
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

TREOSULFAN



TREOSULFAN
C6H14O8S2 MW 278.29
FDA APPROVED 1/21/2025 Grafapex
CAS
299-75-2 |
299-75-2
Treosulphan
Ovastat
Treosulfano
NSC-39069
- Dihydroxybusulfan
- L-threitol-1,4-dimethanesulfonate
[(2S,3S)-2,3-dihydroxy-4-methylsulfonyloxybutyl] methanesulfonate
Trecondi, Treosulfan was authorized for medical use in the European Union in June 2019
For use in combination with fludarabine as a preparative regimen for allogeneic hematopoietic stem cell transplantation for acute myeloid leukemia and myelodysplastic syndrome
Treosulfan, sold under the brand name Trecondi among others, is an alkylating medication given to people before they have a bone marrow transplant from a donor known as allogeneic hematopoietic stem cell transplantation. It is used as a ‘conditioning’ treatment to clear the bone marrow and make room for the transplanted bone marrow cells, which can then produce healthy blood cells.[9][10] It is used together with another medicine called fludarabine in adults and children from one month of age with blood cancers as well as in adults with other severe disorders requiring a bone marrow transplant.[9] It belongs to the family of drugs called alkylating agents.[9] In the body, treosulfan is converted into other compounds called epoxides which kill cells, especially cells that develop rapidly such as bone marrow cells, by attaching to their DNA while they are dividing.[9]
The most common side effects include infections, nausea (feeling sick), stomatitis (inflammation of the lining of the mouth), vomiting, diarrhea, and abdominal pain (belly ache).[9] Tiredness, febrile neutropenia (low white blood cell counts with fever) and high blood levels of bilirubin (a breakdown product of red blood cells) are also seen in more than 1 in 10 adults, and rash also affects more than 1 in 10 children.[9] The most common adverse reactions include musculoskeletal pain, stomatitis, pyrexia, nausea, edema, infection, and vomiting.[7] Selected grade 3 or 4 nonhematological laboratory abnormalities include increased GGT, increased bilirubin, increased ALT, increased AST, and increased creatinine.[7]
Treosulfan was authorized for medical use in the European Union in June 2019,[9] and approved for medical use in the United States in January 2025.[7][11]
Medical Uses
Treosulfan in combination with fludarabine is indicated as part of conditioning treatment prior to allogeneic haematopoietic stem cell transplantation in adults with malignant and non malignant diseases, and in children older than one month with malignant diseases.[7][9]
History
Two main studies showed that treosulfan is at least as effective as busulfan, another medicine used to prepare people for haematopoietic stem cell transplantation.[9]
In one of the studies, involving 570 adults with acute myeloid leukaemia (a blood cancer) or myelodysplastic syndromes (conditions in which large numbers of abnormal blood cells are produced), 64% of patients given treosulfan (with fludarabine) had a successful transplant and were alive and disease-free after 2 years, compared with 51% of patients given busulfan (with fludarabine).[9]
In an additional study in 70 children with blood cancers, 99% of children given treosulfan (with fludarabine) were alive three months after their transplant.[9]
Efficacy was evaluated in MC-FludT.14/L Trial II (NCT00822393), a randomized active-controlled trial comparing treosulfan to busulfan with fludarabine as a preparative regimen for allogeneic transplantation. Eligible patients included adults 18 to 70 years old with AML or MDS, Karnofsky performance status ≥ 60%, and age ≥ 50 years or hematopoietic cell transplantation comorbidity index [HCTCI] score > 2. There were 570 patients randomized to treosulfan (n=280) or busulfan (n=290).
Society and culture
Legal status
Treosulfan was authorized for medical use in the European Union in June 2019,[9] and approved for medical use in the United States in January 2025.[11][12][13]
The US Food and Drug Administration granted orphan drug designation to treosulfan in 1994, for the treatment of ovarian cancer;[14] and in 2015, for conditioning treatment prior to hematopoietic stem cell transplantation in malignant and non-malignant diseases in adults and pediatric patients.[15]
In February 2004, orphan designation (EU/3/04/186) was granted by the European Commission to medac Gesellschaft fuer klinische Spezialpräparate mbH, Germany, for treosulfan for the conditioning treatment prior to haematopoietic progenitor cell transplantation.[16]
Names
Treosulfan is the international nonproprietary name.[17]
Treosulfan is sold under the brand names Trecondi[9] and Grafapex.[7]
SYN
Treosulfan is an active ingredient of the drug Ovastat . Treosulfan is indicated for the treatment of ovarian cancer and belongs to the class of alkylating agents, which prevents the growth and division of cancerous cells.
US3155702 discloses the preparation of Treosulfan by methanesulphonation of (2S,3S)- l,4-dibromobutane-2,3-diol with excess amount of silver methanesulphonate. The presence of free 2,3-diol in the starting material leads to side reactions and formation of undesired by-products which necessitates an additional purification step and thereby results in lower yields. Further, an additional filtration operation is also required to remove silver bromide salt generated during the process and un-reacted silver methanesulphonate, which makes the process less attractive for commercial manufacturing.
US3246012 discloses the preparation of Treosulfan by protection of hydroxyl group of dialkyl tartrates with corresponding aldehyde, ketone or a reactive derivatives to form corresponding cyclic 2,3-O-acetals and 2,3-O-ketals of butanetetrol esters followed by reduction using lithium aluminium hydride to obtain 2,3-O-acetal or ketal protected butanetetrol, which is further methanesulphonated and treated with acid. The use of highly pyrophoric and hazardous reducing agent renders the above process not ideal for industrial production. Organic Syntheses, Coll. Vol. 10, p. 297, 2004 discloses a similar reaction sequence followed by the final de-protection of methanesulphonated 2,3-O-diisopropylidene-L- threitol in methanesulfonic acid at reflux temperature, which leads to a sluggish reaction mixture and a higher number of impurities due to maintaining the reaction mixture for longer time at higher temperature.
IN 1568/MUM/2012 also discloses similar reaction sequence involving reduction of dimethyl-2,3-0-isopropylidene-L-tartrate by sodium-bis(2-methoxyethoxy) aluminium hydride followed by methanesulphonation and final deprotection with formic acid to yield Treosulfan.
KR101367641 describes reduction using lithium borohydride, which requires about 14 hours to complete the reaction and is further extended due to involvement of column chromatography purification. Tetrahedron, vol. 49, no. 30, p. 6645, 1993 describes reduction using sodium borohydride and lithium chloride, followed by flash chromatography purification. Reduction conditions as per Chem. Pharm. Bull. Vol. 42, No. 3, p. 68, 1994, are again not commercially feasible because of lithium aluminium hydride as reducing agent.
Haberland, M., Weber, S., Sharma, A. K., Upadhyay, S., Dua, H., Musmade, S., Singh, G., Lahiri, S., & Cabri, W. (2019). A process for the preparation of Treosulfan (Patent No. WO2019043587A2).
EXAMPLES Detailed experimental parameters suitable for the preparation of Treosulfan or intermediates according to the present invention are provided by the following examples, which are intended to be illustrative and not limiting.
Reference Example 1 (repetition of Tetrahedron, vol. 46, No. 12, p. 4165, 1990):
A reaction mixture of dimethyl-L-tartrate (10. Og), p-toluene sulfonic acid (0.013g) and p- anisaldehydedimethylacetal (l l.Og) in toluene (150ml) was refluxed and the azeotropical mixture of toluene-methanol was continuously removed from the reaction mixture for 3-5 hours. The reaction mixture was cooled to ambient temperature, diluted with dichloromethane (50ml) and neutralised by addition of potassium carbonate (5.0g) followed by stirring for an hour . The reaction mixture was filtered and filtrate was evaporated to give yellow crude compound, which was further dissolved in dichloromethane (25ml) followed by addition of petroleum ether (100ml) and stirred for an hour at ambient temperature. The solid was filtered, washed with petroleum ether (20ml) and dried under vacuum at 35-40°C for 15-20 hours to obtain 16.63g (72.15%) of dimethyl (4R,5R)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5-dicarboxylate having purity 98.4% by HPLC.
Reference Example 2 (repetition of Synthesis, No. 15, p. 2488-90, 2008):
A reaction mixture of dimethyl-L-tartrate (5.0g), p-toluene sulfonic acid (0.0064g) and p- anisaldehyde dimethylacetal (5.35g) in toluene (25ml) was refluxed and the azeotropical mixture of toluene-methanol was continuously removed from the reaction mixture for 3-5 hours. The reaction mixture was cooled to ambient temperature, diluted with dichloromethane (25ml) and neutralised by addition of potassium carbonate (5.0g) followed by stirring for an hour. The reaction mixture was filtered and filtrate was evaporated to give yellow crude residues. The crude was further re-crystallized in petroleum ether (25ml), filtered the solid and washed with petroleum ether (15ml) followed by drying under vacuum at 35-40°C for 15-20 hours to obtain 7.4g (89.15%) of dimethyl (4R,5R)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5-dicarboxylate having purity 98.8% by HPLC. Example-1: Preparation of dimethyl (4R,5R)-2-(4-methoxyphenyl)-l,3-dioxolane- 4,5-dicarboxylate
A reaction mixture of dimethyl-L-tartrate (500g), p-toluene sulfonic acid (5.38g) and p- anisaldehyde dimethylacetal (665g) in toluene (2250ml) was refluxed to 110-115°C. The azeotropical mixture of toluene-methanol was continuously removed from the reaction mixture till the completion of the reaction. The reaction mixture was cooled to ambient temperature and quenched with aq. saturated sodium bicarbonate solution (2500ml), layers were separated. Resulting organic layer was washed with water (2500ml x 2) followed by evaporation of organic layer. Isopropyl alcohol (3500ml) was charged to the residue and heated to 60-70°C followed by cooling at ambient temperature. Reaction mixture was stirred at 0-5°C for 1-2 hours and filtered. The solid thus obtained was washed with pre- cooled isopropyl alcohol and dried under vacuum at 35-40°C for 15-20 hours to obtain 767.0g (92.93%) of dimethyl (4R,5R)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- dicarboxylate having purity 99.97% by HPLC.
Example-2: Preparation of (4S,5S)-2-(4-methoxyphenyl)-l 53-dioxo!ane-4,5- diyifdimethanol
Method-l :To a mixture of dimethyl (4R,5R)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- dicarboxylate (765g), Iodine (13. lg) in tetrahydrofuran (3750ml) and water (76ml), sodium borohydride (146.52g) was added at 0-15°C and stirred for 1 -2 hours at ambient temperature. The reaction was quenched with 30% aq. ammonium chloride (6100ml) solution and dichloromethane (7650ml). The layers were separated and the aqueous layer was extracted by dichloromethane (3800ml x 3) followed by washing of combined organic layers with water (3800ml), The resulting organic layer was evaporated at 35-65°C to obtain 525.0g (83.9%) of (4S,5S)-2-(4-methoxyphenyl)-l,3- dioxolane-4,5-diyl]dimethanol having purity 99.72% by HPLC. Method-2: To a mixture of dimethyl (4R,5R)-2-(4-methoxyphenyl)-l,3-dioxolane- 4,5-dicarboxylate (765g), Iodine (13.10g) in tetrahydrofuran (3750ml) and water (76.5ml), sodium borohydride (146.52g) was added at 0-10°C and stirred for Ihours at 0-5°C and stirred for 3-4 hours at ambient temperature. The reaction was quenched with 30% aq. ammonium chloride (6120ml) solution and dichloromethane (7650ml) at ambient temperature. The layers were separated and the aqueous layer was extracted by dichloromethane (3825m! x 3) followed by washing of combined organic layers with water (3825ml). The resulting organic layer was evaporated at 50-60°C to obtain 525 g (84.7%) of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5-diyl]dirnethaiiol having purity 99.72% by HPLC. Example-3: Preparation of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- diyl]bis(methylene) dimethanesulfonate
Method-l:To a solution of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- diyl]dimethanol (145g) in dichloromethane (2175ml), pyridine (191g) and methanesulphonyl chloride (190. l g) was added at 0-5 °C. The reaction mixture was stirred for 2-3 hours at ambient temperature followed by quenching with water (1450ml). The organic layer was washed with water (1450ml x 4) and evaporated. The resulting residue was added to isopropanol (725ml) and stirred for 1-2 hours at ambient temperature and further for 1-2 hours at 0-5 C. The solid was filtered and washed with pre-cooled isopropanol (145ml). The resulting product was dissolved in acetone (1300ml) followed by addition of isopropanol (2610ml). Resulting reaction mixture was stirred for 1-2 hours at ambient temperature and then cooled at 0-5 °C. The solid thus obtained was filtered and washed with pre-cooled isopropanol (145ml x 2) and dried under vacuum at 30-35°C for 15-20 hours to give 190.8g (79.4%)of (4S,5S)-2-(4- methoxyphenyl)-l,3-dioxolane-4,5-diyl]bis(methylene) dimethanesulfonate. Method-2: To a solution of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- diyl]dimethanol (525, Og) in dichloromethane (7350ml), di-isopropylamine (663. Og) was added at ambient temperature followed by addition of methanesulphonyl chloride solution (624. Og in 525ml dichloromethane) at 0-10°C. The reaction mixture was stirred for 1-2 hours at 0-10 °C followed by stirring for 3-4 hours at ambient temperature. The organic layer was washed with water (2 x 5250ml) and evaporated. The residues were dissolved in acetone (4725ml) followed by addition of isopropanol (9450ml), stirred for about 1-2 hour at ambient temperature and then at 0-5 °C for 1-2 hours. The resulting solid was filtered, washed with pre-cooled isopropanol (525 x 2 ml)and dried under vacuum at 35-45°C for 15-20 hours to give 705.0g (81.45%) of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5-diyl]bis(methylene)
dimethanesulfonate having purity 99.92% by HPLC.
Example-4: Preparation of Treosulfan
Method-1: To a solution of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- diyl]bis(methylene) dimethanesulfonate (745. Og) in methanol (7450ml), concentrated hydrochloric acid (260ml) was added at 15-25°C followed by stirring for 10-15 hours at ambient temperature. The reaction mixture was cooled to 0-5°C and further stirred for 1-2 hours at 0-5°C followed by filtration and washing the solid with pre-cooled methanol (745ml). The solid thus obtained was dissolved in acetone (3725ml) followed by microne filtration. Di-isopropyl ether (7450ml) was added to the filtrate and stirred for 1-2 hours at ambient temperature and then cooled at 0-5°C. The solid thus obtained was filtered and washed with di-isopropyl ether (745ml x 2) followed by drying at 30-35°C for 15-20 hours to obtain 96.5g of Treosulfan having purity 99.9% by HPLC.
XRPD of Treosulfan obtained by above process is shown in Fig. 1. Method-2:To a solution of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- diyl]bis(methylene)dimethanesulfonate (650. Og) in methanol (6500ml), 9N hydrochloric acid (227.5ml) was added at 0-10°C followed by stirring for 6-8 hours at ambient temperature. The reaction mixture was cooled to 0-5°C and further stirred for 1-2 hours followed by filtration and washing the solid with pre-cooled methanol (2 x 650ml). The solid thus obtained was dissolved in acetone (3250ml). Di-isopropyl ether (6500ml) was added to the resulting solution, stirred for 1-2 hours at ambient temperature and then cooled at 0-5°C. The solid thus obtained was filtered and washed with di- isopropyl ether (650ml x 2) followed by drying at 30-35°C for 15-20 hours to obtain 312g (68.4) of Treosulfan having purity 99.81% by HPLC.
PATENT
https://patents.google.com/patent/WO2020064815A1/en
Example 1 – Preparation of form B using water/isopropanol
99.8 mg treosulfan were weighed in a vial (volume 4.0 ml) which was equipped with a PTFE (Polytetrafluoroethylene) sealing and a stirrer. 1.5 ml of a mixture of 80 % by weight water and 20 % by weight isopropanol preheated to 65°C were then added. The resulting solution was completely taken up with a syringe (volume 5 ml) and filtered using a 0.2 pm filter into a second vial (volume 4.0 ml) . The syringe, second vial and filter had been tempered at 65°C before use. The solvents were allowed to evaporate from the open vial at room temperature to dryness which resulted in formation of crystals.
The XRPD pattern of the obtained crystals of form B according to the invention is shown in Figure 1.
PATENT
1568/MUM/2012
Abstract
Abstract: The present invention provides a convenient and cost-effective process for preparation of Treosulfan. The process comprises reduction of dimethyl 2,3-O-isopropylidene-L-tartrate with sodium-bis(2-methoxyethoxy)aluminum hydride to give the alcohol 2,3-O-isopropylidene-L-threitol (III), which on reaction with methanesulfonyl chloride led to 2,3-O-isopropylidene-L-threitol 1,4-bismethanesulfonate of formula (IV) and further treatment of compound (IV) with formic acid gave Treosulfan (I) having desired purity.
Treosulfan (I), chemically known as (2S,3S)-2,3-Dihydroxy-4-memylsidfonyIoxybutylj methanesulfonate is a drug commonly used for treating ovarian cancer. It belongs to the family of anti-cancer medicines called the alkylating agents, which prevent the growth and division of cancerous cells. Treosulfan has been used for bone-marrow ablation before stem-cell transplantation and in the treatment of malignant melanoma and breast cancer.
US 3,155,702 discloses synthesis of Treosulfan by replacement of the halogen function in L-Threitol-l,4-dibromobutane-2,3-diol, by treating with a large excess of an expensive reagent like silver methanesulfonate. Further, the presence of unprotected hydroxyl groups in the starting material inevitably leads to the formation of undesired impurities, which requires additional purification steps for removal of impurities as well for lowering the level of free silver in the active ingredient as per ICH guidelines, which results in lower yields and increases the costs substantially.
Another method reported in US 3,246,012 involves acetal formation of diethyl-L-tartrate with acetone to obtain 2,3-O-isopropylidene-diethyl-L-tartrate, which, when reduced with lithium aluminium hydride gives 2,3-0-methylene-L-threitol. The obtained alcohol was treated with methanesulfonyl chloride to yield the penultimate Treosulfan intermediate, 2,3-O-methylene-L-threitol-1,4-di-(methanesulfonate).
A similar approach which employs tartrate esters in the synthesis of Treosulfan, is disclosed in Organic Syntheses, (1993), Vol.8, p. 155 and Organic .Syntheses, (2004), Coll.Vol.10, p.297. L-tartaric acid is reacted with 2,2-dimethoxypropane in presence of methanol. The resulting methyl ester, dimethyl 2,3-O-isopropylidene-L-tartrate is reduced with lithium aluminium hydride to obtain 2,3-di-O-isopropylidene-L-threitol, which, upon reaction with methanesulfonyl chloride, followed by treatment with methanesulfonic acid yields Treosulfan.
Although these routes involve protection of the diol group and avoid impurities arising out of substitution at those alcohol functionalities, use of a highly pyrophoric, hazardous reagent such as lithium aluminium hydride severely limits their synthetic applicability, especially on commercial scale. Further, the final step involves reaction of 2,3-di-O-isopropylidene-L-threitol with methanesulfonic acid, which is quite sluggish and causes considerable rise in the total number of impurities due to long reaction time.
Thus, there is a need for a convenient, economical process for a commercial scale synthesis of Treosulfan (I), which overcomes the shortcomings of the prior art, does not involve use of hazardous, pyrophoric reagents and yields Treosulfan conforming to regulatory specifications.
The present inventors have developed a novel process for preparation of (2S,3S)-2,3-Dihydroxy-4-methylsulfonyloxybutyl] methanesulfonate (I). The scheme for synthesis comprises reaction of dimethyl 2,3-O-isopropylidene-L-tartrate of formula (II) with sodium-bis(2-methoxyethoxy) aluminum hydride to give the protected diol, 2,3-0-isopropylidene-L-threitoI (III), which on further treatment with methanesulfonyl chloride, followed by reaction of the resultant ester, 2,3-O-isopropyliden-L-threitol 1,4 bismethanesulfonate (IV) with formic acid, yields Treosulfan (I) having desired purity and with impurity levels conforming to ICH guidelines.
Scheme 1; Method embodied in the present invention for the preparation of Treosulfan (I)
In an embodiment, dimethyl 2,3 -O-isopropylidene-L-tartrate of formula (II) was treated with sodium-bis-(2-methoxyethoxy) aluminium hydride in presence of an organic solvent, and in the temperature range of 25 to 80°C, but preferably 60 to 75°C.
The organic solvent was selected from the group of toluene, xylenes, nitrobenzene, hexane, cyclohexane, heptane, N-methyl-2-pyrroIidone, ethers etc.
Upon completion of the reaction, as monitored by TLC, water was carefully added to the reaction mass and the mixture was extracted with a water immiscible organic solvent.
The organic solvent was selected from the group comprising of n-hexane, cyclohexane, heptane, methyl isobutyl ketone, 2-methyl tetrahydrofuran, cyclopentyl methyl ether etc.
The organic layer was separated and concentrated under reduced pressure to give 2,3-0-isopropylidene-L-threitol of formula (III) of desired purity.
It is pertinent to mention that the reaction was quite facile and the desired product was obtained with minimal formation of associated impurities and did not require any subsequent purification.
Further reaction of compound (III) with methanesulfonyl chloride was carried out at 25 to 35°C, in an organic solvent, in presence of an organic base.
The organic solvent was selected from the group comprising of chloroform, ethylene dichloride, dichloromethane, carbon tetrachloride etc., but preferably dichloromethane.
The organic base was selected from triethyl amine, tributyl amine and pyridine.
The reaction mixture was stirred at 25-35°C and after completion of the reaction as monitored by TLC, aqueous solution of sodium bicarbonate was added slowly to the reaction mass. The organic layer was separated, concentrated under reduced pressure and stirred with isopropyl alcohol to obtain the desired compound, 2,3-O-isopropylidene-L-threitol-l,4-bis(methanesulfonate) of formula (IV).
In a further embodiment, compound (TV) was hydrolyzed by treating with formic acid at 25 to 35°C based on TLC. After completion of the reaction, the reaction mass was concentrated and the product Treosulfan (I) was isolated by addition of isopropyl alcohol to the concentrated mass.
It is pertinent to mention that Organic Syntheses (2004), Coll.Vol. 10, p.297 discloses the hydrolysis reaction using methanesulfonic acid in ethanol at reflux temperature. However, the time taken for completion is about ten hours and the procedure is applicable only for laboratory scale reaction. The hydrolysis step disclosed in the present invention is easily scalable and so facile that it takes place at room temperature and within one to two hours. This reduces the time cycle for each batch run and also reduces the possibility of formation of undesired side products.
Dimethyl 2,3-O-isopropylidene-L-tartrate of formula (II) was prepared by the reaction of dimethyl -L-tartrate with acetone by following known synthetic procedures.
The following examples are meant to be illustrative of the present invention. These examples exemplify the invention and are not to be construed as limiting the scope of the invention.
EXAMPLES
Example 1: Synthesis of 2,3-O-isopropylidene-L-threitol (HI)
A solution of dimethyl-2,3-0-isopropylidene-L-tartrate (50.3 g) in toluene (50 ml) was gradually added to the stirred mixture of sodium-bis(2-methoxyethoxy) aluminum hydride (122.8 g) in toluene (50 ml) at 20-40°C. The reaction mixture was heated to 60-80°C, and the reaction was continued till completion, as monitored by TLC. When the reaction was complete, the mass was cooled to 25-3 5°C, quenched with careful addition of water (10ml) and concentrated. Treatment of the resulting residue with methyl tertiary butyl ether, followed by evaporation of the organic layer under reduced pressure afforded 2,3-0-isopropyliden -L-threitol ( III) as pale yellow oil. Yield: 29.8 g (81.2%) [α]D20 + 4.6.°(CHC13, c 5)
Example 2: Synthesis of 2,3-0-isopropylidene-L-threitol-l,4-bis(methanesulfonate)
(IV)
A stirred solution of 2,3-O-isopropylidene-L-threitol (100.2 g), methylene chloride (1250
ml) and pyridine (146.3 g) was cooled to 0-5°C and methanesulfonyl chloride (176.6 g)
was slowly added to it. Temperature of the reaction mixture was raised to 25-35°C and the
reaction was continued at the same temperature till completion of the reaction, as
monitored by HPLC. After completion of the reaction, aqueous sodium bicarbonate
solution was slowly added to the reaction mass and the organic layer was separated.
Aqueous layer from the reaction mixture was extracted with methylene chloride and the
organic layers were combined. Distillation of the organic solvent, optionally followed by
addition of isopropyl alcohol gave the product, 2,3-0-isopropylidene-L-threitol-l,4-
bis(methanesulfonate).
Yield: 160.7 g (79.7%)
[α]D20-21.6°(acetone,c2)
Example 3: Synthesis of Treosulfan (I)
A mixture of formic acid (98%, 1000 ml) and 2,3-0-isopropylidene-L-threitol-l,4-bis(methanesulfonate) (100.5 g) was stirred at room temperature until completion of the desired reaction, as monitored by TLC, When the reaction was complete, the reaction mass was concentrated under reduced pressure..
Treatment of the residue after evaporation with isopropanol yielded the final product Treosulfan, which was optionally subjected to further treatment with acetone and nexanes or petroleum ether, Yield: 74.3 g (85.0%) [α]D20 – 5.3°(acetone, c 2) Purity: > 99 %.
References
- ^ Jump up to:a b “Trecondi APMDS”. Therapeutic Goods Administration (TGA). 11 October 2022. Retrieved 25 January 2025.
- ^ “Updates to the Prescribing Medicines in Pregnancy database”. Therapeutic Goods Administration (TGA). 21 December 2022. Archived from the original on 3 April 2022. Retrieved 2 January 2023.
- ^ “Trecondi (Link Medical Products Pty Ltd T/A Link Pharmaceuticals)”. Therapeutic Goods Administration (TGA). 14 January 2025. Retrieved 25 January 2025.
- ^ “AusPAR: Trecondi”. Therapeutic Goods Administration (TGA). 4 July 2023. Retrieved 25 January 2025.
- ^ “Health product highlights 2021: Annexes of products approved in 2021”. Health Canada. 3 August 2022. Retrieved 25 March 2024.
- ^ “Treosulfan 5g Powder for Solution for Infusion – Summary of Product Characteristics (SmPC)”. (emc). Archived from the original on 20 May 2022. Retrieved 21 April 2020.
- ^ Jump up to:a b c d e f “Grafapex- treosulfan injection, powder, lyophilized, for solution”. DailyMed. 31 January 2025. Retrieved 2 April 2025.
- ^ “Trecondi Product Information” (PDF). European Medicines Agency (EMA). 21 April 2020.
- ^ Jump up to:a b c d e f g h i j k l m “Trecondi EPAR”. European Medicines Agency (EMA). 11 December 2018. Archived from the original on 16 March 2023. Retrieved 21 April 2020. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Romański M, Wachowiak J, Główka FK (October 2018). “Treosulfan Pharmacokinetics and its Variability in Pediatric and Adult Patients Undergoing Conditioning Prior to Hematopoietic Stem Cell Transplantation: Current State of the Art, In-Depth Analysis, and Perspectives”. Clinical Pharmacokinetics. 57 (10): 1255–1265. doi:10.1007/s40262-018-0647-4. PMC 6132445. PMID 29557088.
- ^ Jump up to:a b “FDA approves treosulfan with fludarabine as a preparative regimen for alloHSCT in adult and pediatric patients with AML or MDS”. U.S. Food and Drug Administration (FDA). 6 February 2025. Retrieved 8 March 2025.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Novel Drug Approvals for 2025”. U.S. Food and Drug Administration (FDA). 21 February 2025. Retrieved 9 March 2025.
- ^ “Medexus Announces FDA Approval of Grafapex (treosulfan) for Injection and Provides Business Update” (Press release). Medexus Pharmaceuticals. 22 January 2025. Retrieved 25 January 2025 – via Newsfile.
- ^ “Treosulfan Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 16 May 1994. Retrieved 9 March 2025.
- ^ “Treosulfan Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 8 April 2015. Retrieved 9 March 2025.
- ^ “EU/3/04/186”. European Medicines Agency (EMA). 17 September 2018. Archived from the original on 16 October 2019. Retrieved 21 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ World Health Organization (1972). “International nonproprietary names for pharmaceutical substances (INN). recommended INN: list 12”. WHO Chronicle. 26 (10).
External links
- “Treosulfan”. National Cancer Institute.
- [1]
- Clinical trial number NCT00822393 for “Clinical Phase III Trial Treosulfan-based Conditioning Versus Reduced-intensity Conditioning (RIC)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Trecondi, others |
| Other names | 1,2,3,4-Butanetetrol, 1,4-dimethanesulfonate, Threitol 1,4-dimethanesulfonate, Threitol 1,4-bismethanesulfonate; L-Threitol 1,4-bis(methanesulfonate); Threosulphan; Treosulphan; Tresulfan |
| AHFS/Drugs.com | International Drug Names |
| License data | US DailyMed: Treosulfan |
| Pregnancy category | AU: D[1][2] |
| Routes of administration | By mouth, intravenous |
| ATC code | L01AB02 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)[1][3]<[4]CA: ℞-only[5]UK: POM (Prescription only)[6]US: ℞-only[7]EU: Rx-only[8]In general: ℞ (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 299-75-2 |
| PubChem CID | 9882105 |
| DrugBank | DB11678 |
| ChemSpider | 8057780 |
| UNII | CO61ER3EPI |
| KEGG | C19557D07253 |
| ChEBI | CHEBI:82557 |
| CompTox Dashboard (EPA) | DTXSID0026173 |
| ECHA InfoCard | 100.005.529 |
| Chemical and physical data | |
| Formula | C6H14O8S2 |
| Molar mass | 278.29 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Melting point | 101.5 to 105 °C (214.7 to 221.0 °F) |
| showSMILES | |
| showInChI | |
- Romanski M, Baumgart J, Bohm S, Glowka FK: Penetration of Treosulfan and its Active Monoepoxide Transformation Product into Central Nervous System of Juvenile and Young Adult Rats. Drug Metab Dispos. 2015 Dec;43(12):1946-54. doi: 10.1124/dmd.115.066050. Epub 2015 Oct 1. [Article]
- EMA Summary of Product Characteristics: Trecondi (treosulfan) powder for solution for infusion [Link]
- FDA Approved Drug Products: GRAFAPEX (treosulfan) for injection, for intravenous use [Link]
- EMC Summary of Product Characteristics: Treosulfan 5g Powder for Solution for Infusion [Link]
- NIH LiverTox: Alkylating Agents [Link]
- FDA News Release: FDA approves treosulfan with fludarabine as a preparative regimen for alloHSCT in adult and pediatric patients with AML or MDS [Link]
////////TREOSULFAN, Treosulphan, Ovastat, Treosulfano, Grafapex, acute myeloid leukemia, myelodysplastic syndrome, NSC-39069, Dihydroxybusulfan, L-threitol-1,4-dimethanesulfonate, Trecondi, FSA 2025, APPROVALS 2025, EMA 2019, EU 2019
CS(=O)(=O)OC[C@H](O)[C@@H](O)COS(C)(=O)=O
Bevemipretide




Bevemipretide,
CAS 2356106-71-1 FREE BASE
CAS SBT-272 Trihydrochloride, 2589640-11-7
607.7 g/mol, C31H45N9O4 F553HAL9V8
- SBT-272
- (2R)-2-amino-N-[(2S)-1-[[(1S)-5-amino-1-(3-benzyl-1,2,4-oxadiazol-5-yl)pentyl]amino]-3-(4-hydroxy-2,6-dimethylphenyl)-1-oxopropan-2-yl]-5-(diaminomethylideneamino)pentanamide
- L-Tyrosinamide, D-arginyl-N-[(1S)-5-amino-1-[3-(phenylmethyl)-1,2,4-oxadiazol-5-yl]pentyl]-2,6-dimethyl-
- (2R)-2-amino-N-[(2S)-1-[[(1S)-5-amino-1-(3-benzyl-1,2,4-oxadiazol-5-yl)pentyl]amino]-3-(4-hydroxy-2,6-dimethylphenyl)-1-oxopropan-2-yl]-5-(diaminomethylideneamino)pentanamide
- L-Tyrosinamide, D-arginyl-N-[(1S)-5-amino-1-[3-(phenylmethyl)-1,2,4-oxadiazol-5-yl]pentyl]-2,6-dimethyl-
- (2R)-2-amino-N-[(1S)-1-{[(1S)-5-amino-1-(3-benzyl-1,2,4-oxadiazol-5-yl)pentyl]carbamoyl}-2-(4-hydroxy-2,6-dimethylphenyl)ethyl]-5-carbamimidamidopentanamide
- Originator Stealth BioTherapeutics
- ClassAntidementias; Antiparkinsonians; Neuroprotectants; Peptidomimetics
- Mechanism of Action Adenosine triphosphatase stimulants; Cardiolipin modulators; Reactive oxygen species inhibitors
- Orphan Drug Status Yes – Amyotrophic lateral sclerosis
Phase IAmyotrophic lateral sclerosis
- Preclinical Dry age-related macular degeneration; Frontotemporal dementia; Parkinson’s disease
- No development reported Multiple system atrophy
18 Sep 2024Pharmacodynamics data from a preclinical trial in dry age related macular degenration released by Stealth BioTherapeutics
- 18 Sep 2024Preclinical trials in Dry age-related macular degeneration in USA (Opthalmic)
- 18 Sep 2024Stealth Biotherapeutics plans clinical trial for Dry age related macular degeneration (Topical)
The present technology relates generally to compounds (i.e. peptidomimetics), compositions (e.g. medicaments) and methods for treating, preventing, inhibiting, amelioration or delaying the onset of ophthalmic diseases, disorders or conditions in a mammalian subject. In some embodiments, the ophthalmic disease, disorder or condition is associated with deterioration of the integrity of the ellipsoid zone of one or more eyes of the mammalian subject. For example, the present technology may relate to administering one or more mitochondrial-targeting peptidomimetics (alone, as formulated and/or in combination with other active pharmaceutical ingredients) in effective amounts to treat, prevent, inhibit, ameliorate or delay the onset of ophthalmic diseases, disorders or conditions (e.g., macular degeneration (including (wet or dry) age-related macular degeneration), dry eye, diabetic retinopathy, diabetic macular edema, cataracts, autosomal dominant optic atrophy (DOA), Leber hereditary optic neuropathy (LHON), pigmentary retinopathy, retinitis pigmentosa, glaucoma, ocular hypertension, uveitis, chronic progressive external ophthalmoplegia (often referred to as CPEO or just PEO, e.g., Kearns-Sayre syndrome), and/or Leber congenital amaurosis (LCA)), in mammalian subjects
[0003] The following introduction is provided to assist the understanding of the reader. None of the information provided, or references cited, is admitted as being prior art to the present technology.
[0004] Diseases, disorders and degenerative conditions of the optic nerve and retina are the leading causes of blindness in the world. Many ophthalmic diseases disorders or conditions result from, or are associated with, mitochondrial dysfunction.
[0005] A significant degenerative condition of the retina is age-related macular degeneration (AMD). AMD is the most common cause of blindness in people over the age of 50 in the United States and its prevalence increases with age. AMD is classified as either wet (neovascular) or dry (non-neovascular). The dry form of the disease is more common.
Macular degeneration occurs when the central retina has become distorted and thinned. This change is usually associated with age but also characterized by intra-ocular inflammation and angiogenesis (wet AMD only) and/or intra-ocular infection. The subsequent generation of free radicals, resulting in oxidative tissue damage, local inflammation and production of growth factors (such as VEGF and FGF) and inflammatory mediators, can lead to inappropriate neovascularization in common with the wet form of AMD. Mitochondrial dysfunction is believed to play a role in age-related disorders such as AMD. (Liu et al., Appl. Sci. (2021) 11: 7385). Pieramici & Ehlers have reported that: “RPE mitochondria in AMD eyes undergo more pronounced degenerative changes, with lower mitochondrial density, organelle area and cristae number.” (Pieramici & Ehlers, Presentation at 54th Annual Retina Society Meeting, Sept.30, 2021, slide 3).
[0006] Retinopathy is a leading cause of blindness in type I diabetes and is also common in type II diabetes. The degree of retinopathy depends on the duration of diabetes, and generally begins to occur ten or more years after onset of diabetes. Diabetic retinopathy may be classified as non-proliferative, where the retinopathy is characterized by increased capillary permeability, edema and exudates, or proliferative, where the retinopathy is characterized by neovascularization extending from the retina to the vitreous, scarring, deposit of fibrous tissue and the potential for retinal detachment. Diabetic retinopathy is believed to be caused by the development of glycosylated proteins due to high blood glucose and leads to damage in small blood vessels in the eye. Diabetic retinopathy (often if left untreated) can progress to diabetic macular edema. Diabetic macular edema involves damage to the blood vessels in the retina that progress to a point where they leak fluid into the macula thereby causing the macula to swell and this results in blurred vision. Mitochondrial dysfunction has been linked to the pathogenesis of diabetic retinopathy. (Wu et al. Hindawi Oxidative Medicine and Cellular Longevity, Volume 2018, Article 3420187)
[0007] Glaucoma is made up of a collection of eye diseases that cause vision loss by damage to the optic nerve and retinal ganglion cells (RGCs). An intraocular pressure (IOP) of over 21 mmHg without optic nerve damage is known as ocular hypertension. Elevated IOP due to inadequate ocular drainage is the primary cause of glaucoma. Lowering IOP reduces the risk of progressive RGC loss in glaucoma; however, no currently available treatments directly prevent RGC damage. Glaucoma often develops as the eye ages, or it can occur as the result of an eye injury, inflammation, tumor or in advanced cases of cataract or diabetes. It can also be caused by the increase in IOP caused by treatment with steroids. Drug therapies that are proven to be effective in glaucoma reduce IOP either by decreasing vitreous humor production or by facilitating ocular draining. Such agents are often vasodilators and as such act on the sympathetic nervous system and include adrenergic antagonists. It has been stated that: “… mitochondrial dysfunction plays an important role in the pathogenesis of neurodegenerative diseases…” and “… mitochondrial damage may provide potential strategies for the treatment of glaucoma….” (Liu et al., Appl. Sci. (2021) 11: 7385).
[0008] Autosomal dominant optic atrophy (DOA) is a genetic X-linked neuro-ophthalmic condition characterized by bilateral degeneration of optic nerves. It affects approximately 1 in 10,000 (Denmark) to 1 in 30,000 (worldwide) persons. The nerve damage causes visual loss. It generally begins to manifest itself during the first decade of life and progresses thereafter. The disease itself affects primarily the retinal ganglion nerves. Mutations in the genes known as OPA1 and OPA3, which encode inner mitochondrial membrane proteins (resulting in mitochondrial dysfunction), are generally associated with DOA.
[0009] Leber Hereditary Optic Neuropathy (LHON) is a genetically-based inherited disease that generally starts to manifest itself between the ages of 15 and 35. In LHON, mitochondrial mutations affect complex I subunit genes in the respiratory chain leading to selective degeneration of retinal ganglion cells (RGCs) and optic atrophy generally within a year of disease onset. LHON is caused by mutations in the MT-NDI1, MT-ND4, MT-ND4L and MT-ND6 genes; all of which are associated with mitochondrial genome coding. LHOH affects approximately 1 in 50,000 people worldwide. It generally starts in one eye and progresses quickly to the other eye. Subjects with LHON may eventually become legally or totally blind, often before they turn 50. LHON affects vision needed for tasks such as reading, driving and recognizing others.
[0010] Retinitis pigmentosa (RP) is a group of hereditary retinal degenerative disorders characterized by progressive vision loss. RP is a leading cause of inherited blindness in the developed world. Clinically, RP is manifested by night vision difficulties due to the death of rod photoreceptors followed by the progressive loss of peripheral vision eventually leading to central vision impairment from the secondary loss of cone photoreceptors. RP is caused by mutations of at least 87 genes. The pathogenesis of RP is not well understood. However, mitochondrial dysfunction and oxidative damage are believed to play a key role in the pathogenesis of photoreceptor cell death in RP. (Gopalakrishnan et al., Scientific Reports (2020) 10: 20382)
[0011] Pigmentary retinopathy (PR) is a frequent feature of retinitis pigmentosa.
Pigmentary retinopathy is a non-specific finding that may be found in several mitochondrial diseases, such as Neurogenic weakness, Ataxia, and Retinitis Pigmentosa (NARP). PR is an inherited degenerative disorder of the retina, characterized by progressive photoreceptor damage. The damage leads to atrophy and cell death of the photoreceptors. Patients with PR can follow an autosomal-dominate, autosomal recessive or X-linked recessive pattern. The prevalence is about one in about three to four thousand individuals. Symptoms of the disease include nyctalopia (night blindness), peripheral visual field constriction, and sometimes loss of the central visual acuity or visual field.
[0012] Uveitis is array of intraocular inflammatory diseases of the eye that often results in irreversible visual loss. Uveitis is responsible for an estimated 30,000 new cases of legal blindness annually in the USA. It is believed that this disease is at least in part due to retinal tissue damage caused excessive mitochondrial oxidative stress that triggers a damaging immune response.
[0013] Chronic progressive external ophthalmoplegia (CPEO) is a condition characterized mainly by a loss of the muscle functions including in eye and eyelid movement. The condition typically appears in adults between ages 18 and 40 and slowly worsens over time. CPEO can be caused by genetic changes in any of several genes, which may be located in mitochondrial DNA or nuclear DNA. CPEO can occur as part of other underlying conditions, such as ataxia neuropathy spectrum and Kearns-Sayre syndrome. These conditions may not only involve CPEO, but various additional features that are not shared by most individuals with CPEO.
[0014] Kearns-Sayre syndrome is a condition that affects many parts of the body, especially the eyes. The features of Kearns-Sayre syndrome usually appear before age 20, and the condition is diagnosed by a few characteristic signs and symptoms. People with Kearns-Sayre syndrome have progressive external ophthalmoplegia. Affected individuals also have an eye condition called pigmentary retinopathy, which results from breakdown (degeneration) of the retina that gives it a speckled and streaked appearance.
[0015] Leber congenital amaurosis (LCA) is a rare genetic eye disorder that affects infants. The infants are often blind at birth. LCA can be associated with mitochondrial dysfunction. (Castro-Gago et al., J. Child Neurol. (1996) 11(2):108-11) Children born with LCA have light-gathering cells (rods and cones) of the retina that do not function properly. LCA has been estimated to be 1-2/100,000 births. This disorder affects males and females in equal numbers.
[0016] Drusen are small yellow or white spots between the retinal pigment epithelium and Bruch’s membrane in the retina that can be detected by an ophthalmologist during a dilated eye exam or with retinal photography. Drusen can also be imaged and monitored by optical coherence tomography (OCT). Drusen are made up of lipids and proteins. Drusen are a defining feature of macular degeneration. Drusen can be hard or soft. Larger numbers of drusen, as well as drusen of larger size, indicate higher risk for some vision loss in the future. “Hard” drusen are small and indicate lower risk of future vision loss than “soft” drusen. “Soft” drusen are larger, cluster together, and have edges that are not as clearly defined. Soft drusen are more likely to lead to vision loss.
[0017] Geometric Atrophy (GA) is generally considered part of the later stage of age-related macular degeneration (AMD) and refers to progression of the disease to a point where in regions of the retina, cells begin to waste away and die (i.e. atrophy).
[0018] Best corrected visual acuity (BCVA) is a measure of the best possible vision an eye can achieve with the use of glasses or corrective lenses. It is typically measured using Snellen lines on an eye chart. Repeated testing of the BCVA over time can be used to determine if a subject’s vision is stable, improving or deteriorating.
[0019] Low luminance visual acuity (LLVA) involves standard visual acuity testing under low-light conditions. This is often achieved by adding a neutral density filter in front of the testing eye. It is a useful visual function marker in those with geographic atrophy (GA) and neovascular age-related macular degeneration. Repeated testing of the LLVA over time can be used to determine if a subject’s vision, under low light conditions, is stable, improving or deteriorating.
[0020] Optical coherence tomography (OCT) is a non-invasive imaging method used to generate a picture of the back of the eye (i.e. the retina). OCT uses a low-powered laser to create pictures of the layers of the retina and optic nerve. The cross-sectional images are three-dimensional and color-coded. OCT can measure the thickness of the retina and optic nerve. OCT can be used to diagnose and manage Glaucoma, AMD, diabetes-related retinopathy, cystoid macular edema, macula pucker and macular hole.
[0021] Spectral domain optical coherence tomography (SDOCT) is an interferometric technique that provides depth-resolved tissue structure information encoded in the magnitude and delay of the back-scattered light by spectral analysis of the interference fringe pattern. SDOCT increases axial resolution 2- to 3-fold and scan speed 60- to 110-fold vs conventional (TD) OCT.
[0022] The ellipsoid zone can be mapped using SCOCT and the integrity of (or changes in) the ellipsoid zone can be determined from such mapping/scanning activity. (Itoh et al., Br J Ophthalmol. (2016) 100(3): 295-299). The technology is capable of evaluating the structures of the external limiting membrane (ELM), ellipsoid zone (EZ), interdigitation zone (IZ) and the retinal pigment epithelium (RPE). Id. Use of this technology is capable of accessing EZ integrity and EZ-RPE alterations. Id. The EZ and ELM, in particular, have been linked to visual outcomes and prognosis in numerous macular conditions, such as age-related macular degeneration (AMD) Id. Itoh et al. suggest that the utility of SDOCT as an assessment tool for EZ integrity for clinical trials and disease prognostication/management may prove particularly useful.
[0023] Swept source OCT (SS-OCT) and OCT angiography (OCTA) are relatively new techniques that are capable of better resolution of the retinal pigment epithelium (RPE), Bruch’s membrane (BM) and choriocapillaris (CC) structures. (Zhou et al. Biomedical Optics Express (2020) 11(4): 1834-1850) Using this technology it is possible to generate relative distance and thickness maps of the RPE-BM-CC complex. Id. Use of these techniques may provide a better understanding of the CC in three dimensions, and further
investigate potential functional relationships between RPE, BM and CC, and their involvement in age-related ocular diseases. Id.
[0024] The ellipsoid zone (EZ) of the eye is a mitochondrial rich tissue (Ball et al., Sci. Adv.8, eabn2070 (2022)). The ellipsoid zone can be imaged using optical coherence tomography (Fujita et al., Scientific Reports (2019) 9:12433). The integrity of the EZ can be quantified. (Fugita et al.). There is a clear relationship between the integrity of the ellipsoid zone and visual function. (Fugita et al., Figure.3). Ball et al. suggest that tightly packed mitochondria in the ellipsoid “focus” light for entry into the outer segment and that healthy mitochondria structure (including cristae structure) might be important for producing a Stiles-Crawford effect (SCE) and maintaining visual resolution in mammals. Pieramici & Ehlers describe mapping the ellipsoid zone to thereby observe the ellipsoid zone and possibly monitor changes in the integrity of the ellipsoid zone. (Pieramici & Ehlers, Presentation at 54th Annual Retina Society Meeting, Sept.30, 2021). Pieramici & Ehlers further described the use of Sub-RPE compartment maps as a means to find and monitor drusen formation and RPE atrophy in a subject. In the study being described (which described results from a P2 clinical trial involving treatments with elamipretide), Pieramici & Ehlers concluded, inter alia, that: (i) “Average BCVA and LLVA in NCGA and HRD patients improved significantly at 24 weeks [of treatment with elamipretide]” and (ii) “Baseline higher order OCT parameters, such as EZ integrity, correlated with improved LLVA in Elamipretide-treated eyes” (Pieramici & Ehlers at slide 15).
[0025] In brief, there are many ophthalmic diseases for which there remains a need for treatments/therapies or improved treatments/therapies. For example, there remains a need for treatments/therapies, or improved treatments/therapies, to address ophthalmic diseases, disorders or conditions such as macular degeneration (including (wet or dry) age-related macular degeneration), dry eye, diabetic retinopathy, diabetic macular edema, cataracts, autosomal dominant optic atrophy (DOA), Leber hereditary optic neuropathy (LHON), pigmentary retinopathy, retinitis pigmentosa, glaucoma, ocular hypertension, uveitis, chronic progressive external ophthalmoplegia (e.g., Kearns-Sayre syndrome), and/or Leber congenital amaurosis (LCA). This forgoing discussion addresses these needs.
SCHEME

MAIN

PATENT
WO2023069255
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023069255&_cid=P22-M93M8P-34013-1

Synthesis of (R)-2-amino-N-((S)-1-(((S)-5-amino-1-(3-benzyl-1,2,4-oxadiazol-5- yl)pentyl)amino)-3-(4-hydroxy-2,6-dimethylphenyl)-1-oxopropan-2-yl)-5- guanidinopentanamide (D-Arg-DMT-NH((S)-5-amino-1-(3-benzyl-1,2,4-oxadiazol-5- yl)pent-1-yl), 7a (a.k.a. ((Formula IIa)):
[0134] In some embodiments, Compound 7a (a.k.a. Formula IIa) may be synthesized as illustrated in Scheme 5, below (Also see WO2019/118878, incorporated herein by reference), wherein compound 12a can be prepared as illustrated in Scheme 6, below
[0135] Step a: Synthesis of benzyl (S)-2-((R)-2-((tert-butoxycarbonyl)amino)-5- guanidinopentanamido)-3-(4-hydroxy-2,6-dimethylphenyl)propanoate (3a). To a suspension of 2,6-Dmt-OBn.HCl (2a, 45.0 g, 134 mmol) in ACN (800 mL), NMM (32.7 mL, 298 mmol) was added at 00C. The reaction mixture was stirred until the reaction mixture became transparent. Then Boc-D-Arg-OH.HCl (1a, 46.3 g, 149 mmol) and HOBt.H2O (9.11 g, 59.5 mmol) were added to reaction mixture and stirred for 15 min. Finally, EDC.HCl (38.5 g, 201 mmol) was added and mixture was stirred at 00C for 4 h. Then EtOAc (450 mL), 1N HCl in brine (300 mL) were added. The combined organic extracts were washed with 1N HCl in brine (7×150 mL), NaHCO3/brine (300 mL and until pH of aqueous layer is about pH=6 to 7), dried over Na2SO4, filtered and concentrated to afford 86.0 g (97%) of Boc-D-Arg-DMT- OBn (3a) that was used without further purification.1H-NMR (400 MHz, Methanol-d4) δ 7.33 – 7.18 (m, 5H), 6.43 (s, 2H), 5.06 (s, 2H) 4.71 (t, J=7.8Hz, 1H), 4.07 (t, J=6.7Hz,1H), 3.19 – 3.09 (m, 3H), 3.03-2.97 (m, 1H), 2.23 (s, 6H), 1.72 – 1.65 (m, 1H), 1.54 – 1.43 (m, 3H), 1.45 (s, 9H).
[0136] Step b: Synthesis of (S)-2-((R)-2-((tert-butoxycarbonyl)amino)-5-guanidinopentanamido)-3-(4-hydroxy-2,6-dimethylphenyl)propanoic acid (4a). To a solution of Boc-D-Arg-DM-Tyr-OBn (3a, 84.0 g, 142 mmol) in MeOH (1000 mL) Pd/C (10% w/w, 14.0 g) was added. The hydrogen was purged in reaction mixture at room temperature for 4h. Then reaction mixture was filtrated through filter paper and washed with MeOH (150 mL). The solvent was removed by evaporation. White foam product 4a was obtained (74.0 g, 93%) and used without further purification.1H-NMR (400 MHz, Methanol-d4) δ 6.44 (s, 2H), 4.68 (t, J = 7.2 Hz, 1H), 4.04 (t, J = 6.8 Hz, 1H), 3.15 – 3.09 (m, 3H), 3.02 – 2.94 (m, 1H), 2.29 (s, 6H), 1.74 – 1.59 (m, 1H), 1.54 – 1.43 (m, 1H), 1.45 (s, 9H).
[0137] Step c: Synthesis of tert-butyl ((6R,9S,12S)-1-amino-12-(3-benzyl-1,2,4-oxadiazol-5-yl)-9-(4-hydroxy-2,6-dimethylbenzyl)-1-imino-20,20-dimethyl-7,10,18-trioxo-19-oxa-2,8,11,17-tetraazahenicosan-6-yl)carbamate (6a). DMF (200 mL) was added to 4a (11.17 g, 24 mmol) and stirred at r.t. for 15 min. To the resulting suspension, 12a (10.65 g, 20 mmol) was added and stirred at r.t. for 20 min. After addition of HOBt (612 mg, 4.00 mmol), the suspension was cooled in ice bath. EDC . HCl (5.38 g, 28 mmol) was added in one portion, and the reaction mixture was stirred while cooled in ice bath for 2.5 h and then, for 4.5 h at r.t. The nearly homogeneous reaction mixture was quenched with EtOAc (1500 mL) and the resulting solution was washed for 10 times with brine/aq.0.5 M HCl (1:1; 400 mL). During the 6th and 9th washings, gel in the aqueous phase was formed. After addition of iPrOH (40 mL in each case) and repeated shaking the layers went clear again. Afterwards, the organic phase was washed for 6 times with brine/sat. aq. NaHCO3 (9:1; 400 mL). During the 4th washing, gel in the aqueous phase was formed. After addition of iPrOH (40 mL) and repeated shaking the layers were separated easily. The organic phase was washed with brine (200 mL) and water (100 mL) and the solvent was removed under reduced pressure. No vigorous shaking was performed upon washing with water to avoid difficulties in phase separation. As a result, 16.8 g of the crude product were obtained (6a, 97.0 % purity by HPLC, white amorphous solid).1H-NMR (300 MHz, Methanol-d4) ppm: δ = 7.33–7.16 (m, 5H), 6.38 (s, 2H), 5.18-5.07 (m, 1H), 4.64-4.55 (m, 1H), 4.10 – 3.92 (m, 3H), 3.18-2.77 (m, 6H), 2.20 (s, 6H), 1.97-1.76 (m, 2H), 1.75-1.14 (m, 8H), 1.43 (s, 9H), 1.41 (s, 9H).
[0138] Step d: Synthesis of (R)-2-amino-N-((S)-1-(((S)-5-amino-1-(3-benzyl-1,2,4-oxadiazol-5-yl)pentyl)amino)-3-(4-hydroxy-2,6-dimethylphenyl)-1-oxopropan-2-yl)-5-guanidinopentanamide (7a, but also referred to as (IIa – the tri-hydrochloride salt of Compound I) herein). After 6a (16.8 g) was dissolved in DCM (100 mL) and cooled to 0°C, TFA (20 mL) was added dropwise and the solution was allowed to stir at 0 °C for 10 min, and then at r.t. for 3 h (LC/MS shows no starting material). Then reaction mixture was evaporated (at 0–5 °C) and additionally re-evaporated from DCM (100 mL, at 0–5 °C). The purification by flash chromatography on reverse phase (cartridge C-18, 120G) was performed on crude material divided in 4 parts. Then all solvents were evaporated at reduced pressure at <40oC. White foam was dissolved in isopropanol (100 mL) and 5 mL of HCl in isopropanol (5-6M) was added at 0 oC and evaporated under reduced pressure. This step was repeated 3 times. Additionally, 100 mL of ACN was added and suspension was evaporated one more time. As a result, white powder of 7a was obtained as the tri-hydrochloride salt.1H-NMR (300 MHz, Methanol-d4) δ 7.36 – 7.14 (m, 5H), 6.40 (s, 2H), 5.15 (dd, J = 8.5, 6.3 Hz, 1H), 4.68 (dd, J = 8.7, 7.5 Hz, 1H), 4.07 (s, 2H), 3.97 (t, J = 6.3 Hz, 1H), 3.18 (t, J = 6.9 Hz, 2H), 3.11 (dd, J = 14.2, 8.8 Hz, 1H), 2.95 – 2.84 (m, 3H), 2.22 (s, 6H), 2.02 – 1.59 (m, 6H), 1.57 – 1.28 (m, 4H). MS: EI-MS: m/z 608.4 [M+1].


Synthesis of (S)-1-(3-Benzyl-1,2,4-oxadiazol-5-yl)-5-((tert-Butoxycarbonyl)amino)pentan-1-Aminium 4-Methylbenzenesulfonate (12a)
step a: NH2OH; step b: T3P, NaHCO3; step c: TEA; step d: PTSA
[0139] Step a: Synthesis of N-hydroxy-2-phenylacetimidamide (9a). To a solution of nitrile 8a (1.0 mol) in EtOH (1.2 L) was added NH2OH (50% aqueous solution, 130 g, 2.0 mol).
The solution was heated to reflux and stirred for 12 hours (hrs.). After completion, the reaction mixture was concentrated under reduced pressure. The resulting residue was re-dissolved in EtOH (350 mL) and concentrated under reduced pressure again (this procedure was repeated three times). The resulting solid was triturated in hexane (350 mL), filtered, washed with hexane (100 mL), and then dried to give the desired product 9a as white solid. (10.5 kg; KF = 1295) with good results (purity by HPLC, > 98.9 A%; Assay = 22.2 w%, yield = 91%).1H NMR (300 MHz, DMSO-d6): δ 8.90 (s, 1H), 7.28-7.18 (m, 5H), 5.40 (s, 2H), 3.25 (s, 2H) ppm. MS: (M+H)+: m/z = 151.1
[0140] Step b: Synthesis of (9H-Fluoren-9-yl)methyl tert-Butyl (1-(3-Benzyl-1,2,4-oxadiazol-5-yl)pentane-1,5-diyl) (S)-Dicarbamate (11a). To a solution of protected enantiomerically pure N2-(((9H-fluoren-9-yl)methoxy)carbonyl)-N6-(tert-butoxycarbonyl)-L-lysine (10a, 4.31 kg, 9.2 mol) and hydroxyimidamide 9a (1.1 equivalents “equiv.” or “eq.”) in ethyl acetate was added NaHCO3 (3.0 equiv.). The mixture was stirred at 25 oC for 20 minutes (min.). Then, propane phosphonic acid anhydride (T3P, 50% solution in ethyl acetate, 3.0 equivalents (equiv.)) was added and the reaction mixture was heated to 80 oC and stirred for 4 hrs. (about 60% conversion of compound 10a based on HPLC). Then compound 9a (1.1 equiv.) was added and the reaction mixture was stirred at 80oC for another 20 hr. (about 10% compound 10a remained). The reaction mixture was cooled to room temperature, saturated aqueous NaHCO3 (2.0 L) was added, the mixture was then extracted with ethyl acetate (3x 1.0 L). The combined organic layers were then washed with brine (1 L), dried over anhydrous Na2SO4, filtered and concentrated to give a crude residue, which was generally purified by silica gel column chromatography (Petroleum ether (PE):EtOAc = 5: 1) to give crude product, (9H-fluoren-9-yl)methyl tert-butyl (1-(3-benzyl-1,2,4-oxadiazol-5-yl)pentane-1,5-diyl) (S)-dicarbamate (11a), solution in ACN (19.7 kg, assay = 20%, chiral HPLC purity = 99.12 A%,yield = 73%).1H-NMR (300 MHz, CDCl3): δ 7.78 (d, J = 7.5 Hz, 2H), 7.61 (d, J = 6.3 Hz, 2H), 7.42 (t, J = 7.5 Hz, 2H), 7.35-7.30 (m, 7H), 5.52 (br, 1H), 5.09-5.05 (m, 1H), 4.56-4.37 (m, 3H), 4.22 (t, J = 6.6 Hz, 1H), 4.08 (s, 2H), 1.95-1.86 (m, 2H), 1.48-1.42 (m, 11H) ppm. MS: (M-100+H)+: m/z = 483.2.
[0141] Step c: Synthesis of tert-Butyl (S)-(5-Amino-5-(3-Benzyl-1,2,4-oxadiazol-5-yl)pentyl)-carbamate (5a). To a solution of compound (9H-fluoren-9-yl)methyl tert-butyl (1-(3-benzyl-1,2,4-oxadiazol-5-yl)pentane-1,5-diyl) (S)-dicarbamate (11a) was added TEA (2.5 eq.). The mixture was kept stirring with mechanical stirrer at 20~ 25 °C for 15 h. The reaction mixture was diluted by tap water and MTBE. Separated, aqueous layer was extracted by MTBE for one time. Both MTBE layers were combined, and then washed by NH4Cl. Then anhydrous Na2SO4 was added and that solution stirred for least 2 h, then filtered and washed with MTBE to afford tert-butyl (S)-(5-amino-5-(3-benzyl-1,2,4-oxadiazol-5-yl)pentyl)-carbamate (5a) solution in MTBE (32.9 kg, assay = 6.5%, yield = 88%).1H-NMR (300 MHz, DMSO-d6): δ 7.33-7.25 (m, 5H), 6.78 (br, 1H), 5.09-5.05 (m, 1H), 4.56-4.37 (m, 3H), 4.06 (s, 2H), 3.98 (t, J = 6.6 Hz, 1H), 2.87-2.84 (m, 2H), 2.10 (s, 2H), 1.38-1.34 (m, 2H), 1.24 (s, 9H), 1.20-1.15 (m, 2H) ppm. MS: (M+H)+: m/z = 361.1.
[0142] Step d: Synthesis of (S)-1-(3-Benzyl-1,2,4-oxadiazol-5-yl)-5-((tert-Butoxycarbonyl)-amino)pentan-1-Aminium 4-Methylbenzenesulfonate (12a). p-toluenesulfonic acid (PTSA) was added to solution of crude tert-butyl (S)-(5-amino-5-(3-benzyl-1,2,4-oxadiazol-5-yl)pentyl)-carbamate (5a) in MTBE to afford (S)-1-(3-benzyl-1,2,4-oxadiazol-5-yl)-5-((tert-butoxycarbonyl)amino)pentan-1-aminium 4-methylbenzenesulfonate (12a) (2.7 kg, yield = 85 %, HPLC purity > 99%, ee > 99%) as white solid.1H-NMR (400 MHz, DMSO-d6): δ 8.74 (br, 3H), 7.48 (d, J = 8.0 Hz, 2H), 7.37-7.26 (m, 5H), 7.11 (d, J = 8.0 Hz, 2H), 6.77 (t, J = 5.2 Hz, 1H), 4.82 (t, J = 6.8 Hz, 1H), 4,17 (s, 2H), 2.90-2.86 (m, 2H), 2.29 (s, 3H), 1.39-1.36 (m, 11H), 1.35-1.28 (m, 2H) ppm. MS: (M-172+H)+: m/z = 361.1.
PATENT WO2021016462
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021016462&_cid=P22-M93MJV-41323-1

WO2019118878
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019118878&_cid=P22-M93MNH-43976-1
/////////Bevemipretide, SBT-272 Trihydrochloride, SBT 272, ORHAN DRUG, Stealth BioTherapeutics
Suzetrigine



Suzetrigine
CAS
2649467-58-1 |
Average: 473.4
Monoisotopic: 473.137396951
Chemical Formula
C21H20F5N3O4
FDA 1/30/2025, Journavx
To treat moderate to severe acute pain
Press Release
- 2-Pyridinecarboxamide, 4-[[[(2R,3S,4S,5R)-3-(3,4-difluoro-2-methoxyphenyl)tetrahydro-4,5-dimethyl-5-(trifluoromethyl)-2-furanyl]carbonyl]amino]-
- 4-[(2R,3S,4S,5R)-3-(3,4-difluoro-2-methoxyphenyl)-4,5- dimethyl-5-(trifluoromethyl)oxolane-2- carboxamido]pyridine-2-carboxamide
- 4-[(2R,3S,4S,5R)-3-(3,4-difluoro-2-methoxyphenyl)-4,5-dimethyl-5-(trifluoromethyl)oxolane-2-amido]pyridine2-carboxamide
- 4-[[[(2R,3S,4S,5R)-3-(3,4-Difluoro-2-methoxyphenyl)tetrahydro-4,5-dimethyl-5-(trifluoromethyl)-2-furanyl]carbonyl]amino]-2-pyridinecarboxamide
- CS-0641183
- HY-148800
- VX 548
- VX-548
- VX548
- Management of
Acute, moderate pain
Suzetrigine, sold under the brand name Journavx, is a medication used for the management of pain.[1][2] It is a non-opioid, small-molecule analgesic that works as a selective inhibitor of Nav1.8-dependent pain-signaling pathways in the peripheral nervous system,[3][4] avoiding the addictive potential of opioids. Suzetrigine is taken by mouth.[1]
The most common adverse reactions include itching, muscle spasms, increased blood level of creatine kinase, and rash.[1][2]
It was developed by Vertex Pharmaceuticals,[5] and was approved for medical use in the United States in January 2025.[2][6] Suzetrigine is the first medication to be approved by the US Food and Drug Administration (FDA) in this new class of pain management medicines.[2]
Medical uses
Suzetrigine is indicated for the treatment of moderate to severe acute pain in adults.[1][2]
FDA Approves Novel Non-Opioid Treatment for Moderate to Severe Acute Pain
First Drug Approved in New Class of Non-Opioid Pain Medicines; Agency Continues to Take Steps to Support New Approaches for Pain Management
For Immediate Release:January 30, 2025
Today, the U.S. Food and Drug Administration approved Journavx (suzetrigine) 50 milligram oral tablets, a first-in-class non-opioid analgesic, to treat moderate to severe acute pain in adults. Journavx reduces pain by targeting a pain-signaling pathway involving sodium channels in the peripheral nervous system, before pain signals reach the brain.
Journavx is the first drug to be approved in this new class of pain management medicines.
Pain is a common medical problem and relief of pain is an important therapeutic goal. Acute pain is short-term pain that is typically in response to some form of tissue injury, such as trauma or surgery. Acute pain is often treated with analgesics that may or may not contain opioids.
The FDA has long supported development of non-opioid pain treatment. As part of the FDA Overdose Prevention Framework, the agency has issued draft guidance aimed at encouraging development of non-opioid analgesics for acute pain and awarded cooperative grants to support the development and dissemination of clinical practice guidelines for the management of acute pain conditions.
“Today’s approval is an important public health milestone in acute pain management,” said Jacqueline Corrigan-Curay, J.D., M.D., acting director of the FDA’s Center for Drug Evaluation and Research. “A new non-opioid analgesic therapeutic class for acute pain offers an opportunity to mitigate certain risks associated with using an opioid for pain and provides patients with another treatment option. This action and the agency’s designations to expedite the drug’s development and review underscore FDA’s commitment to approving safe and effective alternatives to opioids for pain management.”
The efficacy of Journavx was evaluated in two randomized, double-blind, placebo- and active-controlled trials of acute surgical pain, one following abdominoplasty and the other following bunionectomy. In addition to receiving the randomized treatment, all participants in the trials with inadequate pain control were permitted to use ibuprofen as needed for “rescue” pain medication. Both trials demonstrated a statistically significant superior reduction in pain with Journavx compared to placebo.
The safety profile of Journavx is primarily based on data from the pooled, double-blind, placebo- and active-controlled trials in 874 participants with moderate to severe acute pain following abdominoplasty and bunionectomy, with supportive safety data from one single-arm, open-label study in 256 participants with moderate to severe acute pain in a range of acute pain conditions.
The most common adverse reactions in study participants who received Journavx were itching, muscle spasms, increased blood level of creatine phosphokinase, and rash. Journavx is contraindicated for concomitant use with strong CYP3A inhibitors. Additionally, patients should avoid food or drink containing grapefruit when taking Journavx.
The application received Breakthrough Therapy, Fast Track and Priority Review designations by the FDA.
The FDA granted approval of Journavx to Vertex Pharmaceuticals Incorporated.
PATENTS
https://patentimages.storage.googleapis.com/08/4f/6e/4f104b27a3772f/US11919887.pdf
https://patentscope.wipo.int/search/en/detail.jsf?docId=US407339565&_cid=P22-M90R90-47554-1



Step 1:
NEt₂ (7.7 mL, 55.2 mmol) was added to a solution of
ethyl 2-diazo-3-oxo-pentanoate (6.69 g, 39.3 mmol) in
DCM (80 mL) with stirring at 0° C. under nitrogen. Trimethylsilyl trifluoromethanesulfonate (8.5 mL, 47.0 mmol)
was added dropwise over 5 mins and the mixture was stirred
for a further 30 mins at 0° C. The reaction mixture was
diluted with pentane (100 mL), the layers separated and the
organic phase washed with dilute aqueous sodium bicarbonate (100 mL) and brine (100 mL). The organic layer was
dried (MgSO4), and concentrated in vacuo to give ethyl
(Z)-2-diazo-3-trimethylsilyloxy-pent-3-enoate (9.4 g, 99%)
as a red oil. H NMR (500 MHz, Chloroform-d) 8 5.33 (q,
J=7.0 Hz, 1H), 4.25 (q, J=7.1 Hz, 2H), 1.67 (d, J=7.0 Hz,
3H), 1.29 (t, J=7.1 Hz, 3H), 0.22 (s, 9H) ppm.
Step 2:
To a solution of 1,1,1-trifluoropropan-2-one (8 mL, 89.4
mmol) in DCM (80 mL) stirring at -78° C. was added TiCl
(70 mL of 1 M in DCM, 70.00 mmol) via cannula. To the
resulting solution, a solution of ethyl (Z)-2-diazo-3-trimethylsilyloxy-pent-3-enoate (36.1 g of 31.3% w/w, 46.6 mmol)
in 40 mL of DCM was added dropwise over 15 mins. After
100 mins the reaction was carefully quenched with water,
allowing the temperature to rise slowly, and then extracted
with DCM. The combined organic layers were dried
(MgSO), filtered, and concentrated in vacuo. Purification
by flash chromatography (330 g SiO₂, 0 to 20% EtOAc in
heptane) gave ethyl 2-diazo-6,6,6-trifluoro-5-hydroxy-4,5-
dimethyl-3-oxo-hexanoate (8.82 g, 67%), which was stored
as a solution in toluene. H NMR (500 MHz, Chloroform-d)
8 4.33 (q, J=7.1 Hz, 2H), 4.14 (q, J=7.0 Hz, 1H), 3.98 (s,
1H), 1.43 (q, J=1.2 Hz, 3H), 1.35 (t, J=7.1 Hz, 3H), 1.31 (dq.
J=7.0, 1.4 Hz, 3H) ppm. ESI-MS m/z calc. 282.08273, found
283.1 (M+1)*; 281.0 (M-1)-.
Step 3:
A solution of rhodium tetraacetate (245 mg, 0.55 mmol)
in benzene (32 mL) was heated at reflux for 10 min before
a solution of ethyl 2-diazo-6,6,6-trifluoro-5-hydroxy-4,5-
dimethyl-3-oxo-hexanoate (10 g, 35.4 mmol) in benzene (13
mL) was added slowly via addition funnel while refluxing
for 60 mins. The mixture was then concentrated in vacuo to
give ethyl rac-(4R, 5R)-4,5-dimethyl-3-oxo-5-(trifluoromethyl)tetrahydrofuran-2-carboxylate (9.0 g, 100%) as a
green coloured residue containing residual catalyst, and as a
mixture of epimers at the position next to the ester. This
material was used without further purification. H NMR
(500 MHz, Chloroform-d) 8 4.83-4.57 (m, 1H), 4.38-4.16
(m, 2H), 2.60 (dddd, J=9.3, 8.2, 5.6, 1.4 Hz, 1H), 1.73-1.63
(m, 3H), 1.30 (t, J=7.1 Hz, 3H), 1.24 (ddq, J=6.4, 4.1, 1.9
Hz, 3H) ppm.
Step 4:
To a stirred solution of ethyl rac-(4R,5R)-4,5-dimethyl- 5
3-oxo-5-(trifluoromethyl)tetrahydrofuran-2-carboxylate (48
g, 188.83 mmol) in DCM (400 mL) stirring at -78° C. was
added DIPEA (29.680 g, 40 mL, 229.64 mmol). A solution
of trifluoromethylsulfonyl trifluoromethanesulfonate
(53.440 g, 32 mL, 189.41 mmol) in DCM (200 mL) was 10
added to the reaction mixture at the same temperature over
1 h. The reaction mixture was stirred for 30 mins at 0° С.
before being quenched with 100 mL saturated aqueous
NaHCO3 solution. The organic layer was separated and
aqueous layer extracted with DCM (160 mL). The combined 15
organic layers were dried (MgSO) and concentrated in
vacuo to give ethyl rac-(4R,5R)-2,3-dimethyl-2-(trifluoromethyl)-4-(trifluoromethylsulfonyloxy)-3H-furan-5-carboxylate (71 g, 97%). H NMR (400 MHz, Chloroform-d) 8
4.38-4.32 (m, 2H), 3.29-3.23 (m, 1H), 1.64 (s, 3H), 1.37- 20
1.33 (m, 6H) ppm.
STEP 5
To stirred a solution of ethyl rac-(4R,5R)-2,3-dimethyl2-(trifluoromethyl)-4-(trifluoromethylsulfonyloxy)-3Hfuran-5-carboxylate (26 g, 67.311 mmol) in toluene (130.00
mL) was added (3,4-difluoro-2-methoxy-phenyl)boronic
acid (14 g, 74.5 mmol) followed by K3PO4 (100 mL of 2 M,
200.00 mmol) under an argon atmosphere. The reaction was
degassed before tetrakis(triphenylphosphine)palladium(0)
(4 g, 3.46 mmol) was added. After further degassing, the
reaction was heated at 100° C. for 2 hours. The reaction was
diluted in water and the aqueous layer extracted with EtOAc
(2×100 mL). The combined organic layers were concentrated in vacuo. Purification by flash chromatography (SiO.
0 to 10% EtOAc in heptane) gave ethyl 4-(3,4-difluoro-2- 35
methoxy-pheny1)-2,3-dimethyl-2-(trifluoromethyl)-3Hfuran-5-carboxylate (24.4 g, 93%) as a 6:1 diastereomeric
mixture, with the major isomer believed to be ethyl rac-(4R,
5R)-4-(3,4-difluoro-2-methoxy-phenyl)-2,3-dimethyl-2-
(trifluoromethyl)-3H-furan-5-carboxylate. Major isomer: H 40
NMR (400 MHz, Chloroform-d) 8 6.88-6.79 (m, 2H), 4.17-
4.09 (m, 2H), 3.90 (s, 3H), 3.46 (q, J=7.4 Hz, 1H), 1.67 (s,
3H), 1.12 (t, J=7.4 Hz, 3H), 1.06 (dd, J=5.4, 2.7 Hz, 3Н)
ppm. Minor isomer ¹H NMR (400 MHz, Chloroform-d) 8
6.88-6.79 (m, 2H), 4.17-4.09 (m, 2H), 3.88 (s, 3H), 3.76- 45
3.71 (m, 1H), 1.51 (s, 3H), 1.12 (t, J=7.4 Hz, 3H), 0.99 (dd,
J=5.4, 2.7 Hz, 3H) ppm. ESI-MS m/z calc. 380.1047, found
381.02 (M+1)+.
Step 6:
To an ice-cooled solution of ethyl 4-(3,4-difluoro-2- 50
methoxy-phenyl)-2,3-dimethyl-2-(trifluoromethyl)-3Hfuran-5-carboxylate (110 g, 243.0 mmol) in DCM (360 mL)
was added BBr, (370 mL of 1 M, 370.0 mmol) dropwise.
Upon completion the mixture was quenched by addition of
water and aqueous sodium bicarbonate solution, the aqueous 55
layer extracted with DCM and the combined organic layers
dried (MgSO) and concentrated in vacuo. The residue was
dissolved in DCM (430 mL) at ambient temperature and
TFA (40 mL, 519.2 mmol) was added, then the reaction was
heated to 45° C. Upon completion, the mixture was
quenched by addition of aqueous sodium bicarbonate solution and the aqueous layer extracted with DCM, dried
(MgSO) and concentrated in vacuo to give the desired
product in a 5:1 mixture of diastereomers. Recrystallization
was carried out by solubilizing the crude in the smallest
possible amount of DCM and adding a layer of heptane on
top of this solution (liquid-liquid diffusion). After approx. 1



https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021113627&_cid=P22-M90RUB-70989-1

Example 6
rel-(2S,3R,5S)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (20), (2S,3R,5R)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)- 5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (21), rel- (2R,3S,5R)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2- carbonyl]amino]pyridine-2-carboxamide (22), and (2R,3S,5S)-4-[[3-(3-chloro-4-fluoro-2-methoxy- phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (23)
[00676] Step 7:
[00677] (4-[[3-(3-Chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (420 mg, 0.8827 mmol) was separated by chiral SFC [(R,R)-Whelk-O1 column, 5 µm particle size, 25 cm x 21.2 mm from Regis Technologies, MeOH, 20 mM NH3], followed by further purification of one or more of the fractions by chiral SFC using a Chiralpak IC column, 5 µm particle size, 25 cm x 20 mm from Daicel or a Chiralpak ID column, 5 µum particle size, 25 cm x 20 mm from Daicel to give:
[00678] First Eluting Isomer: rel-(2S,3R,5S)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (20, 30 mg, 7.1%) (further purified by chiral SFC using Chiralpak IC column). 1H NMR (500 MHz, Chloroform-d) δ 8.92 (s, 1H), 8.47 (d, J = 5.5 Hz, 1H), 8.21 (dd, J = 5.6, 2.1 Hz, 1H), 8.09 (d, J = 2.2 Hz, 1H), 7.87 (d, J = 4.1 Hz, 1H), 7.26 (dd, J = 8.8, 5.8 Hz, 1H), 7.03 (t, J = 8.4 Hz, 1H), 5.87 – 5.82 (m, 1H), 4.77 (d, J = 10.6 Hz, 1H), 3.98 (td, J = 11.2, 8.3 Hz, 1H), 3.88 (s, 3H), 2.51 (dd, J = 13.2, 11.7 Hz, 1H), 2.42 (dd, J = 13.2, 8.3 Hz, 1H), 1.69 (s, 3H) ppm. ESI-MS m/z calc.475.0922, found 476.4 (M+1)+; 474.4 (M-1)-.
[00679] Second Eluting Isomer: (2S,3R,5R)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (21, 29 mg, 6.7%) (further purified by chiral SFC using Chiralpak ID column). 1H NMR (500 MHz, Chloroform-d) δ 8.56 (s, 1H), 8.48 (d, J = 5.5 Hz, 1H), 8.08 (dd, J = 5.5, 2.2 Hz, 1H), 7.98 (d, J = 2.1 Hz, 1H), 7.86 (d, J = 4.4 Hz, 1H), 7.23 (dd, J = 8.8, 5.8 Hz, 1H), 7.01 (t, J = 8.4 Hz, 1H), 5.86 (d, J = 4.2 Hz, 1H), 4.80 (d, J = 9.7 Hz, 1H), 4.10 – 4.00 (m, 1H), 3.93 (s, 3H), 3.52 – 3.48 (m, 1H), 2.86 (dd, J = 13.9, 8.4 Hz, 1H), 2.16 -2.07 (m, 1H), 1.64 (s, 2H) ppm. ESI-MS m/z calc.475.0922, found 476.4 (M+1)+; 474.4 (M-1)-.
[00680] Third Eluting Isomer: rel-(2R,3S,5R)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (22, 42 mg, 9.5%).
1H NMR (500 MHz, Chloroform-d) δ 8.87 (s, 1H), 8.33 (d, J = 5.6 Hz, 1H), 8.08 (dd, J = 5.6, 2.2 Hz, 1H), 7.98 (d, J = 2.2 Hz, 1H), 7.74 (d, J = 4.5 Hz, 1H), 7.12 (dd, J = 8.8, 5.8 Hz, 1H), 6.89 (t, J = 8.4 Hz, 1H), 5.79 (d, J = 4.5 Hz, 1H), 4.63 (d, J = 10.7 Hz, 1H), 3.85 (td, J = 11.2, 8.4 Hz, 1H), 3.74 (s, 3H), 2.37 (dd, J = 13.2, 11.7 Hz, 1H), 2.28 (dd, J = 13.1, 8.4 Hz, 1H), 1.55 (s, 3H) ppm. ESI-MS m/z calc.
475.0922, found 476.4 (M+1)+; 474.4 (M-1)-.
[00681] Fourth Eluting Isomer: (2R,3S,5S)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (23, 40 mg, 8.8%).
1H NMR (500 MHz, Chloroform-d) δ 8.43 (s, 1H), 8.35 (d, J = 5.5 Hz, 1H), 7.95 (dd, J = 5.5, 2.2 Hz, 1H), 7.85 (d, J = 2.2 Hz, 1H), 7.73 (d, J = 4.3 Hz, 1H), 7.10 (dd, J = 8.8, 5.9 Hz, 1H), 6.87 (t, J = 8.4 Hz, 1H), 5.76 – 5.71 (m, 1H), 4.67 (d, J = 9.7 Hz, 1H), 3.97 – 3.87 (m, 1H), 3.80 (s, 3H), 2.73 (dd, J = 13.9, 8.4 Hz, 1H), 1.98 (dd, J = 13.9, 11.6 Hz, 1H), 1.51 (s, 3H) ppm. ESI-MS m/z calc.475.0922, found 476.4 (M+1)+; 474.4 (M-1)-.
[00682] Compound 22 – Solid Form A
Efficacy
When people used suzetrigine in clinical studies conducted through 2024, there was a reduction in pain typically from seven to four on the standard numerical scale used to rate pain.[7][8] Suzetrigine provided pain relief equal to a combination of hydrocodone and paracetamol (acetaminophen) (5 mg of hydrocodone bitartrate and 325 mg of acetaminophen).[8][9]
Suzetrigine suppresses pain at the same level as an opioid, but without the risks of addiction, sedation, or overdose.[10] An alternative to opioids, it is the first pain medication to be approved by the Food and Drug Administration in two decades.[10]
The efficacy of suzetrigine was evaluated in two randomized, double-blind, placebo- and active-controlled trials of acute surgical pain, one following abdominoplasty and the other following bunionectomy.[2] Both trials found that suzetrigine reduced pain more effectively than a placebo.[2]
Contraindications
Concomitant use of suzetrigine with strong CYP3A inhibitors is contraindicated.[1][2]
Adverse effects
Common adverse effects of suzetrigine may include itching, rash, muscle spasms, and increased levels of creatine kinase.[2] Mild side effects may include nausea, constipation, headache, and dizziness.[7][8] As of 2024, long-term safety and side effects remain undetermined.[8]
In preliminary research, suzetrigine had no serious neurological, behavioral, or cardiovascular effects.[3]
Interactions
Consuming grapefruit while using suzetrigine may cause an adverse grapefruit–drug interaction.[1][2]
Mechanism of action
Suzetrigine operates on peripheral nerves, avoiding the addictive potential of opioids which affect the central nervous system.[3][4][7] Unlike opioid medications, which reduce pain signals in the brain, suzetrigine works by closing sodium channels in peripheral nerves, inhibiting pain-signaling nerves from transmitting painful sensations to the brain.[3][4][7]
In pharmacological studies, suzetrigine selectively inhibited Nav1.8 channels, but not other voltage-gated sodium channels, and bound to a unique site on these sodium channels with a novel allosteric mechanism, by binding to the channel’s second voltage sensing domain, thereby stabilizing the closed state, causing tonic inhibition. It exerts its action on dorsal root ganglion.[3]
History
Vertex Pharmaceuticals announced in January 2024 that suzetrigine had successfully met several endpoints in its Phase III clinical trials.[5] The company announced in July 2024 that the FDA had accepted a new drug application for suzetrigine.[11] The FDA granted the application for suzetrigine priority review, fast track, and breakthrough therapy designations.[2][11] In January 2025, the FDA granted approval of Journavx to Vertex Pharmaceuticals.[2]
Society and culture
Legal status
Suzetrigine was approved for medical use in the United States in January 2025.[2]
Names
Suzetrigine is the international nonproprietary name.[12]
Suzetrigine is sold under the brand name Journavx.[1][2]


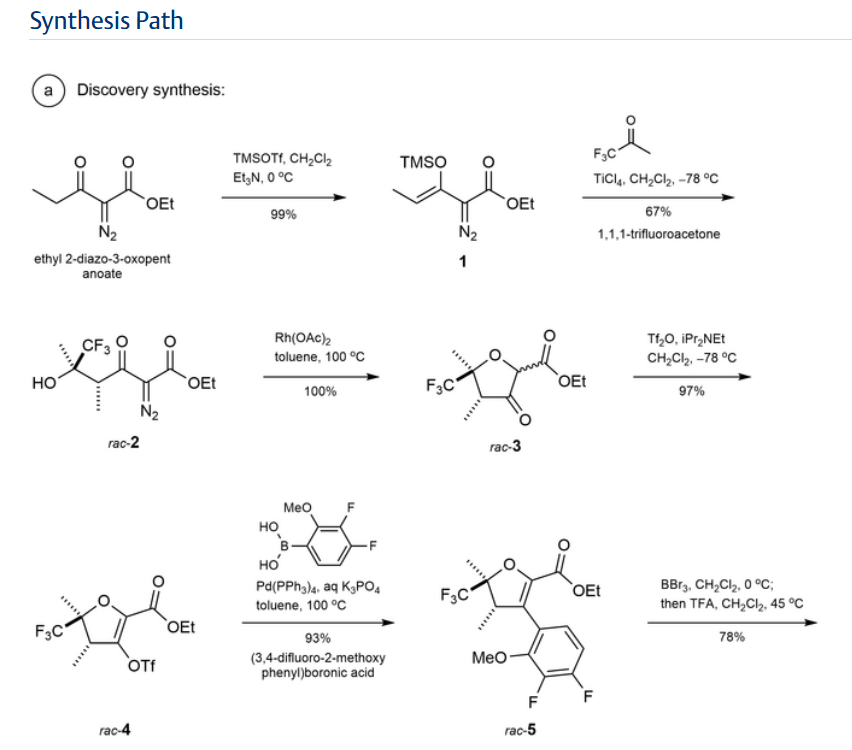


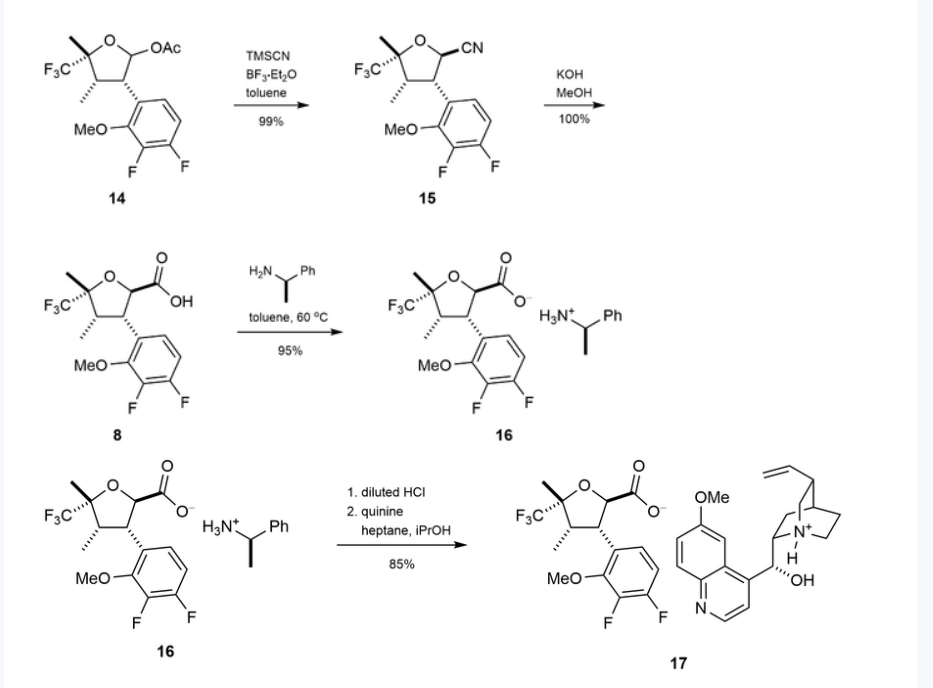
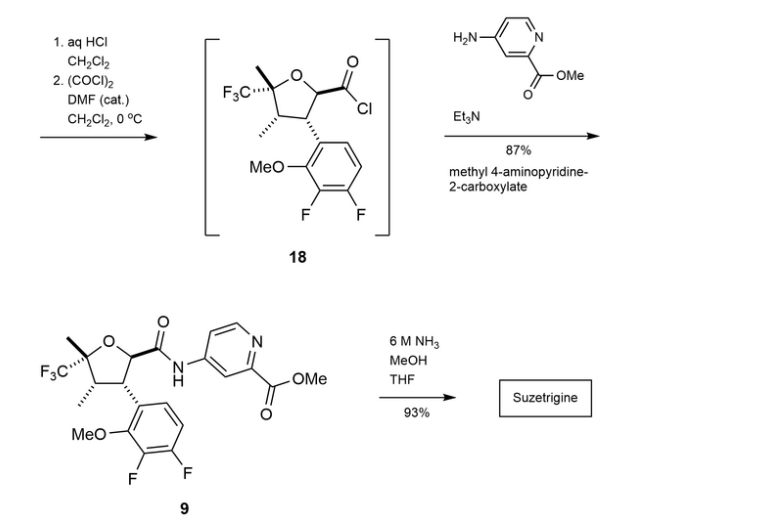

References
a) WO2021113627A1 (Vertex, 10.06.2021; USA-prior. 06.12.2019).
US11834441B2 (Vertex, 05.12.2023; USA-prior. 06.12.2019).
b) WO2022256660A1 (Vertex, 08.12.2022; USA-prior. 04.06.2021).
WO2024123815A1 (Vertex, 13.06.2024; USA-prior. 06.12.2022).
WO2022256708A1 (Vertex, 08.12.2022; USA-prior. 04.06.2021, 02.12.2021).
Source:
Suzetrigine, in Kleemann A., Kutscher B., Reichert D., Bossart M., Pharmaceutical Substances, Thieme. https://pharmaceutical-substances.thieme.com/lexicon/KD-19-0151, accessed: 05-29-2025
| Clinical data | |
|---|---|
| Pronunciation | /suˈzɛtrɪdʒiːn/ soo-ZE-tri-jeen |
| Trade names | Journavx |
| Other names | VX-548 |
| AHFS/Drugs.com | Journavx |
| License data | US DailyMed: Suzetrigine |
| Routes of administration | By mouth |
| Drug class | Nav1.8 sodium channel blocker; Analgesic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2649467-58-1 |
| PubChem CID | 156445116 |
| DrugBank | DB18927 |
| ChemSpider | 128942439 |
| UNII | LOG73M21H5 |
| KEGG | D12860 |
| ChEMBL | ChEMBL5314487 |
| Chemical and physical data | |
| Formula | C21H20F5N3O4 |
| Molar mass | 473.400 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References
- ^ Jump up to:a b c d e f g h “Journavx- suzetrigine tablet, film coated”. DailyMed. 6 February 2025. Retrieved 2 April 2025.
- ^ Jump up to:a b c d e f g h i j k l m n “FDA Approves Novel Non-Opioid Treatment for Moderate to Severe Acute Pain” (Press release). U.S. Food and Drug Administration (FDA). 30 January 2025. Archived from the original on 7 February 2025. Retrieved 30 January 2025.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d e Osteen, Jeremiah D.; Immani, Swapna; Tapley, Tim L.; Indersmitten, Tim; Hurst, Nicole W.; Healey, Tiffany; et al. (January 2025). “Pharmacology and Mechanism of Action of Suzetrigine, a Potent and Selective NaV1.8 Pain Signal Inhibitor for the Treatment of Moderate to Severe Pain”. Pain and Therapy. doi:10.1007/s40122-024-00697-0. PMID 39775738.
- ^ Jump up to:a b c Jones, Jim; Correll, Darin J.; Lechner, Sandra M; Jazic, Ina; Miao, Xiaopeng; Shaw, David; et al. (August 2023). “Selective Inhibition of NaV1.8 with VX-548 for Acute Pain”. The New England Journal of Medicine. 389 (5): 393–405. doi:10.1056/NEJMoa2209870. PMID 37530822. S2CID 260377748.
- ^ Jump up to:a b “Vertex Announces Positive Results From the VX-548 Phase 3 Program for the Treatment of Moderate-to-Severe Acute Pain” (Press release). Vertex. 30 January 2024. Archived from the original on 25 December 2024. Retrieved 31 January 2025 – via Business Wire.
- ^ “Novel Drug Approvals for 2025”. U.S. Food and Drug Administration (FDA). 21 February 2025. Retrieved 9 March 2025.
- ^ Jump up to:a b c d Broadfoot, Marla (20 August 2024). “New Painkiller Could Bring Relief to Millions — without Addiction Risk”. Scientific American. Archived from the original on 30 December 2024. Retrieved 31 January 2025.
- ^ Jump up to:a b c d Hang Kong, Aaron Yik; Tan, Hon Sen; Habib, Ashraf S. (September 2024). “VX-548 in the Treatment of Acute Pain”. Pain Management. 14 (9): 477–486. doi:10.1080/17581869.2024.2421749. PMC 11721852. PMID 39552600.
- ^ Kingwell, Katie (December 2024). “NaV1.8 inhibitor poised to provide opioid-free pain relief”. Nature Reviews. Drug Discovery. 24 (1): 3–5. doi:10.1038/d41573-024-00203-3. PMID 39668193.
- ^ Jump up to:a b Dolgin, Elie (January 2025). “US drug agency approves potent painkiller – the first non-opioid in decades”. Nature. 638 (8050): 304–305. doi:10.1038/d41586-025-00274-1. PMID 39885357.
- ^ Jump up to:a b “Vertex Announces FDA Acceptance of New Drug Application for Suzetrigine for the Treatment of Moderate-to-Severe Acute Pain” (Press release). Vertex. 30 July 2024. Retrieved 31 January 2025 – via Business Wire.
- ^ World Health Organization (2023). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 90”. WHO Drug Information. 37 (3). hdl:10665/373341.
Further reading
- Oliver, Brian; Devitt, Catherine; Park, Grace; Razak, Alina; Liu, Sun Mei; Bergese, Sergio D. (2025). “Drugs in Development to Manage Acute Pain”. Drugs. 85 (1): 11–19. doi:10.1007/s40265-024-02118-0. PMID 39560856.
External links
- “Suzetrigine (Code C199115)”. NCI Thesaurus.
- Clinical trial number NCT05661734 for “A Single-arm Study to Evaluate Safety and Effectiveness of VX-548 for Acute Pain” at ClinicalTrials.gov
- Clinical trial number NCT05558410 for “Evaluation of Efficacy and Safety of VX-548 for Acute Pain After an Abdominoplasty” at ClinicalTrials.gov
//////////Suzetrigine, Journavx, FDA 2025, APPROVALS 2025, CS-0641183, HY-148800, VX 548, VX-548, VX548, Breakthrough Therapy, Fast Track, Priority Review
BENZGALANTAMINE


BENZGALANTAMINE
CAS 224169-27-1
Benzgalantamine gluconate, 1542321-58-3
- 6H-Benzofuro[3a,3,2-ef][2]benzazepin-6-ol, 4a,5,9,10,11,12-hexahydro-3-methoxy-11-methyl-, benzoate (ester), (4aS,6R,8aS)- (9CI)
- Alpha 1062
- GLN 1062
- Memogain
6h-benzofuro(3a,3,2-ef)(2)benzazepin-6-ol, 4a,5,9,10,11,12-hexahydro-3-methoxy-11-methyl-, benzoate (ester), (4as,6r,8as)-
| Formula | C24H25NO4 |
|---|---|
| Molar mass | 391.467 g·mol−1 |


External IDs GLN-1062 gluconate
UNIILN7PMJ4P57
CAS Number1542321-58-3
WeightAverage: 587.622
Monoisotopic: 587.236661015
Chemical FormulaC30H37NO11
Benzgalantamine, sold under the brand name Zunveyl, is a medication used for the treatment of mild to moderate dementia of the Alzheimer’s type.[1] It is a cholinesterase inhibitor.[1] Benzgalantamine is a prodrug of galantamine.[1]
The most common side effects include nausea, vomiting, diarrhea, dizziness, headache, and decreased appetite.[1]
Benzgalantamine was approved for medical use in the United States in July 2024.[1][2][3]
compounds that, in addition to enhancing the sensitivity to acetylcholine and choline, and to their agonists, of neuronal cholinergic receptors, and/or acting as cholinesterase inhibitors and/or neuroprotective agents, have enhanced blood-brain barrier permeability in comparison to their parent compounds. The compounds are derived (either formally by their chemical structure or directly by chemical synthesis) from natural compounds belonging to the class of amaryllidaceae alkaloids e.g., Galantamine, Narwedine and Lycoramine, or from metabolites of said compounds. The compounds of the present invention can either interact as such with their target molecules, or they can act as “pro-drugs”, in the sense that after reaching their target regions in the body, they are converted by hydrolysis or enzymatic attack to the original parent compound and react as such with their target molecules, or both. The compounds of this disclosure may be used as medicaments for the treatment of human brain diseases associated with a cholinergic deficit, including the neurodegenerative diseases Alzheimer’s and Parkinson’s disease and the neurological/psychiatric diseases vascular dementia, schizophrenia and epilepsy. Galantamine derivatives disclosed herein have higher efficacy and lower levels of adverse side effects in comparison to galantamine, in treatment of human brain diseases.
Benzgalantamine is a prodrug of galantamine. Gastrointestinal adverse effects are the most frequently reported side effects in patients undergoing treatment with cholinesterase inhibitors, including galantamine, and are often a reason for treatment discontinuation.2 As a prodrug, benzagalantamine remains inert as it passes through the stomach, thereby avoiding many of the gastrointestinal effects associated with peripheral cholinesterase inhibition.4
Benzgalantamine was approved by the FDA in July 2024 for the treatment of mild-to-moderate dementia in Alzheimer’s patients.3,4
SCHEME


US20090253654
https://patentscope.wipo.int/search/en/detail.jsf?docId=US42863485&_cid=P12-M8ZQT3-74791-1

| O-Benzoyl-galantamine(=(4aS,6R,8aS)-4a,5,9,10,11,12-Hexahydro-3-methoxy-11-methyl-6H-benzofuro[3a,3,2-ef][2]benzazepin-6-ol, benzoate (ester)); yield: 78% |
| O-3,4-Dichlorobenzoyl-galantamine(=(4aS,6R,8aS)-4a,5,9,10,11,12-Hexahydro-3-methoxy-11-methyl-6H-benzofuro[3a,3,2-ef][2]benzazepin-6-ol, 3,4-dichlorobenzoate (ester)); off-white solid; mp. 69-70° C. |
WO2009127218
US20220220121
https://patentscope.wipo.int/search/en/detail.jsf?docId=US368470159&_cid=P12-M8ZR8V-88578-1
Experiment 1
The gluconate salt of Alpha-1062 was created according to the following previously established general scheme:

AND
US20090253654
Medical uses
Benzgalantamine is indicated for the treatment of mild to moderate dementia of the Alzheimer’s type in adults.[1][2]
Side effects
The most common side effects include nausea, vomiting, diarrhea, dizziness, headache, and decreased appetite.[1]
Society and culture
Legal status
Benzgalantamine was approved for medical use in the United States in July 2024.[1][2]
Names
Benzgalantamine is the international nonproprietary name.[4]
References
- ^ Jump up to:a b c d e f g h i “Zunveyl- benzgalantamine tablet, delayed release”. DailyMed. 8 August 2024. Retrieved 15 August 2024.
- ^ Jump up to:a b c https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2024/218549Orig1s000ltr.pdf
- ^ “Alpha Cognition’s Oral Therapy Zunveyl Receives FDA Approval to Treat Alzheimer’s Disease” (Press release). Alpha Cognition. 29 July 2024. Archived from the original on 4 August 2024. Retrieved 4 August 2024 – via Business Wire.
- ^ World Health Organization (2022). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 88”. WHO Drug Information. 36 (3). hdl:10665/363551.
- Baakman AC, ‘t Hart E, Kay DG, Stevens J, Klaassen ES, Maelicke A, Groeneveld GJ: First in human study with a prodrug of galantamine: Improved benefit-risk ratio? Alzheimers Dement (N Y). 2016 Jan 20;2(1):13-22. doi: 10.1016/j.trci.2015.12.003. eCollection 2016 Jan. [Article]
- Bakker C, van der Aart J, Hart EP, Klaassen ES, Bergmann KR, van Esdonk MJ, Kay DG, Groeneveld GJ: Safety, pharmacokinetics, and pharmacodynamics of Gln-1062, a prodrug of galantamine. Alzheimers Dement (N Y). 2020 Oct 13;6(1):e12093. doi: 10.1002/trc2.12093. eCollection 2020. [Article]
- FDA Approved Drug Products: Zunveyl (benzgalantamine) delayed-release tablets for oral use [Link]
- Fierce Pharma: Alpha Cognition’s delayed-release Alzheimer’s drug Zunveyl passes muster with FDA [Link]
- Alpha Cognition: Corporate Presentation Oct 2024 [Link]
External links
- “Benzgalantamine (Code C188656)”. NCI Thesaurus.
| Clinical data | |
|---|---|
| Trade names | Zunveyl |
| Other names | ALPHA-1062 |
| AHFS/Drugs.com | Zunveyl |
| License data | US DailyMed: Benzgalantamine |
| Routes of administration | By mouth |
| Drug class | Cholinesterase inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 224169-27-11542321-58-3 |
| DrugBank | DB19353 |
| UNII | XOI2Q0ZF7GLN7PMJ4P57 |
| KEGG | D12930D12931 |
| ChEMBL | ChEMBL5095056 |
| Chemical and physical data | |
| Formula | C24H25NO4 |
| Molar mass | 391.467 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
//////////BENZGALANTAMINE, Alpha 1062, GLN 1062, Memogain, FDA 2024, APPROVALS 2024, Zunveyl
Tisolagiline


Tisolagiline
CAS 1894207-44-3
PCH79KLX33
(2S)-2-[[4-[4-(trifluoromethyl)phenyl]phenyl]methylamino]propanamide
322.32 g/mol
SCHEME
Tisolagiline (INNTooltip International Nonproprietary Name; developmental code names KDS-2010, SeReMABI) is a potent, highly selective, and reversible monoamine oxidase B (MAO-B) inhibitor which is under development for the treatment of Alzheimer’s disease and obesity.[1][2][3][4] It is taken by mouth.[1] Tisolagiline is being developed by NEUROBiOGEN and Scilex Bio.[1][2] As of December 2024, it is in phase 2 clinical trials for Alzheimer’s disease and obesity.[1][2]
Parkinson’s disease is a progressive disease that ranks second among degenerative neurological diseases, and the incidence rate is estimated to be about 6.3 million patients worldwide, and about 1 in 1,000 people develop Parkinson’s disease. The incidence rate is usually higher in the elderly, but it is now developing in young people as well. Parkinson’s disease is not easy to distinguish from other diseases because the symptoms progress slowly, and it is difficult to detect in the early stages. Clinical characteristics include tremors, rigidity, bradykinesia, postural instability, stooped posture, freezing of gait, depression, sleep disorders, urination disorders, and dementia.
[3]Parkinson’s disease has an unknown cause, but it is known to be a disease that occurs when nerve cells that secrete the neurotransmitter dopamine in the brain are destroyed, resulting in a lack of dopamine. The most widely developed and used drug is levodopa therapy, which is generally administered by administering levodopa, which is converted into dopamine in the body. Levodopa is the most effective treatment for Parkinson’s disease, but there are cases where the drug-related effects decrease or various movement disorders occur during the treatment process. Other drugs used include COMT inhibitors and MAO-B inhibitors, which suppress dopamine metabolism and maintain the concentration of dopamine in the brain.
[4]MAO-B is known to play an important role in dopamine metabolism in the brain and to suppress damage to brain neurons. Although there is no clear evidence that MAO-B inhibitors actually slow down the progression of Parkinson’s disease, it is known that inhibiting MAO-B has an effect of suppressing degeneration or death of dopamine neurons, as it plays an important role in the development of Parkinson’s disease caused by MPTP or similar environmental toxicants. In addition, evidence from animal and clinical trials suggests that MAO-B inhibitors have a brain protective effect, unlike other drugs.
[5]The most representative MAO-B inhibitor approved is selegiline, which is prescribed as a treatment for Parkinson’s disease, but when taken, it is metabolized into amphetamine in the body, causing liver toxicity, and as an irreversible inhibitor, it has various side effects. Azilect, which contains rasagiline, was first marketed in Israel in 2005 and has recently been released in about 50 countries including Europe and the United States. Azilect does not have amphetamine side effects in the body when taken and is said to be more effective than other dopaminergic drugs. However, rasagiline, like selegiline, is an irreversible MAO-B inhibitor, so although it has an excellent MAO-B inhibition effect, it has the disadvantage of safety issues. Therefore, recently, drugs that are effective and can reversibly inhibit activity are being developed as alternatives to complement these shortcomings, but no notable reversible inhibitors have been prescribed to date.
[6]Meanwhile, obesity is a medical condition in which excessive fat accumulates in the body to the extent that it has a negative impact on health. Excessive weight can appear in combination with various diseases as the remaining energy is accumulated excessively due to the difference between energy consumed and energy used.
[7]Previous studies on the hypothalamus in relation to food regulation have focused on neurons that make up a portion of the brain, which has limited our understanding of the brain’s function in controlling food and obesity. Therefore, in order to comprehensively understand brain function, studies on glial cells, which make up the majority, must also be conducted in parallel. In addition, astrocytes, which are the most numerous among glial cells, have recently emerged as cells that can activate or inhibit surrounding neurons by secreting various signaling substances such as GABA (gamma-aminobutyric acid), glutamate, D-serine, and ATP. Astrocytes in the hypothalamus also interact closely with POMC (pro-opiomelanocortin) neurons and express leptin receptors, which can contribute to leptin signaling.
[8]There are two groups of POMC neurons in the hypothalamus: those that induce appetite reduction and those that induce energy consumption. Under normal circumstances, astrocytes help activate nearby POMC neurons that induce energy consumption. However, in obese states, unlike normal astrocytes, they are transformed into reactive astrocytes due to excessive leptin signals, and putrescine is converted into GABA by MAO-B (mono-aminoxidase B) and secreted. In addition, POMC neurons that induce energy consumption express GABAa receptors outside the synapse containing a4, a5, and a6 subunits due to excessive leptin signals, and are affected by persistent GABA secreted from anti-responsive astrocytes. As a result, POMC neurons are inhibited, energy consumption is reduced, and fat accumulation occurs.
[9]At this time, if MAOBI, the causal enzyme of GABA production, is inhibited, GABA production and secretion are inhibited, the inhibition of POMC neurons is relieved, and they are reactivated to promote energy consumption. However, POMC neurons that induce appetite reduction do not express GABAa receptors outside the synapse, so they are not continuously affected by GABA. Therefore, MAOBI inhibitors selectively act on POMC neurons that induce energy consumption and exhibit the effect of obesity treatment. However, most of the existing MAOBI inhibitors are irreversible inhibitors, and there is a problem that they are accompanied by various side effects. Accordingly, drugs that can reversibly inhibit MAOBI are being researched and developed, but no notable reversible MAOBI inhibitor that can effectively act on obesity has been prescribed to date.
REF
Regulatory Toxicology and Pharmacology (2020), 117, 104733
Toxicological Research (Cham, Switzerland) (2023), 39(4), 693-709
Combinatorial Chemistry & High Throughput Screening (2020), 23(9), 836-841
KR2023027416,
WO2023022256
WO2023022256
WO2016052928
PATENT
WO2016052928
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016052928&_cid=P20-M8XX0L-81795-1


Using L-Alaninamide hydrochloride or D-Alaninamide hydrochloride, a reductive amination reaction was performed with the compound of step (a) to obtain an imine compound (step b, reaction scheme 1b), which was then reduced with sodium cyanoborohydride to obtain an amine compound (step c, reaction scheme 1c).
[112]Add 1.2 equivalents of Glycinamide hydrochloride or L-Alaninamide hydrochloride or D-Alaninamide hydrochloride or L-Valinamide hydrochloride or L-Leucinamide hydrochloride to anhydrous methanol at a concentration of 0.92 molar, and then add 1.5 equivalents of triethylamine. When the solution becomes transparent, add 1.0 equivalent of the aldehyde synthesized in step (a). After two hours, wash with ethyl acetate and distilled water. Dry the organic layer with sodium sulfate and concentrate in vacuo. Dissolve the concentrated reaction solution in anhydrous methanol at a concentration of 1.0 molar, and add 4.0 equivalents of sodium cyanoborohydride at 0 ℃. Then, react at room temperature for 18 hours, and after completion of the reaction, wash the reaction solution with ethyl acetate and distilled water. The organic layer was dried over sodium sulfate, concentrated in vacuo, and separated and purified using silica gel column chromatography.
References
- ^ Jump up to:a b c d “KDS 2010”. AdisInsight. 6 February 2025. Retrieved 24 February 2025.
- ^ Jump up to:a b c “Delving into the Latest Updates on KDS-2010 with Synapse”. Synapse. 23 January 2025. Retrieved 24 February 2025.
- ^ Nam MH, Sa M, Ju YH, Park MG, Lee CJ (April 2022). “Revisiting the Role of Astrocytic MAOB in Parkinson’s Disease”. International Journal of Molecular Sciences. 23 (8): 4453. doi:10.3390/ijms23084453. PMC 9028367. PMID 35457272.
4.4. KDS2010 A recently developed KDS2010, which is ~12,500-fold more selective to MAOB than MAOA, differentiates the role of MAOB from MAOA and reports that MAOB does not contribute to DA degradation [39]. KDS2010 is a potent (IC50 = 7.6 nM), and selective MAOB inhibitor named shows no known off-target effect (no other enzymes or channels causing >40% inhibition) or toxicity for 4 weeks of repeated dosing in non-human primates [16,41]. KDS2010 was turned out to be highly effective for alleviating the PD-related motor symptoms and PD-like pathology, including reactive astrogliosis, excessive astrocytic GABA, and nigrostriatal DAergic neuronal loss in multiple rodent models of PD [41]. Its clinical efficacy is still waiting to be tested in future studies.
- ^ Duarte P, Cuadrado A, León R (2021). “Monoamine Oxidase Inhibitors: From Classic to New Clinical Approaches”. Handbook of Experimental Pharmacology. 264: 229–259. doi:10.1007/164_2020_384. ISBN 978-3-030-68509-6. PMID 32852645.
KDS2010 is a novel compound highly potent and selective reversible MAO-B inhibitor (Fig. 2). It has demonstrated learning and memory improvements, promotion of synaptic transmission, and reduction of astrogliosis and astrocytic GABA levels in APP/presenilin 1 mice (Park et al. 2019).
| Clinical data | |
|---|---|
| Other names | KDS-2010; KDS2010; SeReMABI |
| Drug class | Reversible monoamine oxidase B (MAO-B) inhibitor |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1894207-44-3 |
| PubChem CID | 132023446 |
| ChemSpider | 128942408 |
| UNII | PCH79KLX33 |
| ChEMBL | ChEMBL5314546 |
| Chemical and physical data | |
| Formula | C17H17F3N2O |
| Molar mass | 322.331 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
///////////Tisolagiline, PCH79KLX33
Bemfivastatin, PPD 10558, RBx 10558



Bemfivastatin, PPD 10558, RBx 10558
cas 805241-79-6
| Molecular Weight | 588.67 |
|---|---|
| Formula | C34H37FN2O6 |


- PPD-10558 calcium salt
- Ppd-10558(calcium salt)
- 805241-64-9
- ppd-10558 calcium
- 3I8G750MW3
- calcium;(3R,5R)-7-[2-(4-fluorophenyl)-4-[[4-(hydroxymethyl)phenyl]carbamoyl]-3-phenyl-5-propan-2-ylpyrrol-1-yl]-3,5-dihydroxyheptanoate
- C68H72CaF2N4O12
- 1H-PYRROLE-1-HEPTANOIC ACID, 2-(4-FLUOROPHENYL)-.BETA.,.DELTA.-DIHYDROXY-4-(((4-(HYDROXYMETHYL)PHENYL)AMINO)CARBONYL)-5-(1-METHYLETHYL)-3-PHENYL-, CALCIUM SALT (2:1), (.BETA.R,.DELTA.R)-
- calcium;(3R,5R)-7-[2-(4-fluorophenyl)-4-[[4-(hydroxymethyl)phenyl]carbamoyl]-3-phenyl-5-propan-2-ylpyrrol-1-yl]-3,5-dihydroxyheptanoate
- UNII-3I8G750MW3
- 1H-Pyrrole-1-heptanoic acid, 2-(4-fluorophenyl)-beta,delta-dihydroxy-4-(((4-(hydroxymethyl)phenyl)amino)carbonyl)-5-(1-methylethyl)-3-phenyl-, calcium salt (2:1), (betaR,deltaR)-
- Bemfivastatin calcium
- Bemfivastatin hemicalcium
Bemfivastatin (PPD 10558) is an orally active, HMG-CoA Reductase (HMGCR) inhibitor, also known as Statin. Bemfivastatin enhances the activity of liver extraction. Bemfivastatin exhibits little developmental toxicity effects in pregnant rats and rabbits via daily oral doses during organogenesis period. The no observed adverse effect level (NOAEL) are ≥320 mg/kg/day for rats developmental toxicity, 12.5 mg/kg/day for rabbits maternal toxicity, and 25 mg/kg/day for rabbits developmental toxicity, respectively. Bemfivastatin can be used for research on Statin-related hypercholesterolemic myalgia with inability to tolerate statins.
Korean Patent No. 10-1329113 describes a method for preparing (3R,5R)-7-[2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-[(4-hydroxymethylphenylamino)carbonyl]-pyrrol-1-yl]-3,5-dihydroxy-heptanoic acid hemicalcium salt, as shown in the following reaction scheme.




SCHEME

MAIN

PATENT
WO2020040614
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020040614&_cid=P11-M8VDBE-14315-1
Step 3: Preparation of tert-butyl (3R,5R)-7-(2-(4-fluorophenyl)-4-((4-(hydroxymethyl)phenyl)carbamoyl)-5-isopropyl-3-phenyl-1H-pyrrol-1-yl)-3,5-dihydroxyheptanoate
[499]In step 2, tert-butyl 2-((4R,6R)-6-(2-(3-((4-(((tert-butyldimethylsilyl)oxy)methyl)phenyl)carbamoyl)-5-(4-fluorophenyl)-2-isopropyl-4-phenyl-1H-pyrrol-1-yl)ethyl)-2,2-dimethyl-1,3-dioxan-4-yl)acetate (5 g) was dissolved in methanol (37 ml) and THF (37 ml), 1 N HCl aqueous solution (37 ml) was added, and the mixture was stirred at room temperature for 2 hours. EA was added to the reaction solution, diluted, and washed several times with distilled water and brine. The extracted organic layer was dried over Na
2 SO
4 and filtered under reduced pressure. The filtrate was concentrated under reduced pressure, EA and hexane were added, and the mixture was purified by recrystallization to obtain the title compound.
[500]White solid 4.6 g (yield quantitative);
[501]
1H NMR (500 MHz, CDCl 3): 7.24-7.14 (m, 9H), 7.06 (d, J = 8.5 Hz, 2H), 6.99 (t, J = 8.5 Hz, 2H), 6.87 (br s, 1H), 4.57 (s, 2H), 4.45-4.08 (m, 2H), 3.96-3.90 (m, 1H), 3.75-3.71 (m, 1H), 3.58 (sep, J = 7.0 Hz, 1H), 2.32 (d, J = 6.5 Hz, 2H), 1.73-1.65 (m, 1H), 1.64-1.58 (m, 1H), 1.54 (d, J = 7.0 Hz, 6H), 1.45 (s, 9H), 1.27-1.22 (m, 2H), MH+ 645.
Step 4: Preparation of (3R,5R)-7-(2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-((4-hydroxymethylphenylamino)carbonyl)-pyrrol-1-yl)-3,5-dihydroxyheptanoic acid hemicalcium salt
[503]In step 3, tert-butyl (3R,5R)-7-(2-(4-fluorophenyl)-4-((4-(hydroxymethyl)phenyl)carbamoyl)-5-isopropyl-3-phenyl-1H-pyrrol-1-yl)-3,5-dihydroxyheptanoate (4.19 g) obtained was dissolved in MeOH (65 ml) and THF (65 ml), and stirred in an ice bath. NaOH pellets (5 eq, 1.3 g) were added, and the mixture was stirred for 1 more hour at room temperature. After concentrating the reaction solution under reduced pressure, distilled water (44 ml) was added until the formed solid was completely dissolved. After concentrating the reaction solution under reduced pressure, distilled water (430 ml) was added until the solid was completely dissolved. 1 M Ca(OAc)
2 aqueous solution (3.6 ml) was slowly added dropwise, and the mixture was stirred for 15.5 hours at room temperature. After the generated solid was filtered under reduced pressure, it was washed several times with distilled water and the filtered solid was dried in an oven.
[504]2.98 g of white solid (yield 76%);
[505]
1H NMR (500 MHz, DMSO-d 6) δ 9.78 (br s, 1H), 7.46 (d, J = 8.5 Hz, 2H), 7.26-7.23 (m, 2H), 7.19 (t, J = 9.0 Hz, 2H), 7.15 (d, J = 8.5 Hz, 2H), 7.09-7.05 (m, 4H), 7.02-6.98 (m, 1H), 6.41 (br s, 1H), 5.04 (t, J = 5.5 Hz, 1H), 4.75 (br s, 1H), 4.39 (d, J = 5.5 Hz, 2H), 3.98-3.91 (m, 1H), 3.79-3.69 (m, 2H), 3.55-3.50 (m, 1H), 3.22 (sep, J = 7.0 Hz, 1H), 2.03 (dd, J = 15.0 Hz, 4.0 Hz, 1H), 1.90 (dd, J = 15.0 Hz, 8.0 Hz, 1H), 1.63-1.57 (m, 1H), 1.54-1.47 (m, 1H), 1.41-1.36 (m, 1H), 1.37 (d, J = 7.0 Hz, 6H), 1.23-1.16 (m, 1H), MH+ (acid+1) 589.
Step 5: Preparation of (3R,5R)-7-(2-(4-fluorophenyl)-5-isopropyl-3-phenyl-4-((4-hydroxymethylphenylamino)carbonyl)-pyrrol-1-yl)-3,5-dihydroxyheptanoic acid hemicalcium salt
[540]The title compound was prepared in the same manner as in step 4 of Example 15.
[541]
1H NMR (500 MHz, DMSO-d 6) δ 9.78 (br s, 1H), 7.46 (d, J = 8.5 Hz, 2H), 7.26-7.23 (m, 2H), 7.19 (t, J = 9.0 Hz, 2H), 7.15 (d, J = 8.5 Hz, 2H), 7.09-7.05 (m, 4H), 7.02-6.98 (m, 1H), 6.41 (br s, 1H), 5.04 (t, J = 5.5 Hz, 1H), 4.75 (br s, 1H), 4.39 (d, J = 5.5 Hz, 2H), 3.98-3.91 (m, 1H), 3.79-3.69 (m, 2H), 3.55-3.50 (m, 1H), 3.22 (sep, J = 7.0 Hz, 1H), 2.03 (dd, J = 15.0 Hz, 4.0 Hz, 1H), 1.90 (dd, J = 15.0 Hz, 8.0 Hz, 1H), 1.63-1.57 (m, 1H), 1.54-1.47 (m, 1H), 1.41-1.36 (m, 1H), 1.37 (d, J = 7.0 Hz, 6H), 1.23-1.16 (m, 1H), MH+ (acid+1) 589.
KR2001835 63%
KR2016103248
- [1]. Faqi AS, et al. Developmental toxicity of the HMG-CoA reductase inhibitor (PPD10558) in rats and rabbits. Birth Defects Res B Dev Reprod Toxicol. 2012 Feb;95(1):23-37. [Content Brief][2]. Wierzbicki A, et al. Drugs in development for the management of dyslipidaemia[J]. Future Prescriber, 2012, 13(2): 12-15.
/////////Bemfivastatin, PPD 10558, PPD-10558, RBx-10558; PPD10558, RBx10558, PPD 10558, RBx 10558, bemfivastatin CA, RBx 10558
Umifoxolaner, ML 878



Umifoxolaner, ML 878
CAS 2021230-37-3
| Molecular Weight | 643.86 |
|---|---|
| Formula | C26H16ClF10N3O3 |
- 4-[(5S)-5-[3-Chloro-4-fluoro-5-(trifluoromethyl)phenyl]-4,5-dihydro-5-(trifluoromethyl)-3-isoxazolyl]-N-[2-oxo-2-[(2,2,2-trifluoroethyl)amino]ethyl]-1-naphthalenecarboxamide (ACI)
- 4-{(5S)-5-[3-chloro-4-fluoro-5-(trifluoromethyl)phenyl]-5-(trifluoromethyl)-4,5-dihydroisoxazol-3-yl}-N-{2-oxo-2-[(2,2,2-trifluoroethyl)amino]ethyl}naphthalene-1-carboxamide
- ML 878
- 4-[(5S)-5-[3-chloro-4-fluoro-5-(trifluoromethyl)phenyl]-5-(trifluoromethyl)-4H-1,2-oxazol-3-yl]-N-[2-oxo-2-(2,2,2-trifluoroethylamino)ethyl]naphthalene-1-carboxamide
- WHO 11642
umifoxolaner (ML-878) is a γ-aminobutyric acid (GABA) regulated chloride channels antagonist. Umifoxolaner is an anti-parasitic agent
Animals such as mammals and birds are often susceptible to parasite infestations/infections. These parasites may be ectoparasites, such as insects, and endoparasites such as filariae and other worms. Domesticated animals, such as cats and dogs, are often infested with one or more of the following ectoparasites:
– fleas (e.g. Ctenocephalides spp., such as Ctenocephalides felis and the like);
– ticks (e.g. Rhipicephalus spp., Ixodes spp., Dermacentor spp., Amblyomma spp., and the like);
– mites (e.g. Demodex spp., Sarcoptes spp., Otodectes spp., and the like);
– lice (e.g. Trichodectes spp., Cheyletiella spp., Linognathus spp. and the like);
– mosquitoes (Aedes spp., Culex spp., Anopheles spp. and the like); and
– flies (Haematobia spp., Musca spp., Stomoxys spp., Dermatobia spp., Cochliomyia spp. and the like).
Fleas are a particular problem because not only do they adversely affect the health of the animal or human, but they also cause a great deal of psychological stress. Moreover, fleas are also vectors of pathogenic agents in animals and humans, such as dog tapeworm {Dipylidium caninum).
Similarly, ticks are also harmful to the physical and psychological health of the animal or human. However, the most serious problem associated with ticks is that they are the vector of pathogenic agents in both humans and animals. Major diseases which are caused by ticks include borreliosis (Lyme disease caused by Borrelia burgdorferi), babesiosis (or piroplasmosis caused by Babesia spp.) and rickettsioses (also known as Rocky Mountain spotted fever). Ticks also release toxins which cause inflammation or paralysis in the host. Occasionally, these toxins are fatal to the host.
Likewise, farm animals are also susceptible to parasite infestations. For example, cattle are affected by a large number of parasites. A parasite which is very prevalent among farm animals is the tick genus Rhipicephalus {Boophilus), especially those of the species microplus (cattle tick), decolor atus and annulatus. Ticks, such as Rhipicephalus {Boophilus) microplus, are particularly difficult to control because they live in the pasture where farm animals graze.
Animals and humans also suffer from endoparasitic infections including, for example, helminthiasis which is most frequently caused by a group of parasitic worms categorized as cestodes (tapeworm), nematodes (roundworm) and trematodes (flatworm or flukes). These parasites adversely affect the nutrition of the animal and cause severe economic losses in pigs, sheep, horses, and cattle as well as affecting domestic animals and poultry. Other parasites which occur in the gastrointestinal tract of animals and humans include Ancylostoma, Necator, Ascaris, Strongyloides, Trichinella, Capillaria, Toxocara, Toxascaris, Trichuris, Enterobius and parasites which are found in the blood or other tissues and organs such as filarial worms and the extra intestinal stages of Strongyloides, Toxocara and Trichinella.
SCHEME

Patents
WO2017176948
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017176948&_cid=P12-M8S60W-88110-1
Cinchonanium, 9-hydroxy-6′-methoxy-1-[[3,4,5-tris(phenylmethoxy)phenyl]methyl]-, chloride (1:1), (8α,9R)- 2138407-51-7, HYDROXYL AMINE, NAOH, MDC , WATER]


Example 5: Synthesis of (R)-IA-3 using chiral phase transfer catalyst (IIIb-13-1)
Step 1 : Synthesis of intermediate 4-2.
1) The substituted iodobenzene starting material (SM) (200.0 g, 1.0 eq.) and THF (400 ml, 10 volumes) were placed into a 1 L reactor and the mixture was cooled to -10 to -5° C.
2) /‘-PrMgCl (340 ml, 1.1 eq.) added dropwise over 1.5 hours at -10 to -5°C to the mixture. 3) After the addition was complete, the mixture was stirred for 1 h at -10 to -5°C.
4) TLC analysis showed the complete consumption of SM (quenching sample with 1 M HCl).
5) CF3COOMe (94.7 g, 1.2 eq.) was added over an hour at -10~-5°C to the reaction mixture.
6) The mixture was stirred for another 12 hours -10~-5°C.
7) TLC analysis showed the almost complete consumption of intermediate 4-1 (quench with 1M HCl).
8) 1 M HCl 1000 ml was added dropwise to the reaction mixture slowly at 0~5°C over 2 hours.
9) The reaction mixture was extracted with hexane twice (1000 ml, 500 ml).
10) Add ^-toluenesulfonic acid 1.0 g to the organic layer and then the mixture was refluxed for 30 min.
11) The resulting mixture was then concentrated under vacuum at 20~25°C to remove the hexane.
12) Sodium bicarbonate (NaHC03, 300mg) was added and the mixture distilled in vacuum to afford compound 4-2 at 80~82°C, as a red liquid (85.0 grams, purity was 92.5% by HPLC, and the yield was 47.0%).
Step 2: Preparation of the compound of Formula (IIA-3):
4-1 IIA-3
1) Compound 4-2 (70.0 g, 1.0 eq.) and acetonitrile (ACN, 350ml, 5 volumes) were placed into a 1 L reactor. The solid was dissolved completely.
2) Compound 4-1 (70.2 g, 1.2 eq.) was then added to the mixture, and the mixture was heated to 90-95° C.
3) The ACN/water azeotrope was removed by distillation (b.p. 79°C).
4) K2C03 (2.0 g, 0.1 eq.) was then added to the mixture.
5) Distillation was continued to remove ACN/water at 90~95°C for about 6 hours.
6) After this time, about 28% Compound 4-2 remained by HPLC.
7) The mixture was cooled to 15~20°C over 1.5 hours and solid precipitated.
8) Water (50 ml) was added and then the mixture was cooled further to 0° C over 40 min.
9) The mixture was then held at 0° C for 40 minutes.
10) The mixture was filtered and the cake was washed with 100 ml of cold ACN/water (ACN/water, 25:6v/v) to yield 75.0 g of a yellow solid after drying (purity: 95.1%, yield: 50.0%).
Step 3 : Preparation of (R)-IA-3 using chiral phase transfer catalyst IIIb-13-1
1) The Compound of Formula IIA-3 (40.0 g, 1.0 eq.) and DCM (1.2 L, 30 volumes) were placed into a 2 L reactor; the solid was dissolved completely.
2) The mixture was cooled to 0° C and some starting material precipitated out.
3) The catalyst of formula IIIb-13-1 (1.47g, 3% mol) was added to the mixture and the mixture was cooled to -10° C.
4) Hydroxylamine (21. Og, 5.0 eq., 50% in water) was added to a solution of NaOH (15.3 g, 6.0 eq., in 5 volumes of water) in another reactor and stirred for 30 minutes.
5) The hydroxylamine/NaOH solution was then added dropwise to the 2 L reactor over about 4 hours.
6) The resulting reaction mixture was stirred for 16 h at -10°C.
7) In-process samples were taken and analyzed by HPLC until the content of starting material was < 1.0%.
8) When the reaction was complete, the mixture was warmed to 10°C and 200 ml of water was added. The mixture was stirred for 10 minutes.
9) After mixing, the mixture was allowed to stand to separate the aqueous and organic layers and the organic layer was collected.
10) The organic layer was washed with 200 ml of 5% KH2PO4.
11) The two layers were allowed to separate and organic layer was collected.
12) The organic layer was then washed with 200 ml brine, the two layers allowed to separate and the organic layer was again collected.
13) The resulting organic layer was concentrated under vacuum at 25-30°C to about 2 volumes.
14) Toluene (400 ml, 10 volumes) was charged to the vessel and concentration under vacuum was continued at 40~45°C to about 3 volumes. The solvent exchange was repeated twice more using the same procedure.
15) When the solvent exchange was complete, the solution was heated to 55-60°C.
16) The mixture was then cooled to 40° C over 1.5 hours and stirred at 40°C for 3 hours.
17) The mixture was then cooled to 25°C over 2 hours and stirred at 25°C for 3hours.
18) The mixture was finally cooled to 5-10°C over 1 hour and stirred at 8° C for 12 hours.
19) After this time, the mixture was filtered and the filter cake was washed with cold toluene (80 ml, 2 volumes).
20) The product was dried under vacuum at 70~75°C for 12h to yield a white solid (22.0 g, chiral purity: 98.0% by area using the chiral HPLC method described in Example 3, chemical purity: 97.1% by area (HPLC), yield: 48.8%). The 1H MR and LCMS spectra are consistent with the structure of the product.
Example 6: Preparation of (S)-IA-3 using chiral phase transfer catalyst IIIa-13-1
) The compound of Formula IIA-3 (11.6 g, 1.0 eq.) and DCM 360 ml, 30 volumes) were placed into a 1 L reactor; the solid was dissolved completely.
) The mixture was cooled to 0°C and some starting material was precipitated out.
) The catalyst (0.43 g, 3% mol) was added to the resulting mixture, and the mixture was cooled to -10° C.
) Hydroxylamine (6.1 g, 5.0 eq., 50% in water) was added to a solution of NaOH (4.4 g, 6.0 eq., in 5 volumes of water) in another reactor, and the mixture was stirred for 30 minutes.
) The hydroxylamine and NaOH solution was added dropwise to the 1 L reactor over about 2 hours, after which the mixture was stirred for 16 h at -10° C.
) Samples were taken and analyzed by HPLC to monitor the extent of reaction until the content of starting material was < 1.0%.
) When the reaction was complete, the mixture was warmed to 10°C and 50 ml of water was added. The mixture was stirred for 10 minutes.
) The mixture was allowed to settle to separate the aqueous and organic layers and the organic layer was collected.
) The organic layer was washed with 50 ml of 5% KH2PO4.
0) The mixture was allowed to separate and the organic layer was collected.
1) The organic layer was washed with 50 ml brine and the organic layer was again collected. 2) The organic layer was concentrated under vacuum at 25-30°C to about 2 volumes.
3) Toluene (230 ml, 10 volumes) was charged and concentration under vacuum was continued at 40~45°C to about 3 volumes. The solvent exchange was repeated twice more using the same procedure.
14) After the solvent exchange was complete, the solution was heated to 55-60°C.
15) The mixture was then cooled to 40° C over 1.5 hours and stirred at 40° C for 3 hours.
16) The mixture was cooled to 25° C over 2 hours and stirred at 25° C for 3 hours.
17) Finally, the mixture was cooled to 5-10° C over 1 hour and stirred at 8° C for 12 hours, after which the mixture was filtered.
18) The filter cake was washed with cold toluene (25 ml, 2 volumes).
19) The product was dried under vacuum at 85~90°C for 24h, resulting in the product as a white solid (6.8 g, chiral purity: 98.7% by area using the chiral FTPLC method described in Example 3, chemical purity: 99.3% by area (FTPLC), yield: 52.1%).

SEE ALSO US20170239218
//////////Umifoxolaner, ML 878, ML878, CS072E2C38, ML-878, WHO 11642
Bavtavirine



Bavtavirine, CAS 1956373-71-9
- KAJ2CK6ZYE
- 4-((4-Amino-8-(4-((1E)-2-cyanoethenyl)-2,6-dimethylphenyl)-2-quinazolinyl)amino)benzonitrile
- Benzonitrile, 4-((4-amino-8-(4-((1E)-2-cyanoethenyl)-2,6-dimethylphenyl)-2-quinazolinyl)amino)-
C26H20N6 416.48
Benzonitrile, 4-[[4-amino-8-[4-[(1E)-2-cyanoethenyl]-2,6-dimethylphenyl]-2-quinazolinyl]amino]-
Gilead Sciences, Inc.; Institute of Organic Chemistry and Biochemistry of the AS CR, v.v.i.
Bavtavirine is a potent non-nucleoside reverse transcriptase inhibitors (NNRTIs). Bavtavirine is part of highly active antitiretroviral therapy (HAART) treatment regimen. Bavtavirine can be used for HIV disease research.
SCHEME

PATENT
WO2016105564
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016105564&_cid=P11-M8QXHF-67832-1
A mixture of compound 2a (100 mg, 0.30 mmol), 4-cyanoaniline (46 mg, 0.388 mmol, Sigma-Aldrich) and hydrogen chloride solution in 1,4-dioxane (4M, 7 μL, 0.03 mmol) in dry N-methyl-2-pyrrolidone (2 mL) was heated at 120 °C for 2 hours. The reaction mixture was cooled down to room temperature and triethylamine (0.1 mL, 0.72 mmol) was added. After 15 minutes, water (5 mL) was added and the solid product was filtered off and washed with water. The crude residue was taken up in a mixture of dichloromethane and diethyl ether (1:1,5 mL) and then treated in a sonic bath for 3 minutes. The solid compound was filtered off and washed with diethyl ether (5 mL) to afford the title compound 2. 1H NMR (400 MHz, DMSO-d6) δ 9.44 (s, 1H), 8.18 (dd, J = 8.2, 1.5 Hz, 1H), 7.74 (d, J = 16.7 Hz, 1H), 7.70 (d, J = 8.9 Hz, 2H), 7.51 (s, 2H), 7.48 (dd, J = 7.1, 1.3 Hz, 1H), 7.34 (dd, J = 8.2, 7.1 Hz, 1H), 7.26 (d, J = 8.9 Hz, 2H), 6.54 (d, J = 16.7 Hz, 1H), 1.91 (s, 6H). HRMS: (ESI+) calculated for C26H2,N6 [M+H] 417.1822, found 417.1820. LCMS (m/z) 417.2 [M+H], Tr = 4.68 min (LCMS method 1).
////////////Bavtavirine
Uplarafenib


Uplarafenib
1425485-87-5
494.5 g/mol
| Molecular Formula | C22H21F3N4O4S |
| Molecular Weight | 494.487 |
- B-Raf IN 10
- TQU3V7CXC3
- N-[2,4,5-trifluoro-3-(3-morpholin-4-ylquinoxaline-6-carbonyl)phenyl]propane-1-sulfonamide
- B-Raf IN 10; B-Raf IN-10; B-Raf-IN-10
UPLARAFENIB is a small molecule drug with a maximum clinical trial phase of II and has 1 investigational indication. Neupharma, Inc.
There are at least 400 enzymes identified as protein kinases. These enzymes catalyze the phosphorylation of target protein substrates. The phosphorylation is usually a transfer reaction of a phosphate group from ATP to the protein substrate. The specific structure in the target substrate to which the phosphate is transferred is a tyrosine, serine or threonine residue. Since these amino acid residues are the target structures for the phosphoryl transfer, these protein kinase enzymes are commonly referred to as tyrosine kinases or serine/threonine kinases.
The phosphorylation reactions, and counteracting phosphatase reactions, at the tyrosine, serine and threonine residues are involved in countless cellular processes that underlie responses to diverse intracellular signals (typically mediated through cellular receptors), regulation of cellular functions, and activation or deactivation of cellular processes. A cascade of protein kinases often participate in intracellular signal transduction and are necessary for the realization of these cellular processes. Because of their ubiquity in these processes, the protein kinases can be found as an integral part of the plasma membrane or as cytoplasmic enzymes or localized in the nucleus, often as components of enzyme complexes. In many instances, these protein kinases are an essential element of enzyme and structural protein complexes that determine where and when a cellular process occurs within a cell.
The identification of effective small compounds which specifically inhibit signal transduction and cellular proliferation by modulating the activity of tyrosine and serine/threonine kinases to regulate and modulate abnormal or inappropriate cell proliferation, differentiation, or metabolism is therefore desirable. In particular, the identification of compounds that specifically inhibit the function of a kinase which is essential for processes leading to cancer would be beneficial
SCHEME


Patent
WO2022119905 69%
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022119905&_cid=P20-M8O7NY-07177-1

Example 1: Preparation of N-(2,4,5-trifluoro-3-(3-morpholinoquinoxaline-6-carbonyl)phenyl)propane-l-sulfonamide (Compound A)
[181] Step l : To a solution of quinoxalin-2(lH)-one (54.64 g, 374 mmol, 1.0 eq.) in HO Ac (1000 mL) was added a solution of Bn (19.18 mL, 374 mmol, 1.0 eq.) in HOAc (200 mL) dropwise. The resulting mixture was stirred at rt for 12 h, then poured into ice-water. The precipitate was collected by filtration and dried to afford 7-bromoquinoxalin-2(lH)-one as an off-white solid (74 g, 88%).
[182] Step l : To a suspension of 7-bromoquinoxalin-2(lH)-one (224 g, 1 mol, 1.0 eq.) in POCl3 (1000 mL) was added DMF (3.65 g, 0.05 mol, 0.05 eq.). The resulting mixture was stirred at 120 °C for 2 h, then cooled to rt and slowly poured into ice-water with vigorous stirring. The precipitate was collected by filtration and dried to afford 7-bromo-2-chloroquinoxaline as brown solid (180 g, 75%).
[183] Step 3 : To a solution of 7-bromo-2-chloroquinoxaline (50 g, 0.2mol, 1.0 eq.) in CH3CN (200 mL) were added morpholine (89 g, 1.02 mol, 5.0 eq.) and K2CO3 (85 g, 0.61mol, 3.0 eq). The resulting mixture was stirred at 90 °C for 2 h, then cooled and filtered. The filtrate was concentrated and the residue was re-crystallized from EA to afford 4-(7-bromoquinoxalin-2-yl)morpholine (59 g, 98.3%).
[184] Step 4 : To a solution of 4-(7-bromoquinoxalin-2-yl)morpholine (59 g, 0.2 mol, 1.0 eq.) in DMF (500 mL) was added TEA (139 mL, 1.0 mol, 5.0 eq.), EtsSiH (127 mL, 0.8 mol, 4.0 eq) and Pd(dppf)C12.CH2C12 (8.16 g, 0.01 mol, 0.05 eq.). The resulting mixture was stirred at 90 °C for 12h in an autoclave under CO (1 MPa), then cooled and concentrated. The resulting residue was purified by flash column chromatography(EA/PE=l/l) to afford 3-morpholinoquinoxaline-6-carbaldehyde as a yellow solid (40 g, 82.3%).
[185] Step 5 : To a solution of N-(2,4,5-trifluorophenyl)pivalamide (550 mg, 2.4 mmol, E2 eq.) in THF (30 mL) cooled at -78 °C was added LDA (4.1 mL, 4.8mmol, 2.4 eq.) dropwise. The resulting mixture was stirred at -78 °C for 1 h, then a solution of 3-morpholinoquinoxaline-6-carbaldehyde (486 mg, 2.0 mol, 1.0 eq.) in THF (20 mL) was added dropwise. The resulting mixture was stirred at -78 °C for 1 h, then quenched by the addition of NH4CI solution. The mixture was extracted with EA (20 mL X 3) and the combined organic layers were dried over Na2SO4 and concentrated. The resulting residue was purified by flash column chromatography (MeOH/DCM=l/50, v/v) to afford N-(2,4,5-trifluoro-3-(hydroxy(3-morpholinoquinoxalin-6-yl)methyl)phenyl)pivalamide (620 mg, 65.2%).
[186] Step 6 : The solution of N-(2,4,5-trifluoro-3-(hydroxy(3-morpholinoquinoxalin-6-yl)methyl)phenyl)pivalamide (620 mg, 1.3 mmol, 1.0 eq.) in DCM (10 mL) was added MnCb (358 mg, 6.5 mmol, 5.0 eq.). The resulting mixture was stirred at 50 °C overnight, then cooled and filtered. The filtrate was concentrated and the residue was purified by flash column chromatography (PE/EA=l/2,v/v) to afford N-(2,4,5-trifluoro-3-(3-morpholinoquinoxaline-6-carbonyl)phenyl)pivalamide (560 mg, 90%).
[187] Step 7 : To a solution of N-(2,4,5-trifluoro-3-(3-morpholinoquinoxaline-6-carbonyl)phenyl)pivalamide (560 mg , 1.2 mmol, 1.0 eq. ) in HO Ac (10 mL) was added cone. HC1 (50 mL). The mixture was stirred at 110 °C for 4h, then poured onto ice. The mixture was adjusted to pH 10 by the addition of IN NaOH solution, then extracted with DCM (100 mL X 3). The combined organic layers were dried over Na2SO4 and concentrated. The resulting residue was purified by flash column chromatography (PE/EA=l/4,v/v) to afford (3-amino-2,5,6-trifluorophenyl)(3-morpholinoquinoxalin-6-yl)methanone as brown solid (410 mg, 88 % yield).
[188] Step 8 : To a solution of (3-amino-2,5,6-trifluorophenyl)(3-morpholinoquinoxalin-6-yl)methanone (40 mg, 0.1 mmol, 1.0 eq.) in DCM (10 mL) was added TEA (101 mg, 1 mol, 10 eq.) and propane- 1 -sulfonyl chloride (0.5 mL, 0.5 mmol, 5.0 eq.). The resulting mixture was stirred at rt for 1 h, then washed with water and extracted with DCM (lOmL X 3). The combined organic layers were dried over Na2SO4, filtered and concentrated. The resulting residue was purified by flash column chromatography (PE/EA=2/1, v/v) to afford N-(propylsulfonyl)-N-(2,4,5-trifluoro-3-(3-morpholinoquinoxaline-6-carbonyl)phenyl)propane-l-sulfonamide (41 mg, 62.2%).
[189] Step 9 : To a solution of N-(propylsulfonyl)-N-(2,4,5-trifluoro-3-(3-morpholinoquinoxaline-6-carbonyl)phenyl)propane-l -sulfonamide (41 mg, 0.068 mmol, 1.0 eq.) in MeOH/THF (10 mL /10 mL) was added 1 N NaOH (0.15 mmol, 2.2 eq.). The resulting mixture was stirred at rt for 1 h, then concentrated. The resulting residue was purified by flash column chromatography (PE/EA=l/l,v/v) to afford N-(2,4,5-trifluoro-3-(3-morpholinoquinoxaline-6-carbonyl)phenyl)propane-l -sulfonamide (Compound A) (23 mg, 68.9%). LRMS (M+H+) m/z calculated 495.1, found 495.1. XH NMR (CDCh, 400 MHz) 8 8.67 (s, 1 H), 7.98-8.03 (m, 3 H), 7.66-7.73 (m, 1 H), 6.72 (s, 1 H), 3.78-3.88 (m, 8H), 3.12-3.16 (t, 2 H), 1.87-1.92 (q, 2 H), 1.05-1.09 (t, 3 H).
Example 2. Preparation of Crystalline Form I of Compound A
[190] N-(2,4,5-trifluoro-3-(3-morpholinoquinoxaline-6-carbonyl)phenyl)propane-l-sulfonamide (2.53 kg) and ethyl acetate (EA) (9.1 kg) were added to the reactor. The mixture was stirred under refluxing for 2h. The solution was cooled to room temperature. The resulting precipitate was filtered, washed with EA (1 kg), and dried under vacuum at 45 °C to afford Crystalline Form I of N-(2,4,5-trifluoro-3-(3-morpholinoquinoxaline-6-carbonyl)phenyl)propane-1-sulfonamide (1.94 kg, 76.7%).
Example 3. Preparation of Crystalline Form II of Compound A
[191] N-(2,4,5-trifluoro-3-(3-morpholinoquinoxaline-6-carbonyl)phenyl)propane-l-sulfonamide (4.01 kg) was dissolved in EA (60 kg), and water (20 kg) was added. The organic phase was separated and concentrated to 4-6 kg under vacuum at 40-45 °C. The resulting residue was dissolved in EA (6 kg) and stirred for 4 hours at 10-20 oC. The solid was filtered, washed with EA (1.5 kg), and dried under vacuum at 50-55 oC to afford Crystalline Form II of N-(2,4,5-trifluoro-3 -(3 -morpholinoquinoxaline-6-carbonyl)phenyl)propane- 1 -sulfonami de (3.15 kg, 78.6%).
SEE
US20130053384 69%
Avenciguat



Avenciguat, 1579514-06-9
BI-685509, 582.7 g/mol, C34H38N4O5
UNII ZA7KTB4PSP
5-ethoxy-1-[6-[3-methyl-2-[[5-methyl-2-(oxan-4-yl)-3,4-dihydro-1H-isoquinolin-6-yl]methoxy]phenyl]pyridin-2-yl]pyrazole-4-carboxylic acid
Avenciguat (BI-685509) is a potent and orally active sGC activator. Avenciguat restores cyclic guanosine monophosphate (cGMP) and improves functionality of nitric oxide (NO) pathways. Avenciguat can be used in research of chronic kidney disease (CKD) and diabetic kidney disease (DKD).
Avenciguat is under investigation in clinical trial NCT05282121 (A Study to Test Whether BI 685509 Alone or in Combination With Empagliflozin Helps People With Liver Cirrhosis Caused by Viral Hepatitis or Non-alcoholic Steatohepatitis (NASH) Who Have High Blood Pressure in the Portal Vein (Main Vessel Going to the Liver)).
Avenciguat (development name BI 685509) is a soluble guanylate cyclase activator developed by Boehringer Ingelheim for kidney disease,[1][2] and cirrhosis.[3][4][5]
SCHEME

Ref
PAPER
Journal of Pharmacology and Experimental Therapeutics (2023), 384(3), 382-39
PATENT
Boehringer Ingelheim International GmbH
WO2014039434
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014039434&_cid=P12-M29UB4-37937-1

PATENT
US20230293513
WO2020011804
| Clinical data | |
|---|---|
| Other names | BI 685509 |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1579514-06-9 |
| PubChem CID | 89992620 |
| UNII | ZA7KTB4PSP |
| Chemical and physical data | |
| Formula | C34H38N4O5 |
| Molar mass | 582.701 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
^ Cherney, David Z. I.; de Zeeuw, Dick; Heerspink, Hiddo J. L.; Cardona, Jose; Desch, Marc; Wenz, Arne; Schulze, Friedrich; Nangaku, Masaomi (August 2023). “Safety, tolerability, pharmacodynamics and pharmacokinetics of the soluble guanylyl cyclase activator BI 685509 in patients with diabetic kidney disease: A randomized trial”. Diabetes, Obesity and Metabolism. 25 (8): 2218–2226. doi:10.1111/dom.15099. PMID 37232058. S2CID 258909393.
^ Reinhart, Glenn A.; Harrison, Paul C.; Lincoln, Kathleen; Chen, Hongxing; Sun, Peng; Hill, Jon; Qian, Hu Sheng; McHugh, Mark C.; Clifford, Holly; Ng, Khing Jow; Wang, Hong; Fowler, Danielle; Gueneva-Boucheva, Kristina; Brenneman, Jehrod B.; Bosanac, Todd; Wong, Diane; Fryer, Ryan M.; Sarko, Chris; Boustany-Kari, Carine M.; Pullen, Steven S. (March 2023). “The Novel, Clinical-Stage Soluble Guanylate Cyclase Activator BI 685509 Protects from Disease Progression in Models of Renal Injury and Disease”. Journal of Pharmacology and Experimental Therapeutics. 384 (3): 382–392. doi:10.1124/jpet.122.001423. PMID 36507845. S2CID 254387173.
^ Lawitz, Eric J.; Reiberger, Thomas; Schattenberg, Jörn M.; Schoelch, Corinna; Coxson, Harvey O.; Wong, Diane; Ertle, Judith (November 2023). “Safety and pharmacokinetics of BI 685509, a soluble guanylyl cyclase activator, in patients with cirrhosis: A randomized Phase Ib study”. Hepatology Communications. 7 (11). doi:10.1097/HC9.0000000000000276. PMC 10615399. PMID 37889522.
^ Jones, Amanda K.; Chen, Hongxing; Ng, Khing Jow; Villalona, Jorge; McHugh, Mark; Zeveleva, Svetlana; Wilks, James; Brilisauer, Klaus; Bretschneider, Tom; Qian, Hu Sheng; Fryer, Ryan M. (July 2023). “Soluble Guanylyl Cyclase Activator BI 685509 Reduces Portal Hypertension and Portosystemic Shunting in a Rat Thioacetamide-Induced Cirrhosis Model”. Journal of Pharmacology and Experimental Therapeutics. 386 (1): 70–79. doi:10.1124/jpet.122.001532. PMID 37230799. S2CID 258909514.
^ Reiberger, Thomas; Berzigotti, Annalisa; Trebicka, Jonel; Ertle, Judith; Gashaw, Isabella; Swallow, Ros; Tomisser, Andrea (24 April 2023). “The rationale and study design of two phase II trials examining the effects of BI 685509, a soluble guanylyl cyclase activator, on clinically significant portal hypertension in patients with compensated cirrhosis”. Trials. 24 (1): 293. doi:10.1186/s13063-023-07291-3. PMC 10123479. PMID 37095557.
////////////Avenciguat, 1579514-06-9, BI 685509,
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}