mmol) and ethanol (300ml) were heated at 100-1 10 °C for 16 hours. The reaction mixture was concentrated and solid obtained was filtered off and dried to obtain 2-amino-6-hydrazino-9-(2-hydroxyethyl)-7-methyl-purin-8-one (7g, 89 %) as an off white solid.
‘HNMR(400MHz, DMSO d6): δ 3.37 (s, 3H); 3.58-3.61 (m, 2H); 3.71 (t, J=6Hz, 2H); 4.29 (bs, 2H); 4.87 (t, J=5.6Hz, 1H), 6.00 (bs, 2H); 7.63 (s, 1H).
Step-5: N’-[2-Amino-9-(2-hydroxyethyl)-7-methyl-8-oxo-purin-6-yl]furan-2-carbohydrazide
2-amino-6-hydrazino-9-(2-hydroxyethyl)-7-methyl-purin-8-one (4.5g, 18.18mmol) obtained in step 4, 2-furoic acid (2.53g, 22.5mmol), HOBT (2.53g, 18.8 mmol) and N-methylmorpholine were taken in dimethylformamide (40ml). l-Ethyl-3(3′-dimethylaminopropryl)carbodiimide hydrochloride (EDCI.HCl) (5.4g, 28.2mmol) was added to the reaction mixture and stirred at 25-27 °C for 14 hours. The reaction mixture was evaporated and residue was purified by column chromatography to obtain N’-[2-amino-9-(2-hydroxyethyl)-7-methyl-8-oxo-purin-6-yl]furan-2-carbohydrazide (5.3g, 84%) as an off white solid.
‘HNMR (400MHZ, DMSO d6): δ 3.43 (s, 3H); 3.59-3.63 (m, 2H); 3.74 (t, J=6Hz, 2H); 4.88 (t, J=5.6Hz, 1H); 5.98 (bs, 2H); 6.67 (bs, 1H); 7.25 (d, J=3.2Hz, 1H); 7.90 (s, 1H); 8.35 (s, 1H); 10.28 (s, lH).
Step-6: 5-Amino-8-(2-furyl)-3-(2-hydroxyethyl)-l-methyl-[l^,4]triazolo[5,l-flpurin-2-one
A mixture of N’-[2-amino-9-(2-hydroxyethyl)-7-methyl-8-oxo-purin-6-yl]furan-2-carbohydrazide obtained in step 5 (5.3g, 15.9mmol), Ν,Ο-bistrimethylsilylacetamide (27ml, 1 1 1.4mmol) and hexamethyldisilazane (83ml, 397mmol) were heated at 1 10-120 °C for 16 hours. The reaction mixture was quenched with methanol (100ml) and water (100ml) and organic volatiles were evaporated. The solid obtained was filtered off and washed with water (30ml) followed by diethyl ether (100ml) to obtain 5-amino-8-(2-furyl)-3-(2-hydroxyethyl)-l-methyl-[l,2,4]triazolo[5,l-f]purin-2-one (3.50g, 71%) as an off white solid.
‘HNMR (400MHZ, DMSO d6): δ 3.56 (s, 3H); 3.67-3.70 (m, 2H); 3.84-3.87 (m, 2H); 4.88 (t, J=5.6Hz, 1H); 6.73 (bs, 1H); 7.20 (bs, 1H); 7.79 (bs, 2H); 7.94 (bs, 1H).
Step-7: 2-[5-Amino-8-(2-furyl)-l-methyl-2-oxo-[l,2,4]triazolo[5,l-fJpurin-3-yl]ethyl 4-methylbenzenesulfonate
A mixture of 5-amino-8-(2-furyl)-3-(2-hydroxyethyl)-l -methyl-[l,2,4]triazolo[5, l-fJpurin-2-one obtained in step 6 (3.5g, l lmmol), p-toluene sulphonylchloride (5.2 g, 27mmol) were taken in pyridine (30ml)and stirred at 25-27 °C for 16 hours. To the reaction mixture hexane (100ml) was added and solid obtained was filtered off and washed with water (100ml) followed by hexane (100ml) to obtain 2-[5-amino-8-(2-furyl)-l-methyl-2-oxo-[l,2,4]triazolo[5, l-f]purin-3-yl]ethyl 4-methylbenzenesulfonate (4.1g, 78%) as a brown solid. ‘HNMR (400MHz, DMSO d6): δ 2.02 (s, 3H); 3.49 (s, 3H); 3.99 (t, J=4.8Hz, 2H); 4.71 (t, J=4.8Hz, 2H); 6.73-6.75 (m, 1H); 7.01 (d, J=8Hz, 2H); 7.23 (d, J=3.2Hz, 1H); 7.41 (d, J=8.4Hz, 2H); 7.78 (bs, 2H); 7.96 (d, J=1.2Hz, 1H).
Step-8: : 5-Amino-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin-l-yl]ethyl]-l-methyl-[l,2,4]triazolo[5,l-f)purin-2-one
A mixture of 2-[5-amino-8-(2-furyl)-l-methyl-2-oxo-[l ,2,4]triazolo[5, l-f]purin-3-yl]ethyl 4-methylbenzenesulfonate obtained in step 7 (0.25g, 0.533mmol), l-[4-(2-Methoxy-ethoxy)-phenyl]-piperazine (0.188g, 0.799mmol) and DIPEA (0.27ml, 1.599mmol) were taken in DMF (5ml) and stirred at 80 °C for 16 hours. To the reaction mixture water (100ml) was added and solid obtained was filtered off. The crude product was purified by column chromatography to obtain 5-amino-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin- 1 -yl]ethyl]- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -f]purin-2-one (0.135g, 47%) as an off white solid
‘HNMR (400MHz, DMSO d6): δ 2.60 (bs, 4H); 2.68 (t, J=6.4Hz, 2H); 2.96 (bs, 4H); 3.29 (s, 3H); 3.56 (s, 3H); 3.59-3.62 (m, 2H); 3.94-4.00 (m, 4H); 6.71 -6.73 (m, 1H); 6.79-6.86 (m, 4H); 7.19 (dd, J=3.2Hz, 1.2Hz, 1H); 7.80 (bs, 2H); 7.94 (bs, 1H).
ANOTHER……..
Example Gl: 5-Amino-l-ethyl-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyl]piperazin-l-yl]ethyl]-[l,2,4]triazolo[5,l-i]purin-2-one

Step-1 : 2-Amino-6-chloro-7-ethyl-9-(2-hydroxyethyl)purin-8-one
(Procedure is same as step-3 in example Al)
‘HNMR (400MHz, DMSO d6): δ 1.21 (t, J=7.2Hz, 3H); 3.64 (s, 2H); 3.78 (t, J=6Hz, 2H);
3.92 (q, J=7.2Hz, 2H); 4.92 (bs, I H); 6.7 (bs, 2H).
Step-2 : 2-Amino-7-ethyl-6-hydrazino-9-(2-hydroxyethyl)purin-8-one
(Procedure is same as step-4 in example Al)
‘ HNMR (400MHz, DMSO d6): δ 1.07 (t, J=6.8Hz, 3H); 3.59 (q, J=6Hz, 2H); 3.72 (t, J=6Hz,
2H); 3.91 (q, J=6.8Hz, 2H); 4.32 (bs, 2H); 4.86 (t, J=5.6Hz, IH); 5.99 (bs, 2H), 7.55 (bs, IH).
Step-3: N’-[2-Amino-7-ethyl-9-(2-hydroxyethyl)-8-oxo-purin-6-yl]furan- 2carbohydrazide (Procedure is same as step-5 in example Al)
Crude product was used in next step
Step-4: 5-Amino-l-ethyI-8-(2-furyl)-3-(2-hydroxyethyl)-[l,2,4]triazolo[5,l-flpurin-2-one
(Procedure is same as step-6 in example Al)
‘H MR (400MHZ, DMSO d6): δ 1.34 (t, J=7.2Hz, 3H); 3.67 (q, J=5.6Hz, 2H); 3.84 (t, J=5.6Hz, 2H); 4.01 (q, J=7.2Hz, 2H); 4.87 (t, J=6Hz, IH); 6.70 (bs, IH); 7.17 (d, J=2.8Hz, I H); 7.18 (bs, 2H); 7.92 (bs, IH).
Step-5: 2-[5-Amino-l-ethyl-8-(2-furyl)-2-oxo-[l,2,4]triazoIo[5,l-f|purin-3-yl]ethyl 4- methylbenzenesulfonate (procedure is same as step-7 in example Al)
lHNMR (400MHz, DMSO d6): δ 1.35 (t, J=7.2Hz, 3H); 2.00 (s, 3H); 3.95-4.00 (m, 4H); 4.47 (bs, 2H); 6.74 (s, IH); 7.00 (d, J=7.6Hz, 2H); 7.22 (s, IH); 7.42 (d, J=7.6Hz, 2H); 7.78 (bs, 2H); 7.97 (bs, IH).
Step-6: 5-Amino-l-ethyl-8-(2-furyl)-3-[2-[4-[4-(2-methoxyethoxy)phenyi]piperazin-l- yl]ethyl]-[l,2,4]triazolo[5,l-f]purin-2-one (procedure is same as step-8 in example Al)
HNMR(400MHz, DMSO d6): δ 1.35 (t, J=7.2Hz, 3H); 2.60 (bs, 4H); 2.68 (t, J=6.8Hz, 2H); 2.95 (bs, 4H); 3.28(s, 3H);3.61 (t, J=4.4Hz, 2H); 3.94-4.04 (m, 6H); 6.72 (dd, J=2Hz, 3.6Hz, I H); 6.78-6.85 (m, 4H); 7.19 (d, J=3.2Hz, IH); 7.81(bs, 2H); 7.94 (s, IH).
Representative compounds of the present disclosure were tested and had micromolar to nanomolar activity.

 A1 ABOVE
A1 ABOVE
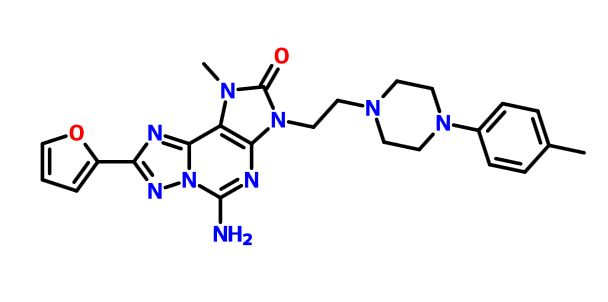
A7 ABOVE
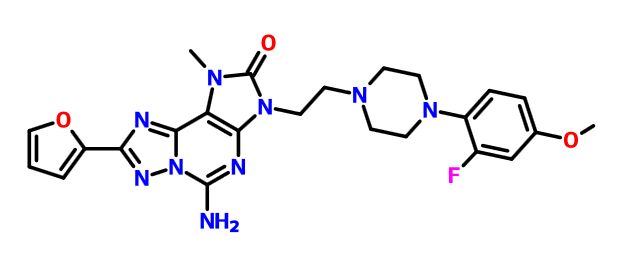
A9 ABOVE
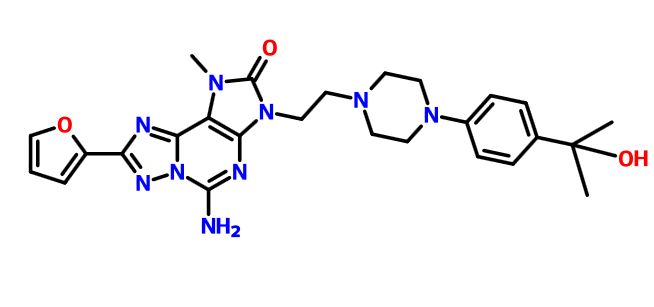
A13 ABOVE
A31 ‘HNMR (400MHz, DMSO d6): δ 2.62 (bs,4H); 2.68 (t, J=6.8Hz, 2H); 2.85 (bs, 4H); 3.28 (s, 3H); 3.57 (s, 3H); 3.59-3.62 (m, 2H); o 3.95 (t, J=6.8Hz, 2H); 4.01-4.04 (m, 2H);
5-Amino-3-[2-[4-[2-fluoro-4-(2- 6.66-6.68 (m, 1H); 6.72 (dd, J=2 Hz,3.6Hz, methoxyethoxy)phenyl]piperazin-l-yl]ethyl]-8- 1H); 6.79 (dd, J=2.8Hz, 14Hz, 1H); 6.92 (t, (2-furyl)- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -f|purin-2- J=9.6Hz, 1H); 7.19 (d, J=3.2Hz, 1 H); 7.93 one (bs, 2H); 7.93-7.94 (m, 1H).
A31 ABOVE
A32 HNM (400MHz, DMSO d6): δ 2.59 (bs,
4H); 2.68(t, J=6.4Hz, 2H); 3.27(t, J=4.8Hz, 4H); 3.56 (s, 3H); 3.96 (t, J=6.4Hz, 2H);
0 6.72(dd, J=2Hz, 3.6Hz, 1H); 6.99 (d, J=8.8Hz,
4-[4-[2-[5-Amino-8-(2-furyl)-l-methyl-2-oxo- 2H); 7.19 (d, J=3.6Hz, 1H);7.56 (d, J=8.8Hz, [ 1 ,2,4]triazolo[5, 1 -f]purin-3-yl]ethyl]piperazin- 2H); 7.80 (bs, 2H); 7.93 (bs, lH).
l-yl]benzonitrile
A32 ABOVE
A36 ‘HNMR(400MHz, CDCI3): δ θ.09 (d,
J=4.4Hz, 2H); 0.50 (d, J=6.8Hz, 2H); 0.82- 0.89 (m, 1H); 2.24 (d, J=6.0Hz, 2H): 2.52- 2.72 (m, 8H); 2.80 (t, J=6.4Hz, 2H); 3.76 (s,
5-Amino-3-[2-[4-(cyclopropylmethyl)piperazin- 3H); 4.07 (t, J=6.8Hz, 2H); 5.89 (bs, 2H); l -yl]ethyl]-8-(2-furyl)-l-methyl- 6.61 (bs, 1H); 7.22 (d, J=2.4Hz, 1H); 7.64 (s, [ 1 ,2,4]triazolo[5, 1 -f]purin-2-one 1H).
A36 ABOVE
A38 ‘HNMR(400MHz, CDCI3): δ 2.62 . (t,
J=4.4Hz, 4H); 2.79 (t, J=6.4Hz, 2H); 2.81 (s, 6H); 3.22 (t, J=4.4Hz, 4H): 3.77 (s, 3H); 4.06 (t, J=6.8Hz, 2H); 5.74 (bs, 2H); 6.60 (dd,
4-[2-[5-Amino-8-(2-fiiryl)- 1 -methyl-2-oxo- J=2.0Hz, 3.2Hz, 1H); 7.24 (d, J=3.6Hz, 1H);
[ 1 ,2,4]triazolo[5, 1 -f]purin-3-yl]ethyl]-N,N- 7.65 (s, 1H).
dimethy l-piperazine- 1 -sulfonamide
A38 ABOVE
A39 ‘HNMR(400MHZ, DMSO d6): δ 1.89-1.94
im, 1H); 2.09-2.18 .(m, 1 H); 2.60 (bs, 4H); 2.67 (t, J=6.4Hz, 2H); 2.96 (bs, 4H); 3.56 (s, 3H); 3.69-3.85 (m, 4H); 3.95 (t, J=6.4Hz,
2H); 4.89 (bs, 1H); 6.72 (dd, J=2.0, 3.2Hz,
5-Amino-8-(2-furyl)-l -methyl-3-[2-[4-(4- 1H); 6.78 (d, J=9.2Hz, 2H); 6.85 (d, J=9.2Hz, tetrahydrofuran-3-yloxyphenyl)piperazin- 1 – 2H): 7.20 (d, J=3.2Hz, 1 H); 7.80 (bs, 2H); yl]ethyl]-[l ,2,4]triazolo[5,l-f]purin-2-one
7.93 (s, 1H).
A39 ABOVE
A42 ‘HNMR(400MHz, CDCI3): δ
2.26 (s,3H); 2.94-2.97 (m, 6H); 3.72 (s, 2H); 3.75 (s, 3H); 4.17 (t, J=6.4Hz, 2H); 5.74 (bs, 2H); 6.59 (dd, J=1.6Hz, 3.6Hz, 1H);7.13 (s, J=3.6Hz, IH); 7.21-7.24 (m, IH); 7.63 (s,
5-Amino-8-(2-furyl)-l-methyl-3-[2-(3-methyl- IH); 8.20 (bs, IH),
7,8-dihydro-5H- 1 ,6-naphthyridin-6-yl)ethyl]- [ 1 ,2,4]triazolo[5, 1 -f]purin-2-one
A42 ABOVE
A57 HNMR(400MHz, DMSO d6): δ 2.95 (t,
J=8Hz, 2H); 3.52 (s, 3H); 3.69 (s, 3H ), 3.97 (t, J=8Hz, 2H); 6.71 (dd, J=2Hz, 3.6Hz, I H );
5-Amino-8-(2-furyl)-3-[2-(4- 6.80 (dd, J=2Hz, 6.8Hz, 2H); 7.10 (d, methoxyphenyl)ethyl]- 1 -methyl- J=8.8Hz, 2H); 7.18 (dd, J=0.8Hz, 3.2Hz, I H );
[ 1 ,2,4]triazolo[5, 1 -f]purin-2-one 7.80 (bs, 2H), 7.94 (dd, J=lHz, 2Hz, I H ).
A57 ABOVE
A58 HNMR(400MHz, DMSO d6): δ 2.61 (bs,
4H); 2.68 (bs, 2H); 3.05(bs, 4H); 3.57 (s, 3H ), 3.96 (bs, 2H); 6.72 (bs, IH); 6.92 (d, J=8Hz, 2H); 7.01 (d, J=10Hz, 2H );7.03(d, J=148Hz, IH); 7.19 (bs , 1 H); 7.80 (bs, 2H); 7.94 (s,
5-amino-3-[2-[4-[4- IH).
(difluoromethoxy)phenyl]piperazin-l-yl]ethyl]- 8-(2-furyl)- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -fjpurin- 2-one
A58 ABOVE
A62 O ‘HNMR (400MHz, DMSO d6): δ 0.66-0.70
(m, 4H); 1.90-1.94 (m, lH); 2.41 (bs, 4H); 2.65 (t, J=6Hz, 2H); 3.38 (bs, 2H); 3.56 (bs, 5H); 3.93 (t, J=6.4 Hz, 2H); 6.71 (bs, 1H );
5-Amino-3-[2-[4- 7.19 (d, J=2.4Hz, 1H); 7.79 (bs, 2H); 7.93 (bs,
(cyclopropanecarbonyl)piperazin- 1 -yl]ethyl]-8- 1H).
(2-furyl)- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -fjpurin-2- one
A62 ABOVE
A63 ‘HNMR (400MHz, DMSO d6): δ 0.07-0.10
(m, 2H); 0.40-0.44 (m, 2H); 0.88-0.94 (m,lH); 2.21 (d, J=6.4Hz, 2H); 2.41-2.45 (m, 4H); 2.64 (t, J=6.4Hz, 2H); 3.38 (bs,4H); 3.56
5-Amino-3-[2-[4-(2- (s, 3H); 3.93 (t, J=6.4Hz, 2H); 6.72 (dd, cyclopropylacetyl)piperazin-l -yl]ethyl]-8-(2- J=2Hz,3.6 Hz, 1H); 7.19-7.20 (m, 1H); 7.80 fury 1)- 1 -methyl-[ 1 ,2,4]triazolo[5, 1 -fJpurin-2- (bs, 2H); 7.93 (d, J=0.8 Hz, 1H).
one
A63 ABOVE
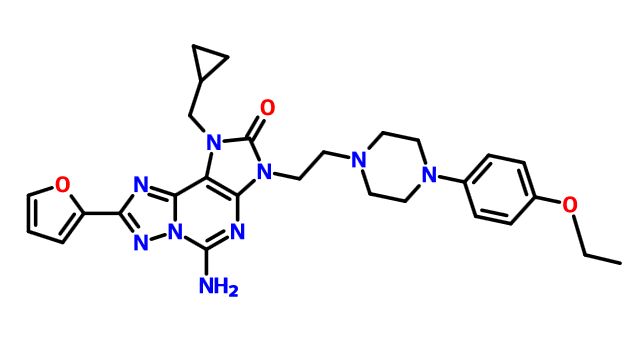
C1 ABOVE

E1 ABOVE

D3 ABOVE
G1 ABOVE
G2
H2
M1
M2
M3
M6
ETC AS IN TABLE……………..


/////////
n21c(nc4c(c1nc(n2)c3occc3)N(C(N4CCN5CCN(CC5)c6ccc(cc6)OCCOC)=O)C)N
CN1C2=C(N=C(N3C2=NC(=N3)C4=CC=CO4)N)N(C1=O)CCN5CCN(CC5)C6=CC=C(C=C6)OCCOC
GKM 001 in pipeline for Diabetes by Advinus




GKM 001……Several probables
Watch out on this post as I get to correct structure………..
![]()

Advinus Therapeutics Private L,
![]()
A glucokinase activator for treatment of type II diabetes
In October 2012, Takeda and Advinus have entered into an agreement to initiate a three-year discovery collaboration program focused on novel targets for inflammation, CNS, and metabolic diseases.
| Company | Advinus Therapeutics Ltd. |
| Description | Activator of glucokinase (GCK; GK) |
| Molecular Target | Glucokinase (GCK) (GK) |
| Mechanism of Action | Glucokinase activator |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Phase I/II |
| Standard Indication | Diabetes |
| Indication Details | Treat Type II diabetes |
Advinus chief executive officer/MD Dr. Rashmi Barbhaiya.
PATENT
https://www.google.co.in/patents/WO2009047798A2?cl=en
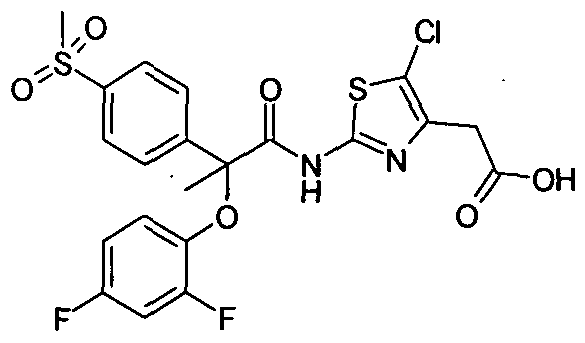
Example Cl : (-)-{5-ChIoro-2-[2-(4-cyclopropanesulfonylphenyI)-2-(2,4- difluorophenoxy)acetylamino]thiazol-4-yl}-acetic acid, ethyl ester


Step I: Preparation of (-)-(4-Cyclopropanesulfonylphenyl)-(2,4- difluorophenoxy)acetic acid (Cl-I):
To a solution of (4-cyclopropanesulfonylphenyl)-(2,4-difluorophenoxy)acetic acid (obtained in example Al -step III) in ethyl acetate was added (S)-(-)-l-phenylethylamine drop wise at -15 °C. After completion of addition the reaction was stirred for 4-6 hours. Solid was filtered and washed with ethyl acetate. The solid was then taken in IN HCl and extracted with ethyl acetate, ethyl acetate layer was washed with brine, dried over anhydrous sodium sulfate. Solvent was removed under reduced pressure to obtain (-)-(4- cyclopropanesulfonylphenyl)-(2,4-difluorophenoxy)acetic acid. Enantiomeric enrichment was done by repeating the diasteriomeric crystallization. [α]23 589 = – 107.1 ° (c = 2%Chloroform) Enantiomeric purity > 99. % (chiral HPLC)
Step II: (-)-{5-Chloro-2-[2-(4-cyclopropanesulfonylphenyl)-2-(2,4- difluorophenoxy)acetyIamino]thiazol-4-yl}-acetic acid ethyl ester : To a solution of (-)-4-cyclopropanesulfonylphenyl)-(2,4-difluorophenoxy)acetic acid (Cl-I) in DCM, was added DMF and cooled to 0 °C, followed by the addition of oxalyl chloride under stirring. Stirring was continued for 1 hour at the same temperature. The resulting mixture was further cooled to -35 °C, and to that, a solution of excess (2- amino-5-chlorothiazol-4-yl)acetic acid ethyl ester in DCM was added drop wise. After completion of reaction, the reaction mixture was poured into IN aqueous HCl under stirring, organic layer was washed with IN HCl, followed by 5% brine, dried over anhydrous sodium sulfate, solvent was removed under reduced pressure to get the crude compound which was purified by preparative TLC to get the title compound. [α]23 589 = – ve (c = 2%Chloroform)
1H NMR(400 MHz, CDCl3): δ 1.06-1.08 (m, 2H), 1.30 (t, J=7.2 Hz, 3H), 1.33-1.38 (m, 2H), 2.42-2.50 (m, IH), 3.73 (d, J=2 Hz, 2H), 4.22 (q, J=7.2 Hz ,2H), 5.75 (s, IH), 6.76- 6.77 (m, IH), 6.83-6.86 (m, IH), 6.90-6.98 (m, IH), 7.73 (d, J=8.4 Hz, 2H), 7.96 (d, J=8.4 Hz, 2H), 9.96 (bs, IH). MS (EI) m/z: 571.1 and 573.1 (M+ 1; for 35Cl and 37Cl respectively).
Examples C2 and C3 were prepared in analogues manner of example (Cl) from the appropriate chiral intermediate:

Example Dl : (+)-{5-Chloro-2-[2-(4-cyclopropanesulfonylphenyl)-2-(2,4- difluorophenoxy)acetylamino]thiazol-4-yl}acetic acid, ethyl ester

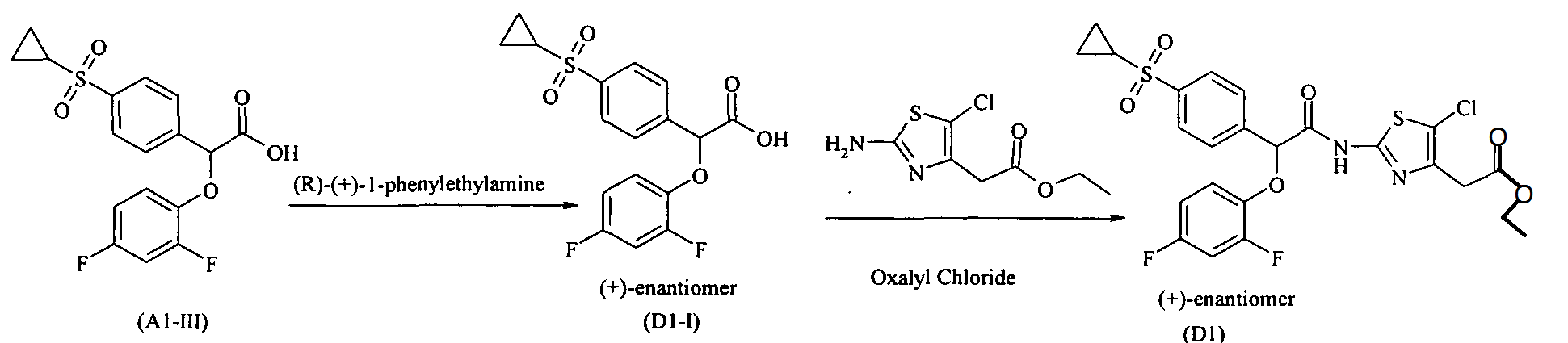
Preparation of (+)-(4-Cyclopropanesulfonylphenyl)-(2,4-difluorophenoxy)acetic acid (Dl-I):
To a solution of (4-cyclopropanesulfonylphenyl)-(2,4-difluorophenoxy)acetic acid (obtained in example Al -step III) in ethyl acetate, was added (R) (+)-l- phenylethylamine drop wise at -15 °C. After completion of addition the reaction was stirred for 4-6 hours. Solid was filtered and washed with ethyl acetate. The solid was then taken in IN HCl and extracted with ethyl acetate, ethyl acetate layer was washed with brine, dried over anhydrous sodium sulfate. Solvent was removed under reduced pressure to obtain (+)-(4-Cyclopropanesulfonylphenyl)-(2,4-difluorophenoxy)acetic acid. Enantiomeric enrichment was done by repeating the diasteriomeric crystallization. [α]23 589 = +93.07° (c = 2%Chloroform) Enantiomeric purity > 99. % (by chiral HPLC)
(+)-(4-CyclopropanesuIfonylphenyI)-(2,4-difluorophenoxy)acetic acid ethyl ester (Dl)
The example Dl was prepared using (+)-4-cyclopropanesulfonylphenyl)-(2,4- difluorophenoxy)acetic acid (Dl-I), and following the same reaction condition for amide coupling as described in example Cl, [ot]23 589 = + ve (c = 2%Chloroform)
PATENT
https://www.google.co.in/patents/WO2008104994A2?cl=en
Synthesis Type-P
Example Pl : {5-Chloro-2-[2-(2,4-difluoro-phenoxy)-2-(4-methanesulfonyl-phenyl)- propionylamino]-thiazol-4-yI}-acetic acid
To a solution of {5-Chloro-2-[2-(2,4-difluoro-phenoxy)-2-(4-methanesulfonyl- phenyl)-propionylamino]-thiazol-4-yl}-acetic acid methyl ester (0.03 g, 0.05 mmol) in THF: Ethanol: water ( ImI + 0.3ml + 0.3 ml) was added lithium hydroxide (0.0046 g, 0.11 mmol). The resulting mixture was stirred for 5 hours at room temperature followed by removal of solvent under reduced pressure. The residue was suspended in water (15 ml), extracted with ethyl acetate to remove impurities. The aqueous layer was acidified with IN HCl (0.5 ml) and extracted with ethyl acetate (2×10 ml), This ethyl acetate layer was washed with water (15 ml), brine (20 ml), dried over anhydrous sodium sulfate and solvent was removed under reduced pressure to give solid product {5-Chloro-2-[2-(2,4-difluoro-phenoxy)-2-(4- methanesulfonyl-phenyl)-propionylamino]-thiazol-4-yl} -acetic acid (9 mg). 1H NMR (400 MHz, CDCl3): δ 1.85 (s, 3H) , 3.07 (s, 3H) , 3.72 ( s, 2H), 6.64-6.69 ( m, 2H ) , 6.89-6.91 (m, IH ), 7.84 ( d, J – 8.4 Hz, 2H), 8.00 ( d, J = 8.8 Hz, 2H). MS (EI) mlz: 530.70 (M + 1), mp: 109-111 0C.
Preparation of {5-Chloro-2-[2-(2,4-difluoro-phenoxy)-2-(4-methanesulfonyl-phenyl)- propionylamino)-thiazol-4-yl}-acetic acid methyl ester used in Example Pl:

To a mixture of 2-(2, 4-Difluoro-phenoxy)-2-(4-methanesulfonyl-phenyl)-propionic acid (0.110 g, 0.22 mmol), (2-Amino-5-chloro-thiazol-4-yl)-acetic acid methyl ester (0.071 g, 0.32 mmol), HOBt (0.052g, 0.38 mmol), and EDCI (0.074 g, 0.38 mmol) in methylene dichloride (10 ml) was added N-methylmorpholine (0.039 g, 0.38 mmol). The resulting mixture was stirred at room temperature for overnight followed by dilution with 10 ml methylene dichloride. The reaction mixture was poured onto water (20 ml), and organic layer separated, washed with water (2x 20 ml), brine (20 ml), dried over sodium sulfate and solvent evaporated to get residue which was purified by preparative TLC using 50% ethyl acetate in hexane as mobile. To give desired compound (0.30 g). 1H NMR (400 MHz, CDCl3): δ 1.45 (t, J = 7.2 Hz, 3H), 1.93 (s, 3H), 3.14 (s, 3H), 3.77 (d, J = 2.8 Hz, IH), 4.26 (q, J = 7.2 Hz, IH), 6.69-6.77(m, 2H), 6.96-7.02 (m, IH), 7.89 (d, J = 8.4 Hz, 2H), 8.07 (d, J= 8.4Hz, IH).; MS (EI) m/z: 559 .00 (M + 1).
PATENT
http://www.google.com/patents/WO2012020357A1?cl=en
4-{2-[2-Cyclopentyloxy-2-(4-cyclopropanesulfonyl-phenyl)-acetylamino]- thiazol-5-yloxy}-benzoic acid, cas 1359151-08-8

Step I: (4-Cyclopropylsulfanyl-phenyl)-oxo-acetic acid ethyl ester:
A1C13 (7.98 g, 48.42 mmole) was suspended in DCM (50 mL) and cooled to 0 C under argon atmosphere. To this suspension was added chlorooxo ethylacetate (4.5 mL, 39.98 mmol) at 0 °C and stirred for 45 min. followed by addition of a solution of cyclopropylsulfanyl-benzene (5 g, 33.28 mmol) in DCM (10 mL) and stirred at 25 °C for 2 hr. Reaction mixture was slowly poured over crushed ice, organic layer was separated and aqueous layer was extracted with DCM (3 X 50 mL), combined organic layer was washed with brine solution, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to obtain (4- cyclopropylsulfanyl-phenyl)-oxo-acetic acid ethyl ester (3.1 g) as an oily product.
*H NMR (400 MHz, CDC13): δ 0.72-0.73 (m, 2H), 1.15-1.17 (m, 2H), 1.40 (t, J = 6.6 Hz, 3H), 2.18-2.21 (m, 1H), 4.41 (q, J = 6.8 Hz, 2H), 7.43 (d, J = 8.0 Hz, 2H), 7.90 (d, J = 8.0 Hz, 2H); MS (EI) m/z: 250.9 (M+l).
Step II: (4-Cyclopropanesulfonyl-phenyl) oxo acetic acid ethyl ester:
(4-Cyclopropylsulfanyl-phenyl)-oxo-acetic acid ethyl ester (3.1 g, 12.53 mmole) in DCM (50 mL) was cooled to 0-5 °C followed by addition of mCPBA (9.8 g , 31.33 mmol) in portion wise at 0 °C. After stirring at 25 °C for 4 hr, the reaction mixture was filtered; filtrate was washed with saturated aq. Na2S203 and satd. aq. sodium bicarbonate solution followed by brine solution, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to give (4-cyclopropanesulfonyl-phenyl) oxo acetic acid ethyl ester (3 g).
*H NMR (400 MHz, CDC13): δ 1.05-1.10 (m, 2H), 1.36-1.39 (m, 2H), 1.40 (t, J = 6.8 Hz, 3H), 2.45-2.50 (m, 1H), 4.42 (q, J = 7.2 Hz, 2H), 8.01 (d, J = 8.4 Hz, 2H), 8.20 (d, J = 8.4 Hz, 2H); MS (EI) m/z: 297.1 (M+NH4).
Step III: p-Toluene sulfonyl hydrazone (4-cyclopropyl sulfonyl) phenyl acetic acid ethyl ester:
A mixture of (4-cyclopropanesulfonyl-phenyl) oxo acetic acid ethyl ester (0.5 g, 1.77 mmole) and p-toluene sulfonyl hydrazide (0.48 g , 2.3 mmol) in toluene (15 mL) was refluxed for 16 hr using a Dean-Stark apparatus. Reaction mixture was concentrated to give the crude product which was purified by column chromatography over silica gel using 20-25% ethyl acetate in hexane as eluent to provide p-toluene sulfonyl hydrazone (4-cyclopropyl sulfonyl) phenyl acetic acid ethyl ester (0.5 g).
MS (EI) m/z 451.0 (M+l).
Step IV: (4-Cyclopropanesulfonyl-phenyl) diazo acetic acid ethyl ester:
To a solution of p-toluene sulfonyl hydrazone (4-cyclopropyl sulfonyl) phenyl acetic acid ethyl ester (0.5 g, 1.23 mmol) in dry DCM (6 mL), was added triethylamine (0.17 mL, 1.35 mmol) and stirred at 25 °C for 1 hr. Reaction mixture was concentrated to provide (4- cyclopropanesulfonyl-phenyl) diazo acetic acid ethyl ester (0.5 g) which was used in next reaction without any purification.
MS (EI) m/z: 295.1 (M+l).
Step V: Cyclopentyloxy-(4-cyclopropanesulfonyl-phenyl)-acetic acid ethyl ester:
(4-Cyclopropanesulfonyl-phenyl) diazo acetic acid ethyl ester (1 g, 3.37 mmol) was dissolved in DCM (16 mL) under argon atmosphere. To this solution, cyclopentanol (0.77 mL, 8.44 mmol) was added followed by rhodium(II)acetate dimer (0.062 g, 0.14 mmol). Mixture was stirred at 25 C for 12 hr. Reaction mixture was diluted with DCM (25 mL), organic layer was washed with water followed by brine solution, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to give a crude product which was purified by column chromatography using 25-35% ethyl acetate in hexane as eluent to provide cyclopentyloxy-(4- cyclopropanesulfonyl-phenyl)-acetic acid ethyl ester (0.35 g).
*H NMR (400 MHz, CDC13): δ 1.02-1.05 (m, 2H), 1.24 (t, J = 6.8 Hz, 3H), 1.35-1.37 (m, 2H), 1.53-1.82 (m, 8H), 2.42-2.50 (m, 1H), 4.02-4.04 (m, 1H), 4.15-4.22 (m, 2H), 5.00 (s, 1H), 7.66 (d, J = 8.0 Hz, 2H), 7.88 (d, J = 8.0 Hz, 2H); MS (EI) m/z: 370.0 (M+18).
Step VI: Cyclopentyloxy-(4-cyclopropanesulfonyl-phenyl)-acetic acid:
To cyclopentyloxy-(4-cyclopropanesulfonyl-phenyl)-acetic acid ethyl ester (0.35 g, 0.99 mmol) was added a solution of lithium hydroxide (0.208 g, 4.97 mmol) in water (4 mL) followed by THF (2 mL) and methanol (1 drop) and stirred for 12 hours at 25 0 C. Organic solvents were evaporated from the reaction mixture and aqueous layer was acidified IN HCl, extracted with ethyl acetate (3 X 10 mL), organic layer was washed with brine solution, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to provide cyclopentyloxy-(4- cyclopropanesulfonyl-phenyl)-acetic acid (0.210 g).
*H NMR (400 MHz, CDC13): δ 1.02-1.07 (m, 2H), 1.34-1.38 (m, 2H), 1.55-1.62 (m, 2H), 1.69- 1.82 (m, 6H), 2.43-2.47 (m, 1H), 4.08-4.10 (m, 1H), 5.02 (s, 1H), 7.65 (d, J = 8.4 Hz, 2H), 7.91 (d, J = 8.4 Hz, 2H); MS (EI) m/z: 342.0 (M+18)
Example Al: 4-{2-[2-Cyclopentyloxy-2-(4-cyclopropanesulfonyl-phenyl)-acetylamino]-

To a mixture of cyclopentyloxy-(4-cyclopropanesulfonyl-phenyl)-acetic acid (Preparation 1) (0.1 g, 0.30 mmol), 4-(2-Amino-thiazol-5-yloxy)-benzoic acid methyl ester (0.085 g, 0.33 mmol), HOBt (0.045 g, 0.33 mmol), and EDCI (0.063 g, 0.33 mmol) in DCM (5 mL), was added N-methyl morpholine (0.033 g, 0.30 mmol). The resulting mixture was stirred at room temperature overnight followed by dilution with methylene chloride (20 mL). The reaction mixture was poured into water; organic layer was washed with water, brine, dried over sodium sulfate, and the organic solvent evaporated to get a residue which was purified by preparative TLC to provide the title compound (0.145 g).
*H NMR (400 MHz, CDC13): δ 1.03-1.05 (m, 2H), 1.34-1.38 (m, 2H), 1.58- 1.65 (m, 2H), 1.76- 1.81 (m, 6H), 2.42-2.45 (m, 1H), 3.89 (s, 3H), 4.05-4.15 (m, 1H), 5.08 (s, 1H), 7.07 (d, J = 8.8 Hz, 2H), 7.15 (s, 1H), 7.68 (d, J = 8.4 Hz, 2H), 7.92 (d, J = 8.4 Hz, 2H), 7.99 (d, J = 8.8 Hz, 2H), 9.72 (s, 1H); MS (EI) m/z: 556.9 (M + 1).
Example Bl: 4-{2-[2-Cyclopentyloxy-2-(4-cyclopropanesulfonyl-phenyl)-acetylamino]- thiazol-5-yloxy}-benzoic acid:
4-{2-[2-Cyclopentyloxy-2-(4-cyclopropanesulfonyl-phenyl)-acetylamino]-thiazol-5-yloxy}- benzoic acid methyl ester (0.145 g, 0.26 mmol, obtained in example Al) was taken in H20: THF (1 :2, 6 mL) to it was added MeOH (1 drop) followed by LiOH (0.054 g, 1.30 mmol) and stirred for 12 hr. After completion of the reaction, organic solvent was removed under reduced pressure. The aqueous layer was washed with diisopropyl ether then acidified with 1 N HC1 to pH 4. The solid formed was filtered, washed with water, diisopropyl ether & dried under vacuum to get the title_compound (0.12 g).
IH NMR- (400 MHz DMSO-ifc):- δ 1.01-1.05 (m, 2H), 1.09-1.13 (m, 2H), 1.22-1.49 (m, 2H), 1.59-1.73 (m, 6H), 2.82-2.86 (m, IH), 3.99-4.01 (m, IH), 5.31 (s, IH), 7.16 (d, J = 8.4 Hz, 2H), 7.37 (s, IH), 7.74 (d, J = 8.4 Hz, 2H), 7.91 (m, 4H), 12.55 (br. s, IH), 12.90 (br.s, IH); MS (EI) m/z: 542.9 (M+l)
CLIPPINGS
Advinus’ GK-activator Achieves Early POC for Diabetes
November 29 2011
Partnership Dialog Actively Underway
Advinus Therapeutics, a research-based pharmaceutical company founded by globally experienced industry executives and promoted by the TATA Group, announced that it has successfully completed a 14-day POC study in 60 Type II diabetic patients on its lead molecule, GKM-001, a glucokinase activator. The results of the trial show effective glucose lowering across all doses tested without any incidence of hypoglycemia or any other clinically relevant adverse events.
The clinical trials on GKM-001 validate the company’s pre-clinical hypothesis that a liver selective Glucokinase activator would not cause hypoglycemia (very low blood sugar), while showing robust efficacy.
“GKM-001 is differentiated from most other GK molecules that are in development, or have been discontinued, due to its novel liver selective mechanism of action. GKM-001 has a prolonged pharmacological effect and a half-life that should support a once a day dosing as both mono and combination therapy.” said Dr. Rashmi Barbhaiya, MD & CEO, Advinus Therapeutics. He added that Advinus is actively exploring partnership options to expedite further development and global marketing of GKM-001.
GKM-001 belongs to a novel class of molecules for treatment of type II diabetes. It is an activator of Glucokinase (GK), a glucose-sensing enzyme found mainly in the liver and pancreas. Being liver selective, GKM-001 mostly activates GK in the liver and not in pancreas, which is its key differentiation from most competitor molecules that activate GK in pancreas as well. The resulting increase in insulin secretion creates a potential for hypoglycemia-a risk GKM-001 is designed to avoid. Advinus has the composition of matter patent on GKM-001 for all major markets globally. Both the Single Ascending Dose data, in healthy and type II diabetics, and the Multiple Ascending Dose Study in Type II diabetics has shown that the molecule shows effective glucose lowering in a dose dependent manner and has excellent safety and tolerability profile over a 40-fold dose range. The pharmacokinetic properties of the molecule support once a day dosing. GKM-001 has the potential to be “First-in-Class” drug to address this large, growing and yet poorly addressed market.
Advinus also has identified a clinical candidate as a back-up to GKM-001, which is structurally different. In its portfolio, the company has a growing pipeline for COPD, sickle cell disease, inflammatory bowel disease, type 2 diabetes, acute and chronic pain and rheumatoid arthritis in various stages of late discovery and pre-clinical development.
About the Diabetes Market:
The present 300 million diabetics population is estimated to jump to 450 million by 2030 worldwide. A large proportion of these patients are poorly controlled despite multiple therapies. Total sales of diabetic prescription products were $32 billion in 2010.
Advinus Therapeutics team discovers novel molecule for treatment of diabetes
- The first glucokinase modulator discovered and developed in India
- A new concept for the management of diabetes for patients, globally
- 100 per cent ‘made in India’ molecule for the treatment of diabetes
- IND approved by DGCI, Phase I clinical trial shows excellent safety and tolerance profiles with efficacy
Bangalore: Advinus Therapeutics (Advinus), the research-based pharmaceutical company founded by leading global pharmaceutical executives and promoted by the Tata group, today, announced the discovery of a novel molecule for the treatment of type II diabetes — GKM-001.The molecule is an activator of glucokinase; an enzyme that regulates glucose balance and insulin secretion in the body.
GKM-001 is a completely indigenously developed molecule and the initial clinical trials have shown excellent results for both safety and efficacy.
“Considering past failures of other companies on this target, our discovery programme primarily focused on identifying a molecule that would be efficacious without causing hypoglycaemia; a side effect associated with most compounds developed for this target.
“Recently completed Phase I data indicate that Advinus’ GKM–001 is a liver selective molecule that has overcome the biggest clinical challenge of hypoglycaemia. GKM-001 is differentiated from most other GK molecules in development due to this novel mechanism of action,” said Dr Rashmi Barbhaiya, MD and CEO, Advinus Therapeutics.
He further added, “We are very proud that GKM-001 is 100 per cent Indian. Advinus’s discovery team in Pune discovered the molecule and entire preclinical development was carried out at our centre in Bangalore. The Investigational New Drug (IND) application was filed with the DGCI for approval to initiate clinical trials in India within 34 months of initiation of the discovery programme. Subsequent to the approval of the IND, we have completed the Phase I Single Ascending Dose study in India within two months.”
GKM-001 is a novel molecule for the treatment of type II diabetes. It is the first glucokinase modulator discovered and developed in India and has potential to be both first or best in class. The success in discovering GKM-001 is attributed to the science-driven efforts in Advinus laboratories and ‘breaking the conventional mold’ for selection of a drug candidate. Advinus has ‘composition of matter’ patent on the molecule for all major markets globally. Glucokinase as a class of target is considered to be novel as currently there is no product in the market or in late clinical trials. The strategy for early clinical development revolved around assessing safety (particularly hypoglycaemia) and early assessment of therapeutic activity (glucose lowering and other biomarkers) in type II diabetics. The Phase I data, in both healthy and type II diabetics, shows excellent safety and tolerability over a 40-fold dose range and desirable pharmacokinetic properties consistent with ‘once a day’ dosing. The next wave of clinical studies planned continues on this strategy of early testing in type II diabetics.
Right behind the lead candidate GKM-001, Advinus has a rich pipeline of back up compounds on the same target. These include several structurally different compounds with diverse potency, unique pharmacology and tissue selectivity. Having discovered the molecule with early indication of wide safety margins, desired efficacy and pharmacokinetic profiles, the company now seeks to out-licence GKM-001 and its discovery portfolio.
Patent
wo 2008104994
| WO2008104994A2 * | 25 Feb 2008 | 4 Sep 2008 | Advinus Therapeutics Private L | 2,2,2-tri-substituted acetamide derivatives as glucokinase activators, their process and pharmaceutical application |
| WO2008104994A2 * | Feb 25, 2008 | Sep 4, 2008 | Advinus Therapeutics Private L | 2,2,2-tri-substituted acetamide derivatives as glucokinase activators, their process and pharmaceutical application |
| WO2009047798A2 * | Oct 7, 2008 | Apr 16, 2009 | Advinus Therapeutics Private L | Acetamide derivatives as glucokinase activators, their process and medicinal applications |
///////GKM 001, pipeline, Diabetes, Advinus, type II diabetes, glucokinase modulator, Rashmi Barbhaiya
Some pics

Annual day party at Advinus !!!with Rashmi Barbhaiya
Dr. Rashmi Barbhaiya, MD & CEO, Advinus Therapeutics Pvt.
 .
.
with Kaushal Joshi, Vishal Pathade, Ramanareddy Jinugu, Mohammed Kakajiwala, Vishal Baxi and Dilip Reddy.
///////
