November 1, 2018
Release
The U.S. Food and Drug Administration today is warning patients and doctors, who use at-home or in-the-office medical devices to monitor levels of…
WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
Home » Uncategorized (Page 32)
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
DRUG REGULATORY AFFAIRS INTERNATIONAL
tatement from FDA Commissioner Scott Gottlieb, M.D., on findings from the romaine lettuce E. coli O157:H7 outbreak investigation and FDA’s efforts to prevent future outbreaks
|
November 1, 2018
Earlier this year, we experienced the largest E. coli
View original post 1,168 more words
DRUG REGULATORY AFFAIRS INTERNATIONAL
FDA warns patients and doctors about risk of inaccurate results from home-use device to monitor blood thinner warfarin
The U.S. Food and Drug Administration today is warning patients and doctors, who use at-home or in-the-office medical devices to monitor levels of the blood thinner, warfarin, that certain test strips used with the devices may provide inaccurate results and should not be relied upon to adjust the drug dosage. Roche Diagnostics issued a voluntary recall of certain test strip lots used with its CoaguChek test meter devices. The recall involves more than 1.1 million packages of CoaguChek XS PT Test Strips that were distributed nationwide from Jan. 12, 2018 to Oct. 29, 2018. Today, the FDA announced this action as…Continue reading
November 1, 2018
The U.S. Food and Drug Administration today is warning patients and doctors, who use at-home or in-the-office medical devices to monitor levels of…
View original post 768 more words

Pirlindole (Lifril, Pyrazidol) is a reversible inhibitor of monoamine oxidase A (RIMA) which was developed and is used in Russia as an antidepressant.[1]:337 It is structurally and pharmacologically related to metralindole.
Biovista is investigating BVA-201, a repurposed oral formulation of pirlindole mesylate, for the potential treatment of multiple sclerosis
SYN 1

SYN 2

PAPER
Khimiko-Farmatsevticheskii Zhurnal (1986), 20(3), 300-3.
PATENT
U.S.S.R. (1986), SU 276060
PAPER
Sudebno-meditsinskaia ekspertiza (1989), 32(4), 49-50
PAPER
Journal of Pharmaceutical and Biomedical Analysis
https://www.sciencedirect.com/science/article/abs/pii/S0731708598002131
PATENT
claiming method for resolving racemic mixture of pirlindole hydrochloride into enantiomerically pure (S)-pirlindole and/or (R)-pirlindole,
Pirlindole, 2, 3, 3a, 4, 5, 6-hexahydro-lH-8-methyl-pyrazine
[3, 2, 1-j , k] carbazole, is a tetracyclic compound of the formula I
(I)
Pirlindole is a reversible monoamine oxidase A inhibitor being up to date useful as a medicament in the treatment of depression.
Pirlindole has an asymmetric carbon atom which implies that there are two enantiomers, (S) -pirlindole and (R) -pirlindole .
The state of the art teaches several methods for the enantiomeric separation of pirlindole. For example, The Journal of Pharmaceutical and Biomedical Analysis, 18(1998) 605- 614, “Enantiomeric separation of pirlindole by liquid chromatography using different types of chiral stationary phases”, Ceccato et al, discloses the enantiomeric separation of pirlindole by liquid chromatography (LC) using three different chiral stationary phases.
Further, The Journal of Pharmaceutical and Biomedical Analysis 27(2002) 447-455, “Automated determination of pirlindole enantiomers in plasma by on-line coupling of a pre-column packed with restricted access material to a chiral liquid chromatographic column”, Chiap et al., discloses the use of a pre-column packed with restricted access material for sample clean up coupled to a column containing a cellulose based chiral stationary phase for separation and quantitative analysis of the enantiomers .
According to the prior art, Chirality 11:261-266 (1999) all attempts to obtain the enantiomers of pirlindole by selective crystallization with optically active acids failed, and it was only possible to obtain at laboratory scale (few grams) as hydrochloride salt, using derivatization technique in conjunction with preparative chromatography.
The characteristics of the process disclosed in the state of the art limit in a definitive way, its implementation on an industrial or semi-industrial scale due to the necessity to use a separation by chromatography on a large scale which makes the process very costly, difficult to implement and with poor reproducibility. .
EXAMPLE 7
(R) -Pirlindole mesylate
Starting from 10 g of (R) -pirlindole (S) -mandelate obtained in Example 1 and following the procedure described in Example 5 using methanesulfonic acid as pharmaceutical acceptable acid, ,
7.4 g (0.023 mole) of (R) -pirlindole mesylate were obtained (yield = 85.2% ). Chiral HPLC (enantiomeric purity = 98.0%).
XAMPLE 9
(S) -pirlindole mesylate
Starting from 10 g of (S) -pirlindole (R) -mandelate obtained in Example 2 and following the procedure described in Example 6 using methanesulfonic acid as pharmaceutical acceptable acid, 6.8 g (0.021 mole) of (S) -pirlindole mesylate were obtained (yield = 77.8%). Chiral HPLC (enantiomeric purity = 98.0%).
PATENT
Process for the preparation of pirlindole . useful for treating depression.
Pirlindole (8-methyl-2,3,3a,4,5,6-hexahydro-lH-pyrazino[3,2,l-jk]carbazole) of formula I
Compound Formula I
also described as Pyrazidole™ represents a new class of original tetracyclic antidepressants, the pyrazinocarbazole derivatives. The drug was synthesized and characterized at the end of the 1960s and was marketed as an anti-depressant in 1975. Current clinical trials have demonstrated to be a highly effective short-acting and safe drug.
[0003] Pirlindole is a selective, reversible inhibitor of MAO-A. In-vitro evidence suggest the catalytic oxidation of Pirlindole into dehydro-pirlindole by MAO-A. Dehydro-pirlindole may be a more potent slowly reversible inhibitor of MAO-A and this might explain the persistence of MAO-A inhibition in-vivo (MAO-The mother of all amine oxidases, John P.M. Finberg et al. 1998, Springer).
[0004] Pirlindole chemical structure is composed of one stereogenic centre which indicates the existence of two enantiomers, the ( ?)-Pirlindole and the (S)-Pirlindole.
[0005] Although Pirlindole pharmacological data and the clinical use were performed on the racemate, recently there have been increasing interest in the pharmacological profile of each enantiomer (WO 2015/171005 Al).
[0006] International patent publication WO 2015/171003A1 filed 9th May 2014 discloses a resolution of racemic pirlindole into optically active pirlindole. The Resolution-Racemization-Recycle (RRR) synthesis described involves derivatization by preparation of pairs of diastereomers in the form of salts from an optically active organic acid. These diastereomers can be separated by conventional techniques such as crystallisation. Although it is a very efficient procedure to prepare laboratorial scale or pre-clinical batch of (/?)- or (S)-Pirlindole, it is not economically convenient at an industrial scale because the process relies on Pirlindole racemate as the starting material.
[0007] Andreeva et al. (Pharmaceutical Chemistry 1992, 26., 365-369) discloses the first isolation of Pirlindole enantiomers in isolated form. ( ?)-Pirlindole of formula II
was isolated as an hydrochloride salt from a racemic base by the fractional crystallization of racemic pirlindole salt with (+)-camphor-10-sulfonic acid. (S)-Pirlindole formula III
was also isolated as an hydrochloride salt although via asymmetric synthesis from the 6-methyl-2,3,4,9-tetrahydro-lH-carbazol-l-one IV
[0008] Compound of formula IV was reacted with chiral auxiliary (S)-(-)-a-methylbenzylamine to afford asymmetric (S)-6-methyl-N-(l-phenylethyl)-2,3,4,9-tetrahydro-lH-carbazol-l-imine V
[0009] Compound of formula V was subjected to stereoselective reduction with sodium borohydride in ethanol. According to Andreeva et al. the reaction might occur through directed intramolecular hydride transfer after formation of a complex between compound of formula V and reducing agent to afford (S)-6-methyl-N-((S)-l-phenylethyl)-2,3,4,9-tetrahydro-lH-carbazol-l-amine VI
[0010] Compound of formula VI is reacted with ethylene glycol ditosylate by ethylene bridge formation under alkaline conditions to yield (S)-8-methyl-3-((S)-l-phenylethyl)-2,3,3a,4,5,6-hexahydro-lH-pyrazino[3,2,l-jk]carbazole VII.
[0011] Alkaline agent is sodium hydride (NaH), in the presence of dimethyl sulfoxide (DMSO) or dimethylformamide (DMF).
[0012] The ratio between alkaline agent, compound of formula VI and ethylene glycol ditosylate is 1.2:1:1.
[0013] The cyclization reaction occurs at room temperature for a period of 4.5 hours. [0014] Compound of formula VII was subjected to catalytic hydrogenolysis conditions to afford the desired hydrochloride salt of compound of formula III.
[0015] The hydrogenolysis reaction was catalysed by Palladium on charcoal (Pd content 0.1 g, 9 mol%) and was conducted in methanol. The conversion of compound of formula VII into compound of formula III was performed under a hydrogen pressure of 1.8-2.0 MPa at 22 °C for a period of 17h.
[0016] The work-up conditions for the hydrogenolysis reaction involved neutralization with ammonia solution followed by benzene recrystallization. The hydrochloride salt of compound of formula III was formed from addition of hydrochloric acid to a solution of free base in ethanol.
[0017] The process yielded (S)-Pirlindole hydrochloride with a final yield of 10% with respect to the intermediate VI.
[0018] The mixture of sodium hydride with DMSO generates dimsyl anion. This anion is very often used in laboratory scale, but because it is unstable its use on large scale should be under specific precautions. Dimsyl anion decomposition is exotermic. It is reported that dimsyl anion decomposition starts even at 20 °C, and above 40 °C it decomposes at an appreciable rate (Lyness, W. I. et ai, U.S. 3,288,860 1966, CI. 260-607).
[0019] The mixture of DMF and sodium hydride is reported in ‘Sax & Lewis’s Dangerous Properties of Industrial Materials’ to give a violent reaction with ignition above 50 °C. Buckey, J. et ai, Chem. Eng. News 1982, 60(28), 5, describes the thermal runaway of a pilot plant reactor containing sodium hydride and DMF from 50 °C. Accelerated Rate Calorimetry (ARC) tests showed exothermic activity as low as 26 °C. Similar behaviour was also seen with DMA. De Wall, G. et ai, Chem. Eng. News 1982, 60(37), 5, reports a similar incident, wherein runaway started at 40 °C, and rose 100 °C in less than 10 minutes, boiling off most of the DMF.
[0020] There exists a need for safe, industrial- and eco-friendly processes for the preparation of Pirlindole enantiomers. These facts are disclosed in order to illustrate the technical problem addressed by the present disclosure.
[0068] In an embodiment, the preparation of (S)-8-methyl-3-((S)-l-phenylethyl)-2,3,3a,4,5,6-hexahydro-lH-pyrazino[3,2,l-jk]carbazole, compound of formula VII was carried out as follow.
[0069] In an embodiment, in a 2 L three necked round bottomed flask equipped with magnetic stirrer, ethylene glycol ditosylate (73 g, 197 mmol) and DMI (240 mL) were loaded. To the resulting clear solution, NaH (60% suspension in mineral oil, 15.8 g, 394 mmol) was added carefully. To the resulting suspension a solution of VI ((S)-6-methyl-N-((S)-l-phenylethyl)-2,3,4,9-tetrahydro-lH-carbazol-l-amine) (30 g, 98.5 mmol) in DMI (60 mL) was added dropwise at 60 °C. The mixture was stirred for 1 h at 60 °C. The mixture was cooled down to room temperature, then MeOH was added slowly with ice-water cooling. A white precipitation appeared, and the resulting suspension was stirred and then filtered. The filtered product was washed with water-MeOH. The product was dried under vacuum to give 24.9 g of compound of formula VII (75.2 mmol, yield: 76%). Purity >99.9area% (HPLC).
[0070] In an embodiment, the preparation of hydrochloride salt of (S)-Pirlindole, compound of formula III, was performed as follow.
[0071] In an embodiment, the free amine VII ((S)-8-methyl-3-((S)-l-phenylethyl)-2,3,3a,4,5,6-hexahydro-lH-pyrazino[3,2,l-jk]carbazole) (8,32 g, 25 mmol) was dissolved in DCM (42 mL) and excess of HCI in MeOH (42 mL) was added. The solvents were evaporated under reduced pressure to dryness to give a yellow oil. The residue was dissolved in MeOH (120 mL) and was added to the dispersion of Pd/C (1,74 g, -50% water) in MeOH (20 mL). The reaction mixture was stirred at 50 °C under a 750 KPa (7.5 bar) pressure of hydrogen for 5h. After completion (HPLC) the suspension was filtered through a celite pad, and the filter cake was washed with MeOH. The pH of the resulting solution was checked (<3) and it was evaporated to give the crude hydrochloride salt of compound of formula III. To the crude material iPrOH was added and the suspension was allowed to stir at reflux. The suspensions were filtered, and the product was dried under vacuum to give the hydrochloride salt of (S)-Pirlindole, compound of formula III (5.11 g, 19.5 mmol, yield: 77%). Purity > 99.5% (HPLC). Enantiomeric purity 99.5% (Chiral HPLC). MS (ESI): m/z 227.2 (M+H)+.
PATENT
Process for the preparation of piperazine ring for the synthesis of pyrazinocarbazole derivatives, such as the antidepressant pirlindole .
Pirlindole hydrochloride is the compound represented in formula I
[0003] It is the common name of 8-methyl-2,3,3a,4,5,6-hexahydro-lH-pyrazino[3,2,l-jk]carbazole hydrochloride which is an active pharmaceutical ingredient marketed with the name Pyrazidol™. The compound is effective as an anti-depressant agent.
[0004] Pirlindole chemical structure belongs to the pyrazinocarbazole group. It is composed of one stereogenic centre which anticipate the existence of two enantiomers, the ( ?)-Pirlindole of formula II and the (S)-Pirlindole of formula III.
[0005] Although Pirlindole pharmacological data and the clinical use were performed on the racemate, recently there have been increasing interest in the pharmacological profile of each enantiomer (WO 2015/171005 Al).
[0006] The document WO 2015/171003Al(Tecnimede group) filed 9th May 2014 discloses a resolution of racemic pirlindole into optically active pirlindole. The Resolution-Racemization-Recycle (RRR) synthesis described involves derivatization by preparation of pairs of diastereomers in the form of salts from an optically active organic acid. These diastereomers can be separated by conventional techniques such as crystallisation. Although it is a very efficient procedure to prepare laboratorial scale or pre-clinical batch of (/?)- or (S)-Pirlindole, it is not economically convenient at an industrial scale because the process relies on Pirlindole racemate as the starting material.
[0007] Processes to prepare Pirlindole involve the formation of a piperazine ring. The state of the art discloses different processes for piperazine ring formation but they are generally a multistep approach, and they are hampered by low yields, expensive reagents, or are reported as unsuccessful (Roderick et al. Journal of Medicinal Chemistry 1966, 9, 181-185).
[0008] The first asymmetric synthesis of Pirlindole enantiomers described by Andreeva et al. (Pharmaceutical Chemistry 1992, 26, 365-369) discloses a one-step process to prepare pyrazinocarbazole piperazine ring system from a tetrahydrocarbazole-amine. The process discloses a very low yield (23.8 %) and employs the use of sodium hydride (NaH) in the presence of dimethyl sulfoxide (DMSO) or dimethyl formamide (DMF), both conditions described as generating exothermic decomposition that can cause reaction ignition or reaction thermal runaway.
[0009] The mixture of sodium hydride with DMSO generates dimsyl anion. This anion is very often used in laboratory scale, but because it is unstable its use on large scale should be under specific precautions. The dimsyl anion decomposition is exothermic. It is reported that dimsyl anion decomposition starts even at 20 °C, and above 40 °C it decomposes at an appreciable rate (Lyness et al. US 3288860).
[0010] The mixture of DMF and sodium hydride is reported in Sax & Lewis’s Dangerous Properties of Industrial Materials to give a violent reaction with ignition above 50 °C. Buckey et al., (Chemical & Engineering News, 1982, 60(28), 5) describes the thermal runaway of a pilot plant reactor containing sodium hydride and DMF from 50 °C. Accelerated Rate Calorimetry (ARC) tests showed exothermic activity as low as 26 °C.
Similar behaviour was also seen with DMA. De Wall et al. (Chem. Eng. News, 1982, 60(37), 5) reports a similar incident, wherein runaway started at 40 °C, and rose 100 °C in less than 10 minutes, boiling off most of the DMF.
[0011] An alternative process for the preparation of a piperazine ring system of a pyrazinocarbazole derivative can involve the formation of a lactam ring in a three steps approach:
1. N-acylation reaction;
2. intramolecular indole acetamide cyclisation to afford a lactam ring;
3. lactam reduction.
[0012] Intramolecular indole chloroacetamide cyclization to yield a lactam ring has been described by Bokanov et al. (Pharmaceutical Chemistry Journal 1988, 23, 12, 1311-1315) particularly in the non-enantioselective synthesis of pyrazinocarbazolone derivatives. Bokanov et al. did not describe the lactam reduction into a piperazine ring.
[0013] Intramolecular indole chloroacetamide cyclization to yield a lactam ring has also been described both by Rubiralta et al. (Journal of Organic Chemistry 54, 23, 5591-5597) and Bennasar, et al. (Journal of Organic Chemistry 1996., 61, 4, 1239-1251), as an unexpected outcome of a photocyclization reaction. The lactam conversion was low (<11% yield).
[0014] Lactam reduction of a pyrazinone into piperazine ring systems is disclosed both by Aubry et al. (Biorganic Medicinal Chemistry Letters 2007, 17, 2598-2602) and Saito et al. (Tetrahedron 1995, 51, 30, 8213-8230) in the total synthesis of alkaloid natural products.
[0015] There exists the need for improved processes for the preparation of piperazine ring derivatives in particular enantioselective processes for the preparation of pyrazinocarbazole intermediates precursors of Pirlindole enantiomers compounds of formula II and III.
Example 1 – Preparation of (S)-8-methyl-3-((S)-l-phenylethyl)-3a,4,5,6-tetrahydro-lH-pyrazino[3,2,l-jk]carbazol-2(3H)-one – Formula IV
[00106] In an embodiment, the preparation of (S)-8-methyl-3-((S)-l-phenylethyl)-3a,4,5,6-tetrahydro-lH-pyrazino[3,2,l-jk]carbazol-2(3H)-one (Formula IV) was carried out as follows. To the solution of VI (S)-6-methyl-N-((S)-l-phenylethyl)-2,3,4,9-tetrahydro-lH-carbazol-l-amine (30 g, 98.5 mmol) in toluene (300 mL), 50 % (w/v) aqueous NaOH (79 g) was added dropwise at 0-5 °C, then the solution of chloroacetyl
chloride (12 mL, 148 mmol, 1.5 equiv.) in toluene (15 mL) was added dropwise at 0-5 °C. The mixture was stirred at 0-5 °C for approximately 2.5 h, and additional chloroacetyl chloride (12 mL, 148 mmol, 1.5 equiv.) in toluene (15 mL) was added dropwise at 0-5 °C. The mixture was stirred at 0-5 °C for approximately 1.5 h. Water was added to the reaction mixture keeping the temperature below 5 °C. The phases were separated, and the aqueous phase was extracted with toluene. The organic phase was treated with 2M aqueous HCI. The resulting suspension was filtered. The filtered solid was identified as the HCI salt of VI, which can be liberated and driven back to the chloroacetylation step. The phases of the mother liquor were separated, and the aqueous phase was extracted with toluene. The organic phase was dried over Na2S04, filtered and concentrated under reduced pressure to about 350 mL as a solution in toluene. The toluene solution of the crude product compound of formula X was reacted in the next step.
[00107] In an embodiment, in the same reaction vessel to the toluene solution of crude intermediate obtained in previous step were added TBAB (0.394 g, 1.22 mmol, 1 w/w% for the theoretical yield of prev. step) and 50 % (w/v) aqueous NaOH (8.1 g, 10 equiv.). The reaction mixture was stirred for 1 h at 65 °C, while the reaction was complete. Water was added to the mixture at 0 °C, and the phases were separated, the organic phase was washed with aqueous HCI, and with water, then dried over Na2S04, filtered and evaporated to give 32.87 g of compound IV (S)-8-methyl-3-((S)-l-phenylethyl)-3a,4,5,6-tetrahydro-lH-pyrazino[3,2,l-jk]carbazol-2(3H)-one (yield: 97% for the two steps) as a brown solid. The crude product was reacted in the next step without further purification.
Example 2 – Preparation of (S)-8-methyl-3-((S)-l-phenylethyl)-2,3,3a,4,5,6-hexahydro-lH-pyrazino[3,2,l-jk]carbazole _ Formula V
[00108] In an embodiment, the preparation of (S)-8-methyl-3-((S)-l-phenylethyl)-2,3,3a,4,5,6-hexahydro-lH-pyrazino[3,2,l-jk]carbazole (Formula V) was performed as follows. To the stirred solution of 32.87 g of IV, (S)-8-methyl-3-((S)-l-phenylethyl)-3a,4,5,6-tetrahydro-lH-pyrazino[3,2,l-jk]carbazol-2(3H)-one (95.4 mmol) in dry THF (170 mL) 66 mL solution of sodium bis(2-methoxyethoxy)aluminium hydride in toluene (70 w/w%, 237 mmol, 2.5 equiv.) was added dropwise. The reaction mixture was warmed to 40 °C, and the end of the addition the mixture was stirred at 50 °C until the total consumption of the starting material. Additional 22 mL of sodium bis(2-methoxyethoxy)aluminium hydride solution (70 w/w%, 79 mmol, 0.8 equiv.) was added dropwise. After completion the mixture was cooled to room temperature and 5% aqueous NaOH was added carefully. Water and DCM were added to the mixture, the phases were separated, and the aqueous phase was extracted with DCM. The organic phase was dried over Na2S04, filtered and the solvent was evaporated to get a brown solid (28.8 g). This crude product was dissolved in DCM and MeOH was added. White solid precipitated. The solid was filtered and washed with MeOH to give V (S)-8-methyl-3-((S)-l-phenylethyl)-2,3,3a,4,5,6-hexahydro-lH-pyrazino[3,2,l-j‘k]carbazole 14.6 g (yield: 46%) as an off-white cotton-like solid.
Example 3 – Preparation of (S)-Pirlindole Hydrochloride – Formula III
[00109] In an embodiment, the preparation of (S)-Pirlindole hydrochloride III was carried out as follows. The free amine V ((S)-8-methyl-3-((S)-l-phenylethyl)-2,3,3a, 4,5,6-hexahydro-lH-pyrazino[3,2,l-jk]carbazole) (8.32 g, 25 mmol) was dissolved in DCM (42 mL) and excess of HCI in MeOH (42 mL) was added. The solvents were evaporated under reduced pressure to dryness to give a yellow oil. The residue was dissolved in MeOH (120 mL) and was added to the dispersion of Pd/C (1.74 g, -50% water) in MeOH (20 mL). The reaction mixture was stirred at 50 °C under 750 KPa (7.5 bar) pressure of hydrogen for 5h. After completion (HPLC) the suspension was filtered through a celite pad, and the filter cake was washed with MeOH. The pH of the resulting solution was checked (<3) and it was evaporated to give the crude hydrochloride salt of compound of formula III. To the crude material iPrOH was added and the suspension was allowed to stir at reflux. The suspensions were filtered, and the product was dried under vacuum to give the hydrochloride salt of (S)-Pirlindole, compound of formula III (5.11 g, 19.5 mmol, yield: 77%). Purity > 99.5% (HPLC). Enantiomeric purity 99.5% (Chiral HPLC). MS (ESI): m/z 227.2 (M+H)+.
[00110] Table 1. Comparative yields
http://www.biomedsearch.com/nih/Pirlindole-in-treatment-depression-meta/21053988.html
General References
 |
|
| Clinical data | |
|---|---|
| Trade names | Pirazidol |
| Routes of administration |
Oral |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 20–30% |
| Protein binding | 95% |
| Metabolism | hepatic |
| Onset of action | 2 to 8 hours |
| Elimination half-life | 185 hours |
| Excretion | urine (50–70%), feces (25–45%) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C15H18N2 |
| Molar mass | 226.32 g/mol |
| 3D model (JSmol) | |
//////////////Pirlindole, Lifril, Pyrazidol, 60762-57-4, DEPRESSION
CC1=CC2=C(C=C1)N3CCNC4C3=C2CCC4
Chemists, engineers, scientists, lend us your ears… Carbon capture, utilisation, and storage (CCUS) is among the largest challenges on the horizon and we need your help. In this perspective, we focus on identifying the critical research needs to make CCUS a reality, with an emphasis on how the principles of green chemistry (GC) and green engineering can be used to help address this challenge. We identify areas where GC principles can readily improve the energy or atom efficiency of processes or reduce the environmental impact. Conversely, we also identify dilemmas where the…
View original post 115 more words


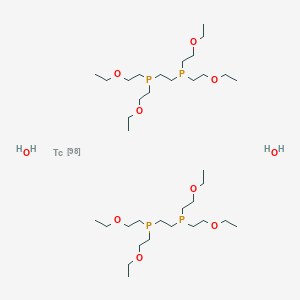
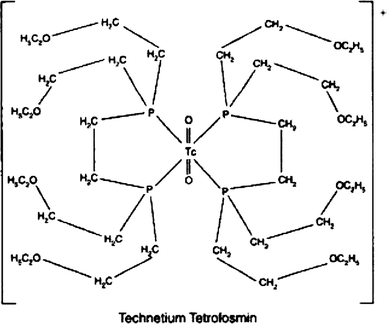
Technetium (99mTc) tetrofosmin, 99mTc-Tetrofosmin
テトロホスミンテクネチウム (99mTc)
| Formula | C36H80O10P4Tc |
|---|---|
| Molar mass | 895.813 g/mol |
| CAS Number |
|
|---|
UNII42FOP1YX93
2-[bis(2-ethoxyethyl)phosphanyl]ethyl-bis(2-ethoxyethyl)phosphane;technetium-98;dihydrate
Technetium Tc 99m tetrofosmin; Technetium Tc-99m tetrofosmin; TECHNETIUM TC-99M TETROFOSMIN KIT; Tc-99m tetrofosmin; Technetium-99 tetrofosmin; Technetium (99mTc) tetrofosmin
Technetium Tc-99m Tetrofosmin is a radiopharmaceutical consisting of tetrofosmin, composed of two bidentate diphosphine ligands chelating the metastable radioisotope technetium Tc-99 (99mTc), with potential imaging activity upon SPECT (single photon emission computed tomography). Upon administration, technetium Tc 99m tetrofosmin is preferentially taken up by, and accumulates in, myocardial cells. Upon imaging, myocardial cells can be visualized and changes in ischemia and/or perfusion can be detected.
Technetium Tc-99m tetrofosmin is a drug used in nuclear myocardial perfusion imaging. The radioisotope, technetium-99m, is chelated by two 1,2-bis[di-(2-ethoxyethyl)phosphino]ethane ligands which belong to the group of diphosphines and which are referred to as tetrofosmin. It is a lipophilic technetium phosphine dioxo cation that was formulated into a freeze-dried kit which yields an injection.[A31592] Technetium Tc-99m tetrofosmin was developed by GE Healthcare and FDA approved on February 9, 1996.
Technetium Tc-99m tetrofosmin is a drug used in nuclear myocardial perfusion imaging. The radioisotope, technetium-99m, is chelated by two 1,2-bis[di-(2-ethoxyethyl)phosphino]ethane ligands which belong to the group of diphosphines and which are referred to as tetrofosmin. It is a lipophilic technetium phosphine dioxo cation that was formulated into a freeze-dried kit which yields an injection.[1] Technetium Tc-99m tetrofosmin was developed by GE Healthcare and FDA approved on February 9, 1996.
Technetium (99mTc) tetrofosmin is a drug used in nuclear medicine cardiac imaging. It is sold under the brand name Myoview (GE Healthcare). The radioisotope, technetium-99m, is chelated by two 1,2-bis[di-(2-ethoxyethyl)phosphino]ethane ligands which belong to the group of diphosphines and which are referred to as tetrofosmin.[1][2]
Tc-99m tetrofosmin is rapidly taken up by myocardial tissue and reaches its maximum level in approximately 5 minutes. About 66% of the total injected dose is excreted within 48 hours after injection (40% urine, 26% feces). Tc-99m tetrofosmin is indicated for use in scintigraphic imaging of the myocardium under stress and rest conditions. It is used to determine areas of reversible ischemia and infarcted tissue in the heart. It is also indicated to detect changes in perfusion induced by pharmacologic stress (adenosine, lexiscan, dobutamine or persantine) in patients with coronary artery disease. Its third indication is to assess left ventricular function (ejection fraction) in patients thought to have heart disease. No contraindications are known for use of Tc-99m tetrofosmin, but care should be taken to constantly monitor the cardiac function in patients with known or suspected coronary artery disease. Patients should be encouraged to void their bladders as soon as the images are gathered, and as often as possible after the tests to decrease their radiation doses, since the majority of elimination is renal. The recommended dose of Tc-99m tetrofosmin is between 5 and 33 millicuries (185-1221 megabecquerels). For a two-dose stress/rest dosing, the typical dose is normally a 10 mCi dose, followed one to four hours later by a dose of 30 mCi. Imaging normally begins 15 minutes following injection.[3]

Amersham (formerly Nycomed Amersham , now GE Healthcare ) has developed and launched 99mTc-tetrofosmin (Myoview) as an injectable nuclear imaging agent for ischemic heart disease in several major territories and for use in detecting breast tumors
Technetium (99mTc) tetrofosmin is a drug used in nuclear medicine cardiac imaging. It is sold under the brand name Myoview (GE Healthcare). The radioisotope, technetium-99m, is chelated by two 1, 2-bis-[bis-(2-ethoxyethyl)phosphino] ethane ligands, which belong to the group of diphosphines and which are referred to as tetrofosmin and has the structural Formula 1 :
Formula 1
99mTc -based radiopharmaceuticals are commonly used in diagnostic nuclear medicine, especially for in vivo imaging (e.g. via immunoscintigraphy or radiolabeling). Usually cold kits are manufactured in advance in accordance with strict requirements of Good Manufacturing Practice (GMP) Guidelines, containing the chemical ingredients (e.g. 99mTc -coordinating ligands, preservatives) in lyophilized form. The radioactive isotope 99mTc (ti/2 = 6h) is added to those kits shortly before application to the patient via intravenous or subcutaneous injection.
Tc-99m tetrofosmin is rapidly taken up by myocardial tissue and reaches its maximum level in approximately 5 minutes. About 66% of the total injected dose is excreted within 48 hours after injection (40% urine, 26% feces). Tc-99m tetrofosmin is indicated for use in scintigraphic imaging of the myocardium under stress and rest conditions. It is used to determine areas of reversible ischemia and infarcted tissue in the heart. It is also indicated to detect changes in perfusion induced by pharmacologic stress (adenosine, lexiscan, dobutamine or persantine) in patients with coronary artery disease. Its third indication is to assess left ventricular function (ejection fraction) in patients thought to have heart disease. No contraindications are known for use of Tc-99m tetrofosmin, but care should be taken to constantly monitor the cardiac function in patients with known or suspected coronary artery disease. Patients should be encouraged to void their bladders as soon as the images are gathered, and as often as possible after the tests to decrease their radiation doses, since the majority of elimination is renal. The recommended dose of Tc-99m tetrofosmin is between 5 and 33 millicuries (185-1221 megabecquerels). For a two-dose stress/rest dosing, the typical dose is normally a 10 mCi dose, followed one to four hours later by a dose of 30 mCi. Imaging normally begins 15 minutes following injection.
99mTc -Tetrofosmin is also described to be useful for tumor diagnostics, in particular of breast cancer and parathyroid gland cancer, and for multidrug resistance (MDR) research.
US5045302 discloses 99mTc-coordinating diphosphine ligands (L), wherein one preferred example thereof is the ether functionalized diphosphine ligand l,2-bis[bis(2-ethoxy- ethyl)phosphino]ethane according to Formula 1, called tetrofosmin (“P53”), that forms a dimeric cationic technetium (V) dioxo phosphine complex, [TCO2L2] with 99mTc, useful as myocardial imaging agent. Example 1 of said patent described the process for preparing tetrofosmin by reacting ethyl vinyl ether, bis(diphosphino)ethane in the presence of a-azo-isobutyronitrile (AIBN) in a fischer pressure-bottle equipped with a teflon stirring bar followed by removal of volatile materials and non-distillable material obtained, as per below mentioned Scheme 1.
Scheme 1
Formula 2 Formula 3 Formula 1
CN 1184225 C discloses tetrofosmin salts containing chloride or bromide or aryl sulfonates as negatively charged counter ions, which can be used for the preparation of a 99mTc- Tetrofosmin radiopharmaceutical composition. According to this patent tetrofosmin hydrochloride is a viscous liquid. Own experiments of the inventors of the present invention revealed that the halide salts of tetrofosmin are hygroscopic oils, which are complicated to handle, e.g. when weighed. The oily and hygrospcopic
properties of tetrofosmin hydrochloride hampers its use in pharmaceutical preparations. Attempts to synthesize the subsalicylate salt of tetrofosmin failed because the starting material sulfosalicylic acid was not soluble in ether in the concentration specified in the patent (3.4 g in 15 ml).
WO2006/064175A1 discloses tetrofosmin was converted to tetrofosmin subsalicylate by reaction with 2.3 to 2.5 molar equivalents of 5-sulfosalicyclic acid at room temperature in ethanol, followed by recrystallisation from ethanol/ether.
WO2015/114002A1 relates to tetrafluoroborate salt of tetrafosmin and its process for the preparation thereof. Further this application also discloses one-vial and two vial kit formulation with tetrafluoroborate salt of tetrafosmin.
The article Proceedings of the International Symposium, 7th, Dresden, Germany, June 18-22, 2000 by Amersham Pharmacia Biotech UK Limited titled “The synthesis of [14C]tetrofosmin, a compound vital to the development of Myoview, Synthesis and Applications of Isotopically Labelled Compounds” disclosed a process for the preparation of tetrofosmin as per below mentioned Scheme 2:
Scheme 2
Formula 1A Formula 7
The starting material was bis(2- ethoxyethyl)benzylphosphine of Formula 4 . This was prepared from benzyl phosphonate, PhCH2P(0)(OEt)2 by reduction with lithium aluminium hydride to give the intermediate benzylphosphine, PhCH2PH2, followed by a photolysis reaction in the presence of ethyl vinyl ether to give compound of Formula 4. The compound of Formula 4 in acetonitrile was treated with dibromo[U-14C]ethane to give compound of Formula 6, further it was treated with excess of 30% aqueous sodium hydroxide in ethanol. The mixture was stirred at room temperature for 24 hours. The solvent was removed and the residue was treated with excess concentrated hydrochloric acid at 0°C. Aqueous work up gave compound of Formula 7. Then compound of Formula 7 in dry benzene was treated with hexachlorodisilane and hydrolysed with excess 30% aqueous sodium hydroxide at 0°C. Aqueous work up followed by flash column chromatography on silica gave [bisphosphinoethane- 1,2-14C]tetrofosmin of formula 1A.
The article Polyhedron (1995), 14(8), 1057-65, titled “Synthesis and characterization of Group 10 metal complexes with a new trifunctional ether phosphine. The X-ray crystal structures of bis[bis(2-ethoxyethyl)benzylphosphine]dichloronickel(II) and bis[bis(2-ethoxyethyl)benzylphosphine]chlorophenylnickel(II)” disclosed the process for the preparation of bis(2-ethoxyethyl)benzylphosphine as per below mentioned Scheme 3:
Scheme 3
Formula 8 Formula 9 Formula 4
The compound bis(2-ethoxyethyl)benzylphosphine of Formula 4 was prepared by first reduction of diethylbenzylphosphonate of Formula 8 using lithium aluminium hydride to obtain benzyl phosphine of Formula 9 followed by radical catalysed coupling reaction with ethyl vinyl ether carried out by using UV photolysis.
Tetrofosmin is extremely sensitive to atmospheric oxygen, which makes synthesis of the substance, as well as manufacturing and handling of the kit complicated as the substance has constantly to be handled in an oxygen free atmosphere.
High purity and stability under dry and controlled conditions are pivotal requirements for chemical compounds used as active ingredients in pharmaceuticals.
The processes disclosed in prior art for the preparation of compound of Formula 4 involves that coupling reaction of benzyl phosphine of Formula 9 with ethyl vinyl ether carried out by using photolytic conditions. Such technology is expensive as it requires separate instruments including isolated facility (to avoid the UV radiation exposure etc.), also it is not suitable for commercial scale production.
PATENT
WO-2018162964
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018162964&tab=PCTDESCRIPTION&maxRec=1000
Example 1
Preparation of benzyl phosphine:
A mixture of lithium aluminium hydride (25 g) in methyl tertiary butyl ether (MTBE) (800 ml) was cooled to 0 to 5°C and added a solution of diethylbenzylphosphonate in methyl tertiary butyl ether (100 g in 200ml). The temperature of reaction mixture was raised to 25 to 30 °C and stirred for 14 to 16 hour. After completion of the reaction, the reaction mixture was cooled to 0 to 5°C and 6N hydrochloric acid was added slowly. Further raised the temperature of reaction mixture to 25 to 30 °C and stirred for 30-45 minutes. The layers were separated, the aqueous layer was extracted with MTBE (250ml) and the combined organic layer was washed with deoxygenated water. The organic layer was dried over sodium sulfate and concentrated to obtain the title compound as non-distillable liquid.
Example 2
Preparation of benzylbis(2-ethoxyethyl)phosphane:
To a mixture of benzyl phosphine (obtained from example 1) and vinyl ethyl ether (250 ml) in pressure RB flask was added a-azo-isobutyronitrile (AIBN) (1.5g). The resulting reaction mixture was maintained at 80 to 90°C for 14 to 16 hours. The mixture was cooled to 20 to 30°C and AIBN (0.5g) added, then continued to heat the reaction mixture at 80 to 90°C for 6 to 7 hours. After completion of the reaction, the reaction mixture was allowed to cool to room temperature and distilled under vacuum to obtain title compound as an oil (107 g).
Example 3
Preparation of Ethane- 1,2-diylbis (benzylbis(2-ethoxyethyl) phosphonium) bromide:
To a mixture of benzylbis(2-ethoxyethyl)phosphane 107.g) in acetonitrile (100ml) in pressure bottle was added 1, 2-dibromoethane (30.5 g). The reaction mixture was maintained at 80 to 90°C for 20 to 25 hours. After completion of the reaction, the reaction mass was cooled to room temperature and stirred for 45 to 60 minutes to obtain the solid. To the solid obtained was added methyl tertiary butyl ether (MTBE) (500ml) and stirred at room temperature for 2 to 3 hour. The reaction mass was filtered, washed with MTBE and suck dried. Further the filtered solid was heated in acetone (400ml) at 50 to 55°C for 2 to 3 hour. Then cooled the reaction mixture to room temperature, stirred, filtered and washed with acetone to obtain the title compound as white solid. (85g)
Example 4
Preparation of Ethane- 1, 2-diylbis (bis (2-ethoxy ethyl) phosphine oxide):
To a mixture of Ethane- 1,2-diylbis (benzylbis(2-ethoxyethyl) phosphonium) bromide (80g) in ethanol (480 ml) was added an aq. solution of sodium hydroxide ( 48g in 160 ml water) at room temperature. The reaction mass was maintained at 25 to 35°C for 10 to 12 hour. After completion of the reaction, the reaction mass was cone, under vacuum to obtained the residue. The residue was dissolved in deoxygenated water (400 ml) and washed with MTBE (400 ml x 2). The layers were separated, the aqueous layer was cooled to 10 to 20°C and 6N hydrochloric acid (200 ml) was added slowly. Then extracted the aqueous layer with dichloromethane (2000 ml), washed the organic layer with deoxygenated water (160 ml), dried the organic layer using sodium sulfate, filtered, and distilled under vacuum to obtain the residue. Further MTBE (160 ml x 2) was added to the residue and continued distillation under vacuum, degassed to obtain the solid. To the obtained solid, MTBE (400 ml) was added and heated at 45 to 50°C for 1-2 hour, further slowly cooled the reaction mass to 25 to 30°C, filtered the solid product. Again MTBE (400 ml) was added to the solid product and heated at 45 to 50°C for 1-2 hour, further slowly cooled the reaction mass to 25 to 30°C, filtered, washed with MTBE and dried under vacuum to obtain the title compound as white solid (32g).
Example 5
Preparation of tetrofosmin free base:
To a mixture of ethane- 1, 2-diylbis (bis (2-ethoxyethyl) phosphine oxide (18g) in toluene (180ml) in pressure RB flask argon/nitrogen gas was purged for 5 minute and hexachlorodisilane (30g) was added. The reaction mixture was heated to 80 to 90°C, stirred for 10 to 12 hour, further slowly cooled to -5 to 0°C and slowly added 30% aqueous sodium hydroxide solution (45g sodium hydroxide in 150 ml deoxygenated water) the temperature of reaction mixture was raised to 25 to 30°C and stirred for 1 to 2 hour. The layers were separated and the aq. layer was extracted with Toluene (180 ml). The combined organic layer was washed with deoxygenated water (180 ml). Further dried the organic layer using sodium sulfate, distilled under vacuum to obtain the residue of tetrofosmin free base (15.5g).
Example 6
Preparation of tetrofosmin disulfosalicylate salt:
To the residue of tetrofosmin free base (15.5g) was added an aq. solution of 5-sulfosalicylic acid dihydrate (21.6g in 75ml deoxygenated water) and stirred at 25 to 30°C for 25 to 30 minutes. Further heated the reaction mass to 55 to 60°C, stirred for 15 to 30 minute, slowly cooled the reaction mass to 10 to 15°C and stirred for 1-2 hour. Filtered, washed with chilled deoxygenated water, and dried under vacuum to obtain the title compound as white solid. (30g).
Example 7
Preparation of Form J of tetrofosmin disulfosalicylate salt:
An aq. solution of 5-sulfosalicylic acid dihydrate (21.6g in 75ml deoxygenated water) was added slowly into tetrofosmin free base (15.5g) and stirred at room temperature for 30 to 40 minutes. The temperature of reaction mixture was further raised to 50 to 60°C, stirred for 20 to 30 minute, cooled the reaction mass to 10 to 15°C and stirred for 1-2 hour. Filtered, washed with chilled deoxygenated water, and dried under vacuum to obtain the title compound.
PATENT
EP337654 ,
PATENT
US9549999
| FDA Orange Book Patents: 1 of 1 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 9549999 |
| Expiration | Mar 10, 2030 |
| Applicant | GE HEALTHCARE |
| Drug Application |
|
 |
|
| Clinical data | |
|---|---|
| Routes of administration |
Intravenous |
| ATC code | |
| Pharmacokinetic data | |
| Bioavailability | N/A |
| Identifiers | |
| CAS Number |
|
| Chemical and physical data | |
| Formula | C36H80O10P4Tc |
| Molar mass | 895.813 g/mol |
Myoview Prescribing Information Page
//////////99mTc-Tetrofosmin, Technetium (99mTc) tetrofosmin, テトロホスミンテクネチウム (99mTc)
CCOCCP(CCOCC)CCP(CCOCC)CCOCC.CCOCCP(CCOCC)CCP(CCOCC)CCOCC.O.O.[Tc]
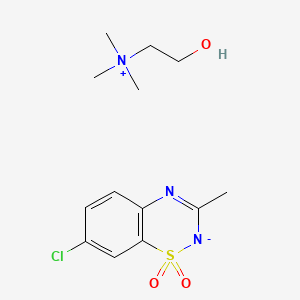
Diazoxide choline,
RN: 1098065-76-9
UNII: 2U8NRZ7P8L
Diazoxide choline; UNII-2U8NRZ7P8L; 2U8NRZ7P8L; YLLWQNAEYILHLV-UHFFFAOYSA-N
| Molecular Formula: | C13H20ClN3O3S |
|---|---|
| Molecular Weight: | 333.831 g/mol |
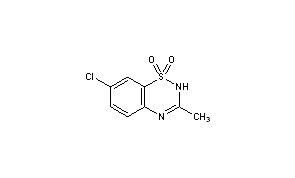
Ethanaminium, 2-hydroxy-N,N,N-trimethyl-, compd. with 7-chloro-3-methyl-2H-1,2,4-benzothiadiazine dioxide (1:1)
7-chloro-3-methyl-1$l^{6},2,4-benzothiadiazin-2-ide 1,1-dioxide;2-hydroxyethyl(trimethyl)azanium
 Diazoxide
Diazoxide
CAS: 364-98-7 FREE FORM
2H-1,2,4-Benzothiadiazine, 7-chloro-3-methyl-, 1,1-dioxide
Diazoxide (INN; brand name Proglycem[1]) is a potassium channel activator, which causes local relaxation in smooth muscle by increasing membrane permeability to potassium ions. This switches off voltage-gated calcium ion channels, preventing calcium flux across the sarcolemma and activation of the contractile apparatus.
In the United States, this agent is only available in the oral form and is typically given in hospital settings.[2]
Diazoxide is used as a vasodilator in the treatment of acute hypertension or malignant hypertension.[3]
Diazoxide also inhibits the secretion of insulin by opening ATP-sensitive potassium channel of beta cells of the pancreas, thus it is used to counter hypoglycemia in disease states such as insulinoma (a tumor producing insulin)[4] or congenital hyperinsulinism.
Diazoxide acts as a positive allosteric modulator of the AMPA and kainate receptors, suggesting potential application as a cognitive enhancer.[5]
The Food and Drug Administration published a Safety Announcement in July 2015 highlighting the potential for development of pulmonary hypertension in newborns and infants treated with this drug.[2]Diazoxide interferes with insulin release through its action on potassium channels.[6] Diazoxide is one of the most potent openers of the K+ ATP channels present on the insulin producing beta cells of the pancreas. Opening these channels leads to hyperpolarization of cell membrane, a decrease in calcium influx, and a subsequently reduced release of insulin.[7] This mechanism of action is the mirror opposite of that of sulfonylureas, a class of medications used to increase insulin release in Type 2 Diabetics. Therefore, this medicine is not given to non-insulin dependent diabetic patients.
SYN
Medicinal Chemistry Research, 12(9), 457-470; 2004

PATENT
WO 2009006483
https://patents.google.com/patent/WO2009006483A1/enIt
PATENT
US 20120238554
PATENT
WO 2013130411
able 16. Characterization of Forms A and B of Diazoxide Choline Salt In
Screening Study
Experiment Form A Form B
*Maj or peaks (2-Θ):
Form A (9.8, 10.5, 14.9, 17.8, 17.9, 18.5, 19.5, 22.1, 22.6, 26.2, 29.6, 31.2);
Form B (8.9, 10.3, 12.0, 18.3, 20.6, 24.1, 24.5, 26.3, 27.1, 28.9).
** Unique FTIR (ATR) absorbances (cm 1):
Form A (2926, 2654, 1592, 1449, 1248);
Form B (3256, 2174, 2890, 1605, 1463, 1235).
6.1.5.1. Solubility Screen in organic solvents.
[00725] Diazoxide choline, prepared in MEK using choline hydroxide as 50 wt % solution in water (see above) displayed some solubility in the following solvents:
acetonitrile, acetone, ethanol, IPA, MEK, DMF, and methanol. These solvents were chosen due to differences in functionality, polarity, and boiling points and their ability to dissolve diazoxide. Other solvents which showed poor ability to dissolve salts were used as antisolvents and in slurry experiments where some solubility was observed: dioxane, MTBE, EtOAc, IP Ac, THF, water, cyclohexane, heptane, CH2C12, and toluene.
[00726] Solvents for crystallizations during screening were chosen based on the solubility screen summarized in Table 17. Crystallizations of diazoxide choline from all conditions afforded a total of two forms, A and B. Forms A and B were found to be anhydrous polymorphs of diazoxide choline. Form B was observed to be generated from most solvents used. It was difficult to isolate pure Form A on large scales (>50 mg) as conditions observed to produce Form A on a smaller scale (approximately 50 mg or less) were found to result in Form B or mixtures of both forms on larger scales. Based on room-temperature slurry experiments, anhydrous Form B was found to be the most thermodynamically stable form in this study. Form A readily converted to Form B in all slurry solvents utilized.
Table 17. Solubility Screen for Diazoxide Choline Salt
Solvent Cmpd Solvent Cone. Temp. Soluble
(mg) (mL) (mg/niL) (°C)
CH2CI2 1.3 5.00 0.26 55 Partially
Toluene 1.4 5.00 0.28 55 No
.1.5.2. Single-Solvent Crystallizations
[00727] Fast cooling procedure: Diazoxide (approximately 20 mg) was weighed out into vials and enough solvent (starting with 0.25 mL) was added until the material completely dissolved at elevated temperature. After hot filtration the vials were placed in a refrigerator (4 °C) for 16 hours. After the cooling-process the samples were observed for precipitates which were isolated by filtration. Vials not demonstrating precipitates were evaporated down to dryness using a gentle stream of nitrogen. All solids were dried in vacuo at ambient temperature and 30 in. Hg.
[00728] Slow cooling procedure: Diazoxide (approximately 30 mg of choline salt) was weighed out into vials and enough solvent was added until the material went into solution at elevated temperature. After hot filtration the vials were then slowly cooled to room temperature at the rate of 20 °C/h and stirred at room temperature for 1-2 hours. All solids were dried in vacuo at ambient temperature and 30 in. Hg.
[00729] Based on the initial solubility study, seven solvents were selected for the fast-cooling crystallization: acetonitrile, acetone, ethanol, IPA, MEK, DMF, and methanol. Table 18 shows a list of the solvents that were used and the amount of solvent needed to dissolve the material. After the cooling-process precipitates were noticed in samples # 2, 3, 5, and 6, the solids were isolated by filtration. The other samples (# 1, 4, and 7) were evaporated down to dryness using a gentle stream of nitrogen. The diazoxide choline salts were found to be consistent with Form A by XRPD analysis for all solids with the exception of sample #2 (consistent with the freeform) and sample #5 (consistent with Form B with preferred orientation observed).
Table 18. Single- Solvent Crystallization of Diazoxide Choline Salt Using Fast- Cooling Procedure
[00730] In accordance with the data obtained from fast-cooling experiments, four solvents which showed precipitation of solids were chosen for the slow-cooling experiments: MeOH, EtOH, MeCN, and IPA (Table 19). All obtained analyzable solids of the choline salt were found to be consistent with Form B by XRPD with the exception of Entry #1 which was consistent with diazoxide freeform and Entry #2 which was not analyzable. Mother liquor of Entry #2 was concentrated to dryness and the residual solids were analyzed by XRPD and found to be Form B material. As a result of obtaining freeform material from the single- solvent crystallizations in methanol, three more alcohols were tested for the single- solvent crystallizations using fast- and slow-cooling procedures. Tables 20 and 21 provide a list of the solvents that were used and the amount of solvent needed to dissolve the material. XRPD patterns of the fast-cooling procedure showed freeform of diazoxide from isobutanol, Form B from isoamyl alcohol, and Form A from tert-amyl alcohol compared to the slow-cooling procedure, which afforded Form B material from all three solvents.
Table 19. Single-Solvent Crystallization of Diazoxide Choline Salt Using Slow- Cooling Procedure
Table 20. Single- Solvent Crystallization of Diazoxide Choline Salt Using Fast- Cooling Procedure
Table 21. Single-Solvent Crystallization of Diazoxide Choline Salt Using Slow- Cooling Procedure
[00731] The results of the choline salt single- solvent fast- and slow-cooling crystallizations (see Tables 19 to 21) indicated that Form A was more likely to be isolated with fast-cooling profiles and Form B with slow-cooling profiles.
6.1.5.3. Binary Solvent Crystallizations
[00732] Binary- solvent crystallizations of the choline salt were performed using four primary solvents (MeOH, EtOH, IPA, and MeCN) and nine cosolvents (MTBE, EtOAc, IPAc, THF, c-hexane, heptane, toluene, CH2CI2, and dioxane) with a fast-cooling profile (supra). XRPD patterns showed that Form B was obtained from mixtures of MeOH with MTBE, EtOAc, IPAc, toluene, and dioxane. As shown in Table 22, Form A was obtained from mixtures of MeOH with THF and with CH2CI2 after evaporating the solvent to dryness. The mixtures of MeOH with cyclohexane and heptane provided the freeform of diazoxide. All solids obtained from fast-cooling procedures with EtOH, IPA, and MeCN as primary solvents provided Form B material.
Table 22. Binary-Solvent Crystallizations of Choline Salt of Diazoxide Using Fast- Cooling Procedure and MeOH as a Primary Solvent
* Solids were dissolved at 62 °C.
** Freeform of diazoxide.
[00733] Binary- solvent recrystallizations of the choline salt with the slow-cooling procedure were performed using two primary solvents (IPA and MeCN) and nine cosolvents (MTBE, EtOAc, IPAc, THF, c-hexane, heptane, toluene, CH2C12, and dioxane). All solids obtained from a slow-cooling procedure with IPA and MeCN as primary solvents provided Form B material based on XRPD analysis. The results of
binary- solvent crystallizations indicated that Form B was the most thermodynamic ally stable form of diazoxide choline.
6.1.5.4. Binary Solvent Crystallizations Using Water as a Cosolvent
[00734] In an attempt to investigate the formation of hydrates of the choline salt, experiments was performed using fast- and slow-cooling procedures and water as a cosolvent.
[00735] The fast cooling procedure (supra) was used with the exception of using different primary solvents which were miscible with water: acetone, acetonitrile, DMF, IPA, i-BuOH, i-AmOH, and t-AmOH. Water was utilized in these crystallizations as a cosolvent. All solids obtained from the fast-cooling procedure with water as the cosolvent provided diazoxide freeform material by XRPD analysis.
[00736] To compare the results obtained from the fast-cooling procedure a set of experiments was performed using a slow-cooling procedure and water as a cosolvent. All obtained solids were analyzed by XRPD and afforded patterns consistent with diazoxide freeform. Without wishing to be bound by theory, these results suggest that the conditions used for crystallization caused dissociation of the choline salt. A small amount of a second crop was obtained in each sample, but only two samples were analyzable by XRPD and indicated that the samples were freeform material. All mother liquors were evaporated to dryness and the residual solids were also analyzed by XRPD to afford patterns consistent with Form B of the choline salt.
6.1.5.5. Metastable Zone Width Estimation
[00737] Form B: To produce a robust process, an understanding of the solubility profiles of the various solid forms under consideration is required. From a practical standpoint, this involves the measurement of the metastable zone width (MSZW) of pure forms, whereby the saturation and supersaturation curves of the different forms are generated over a well defined concentration and temperature range. This knowledge can then be used to design a crystallization protocol that should ideally favor a selective crystal growth of the desired form.
[00738] Form B of diazoxide choline salt showed moderate solubility in a solvent mixture made of MeCN/MeOH/MtBE (10: 1: 12, volume ratios). The wide width of the metastable zone as shown in Table 23 gives many seeding options. During the MSZW measurement, aliquots from the crystallizing material were withdrawn and analyzed by XRPD to ensure that no form conversion occurred during the experiment. Indeed, the material remained unchanged during the test.
Table 23. Meta-Stable Zone Width For Form B Diazoxide Choline Salt in
MeCN/MeOH/MtBE (10:1:12) (v/v).
[00739] Form A: The metastable zone width for Form could not be estimated because this polymorphic form converted during the experiment to Form B.
6.1.5.6. Crystallization of Form A of Diazoxide Choline Salt
[00740] The choline salt of diazoxide (160.3 mg) was dissolved in 1 mL of IPA at 55 °C which was then passed through a Millipore 0.45 μΜ filter into a clean vial. This vial was placed in freezer a -20 °C overnight. Solids were not noticed and the flask was scratched with a micro- spatula. The vial was placed back in the freezer and nucleation was noticed after ten minutes. The solids were collected by vacuum filtration and washed with 1 mL of MtBE. The solids were dried in vacuo at 40 °C and 30 in. Hg to afford 70 mg (43.6% recovery) of Form A as determined by XRPD.
6.1.5.7. 500-mg Scale Crystallization of Form B of Diazoxide Choline Salt
[00741] The choline salt of diazoxide (524.3 mg) was dissolved in 3 mL of IPA at 78 °C and this solution was then cooled to 55 °C for the addition of MtBE. The MtBE (4 mL) was added until nucleation was observed. After nucleation the batch was allowed to cool to room temperature at a rate of 20 °C /h. The solids were collected by vacuum filtration and washed with 1 mL of MtBE. The solids were dried in vacuo at 40 °C and 30 in. of Hg to afford 426.7 mg (81.3% recovery) of Form B as determined by XRPD.
6.1.5.8. 2-g Scale Crystallization of Form B of Diazoxide Choline Salt
[00742] The choline salt of diazoxide (2.0015 g) was dissolved in 5.5 mL of IPA at 78 °C to afford a clear solution. This solution was passed through a Millipore Millex FH 0.45 μΜ filter. This solution was then cooled to 55 °C. MtBE was added in 1 mL portions, with a two minute interval between portions. Nucleation was noted after the second addition of MtBE. This suspension was allowed to cool to room temperature at a rate of 20 °C /h and stirred at this temperature for 16 hours. The solids were collected by vacuum filtration and washed with 1 mL of MtBE. The solids were dried in vacuo at 40 °C and 30 in. of Hg to afford 1.6091 g (80.4% recovery) of Form B as determined by XRPD.
6.1.5.9. Detection of Form Impurities
[00743] Mixtures of diazoxide choline Forms A and B were prepared by adding a minor amount of Form A to Form B. Samples were lightly ground by hands with a mortar and pestle for approximately one minute. Samples were then analyzed by XRPD analysis. XRPD analysis was found to be suitable for detecting 5% of Form A in Form B.
 |
|
| Clinical data | |
|---|---|
| Trade names | Proglycem |
| AHFS/Drugs.com | Monograph |
| Pregnancy category |
|
| Routes of administration |
Oral, intravenous |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Protein binding | 90% |
| Metabolism | Hepatic oxidation and sulfate conjugation |
| Elimination half-life | 21-45 hours |
| Excretion | Renal |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.006.063 |
| Chemical and physical data | |
| Formula | C8H7ClN2O2S |
| Molar mass | 230.672 g/mol |
| 3D model (JSmol) | |
//////////////Diazoxide choline
CC1=NC2=C(C=C(C=C2)Cl)S(=O)(=O)[N-]1.C[N+](C)(C)CCO
CC1=NC2=C(C=C(C=C2)Cl)S(=O)(=O)[N-]1.C[N+](C)(C)CCO


Zoledronic acid
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Zoledronate disodium | 7D7GS1SA24 | 165800-07-7 | IEJZOPBVBXAOBH-UHFFFAOYSA-L |
| Zoledronate trisodium | ARL915IH66 | 165800-08-8 | Not applicable |
| Zoledronic acid hemipentahydrate | 1K9U67HDID | Not Available | AZZILOGHCMYHQY-UHFFFAOYSA-N |
| Zoledronic acid monohydrate | 6XC1PAD3KF | 165800-06-6 | FUXFIVRTGHOMSO-UHFFFAOYSA-N |
Zoledronate (zoledronic acid, marketed by Novartis under the trade names Zometa and Reclast) is a bisphosphonate. Zometa is used to prevent skeletal fractures in patients with cancers such as multiple myeloma and prostate cancer. It can also be used to treat hypercalcemia of malignancy and can be helpful for treating pain from bone metastases.
An annual dose of Zoledronate may also prevent recurring fractures in patients with a previous hip fracture.
Zoledronate is a single 5 mg infusion for the treatment of Paget’s disease of bone. In 2007, the FDA also approved Reclast for the treatment of postmenopausal osteoporosis.
Zoledronic acid, also known as zoledronate, is a medication used to treat a number of bone diseases.[1] This include osteoporosis, high blood calcium due to cancer, bone breakdown due to cancer, and Paget’s disease of bone.[1] It is given by injection into a vein.[1]
Common side effects include fever, joint pain, high blood pressure, diarrhea, and feeling tired.[1] Serious side effects may include kidney problems, low blood calcium, and osteonecrosis of the jaw.[1] Use during pregnancy may result in harm to the baby.[1] It is in the bisphosphonate family of medications.[1] It works by blocking the activity of osteoclast cells and thus decreases the breakdown of bone.[1]
Zoledronic acid was approved for medical use in the United States in 2001.[1] It is on the World Health Organization’s List of Essential Medicines, the most effective and safe medicines needed in a health system.[3] The wholesale cost in the developing world is between 5.73 USD and 26.80 USD per vial.[4] In the United Kingdom, as of 2015, a dose costs the NHS about 220 pounds.[5]
Zoledronic acid is used to prevent skeletalfractures in patients with cancers such as multiple myeloma and prostate cancer, as well as for treating osteoporosis.[6] It can also be used to treat hypercalcemia of malignancy and can be helpful for treating pain from bone metastases.[7]
It can be given at home rather than in hospital. Such use has shown safety and quality-of-life benefits in people with breast cancer and bone metastases.[8]
Zoledronic acid may be given as a 5 mg infusion once per year for treatment of osteoporosis in men and post-menopausal women at increased risk of fracture.[9]
In 2007, the U.S. Food and Drug Administration (FDA) also approved it for the treatment of postmenopausal osteoporosis.[10][11]
A single 5 mg dose of zoledronic acid is used for the treatment of Paget’s disease.[medical citation needed][12]
Side effects can include fatigue, anemia, muscle aches, fever, and/or swelling in the feet or legs. Flu-like symptoms are common after the first infusion, although not subsequent infusions, and are thought to occur because of its potential to activate human γδ T cells(gamma/delta T cells).
There is a risk of severe renal impairment. Appropriate hydration is important prior to administration, as is adequate calcium and vitamin D intake prior to Aclasta therapy in patients with preexisting hypocalcaemia, and for ten days following Aclasta in patients with Paget’s disease of the bone. Monitoring for other mineral metabolism disorders and the avoidance of invasive dental procedures for those who develop osteonecrosis of the jaw is recommended.[14]
Zoledronate is rapidly processed via the kidneys; consequently its administration is not recommended for patients with reduced renal function or kidney disease.[15] Some cases of acute renal failure either requiring dialysis or having a fatal outcome following Reclast use have been reported to the U.S. Food and Drug Administration (FDA).[16] This assessment was confirmed by the European Medicines Agency (EMA), whose Committee for Medicinal Products for Human Use (CHMP) specified new contraindications for the medication on 15 December 2011, which include hypocalcaemia and severe renal impairment with a creatinine clearance of less than 35 ml/min.[17]
A rare complication that has been recently observed in cancer patients being treated with bisphosphonates is osteonecrosis of the jaw. This has mainly been seen in patients with multiple myeloma treated with zoledronate who have had dental extractions.[18]
Atypical fractures : After approving the drug on 8 July 2009, the European Medicines Agency conducted a class review of all bisphosphonates, including Zoledronate, after several cases of atypical fractures were reported.[19] In 2008, the EMA’s Pharmacovigilance Working Party (PhVWP) noted that alendronic acid was associated with an increased risk of atypical fracture of the femur that developed with low or no trauma. In April 2010, the PhVWP noted that further data from both the published literature and post-marketing reports were now available which suggested that atypical stress fractures of the femur may be a class effect. The European Medicines Agency then reviewed all case reports of stress fractures in patients treated with bisphosphonates, relevant data from the published literature, and data provided by the companies which market bisphosphonates. The Agency recommended that doctors who prescribe bisphosphonate-containing medicines should be aware that atypical fractures may occur rarely in the femur, especially after long-term use, and that doctors who are prescribing these medicines for the prevention or treatment of osteoporosis should regularly review the need for continued treatment, especially after five or more years of use.[19]
Zoledronic acid slows down bone resorption, allowing the bone-forming cells time to rebuild normal bone and allowing bone remodeling.[20]
Zoledronic acid has been found to have a direct antitumor effect and to synergistically augment the effects of other antitumor agents in osteosarcoma cells.[21]
Zoledronate has shown significant benefits versus placebo over three years, with a reduced number of vertebral fractures and improved markers of bone density.[22][11] An annual dose of zoledronic acid may also prevent recurring fractures in patients with a previous hip fracture.[9]
Zoledronate also attenuates accumulation of DNA damage in mesenchymal stem cells and protects their function.[23] Given this characteristic, its potential to affect conditions arising from stem-cell dysfunction makes it a promising medicine for a range of age-related diseases[24]
An increase in disease-free survival (DFS) was found in the ABCSG-12 trial, in which 1,803 premenopausal women with endocrine-responsive early breast cancer received anastrozole with zoledronic acid.[25] A retrospective analysis of the AZURE trial data revealed a DFS survival advantage, particularly where estrogen had been reduced.[26]
In a meta-analysis of trials where upfront zoledronic acid was given to prevent aromatase inhibitor-associated bone loss, active cancer recurrence appeared to be reduced.[27]
As of 2010 “The results of clinical studies of adjuvant treatment on early-stage hormone-receptor-positive breast-cancer patients under hormonal treatment – especially with the bisphosphonate zoledronic acid – caused excitement because they demonstrated an additive effect on decreasing disease relapses at bone or other sites. A number of clinical and in vitro and in vivo preclinical studies, which are either ongoing or have just ended, are investigating the mechanism of action and antitumoral activity of bisphosphonates.”[28]
A 2010 review concluded that “adding zoledronic acid 4 mg intravenously every 6 months to endocrine therapy in premenopausal women with hormone receptor-positive early breast cancer … is cost-effective from a US health care system perspective”.[29]
PAPER
J Med Chem 2002,45(17),3721
https://pubs.acs.org/doi/10.1021/jm020819i

Bisphosphonates (BPs) are pyrophosphate analogues in which the oxygen in P−O−P has been replaced by a carbon, resulting in a metabolically stable P−C−P structure. Pamidronate (1b, Novartis), a second-generation BP, was the starting point for extensive SAR studies. Small changes of the structure of pamidronate lead to marked improvements of the inhibition of osteoclastic resorption potency. Alendronate (1c, MSD), with an extra methylene group in the N-alkyl chain, and olpadronate (1h, Gador), the N,N-dimethyl analogue, are about 10 times more potent than pamidronate. Extending one of the N-methyl groups of olpadronate to a pentyl substituent leads to ibandronate (1k, Roche, Boehringer-Mannheim), which is the most potent close analogue of pamidronate. Even slightly better antiresorptive potency is achieved with derivatives having a phenyl group linked via a short aliphatic tether of three to four atoms to nitrogen, the second substituent being preferentially a methyl group (e.g., 4g, 4j, 5d, or 5r). The most potent BPs are found in the series containing a heteroaromatic moiety (with at least one nitrogen atom), which is linked via a single methylene group to the geminal bisphosphonate unit. Zoledronic acid (6i), the most potent derivative, has an ED50 of 0.07 mg/kg in the TPTX in vivo assay after sc administration. It not only shows by far the highest therapeutic ratio when comparing resorption inhibition with undesired inhibition of bone mineralization but also exhibits superior renal tolerability. Zoledronic acid (6i) has thus been selected for clinical development under the registered trade name Zometa. The results of the clinical trials indicate that low doses are both efficacious and safe for the treatment of tumor-induced hypercalcemia, Paget’s disease of bone, osteolytic metastases, and postmenopausal osteoporosis.
SYN 1
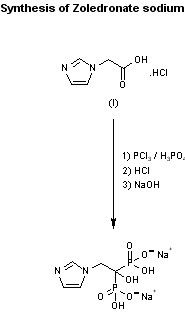
AU 8781453; EP 0275821; JP 1988150291; US 4939130
Zoledronate sodium can be prepared by reaction of 2-(1-imidazolyl)acetic acid hydrochloride (I) with PCl3, with optional presence of phosphoric acid, in refluxing chlorobenzene, followed by hydrolysis with refluxing 9N hydrochloric acid and final formation of the sodium salt by treatment with aqueous NaOH.
SYN
PAPER
https://www.sciencedirect.com/science/article/pii/S0969804311006385

Clip
https://link.springer.com/article/10.1007/s11094-015-1205-0
A One-Pot and Efficient Synthesis of Zoledronic Acid Starting from Tert-butyl Imidazol-1-yl Acetate
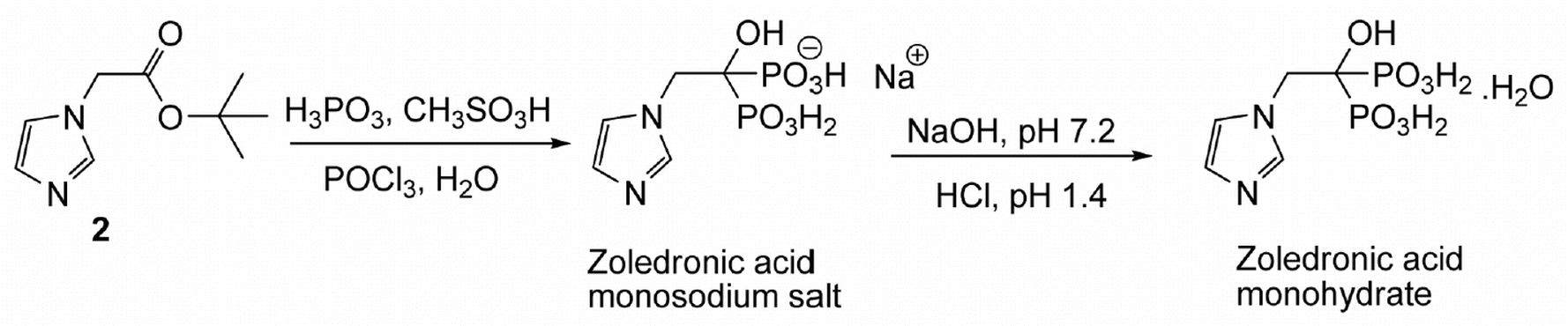
A one-pot synthesis of zoledronic acid in high yield is described. The procedure involves a non-aqueous ester cleavage of the tert-butyl imidazol-1-yl acetate under dry conditions in the presence of methanesulfonic acid as solubilizer and chlorobenzene as solvent to afford in situthe corresponding imidazolium methanesulfonate salt which yields zoledronic acid upon reaction with phosphoric acid and phosphorus oxychloride. A possible chemical mechanism for the synthesis of this acid is described.

Paper
https://www.beilstein-journals.org/bjoc/articles/4/42
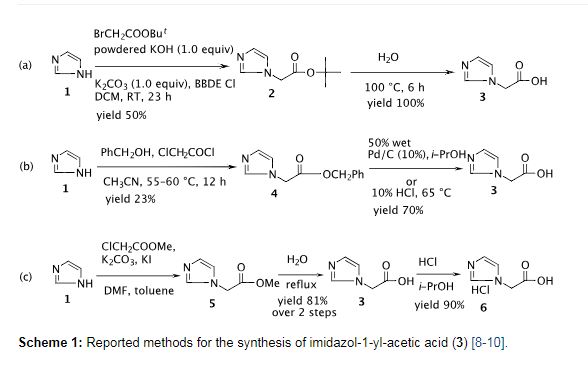


To a solution of imidazole (10.0 g, 0.15 mol) in ethyl acetate (160 mL) was added powdered K2CO3 (29.0 g, 0.21 mol) followed by tert-butyl chloroacetate (25.7 mL, 0.18 mol) at room temperature and the mixture was refluxed for 10.0 h. After completion of the reaction as indicated by TLC (10% MeOH/CHCl3, I2 active), the reaction mass was quenched with cold water (80 mL) and the ethyl acetate layer was separated. The aqueous layer was extracted with ethyl acetate (2 × 80 mL) and the combined ethyl acetate layers were washed with brine, dried with anhydrous sodium sulfate and then concentrated under vacuum. The resulting solid was stirred with hexane (50 mL) at RT, filtered and washed with hexane (2 × 20 mL) to afford the title compound as an off-white solid (20.0 g, 75%). mp: 111.3–113.2 °C (Lit [10]: 111–113 °C). IR (cm−1): 3458, 3132, 3115, 2999, 2981, 2884, 1740, 1508, 1380, 1288, 1236, 1154, 1079, 908, 855, 819, 745, 662, 583; 1H NMR (300 MHz, CDCl3) δ 1.47 (s, 9H), 4.58 (s, 2H), 6.94 (s, 1H), 7.09 (s, 1H), 7.49 (s, 1H); 13C NMR (75 MHz, CDCl3) δ 27.7, 48.6, 82.9, 119.8, 129.2, 137.7, 166.3; MS (m/z) 183.0 [M+1, 100%], 127.0.
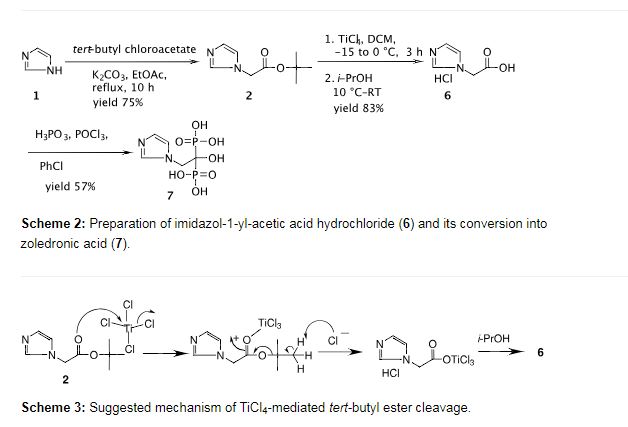
To a solution of imidazol-1-yl-acetic acid tert-butyl ester (2) (10.0 g, 0.05 mol) in dichloromethane (100 mL) was added titanium tetrachloride (8.0 mL, 0.07 mol) dropwise slowly at −15 to −10 °C over 1 h and the mixture was stirred at −5 to 0 °C for 2 h. Isopropyl alcohol (25 mL) was added at 0 to −10 °C over 0.5 h and the reaction mass was stirred at room temperature for 0.5 h. Additional isopropyl alcohol (125 mL) was added dropwise at room temperature over 0.5 h and the mixture was stirred for 1 h. Dichloromethane was distilled out under a low vacuum and the resulting crystalline solid precipitated was filtered to afford the title compound as an off-white crystalline solid (7.4 g, 83%). mp 200.3–202.3 °C; IR (cm−1): 3175, 3125, 3064, 2945, 2869, 2524, 2510, 1732, 1581, 1547, 1403, 1223, 1193, 1081, 780, 650; 1H NMR (300 MHz, D2O + 3-(trimethylsilyl)propionic acid sodium salt) δ 5.1 (s, 3H, -CH2– + HCl), 7.5 (br s, 2H), 8.7 (s, 1H); 13C NMR (75 MHz, D2O + 3-(trimethylsilyl)propionic acid sodium salt) 52.7, 122.4, 125.9, 138.8, 172.8; MS (m/z) 127.0 [M+1, 100%]; HCl-content: found 21.8% (along with 3.25% moisture), calcd 22.43% for C5H6N2O2·HCl.
To a suspension of imidazol-1-yl-acetic acid hydrochloride (6) (7.0 g, 0.043 mol) and phosphorous acid (9.5 g, 0.116 mol) in chlorobenzene (50 mL) was added phosphorous oxychloride (9.6 ml, 0.103 mol) at 80–85 °C over a period of 2 h then heated to 90–95 °C for 2.5 h. The reaction mass was cooled to 60–65 °C and water (100 mL) was added at the same temperature. The aqueous layer was separated, collected and refluxed for 18 h. It was then cooled to room temperature and diluted with methanol (140 mL). The mixture was cooled to 0–5 °C and stirred for 3 h. The precipitated solid was filtered, washed with cold water followed by methanol and then dried under vacuum at 60 °C for 12 h to afford the title compound (6.6 g, 57% yield) as a white solid; mp 237–239 °C (lit [1] 239 °C with decomposition).
PATENT
https://patents.google.com/patent/CN104610357A/en
Sodium Zoledronic (Zoledronate sodium, I), chemical name [1-yl light -2- (lH- imidazol-1-yl) ethylidene] bisphosphonic acid monosodium salt monohydrate, is by the Novartis (Novartis) developed imidazole heterocyclic bisphosphonates, belongs to the third generation of bisphosphonates bisphosphonate drugs, in October 2000, first marketed in Canada. Subsequently approved in the European Union, the United States more than 80 countries or regions, trade name Zometa, for the treatment of hypercalcemia of malignancy (HCM) and multiple myeloma and bone metastases of solid tumors. The drug is effective in treating cancer caused by HCM, advanced bone metastases and Paget’s disease, reduce the incidence of skeletal related events, relieve symptoms and improve quality of life, is also expected to be used to treat osteoporosis. Compared with other similar drugs, high efficacy, dosage, ease of administration, better security, etc., is currently the only FDA-approved for metastatic bone tumor effective bisphosphonate drugs.Currently bisphosphonate drugs in our country is still in the initial stages of clinical applications, but in recent years has made rapid progress, broad market prospect.
[0003] The prior art synthesis reaction conditions zoledronate sodium harsh, toxicity and use methanol, chloroform and chlorobenzene, easily exceeding the amount of residual organic solvents, low yield, low product purity, contamination environment, does not meet the medical criteria, is not conducive to industrial production. Environmental pollution has attracted increasing attention around the world today, the development of new green efficient synthesis of a pharmaceutical drug synthesis is an important issue facing the Institute. In recent years, room temperature ionic liquids as a reaction medium is environmentally friendly, has been widely used in a variety of organic synthesis reactions. Compared with traditional organic solvents, ionic liquids have very low vapor pressure, non-flammable, good thermal stability, both as a reaction medium underway catalysis, can be recycled and many other advantages.

Example 1 of zoledronic alendronate

(1) Synthesis of imidazol-1-yl acetate were added successively imidazole (13.62g, 0.2mol) and [bmim] BF4 (IOOmL) a three-necked flask, heated with stirring warmed to 60 ° C, incubated under reflux was slowly added dropwise chlorination ethyl acetate (24. 51g, 0. 2mol), dropwise addition time is about 2h, dropwise, with stirring maintained at reflux for 16 h, the reaction monitored by TLC showed no starting material end point, completion of the reaction, cooled to room temperature, to give imidazole -1 – ethyl crude, about 24g, crude without purification, was used directly in the next reaction.
[0014] (2) Synthesis of imidazol-1-yl acetate hydrochloride A solution of 24g 1-yl imidazole prepared above was added crude ethyl necked flask, concentrated hydrochloric acid (34 mL), exotherm to 85 ° C, warmed to reflux heating was continued, the reaction was stirred at reflux for 10H, the reaction was completed, the solvent was evaporated under reduced pressure, 20ml of absolute ethanol was added to the residue, vigorously stirred for 2h, filtered off with suction, the filter cake finally at 80 ° C blast pressure and dried to give a white solid imidazol-1-yl acetate hydrochloride about 25. 65g, 79.4% overall yield.
[0015] (3) Synthesis of zoledronic acid monohydrate were added imidazol-1-yl acetate hydrochloride (17. 26g, 0. 137mol) a three-necked flask, in an ionic liquid with stirring – n-butyl-3- methylimidazolium tetrafluoroborate [bmim] BF4 (40mL) and concentration of 85% phosphoric acid solution (16mL), heating to 60 ° C was added dropwise phosphorus trichloride (30mL) , about 4h dropwise, reaction was continued under reflux for 4h at 65 ° C, the reaction was complete, cooled to 40 ° C, filtered off with suction, the filter cake was added to a molar concentration in 80mL 9mol / L hydrochloric acid, heated with stirring state the reaction was refluxed for 6h, the reaction was completed, filtered hot, the filter cake was added to a molar concentration in 80mL 9mol / L hydrochloric acid, the above-described operation is repeated to continue the combined filtrate was evaporated to dryness under reduced pressure to give a yellow oily residue was slowly added to the residue volume ratio of 1: 1 acetone – ethanol mixture 240 mL, was stirred, and the precipitated solid was 15min, filtered off with suction, the filter cake was recrystallized in 30mL of deionized water, suction filtered to give a white solid that is zoledronic acid monohydrate , about 35. 8g, yield 90.1%, determined by HPLC, purity> 98.5%.
After [0016] (4) Synthesis of zoledronic sodium phosphinate obtained above azole zoledronic acid monohydrate (46.4g, 0. 16mol) washed with water (450 mL of) was dissolved, was added sodium hydroxide (5. 6g, 0. IOmol), were refluxed for 30min, cooling and crystallization, filtration, to obtain a crude product zoledronate sodium, crude mother liquor was concentrated and then half with distilled water (410 mL), isopropanol (60 mL), heated to dissolve the combined, activated carbon bleaching, charcoal filtered off, cooling and crystallization, filtration, washed with water, dried at 40-60 ° C to about crystallization water containing one to give zoledronate sodium (42. 4g, 85%), total yield of more than 60% by HPLC assay, purity 99. 8%, mp239 ° C. IR: 711011 ^ 671 (^ 1 is the stretching vibration peak of the PC, 1643〇 ^ 1 = 0 (: stretching vibration peak, 3011〇 ^ 1 = (: – stretching vibration peak 11 ^ 1 is CN 1406〇 the stretching vibration, 1643CHT1 is C = N stretching vibration peak of 3447 (3485 ^^ (^ 1 is the stretching vibration peak of OH, 1459CHT1 symmetrical bending vibration of CH, 2830CHT1 stretching vibration of CH, 1324CHT1 is P = O the stretching vibration, 1094CHT1 stretching vibration peak of .1HNMR PO (400MHz, D20), S: 8.68 (lH, s), 7.48 (lH, s), 7.34 (lH, s), 4.67 (2H, t).
Effects [0017] Example 2 was added dropwise phosphorus trichloride fixed time on the yield other conditions remain unchanged, only the changes of phosphorus trichloride dropwise addition, dropping zoledronic Table 1 Effect of Sodium yield Experimental results show that excessive phosphorus trichloride was added dropwise, and instantly generate a large amount of gas, the reaction is very intense, the liquid splashing, a rapid rise in temperature, resulting in the low yield, if slowly added dropwise, the reaction rate is too slow, consumption too long, and therefore is the best 4h dropping time.

Zoledronic fixed effect of sodium yield other conditions remain unchanged, imidazol-1-yl acetic acid hydrochloride with phosphorus trichloride and phosphoric acid condensation reaction temperature is zoledronic acid monohydrate embodiment the reaction temperature Example 3 Effect yield (Table 2). The results show that, with increasing temperature, increasing the yield, but at higher temperatures to reflux, shows a decreasing trend in yield, due to decomposition sake phosphorus trichloride, resulting in reduction reaction. Further, when the temperature is too high, solvent evaporation and solvent leakage losses will increase, the reflux temperature is low, the reaction rate is slow, the reaction is insufficient, therefore the yield is low, and therefore the optimum reaction temperature is about 65 ° C.

Example 4

Effect of the ionic liquid frequency reuse sodium zoledronic yield of the reaction medium can be recovered and reused important concern is “green chemistry” used in the present embodiment examines the sodium ionic liquid used in the synthesis of zoledronic repeated use, the experiment results shown in Table 3. Seen from Table 3, the ionic liquid after 5 subsequent to use, product yield began to decrease, the ionic liquid may be recovered and reused effectively, and repeated five times using good performance, and therefore is an ionic liquid in this reaction green solvents may be recycled.

Example 5 different ionic liquids zoledronic same impact conditions were examined yield sodium 1-butyl-3-methylimidazolium tetrafluoroborate ionic liquids ([bmim] BF4), N- ethyl pyridinium tetrafluoroborate ([EPy] BF4), l- butyl-3-methylimidazolium hexafluorophosphate ([bmim] PF6), 1- hydroxyethyl-2,3-dimethyl imidazolium chloride (LOH), 1- propyl-3-carbonitrile methylimidazolium chloride (the LCN) and 1-carboxyethyl-3-methyl imidazolium chloride (LOOH) Effects of sodium zoledronic yield the results are shown in Table 4, the test results show little effect on the synthesis of ionic liquids yield.

Clip
Mar 5, 2013 –
Dr. Reddy’s Laboratories announced today that it has launched Zoledronic Acid Injection (4 mg/5 mL), a bioequivalent generic version of Zometa® (zoledronic acid) 4 mg/5 mL Injection in the US market on March 4, 2013, following the approval by the United States Food & Drug Administration (USFDA) of Dr. Reddy’s ANDA for Zoledronic Acid Injection (4 mg/5 mL).
Dr. Reddy’s Zoledronic Acid Injection 4 mg/5mL is available in a single use vial of concentrate.
Zoledronic acid (INN) or zoledronate (marketed by Novartis under the trade names Zometa, Zomera, Aclasta and Reclast) is a bisphosphonate. Zometa is used to prevent skeletal fractures in patients with cancers such as multiple myeloma and prostate cancer, as well as for treating osteoporosis.It can also be used to treat hypercalcemia of malignancy and can be helpful for treating pain from bone metastases.
An annual dose of zoledronic acid may also prevent recurring fractures in patients with a previous hip fracture.
Reclast is a single 5 mg infusion for the treatment of Paget’s disease of bone. In 2007, the U.S. Food and Drug Administration (FDA) also approved Reclast for the treatment of postmenopausal osteoporosis.

Dr. Reddy’s Laboratories Ltd. (NYSE: RDY) is an integrated global pharmaceutical company, committed to providing affordable and innovative medicines for healthier lives. Through its three businesses – Pharmaceutical Services and Active Ingredients, Global Generics and Proprietary Products – Dr. Reddy’s offers a portfolio of products and services including APIs, custom pharmaceutical services, generics, biosimilars, differentiated formulations and NCEs. Therapeutic focus is on gastro-intestinal, cardiovascular, diabetology, oncology, pain management, anti-infective and pediatrics. Major markets include India, USA, Russia and CIS, Germany, UK, Venezuela, S. Africa, Romania, and New Zealand. For more information, log on to: http://www.drreddys.com
Zometa® is a registered trademark of Novartis AG
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Reclast, Zometa, others[2] |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a605023 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
Intravenous |
| Drug class | Bisphosphonate[1] |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Protein binding | 22% |
| Metabolism | Nil |
| Elimination half-life | 146 hours |
| Excretion | Kidney (partial) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| PDB ligand | |
| Chemical and physical data | |
| Formula | C5H10N2O7P2 |
| Molar mass | 272.09 g/mol |
| 3D model (JSmol) | |
Judith Aronhime, Revital Lifshitz-Liron, “Zoledronic acid crystal forms, zoledronate sodium salt crystal forms, amorphous zoledronate sodium salt, and processes for their preparation.” U.S. Patent US20050054616, issued March 10, 2005., US20050054616
/////////////////disodium zoledronate tetrahydrate, zoledronic acid, ZOMETA, CGP-42446, CGP-42446A
OC(CN1C=CN=C1)(P(O)(O)=O)P(O)(O)=O
Bepotastine Besilate
ベポタスチンベシル酸塩
| 10 mg Tablets | For the treatment of allergic rhinitis | 27.03.2017 CDSCO
APPROVED |
USFDA
NDA 22-288 Bepotastine Besilate 1.5% Ophthalmic Solution ISTA Pharmaceuticals, Inc.
https://www.accessdata.fda.gov/drugsatfda_docs/nda/2009/02228s000_ChemR.pdf

Drug Substance Bepotastine besilate is manufactured by Ube Industries and the information for the NDA is submitted through DMF #19966. Bepotastine besilate is a white crystalline powder with no odor and bitter taste. It is very soluble in but sparingly soluble in . It is stable when exposed to light, and optically active. The S-isomer is the active drug and is controlled as an impurity through synthesis. The distribution coefficient in 1-octanol is higher than in aqueous buffer in the pH 5-9 range. There are 10 potential impurities but only one impurity is above 0.1%. Two potential genotoxic impurities are controlled below . Residual is controlled below Bepotastine besilate is stable under long term storage conditions for (25ºC/60% RH) over 5 years
Bepotastine besilate was originally developed as an oral tablet dosage form and got approval in Japan in 2000 for allergic rhinitis. It is a non-sedating anti-allergic drug. The proposed NDA is an ophthalmic solution indicated for allergic conjunctivitis. Bepotastine besilate ophthalmic solution 1.5% is a sterile solution. It is an aqueous solution to be administered as drops at or near physiological pH range of tears. The formulation contains sodium chloride, monobasic sodium phosphate as dihydrate, benzalkonium chloride, sodium hydroxide and purified water; typically these components are used for , preservative action, pH adjustment,
INTRO
Bepotastine is a non-sedating, selective antagonist of the histamine 1 (H1) receptor. Bepotastine was approved in Japan for use in the treatment of allergic rhinitis and uriticaria/puritus in July 2000 and January 2002, respectively, and is marketed by Tanabe Seiyaku Co., Ltd. under the brand name Talion. It is available in oral and opthalmic dosage forms in Japan. The opthalmic solution is FDA approved since Sept 8, 2009 and is under the brand name Bepreve.
Tae Hee Ha, Chang Hee Park, Won Jeoung Kim, Soohwa Cho, Han Kyong Kim, Kwee Hyun Suh, “PROCESS FOR PREPARING BEPOTASTINE AND INTERMEDIATES USED THEREIN.” U.S. Patent US20100168433, issued July 01, 2010., US20100168433
BEPREVE® (bepotastine besilate ophthalmic solution) 1.5% is a sterile, topically administered drug for ophthalmic use. Each mL of BEPREVE contains 15 mg bepotastine besilate.
Bepotastine besilate is designated chemically as (+) -4-[[(S)-p-chloro-alpha -2pyridylbenzyl] oxy]-1-piperidine butyric acid monobenzenesulfonate. The chemical structure for bepotastine besilate is:
 |
Bepotastine besilate is a white or pale yellowish crystalline powder. The molecular weight of bepotastine besilate is 547.06 daltons. BEPREVE ophthalmic solution is supplied as a sterile, aqueous 1.5% solution, with a pH of 6.8.
The osmolality of BEPREVE (bepotastine besilate ophthalmic solution) 1.5% is approximately 290 mOsm/kg.
ベポタスチンベシル酸塩 JP17
Bepotastine Besilate

C21H25ClN2O3▪C6H6O3S : 547.07
[190786-44-8]
Bepotastine (Talion, Bepreve) is a 2nd generation antihistamine.[1] It was approved in Japan for use in the treatment of allergic rhinitisand urticaria/pruritus in July 2000 and January 2002, respectively. It is currently marketed in the United States under the brand-name Bepreve, by ISTA Pharmaceuticals.
Bepotastine besilate is a second-generation antihistamine that was launched in a tablet formulation under a collaboration between Tanabe Seiyaku and Ube in 2000 and in 2002 for the treatment of allergic rhinitis including sneeze, mucus discharge and solidified mucus, and for the treatment of urticaria, respectively. An orally disintegrating tablet was made available in Japan in 2006, while a dry syrup formulation for the treatment of allergic rhinitis was studied in clinical trials at Tanabe Seiyaku for the treatment of allergic rhinitis
Originally developed at Ube, bepotastine besilate was later licensed to Tanabe Seiyaku as part of a collaboration agreement. In 2010, rights were licensed to Dong-A and Mitsubishi Tanabe Pharma in Korea for the treatment of eye disorders.
Bepotastine is available as an ophthalmic solution and oral tablet. It is a direct H1-receptor antagonist that inhibits the release of histamine from mast cells.[2] The ophthalmic formulation has shown minimal systemic absorption, between 1 and 1.5% in healthy adults.[3] Common side effects are eye irritation, headache, unpleasant taste, and nasopharyngitis.[3] The main route of elimination is urinary excretion, 75-90% excreted unchanged.[3]
It is marketed in Japan by Tanabe Seiyaku under the brand name Talion. Talion was co-developed by Tanabe Seiyaku and Ube Industries, the latter of which discovered bepotastine. In 2001, Tanabe Seiyaku granted Senju, now owned by Allergan, exclusive worldwide rights, with the exception of certain Asian countries, to develop, manufacture and market bepotastine for ophthalmic use. Senju, in turn, has granted the United States rights for the ophthalmic preparation to ISTA Pharmaceuticals.
In 2011, ISTA pharmaceuticals experienced a 2.4% increase in net revenues from 2010, which was driven by the sales of Bepreve. Their net revenue for 2011 was $160.3 million.[4] ISTA Pharmaceuticals was acquired by Bausch & Lomb in March 2012 for $500 million.[5] Bausch & Lomb hold the patent for bepotastine besilate (https://www.accessdata.fda.gov/scripts/cder/ob/docs/temptn.cfm. On November 26, 2014, Bausch & Lomb sue Micro Labs USA for patent infringement.[6] Bausch & Lomb was recently bought out by Valeant Pharmaceuticals in May 2013 for $8.57 billion, Valeant’s largest acquisition to date, causing the company’s stock to rise 25% when the deal was announced.[7]
A Phase III clinical trial was carried out in 2010 to evaluate the effectiveness of bepotastine besilate ophthalmic solutions 1.0% and 1.5%.[8] These solutions were compared to a placebo and evaluated for their ability to reduce ocular itchiness. The study was carried out with 130 individuals and evaluated after 15 minutes, 8 hours, or 16 hours. There was a reduction in itchiness at all-time points for both ophthalmic solutions. The study concluded that bepotastine besilate ophthalmic formulations reduced ocular itchiness for at least 8 hours after dosing compared to placebo. Phase I and II trials were carried out in Japan.
Studies have been performed in animals and bepotastine besilate was not found to be teratogenic in rats during fetal development, even at 3,300 times more that typical use in humans.[3] Evidence of infertility was seen in rats at 33,000 times the typical ocular does in humans.[3] The safety and efficacy has not been established in patients under 2 years of age and has been extrapolated from adults for patients under 10 years of age.[3]
SYN
EP 0335586; JP 1989242574; JP 1990025465; JP 1993294929; US 4929618
The reaction of 4-[1-(4-chlorophenyl)-1-(2-pyridyl)methoxy]piperidine (I) with ethyl 4-bromobutyrate (II) by means of K2CO3 in refluxing acetone gives the corresponding condensation product (III), which is then hydrolyzed with NaOH in ethanol/water yielding compound (IV).
SYN 2
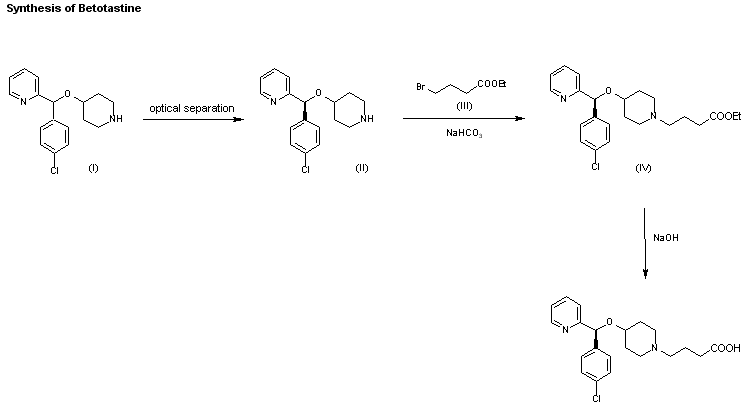
JP 1998237070; JP 2000198784; WO 9829409
A new synthesis of betotastine has been developed: The racemic 4-[1-(4-chlorophenyl)-1-(2-pyridyl)methoxy]piperidine (I) is submitted to optical resolution with N-acyl amino acids such as N-acetyl-L-phenylalanine (preferred), N-acetyl-L-leucine, N-(benzyloxycarbonyl)-L-phenylalanine, N-(benzyloxycarbonyl)-L-valine, N-(benzyloxycarbonyl)-L-threonine, N-(benzyloxycarbonyl)-L-serine or with (2R,3R)-3-(5-chloro-2-nitrophenylsulfanyl)-2-hydroxy-3-(4-methoxyphenyl)propionic acid (preferred) or (2R,3R)-2-hydroxy-3-(4-methoxyphenyl)-3-(2-nitrophenylsulfanyl)propionic acid as chiral intermediates, yielding the (S)-isomer (II). The condensation of (II) with ethyl 4-bromobutyrate (III) by means of a base such as Na2CO3, NaHCO3, K2CO3 or KHCO3 gives the expected 4-(1-piperidinyl)butyric acid ester (IV), which is finally hydrolyzed with NaOH or KOH in aqueous ethanol or methanol.
SYN 3
A new synthesis of betotastine has been developed: The racemic 4-[1-(4-chlorophenyl)-1-(2-pyridyl)methoxy]piperidine (I) is submitted to optical resolution with N-acyl amino acids such as N-acetyl-L-phenylalanine (preferred), N-acetyl-L-leucine, N-(benzyloxycarbonyl)-L-phenylalanine, N-(benzyloxycarbonyl)-L-valine, N-(benzyloxycarbonyl)-L-threonine, N-(benzyloxycarbonyl)-L-serine or with (2R,3R)-3-(5-chloro-2-nitrophenylsulfanyl)-2-hydroxy-3-(4-methoxyphenyl)propionic acid (preferred) or (2R,3R)-2-hydroxy-3-(4-methoxyphenyl)-3-(2-nitrophenylsulfanyl)propionic acid as chiral intermediates, yielding the (S)-isomer (II). The condensation of (II) with ethyl 4-bromobutyrate (III) by means of a base such as Na2CO3, NaHCO3, K2CO3 or KHCO3 gives the expected 4-(1-piperidinyl)butyric acid ester (IV), which is finally hydrolyzed with NaOH or KOH in aqueous ethanol or methanol.
CLIP
A Novel Synthetic Method for Bepotastine, a Histamine H1 Receptor …
file:///C:/Users/91200291/Downloads/B130241_549.pdf


Scheme 1. Synthesis of bepotastine l-menthyl ester N-benzyloxycarbonyl-L-aspartic acid complex (3), bepotastine besilate (4) and bepotastine calcium (5). Reagents and conditions; i) 4-bromobutanoic acid l-menthyl ester, K2CO3, acetone, reflux, 7 h, 95-99%; ii) N-benzyloxycarbonyl-L-aspartic acid (NCbzLAA), ethyl acetate, rt, 12 h, 71-73%; iii) Ethyl acetate/H2O, NaHCO3, 97-99%; iv) EtOH:H2O = 1:1, NaOH, rt, 12 h, 3.0 N-HCl Neutralization, 92- 95%; v) AcOH, reflux, 12 h, racemization 97-100%; vi) Bezensulfonic acid, acetonitrile, rt, 12 h, 64-67%; vii) NaOH, H2O, CaCl2, rt, 12 h, 86-89%.
Synthesis of (S)-Bepotastine Besilate (4). Bepotastine (50 g, 0.13 mol) was dissolved in 500 mL of acetonitrile, and benzenesulfonic acid monohydrate (20 g, 0.11 mol) was added to the reaction mixture. Bepotastine besilate (0.5 g, 1.28 mmol) was seeded in the reaction mixture and stirred at rt for 12 h. The solid precipitate was filtered and dried. The product was obtained 38 g (yield: 64%, optical purity: 99.5% ee) as a pale white crystalline powder. Melting point: 161- 163 o C. Water: 0.2% (Karl-Fischer water determination). MS: m/z 389.1 [M+H]; 1 H-NMR (300 MHz, DMSO-d6) δ 9.2 (br s, 1H), 8.5 (d, J = 4.1 Hz, 1H), 7.8 (t, J = 7.7 Hz, 1H), 7.6 (m, 3H), 7.4 (m, 4H), 7.3 (m, 4H), 5.7 (s, 1H), 3.7 (br s, 2H), 3.3 (br s, 3H), 3.1 (br s, 2H), 2.3 (t, J = 14.1 Hz, 2H), 2.2 (m, 1H), 2.0 (m, 1H), 1.8 (m, 3H), 1.7 (m, 1H); IR (KBr, cm−1 ): 3422, 2996, 2909, 2735, 2690, 2628, 1719, 1592, 1572, 1488, 1470, 1436, 1411, 1320, 1274, 1221, 1160, 1123, 1066, 1031, 1014, 996, 849, 830, 771, 759, 727, 693, 612, 564
| FDA Orange Book Patents: 1 of 3 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 8784789 |
| Expiration | Sep 5, 2024 |
| Applicant | BAUSCH AND LOMB INC |
| Drug Application | N022288 (Prescription Drug: BEPREVE. Ingredients: BEPOTASTINE BESILATE) |
| FDA Orange Book Patents: 2 of 3 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 6780877 |
| Expiration | Sep 19, 2019 |
| Applicant | BAUSCH AND LOMB INC |
| Drug Application | N022288 (Prescription Drug: BEPREVE. Ingredients: BEPOTASTINE BESILATE) |
| FDA Orange Book Patents: 3 of 3 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 8877168 |
| Expiration | Jul 30, 2023 |
| Applicant | BAUSCH AND LOMB INC |
| Drug Application | N022288 (Prescription Drug: BEPREVE. Ingredients: BEPOTASTINE BESILATE) |
 |
|
| Clinical data | |
|---|---|
| Trade names | Bepreve |
| AHFS/Drugs.com | International Drug Names |
| MedlinePlus | a610012 |
| Pregnancy category |
|
| Routes of administration |
Oral, topical (eye drops) |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | High (oral) Minimal (topical) |
| Protein binding | ~55% |
| Excretion | Renal (75–90%) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C21H25ClN2O3 |
| Molar mass | 388.88 g/mol |
| 3D model (JSmol) | |
|url= value (help).////////////Bepotastine Besilate, ベポタスチンベシル酸塩 ,Talion , tau284, TAU-284DS, TAU-284, DA-5206
HL-151 , SNJ-1773
C1CN(CCC1OC(C2=CC=C(C=C2)Cl)C3=CC=CC=N3)CCCC(=O)O.C1=CC=C(C=C1)S(=O)(=O)O
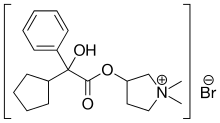

Cas 596-51-0,
CAS FREE FORM OF ABOVE 13283-82-4
Glycopyrrolate, ATC:A03AB02
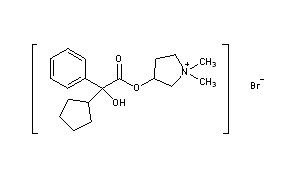
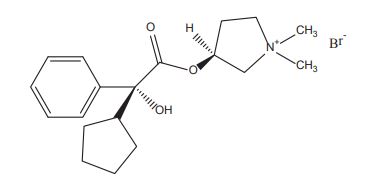
FREE FORM OF ABOVE 740028-90-4
NMR analysis of the diastereomers of glycopyrronium bromide
Finnish Chemical Letters (1975), (3-4), 94-6
Michael Woehrmann, Lara Terstegen, Stefan Biel, Thomas Raschke, Svenja-Kathrin Cerv, Werner Zilz, Sven Untiedt, Thomas Nuebel, Uwe Schoenrock, Heiner Max, Helga Biergiesser, Yvonne Eckhard, Heike Miertsch, Heike Foelster, Cornelia Meier-Zimmerer, Bernd Traupe, Inge Kruse, “GLYCOPYRROLATE IN COSMETIC PREPARATIONS.” U.S. Patent US20090208437, issued August 20, 2009.US20090208437
Glycopyrrolate is a muscarinic antagonist used as an antispasmodic, in some disorders of the gastrointestinal tract, and to reduce salivation with some anesthetics.
Glycopyrronium (as the bromide salt glycopyrrolate) is a synthetic anticholinergic agent with a quaternary ammonium structure. A muscarinic competitive antagonist used as an antispasmodic, in some disorders of the gastrointestinal tract, and to reduce salivation with some anesthetics. In October 2015, glycopyrrolate was approved by the FDA for use as a standalone treatment for Chronic obstructive pulmonary disease (COPD), as Seebri Neohaler.
In anesthesia, glycopyrronium injection can be used as a before surgery in order to reduce salivary, tracheobronchial, and pharyngealsecretions, as well as decreasing the acidity of gastric secretion. It is also used in conjunction with neostigmine, a neuromuscular blocking reversal agent, to prevent neostigmine’s muscarinic effects such as bradycardia.
It is also used to reduce excessive saliva (sialorrhea),[3][4][5] and Ménière’s disease.[6]
It decreases acid secretion in the stomach and so may be used for treating stomach ulcers, in combination with other medications.
It has been used topically and orally to treat hyperhidrosis, in particular, gustatory hyperhidrosis.[7][8]
In inhalable form it is used to treat chronic obstructive pulmonary disease (COPD). Doses for inhalation are much lower than oral ones, so that swallowing a dose will not have an effect.[9][10]
Since glycopyrronium reduces the body’s sweating ability, it can even cause hyperthermia and heat stroke in hot environments. Dry mouth, difficulty urinating, headaches, diarrhea and constipation are also observed side effects of the medication. The medication also induces drowsiness or blurred vision, an effect exacerbated by the consumption of alcohol.
Glycopyrronium blocks muscarinic receptors,[11] thus inhibiting cholinergic transmission.
Glycopyrronium bromide affects the gastrointestinal tracts, liver and kidney but has a very limited effect on the brain and the central nervous system. In horse studies, after a single intravenous infusion, the observed tendencies of glycopyrronium followed a tri-exponential equation, by rapid disappearance from the blood followed by a prolonged terminal phase. Excretion was mainly in urine and in the form of an unchanged drug. Glycopyrronium has a relatively slow diffusion rate, and in a standard comparison to atropine, is more resistant to penetration through the blood-brain barrier and placenta.[12]
It has been studied in asthma.[13][14]

PATENT
https://patents.google.com/patent/CN103819384A/en


PAtent
https://patents.google.com/patent/CN103159659A/en

glycopyrrolate (I)
Methyl ethyl ketone (20mL) IOOmL three-necked flask was added 8 (4.6g, 15mmol) was, at (Γ5 ° C was added dropwise dibromomethane (2.9g, 30mmol) in butanone (5 mL) was added dropwise completed, continued The reaction was stirred for 15min, and a white solid precipitated, was allowed to stand 36h at room temperature, filtered off with suction, the filter cake was sufficiently dried to give crude ketone was recrystallized twice to give a white powdery crystals I (3.9g, 66%) mp 191~193 ° C chromatographic purity 99.8% [HPLC method, mobile phase: lmol / L triethylamine acetate – acetonitrile – water (1: 150: 49); detection wavelength: 230nm, a measurement of the area normalization method] .MS m / z: 318 ( m-BrO 1HNMR (CD3OD) δ:! 1.33~1.38 (m, 2H), 1.55~1.70 (m, 6H), 2.11~2.21 (m, 1H), 2.67~2.80 (m, 1H), 3.02 (m, 1H), 3.06 (s, 3H), 3.23 (s, 3H), 3.59~3.71 (m, 3H), 3.90 (dd, /=13.8,1H), 5.47 (m, 1H), 7.27 (t, 1H) , 7.35 (t, 2H), 7.62 (dd, 2H) .13C bandit R (DMSO) δ: 27.0, 27.4, 28.0, 31.3, 47.8, 53.8, 54.3, 66.0, 71.3, 74.6, 81.1, 126.9,128.7,129.3 , 143.2 17 5.00
Patent
https://patents.google.com/patent/WO2016204998A1/en

PATENT
https://patents.google.com/patent/EP2417106B1/en
It has the following chemical structure:


EXAMPLE Example 1 Preparation of (3S,2’R)- and (3R,2’S)-3-[(cyclopentyl-hydroxyphenylacetyl)-oxy]-1,1-dimethylpyrrolidinium bromide
This process is summarised in the following reaction scheme:

Reference Example 2 Preparation of cyclopentyl-hydroxy-phenyl-acetic acid 1-methyl-pyrrolidin-3yl-ester in toluene
Example 3 Preparation of 2-cyclopentyl-2-hydroxy-1-imidazol-1-yl-2-phenyl-ethanone, the active intermediate
This process is summarised in the following reaction scheme:

The compound was characterised by IR-spectroscopy (measured as a solid film on a BRUKER TENSOR 27 FT-IR spectrometer over a wave number range of 4000-600 cm-1 with a resolution of 4 cm-1). An assignment of the most important bands is given below:
| Wavenumber (cm-1) | Assignments |
| 3300 ∼ 2500 | O-H stretching |
| 3167, 3151, 3120 | Imidazole CH stretching |
| 2956, 2868 | Cyclopentyl CH stretching |
| 1727 | C=O stretching |
| 1600, 1538, 1469 | Aromatic rings stretching |
| 735 | Mono-subst. benzene CH o.o.p. bending |
| 704 | Mono-subst. benzene ring o.o.p. bending |
SYN
PAPER
https://link.springer.com/article/10.1007/s41981-018-0015-4
Journal of Flow Chemistry, pp 1–8| Cite as
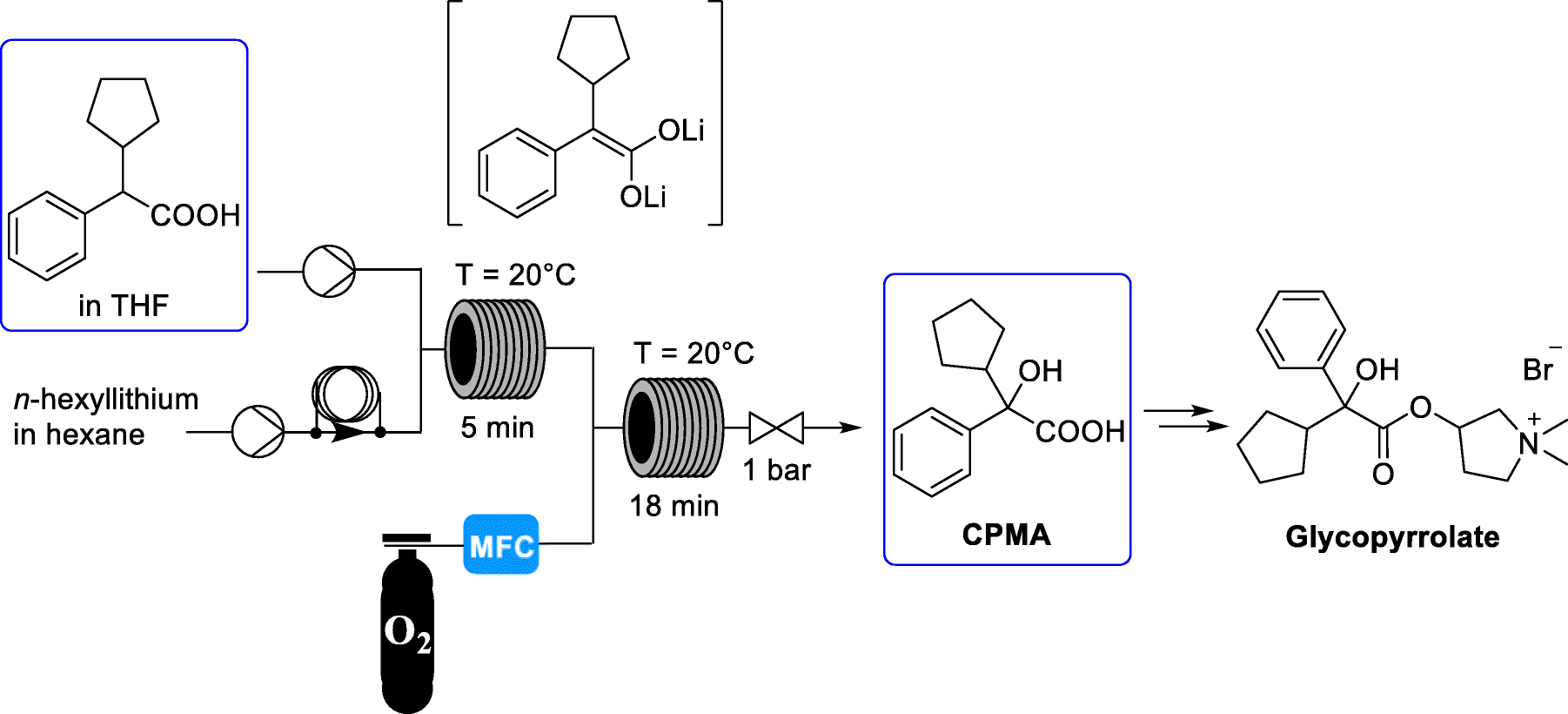
The medicinal properties of glycopyrronium bromide (glycopyrrolate, 4) were first identified in the late 1950s [1]. Glycopyrrolate is an antagonist of muscarinic cholinergic receptors and is used for the treatment of drooling or excessive salivation (sialorrhea) [2], excess sweating (hyperhidrosis) [3], and overactive bladder and for presurgery treatment. In addition, it has recently been introduced as an effective bronchodilator for the treatment of chronic obstructive pulmonary disease (COPD) for asthma patients [4]. Glycopyrrolate displays few side effects because it does not pass through the blood brain barrier. Cyclopentyl mandelic acid (CPMA, 1), or its corresponding ester derivatives, are key intermediates in the synthetic routes to 4. CPMA (1) reacts with 1-methyl-pyrrolidin-3-ol (2) to form tertiary amine 3. N-Methylation of 3 by methyl bromide gives quaternary ammonium salt glycopyrrolate 4 as a racemate (Scheme 1) [5].
Scheme 1
Synthesis of glycopyrrolate 4 from CPMA (1)
CPMA (1) is a synthetically challenging intermediate to prepare (Scheme 2). Routes A to D are most likely to be the commercially applied methods because these procedures are described in patents [5]. The published descriptions for the yields of 1 range from 28 to 56% for routes A to D. Ethyl phenylglyoxylate is reacted with cyclopentyl magnesium bromide to form an ester which is then hydrolyzed (route A) [6]. Phenylglyoxylic acid can be reacted in a similar manner with cyclopentyl magnesium bromide to directly form 1 (route B) [7]. Alternatively, the inverse addition of phenyl-Grignard reagent to cyclopentyl glyoxylic acid ester is reported (route C) [8]. Cyclopentyl glyoxylic acid ester can also be reacted with cyclopentadienyl magnesium bromide which is followed by an additional hydrogenation step with Pd/C and H2 to afford 1 (route D) [9, 10].
Scheme 2
Existing synthetic pathways to CPMA (1)
PATENT
EXA M PL E S
EXAM PL E 1
Scheme 1
ST E P I
To a stirred solution of N-methyl pyrrol i din- 3-ol (2, 1 equiv) and Et3N (1.2 equiv) in dichloromethane was added a solution of 2-cyclopentyl-2-oxoacetyl chloride (1, 1.1 equiv) in DCM at O °C under nitrogen atmosphere for 20 min. The resulting solution was allowed to stir at room temperature over 10h. After completion, the mixture was quenched with water and extracted with diethyl ether to afford the pure product (3A).
Similarly, the product 3A is also obtained by reaction of 2 with other reagents, phenyl oxalic acid, methyl phenyl oxalate, and phenyl hemi-oxaldehyde respectively as shown in Scheme 1.
ST E P II
3A
To a mixture of bromobenzene (2.2 equiv) and Mg metal (2.2 equiv) in TH F (15 mL) was stirred over a period of 30 min at 0 · C. To this mixture, a solution of 1 -methyl pyrrol idin-3-yl 2-cyclopentyl-2-oxoacetate (3, 1 equiv) in T HF was added in portions over a period of 30 min. Up on completion, the reaction mixture was poured into ice water and extracted with ethyl acetate. The organic layer was separated and concentrated in vacuo. The resulting residue was purified by column chromatography to afford the pure product (5).
ST E P III
To a solution of compound 5 (1 equiv) in acetonitrile and chloroform mixture (10 mL, 2:3) was added methyl bromide (4 equiv). The mixture was stirred at room temperature for 72h. The solvents were evaporated, and the resulting residue was washed with diethyl ether to afford the pure product (6) as a white solid.
EXAM PL E 2
Scheme 2
ST E P I
To a stirred solution of N-methyl pyrrol i din- 3-ol (2, 1 equiv) and Et3N (1.2 equiv) in dichloromethane was added a solution of 2- oxo-2- phenyl acetyl chloride (1.1 equiv) in dichloromethane at 0 °C under nitrogen atmosphere for 15 min. The resulting solution was allowed to stir at room temperature over 12h. After completion, the mixture was quenched with water and extracted with diethyl ether to afford the pure product (3B).
Similarly, the product 3B is also obtained by reaction of 2 with other reagents, phenyl oxalic acid, methyl phenyl oxalate, and phenyl hemi-oxaldehyde respectively as shown in Scheme 2.
ST E P II
To a mixture of cyclopentyl bromide (4, 2.2 equiv) and Mg metal (2.2 equiv) in THF (15 mL) was stirred over a period of 30 min at 0 – C. To this mixture, a solution of 1-methylpyrrolidin-3-yl-2-oxo-2-phenylacetate (3B, 1 equiv) in TH F was added in portions over a period of 30 min. Up on completion, the reaction mixture was poured into ice water and extracted with ethyl acetate. The organic layer was separated and concentrated in vacuo. The resulting residue was purified by column chromatography to afford the pure product (5).
ST E P III
To a solution of compound 5 (1 equiv) in acetonitrile and chloroform mixture (10 mL, 2:3) was added methyl bromide (4 equiv). The mixture was stirred at room temperature for 75h. The solvents were evaporated, and the resulting residue was washed with diethyl ether to afford the pure product (6) as a white solid.
The invention has been described in detail with reference to preferred embodiments thereof. However, it will be appreciated by those skilled in the art that changes may be made in these embodiments without departing from the principles and nature of the invention, the scope of which is defined in the appended claims and their equivalents.
 |
|
| Clinical data | |
|---|---|
| Trade names | Robinul, Cuvposa, Seebri, Qbrexza, others |
| License data | |
| Pregnancy category |
|
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChemCID | |
| ChemSpider | |
| UNII | |
| ECHA InfoCard | 100.008.990 |
| Chemical and physical data | |
| Formula | C19H28BrNO3 |
| Molar mass | 398.335 g/mol |
| 3D model (JSmol) | |
 |
|
| Clinical data | |
|---|---|
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a602014 |
| Pregnancy category |
|
| Routes of administration |
By mouth, intravenous, inhalation |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Elimination half-life | 0.6–1.2 hours |
| Excretion | 85% renal, unknown amount in the bile |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| ECHA InfoCard | 100.008.990 |
| Chemical and physical data | |
| Formula | C19H28NO3+ |
| Molar mass | 318.431 g/mol |
| 3D model (JSmol) | |


Iron sucrose (USP);
Ferric oxide, saccharated;
Sucroferric oxyhydroxide;
Venofer (TN)
含糖酸化鉄;
スクロオキシ水酸化鉄
| Molecular Formula: | C12H29Fe5Na2O23 |
|---|---|
| Molecular Weight: | 866.546 g/mol |
|
CAS REGISTRY NUMBER 12134-57-5, 8047-67-4
disodium;(2R,3R,4S,5S,6R)-2-[(2S,3S,4S,5R)-3,4-dihydroxy-2,5-bis(hydroxymethyl)oxolan-2-yl]oxy-6-(hydroxymethyl)oxane-3,4,5-triol;iron(3+);oxygen(2-);hydroxide;trihydrate
Iron sugar; Saccharated iron; Sucroferric oxyhydroxide; Saccharated iron oxide; Saccharated ferric oxide; Ferrivenin
Ferric oxyhydroxide; Ferrihydrite; Iron oxyhydroxide; P-TOL; PA-21; PA21-1; Phosphate binder – Vifor Pharma; suroferric oxyhydroxide tablets; Velphoro
NDC 49230-645-51
Iron saccharate (Sucroferric oxyhydroxide or Iron Sucrose) is used as a source of iron in patients with iron deficiency anemia with chronic kidney disease (CKD), including those who are undergoing dialysis (hemodialysis or peritoneal) and those who do not require dialysis. Due to less side effects than iron dextran, iron saccharate is more preferred in chronic kidney disease patients.
Mixture of polynuclear iron(III)-oxyhydroxide, starch and sucrose
VIFOR FRESENIUS MEDICAL CARE RENAL PHARMA FRANCE
Approved in US
Indicated for the control of serum phosphorus levels in patients with chronic kidney disease on dialysis.
THERAPEUTIC CLAIM Oral phosphate binder, treatement of elevated
phosphate levels in patients undergoing dialysis
CHEMICAL DESCRIPTIONS
1. Ferric hydroxide oxide
2. Mixture of iron(III) oxyhydroxide, sucrose, starches
3. Polynuclear iron(III) oxyhydroxide stabilized with sucrose and starches
structure
O =Fe -OH
MOLECULAR FORMULA FeHO2•xC12H22O11•y(C6H10O5)n
SPONSOR Vifor (International) Inc.
CODE DESIGNATIONS PA21
CAS REGISTRY NUMBER 12134-57-5
Sucroferric oxyhydroxide is a brown, amorphous powder. The drug substance is relatively poorly defined, so that the manufacturing process is particularly important. Sucroferric oxyhydroxide is prepared by basifying a ferric chloride solution, giving a polynuclear iron(III)-oxyhydroxide suspension which is mixed with potato and maize starches and sucrose. Vifor states that the sucrose stabilises the iron core and thus maintain the high phosphate adsorption capacity while the starches function as processing aids, but they are added simultaneously and the drug substance is probably a complex mixture of species.
The solubility of the active moiety, polynuclear iron oxyhydroxide, is evidently low in the gastrointestinal (GI) tract so that iron absorption is low. Aqueous solubility at different pH has been very poorly quantified. Vifor states that the “sucrose part is soluble in water, iron(III)-oxyhydroxide/starch mixture is practically insoluble in water.” While the iron oxide particle size is important in determining the phosphate binding, it is relatively difficult to directly measure. The sucrose/starch “wrapped” drug substance particle size is established and the process is controlled, but it does not correlate well with phosphate adsorption. Sucroferric oxyhydroxide cannot be controlled in the manner of a well-defined molecular drug and some variability between batches is likely. The drug substance specification includes a phosphate adsorption test. Vifor has tested the adsorption of a range of other in vivo chemical species to sucroferric oxyhydroxide and not identified any likely to be strongly bound, or affect phosphate binding, except for oxalate. Some drugs, however, do interact, for example alendronate is strongly absorbed (and the PI warnings in that context should be generalised to all bisphosphonates, not just identify the single drug in class studied)….https://www.tga.gov.au/sites/default/files/auspar-sucroferric-oxyhydroxide-150219.pdf
EMA
| Name | Active substance | Therapeutic area | Date of authorisation / refusal | Has current safety alert | Status | ||||
|---|---|---|---|---|---|---|---|---|---|
| Velphoro | mixture of polynuclear iron(III)-oxyhydroxide, sucrose and starches | HyperphosphatemiaRenal Dialysis | 26/08/2014 | Authorised |
| Name | Velphoro |
|---|---|
| Agency product number | EMEA/H/C/002705 |
| Active substance | mixture of polynuclear iron(III)-oxyhydroxide, sucrose and starches |
| International non-proprietary name(INN) or common name | mixture of polynuclear iron(III)-oxyhydroxide, sucrose and starches |
| Therapeutic area | HyperphosphatemiaRenal Dialysis |
| Anatomical therapeutic chemical (ATC) code | V03AE05 |
| Additional monitoring | This medicine is under additional monitoring. This means that it is being monitored even more intensively than other medicines. For more information, see medicines under additional monitoring. |
| Marketing-authorisation holder | Vifor Fresenius Medical Care Renal Pharma France |
|---|---|
| Revision | 5 |
| Date of issue of marketing authorisation valid throughout the European Union | 26/08/2014 |
Vifor Fresenius Medical Care Renal Pharma France
100-101 Terrasse Boieldieu
Tour Franklin- La Défense 8
92042 Paris la Défense Cedex
France
Sucroferric oxyhydroxide (INN; trade name Velphoro, by Vifor Fresenius Medical Care Renal Pharma) is a non-calcium, iron-based phosphate binder used for the control of serum phosphorus levels in adult patients with chronic kidney disease (CKD) on haemodialysis(HD) or peritoneal dialysis (PD).[1] It is used in form of chewable tablets.
In a healthy person, normal serum phosphate levels are maintained by the regulation of dietary absorption, bone formation and resorption, equilibration with intracellular stores, and renal excretion.[2] When kidney function is impaired, phosphate excretion declines. Without specific treatment, hyperphosphataemia occurs almost universally, despite dietary phosphate restriction and conventional dialysis treatment.[2][3] In patients on dialysis, hyperphosphataemia is an independent risk factor for fractures, cardiovascular disease and mortality.[4][5] Abnormalities in phosphate metabolism such as hyperphosphatemia are included in the definition of the new chronic kidney disease–mineral and bone disorder (CKD-MBD).[5]
Sucroferric oxyhydroxide comprises a polynuclear iron(III)-oxyhydroxide core that is stabilised with a carbohydrate shell composed of sucrose and starch.[6][7] The carbohydrate shell stabilises the iron(III)-oxyhydroxide core to preserve the phosphate adsorption capacity.
Dietary phosphate binds strongly to sucroferric oxyhydroxide in the gastrointestinal (GI) tract. The bound phosphate is eliminated in the faeces and thereby prevented from absorption into the blood. As a consequence of the decreased dietary phosphate absorption, serum phosphorus concentrations are reduced.
Sucroferric oxyhydroxide is approved by the United States Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for the control of serum phosphorus levels in patients with chronic kidney disease (CKD) on dialysis.[1][8]
The most frequently reported adverse drug reactions reported from trials were diarrhoea and discoloured faeces.[1][8] The vast majority of gastrointestinal adverse events occurred early during treatment and abated with time under continued dosing.[1]
Drug-interaction studies and post hoc analyses of Phase 3 studies showed no clinically relevant interaction of sucroferric oxyhydroxide with the systemic exposures to losartan, furosemide, omeprazole, digoxin, and warfarin,[9] the lipid-lowering effects of statins,[10] and oral vitamin D receptor agonists.[11] According to the European label (Summary of Product Characteristics), medicinal products that are known to interact with iron (e.g. doxycycline) or have the potential to interact with Velphoro should be administered at least one hour before or two hours after Velphoro.[1] This allows sucroferric oxyhydroxide to bind phosphate as intended and be excreted without coming into contact with medications in the gut that it might interact with. According to the US prescribing information, Velphoro should not be prescribed with oral levothyroxine.[8] The combination of sucroferric oxyhydroxide and levothyroxine is contraindicated because sucroferric oxyhydroxide contains iron, which may cause levothyroxine to become insoluble in the gut, thereby preventing the intestinal absorption of levothyroxine.[12]
The chewability of sucroferric oxyhydroxide compares well with that of Calcimagon, a calcium containing tablet used as a standard for very good chewability.[13] Tablets of sucroferric oxyhydroxide easily disintegrated in artificial saliva.
Clinical Phase 3 studies showed that sucroferric oxyhydroxide achieves and maintains phosphate levels in compliance with the KDOQI guidelines.[14][15] The reduction in serum phosphate levels of sucroferric oxyhydroxide-treated patients was non-inferior to that in sevelamer-treated patients. The required daily pill burden was lower with sucroferric oxyhydroxide.[14]
Sucroferric oxyhydroxide binds phosphate under empty and full stomach conditions and across the physiologically relevant pH range of the GI tract.[7]
In a retrospective, real-world study, hyperphosphatemic peritoneal dialysis patients who were prescribed to switch to sucroferric oxyhydroxide from sevelamer, lanthanum carbonate, or calcium acetate had significant reductions in serum phosphorus levels, along with a 53% decrease in the prescribed daily pill burden.[16]
Sucroferric oxyhydroxide nonproprietary drug name
The US Food and Drug Administration has given the green light to Vifor Fresenius Medical Care Renal Pharma’s hyperphosphatemia drug Velphoro.
The approval for Velphoro (sucroferric oxyhydroxide), formerly known as PA21, is based on Phase III data demonstrated that the drug successfully controls the accumulation of phosphorus in the blood with the advantage of a much lower pill burden than the current standard of care in patients with chronic kidney disease on dialysis, namely Sanofi’s Renvela (sevelamer carbonate). read this at
http://www.pharmatimes.com/Article/13-11-28/FDA_okays_Vifor_Fresenius_phosphate_binder_Velphoro.aspx
Velphoro (PA21) receives US FDA approval for the treatment of hyperphosphatemia in Chronic Kidney Disease Patients on dialysis
Velphoro (sucroferric oxyhydroxide) has received US Food and Drug Administration (FDA) approval for the control of serum phosphorus levels in patients with Chronic Kidney Disease (CKD) on dialysis. Velphoro will be launched in the US by Fresenius Medical Care North America in 2014.
Velphoro (previously known as PA21) is an iron-based, calcium-free, chewable phosphate binder. US approval was based on a pivotal Phase III study, which met its primary and secondary endpoints. The study demonstrated that Velphoro® successfully controls hyperphosphatemia with fewer pills than sevelamer carbonate, the current standard of care in patients with CKD on dialysis. The average daily dose to control hyperphosphatemia was 3.3 pills per day after 52 weeks.
Velphoro was developed by Vifor Pharma. In 2011, all rights were transferred to Vifor Fresenius Medical Care Renal Pharma, a common company of Galenica and Fresenius Medical Care. In the US, Velphorowill be marketed by Fresenius Medical Care North America, a company with a strong marketing and sales organization, and expertise in dialysis care. The active ingredient of Velphoro is produced by Vifor Pharma in Switzerland.
Hyperphosphatemia, an abnormal elevation of phosphorus levels in the blood, is a common and serious condition in CKD patients on dialysis. Most dialysis patients are treated with phosphate binders. However, despite the availability of a number of different phosphate binders, up to 50% of patients depending on the region are still unable to achieve and maintain their target serum phosphorus levels. In some patients, noncompliance due to the high pill burden and poor tolerability appear to be key factors in the lack of control of serum phosphorus levels. On average, dialysis patients take approximately 19 pills per day with phosphate binders comprising approximately 50% of the total daily pill burden. The recommended starting dose of Velphoro is 3 tablets per day (1 tablet per meal).
Full results from the pivotal Phase III study involving more than 1,000 patients were presented at both the 50th ERA-EDTA (European Renal Association European Dialysis and Transplant Association) Congress in Istanbul, Turkey, in May 2013, and the American Society of Nephrology (ASN) Kidney Week in Atlanta, Georgia, in November 2013. Velphorowas shown to be a potent phosphate binder, with lower pill burden and a good safety profile.
Based on these data, Vifor Fresenius Medical Care Renal Pharma believes that Velphoro offers a new and effective therapeutic option for the control of serum phosphorus levels in patients with chronic kidney disease on dialysis.
The regulatory processes in Europe, Switzerland and Singapore are ongoing and decisions are expected in the first half 2014. Further submissions for approval are being prepared.
PATENT
https://patents.google.com/patent/WO2016038541A1/en
Hyperphosphatemia is associated with significant increase in morbidity and mortality, and may induce severe complications, such as hypocalcemia, decreasing of vitamin-D production and metastatic calcification. Hyperphosphatemia is also contributing to the increased incidence of cardiovascular disease among dialysis-dependent patients. The phosphate binding capacity of iron oxide hydroxides is known in the art. The possible medical application of iron hydroxides and iron oxide hydroxides as phosphate adsorbents is also described.
US 4,970,079 patent discloses a method of controlling serum phosphate level in patients by iron oxy-hydroxides which bind to ingested phosphate. US 5,514,281 patent also discloses a process for the selective elimination of inorganic phosphate from body fluids by using a polynuclear metal oxyhydroxide preferably iron (III) oxyhydroxide.
US 6,174,442 patent describes an adsorbent for phosphate and a process for the preparation thereof, which contains polynuclear β-iron hydroxide stabilized by carbohydrates and/or humic acid.
In order to obtain an iron-based compound which can be used as a pharmaceutical, it is necessary to have an iron-based compound which is stable. It is known that iron oxide- hydroxide is not a stable compound with time ageing occurs. Ageing usually not only involves crystallization but also particle enlargement. Such ageing may alter the phosphate binding of an iron oxide -hydroxide based phosphate adsorbent. Accordingly, there exists a need for a process for manufacturing of an iron containing phosphate adsorbent. The process needs to be scalable, robust and consistently producing an iron containing phosphate adsorbent of the required pharmaceutical grade.
Examples
In examples which are intended to illustrate embodiments of the invention but which are not intended to limit the scope of the invention: ) Method of Making an Iron Containing Phosphate Adsorbent
To a solution of 1.96 kg sodium carbonate dissolved in 12.5 liter water, solution of 2.5 kg iron (III) chloride hexahydrate dissolved in 17.5 liter water was added at a temperature of 5 – 10°C. The resulting mixture was stirred for 90 to 120 minutes at 5 – 10°C. (25.0×3) liter water was added to the reaction mass and raised the temperature at 15 – 20°C with stirring. Stopped the stirring, settled precipitate and the supernatant water was removed. The precipitate was filtered and washed with 1.25 liter water. A suspension of the precipitate was prepared in water. To this, 875.0 gm sucrose and 695.0 gm potato starch were added and stirred for 120 minutes at 25 – 35°C. Cooled the reaction mass at 10 – 15°C and stirred for 90 to 120 minutes. 25.0 liters cold acetone was added to the reaction mass at 10 – 15°C and stirred for 90 to 120 minutes. The final product was filtered and washed with 1.25 liter cold acetone and further dried under vacuum at 30-35°C.
Yield: 2.08 kg ) Large-scale Method of Making an Iron Containing Phosphate Adsorbent
An aqueous solution of sodium carbonate and an aqueous solution of iron (III) chloride hexahydrate were mixed at a temperature of 5 – 10°C, optionally in the presence of solvent- 1. A volume of aqueous solution of sodium carbonate necessary to maintain the pH at about 7.0 to form a colloidal suspension of ferric hydroxide. The resulting mixture was stirred for 90 to 120 minutes at 5 – 10°C. Water was added to the reaction mass with stirring. Stopped the stirring, settled precipitated product and the water was decanted or siphoned. The precipitated product was further filtered and washed with using water. Suspension of the precipitated product was prepared in the water. Subsequently, sucrose and starch were added in to the suspension and stirred for 120 minutes at 25 – 35°C. Cooled the reaction mixture at 10 – 15°C and stirred for 90 to 120 minutes. Solvent-2 was added to the reaction mixture at 10 – 15°C and stirred for 90 to 120 minutes. The product was filtered and washed with the solvent-2 and further dried under vacuum at 30-35°C. Few illustrative examples provided in Table- 1, wherein the iron containing phosphate adsorbents were prepared according to the process of example-2 using the respective combination of Solvent- 1 and Solvent-2 as given in the table:
Table-1

3) Physical Properties of an Iron Containing Phosphate Adsorbents prepared as per above example-2.
> BET active Surface Area:
· Instrument : Surface area analyzer
• Condition : Surface area (m2/gm) at N2.P/P0 = 10%
Table-2

> Phosphate Binding Capacity at pH 3.0:
· Method : Ion Chromatography Instrument : Metrohm IC equipped with pump, Injector, conductivity detector and recorder.
Column Dionex Ion Pac AS-11 (4.0 x 250mm), 13μπι
Guard column Dionex Ion Pac AG-11 (4.0 x 50mm), 13μπι
Buffer preparation Weigh accurately about 2.118g of Sodium carbonate and 180mg of Sodium hydroxide in 1700mL water.
Mobile phase preparation : Buffer and acetonitrile (1700:300).
Results: Phosphate binding of an iron containing phosphate adsorbents obtained by following the process of the present invention found in the range of 30 mg/gm to 60 mg/gm. Particle Size Distribution:
Instrument Model : Malvern Mastersizer 2000 Particle size analyzer
Sampling Unit : Hydro 2000S
Analysis Model : General Purpose
Dispersant : 0.1% Span 85 in n-Hexane
Dispersant RI : 1.380
Stirrer Speed : 2200 RPM
Absorption : 1
Particle RI : 1.5
Obscuration : 10% to 20%
Sample Measurement time : 12 seconds
Background Measurement time : 12 seconds Table-3
Particle size distribution
Example no.
d(0.9) (μηι)
3d 43.67
3e 65.37
3f 37.75

| Clinical data | |
|---|---|
| Trade names | Velphoro |
| AHFS/Drugs.com | Consumer Drug Information |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Oral (chewable tablets) |
| ATC code | |
| Legal status | |
| Legal status |
|
| Chemical and physical data | |
| Formula | Varies |
| FDA Orange Book Patents: 1 of 2 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 6174442 |
| Expiration | Dec 19, 2018 |
| Applicant | VIFOR FRESENIUS |
| Drug Application | N205109 (Prescription Drug: VELPHORO. Ingredients: SUCROFERRIC OXYHYDROXIDE) |
/////////////Sucroferric oxyhydroxide, EU 2014, Iron sugar, Saccharated iron, Sucroferric oxyhydroxide, Saccharated iron oxide, Saccharated ferric oxide, Ferrivenin, 含糖酸化鉄, スクロオキシ水酸化鉄 , NDC 49230-645-51
C(C1C(C(C(C(O1)OC2(C(C(C(O2)CO)O)O)CO)O)O)O)O.O.O.O.[OH-].[O-2].[O-2].[O-2].[O-2].[O-2].[O-2].[O-2].[O-2].[Na+].[Na+].[Fe+3].[Fe+3].[Fe+3].[Fe+3].[Fe+3]