
Home » Uncategorized (Page 141)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
BC-3781, LEFAMULIN……A Pleuromutilin by Nabriva (Austria) in phase 2

BC-3781
Topical pleuromutilin antibiotic agent
Gram-positive, including MRSA, PHASE 2 COMPLETED
Nabriva (Austria)
SEE UPDATED POST AT https://newdrugapprovals.org/2014/10/10/nabrivas-lefamulin-bc-3781-receives-fda-fast-track-status-to-treat-cabp-and-absssi/ ………….C0NTAINS SYNTHESIS
BC-3781
The pleuromutilin BC-3781 belongs to the first generation of pleuromutilins to combine excellent oral
bioavailability with substantial activity against Gram-positive pathogens and atypicals as well as some
Gram-negative pathogens. In particular, BC-3781 is highly active against multi-drug resistant (MDR)
pathogens including methicillin resistant Staphylococcus aureus (MRSA), MDR Streptococcus pneumonia
(i.e. macrolide and quinolone resistance), and vancomycin resistant Enterococcus faecium. It is
characterized by excellent in vivo activities (e.g. pneumonia model), outstanding PK/PD parameters,
allowing once a day dosing, and a novel mode of action. BC-3781 is being developed for both oral and IV
administration and is intended for the treatment of serious multi-drug resistant skin & skin structure
infections (CSSI) and moderate to severe pneumonia (CAP, HAP etc).
Pleuromutilins have been known since 1951, but only entered the market![]() in 2007 with the approval of retapamulin for topical use. Until today, there are no pleuromutilins for systemic use approved in human clinical practice.
in 2007 with the approval of retapamulin for topical use. Until today, there are no pleuromutilins for systemic use approved in human clinical practice.
Nabriva is currently working on the development of new compounds is this class. The lead compound, BC-3781, if approved, will be the first pleuromutilin for systemic use in humans.
The compound shows potent in vitro activity against a large collection of staphylococci, streptococci, andE. faecium. When compared to linezolid and vancomycin, the compound shows greater overall potency againstS. aureus [121]. BC-3781 shows improved activity against most bacteria commonly associated with community-acquired respiratory tract infections, the compound is especially potent against S. pneumoniaincluding penicillin resistant strains. It also shows improved activity against H. influenza, M. catarrhalis, M. pneumoniae and C. pneumoniae.
BC-3781 is undergoing Phase I clinical trials for CAP and in March of 2011 has completed a Phase II clinical study comparing it to vancomycin for treatment of aBSSSI [119,120,121,122,123]. Nabriva Therapeutics AG announced that the cooperation with Forest Laboratories to develop the compound had elapsed, and that Nabriva retained all rights in BC-3781. The company informed that the product was Phase III ready and that it was seeking partners to continue further development [203].
Nabriva is also developing BC-7013 for topical use against Gram-positive infections and working on the discovery of new pleuromutilins [119,124].
Dr William Prince, CMO Nabriva Therapeutics commented:
“This is the first patient study with a systemic pleuromutilin. It will be an important proof of concept
for an exciting new class of antibiotics. The phase II study builds on our extensive preclinical and
phase I data which have demonstrated that BC-3781 can achieve therapeutically relevant blood and
tissue levels in man with excellent tolerability when administered by either oral or intravenous
routes.”
Dr. David Chiswell, CEO Nabriva Therapeutics commented:
“With a worldwide problem due to antibiotic resistant bacteria, there is a very significant need for
new classes of antibiotics with unique modes of action such as the pleuromutilins. The commercial
prospects for BC-3781 as the leading compound of an exciting new class are excellent, especially as it
has an ideal anti-bacterial spectrum for both skin and respiratory infections and is being developed
with both oral and intravenous formulations”
BC-3781 is highly active against key pathogens, including MRSA, associated with skin infections and
community and hospital acquired pneumonia and is more potent than Linezolid and vancomycin. The
compound’s novel mode of action ensures that it overcomes resistance mechanisms affecting all
approved classes of antibiotics. BC-378
About Nabriva Therapeutics
Nabriva Therapeutics is a biotechnology company focused on developing a new class of antibiotics for
the treatment of serious infections caused by resistant pathogens. Nabriva’s lead systemic product,
BC-3781, is being developed for the treatment of serious skin infections and bacterial pneumonia
caused by S. aureus, , S. pneumoniae, H. influenza, Mycoplasma, Legionella and other bacteria,
including drug resistant strains such as MRSA and vancomycin resistant E. faecium. In addition,
Nabriva Therapeutics’ topical pleuromutilin product candidate, BC-7013, is in clinical phase I. Nabriva
Therapeutics has a proven track record in world-class medicinal chemistry, clinical expertise, a
seasoned management team and solid IP. Nabriva Therapeutics is located in Vienna, Austria.
For more information on Nabriva please visit http://www.nabriva.com.
REF
http://www.phase4-partners.com/wp-content/uploads/2013/09/100412.pdf
http://www.glsv-vc.com/downloads/2010-06-02_First%20Patient_PressRelease.pdf
119
Nabriva. Pleuromutilins. Available online: http://www.nabriva.com/programs/pleuromutilins/ (accessed on 7 December 2012).
120
Forest Laboratories. Our pipeline: Solid, and set for further growth. Available online: http://www.frx.com/research/pipeline.aspx (accessed on 13 April 2013).
121
Sader, H.S.; Biedenbach, D.J.; Paukner, S.; Ivezic-Schoenfeld, Z.; Jones, R.N. Antimicrobial activity of the investigational pleuromutilin compound BC-3781 tested against Gram-positive organisms commonly associated with acute bacterial skin and skin structure infections. Antimicrob. Agents Chemother. 2012,56, 1619–1623, doi:10.1128/AAC.05789-11.
122
Sader, H.S.; Paukner, S.; Ivezic-Schoenfeld, Z.; Biedenbach, D.J.; Schmitz, F.J.; Jones, R.N. Antimicrobial activity of the novel pleuromutilin antibiotic BC-3781 against organisms responsible for community-acquired respiratory tract infections (CARTIs). J. Antimicrob. Chemother. 2012, 67, 1170–1175, doi:10.1093/jac/dks001.
123
Nabriva Therapeutics AG. Study comparing the safety and efficacy of two doses of BC-3781 vs. vancomycin in patients with acute bacterial skin and skin structure infection (ABSSSI). Available online: http://www.clinicaltrials.gov/ct2/show/NCT01119105 (accessed on 13 April 2013).
124
Novak, R. Are pleuromutilin antibiotics finally fit for human use? Ann. NY Acad. Sci. 2011, 1241, 71–81, doi:10.1111/j.1749-6632.2011.06219.x.
 valnemulin
valnemulin
 retapamulin
retapamulin
A safe, cheap and effective method for slow-freezing human stem cells
Human pluripotent stem cells (hPSCs) show great potential and versatility in regenerative medicine and new therapeutic approaches to fight disease. Patient-specific, individualized treatments using stem cells have even been generated for a number of diseases. Although further research into hPSCs is needed in order to harness their full potential, preserving the stem cells and storing them in the large numbers required for research has proved difficult.
Teruo Akuta and colleagues at the RIKEN Center for Developmental Biology, together with scientists from the Foundation for Biomedical Research and Innovation, have now developed a cost-effective, efficient and reliable slow-freezing method for preserving hPSCs in large numbers with a high survival rate.
Vitrification, which involves the use of cryoprotectants to chill cells to low temperatures without freezing, and conventional slow-freezing techniques are currently used for the cryopreservation of hPSCs. “Vitrification using liquid nitrogen is a highly skilled task,” notes Akuta, “and is not…
View original post 263 more words
Plazomicin…………against multidrug-resistant Klebsiella pneumoniae and Escherichia coli.

Plazomicin
6′-(hydroxylethyl)-1-(haba)-sisomicin
Plazomicin is a neoglycoside antibiotic with activity against a broad range of Gram-positive and Gram-negive pathogens. Plazomicin showed potent in vitro activity against multidrug-resistant Klebsiella pneumoniae and Escherichia coli.
| Synonyms: O-2-Amino-2,3,4,6-tetradeoxy-6-[(2-hydroxyethyl)amino]-α-D-glycero-hex-4-enopyranosyl-(1→4)-O-[3-deoxy-4-C-methyl-3-(methylamino)-β-L-arabinopyranosyl-(1→6)]-N1-[(2S)-4-amino-2-hydroxy-1-oxobutyl]-2-deoxy-D-streptamine; ACHN 490; |
| CAS Number: 1154757-24-0 |
| Achaogen (USA)Phase II completed |
| Mol. Formula: C25H48N6O10 |
| Aminoglycosides, Broad-spectrum, |
| Mol. Weight: 592.68 |

To continue the development of plazomicin, the company has received a contract option of US$ 60M from the Biomedical Advanced Research and Development Authority (BARDA) to support a global Phase III clinical study. The study will evaluate plazomicin in treating patients with serious Gram-negative bacterial infections due to carbapenem-resistant Enterobacteriaceae. The study is expected to start in the fourth quarter of 2013 [4].
Achaogen is a clinical-stage biopharmaceutical company passionately committed to the discovery, development, and commercialization of novel antibacterials to treat multi-drug resistant, or MDR, gram-negative infections.

Achaogen (a-KAY-o-jen) is developing plazomicin, its lead product candidate, for the treatment of serious bacterial infections due to MDR Enterobacteriaceae, including carbapenem-resistant Enterobacteriaceae, or CRE. In 2013, the Centers for Disease Control and Prevention identified CRE as a “nightmare bacteria” and an immediate public health threat that requires “urgent and aggressive action.” We expect to initiate a Phase 3 superiority trial of plazomicin in the first quarter of 2014.
CRE are one of many types of MDR gram-negative pathogens threatening patients. Bacteria such as Pseudomonas aeruginosa, Acinetobacter baumannii, and extended-spectrum beta-lactamase producing Enterobacteriaceae each pose “serious” resistance threats, according to the CDC, and also drive a great need for new, safe, and effective antibiotics. We have assembled the chemistry and microbiology expertise and capabilities required to develop new agents for the treatment of gram-negative infections. Plazomicin was the first clinical candidate from our gram-negative antibiotic discovery engine. In addition, our research and development pipeline includes two antipseudomonal programs targeting P. aeruginosa—a program to discover and develop small molecule inhibitors of LpxC, which is an enzyme essential for the synthesis of the outer membrane of gram-negative bacteria, and a therapeutic antibody program. We are also pursuing small molecule research programs targeting other essential gram-negative enzymes.
Achaogen has built an exceptional research and development team with deep expertise in the discovery and development of new drugs from research through commercialization. Our executive team has over 60 years of combined industry experience, and a proven track record of leadership, global registration, and lifecycle management for over 20 products. Our facility is located on the shores of the San Francisco Bay, ten minutes from the San Francisco International Airport, and only fifteen minutes from downtown San Francisco.
http://www.google.com/patents/US20100099661
Common Intermediates Sisomicin

Amberlite IRA-400 (OH form) (200 g) was washed with MeOH (3×200 m1). To a stirring suspension of the washed resin in MeOH (150 mL) was added sisomicin sulfate (20.0 g, 0.029 mol) and the mixture was stirred overnight. The resin was then filtered and washed with MeOH (100 mL) and the combined organic layers were concentrated to dryness to yield the desired sisomicin (11.57 g, 0.026 mol, 89.6% yield): MS m/e [M+H]+ calcd 448.3, found 448.1.
Example 1 6′-(2-Hydroxy-ethyl)-1-(4-amino-2(S)-hydroxy-butyryl)-sisomicin

6′-(2-tert-Butyldimethylsililoxy-ethyl)-2′,3,3″-triBoc-1-(N-Boc-4-amino-2(S)-hydroxy-butyryl)-sisomicin
2′,3,3″-triBoc-1-(N-Boc-4-amino-2(S)-hydroxy-butyryl)-sisomicin (0.10 g, 0.105 mmol) was treated with tert-butyldimethylsilyloxy acetaldehyde following Procedure 1-Method A to yield the desired 6′-(2-tert-butyldimethylsilyloxy-ethyl)-2′,3,3″-triBoc-1-(N-Boc-4-amino-2(S)-hydroxy-butyryl)-sisomicin (MS m/e [M+H]+ calcd 1107.6, found 1107.4), which was carried through to the next step without further purification.

6′-(2-Hydroxy-ethyl)-1-(4-amino-2(S)-hydroxy-butyryl)-sisomicin
6′ -(2-tert-butyldimethylsililoxy-ethyl)-2′,3,3″-triBoc-1-(N-Boc-4-amino-2(S)-hydroxy-butyryl)-sisomicin (0.105 mmol) was submitted to Procedure 3-Method B for Boc removal to yield a crude, which was purified by RP HPLC Method 1-Column A to yield 6′-(2-hydroxy-ethyl)-1-(4-amino-2(S)-hydroxy-butyryl)-sisomicin: MS m/e [M+H]+ calcd 593.3, found 593.2, [M+Na]+615.3 ; CLND 97.5% purity.
- Achaogen. Study for the treatment of complicated urinary tract infection and acute pyelonephritis.Available online: http://www.clinicaltrials.gov/ct2/show/NCT01096849 (accessed on 11 April 2013).
- Zhanel, G.G.; Lawson, C.D.; Zelenitsky, S.; Findlay, B.; Schweizer, F.; Adam, H.; Walkty, A.; Rubinstein, E.; Gin, A.S.; Hoban, D.J.; et al. Comparison of the next-generation aminoglycoside plazomicin to gentamicin, tobramycin and amikacin. Expert Rev. Anti-Infect. Ther. 2012, 10, 459–473, doi:10.1586/eri.12.25.
- Endimiani, A.; Hujer, K.M.; Hujer, A.M.; Armstrong, E.S.; Choudhary, Y.; Aggen, J.B.; Bonomo, R.A. ACHN-490, a neoglycoside with potent in vitro activity against multidrug-resistant Klebsiella pneumoniae isolates. Antimicrob. Agents Chemother. 2009, 53, 4504–4507.
- Achaogen. Achaogen pipeline. Available online: http://www.achaogen.com (accessed on 30 August 2012).
- Achaogen. Achaogen Awarded $60M Contract Option by BARDA for the Clinical Development of Plazomicin. Available online: http://www.achaogen.com/news/151/15 (accessed on 19 June 2013).
- Achaogen. Achaogen announces all objectives met in Phase 2 Plazomicin complicated urinary tract infections study and start of first-in-human study with ACHN-975. Available online: http://www.achaogen.com/uploads/news/id148/Achaogen_PressRelease_2012–05–15.pdf (accessed on 10 April 2013).
- Achaogen. Achaogen Announces Agreement with FDA on a Special Protocol Assessment for a Phase 3 Clinical Trial of Plazomicin to Treat Infections Caused by Carbapenem-Resistant Enterobacteriaceae (CRE); Achaogen: San Francisco, CA, USA, 2013.
- Comparison of the next-generation aminoglycoside plazomicin to gentamicin, tobramycin and amikacin
-
4-23-2010ANTIBACTERIAL AMINOGLYCOSIDE ANALOGS

| US8318685 | Nov 14, 2011 | Nov 27, 2012 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8367625 | Apr 7, 2011 | Feb 5, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8372813 | Apr 7, 2011 | Feb 12, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8377896 | Mar 9, 2011 | Feb 19, 2013 | Isis Pharmaceuticals, Inc | Antibacterial 4,6-substituted 6′, 6″ and 1 modified aminoglycoside analogs |
| US8399419 | Mar 9, 2011 | Mar 19, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8481502 | Apr 6, 2012 | Jul 9, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8492354 | Nov 14, 2011 | Jul 23, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8524675 | Nov 14, 2011 | Sep 3, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8524689 | Nov 14, 2011 | Sep 3, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8569264 | Jan 5, 2012 | Oct 29, 2013 | Isis Pharmaceuticals, Inc. | Antibacterial 4,5-substituted aminoglycoside analogs having multiple substituents |
| US8653041 | Oct 15, 2012 | Feb 18, 2014 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8653042 | Nov 14, 2011 | Feb 18, 2014 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8658606 | Nov 14, 2011 | Feb 25, 2014 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
SITAFLOXACIN …………Antibacterial [DNA-gyrase inhibitor]

7-[(4S)-4-Amino-6-azaspiro[2.4]heptan-6-yl]-8-chloro-6-fluoro-1-[(2S)-2-fluorocyclopropyl]-4-oxoquinoline-3-carboxylic acid
(1R-(1a(S*),2a))-7-(7-Amino-5-azaspiro[2.4]hept-5-yl)-8-chloro-6-fluoro-1-(2-fluorocyclopropyl)-1,4-dihydro-4-oxo-3-quinolinecarboxylic Acid
SYNTHESIS……….http://www.drugfuture.com/synth/syndata.aspx?ID=176447
127254-10-8 [RN]
127254-10-8(ACETATE)
- DU 6859A
- DU-6859a
- Sitafloxacin
- UNII-9TD681796G
Sitafloxacin (INN; also called DU-6859a) is a fluoroquinolone antibiotic[1] that shows promise in the treatment of Buruli ulcer. The molecule was identified by Daiichi Sankyo Co., which brought ofloxacin and levofloxacin to the market. Sitafloxacin is currently marketed in Japan by Daiichi Sankyo under the tradename Gracevit.
Sitafloxacin is a new-generation, broad-spectrum oral fluoroquinolone antibiotic.It is very active against many Gram-positive, Gram-negative and anaerobic clinical isolates, including strains resistant to other fluoroquinolones, was recently approved in Japan for the treatment of respiratory and urinary tract infections. Sitafloxacin is active against methicillin-resistant staphylococci, Streptococcus pneumoniae and other streptococci with reduced susceptibility to levofloxacin and other quinolones and enterococci

163253-35-8
-
C19-H18-Cl-F2-N3-O3.3/2H2-O
- 427.833

AU 8933702; EP 0341493; JP 1990231475; JP 1995300416; JP 1999124367; JP 1999124380; US 5587386; US 5767127
The condensation of 3-chloro-2,4,5-trifluorobenzoylacetic acid ethyl ester (I) with (1R,2S)-N-(tert-butoxycarbonyl)-2-fluorocyclopropylamine (III) and ethyl orthoformate (II) in hot acetic anhydride gives (1R,2S)-2-(3-chloro-2,4,5-trifluorobenzoyl)-3-(2-fluorocyclopropylamino)acrylic acid ethyl ester (IV). The cyclization of (IV) by means of NaH yields the quinolone (V), which is hydrolyzed with HCl to the free acid (VI). The condensation of (VI) with 7(S)-(tert-butoxycarbonylamino)-5-azaspiro[2.4]heptane (VII) by means of triethylamine in refluxing acetonitrile affords the protected final product (VIII), which is finally deprotected with trifluoroacetic acid and anisole.

The chiral intermediate (1R,2S)-N-(tert-butoxycarbonyl)-2-fluorocyclopropylamine (III) is obtained as follows: 1) The cyclization of butadiene (IX) with dibromofluoromethane by means of BuONa, followed by oxidation with KMnO4, esterification with ethanol – sulfuric acid and reduction with tributyltin hydride gives 2-fluorocyclopropanecarboxylic acid ethyl ester as a cis/trans mixture (X), which is separated by crystallization. The cis-racemic-isomer (XI) is hydrolyzed with NaOH to the corresponding acid (XII), which is condensed with (R)-alpha-methylbenzylamine (XIII) by means of diphenyl chlorophosphate to give the mixture of diastereomers (XIV). This mixture is separated by crystallization, yielding pure (1S,2S)-2-fluoro-N-[alpha(R)-methylbenzyl]cyclopropanecarboxamide (XV), which is hydrolyzed with HCl to the corresponding free acid (XVI). Finally, this compound is converted into (III) by treatment with diphenylphosphoryl azide in refluxing tert-butanol.

b) The intermediate 7(S)-(tert-Butoxycarbonylamino)-5-azaspiro[2.4]heptane (VII) can also be obtained as follows: 1) The cyclopropanation of ethyl acetoacetate (XXXI) with 1,2-dibromoethane (XXXII) by means of K2CO3 in DMF gives 1-acetylcyclopropane-1-carboxylic acid ethyl ester (XXXIII), which is brominated with Br2 in ethanol yielding the bromoacetyl derivative (XXXIV). The cyclization of (XXXI) with (R)-alpha-methylbenzylamine (XIII) by means of triethylamine affords 5-[1(R)-phenylethyl]-5-azaspiro[2.4]heptane-4,7-dione (XXXV), which by reaction with hydroxylamine is converted into the monooxime (XXXVI). The reduction of (XXXVI) with H2 over RaNi in methanol affords 7-amino-5-[1(R)-phenylethyl]-5-azaspiro[2.4]heptan-4-one as a diastereomeric mixture (XXXVII) + (XXXVIII), which is separated by column chromatography. The reduction of the (7S)-isomer (XXXVIII) with LiAlH4 in THF gives 7(S)-amino-5-[1(R)-phenylethyl]-5-azaspiro[2.4]heptane (XXXIX), which is protected in the usual way to the tert-butoxycarbonyl derivative (XL). Finally, this compound is debenzylated to (VII) by hydrogenation with H2 over Pd/C in ethanol.
The chiral intermediate (1R,2S)-N-(tert-butoxycarbonyl)-2-fluorocyclopropylamine (III) is obtained as follows: 1) The cyclization of butadiene (IX) with dibromofluoromethane by means of BuONa, followed by oxidation with KMnO4, esterification with ethanol – sulfuric acid and reduction with tributyltin hydride gives 2-fluorocyclopropanecarboxylic acid ethyl ester as a cis/trans mixture (X), which is separated by crystallization. The cis-racemic-isomer (XI) is hydrolyzed with NaOH to the corresponding acid (XII), which is condensed with (R)-alpha-methylbenzylamine (XIII) by means of diphenyl chlorophosphate to give the mixture of diastereomers (XIV). This mixture is separated by crystallization, yielding pure (1S,2S)-2-fluoro-N-[alpha(R)-methylbenzyl]cyclopropanecarboxamide (XV), which is hydrolyzed with HCl to the corresponding free acid (XVI). Finally, this compound is converted into (III) by treatment with diphenylphosphoryl azide in refluxing tert-butanol.

b) The intermediate 7(S)-(tert-Butoxycarbonylamino)-5-azaspiro[2.4]heptane (VII) can also be obtained as follows: 2) The reaction of 1-acetylcyclopropane-1-carboxylic acid ethyl ester (XXXIII) with (R)-alpha-methylbenzylamine (XIII) by means of NaOH and ethyl chloroformate gives the corresponding amide (XLI), which by reaction with ethylene glycol and p-toluenesulfonic acid is converted into the ethylene ketal (XLII). The bromination of (XLII) with Br2 in dioxane affords the bromomethyl dioxolane (XLIII), which is finally cyclized to 5-[1(R)-phenylethyl]-5-azaspiro[2.4]heptane-4,7-dione (XXXV), already obtained as an intermediate in the preceding synthesis.

The chiral intermediate (1R,2S)-N-(tert-butoxycarbonyl)-2-fluorocyclopropylamine (III) can also be obtained as follows: 3) A study of the influence of different substituents in the cis/trans ratio of the cyclopropanation process has been performed. The general method is as follows: the reaction of benzylamine (XXIII) with acetaldehyde and trichloromethyl chloroformate gives the N-benzyl-N-vinylcarbamoyl chloride (XXIV), which by treatment with alcohol yields the N-vinylcarbamate (XXV). The cyclopropanation of (XXV) with fluorodiiodomethane and diethyl zinc as before preferentially affords the cis-N-(2-fluorocyclopropyl)carbamate (XXVI), which is purified by crystallization. The hydrogenolysis of (XXVI) with H2 over Pd/C in acetic acid gives cis-racemic-2-fluorocyclopropylamine (XXVII), which is submitted to optical resolution with L-menthyl chloroformate to afford pure (1R,2S)-isomer (XXII). Finally, this compound is converted into (III) with tert-butoxycarbonyl anhydride as before.
References
- Anderson, DL. (Jul 2008). “Sitafloxacin hydrate for bacterial infections.”. Drugs Today (Barc) 44 (7): 489–501. doi:10.1358/dot.2008.44.7.1219561.PMID 18806900.
- Chem Pharm Bull 1998,46(4),587
- J Med Chem 1994,37(20),3344
- Drugs Fut 1994,19(9),827
- 33rd Intersci Conf Antimicrob Agents Chemother (Oct 17-20, New Orleans) 1993,Abst 975
- Tetrahedron Lett 1992,33(24),3487-90
- Keating GM (April 2011). “Sitafloxacin: in bacterial infections”. Drugs 71 (6): 731–44. doi:10.2165/11207380-000000000-00000.PMID 21504249.
- (Japanese) Gracevit グレースビット (PDF) Daiichi Sankyo Co. January 2008.
|
3-7-2012
|
Method for Production of Quinolone-Containing Lyophilized Preparation
|
|
|
12-5-2007
|
Stabilized liquid preparation
|
|
|
8-24-2007
|
PHARMACEUTICAL COMPOSITION
|
|
|
6-29-2007
|
PHARMACEUTICAL COMPOSITION
|
|
|
7-15-2005
|
Pharmaceutical composition
|
|
|
3-2-2005
|
Highly absorptive solid preparation
|
|
|
7-9-2004
|
Highly absorbable solid preparation
|
|
|
2-6-2004
|
Medicinal composition
|
|
|
12-17-1999
|
NOVEL THERAPEUTIC AGENTS THAT MODULATE ENZYMATIC PROCESSES
|
Carrots Cut Men’s Prostate Cancer Risk by 50%:

http://www.ncbi.nlm.nih.gov/pubmed/24519559
Erectile dysfunction can be reversed without medication
Men suffering from sexual dysfunction can be successful at reversing their problem, by focusing on lifestyle factors and not just relying on medication, according to new research at the University of Adelaide.
In a new paper published in the Journal of Sexual Medicine, researchers highlight the incidence of erectile dysfunction and lack of sexual desire among Australian men aged 35-80 years.
Over a five-year period, 31% of the 810 men involved in the study developed some form of erectile dysfunction.
“Sexual relations are not only an important part of people’s wellbeing. From a clinical point of view, the inability of some men to perform sexually can also be linked to a range of other health problems, many of which can be debilitating or potentially fatal,” says Professor Gary Wittert, Head of the Discipline of Medicine at the University of Adelaide and Director of the University’s Freemasons Foundation Centre for…
View original post 255 more words
DS-8587 (Daiichi Sankyo (Japan) a new broad-spectrum antibacterial agent, is in phase I clinical trials for the treatment of bacterial infection.

DS-8587
Daiichi Sankyo (Japan)
7-[3a(R)-Amino-6a(S)-fluoroperhydrocyclopenta[c]pyrrol-2-yl]-6-fluoro-1-[(1R,2S)-2-fluorocyclopropyl]-8-methyl-4-oxo-1,4-dihydroquinoline-3-carboxylic acid hydrochloride dihydrate
7-[(1S,6S)-1-amino-4-oxa-8-azabicyclo[4.3.0]nonan-8-yl]-6-fluoro-1-[(1R,2S)-2-fluorocyclopropan-1-yl]-1,4-dihydro-8-methoxy-4-oxoquinoline-3-carboxylic acid
| C21 H22 F3 N3 O3 . Cl H . 2 H2 O | |
| Mw | 493.904 |
DS-8587, a new broad-spectrum antibacterial agent, is in phase I clinical trials at Daiichi Sankyo for the treatment of bacterial infection.
DS-8587, from Daiichi Sankyo, is a fluoroquinolone with improved activity against both Gram-negative and Gram-positive bacteria. The compound is especially effective against Acinetobacter baumannii but also has improved activity against streptococci, staphylococci, enterococci, E. coli, and anaerobes . The compound is currently under Phase I of clinical development .
DS-8587, a new generation of fluoroquinolone, against Acinetobacter baumannii. The MICs against clinical isolates and inhibitory activity against target enzymes of DS-8587 was superior to ciprofloxacin and levofloxacin. Furthermore, the antibacterial activity of DS-8587 was less affected by adeA/adeB/adeC or abeM efflux pumps and frequency of single-step mutations with DS-8587 was lower as compared to those with ciprofloxacin. DS-8587 might be an effective agent against A. baumanniiinfection.
WO 2008082009 or
http://www.google.com/patents/EP2540715A1?cl=en
- [Reference Example 71]
(3S)-3-[3-(tert-Butyldimethylsilyloxy)-1-propyl]-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester
-


-
[(3S)-3-(3-Hydroxy-1-propyl)-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester (46 g) and imidazole (11.9 g) were dissolved in dimethylformamide (600 mL). After addition of tert-butyldimethylsilyl chloride (23.2 g) under ice-cooling, the mixture was stirred at room temperature for 59.5 hours. The reaction solution was extracted with a 10% citric acid solution and ethyl acetate. Then, the organic layer was sequentially washed with saturated sodium bicarbonate water and brine, dried over anhydrous sodium sulfate, and filtered. Thereafter, the solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 9:1 -> 8:2 -> 2:1) to give 29.7 g of the title compound as a pale yellow oil.
1H-NMR (400 MHz, CDCl3) δ: 7.37-7.22 (5H, m), 5.48 (1H, q, J=7.11 Hz), 3.58 (2H, t, J=6.13 Hz), 3.34 (1H, d, J=10.05 Hz), 3.12 (1H, d, J=10.05 Hz), 2.94 (1H, d, J=16.91 Hz), 2.31 (1H, d, J=17.16 Hz), 1.86-1.74 (1H, m), 1.72-1.62 (1H, m), 1.51 (3H, d, J=7.11 Hz), 1.49-1.24 (2H, m), 1.33 (9H, s), 0.88 (9H, s), 0.03 (6H, s).

[Reference Example 72]
(3S)-3-[3-(tert-Butyldimethylsilyloxy)-1-propyl]-4-fluoro-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester
-

-
(3S)-3-[3-(tert-Butyldimethylsilyloxy)-1-propyl]-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester (30 g) was dissolved in tetrahydrofuran (280 mL), and the atmosphere was replaced with argon. Then, lithium hexamethyldisilazide (1.0 M solution in tetrahydrofuran) (78.0 mL) was added dropwise at -15°C, and the mixture was stirred at -5°C for 30 minutes. After cooling to -15°C again, a solution of N-fluorobenzenesulfonimide (26.6 g) in tetrahydrofuran (220 mL) was added dropwise, and the mixture was stirred at room temperature for 17 hours. The reaction solution was extracted with a 10% citric acid solution and ethyl acetate. Then, the organic layer was washed with brine, dried over anhydrous sodium sulfate, and filtered. Thereafter, the solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 9:1 -> 8:2) to give 8.15 g of the title compound as a pale yellow solid. 1H-NMR (400 MHz, CDCl3) δ: 7.37-7.23 (5H, m), 5.53-5.44 (1H, m), 5.18 (1H, d, J=51.72 Hz), 3.64-3.52 (2H, m), 3.32-3.19 (2H, m), 1.92-1.65 (2H, m), 1.55 (3H, d, J=4.66 Hz), 1.33 (9H, s), 0.88 (9H, s), 0.03 (6H, s).
MS (FAB) m/z: 480 (M+H)+.
IR (ATR) ν: 3421, 2977, 2935, 2877, 1698, 1454, 1369, 1309, 1249, 1153, 1058, 1035, 1006, 842 cm-1.

[Reference Example 73]
(3S)-4-Fluoro-3-(3-hydroxy-1-propyl)-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester
-

-
(3S)-3-[3-(tert-Butyldimethylsilyloxy)-1-propyl]-4-fluoro-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester (8.15 g) was dissolved in tetrahydrofuran (25.0 mL). Acetic acid (22.0 mL) and tetrabutylammonium fluoride (1.0 M solution in tetrahydrofuran) (25.0 mL) were added under ice-cooling, and the mixture was stirred at room temperature for 21.5 hours. The reaction solution was extracted with a 10% citric acid solution and ethyl acetate. Then, the organic layer was sequentially washed with saturated sodium bicarbonate water and brine, dried over anhydrous sodium sulfate, and filtered. Thereafter, the solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 9:1 -> 8:2 -> 1:1) to give 5.77 g of the title compound as a pale yellow oil.
1H-NMR (400 MHz, CDCl3) δ: 7.37-7.22 (5H, m), 5.48 (1H, q, J=7.03 Hz), 5.20 (1H, d, J=51.48 Hz), 3.69-3.59 (2H, m), 3.31-3.21 (2H, m), 1.95-1.72 (2H, m), 1.68-1.43 (2H, m), 1.56 (3H, d, J=7.11 Hz), 1.33 (9H, s).

[Reference Example 74]
(3S)-3-(3-benzenesulfonyloxy-1-propyl)-4-Fluoro-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester
-

-
(3S)-4-Fluoro-3-(3-hydroxy-1-propyl)-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester (12.20 g) was dissolved in dichloromethane (400 mL). Benzenesulfonyl chloride (9.06 mL), triethylamine (10.7 mL), and 4-dimethylaminopyridine (2.04 g) were added under ice-cooling, and the mixture was stirred at room temperature for 12.5 hours. Saturated sodium bicarbonate water was added to the reaction solution, and the mixture was stirred for 30 minutes, followed by extraction with dichloromethane. The organic layer was sequentially washed with a 10% citric acid solution and brine, dried over anhydrous sodium sulfate, and filtered. Thereafter, the solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 8:2 -> 1:1) to give 11.7 g of the title compound as a pale yellow oil.
1H-NMR (400 MHz, CDCl3) δ: 7.94 – 7.87 (2H, m), 7.71-7.63 (1H, m), 7.60-7.53 (2H, m), 7.37-7.23 (5H, m), 5.46 (1H, q, J=7.11 Hz), 5.15 (1H, d, J=51.48 Hz), 4.10-3.98 (2H, m), 3.26-3.15 (2H, m), 1.88-1.50 (4H, m), 1.55 (3H, s), 1.30 (9H, s).

[Reference Example 75]
(1S,5R)-5-Fluoro-4-oxo-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octan-1-ylcarboxylic acid tert-butyl ester
-

-
(3S)-3-(3-benzenesulfonyloxy-1-propyl)-4-Fluoro-5-oxo-1-[(1R)-1-phenylethyl]pyrrolidine-3-carboxylic acid tert-butyl ester (10.9 g) was dissolved in tetrahydrofuran (350 mL), and the atmosphere was replaced with argon. Then, potassium hexamethyldisilazide (0.5 M solution in toluene) (86.5 mL) was added dropwise at – 15°C, and the mixture was stirred at 0°C for 1.5 hours. After cooling to -10°C, saturated aqueous ammonium chloride (100 mL) was added dropwise, and the mixture was stirred at room temperature for 30 minutes. The reaction solution was extracted with a 10% citric acid solution and ethyl acetate. Then, the organic layer was washed with brine, dried over anhydrous sodium sulfate, and filtered. Thereafter, the solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 9:1 -> 7:1) to give 4.36 g of the title compound as a pale yellow solid.
1H-NMR (400 MHz, CDCl3) δ: 7.38-7.25 (5H, m), 5.58-5.49 (1H, m), 3.63 (1H, d, J=10.3 Hz), 2.91 (1H, dd, J=10.3, 3.2 Hz), 2.67-2.56 (1H, m), 2.50-2.38 (1H, m), 2.26-2.09 (1H, m), 2.06-1.94 (1H, m), 1.74-1.66 (1H, m), 1.54 (3H, d, J=7.1 Hz), 1.50-1.40 (1H, m), 1.34 (9H, s).

[Reference Example 76]
(1R,5R)-1-(tert-Butoxycarbonylamino)-5-fluoro-4-oxo-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octane
-


-
(1S,5R)-5-Fluoro-4-oxo-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octan-1-ylcarboxylic acid tert-butyl ester (4.36 g, 12.5 mmol) was dissolved in dichloromethane (70 mL). Trifluoroacetic acid (70 mL) was added dropwise, and the mixture was stirred at room temperature for six hours. The solvent was evaporated under reduced pressure, and then the residue was azeotropically distilled with toluene to give carboxylic acid (3.70 g).
-
The resulting carboxylic acid was dissolved in toluene. Triethylamine (3.51 mL, 25.2 mmol) and diphenylphosphoryl azide (2.98 ml, 13.8 mmol) were added, and the mixture was heated to reflux for five hours. The solvent was evaporated under reduced pressure. Then, 1,4-dioxane (110 ml) and 6N hydrochloric acid (110 mL) were added to the residue, and the mixture was stirred at 60°C for 2.5 hours. After extraction with water and ethyl acetate, the aqueous layer was made alkaline with a saturated sodium hydroxide solution and extracted with chloroform twice. The organic layers were combined, dried over anhydrous sodium sulfate, and filtered, and then the solvent was evaporated under reduced pressure. Di-tert-butyl dicarbonate (11.05 g) was added to the residue, and the mixture was stirred at 75°C for six hours. The reaction solution was concentrated under reduced pressure, and then the residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 9:1 -> 1:1) to give 3.69 g of the title compound as a pale yellow oil.
1H-NMR (400 MHz, CDCl3) δ: 7.37-7.23 (5H, m), 5.50 (1H, q, J=7.1 Hz), 5.22 (1H, brs), 3.34 (2H, s), 2.49-2.37 (1H, m), 2.32-2.03 (3H, m), 2.02-1.90 (1H, m), 1.51 (3H, d, J=7.1 Hz), 1.55-1.48 (1H, m), 1.35 (9H, s).

[Reference Example 77]
(1R,5S)-1-(tert-Butoxycarbonylamino)-5-fluoro-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octane
-

-
(1R,5R)-1-(tert-Butoxycarbonylamino)-5-fluoro-4-oxo-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octane (3.69 g, 10.2 mmol) was dissolved in tetrahydrofuran (200 mL). A 1.20 M solution of a borane-tetrahydrofuran complex in tetrahydrofuran (42.4 mL, 50.9 mmol) was added dropwise under ice-cooling, and the mixture was stirred for two hours while gradually heating to room temperature. The solvent was evaporated under reduced pressure. Under ice-cooling, 90% aqueous ethanol (100 mL) and triethylamine (100 mL) were added to the residue, and the mixture was heated to reflux for two hours. The solvent was evaporated under reduced pressure, and then the residue was extracted with saturated sodium bicarbonate water and dichloromethane. Thereafter, the target substance was extracted from the aqueous layer with dichloromethane. The organic layers were combined, washed with brine, and dried over anhydrous sodium sulfate. After filtration, the solvent was evaporated under reduced pressure, and the resulting residue was subjected to silica gel column chromatography (hexane:ethyl acetate = 95:5 -> 90:10) to give 3.33 g of the title compound as a pale yellow oil.
1H-NMR (400 MHz, CDCl3) δ: 7.32 – 7.18 (5H, m), 5.38 (1H, brs), 3.22 (1H, q, J=6.37 Hz), 2.92-2.57 (4H, m), 2.12-1.86 (4H, m), 1.80-1.67 (1H, m), 1.63-1.52 (3H, m), 1.42 (9H, s), 1.32 (3H, d, J=6.37 Hz)

- [Reference Example 78]
(1R,5S)-1-(tert-Butoxycarbonylamino)-5-fluoro-3-azabicyclo[3.3.0]octane
-

-
(1R,5S)-1-(tert-Butoxycarbonylamino)-5-fluoro-3-[(1R)-1-phenylethyl]-3-azabicyclo[3.3.0]octane (700 mg, 2.0 mmol) was dissolved in ethanol (30 mL). 10% palladium-carbon (50% wet) (1.01 g) was added, and the mixture was stirred in a hydrogen atmosphere at 50°C for 15 hours. The catalyst was removed by filtration, and then the filtrate was concentrated under reduced pressure. The resulting residue was subjected to silica gel column chromatography (dichloromethane:methanol = 98:2 -> 95:5) to give 446 mg of the title compound as a pale yellow solid.
[α]D 23-15° (c=0.100, MeOH).
1H-NMR (400 MHz, CDCl3) δ: 5.29 (1H, brs), 3.47-3.18 (2H, m), 2.93-2.79 (2H, m), 2.15-1.71 (6H, m), 1.45 (9H, s).

- [Example 17]
7-[(1R,5S)-1-Amino-5-fluoro-3-azabicyclo[3.3.0]octan-3-yl]-6-fluoro-1-[(1R,2S)-2-fluorocyclopropane]-1,4-dihydro-8-methyl-4-oxoquinoline-3-carboxylic acid
-

-
Triethylamine (0.215 mL, 1.54 mmol) and 6,7-difluoro-1-[(1R,2S)-2-fluorocyclopropane]-1,4-dihydro-8-methyl-4-oxoquinoline-3-carboxylic acid-BF2 chelate (530 mg, 1.53 mmol) were added to a solution of (1R,5S)-1-(tert-butoxycarbonylamino)-5-fluoro-3-azabicyclo[3.3.0]octane (250 mg, 1.02 mmol) in dimethyl sulfoxide (5.0 mL). The mixture was stirred at room temperature for seven days. Triethylamine (0.215 mL, 1.54 mmol) and 6,7-difluoro-1-[(1R,2S)-2-fluorocyclopropane]-1,4-dihydro-8-methyl-4-oxoquinoline-3-carboxylic acid-BF2 chelate (530 mg, 1.53 mmol) were further added to the reaction solution, and the mixture was stirred at room temperature for seven days. Triethylamine (0.215 mL, 1.54 mmol) and 6,7-difluoro-1-[(1R,2S)-2-fluorocyclopropane]-1,4-dihydro-8-methyl-4-oxoquinoline-3-carboxylic acid-BF2 chelate (530 mg, 1.53 mmol) were further added to the reaction solution, and the mixture was stirred at room temperature for ten days. Ethanol (6.0 mL), water (2.0 mL), and triethylamine (2.0 mL) were added to the reaction solution, and the mixture was stirred at 80°C for one hour. The solvent was evaporated under reduced pressure, and then the residue was extracted with a 10% citric acid solution and ethyl acetate. Then, the organic layer was washed with water twice and brine, dried over anhydrous sodium sulfate, and filtered. Thereafter, the solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography (dichloromethane:methanol = 98:2), and the resulting fraction was concentrated under reduced pressure. Then, the residue was dissolved in concentrated hydrochloric acid (3.5 mL) under ice-cooling, and the solution was stirred at room temperature for one hour. The reaction solution was washed with chloroform five times, and then the aqueous layer was adjusted to pH 12 with a saturated sodium hydroxide solution. The basic solution was adjusted to pH 7.4 with hydrochloric acid, followed by extraction with chloroform. The organic layer was dried over anhydrous sodium sulfate and filtered, and then the solvent was evaporated under reduced pressure. The resulting residue was purified by PTLC (developed with the lower layer of chloroform:methanol:water = 7:3:1). The resulting residue was solidified with isopropanol to give 7.1 mg of the title compound as a pale yellow solid.
-
1H-NMR (400 MHz, 0.1N NaOD) δ: 8.50 (1H, s), 7.77 (1H, d, J=13.73 Hz), 5.80-4.80 (1H, m), 4.22-4.10 (1H, m), 3.99-3.85 (1H, m), 3.68-3.47 (2H, m), 3.43-3.34 (1H, m), 2.68 (3H, s), 2.21-1.98 (3H, m), 1.97-1.56 (4H, m), 1.42-1.23 (1H, m).
MS (FAB); m/z: 422 (M+H)+.
Anal.Calcd C21H22F3N3O3·0.5H2O·0.25IPA: C, 58.65; H, 5.66; F, 12.80; N, 9.43. Found: C, 58.63; H, 5.35; F, 12.35; N, 9.22.
IR (ATR) ν: 2971, 2856, 1722, 1614, 1450, 1432, 1322, 1132, 1097, 987, 954, 929, 887, 856, 804 cm-1.

WO 2012018105
http://www.google.st/patents/WO2012018105A1?cl=en
The following structural formula

compound represented by, 7 – [(1R, 5S) -1 – amino-5 – fluoro-3 – azabicyclo [3.3.0] octan-3 – yl] -6 – fluoro -1 – [(1R, . the 2S) -2 – fluoro-cyclopropane-1 – yl] -1,4 – dihydro-8 – methyl-4 – oxo-3-quinoline -) is referred to as carboxylic acid (hereinafter referred to as Compound A multi-agent containing quinolone resistance including resistant Gram-positive cocci resistant pneumococcus, etc., widely against gram-negative bacteria from Gram-positive bacteria, and, in addition to show strong antibacterial activity, convulsions, which is known in the art as a side effect of the antimicrobial agent of the present system potential cardiotoxicity light and toxicity-inducing activity (photosensitivity), has been reported recently in clinical further (QT prolongation), blood sugar abnormalities, and to express the side effects of delayed-type drug 疹等 is excellent safety low, Then, it is excellent oral absorbability and organ migration properties become apparent, is expected as an antimicrobial agent superior (Patent Document 1).
WO 2008/082009 pamphlet
The compound A, was synthesized according to the method described in Patent Document 1.
Preparation 7 1 hydrochloride dihydrate Ratings (1) Compound A crystalline acid addition salt preparation of acid addition salts of (Example 1) Compound A, and Compound A – [(1R, 5S) – 1 – amino-5 – fluoro-3 – azabicyclo [3.3.0] octan-3 – yl] -6 – fluoro -1 – [(1R, 2S) -2 – fluoro-cyclo-1 – yl] -1 , 4 – dihydro-8 – methyl-4 – oxo-3-quinoline – was added 1mol / L hydrochloric acid (74μL) carboxylic acid (Compound A) (31.3mg,, 0.074mmol) in, and dried under reduced pressure at room temperature.10% aqueous 2 the residue – was added to (100μL) propanol was dissolved by heating at 60 ℃, and allowed to stand day out on the room temperature. Collected by filtration the precipitated crystals, and the 1st air dried, 19.9mg (yield: 54%) was obtained.
Elemental analysis: C 21 H 22 F 3 N 3 O 3 · HCl · 2H 2 O
Theoretical value: C; 51.07, H; 5.51, N; 8.51, F; 11.54, Cl; 7.18
Measured value: C; 50.93, H; 5.40, N; 8.49, F; 11.30, Cl; 7.47
The characteristic diffraction peaks in the powder X-ray diffraction: 2θ = 5.3,7.9,10.6,13.3,21.1,23.0,25.1,27.6 (°)
2 5% aqueous (1001.6mg, 0.746mmol) in the preparation of Compound A one hydrochloride monohydrate (2) Compound A – was added propanol (30mL), was dissolved by heating at 60 ℃. After stirring day out on the room temperature and stirred for 6 hours at 10 ℃. Collected by filtration the precipitated crystals, and the 1st dried air, 839.3 mg (yield: 87%) was obtained.
Elemental analysis: C 21 H 22 F 3 N 3 O 3 · HCl · 1H 2 O
Theoretical value: C; 53.00, H; 5.30, N; 8.83, F; 11.98, Cl; 7.45
Measured value: C; 53.25, H; 5.43, N; 8.51, F; 11.58, Cl; 7.18
The characteristic diffraction peaks in the powder X-ray diffraction: 2θ = 11.3,14.0,20.1,21.4,22.8,24.0,26.0,26.6 (°)
ref
- Higuchi, S.; Onodera, Y.; Chiba, M.; Hoshino, K.; Gotoh, N. Potent in vitro antibacterial activity of DS-8587, a new generation of broad spectrum quinolone, against Acinetobacter baumannii. Antimicrob. Agents Chemother. 2013, doi:10.1128/AAC.02374-12.
- Daiichi Sankyo. Major R&D pipeline as of July, 2013. Available online: http://www.daiichisankyo.com/rd/pipeline/pdf/Pipeline_EN.pdf (accessed on 28 September 2013).
| EP0343524A1 | May 19, 1989 | Nov 29, 1989 | Shionogi Seiyaku Kabushiki Kaisha | Pyridonecarboxylic acids and antibacterial agents |
| JPH0395176A | Title not available | |||
| JPH02231475A | Title not available | |||
| JPH08225567A | Title not available | |||
| JPS6345261A | Title not available | |||
| JPS6456673A | Title not available | |||
| JPS61282382A | Title not available | |||
| US5017708 * | Sep 8, 1989 | May 21, 1991 | Shionogi & Co., Ltd. | Azabicycloalkanes |
| WO1994014794A1 | Dec 28, 1993 | Jul 7, 1994 | Hideki Ao | 8-methoxyquinolonecarboxylic acid derivative |
| WO1995021163A1 | Feb 2, 1995 | Aug 10, 1995 | Katsumi Chiba | Pyridonecarboxylic acid derivative substituted by bicyclic amino group, ester thereof, salt thereof, and bicyclic amine as intermediate therefor |
| WO1996023782A1 | Feb 1, 1996 | Aug 8, 1996 | Daiichi Seiyaku Co | Heterocyclic compounds |

1, nemonoxacin; 2, delafloxacin; 3, finafloxacin; 4, zabofloxacin; 5, JNJ-Q2; 6, DS-8587; 7, KPI-10; 8, ozenoxacin; 9, chinfloxacin; 10, ACH-702.
Ozenoxacin in phase 3……topical formulation in the treatment of impetigo

1-cyclopropyl-8-methyl-7-[5-methyl-6-(methylamino)-3-pyridinyl]-4-oxo-1 ,4-dihydro-3- quinolinecarboxylic acid
1-cyclopropyl-8-methyl-7-{5-methyl-6-[(methylamino)methyl]-3-pyridyl}-4-oxo-1,4-dihydro-3-quinolinecarboxylic acid.
Ferrer Internacional (Spain), phase 3 Gram-positive
Ferrer Internacional has completed one Phase III clinical trial to evaluate the topical formulation of ozenoxacin in the treatment of impetigo [
|
poster……http://landing.quotientbioresearch.com/blog/bid/50380/Ozenoxacin-Activity-against-Atypical-Bacteria
Ozenoxacin is active against a great number of pathogens, such as Propionibacterium acnes, Staphylococcus aureus, methicillin-susceptible Staphylococcus aureus (MSSA), methicillin-resistant Staphylococcus aureus (MRSA) including ciprofloxacin-resistant strains, methicillin-susceptible Staphylococcus epidermidis (MSSE), methicillin-resistant Staphylococcus epidermidis (MRSE), Streptococcus pyogenes, Group G Streptococci, penicillin-resistant Streptococcus pneumoniae, Beta-lactamase positive Haemophilus influenzae, non-typeable strains of Haemophilus influenzae, Beta-lactamase positive Moraxella catarrhalis, Neisseria meningitides, Legionella pneumophila, Mycoplasma pneumoniae, Legionella pneumophila, Mycobacterium tuberculosis, Streptococcus agalactiae group B, Neisseria gonorrhoeae, Chlamydia trachomatis, Mycoplasma hominis, Ureaplasma urealyticum Helicobacter pylori, Bacteroides fragilis, Clostridium perfringens, Escherichia coli, quinolone-resistant Escherichia coli, Salmonella spp., Shigella spp., ciprofloxacin-susceptible Pseudomonas aeruginosa, Clostridium difficile, and Listeria monocytogenes.
Ozenoxacin is a novel non-fluorinated quinolone antibacterial agent. It is currently in late stage phase 3 trials for the topical treatment of impetigo. The bacterial action of ozenoxacin is through the dual inhibition of DNA gyrase and topoisomerase IV. Excellent in vitro and in vivo antibacterial activity has been demonstrated in pre-clinical and clinical studies against a broad range of bacterial organisms. This includes organisms with emerging resistance to quinolones. Phase I and II clinical trials have also shown that ozenoxacin is a safe and effective antibacterial agent. No evidence of adverse effects as linked to topically formulated halogenated quinolones has been shown.
Ozenoxacin (I) was firstly disclosed in US6335447, and equivalent patents. Its chemical name is 1-cyclopropyl-8-methyl-7-[5-methyl-6-(methylamino)-3-pyridinyl]-4-oxo-1 ,4-dihydro-3- quinolinecarboxylic acid. Its chemical formula is: H

Ozenoxacin (I)
Topical application of antimicrobial agents is a useful tool for therapy of skin and skin structures infections, sexually transmitted diseases and genital tract infections and some systemic infections susceptible to topical treatment. Topical antimicrobial therapy has several potential advantages compared with systemic therapy.
Firstly, it can avoid an unnecessary exposure of the gut flora which may exert selection for resistance. Secondly, it is expected that the high local drug concentration in topical application and the negligible systemic absorption should overwhelm many mutational resistances. Thirdly, topical applications are less likely than systemic therapy to cause side effects. Accordingly, some topical compositions comprising ozenoxacin have been reported in the art.
JP2002356426A discloses ointments and gels for skin. An ointment comprising ozenoxacin 1%, N-methyl-2-pyrrolidone 8%, propylene glycol 14.9%, oleic acid 0.9%, diisopropanolamine 2.3%, polyethylene glycol 400 20.2%, polyethylene glycol 4000 50.2%, and water 3.2% is reported in Example 2.
JP2003226643A discloses aqueous solutions comprising ozenoxacin, cyclodextrin, and a viscous agent.
EP1731138A1 discloses fine particle dispersion liquid comprising ozenoxacin to be used in the manufacture of pharmaceutical compositions.
WO2007015453A1 discloses lotions comprising ozenoxacin.
JP2007119456A discloses aqueous suspensions containing nanoparticles and solution granules of ozenoxacin to be used in the manufacture of pharmaceutical compositions. Ophthalmic solutions are mentioned preferably. A combined use of ozenoxacin, magnesium ions, and hydroxypropyl-β-cyclodextrin specially for ophthalmic use is disclosed in Yamakawa, T. et al., Journal of Controlled Release (2003), 86(1 ), 101-103.
Semisolid topical compositions are useful alternatives to liquid compositions, because of their better manipulation and consequent patient preferences. However, in spite of the great diversity of components present in the semisolid compositions disclosed in the art, no quantitative stability studies are available for them.
Thus, there is a need of proved stable semisolid topical compositions comprising ozenoxacin as active ingredient, wherein microbiological and therapeutic activities are warranted because of demonstrated durable and prolonged pharmaceutical stability.
Synthesis
US6335447
http://www.google.co.in/patents/US6335447
EXAMPLE 5
To a solution of 0.80 g of 7-[6-({[(benzyloxy)-carbonyl] (methyl)amino}methyl)-5-methyl-3-pyrdyl]-1-cyclo-propyl-8-methyl-4-oxo-1,4-dihydro-3-quinoline-carboxylic acid in 16 ml of acetic acid was added 0.20 g of 5% (w/w) palladium-carbon and the mixture was stirred at ambient temperature and atmospheric pressure for 2 hours under a hydrogen atmosphere. The reaction mixture was filtered and the solvent was evaporated under reduced pressure. The obtained residue was dissolved in a mixed solvent consisting of 3.8 ml of ethanol and 3.8 ml of water. After adding 3.8 ml of an aqueous 1 mol/l sodium hydroxide solution thereto and adjusting the solution to pH 5.5with 1 mol/l hydrochloric acid, 10 ml of chloroform was added thereto. An organic layer was separated and dried over anhydrous magnesium sulfate and the solvent was evaporated under reduced pressure. Addition of diethyl ether to the obtained residue and filtration of crystals afforded 0.25 g of colorless crystals of 1-cyclopropyl-8-methyl-7-{5-methyl-6-[(methylamino)methyl]-3-pyridyl}-4-oxo-1,4-dihydro-3-quinolinecarboxylic acid.
IR (KBr) cm−1: 3322, 1721; NMR(d1-TFA) δ: 1.2-1.9 (4H, m), 2.94 (3H, s), 3.05 (3H, s), 3.29 (3H, s), 4.6-5.0 (1H, m), 5.12 (2H, s), 7.91 (1H, d, J=8.5 Hz), 8.6-9.0 (2H, m), 9.0-9.3 (1H, brs), 9.75 (1H, s). Melting point: 199° C.
- Ferrer Group. Key development projects. Available online: http://www.ferrergrupo.com/Innovation_Innovacion-Pipeline-de-proyectos-ENG (accessed on 15 April 2013).
- Yamakawa, T.; Mitsuyama, J.; Hayashi, K. In vitro and in vivo antibacterial activity of T-3912, a novel non-fluorinated topical quinolone. J. Antimicrob. Chemother. 2002, 49, 455–465, doi:10.1093/jac/49.3.455.
- Ferrer Internacional. Efficacy and safety of ozenoxacin 1% cream versus placebo in the treatment of patients with impetigo. Available online: http://clinicaltrials.gov/ct2/show/NCT01397461 (accessed on 13 April 2013).
SUROTOMYCIN for Clostridium difficile-associated diarrhea

Surotomycin
Click to access surotomycin.pdf
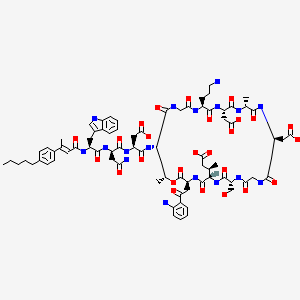
N-[(2E)-3-(4-Pentylphenyl)-2-butenoyl]-D-tryptophyl-D-asparaginyl-N-[(3S,6S,9R,15S,18R,21S,24S,30S,31R)-3-[2-(2-aminophenyl)-2-oxoethyl]-24-(3-aminopropyl)-15,21-bis(carboxymethyl)-6-[(2R)-1-carboxy-2 -propanyl]-9-(hydroxymethyl)-18,31-dimethyl-2,5,8,11,14,17,20,23,26,29-decaoxo-1-oxa-4,7,10,13,16,19,22,25,28-nonaazacyclohentriacontan-30-yl]-L-α-asparagine
MOLECULAR FORMULA C77H101N17O26
MOLECULAR WEIGHT 1680.7
SPONSOR Cubist Pharmaceuticals, Inc.
CODE DESIGNATION CB-183,315
CB-315, CB-183315, CB-183,315
CAS REGISTRY NUMBER 1233389-51-9
U.S. – Fast Track (Treat Clostridium difficile-associated diarrhea (CDAD));
U.S. – Qualified Infectious Disease Program (Treat Clostridium difficile-associated diarrhea (CDAD))
| Company | Cubist Pharmaceuticals Inc. |
| Description | Oral antibacterial lipopeptide |
| Therapeutic Modality | Macrocycle |
| Latest Stage of Development | Phase III |
| Standard Indication | Diarrhea (infectious) |
| Indication Details | Treat Clostridium difficile-associated diarrhea (CDAD) |
EMEA……..
| Name | |||
|---|---|---|---|
| P/0096/2013: EMA decision of 29 April 2013 on the agreement of apaediatric investigation plan and on the granting of a deferral for surotomycin (EMEA-001226-PIP01-11) |
Surotomycin is an investigational oral antibiotic. This antibiotic is under investigation for the treatment of life-threatening Diarrhea, commonly caused by the bacteria Clostridium difficile.[1]
CB-183315 is an investigational antibacterial drug candidate in phase III clinical trials at Cubist for the treatment of Clostridium difficile-associated diarrhea. It is a potent, oral, cidal lipopeptide. In 2012, Qualified Infectious Disease Product Designation was assigned in the U.S. for the treatment of clostridium difficile-associated diarrhea (CDAD).
Surotomycin (CB-315)

 Surotomycin Overview
Surotomycin Overview  Surotomycin Fact Sheet
Surotomycin Fact SheetSurotomycin is an antibacterial lipopeptide discovered by Cubist scientists in our research laboratories in Lexington, Massachusetts. Surotomycin is both bactericidal against Clostridium difficile and more potent than vancomycin in vitro. Surotomycin stays at the site of infection in the bowel, with minimal systemic absorption and it does not interfere with normal bowel flora. Based on its features and its preclinical safety profile, Cubist filed an Investigational New Drug (IND) Application for surotomycin in December 2008.
Following safety and pharmacokinetic studies in healthy human volunteers, Cubist began a Phase 2 study in April 2010 to assess the safety and efficacy of surotomycin in patients with CDAD, in particular to assess its ability to reduce relapse rates. In this trial of 209 patients, two different doses of surotomycin were studied and compared with oral vancomycin. The higher dose demonstrated a high clinical cure rate as evidenced by resolution of diarrhea, comparable to oral vancomycin. The most interesting results in this study, however, relate to recurrence rates. The percent of patients who had an initial response to treatment but who subsequently had a recurrence or relapse was 36 percent in the oral vancomycin arm and was 17 percent in the surotomycin 250mg treatment group — about a 50% reduction in relapse rate, which was statistically significant. In this trial, 32% of patients were infected with the hypervirulent NAP-1 strain of C. difficile. The clinical response rate in the subset of patients infected with the NAP-1 strain was comparable across the surotomycin and oral vancomycin groups. Though not statistically significant, there was a modest reduction in the relapse rates in the subset of surotomycin patients infected with NAP-1 strains.
The ability to reduce relapses is important to both patients and health care providers. In the Phase 2 study we assessed the impact of surotomycin and oral vancomycin on normal bowel flora. Treatment with surotomycin had a very minimal impact on levels of Bacteroides, a key normal bowel bacterial species, compared to oral vancomycin which resulted in a marked depletion of stool levels of these bacteria during treatment. Why does this matter? The reason is — bowel flora like Bacteroides are critical in providing a competitive environment in the bowel that prevents C. difficile overgrowth. We believe that it is this difference in impact on normal bowel flora that helps explain the differences seen in recurrence rates following treatment with Surotomycin versus oral vancomycin.
Surotomycin’s Phase 3 program includes two identical global, randomized, double-blind, active-controlled, multi-center trials. The primary objective is to demonstrate non-inferiority of surotomycin versus the comparator, oral vancomycin, in clinical response at the end of treatment in adult subjects with CDAD, using a non-inferiority margin of 10%. We also have designed this trial to allow us to demonstrate that sustained clinical response to surotomycin at the end of the study is superior to oral vancomycin. Also, we will fully evaluate the safety of surotomycin in the study subjects.
In late 2012 Cubist received from the FDA a Qualified Infectious Disease Product (QIDP) designation for surotomycin. Additionally, in early 2013 Cubist was granted Fast track status for surotomycin. The QIDP designation and subsequent granting of Fast Track status was made possible by the GAIN Act, Title VIII (Sections 801 through 806) of the Food and Drug Administration Safety and Innovation Act. The GAIN Act provides pharmaceutical and biotechnology companies with incentives to develop new antibacterial and antifungal drugs for the treatment of life-threatening infectious diseases caused by drug resistant pathogens. Qualifying pathogens are defined by the GAIN Act to include multi-drug resistant Gram-negative bacteria, including Pseudomonas, Acinetobacter, Klebsiella, and Escherichia coli species; resistant Gram-positive pathogens, including methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Staphylococcus aureus and vancomycin-resistant Enterococcus; multi-drug resistant tuberculosis; and Clostridium difficile.
About CDAD
CDAD is a disease caused by an overgrowth of, and subsequent toxin production by, C. difficile, a resident anaerobic spore-forming Gram-positive bacterium of the lower gastrointestinal tract. This overgrowth is caused by the use of antibiotics for the treatment of common community and hospital acquired infections (HAIs). Although they treat the underlying infection, many antibiotics disrupt the natural gut flora and allow C. difficile to proliferate. C. difficile produces enterotoxin and cytotoxin, which can lead to severe diarrhea, sepsis and even death. While most types of HAIs are declining, the infection caused by C. difficile remains at historically high levels. According to the latest data from the Centers for Disease Control, C. difficile continues to be the leading cause of death associated with gastroenteritis in the US. For CDAD alone, there was more than a five-fold increase in deaths between 1999 and 2007. C. difficile causes diarrhea linked to 14,000 American deaths each year. About 25% of C. difficile infections first show symptoms in hospital patients; 75% first show in nursing home patients or in people recently cared for in doctors’ offices and clinics. C. difficile infections cost at least $1 billion in extra health care costs annually.
SUROTOMYCIN
CB-183,315 is a cyclic lipopeptide antibiotic currently in Phase III clinical trials for the treatment of Clostridium difficile-associated disease (CDAD). As disclosed in International Patent Application WO 2010/075215, herein incorporated by reference in its entirety, CB-183,315 has antibacterial activity against a broad spectrum of bacteria, including drug-resistant bacteria and C. difficile. Further, the CB-183,315 exhibits bacteriacidal activity.
CB-183,315 (Figure 1) can be made by the deacylation of BOC-protected daptomycin, followed by acylation and deprotection as described in International Patent Application WO 2010/075215.
During the preparation and storage of CB-183,315, the CB-183,315 molecule can convert to structurally similar compounds as shown in Figures 2-4, leading to the formation of anhydro-CB-183,315 (Figure 3) and beta-isomer of CB-183,315 (“B- isomer CB183,315” in Figure 2). Accordingly, one measure of the chemical stability of CB- 183 ,315 is the amount of CB- 183 ,315 (Figure 1 ) present in the CB- 183 ,315 composition relative to the amount of structurally similar compounds including anhydro-CB-183,315 (Figure 3) and beta-isomer of CB-1 83,315 (Figure 2). The amount of CB-183,315 relative to the amount of these structurally similar compounds can be measured by high performance liquid chromatography (FIPLC) after reconstitution in an aqueous diluent (e.g., as described in Example 10). In particular, the purity of CB-183,315 and amounts of structurally similar compounds (e.g., Figures 2, 3 and 4) can be determined from peak areas obtained from HPLC (e.g., according to Example 10 herein), and measuring the rate of change in the amounts of CB-183,315 over time can provide a measure of CB-183,315 chemical stability in a solid form.
There is a need for solid CB-183,315 compositions with improved chemical stability in the solid form (i.e., higher total percent CB-183,315 purity over time), providing advantages of longer shelf life, increased tolerance for more varied storage conditions (e.g., higher temperature or humidity) and increased chemical stability.
……………..
WO2010075215A1
http://www.google.com/patents/WO2010075215A1?cl=en ………… copy paste link
Example 1
Preparation of N-{1 -[(E)-3-(4-pentylphenyl)but-2-enoyl]}-L-tryptophyl-D- asparaginyl-L-α-aspartyl-L-threonylglycyl-L-ornithyl-L-α-aspartyl-D-alanyl-L-α- aspartylglycyl-D-seryl-(3R)-3-methyl-L-α-glutamyl-(αS)-α,2-diamino-γ- oxobenzenebutanoic acid (13→4)-lactone (49).


1003 1004

Step 1 : Preparation of (E)-ethyl 3-(4-pentylphenyl) but-2-enoate (1002).
A mixture of commercially available 1-(4-pentylphenyl)ethanone (5 g, 26.3 mmol) and (ethoxycarbonylmethylene)-triphenylphosphorane (18.3 g, 52.5 mmol) was stirred at 150 0C for 48 hours under a nitrogen atmosphere. The reaction mixture was cooled to ambient temperature and diluted with ethyl acetate (50 ml_) and petroleum ether (200 ml_). The suspension was filtered through a fritted funnel. The concentrated filtrate was purified by flash column chromatography with silica gel (petroleum ether : ethyl acetate = 80:1 ) to give the title compound (1.6 g) having the following physical data: 1H NMR (300 MHz, δ, CDCI3) 0.90 (br, 3H), 1.36 (br, 7), 1.63 (br, 2H), 2.58 (s, 3H), 2.63 (br, 2H), 4.22 (q, 2H), 6.15 (s, 1 H), 7.20 (d, 2H), 7.41 (d, 2H).
Step 2: Preparation of (E)-3-(4-pentylphenyl) but-2-enoic acid (1003).
A solution of compound 1002 (1.5 g, 5.77 mmol) in ethanol (50 ml_) and 3N potassium hydroxide (25 ml_) was stirred at 45 0C for 3 hours. The reaction mixture was concentrated and the resulting residue was diluted with water (50 ml_). The aqueous solution was acidified to pH 2 with 1 N hydrochloric acid and extracted with EtOAc (2 * 30 ml_). The combined organic layers were dried with anhydrous sodium sulfate, filtered and concentrated. The residue was purified by flash column chromatography (silica gel, petroleum ether : ethyl acetate = 10:1) to afford the title compound (0.95 g) having the following physical data: 1 H NMR (300 MHz, δ, CDCI3) 0.90 (br, 3H), 1.33 (br, 4H), 1.62 (br, 2H), 2.60 (br, 5H), 6.18 (s, 1 H), 7.18 (d, 2H), 7.42 (d, 2H).
Step 3: Preparation of (E)-3-(4-pentylphenyl)but-2-enoyl chloride (1004).
Oxalyl chloride (3.2 mL, 36.60 mmol) and DMF (50 μl_) were added drop wise to a solution of compound 1003 (5.0 g, 21.52 mmol) in dichloromethane (100 mL) at 0 0C. The reaction solution was warmed up to room temperature and stirred for 4 hours. The reaction mixture was concentrated in vacuum and the residue was dried under hi-vacuum for 3 hours. The crude product was used in the next step without further purification.
Step 4: Preparation of N-{1 -[(E)-3-(4-pentylphenyl)but-2-enoyl]}-L-tryptophyl-D- asparaginyl-L-α-aspartyl-L-threonylglycyl-L-[(N-tert-butoxycarbonyl)-ornithyl]-L-α- aspartyl-D-alanyl-L-α-aspartylglycyl-D-seryl-(3R)-3-methyl-L-α-glutamyl-(αS)-α,2- diamino-γ-oxobenzenebutanoic acid (13-→4)-lactone (1005).
Deacylated BOC-protected daptomycin (3.5Og, 2.23 mmol) and sodium bicarbonate (1.13 g, 61.0 mmol) were dissolved in THF (130 mL) and water (50 mL). The deacylated BOC-protected daptomycin sodium bicarbonate solution was cooled to 0 0C. and a solution of compound 1004 (1.96 g, 7.82 mmol) in THF (20 mL) was then introduced. The reaction mixture was warmed to room temperature and stirred for 4 hours. The mixture was concentrated in vacuum to remove THF. The remaining aqueous solution was loaded on a C18 flash chromatography column (35mηnχ 300mm, Bondesil HF C18 resin purchased from Varian). The column was first washed with water to remove salt and then with methanol to wash out product. Crude compound 1005 (3.46 g) was afforded as a white solid after removal of methanol. MS m/z 1780.8 (M + H)+.
Steps 5-6: Preparation of N-{1-[(E)-3-(4-pentylphenyl)but-2-enoyl]}-L-tryptophyl- D-asparaginyl-L-α-aspartyl-L-threonylglycyl-L-ornithyl-L-α-aspartyl-D-alanyl-L-α- aspartylglycyl-D-seryl-(3R)-3-methyl-L-α-glutamyl-(αS)-α,2-diamino-γ- oxobenzenebutanoic acid (13→4)-lactone (49).
TFA (10 ml_) was added to a solution of compound 1005 (3.46 g) in DCM (50 mL) at room temperature. The reaction mixture was stirred vigorously for 45 minutes and added slowly to vigorously stirring diethyl ether (100 mL). The resulting yellow precipitation was collected by filtration. The crude product was purified by Preparative HPLC to afford the TFA salt of compound 6 (0.75 g). MP carbonate resin (purchased from Biotage) was added to the solution of compound 6 TFA salt (0.70 g, 0.39 mmol) in anhydrous methanol (30.0 mL). The mixture was stirred at room temperature for 4 hours. The resins were removed by filtration and rinsed with methanol. The methanol solution was concentrated under vacuum to give product as off-white solid (408 mg). MS m/z 1680.7 (M + H)+.
Example 1 b
Alternative preparation of N-{1-[(E)-3-(4-pentylphenyl)but-2-enoyl]}- L-tryptophyl-D-asparaginyl-L-α-aspartyl-L-threonylglycyl-L-ornithyl-L-α-aspartyl-D- alanyl-L-α-aspartylglycyl-D-seryl-(3R)-3-methyl-L-α-glutamyl-(αS)-α,2-diamino-γ- oxobenzenebutanoic acid (13→4)-lactone (49).
daptomycin,

1003

A solution of (E)-3-(4-pentylphenyl)but-2-enoic acid (1 100 g, 4.73 mol), Λ/-Ethyl-Λ/’-(3-dimethylaminopropyl)carbodiimide hydrochloride (907 g, 4.73 mol), HOBT (640 g, 4.73 mol) and 4-(dimethylamino)pyridine (22 g, 0.18 mol) in DMF (11 L) was stirred at room temperature for 4 hours at which point the activation of the (E)-3-(4-pentylphenyl)but-2-enoic acid was deemed complete by HPLC.
This reaction mixture was added to a suspension of Deacylated BOC- protected daptomycin (2600 g, 1.66 mol), sodium bicarbonate (804 g, 9.57 mol) in water (11.25 L) and 1 ,4-dioxane (33.75 L). The mixture was stirred at room temperature for 2.5 hours at which time HPLC indicated complete consumption of Deacylated BOC-protected daptomycin. The reaction mixture was diluted with water (22.5 L) and cooled with an ice bath. Concentrated hydrochloric acid (5.25 L) was added while maintaining the internal temperature below 30 0C. After the addition, the solution was stirred at room temperature for 5 days at which time HPLC indicated complete consumption of the Boc protected intermediate.
The reaction mixture was washed with methyl terf-butyl ether (90 L then approximately 60 L then approximately 45 L then approximately 45 L) to remove 1 ,4-dioxane. The remaining solution (approximately 44 L) was adjusted to pH 2.69 with 2N sodium hydroxide (11.3 L) and water (53.4 L). This material was processed by Tangential Flow Filtration (TTF) with a 1 K membrane until the total volume was reduced to 54 L.Water (120 L) was added in two portions and the solution was concentrated to 52 L by continued TTF. The aqueous solution (30 L of 52 L) was purified by chromatography using the following protocol: The aqueous solution was brought to three times of its volume (30 L→90l_) with 20% IPA in aqueous ammonium acetate solution (50 mM). The diluted solution was applied to a 38 L HP20SS resin column at 1.5 L/min. The column was eluted with IPA solution in aqueous 50 mM ammonium acetate (25%→30%→35%, 60 L each concentration).
Fractions (approximately 11 L) were collected and analyzed by HPLC. The fractions with HPLC purity less than 80% were combined and purified again using the same method. The key fractions from both chromatographic separations (with HPLC purity >80%) were combined and acidified with concentrated HCI to pH 2-3. The resulting solution was desalted on an ion exchange column (HP20SS resin, 16 L) which was eluted with WFI (until conductivity = 4.8 μS) followed by IPA in WFI (36 L 10%→ 40 L 60%). The yellow band which was eluted with 60% IPA (approximately 19L) was collected, adjusted to pH 2-3 with concentrated HCI and lyophilized to yield 636.5 g of Compound 49 (HPLC purity of 87.0%). MS m/z 1680.7 (M + H)+.
……………………………..
see formulation
| WO2012162567A1 | May 24, 2012 | Nov 29, 2012 | Cubist Pharmaceuticals, Inc. | Cb-183,315 compositions and related methods |
References
- http://www.cubist.com/downloads/Surotomycin-Fact-Sheet-13013.pdf
-
- Cubist Pharmaceuticals. Cubist products and pipeline. Available online: http://www.cubist.com/products/(accessed on 15 April 2013).
- Cubist Pharmaceuticals. Study of CB-183,315 in patients with Clostridium difficile associated diarrhea.Available online: http://www.clinicaltrials.gov/ct2/show/NCT01597505 (accessed on 15 April 2013).
- Cubist Pharmaceuticals. A study of CB-183,315 in patients with Clostridium difficile associated diarrhea.Available online: http://www.clinicaltrials.gov/ct2/show/NCT01598311 (accessed on 15 April 2013).
- Mascio, C.T.M.; Mortin, L.I.; Howland, K.T.; van, P.A.D.G.; Zhang, S.; Arya, A.; Chuong, C.L.; Kang, C.; Li, T.; Silverman, J.A. In vitro and in vivo characterization of CB-183,315, a novel lipopeptide antibiotic for treatment of Clostridium difficile. Antimicrob. Agents Chemother. 2012, 56, 5023–5030, doi:10.1128/AAC.00057-12.
-
WO2012162567A1 May 24, 2012 Nov 29, 2012 Cubist Pharmaceuticals, Inc. Cb-183,315 compositions and related methods
-
WO2001097851A2 * Jun 18, 2001 Dec 27, 2001 Cubist Pharm Inc Compositions and methods to improve the oral absorption of antimicrobial agents WO2010075215A1 Dec 18, 2009 Jul 1, 2010 Cubist Pharmaceuticals, Inc. Novel antibacterial agents for the treatment of gram positive infections WO2011063419A2 * Nov 23, 2010 May 26, 2011 Cubist Pharmaceuticals Inc. Lipopeptide compositions and related methods
This Little Known Chinese Herb Kills 12,000 Cancer Cells For Every Healthy Cell

 artemisinin
artemisininWormwood
Other common name(s): absinthium, absinth wormwood
Scientific/medical name(s): Artemisia absinthium
Description
Wormwood is a shrubby perennial plant whose upper shoots, flowers, and leaves are used in herbal remedies and as a bitter flavoring for alcoholic drinks. It is native to Europe, northern Africa, and western Asia, and now also grows in North America.
Overview
Available scientific evidence does not support claims that wormwood is effective in treating cancer, the side effects of cancer treatment, or any other conditions. The plant contains a volatile oil with a high level of thujone (see Thuja). There are reports that taking large doses of wormwood internally can cause serious problems with the liver and kidneys. It can also cause nausea, vomiting, stomach pain, headache, dizziness, seizures, numbness of the legs and arms, delirium, and paralysis.
Wormwood, or Artemisia absinthium, should not be confused with sweet wormwood, or Artemisia annua. Although wormwood is related to sweet wormwood, they are used in different ways. Extracts of sweet wormwood have been used in traditional herbal medicine, and an active ingredient, artemisinin, is now used in conventional medical treatment of malaria.
How is it promoted for use?
Wormwood is promoted as a sedative and anti-inflammatory. There are also claims that it can treat loss of appetite, stomach disorders, and liver and gallbladder complaints. In folk medicine it is used for a wide range of stomach disorders, fever, and irregular menstruation. It is also used to fight intestinal worms. Externally, it is applied to poorly healing wounds, ulcers, skin blotches, and insect bites. It is used in Moxibustion treatments for cancer (seeMoxibustion). Available scientific evidence does not support these claims.
What does it involve?
Wormwood is taken in small doses for a short period of time, usually a maximum of 4 weeks. It is available as a capsule and as a liquid that can be added to water to make a tincture. The whole herb is sometimes brewed as a tea. Wormwood oil, washes, or poultices can also be used on the skin. Although pure wormwood is not available, “thujone-free” wormwood extract has been approved by the US Food and Drug Administration (FDA) for use in foods and as a flavoring in alcoholic drinks such as vermouth.
What is the history behind it?
Artemisia absinthium was used by Hippocrates, and the earliest references to wormwood in Western civilization can be found in the Bible. Extract of wormwood was also used in ancient Egypt. The herb is mentioned often in first-century Greek and Roman writings and reportedly was placed in the sandals of Roman soldiers to help soothe their sore feet. It was taken as a treatment for tapeworms as far back as the Middle Ages.
In 1797, Henri Pernod developed absinthe, an alcoholic drink containing distilled spirits of wormwood, fennel, anise and sometimes other herbs. Absinthe became very popular in Europe and the United States in the nineteenth century. It was eventually banned in several countries in the early twentieth century due to its purported ill effects and addictive qualities. More recent analysis has suggested that, when properly prepared and distilled, the thujone content in these drinks was very low. It appears more likely that the addictiveness and other ill effects of absinthe were due to its alcohol content, which is around 60% to 85%. Varying additives or impurities from different distillers may have also produced some of these effects. Even though absinthe is illegal in some countries, various types can be found in some European countries. However, their thujone content is strictly limited. Wormwood is also an ingredient in vermouth and other drinks.
What is the evidence?
Available scientific studies do not support the use of wormwood for the treatment of cancer or the side effects of conventional cancer treatment. There is not enough evidence available to support its use for other conditions. Wormwood oil has been tested in laboratory studies and appears to inhibit the growth of some fungi. However, human tests have not been completed.
Some derivatives of Artemisia annua, or sweet wormwood, a relative of wormwood, have been shown to be effective in the treatment of malaria. In fact, the World Health Organization approved artemisinin for use against malaria in Africa in 2004. These extracts also show some promise in laboratory studies as cancer treatment drugs. Further studies are required to find out whether the anti-cancer results apply to people. It is important to remember that extracted compounds are not the same as the whole herb, and study results are not likely to show the same effects.
Are there any possible problems or complications?
Wormwood should be avoided, especially by women who are pregnant or breast-feeding, by people who have had seizures, and by those with ulcers or stomach irritation. Thujone, a component of wormwood, is known to cause muscle spasms, seizures, and hallucinations if taken internally. In high doses it is known to damage the liver and the kidneys.
Because of its thujone content, large doses of wormwood taken internally can lead to vomiting, stomach and intestinal cramps, headaches, dizziness, nervous system problems, and seizures. Wormwood can also lead to liver failure. The New England Journal of Medicine reported that a man who ordered essential oil of wormwood over the Internet, thinking he had purchased absinthe, suffered liver failure shortly after drinking the oil. Wormwood may also make seizures more likely and may interfere with the anti-convulsant effects of medicines such as phenobarbital.
The plant is a relative of ragweed and daisies. Those with allergies to these types of plants may also be allergic to wormwood. Contact with wormwood can cause rash in some people.
Relying on this type of treatment alone and avoiding or delaying conventional medical care for cancer may have serious health consequences.
