
Home » Uncategorized (Page 29)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Octamoxin, октамоксин , أوكتاموكسين , 奥他莫辛 ,
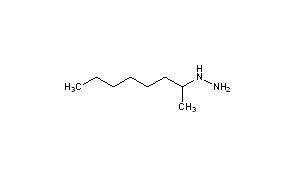
- Molecular FormulaC8H20N2
- Average mass144.258 Da

References
- ^ “Octamoxin – Compound Summary”. USA: National Center for Biotechnology Information. 26 March 2005. Identification and Related Records. Retrieved 31 May 2012.
- ^ “Dictionary of pharmacological agents – Google Books”.
- ^ “13-06781. Octamoxin [Archived]: The Merck Index”.
- ^ Levy J, Michel-Ber E (1966). “[Relations between the antidepressive effects of octamoxine revealed by 3 pharmacological tests and inhibition of cerebral monoamine oxidase in mice]”. Thérapie (in French). 21 (4): 929–45. PMID 5925088.
- ^ Gayral L, Stern H, Puyuelo R (1966). “[Indications and results of the treatment of mental depression by octamoxine (ximaol)]”. Thérapie (in French). 21 (5): 1183–90. PMID 5976767.
 |
|
| Names | |
|---|---|
| Preferred IUPAC name
1-Methylheptylhydrazine[citation needed]
|
|
| Systematic IUPAC name
Octan-2-ylhydrazine[1]
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChemSpider | |
|
PubChem CID
|
|
| UNII | |
| Properties | |
| C8H20N2 | |
| Molar mass | 144.262 g·mol−1 |
| Density | 0.831 g/mL |
| Boiling point | 228 °C (442 °F; 501 K) |
| Pharmacology | |
| Oral | |
| Related compounds | |
|
Related compounds
|
Tuaminoheptane |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
Labetalol Hydrochloride, ラベタロール ,

Labetalol
ラベタロール;
- Molecular FormulaC19H24N2O3
- Average mass328.405 Da
Labetalol hydrochloride, AH-5158A, Sch-15719W, Amipress, Trandate, Normodyne
Labetalol was granted FDA approval on 1 August 1984
Presolol; (RS)-2-Hydroxy-5-{1-hydroxy-2-[(1-methyl-3-phenylpropyl)amino]ethyl}benzamide; 5-[1-Hydroxy-2-[(1-methyl-3-phenyl propyl)amino]ethyl]salicylamide
A salicylamide derivative that is a non-cardioselective blocker of BETA-ADRENERGIC RECEPTORS and ALPHA-1 ADRENERGIC RECEPTORS.
- AH 5158
- Albetol
- EC 253-258-3
- EINECS 253-258-3
- HSDB 6537
- Ibidomide
- Labetalol
- Labetalolum
- Labetalolum [INN-Latin]
- Labetolol
- SCH 15719W
- UNII-R5H8897N95
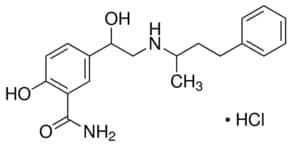
Labetalol hydrochloride
- CAS Number 32780-64-6,
- Empirical Formula (Hill Notation) C19H24N2O3 · HCl,
- Molecular Weight 364.87
REF https://www.accessdata.fda.gov/drugsatfda_docs/anda/98/74787_Labetalol%20Hydrochloride_Chemr.pdf

RR
CAS 75659-07-3
- (R,R)-Labetalol
- Dilevalol
- Dilevalolum
- Dilevalolum [Latin]
- UNII-P6629XE33T
Labetalol is a racemic mixture of 2 diastereoisomers where dilevalol, the R,R’ stereoisomer, makes up 25% of the mixture.8 Labetalol is formulated as an injection or tablets to treat hypertension
Labetalol is a medication used to treat high blood pressure and in long term management of angina.[1][2] This includes essential hypertension, hypertensive emergencies, and hypertension of pregnancy.[2] In essential hypertension it is generally less preferred than a number of other blood pressure medications.[1] It can be given by mouth or by injection into a vein.[1]
Common side effects include low blood pressure with standing, dizziness, feeling tired, and nausea.[1] Serious side effects may include low blood pressure, liver problems, heart failure, and bronchospasm.[1] Use appears safe in the latter part of pregnancy and it is not expected to cause problems during breastfeeding.[2][3] It works by blocking the activation of β-receptors and α-receptors.[1]
Labetalol was patented in 1966 and came into medical use in 1977.[4] It is available as a generic medication.[2] A month supply in the United Kingdom costs the NHS about 8 £ as of 2019.[2] In the United States the wholesale cost of this amount is about US$12.[5] In 2016 it was the 233rd most prescribed medication in the United States with more than 2
Medical uses
Labetalol is effective in the management of hypertensive emergencies, postoperative hypertension, pheochromocytoma-associated hypertension, and rebound hypertension from beta blocker withdrawal. [7]
It has a particular indication in the treatment of pregnancy-induced hypertension which is commonly associated with pre-eclampsia. [8]
It is also used as an alternative in the treatment of severe hypertension.[7]
Special populations
Pregnancy: studies in lab animals showed no harm to the baby. However, a comparable well-controlled study has not been performed in pregnant women.[9]
Nursing: breast milk has been shown to contain small amounts of labetalol (0.004% original dose). Prescribers should be cautious in the use of labetalol for nursing mothers.[9]
Pediatric: no studies have established safety or usefulness in this population.[9]
Geriatric: the elderly are more likely to experience dizziness when taking labetalol. Labetalol should be dosed with caution in the elderly and counseled on this side effect.[9]
Side effects
Common
- Neurologic: headache (2%), dizziness (11%) [9]
- Gastrointestinal: nausea (6%), dyspepsia (3%) [9]
- Cholinergic: nasal congestion (3%), ejaculation failure (2%) [9]
- Respiratory: dyspnea (2%) [9]
- Other: fatigue (5%), vertigo (2%), orthostatic hypotension [9]
Low blood pressure with standing is more severe and more common with IV formulation (58% vs 1%[9]) and is often the reason larger doses of the oral formulation cannot be used.[10]
Rare
- Fever [9]
- Muscle cramps [9]
- Dry eyes [9]
- Heart block [9]
- Hyperkalemia [9]
- Hepatotoxicity [9]
- Drug eruption similar to lichen planus[11]
- Hypersensitivity – which may result in a lethal respiratory distress[9]
Contraindications
Labetalol is contraindicated in people with overt cardiac failure, greater-than-first-degree heart block, severe bradycardia, cardiogenic shock, severe hypotension, anyone with a history of obstructive airway disease including asthma, and those with hypersensitivity to the drug.[12]
Chemistry
The minimum requirement for adrenergic agents is a primary or secondary amine separated from a substituted benzene ring by one or two carbons.[13] This configuration results in strong agonist activity. As the size of the substituent attached to the amine becomes greater, particularly with respect to a t-butyl group, then the molecule typically is found to have receptor affinity without intrinsic activity, and is, therefore, an antagonist.[13] Labetalol, with its 1-methyl-3-phenylpropyl substituted amine, is greater in size relative to a t-butyl group and therefore acts predominantly as an antagonist. The overall structure of labetalol is very polar. This was created by substituting the isopropyl group in the standard beta-blocker structure with an aralkyl group, including a carboxamide group on the meta position, and by adding a hydroxyl group on the para position.[14]
Labetalol has two chiral carbons and consequently exists as four stereoisomers.[15] Two of these isomers, the (S,S)- and (R,S)- forms are inactive. The third, the (S,R)-isomer, is a powerful α1 blocker. The fourth isomer, the (R,R)-isomer which is also known as dilevalol, is a mixed nonselective β blocker and selective α1 blocker.[14] Labetalol is typically given as a racemic mixture to achieve both alpha and beta receptor blocking activity.[16]
| Stereoisomers of labetalol | |
|---|---|
-Labetalol_Structural_Formula_V1.svg) (R,R)-Labetalol CAS number: 75659-07-3 |
-Labetalol_Structural_Formula_V1.svg) (S,S)-Labetalol CAS number: 83167-24-2 |
-Labetalol_Structural_Formula_V1.svg) (R,S)-Labetalol CAS number: 83167-32-2 |
-Labetalol_Structural_Formula_V1.svg) (S,R)-Labetalol CAS number: 83167-31-1 |
Labetalol acts by blocking alpha and beta adrenergic receptors, resulting in decreased peripheral vascular resistance without significant alteration of heart rate or cardiac output.
The β:α antagonism of labetalol is approximately 3:1.[17][18]
It is chemically designated in International Union of Pure and Applied Chemistry (IUPAC) nomenclature as 2-hydroxy-5-[1-hydroxy-2-[(1-methyl-3-phenylpropyl)amino]ethyl]benzamide monohydrochloride.[16][19]
Pharmacology
Mechanism of action
Labetalol’s dual alpha and beta adrenergic antagonism has different physiological effects in short- and long-term situations. In short-term, acute situations, labetalol decreases blood pressure by decreasing systemic vascular resistance with little effect on stroke volume, heart rate and cardiac output.[20] During long-term use, labetalol can reduce heart rate during exercise while maintaining cardiac output by an increase in stroke volume.[21]
Labetalol is a dual alpha (α1) and beta (β1/β2) adrenergic receptor blocker and competes with other Catecholamines for binding to these sites.[22] Its action on these receptors are potent and reversible.[12] Labetalol is highly selective for postsynaptic alpha1- adrenergic, and non-selective for beta-adrenergic receptors. It is about equipotent in blocking both beta1- and beta2- receptors.[14]
The amount of alpha to beta blockade depends on whether labetalol is administered orally or intravenously (IV). Orally, the ratio of alpha to β blockade is 1:3. Intravenously, alpha to β blockade ratio is 1:7.[14][12] Thus, the labetalol can be thought to be a beta-blocker with some alpha-blocking effects.[12][22][23] By comparison, labetalol is a weaker β-blocker than propranolol, and has a weaker affinity for alpha-receptors compared to Phentolamine.[14][22]
Labetalol possesses intrinsic sympathomimetic activity.[23] In particular, it is a partial agonist at beta2- receptors located in the vascular smooth muscle. Labetalol relaxes vascular smooth muscle by a combination of this partial beta2- agonism and through alpha1- blockade.[23][24] Overall, this vasodilatory effect can decrease blood pressure.[25]
Similar to local anesthetics and sodium channel blocking antiarrhythmics, labetalol also has membrane stabilizing activity.[23][26] By decreasing sodium entry, labetalol decreases action potential firing and thus has local anesthetic activity.[27]
Physiological action
The physiological effects of labetalol when administered acutely (intravenously) are not predictable solely by their receptor blocking effect, i.e. blocking beta1- receptors should decrease heart rate, but labetalol does not. When labetalol is given in acute situations, it decreases the peripheral vascular resistance and systemic blood pressure while having little effect on the heart rate, cardiac output and stroke volume, despite its alpha1-, beta1- and beta2- blocking mechanism.[20][21] These effects are mainly seen when the person is in the upright position.[25]
Long term labetalol use also has different effects from other beta-blocking drugs. Other beta-blockers, such as propranolol, persistently reduce cardiac output during exercise. The peripheral vascular resistance decreases when labetalol is first administered. Continuous labetalol use further decreases peripheral vascular resistance. However, during exercise, cardiac output remains the same due to a compensatory mechanism that increases stroke volume. Thus, labetalol is able to reduce heart rate during exercise while maintaining cardiac output by the increase in stroke volume.[21]
Pharmacokinetics
Labetalol, in animal models, was found to cross the blood-brain-barrier in only negligible amounts.[28]
History
Labetalol was the first drug created that combined both alpha- and beta- adrenergic receptor blocking properties. It was created to potentially fix the compensatory reflex issue that occurred when blocking a single receptor subtype, i.e. vasoconstriction after blocking beta-receptors or tachycardia after blocking alpha receptors. Because the reflex from blocking the single receptor subtypes acted to prevent the lowering of blood pressure, it was postulated that weak blocking of both alpha- and beta- receptors could work together to decrease blood pressure.[14][21]
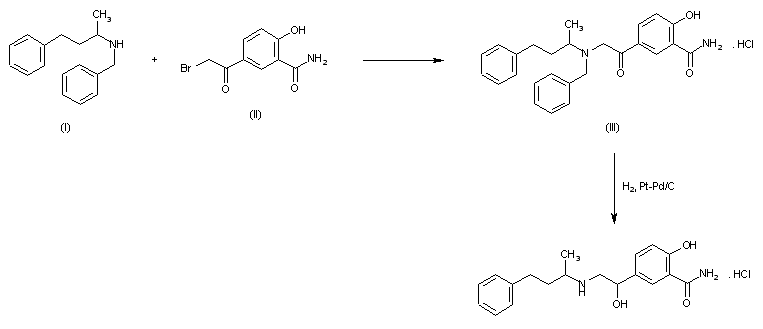
Syn 1
Drugs Fut 1976,1(3),125
DE 1643224; FR 1557677; FR 8010M; GB 1200886; US 3642896; US 3644353; US 3705233
Condensation of 5-bromoacetylsalicylamide (I) with N-benzyl-N-(1-methyl-3-phenylpropyl)amine (II) in refluxing butanone to 5-(N-benzyl-N-(1-methyl-3-phenylpropyl) glycyl)salicylamide hydrochloride (III), m.p. 139-141 C, which is reduced with H2 over Pt-Pd/C in ethanol.
SYN 2
Reductocondensation of 5-(N,N-dibenzylglycyl)salicylamide (IV) and benzylace-tone (V) with H2 over Pd-Pt/C in methanol – acetic acid.
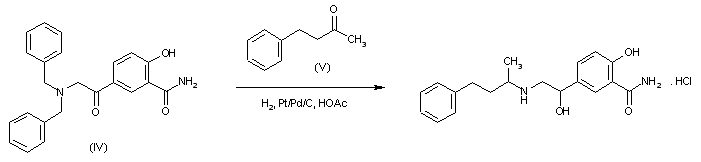
SYN 3
Reaction of methyl 5-(2-amino-1-hydroxyethyl)salicylate hydrochloride (VI) with NH3 to 5-(2-amino-1-hydroxyethyl)salicylamide hydrochloride (VII), m.p. >360 C, which is finally condensed with benzylacetone (V) and reduced with H2 over Pd-Pt/C in methanol.
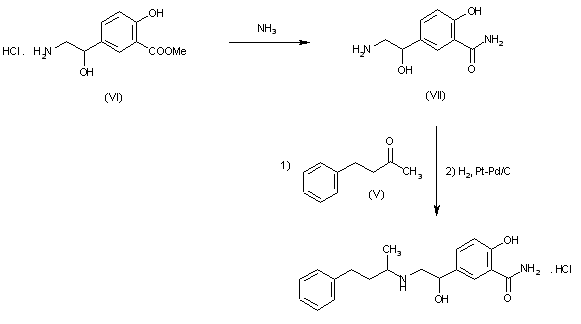
SYN 4

SYN 5
2-hydroxy-5-(1-hydroxy-2-((1-methyl-3-phenylpropyl)amino)ethyl)-, monohydrochloride, could be produced through many synthetic methods.
Following is one of the synthesis routes: 5-Bromoacetylsalicylamide (I) with N-benzyl-N-(1-methyl-3-phenylpropyl)amine (II) is condensed in the presence of refluxing butanone to produce 5-(N-benzyl-N-(1-methyl-3-phenylpropyl) glycyl)salicylamide hydrochloride (III), m.p. 139-141 C, and next the yielding compound is reduced with H2 over Pt-Pd/C in ethanol.

SYN 6
https://patents.google.com/patent/WO2017098520A1/en
aration of Labetaiol Hydrochloride of

Scheme -I illustrates the process for preparation of Labetaiol Hydrochloride of formula (I).

30% NaOH
Step – Sodium borohydride

Pure Labetaiol Hydrochloride (I)
aration of Labetaiol Hydrochloride of
Scheme -I illustrates the process for preparation of Labetaiol Hydrochloride of formula (I).
30% NaOH
Step – Sodium borohydride
Pure Labetaiol Hydrochloride (I)
SYN
https://patents.google.com/patent/EP0009702A1/en
-
The substance labetalol is known from British patent specification 1,266,058 and U.S.P. 4,012,444. Its pharmacological properties are discussed by Farmer et. al. in British Journal of Pharmacology, 45: 660-675 (1972), who designate it AH5158; it is shown to block a- and β-adrenergic receptors, suggesting that it would be useful in the treatment of arrhythmia, hypertension and angina pectoris.
- [0003]
The unique pharmacological properties of labetalol and its use as an antihypertensive agent are said to be largely a function of the exquisite balance of its a- and a-blocking activities. The file history of U.S.P. 4,012,444 indeed indicates that slight changes in the chemical structure of labetalol deleteriously affect this balance, and, even in the few analogous compounds where the balance is retained, the absolute potencies of these compounds are shown to be too low for them to be useful antihypertensive agents. Therefore, in the treatment of hypertension, labetalol is the compound of choice among those disclosed in British patent specification 1,266,058 and U.S.P. 4,012,444.
- [0004]
Labetalol has two asymmetrically substituted carbon atoms and therefore can exist as two diastereoisomers and four optical isomers. Indeed, British patent specification 1,266,058 and U.S.P. 4,012,444 disclose that compounds such as labetalol have optically active forms, but give no example of an optically active form. These patent specifications .teach that “the racemic mixtures may be resolved by conventional methods, for example by salt formation with an optically active acid, followed by fractional crystallization”, but give no method of resolution. Example 14 of each specifi– cation does indeed describe the separation of labetalol into two diastereoisomers “1” and “2”, using benzoic acid, but this is not an optical resolution. In British patent specifications 1,541,932 and 1,541,933, “isomer 1” is designated “diastereoisomer A” and is characterised as that diastereoisomer whose hydrochloride salt has the higher melting point. These two British patent specifications also disclose that diastereoisomer A is a valuable antiarrhythmic agent since it has strongly reduced β-adrenergic blocking activity and is therefore useful in the treatment of people who have suffered myocardial infarction.
- [0005]
We have now discovered that diastereoisomer A is composed of the (S,R) and (R,S) optical isomers of labetalol, whereas diastereoisomer B is composed of the (S,S) and (R,R) optical isomers. We have also-surprisingly found that the novel (R,R) optical isomer of labetalol exhibits, in comparison with labetalol itself, both an unexpectedly high increase in β-adrenergic blocking potency and a decrease in a-adrenergic blocking potency. Thus, when the (R,R) optical isomer is compared with labetalol, the ratio of the β-adrenergic blocking potency to the a-adrenergic blocking potency is found to be greatly and unexpectedly increased. In particular, animal tests have indicated that the (R,R) optical isomer has about twelve times the β-blocking potency of labetalol, but only about one third of the a-blocking potency of labetalol. These. properties could in no way have been predicted theoretically, especially as the β-blocking potency of diastereoisomer B is not significantly different from that of labetalol and the a-blocking potency of diastereoisomer B is half that of labetalol. Indeed, it is clear, when the activities of the four optical isomers of labetalol are compared, that the activities of the diastereoisomers A and B and indeed of labetalol itself cannot be calculated from the activities of their components. One can put this the other way around by saying that the α-and β-blocking activities of the four optical isomers of labetalol do not merely average to give the a- and β-blocking activites of labetalol and of its diastereoisomers A and B. Some of the activities are much greater than could ever have been expected on a simple basis of mathematical proportions, in particular the high β-blocking activity of the (R,R) optical isomer: this activity is much higher than the β-blocking activity of diastereoisomer B so that antagonism evidently exists between the (S,S) and (R,R) optical isomers with respect to the β-blocking activity. This degree of antagonism could in no way have been foreseen. In the absence of this antagonism, the (R,R) optical isomer shows a balance of properties that make it the optical isomer of choice in the treatment of hypertension. In particular, the (R,R) optical isomer possesses potent antihypertensive activity and rapid onset of activity while substantially lacking the undesirable side-effects usually associated with a-blockade, e.g. postural hypotension.
-
The following Table shows the relationships between labetalol, its diastereoisomersA and B and the four pure optical isomers; below each compound are given its potencies as an a-blocking and then as a β-blocking agent, all relative to the values for labetalol (assigned values 1.0 for each blocking activity):

This table clearly shows the unexpectedly high β-blocking activity and ratio of β-:α-blocking activities possessed by the (R,R)-optical isomer. Additionally, the (R,R)–optical isomer has been found to possess greater direct peripheral vasodilation activity than labetalol, and this also contributes to its anti-hypertensive activity. Moreover, the (R,R)-optical isomer is substantially non-toxic at therapeutic doses.
- [0007]
According to the invention therefore we provide the (R,R)-optical isomer of labetalol, namely 5- {(R)–1-hydroxy-2-[(R)-(1-methyl-3-phenylpropyl)amino]ethyl} salicylamide, which can be characterised by means of its hydrochloride salt which is dimorphic with m.pts. of about 133-134°C. and about 192-193.5°C. and an [α]D 26 of about -30.6° (conc. 1 mg./ml., ethanol), said (R,R) optical isomer being substantially free of the corresponding (R,S), (S,R) and (S,S) optical isomers
reaction scheme:

- E. (-)-5- { (R)-l-Hydroxy-2-[(R)-(l-methyl-3-phenylpropyl)-amino]ethyl} salicylamide hydrochloride salt (9)
- [0032]
Treat a solution of 3.0 g. (0.0059 mol.) of 2-0-benzyl-5-{(R) -1-hydroxy-2-[(R)-(1-methyl-3-phenylpropyl)benzylamino]ethyl} salicylamide in 30 ml. of ethyl ether with 2N ethereal hydrogen chloride until no further precipitation occurs. Wash the precipitated 2-0-benzyl-5-{(R)-1-hydroxy-2-[(R)-(1-methyl–3-phenylpropyl)benzylamino]ethyl} salicylamide hydrochloride with ether to remove excess hydrogen chloride and dissolve it in 100 ml. ethanol. To the ethanol solution add 300 mg. of a 20% palladium hydroxide on carbon catalyst and hydrogenate (3 atm.; 3.1 kg. cm.-2) in a Paar apparatus with shaking at room temperature for 3 hours. Filter off the catalyst, evaporate, and triturate the solid residue with isopropanol. Dissolve the solid in 11 ml. of 1N sodium hydroxide, adjust the pH to about 8 and precipitate the free base by bubbling in carbon dioxide. Collect the free base, wash it with water and dry it in vacuo at 40°C. Chromatograph the free base on 450 g. of silica gel and dissolve the pure product in 20 ml. of boiling acetonitrile. Cool the solution and carefully acidify with 2N ethereal HC1 to about pH2. Solidify the gum which precipitates by refluxing the mixture for 10 minutes, filter off the solid, wash it with ethyl ether and recrystallize it from ethanol to obtain analytically pure product (9), m.p. 192-193.5°C.(dec.), [α]D26 = -30.6° (c=1.0, ethanol).
Dilevalol
Synonyms:(R,R)-Labetalol
ATC:C02CB
- Use:α- and β-adrenoceptor antagonist, α- and β-blocker, isomer of labetalol, antihypertensive
- Chemical name:[R-(R*,R*)]-2-hydroxy-5-[1-hydroxy-2-[(1-methyl-3-phenylpropyl)amino]ethyl]benzamide
- Formula:C19H24N2O3
- MW:328.41 g/mol
- CAS-RN:75659-07-3
- LD50:1719 mg/kg (M, p.o.);
1228 mg/kg (R, p.o.)
Derivatives
Monohydrochloride
- Formula:C19H24N2O3 • HCl
- MW:364.87 g/mol
- CAS-RN:75659-08-4
- LD50:1079 mg/kg (M, p.o.);
82 mg/kg (R, i.v.); 1026 mg/kg (R, p.o.)
Synthesis Path
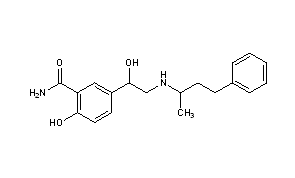
 |
|
| Clinical data | |
|---|---|
| Pronunciation | /ləˈbɛtəlɔːl/ |
| Trade names | Normodyne, Trandate, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a685034 |
| Pregnancy category |
|
| Routes of administration |
By mouth, intravenous |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 25% |
| Protein binding | 50% |
| Metabolism | Liver pass metabolism, |
| Elimination half-life | Tablet: 6-8 hours; IV: 5.5 hours |
| Excretion | Excreted in urine, not removed by hemodialysis |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.048.401 |
| Chemical and physical data | |
| Formula | C19H24N2O3 |
| Molar mass | 328.412 g·mol−1 |
| 3D model (JSmol) | |
| Chirality | Racemic mixture |
References
- ^ Jump up to:a b c d e f “Labetalol Hydrochloride Monograph for Professionals”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 3 March 2019.
- ^ Jump up to:a b c d e British national formulary : BNF 76 (76 ed.). Pharmaceutical Press. 2018. pp. 147–148. ISBN 9780857113382.
- ^ “Labetalol Use During Pregnancy”. Drugs.com. Retrieved 11 March 2019.
- ^ Fischer, Jnos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 463. ISBN 9783527607495.
- ^ “NADAC as of 2019-02-27”. Centers for Medicare and Medicaid Services. Retrieved 3 March 2019.
- ^ “The Top 300 of 2019”. clincalc.com. Retrieved 22 December 2018.
- ^ Jump up to:a b Koda-Kimble, Mary A.; Alldredge, Brian K. (2013). “21”. Koda-Kimble and Young’s Applied Therapeutic: The Clinical Use of Drugs. Philadelphia: Philadelphia: Lippincott Williams & Wilkins. ISBN 978-1-60913-713-7.
- ^ Arulkumaran, N; Lightstone, L (December 2013). “Severe pre-eclampsia and hypertensive crises”. Best Practice & Research. Clinical Obstetrics & Gynaecology. 27 (6): 877–84. doi:10.1016/j.bpobgyn.2013.07.003. PMID 23962474.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “Trandate” (PDF). Prometheus Laboratories Inc. November 2010. Retrieved 3 November 2015.
- ^ “Labetalol hydrochloride” (PDF). Hospira. May 2015. Retrieved 3 November 2015.
- ^ Shiohara T, Kano Y (2007). “Lichen planus and lichenoid dermatoses”. In Bolognia JL (ed.). Dermatology. St. Louis: Mosby. p. 161. ISBN 978-1-4160-2999-1.
- ^ Jump up to:a b c d “Labetalol [package insert]. Spring Valley, NY: Par Pharmaceutical; 2011” (PDF). Retrieved 2015-11-03.
- ^ Jump up to:a b Medicinal Chemistry of Adrenergics and Cholinergics
- ^ Jump up to:a b c d e f Louis, W.J.; McNeill, JJ; Drummer, OH (1988). Doyle, AE (ed.). Labetalol and other vasodilator/Beta-blocking drugs. IN: Handbook of Hypertension. Amsterdam, Netherlands: Elsevier Sciences Publishing Co. pp. 246–273. ISBN 978-0-444-90469-0.
- ^ Riva E, Mennini T, Latini R (December 1991). “The alpha- and beta-adrenoceptor blocking activities of labetalol and its RR-SR (50:50) stereoisomers”. Br. J. Pharmacol. 104 (4): 823–8. doi:10.1111/j.1476-5381.1991.tb12513.x. PMC 1908821. PMID 1687367.
- ^ Jump up to:a b Robertson D, Biaggioni, I. Adrenoceptor Antagonist Drugs. In: Katzung BG, Masters SB, Trevor AJ, eds. Basic & Clinical Pharmacology. 12th ed. San Francisco, CA: McGraw Hill Lange Medical; 2012: 151-168. ISBN 978-0-07-176401-8.
- ^ Katzung, Bertram G. (2006). Basic and clinical pharmacology. New York: McGraw-Hill Medical. p. 170. ISBN 978-0-07-145153-6.
- ^ D A Richards; J Tuckman; B N Prichard (October 1976). “Assessment of alpha- and beta-adrenoceptor blocking actions of labetalol”. Br J Clin Pharmacol. 3 (5): 849–855. doi:10.1111/j.1365-2125.1976.tb00637.x. PMC 1428931. PMID 9968.
- ^ “labetalol | C19H24N2O3 – PubChem”. pubchem.ncbi.nlm.nih.gov. Retrieved 2015-11-04.
- ^ Jump up to:a b MacCarthy, E. P.; Bloomfield, S. S. (1983-08-01). “Labetalol: a review of its pharmacology, pharmacokinetics, clinical uses and adverse effects”. Pharmacotherapy. 3(4): 193–219. doi:10.1002/j.1875-9114.1983.tb03252.x. ISSN 0277-0008. PMID 6310529.
- ^ Jump up to:a b c d Louis, W. J.; McNeil, J. J.; Drummer, O. H. (1984-01-01). “Pharmacology of combined alpha-beta-blockade. I”. Drugs. 28 Suppl 2: 16–34. doi:10.2165/00003495-198400282-00003. ISSN 0012-6667. PMID 6151889.
- ^ Jump up to:a b c Robertson, D; Biaggioni, I (2012). Katzung, BG (ed.). Adrenoceptor Antagonist Drugs IN: Basic & Clinical Pharmacology (12 ed.). San Francisco: McGraw Hill Lange Medical. pp. 151–168. ISBN 978-0-07-176401-8.
- ^ Jump up to:a b c d Westfall, David P (2004). Craig, Charles R (ed.). Adrenoreceptor Antagonists IN: Modern Pharmacology with Clinical Applications (6th ed.). Baltimore, MD: Lippincott Williams & Wilkins. pp. 109–117. ISBN 978-0781737623.
- ^ Lund-Johansen, P. (1988-01-01). “Hemodynamic effects of beta-blocking compounds possessing vasodilating activity: a review of labetalol, prizidilol, and dilevalol”. Journal of Cardiovascular Pharmacology. 11 Suppl 2: S12–17. doi:10.1097/00005344-198800000-00004. ISSN 0160-2446. PMID 2464093.
- ^ Jump up to:a b Lund-Johansen, P. (1984-01-01). “Pharmacology of combined alpha-beta-blockade. II. Haemodynamic effects of labetalol”. Drugs. 28 Suppl 2: 35–50. doi:10.2165/00003495-198400282-00004. ISSN 0012-6667. PMID 6151890.
- ^ Mottram, Allan R.; Erickson, Timothy B. (2009). Field, John (ed.). Toxicology in Emergency Cardiovascular Care IN: The Textbook of Emergency Cardiovascular Care and CPR. Philadelphia, PA: Lippincott WIlliams & Wilkins. pp. 443–452. ISBN 978-0-7817-8899-1.
- ^ Exam Zone (1 January 2009). Elsevier Comprehensive Guide. Elsevier India. pp. 449–. ISBN 978-81-312-1620-0.
- ^ Detlev Ganten; Patrick J. Mulrow (6 December 2012). Pharmacology of Antihypertensive Therapeutics. Springer Science & Business Media. pp. 147–. ISBN 978-3-642-74209-5.
External links
References
-
- EP 9 702 (Schering Corp.; appl. 17.9.1979; USA-prior. 20.9.1978).
-
Improvement of diastereomer separation:
- DOS 2 616 403 (Scherico; appl. 14.4.1976; USA-prior. 17.4.1975).
- US 4 173 583 (Schering Corp.; 6.11.1979; appl. 21.9.1978; prior. 17.4.1975).
-
Synthesis without chromatographic purification:
- EP 92 787 (Schering Corp.; appl. 20.4.1983; USA-prior. 26.4.1982).
-
Chiral reduction of IV:
- Clifton, J.E. et al.: J. Med. Chem. (JMCMAR) 25, 670 (1982).
- Gold, E.H. et al.: J. Med. Chem. (JMCMAR) 25, 1363 (1982).
- EP 382 157 (Schering Corp.; appl. 6.2.1990; USA-prior. 10.2.1989, 26.9.1989).
- US 4 948 732 (Schering Corp.; 14.8.1990; prior. 26.9.1989, 10.2.1989).
Novobiocin, ノボビオシン;

Novobiocin
ノボビオシン;
- Molecular FormulaC31H36N2O11
- Average mass612.624 Da
Monoisotopic: 612.231910004
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Novobiocin sodium | Q9S9NQ5YIY | 1476-53-5 | WWPRGAYLRGSOSU-RNROJPEYSA-M |
Reata Pharmaceuticals Inc
Abgentis is investigating a novobiocin analog, GYR-12 (discovery), as a re-engineered, previously-marketed-but-uncompetitive (undisclosed) antibacterial compound inhibiting ATPase activity of DNA supercoiling GyrB/ParE, for the potential broad-spectrum treatment of bacterial infections, including multi-drug resistant Gram-negative infections. In April 2017, development was underway [1924695].
Novobiocin, also known as albamycin or cathomycin, is an aminocoumarin antibiotic that is produced by the actinomycete Streptomyces niveus, which has recently been identified as a subjective synonym for S. spheroides[1] a member of the order Actinobacteria. Other aminocoumarin antibiotics include clorobiocin and coumermycin A1.[2] Novobiocin was first reported in the mid-1950s (then called streptonivicin).[3][4]
It is active against Staphylococcus epidermidis and may be used to differentiate it from the other coagulase-negative Staphylococcus saprophyticus, which is resistant to novobiocin, in culture.
Novobiocin was licensed for clinical use under the tradename Albamycin (Pharmacia And Upjohn) in the 1960s. Its efficacy has been demonstrated in preclinical and clinical trials.[5][6] The oral form of the drug has since been withdrawn from the market due to lack of efficacy.[7] Novobiocin is an effective antistaphylococcal agent used in the treatment of MRSA.[8]
Mechanism of action
The molecular basis of action of novobiocin, and other related drugs clorobiocin and coumermycin A1 has been examined.[2][9][10][11][12] Aminocoumarins are very potent inhibitors of bacterial DNA gyrase and work by targeting the GyrB subunit of the enzyme involved in energy transduction. Novobiocin as well as the other aminocoumarin antibiotics act as competitive inhibitors of the ATPase reaction catalysed by GyrB. The potency of novobiocin is considerably higher than that of the fluoroquinolones that also target DNA gyrase, but at a different site on the enzyme. The GyrA subunit is involved in the DNA nicking and ligation activity.
Novobiocin has been shown to weakly inhibit the C-terminus of the eukaryotic Hsp90 protein (high micromolar IC50). Modification of the novobiocin scaffold has led to more selective Hsp90 inhibitors.[13] Novobiocin has also been shown to bind and activate the Gram-negative lipopolysaccharide transporter LptBFGC.[14][15]
Structure
Novobiocin is an aminocoumarin. Novobiocin may be divided up into three entities; a benzoic acid derivative, a coumarin residue, and the sugar novobiose.[9] X-ray crystallographic studies have found that the drug-receptor complex of Novobiocin and DNA Gyrase shows that ATP and Novobiocin have overlapping binding sites on the gyrase molecule.[16] The overlap of the coumarin and ATP-binding sites is consistent with aminocoumarins being competitive inhibitors of the ATPase activity.[17]
Structure–activity relationship
In structure activity relationship experiments it was found that removal of the carbamoyl group located on the novobiose sugar lead to a dramatic decrease in inhibitory activity of novobiocin.[17]
Biosynthesis
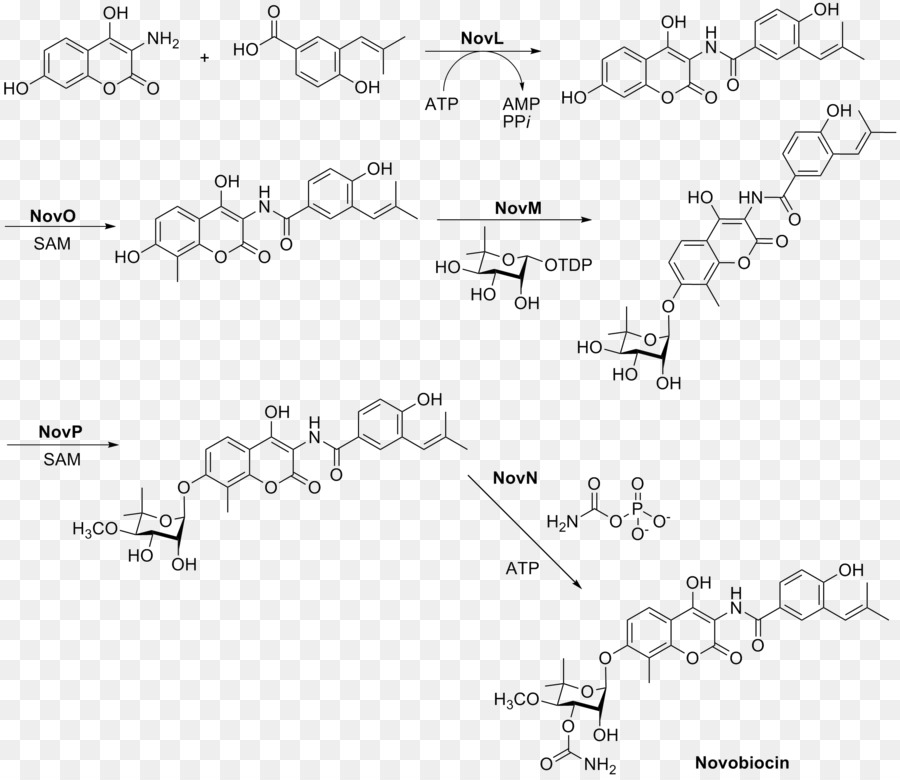
This aminocoumarin antibiotic consists of three major substituents. The 3-dimethylallyl-4-hydroxybenzoic acid moiety, known as ring A, is derived from prephenate and dimethylallyl pyrophosphate. The aminocoumarin moiety, known as ring B, is derived from L-tyrosine. The final component of novobiocin is the sugar derivative L-noviose, known as ring C, which is derived from glucose-1-phosphate. The biosynthetic gene cluster for novobiocin was identified by Heide and coworkers in 1999 (published 2000) from Streptomyces spheroidesNCIB 11891.[18] They identified 23 putative open reading frames (ORFs) and more than 11 other ORFs that may play a role in novobiocin biosynthesis.
The biosynthesis of ring A (see Fig. 1) begins with prephenate which is a derived from the shikimic acid biosynthetic pathway. The enzyme NovF catalyzes the decarboxylation of prephenate while simultaneously reducing nicotinamide adenine dinucleotide phosphate (NADP+) to produce NADPH. Following this NovQ catalyzes the electrophilic substitution of the phenyl ring with dimethylallyl pyrophosphate (DMAPP) otherwise known as prenylation.[19] DMAPP can come from either the mevalonic acid pathway or the deoxyxylulose biosynthetic pathway. Next the 3-dimethylallyl-4-hydroxybenzoate molecule is subjected to two oxidative decarboxylations by NovR and molecular oxygen.[20] NovR is a non-heme iron oxygenase with a unique bifunctional catalysis. In the first stage both oxygens are incorporated from the molecular oxygen while in the second step only one is incorporated as determined by isotope labeling studies. This completes the formation of ring A.

Figure 1. Biosynthetic scheme of benzamide portion of novobiocin (4-hydroxy-3-(3-methylbut-2-en-1-yl)benzoic acid)
The biosynthesis of ring B (see Fig. 2) begins with the natural amino acid L-tyrosine. This is then adenylated and thioesterified onto the peptidyl carrier protein (PCP) of NovH by ATPand NovH itself.[21] NovI then further modifies this PCP bound molecule by oxidizing the β-position using NADPH and molecular oxygen. NovJ and NovK form a heterodimer of J2K2 which is the active form of this benzylic oxygenase.[22] This process uses NADP+ as a hydride acceptor in the oxidation of the β-alcohol. This ketone will prefer to exist in its enol tautomer in solution. Next a still unidentified protein catalyzes the selective oxidation of the benzene (as shown in Fig. 2). Upon oxidation this intermediate will spontaneously lactonize to form the aromatic ring B and lose NovH in the process.

Figure 2. Biosynthesis of 3-amino-4,7-dihydroxy-2H-chromen-2-one component of novobiocin (ring B)
The biosynthesis of L-noviose (ring C) is shown in Fig. 3. This process starts from glucose-1-phosphate where NovV takes dTTP and replaces the phosphate group with a dTDP group. NovT then oxidizes the 4-hydroxy group using NAD+. NovT also accomplishes a dehydroxylation of the 6 position of the sugar. NovW then epimerizes the 3 position of the sugar.[23] The methylation of the 5 position is accomplished by NovU and S-adenosyl methionine (SAM). Finally NovS reduces the 4 position again to achieve epimerization of that position from the starting glucose-1-phosphate using NADH.

Figure 3. Biosynthesis of L-noviose component of novobiocin (ring C)
Rings A, B, and C are coupled together and modified to give the finished novobiocin molecule. Rings A and B are coupled together by the enzyme NovL using ATP to diphosphorylate the carboxylate group of ring A so that the carbonyl can be attacked by the amine group on ring B. The resulting compound is methylated by NovO and SAM prior to glycosylation.[24] NovM adds ring C (L-noviose) to the hydroxyl group derived from tyrosine with the loss of dTDP. Another methylation is accomplished by NovP and SAM at the 4 position of the L-noviose sugar.[25] This methylation allows NovN to carbamylate the 3 position of the sugar as shown in Fig. 4 completing the biosynthesis of novobiocin.

CLIP

CLIP

CLIP

PATENT
US-20190241599
Novel co-crystal forms of novobiocin and its analogs and proline, processes for their preparation and compositions comprising them are claimed. Also claims are methods for inhibiting heat shock protein 90 and treating or preventing neurodegenerative disorders, such as diabetic peripheral neuropathy.
References
- ^ Lanoot B, Vancanneyt M, Cleenwerck I, Wang L, Li W, Liu Z, Swings J (May 2002). “The search for synonyms among streptomycetes by using SDS-PAGE of whole-cell proteins. Emendation of the species Streptomyces aurantiacus, Streptomyces cacaoi subsp. cacaoi, Streptomyces caeruleus and Streptomyces violaceus”. International Journal of Systematic and Evolutionary Microbiology. 52 (Pt 3): 823–9. doi:10.1099/ijs.0.02008-0. PMID 12054245.
- ^ Jump up to:a b Alessandra da Silva Eustáquio (2004) Biosynthesis of aminocoumarin antibiotics in Streptomyces: Generation of structural analogues by genetic engineering and insights into the regulation of antibiotic production. DISSERTATION
- ^ Hoeksema H.; Johnson J. L.; Hinman J. W. (1955). “Structural studies on streptonivicin, a new antibiotic”. J Am Chem Soc. 77 (24): 6710–6711. doi:10.1021/ja01629a129.
- ^ Smith C. G.; Dietz A.; Sokolski W. T.; Savage G. M. (1956). “Streptonivicin, a new antibiotic. I. Discovery and biologic studies”. Antibiotics & Chemotherapy. 6: 135–142.
- ^ Raad I, Darouiche R, Hachem R, Sacilowski M, Bodey GP (November 1995). “Antibiotics and prevention of microbial colonization of catheters”. Antimicrobial Agents and Chemotherapy. 39 (11): 2397–400. doi:10.1128/aac.39.11.2397. PMC 162954. PMID 8585715.
- ^ Raad II, Hachem RY, Abi-Said D, Rolston KV, Whimbey E, Buzaid AC, Legha S (January 1998). “A prospective crossover randomized trial of novobiocin and rifampin prophylaxis for the prevention of intravascular catheter infections in cancer patients treated with interleukin-2”. Cancer. 82 (2): 403–11. doi:10.1002/(SICI)1097-0142(19980115)82:2<412::AID-CNCR22>3.0.CO;2-0. PMID 9445199.
- ^ “Determination That ALBAMYCIN (Novobiocin Sodium) Capsule, 250 Milligrams, Was Withdrawn From Sale for Reasons of Safety or Effectiveness”. The Federal Register. 19 January 2011.
- ^ Walsh TJ, Standiford HC, Reboli AC, John JF, Mulligan ME, Ribner BS, Montgomerie JZ, Goetz MB, Mayhall CG, Rimland D (June 1993). “Randomized double-blinded trial of rifampin with either novobiocin or trimethoprim-sulfamethoxazole against methicillin-resistant Staphylococcus aureus colonization: prevention of antimicrobial resistance and effect of host factors on outcome”. Antimicrobial Agents and Chemotherapy. 37 (6): 1334–42. doi:10.1128/aac.37.6.1334. PMC 187962. PMID 8328783.
- ^ Jump up to:a b Maxwell A (August 1993). “The interaction between coumarin drugs and DNA gyrase”. Molecular Microbiology. 9 (4): 681–6. doi:10.1111/j.1365-2958.1993.tb01728.x. PMID 8231802.
- ^ Maxwell A (February 1999). “DNA gyrase as a drug target”. Biochemical Society Transactions. 27 (2): 48–53. doi:10.1042/bst0270048. PMID 10093705.
- ^ Lewis RJ, Tsai FT, Wigley DB (August 1996). “Molecular mechanisms of drug inhibition of DNA gyrase”. BioEssays. 18 (8): 661–71. doi:10.1002/bies.950180810. PMID 8760340.
- ^ Maxwell A, Lawson DM (2003). “The ATP-binding site of type II topoisomerases as a target for antibacterial drugs”. Current Topics in Medicinal Chemistry. 3 (3): 283–303. doi:10.2174/1568026033452500. PMID 12570764.
- ^ Yu XM, Shen G, Neckers L, Blake H, Holzbeierlein J, Cronk B, Blagg BS (September 2005). “Hsp90 inhibitors identified from a library of novobiocin analogues”. Journal of the American Chemical Society. 127 (37): 12778–9. doi:10.1021/ja0535864. PMID 16159253.
- ^ Mandler MD, Baidin V, Lee J, Pahil KS, Owens TW, Kahne D (June 2018). “Novobiocin Enhances Polymyxin Activity by Stimulating Lipopolysaccharide Transport”. Journal of the American Chemical Society. 140 (22): 6749–6753. doi:10.1021/jacs.8b02283. PMC 5990483. PMID 29746111.
- ^ May JM, Owens TW, Mandler MD, Simpson BW, Lazarus MB, Sherman DJ, Davis RM, Okuda S, Massefski W, Ruiz N, Kahne D (December 2017). “The Antibiotic Novobiocin Binds and Activates the ATPase That Powers Lipopolysaccharide Transport”. Journal of the American Chemical Society. 139 (48): 17221–17224. doi:10.1021/jacs.7b07736. PMC 5735422. PMID 29135241.
- ^ Tsai FT, Singh OM, Skarzynski T, Wonacott AJ, Weston S, Tucker A, Pauptit RA, Breeze AL, Poyser JP, O’Brien R, Ladbury JE, Wigley DB (May 1997). “The high-resolution crystal structure of a 24-kDa gyrase B fragment from E. coli complexed with one of the most potent coumarin inhibitors, clorobiocin”. Proteins. 28 (1): 41–52. doi:10.1002/(sici)1097-0134(199705)28:1<41::aid-prot4>3.3.co;2-b. PMID 9144789.
- ^ Jump up to:a b Flatman RH, Eustaquio A, Li SM, Heide L, Maxwell A (April 2006). “Structure-activity relationships of aminocoumarin-type gyrase and topoisomerase IV inhibitors obtained by combinatorial biosynthesis”. Antimicrobial Agents and Chemotherapy. 50 (4): 1136–42. doi:10.1128/AAC.50.4.1136-1142.2006. PMC 1426943. PMID 16569821.
- ^ Steffensky M, Mühlenweg A, Wang ZX, Li SM, Heide L (May 2000). “Identification of the novobiocin biosynthetic gene cluster of Streptomyces spheroides NCIB 11891”. Antimicrobial Agents and Chemotherapy. 44 (5): 1214–22. doi:10.1128/AAC.44.5.1214-1222.2000. PMC 89847. PMID 10770754.
- ^ Pojer F, Wemakor E, Kammerer B, Chen H, Walsh CT, Li SM, Heide L (March 2003). “CloQ, a prenyltransferase involved in clorobiocin biosynthesis”. Proceedings of the National Academy of Sciences of the United States of America. 100 (5): 2316–21. Bibcode:2003PNAS..100.2316P. doi:10.1073/pnas.0337708100. PMC 151338. PMID 12618544.
- ^ Pojer F, Kahlich R, Kammerer B, Li SM, Heide L (August 2003). “CloR, a bifunctional non-heme iron oxygenase involved in clorobiocin biosynthesis”. The Journal of Biological Chemistry. 278 (33): 30661–8. doi:10.1074/jbc.M303190200. PMID 12777382.
- ^ Chen H, Walsh CT (April 2001). “Coumarin formation in novobiocin biosynthesis: beta-hydroxylation of the aminoacyl enzyme tyrosyl-S-NovH by a cytochrome P450 NovI”. Chemistry & Biology. 8 (4): 301–12. doi:10.1016/S1074-5521(01)00009-6. PMID 11325587.
- ^ Pacholec M, Hillson NJ, Walsh CT (September 2005). “NovJ/NovK catalyze benzylic oxidation of a beta-hydroxyl tyrosyl-S-pantetheinyl enzyme during aminocoumarin ring formation in novobiocin biosynthesis”. Biochemistry. 44 (38): 12819–26. CiteSeerX 10.1.1.569.1481. doi:10.1021/bi051297m. PMID 16171397.
- ^ Thuy TT, Lee HC, Kim CG, Heide L, Sohng JK (April 2005). “Functional characterizations of novWUS involved in novobiocin biosynthesis from Streptomyces spheroides”. Archives of Biochemistry and Biophysics. 436 (1): 161–7. doi:10.1016/j.abb.2005.01.012. PMID 15752721.
- ^ Pacholec M, Tao J, Walsh CT (November 2005). “CouO and NovO: C-methyltransferases for tailoring the aminocoumarin scaffold in coumermycin and novobiocin antibiotic biosynthesis”. Biochemistry. 44 (45): 14969–76. doi:10.1021/bi051599o. PMID 16274243.
- ^ Freel Meyers CL, Oberthür M, Xu H, Heide L, Kahne D, Walsh CT (January 2004). “Characterization of NovP and NovN: completion of novobiocin biosynthesis by sequential tailoring of the noviosyl ring”. Angewandte Chemie. 43 (1): 67–70. doi:10.1002/anie.200352626. PMID 14694473.
External links
- Novobiocin bound to proteins in the PDB
 |
|
 |
|
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Routes of administration |
intravenous |
| ATCvet code | |
| Pharmacokinetic data | |
| Bioavailability | negligible oral bioavailability |
| Metabolism | excreted unchanged |
| Elimination half-life | 6 hours |
| Excretion | renal |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard(EPA) | |
| ECHA InfoCard | 100.005.589 |
| Chemical and physical data | |
| Formula | C31H36N2O11 |
| Molar mass | 612.624 g·mol−1 |
| 3D model (JSmol) | |
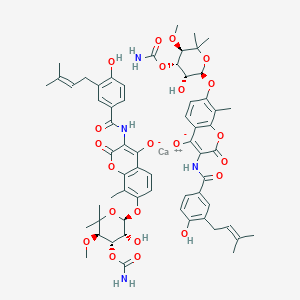
4309-70-0 CAS
calcium;7-[(2R,3R,4S,5R)-4-carbamoyloxy-3-hydroxy-5-methoxy-6,6-dimethyloxan-2-yl]oxy-3-[[4-hydroxy-3-(3-methylbut-2-enyl)benzoyl]amino]-8-methyl-2-oxochromen-4-olate
///////// Novobiocin, ノボビオシン , Antibacterial, Antimicrobial, crystallinic acid, streptonivicin,
History
Novobiocin is a coumarin antibiotic obtained from Streptomyces niveus and other Streptomyces species. Novobiocin is useful primarily in infections involving staphylococci, and other gram-positive organisms. It acts by inhibiting the initiation of DNA replication in bacterial and mammanlian cells. Evidences indicated that Novobiocin blocks prokaryotic DNA gyrase and eukaryotic II topoisomerase, enzymes that relax super-coiled DNA and are crucial for DNA replication.1
Novobiocin

| UIPAC Name | 4-Hydroxy-3-4-hydroxy-3-(3-methylbut-2-enyl)benzamido-8-methylcoumarin-7-yl 3-O-carbamoyl-5,5-di-C-methyl-α-l-lyxofuranoside |
| CAS Number | 303-81-1 |
| Molecular Mass | 612.624 g / mol |
| Chemical Formular | C31H36N2O11 |
Biosynthesis
The substituted coumarin (ring B, red) and the 4-OH benzoyl moiety (ring A, aqua) in novobiocin were derived from -Tyr based on earlier labeling studies. β-OH-Tyr is proposed to be a common intermediate in these two biosynthetic pathways.2

NovH is a -Tyr specific didomain NRPS that generates the
-tyrosyl-S-NovH intermediate. NovH, isolated from E. coli is primed by a PPTase with CoA. The A domain activates
-Tyr as
-tyrosyl-AMP and then transfers the
-tyrosyl group to the HS-pant-PCP domain of NovH through thioester formation.3

-tyrosyl-S-NovH is then function as a cytochrome P450 monooxygenase that hydroxylates the β-carbon of the tethered
-tyrosyl group on NovH. While the substrate
-tyrosyl-S-NovH provides two electrons for a single round of the hydroxylation reaction, the other two electrons needed to reduce the oxygen atom are provided by NADPH via two-electron transfer effected by electron transfer proteins ferrodoxin (Fd) and ferrodoxin reductase (Fd Red).3 The electron transfer route is from NADPH→FAD in Fd Red→Fe–S center in Fd→Heme in NovI→oxygen.

Both NovJ and NovK are similar to 3-keto-ACP reductase and they may form a heterodimer and operate in the reverse direction to oxidize 3-OH to 3-keto. NovO is similar to some quinone C-methyltransferases 3 but the timing of methylation is not clear. NovC resembles flavin-dependent monooxygenases (35 and 32% similarity to dimethylaniline and cyclohexanone monooxygenases, respectively) 3 and is proposed to hydroxylate the ortho position of the phenyl ring. The nucleophilic attack of the ortho hydroxyl group on the thioester carbonyl center would release the coumarin ring and regenerate NovH. Ring B is then synthesized.

Synthesis





Mechanism of action
E.Coli DNA gyrase utilizes ATP to catalyze the negative supercoiling, or under-twisting, of duplex DNA. The energy coupling components of the supercoiling reaction includes 1) the DNA-dependent hydrolysis that converts ATP to ADP and Pi, and 2) the gyrase cleavage reaction that targets the specified DNA site. The two activities are induced by treating the stable gyrase-DNA complex trapped by the inihibitor oxolinic acid with sodium dodecyl sulfate (SDS or Sulphate). 4 Novobiocin competes with ATP in the ATPase and supercoiling assays, hence Novobiocin prevents the ATP from shifting the primary cleavage site on ColE1 DNA by places the site of action of the antibiotics at a reaction step prior to ATP hydrolysis and blocks the binding of ATP. 4 Such a simple mechanism of action represents for all effects of the drugs on DNA gyrase.
Clinical Use
Due to factors as low solubility, poor pharmacokinetics, and limited activity agasinst Gram-negative bacteria, the clinical usage of Novobiocin is not achieved. 5 Therefore, it is of interest to study the novobiocin biosynthetic pathway in order to generate analogs with enhanced solubility and pharmacokinetic properties while maintaining the gyrase inhibitory properties.
References
1 J.C. D’Halluin, M. Milleville, and P. Boulanger. “Effect of Novobiocin on adenovirus DNA synthesis and encapsidation”. Nucleic Acids Research 1980; 8: 1625-1641
2 M. Steffensky, S.M. Li and L. Heide, “Cloning, overexpression, and purification of novobiocic acid synthetase from Streptomyces spheroides ” NCIB 11891. J. Biol. Chem. 275 (2000), pp. 21754–21760.
3 Huawei Chen and Christopher T. Walsh, “Coumarin formation in novobiocin biosynthesis: β-hydroxylation of the aminoacyl enzyme tyrosyl-S-NovH by a cytochrome P450 NovI” Chemistry and Biology; 2001; 8: 301-312
4 K. Scheirer and N. P. Higgins. “The DAN Cleavage Reaction of DNA Gyrase ” The Journal of Biological Chemistry; 1997; 272 (43): 27202-27209
5 N Pi, C. L. F. Meyers, M. Pacholec, C. T. Walsh, and J. A. Leary. “Mass spectrometric characterization of a three-enzyme tandem reacton for assembly and modification of the novobiocin skeleton” PNAS 2004;101;10036-10041
Imipenem, イミペネム水和物
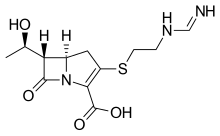
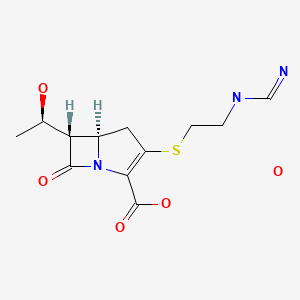
Imipenem
イミペネム水和物
Cas 74431-23-5
- Molecular FormulaC12H19N3O5S
- Average mass317.361 Da
(5R,6S)-3-((2-(Formimidoylamino)ethyl)thio)-6-((R)-1-hydroxyethyl)-7-oxo-1-azabicyclo(3.2.0)hept-2-ene-2-carboxylic acid monohydrate
Antibacterial, Cell wall biosynthesis inhibitor
Imipenem (Primaxin among others) is an intravenous β-lactam antibiotic discovered by Merck scientists Burton Christensen, William Leanza, and Kenneth Wildonger in the mid-1970s.[1] Carbapenems are highly resistant to the β-lactamase enzymes produced by many multiple drug-resistant Gram-negative bacteria,[2] thus play a key role in the treatment of infections not readily treated with other antibiotics.[3]
Imipenem was patented in 1975 and approved for medical use in 1985.[4] It was discovered via a lengthy trial-and-error search for a more stable version of the natural product thienamycin, which is produced by the bacterium Streptomyces cattleya. Thienamycin has antibacterial activity, but is unstable in aqueous solution, so impractical to administer to patients.[5] Imipenem has a broad spectrum of activity against aerobic and anaerobic, Gram-positive and Gram-negative bacteria.[6] It is particularly important for its activity against Pseudomonas aeruginosa and the Enterococcus species. It is not active against MRSA, however.
Medical uses
Spectrum of bacterial susceptibility and resistance
Acinetobacter anitratus, Acinetobacter calcoaceticus, Actinomyces odontolyticus, Aeromonas hydrophila, Bacteroides distasonis, Bacteroides uniformis, and Clostridium perfringens are generally susceptible to imipenem, while Acinetobacter baumannii, some Acinetobacter spp., Bacteroides fragilis, and Enterococcus faecalis have developed resistance to imipenem to varying degrees. Not many species are resistant to imipenem except Pseudomonas aeruginosa (Oman) and Stenotrophomonas maltophilia.[7]
Coadministration with cilastatin
Imipenem is rapidly degraded by the renal enzyme dehydropeptidase 1 when administered alone, and is almost always coadministered with cilastatin to prevent this inactivation[8]
Adverse effects
Common adverse drug reactions are nausea and vomiting. People who are allergic to penicillin and other β-lactam antibiotics should take caution if taking imipenem, as cross-reactivity rates are high. At high doses, imipenem is seizurogenic.[9]
Mechanism of action
Imipenem acts as an antimicrobial through inhibiting cell wall synthesis of various Gram-positive and Gram-negative bacteria. It remains very stable in the presence of β-lactamase (both penicillinase and cephalosporinase) produced by some bacteria, and is a strong inhibitor of β-lactamases from some Gram-negative bacteria that are resistant to most β-lactam antibiotics.
SYM
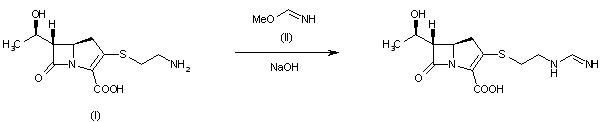
By reaction of thienamycin (I) with methyl formimidate (II) by means of NaOH in water.
| DE 2652679; FR 2332012; GB 1570990; NL 7612939 |
SYN 2
WO 0294828
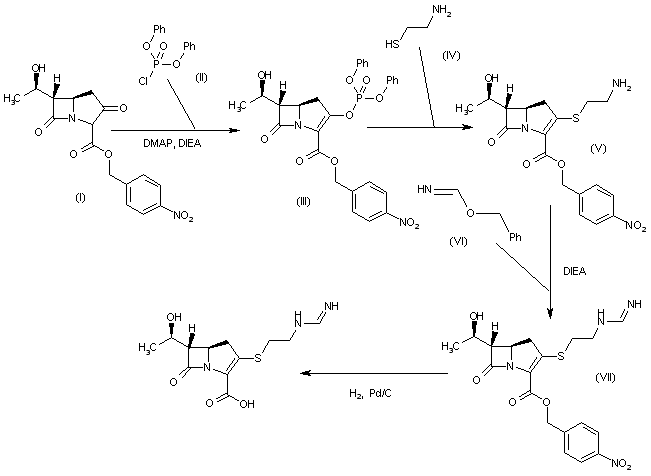
The reaction of (3R,5R,6S)-6-(1(R)-hydroxyethyl)-2-oxo-1-carbapenem-3-carboxylic acid p-nitrobenzyl ester (I) with diphenyl chlorophosphate by (II) means of DMAP and DIEA in DMA/dichloromethane gives the enol phosphate (III), which is condensed with 2-aminoethanethiol (IV) in DMA to yield the 2-aminoethylsulfanyl derivative (V). The reaction of (V) with benzyl formimidate (VI) by means of DIEA in DMA affords the intermediate p-nitrobenzyl ester (VII), which is finally hydrogenated with H2 over Pd/C in water/isopropanol/N-methylmorpholine to provide the target Imipemide.
SYN 3
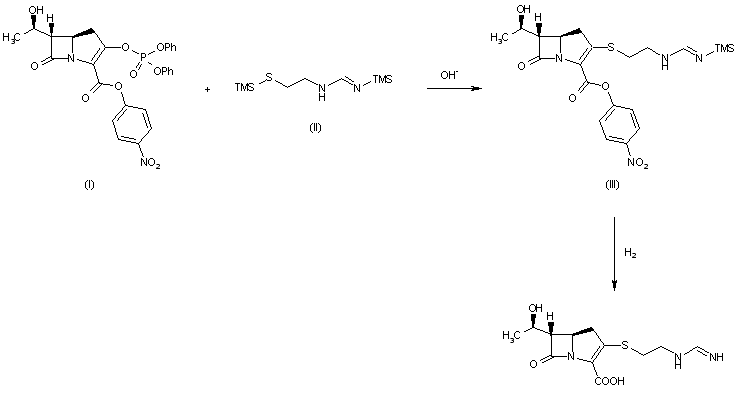
Tetrahedron Lett 1982,23(47),4903
The condensation of 7-oxo-6-(1-hydroxyethyl)-3-(diphenoxyphosphate)-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid p-nitrophenyl ester (I) with the bis(trimethylsilyl) derivative of 2-(iminomethylamino)ethanethiol (II) in the presence of base gives p-nitrophenyl ester of MK-0787, protected with a trimethylsilyl group (III), which is finally deprotected by hydrogenolysis.
CLIP

Synthesis Path
References
- ^ U.S. Patent 4,194,047
- ^ Clissold, SP; Todd, PA; Campoli-Richards, DM (Mar 1987). “Imipenem/cilastatin. A review of its antibacterial activity, pharmacokinetic properties and therapeutic efficacy”. Drugs. 33 (3): 183–241. doi:10.2165/00003495-198733030-00001. PMID 3552595.
- ^ Vardakas, KZ; Tansarli, GS; Rafailidis, PI; Falagas, ME (Dec 2012). “Carbapenems versus alternative antibiotics for the treatment of bacteraemia due to Enterobacteriaceae producing extended-spectrum β-lactamases: a systematic review and meta-analysis”. The Journal of Antimicrobial Chemotherapy. 67 (12): 2793–803. doi:10.1093/jac/dks301. PMID 22915465.
- ^ Fischer, Janos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 497. ISBN 9783527607495.
- ^ Kahan, FM; Kropp, H; Sundelof, JG; Birnbaum, J (Dec 1983). “Thienamycin: development of imipenen-cilastatin”. The Journal of Antimicrobial Chemotherapy. 12 Suppl D: 1–35. doi:10.1093/jac/12.suppl_d.1. PMID 6365872.
- ^ Kesado, Tadataka; Hashizume, Terutaka; Asahi, Yoshinari (1980). “Antibacterial activities of a new stabilized thienamycin, N-formimidoyl thienamycin, in comparison with other antibiotics”. Antimicrobial Agents and Chemotherapy. 17 (6): 912–7. doi:10.1128/aac.17.6.912. PMC 283902. PMID 6931548.
- ^ “Imipenem spectrum of bacterial susceptibility and Resistance” (PDF). Retrieved 4 May 2012.
- ^ “IMIPENEM/CILASTATIN”. livertox.nih.gov. Retrieved 2019-03-08.
- ^ Cannon, Joan P.; Lee, Todd A.; Clark, Nina M.; Setlak, Paul; Grim, Shellee A. (2014-08-01). “The risk of seizures among the carbapenems: a meta-analysis”. Journal of Antimicrobial Chemotherapy. 69 (8): 2043–2055. doi:10.1093/jac/dku111. ISSN 0305-7453.
Further reading
- Clissold, SP; Todd, PA; Campoli-Richards, DM (1987). “Imipenem/cilastatin. A review of its antibacterial activity, pharmacokinetic properties and therapeutic efficacy”. Drugs. 33(3): 183–241. doi:10.2165/00003495-198733030-00001. PMID 3552595.
- Buckley, MM; Brogden, RN; Barradell, LB; Goa, KL (1992). “Imipenem/cilastatin. A reappraisal of its antibacterial activity, pharmacokinetic properties and therapeutic efficacy”. Drugs. 44 (3): 408–44. doi:10.2165/00003495-199244030-00008. PMID 1382937.
External links
- Imipenem bound to proteins in the PDB
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Primaxin |
| AHFS/Drugs.com | International Drug Names |
| MedlinePlus | a686013 |
| Pregnancy category |
|
| Routes of administration |
IM, IV |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Protein binding | 20% |
| Metabolism | Renal |
| Elimination half-life | 38 minutes (children), 60 minutes (adults) |
| Excretion | Urine (70%) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.058.831 |
| Chemical and physical data | |
| Formula | C12H17N3O4S |
| Molar mass | 299.347 g/mol g·mol−1 |
| 3D model (JSmol) | |
-
- Synonyms:Imipemide
- ATC:J01DH51
- Use:carbapenem antibiotic
- Chemical name:[5R-[5α,6α(R*)]]-6-(1-hydroxyethyl)-3-[[2-[(iminomethyl)amino]ethyl]thio]-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid
- Formula:C12H17N3O4S
- MW:299.35 g/mol
- CAS-RN:64221-86-9
- InChI Key:ZSKVGTPCRGIANV-ZXFLCMHBSA-N
- InChI:InChI=1S/C12H17N3O4S/c1-6(16)9-7-4-8(20-3-2-14-5-13)10(12(18)19)15(7)11(9)17/h5-7,9,16H,2-4H2,1H3,(H2,13,14)(H,18,19)/t6-,7-,9-/m1/s1
- EINECS:264-734-5
- LD50:1660 mg/kg (M, i.v.); >5 g/kg (M, p.o.);
1972 mg/kg (R, i.v.); >5 g/kg (R, p.o.)
Derivatives, monohydrate
- Formula:C12H17N3O4S • H2O
- MW:317.37 g/mol
- CAS-RN:74431-23-5
References
-
-
Leanza, W.J. et al.: J. Med. Chem. (JMCMAR) 22, 1435 (1979).
-
a Salzmann, T.L. et al.: J. Am. Chem. Soc. (JACSAT) 102, 6161-6163 (1980).
-
Reider, P.J.; Grabowski, E.J.J.: Tetrahedron Lett. (TELEAY) 23, 2293-2296 (1982).
-
Grabowski, E.J.J.: Chirality (CHRLEP) 17, 249-259 (2005).
-
US 4 194 047 (Merck & Co.; 18.3.1980; prior. 21.11.1975).
-
DOS 2 652 679 (Merck & Co.; appl. 19.11.1976; USA-prior. 21.11.1975).
-
b US 5 998 612 (Merck & Co.; 7.12.1999; appl. 12.6.1992; prior. 23.10.1981).
-
c US 4 981 992 (Takasago; 27.1.1998; appl. 13.5.1996; J-prior. 11.5.1995).
-
US 5 204 460 (Takasago; 20.4.1993; appl. 8.11.1991; J-prior. 8.11.1990).
-
US 5 204 462 (Takasago; 20.4.1993; appl. 8.11.1991; J-prior. 8.11.1990).
-
US 5 712 388 (Takasago; 27.1.1998; appl. 13.5.1996; J-prior. 11.5.1995).
-
US 5 081 239 (Takasago; 14.1.1992; appl. 29.11.1989; J-prior. 29.11.1988).
-
-
Acetoxylation of 2-azetidinones in 4-position:
-
Noyori, R. et al.: J. Am. Chem. Soc. (JACSAT) 111, 9134-9135 (1989).
-
Noyori, R. et al.: Angew. Chem. (ANCEAD) 114, 2108-2123 (2002).
-
US 5 288 862 (Takasago; 22.2.1994; appl. 16.4.1992; J-prior. 18.4.1991).
-
US 5 606 052 (Takasago; 25.2.1997; appl. 16.4.1992; J-prior. 18.4.1991).
-
-
Noyori-catalyst:
-
US 4 739 084 (Takasago; 19.4.1988; appl. 15.4.1987; J-prior. 13.5.1986).
-
-
d process of Nippon Soda (Nisso):
-
US 5 026 844 (Suntory & Nippon Soda; 25.6.1991; appl. 13.10.1989; J-prior. 19.10.1988).
-
US 5 792 861 (Tanabe Seiyaku & Nippon Soda; 11.8.1998; appl. 29.6.1994, 4.11.1996; J-prior. 30.6.1993).
-
US 5 808 055 (Suntory & Nippon Soda; 15.9.1998; appl. 30.3.1993, 5.7.1995; J-prior. 30.3.1993).
-
e US 4 791 198 (Kanegafuchi; 13.12.1988; appl. 1.7.1985, 6.1.1987; J-prior. 5.7.1984, 14.1.1986).
-
US 4 861 877 (Kanegafuchi; 29.8.1989; appl. 1.7.1985, 6.1.1987; J-prior. 5.7.1984, 14.1.1985, 14.1.1986).
-
US 5 061 817 (Kanegafuchi; 29.10.1991; appl. 1.7.1985, 6.1.1987, 31.5.1988; J-prior. 5.7.1984, 14.1.1986).
-
US 4 914 200 (Kanegafuchi; 3.4.1990; appl. 28.4.1987, 14.2.1989; J-prior. 30.4.1986, 13.11.1986, 9.2.1987).
-
-
Enzymatic reduction of alkyl-2-(N-benzoylamino)methyl-3-oxobutyrates with bakers yeast:
-
US 5 463 047 (Ciba-Geigy; 31.10.1995; appl. 15.9.1994; CH-prior. 4.5.1987).
-
-
Further synthesis processes of Merck & Co. for thienamycin:
-
Johnston, D.B.R. et al.: J. Am. Chem. Soc. (JACSAT) 100, 313-315 (1978).
-
Mellilo, D.G. et al.: Tetrahedron Lett. (TELEAY) 21, 2783 (1980).
-
Melillo, D.G. et al.: J. Org. Chem. (JOCEAH) 51, 1498-1504 (1986).
-
Karady, S. et al.: J. Am. Chem. Soc. (JACSAT) 103, 6765-6767 (1981).
-
US 4 269 772 (Merck & Co.; 26.5.1981; appl. 14.1.1980).
-
US 4 282 148 (Merck & Co.; 4.8.1981; appl. 14.1.1980).
-
US 4 287 123 (Merck & Co.; 1.9.1981; appl. 14.1.1980).
-
US 4 290 947 (Merck & Co.; 22.9.1981; appl. 29.5.1980).
-
US 4 360 684 (Merck & Co.; 23.11.1982; appl. 8.4.1981).
-
US 4 206 219 (Merck & Co.; 3.6.1980; appl. 24.10.1978).
-
US 4 348 320 (Merck & Co.; 7.9.1982; appl. 20.8.1980; USA-prior. 19.11.1976).
-
US 4 460 507 (Merck & Co.; 17.7.1984; appl. 29.4.1982; USA-prior. 10.10.1980).
-
US 5 055 573 (Merck & Co.; 8.10.1991, appl. 24.8.1990; USA-prior. 19.11.1976).
-
US 5 037 974 (Merck & Co.; 6.8.1991; appl. 14.8.1990; prior. 23.5.1988, 10.4.1990).
-
-
Review of thienamycin syntheses:
-
Nicolaou, K.C.; Sorensen, E.J.: Classics in Total Synthesis, VCH 1996, Weinheim & New York, chapter 16, p. 249-263.
-
Berks, A.H.: Tetrahedron (TETRAB) 52, 331-375 (1996).
-
-
Alternative 2-azetidinone ring closure with chlorosulfonyl isocyanate:
-
US 4 350 631 (Merck & Co.; 21.9.1982; appl. 18.3.1981; prior. 18.12.1980).
-
-
Thienamycin (by fermentation of S. cattleya):
-
US 3 950 357 (Merck & Co.; 13.4.1976; appl. 25.11.1974).
-
DOS 2 552 638 (Merck & Co.; appl. 24.11.1975; USA-prior. 25.11.1974).
-
-
Combination with cilastatin:
-
EP 48 301 (Merck & Co.; appl. 24.9.1980).
-
/////////////Imipenem, イミペネム水和物 , MK-787,
Quinacillin
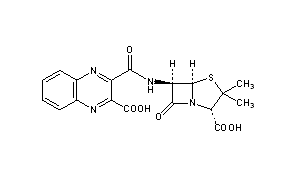
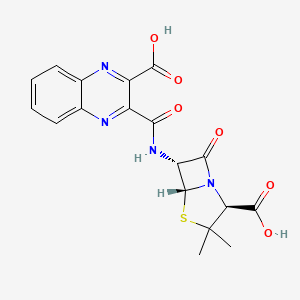
UNII-83NB50X92M
Cas 1596-63-0
83NB50X92M
Quinacilina
MW 416.4 g/mol, MF C18H16N4O6S
(2S,5R,6R)-6-[(3-carboxyquinoxaline-2-carbonyl)amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid
- 4-Thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid, 6-(3-carboxy-2-quinoxalinecarboxamido)-3,3-dimethyl-7-oxo- (7CI,8CI)
- 4-Thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid, 6-[[(3-carboxy-2-quinoxalinyl)carbonyl]amino]-3,3-dimethyl-7-oxo-, [2S-(2α,5α,6β)]-
- (2S,5R,6R)-6-[[(3-Carboxy-2-quinoxalinyl)carbonyl]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid
- 3-Carboxy-2-quinoxalinylpenicillanic acid
- 3-Carboxy-2-quinoxalinylpenicillin
- 6-(3-Carboxy-2-quinoxalinecarboxamido)-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid
- Penicillin, (3-carboxy-2-quinoxalinyl)-
Nicotinamide riboside chloride

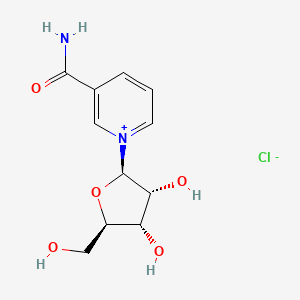

Nicotinamide riboside chloride
CAS 23111-00-4 CHLORIDE
CAS : 1341-23-7 (cation) 23111-00-4 (chloride) 445489-49-6 (Triflate)
3-Carbamoyl-1-((2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)pyridin-1-ium chloride
Nicotinamide ribose chloride
UNII-8XM2XT8VWI
MW 290.7 g/mol
1-[(2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]pyridin-1-ium-3-carboxamide;chloride
C1=CC(=C[N+](=C1)C2C(C(C(O2)CO)O)O)C(=O)N.[Cl-]
Nicotinamide riboside; SRT647; SRT-647; SRT 647; Nicotinamide Riboside Triflate, α/β mixture
EH-301, nicotinamide riboside chloride,AND pterostilbene,, BY Elysium Health Inc
Nicotinamide riboside, also known as NR and SRT647, is a pyridine-nucleoside form of vitamin B3 that functions as a precursor to nicotinamide adenine dinucleotide or NAD+. NR blocks degeneration of surgically severed dorsal root ganglion neurons ex vivo and protects against noise-induced hearing loss in living mice. Nicotinamide riboside prevents muscle, neural and melanocyte stem cell senescence. Increased muscular regeneration in mice has been observed after treatment with nicotinamide riboside, leading to speculation that it might improve regeneration of organs such as the liver, kidney, and heart. Nicotinamide riboside also lowers blood glucose and fatty liver in prediabetic and type 2 diabetic models while preventing the development of diabetic peripheral neuropathy. Note: Nicotinamide Riboside chloride is a α/β mixture
Nicotinamide riboside (NR) is a pyridine–nucleoside form of vitamin B3 that functions as a precursor to nicotinamide adenine dinucleotide or NAD+.[1][2]
Chemistry
While the molecular weight of nicotinamide riboside is 255.25 g/mol,[3] that of its chloride salt is 290.70 g/mol.[4][5] As such, 100 mg of nicotinamide riboside chloride provides 88 mg of nicotinamide riboside.
History
Nicotinamide riboside (NR) was first described in 1944 as a growth factor, termed Factor V, for Haemophilus influenza, a bacterium that lives in and depends on blood. Factor V, purified from blood, was shown to exist in three forms: NAD+, NMN and NR. NR was the compound that led to the most rapid growth of this bacterium.[6] Notably, H. influenza cannot grow on nicotinic acid, nicotinamide, tryptophan or aspartic acid, which were the previously known precursors of NAD+.[7]
In 2000, yeast Sir2 was shown to be an NAD+-dependent protein lysine deacetylase,[8] which led several research groups to probe yeast NAD+ metabolism for genes and enzymes that might regulate lifespan. Biosynthesis of NAD+ in yeast was thought to flow exclusively through NAMN (nicotinic acid mononucleotide).[9][10][11][12][13]
When NAD+ synthase (glutamine-hydrolysing) was deleted from yeast cells, NR permitted yeast cells to grow. Thus, these Dartmouth College investigators proceeded to clone yeast and human nicotinamide riboside kinases and demonstrate the conversion of NR to NMN by nicotinamide riboside kinases in vitro and in vivo. They also demonstrated that NR is a natural product found in cow’s milk.[14][15]
Properties
Although it is a form of vitamin B3, NR exhibits unique properties that distinguish it from the other B3 vitamins—niacin and nicotinamide. In a head-to-head experiment conducted on mice, each of these vitamins exhibited unique effects on the hepatic NAD+ metabolome with unique kinetics, and with NR as the form of B3 that produced the greatest increase in NAD+ at a single timepoint.[16]
Different biosynthetic pathways are responsible for converting the different B3 vitamins into NAD+. The enzyme nicotinamide phosphoribosyltransferase (Nampt) catalyzes the rate-limiting step of the two-step pathway converting nicotinamide to NAD+. Two nicotinamide riboside kinases (NRK1 and NRK2) convert NR to NAD+ via a pathway that does not require Nampt.[14]
Animal studies have demonstrated that these enzymes respond differently to age and stress. In a mouse model of dilated cardiomyopathy, NRK2 mRNA expression increased, while Nampt mRNA expression decreased.[17] A similar increase in NRK1 and NRK2 expression has been observed in injured central and peripheral neurons.[18][19][20][21][22]
Niacin is known for its tendency to cause an uncomfortable flushing of the skin. This flushing is triggered by the activation of the GPR109A G-protein coupled receptor. NR does not activate this receptor,[23] and has not been shown to cause flushing in humans—even at doses as high as 2,000 mg/day.[16][24][25][26]
Despite being an NAD+ precursor, nicotinamide acts as an inhibitor of the NAD+-consuming sirtuin enzymes.[10] When sirtuins consume NAD+, they create nicotinamide and O-acetyl-ADP-ribose as products of the deacetylation reaction. Consistent with high-dose nicotinamide as a sirtuin inhibitor, NR and niacin, but not nicotinamide, have been shown to increase hepatic levels of O-acetyl-ADP-ribose.[16]
Commercialization
In 2004, Dartmouth Medical School researcher Dr. Charles Brenner discovered that NR could be converted to NAD+ via the eukaryotic nicotinamide riboside kinase biosynthetic pathway[14] Dartmouth was subsequently issued patents for nutritional and therapeutic uses of NR, in 2006.[27] ChromaDex licensed these patents in July 2012, and began to develop a commercially viable, full-scale process to bring NR to market.[28]
Human Clinical Testing
There have been five published clinical trials on groups of both men and women testing for safety. One of these trials studied NR in combination with pterostilbene,[29] while the other four examined the effects of NR alone.[16][24][25][26]
The first published clinical trial established the safety and characterized the pharmacokinetics of single doses of NR.[16] Since then, doses as high as 2,000 mg/day have been administered over periods as long as 12 weeks.[25] These studies show that NR can significantly increase levels of NAD+ and some of its associated metabolites in both whole blood and peripheral blood mononuclear cells.[16][24][26]
In a 12 week clinical trial of obese insulin-resistant men using 2000 mg/day, NR appeared safe, but did not improve insulin sensitivity or whole-body glucose metabolism.[26] In a trial of NR 250 mg plus 50 mg of pterostilbene, as well as with double this dose, the combined supplement raised NAD+ levels in a trial of older adults.[29]
PATENT
WO-2019126482
Crystalline form of nicotinamide riboside chloride, useful for treating motor neuron disease or ALS, infertility, kidney damage, and liver damage or fatty liver. Elysium Health in collaboration with Mayo Clinic , is developing EH-301 (clinical, in July 2019), a combination of nicotinamide riboside chloride and pterostilbene for the treatment of amyotrophic lateral sclerosis. See WO2019108878 , claiming use of composition comprising nicotinamide riboside and pterostilbene, for treating obesity.
Nicotinamide riboside is a pyridine-nucleoside form of niacin ( i.e ., vitamin B3) that serves as a precursor to nicotinamide adenine dinucleotide (NAD+). NAD+promotes cellular metabolism, mitochondrial function, and energy production. Currently, nicotinamide riboside is made through synthetic methods or fermentation processes. Because of its significant potential to confer health benefits when used as a dietary supplement, there exists a need to develop highly efficient and scalable processes for the manufacture and purification of nicotinamide riboside.
SUMMARY OF THE INVENTION
In certain aspects, the present invention provides a crystalline form of a compound having the structure of formula (I)
Example 1. Scale-Up Synthesis and Crystallization of Nicotinamide Riboside Chloride
900 kg of nicotinamide riboside triacetate and 2133 kg of methanol were charged to a reactor and mixed, then cooled to 0 °C. 747 kg of 7M mmmonia in methanol (i.e.,“methanolic NH3”) was slowly charged to the reactor at 0 °C. The reaction mixture was passed through a polish filter, then the reaction mixture was stirred for 14 hours. A sample from the reaction mixture was taken to assess reaction progress. Upon completion of the reaction, the reaction mixture was
placed under vacuum, then warmed to 20 °C to 25 °C for 4 hours. Vacuum was applied until solids formed. Once solids were formed, the resultant slurry was filtered on a Nutsche filter dryer. Solids were washed with 1422 kg of ethanol, then 1422 kg of acetone, then 1322 kg of methyl tert butyl ether (MTBE). The resultant solids were then dried at 40 °C. Product was formed with 60% yield. The process flow diagram for this reaction is shown in FIG. 6.
Example 2. Optional Secondary Isolation
The crystalline form may optionally undergo a second isolation process according to the following steps: The solids obtained in Example 1 were dissolved in purified water at 30 °C to 40 °C. Ethanol was slowly added to the solution and mixed for 10 hours, over which time the solids began to precipitate. MTBE was then added and mixed for 2 hours. The mixture was then filtered on a Buchner funnel, and the solids were washed with ethanol, then acetone, then MTBE. Solids were dried at 40 °C.
Example 3. Spectroscopic Data.
The crystalline form made by the process described in Examples 1 and 2 has an XRD spectrum substantially as shown in FIG. 1. The instrument utilized in collecting the XRD data is a Rigaku Smart Lab X-Ray diffraction system.
Specifically, in order to collect the XRD data, The Rigaku Smart-Lab X-ray diffraction system was configured for reflection Bragg-Brentano geometry using a line source X-ray beam. The X-ray source is a Cu Long Fine Focus tube that was operated at 40 kV and 44 mA. That source provides an incident beam profile at the sample that changes from a narrow line at high angles to a broad rectangle at low angles. Beam conditioning slits are used on the line X-ray source to ensure that the maximum beam size is less than 10 mm both along the line and normal to the line. The Bragg-Brentano geometry is a para-focusing geometry controlled by passive divergence and receiving slits with the sample itself acting as the focusing component for the optics. The inherent resolution of Bragg-Brentano geometry is governed in part by the diffractometer radius and the width of the receiving slit used. Typically, the Rigaku Smart-Lab is operated to give peak widths of 0.1 °2Q or less. The axial divergence of the X-ray beam is controlled by 5.0-degree Sober slits in both the incident and diffracted beam paths.
The samples were prepared in a low background Si holder using light manual pressure to keep the sample surface flat and level with the reference surface of the sample holder. The single crystal Si low background holder has a small circular recess (10 mm diameter and about 0.2 mm depth) that held between 20 and 25 mg of the sample. The samples were analyzed from 2 to 40
°2Q using a continuous scan of 6 °20 per minute with an effective step size of 0.02 °20. The data collection procedure used to analyze these samples was not validated. The peak lists were generated using PDXL2 v.2.3.1.0. The figures were created using PlotMon VI.00.
PATENT
WO2019108878 , claiming use of composition comprising nicotinamide riboside and pterostilbene, for treating obesity.
CLIP
https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0186459
CLIP


References
- ^ Bogan, K.L., Brenner, C. (2008). “Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition”. Annu. Rev. Nutr. 28: 115–130. doi:10.1146/annurev.nutr.28.061807.155443. PMID 18429699.
- ^ Chi Y, Sauve AA (November 2013). “Nicotinamide riboside, a trace nutrient in foods, is a vitamin B3 with effects on energy metabolism and neuroprotection”. Curr Opin Clin Nutr Metab Care. 16 (6): 657–61. doi:10.1097/MCO.0b013e32836510c0. PMID 24071780.
- ^ “Nicotinamide riboside”. pubchem.ncbi.nlm.nih.gov.
- ^ “GRAS Notices, GRN No. 635”. http://www.accessdata.fda.gov. Retrieved 18 February 2019.
- ^ “Spherix/ChromaDex GRAS submission” (PDF). FDA.gov. Retrieved 18 February2019.
- ^ Gingrich, W; Schlenk, F (June 1944). “Codehydrogenase I and Other Pyridinium Compounds as V-Factor for Hemophilus influenzae and H. parainfluenzae”. Journal of Bacteriology. 47 (6): 535–50. PMC 373952. PMID 16560803.
- ^ Belenky, P.; et al. (2007). “NAD+ Metabolism in Health and Disease”. Trends in Biochemical Sciences. 32 (1): 12–19. doi:10.1016/j.tibs.2006.11.006. PMID 17161604.
- ^ Imai, S.; et al. (2000). “Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase”. Nature. 403 (6771): 795–800. doi:10.1038/35001622. PMID 10693811.
- ^ Anderson; et al. (2003). “Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae”. Nature. 423 (6936): 181–185. doi:10.1038/nature01578. PMC 4802858. PMID 12736687.
- ^ Jump up to:a b Bitterman; et al. (2002). “Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast Sir2 and human SIRT1”. J. Biol. Chem. 277 (47): 45099–45107. doi:10.1074/jbc.m205670200. PMID 12297502.
- ^ Gallo; et al. (2004). “Nicotinamide clearance by pnc1 directly regulates sir2-mediated silencing and longevity”. Mol. Cell. Biol. 24 (3): 1301–1312. doi:10.1128/mcb.24.3.1301-1312.2004.
- ^ Panozzo, C.; et al. (2002). “Aerobic and anaerobic NAD+ metabolism in Saccharomyces cerevisiae”. FEBS Lett. 517 (1–3): 97–102. doi:10.1016/s0014-5793(02)02585-1. PMID 12062417.
- ^ Sandmeier, JJ; Celic, I; Boeke, JD; Smith, JS (March 2002). “Telomeric and rDNA silencing in Saccharomyces cerevisiae are dependent on a nuclear NAD(+) salvage pathway”. Genetics. 160 (3): 877–89. PMC 1462005. PMID 11901108.
- ^ Jump up to:a b c Bieganowki, P. & Brenner, C. (2004). “Discoveries of Nicotinamide Riboside as a Nutrient and Conserved NRK Genes Establish a Preiss-Handler Independent Route to NAD+ in Fungi and Humans”. Cell. 117 (4): 495–502. doi:10.1016/s0092-8674(04)00416-7. PMID 15137942.
- ^ Hautkooper, R.H.; et al. (2012). “Sirtuins as regulators of metabolism and healthspan”. Nat. Rev. Mol. Cell Biol. 13 (4): 225–238. doi:10.1038/nrm3293. PMC 4872805. PMID 22395773.
- ^ Jump up to:a b c d e f Trammell, Samuel A. J.; Schmidt, Mark S.; Weidemann, Benjamin J.; Redpath, Philip; Jaksch, Frank; Dellinger, Ryan W.; Li, Zhonggang; Abel, E. Dale; Migaud, Marie E.; Brenner, Charles (10 October 2016). “Nicotinamide riboside is uniquely and orally bioavailable in mice and humans”. Nature Communications. 7 (1): 12948. doi:10.1038/ncomms12948. PMC 5062546. PMID 27721479.
- ^ Diguet, Nicolas; Trammell, Samuel A.J.; Tannous, Cynthia; Deloux, Robin; Piquereau, Jérôme; Mougenot, Nathalie; Gouge, Anne; Gressette, Mélanie; Manoury, Boris; Blanc, Jocelyne; Breton, Marie; Decaux, Jean-François; Lavery, Gareth G.; Baczkó, István; Zoll, Joffrey; Garnier, Anne; Li, Zhenlin; Brenner, Charles; Mericskay, Mathias (22 May 2018). “Nicotinamide Riboside Preserves Cardiac Function in a Mouse Model of Dilated Cardiomyopathy”. Circulation. 137 (21): 2256–2273. doi:10.1161/CIRCULATIONAHA.116.026099. PMID 29217642.
- ^ Vaur, Pauline; Brugg, Bernard; Mericskay, Mathias; Li, Zhenlin; Schmidt, Mark S.; Vivien, Denis; Orset, Cyrille; Jacotot, Etienne; Brenner, Charles; Duplus, Eric (December 2017). “Nicotinamide riboside, a form of vitamin B , protects against excitotoxicity-induced axonal degeneration”. The FASEB Journal. 31 (12): 5440–5452. doi:10.1096/fj.201700221RR. PMID 28842432.
- ^ Sasaki, Y.; Araki, T.; Milbrandt, J. (16 August 2006). “Stimulation of Nicotinamide Adenine Dinucleotide Biosynthetic Pathways Delays Axonal Degeneration after Axotomy”. Journal of Neuroscience. 26 (33): 8484–8491. doi:10.1523/JNEUROSCI.2320-06.2006. PMID 16914673.
- ^ Frederick, David W.; Loro, Emanuele; Liu, Ling; Davila, Antonio; Chellappa, Karthikeyani; Silverman, Ian M.; Quinn, William J.; Gosai, Sager J.; Tichy, Elisia D.; Davis, James G.; Mourkioti, Foteini; Gregory, Brian D.; Dellinger, Ryan W.; Redpath, Philip; Migaud, Marie E.; Nakamaru-Ogiso, Eiko; Rabinowitz, Joshua D.; Khurana, Tejvir S.; Baur, Joseph A. (August 2016). “Loss of NAD Homeostasis Leads to Progressive and Reversible Degeneration of Skeletal Muscle”. Cell Metabolism. 24 (2): 269–282. doi:10.1016/j.cmet.2016.07.005. PMC 4985182. PMID 27508874.
- ^ Cantó, Carles; Jiang, Lake Q.; Deshmukh, Atul S.; Mataki, Chikage; Coste, Agnes; Lagouge, Marie; Zierath, Juleen R.; Auwerx, Johan (March 2010). “Interdependence of AMPK and SIRT1 for Metabolic Adaptation to Fasting and Exercise in Skeletal Muscle”. Cell Metabolism. 11 (3): 213–219. doi:10.1016/j.cmet.2010.02.006. PMC 3616265. PMID 20197054.
- ^ Rappou, Elisabeth; Jukarainen, Sakari; Rinnankoski-Tuikka, Rita; Kaye, Sanna; Heinonen, Sini; Hakkarainen, Antti; Lundbom, Jesper; Lundbom, Nina; Saunavaara, Virva; Rissanen, Aila; Virtanen, Kirsi A.; Pirinen, Eija; Pietiläinen, Kirsi H. (March 2016). “Weight Loss Is Associated With Increased NAD /SIRT1 Expression But Reduced PARP Activity in White Adipose Tissue”. The Journal of Clinical Endocrinology & Metabolism. 101 (3): 1263–1273. doi:10.1210/jc.2015-3054. PMID 26760174.
- ^ Cantó, Carles; Houtkooper, Riekelt H.; Pirinen, Eija; Youn, Dou Y.; Oosterveer, Maaike H.; Cen, Yana; Fernandez-Marcos, Pablo J.; Yamamoto, Hiroyasu; Andreux, Pénélope A.; Cettour-Rose, Philippe; Gademann, Karl; Rinsch, Chris; Schoonjans, Kristina; Sauve, Anthony A.; Auwerx, Johan (June 2012). “The NAD+ Precursor Nicotinamide Riboside Enhances Oxidative Metabolism and Protects against High-Fat Diet-Induced Obesity”. Cell Metabolism. 15 (6): 838–847. doi:10.1016/j.cmet.2012.04.022. PMC 3616313. PMID 22682224.
- ^ Jump up to:a b c Airhart, Sophia E.; Shireman, Laura M.; Risler, Linda J.; Anderson, Gail D.; Nagana Gowda, G. A.; Raftery, Daniel; Tian, Rong; Shen, Danny D.; O’Brien, Kevin D.; Sinclair, David A. (6 December 2017). “An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers”. PLOS ONE. 12 (12): e0186459. doi:10.1371/journal.pone.0186459. PMC 5718430. PMID 29211728.
- ^ Jump up to:a b c Dollerup, Ole L; Christensen, Britt; Svart, Mads; Schmidt, Mark S; Sulek, Karolina; Ringgaard, Steffen; Stødkilde-Jørgensen, Hans; Møller, Niels; Brenner, Charles; Treebak, Jonas T; Jessen, Niels (August 2018). “A randomized placebo-controlled clinical trial of nicotinamide riboside in obese men: safety, insulin-sensitivity, and lipid-mobilizing effects”. The American Journal of Clinical Nutrition. 108 (2): 343–353. doi:10.1093/ajcn/nqy132. PMID 29992272.
- ^ Jump up to:a b c d Martens, Christopher R.; Denman, Blair A.; Mazzo, Melissa R.; Armstrong, Michael L.; Reisdorph, Nichole; McQueen, Matthew B.; Chonchol, Michel; Seals, Douglas R. (29 March 2018). “Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD+ in healthy middle-aged and older adults”. Nature Communications. 9 (1): 1286. doi:10.1038/s41467-018-03421-7. PMC 5876407. PMID 29599478.
- ^ Brenner, Charles (20 April 2006). “Nicotinamide riboside kinase compositions and methods for using the same”. Google Patents. Dartmouth College. Retrieved 19 February2019.
- ^ “ChromaDex Licenses Exclusive Patent Rights for Nicotinamide Riboside (NR) Vitamin Technologies”. 2012-07-16. Retrieved 15 February 2019.
- ^ Jump up to:a b Dellinger, Ryan W.; Santos, Santiago Roel; Morris, Mark; Evans, Mal; Alminana, Dan; Guarente, Leonard; Marcotulli, Eric (24 November 2017). “Repeat dose NRPT (nicotinamide riboside and pterostilbene) increases NAD+ levels in humans safely and sustainably: a randomized, double-blind, placebo-controlled study”. NPJ Aging and Mechanisms of Disease. 3 (1): 17. doi:10.1038/s41514-017-0016-9. PMC 5701244. PMID 29184669.
Further reading
- “Press Release: NIH researchers find potential target for reducing obesity-related inflammation”. National Institutes of Health (NIH). 16 November 2015.
- Stipp, David (March 11, 2015). “Guest Blog: Beyond Resveratrol: The Anti-Aging NAD Fad”. Scientific American Blog Network.
- Zhang, H; Ryu, D; Wu, Y; Gariani, K; Wang, X; Luan, P; D’Amico, D; Ropelle, ER; Lutolf, MP; Aebersold, R; Schoonjans, K; Menzies, KJ; Auwerx, J (17 June 2016). “NAD⁺ repletion improves mitochondrial and stem cell function and enhances life span in mice”. Science. 352 (6292): 1436–43. doi:10.1126/science.aaf2693. PMID 27127236.
- Dolopikou CF, Kourtzidis IA, Margaritelis NV, Vrabas IS, Koidou I, Kyparos A, Theodorou AA, Paschalis V, Nikolaidis MG. (2019 Feb 6). Acute nicotinamide riboside supplementation improves redox homeostasis and exercise performance in old individuals: a double-blind cross-over study. doi:10.1007/s00394-019-01919-4.
ADDITIONAL INFORMATION
High dose nicotinic acid is used as an agent that elevates high-density lipoprotein cholesterol, lowers low-density lipoprotein cholesterol and lower free fatty acids through a mechanism that is not completely understood. It was suggested that nicotinamide riboside might possess such an activity by elevating NAD in the cells responsible for reverse cholesterol transport. The discovery that the Wallerian degeneration slow gene encodes a protein fusion with NMN adenylyltransferase 1 indicated that increased NAD+ precursor supplementation might oppose neurodegenerative processes.
ChromaDex acquired intellectual property on uses and synthesis of NR from Dartmouth College, Cornell University, and Washington University and began distributing NR as Niagen in 2013. In November 2015 ChromaDex received New Dietary Ingredient (NDI) status for Niagen from the U.S. Food and Drug Administration (FDA) and the FDA issued a generally recognized as safe (GRAS) No Objection Letter for Nicotinamide Riboside Chloride (NR) on August 3, 2016.
REFERENCES
1: Chi Y, Sauve AA. Nicotinamide riboside, a trace nutrient in foods, is a vitamin B3 with effects on energy metabolism and neuroprotection. Curr Opin Clin Nutr Metab Care. 2013 Nov;16(6):657-61. doi: 10.1097/MCO.0b013e32836510c0. Review. PubMed PMID: 24071780.
2: Bogan KL, Brenner C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu Rev Nutr. 2008;28:115-30. doi: 10.1146/annurev.nutr.28.061807.155443. Review. PubMed PMID: 18429699.
3: Ghanta S, Grossmann RE, Brenner C. Mitochondrial protein acetylation as a cell-intrinsic, evolutionary driver of fat storage: chemical and metabolic logic of acetyl-lysine modifications. Crit Rev Biochem Mol Biol. 2013 Nov-Dec;48(6):561-74. doi: 10.3109/10409238.2013.838204. Review. PubMed PMID: 24050258; PubMed Central PMCID: PMC4113336.
4: Yang Y, Sauve AA. NAD(+) metabolism: Bioenergetics, signaling and manipulation for therapy. Biochim Biophys Acta. 2016 Dec;1864(12):1787-1800. doi: 10.1016/j.bbapap.2016.06.014. Review. PubMed PMID: 27374990.
5: Sauve AA. NAD+ and vitamin B3: from metabolism to therapies. J Pharmacol Exp Ther. 2008 Mar;324(3):883-93. doi: 10.1124/jpet.107.120758. Review. PubMed PMID: 18165311.
6: Kato M, Lin SJ. Regulation of NAD+ metabolism, signaling and compartmentalization in the yeast Saccharomyces cerevisiae. DNA Repair (Amst). 2014 Nov;23:49-58. doi: 10.1016/j.dnarep.2014.07.009. Review. PubMed PMID: 25096760; PubMed Central PMCID: PMC4254062.
7: Gerlach G, Reidl J. NAD+ utilization in Pasteurellaceae: simplification of a complex pathway. J Bacteriol. 2006 Oct;188(19):6719-27. Review. PubMed PMID: 16980474; PubMed Central PMCID: PMC1595515.
8: Srivastava S. Emerging therapeutic roles for NAD(+) metabolism in mitochondrial and age-related disorders. Clin Transl Med. 2016 Dec;5(1):25. doi: 10.1186/s40169-016-0104-7. Review. PubMed PMID: 27465020; PubMed Central PMCID: PMC4963347.
9: Handschin C. Caloric restriction and exercise “mimetics”: Ready for prime time? Pharmacol Res. 2016 Jan;103:158-66. doi: 10.1016/j.phrs.2015.11.009. Review. PubMed PMID: 26658171; PubMed Central PMCID: PMC4970791.
10: Ruggieri S, Orsomando G, Sorci L, Raffaelli N. Regulation of NAD biosynthetic enzymes modulates NAD-sensing processes to shape mammalian cell physiology under varying biological cues. Biochim Biophys Acta. 2015 Sep;1854(9):1138-49. doi: 10.1016/j.bbapap.2015.02.021. Review. PubMed PMID: 25770681.
11: Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014 Aug;24(8):464-71. doi: 10.1016/j.tcb.2014.04.002. Review. PubMed PMID: 24786309; PubMed Central PMCID: PMC4112140.
12: Jaehme M, Slotboom DJ. Structure, function, evolution, and application of bacterial Pnu-type vitamin transporters. Biol Chem. 2015 Sep;396(9-10):955-66. doi: 10.1515/hsz-2015-0113. Review. PubMed PMID: 26352203.
13: Magni G, Di Stefano M, Orsomando G, Raffaelli N, Ruggieri S. NAD(P) biosynthesis enzymes as potential targets for selective drug design. Curr Med Chem. 2009;16(11):1372-90. Review. PubMed PMID: 19355893.
14: Mendelsohn AR, Larrick JW. Partial reversal of skeletal muscle aging by restoration of normal NAD⁺ levels. Rejuvenation Res. 2014 Feb;17(1):62-9. doi: 10.1089/rej.2014.1546. Review. PubMed PMID: 24410488.
15: Penberthy WT. Pharmacological targeting of IDO-mediated tolerance for treating autoimmune disease. Curr Drug Metab. 2007 Apr;8(3):245-66. Review. PubMed PMID: 17430113.
16: Gazzaniga F, Stebbins R, Chang SZ, McPeek MA, Brenner C. Microbial NAD metabolism: lessons from comparative genomics. Microbiol Mol Biol Rev. 2009 Sep;73(3):529-41, Table of Contents. doi: 10.1128/MMBR.00042-08. Review. PubMed PMID: 19721089; PubMed Central PMCID: PMC2738131.
17: Magni G, Amici A, Emanuelli M, Orsomando G, Raffaelli N, Ruggieri S. Enzymology of NAD+ homeostasis in man. Cell Mol Life Sci. 2004 Jan;61(1):19-34. Review. PubMed PMID: 14704851.
18: Magni G, Orsomando G, Raffelli N, Ruggieri S. Enzymology of mammalian NAD metabolism in health and disease. Front Biosci. 2008 May 1;13:6135-54. Review. PubMed PMID: 18508649.
19: Belenky P, Bogan KL, Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci. 2007 Jan;32(1):12-9. Review. Erratum in: Trends Biochem Sci. 2008 Jan;33(1):1. PubMed PMID: 17161604.
20: Niven DF, O’Reilly T. Significance of V-factor dependency in the taxonomy of Haemophilus species and related organisms. Int J Syst Bacteriol. 1990 Jan;40(1):1-4. Review. PubMed PMID: 2145965.
|
|
 |
|
| Names | |
|---|---|
| Other names
1-(β-D-Ribofuranosyl)nicotinamide; N-Ribosylnicotinamide
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChEBI | |
| ChemSpider | |
| KEGG | |
|
PubChem CID
|
|
| Properties | |
| C11H15N2O5+ | |
| Molar mass | 255.25 g/mol |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
///////////// EH-301, EH 301, EH301, Nicotinamide riboside, SRT647, SRT-647, SRT 647, Nicotinamide Riboside Triflate, α/β mixture
C1=CC(=C[N+](=C1)C2C(C(C(O2)CO)O)O)C(=O)N.[Cl-]
Quinupramine, キヌプラミン
Quinupramine
キヌプラミン
- 5-(1-azabicyclo[2.2.2]oct-3-yl)-10,11-dihydro-5H-dibenz[b,f]azepine
- Formula:C21H24N2
- MW:304.44 g/mol
- CAS:31721-17-2
Quinupramine (brand names Kevopril, Kinupril, Adeprim, Quinuprine) is a tricyclic antidepressant (TCA) used in Europe for the treatment of depression.[1][2]
Pharmacologically, quinupramine acts in vitro as a strong muscarinic acetylcholine receptor antagonist (anticholinergic) and H1 receptorantagonist (antihistamine), moderate 5-HT2 receptor antagonist, and weak serotonin and norepinephrine reuptake inhibitor.[3] It has negligible affinity for the α1-adrenergic, α2-adrenergic, β-adrenergic, or D2 receptor.[3]
Clinically, quinupramine is reported to be stimulating similarly to imipramine, desipramine, and demexiptiline.[4] It can be inferred that its in vivo metabolites may have stronger effects on the reuptake of norepinephrine and/or serotonin than quinupramine itself
SYN
References
- ^ Swiss Pharmaceutical Society (2000). Index Nominum 2000: International Drug Directory (Book with CD-ROM). Boca Raton: Medpharm Scientific Publishers. p. 908. ISBN 3-88763-075-0.
- ^ José Miguel Vela; Helmut Buschmann; Jörg Holenz; Antonio Párraga; Antoni Torrens (2007). Antidepressants, Antipsychotics, Anxiolytics: From Chemistry and Pharmacology to Clinical Application. Weinheim: Wiley-VCH. p. 248. ISBN 978-3-527-31058-6.
- ^ Jump up to:a b Sakamoto H, Yokoyama N, Kohno S, Ohata K (December 1984). “Receptor binding profile of quinupramine, a new tricyclic antidepressant”. Japanese Journal of Pharmacology. 36 (4): 455–60. doi:10.1254/jjp.36.455. PMID 6098759.
- ^ Kent, Angela; M. Billiard (2003). Sleep: physiology, investigations, and medicine. New York: Kluwer Academic/Plenum. p. 233. ISBN 0-306-47406-9.
-
- DOS 2 030 492 (Sogeras; appl. 20.6.1970; GB-prior. 20.6.1969).
- GB 1 252 320 (Sogeras; valid from 29.5.1970; prior. 20.6.1969).
 |
|
| Clinical data | |
|---|---|
| Routes of administration |
Oral |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Elimination half-life | 33 hours |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ECHA InfoCard | 100.046.149 |
| Chemical and physical data | |
| Formula | C21H24N2 |
| Molar mass | 304.43 g/mol g·mol−1 |
//////////////Quinupramine, キヌプラミン
TENAPANOR


Tenapanor
Molecular FormulaC50H66Cl4N8O10S2
Average mass1145.049 Da
1234423-95-0 [RN]
1234423-95-0 (free base) 1234365-97-9 (2HCl)
9652
3-((S)-6,8-dichloro-2-methyl-1,2,3,4-tetrahydroisoquinolin-4-yl)-N-(26-((3-((S)-6,8-dichloro-2-methyl-1,2,3,4-tetrahydroisoquinolin-4-yl)phenyl)sulfonamido)-10,17-dioxo-3,6,21,24-tetraoxa-9,11,16,18-tetraazahexacosyl)benzenesulfonamide
Benzenesulfonamide, N,N’-(10,17-dioxo-3,6,21,24-tetraoxa-9,11,16,18-tetraazahexacosane-1,26-diyl)bis[3-[(4S)-6,8-dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]-
12,15-Dioxa-2,7,9-triazaheptadecanamide, 17-[[[3-[(4S)-6,8-dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulfonyl]amino]-N-[2-[2-[2-[[[3-[(4S)-6,8-dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulfonyl]amino]ethoxy]ethoxy]ethyl]-8-oxo-
1-[2-[2-[2-[[3-[(4S)-6,8-dichloro-2-methyl-3,4-dihydro-1H-isoquinolin-4-yl]phenyl]sulfonylamino]ethoxy]ethoxy]ethyl]-3-[4-[2-[2-[2-[[3-[(4S)-6,8-dichloro-2-methyl-3,4-dihydro-1H-isoquinolin-4-yl]phenyl]sulfonylamino]ethoxy]ethoxy]ethylcarbamoylamino]butyl]urea
17-[[[3-[(4S)-6,8-Dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulfonyl]amino]-N-[2-[2-[2-[[[3-[(4S)-6,8-dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulfonyl]amino]ethoxy]ethoxy]ethyl]-8-oxo-12,15-dioxa-2,7,9-triazaheptadecanamide
Tenapanor, also known as AZD-1722 and RDX 5791, is an inhibitor of the sodium-proton (Na(+)/H(+)) exchanger NHE3, which plays a prominent role in sodium handling in the gastrointestinal tract and kidney. Tenapanor possesses an excellent preclinical safety profile and, as of now, there are no serious concerns about its side effects.
Tenapanor is a drug developed by Ardelyx, which acts as an inhibitor of the sodium-proton exchanger NHE3. This antiporterprotein is found in the kidney and intestines, and normally acts to regulate the levels of sodium absorbed and secreted by the body. When administered orally, tenapanor selectively inhibits sodium uptake in the intestines, limiting the amount absorbed from food, and thereby reduces levels of sodium in the body.[1] This may make it useful in the treatment of chronic kidney disease and hypertension, both of which are exacerbated by excess sodium in the diet.[2]
Ardelyx and licensees Kyowa Hakko Kirin and Fosun Pharma are developing tenapanor, an NHE3 (Na+/H+ exchange-3) inhibitor that increases fluid content in the GI tract and which also reduces GI tract pain via an unknown TRPV-1-dependent pathway, for treating constipation-predominant irritable bowel syndrome (IBS-C) and hyperphosphatemia in patients with end stage renal disease (ESRD).
Syn

PATENT
WO2010078449
PATENT
WO-2019091503
A novel crystalline form of tenapanor free base, process for its preparation, composition comprising it and its use for the preparation of tenapanor with chemical purity >98.8% is claimed. Also claimed are salt forms of tenapanor, preferably tenapanor phosphate and their use for treating irritable bowel syndrome, constipation, hyperphosphatemia, final stage renal failure, chronic kidney disease and preventing excess sodium in patients with kidney and heart conditions. Further claimed are processes for the preparation of tenapanor comprising the steps of reaction of a diamine compound with 1,4-diisocyanatobutane, followed by deprotection and condensation to obtain tenapanor. Novel intermediates of tenapanor and their use for the preparation of tenapanor are claimed. Tenapanor is known to be a sodium hydrogen exchanger 3 inhibitor and analgesic.
enapanor, having the chemical name 17-[[[3-[(4S)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulphonyl]amino]-N-[2-[2-[2-[[[3-[(4S)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl -4-isoquinolinyl] phenyl] sulphonyl] amino] ethoxy] ethoxy ] ethyl] – 8 -oxo- 12,15 -dioxa-2 ,7,9-triazaheptadecaneamide, is a selective inhibitor of the sodium protonic NHE3 antiporter. Orally administered tenapanor selectively inhibits the absorption of sodium in the intestine. This leads to an increase of water content in the digestive tract, improved bowel flow and normalization of the frequency of bowel movement and stool consistency. At the same time it exhibits antinociceptive activity and ability to lower serum phosphate levels. Because of these properties, it is clinically tested for the treatment of irritable bowel syndrome, especially when accompanied by constipation, treatment of hyperphosphatemia, especially in patients with dialysis with final stage renal failure, treatment of chronic kidney disease, and prevention of excess sodium in patients with kidney and heart conditions. The tenapanor molecule, which was first described in the international patent application WO 2010/078449, has the following structural formula:
In this document, tenapanor was prepared as bishydrochloride salt. The bishydrochloride salt was prepared only in the form of an amorphous foam, which, after solidification, required grinding for further processing. However, the thus obtained particles are of varying sizes, while a narrow particle size distribution is required for pharmaceutical use in order to ensure uniform behavior. The amorphous foam obtained in the said document is essentially a thickened reaction mixture or a slightly purified reaction mixture containing, in addition to tenapanor, various impurities. The possibilities to purify the reaction mixtures are limited. Moreover, amorphous foams tend to adsorb solvents, and it is usually difficult to remove (or dry out) the residual solvents from the amorphous foam. This is undesirable for pharmaceutical use. A typical feature of amorphous foams is a large specific surface, resulting in a greater interaction of the substance with the surrounding environment. This significantly increases the risk of decomposition of the substance, for example through air oxygen, moisture or light. The present invention aims at overcoming these problems.
It would be advantageous to provide tenapanor solid forms (tenapanor free base or tenapanor salts) which are precipitated in solid forms, thus allowing to filter off the liquid reaction mixture containing the impurities. This results in a significantly improved purity.
The process used in WO 2010/078449 for the preparation of bishydrochloride salt of tenapanor was based on the preparation of 3-(6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinolin-4-yl)benzene-l-sulfonyl chloride of formula III from 4-(3-bromophenyl)-6,8-dichloro-2-methyl-l, 2,3,4-te
Scheme 1
The said document also discloses resolution of the starting tetrahydroisoquinoline of formula II by L-or D-dibenzoylt
(II) (S-II) (R-II)
Scheme 2
WO 2010/078449 discloses further steps of preparation of tenapanor, as shown in Scheme 3.
(V) (I)
Scheme 3
Individual synthetic steps described in Scheme 3 result in low yields: 42% for the reaction of the chloride of formula III with 2-(2-(2-aminoethoxy)ethoxy)ethylamine of formula IV, and 59% for the subsequent reaction with 1,4-diisocyanatobutane of formula V. The products of both synthetic steps are isolated by preparative chromatography which is technologically an unsuitable isolation and purification technique. The low yields and the need to use preparative chromatography for the isolation are caused by an abundance of side products and impurities and by the inability of the intermediates as well as of the product to provide a crystalline form.
Thus present invention thus further aims at providing a method of preparation of tenapanor which would be economically effective, in particular in relation to the expensive starting compound 4-(3-bromophenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline, and which would also enable industrial scale production, in particular by removing steps which cannot be scaled up effectively or which cannot be scaled up at all. Furthermore, the method of preparation of tenapanor should provide tenapanor in a form which is useful for use in pharmaceutical forms and does not have the disadvantages of an amorphous foam.
Tenapanor free base in the form of an amorphous solid foam was prepared by the procedure disclosed in patent application WO 2010/078449, Example 202. The chemical purity of the tenapanor prepared by this procedure was 96.5% (HPLC). The structure of tenapanor was verified by MS and H and 13C NMR spectra.
Step A
Preparation of (5)- -(3-(benzylthio)phenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline
Potassium carbonate (9.30 g) and anhydrous xylene (500 ml) were added to the reaction vessel. Benzyl mercaptane (25 g) was added dropwise to the stirred mixture under ice -cooling. The resulting mixture was stirred at 25 °C for lh.
(S)-4-(3-bromophenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline 50 g in anhydrous xylene (500 ml), Pd2(dba)3 (3 g) and Xantphos (3 g). The resulting solution was stirred at 25 °C for 30 minutes and then added to a solution of benzyl mercaptane. The resulting reaction mixture was maintained at 140 °C for 16 h. The mixture was then concentrated and the residue was subjected to preparative chromatography on silica gel with the mobile phase ethyl acetate / petroleum ether (1: 100-1 :50). 20 g of product are obtained as a yellow oil (36% yield).
Ste B
Preparation of (5) -3 -(6 , 8 -dichloro-2 -methyl- 1,2,3 ,4-tetr ahydroisoquinolin-4-yl)benzenesulf onyl chloride hydrochloride
(S)-4-(3-(benzylthio)phenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline (16 g) was dissolved in the reaction vessel in acetic acid/water (160 mL: 16 mL) mixture. The mixture was cooled in an ice bath and then gaseous Cl2 was introduced into the well stirred mixture. After disappearance of the starting material, the reaction mixture was purged with nitrogen and concentrated in vacuo. A product (10 g, 66.6%) was obtained as a colorless substance.
Step C
Preparation of (S)-N-(2-(2-(2-aminoethoxy)ethoxy)ethyl)-3-(6,8-dichloro-2-methyl-l, 2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonamide
2-(2-(2-Aminoethoxy)ethoxy)ethylamine HC1 (30 g; 0.2 mol) and triethylamine (5.2 g; 52 mmol) were dissolved in dichloromethane (500 ml) and the mixture was chilled in an ice bath. (S)-3-(6,8-Dichloro-2-methyl-l,2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonyl chloride hydrochloride (10 g; 26 mmol) was added in parts during 40 minutes to the chilled reaction mixture. The ice bath was removed and the reaction mixture was stirred at laboratory temperature for additional 30 minutes.
The dichloromethane solution was extracted three times by brine (2x 250 ml), dried over sodium sulphate, and concentrated in vacuo. The residue was purified using preparative chromatography on silica gel with dichloromethane-methanol mobile phase.
Yield 7.2 g. HRMS 502.1247 [M+H]+, C22H29CI2N3O4S.
Step D
Preparation of 17-[[[3-[(4S)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulphonyl]amino]-N-[2-[2-[2-[[[3-[(4S)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl -4-isoquinolinyl] phenyl] sulphonyl] amino] ethoxy] ethoxy ] ethyl] – 8 -oxo- 12,15 -dioxa-2 ,7,9-triazah
(S)-N-(2-(2-(2-aminoethoxy)ethoxy)ethyl)-3-(6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonamide (5g; 10 mmol) prepared in step A was dissolved in dichloromethane (50 ml). Triethylamine (1.5 g; 14.9 mmol) and 1 ,4-diisocyanatobutane (0.48 g; 3.4 mmol) were added to the solution. The reaction mixture was cooled using ice and stirred overnight. The resulting fine suspension was filtered off, the filtrate was concentrated and the obtained product was purified by preparative chromatography on on silica gel with dichloromethane-methanol mixture as a mobile phase
Yield: 2 g of tenapanor in the form of amorphous solid foam. HPLC purity 96.5 %.
HRMS 1143.3186 [M+H]+, C5oH66Cl4N8010S2. *H NMR (500MHz, DMSO, ppm):7.69-7.66 (m, 6H), 7.54-7.50 (m, 6H), 6.89 (bs, 2H), 5.9 (t, 2H), 5.79 (t, 2H), 4.4 (dd, 2H), 3.7 (dd, 4H), 3.44-3.44 (m, 8H), 3.35 (dd, 8H), 3.12 (dd, 4H), 2.96-2.64 (m, 12H), 2.37 (s, 6H), 1.31 (bs, 4H).
Ste E
Preparation of bishydrochloride salt of tenapanor
Tenapanor free base (1 g; 0.85 mmol) prepared in step B was dissolved in a mixture of methanol (10 ml) and 4M aqueous HCl (0.5 ml; 2 mmol) under mild reflux. The solution was concentrated on rotary vacuum evaporator, and the title product was obtained in the yield of 1 g of amorphous solid foam.
Example 1
Preparation of tenapanor, crystalline form I
Tenapanor free base (200 mg, 0.17 mmol), prepared as in step D of the comparative example, was dissolved in 0.4 ml acetonitrile under mild reflux. The clear solution was cooled at the rate of 1 °C/min with stirring to laboratory temperature (i.e., range from 22 °C to 26 °C) and then stirred for additional 2 hours at this temperature. The resulting crystals were isolated by filtration on sintered glass filter and dried for 6 hours in a vacuum oven at 40 °C. Crystallization yield was 170 mg of crystalline form I of tenapanor. HPLC showed a purity of 99.5%.
Examples 4 to 9 illustrate the inventive method of preparation of crystalline tenapanor.
Example 4
Preparation of (5)- -(3-(benzylthio)phenyl)-6,8-dichloro-2-methyl-l ,2,3,4-tetrahydroisoquinoline
DIPEA (9.6 mL) and anhydrous dioxane (100 mL) were added to a reaction vessel. Benzyl mercaptan (8.1 ml) was added dropwise to the stirred mixture under ice -cooling. The resulting mixture was stirred at 25 °C for lh.
In a second reaction vessel, (S)-4-(3-bromophenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline (21.2 g) in anhydrous dioxane (140 mL), Pd2(dba)3 (835 mg)and Xantphos (835 mg) were mixed. The resulting solution was stirred at 25 °C for 30 minutes and then added to the solution of benzyl mercaptan. The resulting reaction mixture was maintained at gentle reflux for 3 hours.
After cooling, the suspension obtained was filtered through a thin layer of celite. HC1 was added to the filtrate. The precipitated hydrochloride was isolated by filtration, washed well and dried. 21 g of pinkish product were obtained (81.6% yield).
Example 5
Preparation of (5) -3 -(6 , 8 -dichloro-2 -methyl- 1,2,3 ,4-tetr ahydroisoquinolin-4-yl)benzenesulf onyl chloride hydrochlorid
(S)-4-(3-(benzylthio)phenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline hydrochloride (11.1 g) was stirred in DCM/2M HC1 (70 mL:6 mL) mixture in a reaction vessel. The mixture was cooled in an ice bath and then gaseous Cl2 was introduced into the vigorously stirred mixture. After disappearance of the starting material, the resulting suspension was bubbled through by nitrogen and the product was filtered off and washed with DCM. 9.2 g of white product was obtained (82.7% yield).
Example 6
In the reaction vessel, t-butyl 2-(2-(2-amionoethoxy)ethoxy)ethylcarbamate (21.8 g) was stirred in DCM. The mixture was cooled in an ice bath under an inert atmosphere. To the cooled solution was
added 1 ,4-diisocyanatobutane (6.14 g) and TEA (0.1 mL). The cooling bath was removed and the reaction mixture was further stirred for 2 h.
35% HCl was added to the reaction mixture and the mixture was stirred under gentle reflux overnight.
After cooling, the precipitated product was filtered off and washed with DCM.
The product was recrystallized from propan-2-ol. 22.3 g of white product was obtained (80% yield).
Example 7
Preparation of (5)-N-(2-(2-(2-aminoethoxy)ethoxy)ethyl)-3-(6,8-dichloro-2-methyl-l , 2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonamide
(S)-3-(6,8-dichloro-2-methyl-l ,2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonyl chloride hydrochloride (11.7 g) prepared in Example 2 was stirred in dichloromethane (100 ml) and the suspension was cooled in an ice bath. To the cooled suspension was added a solution of t-butyl 2-(2-(2-amionoethoxy)ethoxy)ethylcarbamate (6.8 g) and DIPEA (14 ml) in DCM (50 ml). The resulting solution was stirred for 2 hours in an ice bath. The reaction mixture was extracted twice with water. Concentrated HCl (15 mL) was added to the dichloromethane solution and the mixture heated at gentle reflux for 2 h.
The precipitated product, after cooling, was extracted into water. The aqueous phase was separated and basified with Na2C03. The product as the free base was extracted into DCM and the dichloromethane solution was dried over sodium sulfate and concentrated in vacuo. 12.9 g of product were obtained.
Yield 93.4%. HRMS 502.1247 [M+H]+, C22H29CI2N3O4S.
Example 8
Preparation of 17-[[[3-[(45)-6,8-dichloro-l ,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl] sulfonyl]amino]-N-[2-[2-[2-[[[3-[(45)-6,8-dichloro-l ,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl] phenyl] sulf onyl] amino] ethoxy ] ethoxy ] ethyl] – 8 -oxo- 12,15 -dioxa-2 ,7 ,9-triazaheptadecanamide (tenapanor free base)
(S)-N-(2-(2-(2-aminoethoxy)ethoxy)ethyl)-3-(6,8-dichloro-2-methyl-l,2,3,4- tetrahydroisoquinolin- 4-yl)benzenesulfonamide (12.9 g) prepared in Example 4 was dissolved in dichloromethane (150 ml). To the solution was added triethylamine (0.3 ml) and 1,4-diisocyanatobutane (1.7 g). The reaction mixture was stirred at 25 °C for 2 h. The resulting reaction mixture was extracted with water and aqueous Na2C03. The dichloromethane solution of the product was dried over sodium sulfate and concentrated to a solid foam. Yield 13.9 g. The crude product was taken up in acetone (100 ml) and then recrystallized from methanol (80 ml). 7.3 g of white crystalline product was obtained. Yield 49.8%.
HRMS 1143.3186 [M+H]+, C5oH66Cl4N8010S2. !H NMR (500MHz, DMSO, ppm):7.69-7.66 (m, 6H), 7.54-7.50 (m, 6H), 6.89 (bs, 2H), 5.9 (t, 2H), 5.79 (t, 2H), 4.4 (dd, 2H), 3.7 (dd, 4H), 3.44-3.44 (m, 8H), 3.35 (dd, 8H), 3.12 (dd, 4H), 2.96-2.64 (m, 12H), 2.37 (s, 6H), 1.31 (bs, 4H)
Example 9
Preparation of 17-[[[3-[(45)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl] sulfonyl]amino]-N-[2-[2-[2-[[[3-[(45)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl] phenyl] sulf onyl] amino] ethoxy ] ethoxy ] ethyl] – 8 -oxo- 12,15 -dioxa-2 ,7 ,9-
(S)-3-(6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonyl chloride hydrochloride (0.81 g) prepared in Example 2 and l,l’-(butane-l,4-diyl)bis(3-(2-(2-(2-aminoethoxy)ethoxy)ethyl)urea) dihydrochloride prepared according to Example 3 (0.48 g) were stirred in anhydrous ΝΜΡ (10 ml). To the suspension was added DIPEA (2 mL) and the resulting solution was stirred at 60 °C for 1.5 h. Water (10 mL) was added dropwise to the reaction mixture and the mixture was cooled to 5 °C. The precipitated product was isolated and stirred in acetone at 5 °C overnight. The beige product was filtered off (0.67 g) and recrystallized from methanol (12 ml).
0.53 g of a colorless crystalline product was obtained.
Yield 78.7 %. HRMS 502.1247 [M+H]+, C22H29CI2N3O4S. DSC analysis showed the melting temperature of 130.5 °C.
Example 10
Tenapanor (1.48 g, 1.3 mmol) is dissolved in 10 ml of tetrahydrofurane (THF). From the thus prepared solution, 1 ml is taken and phosphoric acid (0.4 mmol) is added. The mixture is stirred at room temperature for 24 hours. Salt of tenapanor with phosphoric acid precipitated from the solution in solid stable form, the salt was filtered off, washed with THF and dried by stream of inert gas. XRPD confirmed amorphousness of the product.
Example 11
Tenapanor (1.48 g, 1.3 mmol) is dissolved in 10 ml of tetrahydrofurane (THF). From the thus prepared solution, 1 ml is taken and hydrobromic acid (0.4 mmol) is added. The mixture is stirred at room temperature for 24 hours. Salt of tenapanor with hydrobromic acid precipitated from the solution in solid stable form, the salt was filtered off, washed with THF and dried by stream of inert gas. XRPD confirmed amorphousness of the product.
Example 12
Tenapanor (1.48 g, 1.3 mmol) is dissolved in 10 ml of acetone. From the thus prepared solution, 1 ml is taken and phosphoric acid (0.4 mmol) is added. The mixture is stirred at room temperature for 24 hours. Salt of tenapanor with phosphoric acid precipitated from the solution in solid stable form, the salt was filtered off, washed with acetone and dried by stream of inert gas. XRPD confirmed amorphousness of the product.
Example 13
Tenapanor (1.48 g, 1.3 mmol) is dissolved in 10 ml of acetone. From the thus prepared solution, 1 ml is taken and citric acid (0.4 mmol) is added. The mixture is stirred at room temperature for 24 hours. Salt of tenapanor with citric acid precipitated from the solution in solid stable form, the salt was filtered off, washed with acetone and dried by stream of inert gas. XRPD confirmed amorphousness of the product.
Other pharmaceutically acceptable acids were tested by the procedures shown in Examples 10-13, but did not yield salts which would precipitate in amorphous stable solid form from the solution. The tested acids were: methanesulfonic acid, benzenesulfonic acid, oxalic acid, maleinic acid, tartaric acid, fumaric acid, trichloroacetic acid.
Example 14
Tenapanor (500 mg, 0.44 mmol) is dissolved in 20 ml of THF at 45 °C. To this clear solution, a solution of phosphoric acid in THF (50 μ1/5 ml) is added dropwise during 10 minutes. The resulting suspension is stirred at room temperature for 30 minutes. The precipitated salt of tenapanor with phosph (79 %) oric is filtered off, washed with 3 ml of THF and dried by stream of inert gas. Yield: 430 mg of colourless salt of tenapanor with phosphoric acid. XRPD showed amorphousness of the product.
Example 15
Tenapanor (500 mg, 0.44 mmol) is dissolved in 20 ml of THF at 45 °C. To this clear solution, hydrobromic acid (48%; 100 μΐ) is added dropwise during 10 minutes. A fine precipitate forms already during the dropwise addition of HBr, and the suspension is stirred at room temperature for 30 minutes. The precipitated salt of tenapanor with HBr is filtered off, washed with 3 ml of THF and dried by stream of inert gas. Yield: 397 mg (69 %) of colourless salt of tenapanor with HBr (1 :2). XRPD showed amorphousness of the product.
References
- ^ Spencer AG, Labonte ED, Rosenbaum DP, Plato CF, Carreras CW, Leadbetter MR, Kozuka K, Kohler J, Koo-McCoy S, He L, Bell N, Tabora J, Joly KM, Navre M, Jacobs JW, Charmot D (2014). “Intestinal inhibition of the na+/h+ exchanger 3 prevents cardiorenal damage in rats and inhibits na+ uptake in humans”. Sci Transl Med. 6 (227): 227ra36. doi:10.1126/scitranslmed.3007790. PMID 24622516.
- ^ Salt-buster drug cuts sodium absorbed from food. New Scientist, 14 March 2014
REFERENCES
1: Johansson SA, Knutsson M, Leonsson-Zachrisson M, Rosenbaum DP. Effect of Food Intake on the Pharmacodynamics of Tenapanor: A Phase 1 Study. Clin Pharmacol Drug Dev. 2017 Mar 24. doi: 10.1002/cpdd.341. [Epub ahead of print] PubMed PMID: 28339149.
2: Johansson S, Rosenbaum DP, Ahlqvist M, Rollison H, Knutsson M, Stefansson B, Elebring M. Effects of Tenapanor on Cytochrome P450-Mediated Drug-Drug Interactions. Clin Pharmacol Drug Dev. 2017 Mar 16. doi: 10.1002/cpdd.346. [Epub ahead of print] PubMed PMID: 28301096.
3: Chey WD, Lembo AJ, Rosenbaum DP. Tenapanor Treatment of Patients With Constipation-Predominant Irritable Bowel Syndrome: A Phase 2, Randomized, Placebo-Controlled Efficacy and Safety Trial. Am J Gastroenterol. 2017 Feb 28. doi: 10.1038/ajg.2017.41. [Epub ahead of print] PubMed PMID: 28244495.
4: Carney EF. Dialysis: Efficacy of tenapanor in hyperphosphataemia. Nat Rev Nephrol. 2017 Apr;13(4):194. doi: 10.1038/nrneph.2017.27. PubMed PMID: 28239171.
5: Block GA, Rosenbaum DP, Leonsson-Zachrisson M, Åstrand M, Johansson S, Knutsson M, Langkilde AM, Chertow GM. Effect of Tenapanor on Serum Phosphate in Patients Receiving Hemodialysis. J Am Soc Nephrol. 2017 Feb 3. pii: ASN.2016080855. doi: 10.1681/ASN.2016080855. [Epub ahead of print] PubMed PMID: 28159782.
6: Koliani-Pace J, Lacy BE. Update on the Management of Chronic Constipation. Curr Treat Options Gastroenterol. 2017 Mar;15(1):126-134. doi: 10.1007/s11938-017-0118-2. Review. PubMed PMID: 28116695.
7: Charoenphandhu N, Kraidith K, Lertsuwan K, Sripong C, Suntornsaratoon P, Svasti S, Krishnamra N, Wongdee K. Na(+)/H(+) exchanger 3 inhibitor diminishes hepcidin-enhanced duodenal calcium transport in hemizygous β-globin knockout thalassemic mice. Mol Cell Biochem. 2017 Mar;427(1-2):201-208. doi: 10.1007/s11010-016-2911-y. PubMed PMID: 27995414.
8: Thammayon N, Wongdee K, Lertsuwan K, Suntornsaratoon P, Thongbunchoo J, Krishnamra N, Charoenphandhu N. Na(+)/H(+) exchanger 3 inhibitor diminishes the amino-acid-enhanced transepithelial calcium transport across the rat duodenum. Amino Acids. 2017 Apr;49(4):725-734. doi: 10.1007/s00726-016-2374-1. PubMed PMID: 27981415.
9: Afsar B, Vaziri ND, Aslan G, Tarim K, Kanbay M. Gut hormones and gut microbiota: implications for kidney function and hypertension. J Am Soc Hypertens. 2016 Dec;10(12):954-961. doi: 10.1016/j.jash.2016.10.007. Review. PubMed PMID: 27865823.
10: Johansson S, Leonsson-Zachrisson M, Knutsson M, Spencer AG, Labonté ED, Deshpande D, Kohler J, Kozuka K, Charmot D, Rosenbaum DP. Preclinical and Healthy Volunteer Studies of Potential Drug-Drug Interactions Between Tenapanor and Phosphate Binders. Clin Pharmacol Drug Dev. 2016 Sep 22. doi: 10.1002/cpdd.307. [Epub ahead of print] PubMed PMID: 27654985.
11: Ketteler M, Liangos O, Biggar PH. Treating hyperphosphatemia – current and advancing drugs. Expert Opin Pharmacother. 2016 Oct;17(14):1873-9. doi: 10.1080/14656566.2016.1220538. Review. PubMed PMID: 27643443.
12: Johansson S, Rosenbaum DP, Knutsson M, Leonsson-Zachrisson M. A phase 1 study of the safety, tolerability, pharmacodynamics, and pharmacokinetics of tenapanor in healthy Japanese volunteers. Clin Exp Nephrol. 2016 Jul 1. [Epub ahead of print] PubMed PMID: 27368672.
13: Block GA, Rosenbaum DP, Leonsson-Zachrisson M, Stefansson BV, Rydén-Bergsten T, Greasley PJ, Johansson SA, Knutsson M, Carlsson BC. Effect of Tenapanor on Interdialytic Weight Gain in Patients on Hemodialysis. Clin J Am Soc Nephrol. 2016 Sep 7;11(9):1597-605. doi: 10.2215/CJN.09050815. PubMed PMID: 27340281; PubMed Central PMCID: PMC5012484.
14: Nusrat S, Miner PB Jr. New pharmacological treatment options for irritable bowel syndrome with constipation. Expert Opin Emerg Drugs. 2015;20(4):625-36. doi: 10.1517/14728214.2015.1105215. Review. PubMed PMID: 26548544.
15: Spencer AG, Greasley PJ. Pharmacologic inhibition of intestinal sodium uptake: a gut centric approach to sodium management. Curr Opin Nephrol Hypertens. 2015 Sep;24(5):410-6. doi: 10.1097/MNH.0000000000000154. Review. PubMed PMID: 26197202.
16: Zielińska M, Wasilewski A, Fichna J. Tenapanor hydrochloride for the treatment of constipation-predominant irritable bowel syndrome. Expert Opin Investig Drugs. 2015;24(8):1093-9. doi: 10.1517/13543784.2015.1054480. Review. PubMed PMID: 26065434.
17: Thomas RH, Luthin DR. Current and emerging treatments for irritable bowel syndrome with constipation and chronic idiopathic constipation: focus on prosecretory agents. Pharmacotherapy. 2015 Jun;35(6):613-30. doi: 10.1002/phar.1594. Review. PubMed PMID: 26016701.
18: Gerritsen KG, Boer WH, Joles JA. The importance of intake: a gut feeling. Ann Transl Med. 2015 Mar;3(4):49. doi: 10.3978/j.issn.2305-5839.2015.03.21. PubMed PMID: 25861604; PubMed Central PMCID: PMC4381464.
19: Labonté ED, Carreras CW, Leadbetter MR, Kozuka K, Kohler J, Koo-McCoy S, He L, Dy E, Black D, Zhong Z, Langsetmo I, Spencer AG, Bell N, Deshpande D, Navre M, Lewis JG, Jacobs JW, Charmot D. Gastrointestinal Inhibition of Sodium-Hydrogen Exchanger 3 Reduces Phosphorus Absorption and Protects against Vascular Calcification in CKD. J Am Soc Nephrol. 2015 May;26(5):1138-49. doi: 10.1681/ASN.2014030317. PubMed PMID: 25404658; PubMed Central PMCID: PMC4413764.
20: Spencer AG, Labonte ED, Rosenbaum DP, Plato CF, Carreras CW, Leadbetter MR, Kozuka K, Kohler J, Koo-McCoy S, He L, Bell N, Tabora J, Joly KM, Navre M, Jacobs JW, Charmot D. Intestinal inhibition of the Na+/H+ exchanger 3 prevents cardiorenal damage in rats and inhibits Na+ uptake in humans. Sci Transl Med. 2014 Mar 12;6(227):227ra36. doi: 10.1126/scitranslmed.3007790. PubMed PMID: 24622516.
 |
|
| Clinical data | |
|---|---|
| Routes of administration |
Oral |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.243.471 |
| Chemical and physical data | |
| Formula | C50H66Cl4N8O10S2 |
| Molar mass | 1145.046 g/mol g·mol−1 |
| 3D model (JSmol) | |
//////////////Tenapanor, AZD 1722, RDX 5791, chronic kidney disease, hypertension
CN1CC(C2=CC(=CC(=C2C1)Cl)Cl)C3=CC(=CC=C3)S(=O)(=O)NCCOCCOCCNC(=O)NCCCCNC(=O)NCCOCCOCCNS(=O)(=O)C4=CC=CC(=C4)C5CN(CC6=C(C=C(C=C56)Cl)Cl)C
Etosalamide, этосаламид , إيتوسالاميد , 依托柳胺 ,
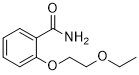
Cas 15302-15-5
Chemical Formula: C11H15NO3
Molecular Weight: 209.245
o-(2-Ethoxyethoxy)benzamide
Etosalamide, also known as Ethosalamide, is an antipyretic and analgesics agent
SYN

OR

CAS:592-55-2, 2-Bromoethyl ethyl ether
Cas, 611-20-1, 2-Hydroxybenzonitrile

PATENT
DE 1013643
PATENT
GB 774635
PATENT
US2822391
78 – 79 MP
PAPER
Journal of Chemical and Engineering Data (1962), 7, 265-6
70 – 71.5 MP
PATENT
WO 2004003198
US 20100226943
/////////Etosalamide, этосаламид , إيتوسالاميد , 依托柳胺 , ethosalamide
O=C(N)C1=CC=CC=C1OCCOCC
KETOROLAC
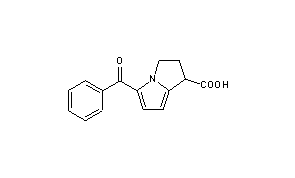

Ketorolac
- Molecular FormulaC15H13NO3
- Average mass255.269 Da
Ketorolac, sold under the brand name Toradol among others, is a nonsteroidal anti-inflammatory drug (NSAID) used to treat pain.[1]Specifically it is recommended for moderate to severe pain.[2] Recommended duration of treatment is less than six days.[1] It is used by mouth, by injection into a vein or muscle, and as eye drops.[1][2] Effects begin within an hour and last for up to eight hours.[1]
Common side effects include sleepiness, dizziness, abdominal pain, swelling, and nausea.[1] Serious side effects may include stomach bleeding, kidney failure, heart attacks, bronchospasm, heart failure, and anaphylaxis.[1] Use is not recommended during the last part of pregnancy or during breastfeeding.[1] Ketorolac works by blocking cyclooxygenase 1 and 2 (COX1 and COX2) thereby decreasing prostaglandins.[1][3]
Ketorolac was patented in 1976 and approved for medical use in 1989.[4][1] It is avaliable as a generic medication.[2] In the United Kingdom it costs the NHS less than a £ per injectable dose as of 2019.[2] In the United States the wholesale cost of this amount is about 1.50 USD.[5] In 2016 it was the 296th most prescribed medication in the United States with more than a million prescriptions.[6]
Medical uses
Ketorolac is used for short-term management of moderate to severe pain.[7]It is usually not prescribed for longer than five days.[8][9][10][11] Ketorolac is effective when administered with paracetamol to control pain in neonates because it does not depress respiration as do opioids.[12] Ketorolac is also an adjuvant to opioid medications and improves pain relief. It is also used to treat dysmenorrhea.[11] Ketorolac is used to treat idiopathic pericarditis, where it reduces inflammation.[13]
Ketorolac is used for short-term pain control not lasting longer than five days, and can be administered orally, by intramuscular injection, intravenously, and by nasal spray.[8] Ketorolac is initially administered by intramuscular injection or intravenously.[7] Oral therapy is only used as a continuation from the intramuscular or intravenous starting point.[8][12]
Ketorolac is used during eye surgery help with pain.[14] Ketorolac is effective in treating ocular itching.[15] The ketorolac ophthalmic formulation is associated with a decreased development of macular edema after cataract surgery and is more effective alone rather than as an opioid/ketorolac combination treatment.[16][17] Ketorolac has also been used to manage pain from corneal abrasions.[18]
During treatment with ketorolac, clinicians monitor for the manifestation of adverse effects and side effects. Lab tests, such as liver function tests, bleeding time, BUN, serum creatinine and electrolyte levels are often used and help to identify potential complications.[8][9]
Contraindications
Ketorolac is contraindicated in those with hypersensitivity, allergies to the medication, cross-sensitivity to other NSAIDs, prior to surgery, history of peptic ulcer disease, gastrointestinal bleeding, alcohol intolerance, renal impairment, cerebrovascular bleeding, nasal polyps, angioedema, and asthma.[8][9] Recommendations exist for cautious use of ketorolac in those who have experienced cardiovascular disease, myocardial infarction, stroke, heart failure, coagulation disorders, renal impairment, and hepatic impairment.[8][9]
Adverse effects
Though uncommon, potentially fatal adverse effects are stroke, myocardial infarction, GI bleeding, Stevens-Johnson Syndrome, toxic epidermal necrolysis and anaphylaxis. A less serious and more common (>10%) side effect is drowsiness. Infrequent (<1%) side effects are paresthesia, prolonged bleeding time, injection site pain, purpura, sweating, abnormal thinking, increased production of tears, edema, pallor, dry mouth, abnormal taste, urinary frequency, increased liver enzymes, itching and others. Ketorolac can cause premature constriction of the ductus arteriosis in an infant during the third trimester of pregnancy.[8][9] Platelet function is decreased related to the use of ketorolac.[19]
The practice of restricting treatment with ketorolac is due to its potential to cause kidney damage.[20]
Interactions
Ketorolac can interact with other medications. Probenecid can increase the probability of having an adverse reaction or experiencing a side effect when taken with ketorolac. Pentoxifylline can increase the risk of bleeding. When aspirin is taken at the same time as ketorolac, the effectiveness is decreased. Problematic GI effects are additive and become more likely if potassium supplements, aspirin, other NSAIDS, corticosteroids, or alcohol is taken at the same time. The effectiveness of antihypertensives and diuretics can be lowered. The use of ketorolac can increase serum lithium levels to the point of toxicity. Toxicity to methotrexate is more likely if ketorolac is taken at the same time. The risk of bleeding increases with the concurrent medications clopidogrel, cefoperazone, valproic acid, cefotetan, eptifibatide, tirofiban, and copidine. Anticoagulants and thrombolytic medications also increase the likelihood of bleeding. Medications used to treat cancer can interact with ketorolac along with radiation therapy. The risk of toxicity to the kidneys increases when ketorolac is taken with cyclosporine.[8][9]
Interactions with ketorolac exist with some herbal supplements. The use of Panax ginseng, clove, ginger, arnica, feverfew, dong quai, chamomile, and Ginkgo biloba increases the risk of bleeding.[8][9]
Mechanism of action
The primary mechanism of action responsible for ketorolac’s anti-inflammatory, antipyretic and analgesic effects is the inhibition of prostaglandin synthesis by competitive blocking of the enzyme cyclooxygenase (COX). Ketorolac is a non-selective COX inhibitor.[21] Ketorolac has been assessed to be a relatively higher risk NSAID when compared to aceclofenac, celecoxib, and ibuprofen.[13] It is considered a first-generation NSAID.[19]
History
In the US, ketorolac was the only widely available intravenous NSAID for many years; an IV form of paracetemol, which is not an NSAID, became available in Europe in 2009 and then in the US.[12]
The Syntex company, of Palo Alto, California developed the ophthalmic solution Acular around 2006.[citation needed]
In 2007, there were concerns about the high incidence of reported side effects. This led to restriction in its dosage and maximum duration of use. In the UK, treatment was initiated only in a hospital, although this was not designed to exclude its use in prehospital care and mountain rescue settings.[7] Dosing guidelines were published at that time.[22]
Concerns over the high incidence of reported side effects with ketorolac trometamol led to its withdrawal (apart from the ophthalmic formulation) in several countries, while in others its permitted dosage and maximum duration of treatment have been reduced. From 1990 to 1993, 97 reactions with a fatal outcome were reported worldwide.[23]
The eye-drop formulation was approved by the FDA in 1992.[24] An intranasal formulation was approved by the FDA in 2010[25] for short-term management of moderate to moderately severe pain requiring analgesia at the opioid level.
Synthesis
DOI: 10.1021/jo00348a014

1H-Pyrrolizine-1-carboxylic acid, 2,3-dihydro-5-benzoyl-, (+-)-, could be produced through many synthetic methods.
Following is one of the reaction routes:

2-Methylthiopyrrole (I) is benzoylated with N,N-dimethylbenzamide (II) to produce 5-benzoyl-2-methylthiopyrrole (III) in the presence of POCl3 in refluxing CH2Cl2, and the yielding product is condensed with spiro[2.5]-5,7-dioxa-6,6-dimethyloctane-4,8-dione (IV) in the presence of NaH in DMF giving compound (V). The oxidation of (V) with m-chloroperbenzoic acid in CH2Cl2affords the sulfone (VI), which is submitted to methanolysis with methanol and HCl giving 1-(3,3-dimethoxycarbonylpropyl)-2-methanesulfonyl-5-benzoylpyrrole (VII). The cyclization of (VII) with NaH in DMF yields dimethyl 5-benzoyl-1,2-dihydro-3H-pyrrolo[1,2-a]pyrrole-1,1-dicarboxylate (VIII), which is finally hydrolyzed and decarboxylated with KOH in refluxing methanol.Compound (III) can be oxidized with m-chloroperbenzoic acid as before giving 2-methanesulfonyl-5-benzoylpyrrole (IX), which is then condensed with spiro compound (IV) as before to afford compound (VI), already obtained.
SYN
DE 2731678; ES 460706; ES 470214; FR 2358406; FR 2375234; GB 1554075
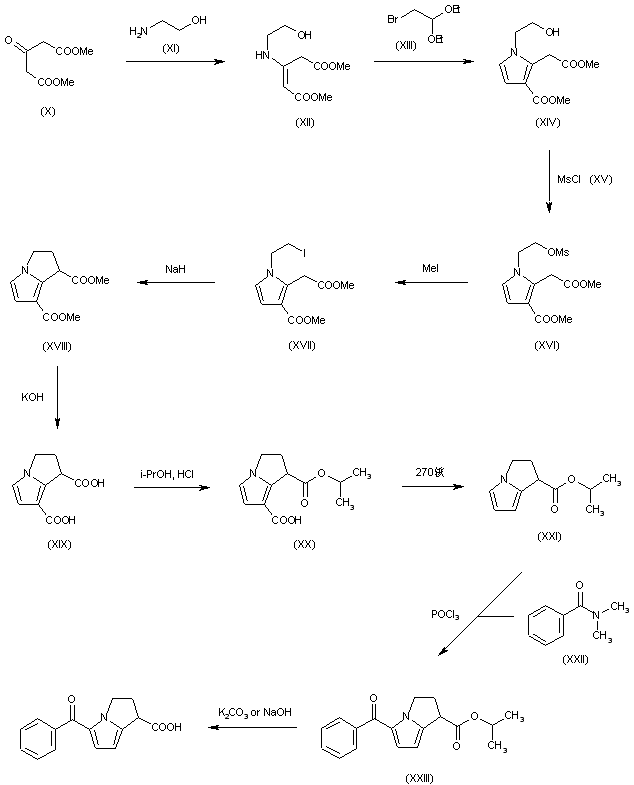
The condensation of dimethylacetone-1,3-dicarboxylate (X) with ethanolamine (XI) yields methyl 3-(methoxycarbonylmethyl)-3-(2-hydroxyethylamino)acrylate (XII), which is cyclized with bromoacetaldehyde diethylacetal (XIII) affording methyl 1-(2-hydroxyethyl)-3-methoxycarbonylpyrrol-2-acetate (XIV). Acylation of (XIV) with methanesulfonyl chloride (XV) and triethylamine in CH2Cl2 yields the corresponding mesylate (XVI), which by treatment with methyl iodide in refluxing acetonitrile is converted into methyl 1-(2-iodoethyl)-3-methoxycarbonylpyrrole-2-acetate (XVII). The cyclization of (XVII) with NaH in DMF yields dimethyl 1,2-dihydro-3H-pyrrolo[1,2-a]pyrrole-1,7-dicarboxylate (XVIII), which is hydrolyzed with KOH in refluxing methanol – water to the corresponding diacid (XIX). Partial esterification of (XIX) with isopropanol and HCl gives isopropyl 1,2-dihydro-3H-7-carboxypyrrolo[1,2-a]pyrrole-1-carboxylate (XX), which is decarboxylated by heating at 270 C affording isopropyl 1,2-dihydro-3H-pyrrolo[1,2-a]pyrrole-1-carboxylate (XXI). Benzoylation of (XXI) with N,N-dimethylbenzamide (XXII) and POCl3 in refluxing CH2Cl2 yields isopropyl 5-benzoyl-1,2-dihydro-3H-pyrrolo[1,2-a]pyrrole-1-carboxylate (XXIII), which is finally hydrolyzed with K2CO3 or NaOH in methanol – water.
SYN2
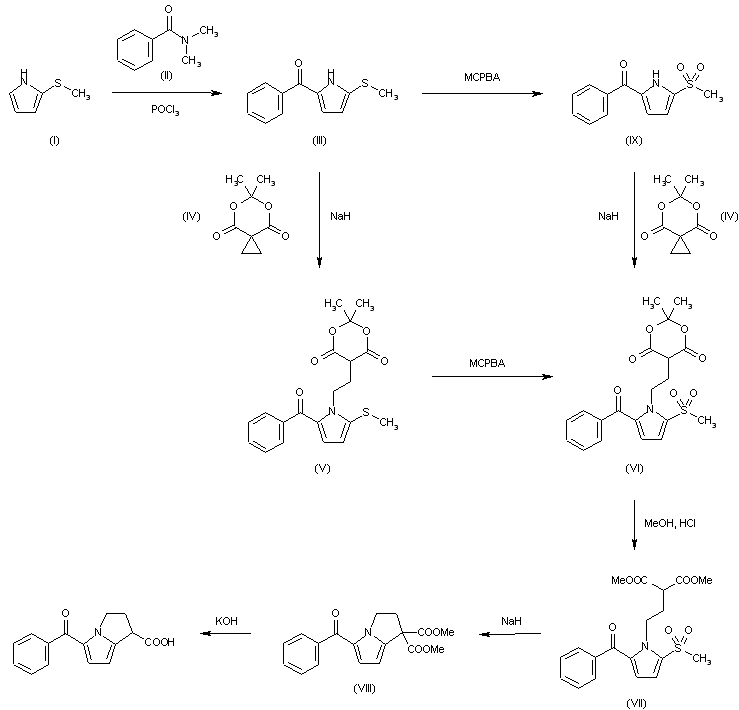
The benzoylation of 2-methylthiopyrrole (I) with N,N-dimethylbenzamide (II) by means of POCl3 in refluxing CH2Cl2 gives 5-benzoyl-2-methylthiopyrrole (III), which is condensed with spiro[2.5]-5,7-dioxa-6,6-dimethyloctane-4,8-dione (IV) by means of NaH in DMF yielding compound (V). The oxidation of (V) with m-chloroperbenzoic acid in CH2Cl2 affords the sulfone (VI), which is submitted to methanolysis with methanol and HCl giving 1-(3,3-dimethoxycarbonylpropyl)-2-methanesulfonyl-5-benzoylpyrrole (VII). The cyclization of (VII) with NaH in DMF yields dimethyl 5-benzoyl-1,2-dihydro-3H-pyrrolo[1,2-a]pyrrole-1,1-dicarboxylate (VIII), which is finally hydrolyzed and decarboxylated with KOH in refluxing methanol. Compound (III) can be oxidized with m-chloroperbenzoic acid as before giving 2-methanesulfonyl-5-benzoylpyrrole (IX), which is then condensed with spiro compound (IV) as before to afford compound (VI), already obtained.
References
- ^ Jump up to:a b c d e f g h i “Ketorolac Tromethamine Monograph for Professionals”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 13 April 2019.
- ^ Jump up to:a b c d British national formulary : BNF 76 (76 ed.). Pharmaceutical Press. 2018. pp. 1144, 1302–1303. ISBN 9780857113382.
- ^ “DailyMed – ketorolac tromethamine tablet, film coated”. dailymed.nlm.nih.gov. Retrieved 14 April 2019.
- ^ Fischer, Jnos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 521. ISBN 9783527607495.
- ^ “NADAC as of 2019-02-27”. Centers for Medicare and Medicaid Services. Retrieved 3 March 2019.
- ^ “The Top 300 of 2019”. clincalc.com. Retrieved 22 December 2018.
- ^ Jump up to:a b c Mallinson, Tom (2017). “A review of ketorolac as a prehospital analgesic”. Journal of Paramedic Practice. 9 (12): 522–526. doi:10.12968/jpar.2017.9.12.522. Retrieved 2 June 2018.
- ^ Jump up to:a b c d e f g h i Vallerand, April H. (2017). Davis’s Drug Guide for Nurses. Philadelphia: F.A. Davis Company. p. 730. ISBN 9780803657052.
- ^ Jump up to:a b c d e f g Physician’s Desk Reference 2017. Montvale, New Jersey: PDR, LLC. 2017. pp. S–474–5. ISBN 9781563638381.
- ^ “Ketorolac-tromethamine”. The American Society of Health-System Pharmacists. Retrieved 3 April 2011.
- ^ Jump up to:a b Henry, p. 291.
- ^ Jump up to:a b c Martin, Lizabeth D; Jimenez, Nathalia; Lynn, Anne M (2017). “A review of perioperative anesthesia and analgesia for infants: updates and trends to watch”. F1000Research. 6: 120. doi:10.12688/f1000research.10272.1. ISSN 2046-1402. PMC 5302152. PMID 28232869.
- ^ Jump up to:a b Schwier, Nicholas; Tran, Nicole (2016). “Non-Steroidal Anti-Inflammatory Drugs and Aspirin Therapy for the Treatment of Acute and Recurrent Idiopathic Pericarditis”. Pharmaceuticals. 9 (2): 17. doi:10.3390/ph9020017. ISSN 1424-8247. PMC 4932535. PMID 27023565.
- ^ Saenz-de-Viteri, Manuel; Gonzalez-Salinas, Roberto; Guarnieri, Adriano; Guiaro-Navarro, María Concepción (2016). “Patient considerations in cataract surgery – the role of combined therapy using phenylephrine and ketorolac”. Patient Preference and Adherence. 10: 1795–1801. doi:10.2147/PPA.S90468. ISSN 1177-889X. PMC 5029911. PMID 27695298.
- ^ Karch, Amy (2017). Focus on nursing pharmacology. Philadelphia: Wolters Kluwer. p. 272. ISBN 9781496318213.
- ^ Lim, Blanche X; Lim, Chris HL; Lim, Dawn K; Evans, Jennifer R; Bunce, Catey; Wormald, Richard; Wormald, Richard (2016). “Prophylactic non-steroidal anti-inflammatory drugs for the prevention of macular oedema after cataract surgery”. Cochrane Database Syst Rev. 11: CD006683. doi:10.1002/14651858.CD006683.pub3. PMID 27801522.
- ^ Sivaprasad, Sobha; Bunce, Catey; Crosby-Nwaobi, Roxanne; Sivaprasad, Sobha (2012). “Non-steroidal anti-inflammatory agents for treating cystoid macular oedema following cataract surgery”. Cochrane Database Syst Rev (2): CD004239. doi:10.1002/14651858.CD004239.pub3. PMID 22336801.
- ^ Wakai A, Lawrenson JG, Lawrenson AL, Wang Y, Brown MD, Quirke M, Ghandour O, McCormick R, Walsh CD, Amayem A, Lang E, Harrison N (2017). “Topical non-steroidal anti-inflammatory drugs for analgesia in traumatic corneal abrasions”. Cochrane Database Syst Rev. 5: CD009781. doi:10.1002/14651858.CD009781.pub2. PMID 28516471.
- ^ Jump up to:a b Henry, p. 279.
- ^ Henry, p. 280.
- ^ Lee, I. O.; Seo, Y. (2008). “The Effects of Intrathecal Cyclooxygenase-1, Cyclooxygenase-2, or Nonselective Inhibitors on Pain Behavior and Spinal Fos-Like Immunoreactivity”. Anesthesia & Analgesia. 106 (3): 972–977, table 977 contents. doi:10.1213/ane.0b013e318163f602. PMID 18292448.
- ^ MHRA Drug Safety Update October 2007, Volume 1, Issue 3, pp 3-4.
- ^ Committee on the Safety of Medicines, Medicines Control Agency: Ketorolac: new restrictions on dose and duration of treatment. Current Problems in Pharmacovigilance:June 1993; Volume 19 (pages 5-8).
- ^ “Ketorolac ophthalmic medical facts from”. Drugs.com. Retrieved 2013-10-06.
- ^ “Sprix Information from”. Drugs.com. Retrieved 2013-10-06.
Bibliography
- AHFS drug information. Bethesda, MD: American Society of Health-System Pharmacists. 2011. ISBN 9781585282609.
- Hamilton, Richart (2015). Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. p. 9. ISBN 9781284057560.
- Handley, Dean A.; Cervoni, Peter; McCray, John E.; McCullough, John R. (1998). “Preclinical enantioselective pharmacology of (R)- and (S)- ketorolac”. J Clin Pharmacol. 38 (2 Suppl): 25S–35S. doi:10.1002/j.1552-4604.1998.tb04414.x. ISSN 0091-2700. PMID 9549656.
- Henry, Norma (2016). RN pharmacology for nursing : review module. Overland Park, KS: Assessment Technologies Institute. ISBN 9781565335738.
- Kizior, Robert (2017). Saunders nursing drug handbook 2017. St. Louis, MO: Elsevier. ISBN 9780323442916.
External links
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Toradol, Acular, Sprix, others |
| Synonyms | Ketorolac tromethamine |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a693001 |
| License data | |
| Pregnancy category |
|
| Routes of administration |
By mouth, IM, IV, eye drops |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 100% (All routes) |
| Metabolism | Liver |
| Elimination half-life | 3.5 h to 9.2 h, young adults; 4.7 h to 8.6 h, elderly (mean age 72) |
| Excretion | Kidney: 91.4% (mean) Biliary: 6.1% (mean) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| PDB ligand | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.110.314 |
| Chemical and physical data | |
| Formula | C15H13NO3 |
| Molar mass | 255.27 g/mol g·mol−1 |
| 3D model (JSmol) | |
| Chirality | Racemic mixture |
//////////Ketorolac,
