PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Trecondi, Treosulfan was authorized for medical use in the European Union in June 2019
For use in combination with fludarabine as a preparative regimen for allogeneic hematopoietic stem cell transplantation for acute myeloid leukemia and myelodysplastic syndrome
Treosulfan, sold under the brand name Trecondi among others, is an alkylating medication given to people before they have a bone marrow transplant from a donor known as allogeneic hematopoietic stem cell transplantation. It is used as a ‘conditioning’ treatment to clear the bone marrow and make room for the transplanted bone marrow cells, which can then produce healthy blood cells.[9][10] It is used together with another medicine called fludarabine in adults and children from one month of age with blood cancers as well as in adults with other severe disorders requiring a bone marrow transplant.[9] It belongs to the family of drugs called alkylating agents.[9] In the body, treosulfan is converted into other compounds called epoxides which kill cells, especially cells that develop rapidly such as bone marrow cells, by attaching to their DNA while they are dividing.[9]
The most common side effects include infections, nausea (feeling sick), stomatitis (inflammation of the lining of the mouth), vomiting, diarrhea, and abdominal pain (belly ache).[9] Tiredness, febrile neutropenia (low white blood cell counts with fever) and high blood levels of bilirubin (a breakdown product of red blood cells) are also seen in more than 1 in 10 adults, and rash also affects more than 1 in 10 children.[9] The most common adverse reactions include musculoskeletal pain, stomatitis, pyrexia, nausea, edema, infection, and vomiting.[7] Selected grade 3 or 4 nonhematological laboratory abnormalities include increased GGT, increased bilirubin, increased ALT, increased AST, and increased creatinine.[7]
Treosulfan was authorized for medical use in the European Union in June 2019,[9] and approved for medical use in the United States in January 2025.[7][11]
Medical Uses
Treosulfan in combination with fludarabine is indicated as part of conditioning treatment prior to allogeneic haematopoietic stem cell transplantation in adults with malignant and non malignant diseases, and in children older than one month with malignant diseases.[7][9]
History
Two main studies showed that treosulfan is at least as effective as busulfan, another medicine used to prepare people for haematopoietic stem cell transplantation.[9]
In one of the studies, involving 570 adults with acute myeloid leukaemia (a blood cancer) or myelodysplastic syndromes (conditions in which large numbers of abnormal blood cells are produced), 64% of patients given treosulfan (with fludarabine) had a successful transplant and were alive and disease-free after 2 years, compared with 51% of patients given busulfan (with fludarabine).[9]
In an additional study in 70 children with blood cancers, 99% of children given treosulfan (with fludarabine) were alive three months after their transplant.[9]
Efficacy was evaluated in MC-FludT.14/L Trial II (NCT00822393), a randomized active-controlled trial comparing treosulfan to busulfan with fludarabine as a preparative regimen for allogeneic transplantation. Eligible patients included adults 18 to 70 years old with AML or MDS, Karnofsky performance status ≥ 60%, and age ≥ 50 years or hematopoietic cell transplantation comorbidity index [HCTCI] score > 2. There were 570 patients randomized to treosulfan (n=280) or busulfan (n=290).
Society and culture
Legal status
Treosulfan was authorized for medical use in the European Union in June 2019,[9] and approved for medical use in the United States in January 2025.[11][12][13]
The US Food and Drug Administration granted orphan drug designation to treosulfan in 1994, for the treatment of ovarian cancer;[14] and in 2015, for conditioning treatment prior to hematopoietic stem cell transplantation in malignant and non-malignant diseases in adults and pediatric patients.[15]
In February 2004, orphan designation (EU/3/04/186) was granted by the European Commission to medac Gesellschaft fuer klinische Spezialpräparate mbH, Germany, for treosulfan for the conditioning treatment prior to haematopoietic progenitor cell transplantation.[16]
Treosulfan is sold under the brand names Trecondi[9] and Grafapex.[7]
SYN
Treosulfan is an active ingredient of the drug Ovastat . Treosulfan is indicated for the treatment of ovarian cancer and belongs to the class of alkylating agents, which prevents the growth and division of cancerous cells.
US3155702 discloses the preparation of Treosulfan by methanesulphonation of (2S,3S)- l,4-dibromobutane-2,3-diol with excess amount of silver methanesulphonate. The presence of free 2,3-diol in the starting material leads to side reactions and formation of undesired by-products which necessitates an additional purification step and thereby results in lower yields. Further, an additional filtration operation is also required to remove silver bromide salt generated during the process and un-reacted silver methanesulphonate, which makes the process less attractive for commercial manufacturing.
US3246012 discloses the preparation of Treosulfan by protection of hydroxyl group of dialkyl tartrates with corresponding aldehyde, ketone or a reactive derivatives to form corresponding cyclic 2,3-O-acetals and 2,3-O-ketals of butanetetrol esters followed by reduction using lithium aluminium hydride to obtain 2,3-O-acetal or ketal protected butanetetrol, which is further methanesulphonated and treated with acid. The use of highly pyrophoric and hazardous reducing agent renders the above process not ideal for industrial production. Organic Syntheses, Coll. Vol. 10, p. 297, 2004 discloses a similar reaction sequence followed by the final de-protection of methanesulphonated 2,3-O-diisopropylidene-L- threitol in methanesulfonic acid at reflux temperature, which leads to a sluggish reaction mixture and a higher number of impurities due to maintaining the reaction mixture for longer time at higher temperature.
IN 1568/MUM/2012 also discloses similar reaction sequence involving reduction of dimethyl-2,3-0-isopropylidene-L-tartrate by sodium-bis(2-methoxyethoxy) aluminium hydride followed by methanesulphonation and final deprotection with formic acid to yield Treosulfan.
KR101367641 describes reduction using lithium borohydride, which requires about 14 hours to complete the reaction and is further extended due to involvement of column chromatography purification. Tetrahedron, vol. 49, no. 30, p. 6645, 1993 describes reduction using sodium borohydride and lithium chloride, followed by flash chromatography purification. Reduction conditions as per Chem. Pharm. Bull. Vol. 42, No. 3, p. 68, 1994, are again not commercially feasible because of lithium aluminium hydride as reducing agent.
Haberland, M., Weber, S., Sharma, A. K., Upadhyay, S., Dua, H., Musmade, S., Singh, G., Lahiri, S., & Cabri, W. (2019). A process for the preparation of Treosulfan (Patent No. WO2019043587A2).
EXAMPLES Detailed experimental parameters suitable for the preparation of Treosulfan or intermediates according to the present invention are provided by the following examples, which are intended to be illustrative and not limiting.
Reference Example 1 (repetition of Tetrahedron, vol. 46, No. 12, p. 4165, 1990):
A reaction mixture of dimethyl-L-tartrate (10. Og), p-toluene sulfonic acid (0.013g) and p- anisaldehydedimethylacetal (l l.Og) in toluene (150ml) was refluxed and the azeotropical mixture of toluene-methanol was continuously removed from the reaction mixture for 3-5 hours. The reaction mixture was cooled to ambient temperature, diluted with dichloromethane (50ml) and neutralised by addition of potassium carbonate (5.0g) followed by stirring for an hour . The reaction mixture was filtered and filtrate was evaporated to give yellow crude compound, which was further dissolved in dichloromethane (25ml) followed by addition of petroleum ether (100ml) and stirred for an hour at ambient temperature. The solid was filtered, washed with petroleum ether (20ml) and dried under vacuum at 35-40°C for 15-20 hours to obtain 16.63g (72.15%) of dimethyl (4R,5R)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5-dicarboxylate having purity 98.4% by HPLC.
Reference Example 2 (repetition of Synthesis, No. 15, p. 2488-90, 2008):
A reaction mixture of dimethyl-L-tartrate (5.0g), p-toluene sulfonic acid (0.0064g) and p- anisaldehyde dimethylacetal (5.35g) in toluene (25ml) was refluxed and the azeotropical mixture of toluene-methanol was continuously removed from the reaction mixture for 3-5 hours. The reaction mixture was cooled to ambient temperature, diluted with dichloromethane (25ml) and neutralised by addition of potassium carbonate (5.0g) followed by stirring for an hour. The reaction mixture was filtered and filtrate was evaporated to give yellow crude residues. The crude was further re-crystallized in petroleum ether (25ml), filtered the solid and washed with petroleum ether (15ml) followed by drying under vacuum at 35-40°C for 15-20 hours to obtain 7.4g (89.15%) of dimethyl (4R,5R)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5-dicarboxylate having purity 98.8% by HPLC. Example-1: Preparation of dimethyl (4R,5R)-2-(4-methoxyphenyl)-l,3-dioxolane- 4,5-dicarboxylate
A reaction mixture of dimethyl-L-tartrate (500g), p-toluene sulfonic acid (5.38g) and p- anisaldehyde dimethylacetal (665g) in toluene (2250ml) was refluxed to 110-115°C. The azeotropical mixture of toluene-methanol was continuously removed from the reaction mixture till the completion of the reaction. The reaction mixture was cooled to ambient temperature and quenched with aq. saturated sodium bicarbonate solution (2500ml), layers were separated. Resulting organic layer was washed with water (2500ml x 2) followed by evaporation of organic layer. Isopropyl alcohol (3500ml) was charged to the residue and heated to 60-70°C followed by cooling at ambient temperature. Reaction mixture was stirred at 0-5°C for 1-2 hours and filtered. The solid thus obtained was washed with pre- cooled isopropyl alcohol and dried under vacuum at 35-40°C for 15-20 hours to obtain 767.0g (92.93%) of dimethyl (4R,5R)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- dicarboxylate having purity 99.97% by HPLC.
Example-2: Preparation of (4S,5S)-2-(4-methoxyphenyl)-l 53-dioxo!ane-4,5- diyifdimethanol
Method-l :To a mixture of dimethyl (4R,5R)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- dicarboxylate (765g), Iodine (13. lg) in tetrahydrofuran (3750ml) and water (76ml), sodium borohydride (146.52g) was added at 0-15°C and stirred for 1 -2 hours at ambient temperature. The reaction was quenched with 30% aq. ammonium chloride (6100ml) solution and dichloromethane (7650ml). The layers were separated and the aqueous layer was extracted by dichloromethane (3800ml x 3) followed by washing of combined organic layers with water (3800ml), The resulting organic layer was evaporated at 35-65°C to obtain 525.0g (83.9%) of (4S,5S)-2-(4-methoxyphenyl)-l,3- dioxolane-4,5-diyl]dimethanol having purity 99.72% by HPLC. Method-2: To a mixture of dimethyl (4R,5R)-2-(4-methoxyphenyl)-l,3-dioxolane- 4,5-dicarboxylate (765g), Iodine (13.10g) in tetrahydrofuran (3750ml) and water (76.5ml), sodium borohydride (146.52g) was added at 0-10°C and stirred for Ihours at 0-5°C and stirred for 3-4 hours at ambient temperature. The reaction was quenched with 30% aq. ammonium chloride (6120ml) solution and dichloromethane (7650ml) at ambient temperature. The layers were separated and the aqueous layer was extracted by dichloromethane (3825m! x 3) followed by washing of combined organic layers with water (3825ml). The resulting organic layer was evaporated at 50-60°C to obtain 525 g (84.7%) of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5-diyl]dirnethaiiol having purity 99.72% by HPLC. Example-3: Preparation of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- diyl]bis(methylene) dimethanesulfonate
Method-l:To a solution of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- diyl]dimethanol (145g) in dichloromethane (2175ml), pyridine (191g) and methanesulphonyl chloride (190. l g) was added at 0-5 °C. The reaction mixture was stirred for 2-3 hours at ambient temperature followed by quenching with water (1450ml). The organic layer was washed with water (1450ml x 4) and evaporated. The resulting residue was added to isopropanol (725ml) and stirred for 1-2 hours at ambient temperature and further for 1-2 hours at 0-5 C. The solid was filtered and washed with pre-cooled isopropanol (145ml). The resulting product was dissolved in acetone (1300ml) followed by addition of isopropanol (2610ml). Resulting reaction mixture was stirred for 1-2 hours at ambient temperature and then cooled at 0-5 °C. The solid thus obtained was filtered and washed with pre-cooled isopropanol (145ml x 2) and dried under vacuum at 30-35°C for 15-20 hours to give 190.8g (79.4%)of (4S,5S)-2-(4- methoxyphenyl)-l,3-dioxolane-4,5-diyl]bis(methylene) dimethanesulfonate. Method-2: To a solution of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- diyl]dimethanol (525, Og) in dichloromethane (7350ml), di-isopropylamine (663. Og) was added at ambient temperature followed by addition of methanesulphonyl chloride solution (624. Og in 525ml dichloromethane) at 0-10°C. The reaction mixture was stirred for 1-2 hours at 0-10 °C followed by stirring for 3-4 hours at ambient temperature. The organic layer was washed with water (2 x 5250ml) and evaporated. The residues were dissolved in acetone (4725ml) followed by addition of isopropanol (9450ml), stirred for about 1-2 hour at ambient temperature and then at 0-5 °C for 1-2 hours. The resulting solid was filtered, washed with pre-cooled isopropanol (525 x 2 ml)and dried under vacuum at 35-45°C for 15-20 hours to give 705.0g (81.45%) of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5-diyl]bis(methylene)
dimethanesulfonate having purity 99.92% by HPLC.
Example-4: Preparation of Treosulfan
Method-1: To a solution of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- diyl]bis(methylene) dimethanesulfonate (745. Og) in methanol (7450ml), concentrated hydrochloric acid (260ml) was added at 15-25°C followed by stirring for 10-15 hours at ambient temperature. The reaction mixture was cooled to 0-5°C and further stirred for 1-2 hours at 0-5°C followed by filtration and washing the solid with pre-cooled methanol (745ml). The solid thus obtained was dissolved in acetone (3725ml) followed by microne filtration. Di-isopropyl ether (7450ml) was added to the filtrate and stirred for 1-2 hours at ambient temperature and then cooled at 0-5°C. The solid thus obtained was filtered and washed with di-isopropyl ether (745ml x 2) followed by drying at 30-35°C for 15-20 hours to obtain 96.5g of Treosulfan having purity 99.9% by HPLC.
XRPD of Treosulfan obtained by above process is shown in Fig. 1. Method-2:To a solution of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- diyl]bis(methylene)dimethanesulfonate (650. Og) in methanol (6500ml), 9N hydrochloric acid (227.5ml) was added at 0-10°C followed by stirring for 6-8 hours at ambient temperature. The reaction mixture was cooled to 0-5°C and further stirred for 1-2 hours followed by filtration and washing the solid with pre-cooled methanol (2 x 650ml). The solid thus obtained was dissolved in acetone (3250ml). Di-isopropyl ether (6500ml) was added to the resulting solution, stirred for 1-2 hours at ambient temperature and then cooled at 0-5°C. The solid thus obtained was filtered and washed with di- isopropyl ether (650ml x 2) followed by drying at 30-35°C for 15-20 hours to obtain 312g (68.4) of Treosulfan having purity 99.81% by HPLC.
Example 1 – Preparation of form B using water/isopropanol
99.8 mg treosulfan were weighed in a vial (volume 4.0 ml) which was equipped with a PTFE (Polytetrafluoroethylene) sealing and a stirrer. 1.5 ml of a mixture of 80 % by weight water and 20 % by weight isopropanol preheated to 65°C were then added. The resulting solution was completely taken up with a syringe (volume 5 ml) and filtered using a 0.2 pm filter into a second vial (volume 4.0 ml) . The syringe, second vial and filter had been tempered at 65°C before use. The solvents were allowed to evaporate from the open vial at room temperature to dryness which resulted in formation of crystals.
The XRPD pattern of the obtained crystals of form B according to the invention is shown in Figure 1.
PATENT
1568/MUM/2012
Abstract
Abstract: The present invention provides a convenient and cost-effective process for preparation of Treosulfan. The process comprises reduction of dimethyl 2,3-O-isopropylidene-L-tartrate with sodium-bis(2-methoxyethoxy)aluminum hydride to give the alcohol 2,3-O-isopropylidene-L-threitol (III), which on reaction with methanesulfonyl chloride led to 2,3-O-isopropylidene-L-threitol 1,4-bismethanesulfonate of formula (IV) and further treatment of compound (IV) with formic acid gave Treosulfan (I) having desired purity.
Treosulfan (I), chemically known as (2S,3S)-2,3-Dihydroxy-4-memylsidfonyIoxybutylj methanesulfonate is a drug commonly used for treating ovarian cancer. It belongs to the family of anti-cancer medicines called the alkylating agents, which prevent the growth and division of cancerous cells. Treosulfan has been used for bone-marrow ablation before stem-cell transplantation and in the treatment of malignant melanoma and breast cancer.
US 3,155,702 discloses synthesis of Treosulfan by replacement of the halogen function in L-Threitol-l,4-dibromobutane-2,3-diol, by treating with a large excess of an expensive reagent like silver methanesulfonate. Further, the presence of unprotected hydroxyl groups in the starting material inevitably leads to the formation of undesired impurities, which requires additional purification steps for removal of impurities as well for lowering the level of free silver in the active ingredient as per ICH guidelines, which results in lower yields and increases the costs substantially. Another method reported in US 3,246,012 involves acetal formation of diethyl-L-tartrate with acetone to obtain 2,3-O-isopropylidene-diethyl-L-tartrate, which, when reduced with lithium aluminium hydride gives 2,3-0-methylene-L-threitol. The obtained alcohol was treated with methanesulfonyl chloride to yield the penultimate Treosulfan intermediate, 2,3-O-methylene-L-threitol-1,4-di-(methanesulfonate).
A similar approach which employs tartrate esters in the synthesis of Treosulfan, is disclosed in Organic Syntheses, (1993), Vol.8, p. 155 and Organic .Syntheses, (2004), Coll.Vol.10, p.297. L-tartaric acid is reacted with 2,2-dimethoxypropane in presence of methanol. The resulting methyl ester, dimethyl 2,3-O-isopropylidene-L-tartrate is reduced with lithium aluminium hydride to obtain 2,3-di-O-isopropylidene-L-threitol, which, upon reaction with methanesulfonyl chloride, followed by treatment with methanesulfonic acid yields Treosulfan. Although these routes involve protection of the diol group and avoid impurities arising out of substitution at those alcohol functionalities, use of a highly pyrophoric, hazardous reagent such as lithium aluminium hydride severely limits their synthetic applicability, especially on commercial scale. Further, the final step involves reaction of 2,3-di-O-isopropylidene-L-threitol with methanesulfonic acid, which is quite sluggish and causes considerable rise in the total number of impurities due to long reaction time. Thus, there is a need for a convenient, economical process for a commercial scale synthesis of Treosulfan (I), which overcomes the shortcomings of the prior art, does not involve use of hazardous, pyrophoric reagents and yields Treosulfan conforming to regulatory specifications. The present inventors have developed a novel process for preparation of (2S,3S)-2,3-Dihydroxy-4-methylsulfonyloxybutyl] methanesulfonate (I). The scheme for synthesis comprises reaction of dimethyl 2,3-O-isopropylidene-L-tartrate of formula (II) with sodium-bis(2-methoxyethoxy) aluminum hydride to give the protected diol, 2,3-0-isopropylidene-L-threitoI (III), which on further treatment with methanesulfonyl chloride, followed by reaction of the resultant ester, 2,3-O-isopropyliden-L-threitol 1,4 bismethanesulfonate (IV) with formic acid, yields Treosulfan (I) having desired purity and with impurity levels conforming to ICH guidelines.
Scheme 1; Method embodied in the present invention for the preparation of Treosulfan (I) In an embodiment, dimethyl 2,3 -O-isopropylidene-L-tartrate of formula (II) was treated with sodium-bis-(2-methoxyethoxy) aluminium hydride in presence of an organic solvent, and in the temperature range of 25 to 80°C, but preferably 60 to 75°C. The organic solvent was selected from the group of toluene, xylenes, nitrobenzene, hexane, cyclohexane, heptane, N-methyl-2-pyrroIidone, ethers etc. Upon completion of the reaction, as monitored by TLC, water was carefully added to the reaction mass and the mixture was extracted with a water immiscible organic solvent. The organic solvent was selected from the group comprising of n-hexane, cyclohexane, heptane, methyl isobutyl ketone, 2-methyl tetrahydrofuran, cyclopentyl methyl ether etc. The organic layer was separated and concentrated under reduced pressure to give 2,3-0-isopropylidene-L-threitol of formula (III) of desired purity. It is pertinent to mention that the reaction was quite facile and the desired product was obtained with minimal formation of associated impurities and did not require any subsequent purification.
Further reaction of compound (III) with methanesulfonyl chloride was carried out at 25 to 35°C, in an organic solvent, in presence of an organic base. The organic solvent was selected from the group comprising of chloroform, ethylene dichloride, dichloromethane, carbon tetrachloride etc., but preferably dichloromethane. The organic base was selected from triethyl amine, tributyl amine and pyridine. The reaction mixture was stirred at 25-35°C and after completion of the reaction as monitored by TLC, aqueous solution of sodium bicarbonate was added slowly to the reaction mass. The organic layer was separated, concentrated under reduced pressure and stirred with isopropyl alcohol to obtain the desired compound, 2,3-O-isopropylidene-L-threitol-l,4-bis(methanesulfonate) of formula (IV). In a further embodiment, compound (TV) was hydrolyzed by treating with formic acid at 25 to 35°C based on TLC. After completion of the reaction, the reaction mass was concentrated and the product Treosulfan (I) was isolated by addition of isopropyl alcohol to the concentrated mass. It is pertinent to mention that Organic Syntheses (2004), Coll.Vol. 10, p.297 discloses the hydrolysis reaction using methanesulfonic acid in ethanol at reflux temperature. However, the time taken for completion is about ten hours and the procedure is applicable only for laboratory scale reaction. The hydrolysis step disclosed in the present invention is easily scalable and so facile that it takes place at room temperature and within one to two hours. This reduces the time cycle for each batch run and also reduces the possibility of formation of undesired side products. Dimethyl 2,3-O-isopropylidene-L-tartrate of formula (II) was prepared by the reaction of dimethyl -L-tartrate with acetone by following known synthetic procedures.
The following examples are meant to be illustrative of the present invention. These examples exemplify the invention and are not to be construed as limiting the scope of the invention. EXAMPLES Example 1: Synthesis of 2,3-O-isopropylidene-L-threitol (HI) A solution of dimethyl-2,3-0-isopropylidene-L-tartrate (50.3 g) in toluene (50 ml) was gradually added to the stirred mixture of sodium-bis(2-methoxyethoxy) aluminum hydride (122.8 g) in toluene (50 ml) at 20-40°C. The reaction mixture was heated to 60-80°C, and the reaction was continued till completion, as monitored by TLC. When the reaction was complete, the mass was cooled to 25-3 5°C, quenched with careful addition of water (10ml) and concentrated. Treatment of the resulting residue with methyl tertiary butyl ether, followed by evaporation of the organic layer under reduced pressure afforded 2,3-0-isopropyliden -L-threitol ( III) as pale yellow oil. Yield: 29.8 g (81.2%) [α]D20 + 4.6.°(CHC13, c 5) Example 2: Synthesis of 2,3-0-isopropylidene-L-threitol-l,4-bis(methanesulfonate) (IV) A stirred solution of 2,3-O-isopropylidene-L-threitol (100.2 g), methylene chloride (1250 ml) and pyridine (146.3 g) was cooled to 0-5°C and methanesulfonyl chloride (176.6 g) was slowly added to it. Temperature of the reaction mixture was raised to 25-35°C and the reaction was continued at the same temperature till completion of the reaction, as monitored by HPLC. After completion of the reaction, aqueous sodium bicarbonate solution was slowly added to the reaction mass and the organic layer was separated. Aqueous layer from the reaction mixture was extracted with methylene chloride and the organic layers were combined. Distillation of the organic solvent, optionally followed by addition of isopropyl alcohol gave the product, 2,3-0-isopropylidene-L-threitol-l,4- bis(methanesulfonate). Yield: 160.7 g (79.7%) [α]D20-21.6°(acetone,c2)
Example 3: Synthesis of Treosulfan (I) A mixture of formic acid (98%, 1000 ml) and 2,3-0-isopropylidene-L-threitol-l,4-bis(methanesulfonate) (100.5 g) was stirred at room temperature until completion of the desired reaction, as monitored by TLC, When the reaction was complete, the reaction mass was concentrated under reduced pressure.. Treatment of the residue after evaporation with isopropanol yielded the final product Treosulfan, which was optionally subjected to further treatment with acetone and nexanes or petroleum ether, Yield: 74.3 g (85.0%) [α]D20 – 5.3°(acetone, c 2) Purity: > 99 %.
References
^ Jump up to:ab“Trecondi APMDS”. Therapeutic Goods Administration (TGA). 11 October 2022. Retrieved 25 January 2025.
^ Jump up to:abcdefghijklm“Trecondi EPAR”. European Medicines Agency (EMA). 11 December 2018. Archived from the original on 16 March 2023. Retrieved 21 April 2020. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
Clinical trial number NCT00822393 for “Clinical Phase III Trial Treosulfan-based Conditioning Versus Reduced-intensity Conditioning (RIC)” at ClinicalTrials.gov
Romanski M, Baumgart J, Bohm S, Glowka FK: Penetration of Treosulfan and its Active Monoepoxide Transformation Product into Central Nervous System of Juvenile and Young Adult Rats. Drug Metab Dispos. 2015 Dec;43(12):1946-54. doi: 10.1124/dmd.115.066050. Epub 2015 Oct 1. [Article]
EMA Summary of Product Characteristics: Trecondi (treosulfan) powder for solution for infusion [Link]
FDA Approved Drug Products: GRAFAPEX (treosulfan) for injection, for intravenous use [Link]
EMC Summary of Product Characteristics: Treosulfan 5g Powder for Solution for Infusion [Link]
FDA News Release: FDA approves treosulfan with fludarabine as a preparative regimen for alloHSCT in adult and pediatric patients with AML or MDS [Link]
ABBV-744 is a highly BDII-selective BET bromodomain inhibitor, used in the research of inflammatory diseases, cancer, and AIDS.
Acute Myeloid Leukemia (AML)
Phase I, AbbVie is evaluating oral agent ABBV-744 in early clinical trials for the treatment of metastatic castration resistant prostate cancer (CRPC) and for the treatment of relapsed or refractory acute myeloid leukemia (AML).
Bromodomains refer to conserved protein structural folds which bind to N-acetylated lysine residues that are found in some proteins. The BET family of bromodomain containing proteins comprises four members (BRD2, BRD3, BRD4 and BRDt) . Each member of the BET family employs two bromodomains to recognize N-acetylated lysine residues typically, but not exclusively those found on transcription factors (Shi, J., et al. Cancer Cell 25 (2) : 210-225 (2014) ) or on the amino-terminal tails of histone proteins. Numbering from the N-terminal end of each BET protein the tandem bromodomains are typically labelled Binding Domain I (BDI) and Binding Domain II (BDII) . These interactions modulate gene expression by recruiting transcription factors to specific genome locations within chromatin. For example, histone-bound BRD4 recruits the transcription factor P-TEFb to promoters, resulting in the expression of a subset of genes involved in cell cycle progression (Yang et al., Mol. Cell. Biol. 28: 967-976 (2008) ) . BRD2 and BRD3 also function as transcriptional regulators of growth promoting genes (LeRoy et al., Mol. Cell 30: 51-60 (2008) ) . BET family members were recently established as being important for the maintenance of several cancer types (Zuber et al., Nature 478: 524-528 (2011) ; Mertz et al; Proc. Nat’l. Acad. Sci. 108: 16669-16674 (2011) ; Delmore et al., Cell 146: 1-14, (2011) ; Dawson et al., Nature 478: 529-533 (2011) ) . BET family members have also been implicated in mediating acute inflammatory responses through the canonical NF-KB pathway (Huang et al., Mol. Cell. Biol. 29: 1375-1387 (2009) ) resulting in the upregulation of genes associated with the production of cytokines (Nicodeme et al., Nature 468: 1119-1123, (2010) ) . Suppression of cytokine induction by BET bromodomain inhibitors has been shown to be an effective approach to treat inflammation-mediated kidney disease in an animal model (Zhang, et al., J. Biol. Chem. 287: 28840-28851 (2012) ) . BRD2 function has been linked to pre-disposition for dyslipidemia or improper regulation of adipogenesis, elevated inflammatory profiles and increased susceptibility to autoimmune diseases (Denis, Discovery Medicine 10: 489-499 (2010) ) . The human immunodeficiency virus utilizes BRD4 to initiate transcription of viral RNA from stably integrated viral DNA (Jang et al., Mol. Cell, 19: 523-534 (2005) ) . BET bromodomain inhibitors have also been shown to reactivate HIV transcription in models of latent T cell infection and latent monocyte infection (Banerjee, et al, J. Leukocyte Biol. doi: 10.1189/jlb. 0312165) . BRDt has an important role in spermatogenesis that is blocked by BET bromodomain inhibitors (Matzuk, et al., Cell 150: 673-684 (2012) ) . Thus, compounds that inhibit the binding of BET family bromodomains to their cognate acetylated lysine proteins are being pursued for the treatment of cancer, inflammatory diseases, kidney diseases, diseases involving metabolism or fat accumulation, and some viral infections, as well as for providing a method for male contraception. Accordingly, there is an ongoing medical need to develop new drugs to treat these indications.
Phase 1 Clinical, Acute myelogenous leukemia, Protein cereblon modulator
Useful for treating chronic lymphocytic leukemia, chronic myelocytic leukemia, acute lymphoblastic leukemia or acute myeloid leukemia.
Celgene is developing CC-90009, a cereblon E3 ligase modulator, for treating AML; in January 2019, data from a phase I trial were expected later that year.
0iginator Celgene Corporation
Class Antineoplastics
Mechanism of Action CRBN protein modulators; Ubiquitin protein ligase complex modulators
Phase I Acute myeloid leukaemia
28 Mar 2019 No recent reports of development identified for clinical-Phase-Unknown development in Acute-myeloid-leukaemia in USA (IV)
01 Sep 2016 Phase-I clinical trials in Acute myeloid leukaemia (Second-line therapy or greater) in Canada (IV) (NCT02848001)
04 Aug 2016 Celgene plans a phase I trial for Acute Myeloid Leukaemia in USA and Canada (NCT02848001)
In September 2016, Celgene initiated a phase I dose-finding trial of CC 90009 in patients with relapsed or refractory acute myeloid leukaemia (NCT02848001; CC-90009-AML-001). The open-label study intends to enrol 60 patients in the US and Canada
CC-90009 is a cereblon modulator. CC-90009 specifically binds to CRBN, thereby affecting the activity of the ubiquitin E3 ligase complex. This leads to the ubiquitination of certain substrate proteins and induces the proteasome-mediated degradation of certain transcription factors, including Ikaros (IKZF1) and Aiolos (IKZF3), which are transcriptional repressors in T-cells. This reduces the levels of these transcription factors, and modulates the activity of the immune system, which may include the activation of T-lymphocytes. .
Development Overview
cereblon modulator CC-90009A modulator of cereblon (CRBN), which is part of the cullin 4-RING E3 ubiquitin ligase complex (CRL4-CRBN E3 ubiquitin ligase; CUL4-CRBN E3 ubiquitin ligase), with potential immunomodulating and pro-apoptotic activities. Upon administration, CC-90009 specifically binds to CRBN, thereby affecting the activity of the ubiquitin E3 ligase complex. This leads to the ubiquitination of certain substrate proteins and induces the proteasome-mediated degradation of certain transcription factors, including Ikaros (IKZF1) and Aiolos (IKZF3), which are transcriptional repressors in T-cells. This reduces the levels of these transcription factors, and modulates the activity of the immune system, which may include the activation of T-lymphocytes. In addition, this downregulates the expression of other proteins, including interferon regulatory factor 4 (IRF4) and c-myc, which plays a key role in the proliferation of certain cancer cell types. CRBN, the substrate recognition component of the E3 ubiquitin ligase complex, plays a key role in the ubiquitination of certain proteins. Check for active clinical trials using this agent. (NCI Thesaurus)
Provided herein are methods of treating, preventing, managing, and/or ameliorating a hematologic malignancy with 2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide or a stereoisomer or a mixture of
stereoisomers, an isotopologue, pharmaceutically acceptable salt, tautomer, solvate, hydrate, co-crystal, clathrate, or polymorph thereof. Further provided is a compound for use in methods of treating, preventing, managing, and/or ameliorating a hematologic malignancy, wherein the compound is 2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide or a stereoisomer or a mixture of stereoisomers, an isotopologue, pharmaceutically acceptable salt, tautomer, solvate, hydrate, co-crystal, clathrate, or polymorph thereof.
The term Compound 1 refers to”2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide” having the structure:
and its stereoisomers or mixture of stereoisomers, isotopologues, pharmaceutically acceptable salts, tautomers, solvates, hydrates, co-crystals, clathrates, or polymorphs thereof. In certain embodiments, Compound 1 refers to 2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide and its tautomers. In certain embodiments, Compound 1 refers to a polymorph of 2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-
oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide. In certain embodiments, Compound 1 refers to polymorph Form C of 2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide. In one embodiment, the stereoisomer is an enantiomer.
PATENT
WO-2019136016
Novel isotopologs of the compound presumed to be CC-90009 , processes for their preparation and compositions comprising them are claimed.
SOLID FORMS OF 2-(4-CHLOROPHENYL)-N-((2-(2,6-DIOXOPIPERIDIN-3-YL)-1-OXOISOINDOLIN-5-YL)METHYL)-2,2-DIFLUOROACETAMIDE, AND THEIR PHARMACEUTICAL COMPOSITIONS AND USES
Enasidenib (AG-221) is an experimental drug in development for treatment of cancer. It is a small molecule inhibitor of IDH2 (isocitrate dehydrogenase 2). It was developed by Agios Pharmaceuticals and is licensed to Celgene for further development.
An orally available inhibitor of isocitrate dehydrogenase type 2 (IDH2), with potential antineoplastic activity. Upon administration, AG-221 specifically inhibits IDH2 in the mitochondria, which inhibits the formation of 2-hydroxyglutarate (2HG). This may lead to both an induction of cellular differentiation and an inhibition of cellular proliferation in IDH2-expressing tumor cells. IDH2, an enzyme in the citric acid cycle, is mutated in a variety of cancers; It initiates and drives cancer growth by blocking differentiation and the production of the oncometabolite 2HG.
Isocitrate dehydrogenases (IDHs) catalyze the oxidative decarboxylation of isocitrate to 2-oxoglutarate (i.e., a-ketoglutarate). These enzymes belong to two distinct subclasses, one of which utilizes NAD(+) as the electron acceptor and the other NADP(+). Five isocitrate dehydrogenases have been reported: three NAD(+)-dependent isocitrate dehydrogenases, which localize to the mitochondrial matrix, and two NADP(+)-dependent isocitrate dehydrogenases, one of which is mitochondrial and the other predominantly cytosolic. Each NADP(+)-dependent isozyme is a homodimer.
IDH2 (isocitrate dehydrogenase 2 (NADP+), mitochondrial) is also known as IDH; IDP; IDHM; IDPM; ICD-M; or mNADP-IDH. The protein encoded by this gene is the
NADP(+)-dependent isocitrate dehydrogenase found in the mitochondria. It plays a role in intermediary metabolism and energy production. This protein may tightly associate or interact with the pyruvate dehydrogenase complex. Human IDH2 gene encodes a protein of 452 amino acids. The nucleotide and amino acid sequences for IDH2 can be found as GenBank entries NM_002168.2 and NP_002159.2 respectively. The nucleotide and amino acid sequence for human IDH2 are also described in, e.g., Huh et al., Submitted (NOV-1992) to the
EMBL/GenBank/DDBJ databases; and The MGC Project Team, Genome Res.
14:2121-2127(2004).
Non-mutant, e.g., wild type, IDH2 catalyzes the oxidative decarboxylation of isocitrate to a-ketoglutarate (a- KG) thereby reducing NAD+ (NADP+) to NADH (NADPH), e.g., in the forward reaction:
It has been discovered that mutations of IDH2 present in certain cancer cells result in a new ability of the enzyme to catalyze the NAPH-dependent reduction of α-ketoglutarate to R(-)-2-hydroxyglutarate (2HG). 2HG is not formed by wild- type IDH2. The production of 2HG is believed to contribute to the formation and progression of cancer (Dang, L et al, Nature 2009, 462:739-44).
The inhibition of mutant IDH2 and its neoactivity is therefore a potential therapeutic treatment for cancer. Accordingly, there is an ongoing need for inhibitors of IDH2 mutants having alpha hydroxyl neoactivity.
Mechanism of action
Isocitrate dehydrogenase is a critical enzyme in the citric acid cycle. Mutated forms of IDH produce high levels of 2-hydroxyglutarate and can contribute to the growth of tumors. IDH1 catalyzes this reaction in the cytoplasm, while IDH2 catalyzes this reaction in mitochondria. Enasidenib disrupts this cycle.[1][2]
Development
The drug was discovered in 2009, and an investigational new drug application was filed in 2013. In an SEC filing, Agios announced that they and Celgene were in the process of filing a new drug application with the FDA.[3] The fast track designation allows this drug to be developed in what in markedly less than the average 14 years it takes for a drug to be developed and approved.[4]
Example 1, Step 1: preparation of 6-trifluoromethyl-pyridine-2-carboxylic acid
Diethyl ether (4.32 L) and hexanes (5.40 L) are added to the reaction vessel under N2 atmosphere, and cooled to -75 °C to -65 °C. Dropwise addition of n-Butyl lithium (3.78 L in 1.6 M hexane) under N2 atmosphere at below -65 °C is followed by dropwise addition of dimethyl amino ethanol (327.45 g, 3.67 mol) and after 10 min. dropwise addition of 2-trifluoromethyl pyridine (360 g, 2.45 mol). The reaction is stirred under N2 while maintaining the temperature below -65 °C for about 2.0-2.5 hrs. The reaction mixture is poured over crushed dry ice under N2, then brought to a temperature of 0 to 5 °C while stirring (approx. 1.0 to 1.5 h) followed by the addition of water (1.8 L). The reaction mixture is stirred for 5-10 mins and allowed to warm to 5-10 °C. 6N HC1 (900 mL) is added dropwise until the mixture reached pH 1.0 to 2.0, then the mixture is stirred for 10-20 min. at 5-10 °C. The reaction mixture is diluted with ethyl acetate at 25-35 °C, then washed with brine solution. The reaction is concentrated and rinsed with n-heptane and then dried to yield 6-trifluoromethyl-pyridine-2-carboxylic acid.
Example 1, Step 2: preparation of 6-trifluoromethyl-pyridine-2-carboxylic acid methyl ester Methanol is added to the reaction vessel under nitrogen atmosphere. 6-trifluoromethyl- pyridine-2-carboxylic acid (150 g, 0.785 mol) is added and dissolved at ambient temperature. Acetyl chloride (67.78 g, 0.863 mol) is added dropwise at a temperature below 45 °C. The reaction mixture is maintained at 65-70 °C for about 2-2.5 h, and then concentrated at 35-45 °C under vacuum and cooled to 25-35 °C. The mixture is diluted with ethyl acetate and rinsed with saturated NaHC03 solution then rinsed with brine solution. The mixture is concentrated at temp 35-45 °C under vacuum and cooled to 25-35 °C, then rinsed with n-heptane and concentrated at temp 35-45 °C under vacuum, then degassed to obtain brown solid, which is rinsed with n-heptane and stirred for 10-15 minute at 25-35 °C. The suspension is cooled to -40 to -30 °C while stirring, and filtered and dried to provide 6-trifluoromethyl-pyridine-2-carboxylic acid methyl ester.
Example 1, Step 3: preparation of 6-(6-Trifluoromethyl-pyridin-2-yl)-lH-l,3,5-triazine-2,4-dione
1 L absolute ethanol is charged to the reaction vessel under N2 atmosphere and Sodium Metal (11.2 g, 0.488 mol) is added in portions under N2 atmosphere at below 50 °C. The reaction is stirred for 5-10 minutes, then heated to 50-55 °C. Dried Biuret (12.5 g, 0.122 mol) is added to the reaction vessel under N2 atmosphere at 50-55 °C temperature, and stirred 10-15 minutes. While maintaining 50-55 °C 6-trifluoromethyl-pyridine-2-carboxylic acid methyl ester (50.0 g, 0.244 mol) is added. The reaction mixture is heated to reflux (75-80 °C) and maintained for 1.5-2 hours. Then cooled to 35-40 °C, and concentrated at 45-50 °C under vacuum. Water is added and the mixture is concentrated under vacuum then cooled to 35-40 °C more water is added and the mixture cooled to 0 -5 °C. pH is adjusted to 7-8 by slow addition of 6N HC1, and solid precipitated out and is centrifuged and rinsed with water and centrifuged again. The off white to light brown solid of 6-(6-Trifluoromethyl-pyridin-2-yl)-lH-l,3,5-triazine-2,4-dione is dried under vacuum for 8 to 10 hrs at 50 °C to 60 °C under 600mm/Hg pressure to provide 6-(6-Trifluoromethyl-pyridin-2-yl)-lH-l,3,5-triazine-2,4-dione.
Example 1, Step 4: preparation of 2, 4-Dichloro-6-(6-trifluoromethyl-pyridin-2-yl)-l, 3, 5-triazine
POCI3 (175.0 mL) is charged into the reaction vessel at 20- 35 °C, and 6-(6-Trifluoromethyl-pyridin-2-yl)-lH-l,3,5-triazine-2,4-dione (35.0 g, 0.1355 mol) is added in portions at below 50 °C. The reaction mixture is de-gassed 5-20 minutes by purging with N2 gas. Phosphorous pentachloride (112.86 g, 0.542 mol) is added while stirring at below 50 °C and the resulting slurry is heated to reflux (105-110 °C) and maintained for 3-4 h. The reaction mixture is cooled to 50-55 °C, and concentrated at below 55 °C then cooled to 20-30 °C. The reaction mixture is rinsed with ethyl acetate and the ethyl acetate layer is slowly added to cold water (temperature ~5 °C) while stirring and maintaining the temperature below 10 °C. The mixture is stirred 3-5 minutes at a temperature of between 10 to 20 °C and the ethyl acetate layer is collected. The reaction mixture is rinsed with sodium bicarbonate solution and dried over anhydrous sodium sulphate. The material is dried 2-3 h under vacuum at below 45 °C to provide 2, 4-Dichloro-6-(6-trifluoromethyl-pyridin-2-yl)-l, 3, 5-triazine. Example 1, Step 5: preparation of 4-chloro-6-(6-(trifluoromethyl)pyridin-2-yl)-N-(2-(trifluoro-methyl)- pyridin-4-yl)-l,3,5-triazin-2-amine
A mixture of THF (135 mL) and 2, 4-Dichloro-6-(6-trifluoromethyl-pyridin-2-yl)-l, 3, 5-triazine (27.0 g, 0.0915 mol) are added to the reaction vessel at 20 – 35 °C, then 4-amino-2-(trifluoromethyl)pyridine (16.31 g, 0.1006 mol) and sodium bicarbonate (11.52 g, 0.1372 mol) are added. The resulting slurry is heated to reflux (75-80 °C) for 20-24 h. The reaction is cooled to 30-40 °C and THF evaporated at below 45 °C under reduced pressure. The reaction mixture is cooled to 20-35 °C and rinsed with ethyl acetate and water, and the ethyl acetate layer collected and rinsed with 0.5 N HC1 and brine solution. The organic layer is concentrated under vacuum at below 45 °C then rinsed with dichloromethane and hexanes, filtered and washed with hexanes and dried for 5-6h at 45-50 °C under vacuum to provide 4-chloro-6-(6-(trifluoromethyl)pyridin-2-yl)-N-(2-(trifluoro-methyl)- pyridin-4-yl)-l,3,5-triazin-2-amine.
Example 1, Step 6: preparation of 2-methyl-l-(4-(6-(trifluoromethyl)pyridin-2-yl)-6-(2-(trifluoromethyl)- pyridin-4-ylamino)-l,3,5-triazin-2-ylamino)propan-2-ol
THF (290 mL), 4-chloro-6-(6-(trifluoromethyl)pyridin-2-yl)-N-(2-(trifluoro-methyl)-pyridin-4-yl)-l,3,5-triazin-2-amine (29.0 g, 0.06893 mol), sodium bicarbonate (8.68 g, 0.1033 mol), and 1, 1-dimethylaminoethanol (7.37 g, 0.08271 mol) are added to the reaction vessel at 20-35 °C. The resulting slurry is heated to reflux (75-80 °C) for 16-20 h. The reaction is cooled to 30-40 °C and THF evaporated at below 45 °C under reduced pressure. The reaction mixture is cooled to 20-35 °C and rinsed with ethyl acetate and water, and the ethyl acetate layer collected. The organic layer is concentrated under vacuum at below 45 °C then rinsed with dichlorom ethane and hexanes, filtered and washed with hexanes and dried for 8-1 Oh at 45-50 °C under vacuum to provide 2-methyl-l-(4-(6-(trifluoromethyl)pyridin-2-yl)-6-(2-(trifluoromethyl)- pyridin-4-ylamino)-l,3,5-triazin-2-ylamino)propan-2-ol.
The U.S. Food and Drug Administration today approved Idhifa (enasidenib) for the treatment of adult patients with relapsed or refractory acute myeloid leukemia (AML) who have a specific genetic mutation. The drug is approved for use with a companion diagnostic, the RealTime IDH2 Assay, which is used to detect specific mutations in the IDH2 gene in patients with AML.
The U.S. Food and Drug Administration today approved Idhifa (enasidenib) for the treatment of adult patients with relapsed or refractory acute myeloid leukemia (AML) who have a specific genetic mutation. The drug is approved for use with a companion diagnostic, the RealTime IDH2 Assay, which is used to detect specific mutations in the IDH2 gene in patients with AML.
“Idhifa is a targeted therapy that fills an unmet need for patients with relapsed or refractory AML who have an IDH2 mutation,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “The use of Idhifa was associated with a complete remission in some patients and a reduction in the need for both red cell and platelet transfusions.”
AML is a rapidly progressing cancer that forms in the bone marrow and results in an increased number of abnormal white blood cells in the bloodstream and bone marrow. The National Cancer Institute at the National Institutes of Health estimates that approximately 21,380 people will be diagnosed with AML this year; approximately 10,590 patients with AML will die of the disease in 2017.
Idhifa is an isocitrate dehydrogenase-2 inhibitor that works by blocking several enzymes that promote cell growth. If the IDH2 mutation is detected in blood or bone marrow samples using the RealTime IDH2 Assay, the patient may be eligible for treatment with Idhifa.
The efficacy of Idhifa was studied in a single-arm trial of 199 patients with relapsed or refractory AML who had IDH2 mutations as detected by the RealTime IDH2 Assay. The trial measured the percentage of patients with no evidence of disease and full recovery of blood counts after treatment (complete remission or CR), as well as patients with no evidence of disease and partial recovery of blood counts after treatment (complete remission with partial hematologic recovery or CRh). With a minimum of six months of treatment, 19 percent of patients experienced CR for a median 8.2 months, and 4 percent of patients experienced CRh for a median 9.6 months. Of the 157 patients who required transfusions of blood or platelets due to AML at the start of the study, 34 percent no longer required transfusions after treatment with Idhifa.
Common side effects of Idhifa include nausea, vomiting, diarrhea, increased levels of bilirubin (substance found in bile) and decreased appetite. Women who are pregnant or breastfeeding should not take Idhifa because it may cause harm to a developing fetus or a newborn baby.
The prescribing information for Idhifa includes a boxed warning that an adverse reaction known as differentiation syndrome can occur and can be fatal if not treated. Sign and symptoms of differentiation syndrome may include fever, difficulty breathing (dyspnea), acute respiratory distress, inflammation in the lungs (radiographic pulmonary infiltrates), fluid around the lungs or heart (pleural or pericardial effusions), rapid weight gain, swelling (peripheral edema) or liver (hepatic), kidney (renal) or multi-organ dysfunction. At first suspicion of symptoms, doctors should treat patients with corticosteroids and monitor patients closely until symptoms go away.
Idhifa was granted Priority Review designation, under which the FDA’s goal is to take action on an application within six months where the agency determines that the drug, if approved, would significantly improve the safety or effectiveness of treating, diagnosing or preventing a serious condition. Idhifa also received Orphan Drugdesignation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Idhifa to Celgene Corporation. The FDA granted the approval of the RealTime IDH2 Assay to Abbott Laboratories
The U.S. Food and Drug Administration today approved Rydapt (midostaurin) for the treatment of adult patients with newly diagnosed acute myeloid leukemia (AML) who have a specific genetic mutation called FLT3, in combination with chemotherapy. The drug is approved for use with a companion diagnostic, the LeukoStrat CDx FLT3 Mutation Assay, which is used to detect the FLT3 mutation in patients with AML.
April 28, 2017
Release
The U.S. Food and Drug Administration today approved Rydapt (midostaurin) for the treatment of adult patients with newly diagnosed acute myeloid leukemia (AML) who have a specific genetic mutation called FLT3, in combination with chemotherapy. The drug is approved for use with a companion diagnostic, the LeukoStrat CDx FLT3 Mutation Assay, which is used to detect the FLT3 mutation in patients with AML.
AML is a rapidly progressing cancer that forms in the bone marrow and results in an increased number of white blood cells in the bloodstream. The National Cancer Institute estimated that approximately 19,930 people would be diagnosed with AML in 2016 and 10,430 were projected to die of the disease.
“Rydapt is the first targeted therapy to treat patients with AML, in combination with chemotherapy,” said Richard Pazdur, M.D., acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research and director of the FDA’s Oncology Center of Excellence. “The ability to detect the gene mutation with a diagnostic test means doctors can identify specific patients who may benefit from this treatment.”
Rydapt is a kinase inhibitor that works by blocking several enzymes that promote cell growth. If the FLT3 mutation is detected in blood or bone marrow samples using the LeukoStrat CDx FLT3 Mutation Assay, the patient may be eligible for treatment with Rydapt in combination with chemotherapy.
The safety and efficacy of Rydapt for patients with AML were studied in a randomized trial of 717 patients who had not been treated previously for AML. In the trial, patients who received Rydapt in combination with chemotherapy lived longer than patients who received chemotherapy alone, although a specific median survival rate could not be reliably estimated. In addition, patients who received Rydapt in combination with chemotherapy in the trial went longer (median 8.2 months) without certain complications (failure to achieve complete remission within 60 days of starting treatment, progression of leukemia or death) than patients who received chemotherapy alone (median three months).
Common side effects of Rydapt in patients with AML include low levels of white blood cells with fever (febrile neutropenia), nausea, inflammation of the mucous membranes (mucositis), vomiting, headache, spots on the skin due to bleeding (petechiae), musculoskeletal pain, nosebleeds (epistaxis), device-related infection, high blood sugar (hyperglycemia) and upper respiratory tract infection. Rydapt should not be used in patients with hypersensitivity to midostaurin or other ingredients in Rydapt. Women who are pregnant or breastfeeding should not take Rydapt because it may cause harm to a developing fetus or a newborn baby. Patients who experience signs or symptoms of lung damage (pulmonary toxicity) should stop using Rydapt.
Rydapt was also approved today for adults with certain types of rare blood disorders (aggressive systemic mastocytosis, systemic mastocytosis with associated hematological neoplasm or mast cell leukemia). Common side effects of Rydapt in these patients include nausea, vomiting, diarrhea, swelling (edema), musculoskeletal pain, abdominal pain, fatigue, upper respiratory tract infection, constipation, fever, headache and shortness of breath.
The FDA granted the approval of Rydapt to Novartis Pharmaceuticals Corporation. The FDA granted the approval of the LeukoStrat CDx FLT3 Mutation Assay to Invivoscribe Technologies Inc.
Midostaurin is an inhibitor of tyrosine kinase, protein kinase C, and VEGF. Midostaurin inhibits cell growth and phosphorylation of FLT3, STAT5, and ERK. It is a potent inhibitor of a spectrum of FLT3 activation loop mutations.
it is prepared by acylation of the alkaloid staurosporine (I) with benzoyl chloride (II) in the presence of diisopropylethylamine in chloroform.
Midostaurin is a synthetic indolocarbazole multikinase inhibitor with potential antiangiogenic and antineoplastic activities. Midostaurin inhibits protein kinase C alpha (PKCalpha), vascular endothelial growth factor receptor 2 (VEGFR2), c-kit, platelet-derived growth factor receptor (PDGFR) and FMS-like tyrosine kinase 3 (FLT3) tyrosine kinases, which may result in disruption of the cell cycle, inhibition of proliferation, apoptosis, and inhibition of angiogenesis in susceptible tumors.
MIDOSTAURIN
Derivative of staurosporin, orally active, potent inhibitor of FLT3 tyrosine kinase (fetal liver tyrosine kinase 3). In addition Midostaurin inhibits further molecular targets such as VEGFR-1 (Vascular Endothelial Growth Factor Receptor 1), c-kit (stem cell factor receptor), H-and K-RAS (Rat Sarcoma Viral homologue) and MDR (multidrug resistance protein).
Midostaurin inhibits both wild-type FLT3 and FLT3 mutant, wherein the internal tandem duplication mutations (FLT3-ITD), and the point mutation to be inhibited in the tyrosine kinase domain of the molecule at positions 835 and 836.Midostaurin is tested in patients with AML.
Midostaurin, a protein kinase C (PKC) and Flt3 (FLK2/STK1) inhibitor, is in phase III clinical development at originator Novartis for the oral treatment of acute myeloid leukemia (AML).
Novartis is conducting phase III clinical trials for the treatment of aggressive systemic mastocytosis or mast cell leukemia. The National Cancer Institute (NCI) is conducting phase I/II trials with the drug for the treatment of chronic myelomonocytic leukemia (CMML) and myelodysplastic syndrome (MDS).
Massachusetts General Hospital is conducting phase I clinical trials for the treatment of adenocarcinoma of the rectum in combination with radiation and standard chemotherapy.
After successful Phase II clinical trials, a Phase III trial for AML has started in 2008. It is testing midostaurin in combination with daunorubicin and cytarabine.[2] In another trial, the substance has proven ineffective in metastaticmelanoma.[3]
Midostaurin has also been studied at Johns Hopkins University for the treatment of age-related macular degeneration (AMD), but no recent progress reports for this indication have been made available. Trials in macular edema of diabetic origin were discontinued at Novartis.
In 2004, orphan drug designation was received in the E.U. for the treatment of AML. In 2009 and 2010, orphan drug designation was assigned for the treatment of acute myeloid leukemia and for the treatment of mastocytosis, respectively, in the U.S. In 2010, orphan drug designation was assigned in the E.U. for the latter indication.
MIDOSTAURIN
References
Fischer, T.; Stone, R. M.; Deangelo, D. J.; Galinsky, I.; Estey, E.; Lanza, C.; Fox, E.; Ehninger, G.; Feldman, E. J.; Schiller, G. J.; Klimek, V. M.; Nimer, S. D.; Gilliland, D. G.; Dutreix, C.; Huntsman-Labed, A.; Virkus, J.; Giles, F. J. (2010). “Phase IIB Trial of Oral Midostaurin (PKC412), the FMS-Like Tyrosine Kinase 3 Receptor (FLT3) and Multi-Targeted Kinase Inhibitor, in Patients with Acute Myeloid Leukemia and High-Risk Myelodysplastic Syndrome with Either Wild-Type or Mutated FLT3”. Journal of Clinical Oncology28 (28): 4339–4345. doi:10.1200/JCO.2010.28.9678. PMID20733134. edit
Discovery of 3-(2,6-dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase.
Midostaurin according to the invention is N-[(9S,10R,11R,13R)-2,3,10,11,12,13-hexahydro-10-methoxy-9-methyl-1-oxo-9,13-epoxy-1H,9H-diindolo[1,2,3-gh:3′,2′,1′-lm]pyrrolo[3,4-j][1,7]benzodiazonin-11-yl]-N-methylbenzamide of the formula (II):
or a salt thereof, hereinafter: “Compound of formula II or midostaurin”.
Compound of formula II or midostaurin [International Nonproprietary Name] is also known as PKC412.
Midostaurin is a derivative of the naturally occurring alkaloid staurosporine, and has been specifically described in the European patent No. 0 296 110 published on Dec. 21, 1988, as well as in U.S. Pat. No. 5093330 published on Mar. 3, 1992, and Japanese Patent No. 2 708 047.
The nomenclature of the products is, on the complete structure of staurosporine ([storage]-NH-CH ₃derived, and which is designated by N-substituent on the nitrogen of the methylamino group
Example 18:
N-Benzoyl-staurospor
A solution of 116.5 mg (0.25 mmol) of staurosporine and 0.065 ml (0.38 mmol) of N, N-diisopropylethylamine in 2 ml of chloroform is added at room temperature with 0.035 ml (0.3 mmol) of benzoyl chloride and 10 stirred minutes.The reaction mixture is diluted with chloroform, washed with sodium bicarbonate, dried over magnesium sulfate and evaporated. The crude product is chromatographed on silica gel (eluent methylene chloride / ethanol 30:1), mp 235-247 ° with brown coloration.
cut paste may not be ok below
Staurosporine the formula [storage]-NH-CH ₃ (II) (for the meaning of the rest of [storage] see above) as the basic material of the novel compounds was already in 1977, from the cultures of Streptomyces staurosporeus AWAYA, and TAKAHASHI
O ¯
MURA, sp. nov. AM 2282, see Omura, S., Iwai, Y., Hirano, A., Nakagawa, A.; awayâ, J., Tsuchiya, H., Takahashi, Y., and Masuma, R. J. Antibiot. 30, 275-281 (1977) isolated and tested for antimicrobial activity. It was also found here that the compound against yeast-like fungi and microorganisms is effective (MIC of about 3-25 mcg / ml), taking as the hydrochloride = having a LD ₅ ₀ 6.6 mg / kg (mouse, intraperitoneal). Stagnated recently it has been shown in extensive screening, see Tamaoki, T., Nomoto, H., Takahashi, I., Kato, Y, Morimoto, M. and Tomita, F.: Biochem. and Biophys. Research Commun. 135 (No. 2), 397-402 (1986) that the compound exerts a potent inhibitory effect on protein kinase C (rat brain)
0.035 ml (0.3 mmol) of benzoyl chloride is added at room temperature to a solution of 116.5 mg (0.25 mmol) of staurosporine and 0.065 ml (0.38 mmol) of N,N-diisopropylethylamine in 2 ml of chloroform and the whole is stirred for 10 minutes. The reaction mixture is diluted with chloroform, washed with sodium bicarbonate solution, dried over magnesium sulphate and concentrated by evaporation. The crude product is chromatographed on silica gel (eluant:methylene chloride/ethanol 30:1); m.p. 235
A variety of PKC inhibitors are available in the art for use in the invention. These include bryostatin (U.S. Patent 4,560,774), safinogel (WO 9617603), fasudil (EP 187371), 7- hydoxystaurosporin (EP 137632B), various diones described in EP 657458, EP 657411 and WO9535294, phenylmethyl hexanamides as described in WO9517888, various indane containing benzamides as described in WO9530640, various pyrrolo [3,4-c]carbazoles as described in EP 695755, LY 333531 (IMSworld R & D Focus 960722, July 22, 1996 and Pharmaprojects Accession No. 24174), SPC-104065 (Pharmaprojects Accession No. 22568), P-10050 (Pharmaprojects Accession No. 22643), No. 4432 (Pharmaprojects Accession No. 23031), No. 4503 (Pharmaprojects Accession No. 23252), No. 4721 (Pharmaprojects Accession No. 23890), No. 4755 (Pharmaprojects Accession No. 24035), balanol (Pharmaprojects Accession No. 20376), K-7259 (Pharmaprojects Accession No. 16649), Protein kinase C inhib, Lilly (Pharmaprojects Accession No. 18006), and UCN-01 (Pharmaprojects Accession No. 11915). Also see, for example, Tamaoki and Nakano (1990) Biotechnology 8:732-735; Posada et al. (1989) Cancer Commun. 1:285-292; Sato et al. (1990) Biochem Biophys. Res. Commun. 173:1252-1257; Utz et al. (1994) Int. J. Cancer 57:104-110; Schwartz et al. (1993) J. Na . Cancer lnst. 85:402-407; Meyer et al. (1989) Int. J. Cancer 43:851-856; Akinaga et al. (1991) Cancer Res. 51:4888-4892, which disclosures are herein incorporated by reference. Additionally, antisense molecules can be used as PKC inhibitors. Although such antisense molecules inhibit mRNA translation into the PKC protein, such antisense molecules are considered PKC inhibitors for purposes of this invention. Such antisense molecules against PKC inhibitors include those described in published PCT patent applications WO 93/19203, WO 95/03833 and WO 95/02069, herein incorporated by reference. Such inhibitors can be used in formulations for local delivery to prevent cellular proliferation. Such inhibitors find particular use in local delivery for preventing rumor growth and restenosis.
N-benzoyl staurosporine is a benzoyl derivative of the naturally occurring alkaloid staurosporine. It is chiral compound ([a]D=+148.0+-2.0°) with the formula C35H30R1O4 (molecular weight 570.65). It is a pale yellow amorphous powder which remains unchanged up to 220°C. The compound is very lipophilic (log P>5.48) and almost insoluble in water (0.068 mg/1) but dissolves readily in DMSO.
……………………….
staurosporine
Staurosporine (antibiotic AM-2282 or STS) is a natural product originally isolated in 1977 from the bacterium Streptomyces staurosporeus. It was the first of over 50 alkaloids to be isolated with this type of bis-indole chemical structure. The chemical structure of staurosporine was elucidated by X-ray analysis of a single crystal and the absolute stereochemical configuration by the same method in 1994.
Staurosporine was discovered to have biological activities ranging from anti-fungal to anti-hypertensive. The interest in these activities resulted in a large investigative effort in chemistry and biology and the discovery of the potential for anti-cancer treatment
Staurosporine is the precursor of the novel protein kinase inhibitormidostaurin(PKC412). Besides midostaurin, staurosporine is also used as a starting material in the commercial synthesis of K252c (also called staurosporine aglycone). In the natural biosynthetic pathway, K252c is a precursor of staurosporine.
Indolocarbazoles belong to the alkaloid sub-class of bisindoles. Of these carbazoles the Indolo(2,3-a)carbazoles are the most frequently isolated; the most common subgroup of the Indolo(2,3-a)carbazoles are the Indolo(2,3-a)pyrrole(3,4-c)carbazoles which can be divided into two major classes – halogenated (chlorinated) with a fully oxidized C-7 carbon with only one indole nitrogen containing a β-glycosidic bond and the second class consists of both indole nitrogen glycosilated, non-halogenated, and a fully reduced C-7 carbon. Staurosporine is part of the second non-halogenated class.
The biosynthesis of staurosporine starts with the amino acid L-tryptophan in its zwitterionic form. Tryptophan is converted to an imineby enzyme StaO which is an L-amino acid oxidase (that may be FAD dependent). The imine is acted upon by StaD to form an uncharacterized intermediate proposed to be the dimerization product between 2 imine molecules. Chromopyrrolic acid is the molecule formed from this intermediate after the loss of VioE (used in the biosynthesis of violacein – a natural product formed from a branch point in this pathway that also diverges to form rebeccamycin. An aryl aryl coupling thought to be catalyzed by a cytochrome P450enzyme to form an aromatic ring system occurs
This is followed by a nucleophilic attack between the indole nitrogens resulting in cyclization and then decarboxylation assisted by StaC exclusively forming staurosporine aglycone or K252c. Glucose is transformed to NTP-L-ristoamine by StaA/B/E/J/I/K which is then added on to the staurosporine aglycone at 1 indole N by StaG. The StaN enzyme reorients the sugar by attaching it to the 2nd indole nitrogen into an unfavored conformation to form intermediated O-demethyl-N-demethyl-staurosporine. Lastly, O-methylation of the 4’amine by StaMA and N-methylation of the 3′-hydroxy by StaMB leads to the formation of staurosporine
A LSD1 inhibitor potentially for the treatment of small cell lung cancer and acute myeloid leukemia.
GSK2879552 is an orally available, irreversible, inhibitor of lysine specific demethylase 1 (LSD1), with potential antineoplastic activity. Upon administration, GSK2879552 binds to and inhibits LSD1, a demethylase that suppresses the expression of target genes by converting the dimethylated form of lysine at position 4 of histone H3 (H3K4) to mono- and unmethylated H3K4. LSD1 inhibition enhances H3K4 methylation and increases the expression of tumor-suppressor genes. This may lead to an inhibition of cell growth in LSD1-overexpressing tumor cells. LSD1, overexpressed in certain tumor cells, plays a key role in tumor cell growth and survival. Check for active clinical trials or closed clinical trials using this agent.
Formula: C23H29ClN2O2 M.Wt: 400.94
CAS 1902123-72-1
Molecular Weight:
437.41
Formula:
C23H28N2O2.2HCl
Chromatin modification plays an essential role in transcriptional regulation (T. Kouzarides, 2007, Cell 128: 693-705). These modifications, which include DNA methylation, histone acetylation and hsitone methylation, are disregulated in tumors. This epigenetic disregulation plays an important role in the silencing of tumor suppressors and overexpression of oncogenes in cancer (M. Esteller, 2008, N Engl J Med 358: 1148-59. P. Chi et al, 2010, Nat Rev Cane 10:457-469.). The enzymes that regulate histone methylation are the histone methyl transferases and the histone demethylases.
Lysine-specific demethylase 1 (LSDl; also known as BHC110) is a histone lysine demethylase reported to demethylate H3K4mel/2 (Y. Shi et al, 2004, Cell 119: 941-953) and H3K9mel/2 (R. Schule et al.,2005, Nature 437: 436-439). LSDl is overexpressed in multiple human cancers, including prostate where it is associated with more frequent relapse (P. Kahl et al, 2006, Cane. Res. 66: 11341-11347), breast (J. Kirfel et al, 2010, Carcinogenesis 31: 512-520) neuroblastoma (J. Kirfel et al, 2009, Cane. Res. 69: 2065-2071. G. Sun et al, 2010, Mol. Cell. Biol. 28: 1997-2000). LSDl is essential for transcriptional regulation mediated by a number of nuclear hormone receptors, including androgen receptor in prostate cancer (R. Schuele et al, 2005, Nature 437: 436-439. R. Schuele et al, 2007, Nat. Cell Biol. 9: 347-353. R. Schuele et al, 2010, Nature 464: 792-796), estrogen receptor in breast carcinomas (M.G. Rosenfeld et al, 2007, Cell 128: 505-518), and TLX receptor in neuorblastoma (S. Kato et al, 2008, Mol. Cell. Biol. 28: 3995-4003). These studies have shown that knockdown of LSDl expression results in decreased cancer cell proliferation. Additionally, LSDl is overexpressed in multiple cancer types that are nuclear hormone receptor-independent. Those tumors include ER-negative breast (J. Kirfel et al, 2010, Carcinogenesis 31: 512-520), small-cell lung, bladder, head & neck, colon, serous ovary, and kidney Wilm’s tumor. Therefore, potent selective small molecule inhibitors of LSDl may be useful for treatment of cancers that are nuclear hormone receptor-dependent and/or nuclear hormone receptor-independent.
The compositions and methods provided herein can potentially be useful for the treatment of cancer including tumors such as skin, breast, brain, cervical carcinomas, testicular carcinomas, etc. More particularly, cancers that may be treated by the compositions and methods of the invention include, but are not limited to tumor types such as astrocytic, breast, cervical, colorectal, endometrial, esophageal, gastric, head and neck, hepatocellular, laryngeal, lung, oral, ovarian, prostate and thyroid carcinomas and sarcomas. More specifically, these compounds can potentially be used to treat: Cardiac: sarcoma (angiosarcoma, fibrosarcoma, rhabdomyosarcoma, liposarcoma), myxoma, rhabdomyoma, fibroma, lipoma and teratoma; Lung: bronchogenic carcinoma (squamous cell, undifferentiated small cell, undifferentiated large cell, adenocarcinoma), alveolar (bronchiolar) carcinoma, bronchial adenoma, sarcoma, lymphoma, chondromatous hamartoma, mesothelioma; Gastrointestinal: esophagus (squamous cell carcinoma, adenocarcinoma, leiomyosarcoma, lymphoma), stomach (carcinoma, lymphoma, leiomyosarcoma), pancreas (ductal adenocarcinoma, insulinoma, glucagonoma, gastrinoma, carcinoid tumors, vipoma), small bowel (adenocarcinoma, lymphoma, carcinoid tumors, Kaposi’s sarcoma, leiomyoma, hemangioma, lipoma, neurofibroma, fibroma), large bowel (adenocarcinoma, tubular adenoma, villous adenoma, hamartoma, leiomyoma); Genitourinary tract: kidney (adenocarcinoma, Wilm’s tumor
(carcinoma); Hematologic: blood (myeloid leukemia (acute and chronic), acute lymphoblastic leukemia, chronic lymphocytic leukemia, myeloproliferative diseases, multiple myeloma, myelodysplasia syndrome), Hodgkin’s disease, non-Hodgkin’s lymphoma (malignant lymphoma); Skin: malignant melanoma, basal cell carcinoma, squamous cell carcinoma, Kaposi’s sarcoma, moles dysplastic nevi, lipoma, angioma, dermatofibroma, keloids, psoriasis; and Adrenal glands: neuroblastoma. Thus, the term “cancerous cell” as provided herein, includes a cell afflicted by any one of or related to the above identified conditions.
To the solution of 2,2,2-trifluoro-N-(trans-2-phenylcyclopropyl)-N-(piperidin-4-ylmethyl)acetamide (200 mg, 0.613 mmol, Example l ib) and 4-(bromomethyl)benzoic acid (198 mg, 0.919 mmol) in acetonitrile (6 mL) was added potasium carbonate (254 mg, 1.838 mmol). The reaction mixture was stirred for 3 hours at the 90 °C. The reaction mixture was then filtered and evaporated. The crude oil was mixed with 10 mL of 10 % acetic acid and 10 mL of ethyl acetate. Layers were separated, and the organic layer was discharged. Aqueous layer was neutralized with 1 M Na2C03, and the product was extracted into 10 mL of ethyl acetate. The organic layer was washed with brine, dried over MgS04, filtered and evaporated. The oil was dissolved in 6 ml of EtOH and 3 ml of 1 M NaOH. The reaction mixture was stirred for 20 min, and then it was concentrated. The solution was then partioned between 2 ml of water and 5 mL of ethyl acetate. The organic layer was separated and evaporated. The oil was purified on preparatory HPLC (2 to 10 % AcCN: H20 with 0.1 % formic acid modifier). The fractions were collected. To each
fraction was added 1 ml of 1 M HCl, and the fractions were evaporated to dryness. 4-((4-(((trans-2-phenylcyclopropyl)amino)methyl)piperidin-l-yl)methyl)benzoic acid (50 mg, 0.118 mmol, 19.33 % yield) was isolated as a white solid. 1H NMR (400 MHz,
tert-Butyl 4-(bromomethyl)benzoate (1 g, 3.13 mmol) and piperidin-4-ylmethanol (0.361 g, 3.13 mmol) were dissolved in acetonitrile (25 mL). K2CO3 (1.300 g, 9.40 mmol) was added and the reaction mixture was heated to reflux for 20 min. The reaction mixture was cooled down to room temperature, filtered and evaporated. The resulting solid was partitioned between ethyl acetate (50mL) and 1 M HC1 (50 mL). The layers were separated and the aqueous layer was washed with ethyl acetate and the organic layers were discarded. The aqueous layer was basified with 8 M NaOH to pH -10 and extracted 2 times with 50 mL of ethyl acetate. The organic layers were combined, washed with brine and dried over MgSC^, filtered and evaporated. tert-Butyl 4-((4- (hydroxymethyl)piperidin-l-yl)methyl)benzoate (0.95 g, 2.99 mmol, 95 % yield) was isolated as yellow oil. 1H NMR (400 MHz, CHLOROFORM-d) δ 7.95 (d, J= 8.34 Hz, 2H), 7.39 (d, J = 8.08 Hz, 2H), 3.56 (s, 2H), 3.51 (d, J = 6.57 Hz, 2H), 2.90 (d, J= 11.37 Hz, 2H), 1.94 – 2.04 (m, 2H), 1.73 (d, J= 14.15 Hz, 2H), 1.61 (s, 9H), 1.40 – 1.56 (m, 2H), 1.30 – 1.37 (m, 2H); LC-MS Rt = 0.67 min; MS (ESI): 306.2 [M+H]+.
To a solution of oxalyl chloride (0.408 mL, 4.67 mmol) in dichloromethane (5 mL) at -60 °C was added a solution of DMSO (0.508 mL, 7.15 mmol) in 15 mL of dichloromethane over 30 minutes. The reaction was stirred for 30 minutes at -60 °C A solution of tert-butyl 4-((4-(hydroxymethyl)piperidin-l-yl)methyl)benzoate (950 mg, 3.11 mmol) in 5 mL of dichloromethane was added over 10 minutes at -60 °C. The reaction mixture was stirred for 3 hours at – 60 °C, then triethylamine (2.168 mL, 15.55 mmol) was added and after 10 minutes 10 mL of water was added. The reaction mixture was allowed to warm up to the room temperature. The layers were separated. The pH of the water layer was adjusted to ~7 with 1 M HC1 and then extracted with 20 mL of dichloromethane. The combined organic layers were washed with water and brine, then dried over MgSO, filtered and evaporated. The resulting oil was purified on a silica column eluting with EtOAc to yield tert-butyl 4-((4-formylpiperidin-l-yl)methyl)benzoate (550 mg, 1.722 mmol, 55.4 % yield) as a yellow oil. 1H NMR (400 MHz, CHLOROFORM-d) δ 9.67 (d, J= 1.26 Hz, 1H), 7.96 (d, J= 8.34 Hz, 2H), 7.38 (d, J= 8.34 Hz, 2H), 3.56 (s, 2H), 2.75 – 2.92 (m, 2H), 2.21 – 2.35 (m, 1H), 2.14 (t, J= 10.48 Hz, 2H), 1.91 (dd, J= 2.78, 13.14 Hz, 2H), 1.65 – 1.81 (m, 2H), 1.58 – 1.64 (m, 9H); LC-MS Rt = 0.69 min; MS (ESI): 304.2
To a solution of tert-butyl 4-((4-formylpiperidin-l-yl)methyl)benzoate (6.7 g, 22.08 mmol) in methanol (50 mL) was added (lR,2S)-2-phenylcyclopropanamine (3.53 g, 26.5 mmol). The reaction mixture was refluxed for 5 minutes then cooled down to the room temperature. Sodium cyanotrihydroborate (2.082 g, 33.1 mmol) was added. The reaction mixture was stirred 1 hour at room temperature. Water (50 mL) was added. The reaction was concentrated and 50 mL of dichloromethane was added. The layers were separated. The organics were washed with 10 % acetic acid (50 mL). The layers were separated and 50 mL of brine was added slowly as a solid crashed out. The solid was filtered and suspended in isopropanol. The suspension was sonicated and filtered. tert-Butyl 4-((4-(((( 1 R,2S)-2-phenylcyclopropyl)amino)methyl)piperidin- 1 -yl)methyl)benzoate (5.8 g, 13.65 mmol, 61.8 % yield) was isolated as a white solid. 1H NMR (400 MHz,
Axl receptor tyrosine kinase inhibitors, Fms-like tyrosine kinase 3 inhibitors, Proto oncogene protein c-kit inhibitors
Who Atc Codes
L01X-E (Protein kinase inhibitors)
Ephmra Codes
L1H (Protein Kinase Inhibitor Antineoplastics)
Indication
Cancer, Hepatic impairment
Gilteritinib(ASP-2215) is a potent FLT3/AXL inhibitor with IC50 of 0.29 nM/<1 nM respectively; shows potent antileukemic activity against AML with either or both FLT3-ITD and FLT3-D835 mutations. IC50 value: 0.29 nM(FLT3); <1 nM(Axl kinase) Target: FLT3/AXL inhibitor
ASP2215 inhibited the growth of MV4-11 cells, which harbor FLT3-ITD, with an IC50 value of 0.92 nM, accompanied with inhibition of pFLT3, pAKT, pSTAT5, pERK, and pS6. ASP2215 decreased tumor burden in bone marrow and prolonged the survival of mice intravenously transplanted with MV4-11 cells. ASP2215 may have potential use in treating AML.
Compound A is 6-ethyl-3 – ({3-methoxy-4- [4- (4-methylpiperazin-1-yl) piperidin-1-yl] phenyl} amino) -5- a (tetrahydro -2H- pyran-4-ylamino) pyrazine-2-carboxamide, its chemical structure is shown below.
[Formula 1]
Astellas Inititaties Phase 3 Registration Trial of gilteritinib (ASP2215) in Relapsed or Refractory Acute Myeloid Leukemia Patients
TOKYO, Japan I October 28, 2015 I Astellas Pharma Inc. (TSE:4503) today announced dosing of the first patient in a randomized Phase 3 registration trial of gilteritinib (ASP2215)versus salvage chemotherapy in patients with relapsed or refractory (R/R) acute myeloid leukemia (AML). The primary endpoint of the trial is overall survival (OS).
Gilteritinibis a receptor tyrosine kinase inhibitor of FLT3 and AXL, which are involved in the growth of cancer cells. Gilteritinibhas demonstrated inhibitory activity against FLT3 internal tandem duplication (ITD) as well as tyrosine kinase domain (TKD), two common types of FLT3 mutations that are seen in up to one third of patients with AML.
The gilteritinib Phase 3 trial follows a Phase 1/2 trial, which evaluated doses from 20 to 450 mg once daily. A parallel multi-dose expansion cohort was initiated based on the efficacy seen in the dose escalation phase. Preliminary data from the Phase 1/2 trial presented at the 2015 American Society of Clinical Oncology annual meeting demonstrated a 57.5 percent overall response rate and a 47.2 percent composite Complete Response (CR) rate (CR + CR with incomplete platelet recovery + CR with incomplete hematologic recovery) in 106 patients with FLT3 mutations who received 80 mg and higher doses. Median duration of response was 18 weeks across all doses and median OS was approximately 27 weeks at 80 mg and above in FLT3 mutation positive patients. Common drug-related adverse events (> 10%) observed in the study were diarrhea (13.4%), fatigue (12.4%) and AST increase (11.3%). At the 450 mg dose, two patients reached dose-limiting toxicity (grade 3 diarrhea and ALT/AST elevation) and the maximum tolerated dose was determined to be 300 mg.
On October 27, 2015, the Japanese Ministry of Health, Labor and Welfare (MHLW) announced the selection of gilteritinib as one of the first products designated for SAKIGAKE.
About the Phase 3 Study
The Phase 3 trial is an open-label, multicenter, randomized study of gilteritinib versus salvage chemotherapy in patients with Acute Myeloid Leukemia (AML). The study will enroll 369 patients with FLT3 activating mutation in bone marrow or whole blood, as determined by central lab, AML who are refractory to or have relapsed after first-line AML therapy. Subjects will be randomized in a 2:1 ratio to receive gilteritinib (120 mg) or salvage chemotherapy consisting of LoDAC (low-dose cytarabine), azacitidine, MEC (mitoxantrone, etoposide, and intermediate-dose cytarabine), or FLAG-IDA (fludarabine, cytarabine, and granulocyte colony-stimulating factor with idarubicin). The primary endpoint of the trial is OS. For more information about this trial go to http://www.clinicaltrials.gov, trial identifier NCT02421939.
Gilteritinib was discovered through a research collaboration with Kotobuki Pharmaceutical Co., Ltd., and Astellas has exclusive global rights to develop, manufacture and potentially commercialize gilteritinib.
About Acute Myeloid Leukemia
Acute myeloid leukemia is a cancer that impacts the blood and bone marrow and most commonly experienced in older adults. According to the//www.cancer.org/acs/groups/content/@editorial/documents/document/acspc-044552.pdf” target=”_blank” rel=”nofollow”>American Cancer Society, in 2015, there will be an estimated 20,830 new cases of AML diagnosed in the United States, and about 10,460 cases will result in death.
About SAKIGAKE
The SAKIGAKE designation system can shorten the review period in the following three approaches: 1.) Prioritized Consultation 2.) Substantial Pre-application Consultation and 3.) Prioritized Review. Also, the system will promote development with the following two approaches: 4.) Review Partner System (to be conducted by the Pharmaceuticals and Medical Devices Agency) and 5.) Substantial Post-Marketing Safety Measures.
About Astellas
Astellas Pharma Inc., based in Tokyo, Japan, is a company dedicated to improving the health of people around the world through the provision of innovative and reliable pharmaceutical products. We focus on Urology, Oncology, Immunology, Nephrology and Neuroscience as prioritized therapeutic areas while advancing new therapeutic areas and discovery research leveraging new technologies/modalities. We are also creating new value by combining internal capabilities and external expertise in the medical/healthcare business. Astellas is on the forefront of healthcare change to turn innovative science into value for patients. For more information, please visit our website at http://www.astellas.com/en.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....














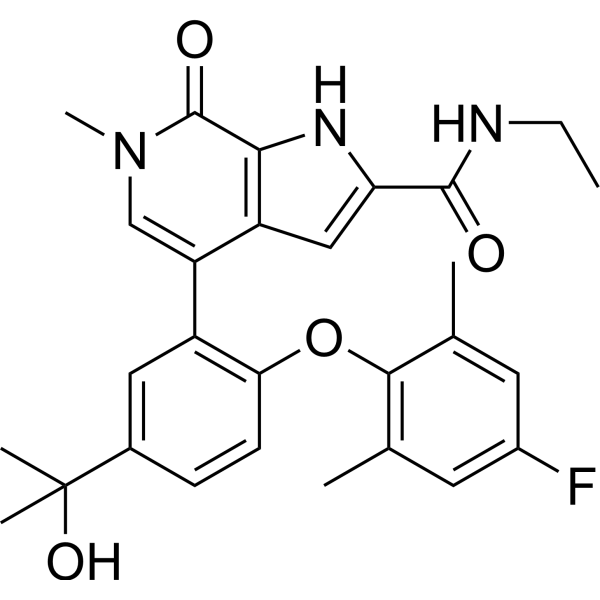



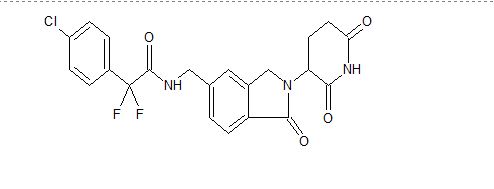
![2-(4-Chlorophenyl)-N-[[2-(2,6-dioxopiperidin-3-yl)-1-oxo-3H-isoindol-5-yl]methyl]-2,2-difluoroacetamide.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=118647211&t=l)

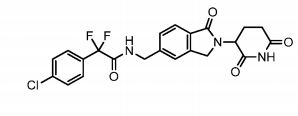




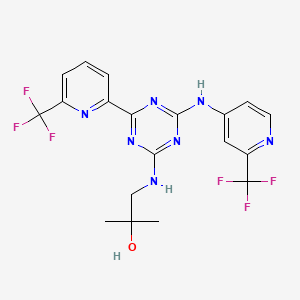


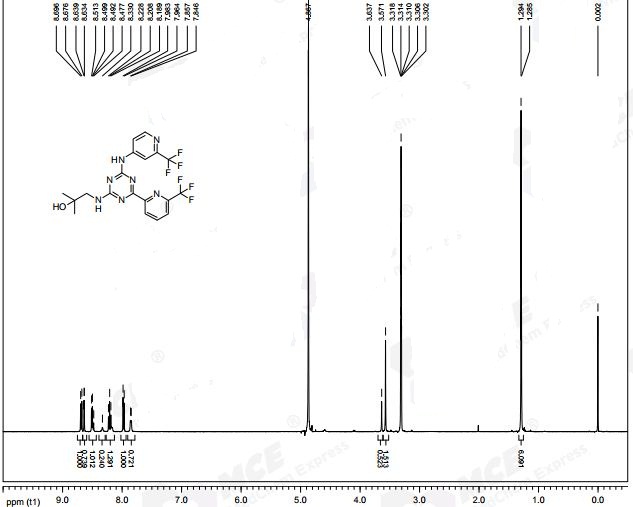











 MIDOSTAURIN
MIDOSTAURIN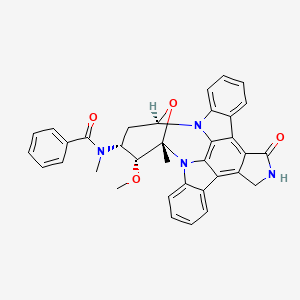 MIDOSTAURIN
MIDOSTAURIN







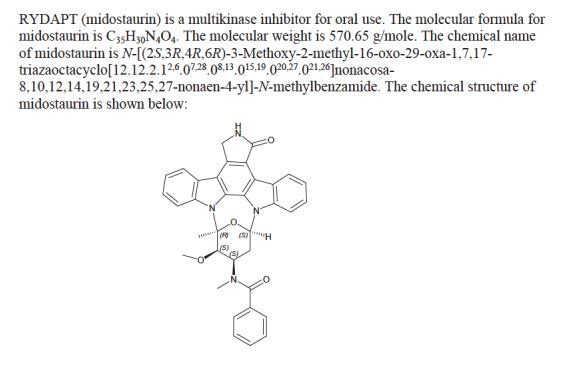




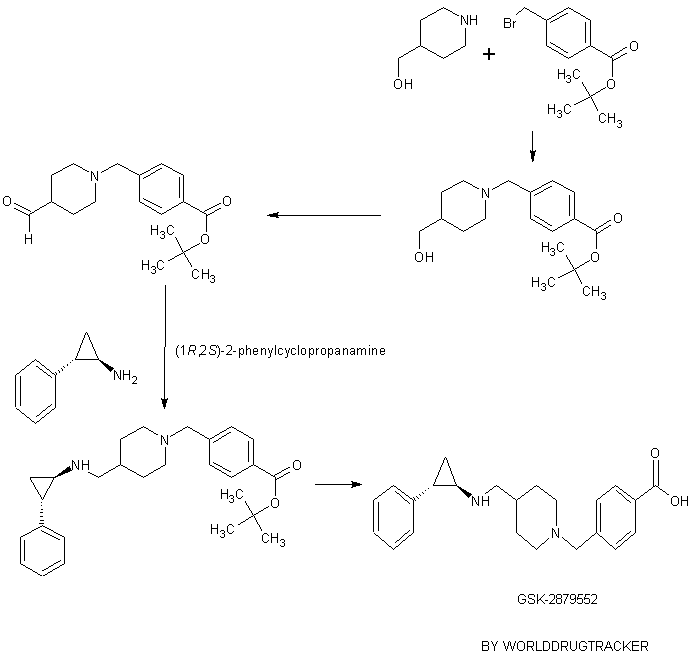










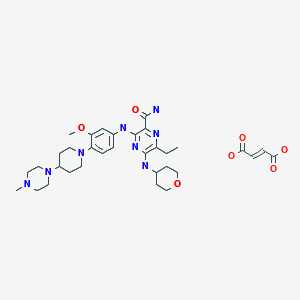
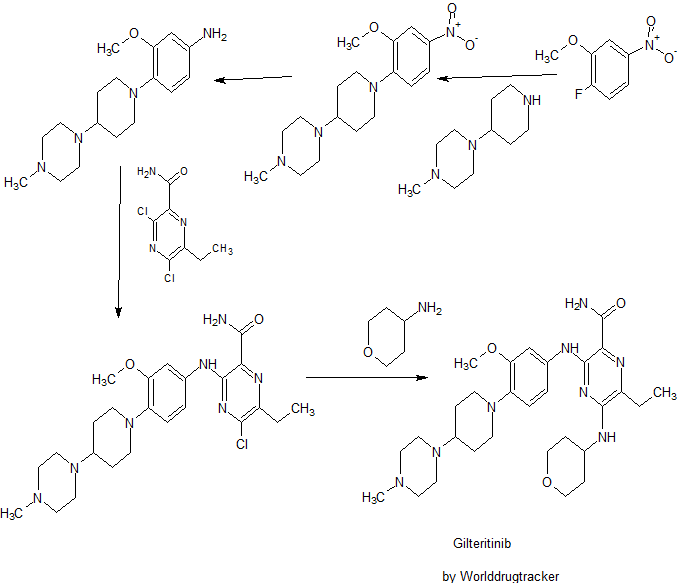
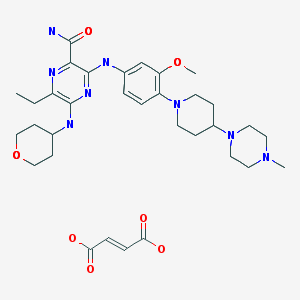
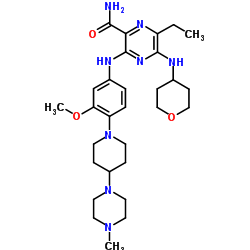


{kind=link}