



| Publication Number | Publication Date | IPCR Assignee/Applicant | Structure hits | Tools | |
|---|---|---|---|---|---|
|
1.
WO-2015178020-A1 |
2015-11-26 |
EN
|
|
||
|
2.
WO-2015174098-A1 |
2015-11-19 |
EN
|
|
||
|
3.
US-9187463-B2 |
2015-11-17 |
|
|||
|
4.
US-20150322055-A1 |
2015-11-12 |
|
|||
|
5.
EP-2922849-A1 |
2015-09-30 |
EN
|
|
||
|
6.
EP-2710002-A4 |
2014-10-01 |
EN
|
|
||
|
7.
US-8816090-B2 |
2014-08-26 |
|
|||
|
8.
EP-1856114-B1 |
2014-08-20 |
EN
|
|
||
|
9.
US-20140187583-A1 |
2014-07-03 |
|
|||
|
10.
WO-2014080633-A1 |
2014-05-30 |
EN
|
|
||
|
11.
EP-2710002-A1 |
2014-03-26 |
EN
|
|
||
|
12.
US-20140051726-A1 |
2014-02-20 |
|
|||
|
13.
EP-2688648-A1 |
2014-01-29 |
EN
|
|
||
|
14.
WO-2012157288-A1 |
2012-11-22 |
EN
|
|
||
|
15.
WO-2012127878-A1 |
2012-09-27 |
EN
|
|
||
|
16.
US-20080207690-A1 |
2008-08-28 |
|
|||
|
17.
EP-1856114-A1 |
2007-11-21 |
EN
|
|
||
|
18.
WO-2006090224-A1 |
2006-08-31 |
EN
|
|
Home » Uncategorized (Page 80)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
WCK ? New molecules from Wochkardt to treat bacterial infections

(2S, 5R)-7-OXO-N-[(3S)-PYRROLIDIN-3-YLOXY]-6-(SULFOOXY)-1,6-DIAZABICYCLO [3.2.1]OCTANE-2-CARBOXAMIDE
- (2S,5R)-7-Oxo-N-((3S)-pyrrolidin-3-yloxy)-6-(sulfooxy)-1,6-diazabicyclo[3.2.1]octane-2-carboxamide
- C11 H18 N4 O7 S, 350.35
- Sulfuric acid, mono[(1R,2S,5R)-7-oxo-2-[[[(3S)-3-pyrrolidinyloxy]amino]carbonyl]-1,6-diazabicyclo[3.2.1]oct-6-yl] ester
CAS 1452458-72-8
KEEP WATCHING THIS POST
SODIUM SALT CAS 1629221-44-8
Sulfuric acid, mono[(1R,2S,5R)-7-oxo-2-[[[(3S)-3-pyrrolidinyloxy]amino]carbonyl]-1,6-diazabicyclo[3.2.1]oct-6-yl] ester, sodium salt (1:1)
Patent
http://www.google.com/patents/WO2015110886A1?cl=en

Formula (II) Formula (III) Formula (IV)
Hydrogenolysis

Formula (I)
Scheme – 1

Formula (VII) Formula (VIII)
Hydrazine hydrate

Formula I
Scheme – 2
Example 1
Synthesis of tert-butyl (3S)-2-(aminooxy)pyrrolidine-l-carboxylate (III):
Step 1; Preparation of 3-(R)-hydroxypyrrolidine hydrochloride (VIII):
To a stirred suspension of commercially available (25, 4i?)-4-hydroxy-2-pyrrolidinecarboxylic acid (L-hydroxyproline) (VII) (100 g, 0.762 mol) in anhydrous cyclohexanol (500 ml), was added 2-cyclohexen-l-one (5 ml). The resulting mixture was heated under reflux at about 154°C for about 48 hour. The obtained clear solution was allowed to cool to room temperature and then was cooled further to 10°C. To this, about 15 % solution of hydrochloric acid in ethanol (234 ml) was added and then stirred for 30 minutes. The separated solid was filtered under suction and washed with ethyl acetate (2 x 100 ml). The solid was dried under reduced pressure to obtain 47.5 g of 3-(R)-hydroxypyrrolidine hydrochloride (VIII) in 51 % yield. The solid was used without further purification in the next step.
Analysis:
Mass: 87.8 (M+l) as free base; for Molecular weight of 123.57 and Molecular Formula of C4Hi0ClNO; and
1H NMR (400MHz, DMSO): 5 9.58 – 9.32 (brd, 2H), 5.36 (brs, 1H), 4.36 – 3.39 (brs, 1H), 3.17 (brs, 2H), 3.11-2.96 (dd, 2H), 1.90 – 1.81 (m, 2H).
Step 2: Preparation of (3R)-l-(tert-butoxycarbonyl)-3-hydroxypyrrolidine (IX):
To a stirred suspension of 3-(i?)-hydroxypyrrolidine hydrochloride (VIII) (110 g, 0.9 mol) in dichloromethane (1100 ml), triethylamine (273 g, 2.7 mol) was added at 0-5°C. After 5 minute of stirring di-feri-butyldicarbonate [(Boc)20] (245 g, 1.125 mol) was added to the reaction mixture in small portions, followed by 4-dimethylaminopyridine (10.99 g, 0.09 mol). The reaction mixture was stirred for 2 hour and then poured in to water (1100 ml). The organic layer was separated and washed with saturated ammonium chloride solution (1×1100 ml) and water (1100 ml). The organic layer was dried over anhydrous sodium sulphate and the solvent evaporated under reduced pressure. The residue was purified by silica gel (60-120 mesh) column chromatography using 1-5% mixtures of acetone: hexane as an eluent. The combined fractions were evaporated, to obtain the 118 g of (3i?)-l-(ieri-butoxycarbonyl)-3-hydroxypyrrolidine (IX), as a white solid, in 71 % yield.
Analysis:
Melting point: 55 – 58°C;
Mass: 188 (M+l); for Molecular Weight of 187.24 and Molecular Formula of C9H17N03; and
1H NMR (400MHz, CDC13): 54.428 – 4.424 (s, 1H), 3.46 – 3.43 (m, 2H), 3.37 -3.28 (m, 2H), 2.36 – 2.30 (d, 1H), 2.00 – 1.86 (m, 2H), 1.44 (s, 9H).
Step 3: Preparation of (5)-3-[(l,3-dihydro-l,3-dioxo-isoindol-2-yl)oxy]pyrrolidine-l-carbox lic acid tert- butyl ester (X):
To a stirred solution of di-isopropyl azodicarboxylate (97.17 g, 0.481 mol) in tetrahydrofuran (1200 ml), a solution triphenyl phosphine (125.9 g, 0.481 mol) in tetrahydrofuran (300 ml) was added at temperature below -10°C. The resulting reaction mixture was stirred for further 45 minute at the same condition and a solution of (3i?)-l-(ieri-butoxycarbonyl)-3-hydroxypyrrolidine (IX) (60 g, 0.3204 mol) in tetrahydrofuran (300 ml) was added over a period of 15 minute. After another 45 minute of stirring, N-hydroxy phthalimide (52.4 g, 0.3204mol) was added in one portion to the reaction mass. The reaction mixture was allowed to warm to room temperature and stirred for 16 hour.
The completion of the reaction was monitored by thin layer chromatography. After completion of reaction, the solvent was evaporated under reduced pressure. The residue thus obtained was stirred with di-isopropyl ether (600 ml). The precipitate formed was filtered under suction. The filtrate was concentrated under reduced pressure and the residual mass was purified by silica gel (60-120 mesh) column chromatography using 1-5 % mixtures of acetone: hexane as an eluent. The solvent from the combined fractions was evaporated to obtain 63 g of (5)-3-[(l,3-dihydro-l,3-dioxo-isoindol-2-yl)oxy]pyrrolidine-1-carboxylic acid tert-buty\ ester (X), as a white solid, in 59% yield.
Analysis:
Melting point: 112-115°C;
Mass: 333.2 (M+l); for Molecular Weight of 332.36 and Molecular Formula of ![]()
1H NMR (400 MHz, CDC13): 57.86-7.83 (m, 2H), 7.78-7.75 (m, 2H), 4.99 – 4.94 (d, 1H), 3.80 – 3.68 (m, 2H), 3.60 – 3.53 (m, 2H), 2.28-2.25 (m, 1H), 2.02 (m, 1H), 1.48 (s, 9H).
Step 4: Preparation of tert-butyl (35)-2-(aminooxy)pyrrolidine-l-carboxylate (III):
To a stirred suspension of the (5)-3-[(l,3-dihydro-l,3-dioxo-isoindol-2-yl) oxy]pyrrolidine-l-carboxylic acid tert-buty\ ester (X) (12.68 g, 0.0381 mol) in dichloromethane (200 ml) was added 99% hydrazine hydrate (3.81 g, 0.0762 mol) drop-wise over a period of 30 minutes, at 25°C. After 2 hour of stirring, the separated solid was filtered and washed with dichloromethane (2 x 50 ml). The filtrate and washings were combined and washed with water (2 x 65 ml) and finally with brine (1 x 65 ml). The organic layer was dried over anhydrous sodium sulphate and the solvent was evaporated under reduced pressure to obtain 7.71 g of tert-buty\ (3S)-2-(aminooxy pyrrolidine- 1-carboxylate (III) as pale yellow oil.
Analysis:
Mass: 203 (M+l); for Molecular Weight of 202.26 and Molecular Formula of C9H18N203.
Example 2
Synthesis of (25, 5R)-7-oxo-N-r(35)-pyrrolidin-3-yl-oxyl-6-(sulfooxy)-l,6-diaza bicyclor3.2. lloctane-2-carboxamide (I) :
Step 1: Preparation of fert-butyl-(35)-3-[({[25, 5R)-6-(benzyloxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]pyrrolidine-l-carboxylate (IV):
To a clear, stirred solution of sodium (25, 5i?)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxylate (II) (11.38 g, 0.0382 mol) in water (114 ml), was added EDC.HC1 (18.24 g, 0.0955 mol) at 15°C, in small portions. After 10 minutes, a solution of feri-butyl-(35)-3-(aminooxy) pyrrolidine- 1-carboxylate (III, 7.72 g, 0.0382 mol), prepared as per the literature procedure: US5233053, Chemistry Letters, 893-896, (1986) and depicted in scheme 2), in dimethylformamide (24 ml) was added drop wise, to the above stirred solution, at about 10°C. The reaction mass was allowed to warm to 25°C and HOBt (5.15 g, 0.0382 mol) was added in small portions over a period of 15 minutes and the reaction mixture was stirred further at room temperature for 16 hour. After completion of the reaction (monitored by thin layer chromatography using solvent system acetone: hexane (35:65)) the resulting mixture was filtered and the residue was washed with water (120 ml). The residual white solid was suspended in fresh water (120 ml) and the mixture stirred at 50°C, for 3 hour. The resulting suspension was filtered and the residual solid dried under reduced pressure to obtain 16.1 g of tert-buty\ (35)-3-[({ [25,5i?)-6-(benzyloxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino) oxy]pyrrolidine- 1-carboxylate (IV) as off white solid in 92% yield.
Analysis:
Mass: 461.3 (M+l); for Molecular weight of 460.53 and Molecular formula of ![]()
1H NMR (400MHz, CDC13): δ 9.08-9.03 (d, 1H), 7.43-7.36 (m, 5H), 5.06-4.88 (dd, 2H), 4.63-4.57 (d, 1H), 3.97-.396 (d, 1H), 3.64-3.53 (m, 2H), 3.47-3.37 (m, 2H), 3.31 (s, 1H), 3.02-2.99 (d, 1H), 2.75-2.73 (d, 1H), 2.29(m, 2H), 2.18-2.15 (m, 1H), 2.01-1.90 (m, 3H), 1.66 (m, 1H), 1.46 (s, 9H).
Step 2: Preparation of tert-butyl-(35)-3-[({[25,5R)-6-hydroxy-7-oxo-l,6-diazabicylco
[3.2.1]oct-2-yl]carbonyl}amino)oxy]pyrrolidine-l-carboxylate (V):
ieri-Butyl-(35)-3-[({ [25,5R)-6-(benzyloxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]pyrrolidine-l-carboxylate (IV) (10 g, 0.02171 mol) was dissolved in a mixture of dimethylformamide and dichloromethane ( 1 : 1 , 50 ml : 50 ml) to obtain a clear solution. To this solution, was added 10% palladium on carbon (2.5 g, 50% wet) catalyst. The suspension was stirred for 4 hour, at 50 psi hydrogen atmosphere, at 25°C. After completion of the reaction (monitored by thin layer chromatography), the resulting mixture was filtered through a celite pad. The residue was washed with dichloromethane (50 ml). The solvent from the combined filtrate was evaporated under reduced pressure to obtain 8.04 g of ieri-butyl(35)-3-[({ [25,5i?)-6-hydroxy-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl
amino)oxy]pyrrolidine-l-carboxylate (V) as oil. This was used as such for the next reaction without further purification.
Analysis:
Mass: 371.2 (M+l); for Molecular Weight of 370.4 and Molecular Formula of
Step 3: Preparation of tert-butyl-(35)-3-[({[25,5R)-6-(sulfooxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]pyrrolidine-l-carboxylate, tetrabutyl ammonium salt (VI):
To a stirred solution of ieri-butyl(35)-3-[({ [25,5i?)-6-hydroxy-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]pyrrolidine-l-carboxylate (V) (8.04 g, 0.0217 mol) in dimethylformamide (50 ml), was added sulfur trioxide dimethyl formamide complex (3.98 g, 0.0260 mol) in one portion, at about 10°C. The stirring was continued further for 30 minute and then the reaction mixture was allowed to warm to room temperature. After 2 hour, a solution of tetrabutylammonium acetate (7.83 g, 0.0260 mol) in water (25.8 ml) was added to the resulting reaction mass under stirring. After additional 2 hour of stirring, the solvent from the reaction mixture was evaporated under reduced pressure to obtain an oily residue. The oily mass was co-evaporated with xylene (2 x 20 ml) to obtain thick mass. This mass was partitioned between dichloromethane (100 ml) and water (100 ml). The organic layer was separated and the aqueous layer re-extracted with dichloromethane (50 ml). The combined organic extracts were washed with water (3 x 50 ml), dried over anhydrous sodium sulphate and the solvent evaporated under reduced pressure. The residual oily mass was triturated with ether (3 x 50 ml), each time the ether layer was decanted and finally the residue was concentrated under reduced pressure to obtain 11.3 g of tert-butyl(3S)-3-[({ [2S,5R)-6-(sulfooxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy] pyrrolidine- 1-carboxylate, tetrabutylammonium salt (VI), as a white foam, in 75 % yield.
Analysis:
Mass: 449.3 (M-l, without TBA); for Molecular weight of 691.94 and Molecular formula of C32H61N5O9S; and
1H NMR (400MHz, CDC13): 59.14-9.10 (d, 1H), 4.63 (s, 1H), 4.35 (s, 1H), 3.94-3.92 (d, 1H), 3.66-3.35 (m, 5H), 3.29-3.27 (m, 8H), 2.83-2.80 (d, 1H), 2.35-2.17 (m, 3H), 1.98-1.87 (m, 2H), 1.73 (m, 1H), 1.70-1.62 (m, 8H), 1.49-1.40 (m, 17H), 1.02-0.99 (t, 12H).
Step 4: Preparation of (25,5R)-7-oxo-iV-[(35)-pyrrolidin-2-yl-oxy]-6-(sulfooxy)-l,6-diazabicyclo [3.2.1]octane-2-carboxamide (I):
To a stirred solution of ieri-butyl(35)-3-[({ [25,5i?)-6-(sulfooxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]pyrrolidine-l-carboxylate tetrabutyl ammonium salt (VI) (11 g, 0.0158 mol) in dichloromethane (55 ml), was added trifluoroacetic acid (55 ml) drop wise at about -10 °C over a period of 1 hour. After 1 hour of stirring, the resulting mixture was poured into hexane (550 ml), stirred well for 30 minute and the separated oily layer was collected. This procedure was repeated one more time and finally the combined oily layer was added to diethyl ether (110 ml) under vigorous stirring, at about 25 °C. The ether layer was removed by decantation from the precipitated solid. This procedure was repeated twice again with diethyl ether (2 x 110 ml). The solid thus obtained was stirred with fresh dichloromethane (110 ml) for 30 minutes and filtered. The residual solid was dried at about 45 °C under reduced pressure to obtain 5.7 g of (25,5i?)-7-oxo-N-[(35)-pyrrolidin-2-yl-oxy]-6-(sulfo-oxy)- l,6-diaza bicyclo[3.2.1] octane-2-carboxamide (I), as a white amorphous solid having XRPD as shown in Figure 1.
Analysis:
Mass: 349.2 (M-l); for Molecular Weight of 350.35 and Molecular Formula of ![]()
1H NMR (400MHz, DMSO-D6): δ 11.44 (brs, 1H), 8.80 (brs, 2H), 4.64-4.63 (m, 1H), 4.00 (s, 1H), 3.78-3.77 (d, 1H), 3.38-3.23 (m, 4H), 3.03-2.93 (dd, 2H), 2.48-2.11 (m, 1H), 2.00- 1.94 (m, 2H), 1.88- 1.86 (m, 1H), 1.71-1.65 (m, 2H).
Example 3
Preparation of Crystalline Form I of (25,5R)-7-oxo-jV-r(35)-pyrrolidin-2-yl-oxyl-6-(sulfooxy)-l,6-diaza bicyclor3.2.11 octane-2-carboxamide:
The solid (5 g) obtained in Step 4 of Example 2 was dissolved in water (30 ml) with stirring. To this solution, Isopropanol (210 ml) was slowly added at 25 °C and stirred for 12 hours. The separated solid was filtered and washed with additional isopropanol ( 10 ml) and dried under reduced pressure to obtain 3.9 g of (25,5i?)-7-oxo-N-[(35)-pyrrolidin-2-yl-oxy]-6-(sulfo-oxy)-l,6-diazabicyclo[3.2.1]octane-2-carboxamide as crystalline Form I, having XRPD as shown in Figure 2, in 78 % yield.
Analysis:
Purity as determined by HPLC: 95.56 %; and
X-ray powder diffraction pattern comprising peak at (2 Theta Values): 10.57 (± 0.2), 12.01 (± 0.2), 13.61 (± 0.2), 15.47 (± 0.2), 17.86 (± 0.2), 18.34 (± 0.2), 19.09 (± 0.2), 19.81 (± 0.2), 22.69 (± 0.2), 24.79 (± 0.2), 27.22 (± 0.2) and 33.41 (± 0.2)
//////
WCK ? , WCK Series by Wockhardt for treating the bacterial infection

(2S,5R)-7-0X0-N-[(2S)-PYRROLLIDIN-2-YL-METHYLOXY]-6-(SULFOOXY)-1,6-DIAZABICYCLO[3.2.1 ]OCTANE-2-CARBOXAMIDE
(2S,5R)-7-Oxo-N-((2S)-pyrrolidin-2-ylmethyloxy)-6-(sulfooxy)-1,6-diazabicyclo[3.2.1]octane-2-carboxamide
Sulfuric acid, mono[(1R,2S,5R)-7-oxo-2-[[[(2S)-2-pyrrolidinylmethoxy]amino]carbonyl]-1,6-diazabicyclo[3.2.1]oct-6-yl] ester
KEEP WATCHING THIS POST
MW 364.37, C12 H20 N4 O7 S
CAS 1452459-04-9 FREE FORM
CAS Na SALT 1572988-44-3
Sulfuric acid, mono[(1R,2S,5R)-7-oxo-2-[[[(2S)-2-pyrrolidinylmethoxy]amino]carbonyl]-1,6-diazabicyclo[3.2.1]oct-6-yl] ester, sodium salt (1:1)
PATENTS, WO 2015079329, WO 2015079389 , WO 2015063714, US 20130225554
Emergence of bacterial resistance to known antibacterial agents is becoming a major challenge in treating bacterial infections. One way forward to treat bacterial infections, and especially those caused by resistant bacteria, is to develop newer antibacterial agents that can overcome the bacterial resistant. Coates et al. (Br. J. Pharmacol. 2007; 152(8), 1147-1154.) have reviewed novel approaches to developing new antibiotics. However, the development of new antibacterial agents is a challenging task. For example, Gwynn et al. (Annals of the New York Academy of Sciences, 2010, 1213: 5-19) have reviewed the challenges in discovery of antibacterial agents.
Several compounds have been described in the prior art for use in treatment of bacterial infections (for example, see Patent Application Nos. PCT/IB2012/054296, PCT/IB2012/054290, US20130225554, PCT/US2010/060923, PCT/EP2010/067647, PCT/US2010/052109, PCT/US2010/048109, PCT/GB2009/050609, PCT/EP2009/056178, PCT/US2009/041200, PCT/US2013/034562, PCT/US2013/034589, PCT/IB2013/053092 and PCT/IB2012054706). However, there remains a need for potent antibacterial agents for preventing and/or treating bacterial infections, including those caused by bacteria that are resistant to known antibacterial agents.

PATENT
https://encrypted.google.com/patents/WO2015079329A2?cl=en



Formula (I)
Scheme -1

Formula (VII) Formula (VIII)

Formula (III) Formula (X) Formula (IX)
Scheme 2
Example 1
Synthesis of fert-butyl (25)-2-r(aminooxy)methyllpyrrolidine-l-carboxylate
Step 1: Synthesis of l-(tert-butoxycarbonyl)-(25)-pyrrolidine-2-carboxylic acid (VII):
To a stirred suspension of (2S)-pyrrolidine-2-carboxylic acid (L-proline) (200 g, 1.73 mol) in 1,4-dioxan and water mixture (1: 1, 1000 ml : 1000 ml) was added a solution of sodium hydroxide (138.97 g, 3.47 mol in 740 ml water) over a period of 20 minutes at 0 °C. Bi-feri-butyl dicarbonate (415.3 ml, 1.9 mol in 400 ml 1,4-dioxan) was added to the resulting clear solution over a period of 30 minutes, at temperature of about 0-5 °C. The reaction mixture was allowed to warm to room temperature and stirred for 16 hours. After completion of the reaction (monitored by thin layer chromatography), the reaction mixture was concentrated to 40 % of the initial volume under reduced pressure at 40-50 °C. The pH of the residual mixture was adjusted to 2 – 2.5 using 30 % aqueous potassium hydrogen sulphate at 15 °C under continuous stirring. The separated solid was filtered under suction and washed with water (2×400 ml) and dried under reduced pressure (4 mm Hg), to obtain 370 g of l-(ieri-butoxycarbonyl)-(25)-pyrrolidine-2-carboxylic acid (VII) as white solid.
Analysis:
Mass: 216 (M+l), for Molecular Weight: 215.24 and Molecular Formula:
1H NMR (400 MHz, CDC13): δ 10.60 (s, 1H), 4.35-4.24 (dd, 1H), 3.54-.3.34 (M, 2H), 2.27-1.91 (unresolved, 4H), 1.47-1.41 (d, 9H);
Purity as determined by HPLC: 99.92 %.
Step 2: Synthesis of tert-iutyl-(25)-2-(hydroxymethyl)-pyrrolidine-l-carboxylate (IX):
N-Methylmorpholine (113 ml, 1.114 mol) was added to the suspension of \-{tert-butoxycarbonyl)-(25)-pyrrolidine-2-carboxylic acid (VII, 30 g, 139 mmol) in tetrahydrofuran (2000 ml) under stirring at temperature of about 0 °C. Ethyl chloroformate (106 ml, 1.114 mol) was added drop- wise to the above obtained clear solution over a period of 30 minutes. After stirring for 1 hour, the resulting suspension was filtered over celite and the residue was washed with tetrahydrofuran (2×200 ml). To the combined filtrate was added dropwise a solution of sodium borohydride (42.1 g, 1.114 mol) in 210 ml water, containing a catalytic amount of sodium hydroxide, at temperature of about -10 °C over a period of 1-2 hours under stirring. The reaction mixture was allowed to warm to room temperature and stirred further for an hour. The reaction mixture was filtered through celite bed and the filtrate concentrated under reduced pressure to yield 180 g of ieri-butyl(25)-2-(hydroxymethyl)-pyrrolidene-l-carboxylate (IX) as colorless oil.
Analysis:
Mass: 202 (M+l), for Molecular Weight: 201.2 and Molecular Formula: C10H19NO3;
1H NMR (400 MHz, CDC13): δ 3.94-.3.92 (m, 1H), 3.80 (board, 1H), 3.63-3.54 (m, 2H), 3.45-3.40 (m, 1H), 3.32-3.28 (m, 1H), 2.01-1.96 (m, 1H), 1.84-1.75 (m, 2H), 1.63 (m, 1H), 1.45 (s, 9H);
Purity as determined by HPLC: 87.7 %.
Step 3: Synthesis of fert-butyl (25)-2-[[(l,3-dihydro-l,3-dioxo-2H-isoindol-2-yl)oxy] methyl] -pyrrolidine-1 -carboxylate (X) :
Triphenylphosphine (328.4 g, 1.253 mol) in tetrahydrofuran (1260 ml) was added to solution of Diisopropyl azodicarboxylate (253.3 g, 1.253 mol) in tetrahydrofuran at temperature of -15 °C under stirring. After stirring for an hour, N-feri-butoxylcarbonyl-L-prolinol (IX) (180 g, 0.895 mol) in tetrahydrofuran (540 ml) was added to the resulting mixture over a period of 15 minutes. After stirring the mixture for 45 minutes, N-Hydroxy phthalimide (146 g, 0.895 mol) was added and the mixture was allowed to warm to room temperature and stirred further for 16 hours. The solvent was evaporated under reduced pressure and residual oil was dissolved in dichloromethane (5000 ml) and washed with an aqueous 5 % sodium hydrogen carbonate solution (2×300 ml). The organic layer was dried over anhydrous sodium sulfate and the solvent evaporated under reduced pressure to obtain viscous oil. Diisopropyl ether (720 ml) was added to the oil, the mixture was stirred well and separated solid was filtered under suction. The filtrate was concentrated under reduced pressure and the residue was further purified by chromatography over a silica gel column (60 -120 mesh) and eluted with mixtures of ethyl acetate and hexane. Upon concentration of the combined eluted fractions, 230 g of teri-butyl (25)-2-[[( l,3-dihydro- l,3-dioxo-2H-isoindol-2-yl)oxy]methyl]-pyrrolidine- l-carboxylate (X) was obtained as yellow oil.
Analysis:
Mass: 347.3 (M+l), for Molecular Weight: 346.39 and Molecular Formula: ![]()
1H NMR (400 MHz, CDCI3): δ 7.80-7.78 (m, 2H), 7.72-7.70 (m, 2H), 4.32 (brs, 1H), 4.05 (brs, 2H), 3.36-3.31 (m, 2H), 2.27-2.25 (m, 1H), 2.08(m, 1H), 1.88-1.87 (m, 2H), 1.43 (s, 9H).
Step 4: Synthesis of fert-butyl (25)-2-[(aminooxy)methyl]pyrrolidine-l-carboxylate (HI):
To a stirred solution of the compound of Formula (X) ( 100 g, 0.288 mol) in dichloromethane (2000 ml) was added 99 % hydrazine hydrate (28.9 g, 0.577 mol) drop-wise over a period of 30 minutes at temperature of about 25 °C. The stirring was continued further for a period of 3 hours. The separated solid was filtered and the solid washed with additional dichloromethane (2 x 500 ml). The combined organic layer was collected and washed with water (2 x 500 ml). The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain 62.4 g of tert-butyl (25)-2-[(aminooxy)methyl]pyrrolidine-l-carboxylate (III) as a colorless oil. This was used as such for the next reaction without further purification.
Analysis:
Mass: 215.1 (M- l), for Molecular Weight: 216.2 and Molecular Formula:
Example 2
Synthesis of (25,5R)-7-oxo-N-r(25)-pyrrolidin-2-yl-methyloxyl-6-(sulfooxy)-l,6- diazabicvclor3.2.11octane-2-carboxamide (I)
Step 1: Synthesis of tert-butyl (25)-2-{[({[25,5R)-6-(benzyloxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]methyl}pyyrolidine-l-carboxylate
(IV):
Sodium(25,5i?)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxylate (II, 77.4 g, 0.259 mol; prepared according to the procedure disclosed in Indian patent application No. 699/MUM/2013) was dissolved in water (774 ml) to obtain a clear solution. To the clear solution was added EDC.HC1 (120.8 g, 0.632 mol) at temperature of about 15°C and after 10 minutes a solution of tert-buty\ (25)-2-[(aminooxy)methyl]pyrrolidine-l-carboxylate (III, 62.4 g, 0.288 moles prepared as per the literature procedure depicted in scheme 2) in dimethylformamide (125 ml) was added drop wise under continuous stirring at temperature of about 10 °C. The reaction mass was allowed to warm to temperature of about 25°C and then HOBt (38.96 g, 0.288 mol) was added in small portions over a period of 15 minutes and the resulting mixture was further stirred at room temperature for 16 hours. The reaction progress was monitored using thin layer chromatography using mixture of acetone and hexane (35: 65) as solvent system. The resulting suspension was filtered and the residue was washed with water (200 ml). The residual white solid was suspended in water (200 ml) and the mixture stirred with heating at temperatyre of about 50 °C for 3 hours. The resulting suspension was filtered, the residue dried at atmospheric temperature and then dried under vacuum to obtain 105 g of ierr-Butyl(25)-2- { [( { [25,5R)-6-(benzyloxy)-7-oxo- l,6-diazabicylco[3.2. l]oct-2-yl]carbonyl} amino)oxy]methyl}pyyrolidine-l-carboxylate (IV) as off white solid.
Analysis:
Mass: 475.4 (M+l), for Molecular Weight of 474.56 and Molecular Formula of ![]()
1H NMR (400 MHz, CDCI3): δ 10.16 (br s, 1H), 7.43-7.35 (m, 5H), 5.06-4.88 (dd, 2H), 4.12 (s, 1H), 3.94-.393 (d, 2H), 3.83 (unresolved s, 1H), 3.75-3.73 (m, 1H), 3.37-3.28 (dt, 2H), 3.02-2.86 (dd, 2H), 2.31-2.26 (m, 1H), 2.02-1.84 (m, 6H), 1.71-1.68 (m, 1H), 1.45 (s, 9H).
Step 2: Synthesis of tert-butyl(25)-2-{[({[25,5R)-6-hydroxy-7-oxo-l,6-diazabicylco
[3.2.1]oct-2-yl]carbonyl}amino)oxy]methyl}pyyrolidine-l-carboxylate (V):
tert-butyl(25)-2-{ [({ [25,5R)-6-(benzyloxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl] carbonyl}amino)oxy]methyl}pyyrolidine-l-carboxylate (IV, 85 g, 0.179 mol) was dissolved in a mixture of dimethylformamide and dichloro methane (1: 1, 425 ml : 425 ml) to obtain a clear solution. To this solution was added 10 % Pd-C (17 g, 50 % wet) catalyst. The suspension was stirred for 4 hours under 7 psi hydrogen atmosphere at temperature of about 25 °C. The resulting mixture was filtered through celite under suction. The residue was washed with dichloromethane (170 ml). The solvent from the filtrate was evaporated under reduced pressure to furnish 68.8 g of tert-buty\(2S)-2-{ [( { [25,5i?)-6-hydroxy-7-oxo- l,6-diazabicylco[3.2. l]oct-2-yl]carbonyl} amino)oxy] methyl}pyyrolidine-l-carboxylate (V) as oil. The obtained product was used as such for the next reaction without further purification.
Analysis:
Mass: 385.4 (M+l), for Molecular Weight of 384.4 and Molecular Formula of C17H28N406.
Step 3: Synthesis of tert-butyl(25)-2-{[({[25,5R)-6-(sulfooxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]methyl}pyyrolidine-l-carboxylate, tetra butyl ammonium salt (VI):
To solution of ieri-butyl(25)-2-{ [({ [25,5i?)-6-hydroxy7-oxo-l,6-diazabicylco [3.2.1]oct-2-yl]carbonyl}amino)oxy]methyl}pyyrolidine-l-carboxylate (V, 68.8 g, 0.178 mol) in dimethylformamide, (345 ml) was added sulfur trioxide dimethylformamide complex (30 g, 0.196 mol) under stirring at temperature of about 10 °C. The reaction mass was stirred at the same temperature for 30 minutes and then allowed to warm to room temperature. After 2 hours solution of tetra butyl ammonium acetate (59.09 g, 0.196 mol) in water (178 ml) was added to the reaction mixture under stirring. After 2 hours, the solvent from the reaction mixture was evaporated under reduced pressure to obtain an oily residue. The oily mass was co-evaporated with xylene (2×140 ml) to obtain thick mass. This mass was partitioned between dichloromethane (690 ml) and water (690 ml). The organic layer was separated and the aqueous layer re-extracted with dichloromethane (345 ml). The combined organic extracts were washed with water (3×345 ml) and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure and the resulting oily mass was triturated with ether (3×140 ml), each time the ether layer was decanted and finally the residue was concentrated under reduced pressure to obtain 102 g of ieri-butyl(25)-2- { [({ [25,5i?)-6-(sulfooxy)-7-oxo- l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]methyl}pyyrolidine- l-carboxylate, tetrabutyl ammonium salt (VI) as fluffy material.
Analysis:
Mass: 463.4 (M- l without TBA), for Molecular Weight of 705.96 and Molecular Formula of C33H63N5O9 S;
1H NMR (400 MHz, CDCI3): δ 10.2 (s, 1H), 4.35 (s, 1H), 4.14 (s, 1H), 3.91 -3.92 (d, 2H), 3.74 (m, 1H), 3.36-3.27 (m, 10H), 2.96-2.88 (dd, 2H), 2.31-2.26 (m, 2H), 2.19-1.98 (m, 2H), 1.95-1.70 (m, 4H), 1.68- 1.62 (p, 8H), 1.49- 1.40 (m, 17H), 1.02-0.98 (t, 12H).
Step 4: (25,5R)-7-oxo-N-[(25)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)-l,6-diaza bicyclo [3.2.1]octane-2-carboxamide (I):
feri-butyl(25)-2-{ [( { [25,5i?)-6-(sulfooxy)-7-oxo-l ,6-diazabicylco[3.2.1]oct-2-yl] carbonyl}amino)oxy]methyl}pyyrolidine- l-carboxylate, tetrabutylammonium salt (VI) (50 g, 0.0708 mol) was dissolved in dichloromethane (250 ml) and to the clear solution was slowly added trifluoroacetic acid (250 ml) at temperature of about -10 °C over a period of 1 hour under stirring. After stirring for an hour, the resulting mixture was poured into hexane (2500 ml) and the oily layer was separated. This procedure was repeated one more time and finally the separated oily layer was added to diethyl ether (500 ml) under vigorous stirring at temperature of about 25 °C. The ether layer was removed by decantation from the precipitated solid. This procedure was repeated twice again with diethyl ether (2x500ml). The solid thus obtained was stirred with fresh dichloromethane (500 ml) for 30 minutes and filtered. The residual solid was dried at temperature of about 45 °C under reduced pressure to yield 25 g of (25,5i?)-7-Oxo-N-[(25)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)- l,6-diazabicyclo[3.2.1]octane-2-carboxamide (I) in amorphous form. The XRD of the obtained amorphous form is shown in Figure 1.
Analysis:
Mass: 363.2 (M- l), for Molecular Weight: 364.37 and Molecular Formula: C12H2oN407S;
1H NMR (400 MHz, DMSO-D6): δ 1 1.73 (s, 1H), 8.62-8.83 (d, 2H), 3.88-4.00 (m, 3H), 3.74-3.81 (m, 2H), 3.19 (t, 2H), 2.94-3.04 (dd, 2H), 1.96-2.03 (m, 2H), 1.80-1.92 (m, 3H), 1.54- 1.73 (m, 3H);
Purity as determined by HPLC: 90.30 %.
Example 3
Preparation of Crystalline Form I of (25,5R)-7-oxo-N-r(25)-pyrrolidin-2-yl- methyloxyl-6-(sulfooxy)-l,6-diaza bicvclor3.2.11octane-2-carboxamide
The amorphous solid obtained in the Step 4 of Example 2 was dissolved in water (75 ml) and to this solution isopropanol (200 ml) was slowly added at temperature of about 25 °C. The solution was further stirred for 12 hours. The separated solid thus obtained was filtered and washed with additional isopropanol (25 ml) and dried under reduced pressure to obtain 19 g of (25,5i?)-7-Oxo-N-[(25)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)-l ,6-diazabicyclo[3.2.1]octane-2-carboxamide as crystalline Form I. The XRD of the obtained crystalline Form I is shown in Figure 2.
X-ray powder diffraction pattern comprising peak at (2 Theta Values): 8.08 (± 0.2), 1 1.45 (± 0.2), 16.26 (± 0.2), 17.89 (± 0.2), 18.15 (± 0.2), 19.66 (± 0.2), 21.15 (± 0.2), 23.55 (± 0.2), 24.23 (± 0.2), 24.94 (± 0.2), 25.66 (± 0.2) and 29.41 (± 0.2).
Typical X-ray analysis was performed as follows. Pass the test substance through sieve #100 BSS or gently grind it with a mortar and pestle. Place the test substance uniformly on a sample holder having cavity surface on one side, press the sample and cut into thin uniform film using a glass slide in such a way that the surface of the sample should be smooth and even. Record the X-ray diffractogram using the following instrument parameters:
Instrument : X-Ray Diffractometer
(PANalytical, Model X’Pert Pro
MPD)
Target source : CuK(a)
Antiscattering slit (Incident beam) : 1°
Programmable Divergent slit : 10 mm (fixed)
Anti- scattering slit (Diffracted beam) : 5.5 mm
Step width : 0.02°
Voltage : 40 kV
Current : 40 mA
Time per step : 30 seconds
Scan range : 3 to 40°
Example 4
Preparation of Pure (25,5R)-7-oxo-N-r(25)-pyrrolidin-2-yl-methyloxyl-6-(sulfooxy)- l,6-diazabicyclor3.2.11octane-2-carboxamide
(25,5i?)-7-Oxo-N-[(25)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)- l,6-diazabicyclo [3.2.1] octane-2-carboxamide (5 g) was slowly dissolved in water (50 ml) under stirring until clear solution appears. To this clear solution 350 ml of isopropanol was added drop wise under stirring over the period of 2 hours. Formation of fine white precipitates was observed after the completion of the addition of isopropanol. The resulted fine suspension was stirred at temperature of about 25 °C for 20 hours. The formed white precipitates were filtered and vacuum dried at temperature of about 30-40 °C, under reduced pressure (2 mm Hg) to get 4.4 g of (2S,5i?)-7-oxo-N-[(2S)-pyrroMin-2-yl-methyloxy]-6-(sulfooxy)-l,6-diazabicyclo [3.2.1] octane-2-carboxamide.
The above obtained (25,5i?)-7-oxo-N-[(25)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)-l,6-diazabicyclo[3.2.1]octane-2-carboxamide (3.4 gm) was dissolved in 34 ml of water to get clear solution. To the obtained clear solution 170 ml of isopropanol was added drop wise over a period of 1 hour. Formation of fine oily globules was observed and allowed to stand still for 15 minutes. The upper clear water and isopropanol layer was decanted from the oily mass. The clear decanted solution was allowed to stand at temperature of about 25 °C for 48 hours. Formation of crystals was observed and were collected by filtration. The collected crystals were dried at temperature of about 30-40 °C, under reduced pressure (2 mm Hg) to get 2 g of (2S,5i?)-7-oxo-N-[(2S)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)- 1 ,6-diazabicyclo[3.2.1 ]octane-2-carboxamide which was analyzed for content of various components using HPLC and the results are described in Table 1.
The relative % content of (25,5i?)-7-oxo-N-[(25)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)-l,6-diazabicyclo[3.2.1]octane-2-carboxamide with other substances (Table 1) was determined using HPLC (Agilent 1100 or equivalent). The HPLC column having 250 mm length and 4.6 mm ID packed with 5 μ particles of octa-decyl silane (ODS) was used. Mobile phase A used was a mixture of buffer (0.02 M potassium dihydrogen phosphate in HPLC grade water, pH adjusted to 2.5 with orthophosphoric acid and again readjusted to 7.0 with dilute ammonia), HPLC grade water and acetonitrile in a ratio of 40 : 60 : 0.2; v/v/v. Mobile phase B was mixture of buffer and acetonitrile in a ratio of 40 : 60; v/v. Mobile phase was run in gradient mode. Initially mobile phase A and B was run at 100 : 0 for 15 minutes, slowly ratio of mobile phase B was raised to 100 % in 10 minutes, held for 10 minutes at this concentration and brought back to initial condition in next 5 minutes and held for 10 minutes before next run. Flow rate of mobile phase was maintained at 1.0 ml/min. Column temperature was maintained at temperature of about 30°C. Detection was carried out using UV detector at wavelength 225 nm. Test solutions were prepared in mobile phase A. The method is capable of resolving diastereomers (Table 1, Sr. No. 1 and 2) with resolution of not less than 2.0.

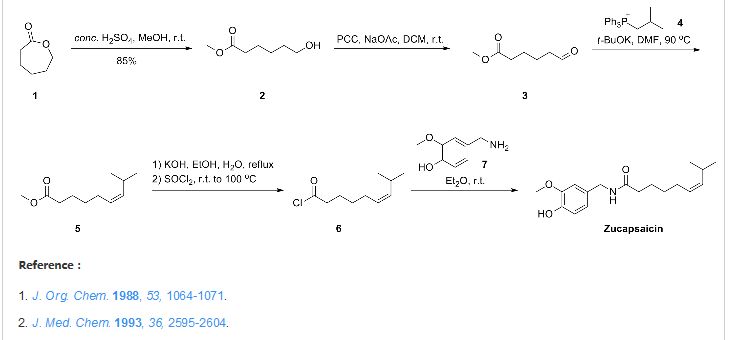
Zucapsaicin for osteoarthritis

Zucapsaicin (珠卡赛辛)
cis-Capsaicin; (Z)-Capsaicin
Zucapsaicin; Civamide; Cis-Capsaicin; 25775-90-0; (Z)-Capsaicin; (Z)-N-(4-Hydroxy-3-methoxybenzyl)-8-methylnon-6-enamide;
(Z)-N-[(4-Hydroxy-3-methoxyphenyl)methyl]-8-methylnon-6-enamide
CAS No. 25775-90-0
| MF C18H27NO3 | |
| Molecular Weight: | 305.41188 g/mol |
|---|
WINSTON INNOVATOR
SANOFI
(Zuacta®/Civanex®
A medication used to treat osteoarthritis of the knee and other neuropathic pain.TRPV1 CHANNEL AGONIST


Zucapsaicin (Civanex) is a medication used to treat osteoarthritis of the knee and other neuropathic pain. It is applied three times daily for a maximum of three months. It reduces pain, and improves articular functions. It is the cis-isomer of capsaicin. Civamide, manufactured by Winston Pharmaceuticals, is produced in formulations for oral, nasal, and topical use (patch and cream).[1]
Zucapsaicin has been tested for treatment of a variety of conditions associated with ongoing nerve pain. This includes herpes simplex infections; cluster headaches and migraine; and knee osteoarthritis.[2]
Civanex (zucapsaicin) cream is a TRPV-1 modulator in development for the treatment of signs and symptoms of osteoarthritis of the knee.
Zucapsaicin, the cis-isomer of the natural product capsaicin, is a
topical analgesic that was initially developed by Winston Pharmaceuticals
and approved in Canada in July 2010 for the treatment of
severe pain in adults with osteoarthritis of the knee.
Bronson, J.; Dhar, M.; Ewing, W.; Lonberg, N. In Annual Reports in MedicinalChemistry; John, E. M., Ed.; Academic Press, 2011; Vol. 46, p 433.
The advantagesof zucapsaicin compared with naturally-occurring capsaicin, are reported to be a lesser degree of local irritation (stinging, burning,
erythema) in patients and a greater degree of efficacy in preclinical
animal models of pain.
Bernstein, J. E. U.S. 5063060, 1991.
Bernstein, J. E. U.S. 20050084520 A1, 2005.
The analgesic action of both
zucapsaicin and capsaicin is mediated through the transient receptor
potential vanilloid type 1 (TRPV1) channel, a ligand-gated ion
channel expressed in the spinal cord, brain, and localized on neurons
in sensory projections to the skin, muscles, joints, and
gut.
Westaway, S. M. J. Med. Chem. 2007, 50, 2589.
The scale preparation of zucapsaicin likely parallels the original
approach described by Gannett and co-workers involving the
coupling of vanillylamine with (Z)-8-methylnon-6-enoyl chloride.
Gannett, P. M.; Nagel, D. L.; Reilly, P. J.; Lawson, T.; Sharpe, J.; Toth, B. J. Org.Chem. 1988, 53, 1064.
Orito and co-workers elaborated this original approach in
an effort to prepare both capsaicin and zucapsaicin on gram-scale,
Kaga, H.; Miura, M.; Orito, K. J. Org. Chem. 1989, 54, 3477.
References
- 1 Winston Pharmaceuticals website http://www.winstonlabs.com/productdevelopment/civamide.asp
- 2 Zucapsaicin information from the National Library of Medicine http://druginfo.nlm.nih.gov/drugportal
Janusz, John M.; Buckwalter, Brian L.; Young, Patricia A.; LaHann, Thomas R.; Farmer, Ralph W.; et al. Journal of Medicinal Chemistry, 1993 , vol. 36, # 18 p. 2595 – 2604
Journal of Organic Chemistry, , vol. 53, # 5 p. 1064 – 1071
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(Z)-N-[(4-Hydroxy-3-methoxyphenyl)methyl]-8-methylnon-6-enamide
|
|
| Clinical data | |
| Trade names | Civanex |
| Routes of administration |
Topical |
| Identifiers | |
| CAS Number | 25775-90-0 |
| ATC code | M02AB02 |
| PubChem | CID: 1548942 |
| ChemSpider | 1265956 |
| UNII | 15OX67P384 |
| Synonyms | Civamide; (Z)-Capsaicin; cis-Capsaicin |
| Chemical data | |
| Formula | C18H27NO3 |
| Molecular mass | 305.41188 g/mol |
////Zucapsaicin
Oc1ccc(cc1OC)CNC(CCCC\C=C/C(C)C)=O
see………..http://apisynthesisint.blogspot.in/2015/12/zucapsaicin-for-osteoarthritis.html
RQ 00000010 for the treatment of GERD, functional dyspepsia and chronic constipation.

RQ 00000010
CAS 907607-22-1
| Molecular Formula: | C22H27F3N2O6 |
|---|---|
| Molecular Weight: | 472.45479 g/mol |
HSMMHNBGQLGCBY-UHFFFAOYSA-N;
RaQualia Pharma Inc
PFIZER INNOVATOR
RQ-00000010; RQ-10
4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid
4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid
ΦirΦfff^fΣ^^-TrifluoroethoxyVi.a-benzisoxazol-S-vnoxylmethvπpiperidin-i-vπmethylltetrahydro-2H-pyran-4-carboxylic acid
4-[[4-[[4-(2,2,2-trifluoroethoxy)-1,2-benzoxazol-3-yl]oxymethyl]piperidin-1-yl]methyl]oxane-4-carboxylic acid
PHASE 1 for the treatment of GERD, functional dyspepsia and chronic constipation.
Useful for treating diseases mediated by 5-HT4 receptor activity eg such as gastroesophageal reflux disease (GERD), gastric motility disorder, dyspepsia, constipation, esophagitis, diabetes, CNS and cardiovascular diseases.
RaQualia, following its spin-out from Pfizer, is developing RQ-00000010, a 5-HT4 receptor partial agonist, for the treatment of gastric motility disorders, including gastroparesis associated with Parkinson’s disease.
In November 2015, the drug was reported to be in phase 1 clinical development. RaQualia and licensee CJ CheilJedang are investigating the drug for the treatment of GERD, functional dyspepsia and chronic constipation.
4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid is disclosed in PL1 as a 5-HT4 receptor agonist, which is useful in the treatment or alleviation of disease conditions mediated by 5-HT4 receptor activity; in particular 5-HT4 receptor agonistic activity, such as gastroesophageal reflux disease (GERD), gastrointestinal disease, gastric motility disorder, non-ulcer dyspepsia, functional dyspepsia (FD), irritable bowel syndrome (IBS), constipation, dyspepsia, esophagitis, gastroesophageal disease, gastritis, nausea, central nervous system disease, Alzheimer’s disease, cognitive disorder, emesis, migraine, neurological disease, pain, cardiovascular disorders, cardiac failure, heart arrhythmia, diabetes, and apnea syndrome (See NPL 1 to 13 and PL 2 to 7).
Simply an white solid has been produced in the previously known methods of preparing 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid, described in PL 1. A generic disclosure of pharmaceutically-acceptable salts of 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid of the instant application is disclosed, and the free base of the compound of the instant invention is disclosed and claimed, in PL 1 having an international filing date of December 6, 2006, assigned to the assignee hereof. Thus any salts of the compound have been neither pacifically described nor synthesized in prior art.
It has been found that HCl-salt, HBr-salt, pTSA-salt and EDSA-salt of 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid shown below, can be isolated as a crystalline form which has advantageous properties such as ease of making a formulation, high solubility, and good stability. In addition the salts of the present invention are more easily purified than a non-crystalline form disclosed in PL 1 (WO2006/090224) and crystalline form disclosed in PL 3 (WO2012/157288).
Patent Literature
{PL 1} WO2006/090224.
{PL 2} US Patent No. 6,106,864.
{PL 3} WO2012/157288
{PL 4} WO00/35298.
{PL 5} WO91/11172.
{PL 6} WO94/02518.
{PL 7} WO98/55148.
Non Patent Literature
{NPL 1} Bockaert J. et al., TiPs 13; 141-145, 1992.
{NPL 2} Ford A. P et al., Med. Res. Rev. 13: 633-662, 1993.
{NPL 3} Gullikson G. W. et al., Drug Dev. Res. 26; 405-417, 1992.
{NPL 4} Richard M. Eglen et al., TiPs 16; 391-398, 1995.
{NPL 5} Bockaert J. et al., CNS Drugs 1; 6-15, 1994.
{NPL 6} Romanelli M. N. et al., Arzheim Forsch./Drug Res., 43; 913-918, 1993.
{NPL 7} Kaumann A. J. et al., Naunyn-Schmiedebergs Arch Pharmacol., 344; 150-159, 1991.
{NPL 8} Remington’s Pharmaceutical Sciences, 19th Edition (Mack Publishing Company, 1995).
{NPL 9} Expert Opinion in Therapeutic Patents, H (6), 981-986, by Liang and Chen (2001).
{NPL 10} Tablets, Vol. 1, by H. Lieberman and L. Lachman (Marcel Dekker, New York, 1980).
{NPL 11} Pharmaceutical Technology On-line, 25(2), 1-14, by Verma et al. (2001).
{NPL 12} J Pharm Sci, 88 (10), 955-958, by Finnin and Morgan (October 1999).
{NPL 13} Evrard, B., et al., Journal of Controlled Release 96 (3), pp. 403-410, 2004.
{NPL 14} Byrn S. R. et al., Solid-State Chemistry of Drugs 2nd ed., pp 3-43 and 461-503, 1999, SSCI, Inc.

PATENT
WO2006090224
| PFIZER JAPAN INC. |
EXAMPLE 1 :
ΦirΦfff^fΣ^^-TrifluoroethoxyVi.a-benzisoxazol-S-vnoxylmethvπpiperidin-i-vπmethylltetrahydro-2H-pyran-4-carboxylic acid
Step 1. Methyl 2-hvdroxy-6-(2,2,2-trifluoroethoxy)benzoate
A mixture of 5-hydroxy-2,2-dimethyl-4tø-1 ,3-benzodioxin-4-one (123 g, 633 mmol, Synth. Commun.
1994, 24t 1025), potassium carbonate (262 g, 1.9 mol) and 2,2,2-trifluoroethyl trifluoromethanesulfonate (95.8 mL, 665 mmol) in Λ/,Λ/-dimethylformamide (600 mL) was stirred at 50 0C for 30 min. Then methanol (300 ml_) was added to the mixture, and stirring was continued for 5 h at that temperature. After cooling to room temperature, the mixture was diluted with water (500 ml_) and neutralized with 2Λ/ hydrochloric acid. Product was extracted with a mixture of ethyl acetate-hexane (5:1 , 500 mL x 3). Combined organic layers were washed with water (500 mL), dried over magnesium sulfate and concentrated under reduced pressure. The residual solid was recrystallized from methanol-water to afford 125 g (79%) of the desired product as colorless crystals.
1H-NMR (CDCI3) δ: 11.47 (1 H, s), 7.36 (1 H, t, J = 8.4 Hz), 6.72 (1 H, dd, J = 1.1 , 8.4 Hz), 6.38 (1 H, q, J = 8.1 Hz), 4.36 (2 H, q, J= 8.0 Hz), 3.96 (3 H, s).
MS (ESI) m/z: 251 (M+H) +, 249 (M-H) \
Step 2. 4-(2,2,2-Trifluoroethoxy)-1 ,2-benzisoxazol-3-ol
To a solution of hydroxylamine sulfate (120 g, 732 mmol) in water (360 mL) was added potassium carbonate (121 g, 875 mmol) at 0 0C. After 30 min of stirring, sodium sulfite (3.74 g, 29.7 mmol) and a methanolic solution of methyl 2-hydroxyl-6-(2,2,2-trifluoroethoxy)benzoate (36.4 g, 146 mmol, EXAMPLE 1 , step 1 , in 360 mL of methanol) were added to the mixture. Then the mixture was warmed to 50 °C and stirred for 30 h. After cooling to room temperature, reaction mixture was partially concentrated to approx. 2/3 volume and acidified with 2Λ/ hydrochloric acid. Product was extracted three times with ethyl acetate. Combined organic layer was washed with brine, dried over sodium sulfate and concentrated under reduced pressure to afford the desired product as a crystalline solid. Crude product (36.3 g) was used for the next step without further purification.
The described above crude product (5.56 g, 22.14 mmol) was suspended in tetrahydrofuran (22.0 mL) and heated at 50 °C. 1 ,1 ‘-carbonyldiimidazole (7.54 g, 46.48 mmol) was added to the suspension at 50 °C. After addition, the mixture was stirred at 50 0C for 14 h, the mixture was cooled to room temperature. 2Λ/ hydrochloric acid was added to the mixture and extracted with ethyl acetate. The organic layer was extracted with 10% aq. potassium carbonate (100 mL x 5). The water layers were acidified with 2Λ/ hydrochloric acid and extracted with ethyl acetate (200 mL x 2). The extracts were combined and dried over sodium sulfate and concentrated in vacuo to give brown solid. The residual solid was recrystallized from ethyl acetate/hexane to give 3.21 g (61 %) of the title compound as colorless needles.
1H-NMR (CDCl3) δ: 7.53 (1 H1 1, J = 8.5 Hz), 7.14 (1 H, d, J= 8.5 Hz), 6.73 (1 H, d, J = 7.9 Hz), 4.63 (2 H, q, J= 8.0 Hz), 3.83 (1 H, br).
MS (ESI) m/z: 234 (M+H) +, 232 (M-H) “.
Step 3. rMethoxy(tetrahydro-4H-pyran-4-ylidene)methoxyKtrimethyl)silane
To a stirred solution of diisopropylamine (5.2 mL, 37 mmol) in tetrahydrofuran (15 mL) was added dropwise n-butyllithium (1.6 M in hexane, 21 mL, 34 mmol) at 0 0C and stirred for 20 min. A mixture of methyl tetrahydro-2W-pyran-4-carboxylate (4.5 g, 31 mmol) and trimethylsilyl chloride (4.3 mL, 34 mmol) was added to the mixture at -40 0C, then trimethylsilyl chloride (0.4 mL, 0.3 mmol) was added to the mixture. The mixture was stirred at room temperature for 2 h. The volatile components were removed by evaporation and the residual mixture was filtered through a pad of celite washing with hexane. The filtrate was evaporated to give 6.9 g (quant.) of the title compound as a clear yellow oil.
1H-NMR (CDCI3) δ: 3.64-3.59 (4 H, m), 3.52 (3 H, s), 2.24 (2 H, t, J = 5.6 Hz), 2.15 (2 H, t, J = 5.4 Hz), 0.22 (9 H, s).
Step 4. Methyl 4-{f4-(hvdroxymeth’vDpiperidin-1 -yllmethylltetrahvdro^rt-pyran^-carboxylate
To a stirred mixture of piperidin-4-ylmethanol (5.0 g, 43.4 mmol), f-butyldimethylsilylchloride (7.2 g, 47.8 mmol), and triethylamine (7.3 ml_, 52.1 mmol) in dichloromethane (50 mL) was added 4-dimethylaminopyridine (530 mg, 4.3 mmol) at 0 0C. After being stirred at 0 0C for 2 h, 50 mL of water was added to the mixture. The mixture was extracted with dichloromethane (50 mL x 3) and the extracts were combined, dried over sodium sulfate, and concentrated in vacuo to give 10.2 g of a crude oil. The residual oil was dissolved with 86 mL of ethanol, and potassium carbonate (7.2 g, 52.1 mmol) and paraformaldehyde (1.56 g, 52.1 mmol) were added to the solution. After being stirred at room temperature for 2 days, the mixture was filtered and the filtrate was concentrated in vacuo to give a yellow oil. The residual oil was dissolved with 45 mL of acetonitrile and magnesium chloride (414 mg, 4.3 mmol) was added to the solution. [methoxy(tetrahydro-4H-pyran-4-ylidene)methoxy](trimethyl)silane (11.3 g, 52.1 mmol, EXAMPLE 1 , step 3) was added to the mixture at 0 0C. After being stirred at 0 0C for 20 h, 100 mL of 2Λ/ hydrochloric acid was added to the mixture. The mixture was stirred for 30 min and washed with diethyl ether (100 mL x 2). The water layer was neutralized with aq. ammonia and extracted with ethyl acetate (100 mL x 2). The extracts were combined and dried over sodium sulfate and concentrated in vacuo to give a yellow oil. The residual oil was purified by silica gel column chromatography (dichloromethane/methanol/aq. ammonia 400: 10: 1 ) to give 6.8 g (41%) of the title compound as a colorless waxy solid.
1H-NMR (CDCI3) δ: 3.75-3.90 (2 H, m), 3.71 (3 H, s), 3.40-3.55 (4 H, m), 2.73 (2 H, m), 2.49 (2 H, m), 2.10-2.25 (2 H, m), 1.95-2.10 (2 H, m), 1.50-1.70 (4 H, m), 1.30-1.50 (2 H, m), 1.10-1.30 (2 H, m).
MS (ESI) m/z: 272 (M+H) +.
Step 5. Methyl 4-{r4-((r4-(2,2,2-trifluoroethoxy)-1 ,2-benzisoxazol-3-vπoxy)methyl)piperidin-1 -yllmethyll-tetrahydro-2H-pyran-4-carboxylate
A mixture of 4-(2,2,2-trifluoroethoxy)-1 ,2-benzisoxazol-3-ol (230 mg, 1 mmol, EXAMPLE 1 , step
2), methyl 4-{[4-(hydroxymethyl)piperidin-1 -yl]methyl}tetrahydro-2/-/-pyran-4-carboxylate (270 mg, 1 mmol, EXAMPLE 1 , step 4), and cyanomethyltributylphosphorane (400 mg, 1.5 mmol) in toluene (1.0 mL) was stirred at 100 0C for 16 h. After cooling, the mixture was concentrated in vacuo to give a dark brown oil. The residual oil was purified by silica gel column chromatography (hexane/ethyl acetate 2 : 1 ) to give 250 mg (51 %) of the title compound as a white solid.
1H-NMR (CDCl3) δ: 7.44 (1 H, dd, J= 7.9, 8.4 Hz), 7.12 (1 H, d, J= 8.4 Hz), 6.61 (1 H, d, J= 7.9 Hz), 4.49 (2 H, q, J= 8.1 Hz), 4.24 (2 H, d, J= 6.4 Hz), 3.88-3.78 (2 H, m), 3.72 (3 H, s), 3.54-3.41 (2 H, m), 2.83-2.71 (2 H, m), 2.52 (2 H, s), 2.35-1.29 (11 H, m).
MS (ESI) m/z: 487 (M+H) +.
Step 6. 4-(r4-(ir4-(2,2,2-Trifluoroethoxy)-1 ,2-benzisoxazol-3-vπoxy)methyl)piperidin-1 -ylimethylltetrahydro-2H-pyran-4-carboxylic acid
A mixture of methyl 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1 ,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2/+pyran-4-carboxylate (89 mg, 0.18 mmol, EXAMPLE 1 , Step 5) in tetrahydrofuran (1 mL), methanol (1 ml_) and 2 Λ/ aq. sodium hydroxide (1 ml_) was stirred at 70 °C for 17 h. The mixture was neutralized with 2 N hydrochloric acid (1 mL) and formed precipitate was filtered.
The precipitate was triturated with diethylether to give 50 mg (58%) of the title compound as a white solid.
1H-NMR (DMSO-d6) δ: 7.59 (1 H1 dd, J= 8.1 , 8.4 Hz), 7.25 (1 H, d, J = 8.4 Hz), 6.94 (1 H, d, J = 8.1 Hz), 4.93 (2 H, q, J= 8.7 Hz), 4.19 (2 H, d, J= 5.9 Hz), 3.75-3.62 (2 H, m), 3.48-3.30 (2 H, m), 2.90-2.74 (2 H, m), 2.50 (2 H, s), 2.29-2.13 (2 H, m), 1.94-1.23 (9 H, m).
A signal due to CO2H was not observed.
MS (ESI) m/z: 473 (M+H) +, 471 (M-H) “.
m.p.: 171.7 °C.
IR (KBr) v: 2950, 1617, 1527, 1188, 1113 cm”1.
Anal. Calcd for C22H27N2O6F3: C, 55.93; H, 5.76; N, 5.93. Found: C, 55.72; H, 5.78; N, 5.80.
PATENT
WO2015174098
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015174098
PATENT
WO2014080633
http://www.google.com/patents/WO2014080633A1?cl=en
PATENT
WO 2015178020
The present invention relates to novel salts of 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid. More particularly, the invention relates to salt forms (HCl-salt, HBr-salt, p-toluenesulfonate salt and ethanedisulfonate salt), and to processes for the preparation of, compositions containing and to uses of, such salt forms.
EXAMPLE 1
Preparation of
4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}tetrahydro-2H-pyran-4-carboxylic acid according to the conventional process
A slurry of 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]-methyl}tetrahydro-2H-pyran-4-carboxylic acid (1.326 kg, 2.807 mol, a white solid) in ethyl acetate (18.564 L) is dissolved at 70 oC. The solution is cooled to 64 oC during 35 min and 200 mg of seed crystal (0.423 mmol) is seeded to the mixture. The mixture is cooled to 40 oC over 5 h period and stirred at this temperature for 14.5 h. The slurry is gradually cooled to 19 oC during 6 h period and the mixture is stirred at this temperature for 46 h. The formed precipitate is collected by filtration and the filter cake is washed with 2.0 L of ethyl acetate. The filter cake is dried under reduced pressure at 50 oC to afford 1.140 kg of the desired crystalline form of 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]-
methyl}tetrahydro-2H-pyran-4-carboxylic acid (86%).
1H-NMR (DMSO-d6) delta: 7.59 (1 H, dd, J = 8.1, 8.4 Hz), 7.25 (1 H, d, J = 8.4 Hz), 6.94 (1 H, d, J = 8.1 Hz), 4.93 (2 H, q, J = 8.7 Hz), 4.19 (2 H, d, J = 5.9 Hz), 3.75-3.62 (2 H, m), 3.48-3.30 (2 H, m), 2.90-2.74 (2 H, m), 2.50 (2 H, s), 2.29-2.13 (2 H, m), 1.94-1.23 (9 H, m).
A signal due to CO2H is not observed.
m.p. (DSC onset): 169 oC.
The temperature has a margin of error of +/- 1 oC.
Crystallinity by PXRD: Crystal (Figure 1): Main peaks at 2-Theta: 5.9, 9.3, 9.8, 11.9, 13.7, 14.3, 15.0, 17.8, 18.2-19.3, 19.7, 22.6, 23.4-24.5 and 24.9 (o ). Each peak has a margin of error of +/- 0.2.
IR nu (diffuse reflection) (Figure 6): 4389-4383, 3426, 2943-2937, 2120, 1904, 1724, 1614, 1535, 1508, 1437, 1420, 1287, 1261, 1221, 1180, 1121, 1094, 1059, 1022, 991, 974, 957, 934, 918, 868, 827, 783, 746, 731, 654, 638, 615, 588, 554, 542 and 507 cm-1. Each peak has a margin of error of +/- 2 cm-1.
Anal. Calcd for C22H27N2O6F3: C, 55.93; H, 5.76; N, 5.93. Found: C, 55.76; H, 5.74; N, 5.85.
PATENT
WO2012/157288
http://www.google.co.in/patents/WO2012157288A1?cl=pt-PT
EXAMPLE 1
Preparation of
4-{[4-({[4-(2,2,2-Trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}-tetrahydro-2H-pyran-4-carboxylic acid according to the conventional process
A mixture of methyl 4-{[4-({[4-(2,2,2-trifluoroethoxy)-1,2-benzisoxazol-3-yl]oxy}methyl)piperidin-1-yl]methyl}-tetrahydro-2H-pyran-4-carboxylate (89 mg, 0.18 mmol, PCT WO2006090224 EXAMPLE 1, Step 5) in tetrahydrofuran (1 mL), methanol (1 mL) and 2 N aq. sodium hydroxide (1 mL) is stirred at 70 oC for 17 h. The mixture is neutralized with 2 N hydrochloric acid (1 mL) and formed precipitate is filtered. The precipitate is triturated with diethylether to give 50 mg (58%) of the title compound as a white solid.
1H-NMR (DMSO-d6) delta: 7.59 (1 H, dd, J = 8.1, 8.4 Hz), 7.25 (1 H, d, J = 8.4 Hz), 6.94 (1 H, d, J = 8.1 Hz), 4.93 (2 H, q, J = 8.7 Hz), 4.19 (2 H, d, J = 5.9 Hz), 3.75-3.62 (2 H, m), 3.48-3.30 (2 H, m), 2.90-2.74 (2 H, m), 2.50 (2 H, s), 2.29-2.13 (2 H, m), 1.94-1.23 (9 H, m).
A signal due to CO2H is not observed.
MS (ESI) m/z: 473 (M+H) +, 471 (M-H) –.
Anal. Calcd for C22H27N2O6F3: C, 55.93; H, 5.76; N, 5.93. Found: C, 55.72; H, 5.78; N, 5.80.
| Patent | Submitted | Granted |
|---|---|---|
| Benzisoxazole Derivatives [US2008207690] | 2008-08-28 | |
| 5-HT4 Receptor Agonist as a Prokinetic Agent [US2014051726] | 2012-03-23 | 2014-02-20 |
| Polymorph Form of 4-methyl)piperidin-1-yl]methyl}-tetrahydro-2H-pyran-4-carboxylic acid [US2014187583] | 2012-05-18 | 2014-07-03 |
see……….http://apisynthesisint.blogspot.in/2015/12/rq-00000010-for-treatment-of-gerd.html
/////c12c(cccc1onc2OCC3CCN(CC3)CC4(CCOCC4)C(=O)O)OCC(F)(F)F
C1CN(CCC1COC2=NOC3=C2C(=CC=C3)OCC(F)(F)F)CC4(CCOCC4)C(=O)O
Lefucoxib (乐福昔布)

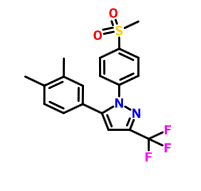
Lefucoxib (乐福昔布)
5-(3,4-dimethyl-phenyl)-1-methanesulfonyl-3-trifluoromethol-pyrazole
1 [4- (methylsulfonyl) phenyl] -3-trifluoromethyl-5- (3,4-dimethylphenyl) – pyrazole
CAS 849048-84-6
![]()
| Molecular Formula: | C19H17F3N2O2S |
|---|---|
| Molecular Weight: | 394.41069 g/mol |
IND FILED
Prostaglandin G/H Synthase 2 (PTGS2; COX-2) Inhibitors
A COX-2 inhibitor potentially for the treatment of rheumatoid arthritis.
cyclooxygenase-2 (COX-2) inhibitor
National Center of Biomedical Analysis
![]()
 CHINA FLAG
CHINA FLAG

PATENT
CN 1468854
http://www.google.com/patents/CN1468854A?cl=en
Example 1
1 [4- (methylsulfonyl) phenyl] -3-trifluoromethyl-5- (3,4-dimethylphenyl) – pyrazole (I1)
1- (3,4- two toluene-yl) -4,4,4-trifluoro-methyl – D-1,3-dione (IV1) of sodium metal was weighed 2.3g (0.1mol) was added 50ml of anhydrous toluene to prepare a sodium sand. After cooling, ethanol was added dropwise 12ml, and then heated at 60 ℃, complete reaction of sodium metal. After cooling to room temperature, was added 3,4-dimethylphenyl ethanone 23.8g (0.1mol) and trifluoroacetic ethyl acetate 20ml (0.2mol), reacted at 100 ℃ 5 hours. Toluene was distilled off under reduced pressure, a 10% aqueous hydrochloric acid was added, the pH was adjusted to 2-3, extracted with ethyl acetate, washed with water, dried over anhydrous MgSO4, ethyl acetate was distilled off under reduced pressure. Then under reduced pressure, distillation, collecting fractions 105-107 ℃ / 0.7mmHg, was 14.6g, 60% yield.
1- [4- (methylsulfonyl) phenyl] -3-trifluoromethyl-5- (3,4-dimethylphenyl) – pyrazole (I1) take the above-prepared substituted (IV1) 2.38g (0.01mol ), 15ml of ethanol, then added p-methanesulfonyl phenyl hydrazine salt alkoxide 2.3g (0.01ml). Was refluxed for 15 hours. Place the refrigerator overnight, the crystals were collected by filtration, recrystallized from ethanol, mp 129-31 ℃, to give 3.1 g.
Elemental analysis: C19H17F3N2O2S Calculated: C, 57.86; H, 4.34; N, 7.10 Found: C, 57.97; H, 4.29; N, 7.20MS (m / z): 395 (M + 1)

References
Cheng, Feixiong, Edited by Lee, Philip W, From Handbook of Metabolic Pathways of Xenobiotics (2014), 4, 1655-1656
Bi, X.; Meng, Z.; Chen, H.; Zhu, X.; Dou, G.
In vivo and in vitro metabolism of lefucoxib in rats, J Pharm Biomed Anal. 2008 Sep 10;48(1):134-9. doi: 10.1016/j.jpba.2008.04.024. Epub 2008 Apr 30.
Bi, X.; Meng, Z.; Dou, G. Determination of lefucoxib in rat plasma, urine, and feces by high-performance liquid chromatography with fluorescence detection: Application in pharmacokinetic studies
J Chromatogr B Anal Technol Biomed Life Sci 2007, 850(1-2): 199
Talanta (2011), 85(1), 8-27
Jiefangjun Yaoxue Xuebao (2009), 25(6), 496-498.
Yaowu Fenxi Zazhi (2006), 26(9), 1222-1224.
Zhongguo Yaolixue Yu Dulixue Zazhi (2007), 21(2), 147-151.
| CN101497585B | Jan 31, 2008 | Jan 12, 2011 | 中国科学院理化技术研究所 | Method for photocatalytic synthesis of 1,3,5-trisubstituted-2-pyrazole derivative |

 .
.
//////////c1c(ccc(c1C)C)c2n(nc(c2)C(F)(F)F)c3ccc(cc3)S(=O)(=O)C
CC1=C(C=C(C=C1)C2=CC(=NN2C3=CC=C(C=C3)S(=O)(=O)C)C(F)(F)F)C
DRL 17822 from Reddy US Therapeutics/Dr Reddy’s



CAS 920493-71-6 and CAS 898911-09-6



DRL 17822
MW 603.6045, MFC30 H31 F6 N7
| Molecular Formula: | C30H31F6N7 |
|---|---|
| Molecular Weight: | 603.604459 g/mol |
Cas 898911-09-6, 1454689-50-9
3-([[3,5-Bis(trifluoromethyl)benzyl](2-methyl-2H-tetrazol-5-yl)amino]methyl)-N,N-bis(cyclopropylmethyl)-8-methylquinolin-2-amine
3-Quinolinemethanamine, 2-[bis(cyclopropylmethyl)amino]-N-[[3,5-bis(trifluoromethyl)phenyl]methyl]-8-methyl-N-(2-methyl-2H-tetrazol-5-yl)-
3-(((3,5-bis(trifluoromethyl)benzyl)(2- methyl-2H-tetrazol-5-yl)amino)methyl)-N,N-bis(cyclopropylmethyl)-8- methylquinolin-2-amine
(3-{ [3,5-bis-trifluoromethyl-benzyl)-(2-methyl-2H-tetrazoIe-5-yl)- amino]-methyl}-8-methyl-quinolin-2-yl)-bis-cyclopropylmethyl-amine
Reddy US Therapeutics (Innovator)




Treatment of Atherosclerosis Therapy Lipoprotein Disorders,
CETP inhibitor (dyslipidemia/atherosclerosis/cardiovascular diseases), Dr Reddy’s
Selective inhibitor of cholesteryl ester transfer protein (CETP)
- 30 Jun 2012Dr Reddy’s Laboratories completes a phase II trial in Hypercholesterolaemia in Italy, Poland and Ukraine (NCT01388816)
- 09 Mar 2012Dr Reddy’s Laboratories completes enrolment in its phase II trial for Hypercholesterolaemia in Italy, Poland, and Ukraine (NCT01388816)
- 02 Sep 2011Phase-II clinical trials in Hypercholesterolaemia in Ukraine (PO)
CLINICAL TRIALS…..Type II Hyperlipidemia PHASE 2…………https://clinicaltrials.gov/ct2/show/NCT01388816
Cardiovascular disease is a leading cause of death worldwide. Among cardiovascular disorders, coronary heart disease (CHD) caused by atherosclerosis is the most common cause of morbidity and mortality. Prevention, stabilization and regression of atherosclerotic plaques may have a major impact on reducing the risk of acute coronary events.
LDL-C lowering agents, primarily the statins, are the current mainstay in the pharmacologic management of dyslipidemia. However even with stain use, residual CHD risk from dyslipidemia remains. Epidemiologic and observational studies have shown that HDL-C is also a strong independent predictor of CHD, suggesting that raising HDL-C levels might afford clinical benefit in the reduction of cardiovascular risk.
Presently only niacin is approved by the FDA for HDL-C elevation and can raise HDL-C levels by 20-30%. However its use can be limited by a high incidence of flushing and, less commonly, by elevation of blood glucose and potential hepatic toxicity.
Cholesteryl ester transfer protein (CETP) inhibitors are being explored for their ability to elevate HDL-C. A small molecule CETP inhibitor, torcetrapib, has been demonstrated to elevate HDL-C by 60-100%. However, a large clinical trial (ILLUMINATE) where it increased HDL-C by a mean of 72% compared to baseline was halted as it failed to show benefit. Post-hoc analysis of this study implicated an off-target increase in blood pressure as potentially counteracting any anti-atherosclerotic benefits. Post-hoc subgroup analysis showed that patients in the highest HDL-C quartile had a 57% reduction in the risk of cardiovascular events.
Increased blood pressure appears to be specifically related to torcetrapib as two other small molecule CETP inhibitors, anacetrapib and dalcetrapib, have not shown this in clinical trials and have been well tolerated. DRL-17822 has also not shown elevation of blood pressure in either animals or in normal volunteers.
This study will investigate the efficacy and tolerability of DRL-17822 as dyslipidemia monotherapy in patients with Type II hyperlipidemia.
Hyperlipidemia or an elevation in serum lipids is associated with an increase incidence of cardiovascular disease and atherosclerosis. Primary hyperlipidemia is a term used to describe a defect in lipoprotein metabolism. The lipoproteins commonly affected are low density lipoprotein (LDL) cholesterol, which transports mainly cholesterol, and very low density lipoprotein-cholesterol (VLDL-cholesterol), which transports mainly triglycerides (TG). Most subjects with hyperlipidemia have a defect in LDL metabolism, characterized by raised cholesterol, LDL-C levels, with or without raised triglyceride levels; such subjects are termed hypercholesterolemic (Fredrickson Type II). Familial hypercholesterolemia (FH) is caused by any one of a number of genetically-determined defects in the LDL receptor, which is important for the entry of cholesterol into cells. The condition is characterized by a reduced number of functional LDL receptors, and is therefore associated with raised serum LDL-C levels due to an increase in LDL.

It is reasonably known in the art that the likelihood of cardiovascular disease can be decreased, if the serum lipids, and in particular LDL-C, can be reduced. It is further known that the progression of atherosclerosis can be retarded or the regression of atherosclerosis can be induced if serum lipids can be lowered. In such cases, individuals diagnosed with hyperlipidemia or hypercholesteremia should consider lipid-lowering therapy to retard the progression or induce the regression of atherosclerosis for purposes of reducing their risk of cardiovascular disease, and in particular coronary artery disease.
Cholesteryl ester-transfer protein (CETP) is an important player in metabolism of lipoproteins, such as, for example, a high density lipoprotein (HDL). CETP is a 70 kDa plasma glycoprotein that is physically associated with HDL particles. It facilitates the transport of cholesteryl ester from HDL to apolipoprotein B-containing lipoproteins. This transfer is accompanied by transfer of triglycerides in the opposite direction. Thus, a decrease in CETP activity can result in an increase in the level of HDL cholesterol and a decrease in the level of very low density lipoprotein (VLDL) and low density lipoprotein (LDL). CETP can therefore simultaneously affect the concentrations of pro-atherogenic (for example, LDL) and anti-atherogenic (for example, HDL) lipoproteins.
Several CETP inhibitors are currently in various clinical phases of development for treating various aforementioned disorders. In spite of having various advantages, CETP inhibitors are proven to be difficult to formulate for oral administration. CETP inhibitors are of a highly lipophilic nature and have extremely low solubility in water. Due to their poor solubility, bioavailability of conventional oral compositions is very poor. The lipophilic nature of CETP inhibitors not only leads to low solubility but also tends to poor wettability, further reducing their tendency to be absorbed from the gastrointestinal tract. In addition to the low solubility, CETP inhibitors also tend to have significant, “food effect”, where a significant difference in rate and amount of drug absorption is observed when the drug is administered with or without a meal. This “food effect”, often complicates the dosing regimen and may require high dosing to achieve the desired therapeutic effect, resulting in potentially unwanted side effects.
Several attempts have been made to improve the solubility of CETP inhibitors, but have generally ended up with limited success. At the outset, most methods aimed at enhancing aqueous concentration and bioavailability of low-solubility drugs only offer moderate improvements. References describing improving the dissolution of poorly soluble drugs include: U.S. Patent Nos. 5,456,923, 5,993,858, 6,057,289, 6,096,338, 6,267,985, 6,280,770, 6,436,430, 6,451,339, 6,531,139, 6,555,558, 6,638,522, 6,962,931 and 7,374,779.
PATENT
WO 2014128564
https://www.google.co.in/patents/WO2014128564A2?cl=en
WO-2014076568
http://www.google.com/patents/WO2014076568A2?cl=en
EXAMPLES
In the following Examples 1-17, various compositions in accordance with the present application were prepared comprising 3-(((3,5-bis(trifluoromethyl)benzyl)(2- methyl-2H-tetrazol-5-yl)amino)methyl)-N,N-bis(cyclopropylmethyl)-8- methylquinolin-2-amine as the CETP inhibitor.:
EXAMPLE 1 :

1. 3-(((3,5-bis(trifluoromethyl)benzyl)(2-methyl-2H-tetrazol-5-yl)amino)methyl)- N,N-bis(cyclopropylmethyl)-8-methylquinolin-2-amineand hydroxypropyl methyl cellulose acetate succinate were mixed together in given solvent mixture to form clear solution.
2. To the solution of step I, Polyoxyl 35 castor oil and talc were added to form a homogenous suspension.
3. The suspension of step 2 was sprayed over inert sugar spheres and dried.
4. The drug layered spheres of step 3 were coated with dispersion made from given seal layer ingredients.
5. The coated spheres of step 4 were formulated further as capsule dosage form.
PATENT
WO 2013046045
https://www.google.co.in/patents/WO2013046045A1?cl=en
PATENT
WO 2013024358
PATENT
WO 2007075194
https://www.google.co.in/patents/WO2007075194A1?cl=en
Syntheis construction





Example 1
Synthesis of (3-{[3,5-bis trifluoromethyl-benzyl )-(2-cyclopropyImethyI-2H- tetrazole -5-yl)-amino]-methyl-}-8-methyI-quinolme-2-yl)-bis- cyclopropylmethyl-amine Step (i): Synthesis of 2~chloro-8-methyl-quinoline-3-carbaldehyde
DMF (1.22 g, 16.7 mmol) was taken in a flask equipped with a drying tube and POCl3 (7.32 g, 46.7 mmol) was added dropwise with stirring at 0° C. To this solution, TV-o-Tolyl acetamide (1.00 g, 6.7 mmol) was added and the solution was refluxed for 6 h at 90° C. The excess POCl3 was distilled off, water was added to the residue and this was stirred at room temperature for 10 min. The solid was filtered and dried under vacuum..This crude compound was purified over silica gel (100-200 mesh) using 6% ethyl acetate and petroleum ether to give the product as a yellowish solid (yield: 78%). 1H NMR (CDCl3, 200 MHz): δ 10.5 (s, IH)5 8.71 (s, IH), 7.83- 7.79 (m, IH), 7.74- 7.70 (m, IH), 7.56-7.49 (m, IH), 2.79 (s, 3H); m/z (EI-MS): 206 (M+, 100%). Step (ϋ): Synthesis of 2-(bis(cyclopropylmethyl)amino)-8-methylquinoline-3- carbaldehyde:
2-Chloro-8-methyl-quinoline-3-carbaldehyde (.115 g, 0.559 mmol), and potassium carbonate (0.231 g, 1.67 mmol) were put in a 25 mL two necked RB flask. To this, 3 mL of DMF was added followed by dropwise addition of bis- cyclopropylmethyl amine (0.083 g, 0.67 mmol). The reaction mixture was refluxed for 2 h and was cooled to RT. It was then poured on crushed ice (10 mL) and extracted with EtOAc (3 x 10 mL). The organic layer was washed with brine and dried over sodium sulphate. The solvent was evaporated under vacuum to give a yellow colored oil (0.081 g, 50%).
1H NMR (CDCl3, 400 MHz): δ 10.5 (s, IH), 8.71 (s, IH), 7.83- 7.79 (m, IH),
7.74-7.70 (m, IH), 7.56-7.49 (m, IH), 3.55-3.47 (m, 4H), 2.79 (s, 3H), 1.73-1.72
(m, 2H), 1.70-1.46 (m, 4H), 1.20-1.11 (m, 4H); m/z (ES-MS ): 295 (M+H-I5
100%); IR (neat, cm“1): 3385, 2948, 1691.
Step (iii): Synthesis of 3-((3,5-bis(trifluoromethyl)benzylamino)methyl)-N,N- bis(cyclopropylmethyl)-8-methylquinolin-2-amine
2-(Bis(cyclopropylmethyl)amino)-8-methylquinoline-3-carbaldehyde (0.081 g, 0.39 mmol), 3,5-bis-trifluoromethylbenzylamine (0.096 g, 0.39 mmol) and acetic acid (0.047 g, 0.78 mmol) were put in a 25 mL RB flask. To this, 2 rnL of methanol was added and stirred at RT for 15 min. Sodium cyanoborohydri.de (0.075 g, 0.77 mmol) was added portionwise and stirring was continued at RT for another 1 h. Methanol was removed from the reaction mixture under vacuum, water was added to this crude and was extracted with ethyl acetate (3 x 50 mL). The organic layer was washed with saturated NaHCO3 solution, brine and dried over sodium sulphate. The solvent was evaporated and the crude residue was purified by column chromatography over silica gel (100-200 mesh) eluting with 4% ethyl acetate in petroleum ether to give the title amine (0.142 g, yield: 99%). 1R NMR (CDCl3, 400 MHz): δ 7.89-7.86 (m, IH), 7.80 (m, IH), 7.75-7.74 (m, IH), 7.60-7.40 (m, 3H), 7.30-7.26 (m,lH), 4.12 (s, 2H), 3.88 (s, 2H), 3.24-3.22 (m, 4H), 2.72 (s, 3H), 0.99-0.92 (m, 2H), 0.44-0.35 (m, 4H), 0.11-0.05 (m, 4H); m/z (EI-MS ): 522 (M++l, 100%); IR (neat, cm“1): 3357, 2929, 2851.
Step (iv): Synthesis of N-(3,5-bis(trifluoromethyl)benzyl)-N-((2- (bis(cyclopropylmethyl)amino)-8-methylqumolin-3-yl)methyl)cyanamide
To a solution of 3-((3,5~bis(trifluoromethyl)benzylamino)methyl)-N,N- bis(cyclopropylmethyl)-8-methylquinolin-2-amine (0.176 g , 0.33 mmol ), obtained in step (iii) , in MeOH (4 mL) under N2 atmosphere was added sodium bicarbonate (0.056 g, 0.67 mmol ) followed by the addition of cyanogen bromide (0.063 g, 0.60 mmol). The reaction mixture was stirred at RT for 2 h. The solvent was removed under vacuum to give the crude residue which was dissolved in water, extracted with ethyl acetate and dried over sodium sulphate. The solvent was evaporated and concentrated in vacuo to afford N-(3,5-bis(trifluoromethyl)benzyl)- N-((2-(bis(cyclopropylmethyl)amino)-8-methylquinolin-3-yl)methyl)cyanamide (0.118 g, 64%).
1H NMR (CDCl3, 400 MHz ): δ 8.07 (s, IH) , 7.82 (s, IH), 7.70 (s, 2H), 7.56-7.55 (m, IH), 7.50-7.49 (m, IH), 4.49 (s, 2H), 4.23 (s, 2H), 3.17 -3.15 (m, 4H), 2.71 (s, 3H), 0.097-0.085 (m, 2H), 0.405-0.401 (m, 4H), 0.385-0.381 (m, 4H); m/z (ES- MS): 547 (M++l, 100%); IR(KBr ,Cm“1 ) : 2273, 1280.
Step (v): Synthesis of (3-{[(3,5-bistrifluoromethyl-benzyl)-(2H-tetrazol-5-yl)- amino]-methyl}-8-methyl-quinolin-2-yl)-bis-cyclopropylmethyl-amine
7V-(3,5-Bis(tiifluoromethyl)benzyl)-N-((2-(bis(cyclopropylmethyl)amino)- 8-methylqumolin-3-yl)methyl)cyanamide (0.118 g, 0.216 mmol), sodium azide (0.70 g 1.08 mmol) and ammonium chloride (.058 g, 1.08 mmol) were put in a RB flask under N2atmosphere. To this reaction mixture, DMF (2 mL) was added and was refluxed for 1 h. The reaction mixture was cooled to RT and ice was added to this and extracted with ethylacetate (3×10 mL). The combined organic layer was washed with brine, dried over sodium sulphate and then concentrated under vacuum to afford of (3-{[(3,5-bistrifluoromethyl-benzyl)-(2H-tetrazol-5-yl)- amino]-methyl}-8-methyl-quinolin-2-yl)-bis-cyclopropylmethyl-amine as a yellow solid (0.125 g, 99%).
1H NMR (CDCl3, 400 MHz ): δ 7.99 (s, IH) , 7.79 -7.74 (m, 4H ), 7.41-7.40 (m,
IH ), 7.33-7.31 (m, IH), 4.99 (s, 2H), 4.80 (s, 2H), 3.68 (s, 4H), 2.16 (s, IH) 1.56-
1.06 (m, HH); m/z (ES-MS): 578 (M++l, 100%); IR (KBr , cm“1) 3680 , 2922 ,
1660 , 1616.
METHYLATION SHOULD GIVE THE PRODUCT
Scheme 1


PATENT
WO 2006073973
http://www.google.co.in/patents/WO2006073973A2?cl=en
Example 47
Synthesis of [2-(bis-cycIopropylmethyI-amino)-8-methyl-quinolin-3-ylmethyI]-(3,5- bis-trifluoromethyl-benzyl)-carbamic acid methyl ester
Step (i): Synthesis of bis-cyclopropylmethyl-amine
(i) a. Synthesis of cyclopropanecarboxylic acid cyclopropylmethyl-amide:

Cyclopropyl carboxylic acid (1.0 g, 11.63 mmol) was added to a 50 mL two neck round bottom flask, along with DCM (25 mL). This mixture was cooled to 0° C, EDCI (4.15 g, 13.95 mmol) was added portionwise to the mixture with stirring under nitrogen atmosphere, and the temperature was maintained for 0.5 h. After this time, hydroxybenzotriazole (1.88 g, 13.95 mmol) was added to the 0° C mixture which was stirred for 10 min, then triethylamine (1.7 g, 11.63 mmol) was added, and stirring of the mixture was continued at the same temperature for another 0.5 h. Then, cyclopropylmethylamine (0.825 g, 11.63 mmol) was added, and the reaction was allowed to reach RT, and stirring was continued overnight. The solvent was then removed in vacuo, and the crude residue was purified by passing through a column over 60-120 silica gel, eluting with dichloromethane, to afford the title compound (1.6 g), yield: 87%. 1H NMR (CDCl3, 200 MHz): d 5.75 (br s, NH, D2O exchangeable), 3.17-3.16 (m, 2H), 1.00-0.80 (m, 4H), 0.77-0.67 (m, 2H), 0.56-0.43 (m, 2H), 0.24-0.16 (m, 2H) m/z (CI-MS): 139 (M+, 100%) (i) b. Synthesis of bis-cyclopropylmethyl-amine

To a suspension of lithium aluminum hydride (1.3 g, 9.35mmol) in 10 mL dry ether, a solution of N-cyclopentenoyl-ethylamine (1.7 g, 13.3 mmol) in dry ether (10 mL) was added under a nitrogen atmosphere. This reaction was stirred at RT for 8 h and the reaction mixture was then quenched with saturated sodium sulfate solution, filtered, and the precipitate was washed with diethyl ether. The filtrate was concentrated to afford the title amine (0.8 g), yield: 69%.
1H NMR (CDCl3, 200 MHz): d 5.75 (br s, NH, D2O exchangeable), 3.16-3.09 (m, 2H), 2.50-2.4 (m, 2H), 0.56-0.43 (m, 4H), 0.24-0.21 (m, 3H), 0.21-0.13 (m, 3H) m/z (ES-MS): 139 (M^+14, 100%)
Step (ii): Synthesis of [2-(bis-cyclopropylmethyl-amino)-8-methyl-quinolin-3-ylmethyl]- (S^-bis-trifluoromethyl-benzy^-carbamicacid methyl ester

The title compound was synthesized by using the same procedure as in Example 35, except using o-tolyl acetanilide in step (i) instead of acetanilide and bis- cyclopropylmethyl amine in step (iii), which yielded the desired product as a light yellow, viscous liquid (0.05 g), yield:40%, of purity 98.8% (HPLC: Symmetry Shield RP8, [0.01M KH2PO4: CH3CN], 217 nM, Rt12.719 min).
1H NMR (CDCl3, 400 MHz): d 7.7 (s, IH), 7.68-7.44 (m, 3H), 7.27-7.24 (m, 2H), 4.78- 4.65 (m, 2H), 4.47-4.4 (m, 2H), 3.8 (s, 3H), 3.16-3.14 (d, J=7Hz, 2H), 2.7 (s, 3H), 1.55 (s, 3H), 1.01-0.9(m, IH), 0.38-0.34 (m, 4H), 0.07-0.05 (m, 4H); m/z (CI-MS): 579 (M+, 100%)
Example 57
Synthesis of (3-{ [3,5-bis-trifluoromethyl-benzyl)-(2-methyl-2H-tetrazoIe-5-yl)- amino]-methyl}-8-methyl-quinolin-2-yl)-bis-cyclopropylmethyl-amine
The title compound was prepared as an oil by following the same synthetic procedures as in Example 52, except using {3-[(3,5-bis-trifluoromethyl-benzylamino)- methyl]-8-methyl-quinolin-2-yl}-bis-cyclopropylmethyl-amine in step (i) instead of {3- [(3,5-bis-trifluoromethyl-benzylamino)-methyl]-quinolin-2-yl}-cyclopentylmethyl-ethyl- amine (0.07 g), yield: 52%.
Purity: 95.53% (HPLC: Symmetry Shield RP8, [0.01M KH2PO4: CH3CN], 217 nM, Rt 9.538 min).
IR (neat, cm4) 3079, 2925, 1582;
1H NMR (CDCl3, 400 MHz): d 7.82 (s, IH), 7.69-7.67 (m, 2H), 7.44-7.41 (m, IH), 7.23- 7.2 (m, 3H), 4.91 (s, 2H), 4.65 (s, 2H), 4.21 (s, 3H), 3.29 -3.19 (m, 4H)5 2.71 (s, 3H), 1.01-1.00 (m, 2H), 0.99-0.83 (m, 2H), 0.39-0.34 (m, 3H), 0.08-0.07 (m, 3H). m/z (ES-MS): 604 (M++!, 100%)
Dr. Reddy’s announces start of Phase II study with the CETP inhibitor, DRL-17822 in dyslipidemia patients
Hyderabad, India, September 02, 2011: Dr Reddy’s Laboratories (NYSE: RDY) announced the initiation of dosing with DRL-17822 in patients with diagnosis of type II dyslipidaemia. DRL-17822, is a selective, orally bioavailable inhibitor of cholesteryl ester transfer protein (CETP), for the treatment and/or prevention of dyslipidaemia, atherosclerosis and associated cardiovascular disease.
The current study is being conducted under a CTA in a number of countries in Europe. The objective of the study is to evaluate the efficacy and safety of DRL-17822 in patients with Type-II dyslipidemia. This is a randomized, double blind, placebo controlled, parallel group study in 160 subjects. The primary outcome measure is to assess the elevation in HDL cholesterol and reduction in LDL cholesterol from baseline to end of treatment compared to placebo. Three doses (50, 150 & 300 mg) of DRL-17822 given once daily for 4 weeks will be evaluated during this study.
Three human Phase I studies with DRL-17822 had already been conducted in Europe, where DRL-17822 was shown to be safe and well tolerated. In these studies, the proof of mechanism had been demonstrated by dose-dependent inhibition of plasma CETP activity as well as by significant increase in HDL cholesterol & decrease in LDL cholesterol levels.
Cardiovascular disease is a leading cause of death among men and women worldwide. Among cardiovascular disorders, coronary heart disease (CHD), caused by atherosclerosis is the most common cause of morbidity and mortality. Stabilization and/or regression of atherosclerotic plaques may have a major impact on reducing the risk of acute coronary events. Low-density lipoprotein cholesterol lowering agents, primarily the statins, are the current mainstay in the pharmacological management of dyslipidaemia. However, significant residual cardiovascular risk remains despite use of statins.
Epidemiological and observational studies demonstrate that reduced high density lipoprotein cholesterol levels are a strong, independent predictor of CHD, suggesting that raising HDL cholesterol levels might afford clinical benefit in the reduction of cardiovascular risk. One approach to raise HDL level has been inhibition of CETP activity. Currently it is believed that, raising HDL cholesterol and lowering LDL cholesterol through CETP inhibition would lead to a significant benefit in terms of CHD risk reduction.
Dr. K. Anji Reddy, Founder Chairman, Dr. Reddy’s Laboratories added, “We are committed to delivering products of differentiated value in this area of high global clinical unmet need. We are excited to continue to advance our CETP program and look forward to the data from our Phase II study. This class of therapy could transform the treatment of CHD and DRL 17822 is in a position to be one of the front-running products in the class”.
Disclaimer
This press release includes forward-looking statements, as defined in the U.S. Private Securities Litigation Reform Act of 1995. We have based these forward-looking statements on our current expectations and projections about future events. Such statements involve known and unknown risks, uncertainties and other factors that may cause actual results to differ materially. Such factors include, but are not limited to, changes in local and global economic conditions, our ability to successfully implement our strategy, the market acceptance of and demand for our products, our growth and expansion, technological change and our exposure to market risks. By their nature, these expectations and projections are only estimates and could be materially different from actual results in the future.
About Dr. Reddy’s
Dr. Reddy’s Laboratories Ltd. (NYSE: RDY) is an integrated global pharmaceutical company, committed to providing affordable and innovative medicines for healthier lives. Through its three businesses – Pharmaceutical Services and Active Ingredients, Global Generics and Proprietary Products – Dr. Reddy’s offers a portfolio of products and services including APIs, custom pharmaceutical services, generics, biosimilars, differentiated formulations and NCEs. Therapeutic focus is on gastro-intestinal, cardiovascular, diabetology, oncology, pain management, anti-infective and pediatrics. Major markets include India, USA, Russia and CIS, Germany, UK, Venezuela, S. Africa, Romania, and New Zealand. For more information, log on to: www.drreddys.com
For more information please contact:
Investors and Financial Analysts:
Kedar Upadhye at kedaru@drreddys.com / +91-40-66834297
Raghavender R at raghavenderr@drreddys.com / +91-40-49002135
Milan Kalawadia (USA) at mkalawadia@drreddys.com / +1 908-203-4931
Media:
S Rajan at rajans@drreddys.com / +91-40- 49002445
| WO2005011634A1 * | Jul 21, 2004 | Feb 10, 2005 | William John Curatolo | Dosage forms providing controlled release of cholesteryl ester transfer protein inhibitors and immediate release of hmg-coa reductase inhibitors |
| WO2006073973A2 * | Dec 28, 2005 | Jul 13, 2006 | Reddy Us Therapeutics Inc | Novel benzylamine derivatives as cetp inhibitors |
| WO2006129167A1 * | May 22, 2006 | Dec 7, 2006 | Pfizer Prod Inc | PHARMACEUTICAL COMPOSITIONS OF CHOLESTERYL ESTER TRANSFER PROTEIN INHIBITORS AND HMG-CoA REDUCTASE INHIBITORS |
| WO2007128568A1 * | May 8, 2007 | Nov 15, 2007 | Novartis Ag | Bicyclic derivatives as cetp inhibitors |
| EP0298666A2 * | Jul 1, 1988 | Jan 11, 1989 | American Home Products Corporation | Spray dried ibuprofen compositions |
| EP1741424A2 * | Jul 27, 1998 | Jan 10, 2007 | Pfizer Products Inc. | Solid pharmaceutical dispersions with enhanced bioavailabilty |
| US5456923 | Dec 23, 1993 | Oct 10, 1995 | Nippon Shinyaku Company, Limited | Method of manufacturing solid dispersion |
| US5474989 | Mar 1, 1994 | Dec 12, 1995 | Kurita Water Industries, Ltd. | Drug composition |
| US5985326 | May 30, 1996 | Nov 16, 1999 | Icos Corporation | Method of producing a solid dispersion of a poorly water soluble drug |
| US6350786 | Sep 7, 1999 | Feb 26, 2002 | Hoffmann-La Roche Inc. | Stable complexes of poorly soluble compounds in ionic polymers |
| US6548555 | Jan 31, 2000 | Apr 15, 2003 | Pfizer Inc | Basic drug compositions with enhanced bioavailability |
| US6638522 | Dec 10, 1999 | Oct 28, 2003 | Pharmasolutions, Inc. | Microemulsion concentrate composition of cyclosporin |
| US6730679 | Mar 20, 1997 | May 4, 2004 | Smithkline Beecham Corporation | Pharmaceutical formulations |
| US7008640 | Jul 16, 2001 | Mar 7, 2006 | Yamanouchi Pharmaceutical Co., Ltd. | Pharmaceutical composition for oral use with improved absorption |
| US7034013 | Mar 19, 2002 | Apr 25, 2006 | Cydex, Inc. | Formulations containing propofol and a sulfoalkyl ether cyclodextrin |
| US7037528 | Jun 5, 2001 | May 2, 2006 | Baxter International Inc. | Microprecipitation method for preparing submicron suspensions |
| US7078057 | Dec 19, 2000 | Jul 18, 2006 | Kerkhof Nicholas J | Process for producing nanometer particles by fluid bed spray-drying |
| US7081255 | Aug 14, 2002 | Jul 25, 2006 | Janssen Pharmaceutica, N.V. | Antifungal compositions with improved bioavailability |
| US8030359 | Feb 9, 2007 | Oct 4, 2011 | Merck Sharp & Dohme Corp. | Polymer formulations of CETP inhibitors |
| US20060178514 | Dec 28, 2005 | Aug 10, 2006 | Anima Baruah | Novel benzylamine derivatives as CETP inhibitors |
| Reference | ||
|---|---|---|
| 1 | * | EINFAL T ET AL: “Methods of amorphization and investigation of the amorphous state“, ACTA PHARMACEUTICA 20130901 CROATIAN PHARMACEUTICAL SOCIETY DEU, vol. 63, no. 3, 1 September 2013 (2013-09-01), pages 305-334, XP002721717, ISSN: 1330-0075 |
| 2 | * | KAI TOSHIYA ET AL: “Oral absorption improvement of poorly soluble drug using solid dispersion technique“, CHEMICAL AND PHARMACEUTICAL BULLETIN (TOKYO), vol. 44, no. 3, 1996, pages 568-571, XP002721716, ISSN: 0009-2363 |
| 3 | * | KIM TAE-WAN ET AL: “Characterization of dual layered pellets for sustained release of poorly water-soluble drug“, CHEMICAL & PHARMACEUTICAL BULLETIN (TOKYO), vol. 55, no. 7, July 2007 (2007-07), pages 975-979, XP002721715, ISSN: 0009-2363 |
| 4 | * | KIM TAE-WAN ET AL: “Modified release of coated sugar spheres using drug-containing polymeric dispersions.“, ARCHIVES OF PHARMACAL RESEARCH JAN 2007, vol. 30, no. 1, January 2007 (2007-01), pages 124-130, XP002721714, ISSN: 0253-6269 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014128564A2 * | Feb 21, 2014 | Aug 28, 2014 | Dr. Reddy’s Laboratories Ltd. | Pharmaceutical compositions of cetp inhibitors |
| Patent | Submitted | Granted |
|---|---|---|
| Novel benzylamine derivatives and their utility as cholesterol ester-transfer protein inhibitors [US2007015758] | 2007-01-18 | |
| Novel benzylamine derivatives as CETP inhibitors [US2006178514] | 2006-08-10 |
| Publication Number | Publication Date | IPCR Assignee/Applicant | Structure hits | Tools | |
|---|---|---|---|---|---|
|
1.
EP-2919765-A2 |
2015-09-23 |
EN
|
|
||
|
2.
US-20150216866-A1 |
2015-08-06 |
|
|||
|
3.
US-9040558-B2 |
2015-05-26 |
|
|||
|
4.
WO-2014128564-A2 |
2014-08-28 |
EN
|
|
||
|
5.
WO-2014076568-A2 |
2014-05-22 |
EN
|
|
||
|
6.
US-20140134235-A1 |
2014-05-15 |
|
|||
|
7.
US-20070015758-A1 |
2007-01-18 |
|
SEE
http://circ.ahajournals.org/cgi/content/meeting_abstract/122/21_MeetingAbstracts/A13981
////////
Cn1nc(nn1)N(Cc2cc5cccc(C)c5nc2N(CC3CC3)CC4CC4)Cc6cc(cc(c6)C(F)(F)F)C(F)(F)F
CC1=CC=CC2=CC(=C(N=C12)N(CC3CC3)CC4CC4)CN(CC5=CC(=CC(=C5)C(F)(F)F)C(F)(F)F)C6=NN(N=N6)C
Pfizer’s PF 04991532 a Hepatoselective Glucokinase Activator Clinical Candidate for Treating Type 2 Diabetes Mellitus

PF 04991532
GKA PF-04991532
(S)-6-{3-cyclopentyl-2-[4-(trifluoromethyl)-1H-imidazol-1-yl]propanamido}nicotinic acid
(S)-6-(3-Cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinic Acid
(S)-6-(3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinic acid
MW 396.36, MF C18 H19 F3 N4 O3
CAS 1215197-37-7
3-Pyridinecarboxylic acid, 6-[[(2S)-3-cyclopentyl-1-oxo-2-[4-(trifluoromethyl)-1H-imidazol-1-yl]propyl]amino]-
http://www.biochemj.org/content/441/3/881
Type 2 diabetes mellitus (T2DM) is a rapidly expanding public epidemic affecting over 300 million people worldwide. This disease is characterized by elevated fasting plasma glucose (FPG), insulin resistance, abnormally elevated hepatic glucose production (HGP), and reduced glucose-stimulated insulin secretion (GSIS). Moreover, long-term lack of glycemic control increases risk of complications from neuropathic, microvascular, and macrovascular diseases.
The standard of care for T2DM is metformin followed by sulfonylureas, dipeptidyl peptidase-4 (DPP-IV) inhibitors, and thiazolidinediones (TZD) as second line oral therapies. As disease progression continues, patients typically require injectable agents such as glucagon-like peptide-1 (GLP-1) analogues and, ultimately, insulin to help maintain glycemic control. Despite these current therapies, many patients still remain unable to safely achieve and maintain tight glycemic control, placing them at risk of diabetic complications and highlighting the need for novel therapeutic options.

Glucokinase (hexokinase IV) continues to be a compelling target for the treatment of type 2 diabetes given the wealth of supporting human genetics data and numerous reports of robust clinical glucose lowering in patients treated with small molecule allosteric activators. Recent work has demonstrated the ability of hepatoselective activators to deliver glucose lowering efficacy with minimal risk of hypoglycemia.
While orally administered agents require a considerable degree of passive permeability to promote suitable exposures, there is no such restriction on intravenously delivered drugs. Therefore, minimization of membrane diffusion in the context of an intravenously agent should ensure optimal hepatic targeting and therapeutic index.
Diabetes is a major public health concern because of its increasing prevalence and associated health risks. The disease is characterized by metabolic defects in the production and utilization of carbohydrates which result in the failure to maintain appropriate blood glucose levels. Two major forms of diabetes are recognized. Type I diabetes, or insulin-dependent diabetes mellitus (IDDM), is the result of an absolute deficiency of insulin. Type II diabetes, or non-insulin dependent diabetes mellitus (NIDDM), often occurs with normal, or even elevated levels of insulin and appears to be the result of the inability of tissues and cells to respond appropriately to insulin. Aggressive control of NIDDM with medication is essential; otherwise it can progress into IDDM.
As blood glucose increases, it is transported into pancreatic beta cells via a glucose transporter. Intracellular mammalian glucokinase (GK) senses the rise in glucose and activates cellular glycolysis, i.e. the conversion of glucose to glucose-6-phosphate, and subsequent insulin release. Glucokinase is found principally in pancreatic β-cells and liver parenchymal cells. Because transfer of glucose from the blood into muscle and fatty tissue is insulin dependent, diabetics lack the ability to utilize glucose adequately which leads to undesired accumulation of blood glucose (hyperglycemia). Chronic hyperglycemia leads to decreases in insulin secretion and contributes to increased insulin resistance. Glucokinase also acts as a sensor in hepatic parenchymal cells which induces glycogen synthesis, thus preventing the release of glucose into the blood. The GK processes are thus critical for the maintenance of whole body glucose homeostasis.
It is expected that an agent that activates cellular GK will facilitate glucose-dependent secretion from pancreatic beta cells, correct postprandial hyperglycemia, increase hepatic glucose utilization and potentially inhibit hepatic glucose release. Consequently, a GK activator may provide therapeutic treatment for NIDDM and associated complications, inter alia, hyperglycemia, dyslipidemia, insulin resistance syndrome, hyperinsulinemia, hypertension, and obesity.
Several drugs in five major categories, each acting by different mechanisms, are available for treating hyperglycemia and subsequently, NIDDM (Moller, D. E., “New drug targets for Type II diabetes and the metabolic syndrome” Nature 414; 821-827, (2001)): (A) Insulin secretogogues, including sulphonyl-ureas (e.g., glipizide, glimepiride, glyburide) and meglitinides (e.g., nateglidine and repaglinide) enhance secretion of insulin by acting on the pancreatic beta-cells. While this therapy can decrease blood glucose level, it has limited efficacy and tolerability, causes weight gain and often induces hypoglycemia. (B) Biguanides (e.g., metformin) are thought to act primarily by decreasing hepatic glucose production. Biguanides often cause gastrointestinal disturbances and lactic acidosis, further limiting their use. (C) Inhibitors of alpha-glucosidase (e.g., acarbose) decrease intestinal glucose absorption. These agents often cause gastrointestinal disturbances. (D) Thiazolidinediones (e.g., pioglitazone, rosiglitazone) act on a specific receptor (peroxisome proliferator-activated receptor-gamma) in the liver, muscle and fat tissues. They regulate lipid metabolism subsequently enhancing the response of these tissues to the actions of insulin. Frequent use of these drugs may lead to weight gain and may induce edema and anemia. (E) Insulin is used in more severe cases, either alone or in combination with the above agents.
Ideally, an effective new treatment for NIDDM would meet the following criteria: (a) it would not have significant side effects including induction of hypoglycemia; (b) it would not cause weight gain; (c) it would at least partially replace insulin by acting via mechanism(s) that are independent from the actions of insulin; (d) it would desirably be metabolically stable to allow less frequent usage; and (e) it would be usable in combination with tolerable amounts of any of the categories of drugs listed herein.
Substituted heteroaryls, particularly pyridones, have been implicated in mediating GK and may play a significant role in the treatment of NIDDM. For example, U.S. Patent publication No. 2006/0058353 and PCT publication Nos. WO2007/043638, WO2007/043638, and WO2007/117995 recite certain heterocyclic derivatives with utility for the treatment of diabetes. Although investigations are on-going, there still exists a need for a more effective and safe therapeutic treatment for diabetes, particularly NIDDM.

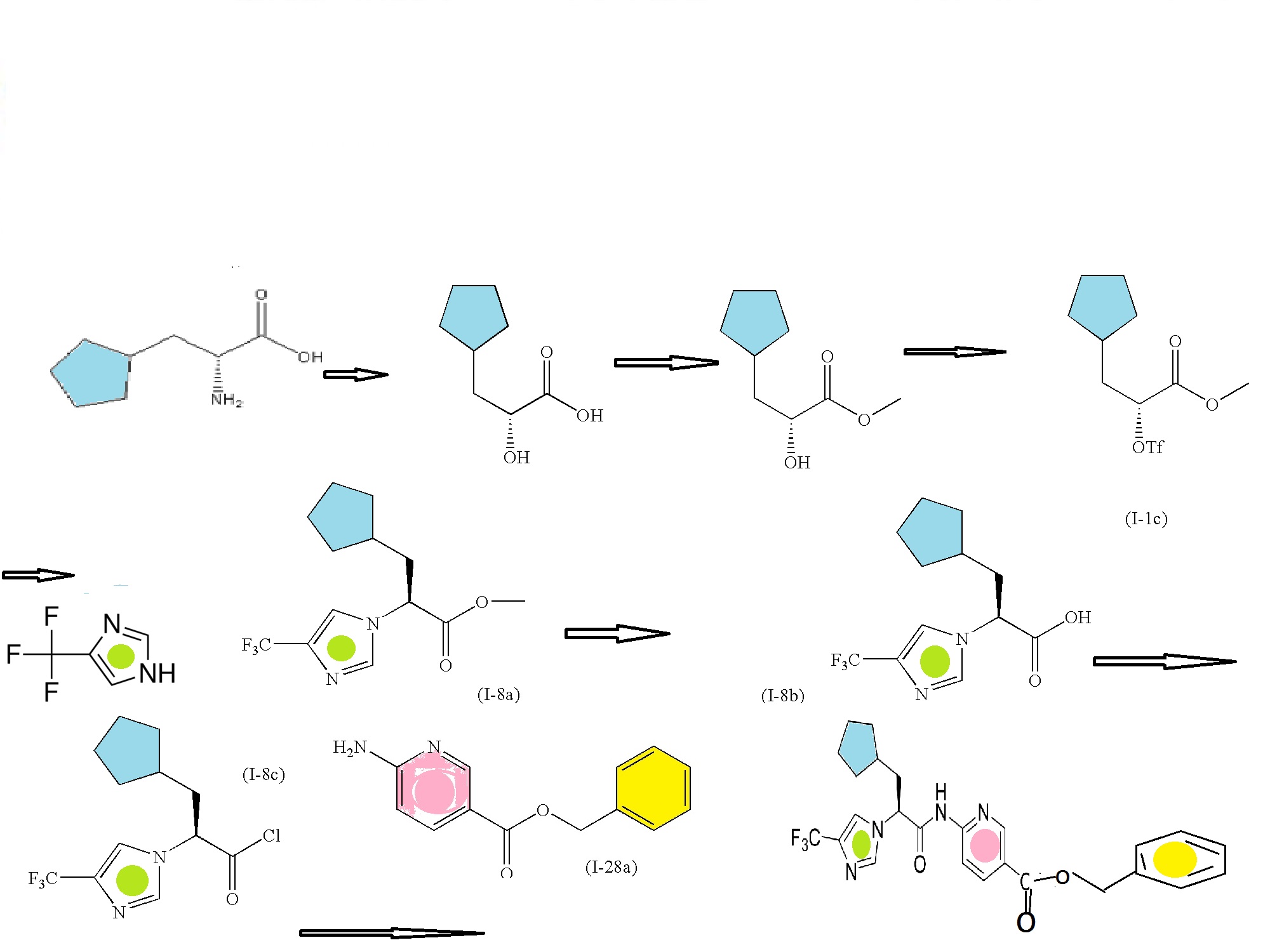
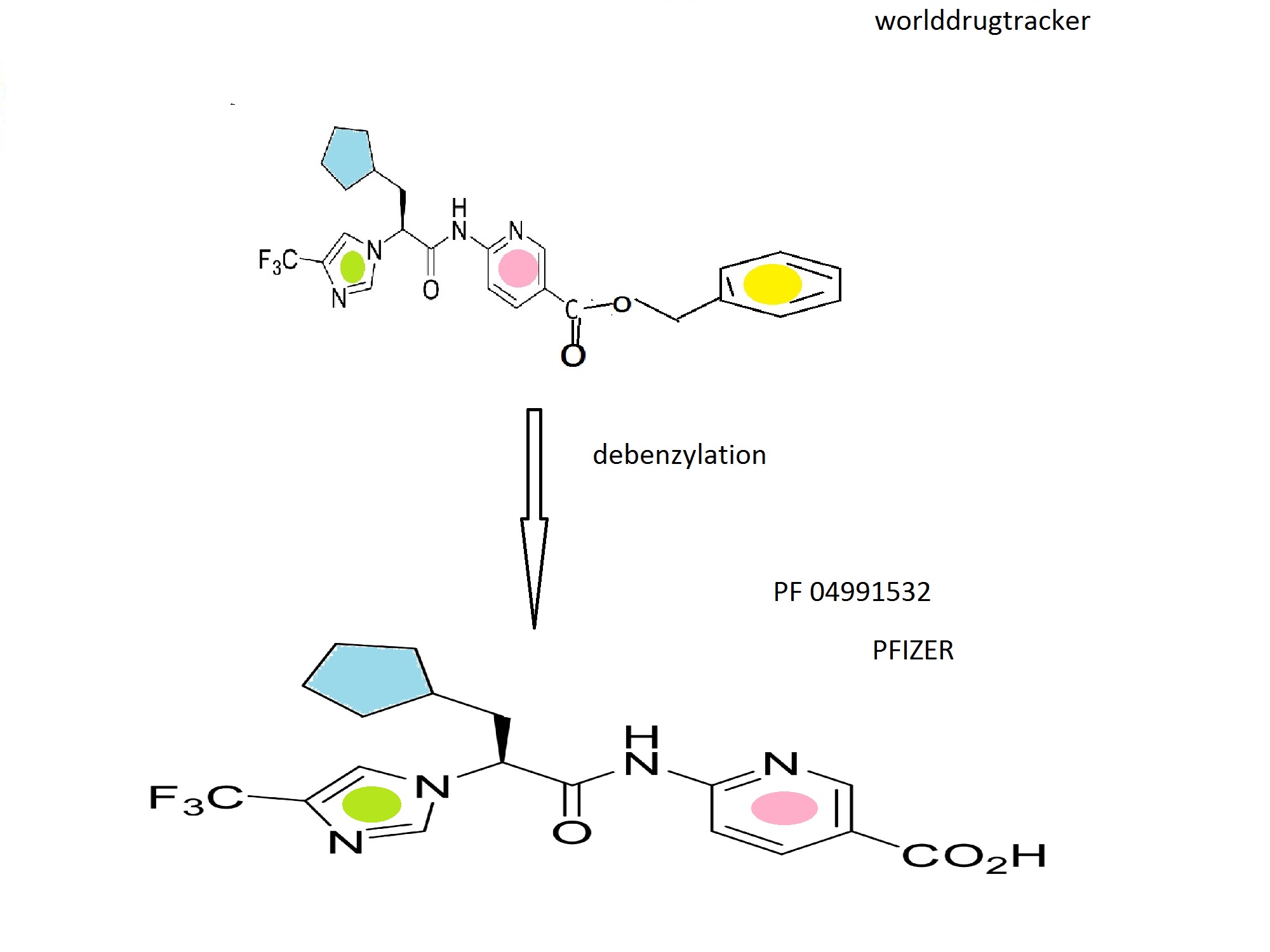
PATENT
US 20100063063
http://www.google.com/patents/US20100063063
SYNTHESIS CONSTRUCTION

6-aminonicotinic acid

BENZYL BROMIDE

FIRST KEY INTERMEDIATE
SECOND SERIES FOR NEXT INTERMEDIATE

(R)-2-amino-3-cyclopentylpropanoic acid

(R)-methyl 3-cyclopentyl-2-hydroxypropanoic acid (I-1a)

(R)-methyl 3-cyclopentyl-2-hydroxypropanoate (I-1b)
Trifluoromethanesulfonic acid anhydride

(R)-methyl 3-cyclopentyl-2-(trifluoromethylsulfonyloxy)propanoate (I-1c)
CONDENSED WITH

4-Trifluoromethyl-1H-imidazole
TO GIVE PRODUCT SHOWN BELOW

(S)-methyl 3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoate (I-8a)

(S)-3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoic acid (I-8b)

CONVERTED TO ACID CHLORIDE, (S)-3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoyl chloride (I-8c)
AND CONDENSED WITH
WILL GIVE BENZYL DERIVATIVE AS BELOW

THEN DEBENZYLATION TO FINAL PRODUCT
Intermediate: (R)-methyl 3-cyclopentyl-2-hydroxypropanoic acid (I-1a)
To a stirred solution of (R)-2-amino-3-cyclopentylpropanoic acid (5.0 grams; Chem-Impex International, Inc., Wood Dale, Ill.) and 1 M H2SO4 (45.1 mL) at 0° C., was added a solution of NaNO2 (3.12 g) in H2O (15.6 mL) drop wise over 10 minutes. The reaction mixture was stirred for 3 hours at 0° C., then for 2 hours at room temperature. The solution was then extracted (3 times) with diethyl ether. The combined organic extracts were dried over MgSO4, filtered, and the filtrate concentrated to afford 2.36 g of (I-1a). 1H NMR (400 MHz, CDCl3) δ 4.26-4.28 (1H), 1.99-2.07 (1H), 1.76-1.81 (4H), 1.60-1.62 (4H), 1.12-1.16 (2H); LCMS for C8H14O3 m/z 157.1 (M−H)−.
Intermediate: (R)-methyl 3-cyclopentyl-2-hydroxypropanoate (I-1b)
To a stirred solution of 2.36 g of (I-1a) in anhydrous methanol (15 mL) at room temperature was added SOCl2(1.64 mL). The resulting mixture was heated at reflux for 2 hours. It was then cooled and concentrated under reduced pressure. The residue was partitioned between ethyl acetate and aqueous saturated NaHCO3 solution. The biphasic mixture was separated and the aqueous portion was extracted with ethyl acetate. The combined extracts were dried over MgSO4, filtered, and the filtrate concentrated under reduced pressure. The resulting residue was purified by flash column chromatography (silica gel, heptanes/ethyl acetate) to afford 1.5 g of (I-1b) as a clear oil.1H NMR (400 MHz, CDCl3) δ 4.15-4.20 (1H), 3.77 (3H), 2.62-2.63 (1H), 1.97-2.05 (1H), 1.49-1.86 (8H), 1.06-1.17 (2H); LCMS for C9H16O3 m/z 171.6 (M)+. Intermediate (I-1b) can alternatively be prepared by the method described below.
A 0.2M solution of Li2CuCl4 was prepared as follows: Anhydrous CUCl2 (26.9 g, 200 mol) and anhydrous LiCl (17.0 g, 400 mmol) were dissolved in THF (1000 mL). The mixture required gentle heating to completely dissolve the solids. After cooling the solution is ready for use.
A solution of Li2CuCl4 (0.2 M in THF, 125 mL, 25.0 mmol) was added slowly to a suspension of cyclopentylmagnesium bromide (2 M in diethyl ether, 135 mL, 270 mmol; Aldrich Chemical Company, Inc., Milwaukee, Wis.) and THF (500 mL) at −50° C. over 2-3 mins. The pale grey/brown suspension was then allowed to warm slowly to −10° C. over 30 mins, by which time the color had developed to a dark grey. The mixture was re-cooled to −78° C. and (R)-methyl oxirane-2-carboxylate (25.0 g, 245 mmol; Aldrich Chemical Company, Inc., Milwaukee, Wis.) was added neat via syringe over 90 seconds. The reaction was then stirred at −78° C. for 20 mins, before removing the ice-bath and allowing to warm to approximately −50° C. over 30 mins. Saturated NH4Cl (aq, 700 mL) was then added and the mixture stirred for 30 mins. The organic layer was collected and the aqueous layer extracted with diethyl ether (2×250 mL). The combined organics were washed with saturated NH4Cl (aq, 350 mL), dried over MgSO4, and evaporated. Distillation of the crude residue (68-70° C. at 0.8 mbar) yielded 65-70% of (I-1b) as a pale yellow oil. A small amount of less volatile material remained in the still pot. 1H NMR (400 MHz; CDCl3): δ 4.17(1H), 3.76 (3H), 2.67 (1H), 2.01 (1H), 1.48-1.88 (8H), 1.11 (2H).
Intermediate: (R)-methyl 3-cyclopentyl-2-(trifluoromethylsulfonyloxy)propanoate (I-1c)
Intermediate: (R)-methyl 3-cyclopentyl-2-(trifluoromethylsulfonyloxy)propanoate (I-1c
Intermediate (I-1b) (6.37 g, 37.0 mmol) was dissolved in dry dichloromethane (260 mL) and stirred under nitrogen in an ice bath. 2,6-Lutidine (9.0 mL, 77 mmol) was added. Trifluoromethanesulfonic acid anhydride (11 mL, 65 mmol) in dry dichloromethane (75 mL) was added dropwise. The reaction was stirred in the ice bath for 60 minutes, concentrated under reduced pressure, and taken up in 1N HCl and methyl t-butyl ether. The aqueous layer was separated, and the organic layer was washed with additional 1N HCl to insure the removal of all the lutidine. The combined organic layer was then washed with brine, dried over sodium sulfate, filtered, concentrated under reduced pressure, and dried under high vacuum to afford (I-1c) (11.3 g, 37 mmol, 100%), which was used immediately without further purification; 1H NMR (400 MHz, CDCl3) δ 5.10-5.14 (1H), 3.82 (3H), 2.02-2.12 (1H), 1.79-1.98 (4H), 1.51-1.66 (4H), 1.08-1.18 (2H).
Intermediate (I-1b) (6.37 g, 37.0 mmol) was dissolved in dry dichloromethane (260 mL) and stirred under nitrogen in an ice bath. 2,6-Lutidine (9.0 mL, 77 mmol) was added. Trifluoromethanesulfonic acid anhydride (11 mL, 65 mmol) in dry dichloromethane (75 mL) was added dropwise. The reaction was stirred in the ice bath for 60 minutes, concentrated under reduced pressure, and taken up in 1N HCl and methyl t-butyl ether. The aqueous layer was separated, and the organic layer was washed with additional 1N HCl to insure the removal of all the lutidine. The combined organic layer was then washed with brine, dried over sodium sulfate, filtered, concentrated under reduced pressure, and dried under high vacuum to afford (I-1c) (11.3 g, 37 mmol, 100%), which was used immediately without further purification; 1H NMR (400 MHz, CDCl3) δ 5.10-5.14 (1H), 3.82 (3H), 2.02-2.12 (1H), 1.79-1.98 (4H), 1.51-1.66 (4H), 1.08-1.18 (2H)
Intermediate: (S)-methyl 3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoate (I-8a)
4-Trifluoromethyl-1H-imidazole (5.0 g, 37.0 mmol; Apollo Scientific Ltd., Bredbury, Cheshire, UK) was stirred in dry THF (180 mL) under nitrogen at room temperature. Lithium hexamethyldisilazide (1M in THF, 33.4 mL, 33.4 mmol) was added dropwise via addition funnel. The mixture was stirred at room temperature for 50 minutes and then chilled in an ice bath. A solution of (I-1c) (11.3 g, 37 mmol) in dry THF (45 mL), which had been chilled in an ice bath, was added in one portion. The reaction was allowed to warm to room temperature, stirred for 2 hours, quenched with saturated aqueous ammonium chloride solution (20 mL) and allowed to stir overnight. The aqueous layer was separated, and the organic layer was concentrated and then diluted with water and ethyl acetate. The organic layer was washed in series with dilute aqueous phosphoric acid, aqueous 10% potassium carbonate, and brine. The organic layer was then dried over sodium sulfate, filtered, and concentrated under reduced pressure to a brown oil. The crude material, containing the undesired regioisomer as a small impurity, was purified by chromatography on a 330 g pre-packed silica gel column, eluting with 10% ethyl acetate/heptane, linear gradient to 70% ethyl acetate/heptane. The product fractions were located by spotting on a silica TLC plate and visualizing with KMnO4 stain. TLC (1:1 ethyl acetate/heptane, developed in potassium permanganate) located the pure and mixed fractions. The clean product fractions were combined, evaporated, and dried under high vacuum to afford (I-8a) as a clear oil (6.61 g, 22.4 mmol, 67%). 1H NMR (400 MHz, CDCl3) δ 7.57 (1H), 7.38 (1H), 4.71-4.74 (1H), 3.76 (3H), 2.01-2.14 (2H), 1.45-1.79 (7H), 1.03-1.18 (2H); m/z 291.4 (M+H)+.
Intermediate: (S)-3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoic acid (I-8b
6N HCl (140 mL) was added to (I-8a) (6.61 g, 22.4 mmol) and the mixture was warmed to 95° C. for 16 hours and then allowed to cool. Solid potassium carbonate (58 g) was added in portions to bring the pH to about 4. A precipitate crashed out. Ethyl acetate was added, and the mixture was stirred until everything dissolved. The aqueous layer was extracted once with ethyl acetate. The combined organics were washed with brine, dried over sodium sulfate, filtered, concentrated under reduced pressure, and dried under high vacuum to afford (I-8b) as a clear glass (6.15 g, 21.9 mmol, 98%). 1H NMR (400 MHz, CDCl3) δ 7.73 (1H), 7.34 (1H), 6.85-7.15 (1H), 4.66-4.70 (1H), 1.98-2.17 (2H), 1.41-1.75 (7H), 1.01-1.19 (2H); m/z 277.4 (M+H)+.
Intermediate: (S)-3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoyl chloride (I-8c)
To a suspension of intermediate (I-8b) (0.25 g, 0.9 mmol) in dichloromethane (5 mL) was added oxalyl chloride (0.35 g, 2.7 mmol) and N,N-dimethylformamide (1 drop) at room temperature. The mixture was stirred for 2 hours at room temperature. The reaction mixture was concentrated in vacuo, and the residue was chased with dichloromethane two times and concentrated in vacuo to afford (I-8c) (0.27 g, 100%) as an oil, which was used in the next step directly.
Intermediate: (S)-6-(3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido) nicotinoyl chloride (I-21a)
-
 Thionyl chloride (225 mg, 1.89 mmol) was added to a solution of the compound of Example 48 (150 mg, 0.387 mmol) in dichloromethane (1.5 mL) and the reaction stirred at room temperature for 1 hour. LCMS of an aliquot in methanol showed ˜67% methyl ester. To the reaction mixture was added another 25 uL of thionyl chloride and this was stirred at room temp for another 30 minutes. Solvents were evaporated to afford 157 mg (100%) of (I-21a) as a grayish-white solid. LCMS in methanol to generate the methyl ester gave m/z 395.9 (M+H)+.
Thionyl chloride (225 mg, 1.89 mmol) was added to a solution of the compound of Example 48 (150 mg, 0.387 mmol) in dichloromethane (1.5 mL) and the reaction stirred at room temperature for 1 hour. LCMS of an aliquot in methanol showed ˜67% methyl ester. To the reaction mixture was added another 25 uL of thionyl chloride and this was stirred at room temp for another 30 minutes. Solvents were evaporated to afford 157 mg (100%) of (I-21a) as a grayish-white solid. LCMS in methanol to generate the methyl ester gave m/z 395.9 (M+H)+.

(I-8b
Intermediate: (S)-3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoic acid (I-8b)
6N HCl (140 mL) was added to (I-8a) (6.61 g, 22.4 mmol) and the mixture was warmed to 95° C. for 16 hours and then allowed to cool. Solid potassium carbonate (58 g) was added in portions to bring the pH to about 4. A precipitate crashed out. Ethyl acetate was added, and the mixture was stirred until everything dissolved. The aqueous layer was extracted once with ethyl acetate. The combined organics were washed with brine, dried over sodium sulfate, filtered, concentrated under reduced pressure, and dried under high vacuum to afford (I-8b) as a clear glass (6.15 g, 21.9 mmol, 98%). 1H NMR (400 MHz, CDCl3) δ 7.73 (1H), 7.34 (1H), 6.85-7.15 (1H), 4.66-4.70 (1H), 1.98-2.17 (2H), 1.41-1.75 (7H), 1.01-1.19 (2H); m/z 277.4 (M+H)+.
(I-28a
Intermediate: benzyl 6-aminonicotinate (I-28a)
To a stirred suspension of 6-aminonicotinic acid (100 g, 0.72 mol; Aldrich Chemical Company, Inc., Milwaukee, Wis.) in N,N-dimethylformamide (700 mL) with brisk mechanical stirring was added potassium carbonate (150 g, 1.08 mol) and the reaction was stirred for 10 min before the portionwise addition of benzyl bromide (95 mL, 0.80 mol). The reaction was stirred at room temperature overnight, then the solids were filtered off and washed thoroughly with ethyl acetate, and the solvent was removed under vacuum. The filter cake was dissolved in water and extracted with ethyl acetate. The residue after evaporation of N,N-dimethylformamide was combined with the ethyl acetate extracts (total volume 2 L of ethyl acetate) and the combined organic extracts washed with brine (5×500 mL), dried (MgSO4) and the solvent removed under reduced pressure. The crude product was refluxed with 1:1 diethyl ether:hexane for 30 min then the solids filtered off (warm), washed with diethyl ether:hexane (1:1), and dried. This solid was precipitated from hot toluene (hot filtration required to remove dibenzylated material) and dried to afford (I-28a) (107.2 g, 65%) as an off-white solid; 1H NMR (DMSO-d6): δ 8.50 (1H), 7.82 (1H), 7.34-7.29 (5H), 6.84 (2H), 6.43 (1H), 5.23 (2H); m/z 229.4 (M+H)+.
Example 47
(S)-benzyl 6-(3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinate
Formula (1A-4) wherein R4 is
To Intermediate (I-8b) (16.28 g, 59.8 mmol) stirring in dry dichloromethane (400 mL) at room temperature under nitrogen was added 2 drops of DMF. Oxalyl chloride (11 mL, 130 mmol) was added dropwise. After the bubbling subsided the reaction was left stirring for 90 minutes and then concentrated under reduced pressure. Two successive portions of 1,2-dichloroethane were added and evaporated to remove all excess oxalyl chloride. The crude acid chloride was taken up in dichloromethane (150 mL) and stirred at room temperature. Intermediate (I-28a) (14.3 g, 62.5 mmol) and pyridine (10 mL, 130 mmol) were stirred in 400 mL dry dichloromethane. This was added to the acid chloride solution, using another 50 mL dry dichloromethane to complete the transfer. The mixture was left stirring at room temperature under nitrogen for 18 hours. The reaction was diluted with dichloromethane and water, and 1M aqueous phosphoric acid was added. The organic layer was separated and washed sequentially with dilute aqueous potassium carbonate, and brine. This was then dried over sodium sulfate, filtered, and concentrated under reduced pressure to a glass, which was taken up in hot ethyl acetate and stirred at room temperature. A precipitate appeared at about 30 minutes. The mixture was stirred for 16 hours and then filtered. The precipitate was washed with ethyl acetate and then diethyl ether and dried under high vacuum at 60° C. to afford the title compound as a white solid (17.8 g, 36.6 mmol, 61%). The mother liquor was evaporated and purified by silica gel chromatography on a 120 g pre-packed column, eluting with 40% ethyl acetate/heptane. The product fractions were combined, concentrated under reduced pressure, dried under high vacuum to a glass, and converted as previously described to additional product (3.5 g, 7.2 mmol, 12%, total yield 73%). 1H NMR (400 MHz, DMSO-d6) δ 11.50 (1H), 8.87-8.88 (1H), 8.29-8.32 (1H), 8.12-8.14 (1H), 7.93-7.94 (2H), 7.39-7.46 (2H), 7.30-7.37 (3H), 5.32 (2H), 5.21-5.25 (1H), 2.06-2.19 (2H), 1.26-1.63 (8H), 1.01-1.06 (1H); m/z 487.5 (M+H)+.
Example 48
(S)-6-(3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinic acid
Formula (1A-4) wherein R4 is

The compound of Example 47 (4.07 g, 8.35 mmol) was added to a 500 mL Parr bottle, followed by ethyl acetate (50 mL) and ethanol (100 mL). The mixture was warmed until all of the solid dissolved, and then cooled to room temperature. 10% Pd/C (450 mg) was added, and the mixture was shaken under 50 psi hydrogen for 90 minutes. The reaction was filtered through a microfiber filter. The filtrate was concentrated under reduced pressure and dried under high vacuum at 50° C. to afford product as a glassy solid (3.0 g, 7.75 mmol, 90.6%). The glassy solid was stirred overnight in diethyl ether. The white solid precipitate was filtered, washed with diethyl ether, suction dried, and dried under high vacuum at 50° C. to afford the title compound as a white solid.
1H NMR (400 MHz, DMSO-d6) δ 13.10-13.25 (1H), 11.44 (1H), 8.83 (1H), 8.23-8.26 (1H), 8.09-8.12 (1H), 7.94-7.95 (2H), 5.22-5.26 (1H), 2.06-2.17 (2H), 1.29-1.64 (8H), 1.04-1.07 (1H);
m/z 397.3 (M+H)+.
THIS NMR IS FROM SUPPORTING INFO OF A JOURNAL
PAPER
Organic Process Research & Development (2012), 16(10), 1635-1645
http://pubs.acs.org/doi/abs/10.1021/op300194c

This work describes the process development and manufacture of early-stage clinical supplies of a hepatoselective glucokinase activator, a potential therapy for type 2 diabetes mellitus. Critical issues centered on challenges associated with the synthesis of intermediates and API bearing a particularly racemization-prone α-aryl carboxylate functionality. In particular, a T3P-mediated amidation process was optimized for the coupling of a racemization-prone acid substrate and a relatively non-nucleophilic amine. Furthermore, an unusually hydrolytically-labile amide in the API also complicated the synthesis and isolation of drug substance. The evolution of the process over multiple campaigns is presented, resulting in the preparation of over 110 kg of glucokinase activator.
(S)-6-(3-Cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinic Acid (1)
Pressure Hydrogenation
1 (89% yield) as a white solid:
mp 187–189 °C;
1H NMR (400 MHz, d6-DMSO) δ 13.23 (s, 1H), 11.49 (s, 1H), 8.86 (dd, J = 0.4, 2.4 Hz, 1H), 8.27 (dd, J = 2.4, 8.8 Hz, 1H), 8.13 (d, J = 8.8 Hz, 1H), 7.97–7.99 (m, 2H), 5.27 (dd, J = 5.6, 10.0 Hz, 1H), 2.20 (ddd, J = 6.0, 10.0, 14.0, 1H), 2.10 (ddd, J = 5.6, 8.4, 14.0, 1H), 1.27–1.69 (m, 8H), 1.03–1.12 (m, 1H);
13C NMR (100 MHz, d6-DMSO) δ 168.8, 165.7, 154.3, 149.7, 139.6, 138.8, 129.9 (q, JCF = 38 Hz), 122.6, 122.0 (q, JCF = 265 Hz), 120.0 (q, JCF = 4 Hz), 112.8, 60.0, 37.6, 36.2, 32.0, 30.8, 24.6, 24.4;
19F NMR (376 MHz, d6-DMSO) δ −60.7.
HRMS-ESI m/z: [M + H]+ calcd for C18H19F3N4O3, 397.1482; found, 397.1481.
Achiral HPLC: rt 4.6 min. Chiral SFC: rt 4.1 min (1), 3.1 min (ent-1).
PAPER
Journal of Medicinal Chemistry (2012), 55(3), 1318-1333
http://pubs.acs.org/doi/abs/10.1021/jm2014887

Glucokinase is a key regulator of glucose homeostasis, and small molecule allosteric activators of this enzyme represent a promising opportunity for the treatment of type 2 diabetes. Systemically acting glucokinase activators (liver and pancreas) have been reported to be efficacious but in many cases present hypoglycaemia risk due to activation of the enzyme at low glucose levels in the pancreas, leading to inappropriately excessive insulin secretion. It was therefore postulated that a liver selective activator may offer effective glycemic control with reduced hypoglycemia risk. Herein, we report structure–activity studies on a carboxylic acid containing series of glucokinase activators with preferential activity in hepatocytes versus pancreatic β-cells. These activators were designed to have low passive permeability thereby minimizing distribution into extrahepatic tissues; concurrently, they were also optimized as substrates for active liver uptake via members of the organic anion transporting polypeptide (OATP) family. These studies lead to the identification of 19 as a potent glucokinase activator with a greater than 50-fold liver-to-pancreas ratio of tissue distribution in rodent and non-rodent species. In preclinical diabetic animals, 19 was found to robustly lower fasting and postprandial glucose with no hypoglycemia, leading to its selection as a clinical development candidate for treating type 2 diabetes.
(S)-6-(3-Cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinic Acid (19)
afford 19 as a white solid (3.22 g, 71%).
1H NMR (400 MHz, DMSO-d6) δ 11.47 (s, 1H), 8.86 (d, J = 1.95 Hz, 1H), 8.27 (dd, J = 2.24, 8.68 Hz, 1H), 8.13 (d, J = 8.78 Hz, 1H), 7.97 (d, J = 4.88 Hz, 2H), 5.27 (dd, J = 5.37, 9.66 Hz, 1H), 2.04–2.26 (m, 2H), 1.38–1.72 (m, 7H), 1.26–1.37 (m, 1H), 1.08 (td, J = 7.88, 11.75 Hz, 1H);
LCMS m/z 397.5 (M + H)+.
HPLC purity (method A): tR = 7.690 min, 100%.

PAPER
Bioorganic & Medicinal Chemistry Letters (2013), 23(24), 6588-6592
http://www.sciencedirect.com/science/article/pii/S0960894X13012638


Figure 1.
Structure of Hepatoselective GKA PF-04991532 (1).
References
Drug Metabolism & Disposition (2015), 43(2), 190-198
PLoS One (2014), 9(5), e97139/1-e97139/9,
Journal of Biological Chemistry (2012), 287(17), 13598-13610
Drug Discovery Today (2012), 17(9-10), 528-529
Biochemical Journal (2012), 441(3), 881-887.
///////////

Figure 1. Representative structures of glucokinase activators.
Pfizer’s PF 04937319 glucokinase activators for the treatment of Type 2 diabetes

PF 04937319
N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide
MW 432.43
MF C22 H20 N6 O4
CAS 1245603-92-2
2-Pyrimidinecarboxamide, N,N-dimethyl-5-[[2-methyl-6-[[(5-methyl-2-pyrazinyl)amino]carbonyl]-4-benzofuranyl]oxy]-
N,N-Dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)carbamoyl)-benzofuran-4- yloxy)pyrimidine-2-carboxamide
Pfizer Inc. clinical candidate currently in Phase 2 development.
CLINICAL TRIALS
A trial to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of single doses of PF-04937319 in subjects with type 2 diabetes mellitus (NCT01044537)
Multiple dose study of PF-04937319 in patients with type 2 diabetes (NCT01272804)
Phase 2 study to evaluate safety and efficacy of investigational drug – PF04937319 in patients with type 2 diabetes (NCT01475461)
SYNTHESIS

Glucokinase is a key regulator of glucose homeostasis and small molecule activators of this enzyme represent a promising opportunity for the treatment of Type 2 diabetes. Several glucokinase activators have advanced to clinical studies and demonstrated promising efficacy; however, many of these early candidates also revealed hypoglycemia as a key risk. In an effort to mitigate this hypoglycemia risk while maintaining the promising efficacy of this mechanism, we have investigated a series of substituted 2-methylbenzofurans as “partial activators” of the glucokinase enzyme leading to the identification of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide as an early development candidate.
Diabetes is a major public health concern because of its increasing prevalence and associated health risks. The disease is characterized by metabolic defects in the production and utilization of carbohydrates which result in the failure to maintain appropriate blood glucose levels. Two major forms of diabetes are recognized. Type I diabetes, or insulin-dependent diabetes mellitus (IDDM), is the result of an absolute deficiency of insulin. Type Il diabetes, or non-insulin dependent diabetes mellitus (NIDDM), often occurs with normal, or even elevated levels of insulin and appears to be the result of the inability of tissues and cells to respond appropriately to insulin. Aggressive control of NIDDM with medication is essential; otherwise it can progress into IDDM. As blood glucose increases, it is transported into pancreatic beta cells via a glucose transporter. Intracellular mammalian glucokinase (GK) senses the rise in glucose and activates cellular glycolysis, i.e. the conversion of glucose to glucose-6-phosphate, and subsequent insulin release. Glucokinase is found principally in pancreatic β-cells and liver parenchymal cells. Because transfer of glucose from the blood into muscle and fatty tissue is insulin dependent, diabetics lack the ability to utilize glucose adequately which leads to undesired accumulation of blood glucose (hyperglycemia). Chronic hyperglycemia leads to decreases in insulin secretion and contributes to increased insulin resistance. Glucokinase also acts as a sensor in hepatic parenchymal cells which induces glycogen synthesis, thus preventing the release of glucose into the blood. The GK processes are thus critical for the maintenance of whole body glucose homeostasis.
It is expected that an agent that activates cellular GK will facilitate glucose-dependent secretion from pancreatic beta cells, correct postprandial hyperglycemia, increase hepatic glucose utilization and potentially inhibit hepatic glucose release. Consequently, a GK activator may provide therapeutic treatment for NIDDM and associated complications, inter alia, hyperglycemia, dyslipidemia, insulin resistance syndrome, hyperinsulinemia, hypertension, and obesity. Several drugs in five major categories, each acting by different mechanisms, are available for treating hyperglycemia and subsequently, NIDDM (Moller, D. E., “New drug targets for Type 2 diabetes and the metabolic syndrome” Nature 414; 821 -827, (2001 )): (A) Insulin secretogogues, including sulphonyl-ureas (e.g., glipizide, glimepiride, glyburide) and meglitinides (e.g., nateglidine and repaglinide) enhance secretion of insulin by acting on the pancreatic beta-cells. While this therapy can decrease blood glucose level, it has limited efficacy and tolerability, causes weight gain and often induces hypoglycemia. (B) Biguanides (e.g., metformin) are thought to act primarily by decreasing hepatic glucose production. Biguanides often cause gastrointestinal disturbances and lactic acidosis, further limiting their use. (C) Inhibitors of alpha-glucosidase (e.g., acarbose) decrease intestinal glucose absorption. These agents often cause gastrointestinal disturbances. (D) Thiazolidinediones (e.g., pioglitazone, rosiglitazone) act on a specific receptor (peroxisome proliferator-activated receptor-gamma) in the liver, muscle and fat tissues. They regulate lipid metabolism subsequently enhancing the response of these tissues to the actions of insulin. Frequent use of these drugs may lead to weight gain and may induce edema and anemia. (E) Insulin is used in more severe cases, either alone or in combination with the above agents. Ideally, an effective new treatment for NIDDM would meet the following criteria: (a) it would not have significant side effects including induction of hypoglycemia; (b) it would not cause weight gain; (c) it would at least partially replace insulin by acting via mechanism(s) that are independent from the actions of insulin; (d) it would desirably be metabolically stable to allow less frequent usage; and (e) it would be usable in combination with tolerable amounts of any of the categories of drugs listed herein.
Substituted heteroaryls, particularly pyridones, have been implicated in mediating GK and may play a significant role in the treatment of NIDDM. For example, U.S. Patent publication No. 2006/0058353 and PCT publication No’s. WO2007/043638, WO2007/043638, and WO2007/117995 recite certain heterocyclic derivatives with utility for the treatment of diabetes. Although investigations are on-going, there still exists a need for a more effective and safe therapeutic treatment for diabetes, particularly NIDDM.
Designing glucokinase activators with reduced hypoglycemia risk: discovery of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide as a clinical candidate for the treatment of type 2 diabetes mellitus
*Corresponding authors
aPfizer Worldwide Research & Development, Eastern Point Road, Groton
E-mail: jeffrey.a.pfefferkorn@pfizer.com
Tel: +860 686 3421
E-mail: jeffrey.a.pfefferkorn@pfizer.com
Tel: +860 686 3421
Med. Chem. Commun., 2011,2, 828-839
DOI: 10.1039/C1MD00116G
http://pubs.rsc.org/en/content/articlelanding/2011/md/c1md00116g/unauth#!divAbstract
http://www.rsc.org/suppdata/md/c1/c1md00116g/c1md00116g.pdf
N,N-Dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)carbamoyl)-benzofuran-4- yloxy)pyrimidine-2-carboxamide (28). To a solution of the 5-methyl-2-aminopyrazine (38.9 g, 356 mmol) in dimethoxyethane (315 mL) in a 3-neck flask equipped with overhead stirring and a condenser at 0 o C was added Me2AlCl (1 M solution in hexanes) (715 mL). The mixture was warmed to room temperature and stirred for 1.5 h. In a separate flask, 26 (52.6 g, 142.5 mmol) was dissolved in dimethoxyethane (210 mL). This mixture was then added to the amine mixture. A gum precipitated and upon scratching the flask it dissipated into a solid. The reaction was refluxed for 3.5 h. Aq. Rochelle’s salt (5 L) and 2-MeTHF (2 L) was added to the mixture and this was allowed to stir with overhead stirring for 14 h, after which time, a yellow solid precipitated. The solid was collected by filtration, washing with 2-MeTHF. The resulting solid was dried in a vacuum oven overnight to afford the desired material (50.0g) in 81% yield.
1 H NMR (400MHz, CDCl3) δ 9.54 (d, J = 1.56 Hz, 1H), 8.50 (s, 2H), 8.37 (s, 1H), 8.14 (d, J = 0.78 Hz, 1H), 7.88 – 7.92 (m, 1H), 7.52 (d, J = 1.37 Hz, 1H), 6.28 (t, J = 0.98 Hz, 1H), 3.14 (s, 3H), 2.98 (s, 3H), 2.55 (s, 3H), 2.49 (d, J = 1.17 Hz, 3H);
MS(ES+ ): m/z 433.4 (M+1), MS(ES- ): m/z 431.3 (M-1).
PAPER

http://pubs.rsc.org/en/content/articlelanding/2013/md/c2md20317k#!divAbstract
PAPER
Bioorganic & Medicinal Chemistry Letters (2013), 23(16), 4571-4578
http://www.sciencedirect.com/science/article/pii/S0960894X13007452

Figure 1.
Glucokinase activators 1 and 2.
PATENT
WO 2010103437
https://www.google.co.in/patents/WO2010103437A1?cl=en
Scheme I outlines the general procedures one could use to provide compounds of the present invention having Formula (I).


Preparations of Starting Materials and Key Intermediates
Preparation of Intermediate (E)-3-(ethoxycarbonyl)-4-(5-methylfuran-2-yl)but- 3-enoic acid (I- 1a):

(Ma) To a vigorously stirred solution of 5-methyl-2-furaldehyde (264 ml_, 2650 mmol) and diethyl succinate (840 ml_, 5050 mmol) in ethanol (1.820 L) at room temperature was added sodium ethoxide (0.93 L of a 21 weight % solution in ethanol) in one portion. The reaction mixture was then heated at reflux for 13 hours. After cooling to room temperature, the mixture was concentrated in vacuo (all batches were combined at this point). The resulting residue was partitioned between ethyl acetate (1 L) and hydrochloric acid (1 L of a 2M aqueous solution). After separation, the aqueous layer was extracted with ethyl acetate (2 x 1 L). The combined organic extracts were then extracted with sodium hydrogen carbonate (2 x 1 L of a saturated aqueous solution). These aqueous extracts were combined and adjusted to pH 2 with hydrochloric acid (2M aqueous solution) then extracted with ethyl acetate (2 x 1 L). These organic extracts were combined and concentrated in vacuo to give desired (E)-3-(ethoxycarbonyl)-4-(5-methylfuran-2-yl)but-3-enoic acid (J1 Ia: 34.34 g, 5%). The original organic extract was extracted with sodium hydroxide (2 L of a 2M aqueous solution). This aqueous extract was adjusted to pH 2 with hydrochloric acid (2M aqueous solution) then extracted with ethyl acetate (2 x 1 L). These organic extracts were combined and concentrated in vacuo to give additional desired materials (395.2 gram, 63%) as red liquid. 1H NMR (CDCI3, 300 MHz) δ ppm 7.48 (s, 1 H), 6.57 (d, 1 H), 6.09 (d, 1 H), 4.24 (q, 2H), 3.87 (s, 2H), 2.32 (s, 3H), 1.31 (t, 3H).
Preparation of Intermediate ethyl 4-acetoxy-2-methylbenzofuran-6- carboxylate (1-1 b):

(M b) To a vigorously stirred solution of (E)-3-(ethoxycarbonyl)-4-(5- methylfuran-2-yl)but-3-enoic acid (1-1 a: 326.6 g, 1 .371 mol) in acetic anhydride (1 .77 L, 18.72 mol) at room temperature was added sodium acetate (193 g, 2350 mmol) in one portion. The reaction mixture was then heated at reflux for 2.5 hours. After cooling to room temperature, the mixture was concentrated in vacuo (all batches were combined at this point). The resulting residue was suspended in dichloromethane (1 .5 L) and filtered, washing the solids with dichloromethane (3 x 500 ml_). The combined filtrate and washings were then washed with sodium hydrogencarbonate (2 x 1 L of a saturated aqueous solution) and brine (2 L), then concentrated in vacuo to give desired ethyl 4-acetoxy-2-methylbenzofuran-6-carboxylate (H b: 549.03 g, quantitative). 1H NMR (CDCI3, 300 MHz) δ ppm 8.00-7.99 (m, 1 H), 7.64 (d, 1 H), 6.32-6.32 (m, 1 H), 4.38 (q, 2H), 2.47 (d, 3H), 2.37 (s, 3H), 1 .39 (t, 3H).
Preparation of Intermediate ethyl 4-hydroxy-2-methylbenzofuran-6- carboxylate (1- 1 c):

(He) To a stirred solution of ethyl 4-acetoxy-2-methylbenzofuran-6- carboxylate (Hb: 549.03 g, 1 .37 mol) in ethanol (4.00 L) at room temperature was added potassium carbonate (266 g, 1 .92 mol) in one portion. The reaction mixture was then heated at 600C for 3 hours. Potassium carbonate (100 g, 0.720 mol) was then added in one portion and the reaction mixture was heated at 600C for a further 3 hours. After cooling to room temperature the mixture was diluted with dichloromethane (2 L) and the suspension filtered, washing the solids with dichloromethane (2 x 1 L) (all batches were combined at this point). The combined filtrate and washings were then washed with citric acid (2.5 L of a 1 M aqueous solution), then concentrated in vacuo and the resulting residue purified by dry flash chromatography (hexane then 2:1 hexane:ethyl acetate). All fractions containing the desired product were combined and concentrated in vacuo. The resulting residue, which solidified on standing, was slurried with cold toluene and filtered. The solids were then stirred with hot toluene and decolourising charcoal for 1 hour, followed by filtration of the hot mixture through a pad of celite. The filtrate was allowed to cool and the resulting precipitate isolated by filtration to give desired ethyl 4-hydroxy-2- methylbenzofuran-6-carboxylate (1-1 c: 360 g, 90%) as orange powder.
1H NMR (CDCI3, 300 MHz) δ ppm 7.73-7.73 (m, 1 H), 7.45 (d, 1 H), 6.51 -6.50 (m, 1 H), 5.85 (s, 1 H), 4.39 (q, 2H), 2.48 (d, 3H), 1.40 (t, 3H). LCMS (liquid chromatography mass spectrometry): m/z 221.06 (96.39 % purity).
Preparation of SM-25-bromo-N,N-dimethylpyrimidine-2-carboxamide (SM-
£1:

(SM-2) Oxalyl chloride (47.4g, 369mmol) was added to a suspension of 5-
Bromo-pyrimidine-2-carboxylic acid (5Og, 250mmol) in dichloromethane (821 ml) at room temperature followed by 1 -2 drop of dimethylformamide. The reaction mixture was stirred under nitrogen for 2 hours LCMS in methanol indicated the presence of the methyl ester and some acid. Dimethylformamide (0.2ml) was added to the reaction mixture. The acid dissolved after 30 minutess. LCMS showed corresponding methyl ester and no starting material peak was observed. The solvent was removed and dried in vacuo to afford the crude 5-Bromo-pyrimidine-2-carbonyl chloride (55g, 100%). The 5-Bromo-pyrimidine-2-carbonyl chloride (55g, 250mmol) was dissolved in tetrahydrofuran (828ml) and dimethyl-amine (2M solution in tetrahydrofuran) (373ml, 745mmol) was added portionwise at room temperature. The reaction was stirred at room temperature under nitrogen for 16 hours, after which time, LCMS indicated completion. The mixture was diluted with ethyl acetate (500ml) and washed with H2O (500ml). The water layer was further extracted with CH2CI2 (5x500ml), all organics combined, and dried over magnesium sulfate. The filtrate was concentrated in vacuo and then suspended in methyl-/-butylether (650ml). The solution was then heated to reflux. The hot solution was allowed to cool overnight to afford pink crystals. The crystals were filtered and washed with cold methyl-t-butylether (100ml) the solid was dried in a vacuum oven at 550C for 12 hourrs to afford the title compound 5-bromo-N,N-dimethylpyhmidine-2-carboxamide (SM-2: 44g, 77%) as a pink solid.
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 2.94 (s, 3 H) 3.13 (s, 3 H) 8.85 (s, 2 H) m/z (M+1 ) = 232.
Preparation of Intermediate Ethyl 4-(2-(dimethylcarbamoyl)Dyrimidin-5- yloxy)-2-methylbenzofuran-6-carboxylate (l-2a):

A mixture of Cs2CO3 (62.1 g, 191 mmol), 5-bromo-N,N- dimethylpyrimidine-2-carboxamide (SM-2: 24g, 104mmol) and ethyl 4- hydroxy-2-methylbenzofuran-6-carboxylate (1-1 c: 2Og, 91 mmol); 1 ,10- phenanthroline (1.64g, 9.07mmol) and copper iodide (864mg, 4.54mmol) in dimethylformamide (200ml) was purged with N2 gas and then heated to 90°C using a mechanical stirrer. The heterogeneous reaction mixture was stirred at this temperature for 18 hours. HPLC indicated near completion. The reaction mixture was cooled to 350C and diluted with ethyl acetate (300ml). The mixture was filtered to remove any cesium carbonate. The filtrate was then partitioned between water (500ml) and ethyl acetate (500ml); however, no separation was observed. Concentrated HCL (20ml) was added to the mixture. When the aqueous phase was about pH1 , the phases separated. The organics were separated and the aqueous layer reextracted with ethyl acetate (2x500ml). All organics were combined and back extracted with water (200ml) and brine (500ml). The organics were separated and treated with activated charcoal (10g) and magnesium sulfate. The mixture was allowed to stir for 10 minutes and then filtered through a plug of celite to afford a crude yellow solution. The filter cake was washed with ethyl acetate (100 ml_). The organics were concentrated in vacuo to afford a crude solid this was dried under high vacuum for 4 days. The dry crude solid was triturated using methanol (80 ml_). The solids were dispersed into a fine light orange crystalline powder with a red liquor. The solids were isolated by filtration and rinsed with methanol (20 ml_). The solid was dried in the vacuum oven at 550C for 12 hours to afford ethyl 4-(2- (dimethylcarbamoyl)pyrimidin-5-yloxy)-2-methylbenzofuran-6-carboxylate (J1 2a) as a yellow solid (18.2g, 54%)
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.41 (t, J=7.12 Hz, 3 H) 2.50 (d, J=0.98 Hz, 3 H) 3.00 (s, 3 H) 3.17 (s, 3 H) 4.41 (d, J=7.22 Hz, 2 H) 6.29 (s, 1 H) 7.62 (d, J=1.17 Hz, 1 H) 8.06 (s, 1 H) 8.50 (s, 2 H). m/z (M+1 ) = 370.5
Preparation of Starting material 5-bromo-N-ethyl-N-methylpyrimidine-2- carboxamide (SM-3):

(SM-3) Oxalyl chloride (1 .45g, 1 1 .1 mmol) was added to a suspension of 5-
Bromo-pyrimidine-2-carboxylic acid (1 .5g, 7.4mmol) in dichloromethane (50ml) at room temperature followed by 1 -2 drop of dimethylformamide. The reaction mixture was stirred under nitrogen for 2 hours LCMS in methanol indicated the presence of the methyl ester and some acid. Dimethylformamide (0.2ml) was added to the reaction mixture and all of the acid dissolved after 30 minutes. LCMS showed corresponding methyl ester and no starting material peak was observed. The solvent was removed and dried in vacuo to afford the crude 5-Bromo-pyrimidine-2-carbonyl chloride (1 -6g). 5-Bromo-pyrinnidine-2-carbonyl chloride (1600mg, 7.225mnnol) was dissolved in dichloromethane (25ml) and triethylamine (4.03ml, 28.9mmol) was added followed by ethyl-methyl-amine (0.68 mL, 7.92 mmol). The reaction was stirred at room temperature under nitrogen for 16 ours, after which time, LCMS indicated completion. The mixture was diluted with dichloromethane (50ml) and washed with water (50ml) followed by 10% citric acid (50ml) and brine (50ml). The organic layer was separated and dried over MgSO4, the residue was filtered and the solvent was removed in vacuo to afford the title compound 5-bromo-N-ethyl-N-methylpyrimidine-2- carboxamide (SM-3): (1.4g, 79.4%) as a brown oil.
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.08 – 1.31 (m, 3 H) 2.99 (d, J=79.05 Hz, 3 H) 3.19 (q, J=7.22 Hz, 1 H) 3.59 (q, J=7.22 Hz, 1 H) 8.84 (d, J=3.12 Hz, 2 H)
Example 2
Preparation of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2- yl)carbamoyl)-benzofuran-4-yloxy)Dyrimidine-2-carboxamide (2):

(2)
To a solution of the 5-methyl-2-aminopyrazine (38.9 g, 356 mmol) in dimethylether (315 ml_) in a 3-neck flask equipped with overhead stirring and a condensor at O0C was added Me2AICI (1 M solution in hexanes) (715 ml_). The mixture was warmed at room temperature and stirred for 1.5 hours. In a separate flask, ethyl 4-(2-(dimethylcarbamoyl)pyrimidin-5-yloxy)-2- methylbenzofuran-6-carboxylate (l-2a: 52.6g, 142.5mmol) was dissolved in dimethylether (210 ml_). This mixture was then added to the complexed amine. A gum precipitated upon scratching the flask and dissipated into a solid. The resultant reaction was refluxed for 3.5 hours HPLC indicated 93% complete. Five liters of Rochelles salt made up in water and 2 liters of 2- methyltetrahydrofuran was added to the mixture. The reaction mixture was then poured into the biphasic system. The mixture was allowed to stir with overhead stirring for 14 hours, after which time, a yellow solid precipitated. The solid was collected through filteration. The solid retained was washed with 2-methyltetrahydrofuran. The resultant solid was dried in vacuo oven overnight to afford the title compound N,N-dimethyl-5-(2-methyl-6-((5- methylpyrazin-2-yl)carbamoyl)benzofuran-4-yloxy)pyhmidine-2-carboxamide (2): (49.98g, 81 %)
1H NMR (400 MHz, CHLOROFORM-d) d ppm 2.49 (d, J=1 .17 Hz, 3H) 2.55 (s, 3H) 2.98 (s, 3 H) 3.14 (s, 3 H) 6.28 (t, J=0.98 Hz, 1 H) 7.52 (d, J=1 .37 Hz, 1 H) 7.88 – 7.92 (m, 1 H) 8.14 (d, J=0.78 Hz, 1 H) 8.37 (s, 1 H) 8.50 (s, 2 H) 9.54 (d, J=1 .56 Hz, 1 H).
m/z (M+1 ) = 433.4, m/z (M-1 )= 431 .5
REFERENCES
Beebe, D.A.; Ross, T.T.; Rolph, T.P.; Pfefferkorn, J.A.; Esler, W.P.
The glucokinase activator PF-04937319 improves glycemic control in combination with exercise without causing hypoglycemia in diabetic rats
74th Annu Meet Sci Sess Am Diabetes Assoc (ADA) (June 13-17, San Francisco) 2014, Abst 1113-P
Amin, N.B.; Aggarwal, N.; Pall, D.; Paragh, G.; Denney, W.S.; Le, V.; Riggs, M.; Calle, R.A.
Two dose-ranging studies with PF-04937319, a systemic partial activator of glucokinase, as add-on therapy to metformin in adults with type 2 diabetes
Diabetes Obes Metab 2015, 17(8): 751
Study to compare single dose of three modified release formulations of PF-04937319 with immediate release material-sparing-tablet (IR MST) formulation previously studied in adults with type 2 diabetes mellitus (NCT02206607)
OTHERS

///////////Pfizer , PF 04937319, glucokinase activators, Type 2 diabetes
TARANABANT

Taranabant ( MK-0364)
701977-09-5
N-[3-(4-Chlorophenyl)-2(S)-(3-cyanophenyl)-1(S)-methylpropyl]-2-methyl-2-[5-(trifluoromethyl)pyridin-2-yloxy]propionamide
N-[(2S,3S)-4-(4-chlorophenyl)-3-(3-cyanophenyl)butan-2-yl]-2-methyl-2-[5-(trifluoromethyl)pyridin-2-yl]oxypropanamide
Taranabant (codenamed MK-0364) is a cannabinoid receptor type 1 inverse agonist being investigated as a potential treatment forobesity due to its anorectic effects.[1][2] It was discovered by Merck & Co.
In October 2008, Merck has stopped its phase III clinical trials with the drugs due to high level of central side effects, mainlydepression and anxiety.[3][4][5][6]
The compound had also been in clinical evaluation in chronic cigarette smokers as an aid for smoking cessation.
Paper
 .
.
http://pubs.rsc.org/en/content/articlelanding/2013/cs/c2cs35410a#!divAbstract
PATENT
WO 2003077847
http://www.google.co.in/patents/WO2003077847A2?cl=en
PAPERS
Convenient total synthesis of taranabant (MK-0364), a novel cannabinoid-1 receptor inverse agonist as an anti-obesity agent
Tetrahedron 2007, 63(52): 12845
Wallace, D.J.; Campos, K.R.; Shultz, S.; Klapars, A.; et al.
New efficient asymmetric synthesis of taranabant, a CB1R inverse agonist for the treatment of obesity
Org Process Res Dev 2009, 13(1): 84
Lin, L.S.; Lanza, T.J. Jr.; Jewell, J.P.; Liu, P.; Shah, S.K.; Qi, H.; Tong, X.; Wang, J.; Xu, S.S.; Fong, T.M.; Shen, C.P.; Lao, J.; Xiao, J.C.; Shearman, L.P.; Stribling, D.S.; Rosko, K.; Strack, A.; Marsh, D.J.; Feng, Y.; Kumar, S.; Samuel, K.; Yin, W.; Ploeg, L.H.; Goulet, M.T.; Hagmann, W.K.
Discovery of N-[(1S,2S)-3-(4-Chlorophenyl)-2- (3-cyanophenyl)-1-methylpropyl]-2-methyl-2- [[5-(trifluoromethyl)pyridin-2-yl]oxy]propanamide (MK-0364), a novel, acyclic cannabinoid-1 receptor inverse agonist for the treatment of obesity
J Med Chem 2006, 49(26): 7584
Cole, P.; Serradell, N.; Rosa, E.; Bolos, J. Taranabant Drugs Fut 2008, 33(3): 206
PAPER
Chen, C.-Y.; Frey, L.F.; Shultz, S.; et al. Catalytic, enantioselective synthesis of taranabant, a novel, acyclic cannabinoid-1 receptor inverse agonist for the treatment of obesity
Org Process Res Dev 2007, 11(3): 616
http://pubs.acs.org/doi/abs/10.1021/op700026n

Chiral amide 1 (MK-0364, taranabant) is a potent, selective, and orally bioavailable cannabinoid-1 receptor (CB-1R) inverse agonist indicated for the treatment of obesity. An asymmetric synthesis featuring a dynamic kinetic resolution via hydrogenation for the preparation of the bromo alcohol 5 is disclosed. Conversion of the alcohol intermediate to the chiral amide 1 is accomplished in good overall yield.
N-[(1S,2S)-3-(4-Chlorophenyl)-2-(3-cyanophenyl)-1-methylpropyl]-2-methyl-2-{[5-(trifluoromethyl)pyridin-2-yl]oxy}propanamide (1, MK-0364). hemisolvate (approximately 94 wt %, 94% isolated yield from amine salt).
1H NMR (CDCl3): δ 8.35 (s, 1H), 7.83 (dd, J = 2.38, 8.70 Hz, 1H), 7.45 (d, J = 7.57 Hz, 1H), 7.31 (t, J = 7.99 Hz, 1H), 7.24 (m, 2H), 7.07 (d, J = 8.34 Hz, 2H), 6.88 (d, J = 8.63 Hz, 1H), 6.72 (d, J = 8.33 Hz, 2H), 5.88 (d, J = 8.95 Hz, 1H), 4.34 (m, 1H), 3.13 (dd, J = 3.04, 12.72 Hz, 1H), 2.82 (m, 2H), 1.76 (s, 3H), 1.72 (s, 3H), 0.87 (d, J = 6.72 Hz, 3H).
13C NMR (CDCl3): δ 173.4, 163.9, 144.5 (q, J = 4.30 Hz), 142.4, 137.5, 136.3 (q, J = 3.02 Hz), 133.0, 132.2, 132.0, 130.7, 130.0, 129.3, 128.5, 123.7 (q, J = 271.45 Hz), 121.1 (q, J = 33.32 Hz), 118.6, 112.7, 112.6, 82.1, 53.6, 48.6, 38.2, 25.4, 25.1, 18.4.
Anal. Calcd for C27H25ClF3N3O2: C 62.85; H 4.88; N 8.14. Found: C 62.95; H 4.74; N 8.00.
References
- Armstrong HE, Galka A, Lin LS, Lanza TJ Jr, Jewell JP, Shah SK, et al. “Substituted acyclic sulfonamides as human cannabinoid-1 receptor inverse agonists.” Bioorganic & Medicinal Chemistry Letters. 2007 Apr 15;17(8):2184-7. PMID 17293109. doi:10.1016/j.bmcl.2007.01.087
- Fong TM, Guan XM, Marsh DJ, Shen CP, Stribling DS, Rosko KM, et al. “Antiobesity efficacy of a novel cannabinoid-1 receptor inverse agonist, N-[(1S,2S)-3-(4-chlorophenyl)-2-(3-cyanophenyl)-1-methylpropyl]-2-methyl-2-[[5-(trifluoromethyl)pyridin-2-yl]oxy]propanamide (MK-0364), in rodents.” Journal of Pharmacology and Experimental Therapeutics. 2007 Jun;321(3):1013-22. PMID 17327489.doi:10.1124/jpet.106.118737
- “Press release by Merck”. Retrieved October 2008.
- Aronne LJ, Tonstad S, Moreno M, Gantz I, Erondu N, Suryawanshi S, Molony C, Sieberts S, Nayee J, Meehan AG, Shapiro D, Heymsfield SB, Kaufman KD, Amatruda JM (May 2010). “A clinical trial assessing the safety and efficacy of taranabant, a CB1R inverse agonist, in obese and overweight patients: a high-dose study”. International Journal of Obesity (2005) 34 (5): 919–35. doi:10.1038/ijo.2010.21.PMID 20157323.
- Kipnes MS, Hollander P, Fujioka K, Gantz I, Seck T, Erondu N, Shentu Y, Lu K, Suryawanshi S, Chou M, Johnson-Levonas AO, Heymsfield SB, Shapiro D, Kaufman KD, Amatruda JM (June 2010). “A one-year study to assess the safety and efficacy of the CB1R inverse agonist taranabant in overweight and obese patients with type 2 diabetes”. Diabetes, Obesity & Metabolism 12 (6): 517–31. doi:10.1111/j.1463-1326.2009.01188.x. PMID 20518807.
- Proietto J, Rissanen A, Harp JB, Erondu N, Yu Q, Suryawanshi S, Jones ME, Johnson-Levonas AO, Heymsfield SB, Kaufman KD, Amatruda JM (August 2010). “A clinical trial assessing the safety and efficacy of the CB1R inverse agonist taranabant in obese and overweight patients: low-dose study”. International Journal of Obesity (2005) 34 (8): 1243–54. doi:10.1038/ijo.2010.38. PMID 20212496.
| Radiolabeled cannabinoid-1 receptor modulators [US7754188] | 2006-06-01 | 2010-07-13 |
| Combination therapy for the treatment of obesity [US2006270650] | 2006-11-30 | |
| CERTAIN CRYSTALLINE DIPHENYLAZETIDINONE HYDRATES, PHARMACEUTICAL COMPOSITIONS THEREOF AND METHODS FOR THEIR USE [US8003636] | 2009-08-13 | 2011-08-23 |
| NOVEL DIPHENYLAZETIDINONE SUBSTITUTED BY PIPERAZINE-1-SULFONIC ACID AND HAVING IMPROVED PHARMACOLOGICAL PROPERTIES [US2009264402] | 2009-10-22 | |
| Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, process for preparing them, medicaments comprising these compounds, and their use [US7759366] | 2009-08-27 | 2010-07-20 |
| COMPOUNDS WITH A COMBINATION OF CANNABINOID CB1 ANTAGONISM AND SEROTONIN REUPTAKE INHIBITION [US8138174] | 2008-09-04 | 2012-03-20 |
| Substituted imidazoline-2,4-diones, process for preparation thereof, medicaments comprising these compounds and use thereof [US2011112097] | 2011-05-12 | |
| Novel phenyl-substituted imidazolidines, process for preparation thereof, medicaments comprising said compounds and use thereof [US2011178134] | 2011-07-21 | |
| HETEROCYCLIC COMPOUNDS, PROCESSES FOR THEIR PREPARATION, MEDICAMENTS COMPRISING THESE COMPOUNDS, AND THE USE THEREOF [US2011183998] | 2011-07-28 | |
| Cyclic pyridyl-N-[1,3,4]-thiadiazol-2-yl-benzene sulfonamides, processes for their preparation and their use as pharmaceuticals [US2011224263] | 2011-09-15 |
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-[(2S,3S)-4-(4-chlorophenyl)-3-(3-cyanophenyl)-2-butanyl]-2-methyl-2-{[5-(trifluoromethyl)-2-pyridinyl]oxy}propanamide
|
|
| Clinical data | |
| Routes of administration |
Oral |
| Identifiers | |
| CAS Number | 701977-09-5 |
| ATC code | A08AX |
| PubChem | CID: 11226090 |
| UNII | X9U622S114 |
| Chemical data | |
| Formula | C27H25ClF3N3O2 |
| Molecular mass | 515.95 g/mol |
///////////CC(C(CC1=CC=C(C=C1)Cl)C2=CC=CC(=C2)C#N)NC(=O)C(C)(C)OC3=NC=C(C=C3)C(F)(F)F
C[C@@H]([C@@H](CC1=CC=C(C=C1)Cl)C2=CC=CC(=C2)C#N)NC(=O)C(C)(C)OC3=NC=C(C=C3)C(F)(F)F
Revised USP Chapter on Visual Inspection published
DRUG REGULATORY AFFAIRS INTERNATIONAL

After the long-awaited Chapter <1790> on visual inspection of injections was first published in the PF 41(1) as a draft the USP has now submitted a revised draft in the PF41 (6). Read more about the revised draft of the USP Chapter <1790>.
After the long-awaited Chapter <1790> Visual Inspection of Injections was first published in the Pharmacopeial Forum 41(1) as a draft the USP has now submitted a revised draft in the PF41 (6). Through its number >1000, the monograph <1790> is not binding but rather offers an explanation to Chapter <790> Visible Particulates in Injections.
With regard to the content, several comments recommended by the industry have been included. The new Chapter 9 represents the biggest change and describes the evaluation of marketed products where anomalies had been observed regarding particles. The test procedure for it is described in Chapter <790>. Yet, this topic was missing…
View original post 149 more words