

| Record ID | Title | Status | Phase |
|---|---|---|---|
| NCT03041311 | Carboplatin, Etoposide, and Atezolizumab With or Without Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Extensive Stage Small Cell Lung Cancer (SCLC) | Recruiting | 2 |
| NCT02978716 | Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Combination With Gemcitabineand Carboplatin in Metastatic Triple Negative Breast Cancer (mTNBC) | Recruiting | 2 |
| NCT02514447 | Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Patients With Previously Treated Extensive Stage SCLC Receiving Topotecan Chemotherapy | Recruiting | 2 |
| NCT02499770 | Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Combination With Etoposide and Carboplatin in Extensive Stage Small Cell Lung Cancer (SCLC) | Active, not recruiting | 2 |
Home » Phase2 drugs (Page 8)
Category Archives: Phase2 drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
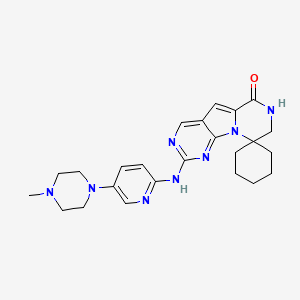
TRILACICLIB, G1T28
Trilaciclib
update 2021/2/12 US FDA APPROVED COSELA
- Molecular FormulaC24H30N8O
- Average mass446.548 Da
- G1T 28
CAS 1374743-00-6
2′-{[5-(4-Methyl-1-piperazinyl)-2-pyridinyl]amino}-7′,8′-dihydro-6’H-spiro[cyclohexane-1,9′-pyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyrimidin]-6′-one
G1T28, SHR 6390
Spiro[cyclohexane-1,9′(6’H)-pyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyrimidin]-6′-one, 7′,8′-dihydro-2′-[[5-(4-methyl-1-piperazinyl)-2-pyridinyl]amino]-
- 7′,8′-Dihydro-2′-[[5-(4-methyl-1-piperazinyl)-2-pyridinyl]amino]spiro[cyclohexane-1,9′(6’H)-pyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyrimidin]-6′-one
- 2′-[[5-(4-Methylpiperazin-1-yl)pyridin-2-yl]amino}-7′,8′-dihydro-6’H-spiro[cyclohexane-1,9′-pyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyrimidin]-6′-one
UNII:U6072DO9XG
Reduction of Chemotherapy-Induced Myelosuppression
Trilaciclib dihydrochloride
1977495-97-8
In phase II clinical development as a chemoprotectant at G1 Therapeutics for first- or second-line treatment in patients with metastatic triple negative breast cancer, in combination with gemcitabine and carboplatin
![]()
PATENT, WO 2014144326, Compound 89 (also referred to as Compound T)
| WO2014144847A3 | |
| Inventors | Norman E. Sharpless, Jay Copeland Strum, John Emerson Bisi, Patrick Joseph Roberts, Francis Xavier Tavares |
| Applicant | G1 Therapeutics, Inc. |
| Norman Sharpless | |
|---|---|
 |
|
| Born | Norman Edward Sharpless September 20, 1966 Greensboro, North Carolina |
| Nationality | American |
| Other names | Ned Sharpless |
| Occupation | Director, Lineberger Comprehensive Cancer Center Founder, G1 Therapeutics ($GTHX) |
| Notable work | Wellcome Distinguished Professor, American Society of Clinical Investigation Member, Association of American Cancer Institute board of directors, |
NCI Director Dr. Norman E. Sharpless

NCI Director Dr. Norman E. Sharpless, Credit: National Institutes of Health
Norman E. “Ned” Sharpless, M.D., was officially sworn in as the 15th director of the National Cancer Institute (NCI) on October 17, 2017. Prior to his appointment, Dr. Sharpless served as the director of the University of North Carolina (UNC) Lineberger Comprehensive Cancer Center, a position he held since January 2014.
Dr. Sharpless was a Morehead Scholar at UNC–Chapel Hill and received his undergraduate degree in mathematics. He went on to pursue his medical degree from the UNC School of Medicine, graduating with honors and distinction in 1993. He then completed his internal medicine residency at the Massachusetts General Hospital and a hematology/oncology fellowship at Dana-Farber/Partners Cancer Care, both of Harvard Medical School in Boston.
After 2 years on the faculty at Harvard Medical School, he joined the faculty of the UNC School of Medicine in the Departments of Medicine and Genetics in 2002. He became the Wellcome Professor of Cancer Research at UNC in 2012.
Dr. Sharpless is a member of the Association of American Physicians as well as the American Society for Clinical Investigation (ASCI), the nation’s oldest honor society for physician–scientists, and served on the ASCI council from 2011 to 2014. Dr. Sharpless was an associate editor of Aging Cell and deputy editor of the Journal of Clinical Investigation. He has authored more than 150 original scientific papers, reviews, and book chapters, and is an inventor on 10 patents. He cofounded two clinical-stage biotechnology companies: G1 Therapeutics and HealthSpan Diagnostics.
In addition to serving as director of NCI, Dr. Sharpless continues his research in understanding the biology of the aging process that promotes the conversion of normal self-renewing cells into dysfunctional cancer cells. Dr. Sharpless has made seminal contributions to the understanding of the relationship between aging and cancer, and in the preclinical development of novel therapeutics for melanoma, lung cancer, and breast cancer.
Synthesis
WO 2016040858


Trilaciclib (G1T28)
Trilaciclib is a potential first-in-class short-acting CDK4/6 inhibitor in development to preserve hematopoietic stem cells and enhance immune system function during chemotherapy. Trilaciclib is administered intravenously prior to chemotherapy and has the potential to significantly improve treatment outcomes.
G1 is currently evaluating trilaciclib in four Phase 2 clinical trials: three studies in patients with small-cell lung cancer (SCLC), and one study in patients with triple-negative breast cancer (TNBC). Preliminary data from the SCLC trials were presented at the American Society of Clinical Oncology 2017 Annual Meeting and at the 2016 World Conference on Lung Cancer.
Data from a Phase 1 trial in healthy volunteers were presented at the American Society of Clinical Oncology 2015 Annual Meeting and published in Science Translational Medicine. Trilacicilib has been extensively studied in animals; these preclinical data have been presented at several scientific meetings and published in Molecular Cancer Therapeutics, Science Translational Medicine, and Cancer Discovery.
Trilaciclib is a small molecule, competitive inhibitor of cyclin dependent kinases 4 and 6 (CDK4/6), with potential antineoplastic and chemoprotective activities. Upon intravenous administration, trilaciclib binds to and inhibits the activity of CDK4/6, thereby blocking the phosphorylation of the retinoblastoma protein (Rb) in early G1. This prevents G1/S phase transition, causes cell cycle arrest in the G1 phase, induces apoptosis, and inhibits the proliferation of CDK4/6-overexpressing tumor cells. In patients with CDK4/6-independent tumor cells, G1T28 may protect against multi-lineage chemotherapy-induced myelosuppression (CIM) by transiently and reversibly inducing G1 cell cycle arrest in hematopoietic stem and progenitor cells (HSPCs) and preventing transition to the S phase. This protects all hematopoietic lineages, including red blood cells, platelets, neutrophils and lymphocytes, from the DNA-damaging effects of certain chemotherapeutics and preserves the function of the bone marrow and the immune system. CDKs are serine/threonine kinases involved in the regulation of the cell cycle and may be overexpressed in certain cancer cell types. HSPCs are dependent upon CDK4/6 for proliferation.
Trilaciclib (G1T28) is a CDK4/6 inhibitor in phase II clinical development as a chemoprotectant at G1 Therapeutics for first- or second-line treatment in patients with metastatic triple negative breast cancer, in combination with gemcitabine and carboplatin. Also, phase II trials are ongoing in newly diagnosed, treatment-naive small-cell lung cancer patients, in combination with carboplatin, etoposide, and atezolizumab and phase I trials in previously treated small-cell lung cancer patients, in combination with topotecan.
U.S. Patent Nos. 8,822,683; 8,598,197; 8,598,186, 8,691,830, 8,829,102, 8,822,683, 9, 102,682, 9,499,564, 9,481,591, and 9,260,442, filed by Tavares and Strum and assigned to Gl Therapeutics describe a class of N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amine cyclin dependent kinase inhibitors including those of the formula with variables as defined therein):

U.S. Patent Nos. 9,464,092, 9,487,530, and 9,527,857 which are also assigned to Gl Therapeutics describe the use of the above pyrimidine-based agents in the treatment of cancer.
These patents provide a general synthesis of the compounds that is based on a coupling reaction of a fused chloropyrimidine with a heteroaryl amine to form the central disubstituted amine. Such coupling reactions are sometimes referred to as Buchwald coupling (see WO Ί56 paragraph 127; reference WO 2010/020675). The lactam of the fused chloropyrimidine, for example, a 2-chloro-spirocyclo-pyrrolo[2,3-d]pyrimidine-one such as Intermediate K as shown below can be prepared by dehydration of the corresponding carboxylic acid. The reported process to prepare intermediate IK requires seven steps.

(Intermediate IK; page 60, paragraph 215 of WO Ί56)
WO 2013/148748 (U.S. S.N. 61/617,657) entitled “Lactam Kinase Inhibitors” filed by Tavares, and also assigned to Gl Therapeutics likewise describes the synthesis of N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amines via the coupling reaction of a fused chloropyrimidine with a heteroaryl amine to form the central disubstituted amine.
WO 2013/163239 (U.S. S.N. 61/638,491) “Synthesis of Lactams” describes a method for the synthesis of this class of compounds with the variation that in the lactam preparation step, a carboxylic acid can be cyclized with a protected amine in the presence of a strong acid and a dehydrating agent, which can be together in one moiety as a strong acid anhydride. The purported improvement is that cyclization can occur without losing the protecting group on the amine before cyclization. The typical leaving group is “tBOC” (t-butoxycarbonyl). The application teaches (page 2 of WO 2013/163239) that the strong acid is, for example, trifluoroacetic acid anhydride, tribromoacetic acid anhydride, trichloroacetic acid anhydride or mixed anhydrides. An additional step may be necessary to take off the N-protecting group. The dehydrating agent can be a carbodiimide-based compound such as DCC (Ν,Ν-dicyclohexylcarbodiimide), EDC (l-ethyl-3-(3-dimethylaminopropyl)carbodiimide, or DIC (Ν,Ν-diisopropylcarbodiimide). DCC and DIC are in the same class of reagents-carbodiimides. DIC is sometimes considered better because it is a liquid at room temperature, which facilitates reactions.
WO 2015/061407 filed by Tavares and licensed to Gl Therapeutics also describes the synthesis of these compounds via the coupling of a fused chloropyrimidine with a heteroaryl amine to form the central disubstituted amine. WO ‘407 focuses on the lactam production step and in particular describes that the fused lactams of these compounds can be prepared by treating the carboxylic acid with an acid and a dehydrating agent in a manner that a leaving group on the amine is not removed during the amide-forming ring closing step.
Other publications that describe compounds of this general class include the following. WO 2014/144326 filed by Strum et al. and assigned to Gl Therapeutics describes compounds and methods for protection of normal cells during chemotherapy using pyrimidine based CDK4/6 inhibitors. WO 2014/144596 filed by Strum et al. and assigned to Gl Therapeutics describes compounds and methods for protection of hematopoietic stem and progenitor cells against ionizing radiation using pyrimidine based CDK4/6 inhibitors. WO 2014/144847 filed by Strum et al. and assigned to Gl Therapeutics describes HSPC-sparing treatments of abnormal cellular proliferation using pyrimidine based CDK4/6 inhibitors. WO2014/144740 filed by Strum et al. and assigned to Gl Therapeutics describes highly active anti -neoplastic and anti-proliferative pyrimidine based CDK 4/6 inhibitors. WO 2015/161285 filed by Strum et al. and assigned to Gl Therapeutics describes tricyclic pyrimidine based CDK inhibitors for use in radioprotection. WO 2015/161287 filed by Strum et al. and assigned to Gl Therapeutics describes analogous tricyclic pyrimidine based CDK inhibitors for the protection of cells during chemotherapy. WO 2015/161283 filed by Strum et al. and assigned to Gl Therapeutics describes analogous tricyclic pyrimidine based CDK inhibitors for use in HSPC-sparing treatments of RB-positive abnormal cellular proliferation. WO 2015/161288 filed by Strum et al. and assigned to Gl Therapeutics describes analogous tricyclic pyrimidine based CDK inhibitors for use as anti -neoplastic and anti-proliferative agents. WO 2016/040858 filed by Strum et al. and assigned to Gl Therapeutics describes the use of combinations of pyrimidine based CDK4/6 inhibitors with other anti-neoplastic agents. WO 2016/040848 filed by Strum et al. and assigned to Gl Therapeutics describes compounds and methods for treating certain Rb-negative cancers with CDK4/6 inhibitors and topoisomerase inhibitors.
Other biologically active fused spirolactams and their syntheses are described, for example, in the following publications. Griffith, D. A., et al. (2013). “Spirolactam-Based Acetyl-CoA Carboxylase Inhibitors: Toward Improved Metabolic Stability of a Chromanone Lead Structure.” Journal of Medicinal Chemistry 56(17): 7110-7119, describes metabolically stable spirolactams wherein the lactam resides on the fused ring for the inhibition of acetyl-CoA carboxylase. WO 2013/169574 filed by Bell et al. describes aliphatic spirolactams as CGRP receptor antagonists wherein the lactam resides on the spiro ring. WO 2007/061677 filed by Bell et al. describes aryl spirolactams as CGRP receptor antagonists wherein the lactam resides on the spiro ring. WO 2008/073251 filed by Bell et al. describes constrained spirolactam compounds wherein the lactam resides on the spiro ring as CGRP receptor antagonists. WO 2006/031606 filed by Bell et al. describes carboxamide spirolactam compounds wherein the spirolactam resides on the spiro ring as CGRP receptor antagonists. WO 2006/031610, WO 2006/031491, and WO 2006/029153 filed by Bell et al. describe anilide spirolactam compounds wherein the spirolactam resides on the spiro ring; WO 2008/109464 filed by Bhunai et al. describes spirolactam compounds wherein the lactam resides on the spiro ring which is optionally further fused.
Given the therapeutic activity of selected N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amines, it would be useful to have additional methods for their preparation. It would also be useful to have new intermediates that can be used to prepare this class of compounds.
PATENT
WO 2014144596
PATENT
Compound 89 (also referred to as Compound T)
| WO2014144847A3 | |
| Inventors | Norman E. Sharpless, Jay Copeland Strum, John Emerson Bisi, Patrick Joseph Roberts, Francis Xavier Tavares |
| Applicant | G1 Therapeutics, Inc. |
EXAMPLES
Intermediates B, E, K, L, 1A, IF and 1CA were synthesized according to US 8,598,186 entitled CDK Inhibitors to Tavares, F.X. and Strum, J.C..
The patents WO 2013/148748 entitled Lactam Kinase Inhibitors to Tavares, F.X., WO 2013/163239 entitled Synthesis of Lactams to Tavares, F.X., and US 8,598,186 entitled CDK Inhibitors to Tavares, F.X. and Strum, J.C. are incorporated by reference herein in their entirety. Example 1
Synthesis of tert-butyl N- [2- [(5-bromo-2-chloro-pyrimidin-4yl)amino] ethyl] carbamate, Compound 1

To a solution of 5-bromo-2,4-dichloropyrimidine (3.2 g, 0.0135 mol) in ethanol (80 mL) was added Hunig’s base (3.0 mL) followed by the addition of a solution of N-(tert- butoxycarbonyl)-l,2-diaminoethane (2.5 g, 0.0156 mole) in ethanol (20 mL). The contents were stirred overnight for 20 hrs. The solvent was evaporated under vacuum. Ethyl acetate (200 mL) and water (100 mL) were added and the layers separated. The organic layer was dried with magnesium sulfate and then concentrated under vacuum. Column chromatography on silica gel using hexane/ethyl acetate (0- 60%) afforded tert-butyl N-[2-[(5-bromo-2-chloro-pyrimidin-4- yl)amino]ethyl]carbamate. 1HNMR (d6-DMSO) δ ppm 8.21 (s, 1H), 7.62 (brs, 1H), 7.27 (brs, 1H), 3.39 (m, 2H), 3.12 (m, 2H), 1.34 (s, 9H). LCMS (ESI) 351 (M + H).
Example 2
Synthesis of tert-butyl N-[2-[[2-chloro-5-(3,3-diethoxyprop-l-ynyl)pyrimidin-4- yl] amino] ethyl] carbamate, Compound 2

To tert-butyl N-[2-[(5-bromo-2-chloro-pyrimidin-4-yl)amino]ethyl]carbamate (1.265 g, 6 mmol) in THF (10 mL) was added the acetal (0.778 mL, 5.43 mmol), Pd(dppf)CH2Cl2 (148 g), and triethylamine (0.757 mL, 5.43 mmol). The contents were degassed and then purged with nitrogen. To this was then added Cul (29 mg). The reaction mixture was heated at reflux for 48 hrs. After cooling, the contents were filtered over CELITE™ and concentrated. Column chromatography of the resulting residue using hexane/ethyl acetate (0- 30%) afforded tert-butyl N- [2- [ [2-chloro-5 -(3 ,3 -diethoxyprop- 1 -ynyl)pyrimidin-4-yl]amino] ethyl] carbamate. 1HNMR (d6-DMSO) δ ppm 8.18 (s, 1H), 7.63 (brs, 1H), 7.40 (brs, 1H), 5.55 (s, 1H), 3.70 (m, 2H), 3.60 (m, 2H), 3.42 (m, 2H), 3.15 (m, 2H), 1.19 – 1.16 (m, 15H). LCMS (ESI) 399 (M + H).
Example 3
Synthesis of tert-butyl N-[2-[2-chloro-6-(diethoxymethyl)pyrrolo[2,3-d]pyrimidin-7- yl] ethyl] carbamate, Compound 3

To a solution of the coupled product (2.1 g, 0.00526 mole) in THF (30 mL) was added TBAF solid (7.0 g). The contents were heated to and maintained at 65 degrees for 2 hrs. Concentration followed by column chromatography using ethyl acetate/hexane (0-50%) afforded tert-butyl N-[2-[2-chloro-6-(diethoxymethyl)pyrrolo[2,3-d]pyrimidin-7-yl]ethyl]carbamate as a pale brown liquid (1.1 g). 1FiNMR (d6-DMSO) δ ppm 8.88 (s, 1H), 6.95 (brs, 1H), 6.69 (s, 1H), 5.79 (s, 1H), 4.29 (m, 2H), 3.59 (m, 4H), 3.34 (m, 1H), 3.18 (m, 1H), 1.19 (m, 9H), 1.17 (m, 6H). LCMS (ESI) 399 (M + H).
Example 4
Synthesis of tert-buty\ N-[2-(2-chloro-6-formyl-pyrrolo [2,3-d] pyrimidin-7- yl)ethyl] carbamate, Compound 4

To the acetal (900 mg) from the preceeding step was added AcOH (8.0 mL) and water
(1.0 mL). The reaction was stirred at room temperature for 16 hrs. Cone, and column chromatography over silica gel using ethyl acetate/hexanes (0- 60%) afforded tert-butyl N-[2-(2- chloro-6-formyl-pyrrolo[2,3-d]pyrimidin-7-yl)ethyl]carbamate as a foam (0.510 g). 1HNMR (d6-DMSO) δ ppm 9.98 (s, 1H), 9.18 (s, 1H), 7.66 (s, 1H), 6.80 (brs, 1H), 4.52 (m, 2H), 4.36 (m, 2H), 1.14 (s, 9H). LCMS (ESI) 325 (M + H).
Example 5
Synthesis of 7- [2-(teri-butoxycarbonylamino)ethyl] -2-chloro-pyrrolo [2,3-d] pyrimidine-6- carboxylic acid, Compound 5

To the aldehyde (0.940 g) from the preceeding step in DMF (4 mL) was added oxone (1.95 g, 1.1 eq). The contents were stirred at room temp for 7 hrs. Silica gel column chromatography using hexane/ethyl acetate (0- 100%) afforded l-\2-(tert- butoxycarbonylamino)ethyl]-2-chloro-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid (0.545 g). 1HNMR (d6-DMSO) δ ppm 9.11 (s, 1H), 7.39 (s, 1H), 4.38 (m, 2H), 4.15 (m, 2H), 1.48 (m, 9H). LCMS (ESI) 341(M + H).
Example 6
Synthesis of methyl 7-[2-(teri-butoxycarbonylamino)ethyl]-2-chloro-pyrrolo[2,3- d]pyrimidine-6-carboxylate, Compound 6

To a solution of 2-chloro-7-propyl-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid (0.545 g, 0.00156 mole) from the preceeding step in toluene (3.5 mL) and MeOH (1 mL) was added TMS- diazomethane (1.2 mL). After stirring overnight at room temperature, the excess of TMS- diazomethane was quenched with acetic acid (3 mL) and the reaction was concentrated under vacuum. The residue was purified by silica gel column chromatography with hexane/ethyl acetate (0- 70%) to afford methyl 7-[2-(tert-butoxycarbonylamino)ethyl]-2-chloro-pyrrolo[2,3- d]pyrimidine-6-carboxylate as an off white solid (0.52 g). 1HNMR (d6-DMSO) δ ppm 9.10 (s, 1H), 7.45 (s, 1H), 6.81 (brs, 1H) 4.60 (m, 2H), 3.91 (s, 3H), 3.29 (m, 2H), 1.18 (m, 9H) LCMS (ESI) 355 (M + H).
Example 7
Synthesis of Chloro tricyclic amide, Compound 7

To methyl 7- [2-(tert-butoxycarbonylamino)ethyl] -2-chloro-pyrrolo [2,3 -d]pyrimidine-6- carboxylate (0.50 g, 0.0014 mole) from the preceeding step in dichloromethane (2.0 mL) was added TFA (0.830 mL). The contents were stirred at room temperature for 1 hr. Concentration under vacuum afforded the crude amino ester which was suspended in toluene (5 mL) and Hunig’s base (0.5 mL). The contents were heated at reflux for 2 hrs. Concentration followed by silica gel column chromatography using hexane/ethyl acetate (0- 50%) afforded the desired chloro tricyclic amide (0.260 g). 1HNMR (d6-DMSO) δ ppm 9.08 (s, 1H), 8.48 (brs, 1H), 7.21 (s, 1H) 4.33 (m, 2H), 3.64 (m, 2H). LCMS (ESI) 223 (M + H).
Example 8
Synthesis of chloro-N-methyltricyclic amide, Compound 8

To a solution of the chloro tricycliclactam, Compound 7, (185 mg, 0.00083 mole) in DMF (2.0 mL) was added sodium hydride (55% dispersion in oil, 52 mg). After stirring for 15 mins, methyl iodide (62 μί, 1.2 eq). The contents were stirred at room temperature for 30 mins. After the addition of methanol (5 mL), sat NaHCOs was added followed by the addition of ethyl acetate. Separation of the organic layer followed by drying with magnesium sulfate and concentration under vacuum afforded the N-methylated amide in quantitative yield. 1FiNMR (d6-DMSO) δ ppm 9.05 (s, 1H), 7.17 (s, 1H) 4.38 (m, 2H), 3.80 (m, 2H), 3.05 (s, 3H). LCMS (ESI) 237 (M + H). Example 9
Synthesis of l-methyl-4-(6-nitro-3-pyridyl)piperazine, Compound 9

To 5-bromo-2-nitropyridine (4.93 g, 24.3 mmole) in DMF (20 mL) was added N- methylpiperazine (2.96 g, 1.1 eq) followed by the addition of DIPEA (4.65 mL, 26.7 mmole). The contents were heated at 90 degrees for 24 hrs. After addition of ethyl acetate (200 mL), water (100 mL) was added and the layers separated. Drying followed by concentration afforded the crude product which was purified by silica gel column chromatography using (0-10%) DCM/Methanol. 1HNMR (d6-DMSO) δ ppm 8.26 (s, 1H), 8.15 (1H, d, J = 9.3 Hz), 7.49 (1H, d, J = 9.4 Hz), 3.50 (m, 4H), 2.49 (m, 4H), 2.22 (s, 3H).
Example 10
Synthesis of 5-(4-methylpiperazin-l-yl)pyridin-2-amine, Compound 10

To l-methyl-4-(6-nitro-3-pyridyl)piperazine (3.4 g) in ethyl acetate (100 mL) and ethanol (100 mL) was added 10%> Pd/C (400 mg) and then the reaction was stirred under hydrogen (10 psi) overnight. After filtration through CELITE™, the solvents were evaporated and the crude product was purified by silica gel column chromatography using DCM/ 7N ammonia in MeOH (0- 5%) to afford 5-(4-methylpiperazin-l-yl)pyridin-2-amine (2.2 g). 1HNMR (d6-DMSO) δ ppm 7.56 (1H, d, J = 3 Hz), 7.13 (1H, m), 6.36 (1H, d, J = 8.8 Hz), 5.33 (brs, 2H), 2.88 (m, 4H), 2.47 (m, 4H), 2.16 (s, 3H).
Example 11
Synthesis of tert-butyl 4-(6-amino-3-pyridyl)piperazine-l-carboxylate, Compound 11

This compound was prepared as described in WO 2010/020675 Al .
Synthesis of Compound 89 (also referred to as Compound T)

Compound 89 was synthesized in a similar manner to that described for compound 78 and was converted to an HCl salt. 1HNMR (600 MHz, DMSO-d6) δ ppm 1.47 (br. s., 6 H) 1.72 (br. s., 2 H) 1.92 (br. s., 2 H) 2.77 (br. s., 3 H) 3.18 (br. s., 2 H) 3.46 (br. s., 2 H) 3.63 (br. s., 2 H) 3.66 (d, J=6.15 Hz, 2 H) 3.80 (br. s., 2 H) 7.25 (s, 1 H) 7.63 (br. s., 2 H) 7.94 (br. s., 1 H) 8.10 (br. s., 1 H) 8.39 (br. s., 1 H) 9.08 (br. s., 1 H) 11.59 (br. s., 1 H). LCMS (ESI) 447 (M + H)
PATENT
WO 2014144740
PATENT
Preparation of Active Compounds
Syntheses
The disclosed compounds can be made by the following general schemes:

Scheme 1
In Scheme 1, Ref-1 is WO 2010/020675 Al; Ref-2 is White, J. D.; et al. J. Org. Chem. 1995, 60, 3600; and Ref-3 Presser, A. and Hufher, A. Monatshefte fir Chemie 2004, 135, 1015.

Scheme 2
In Scheme 2, Ref-1 is WO 2010/020675 Al; Ref-4 is WO 2005/040166 Al; and Ref-5 is Schoenauer, K and Zbiral, E. Tetrahedron Letters 1983, 24, 573.

92


93 
3) Pd/C/H2 ![]()
Scheme 6

Scheme 7
NHfOH

Scheme 8
In Scheme 8, Ref-1 is WO 2010/020675 Al; Ref-2 is WO 2005/040166 Al; and Ref-3 is Schoenauer, K and Zbiral, E. Tetrahedron Letters 1983, 24, 573.
Alternatively, the lactam can be generated by reacting the carboxylic acid with a protected amine in the presence of a strong acid and a dehydrating agent, which can be together in one moiety as a strong acid anhydride. Examples of strong acid anhydrides include, but are not limited to, trifluoroacetic acid anhydride, tribromoacetic acid anhydride, trichloroacetic acid anhydride, or mixed anhydrides. The dehydrating agent can be a carbodiimide based compound such as but not limited to DCC (Ν,Ν-dicyclohexylcarbodiimide), EDC (l-ethyl-3-(3-
dimethylaminopropyl)carbodiimide or DIC (Ν,Ν-diisopropylcarbodiimide). An additional step may be necessary to take off the N-protecting group and the methodologies are known to those skilled in the art.
Alternatively, the halogen moiety bonded to the pyrimidine ring can be substituted with any leaving group that can be displaced by a primary amine, for example to create an intermediate for a final product such as Br, I, F, SMe, SO2Me, SOalkyl, SO2alkyl. See, for Exmaple PCT /US2013/037878 to Tavares.
Other amine intermediates and final amine compounds can be synthesized by those skilled in the art. It will be appreciated that the chemistry can employ reagents that comprise reactive functionalities that can be protected and de-protected and will be known to those skilled in the art at the time of the invention. See for example, Greene, T.W. and Wuts, P.G.M., Greene’s Protective Groups in Organic Synthesis, 4th edition, John Wiley and Sons.

Scheme 9
CDK4/6 Inhibitors of the present invention can be synthesized according to the generalized Scheme 9. Specific synthesis and characterization of the Substituted 2-aminopyrmidines can be found in, for instance, WO2012/061156.
Compounds T, Q, GG, and U were prepared as above and were characterized by mass spectrometry and NMR as shown below:
Compound T
1H NMR (600 MHz, DMSO- d6) ppm 1.47 (br. s., 6 H) 1.72 (br. s., 2 H) 1.92 (br. s., 2 H) 2.77 (br. s., 3 H) 3.18 (br. s., 2 H) 3.46 (br. s., 2 H) 3.63 (br. s., 2 H) 3.66 (d, J=6.15 Hz, 2 H) 3.80 (br. s., 2 H) 7.25 (s, 1 H) 7.63 (br. s., 2 H) 7.94 (br. s., 1 H) 8.10 (br. s., 1 H) 8.39 (br. s., 1 H) 9.08 (br. s., 1 H) 11.59 (br. s., 1 H). LCMS ESI (M + H) 447.
PATENT
Synthesis of N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amines. The application appears to be particularly focused on methods for the preparation of trilaciclib and an analog of it. Trilaciclib is the company’s lead CDK4/6 inhibitor presently in phase II trials against small-cell lung cancer and triple negative breast cancer. Interestingly, the company is working on a second CDK4/6 inhibitor, G1T38 , which is in a phase II trial against breast cancer.
GENERAL METHODS
The structure of starting materials, intermediates, and final products was confirmed by standard analytical techniques, including NMR spectroscopy and mass spectrometry. Unless otherwise noted, reagents and solvents were used as received from commercial suppliers. Proton nuclear magnetic resonance spectra were obtained on a Bruker AVANCE 500 at 500 MHz in DMSO-dis. HPLC analyses were performed on a Waters HPLC using the below HPLC method.
HPLC Method
Column: Atlantis T3 (150 χ 4.6, 3 μιη)
Column Temperature: 40°C
Flow Rate: 1 mL/min
Detection: UV @ 275 nm
Analysis Time: 36 min
Mobile Phase A: Water (with 0.1% Trifluoroacetic Acid)
Mobile Phase B : Acetonitrile (with 0.1% Trifluoroacetic Acid)
Sample preparation: dissolve PC sample, wet or dry solid (~1 mg of active compound) in acetonitrile/water (1/1) to achieve complete dissolution.
HPLC Method Gradient

Example 1. General Routes of Synthesis

Scheme 1-1 : Starting from an appropriately substituted halo pyrimidine, compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a substituted spirolactam. In Step 2 the appropriately substituted spirolactam is protected with a group selected from R2. In Step 3 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 3, Step 4, Step 5, or Step 6. Oxidation prior to Step 3 results in undesired byproducts. In Step 4 the hydroxyl group of the fused spirolactam is converted to a leaving group.
In Step 5 the leaving group is dehydrated to afford a compound of Formula IV. In Step 6 the compound of Formula IV is optionally deprotected.

Scheme 1-2: Starting from an appropriately substituted halo pyrimidine compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a substituted spirolactam. In Step 2 the appropriately substituted spirolactam is protected with a group selected from R2. In Step 3 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam of Formula IV. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 3 or Step 4. Oxidation prior to Step 3 results in undesired byproducts. In Step 4 the compound of Formula IV is optionally deprotected.

Scheme 1-3 : Starting from an appropriately substituted alkyl glycinate, compounds of the present invention can be prepared. In Step 1 the appropriately substituted alkyl glycinate is subjected to cyclohexanone and TMSCN in the presence of base to afford a cyano species. In Step 2 the appropriately substituted cyanospecies is reduced and subsequently cyclized to afford a compound of Formula I.
Scheme 1-4

Scheme 1-4: Starting from an appropriately substituted l-(aminomethyl)cyclohexan-l-amine, compounds of the present invention can be prepared. In Step 1 the appropriately substituted l-(aminomethyl)cyclohexan-l -amine is reductively aminated with an aldehyde. In Step 2 the appropriately substituted cyclohexane amine is optionally deprotected (i.e.: the group selected from R2 if not H is optionally replaced by H). In Step 3 the cyclohexane amine is cyclized to afford a compound of Formula I. In Step 4 the compound of Formula I is optionally protected.
1-5

Conversion

Scheme 1-5: Starting from an appropriately substituted halo pyrimidine, compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a
substituted spirolactam. In Step 2 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 2, Step 3, Step 4, or Step 5. Oxidation prior to Step 2 results in undesired byproducts. In Step 3 the hydroxyl group of the fused spirolactam is converted to a leaving group. In Step 4 the leaving group is dehydrated to afford a compound of Formula IV. In Step 5 the compound of Formula IV is optionally deprotected.
S

Scheme 1-6: Starting from an appropriately substituted halo pyrimidine compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a substituted spirolactam. In Step 2 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam of Formula IV. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 2 or Step 3. Oxidation prior to Step 2 results in undesired byproducts. In Step 3 the compound of Formula IV is optionally deprotected.

Scheme 1-7: Starting from compound of Formula IV a CDK4/6 inhibitor can be prepared. In Step 1 a heteroaryl amine is subjected to a base and a compound of Formula IV is added slowly under chilled conditions to afford a nucleophilic substitution reaction. The compound of Formula IV can previously be prepared as described in the schemes herein.
Example 2. Representative Routes of Synthesis
Scheme 2-1

quant, yield 2 steps
isolated

70% yield 2 steps 75% yield 95% yield
isolated isolated isolated
Scheme 2-1 : An ester route is one embodiment, of the present invention. Ideally, the best synthesis scheme would afford crystalline intermediates to provide material of consistent purity without column chromatography, and high yielding steps while using safe and cost effective reagents when possible.
The first step in the ester route is a SNAr nucleophilic substitution of CI group in commercially available ester 3 using spirolactam 4. Due to low reactivity of 4, a reaction temperature of 85-95 °C was required. Because of the temperature requirements, DIPEA and dimethylacetamide were selected as the base and solvent, respectively. The reaction follows second-order kinetics and usually stalls after -85% conversion. Therefore, the reaction was typically stopped after 60 hours by first cooling it to room temperature at which point solid formation was observed. The mixture was then partitioned between MTBE and water and product was filtered with excellent purity with -53% yield of the desired product 5. The obtained
compound 5 was protected with a Boc group using Boc anhydride and DMAP as the catalyst and dichloromethane as the solvent. The intermediate 6 was obtained in a quantitative yield. Due to the semi-solid nature of compound 6, the material was taken to the next step without further purification. The Dieckmann condensation was initially performed with strong bases such as LiHMDS and tBuOK. A similar result to the aldehyde route (Scheme 2-2) was obtained: a partial deprotection of Boc group was observed that required column chromatography. However, the best results were obtained when DBU was used as base and THF as solvent. The reaction outcome was complete, clean conversion of 6 to 7. Moreover, the product crystallized from the reaction mixture upon seeding, and a quantitative yield was obtained for the two steps.
The hydroxyl group of 7 was removed via a two-step procedure. First, compound 7 was converted completely into triflate 8 using triflic anhydride and triethylamine in dichloromethane. The reaction was found to proceed well at 0°C. Due to the potential instability of the triflate intermediate, it was not isolated. It was immediately taken to the next step and reduced with triethylsilane and palladium tetrakis to afford the product 9 after ethyl acetate crystallization in -70% yield. The Boc group of 9 was removed using trifluoroacetic acid in dichloromethane to afford 10. Intermediate 10 was converted into the final sulfone 11 using Oxone™ in acetonitrile/water solvent system.
The obtained sulfone 11 was use-tested in the coupling step and was found to perform well. In conclusion, the route to sulfone 11 was developed which eliminated the use of column chromatography with good to excellent yields on all steps.
Scheme 2-2

Molecular Weight: 421 
Scheme 2-2: The first step of Scheme 2-2 consistently afforded product 13 contaminated with one major impurity found in substantial amount. Thorough evaluation of the reaction impurity profile by LC-MS and 2D MR was performed, which showed the impurity was structurally the condensation of two aldehyde 12 molecules and one molecule of lactam 4. Therefore, column chromatography was required to purify compound 13, which consistently resulted in a modest 30% yield. A solvent screen revealed that sec-butanol, amyl alcohol, dioxane, and tert-butanol can all be used in the reaction but a similar conversion was observed in each case. However, tert-butanol provided the cleanest reaction profile, so it was selected as a solvent for the reaction. Assessing the impact of varying the stoichiometric ratio of 4 and 12 on the reaction outcome was also investigated. The reaction was performed with 4 equivalents of amine 4 in an attempt to disrupt the 2: 1 aldehyde/amine composition of the impurity. The result was only a marginal increase in product 13 formation. The temperature impact on the reaction outcome was evaluated next. The coupling of aldehyde 12 and 4 was investigated at two different temperatures: 50 °C and 40 °C with 1 : 1 ratio of aldehyde/amine. Reactions were checked at 2 and 4 hours and then every 12 hours. The reaction progress was slow at 50°C and was accompanied by growth of other impurities. The reaction at 40°C was much cleaner; however the conversion was lower in the same time period. The mode of addition of the reagents was investigated as well at 80°C with a slow addition (over 6 hours) of either aldehyde 12 or amine 4 to the reaction mixture. The product distribution did not change and an about 1 to 1 ratio was observed between product and impurity when amine 4 was added slowly to the reaction mixture containing aldehyde 12 and
DIPEA at reflux. The product distribution did change when aldehyde 12 was added slowly to the mixture of amine 4 and DIPEA. However, the major product of the reaction was the undesired impurity. Other organic bases were tried as well as different ratios of DIPEA. No product was observed when potassium carbonate was used as a base. The results of the experiments are presented in Table 1 below.
Table 1

Compound 13 was successfully formed in three cases: triethylamine, 2,6-lutidine and DIPEA, with the DIPEA result being the best. The use of Boc protected spirolactam 4 had no effect on the impurity formation as well. Its utilization was speculated to be beneficial in performing the coupling step together with the following step, preparation of compound 14.
The major impurity formed during Step 1 of Scheme 2-2 is:

Chemical Formula:€2)Η;Μ(¾ 6( 2ί>2
Molecular Weight: 527.4903
The second step (Boc protection of the free lactam) proceeded well using DMAP as a catalyst in dichloromethane at room temperature. The product 14 is a thick oil, and, therefore, cannot be purified by crystallization. The Boc protected intermediate 14 was cyclized successfully into the desired pentacyclic structure 10 upon treatment with a strong base such as LiHMDS or tBuOK. Surprisingly, the Boc group was partially removed during the reaction. The level of deprotection was independent from the internal reaction temperature and was positively correlated with excess of base used. Therefore the mixture of the desired product 10 and 10-Boc compound was treated with acid to completely deprotect Boc group. The conversion of methyl sulfide into the final sulfone 11 was carried out with Oxone™. Initially a mixture of methanol and water was used for the reaction. As the result, a partial displacement of sulfone by methoxy group was detected. The methanol was replaced with acetonitrile and the sulfone displacement was eliminated.
In summary, the ester route (Scheme 2-1) is preferred because:
1. Formation of the impurity during the first step of Scheme 2-2 was unavoidable and resulted in yields of < 35%.
2. Column purification was required to isolate intermediate 14.
3. The aldehyde starting material was not commercially available and required two synthetic steps from the corresponding ester.

Scheme 2-3 : Starting with cyclohexanone, compounds of the present invention can be prepared. In Step 1 the methyl glycinate is subjected to cyclohexanone and TMSCN in the presence of tri ethyl amine in DCM to afford 15. In Step 2 15 hydrogenated with hydrogen gas in the presence of catalytic platinum oxide and subsequently undergoes an intramolecular cyclization to afford compound 16 which is used in the schemes above.

Scheme 2-4: Starting with compound 17, compounds of the present invention can be prepared. In Step 1 compound 17 is subjected to ethyl 2-oxoacetate in the presence platinum on carbon and hydrogen gas to afford compound 18. In Step 2 compound 18 is Boc-deprotected with hydrochloric acid. In Step 3 compound 18 is cyclized to afford compound 16 which is used in the schemes above.
Scheme 2-5

11 19
Scheme 2-5: Starting from compound 11 the CDK 4/6 inhibitor 19 can be prepared. In Step 1 5-(4-methylpiperazin-l-yl)pyridin-2-amine is subjected to LiHMDS and compound 11 is added slowly under chilled conditions to afford a nucleophilic substitution reaction and compound 19. Compound 11 can be prepared as described in the schemes herein.

Scheme 2-6: Starting from compound 11 the CDK 4/6 inhibitor 20 can be prepared. In Step 1 5-(4-isopropylpiperazin-l-yl)pyridin-2-amine is subjected to LiHMDS and compound 11 is added slowly under chilled conditions to afford a nucleophilic substitution reaction and compound 20. Compound 11 can be prepared as described in the schemes herein.
Preparation of Compound 5:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet, and reflux condenser was charged with ethyl 4-chloro-2-(methylthio)pyrimidine-5-carboxylate 3 (49.2 g, 0.21 mol, 1.00 equiv.), spirolactam 4 (39.2 g, 0.23 mol, 1.10 equiv.), DIPEA (54.7 g, 0.42 mol, 2.00 equiv.), and DMAc (147.6 mL, 3 vol). The batch was heated to 90-95 °C, and after 60 h, IPC confirmed -14% (AUC) of ethyl 4-chloro-2-(methylthio)pyrimidine-5-carboxylate remained. The batch was cooled to RT, and precipitate formation was observed. The suspension was diluted with MTBE (100 mL, 2 vol) and water (442 mL, 9 vol) and stirred for 2 h at RT. The product was isolated by vacuum filtration and washed with MTBE (49 mL, 1 vol). The solid cake was conditioned for 1 h and dried under vacuum at 40 °C for 16 h to afford compound 5 [41.0 g, 53% yield] as an off-white solid with a purity of >99% AUC. ¾ MR (CDCh): δ 8.76 (d, J = 2.0 Hz, 1H), 6.51-6.29 (br, 1H), 4.33 (q, J = 7.0 Hz, 2H), 3.78 (s, 2H), 3.58 (s, 2H), 2.92 (s, 2H), 2.53 (s, 3H), 1.63-1.37 (m, 12H). LCMS (ESI, m/z = 365.3 [M+H]).
Preparation of Compound 6:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 5 [41.0 g, 0.11 mol, 1.00 equiv.], Boc-anhydride (36.8 g, 0.17 mol, 1.50 equiv.), DMAP (1.37 g, 0.01 mol, 0.10 equiv.), and dichloromethane (287 mL, 7 vol). The batch was stirred for 3 h at RT. IPC confirmed no starting material remained (AUC). The batch was concentrated into a residue under reduced pressure and taken to the next step (a quantitative yield is assumed for this step). An aliquot (200 mg) was purified by column chromatography (heptanes/ethyl acetate 0 to 100%) to afford compound 6. 1H MR (CDCh): δ 8.64 (s, 1H), 4.31 (q, J = 7.0 Hz, 2H), 4.07 (s, 2H), 3.83 (S, 2H), 3.15 (m, 2H), 2.56 (s, 3H), 172 (m, 3H), 1.59 (m, 15H), 1.42 (t, J= 7.0 Hz, 3H). LCMS (ESI, m/z = 465.2 [M+H]).
Preparation of Compound 7:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with compound 6 [residue from a previous step, quantitative yield assumed, 52.2 g, 0.11 mol, 1.00 equiv.], and THF (261 mL, 5 vol). The batch was cooled to 0°C and 1,8-diazabicyclo[5.4.0]un-dec-7-ene (17.1 g, 0.11 mmol, 1.00 equiv.) was added keeping the internal temperature in 0-10°C range. After the addition was complete, the cooling bath was removed and the reaction mixture was allowed to warm up to RT and after 2 h, IPC confirmed no starting material remained. The batch was seeded with the product (1.0 g) and was cooled to 0°C. The slurry was stirred at 0°C for 2 h. The product was isolated by vacuum filtration and washed with cold (0°C) THF (50 mL, 1 vol). The solid cake was conditioned for 1 h and dried under vacuum at 40°C for 16 h to afford 7 [47 g, quantitative yield] as a light orange solid with a purity of >99% AUC. The color of the product changed into yellow once the batch was exposed to air for an extended period of time (~ 1 day). Material was isolated with substantial amount DBU, according to proton NMR. However, it did not interfere with the next step. 1H MR (CDCh): δ 8.71 (s, 1H), 4.03 (s, 2H), 2.57 (s, 3H), 1.85 (m, 10H), 1.51 (s, 9H). LCMS (ESI, m/z = 419.2 [M+H]).
Preparation of Compound 8:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 7 [40.8 g, 0.10 mol, 1.00 equiv.], triethylamine (31.5 g, 0.31 mol, 3.20 equiv.), and dichloromethane (408 mL, 10 vol). The batch was purged with N2 for 15 min and was cooled to 0°C. Triflic anhydride (44.0 g, 0.16 mol, 1.60 equiv.) was added keeping the
internal temperature in 0-10°C range. The batch was stirred at 0°C and after 3 h, IPC confirmed -7.0% (AUC) of 7 remained. [It was speculated that the product was hydrolyzing back into starting material during the analysis.] Once the reaction was deemed complete, the batch was transferred to a 1 L, separatory funnel and was washed with 50% saturated sodium bicarbonate (200 mL, 5 vol). [It was prepared by mixing saturated sodium bicarbonate (100 mL) with water (100 mL)).] The aqueous layer was separated and was extracted with DCM (2×40 mL, 1 vol). The organic layers were combined and concentrated into a residue under reduced pressure and taken to the next step. LCMS (ESI, m/z = 551.6 [M+H]).
Preparation of Compound 9:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with compound 8 [residue from a previous step, quantitative yield assumed, 53.7 g, 0.10 mol, 1.00 equiv.], and THF (110 mL, 2 vol). The solvent was removed under vacuum distillation and the procedure was repeated two times. The flask was charged with triethylsilane (22.7 g, 0.20 mol, 2.00 equiv.), and DMF (268 mL, 5 vol). The batch was degassed by five cycles of evacuation, followed by backfilling with nitrogen. The flask was charged with tetrakis(triphenylphosphine)palladium(0) (11.3 g, 0.01 mol, 0.1 equiv.). The batch was heated to 45-50°C, and after 14 h, IPC confirmed no starting material remained. The batch was transferred to a 500 mL, separatory funnel while still warm. The reaction was partitioned between water (5 vol) and ethyl acetate (5 vol). The aqueous layer was extracted with ethyl acetate (3 x3 vol). The organic layers were combined and concentrated down to 2 volumes. The precipitate was filtered and washed with ethyl acetate (2x 1 vol). The solid cake was conditioned for 1 h and dried under vacuum at 40°C for 16 h to afford 9 [27.5 g, 70% yield] as a yellow solid with a purity of -98% AUC. Proton NMR showed some triphenylphosphine oxide present. ¾ NMR (DMSO-i¾):5 9.01 (s, 1H), 7.40 (s, 1H), 4.30 (s, 2H), 2.58 (m, 2H), 2.58 (s, 3H), 1.81 (m, 5H), 1.51 (s, 9H). LCMS (ESI, m/z = 403.4 [M+H]).
Preparation of Compound 10 from the Scheme 2-1 route:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged 9 (12.8 g, 31.8 mmol, 1.00 equiv.) and dichloromethane (64 mL, 5 vol). Trifluoroacetic acid (18.2 g, 159 mmol, 5.00 equiv.) was added over 20 min and the solution was stirred for 2 h at RT. IPC confirmed reaction was complete. The batch was transferred to a 500 mL, separatory funnel and washed with saturated sodium bicarbonate (200 mL). The aqueous layer was extracted with dichlorom ethane (3 x3 vol). The organic layers were combined and concentrated down to 1 volume. The precipitate was filtered and conditioned for 1 h and dried under vacuum at 40 °C for 16 h to afford 9 [6.72 g, 70% yield] as an off-white solid with a purity of 99.1% AUC. ¾ NMR (DMSO-dis): δ 8.95 (s, 1H), 8.32 (s, 1H), 7.15 (s, 1H), 3.68 (d, J = 1.0 Hz, 2H), 2.86 (m, 2H), 2.57 (s, 3H), 1.92 (m, 2H), 1.73 (m, 3H), 1.39 (m, 3H). LCMS, ESI, m/z = 303.2 [M+H]).
Preparation of Compound 10 from Scheme 2-2 route:
A 50 mL, three-neck flask equipped with a magnetic stirring bar, thermocouple, N2 inlet was charged 14 (680 mg, 1.62 mmol, 1.00 equiv.) and THF (6.8 mL, 10 vol). A I M solution of potassium tert-butoxide (3.2 mL, 3.24 mmol, 2.00 equiv.) in THF was added over 10 min and the solution was stirred for 2 h at RT. IPC confirmed reaction was complete. The batch was acidified with 4 N hydrogen chloride solution in dioxane (2.4 mL, 9.72 mmol, 6.00 equiv.) and stirred for additional 1 h. The batch was transferred to a 500 mL, separatory funnel and washed with saturated sodium bicarbonate (100 mL). The aqueous layer was extracted with ethyl acetate (3 x20 vol). The organic layers were combined and concentrated down to 3volumes and product precipitated. The precipitate was filtered and conditioned for 1 h and dried under vacuum at 40 °C for 16 h to afford 9 [489 mg, quantitative yield] as an off-white solid.
Preparation of Compound 11 :
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 10 (9.00 g, 29.8 mmol, 1.00 equiv.), and acetonitrile (180 mL, 20 vol). A solution of Oxone™ (45.9 g, 0.15 mol, 5.00 equiv.) in water (180 mL, 20 vol) was added to the batch over 20 min. The batch was stirred for 2 h and IPC confirmed the reaction was complete. The batch was concentrated down to ½ of the original volume and was extracted with dichloromethane DCM (4x 10 vol). The organic layers were combined; polish filtered and concentrated down to -10 vol of DCM. The product was slowly crystallized out by addition of heptanes (-30 vol). The mixture was cooled to 0°C and the product was filtered and dried under vacuum at 40 °C for 16 h to afford 11 [9.45 g, 95% yield] as an off-white solid with a purity of >99% AUC. ¾ NMR (CDCb): 5 9.24 (s, 1H), 7.78 (s, 1H), 7.46 (s, 1H), 3.89 (d, J= 2.0 Hz, 2H), 3.43 (s, 3H), 2.98 (m, 2H), 2.10 (m, 2H), 1.86 (m, 3H), 1.50 (m, 3H). LCMS (ESI, m/z = 335.2 [M+H]).
Preparation of Compound 13:
A 250 mL, single-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet, and reflux condenser was charged with 4-chloro-2-(methylthio)pyrimidine-5-carbaldehyde (2.00 g, 10.6 mmol, 1.00 equiv.), spirolactam 4 (1.96 g, 11.7 mmol, 1.10 equiv.), DIPEA (2.74 g, 21.2 mmol, 2.00 equiv.), and fert-butanol (20 mL, 10 vol). The batch was heated to 80-85 °C, and after 24 h, IPC confirmed no aldehyde 12 remained. The batch was cool to RT and concentrated into a residue, which was loaded on silica gel column. The product was eluted with ethyl acetate/heptanes (0% to 100%). The product containing fractions were pulled out and concentrated to afford 13 [0.98 g, 29% yield] as an off-white solid.
Preparation of Compound 14:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 13 [0.98 g, 3.00 mmol, 1.00 equiv.], Boc-anhydride (4.90 g, 21.5 mmol, 7.00 equiv.), DMAP (36 mg, 0.30 mmol, 0.10 equiv.), and dichloromethane (7 mL, 7 vol). The batch was stirred for 3 h at RT. IPC confirmed no starting material remained. The batch was cool to RT and concentrated into a residue, which was loaded on silica gel column. The product was eluted with ethyl acetate/heptanes (0% to 100%). The product containing fractions were pulled out and concentrated to afford 14 [0.98 g, 29% yield] as an off-white solid.
Preparation of Compound 15:
To a suspension of methyl glycinate (500 g, 3.98 mol, 1 eq) in DCM (10 L) was added
TEA dropwise at rt under nitrogen atmosphere, followed by the addition of cyclohexanone (781 g, 7.96 mol, 2 eq) dropwise over 15 min. To the resulting mixture was added TMSCN (591 g, 5.97 mol, 1.5 eq) dropwise over 1 hour while maintaining the internal reaction temperature below 35
°C. After stirred at rt for 2 hrs, the suspension became a clear solution. The progress of the reaction was monitored by H- MR.
When the methyl glycinate was consumed completely as indicated by H-NMR analysis, the reaction was quenched by water (5 L). The layers were separated. The aqueous layer was extracted with DCM (1 L). The combined organic phase was washed with water (5 L X 2) and
dried over Na2S04 (1.5 Kg). After filtration and concentration, 1.24 Kg of crude 15 was obtained as oil.
The crude 15 was dissolved in IPA (4 L). The solution was treated with HC1/IPA solution (4.4 mol/L, 1.1L) at RT. A large amount of solid was precipitated during the addition. The resulting suspension was stirred for 2 hrs. The solid product was collected by vacuum filtration and rinsed with MTBE (800 mL). 819 g of pure 15 was obtained as a white solid. The yield was 88.4%. ¾- MR (300 MHz, CD3OD) 4.20 (s, 2H), 3.88 (s, 3H), 2.30-2.40 (d, J = 12 Hz, 2H), 1.95-2.02 (d, J = 12 Hz, 2H), 1.55-1.85 (m, 5H), 1.20-1.40 (m, 1H).
Preparation of Compound 16:
To a solution of 15 (10 g, 43 mmol) in MeOH (100 mL) was added methanolic hydrochloride solution (2 .44 mol/L, 35.3 mL, 2 eq) and Pt02 (0.5 g, 5 wt %). The reaction suspension was stirred with hydrogen bubble at 40 °C for 6 hours. H- MR analysis showed consumption of 15. To the reaction mixture was added K2CO3 (15 g, 108 mmol, 2.5 eq) and the mixture was stirred for 3 hrs. The suspension was filtered and the filtrate was concentrated to dryness. The residual oil was diluted with DCM (100 mL) and resulting suspension was stirred for 3 hrs. After filtration, the filtrate was concentrated to provide crude 16 (6.6 g) as an oil. The crude 16 was diluted with EtOAc/hexane (1 : 1, 18 mL) at rt for 2 hrs. After filtration, 16 (4 g) was isolated. The obtained 16 was dissolved in DCM (16.7 mL) and hexane (100 mL) was added dropwise to precipitate the product. After further stirred for 1 h, 2.8 g of the pure 16 was isolated as a white solid. The yield was 39%. HPLC purity was 98.3%; MS (ESI): 169.2 (MH+); 1 H-NMR (300 MHz, D2O) 3.23 (s, 3H), 3.07 (s, 3H), 1.37-1.49 (m, 10H).
Preparation of compound 19:
5-(4-methylpiperazin-l-yl)pyridin-2-amine (803.1 g; 3.0 equivalents based on sulfone 11) was charged to a 22 L flask. The flask was blanketed with N2 and anhydrous THF added (12.4 kg). The resulting black-purple solution was cooled in an ice bath to < 5°C. 1M LiHMDS (4.7 L; 1.2 equivalents based on sulfone 11) was added via an addition funnel in three equal additions to keep the temperature below 10°C. Upon the completion of the addition, the reaction mixture was warmed to 16°C. The sulfone 11 (455.1 g; 1.00 equivalents) was added in five additions. Reaction proceeded until HPLC analysis of an IPC sample indicated less than 3% of sulfone 11 remained.
To quench the reaction, the contents of the 22L flask were transferred to a 100 L flask containing water. After stirring for 30 minutes at <30°C, the crude product was collected by filtration, washed with water and dried to afford 19 (387 g, 99.1% purity, 63.7% yield).
Preparation of compound 20:
5-(4-isopropylpiperazin-l-yl)pyridin-2-amine (1976.2 g; 3.0 equivalents based on sulfone 11) was charged to a 50 L flask. The flask was blanketed with N2 and anhydrous THF added (10.7 kg). The resulting black-purple solution was cooled in an ice bath to < 5°C. 1M LiHMDS (9.6 kg; 3.6 equivalents based on sulfone) was added via an addition funnel at a rate to keep the temperature below 10°C. Upon the completion of the addition, the reaction mixture was warmed to 16°C over 120 minutes by removing the ice bath. The sulfone (1000 g; 1.00 mol) was added in five additions. The reaction proceeded until HPLC analysis of an IPC sample indicated less than 1% of sulfone 11 remained. After completion of the reaction, ammonium chloride was added to the reaction mixture. The mixture stirred at < 32°C for at least 30 minutes and the solids collected by filtration to afford 20 (900 g, 99.1% purity, 64.2% yield).
Alternate Route to Spirolactam via cyclohexanone:
Scheme 2-7

26
In one embodiment the spirolactam is made via the synthetic scheme above. By reducing the nitrile group before addition of the glycinate group the reaction sequence proceeds in higher yield. The chemistry used in Step 1 is described in the literature (J. Org. Chem. 2005, 70,8027-8034), and was performed on a kilogram scale. The chemistry to convert Compound 24 into the
spirolactam was also demonstrated on kilogram scale. The Boc protection of Compound 23, is carried out at -70°C in order to limit formation of the di-Boc protected product. Experimental details of a 200 g pilot run are described below.
Step 1

200 g of cyclohexanone 21 was converted to 22 using Ti(Oi-Pr)4 /TMSCN/NH3. After work-up, 213 g of 22 was obtained. The H- MR was clean. The yield was 84%. The titanium salts were removed by vacuum filtration. In one embodiment, the titanium salts are removed by centrifugation or Celite filtration.
Step 2

190 g of 22 was mixed with LAH (2 eq) in MTBE for 30 minutes at 45°C. After work-up, 148 g of crude 23 was obtained.
Step 3

136 g of the crude 23 from step 2 was converted to 24 with 0.9 eq of B0C2O at -70°C. The reaction was completed and worked up. After concentration, 188 g of 24 was obtained. The yield was 86%. The H-NMR and C-NMR spectra confirmed that the compound was pure.
Step 4

188 g of 24 was subjected to methyl 2-bromoacetate and K2CO3 in acetonitrile to afford 25. 247 g of crude 25 was obtained.
Step 5

247 g of 25 was subjected to TFA in DCE heated to reflux to afford 26. After work-up, 112 g of 6 as TFA salt was obtained. H- MR was clean.
Step 6

26 27
Compound 26 was stirred in EtOH in the presence at room temperature overnight to afford 27. In one embodiment DCM is used as the solvent instead of EtOH.
Example 3. Purge of residual palladium from Step 5 Scheme 2-1:
Since palladium was used in Step 5 of Scheme 2-1, the levels of residual Pd present in the subsequent synthetic steps was determined. Table 2 below and Figure 3 show the palladium levels in the isolated solids.
Table 2

The material after Step 5 was isolated containing 1.47% (14700 ppm) of residual palladium. This data represents the highest level of palladium in the worst case scenario. The workup conditions of the latter steps purged nearly all of the palladium and the final product, 19 bis HC1 salt, contained 14 ppm of Pd, which is below the standard 20 ppm guidline. The Pd levels will likely be even lower once the catal st loading is optimized in Step 5.

19
The process developed in this route was a significant improvement over the one used for the first generation synthesis. Overall, the scheme consists of seven steps with five isolations, all by crystallization. No silica column chromatography is employed in the synthesis, which makes the process highly scalable. The process workup conditions can successfully purge the 1.47% of residual palladium after step 5 of Scheme 2-1.
///////////////TRILACICLIB, G1T28, G1T 28, SHR 6390, PHASE 2, G1 Therapeutics, Inc.
CN1CCN(CC1)C2=CN=C(C=C2)NC3=NC=C4C=C5C(=O)NCC6(N5C4=N3)CCCCC6
BMS-986020

BMS-986020
AM-152; BMS-986020; BMS-986202
cas 1257213-50-5
Chemical Formula: C29H26N2O5
Molecular Weight: 482.536
(R)-1-(4′-(3-methyl-4-(((1-phenylethoxy)carbonyl)amino)isoxazol-5-yl)-[1,1′-biphenyl]-4-yl)cyclopropane-1-carboxylic acid
Cyclopropanecarboxylic acid, 1-(4′-(3-methyl-4-((((1R)-1-phenylethoxy)carbonyl)amino)-5-isoxazolyl)(1,1′-biphenyl)-4-yl)-
1-(4′-(3-Methyl-4-(((((R)-1-phenylethyl)oxy)carbonyl)amino)isoxazol-5-yl)biphenyl-4-yl)cyclopropanecarboxylic acid
UNII: 38CTP01B4L
For treatment for pulmonary fibrosis, phase 2, The lysophosphatidic acid receptor, LPA1, has been implicated as a therapeutic target for fibrotic disorders
Lysophospholipids (LPs), including lysophosphatidic acid (LPA), sphingosine 1-phospate (S1P), lysophosphatidylinositol (LPI), and lysophosphatidylserine (LysoPS), are bioactive lipids that transduce signals through their specific cell-surface G protein-coupled receptors, LPA1-6, S1P1-5, LPI1, and LysoPS1-3, respectively. These LPs and their receptors have been implicated in both physiological and pathophysiological processes such as autoimmune diseases, neurodegenerative diseases, fibrosis, pain, cancer, inflammation, metabolic syndrome, bone formation, fertility, organismal development, and other effects on most organ systems.

- Originator Amira Pharmaceuticals
- DeveloperB ristol-Myers Squibb; Duke University
- Class Antifibrotics; Azabicyclo compounds; Carboxylic acids; Small molecules; Tetrazoles
- Mechanism of Action Lysophosphatidic acid receptor antagonists
- Orphan Drug Status Yes – Fibrosis
- Phase II Idiopathic pulmonary fibrosis
- Phase IPsoriasis
Most Recent Events
- 05 May 2016 Bristol-Myers Squibb plans a phase I trial for Psoriasis in Australia (PO, Capsule, Liquid) (NCT02763969)
- 01 May 2016 Preclinical trials in Psoriasis in USA (PO) before May 2016
- 14 Mar 2016 Bristol-Myers Squibb withdraws a phase II trial for Systemic scleroderma in USA, Canada, Poland and United Kingdom (PO) (NCT02588625)
BMS-986020, also known as AM152 and AP-3152 free acid, is a potent and selective LPA1 antagonist. BMS-986020 is in Phase 2 clinical development for treating idiopathic pulmonary fibrosis. BMS-986020 selectively inhibits the LPA receptor, which is involved in binding of the signaling molecule lysophosphatidic acid, which in turn is involved in a host of diverse biological functions like cell proliferation, platelet aggregation, smooth muscle contraction, chemotaxis, and tumor cell invasion, among others
![]()



PRODUCT PATENT
GB 2470833, US 20100311799, WO 2010141761
Hutchinson, John Howard; Seiders, Thomas Jon; Wang, Bowei; Arruda, Jeannie M.; Roppe, Jeffrey Roger; Parr, Timothy
Assignee: Amira Pharmaceuticals Inc, USA

John Hutchinson
PATENTS
WO 2011159632
WO 2011159635
PATENT
WO 2013025733
Synthesis of Compound 74
Synthetic Route (Scheme XLV)

Compound 74 Compound 74a
[0562] Compound XLV-1 was prepared by the same method as described in the synthesis of compound 1-4 (Scheme 1-A).
[0563] To a solution of compound XLV-1 (8 g, 28.08 mmol) in dry toluene (150 mL) was added compound XLV-2 (1.58 g, 10.1 mmol), triethylamine (8.0 mL) and DPPA (9.2 g, 33.6 mmol). The reaction mixture was heated to 80 °C for 3 hours. The mixture was diluted with EtOAc (50 mL), washed with brine, dried over Na2S04, filtered and concentrated. The residue was purified by column chromatography (PE/EA = 10 IX) to give compound XLV-3 (9.4 g, yield: 83 %). MS (ESI) m/z (M+H)+402.0.
[0564] Compound 74 was prepared analogously to the procedure described in the synthesis of Compound 28 and was carried through without further characterization.
[0565] Compound 74a was prepared analogously to the procedure described in the synthesis of Compound 44a. Compound 74a: 1HNMR (DMSO-d6 400MHz) δ 7.81 (d, J = 8.4 Hz, 2H), 7.41 (d, J = 8.4 Hz, 2H), 7.52 (d, J = 8.4 Hz, 2H), 7.29-7.32 (m, 7 H), 5.78 (q, 1 H), 2.15 (s, 3 H), 1.52 (d, J = 6.0 Hz, 3H), 1.28 (br, 2 H), 0.74 (br, 2 H). MS (ESI) m/z (M+H)+ 483.1.


Paper
Development of a Concise Multikilogram Synthesis of LPA-1 Antagonist BMS-986020 via a Tandem Borylation–Suzuki Procedure
Michael J. Smith*  , Michael J. Lawler, Nathaniel Kopp, Douglas D. Mcleod, Akin H. Davulcu, Dong Lin, Kishta Katipally , and Chris Sfouggatakis
, Michael J. Lawler, Nathaniel Kopp, Douglas D. Mcleod, Akin H. Davulcu, Dong Lin, Kishta Katipally , and Chris Sfouggatakis
, Michael J. Lawler, Nathaniel Kopp, Douglas D. Mcleod, Akin H. Davulcu, Dong Lin, Kishta Katipally , and Chris SfouggatakisChemical and Synthetic Development, Bristol-Myers Squibb Company, One Squibb Drive, New Brunswick, New Jersey 08903, United States
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.7b00301
http://pubs.acs.org/doi/10.1021/acs.oprd.7b00301
*E-mail: michael.smith2@bms.com.

The process development for the synthesis of BMS-986020 (1) via a palladium catalyzed tandem borylation/Suzuki reaction is described. Evaluation of conditions culminated in an efficient borylation procedure using tetrahydroxydiboron followed by a tandem Suzuki reaction employing the same commercially available palladium catalyst for both steps. This methodology addressed shortcomings of early synthetic routes and was ultimately used for the multikilogram scale synthesis of the active pharmaceutical ingredient 1. Further evaluation of the borylation reaction showed useful reactivity with a range of substituted aryl bromides and iodides as coupling partners. These findings represent a practical, efficient, mild, and scalable method for borylation.
1H NMR (500 MHz, DMSO-d6) δ 1.19 (dd, J = 6.8, 3.8 Hz, 2H), 1.50 (dd, J = 6.8, 3.8 Hz, 2H), 1.56 (br s, 3H), 2.14 (br s, 3H), 5.78 (br s, 1H), 6.9–7.45 (br, 5H), 7.45 (br d, J = 8.3 Hz, 2H), 7.65 (d, J = 8.3 Hz, 2H), 7.79 (br d, 2H), 7.82 (br d, 2H), 8.87 (br s, 0.8H), 9.29 (s, 0.2H), 12.39 (br s, 1H). 13C NMR (126 MHz, DMSO-d6) δ 9.2, 15.8, 22.4, 28.3, 72.8, 113.8, 125.4, 125.6, 126.2, 126.3, 127.1, 127.7, 128.4, 130.9, 137.4, 140.0, 141.5, 142.2, 154.4, 159.6, 160.8, 175.2. HRMS (ESI+) Calculated M + H 483.19145, found 483.19095.
REFERENCES
1: Kihara Y, Mizuno H, Chun J. Lysophospholipid receptors in drug discovery. Exp
Cell Res. 2015 May 1;333(2):171-7. doi: 10.1016/j.yexcr.2014.11.020. Epub 2014
Dec 8. Review. PubMed PMID: 25499971; PubMed Central PMCID: PMC4408218.
//////////////BMS-986020, AM 152, BMS 986020, BMS 986202, Orphan Drug, BMS, Amira Pharmaceuticals, Bristol-Myers Squibb, Duke University, Antifibrotics, PHASE 2, pulmonary fibrosis
O=C(C1(C2=CC=C(C3=CC=C(C4=C(NC(O[C@H](C)C5=CC=CC=C5)=O)C(C)=NO4)C=C3)C=C2)CC1)O
ELAMIPRETIDE

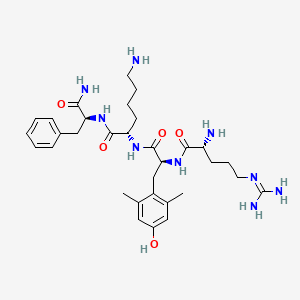
Elamipretide

H-D-Arg-Tyr(2,6-diMe)-Lys-Phe-NH2
D-arginyl-2,6-dimethyl-L-tyrosyl-L-lysyl-L-phenylalaninamide
(2S)-6-amino-2-[[(2S)-2-[[(2R)-2-amino-5-(diaminomethylideneamino)pentanoyl]amino]-3-(4-hydroxy-2,6-dimethylphenyl)propanoyl]amino]-N-[(2S)-1-amino-1-oxo-3-phenylpropan-2-yl]hexanamide
CAS 736992-21-5
Chemical Formula: C32H49N9O5
Molecular Weight: 639.8
-
A free radical scavenger and antioxidant that localizes in the inner mitochondrial membrane.
-
Mitochondrial Protective Agent to Improve Cell Viability
- Elamipretide
- bendavia
- UNII-87GWG91S09
- 736992-21-5
- MTP 131
- RX 31
- SS 31
- 87GWG91S09
- L-Phenylalaninamide, D-arginyl-2,6-dimethyl-L-tyrosyl-L-lysyl-
- SS-31 peptide
- Arg-Dmt-Lys-Phe-NH2
- D-Arg-Dmt-Lys-Phe-NH2
- SS31 peptide
- Elamipretide [USAN:INN]
- MTP-131
- Elamipretide (USAN/INN)
- arginyl-2,’6′-dimethyltyrosyl-lysyl-phenylalaninamide
- CHEMBL3833370
- SCHEMBL15028020
- CTK2H1007
Elamipretide is a cardiolipin peroxidase inhibitor and mitochondria-targeting peptide, Improves Left Ventricular and Mitochondrial Function. In vitro: Elamipretide significantly increases enzymatic activities of both complexes to near normal levels.
Background Information
| Elamipretide is a cardiolipin peroxidase inhibitor and mitochondria-targeting peptide, Improves Left Ventricular and Mitochondrial Function. In vitro: Elamipretide significantly increases enzymatic activities of both complexes to near normal levels. long-term therapy with elamipretide reduces ROS formation, attenuated mPTP openings, and significantly decreases the levels of cytosolic cytochrome c and active caspase-3, thus suppressing a major signaling pathway for apoptosis. Elamipretide represents a new class of compounds that can improve the availability of energy to failing heart and reduce the burden of tissue injury caused by excessive ROS production. [1] In vivo: Fourteen dogs with microembolization-induced HF are randomized to 3 months monotherapy with subcutaneous injections of elamipretide (0.5 mg/kg once daily. Elamipretide has been shown to enhance ATP synthesis in multiple organs, including heart, kidney, neurons, and skeletal muscle. [1] ……by MedChemexpress Co., Ltd. |
Elamipretide (also known as SS-31 and Bendavia)[1][2] is a small mitochondrially-targeted tetrapeptide (D-Arg-dimethylTyr-Lys-Phe-NH2) that appears to reduce the production of toxic reactive oxygen species and stabilize cardiolipin.[3]
Stealth Peptides, a privately held company, was founded in 2006 to develop intellectual property licensed from several universities including elamipretide; it subsequently changed its name to Stealth BioTherapeutics.[4][5]
Acute coronary syndrome; Age related macular degeneration; Cardiac failure; Corneal dystrophy; Diabetic macular edema; Lebers hereditary optic atrophy
- Originator Stealth Peptides
- Developer Stealth BioTherapeutics
- Class Eye disorder therapies; Ischaemic heart disorder therapies; Oligopeptides; Peptides; Small molecules
- Mechanism of Action Free radical scavengers; Mitochondrial permeability transition pore inhibitors
- Phase II/III Barth syndrome
-
- Phase II Acute kidney injury; Corneal disorders; Heart failure; Leber’s hereditary optic atrophy; Mitochondrial disorders; Reperfusion injury
- Phase I/II Diabetic macular oedema; Dry age-related macular degeneration; Mitochondrial myopathies
- Phase I Age-related macular degeneration
- No development reported Chronic heart failure; Diabetes mellitus; Eye disorders; Neurodegenerative disorders
Most Recent Events
- 29 Jun 2017 Initial efficacy and adverse events data from phase II MMPOWER-2 trial in Mitochondrial-myopathies released by Stealth
- 02 Jun 2017 Stealth BioTherapeutics completes a phase II trial in Heart failure in Germany and Serbia (SC) (NCT02814097)
- 01 May 2017 Phase-II/III clinical trials in Barth syndrome (In children, In adolescents, In adults, In the elderly) in USA (SC) (NCT03098797)
Novel crystalline salt (eg hydrochloride, mesylate and tosylate salts) forms of D-Arg-Dmt-Lys-Phe-NH2 (referred to as MTP-131 or elamipretide ) and composition comprising them are claimed. See WO2016190852 , claiming therapeutic compositions including chromanyl compounds, variants and analogues and uses thereof. Stealth BioTherapeutics (formerly known as Stealth Peptides) is developing elamipretide, which targets mitochondria, for the potential iv/sc treatment of cardiac reperfusion injury, acute coronary syndrome, acute kidney injury, mitochondrial myopathy, skeletal muscle disorders and congestive heart failure.
Also, the company is developing an oral formulation of elamipretide , which targets mitochondria and reduces the production of excess reactive oxygen species, for treating chronic heart failure. In January 2015, a phase II trial was ongoing . In July 2016, a phase II trial was initiated in Latvia, Spain and Hungary .
Further, the company is developing an ophthalmic formulation of elamipretide , a mitochondria targeting peptide, for treating ocular diseases including diabetic macular edema, age-related macular degeneration and fuchs’ corneal endothelial dystrophy and Leber’s hereditary optic neuropathy.
In April 2016, a phase II trial was initiated for LHON . Family members of the product case of elamipretide ( WO2007035640 ) hold protection in the EU until 2026 and expires in the US in 2027 with US154 extension.
Acute coronary syndrome; Age related macular degeneration; Cardiac failure; Corneal dystrophy; Diabetic macular edema; Lebers hereditary optic atrophy
SYNTHESIS

NEXT………………………



PATENT 2
ELAMIPRETIDE BY STEALTH
WO-2017156403

; MTP-131; D-Arg-Dmt-Lys-Phe-Nth). Compound
1 has been shown to affect the mitochondrial disease process by helping to protect organs from oxidative damage caused by excess ROS production and to restore normal ATP production.
PATENT
US 20110082084
WO 2011091357
WO 2012129427
WO 2013059071
WO 2013126775
US 20140378396
US 20140093897
WO 2015134096
WO 2015100376
WO 2015060462
US 20150010588
PATENNT
WO 2015197723
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015197723
PROCESS FOR PREPARING
D-ARGINYL-2,6-DIMETHYL-L-TYROSYL-L-LYSYL-L-PHENYLALANINAMIDE
TECHNICAL FIELD
The invention relates to a process for solution-phase synthesis of D- Arginyl-2,6-dimethyl-L-tyrosyl-L-lysyl-L-phenylalaninamide (abbreviated H-D-Arg-(2,6-Dimethyl)Tyr-L-Lys-L-Phe-NH2, development code SS-31 , MTP-131 , X-31) of Formula (I), an active ingredient developed by Stealth BioTherapeutics under the investigational drug brand names Bendavia® and Ocuvia®, for both common and rare diseases including a mitochondrial targeted therapy for ischemia reperfusion injury.

Formula (I)
BACKGROUND
The product belongs to the class of so-called “Szeto-Schiller peptides”. Szeto-Sciller peptides or “SS peptides” are small, aromatic-cationic, water soluble, highly polar peptides, such as disclosed in US 6703483 and US 7576061 , which can readily penetrate cell membranes. The aromatic-cationic peptides include a minimum of two amino acids, and preferably include a minimum of four amino acids, covalently joined by peptide bonds. The maximum number of amino acids is about twenty amino acids covalently joined by peptide bonds. As described by EP 2012/2436390, optimally, the number of amino acids present in the SS peptides is four.
Bendavia® is being tested for the treatment of ischemia reperfusion injury in patients with acute myocardial infarction (AMI), for the treatment of acute kidney injury (AKI) and renal microvascular dysfunction in hypertension, for the treatment of skeletal muscle dysfunction, for the treatment of mitochondrial myopathy and for the treatment of chronic heart failure. Trials are ongoing to assess the Ocuvia’s potential to treat Leber’s Hereditary Optic Neuropathy (LHON) a devastating inherited disease that causes sudden blindness, often in young adults.
Mitochondria are the cell’s powerhouse, responsible for more than 90% of the energy our bodies need to sustain life and support growth. The energetics from mitochondria maintains healthy physiology and prevents disease. In many common and rare diseases, dysfunctional mitochondria are a key component of disease progression.
D-Arginyl-2,6-dimethyl-L-tyrosyl-L-lysyl-L-phenylalaninamide is a cell-permeable and mitochondria-targeted peptide that showed antioxidant activity and was concentrated in the inner mitochondrial membrane. Compound (< 1 nM) significantly reduced intracellular reactive oxygen species, increased mitochondrial potential and prevented tBHP-induced apoptosis in both N2A and SH-SY5Y neuronal cell lines. In rats, intraperitoneal treatment (1 and 3 mg/kg) 1 day prior to unilateral ureteral obstruction and every day thereafter for 14 days significantly decreased tubular damage, macrophage infiltration and interstitial fibrosis. Compound (3 mg/kg i.p. qd for 2 weeks) also prevented apoptosis and insulin reduction in mouse pancreatic islets caused by streptozotocin.
Further studies performed in a G93A mouse model of amyotrophic lateral sclerosis (ALS) demonstrated that the compound (5 mg/kg/day i.p. starting at 30 days of age) led to a significant delay in disease onset.
Potentially useful for the treatment of ALS and may be beneficial in the treatment of aging and diseases associated with oxidative stress.
In the last few years the peptide H-D-Arg-(2,6-Dimethyl)Tyr-L-Lys-L-Phe-NH2, shown in Fig 1 , and its therapeutic activity have been disclosed and
claimed by in several patent applications.
EP 2436390, US 201 10245182 and US 201 10245183 claim topical anesthetic compositions for application to the skin for pain management or anti-skin aging agents, respectively, comprising Szeto-Schiller peptides; SS-31 is specifically claimed as active ingredient. Sequence of solid-phase synthesis is indicated as the preferred preparation process.
US 7718620 claims a process of treating or preventing ischemia-reperfusion injury of the kidney in a mammal by administrating an effective amount of an aromatic-cationic peptide. SS-31 is specifically claimed as active ingredient.
WO2005/001023 discloses a generical process and carrier complexes for delivering molecules to cells comprising a molecule and an aromatic cationic peptide of type D-Arg-Dmt-Lys-Phe-NH2. The tetrapeptide SS-31 is
specifically claimed as product useful for the process at claim 18.
WO2012/1741 17 and WO2014/210056 claim therapeutic compositions based on SS peptides and the aromatic-cationic peptide D-Arg-Dmt-Lys-Phe-NH2 as active agent.
WO 2013/086020, WO 2004/070054 and WO 2005/072295 provide processes for preventing mithochondrial permeability transition and reducing oxidative damage in a mammal, a removed organ, or a cell in need thereof and specifically claims the process wherein the peptide does not have mu-opioid receptor agonist activity, i.e., D-Arg-Dmt-Lys-Phe-NH2.
WO 2009/108695 discloses a process for protecting a kidney from renal injury which may be associated with decreased or blocked blood flow in the subject’s kidney or exposure to a nephrotoxic agent, such as a radiocontrast dye. The processes include administering to the subject an effective amount of an aromatic-cationic peptide to a subject in need thereof and one of the selected peptide is D-Arg-Dmt-Lys-Phe-NH2.
US 6703483 discloses a detailed procedure for the preparation of novel analogs of DALDA [H-Tyr-D-Arg-Phe-Lys-NH2], namely H-Dmt-D-Arg-Phe-Lys-NH2 using the solid-phase techniques and /?-methylbenzhydrylamine
resin and protocols that have been extensively used by inventor’s laboratory.
Most prior art processes for preparing the compound typically comprise conventionally performed peptide solid-phase synthesis with further purification by chromatography in order to obtain the requested purity for therapeutic use.
It is well known that solid-phase synthesis followed by chromatographic purification is time consuming, very expensive and very difficult to be scaled up on industrial scale, so the need of developing a process for large scale production is obvious. The compound is isolated as organic acid salt, as acetate or trifluoro acetate.
eddy et al., Adv. Exp. Med. Biol, 2009, 61 1 , 473 generally describes the liquid-phase synthesis of antioxidant peptides of Figure 1 and similar others (SS-02, SS-20), involving routinely used side chain protecting groups for amino acid building blocks. The guanidine group was protected with NO2 and the ε-ΝΗ2 of Lys was protected by Cbz or 2-Cl-Cbz. These peptides were
synthesized using Boc/Cbz chemistry and BOP reagent coupling. Starting with the C-terminal Lys residue protected as H-Lys(2-Cl-Cbz)-NH2, (prepared
from the commercially available Boc-Lys(2-Cl-Cbz)-OH in two steps by amidation with NH4HCO3 in the presence of DCC/HOBt following a literature procedure [Ueyama et all, Biopolymers, 1992, 32, 1535, PubMed: 1457730], followed by exposure to TFA). Selective removal of the 2-Cl-Cbz in the
presence of the NO2 group was accomplished using catalytic transfer hydrogenolysis (CTH) [Gowda et al., Lett. Pept. Sci., 2002, 9, 153].
A stepwise procedure by standard solution peptide synthesis for preparation of potent μ agonist [DmtJDALDA and its conversion into a potent δ antagonist H-Dmt-Tic-Phe-Lys(Z)-OH by substitution of D-Arg with Tic to enhance the δ opioid agonist activity is described by Balboni et al., J. Med.
Chem., 2005, 48, 5608. A general synthetic procedure for a similar tetrapeptide ([Dmt-D-Arg-Phe-Lys-NH2 is described by Ballet et al., J. Med.
Chem. 2011, 54, 2467.
Similar DALDA analog tetrapeptides were prepared by the manual solid-phase technique using Boc protection for the a-amino group and DIC/HOBt or HBTU/DIEA as coupling agent [Berezowska et al., J. Med. Chem., 2009, 52, 6941 ; Olma et al., Acta Biochim. Polonica, 2001, 48, 4, 1 121 ; Schiller at al., Eur. J. Med. Chem., 2000, 35, 895].
Despite the high overall yield in the solid-phase approach, it has several drawbacks for the scale-up process such as:
a. the application of the highly toxic and corrosive hydrogen fluoride for cleavage of the peptide from the resin,
b. low loading (0.3-0.35 mmol/g of resin) proved necessary for successful end-step, and
c. use of excess amounts of reagents (3-fold of DIC, 2.4-fold of HOBt, etc.) on each step [ yakhovsky et al., Beilstein J. Org. Chem., 2008, 4(39), 1 , doi: 10.376/bjoc.4.39]
SUMMARY
The invention relates to a more efficient process avoiding either solid-phase synthesis or chromatographic purification, more suitable for large scale production. The process of the invention is described in Scheme A.
The following abbreviations are used:
Dmt = 2,6-dimethyl tyrosine; Z= benzyloxycarbonyl; MeSO3H = methane sulphonic acid; Boc = Tert-butyloxycarbonyl; NMM = N-methyl morpholine; TBTU= N,N,N’,N’-Tetramethyl-O-(benzotriazol- l-yl)uronium tetrafluoroborate; DMF = dimethyl formamide; TFA = trifluoroacetic acid
Scheme A shows the process for the solution phase synthesis of peptide
1 for assembly of the tetrapeptide backbone using O-Benzyl (Bzl) group and benzyloxycarbonyl (Z) group respectively, as the temporary protection for amino acids’ N-termini (Scheme Figure 2), followed by a final catalytic hydrogenolysis. The final product is isolated as organic acid salt, for example, acetic acid salt.
H-Phe-NH 2 + Boc-Lys(Z)-OH

Boc-Lys(Z)-Phe-NH 2
(IV)
(V) I MeS03H/CH2CI2
Boc-DMTyr(Bzl)-OH + MeS03H.H-Lys(Z)-Phe-NH 2
(

Boc-DMTyr(Bzl)-Lys(Z)-Phe-NH 2
(VIII)
I MeS03H/CH2CI2
Z-D-Arg-ONa + H-DMTyr(Bzl)-Lys(Z)-Phe-NH 2.MeS03H
(X) (IX)
TBTU/NMM/DMF
Z-D-Arg-DMTyr(Bzl)-Lys(Z)-Phe-NH
(XI)
I H2, Pd/C
X ACOH
H-D-Arg-DMTyr-Lys-Phe-NH
(I)
Scheme A
This process is a notable improvement with respect to the prior art and its advantages can be summarized as follows:
• The synthesis is performed in liquid phase allowing the scale up on industrial scale without need of special equipment; · The selection of the protecting group in the building blocks allows a straightforward synthesis with very simple deprotection at each step and minimize the formation of undesired by-product;
• Each intermediate can be crystallized allowing removal of impurities which are not transferred to the following step;
· The purity of each intermediate is very high and usually close to
99%.
EXAMPLES
Example 1: Preparation of Boc-Lys(Z)-Phe-NH2
Charge 200 mL of DMF, 44 g of Boc-Lys(Z)-OH and 15.6 g of H-Phe-NH2 in a flask. Stir the mixture at room temperature for 10 min. Add 19.2 g of
N-methylmorpholine and 32.1 g of TBTU successively at room temperature. Stir the mixture at room temperature for 1 h. Add 500 mL of water into the reaction mixture to precipitate the product at room temperature. Filter the mixture to isolate the solid product and wash the filter cake with water.
Transfer the filter cake into a flask containing 360 mL of ethyl acetate and heat the mixture at 50°C till all the solid is dissolved. Separate the organic phase of product and discard the small aqueous phase. Concentrate the organic phase at 40~45°C and under vacuum to remove the solvent till lots of solid is formed. Filter the residue to isolate the solid product. Transfer the filter cake into a flask containing 2000 mL of MTBE and heat the mixture at refluxing for 20 min. Then, cool down the mixture to room temperature. Filter the mixture to isolate the solid product. Dry the filter cake at 30 °C and under vacuum to give 35 g of solid product.
Example 2: Preparation of H-Lys(Z)-Phe-NH2.MeSC>3H
Charge 26.3 g of Boc-Lys(Z)-Phe-NH2, 200 mL of methylene chloride
and 9.6 g of methanesulfonic acid. Stir the mixture at 15-20 °C for 18 h. Add 100 mL of MTBE into the mixture and stir at 15-20 °C for 1 h. Filter the mixture to isolate the solid product. Dry the wet cake in air at room temperature to give 26.4 g of white solid product.
Example 3: Preparation of Boc-DMeTyr(Bzl)-Lys(Z)-Phe-NH2
Charge 8.4 g of Boc-DMeTyr(Bzl)-OH, 1 1 g of H-Lys(Z)-Phe-NH2.MeSO3H, 7.4 g of TBTU and 80 mL of THF in a flask. Stir the mixture
at room temperature for 15 min, and then cool down to 10°C. Add 6.36 g of N-methylmorpholine and stir the mixture at 20-25°C for 3 h. Add the reaction mixture into a flask containing 240 mL of water. Add 32 mL of methylene chloride into the mixture obtained in the previous operation of. Stir the resultant mixture at room temperature for 20 min. Filter the mixture to isolate the solid product and wash the filter cake with acetone (300 mL X 2). Dry the filter cake in air at room temperature to give 14.3 g of white solid product.
Example 4: Preparation of H-DMeTyr(Bzl)-Lys(Z)-Phe-NH2.MeS03H
Charge 14 g of Boc-BMeTyr(Bzl)-Lys(Z)-Phe-NH2, 280 mL of methylene chloride and 3.3 g of methanesulfonic acid in a flask. Stir the mixture at 18 ~ 22 °C for 10 h. Add 560 mL of heptanes into the mixture and stir the mixture at room temperature for 30 min. Filter the mixture to isolate the solid product. Dry the wet cake in air at room temperature to give 14 g of white solid product.
Example 5: Preparation of Z-D-Arg-DMeTyr(Bzl)-Lys(Z)-Phe-NH2
Charge 6.34 g of Z-D-Arg-ONa, 100 mL of DMF and 2.0 g of methanesulfonic acid in a flask. Stir the mixture at room temperature till a clear solution was formed. Add 14 g of H-DMeTyr(Bzl)-Lys(Z)-Phe-NH2.MeSO3H and cool down the mixture to 10°C. Add 6.15 g of TBTU and
9.67 g of N-methylmorpholine successively. Stir the mixture at room temperature for 4 h. Add aqueous solution of LiOH prepared by dissolving 2.9 g of LiOH.L O in 8 mL of water. Stir the mixture for 30 min. Add the resultant mixture slowly into a flask containing 420 mL of water under stirring. Add 56 mL of methylene chloride into the mixture. Filter the mixture to isolate the solid product. Transfer the filter cake into a flask containing 150 mL of acetic acid, and heat the mixture at 35-40 °C till most of the solid was dissolved. Add 450 mL of MTBE into the mixture and cool down the mixture under stirring to room temperature. Filter the mixture to isolate the solid product. Dry the filter cake in air at room temperature to give 17.3 g of the white solid product.
Example 6 Preparation of H-D-Arg-DMeTyr-Lys-Phe-NH2.3AcOH
Charge 2.0 g of Z-D-Arg-DMeTyr(Bzl)-Lys(Z)-Phe-NH2, 20 mL of acetic acid and 5% Pd/C catalyst (which is obtained by washing 5.0 g of 5% Pd/C containing 60% of water with 30 mL of acetic acid) in a flask. Change the atmosphere of the flask with hydrogen. Stir the mixture at room temperature and pressure of 1 atm of hydrogen for 2 h. Filter the mixture to remove the Pd/C catalyst and wash the filter cake with 10 mL of acetic acid. Combine the filtrate and washing solution and concentrate the solution at 20°C and under vacuum to remove most the solvent. Add 100 mL of acetonitrile into the residue and stir the mixture at room temperature for 20 min. Filter the mixture to isolate the solid product. Dry the filter cake at room temperature and under vacuum to give 0.7 g of the white product.
PATENT
WO 2016001042
References
- Jump up^ “Recommended INN List 75” (PDF). WHO Drug Information. 30 (1): 111. 2016.
- Jump up^ “Elamipretide”. AdisInsight. Retrieved 24 April 2017.
- Jump up^ Kloner, RA; Shi, J; Dai, W (February 2015). “New therapies for reducing post-myocardial left ventricular remodeling.”. Annals of translational medicine. 3 (2): 20. PMC 4322169
 . PMID 25738140.
. PMID 25738140. - Jump up^ Valigra, Lori (April 9, 2012). “Stealth Peptides sees positive results from Bendavia”. Boston Business Journal.
- Jump up^ Dolgin, Elie (11 February 2016). “New drugs offer hope for mitochondrial disease”. STAT.
| Patent ID | Patent Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2014294796 | AROMATIC-CATIONIC PEPTIDES AND USES OF SAME | 2012-12-05 | 2014-10-02 |
| US2016264623 | TETRAPEPTIDE COMPOUND AND METHOD FOR PRODUCING SAME | 2014-10-23 | 2016-09-15 |
| US2017081363 | PHARMACEUTICALLY RELEVANT AROMATIC-CATIONIC PEPTIDES | 2014-12-23 | |
| US2016340389 | PHARMACEUTICALLY RELEVANT AROMATIC-CATIONIC PEPTIDES | 2014-12-23 | |
| US2017129920 | Process for Preparing D-Arginyl-2, 6-Dimethyl-L-Tyrosyl-L-Lysyl-L-Phenylalaninamide | 2015-06-24 |
REFERENCES
1: Alam NM, Mills WC 4th, Wong AA, Douglas RM, Szeto HH, Prusky GT. A mitochondrial therapeutic reverses visual decline in mouse models of diabetes. Dis Model Mech. 2015 Jul 1;8(7):701-10. doi: 10.1242/dmm.020248. Epub 2015 Apr 23. PubMed PMID: 26035391; PubMed Central PMCID: PMC4486862.
2: Szeto HH, Birk AV. Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clin Pharmacol Ther. 2014 Dec;96(6):672-83. doi: 10.1038/clpt.2014.174. Epub 2014 Sep 4. Review. PubMed PMID: 25188726; PubMed Central PMCID: PMC4267688.
3: Dai W, Shi J, Gupta RC, Sabbah HN, Hale SL, Kloner RA. Bendavia, a mitochondria-targeting peptide, improves postinfarction cardiac function, prevents adverse left ventricular remodeling, and restores mitochondria-related gene expression in rats. J Cardiovasc Pharmacol. 2014 Dec;64(6):543-53. PubMed PMID: 25165999.
4: Eirin A, Ebrahimi B, Zhang X, Zhu XY, Woollard JR, He Q, Textor SC, Lerman A, Lerman LO. Mitochondrial protection restores renal function in swine atherosclerotic renovascular disease. Cardiovasc Res. 2014 Sep 1;103(4):461-72. doi: 10.1093/cvr/cvu157. Epub 2014 Jun 19. PubMed PMID: 24947415; PubMed Central PMCID: PMC4155472.
5: Liu S, Soong Y, Seshan SV, Szeto HH. Novel cardiolipin therapeutic protects endothelial mitochondria during renal ischemia and mitigates microvascular rarefaction, inflammation, and fibrosis. Am J Physiol Renal Physiol. 2014 May 1;306(9):F970-80. doi: 10.1152/ajprenal.00697.2013. Epub 2014 Feb 19. PubMed PMID: 24553434.
6: Brown DA, Hale SL, Baines CP, del Rio CL, Hamlin RL, Yueyama Y, Kijtawornrat A, Yeh ST, Frasier CR, Stewart LM, Moukdar F, Shaikh SR, Fisher-Wellman KH, Neufer PD, Kloner RA. Reduction of early reperfusion injury with the mitochondria-targeting peptide bendavia. J Cardiovasc Pharmacol Ther. 2014 Jan;19(1):121-32. doi: 10.1177/1074248413508003. Epub 2013 Nov 28. PubMed PMID: 24288396; PubMed Central PMCID: PMC4103197.
7: Birk AV, Chao WM, Bracken C, Warren JD, Szeto HH. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br J Pharmacol. 2014 Apr;171(8):2017-28. doi: 10.1111/bph.12468. PubMed PMID: 24134698; PubMed Central PMCID: PMC3976619.
8: Szeto HH. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol. 2014 Apr;171(8):2029-50. doi: 10.1111/bph.12461. Review. PubMed PMID: 24117165; PubMed Central PMCID: PMC3976620.
9: Zhao WY, Han S, Zhang L, Zhu YH, Wang LM, Zeng L. Mitochondria-targeted antioxidant peptide SS31 prevents hypoxia/reoxygenation-induced apoptosis by down-regulating p66Shc in renal tubular epithelial cells. Cell Physiol Biochem. 2013;32(3):591-600. doi: 10.1159/000354463. Epub 2013 Sep 6. PubMed PMID: 24021885.
10: Dai DF, Hsieh EJ, Chen T, Menendez LG, Basisty NB, Tsai L, Beyer RP, Crispin DA, Shulman NJ, Szeto HH, Tian R, MacCoss MJ, Rabinovitch PS. Global proteomics and pathway analysis of pressure-overload-induced heart failure and its attenuation by mitochondrial-targeted peptides. Circ Heart Fail. 2013 Sep 1;6(5):1067-76. doi: 10.1161/CIRCHEARTFAILURE.113.000406. Epub 2013 Aug 9. PubMed PMID: 23935006; PubMed Central PMCID: PMC3856238.
/////////////////////Elamipretide, SS-31, Bendavia, PEPTIDE
CC1=CC(=CC(=C1CC(C(=O)NC(CCCCN)C(=O)NC(CC2=CC=CC=C2)C(=O)N)NC(=O)C(CCCN=C(N)N)N)C)O
Funapide, TV 45070, XEN-402, фунапид فونابيد 呋纳匹特

Funapide TV 45070, XEN-402, Funapide, (+)-
фунапид
فونابيد
呋纳匹特
- Molecular FormulaC22H14F3NO5
- Average mass429.345 Da
(S)-1′-[(5-Methyl-2-furyl)methyl]spiro[6H-furo[3,2-f][1,3]benzodioxole-7,3′-indoline]-2′-one
Spiro(furo(2,3-F)-1,3-benzodioxole-7(6H),3′-(3H)indol)-2′(1’H)-one, 1′-((5-(trifluoromethyl)-2-furanyl)methyl)-, (3’S)-
(3’S)-1′-((5-(Trifluoromethyl)furan-2-yl)methyl)-2H,6H-spiro(furo(2,3-F)(1,3)benzodioxole-7,3′-indol)-2′(1’H)-one
Spiro[furo[2,3-f]-1,3-benzodioxole-7(6H),3′-[3H]indol]-2′(1’H)-one, 1′-[[5-(trifluoromethyl)-2-furanyl]methyl]-, (7S)-
TV-45070
UNII-A5595LHJ2L
XEN-401-S
XEN402
(3’S)-1′-{[5-(trifluoromethyl)furan-2-yl]methyl}-2H-6H-spiro[furo[2,3-f]-1,3-benzodioxole-7,3′-indol]-2′(1’H)-one
(7S)-1′-{[5-(Trifluoromethyl)-2-furyl]methyl}spiro[furo[2,3-f][1,3]benzodioxole-7,3′-indol]-2′(1’H)-one
1259933-16-8 CAS
UNII-A5595LHJ2L
Phase II clinical trials for Postherpetic neuralgia (PHN)
Treatment of Neuropathic Pain
- Originator Xenon Pharmaceuticals
- Developer Teva Pharmaceutical Industries; Xenon Pharmaceuticals
- Class Benzodioxoles; Fluorobenzenes; Furans; Indoles; Non-opioid analgesics; Small molecules; Spiro compounds
- Mechanism of Action Nav1.7-voltage-gated-sodium-channel-inhibitors; Nav1.8 voltage-gated sodium channel inhibitors
- Orphan Drug Status Yes – Erythromelalgia
Highest Development Phases
- Phase II Erythromelalgia; Postherpetic neuralgia
- No development reported Dental pain; Pain
- Discontinued Musculoskeletal pain
Most Recent Events
- 09 May 2017 Teva Pharmaceutical Industries completes a phase IIb trial for Postherpetic neuralgia in USA (Topical) (NCT02365636)
- 26 Sep 2016 Adverse events data from a phase II trial in Musculoskeletal pain presented at the 16th World Congress on Pain (PAN – 2016)
- 19 Aug 2015 No recent reports of development identified – Phase-I for Pain (In volunteers) in Canada (PO)
MP 100 – 102 DEG CENT EP2538919
S ROT ALPHA 0.99 g/100ml, dimethyl sulfoxide, 14.04, US 20110087027
Funapide (INN) (former developmental code names TV-45070 and XEN402) is a novel analgesic under development by Xenon Pharmaceuticals in partnership with Teva Pharmaceutical Industries for the treatment of a variety of chronic pain conditions, including osteoarthritis, neuropathic pain, postherpetic neuralgia, and erythromelalgia, as well as dental pain.[1][2][3][4] It acts as a small-moleculeNav1.7 and Nav1.8 voltage-gated sodium channel blocker.[1][2][4] Funapide is being evaluated in humans in both oral and topicalformulations, and as of July 2014, has reached phase IIb clinical trials.[1][3]

Sodium channels play a diverse set of roles in maintaining normal and pathological states, including the long recognized role that voltage gated sodium channels play in the generation of abnormal neuronal activity and neuropathic or pathological pain. Damage to peripheral nerves following trauma or disease can result in changes to sodium channel activity and the development of abnormal afferent activity including ectopic discharges from axotomised afferents and spontaneous activity of sensitized intact nociceptors. These changes can produce long-lasting abnormal hypersensitivity to normally innocuous stimuli, or allodynia. Examples of neuropathic pain include, but are not limited to, post-herpetic neuralgia, trigeminal neuralgia, diabetic neuropathy, chronic lower back pain, phantom limb pain, and pain resulting from cancer and chemotherapy, chronic pelvic pain, complex regional pain syndrome and related neuralgias.
There have been some advances in treating neuropathic pain symptoms by using medications, such as gabapentin, and more recently pregabalin, as short-term, first-line treatments. However, pharmacotherapy for neuropathic pain has generally had limited success with little response to commonly used pain reducing drugs, such as NSAIDS and opiates. Consequently, there is still a considerable need to explore novel treatment modalities.
There remain a limited number of potent effective sodium channel blockers with a minimum of adverse events in the clinic. There is also an unmet medical need to treat neuropathic pain and other sodium channel associated pathological states effectively and without adverse side effects. PCT Published Patent Application No. WO 2006/110917, PCT Published Patent Application No. WO 2010/045251 , PCT Published Patent Application No. WO 2010/045197, PCT Published Patent Application No. WO 2011/047174 and PCT Published Patent Application No. WO 2011/002708 discloses certain spiro-oxindole compounds. These compounds are disclosed therein as being useful for the treatment of sodium channel-mediated diseases, preferably diseases related to pain, central nervous conditions such as epilepsy, anxiety, depression and bipolar disease;
cardiovascular conditions such as arrhythmias, atrial fibrillation and ventricular fibrillation; neuromuscular conditions such as restless leg syndrome; neuroprotection against stroke, neural trauma and multiple sclerosis; and channelopathies such as erythromelalgia and familial rectal pain syndrome.
Methods of preparing these compounds and pharmaceutical compositions containing them are also disclosed in PCT Published Patent Application No. WO 2006/110917, PCT Published Patent Application No. WO 2010/045251 , PCT
Published Patent Application No. WO 2010/045197, PCT Published Patent Application No. WO 2011/047174 and PCT Published Patent Application No. WO 2011/002708.
Postherpetic neuralgia (PHN) is a rare disorder that is defined as significant pain or abnormal sensation 120 days or more after the presence of the initial rash caused by shingles. This pain persists after the healing of the associated rash. Generally, this affliction occurs in older individuals and individuals suffering from immunosuppression. There are about one million cases of shingles in the US per year, of which 10–20% will result in PHN.
Topical analgesics such as lidocaine and capsaicin are traditionally used to treat this disorder. Both lidocaine and TV-45070 have a mechanism of action that involves the inhibition of voltage-gated sodium ion channels.
TV-45070 (formerly XEN-402) was in-licensed by Teva from Xenon Pharmaceuticals and is reported to be an antagonist of the Nav1.7 sodium ion channel protein.
It is currently in Phase II clinical trials for PHN. Interestingly, the loss of function of the Nav1.7 sodium ion channel was reported to result in the inability to experience pain as a hereditary trait in certain individuals.
Primary erythromelalgia is another rare disease where alterations in Nav1.7 or mutations in the corresponding encoding gene SCN9A have been reported to result in chronic burning pain that can last for hours or even days. Thus, compounds which regulate this protein have potential therapeutic value as analgesics for chronic pain.

| Inventors | Mikhail Chafeev, Jianmin Fu, Jean-Jacques Cadieux |
| Original Assignee | Xenon Pharmaceuticals Inc. |
PATENT
US 20100331386
WO 2011106729
US 20110087027
US 20110086899
US 20130143941
US 20130210884
WO 2013154712
US 20150216794
WO 2016127068
WO 2016109795
CN 106518886
US 20170239183
SYNTHESIS
WO 2013154712

CONTD…….

Synthesis
CN 106518886

PATENT
US 20100331386
Preparation of the (S)-Enantiomer of the Invention
The (S)-enantiomer of the invention and the corresponding (R)-enantiomer are prepared by the resolution of the compound of formula (I), as set forth above in the Summary of the Invention, using either chiral high pressure liquid chromatography methods or by simulated moving bed chromatography methods, as described below in the following Reaction Scheme wherein “chiral HPLC” refers to chiral high pressure liquid chromatography and “SMB” refers to simulated moving bed chromatography:

The compound of formula (I) can be prepared by the methods disclosed in PCT Published Patent Application No. WO 2006/110917, by methods disclosed herein, or by methods known to one skilled in the art.
One of ordinary skill in the art would recognize variations in the above Reaction Scheme which are appropriate for the resolution of the individual enantiomers.
Alternatively, the (S)-enantiomer of formula (I-S) and the (R)-enantiomer of formula (I-R), can be synthesized from starting materials which are known or readily prepared using process analogous to those which are known.
Preferably, the (S)-enantiomer of the invention obtained by the resolution methods disclosed herein is substantially free of the (R)-enantiomer or contains only traces of the (R)-enantiomer.
The following Synthetic Examples serve to illustrate the resolution methods disclosed by the above Reaction Schemes and are not intended to limit the scope of the invention.
Synthetic Example 1Synthesis of 1-{[5-(trifluoromethyl)furan-2-yl]methyl}spiro[furo[2,3-f][1,3]benzodioxole-7,3′-indol]-2′(1′H)-one (Compound of formula (I))

To a suspension of spiro[furo[2,3-f][1,3]benzodioxole-7,3′-indol]-2′(1′H)-one (1.0 g, 3.6 mmol), which can be prepared according to the methods disclosed in PCT Published Patent Application No. WO 2006/110917, and cesium carbonate (3.52 g, 11 mmol) in acetone (50 mL) was added 2-bromomethyl-5-trifluoromethylfuran (1.13 g, 3.9 mmol) in one portion and the reaction mixture was stirred at 55-60° C. for 16 hours. Upon cooling to ambient temperature, the reaction mixture was filtered and the filtrate was evaporated under reduced pressure. The residue was subjected to column chromatography, eluting with ethyl acetate/hexane (1/9-1/1) to afford 1′-{[5-(trifluoromethyl)furan-2-yl]methyl}spiro[furo[2,3-f][1,3]benzodioxole-7,3′-indol]-2′(1 ′H)-one, i.e., the compound of formula (I), (1.17 g, 76%) as a white solid: mp 139-141° C.;
1H NMR (300 MHz, CDCl3) δ 7.32-6.97 (m, 5H), 6.72 (d, J=3.3 Hz, 1H), 6.66 (s, 1H), 6.07 (s, 1H), 5.90-5.88 (m, 2H), 5.05, 4.86 (ABq, JAB=16.1 Hz, 2H), 4.91 (d, J=9.0 Hz, 1H), 4.66 (d, J=9.0 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 176.9, 155.7, 153.5, 148.8, 142.2, 141.9, 140.8, 140.2, 139.7, 139.1, 132.1, 129.2, 124.7, 124.1, 123.7, 121.1, 120.1, 117.6, 114.5, 114.4, 110.3, 109.7, 103.0, 101.9, 93.8, 80.0, 57.8, 36.9;
MS (ES+) m/z 430.2 (M+1), 452.2 (M+23); Cal’d for C22H14F3NO5: C, 61.54%; H, 3.29%; N, 3.26%; Found: C, 61.51%; H, 3.29%; N, 3.26%.
Synthetic Example 2Resolution of Compound of Formula (I) by Chiral HPLC
The compound of formula (I) was resolved into the (S)-enantiomer of the invention and the corresponding (R)-enantiomer by chiral HPLC under the following conditions:
Column: Chiralcel® OJ-RH; 20 mm I.D.×250 mm, 5 mic; Lot: OJRH CJ-EH001 (Daicel Chemical Industries, Ltd)
Eluent: Acetonitrile/Water (60/40, v/v, isocratic)
Flow rate: 10 mL/min
Run time: 60 min
Loading: 100 mg of compound of formula (I) in 1 mL of acetonitrileTemperature: Ambient
Under the above chiral HPLC conditions, the (R)-enantiomer of the compound of formula (I), i.e., (R)-1′-{[5-(trifluoromethyl)furan-2-yl]methyl}spiro[furo[2,3-f][1,3]-benzodioxole-7,3′-indol]-2′(1′H)-one, was isolated as the first fraction as a white solid; ee (enantiomeric excess)>99% (analytical OJ-RH, 55% acetonitrile in water); mp 103-105° C.; 1H NMR (300 MHz, DMSO-d6) δ 7.32-6.99 (m, 5H), 6.71 (d, J=3.4 Hz, 1H), 6.67 (s, 1H), 6.05 (s, 1H), 5.89 (d, J=6.2 Hz, 2H), 5.13, 5.02 (ABq, JAB=16.4 Hz, 2H), 4.82, 4.72 (ABq, JAB=9.4 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 177.2, 155.9, 152.0, 149.0, 142.4, 142.0, 141.3, 132.0, 129.1, 123.9, 120.6, 119.2, 117.0, 112.6, 109.3, 108.9, 103.0, 101.6, 93.5, 80.3, 58.2, 36.9; MS (ES+) m/z 430.2 (M+1), [α]D−17.46° (c 0.99, DMSO).
The (S)-enantiomer of the compound of formula (I), i.e., (S)-1′-{[5-(trifluoromethypfuran-2-yl]methyl}spiro-[furo[2,3-f][1,3]benzodioxole-7,3′-indol]-2′(1′H)-one was isolated as the second fraction as a white solid; ee >99% (analytical OJ-RH, 55% acetonitrile in water); mp 100-102° C.; 1H NMR (300 MHz, DMSO-d6) δ 7.32-6.99 (m, 5H), 6.71 (d, J=3.4 Hz, 1H), 6.67 (s, 1H), 6.05 (s, 1H), 5.89 (d, J=6.3 Hz, 2H), 5.12, 5.02 (ABq, JAB=16.4 Hz, 2H), 4.82, 4.72 (ABq, JAB=9.4 Hz, 2H); 13C NMR (75MHz, CDCl3) δ 177.2, 155.9, 152.0, 149.0, 142.4, 142.0, 141.3, 132.0, 129.1, 123.9, 120.6, 119.2, 117.0, 112.6, 109.3, 108.9, 103.0, 101.6, 93.5, 80.3, 58.2, 36.9; MS (ES+) m/z 430.2 (M+1), [α]D+14.04° (c 0.99, DMSO)
Synthetic Example 3Resolution of Compound of Formula (I) by SMB Chromatography
The compound of formula (I) was resolved into the (S)-enantiomer of the invention and the corresponding (R)-enantiomer by SMB chromatography under the following conditions:
Extract: 147.05 mL/min, Raffinate: 76.13 mL/min Eluent: 183.18 mL/min Feed: 40 mL/min Recycling: 407.88 mL/min Run Time: 0.57 min Temperature: 25° C. Pressure: 46 bar
The feed solution (25 g of compound of formula (I) in 1.0 L of mobile phase (25:75:0.1 (v:v:v) mixture of acetonitrile/methanol/trifluoroacetic acid)) was injected continuously into the SMB system (Novasep Licosep Lab Unit), which was equipped with eight identical columns in 2-2-2-2 configuration containing 110 g (per column, 9.6 cm, 4.8 cm I.D.) of ChiralPAK-AD as stationary phase. The first eluting enantiomer (the (R)-enantiomer of the compound of formula (I)) was contained in the raffinate stream and the second eluting enantiomer (the (S)-enantiomer of the compound of formula (I)) was contained in the extract stream. The characterization data of the (S)-enantiomer and the (R)-enantiomer obtained from the SMB resolution were identical to those obtained above utilizing chiral HPLC.
The compound of formula (I) was resolved into its constituent enantiomers on a Waters preparative LCMS autopurification system. The first-eluting enantiomer from the chiral column was brominated (at a site well-removed from the stereogenic centre) to give the corresponding 5′-bromo derivative, which was subsequently crystallized to generate a single crystal suitable for X-ray crystallography. The crystal structure of this brominated derivative of the first-eluting enantiomer was obtained and its absolute configuration was found to be the same as the (R)-enantiomer of the invention. Hence, the second-eluting enantiomer from the chiral column is the (S)-enantiomer of the invention. Moreover, the material obtained from the extract stream of the SMB resolution had a specific optical rotation of the same sign (positive, i.e. dextrorotatory) as that of the material obtained from the aforementioned LC resolution.
Patent
EXAMPLE 8
Synthesis of (7S)-1 ‘-{[5-(trifluoromethyl)furan-2- yllmethylJspirotfurop.S-flll .Sl enzoclioxole-y.S’-indoll-Zil ‘Wi-one
Compound of formula (ia1 )

To a cooled (0 °C) solution of (3S)-3-(6-hydroxy-1 ,3-benzodioxol-5-yl)-3- (hydroxymethyl)-1-{[5-(trifluoromethyl)furan-2-yl]methyl}-1 ,3-dihydro-2H-indol-2-one prepared according to the procedure described in Example 7 (16.4 mmol) and 2- (diphenylphosphino)pyridine (5.2 g, 20 mmol) in anhydrous tetrahydrofuran (170 mL) was added di-ferf-butylazodicarboxylate (4.5 g, 20 mmol). The mixture was stirred for 2 h at 0 °C, then the reaction was diluted with ethyl acetate (170 mL), washed with 3 N hydrochloric acid (7 x 50 mL) and brine (2 x 100 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was dissolved in ethanol (80 mL), decolorizing charcoal (15 g) was added and the mixture was heated at reflux for 1 h. The mixture was filtered while hot through a pad of diatomaceous earth. The filtrate was concentrated in vacuo and the residue triturated in a mixture of diethyl ether/hexanes to afford (7S)-1 ‘-{[5-(trifluoromethyl)furan-2-yl]methyl}spiro- [furo[2,3-/][1 ,3]benzodioxole-7,3’-indol]-2′(1 ‘H)-one (1.30 g) as a colorless solid in 18% yield. The mother liquor from the trituration was concentrated in vacuo, trifluoroacetic acid (20 mL) was added and the mixture stirred for 3 h at ambient temperature. The mixture was diluted with ethyl acetate (100 mL), washed with saturated aqueous ammonium chloride (100 mL), 3 N hydrochloric acid (4 x 60 mL) and brine (2 x 100 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was purified by column chromatography, eluting with a gradient of ethyl acetate in hexanes to afford further (7S)-1 ‘-{[5-(trifluoromethyl)furan-2-yl]methyl}spiro- [furo[2,3- ][1 ,3]benzodioxole-7,3’-indol]-2′(1 ‘H)-one (2.6 g) as a colorless solid (37% yield, overall yield 55% over 2 steps): H NMR (300 MHz, CDCI3) £7.29-6.96 (m, 4H), 6.73 (s, 1 H), 6.50 (s, 1 H), 6.38 (s, 1 H), 6.09 (s, 1 H), 5.85 (br s, 2H), 5.06 (d, J = 16.0 Hz, 1 H), 4.93-4.84 (m, 2H), 4.68-4.65 (m, 1 H); MS (ES+) m/z 429.8 (M + 1 ); ee (enantiomeric excess) >99.5% (HPLC, Chiralpak IA, 2.5% acetonitrile in methyl tert- butyl ether).
EXAMPLE 9
Synthesis of 1-(diphenylmethyl)-1 H-indole-2,3-dione
Compound of formula (15a)

A. To a suspension of hexanes-washed sodium hydride (34.0 g, 849 mmol) in anhydrous Λ/,/V-dimethylformamide (400 mL) at 0 °C was added a solution of isatin (99.8 g, 678 mmol) in anhydrous Λ/,/V-dimethylformamide (400 mL) dropwise over 30 minutes. The reaction mixture was stirred for 1 h at 0 °C and a solution of benzhydryl bromide (185 g, 745 mmol) in anhydrous N-dimethylformamide (100 mL) was added dropwise over 5 minutes. The reaction mixture was allowed to warm to ambient temperature, stirred for 16 h and heated at 60 °C for 2 h. The mixture was cooled to 0 °C and water (500 mL) was added. The mixture was poured into water (2 L), causing a precipitate to be deposited. The solid was collected by suction filtration and washed with water (2000 mL) to afford 1-(diphenylmethyl)-1H-indole-2,3- dione (164 g) as an orange solid in 77% yield.
B. Alternatively, to a mixture of isatin (40.0 g, 272 mmol), cesium carbonate (177 g, 543 mmol) and A/./V-dimethylformamide (270 mL) at 80 °C was added dropwise a solution of benzhydryl bromide (149 g, 544 mmol) in N,N- dimethyiformamide (200 mL) over 30 minutes. The reaction mixture was heated at 80 °C for 3 h, allowed to cool to ambient temperature and filtered through a pad of diatomaceous earth. The pad was rinsed with ethyl acetate (1000 mL). The filtrate was washed with saturated aqueous ammonium chloride (4 x 200 mL), 1 N
hydrochloric acid (200 mL) and brine (4 x 200 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was triturated with diethyl ether to afford 1 -(diphenylmethyl)-1 H-indole-2,3-dione (59.1 g) as an orange solid in 69% yield. The mother liquor from the trituration was concentrated in vacuo and the residue triturated in diethyl ether to afford a further portion of 1-(diphenylmethyl)-1 H- indole-2,3-dione (8.2 g) in 10% yield: 1H NMR (300 MHz, CDCI3) £7.60 (d, J = 7.4 Hz, 1 H), 7.34-7.24 (m, 1 1 H), 7.05-6.97 (m, 2H), 6.48 (d, J = 8.0 Hz, 1 H); MS (ES+) m/z 313.9 (M + 1 ).
C. Alternatively, a mixture of isatin (500 g, 3.4 mol) and anhydrous N,N- dimethylformamide (3.5 L) was stirred at 15-35 °C for 0.5 h. Cesium carbonate (2.2 kg, 6.8 mol) was added and the mixture stirred at 55-60 °C for 1 h. A solution of benzhydryl bromide (1.26 kg, 5.1 mol) in anhydrous N, A/-dimethylformamide (1.5 L) was added and the resultant mixture stirred at 80-85 °C for 1 h, allowed to cool to ambient temperature and filtered. The filter cake was washed with ethyl acetate (12.5 L). To the combined filtrate and washes was added 1 N hydrochloric acid (5 L). The phases were separated and the aqueous phase was extracted with ethyl acetate (2.5 L). The combined organic extracts were washed with 1 N hydrochloric acid (2 * 2.5 L) and brine (3 χ 2.5 L) and concentrated in vacuo to a volume of approximately 750 mL. Methyl ferf-butyl ether (2 L) was added and the mixture was cooled to 5-15 °C, causing a solid to be deposited. The solid was collected by filtration, washed with methyl ferf- butyl ether (250 mL) and dried in vacuo at 50-55 °C for 16 h to afford 1- (diphenylmethyl)-1 H-indole-2,3-dione (715 g) as an orange solid in 67% yield: 1H NMR (300 MHz, CDCI3) 7.60 (d, J = 7.4 Hz, H), 7.34-7.24 (m, 1 H), 7.05-6.97 (m, 2H), 6.48 (d, J = 8.0 Hz, 1 H); MS (ES+) m/z 313.9 (M + 1 ).
EXAMPLE 10
Synthesis of 1-(diphenylmethyl)-3-hydroxy-3-(6-hydroxy-1 ,3-benzodioxol-5-yl)-1 ,3- dihydro-2H-indol-2-one
Compound of formula (16a1 )

A. To a solution of sesamol (33.1 g, 239 mmol) in anhydrous
tetrahydrofuran (500 mL) at 0 °C was added dropwise a 2 M solution of
isopropylmagnesium chloride in tetrahydrofuran (104 mL, 208 mmol), followed by 1 – (diphenylmethyl)-1H-indole-2,3-dione (50.0 g, 160 mmol) and tetrahydrofuran (100 mL). The reaction mixture was stirred at ambient temperature for 5 h, diluted with ethyl acetate (1500 mL), washed with saturated aqueous ammonium chloride (400 mL) and brine (2 x 400 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was triturated with a mixture of diethyl ether and hexanes to afford 1- (diphenylmethyl)-3-hydroxy-3-(6-hydroxy-1 ,3-benzodioxol-5-yl)-1 ,3-dihydro-2H-in
2- one (70.7 g) as a colorless solid in 98% yield: 1H NMR (300 MHz, CDCI3) <59.12 (br s, 1 H), 7.45-7.43 (m, 1 H), 7.30-7.22 (m, 10H), 7.09-7.07 (m, 2H), 6.89 (s, 1 H), 6.56- 6.55 (m, 1 H), 6.47-6.46 (m, 1 H), 6.29-6.28 (m, 1 H), 5.86 (s, 2H), 4.52 (br s, 1 H); MS (ES+) m/z 433.7 (M – 17).
B. Alternatviely, a mixture of sesamol (0.99 kg, 7.2 mol) and anhydrous tetrahydrofuran (18 L) was stirred at 15-35 °C for 0.5 h and cooled to -5-0 °C.
Isopropyl magnesium chloride (2.0 M solution in tetrahydrofuran, 3.1 L, 6.2 mol) was added, followed by 1-(diphenylmethyl)-1 H-indole-2,3-dione (1.50 kg, 4.8 mol) and further anhydrous tetrahydrofuran (3 L). The mixture was stirred at 15-25 °C for 5 h. Ethyl acetate (45 L) and saturated aqueous ammonium chloride (15 L) were added. The mixture was stirred at 15-25 °C for 0.5 h and was allowed to settle for 0.5 h. The phases were separated and the organic phase was washed with brine (2.3 L) and concentrated in vacuo to a volume of approximately 4 L. Methyl ferf-butyl ether (9 L) was added and the mixture concentrated in vacuo to a volume of approximately 4 L. Heptane (6 L) was added and the mixture was stirred at 15-25 °C for 2 h, causing a solid to be deposited. The solid was collected by filtration, washed with methyl tert- butyl ether (0.3 L) and dried in vacuo at 50-55 °C for 7 h to afford 1-(diphenylmethyl)-3- hydroxy-3-(6-hydroxy-1 ,3-benzodioxol-5-yl)-1 ,3-dihydro-2H-indol-2-one (2.12 kg) as an off-white solid in 98% yield: 1H NMR (300 MHz, CDCI3) 9.12 (br s, 1 H), 7.45-7.43 (m, 1 H), 7.30-7.22 (m, 10H), 7.09-7.07 (m, 2H), 6.89 (s, 1 H), 6.56-6.55 (m, 1 H), 6.47-6.46 (m, 1 H), 6.29-6.28 (m, 1 H), 5.86 (s, 2H), 4.52 (br s, 1 H); MS (ES+) m/z 433.7 (M – 17).
EXAMPLE 1 1
Synthesis of 3-[6-(benzyloxy)-1 ,3-benzodioxol-5-yl]-1-(diphenylmethyl)-3-hydroxy-1 ,3- dihydro-2H-indol-2-one
Compound of formula (17a1)

A. A mixture of 1-(diphenylmethyl)-3-hydroxy-3-(6-hydroxy-1 ,3- benzodioxol-5-yl)-1 ,3-dihydro-2H-indol-2-one (30.0 g, 66.5 mmol), benzyl bromide (8.3 mL, 70 mmol), and potassium carbonate (18.4 g, 133 mmol) in anhydrous N,N- dimeihylformamide (100 mL) was stirred at ambient temperature for 16 h. The reaction mixture was filtered and the solid was washed with /V,A/-dimethylformamide (100 mL). The filtrate was poured into water (1000 mL) and the resulting precipitate was collected by suction filtration and washed with water to afford 3-[6-(benzyloxy)-1 ,3-benzodioxol- 5-yl]-1-(diphenylmethyl)-3-hydroxy-1 ,3-dihydro-2H-indol-2-one (32.0 g) as a beige solid in 83% yield: 1H NMR (300 MHz, CDCI3) 7.42-7.28 (m, 9H), 7.22-7.14 (m, 6H), 7.10- 6.93 (m, 3H), 6.89-6.87 (m, 2H), 6.53 (d, J = 7.6 Hz, 1 H), 6.29 (br s, 1 H), 5.88 (s, 1 H), 5.85 (s, 1 H), 4.66 (d, J = 14.2 Hz, 1 H), 4.51 (d, J = 14.1 Hz, 1 H), 3.95 (s, 1 H); MS (ES+) m/z 542.0 (M + 1), 523.9 (M – 17).
B. Alternatively, to a solution of 1-(diphenylmethyl)-3-hydroxy-3-(6- hydroxy-1 ,3-benzodioxol-5-yl)-1 ,3-dihydro-2H-indol-2-one (2.1 kg, 4.6 mol) in anhydrous A/,A/-dimethylformamide (8.4 L) at 20-30 °C was added potassium carbonate (1.3 kg, 9.2 mol), followed by benzyl bromide (0.58 L, 4.8 mol). The mixture was stirred at 20-30 °C for 80 h and filtered. The filter cake was washed with
A/,/V-dimethylformamide (0.4 L) and the filtrate was poured into water (75 L), causing a solid to be deposited. The mixture was stirred at 15-25 °C for 7 h. The solid was collected by filtration, washed with water (2 L) and dried in vacuo at 50-60 °C for 48 h to afford 3-[6-(benzyloxy)-1 ,3-benzodioxol-5-yl]-1-(diphenylmethyl)-3-hydroxy-1 ,3- dihydro-2H-indol-2-one (2.1 1 kg) as an off-white solid in 84% yield; 1H NMR (300
MHz, CDCI3) £7.42-7.28 (m, 9H), 7.22-7.14 (m, 6H), 7.10-6.93 (m, 3H), 6.89-6.87 (m, 2H), 6.53 (d, J = 7.6 Hz, 1 H), 6.29 (br s, 1 H), 5.88 (s, 1 H), 5.85 (s, 1 H), 4.66 (d, J = 14.2 Hz, 1 H), 4.51 (d, J = 14.1 Hz, 1 H), 3.95 (s, 1 H); MS (ES+) m/z 542.0 (M + 1 ).
EXAMPLE 12
Synthesis of 3-[6-(benzyloxy)-1 ,3-benzodioxol-5-yl]-1 -(diphenylmethyl)-l ,3-dihydro-2H- indol-2-one
Compound of formula (18a1 )

A. To a solution of 3-[6-(benzyloxy)-1 ,3-benzodioxol-5-yl]-1- (diphenylmethyl)-3-hydroxy-1 ,3-dihydro-2H-indol-2-one (32.0 g, 57.7 mmol) in dichloromethane (100 mL) was added trifluoroacetic acid (50 mL) followed by triethylsilane (50 mL). The reaction mixture was stirred at ambient temperature for 2 h and concentrated in vacuo. The residue was dissolved in ethyi acetate (250 mL), washed with saturated aqueous ammonium chloride (3 x 100 mL) and brine (3 x 100 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was triturated with diethyl ether to afford 3-[6-(benzyloxy)-1 ,3-benzodioxol-5- yl]-1-(diphenylmethyl)-1 ,3-dihydro-2H-indol-2-one (19.0 g) as a colorless solid in 61 % yield: 1H NMR (300 MHz, CDCI3) 7.31 -7.23 (m, 15H), 7.10-6.88 (m, 4H), 6.50-6.45 (m, 3H), 5.86 (s, 2H), 4.97-4.86 (m, 3H); MS (ES+) m/z 525.9 (M + 1).
B. Alternatively, to a solution of 3-[6-(benzyloxy)-1 ,3-benzodioxol-5-yl]-1- (diphenylmethyl)-3-hydroxy-1 ,3-dihydro-2H-indol-2-one (2.0 kg, 3.7 mol) in
dichloromethane (7 L) at 20-30 °C was added trifluoracetic acid (2.5 L), followed by triethylsilane (3.1 L). The mixture was stirred at 15-35 °C for 4 h and concentrated in vacuo to dryness. To the residue was added ethyl acetate (16 L) and the mixture was stirred at 15-35 °C for 0.5 h, washed with saturated aqueous ammonium chloride (3 x 7 L) and brine (3 χ 7 L) and concentrated in vacuo to a volume of approximately 7 L. Methyl ferf-butyl ether (9 L) was added and the mixture concentrated in vacuo to a volume of approximately 9 L and stirred at 10-20 °C for 2.5 h, during which time a solid was deposited. The solid was collected by filtration, washed with methyl te/t-butyl ether (0.4 L) and dried in vacuo at 50-55 °C for 7 h to afford 3-[6-(benzyloxy)-1 ,3- benzodioxol-5-yl]-1-(diphenylmethyl)-1 ,3-dihydro-2H-indol-2-one (1 .26 kg) as an off-white solid in 65% yield: 1H NMR (300 MHz, CDCI3) £7.31 -7.23 (m, 15H), 7.10- 6.88 (m, 4H), 6.50-6.45 (m, 3H), 5.86 (s, 2H), 4.97-4.86 (m, 3H); MS (ES+) m/z 525.9 (M + 1).
EXAMPLE 13
Synthesis of (3S)-3-[6-(benzyloxy)-1 ,3-benzodioxol-5-yl]-3-[(benzyloxy)methyl]-1 –
(diphenylmethyl)-1 ,3-dihydro-2H-indol-2-one
Compound of formula (19a1 )

A. To a nitrogen-degassed mixture of 50% w/w aqueous potassium hydroxide (69.6 mL, 619 mmol), toluene (100 mL), and (9S)-1 -(anthracen-9- ylmethyl)cinchonan-1 -ium-9-ol chloride (0.50 g, 0.95 mmol) cooled in an ice/salt bath to an internal temperature of -18 °C was added a nitrogen-degassed solution of 3-[6- (benzyloxy)-l ,3-benzodioxol-5-yl]-1 -(diphenylmethyl)-l ,3-dihydro-2H-indol-2-one (10.0 g, 19.0 mmol) and benzyl chloromethyl ether (2.9 mL, 21 mmol) in
toluene/tetrahydrofuran (1 :1 v/v, 80 mL) dropwise over 1 h. The reaction mixture was stirred for 3.5 h and diluted with ethyl acetate (80 mL). The organic phase was washed with 1 N hydrochloric acid (3 x 150 mL) and brine (2 x 100 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo to afford (3S)-3-[6-(benzyloxy)-1 ,3- benzodioxol-5-yl]-3-[(benzyloxy)methyl]-1-(diphenylmethyl)-1 ,3-dihydro-2/-/-indol-2-one (12.6 g) as a colorless solid in quantitative yield: 1H NMR (300 MHz, CDCI3) 7.42 (d, 2H), 7.24-6.91 (m, 21 H), 6.69-6.67 (m, 2H), 6.46 (d, J = 7.7 Hz, 1 H), 6.15 (s, 1 H), 5.83- 5.81 (m, 2H), 4.53-4.31 (m, 3H), 4.17-4.09 (m, 3H); MS (ES+) m/z 646.0 (M + 1); ee (enantiomeric excess) 90% (HPLC, Chiralpak IA, 2.5% acetonitrile in methyl tert-butyl ether).
B. Alternatively, a mixture of 50% w/v aqueous potassium hydroxide (4.2 kg), toluene (12 L) and (9S)-1 -(anthracen-9-ylmethyl)cinchonan-1 -ium-9-ol chloride (0.06 kg, 0.1 mol) was degassed with dry nitrogen and cooled to -18 to -22 °C. To this mixture was added a cold (-18 to -22 °C), nitrogen-degassed solution of 3-[6-
(benzyloxy)-l ,3-benzodioxol-5-yl]-1 ~(diphenylmethyl)-1 ,3-dihydro-2H-indol-2-one (1.2 kg, 2.3 mol) and benzyl chloromethyl ether (0.43 kg, 2.8 mol) in toluene (10 L) and tetrahydrofuran (10 L) at -18 to 22 °C over 3 h. The mixture was stirred at -18 to -22 °C for 5 h, allowed to warm to ambient temperature and diluted with ethyl acetate (10 L). The phases were separated and the organic layer was washed with 1 N
hydrochloric acid (3 χ 8 L) and brine (2 χ 12 L) and concentrated in vacuo to dryness to afford (3S)-3-[6-(benzyloxy)-1 ,3-benzodioxol-5-yl]-3-[(benzyloxy)methyl]-1- (diphenylmethyl)-1 ,3-dihydro-2H-indol-2-one (1.5 kg) as a colorless solid in quantitative yield: 1H NMR (300 MHz, CDCI3) £7.42 (d, 2H), 7.24-6.91 (m, 21 H), 6.69-6.67 (m, 2H), 6.46 (d, J = 7.7 Hz, 1 H), 6.15 (s, 1 H), 5.83-5.81 (m, 2H), 4.53-4.31 (m, 3H), 4.17- 4.09 (m, 3H); MS (ES+) m/z 646.0 (M + 1); ee (enantiomeric excess) 90% (HPLC, ChiralPak IA). EXAMPLE 14
Synthesis of (3S)-1-(diphenylmethyl)-3-(6-hydroxy-1 ,3-benzodioxol-5-yl)-3- (hydroxymethyl)-1 ,3-dihydro-2/-/-indol-2-one
Compound of formula (20a1)

A. A mixture of (3S)-3-[6-(benzyloxy)-1 ,3-benzodioxol-5-yl]-3- [(benzyloxy)methyl]-1 -(diphenylmethyl)-1 ,3-dihydro-2/-/-indol-2-one (8.8 g, 14 mmol), 10% w/w palladium on carbon (50% wetted powder, 3.5 g, 1.6 mmol), and acetic acid (3.9 ml_, 68 mmol) in a nitrogen-degassed mixture of ethanol/tetrahydrofuran (1 : 1 v/v, 140 mL) was stirred under hydrogen gas (1 atm) at ambient temperature for 4 h. The reaction mixture was filtered through a pad of diatomaceous earth and the pad was rinsed with ethyl acetate (100 mL). The filtrate was concentrated in vacuo to afford (3S)-1-(diphenylmethyl)-3-(6-hydroxy-1 ,3-benzodioxol-5-yl)-3-(hydroxymethyl)-1 ,3- dihydro-2H-indol-2-one as a colorless solid that was carried forward without further purification: H NMR (300 MHz, CDCI3) 9.81 (br s, 1 H), 7.35-7.24 (m, 1 1 H), 7.15- 7.01 (m, 3H), 6.62 (s, 1 H), 6.54-6.47 (m, 2H), 5.86-5.84 (m, 2H), 4.76 (d, J = 1 1.0 Hz, 1 H), 4.13-4.04 (m, 1 H), 2.02 (s, 1 H); MS (ES+) m/z 465.9 (M + 1); ee (enantiomeric excess) 93% (HPLC, Chiralpak IA, 2.5% acetonitrile in methyl ie t-butyl ether).
B. Alternatively, a glass-lined hydrogenation reactor was charged with (3S)-3-[6-(benzyloxy)-1 ,3-benzodioxol-5-yl]-3-[(benzyloxy)methyl]-1 -(diphenylmethyl)- 1 ,3-dihydro-2H-indol-2-one (0.1 kg, 0.15 mol), tetrahydrofuran (0.8 L), ethanol (0.4 L), acetic acid (0.02 L) and 20% w/w palladium (li) hydroxide on carbon (0.04 kg). The reactor was purged three times with nitrogen. The reactor was then purged three times with hydrogen and was then pressurized to 50-55 lb/in2 with hydrogen. The mixture was stirred at 20-30 °C for 5 h under a 50-55 lb/in2 atmosphere of hydrogen. The reactor was purged and the mixture was filtered. The filtrate was concentrated in vacuo to a volume of approximately 0.2 L and methyl te/t-butyl ether (0.4 L) was added. The mixture was concentrated in vacuo to a volume of approximately 0.2 L and methyl ie/t-butyl ether (0.2 L) was added, followed by heptane (0.25 L). The mixture was stirred at ambient temperature for 2 h, during which time a solid was deposited. The solid was collected by filtration, washed with heptane (0.05 L) and dried in vacuo at a temperature below 50 °C for 8 h to afford (3S)-1 -(diphenylmethyl)-3-(6-hydroxy- 1 ,3-benzodioxol-5-yl)-3-(hydroxymethyl)-1 ,3-dihydro-2H-indol-2-one (0.09 kg) as a colorless solid in 95% yield: 1H NMR (300 MHz, CDCI3) 9.81 (br s, 1 H), 7.35-7.24 (m, 1 1 H), 7.15-7.01 (m, 3H), 6.62 (s, 1 H), 6.54-6.47 (m, 2H), 5.86-5.84 (m, 2H), 4.76 (d, J = 1 1.0 Hz, 1 H), 4.13-4.04 (m, 1 H), 2.02 (s, 1 H); MS (ES+) m/z 465.9 (M + 1); ee (enantiomeric excess) 91% (HPLC, ChiralPak IA).
EXAMPLE 15
Synthesis of (7S)-1′-(diphenylmethyl)spiro[furo[2,3-/][1 ,3]benzodioxole-7,3′-indol]-
2′(1 ‘tf)-one
Compound of formula (21 a1 )

A. To a cooled (0 °C) solution of (3S)-1 -(diphenylmethyl)-3-(6-hydroxy-1 ,3- benzodioxol-5-yl)-3-(hydroxymethyl)-1 ,3-dihydro-2H-indol-2-one prepared according to the procedure described in Example 14 (13.6 mmol) and 2-
(diphenylphosphino)pyridine (4.3 g, 16 mmol) in anhydrous tetrahydrofuran (140 mL) was added di-tert-butylazodicarboxylate (3.8 g, 17 mmol). The reaction mixture was stirred at 0 °C for 3 h, diluted with ethyl acetate (140 mL), washed with 3 N
hydrochloric acid (6 * 50 mL) and brine (2 χ 100 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was triturated with a mixture of diethyl ether and hexanes to afford (7S)-1 ‘-(diphenylmethyl)spiro[furo[2,3- ][1 ,3]benzodioxole-7,3’-indol]-2′(1 ‘H)-one (4.55 g) as a colorless solid in a 75% yield over 2 steps: 1H NMR (300 MHz, CDCI3) 7.34-7.24 (m, 10H), 7.15-7.13 (m, 1 H), 7.04 (s, 1 H), 6.99-6.95 (m, 2H), 6.50-6.48 (m, 2H), 6.06 (s, 1 H), 5.85-5.83 (m, 2H), 4.96 (d, J = 8.9 Hz, 1 H), 4.69 (d, J = 8.9 Hz, 1 H); MS (ES+) m/z 447.9 (M + 1); ee
(enantiomeric excess) 93% (HPLC, Chiraipak IA, 2.5% acetonitrile in methyl te/f-butyl ether).
B. Alternativel, to a cooled (0-5 °C) solution of (3S)-1-(diphenylmethyl)-3- (6-hydroxy-1 ,3-benzodioxol-5-yl)-3-(hydroxymethyl)-1 ,3-dihydro-2 -/-indol-2-one (1 .0 kg, 2.1 mol) and 2-(diphenylphosphino)pyridine (0.66 kg, 2.5 mol) in anhydrous tetrahydrofuran (20 L) was added over 2 h a solution of di-terf-butylazodicarboxylate (0.62 kg, 2.7 mmol) in anhydrous tetrahydrofuran (5 L). The mixture was stirred for 4 h at 0-5 °C and was allowed to warm to ambient temperature. The mixture was diluted with ethyl acetate (20 L), washed with 3 N hydrochloric acid (6 * 8 L) and brine (2 x 12 L) and concentrated in vacuo to a volume of approximately 1.5 L. Methyl rert-butyl ether (4 L) was added and the mixture concentrated in vacuo to a volume of
approximately 1.5 L. Methyl terf-butyl ether (2 L) and heptane (2 L) were added and the mixture was stirred at ambient temperature for 2 h, during which time a solid was deposited. The solid was collected by filtration, washed with heptane (0.5 L) and dried in vacuo below 50 °C for 8 h to afford (7S)-1′-(diphenylmethyl)spiro[furo[2,3- f][1 ,3]benzodioxole-7,3′-indol]-2′(1’H)-one (0.76 kg) as a colorless solid in 79% yield: 1H NMR (300 MHz, CDCI3) 7.34-7.24 (m, 10H), 7.15-7.13 (m, 1 H), 7.04 (s, 1 H), 6.99- 6.95 (m, 2H), 6.50-6.48 (m, 2H), 6.06 (s, 1 H), 5.85-5.83 (m, 2H), 4.96 (d, J = 8.9 Hz, 1 H), 4.69 (d, J = 8.9 Hz, 1 H); MS (ES+) m/z 447.9 (M + 1 ); ee (enantiomeric excess) 92% (HPLC, ChiralPak IA).
EXAMPLE 16
Synthesis of (7S)-spiro[furo[2,3-f][1 ,3]benzodioxole-7,3′-indol]-2′(1 ‘H)-one
Compound of formula (22a1)

A. To a solution of (7S)-1′-(diphenylmethyl)spiro[furo[2,3- f][1 ,3]benzodioxole-7,3′-indol]-2′(1’H)-one (4.55 g, 10.2 mmol) in trifluoroacetic acid (80 ml_) was added triethylsilane (7 ml_). The reaction mixture was heated at reflux for 2.5 h, allowed to cool to ambient temperature and concentrated in vacuo. The residue was triturated with a mixture of diethyl ether and hexanes to afford
(7S)-spiro[furo[2,3-/][1 ,3]benzodioxole-7,3,-indol]-2′(1’W)-one (2.30 g) as a colorless solid in 80% yield: 1H NMR (300 MHz, CDCI3) £8.27 (br s, 1 H), 7.31-7.26 (m, 1 H), 7.17-7.15 (m, 1 H), 7.07-7.02 (m, 1 H), 6.96-6.94 (m, 1 H), 6.53-6.52 (m, 1 H), 6.24-6.23 (m, 1 H), 5.88-5.87 (m, 2H), 4.95 (d, J = 8.6 Hz, 1 H), 4.68 (d, J = 8.9 Hz, 1 H); MS (ES+) m/z 281.9 (M + 1 ); ee (enantiomeric excess) 99% (HPLC, Chiralpak IA, 2.5% acetonitrile in methyl fert-butyl ether). B. Alternatively, a mixture of (7S)-1 ‘-(diphenylmethyl)spiro[furo[2,3- /Kl^benzodioxole^-indol^ r^-one (0.70 kg, 1.6 mol), trifluoroacetic acid (12 L) and triethylsilane (1.1 L) was heated at reflux under nitrogen atmosphere for 3 h, allowed to cool to ambient temperature and concentrated in vacuo to dryness. To the residue was added ethyl acetate (0.3 L), methyl fert-butyl ether (1 L) and heptane (3.5 L), causing a solid to be deposited. The solid was collected by filtration, taken up in dichloromethane (3 L), stirred at ambient temperature for 1 h and filtered. The filtrate was concentrated in vacuo to dryness. The residue was taken up in ethyl acetate (0.3 L), methyl ferf-butyl ether (1 L) and heptane (3.5 L), causing a solid to be deposited. The solid was collected by filtration and dried in vacuo below 50 °C for 8 h to afford (7S)-spiro[furo[2,3- ][1 ,3]benzodioxole-7,3’-indol]-2′(1 ‘ -/)-one (0.40 kg) as a colorless solid in 91 % yield: 1H NMR (300 MHz, CDCI3) 8.27 (br s, 1 H), 7.31-7.26 (m, 1 H), 7.17-7.15 (m, 1 H), 7.07-7.02 (m, 1 H), 6.96-6.94 (m, 1 H), 6.53-6.52 (m, 1 H), 6.24-6.23 (m, 1 H), 5.88-5.87 (m, 2H), 4.95 (d, J = 8.6 Hz, 1 H), 4.68 (d, J = 8.9 Hz, 1 H); MS (ES+) m/z 281.9 (M + 1); ee (enantiomeric excess) 98.6% (HPLC, ChiralPak IA).
EXAMPLE 17
Synthesis of of (7S)-1 ‘-{[5-(trifluoromethyl)furan-2- yl]methyl}spiro[furo[2,3- ][1 ,3]benzodioxole-7,3’-indol]-2′(rH)-one
Compound of formula (Ia1)

A. To a mixture of (7S)-6H-spiro[[1 ,3]dioxolo[4,5-f]benzofuran-7,3′-indolin]- 2′-one (1.80 g, 6.41 mmol) and 2-(bromomethyl)-5-(trifluoromethyl)furan (1.47 g, 6.41 mmol) in acetone (200 mL) was added cesium carbonate (3.13 g, 9.61 mmol). The reaction mixture was heated at reflux for 2 h and filtered while hot through a pad of diatomaceous earth. The filtrate was concentrated in vacuo to afford (7S)-1′-{[5- (trifluoromethyOfuran^-yllmethy^spiroIfurop.S- ltl .Slbenzodioxole^.S’-indol^ rH)- one (2.71 g) as a colorless solid in quantitative yield (97% purity by HPLC). The product was crystallized from a mixture of methanol and hexanes to afford (7S)-1 ‘-{[5- (trifluoromethy furan^-yllmethylJspirotfuro^.S- lfl .Slbenzodioxole^.S’-indoll^ rH)- one (1.46 g) as colorless needles in 53% yield. The mother liquor was concentrated in vacuo and subjected to a second crystallization in methanol and hexanes to afford further (7S)-1 ‘-{[5-(trifluoromethyl)furan-2-yl]methyl}spiro[furo[2,3-/][1 ,3]benzodioxole- 7,3’-indol]-2′(1 ‘H)-one (0.469 g) as a colorless solid in 17% yield (total yield 70%): 1H NMR (300 MHz, CDCI3) δ 7.29-6.96 (m, 4H), 6.73 (s, 1 H), 6.50 (s, 1 H), 6.38 (s, 1 H), 6.09 (s, 1 H), 5.85 (br s, 2H), 5.06 (d, J = 16.0 Hz, 1 H), 4.93-4.84 (m, 2H), 4.68-4.65 (m, 1 H); MS (ES+) m/z 429.8 (M + 1); ee (enantiomeric excess) >99.5% (HPLC, Chiralpak IA, 2.5% acetonitrile in methyl tert-butyl ether).
B. Alternatively, to a solution of (7S)-spiro[furoI2,3-f][1 ,3]benzodioxole-7,3′- indol]-2′(1’H)-one (0.40 kg, 1.4 mol) in anhydrous N, W-dimethylformamide (5 L) was added cesium carbonate (1.2 kg, 3.4 mol), followed by 2-(bromomethyl)-5- (trifluromethyl)furan (0.24 L, 1.7 mol). The mixture was heated at 80-85 °C for 3 h, allowed to cool to ambient temperature and filtered through a pad of diatomaceous earth. The pad was washed with ethyl acetate (8 L). The combined filtrate and washes were washed with water (4 L), saturated aqueous ammonium chloride (2 * 4 L) and brine (2 * 4 L) and concentrated in vacuo to dryness. The residue was purified by recrystallization from te/t-butyl methyl ether (0.4 L) and heptane (0.8 L), followed by drying of the resultant solid in vacuo at 40-50 °C for 8 h to afford (7S)-1 ‘-{[5- (trifluoromethyl)furan-2-yl]methyl}spiro[furo[2,3-f][1 ,3]benzodioxole-7,3’-indol]-2′(1 ‘H)- one (0.37 kg) as a colorless solid in 61% yield: 1H NMR (300 MHz, CDCI3) δ 7.29-6.96 (m, 4H), 6.73 (s, 1 H), 6.50 (s, 1 H), 6.38 (s, 1 H), 6.09 (s, 1 H), 5.85 (br s, 2H), 5.06 (d, J = 16.0 Hz,1 H), 4.93-4.84 (m, 2H), 4.68-4.65 (m, 1 H); MS (ES+) m/z 429.8 (M + 1 ); ee (enantiomeric excess) > 99% (HPLC, Chiralpak IA).
PATENT
Cadieux, J.-J.; Chafeev, M.; Chowdhury, S.; Fu, J.; Jia, Q.; Abel, S.; El-Sayed, E.; Huthmann, E.; Isarno, T. Synthetic Methods For Spiro-Oxindole Compounds. U.S. Patent 8,445,696, May 21, 2013.
PATENT
Sun, S.; Fu, J.; Chowdhury, S.; Hemeon, I. W.; Grimwood, M. E.; Mansour, T. S. Asymmetric Syntheses of Spiro-Oxindole Compounds Useful As Therapeutic Agents. U.S. Patent 9,487,535, Nov 08, 2016.
PAPER

TV-45070 is a small-molecule lactam containing a chiral spiro-ether that has been reported as a potential topical therapy for pain associated with the Nav1.7 sodium ion channel encoded by the gene SCN9A. A pilot-scale synthesis is presented that is highlighted by an asymmetric aldol coupling at ambient temperature, used to create a quaternary chiral center. Although only a moderate ee is obtained, the removal of the undesired isomer is achieved through preferential precipitation of a near racemic mixture from the reaction, leaving the enantiopure isomer in solution. Cyclization to form the final API uses an uncommon diphenylphosphine-based leaving group which proved successful on the neopentyl system when other traditional leaving groups failed.
The First Asymmetric Pilot-Scale Synthesis of TV-45070
Joseph A. Sclafani*†§ , Jian Chen†∥, Daniel V. Levy†§, Harlan Reese†⊥, Mina Dimitri†, Partha Mudipalli†, Michael Christie†, Christopher J. Neville‡, Mark Olsen‡, and Roger P. Bakale†#
, Jian Chen†∥, Daniel V. Levy†§, Harlan Reese†⊥, Mina Dimitri†, Partha Mudipalli†, Michael Christie†, Christopher J. Neville‡, Mark Olsen‡, and Roger P. Bakale†#†Chemical Process Research and Development, ‡Analytical Research and Development, Teva Branded Pharmaceutical Products R&D Inc., 383 Phoenixville Pike, Malvern, Pennsylvania 19355, United States
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.7b00237
Publication Date (Web): September 8, 2017
Copyright © 2017 American Chemical Society
*E-mail: jasclafan@yahoo.com.
(S)-1′-[(5-Methyl-2-furyl)methyl]spiro[6H-furo[3,2-f][1,3]benzodioxole-7,3′-indoline]-2′-one (1)
1H NMR (DMSO, 400 MHz) δ 7.32 (t, J = 7.7 Hz, 1H), 7.20 (m, 3H), 7.07 (t, J = 7.3 Hz, 1H), 6.77 (d, J= 3.3 Hz, 1H), 6.72 (s, 1H), 6.10 (s, 1H), 5.94 (d, J = 9.1 Hz, 1H), 5.94 (d, J = 9.1 Hz, 1H), 5.13 (d, J = 16.5 Hz, 1H), 5.02 (d, J = 16.5 Hz, 1H), 4.82 (d, J = 9.5 Hz, 1H), 4.73 (d, J = 9.5 Hz, 1H).
13C NMR (100 MHz, DMSO-d6): 176.48, 155.28, 153.02, 148.40, 141.80, 141.51, 139.54 (q, JCF = 41.9 Hz), 131.63, 128.79, 123.64, 123.29, 119.69, 118.92 (q, JCF = 266.4 Hz), 114.01 (q, JCF = 2.9 Hz) 109.86, 109.21, 102.55, 101.44, 93.31, 79.52, 57.41, 36.44.



References
- ^ Jump up to:a b c Bagal, Sharan K.; Chapman, Mark L.; Marron, Brian E.; Prime, Rebecca; Ian Storer, R.; Swain, Nigel A. (2014). “Recent progress in sodium channel modulators for pain”. Bioorganic & Medicinal Chemistry Letters. 24 (16): 3690–9. ISSN 0960-894X. PMID 25060923. doi:10.1016/j.bmcl.2014.06.038.
- ^ Jump up to:a b Stephen McMahon; Martin Koltzenburg; Irene Tracey; Dennis C. Turk (1 March 2013). Wall & Melzack’s Textbook of Pain: Expert Consult – Online. Elsevier Health Sciences. p. 508. ISBN 0-7020-5374-0.
- ^ Jump up to:a b Xenon Pharma. “TV-45070: A Small Molecule for the Treatment of the Orphan Disease EM and Other Pain Disorders”.
- ^ Jump up to:a b Xenon Pharma (2012). “Teva and Xenon Announce Teva’s World Wide License of Xenon’s Pain Drug XEN402”.
External links
PATENT
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2006110917A2 | 11 Apr 2006 | 19 Oct 2006 | Xenon Pharmaceuticals Inc. | Spiro-oxindole compounds and their uses as therapeutic agents |
| WO2010045197A1 | 13 Oct 2009 | 22 Apr 2010 | Xenon Pharmaceuticals, Inc. | Spiro-oxindole compounds and their use as therapeutic agents |
| WO2010045251A2 | 13 Oct 2009 | 22 Apr 2010 | Xenon Pharmaceuticals, Inc. | Spiro-oxindole compounds and their use as therapeutic agents |
| WO2010104525A1 | 1 Jun 2009 | 16 Sep 2010 | Telcordia Technologies, Inc. | Scalable disruptive-resistant communication method |
| WO2011002708A1 | 28 Jun 2010 | 6 Jan 2011 | Xenon Pharmaceuticals Inc. | Enantiomers of spiro-oxindole compounds and their uses as therapeutic agents |
| WO2011047173A2 | 14 Oct 2010 | 21 Apr 2011 | Xenon Pharmaceuticals Inc. | Pharmaceutical compositions for oral administration |
| WO2011047174A1 | 14 Oct 2010 | 21 Apr 2011 | Xenon Pharmaceuticals Inc. | Synthetic methods for spiro-oxindole compounds |
| WO2011106729A2 | 25 Feb 2011 | 1 Sep 2011 | Xenon Pharmaceuticals Inc. | Pharmaceutical compositions of spiro-oxindole compound for topical administration and their use as therapeutic agents |
| Reference | ||
|---|---|---|
| 1 | * | DEHMLOW E V ET AL: “Monodeazacinchona alkaloid derivatives: synthesis and preliminary applications as phase-transfer catalysts“, EUROPEAN JOURNAL OF ORGANIC CHEMISTRY, WILEY – V C H VERLAG GMBH & CO. KGAA, DE, vol. 13, 1 January 2002 (2002-01-01), pages 2087 – 2093, XP002399953, ISSN: 1434-193X, DOI: 10.1002/1099-0690(200207)2002:13<2087::AID-EJOC2087>3.0.CO;2-Z |
| 2 | E.J. COREY; M.C. NOE, ORG. SYNTH., vol. 80, 2003, pages 38 – 45 | |
| 3 | GARST, J. F.; UNGVARY, F.: “Grignard Reagents”, 2000, JOHN WILEY & SONS, article “Mechanism of Grignard reagent formation“, pages: 185 – 275 | |
| 4 | GREENE, T.W.; P.G.M. WUTS: “Greene’s Protective Groups in Organic Synthesis, 4th Ed.,“, 2006, WILEY | |
| 5 | GREENE, T.W.; WUTS, P.G.M.: “Greene’s Protective Groups in Organic Synthesis, 4th Ed.“, 2006, WILEY | |
| 6 | HUGHES, D.L., ORG. PREP., vol. 28, 1996, pages 127 – 164 | |
| 7 | KUMARA SWAMY, K.C. ET AL.: “Mitsunobu and Related Reactions: Advances and Applications“, CHEM. REV., vol. 109, 2009, pages 2551 – 2651, XP055023394, DOI: doi:10.1021/cr800278z | |
| 8 | MERSMANN, A.: “Crystallization Technology Handbook; 2nd ed.“, 2001, CRC | |
| 9 | SMITH, M.; BAND J. MARCH: “Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 5th edition“, December 2000, WILEY | |
| 10 | SMITH, M.B.; J. MARCH: “Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 5th edition“, December 2000, WILEY | |
| 11 | * | TAKASHI OOI ET AL: “Recent Advances in Asymmetric Phase-Transfer Catalysis“, ANGEWANDTE CHEMIE INTERNATIONAL EDITION, vol. 46, no. 23, 4 June 2007 (2007-06-04), pages 4222 – 4266, XP055060024, ISSN: 1433-7851, DOI: 10.1002/anie.200601737 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2016109795A1 | 31 Dec 2015 | 7 Jul 2016 | Concert Pharmaceuticals, Inc. | Deuterated funapide and difluorofunapide |
| US8742109 | 14 Sep 2012 | 3 Jun 2014 | Xenon Pharmaceuticals Inc. | Synthetic methods for spiro-oxindole compounds |
| US8883840 | 14 Sep 2012 | 11 Nov 2014 | Xenon Pharmaceuticals Inc. | Enantiomers of spiro-oxindole compounds and their uses as therapeutic agents |
| US8916580 | 6 Mar 2013 | 23 Dec 2014 | Xenon Pharmaceuticals Inc. | Spiro-oxindole compounds and their use as therapeutic agents |
| US9260446 | 7 May 2014 | 16 Feb 2016 | Xenon Pharmaceuticals Inc. | Synthetic methods for spiro-oxindole compounds |
| US9458178 | 14 Nov 2014 | 4 Oct 2016 | Xenon Pharmaceuticals Inc. | Spiro-oxindole compounds and their use as therapeutic agents |
| US9480677 | 9 Oct 2014 | 1 Nov 2016 | Xenon Pharmaceuticals Inc. | Enantiomers of spiro-oxindole compounds and their uses as therapeutic agents |
| US9487535 | 11 Mar 2013 | 8 Nov 2016 | Xenon Pharmaceuticals Inc. | Asymmetric syntheses for spiro-oxindole compounds useful as therapeutic agents |
| US9504671 | 25 Feb 2011 | 29 Nov 2016 | Xenon Pharmaceuticals Inc. | Pharmaceutical compositions of spiro-oxindole compound for topical administration and their use as therapeutic agents |
| US9682033 | 5 Feb 2016 | 20 Jun 2017 | Teva Pharmaceuticals International Gmbh | Methods of treating postherpetic neuralgia with a topical formulation of a spiro-oxindole compound |
| US9695185 | 6 Jan 2016 | 4 Jul 2017 | Xenon Pharmaceuticals Inc. | Synthetic methods for spiro-oxindole compounds |
 |
|
| Clinical data | |
|---|---|
| Routes of administration |
By mouth, topical |
| ATC code |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| Chemical and physical data | |
| Formula | C22H14F3NO5 |
| Molar mass | 429.34547 g/mol |
| 3D model (JSmol) | |
//////////TV 45070, XEN 402, TEVA, XENON, Postherpetic neuralgia, PHN, PHASE 2, Funapide, фунапид , فونابيد , 呋纳匹特 , Orphan Drug Status
C1C2(C3=CC=CC=C3N(C2=O)CC4=CC=C(O4)C(F)(F)F)C5=CC6=C(C=C5O1)OCO6
Riamilovir, Triazavirin

![[1,2,4]Triazolo[5,1-c][1,2,4]triazin-4(1H)-one, 7-(methylthio)-3-nitro-.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=3113817&t=l)
Riamilovir, Triazavirin
Riamilovir sodium dihydrate, CAS 928659-17-0,
Riamilovir CAS: 123606-06-4
Chemical Formula: C5H4N6O3S
Molecular Weight: 228.19
[1,2,4]Triazolo[5,1-c][1,2,4]triazin-4(1H)-one, 7-(methylthio)-3-nitro- (9CI)
7-(Methylthio)-3-nitro[1,2,4]triazolo[5,1-c][1,2,4]triazin-4(6H)-one
1,2,4]Triazolo[5,1-c][1,2,4]triazin-4(6H)-one, 7-(methylthio)-3-nitro-
7-(methylsulfanyl)-3-nitro[1,2,4]triazolo[5,1-c][1,2,4]triazin- 4(1H)-one
7-thio-substituted-3-nitro-1,2,4-triazolo[5,1-c]-1,2,4-triazin-4(1H)-one
Riamilovir sodium CAS 116061-59-7
Riamilovir sodium dihydrate, CAS 928659-17-0, Triazavirin
Flavivirus infection; Zika virus infection

Zika virus

Flavivirus
Anti-viral drug

http://apps.who.int/medicinedocs/documents/s23256en/s23256en.pdf

Triazavirin (TZV) is a broad-spectrum antiviral drug developed in Russia through a joint effort of Ural Federal University, Russian Academy of Sciences, Ural Center for Biopharma Technologies and Medsintez Pharmaceutical.

It has an azoloazine base structure, which represents a new structural class of non-nucleoside antiviral drugs.[1]
It was originally developed as a potential treatment for pandemic influenza strains such as H5N1, and most of the testing that has been done has focused on its anti-influenza activity.[2][3][4]
However triazavirin has also been found to have antiviral activity against a number of other viruses including tick-borne encephalitis,[5]and is also being investigated for potential application against Lassa fever and Ebola virus disease.[6][7][8][9][10]
Yunona Holdings, was investigating riamilovir sodium dihydrate (triazavirin), a novel nucleoside inhibitor of human influenza virus A and B replication, for the potential oral treatment of influenza virus infection.
In November 2009, the company was seeking to outlicense the drug for development in the EU, presumed to be for use as a prescription medicine .
The Ural Branch of the Russian Academy of Sciences had previously developed, and Yunona Holdings registered and launched, triazavirinin in Russia as an OTC product .
Negative-sense, single-stranded RNA viruses (ssRNA), such as ssRNA viruses belonging to the Order Mononegavirales such as viruses in the Rhabdoviridae family, in particular the Rabies virus, the Filoviridae family, in particular the Ebolavirus, and the Paramyxoviridae family, in particular the Measles virus, other ssRNA viruses belonging to unassigned families such as notably the
Arenaviridae family, the Bunyaviridae family and the Orthomyxoviridae family and other unassigned ssRNA viruses such as notably the Deltavirus, cause many diseases in wildlife, domestic animals and humans. These ssRNA viruses belonging to different families are genetically and antigenically diverse, exhibiting broad tissue tropisms and a wide pathogenic potential.
For example, the Filoviridae viruses belonging to the Order
Mononegavirales, in particular the Ebolaviruses and Marburgviruses, are among the most lethal and most destructive viruses in the world. Filoviridae viruses are of particular concern as possible biological weapons since they have the potential for aerosol dissemination and weaponization.
The Ebolavirus includes five species: the Zaire, Sudan, Reston, Ta‘i Forest and Bundibugyo Ebolaviruses. In particular the Zaire, Sudan and Bundibugyo Ebolavirus cause severe, often fatal, viral hemorraghic fevers in humans and nonhuman primates.
For more than 30 years, the Ebolavirus has been associated with periodic episodes of hemorrhagic fever in Central Africa that produce severe disease in
infected patients. Mortality rates in outbreaks have ranged from 50% for the Sudan species of the Ebolavirus to up to 90% for the Zaire species of the Ebolavirus ((Sanchez et al., Filoviridae: Marburg and Ebola Viruses, in Fields Virology, pages 1409-1448 (Lippincott Williams & Wilkins, Philadelphia)). In November 2007, during an outbreak in the Bundibugyo district of Uganda, near the border with the Democratic Republic of the Congo the fifth species of the Ebolavirus was discovered, the Bundibugyo species. Said outbreak resulted in a fatality rate of about 25% (Towner et al., PLoS Pathog., 4(11 ) :e1000212 (2008)). The Zaire species of the Ebolavirus has also decimated populations of wild apes in this same region of Africa (Walsh et al., Nature, 422:611-614 (2003)).
When infected with the Ebolavirus, the onset of illness is abrupt and is characterized by high fever, headaches, joint and muscle aches, sore throat, fatigue, diarrhea, vomiting, and stomach pain. A rash, red eyes, hiccups and internal and external bleeding may be seen in some patients. Within one week of becoming infected with the virus, most patients experience chest pains and multiple organ failure, go into shock, and die. Some patients also experience blindness and extensive bleeding before dying.
Another example of a negative sense single-stranded RNA envelope virus is the Morbilllivirus such as the Measles virus which is associated with Measles and the Lyssavirus such as the Rabies virus.
The Lyssavirus, belonging to the family Rhabdoviridae, includes eleven recognized species, in particular the Rabies virus which is known to cause Rabies. Rabies is an ancient disease with the earliest reports possibly dated to the Old World before 2300 B.C and remains a world health threat due to remaining lack of effective control measures in animal reservoir populations and a widespread lack of human access to vaccination. The Rabies virus is distributed worldwide among mammalian reservoirs including carnivores and bats. Each year there are many reported cases of transmission of the Rabies virus from animals to humans (e.g. by an animal bite). More than 50,000 people annually die of Rabies, particularly in Asia and Africa.
Thus, there remains a need for antiviral compounds which are effective for use in the treatment of the ssRNA virus infections different from the Influenza A and Influenza B virus infections
SYNTHESIS CONTRUCTED WITH 3 ARTICLES AS BELOW
RU 2340614 C2 20081210,
e-EROS Encyclopedia of Reagents for Organic Synthesis, 1-7; 2009,
European Journal of Medicinal Chemistry, 113, 11-27; 2016


Khimiya Geterotsiklicheskikh Soedinenii (1989), (2), 253-7.
Khimiya Geterotsiklicheskikh Soedinenii (1992), (11), 1555-9.
Zhurnal Organicheskoi Khimii (1996), 32(5), 770-776
PAPER
Russian Journal of Organic Chemistry (Translation of Zhurnal Organicheskoi Khimii) (2002), 38(2), 272-280.
https://link.springer.com/article/10.1023%2FA%3A1015538322029
Adamantylation of 3-Nitro- and 3-Ethoxycarbonyl-1,2,4-triazolo[5,1-c]-1,2,4-triazin-4-ones
Abstract
Reaction of 3-nitro- and 3-ethoxycarbonyl-1,2,4-triazolo[5,1-c]-1,2,4-triazin-4-ones with 1-adamantanol (or 1-adamantyl nitrate) in concentrated sulfuric acid or with 1-bromoadamantane in sulfolane affords N-adamantyl derivatives. The adamantylation of 3-nitro-1,4-dihydro-7H-1,2,4-triazolo[5,1-c]-1,2,4-triazin-4-one yields a mixture of N8– and N1-isomers that undergo interconversion in concentrated sulfuric acid along intermolecular mechanism.
PATENT
RU 2340614 C2 20081210,
PAPER
Russian Chemical Bulletin (2010), 59(1), 136-143.
Synthesis and antiviral activity of nucleoside analogs based on 1,2,4-triazolo[3,2-c][1,2,4]triazin-7-ones
Abstract
Nucleoside analogs containing hydroxybutyl, hydroxyethoxymethyl, allyloxymethyl, and propargyloxymethyl fragments were synthesized based on 1,2,4-triazolo[3,2-c][1,2,4]triazin-7-ones isosteric to purine bases. Some of the compounds obtained inhibit in vitro reproduction of influenza and respiratory syncytial virus infection.
PATENT
WO 2015117016
PAPER
Chemistry of Heterocyclic Compounds (New York, NY, United States) (2015), 51(3), 275-280.
https://link.springer.com/article/10.1007%2Fs10593-015-1695-4

The nucleophilic susbstitution of nitro group in [1,2,4]triazolo[5,1-c][1,2,4]triazinones upon treatment with cysteine and glutathione was studied as a model for the interaction with thiol groups of virus proteins, which mimics the metabolic transformations of antiviral drug Triazavirin® and its derivatives.
Nucleophilic substitution of nitro group in nitrotriazolotriazines as a model of potential interaction with cysteine-containing proteins
PATENT
Example 1 : One pot synthesis of the sodium salt of 7-methylthio-3-nitro [1 ,
2, 4] triazolo [5,1 -c] [1, 2, 4] triazin -4 (1H)-one
Step 1 : Diazotization of compound (B): A solution (solution [1], herein after) was prepared of 5.8 g (0.05 ) of 5-amino-3-mercapto-1 ,2,4-triazole in 6.7 ml of nitric acid (15 M) and 12 ml of water. Said solution [1] was refrigerated to -7°C . Then a 40% sodium nitrite solution was added to the solution [1] in portions of 0.5 mL to obtain a total amount of sodium nitrite equal to 3.8 g in the mixture.
Step 2: Condensation of diazonium compound with an a-nitroester:
To the resulting diazonium salt of step 1 , 8.54 ml of diethyl nitromaionate was added. After holding for five minutes, a cooled solution of sodium hydroxide was slowly added dropwise to the reaction mixture until the pH was between pH 9 and pH 10 (solution [2], herein after). The resulting solution [2] was stirred at 0°C for 1 hour and at room temperature for 2 hours.
Step 3: alkylation: To the solution [2] of step 2, 6.23 ml (0.1 moi) of methyl iodide was added. The mixture was stirred for 1 hour at room temperature and filtered. The resulting precipitate was successively crystallized from water and dried in air. The reaction scheme is depicted below in Scheme 1.

SCHEME 1
The yield was 9.87 g (69%).
Physical and chemical characteristics of the sodium salt of 7-methylthio-3-nitro
[1, 2, 4] triazolo [5,1-c] [1, 2, 4] triazin -4 (1H)-one sodium salt: yellow crystalline powder, soluble in water, acetone, dimethylsulfoxide, dimethylformamide. insoluble in chloroform; Tmelt = 300°C, H NMR spectrum, δ, ppm, solvent DMSO-d6: 2.62 (3H, s, SCH3); IR spectrum, n, cm“1: 3535 (OH), 1649 (C=0), 1505 (N02), 1367 (N02); found.. %: C – 20.86, H 2.51 , N 29.28;
C5H;N6Na05S; Calculated, %: C – 20.98, H 2.47, N 29.36.
Example 2: Synthesis of the sodium salt of 7-methylthio-3-nitro [1, 2, 4] triazolo [5,1-c] [1, 2, 4] triazin -4 (1H)-one sodium salt
In this example the synthesis comprises 3 steps: in the first step 5-amino-3-mercapto-1.2,4-triazole (i.e. compound (B)) was prepared by condensation of aminoguanidine with a thio-derivative (thio ester) of formic acid, HC(=0)S-R, wherein -R was: methyl. In the second step 5-amino-3-mercapto-1 , 2,4-triazole was converted to the corresponding diazonium salt. In the third step this diazonium salt was reacted with an a-nitroester, 2-nitroacetoacetic ester, to form the 7-methylthio-3-nitro [1, 2, 4] triazolo [5,1-c] [1, 2, 4] triazin -4 (1H)-one. The different steps are explained in more detail below.
Step 1 : Synthesis of compound (B): In a reaction flask equipped with a stirrer, reflux condenser, under inert gas (nitrogen, argon), 20 g (0.1 mol) of aminoguanidine and 7.6 g (0.1 mol) methylthio-formate was added to 400 ml of absolute pyridine. The reaction mixture was boiled for 4 hours at 115°C.
Subsequently the reaction mixture was transferred into distilled water and washed several times with water. The washed mixture was dried over a Nutsche filter under vacuum. Recrystallization was carried out from ethanol. The reaction scheme is depicted below in Scheme 2.

SCHEME 2
The yield was 19.3 g (70%)
Step 2: Diazotation of compound (B): A solution (solution [3], herein after) was prepared of 26 g (0.1 mol) of 5-amino-3-mercapto-1 ,2,4-triazole (as obtained in step 1) in 32 ml of nitric acid (0.1 mol) and 200 ml of water. The solution was mixed and cooled to -5°C. In a separate recipient, a 0.1 M solution of sodium nitrite was prepared by dissolving 16 g of sodium nitrite in 100 ml of water. The sodium nitrite solution was put in the freezer until there was ice formation and subsequently the ice was crushed. Thereafter, the solution [3] and the sodium nitrite crushed ice were transferred into a 1 L reactor and stirred for 1 hour while the reactor temperature was kept at 0°C. The low temperature and the fact that the two reaction components are in different phases (i.e. liquid and solid) ensured a slow gradual progress of diazotization reaction at the phase interface. The end of the diazotization process was controlled by a iodine starch test (proof of the absence of sodium nitrite in a free state).
The rea

SCHEME 3
Step 3: Condensation of the diazonium compound with an α-nitroester: A solution (solution [4], herein after) was prepared by mixing 17.5 g of methyl 2- nitro-acetoacetate in 300 mL of isopropanoi. The solution [4] was mixed with the diazonium salt of step 2. The mixture was cooled to 0°C. At 0°C, a 10% sodium hydroxide solution was added to the reaction mixture (to neutralize residual nitrite and acetate) until there was a marked alkaline reaction (pH between 8 and 9). The temperature was controlled and was kept below +5°C. The resulting mixture was stirred for 1 hour. The precipitate was filtered off and dried in air. The yield was 78%.
The reaction scheme is depicted in Scheme 4

SCHEME 4
Example 3: Synthesis of the sodium salt of 7-methylthio-3-nitro [1, 2, 4] triazolo [5,1-c] [1, 2, 4] triazin -4 (1H)-one
The synthesis of the sodium salt of 7-methylthio-3-nitro-1 ,2,4-triazolo [5,1-c]-1 ,2,4-triazin-7-one may be carried out as in Example 2, only in step 2 the aqueous alcohol solution is replaced by an alcohol with alkali (such as sodium hydroxide). The yield of the antiviral compound (A) (sodium salt of 7-methylthio-3-nitro [1 2, 4] triazolo [5,1-c] [1 , 2. 4] triazin -4 (1 H)-one) may increase to 83%. The reaction scheme is depicted below in Scheme 5:

SCHEME 5
PATENT
Process for the preparation of 7-thio-substituted-3-nitro-1,2,4-triazolo[5,1-c]-1,2,4-triazin-4(1H)-one i.e. riamilovir sodium dihydrate is claimed. Also claimed are use of triazolo compounds for the treatment of ssRNA virus infections such as Zika virus and flavivirus, ssRNA viruses different from the Influenza A and Influenza B viruses and compositions comprising them. Along with concurrently published WO2017144709 claiming similar derivatives. Represents new area of interest from Doring International Gmbh and the inventors on this moiety.
Example 1 : One pot synthesis of the sodium salt of 7-methylthio-3-nitro [1, 2, 4] triazolo [5,1 -cj [1, 2, 4] triazin -4 (1H)-one
Step 1: Diazotization of compound (B): A solution (solution [1], herein after) was prepared of 5.8 g (0.05 M) of 5-amino-3-mercapto-1 ,2,4-triazole in 6.7 ml of nitric acid (15 M) and 12 ml of water. Said solution [1] was refrigerated to -7°C . Then a 40% sodium nitrite solution was added to the solution [1] in portions of 0.5 mL to obtain a total amount of sodium nitrite equal to 3.8 g in the mixture.
Step 2: Condensation of diazonium compound with an a-nitroester: To the resulting diazonium salt of step 1 , 8.54 ml of diethyl nitromalonate was added. After holding for five minutes, a cooled solution of sodium hydroxide was slowly added dropwise to the reaction mixture until the pH was between pH 9 and pH 10 (solution [2], herein after). The resulting solution [2] was stirred at 0°C for 1 hour and at room temperature for 2 hours.
Step 3: alkylation: To the solution [2] of step 2, 6.23 ml (0.1 mol) of methyl iodide was added. The mixture was stirred for 1 hour at room temperature and
filtered. The resulting precipitate was successively crystallized from water and dried in air. The reaction scheme is depicted below in Scheme 1.

SCHEME 1
The yield was 9.87 g (69%).
Physical and chemical characteristics of the sodium salt of 7-methylthio-3-nitro
[1, 2, 4] triazolo [5,1-c] [1, 2, 4] triazin -4 (1H)-one sodium salt: yellow crystalline powder, soluble in water, acetone, dimethylsulfoxide, dimethylformamide, insoluble in chloroform; Tmei, = 300°C, 1H NMR spectrum, δ, ppm, solvent DMSO-d6: 2.62 (3H, s, SCH3); IR spectrum, n, cm“1: 3535 (OH), 1649 (CO), 1505 (N02), 1367 (N02); found, %: C – 20.86, H 2.51 , N 29.28; C5H7N6Na05S; Calculated, %: C – 20.98, H 2.47, N 29.36.
Example 2: Synthesis of the sodium salt of 7-methylthio-3-nitro [1, 2,
4] triazolo [5,1-c] [1, 2, 4] triazin -4 (1H)-one sodium salt
In this example the synthesis comprises 3 steps: in the first step 5-amino-3-mercapto-1 ,2,4-triazole (i.e. compound (B)) was prepared by condensation of aminoguanidine with a thio-derivative (thio ester) of formic acid, HC(=0)S-R, wherein -R was: methyl. In the second step 5-amino-3-mercapto-1 ,2,4-triazole was converted to the corresponding diazonium salt. In the third step this diazonium salt was reacted with an a-nitroester, 2-nitroacetoacetic ester, to form the 7-methylthio-3-nitro [1, 2, 4] triazolo [5,1-c] [1, 2, 4] triazin -4 (1H)-one. The different steps are explained in more detail below.
Step 1 : Synthesis of compound (B): In a reaction flask equipped with a stirrer, reflux condenser, under inert gas (nitrogen, argon), 20 g (0.1 mo!) of
aminoguanidine and 7.6 g (0.1 mol) methylthio-formate was added to 400 ml of absolute pyridine. The reaction mixture was boiled for 4 hours at 115°C. Subsequently the reaction mixture was transferred into distilled water and washed several times with water. The washed mixture was dried over a Nutsche filter under vacuum. Recrystallization was carried out from ethanol. The reaction scheme is depicted below in Scheme 2.

SCHEME 2
The yield was 19.3 g (70%)
Step 2: Diazotation of compound (B): A solution (solution [3], herein after) was prepared of 26 g (0.1 mol) of 5-amino-3-mercapto-1 ,2,4-triazole (as obtained in step 1 ) in 32 ml of nitric acid (0.1 mol) and 200 ml of water. The solution was mixed and cooled to -5°C. In a separate recipient, a 0.1 M solution of sodium nitrite was prepared by dissolving 16 g of sodium nitrite in 100 ml of water. The sodium nitrite solution was put in the freezer until there was ice formation and subsequently the ice was crushed. Thereafter, the solution [3] and the sodium nitrite crushed ice were transferred into a 1 L reactor and stirred for 1 hour while the reactor temperature was kept at 0°C. The low temperature and the fact that the two reaction components are in different phases (i.e. liquid and solid) ensured a slow gradual progress of diazotization reaction at the phase interface. The end of the diazotization process was controlled by a iodine starch test (proof of the absence of sodium nitrite in a free state).
The rea

SCHEME 3
Step 3: Condensation of the diazonium compound with an a-nitroester: A solution (solution [4], herein after) was prepared by mixing 17.5 g of methyl 2-nitro-acetoacetate in 300 mL of isopropanol. The solution [4] was mixed with the diazonium salt of step 2. The mixture was cooled to 0°C. At 0°C, a 10% sodium hydroxide solution was added to the reaction mixture (to neutralize residual nitrite and acetate) until there was a marked alkaline reaction (pH between 8 and 9). The temperature was controlled and was kept below +5°C. The resulting mixture was stirred for 1 hour. The precipitate was filtered off and dried in air. The yield was 78%.
The reaction scheme is depicted in Scheme 4

SCHEME 4
Example 3: Synthesis of the sodium salt of 7-methylthio-3-nitro [1, 2, 4] triazolo [5,1-c] [1, 2, 4] triazin -4 (1H)-one
The synthesis of the sodium salt of 7-methy I th io-3-nitro- 1 ,2, 4-triazolo [5,1-c]-1 ,2,4-triazin-7-one may be carried out as in Example 2, only in step 2 the aqueous alcohol solution is replaced by an alcohol with alkali (such as sodium hydroxide). The yield of the antiviral compound (A) (sodium salt of 7-methylthio-3-nitro [1 2, 4] triazolo [5,1-c] [1 , 2, 4] triazin -4 (1 H)-one) may increase to 83%.
The reaction scheme is depicted below in Scheme 5:

SCHEME 5
References
- Jump up^ Rusinov VL, Sapozhnikova IM, Ulomskii EN, Medvedeva NR, Egorov VV, Kiselev OI, Deeva EG, Vasin AV, Chupakhin ON. Nucleophilic substitution of nitro group in nitrotriazolotriazines as a model of potential interaction with cysteine-containing proteins. Chemistry of Heterocyclic Compounds 2015;51(3):275-280. doi 10.1007/s10593-015-1695-4
- Jump up^ Loginova SIa, Borisevich SV, Maksimov VA, Bondarev VP, Kotovskaia SK, Rusinov VL, Charushin VN. Investigation of triazavirin antiviral activity against influenza A virus (H5N1) in cell culture. (Russian) Antibiotiki i Khimioterapiia. 2007;52(11-12):18-20. PMID 19275052
- Jump up^ Karpenko I, Deev S, Kiselev O, Charushin V, Rusinov V, Ulomsky E, Deeva E, Yanvarev D, Ivanov A, Smirnova O, Kochetkov S, Chupakhin O, Kukhanova M. Antiviral properties, metabolism, and pharmacokinetics of a novel azolo-1,2,4-triazine-derived inhibitor of influenza A and B virus replication. Antimicrobial Agents and Chemotherapy. 2010 May;54(5):2017-22. doi: 10.1128/AAC.01186-09 PMID 20194696
- Jump up^ Kiselev OI, Deeva EG, Mel’nikova TI, Kozeletskaia KN, Kiselev AS, Rusinov VL, Charushin VN, Chupakhin ON. A new antiviral drug Triazavirin: results of phase II clinical trial. (Russian). Voprosy Virusologii. 2012 Nov-Dec;57(6):9-12. PMID 23477247
- Jump up^ Loginova SIa, Borisevich SV, Rusinov VL, Ulomskiĭ UN, Charushin VN, Chupakhin ON. Investigation of Triazavirin antiviral activity against tick-borne encephalitis pathogen in cell culture. (Russian). Antibiotiki i Khimioterapiia. 2014;59(1-2):3-5. PMID 25051708
- Jump up^ “Target: Ebola”. Pravda. Retrieved 18 January 2015.
- Jump up^ “Yekaterinburg pharmacies to sell domestic antiviral drug”. Retrieved 18 January 2015.
- Jump up^ “Ebola crisis: Vaccine ‘too late’ for outbreak. BBC News, 17 October 2014”. BBC News.
- Jump up^ Kukil Bora. Russia Will Begin Testing Triazavirin, Used For Lassa Fever, And Other Drugs On Ebola: Health Ministry. International Business Times, 12 November 2014
- Jump up^ Darya Kezina. New antiviral drug from Urals will help fight Ebola and other viruses. Russia Beyond the Headlines, 12 November 2014
 |
|
| Legal status | |
|---|---|
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| ECHA InfoCard | 100.217.074 |
| Chemical and physical data | |
| Formula | C5H4N6O3S |
| Molar mass | 228.189 |
| 3D model (JSmol) | |
///////////riamilovir sodium dihydrate, Riamilovir , ANTIVIRAL, Triazavirin, Flavivirus infection, Zika virus infection
O=C1N2C(NN=C1[N+]([O-])=O)=NC(SC)=N2
Prexasertib , прексасертиб , بريكساسيرتيب , 普瑞色替 ,
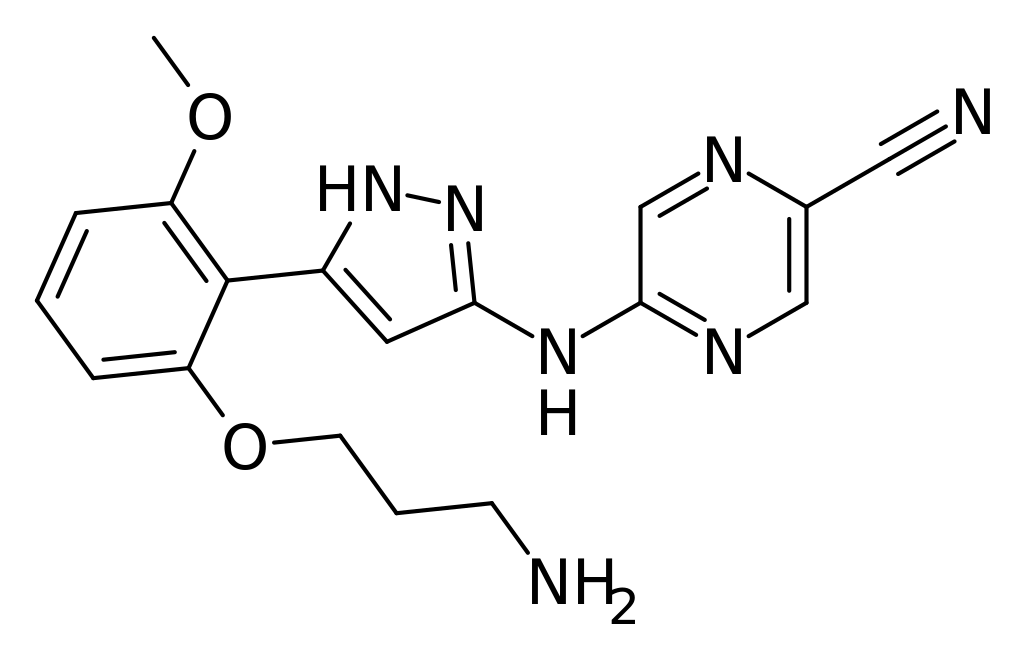
Prexasertib
Captisol® enabled prexasertib; CHK1 Inhibitor II; LY 2606368; LY2606368 MsOH H2O
5-(5-(2-(3-aminopropoxy)-6-methoxyphenyl)-1H-pyrazol-3-ylamino)pyrazine-2-carbonitrile
2-Pyrazinecarbonitrile, 5-[[5-[2-(3-aminopropoxy)-6-methoxyphenyl]-1H-pyrazol-3-yl]amino]-
| Name | Prexasertib |
|---|---|
| Lab Codes | LY-2606368 |
| Chemical Name | 5-({5-[2-(3-aminopropoxy)-6-methoxyphenyl]-1H-pyrazol-3-yl}amino)pyrazine-2-carbonitrile |
| Chemical Structure | |
| Molecular Formula | C18H19N7O2 |
| UNII | UNII:820NH671E6 |
| Cas Registry Number | 1234015-52-1 |
| OTHER NAMES |
прексасертиб [Russian] [INN]
|
| Originator | Array BioPharma |
| Developer | Eli Lilly, National Cancer Institute |
| Mechanism Of Action | Checkpoint kinase inhibitors, Chk-1 inhibitors |
| Who Atc Codes | L01X-E (Protein kinase inhibitors) |
| Ephmra Codes | L1H (Protein Kinase Inhibitor Antineoplastics) |
| Indication | Breast cancer, Ovarian cancer, Solid tumor, Head and neck cancer, Leukemia, Neoplasm Metastasis, Colorectal Neoplasms, Squamous Cell Carcinoma |


 2100300-72-7 CAS
2100300-72-7 CAS

Prexasertib mesylate hydrate
CAS#: 1234015-57-6 (mesylate hydrate)
Chemical Formula: C19H25N7O6S
Molecular Weight: 479.512, CODE LY-2940930
LY-2606368 (free base)

Prexasertib mesylate ANHYDROUS
CAS#: 1234015-55-4 (mesylate)
Chemical Formula: C19H23N7O5S
Molecular Weight: 461.497
Prexasertib dihydrochloride
1234015-54-3. MW: 438.3169
LY2606368 is a small-molecule Chk-1 inhibitors invented by Array and being developed by Eli Lilly and Company. Lilly is responsible for all clinical development and commercialization activities. Chk-1 is a protein kinase that regulates the tumor cell’s response to DNA damage often caused by treatment with chemotherapy. In response to DNA damage, Chk-1 blocks cell cycle progression in order to allow for repair of damaged DNA, thereby limiting the efficacy of chemotherapeutic agents. Inhibiting Chk-1 in combination with chemotherapy can enhance tumor cell death by preventing these cells from recovering from DNA damage.
Originator Array BioPharma; Eli Lilly
Developer Eli Lilly; National Cancer Institute (USA)
Class Antineoplastics; Nitriles; Pyrazines; Pyrazoles; Small molecules
Mechanism of Action Checkpoint kinase 1 inhibitors; Checkpoint kinase 2 inhibitors
Highest Development Phases
- Phase II Breast cancer; Ovarian cancer; Small cell lung cancer; Solid tumours
- Phase I Acute myeloid leukaemia; Colorectal cancer; Head and neck cancer; Myelodysplastic syndromes; Non-small cell lung cancer
Most Recent Events
- 10 Apr 2017 Eli Lilly completes a phase I trial for Solid tumours (Late-stage disease, Second-line therapy or greater) in Japan (NCT02514603)
- 10 Mar 2017 Phase-I clinical trials in Solid tumours (Combination therapy, Metastatic disease, Inoperable/Unresectable) in USA (IV) (NCT03057145)
- 22 Feb 2017 Khanh Do and AstraZeneca plan a phase H trial for Solid tumour (Combination therapy, Metastatic disease, Inoperable/Unresectable) in USA (NCT03057145)
Prexasertib (LY2606368) is a small molecule checkpoint kinase inhibitor, mainly active against CHEK1, with minor activity against CHEK2. This causes induction of DNA double-strand breaks resulting in apoptosis. It is in development by Eli Lilly. [1]
A phase II clinical trial for the treatment of small cell lung cancer is expected to be complete in December 2017.[2]
an aminopyrazole compound, or a pharmaceutically acceptable salt thereof or a solvate of the salt, that inhibits Chkl and is useful for treating cancers characterized by defects in deoxyribonucleic acid (DNA) replication, chromosome segregation, or cell division.
Chkl is a protein kinase that lies downstream from Atm and/or Atr in the DNA damage checkpoint signal transduction pathway. In mammalian cells, Chkl is phosphorylated in response to agents that cause DNA damage including ionizing radiation (IR), ultraviolet (UV) light, and hydroxyurea. This phosphorylation which activates Chkl in mammalian cells is dependent on Atr. Chkl plays a role in the Atr dependent DNA damage checkpoint leading to arrest in S phase and at G2M. Chkl phosphorylates and inactivates Cdc25A, the dual-specificity phosphatase that normally dephosphorylates cyclin E/Cdk2, halting progression through S-phase. Chkl also phosphorylates and inactivates Cdc25C, the dual specificity phosphatase that dephosphorylates cyclin B/Cdc2 (also known as Cdkl) arresting cell cycle progression at the boundary of G2 and mitosis (Fernery et al, Science, 277: 1495-1, 1997). In both cases, regulation of Cdk activity induces a cell cycle arrest to prevent cells from entering mitosis in the presence of DNA damage or unreplicated DNA. Various inhibitors of Chkl have been reported. See for example, WO 05/066163,
WO 04/063198, WO 03/093297 and WO 02/070494. In addition, a series of aminopyrazole Chkl inhibitors is disclosed in WO 05/009435.
However, there is still a need for Chkl inhibitors that are potent inhibitors of the cell cycle checkpoints that can act effectively as potentiators of DNA damaging agents. The present invention provides a novel aminopyrazole compound, or a pharmaceutically acceptable salt thereof or solvate of the salt, that is a potent inhibitor of Chkl . The compound, or a pharmaceutically acceptable salt thereof or a solvate of the salt, potently abrogates a Chkl mediated cell cycle arrest induced by treatment with DNA damaging agents in tissue culture and in vivo. Furthermore, the compound, or a pharmaceutically acceptable salt thereof or a solvate of the salt, of the present invention also provides inhibition of Chk2, which may be beneficial for the treatment of cancer. Additionally, the lack of inhibition of certain other protein kinases, such as CDKl, may provide a -2- therapeutic benefit by minimizing undesired effects. Furthermore, the compound, or a pharmaceutically acceptable salt thereof or a solvate of the salt, of the present invention inhibits cell proliferation of cancer cells by a mechanism dependent on Chkl inhibition.
| Inventors | Francine S. Farouz, Ryan Coatsworth Holcomb, Ramesh Kasar, Steven Scott Myers |
| Applicant | Eli Lilly And Company |
Preparation 8
tert-Butyl 3-(2-(3-(5-cyanopyrazin-2-ylamino)-lH-pyrazol-5-yl)-3- methoxyphenoxy)propylcarbamate

A solution of tert-butyl 3-(2-(3-(5-bromopyrazin-2-ylamino)-lH-pyrazol-5-yl)-3- methoxyphenoxy)propylcarbamate (0.378 g, 0.730 mmol) and zinc cyanide (0.10 g, 0.870 mmol) in DMF (10 mL) is degassed with a stream of nitrogen for one hour and then -25- heated to 80 0C. To the reaction is added Pd(Ph3P)4 (0.080 g, 0.070 mmol), and the mixture is heated overnight. The reaction is cooled to room temperature and concentrated under reduced pressure. The residue is purified by silica gel chromatography (CH2Cl2/Me0H) to give 0.251 g (73%) of the title compound.
Preparation 12 tert-Butyl 3-(2-(3-(5-cyanopyrazin-2-ylamino)-lH-pyrazol-5-yl)-3- methoxyphenoxy)propylcarbamate

A 5 L flange-neck round-bottom flask equipped with an air stirrer rod and paddle, thermometer, pressure-equalizing dropping funnel, and nitrogen bubbler is charged with 5-(5-(2-hydroxy-6-methoxy-phenyl)-lH-pyrazol-3-ylamino)-pyrazine-2-carbonitrile (47.0 g, 152 mmol) and anhydrous THF (1.2 L). The stirred suspension, under nitrogen, is cooled to 0 0C. A separate 2 L 3 -necked round-bottom flask equipped with a large -28- magnetic stirring bar, thermometer, and nitrogen bubbler is charged with triphenylphosphine (44.0 g; 168 mmol) and anhydrous THF (600 mL). The stirred solution, under nitrogen, is cooled to 0 0C and diisopropylazodicarboxylate (34.2 g; 169 mmol) is added and a milky solution is formed. After 3-4 min, a solution of7-butyl-N-(3- hydroxypropyl)-carbamate (30.3 g, 173 mmol) in anhydrous THF (100 mL) is added and the mixture is stirred for 3-4 min. This mixture is then added over 5 min to the stirred suspension of starting material at 0 0C. The reaction mixture quickly becomes a dark solution and is allowed to slowly warm up to room temperature. After 6.5 h, more reagents are prepared as above using PPh3 (8 g), DIAD (6.2 g) and carbamate (5.4 g) in anhydrous THF (150 mL). The mixture is added to the reaction mixture, cooled to -5 0C and left to warm up to room temperature overnight. The solvent is removed in vacuo. The resulting viscous solution is loaded onto a pad of silica and product is eluted with ethyl acetate. The concentrated fractions are separately triturated with methanol and resulting solids are collected by filtration. The combined solids are triturated again with methanol (400 mL) and then isolated by filtration and dried in vacuo at 50 0C overnight to give 31.3 g of desired product. LC-ES/MS m/z 466.2 [M+ 1]+.
Example 2
5 -(5 -(2-(3 -Aminopropoxy)-6-methoxyphenyl)- 1 H-pyrazol-3 -ylamino)pyrazine-2- carbonitrile dihydrogen chloride salt
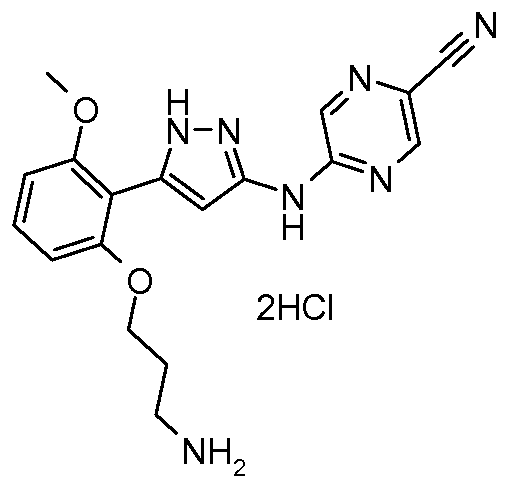
A 5 L flange-neck, round-bottom flask equipped with an air stirrer rod and paddle, thermometer, and air condenser with bubbler attached, is charged with tert-bvXyl 3-(2-(3- (5-cyanopyrazin-2-ylamino)-lH-pyrazol-5-yl)-3-methoxyphenoxy)propylcarbamate (30.9 g, 66.3 mmol) and ethyl acetate (3 L). The mechanically stirred yellow suspension is cooled to just below 10 0C. Then hydrogen chloride from a lecture bottle is bubbled in -29- vigorously through a gas inlet tube for 15 min with the ice-bath still in place. After 5 h the mixture is noticeably thickened in appearance. The solid is collected by filtration, washed with ethyl acetate, and then dried in vacuo at 60 0C overnight to give 30.0 g. 1H NMR (400 MHz, DMSO-d6) δ 2.05 (m, 2H), 2.96 (m, 2H), 3.81 (s, 3H), 4.12 (t, J = 5.8 Hz, 2H), 6.08 (br s, 3H), 6.777 (d, J = 8.2 Hz, IH), 6.782 (d, J = 8.2 Hz, IH), 6.88 (br s, IH), 7.34 (t, J = 8.2 Hz, IH), 8.09 (br s, IH), 8.55 (br s, IH), 8.71 (s, IH), 10.83 (s, IH), 12.43 (br s, IH). LC-ES/MS m/z 366.2 [M+lf.
Example 3 5 -(5 -(2-(3 -Aminopropoxy)-6-methoxyphenyl)- 1 H-pyrazol-3 -ylamino)pyrazine-2- carbonitrile

5-(5-(2-(3-Aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3-ylamino)pyrazine-2- carbonitrile dihydrogen chloride salt (3.0 g, 6.84 mmol) is suspended in 200 mL of CH2Cl2. 1 N NaOH is added (200 mL, 200 mmol). The mixture is magnetically stirred under nitrogen at room temperature for 5 h. The solid is collected by filtration and washed thoroughly with water. The filter cake is dried in vacuo at 50 0C overnight to give 2.26 g (90%) of the free base as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 1.81 (m, 2H), 2.73 (t, J = 6.2 Hz, 2H), 3.82 (s, 3H), 4.09 (t, J = 6.2 Hz, 2H), 6.76 (t, J = 8.2 Hz, 2H), 6.93 (br s, IH), 7.31 (t, J = 8.2 Hz, IH), 8.52 (br s, IH), 8.67 (s, IH). LC- MS /ES m/z 366.2 [M+ 1]+.
Example 4
5 -(5 -(2-(3 -Aminopropoxy)-6-methoxyphenyl)- 1 H-pyrazol-3 -ylamino)pyrazine-2- carbonitrile methanesulfonic acid salt -30-

5-(5-(2-(3-aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3-ylamino)pyrazine-2- carbonitrile (1.0 g, 2.74 mmol) is suspended in MeOH (100 mL). A I M solution of methanesulfonic acid in MeOH (2.74 mL, 2.74 mmol) is added to the mixture dropwise with stirring. The solid nearly completely dissolves and is sonicated and stirred for 15 min, filtered, and concentrated to 50 mL. The solution is cooled overnight at -15 0C and the solid that forms is collected by filtration. The solid is dried in a vacuum oven overnight to give 0.938 g (74%) of a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 1.97 (m, 2H), 2.28 (s, 3H), 2.95 (m, 2H), 3.79 (s, 3H), 4.09 (t, J = 5.9 Hz, 2H), 6.753 (d, J = 8.4 Hz, IH), 6.766 (d, J = 8.4 Hz, IH), 6.85 (br s, IH), 7.33 (t, J = 8.4 Hz, IH), 7.67 (br s, 3H), 8.49 (br s, IH), 8.64 (s, IH), 10.70 (s, IH), 12.31 (s, IH). LC-ES/MS m/z 366.2 [M+l]+.
Preparation 18 tert-Butyl 3-(2-(3-(5-cyanopyrazin-2-ylamino)-lH-pyrazol-5-yl)-3- methoxyphenoxy)propylcarbamate

5-(5-(2-Hydroxy-6-methoxyphenyl)-lH-pyrazol-3-ylamino)pyrazine-2- carbonitrile (618 g, 1.62 mol) is slurried in tetrahydrofuran (6.18 L, 10 volumes) and chilled to -5 to 0 0C with an acetone/ice bath. Triethylamine (330 g, 3.25 mol) is added through an addition funnel over 30 – 40 min at -5 to 5 0C. The resulting slurry is stirred at -5 to 5 0C for 60 – 90 min. The insoluble triethylamine hydrochloride is filtered and the solution of the phenol ((5-(2-hydroxy-6-methoxyphenyl)-lH-pyrazol-3- ylamino)pyrazine-2-carbonitrile) collected in an appropriate reaction vessel. The cake is rinsed with THF (1.24 L). The THF solution of the phenol is held at 15 to 20 0C until needed.
Triphenylphosphine (1074 g, 4.05 mol) is dissolved at room temperature in THF (4.33 L). The clear colorless solution is cooled with an acetone/ice bath to -5 to 5 0C. Diisopropylazodicarboxylate (795 g, 3.89 mol) is added dropwise through an addition funnel over 40 – 60 min, keeping the temperature below 10 0C. The resulting thick white slurry is cooled back to -5 to 0 0C. tert-Butyl 3-hydroxypropylcarbamate (717g, 4.05 moles) is dissolved in a minimum of THF (800 mL). The tert-butyl 3- hydroxypropylcarbamate/THF solution is added, through an addition funnel, over 20 – 30 -35- min at -5 to 5 0C to the reagent slurry. The prepared reagent is stirred in the ice bath at -5 to 0 0C until ready for use.
The prepared reagent slurry (20%) is added to the substrate solution at 15 to 20 0C. The remaining reagent is returned to the ice bath. The substrate solution is stirred at ambient for 30 min, then sampled for HPLC. A second approximately 20% portion of the reagent is added to the substrate, stirred at ambient and sampled as before. Addition of the reagent is continued with monitoring for reaction completion by HPLC. The completed reaction is concentrated and triturated with warm methanol (4.33 L, 50 – 60 0C) followed by cooling in an ice bath. The resulting yellow precipitate is filtered, rinsed with cold MeOH (2 L), and dried to constant weight to provide 544 g (72%) of the title compound, mp 214 – 216 0C; ES/MS m/z 466.2 [M+l]+.
Example 5
2-Pyrazinecarbonitrile, 5-[[5-[-[2-(3-aminopropyl)-6-methoxyphenyl]-lH-pyrazol-3- yl]amino] monomesylate monohydrate (Chemical Abstracts nomenclature)

tert-Butyl 3-(2-(3-(5-cyanopyrazin-2-ylamino)-lH-pyrazol-5-yl)-3- methoxyphenoxy)propylcarbamate (1430 g, 3.07 mol) is slurried with acetone (21.5 L) in a 30 L reactor. Methanesulfonic acid (1484 g, 15.36 mol) is added through an addition funnel in a moderate stream. The slurry is warmed to reflux at about 52 0C for 1 to 3 h and monitored for reaction completion by HPLC analysis. The completed reaction is cooled from reflux to 15 to 20 0C over 4.5 h. The yellow slurry of 2-pyrazinecarbonitrile, 5-[[5-[-[2-(3-aminopropyl)-6-methoxyphenyl]-lH-pyrazol-3-yl]amino] dimesylate salt is filtered, rinsed with acetone (7 L) and dried in a vacuum oven. The dimesylate salt, (1608 g, 2.88 mol) is slurried in water (16 L). Sodium hydroxide (aqueous 50%, 228 g, 2.85 mol) is slowly poured into the slurry. The slurry is -36- heated to 60 0C and stirred for one hour. It is then cooled to 16 0C over 4 h and filtered. The wet filter cake is rinsed with acetone (4 L) and dried to constant weight in a vacuum oven at 40 0C to provide 833 g (94%) of 2-pyrazinecarbonitrile, 5-[[5-[-[2-(3- aminopropyl)-6-methoxyphenyl]-lH-pyrazol-3-yl]amino] monomesylate monohydrate. mp 222.6 0C; ES/MS m/z 366.2 [M+l]+.
Example 5a
2-Pyrazinecarbonitrile, 5-[[5-[-[2-(3-aminopropyl)-6-methoxyphenyl]-lH-pyrazol-3- yl] amino] monomesylate monohydrate (Chemical Abstracts nomenclature)
Crude 2-pyrazinecarbonitrile, 5 -[ [5 – [- [2-(3 -aminopropyl)-6-methoxyphenyl]- IH- pyrazol-3-yl] amino] monomesylate monohydrate is purified using the following procedure. The technical grade 2-pyrazinecarbonitrile, 5-[[5-[-[2-(3-aminopropyl)-6- methoxyphenyl]-lH-pyrazol-3-yl] amino] mono mesylate mono hydrate (1221 g, 2.55 mol) is slurried in a solvent mixture of 1: 1 acetone/water (14.7 L). The solid is dissolved by warming the mixture to 50 – 55 0C. The solution is polish filtrated while at 50 – 55 0C through a 0.22μ cartridge filter. The solution is slowly cooled to the seeding temperature of about 42 – 45 0C and seeded. Slow cooling is continued over the next 30 – 60 min to confirm nucleation. The thin slurry is cooled from 38 to 15 0C over 3 h. A vacuum distillation is set up and the acetone removed at 110 – 90 mm and 20 – 30 0C. The mixture is cooled from 30 to 15 0C over 14 h, held at 15 0C for 2 h, and then filtered. The recrystallized material is rinsed with 19: 1 water/acetone (2 L) and then water (6 L) and dried to constant weight in a vacuum oven at 40 0C to provide 1024 g (83.9%) of the title compound, mp 222.6 0C; ES/MS m/z 366.2 [M+l]+. X-ray powder diffraction (XRPD) patterns may be obtained on a Bruker D8
Advance powder diffractometer, equipped with a CuKa source (λ=l.54056 angstrom) operating at 40 kV and 40 mA with a position-sensitive detector. Each sample is scanned between 4° and 35° in °2Θ ± 0.02 using a step size of 0.026° in 2Θ ± 0.02 and a step time of 0.3 seconds, with a 0.6 mm divergence slit and a 10.39 mm detector slit. Primary and secondary Soller slits are each at 2°; antiscattering slit is 6.17 mm; the air scatter sink is in place. -37-
Characteristic peak positions and relative intensities:

Differential scanning calorimetry (DSC) analyses may be carried out on a Mettler- Toledo DSC unit (Model DSC822e). Samples are heated in closed aluminum pans with pinhole from 25 to 350 0C at 10 °C/min with a nitrogen purge of 50 mL/min. Thermogravimetric analysis (TGA) may be carried out on a Mettler Toledo TGA unit (Model TGA/SDTA 85Ie). Samples are heated in sealed aluminum pans with a pinhole from 25 to 350 0C at 10 0C /min with a nitrogen purge of 50 mL/min.
The thermal profile from DSC shows a weak, broad endotherm form 80 – 1400C followed by a sharp melting endotherm at 222 0C, onset (225 0C, peak). A mass loss of 4% is seen by the TGA from 25 – 140 0C.
PATENT
US 20110144126
WO 2017015124
WO 2017100071
WO 2017105982
WO 2016051409
PATENT
Preparation 1
tert-Butyl (E)-(3-(2-(3-(dimethylamino)ac^’loyl)-3-me1hoxyphenox50propyl)carbamate

L _l H
Combine l-(2-hydroxy-6-methox>’phenyl)e1han-l-one (79.6 kg, 479 mol) and 1,1-<iimethoxy-N,N-dimemylmethanamino (71.7 kg, 603.54 mol) with DMF (126 kg). Heat to 85-90 °C for 12 hours. Cool the reaction mixture containing intermediate (E)-3-(dimethylamino)-l-(2-hydroxy-6-methoxyphenyl)prop-2-en-l-one (mp 84.74 °C) to ambient temperature and add anhydrous potassium phosphate (136 kg, 637.07 mol) and tert-butyl (3-bromopropyl)carbamate (145 kg, 608.33 mol). Stir the reaction for 15 hours at ambient temperature. Filter the mixture and wash the filter cake with ΜΓΒΕ (3 χ , 433 kg, 300 kg, and 350 kg). Add water (136 kg) and aqueous sodium chloride (25% solution, 552 kg) to the combined MTBE organic solutions. Separate the organic and aqueous phases. Back-extract the resulting aqueous phase with MTBE (309 kg) and add the MTBE layer to the organic solution. Add an aqueous sodium chloride solution (25% solution, 660 kg) to the combined organic extracts and separate the layers. Concentrate the organic extracts to 1,040 kg – 1,200 kg and add water (400 kg) at 30-35 °C to the residue. Cool to ambient temperature and collect material by filtration as a wet cake to give the title product (228.35 kg, 90%). ES/MS (m/z): 379.22275 (M+l).
Preparation 2
tert-Butyl (3-(2-(2-cyanoacetyl)-3-methoxyphenoxy)propyl)carbamate
“9 o

![]()
![]()

Combine ethanol (1044 kg), hydroxyl amino hydrochloride (30 kg, 431.7 mol), and terr-butyl (E)-(3-(2-(3-(^me%lamino)acryloyl)-3-
methoxyphenoxy)propyl)carbamate (228.35 kg, 72% as a wet water solid, 434.9 mol) to form a solution. Heat the solution to 35 – 40 °C for 4-6 hours. Cool the reaction to ambient temperature and concentrate to a residue. Add MTBE (300 kg) to the residue and concentrate the solution to 160 kg – 240 kg. Add MTBE (270 kg) and concentrate the solution. Add MTBE (630 kg), water (358 kg), and sodium chloride solution (80 kg, 25% aqueous) and stir for 20 minutes at ambient temperature. Let the mixture stand for 30 minutes. Separate the aqueous layer. Add water (360 kg) and sodium chloride solution (82 kg, 25% sodium chloride) to the organic phase. Stir for 20 minutes at ambient temperature. Let the mixture stand for 30 minutes. Separate the aqueous portion. Add sodium chloride solution (400 kg, 25 % aqueous) to the organic portion. Stir for 20 minutes at ambient temperature. Let the mixture stand for 30 minutes at ambient temperature. Separate the aqueous portion. Concentrate the organic portion to 160 kg – 240 kg at 40 °C. Add ethanol (296 kg) to the organic portion. Concentrate the solution to 160 kg to 240 kg at 40 °C to provide an intermediate of tert-butyl (3-(2-(isoxazol-5-yl)-3-methox>’phenoxy)propyl)carbamate. Add ethanol (143 kg) and water (160 kg) to the concentrated solution. Add potassium hydroxide (31.8 kg) at 40 °C. Add ethanol (80 kg) and adjust the temperature to 45-50 °C. Stir for 4-6 hours at 45-50 °C and concentrate to 160 kg – 240 kg at 40 °C. Add water to the concentrate (160 kg) and acetic acid (9.0 kg) drop-wise to adjust the pH to 10-12 while mamtaining the temperature of the solution at 25 to 35 °C. Add ethyl acetate (771 kg) and acetic acid drop-wise to adjust the pH to 5-7 while maintaining the temperature of the solution at 25-35 °C. Add sodium chloride solution (118 kg, 25% aqueous solution). Stir the mixture for 20 minutes at ambient temperature. Let the solution stand for 30 minutes at ambient temperature. Separate Ihe aqueous portion. Heat the organic portion to 30-35 °C. Add water (358 kg) drop-wise. Stir the solution for 20 minutes while maintaining the temperature at 30 to 35 °C. Let the mixture stand for 30 minutes and separate the aqueous portion. Wash the organic portion with sodium chloride solution (588 kg, 25% aqueous) and concentrate the organic portion to 400 kg – 480 kg at 40-50 °C. Heat the concentrated solution to 50 °C to form a solution. Maintain the solution at 50 °C and add M-heptane (469 kg) drop-wise. Stir the solution for 3 hours at 50 °C before slowly cooling to ambient temperature to crystallize the product. Stir at ambient temperature for 15 hours and filter the crystals. Wash the crystals with ethanol/«-heptane (1 :2, 250 kg) and dry at 45 °C for 24 hours to provide the title compound (133.4 kg, 79.9%), rap. 104.22 °C,
Example 1
5-(5-(2-(3-Ammopropoxy)-6-memoxyphenyl)-lH-pyrazol-3-ylammo)pyrazine-2- carbonitrile (S)-lactate monohydrate

Combine a THJF solution (22%) of fcrt-butyl (3-(2-(2-cyanoacetyl)-3-memoxyphenoxy)propyl)carbamate (1.0 eqv, this is define as one volume) with hydrazine (35%, 1.5 eqv), acetic acid (glacial, 1.0 eqv), water (1 volume based on the THF solution) and methanol (2 volumes based on the THF solution). This is a continuous operation. Heat the resulting mixture to 130 °C and 1379 kPa with a rate of V/Q = 70 minutes, tau = 60. Extract the solution with toluene (4 volumes), water (1 volume), and sodium carbonate (10% aqueous, 1 eqv). Isolate Ihe toluene layer and add to DMSO (0.5 volumes). Collect a solution of the intermediate compound tert-butyl (3-(2-(3-amino-lH-pyrazol-5-yl)-3-methoxyphenoxy) propyl)carbamate (26.59 kg, 91%) in 10 days, mp = 247.17 °C as a DMSO solution (3 volumes of product). N-Eftylmorpholine (1.2 eqv) and 5-chloropyrazine-2-carbonitrile (1.15 eqv) in 2 volumes of DMSO is combined in a tube reactor at 80 °C, V/Q = 3 and tau = 170 minutes at ambient pressure. Add the product stream to methanol (20 vol). As a continuous process, filter the mixture and wash with methanol followed by MTBE. Air dry the material on the filter to give tert-butyl (3-(2-(3-((5-cyanopyrazm-2-yl)arnino)-lH-pyrazol-5-yl)-3-methox>’phenoxy) propyl)carbamate in a continuous fashion (22.2 kg, 88.7%, 8 days). Dissolve a solution of fcrt-butyl (3-(2-(3-((5-cyanopyrazin-2-yl)amino)-lH-pyrazol-5-yl)-3-methoxyphenoxy) propyl)carbamate in formic acid (99%, 142 kg) at ambient temperature and agitate for 4 hours to provide an intermediate of 5-((5-(2-(3-aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3-yl)amino)pyrazine-2-carbonitrile formate. Dilute the solution with water (55 kg), (S)-lactic acid (30%, 176 kg) and distill the resulting mixture until < 22 kg formic acid remains. Crystallize the resulting residue from THF and wash with a THF -water (0.5% in THF) solution. Dry the wet cake at 30 °C at >10% relative humidity to give the title product as a white to yellow solid (24.04 kg, 85-90%), mp. 157 °C.
Alternate Preparation Example 1
5-(5-(2-(3-Ammopropoxy)-6-memoxyphenyl)-lH-pyrazol-3-ylammo)pyrazine-2- carbonitrile (S)-lactate monohydrate
Add 5-({3-[2-(3-aminopropoxy)-6-methoxyphenyl]-lH-pyrazol-5-yl}ammo)pyrazine-2-carbonitrile (4.984 g, 13.33 mmol, 97.7 wt%) to n-PrOH (15.41 g, 19.21 mL) to form a slurry. Heat the slurry to 60 °C. Add (S)-lactic acid (1.329 g, 14.75 mmol) to water (19.744 mL) and add this solution to the slurry at 58 °C. Heat the solution to 60 °C and add n-PrOH (21.07 g, 26.27 mL). Seed the solution with 5-((5-(2-(3-aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3-yl)ammo)pyrazme-2-carbom^ (S)-lactate monohydrate (48.8 mg, 0.1 mmol) and cool the solution to 40 °C over 35 minutes. Add H-PrOH (60.5 mL) to the slurry at 40 °C via a syringe pump over 2 hours and maintain the temperature at 40 °C. Once complete, air cool the slurry to ambient temperature for 2 hours, the cool the mixture in ice-water for 2 hours. Filter the product, wash the wet cake with 6:1 (v/v) rc-PrOH : H20 (15 mL), followed by n-PrOH (15 mL) and dry the wet cake for 20 minutes. Dry the solid overnight at 40 °C in vacuo to give the title compound as a white to yellow solid (5.621 g, 89.1%), m.p. 157 °C.
Crystalline Example 1
Crystalline 5-(5-(2-(3-aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3- ylamino)pyrazine-2-carbonitrile (S)-lactate monohydrate Prepare a slurry having 5-(5-(2-(3-aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3 -y lamino)py razine-2-carbonitrile (368 mg, 1.0 mmol) in a 10:1 THF-water (5 mL) solution and stir at 55 °C. Add (S)-lactic acid (110 mg, 1.22 mmol) dissolved in THF (1 mL). A clear solution forms. Stir for one hour. Reduce Ihe temperature to 44 °C and stir until an off-white precipitate forms. Filter the material under vacuum, rinse with THF, and air dry to give the title compound (296 mg, 80%).
X-Ray Powder Diffraction, Crystalline Example 1 Obtain the XRPD patterns of the crystalline solids on a Bruker D4 Endeavor X-ray powder diffractometer, equipped with a CuKa source (λ = 1.54060 A) and a Vantec detector, operating at 35 kV and 50 mA. Scan the sample between 4 and 40° in 2Θ, with a step size of 0.0087° in 2Θ and a scan rate of 0.5 seconds/step, and with 0.6 mm divergence, 5.28mm fixed anti-scatter, and 9.5 mm detector slits. Pack the dry powder on a quartz sample holder and obtain a smooth surface using a glass slide. It is well known in the crystallography art that, for any given crystal form, the relative intensities of the diffraction peaks may vary due to preferred orientation resulting from factors such as crystal morphology and habit. Where the effects of preferred orientation are present, peak intensities are altered, but the characteristic peak positions of the polymorph are unchanged. See, e.g. The U. S. Pharmacopeia 35 – National Formulary 30 Chapter <941> Characterization of crystalline and partially crystalline solids by XRPD Official December 1, 2012-May 1, 2013. Furthermore, it is also well known in the
crystallography art that for any given crystal form the angular peak positions may vary slightly. For example, peak positions can shift due to a variation in the temperature or humidity at which a sample is analyzed, sample displacement, or the presence or absence of an internal standard. In the present case, a peak position variability of ± 0.2 in 2Θ will take into account these potential variations without hindering the unequivocal identification of the indicated crystal form Confirmation of a crystal form may be made based on any unique combination of distinguishing peaks (in units of ° 2Θ), typically the more prominent peaks. The crystal form diffraction patterns, collected at ambient temperature and relative humidity, were adjusted based on NIST 675 standard peaks at 8.85 and 26.77 degrees 2-theta,
Characterize a prepared sample of crystalline 5-(5-(2-(3-aminopropoxy)-6-methoxyphenyl)- lH-pyrazol-3-ylamino)pyrazine-2-carbonitrile (S)-lactate monohydrate by an XPRD pattern using CuKa radiation as having diffraction peaks (2-theta values) as described in Table 1 below. Specifically the pattern contains a peak at 12.6 in
combination with one or more of the peaks selected from the group consisting of 24.8, 25.5, 8.1, 6.6, 12.3, and 16.3 with a tolerance for the diffraction angles of 0.2 degrees.
PATENT
Example 1
5-(5-(2-(3-Aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3-ylamino)pyrazine-2- carbonitrile S)-lactate monohydrate

Combine a THF solution (22%) of i<?ri-butyl (3-(2-(2-cyanoacetyl)-3-methoxyphenoxy)propyl)carbamate (1.0 eqv, this is define as one volume) with hydrazine (35%, 1.5 eqv), acetic acid (glacial, 1.0 eqv), water (1 volume based on the THF solution) and methanol (2 volumes based on the THF solution). As this is a continuous operation, grams or kg is irrelevant in this processing methodology. Heat the resulting mixture to 130 °C and 1379 kPa with a rate of V/Q = 70 minutes (where V refers to the volume of the reactor and Q refers to flow rate), tau = 60. Extract the solution with toluene (4 volumes), water (1 volume), and sodium carbonate (10% aqueous, 1 eqv). Isolate the toluene layer and add to DMSO (0.5 volumes). Collect a solution of the intermediate compound i<?ri-butyl (3-(2-(3-amino- lH-pyrazol-5-yl)-3-methoxyphenoxy)
propyl)carbamate (26.59 kg, 91%) in 10 days, mp = 247.17 °C as a DMSO solution (3 volumes of product). N-ethylmorpholine (1.2 eqv) and 5-chloropyrazine-2-carbonitrile (1.15 eqv) in 2 volumes of DMSO is combined in a tube reactor at 80 °C, V/Q = 3 and tau = 170 minutes at ambient pressure. Add the product stream to methanol (20 vol). As a continuous process, filter the mixture and wash with methanol followed by MTBE. Air dry the material on the filter to give i<?ri-butyl (3-(2-(3-((5-cyanopyrazin-2-yl)amino)-lH-pyrazol-5-yl)-3-methoxyphenoxy) propyl)carbamate in a continuous fashion (22.2 kg, 88.7%, 8 days). Dissolve a solution of i<?ri-butyl (3-(2-(3-((5-cyanopyrazin-2-yl)amino)-lH-pyrazol-5-yl)-3-methoxyphenoxy) propyl)carbamate in formic acid (99%, 142 kg) at ambient temperature and agitate for 4 hours to provide an intermediate of 5-((5-(2-(3-aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3-yl)amino)pyrazine-2-carbonitrile formate. Dilute the solution with water (55 kg), (S)-lactic acid (30%, 176 kg) and distill the resulting mixture until < 22 kg formic acid remains. Crystallize the resulting residue from THF and wash with a THF -water (0.5% in THF) solution. Dry the wet cake at 30 °C at >10% relative humidity to give the title product as a white to yellow solid (24.04 kg, 85-90%), m.p. 157 °C.
Alternate Preparation Example 1
5-(5-(2-(3-Aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3-ylamino)pyrazine-2- carbonitrile (S)-lactate monohydrate
Add 5-({3-[2-(3-aminopropoxy)-6-methoxyphenyl]-lH-pyrazol-5-yl}amino)pyrazine-2-carbonitrile (4.984 g, 13.33 mmol, 97.7 wt%) to n-PrOH (15.41 g, 19.21 mL) to form a slurry. Heat the slurry to 60 °C. Add (S)-lactic acid (1.329 g, 14.75 mmol) to water (19.744 mL) and add this solution to the slurry at 58 °C. Heat the solution to 60 °C and add n-PrOH (21.07 g, 26.27 mL). Seed the solution with 5-((5-(2-(3-aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3-yl)amino)pyrazine-2-carbonitrile (S)-lactate monohydrate (48.8 mg, 0.1 mmol) and cool the solution to 40 °C over 35 minutes. Add ft-PrOH (60.5 mL) to the slurry at 40 °C via a syringe pump over 2 hours and maintain the temperature at 40 °C. Once complete, air cool the slurry to ambient temperature for 2 hours, then cool the mixture in ice-water for 2 hours. Filter the product, wash the wet cake with 6:1 (v/v) n-PrOH : H20 (15 mL), followed by n-PrOH (15 mL)
and dry the wet cake for 20 minutes. Dry the solid overnight at 40 °C in vacuo to give the title compound as a white to yellow solid (5.621 g, 89.1%), m.p. 157 °C.
Clip
science 2017, 356, 1144
Kilogram-Scale Prexasertib Monolactate Monohydrate Synthesis under Continuous-Flow CGMP Conditions

A multidisciplinary team from Eli Lilly reports the development and implementation of eight continuous unit operations for the synthesis of ca. 3 kg API per day under CGMP conditions (K. P. Cole et al., Science 2017, 356, 1144). The recent drive toward more potent APIs that have a low annual demand (<100 kg) has made continuous synthesis a viable alternative to traditional batch processes with advantages which include reducing equipment footprint and worker exposure. In this report the authors describe the enablement of three continuous synthetic steps followed by a salt formation, using surge tanks between steps to allow each step to be taken offline if online PAT detects a loss in reaction performance. A combination of MSMPRs (mixed-suspension, mixed-product removal) vessels, plug-flow reactors, and dissolve-off filters were used to perform the chemistry, with an automated 20 L rotary evaporator used to concentrate process streams and perform solvents swaps. This paper gives an excellent account of the potential solutions to continuous API synthesis and is well worth a read for anyone contemplating such methodology.




Integrated flow synthesis and purification process for prexasertib meets high industry standards

Eli Lilly has taken an important step away from traditional batch process drug manufacturing by using an industry-first continuous process to make a compound for phase I and II clinical trials. Workers at Lilly’s Kinsale site in Ireland, did three steps involved in producing cancer drug candidate prexasertib continuously, under current good manufacturing practice (CGMP) standards that ensure safety for human consumption.
Continuous processing relies on chemical and physical changes happening as substances flow through pipes. Isolated steps of this type are already well-established in the pharmaceutical industry. However, Lilly principal research scientist Kevin Cole stresses that a series including reaction and purification steps like this has not been demonstrated before. And the company wants to go much further.
‘We envision entire synthetic routes consisting of many reaction and separation unit operations being executed simultaneously in flow, with heavy reliance on design space understanding, process analytical technologies and process modelling to ensure quality,’ Cole says. ‘We think this will drastically change the environment for pharmaceutical manufacturing.’
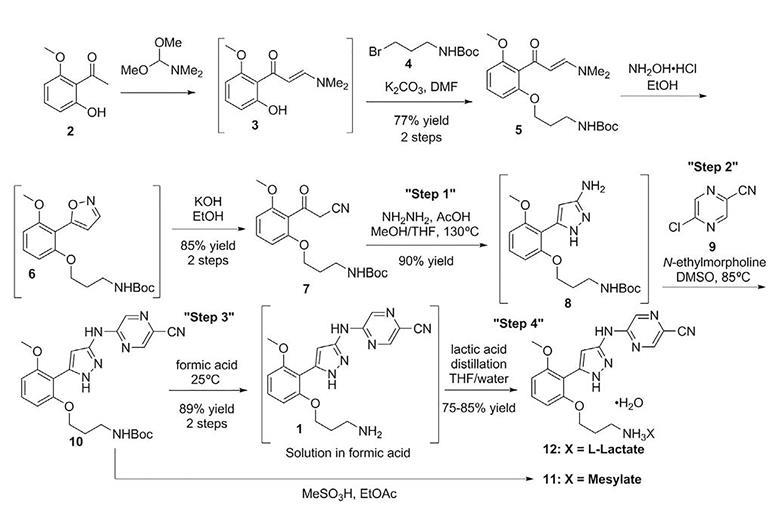
In batch processes different chemical reaction and purification steps are typically done in large, costly vessels. However, this can be uneconomical when small amounts of drug molecules are needed for early stage clinical trials and, because drugs are getting more potent, increasingly in mainstream production.
By contrast, small volume continuous flow processing runs in more compact equipment in fume hoods. Flow systems can adapt to different processes, with cheap parts that can either be dedicated to specific drugs or readily replaced. The US Food and Drug Administration (FDA) has also been promoting continuous manufacturing because it integrates well with advanced process analytical technology. This helps pharmaceutical companies make high quality drugs with less FDA oversight.
Lilly chose prexasertib as its test case for such a process because it’s challenging to make. It is a chain of three aromatic rings, and one challenge comes because its central ring is formed using hydrazine. Hydrazine is used as a component in rocket fuel, and is also highly toxic. A second challenge comes from prexasertib itself, which, as a potent kinase inhibitor, is toxic to healthy cells, as well as cancerous ones, even at low doses. Lilly therefore wants to minimise its workers’ exposure.
Feeding the plant
Cole and his colleagues at Lilly’s labs in Indianapolis, US, have developed flow processes for three of the seven steps involved in prexasertib production. They start with the hydrazine step, which they could safely speed up by super-heating in the continuous process. After aqueous workup purification the solution of the two-ring intermediate solution runs into a ‘surge tank’. From there the solution flows intermittently into a rotary evaporator that removes solvents to concentrate it.
The second continuous flow step adds the third of prexasertib’s rings. In this case, the Lilly team purified the intermediate by crystallising it and filtering it out, washing away impurities. They could then redissolve the pure intermediate in formic acid, which also removes a protecting group, giving the desired prexasertib molecule. Automating this was probably the hardest part, Cole says. ‘Development of a predictive filtration model, equipment design and identification of formic acid as the solvent were keys to success,’ he explains. The final flow step then starts converting prexasertib to its final lactate salt form.

After developing the processes and systems in Indianapolis, Lilly shipped them to be equipped in an existing facility at its Kinsale manufacturing site at the cost of €1 million (£870,000). Once the prexasertib system was installed, the company was able to make 3kg of raw material per day for clinical trials. Cole describes the level of manual intervention needed as ‘moderate’.
Klavs Jensen from the Massachusetts Institute of Technology calls the paper describing the work ‘terrific’. ‘This work marks an important milestone in the continuous manufacturing of pharmaceuticals by demonstrating the feasibility of producing a modern kinase inhibitor under CGMP conditions,’ he says.
Likewise, Brahim Benyahia from Loughborough University, UK, calls this achievement ‘very interesting’. ‘The paper is another example that demonstrates the benefits and feasibility of the integrated continuous approach in pharma,’ he says.
Cole adds that Lilly has several other similar projects in advanced stages of development intended for the €35 million small-volume continuous plant it recently built in Kinsale. ‘We are committed to continuous manufacturing as well as full utilisation of our new facility,’ he says.
Correction: This article was updated on 16 June 2017 to clarify the chronology of the completion of the Kinsale, Ireland plant
References
K Cole et al, Science, 2017, DOI: 10.1126/science.aan0745
References
REFERENCES
1: Lowery CD, VanWye AB, Dowless M, Blosser W, Falcon BL, Stewart J, Stephens J, Beckmann RP, Bence Lin A, Stancato LF. The Checkpoint Kinase 1 Inhibitor Prexasertib Induces Regression of Preclinical Models of Human Neuroblastoma. Clin Cancer Res. 2017 Mar 7. pii: clincanres.2876.2016. doi: 10.1158/1078-0432.CCR-16-2876. [Epub ahead of print] PubMed PMID: 28270495.
2: Zeng L, Beggs RR, Cooper TS, Weaver AN, Yang ES. Combining Chk1/2 inhibition with cetuximab and radiation enhances in vitro and in vivo cytotoxicity in head and neck squamous cell carcinoma. Mol Cancer Ther. 2017 Jan 30. pii: molcanther.0352.2016. doi: 10.1158/1535-7163.MCT-16-0352. [Epub ahead of print] PubMed PMID: 28138028.
3: Ghelli Luserna Di Rorà A, Iacobucci I, Imbrogno E, Papayannidis C, Derenzini E, Ferrari A, Guadagnuolo V, Robustelli V, Parisi S, Sartor C, Abbenante MC, Paolini S, Martinelli G. Prexasertib, a Chk1/Chk2 inhibitor, increases the effectiveness of conventional therapy in B-/T- cell progenitor acute lymphoblastic leukemia. Oncotarget. 2016 Aug 16;7(33):53377-53391. doi: 10.18632/oncotarget.10535. PubMed PMID: 27438145; PubMed Central PMCID: PMC5288194.
REFERENCES
1: Zeng L, Beggs RR, Cooper TS, Weaver AN, Yang ES. Combining Chk1/2 inhibition with cetuximab and radiation enhances in vitro and in vivo cytotoxicity in head and neck squamous cell carcinoma. Mol Cancer Ther. 2017 Jan 30. pii: molcanther.0352.2016. doi: 10.1158/1535-7163.MCT-16-0352. [Epub ahead of print] PubMed PMID: 28138028.
2: Ghelli Luserna Di Rorà A, Iacobucci I, Imbrogno E, Papayannidis C, Derenzini E, Ferrari A, Guadagnuolo V, Robustelli V, Parisi S, Sartor C, Abbenante MC, Paolini S, Martinelli G. Prexasertib, a Chk1/Chk2 inhibitor, increases the effectiveness of conventional therapy in B-/T- cell progenitor acute lymphoblastic leukemia. Oncotarget. 2016 Aug 16;7(33):53377-53391. doi: 10.18632/oncotarget.10535. PubMed PMID: 27438145; PubMed Central PMCID: PMC5288194.
3: King C, Diaz HB, McNeely S, Barnard D, Dempsey J, Blosser W, Beckmann R, Barda D, Marshall MS. LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms. Mol Cancer Ther. 2015 Sep;14(9):2004-13. doi: 10.1158/1535-7163.MCT-14-1037. PubMed PMID: 26141948.
4: Hong D, Infante J, Janku F, Jones S, Nguyen LM, Burris H, Naing A, Bauer TM, Piha-Paul S, Johnson FM, Kurzrock R, Golden L, Hynes S, Lin J, Lin AB, Bendell J. Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients With Advanced Cancer. J Clin Oncol. 2016 May 20;34(15):1764-71. doi: 10.1200/JCO.2015.64.5788. PubMed PMID: 27044938.
 |
|
| Clinical data | |
|---|---|
| Pregnancy category |
|
| ATC code |
|
| Identifiers | |
| CAS Number | |
| ChemSpider | |
| UNII | |
| Chemical and physical data | |
| Formula | C18H19N7O2 |
| Molar mass | 365.40 g·mol−1 |
| 3D model (JSmol) | |
////////////Prexasertib, прексасертиб , بريكساسيرتيب , 普瑞色替 , PHASE 2, LY-2606368, LY 2606368
| N#CC1=NC=C(NC2=NNC(C3=C(OC)C=CC=C3OCCCN)=C2)N=C1 |
GSK 2330672, LINERIXIBAT

GSK 2330672
LINERIXIBAT
GSK 672; GSK-2330672
RN: 1345982-69-5
UNII: 386012Z45S
CAS: 1345982-69-5
Chemical Formula: C28H38N2O7S
Molecular Weight: 546.68
Pentanedioic acid, 3-((((3R,5R)-3-butyl-3-ethyl-2,3,4,5-tetrahydro-7-methoxy-1,1-dioxido-5-phenyl-1,4-benzothiazepin-8-yl)methyl)amino)-
Pentanedioic acid, 3-((((3R,5R)-3-butyl-3-ethyl-2,3,4,5-tetrahydro-7-methoxy-1,1-dioxido-5-phenyl-1,4-benzothiazepin-8-yl)methyl)amino)-
3-({[(3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-1 ,1 -dioxido-5-phenyl- 2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8-yl]methyl}amino)pentanedioic acid
3-[[[(3R,5R)-3-Butyl-3-ethyl-2,3,4,5-tetrahydro-7-methoxy-1,1-dioxido-5-phenyl-1,4-benzothiazepin-8-yl]methyl]amino]pentanedioic acid
- Originator GlaxoSmithKline
- Class Antihyperglycaemics
- Mechanism of Action Sodium-bile acid cotransporter-inhibitors
Highest Development Phases
- Phase II Primary biliary cirrhosis; Pruritus; Type 2 diabetes mellitus
- Phase I Cholestasis
Most Recent Events
- 01 Jan 2017 Phase-II clinical trials in Pruritus in USA (PO) (NCT02966834)
- 14 Nov 2016 GlaxoSmithKline completes a phase I trial for Cholestasis in Healthy volunteers in Japan (PO, Tablet) (NCT02801981)
- 11 Nov 2016 Efficacy, safety and pharmacodynamic data from a phase II trial in Primary biliary cirrhosis and Pruritus presented at The Liver Meeting® 2016: 67th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD-2016)
| Inventors | Christopher Joseph Aquino, Jon Loren Collins, David John Cowan, Yulin Wu |
| Applicant | Glaxosmithkline Llc |

GSK2330672 , an ileal bile acid transport (iBAT) inhibitor indicated for diabetes type II and cholestatic pruritus, is currently under Phase IIb evaluation in the clinic. The API is a highly complex molecule containing two stereogenic centers, one of which is quaternary
GSK-2330672 is highly potent, nonabsorbable apical sodium-dependent bile acid transporter inhibitor for treatment of type 2 diabetes.
More than 200 million people worldwide have diabetes. The World Health Organization estimates that 1 .1 million people died from diabetes in 2005 and projects that worldwide deaths from diabetes will double between 2005 and 2030. New chemical compounds that effectively treat diabetes could save millions of human lives.
Diabetes refers to metabolic disorders resulting in the body’s inability to effectively regulate glucose levels. Approximately 90% of all diabetes cases are a result of type 2 diabetes whereas the remaining 10% are a result of type 1 diabetes, gestational diabetes, and latent autoimmune diabetes of adulthood (LADA). All forms of diabetes result in elevated blood glucose levels and, if left untreated chronically, can increase the risk of macrovascular (heart disease, stroke, other forms of cardiovascular disease) and microvascular [kidney failure (nephropathy), blindness from diabetic retinopathy, nerve damage (diabetic neuropathy)] complications.
Type 1 diabetes, also known as juvenile or insulin-dependent diabetes mellitus (IDDM), can occur at any age, but it is most often diagnosed in children, adolescents, or young adults. Type 1 diabetes is caused by the autoimmune destruction of insulin-producing beta cells, resulting in an inability to produce sufficient insulin. Insulin controls blood glucose levels by promoting transport of blood glucose into cells for energy use. Insufficient insulin production will lead to decreased glucose uptake into cells and result in accumulation of glucose in the bloodstream. The lack of available glucose in cells will eventually lead to the onset of symptoms of type 1 diabetes: polyuria (frequent urination), polydipsia (thirst), constant hunger, weight loss, vision changes, and fatigue. Within 5-10 years of being diagnosed with type 1 diabetes, patient’s insulin-producing beta cells of the pancreas are completely destroyed, and the body can no longer produce insulin. As a result, patients with type 1 diabetes will require daily administration of insulin for the remainder of their lives.
Type 2 diabetes, also known as non-insulin-dependent diabetes mellitus (NIDDM) or adult-onset diabetes, occurs when the pancreas produces insufficient insulin and/or tissues become resistant to normal or high levels of insulin (insulin resistance), resulting in excessively high blood glucose levels. Multiple factors can lead to insulin resistance including chronically elevated blood glucose levels, genetics, obesity, lack of physical activity, and increasing age. Unlike type 1 diabetes, symptoms of type 2 diabetes are more salient, and as a result, the disease may not be diagnosed until several years after onset with a peak prevalence in adults near an age of 45 years. Unfortunately, the incidence of type 2 diabetes in children is increasing.
The primary goal of treatment of type 2 diabetes is to achieve and maintain glycemic control to reduce the risk of microvascular (diabetic neuropathy, retinopathy, or nephropathy) and macrovascular (heart disease, stroke, other forms of cardiovascular disease) complications. Current guidelines for the treatment of type 2 diabetes from the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) [Diabetes Care, 2008, 31 (12), 1 ] outline lifestyle modification including weight loss and increased physical activity as a primary therapeutic approach for management of type 2 diabetes. However, this approach alone fails in the majority of patients within the first year, leading physicians to prescribe medications over time. The ADA and EASD recommend metformin, an agent that reduces hepatic glucose production, as a Tier 1 a medication; however, a significant number of patients taking metformin can experience gastrointestinal side effects and, in rare cases, potentially fatal lactic acidosis. Recommendations for Tier 1 b class of medications include sulfonylureas, which stimulate pancreatic insulin secretion via modulation of potassium channel activity, and exogenous insulin. While both medications rapidly and effectively reduce blood glucose levels, insulin requires 1 -4 injections per day and both agents can cause undesired weight gain and potentially fatal hypoglycemia. Tier 2a recommendations include newer agents such as thiazolidinediones (TZDs pioglitazone and rosiglitazone), which enhance insulin sensitivity of muscle, liver and fat, as well as GLP-1 analogs, which enhance postprandial glucose-mediated insulin secretion from pancreatic beta cells. While TZDs show robust, durable control of blood glucose levels, adverse effects include weight gain, edema, bone fractures in women, exacerbation of congestive heart failure, and potential increased risk of ischemic cardiovascular events. GLP-1 analogs also effectively control blood glucose levels, however, this class of medications requires injection and many patients complain of nausea. The most recent addition to the Tier 2 medication list is DPP-4 inhibitors, which, like GLP-1 analogs, enhance glucose- medicated insulin secretion from beta cells. Unfortunately, DPP-4 inhibitors only modestly control blood glucose levels, and the long-term safety of DPP-4 inhibitors remains to be firmly established. Other less prescribed medications for type 2 diabetes include a-glucosidase inhibitors, glinides, and amylin analogs. Clearly, new medications with improved efficacy, durability, and side effect profiles are needed for patients with type 2 diabetes.
GLP-1 and GIP are peptides, known as incretins, that are secreted by L and K cells, respectively, from the gastrointestinal tract into the blood stream following ingestion of nutrients. This important physiological response serves as the primary signaling mechanism between nutrient (glucose/fat) concentration in the
gastrointestinal tract and other peripheral organs. Upon secretion, both circulating peptides initiate signals in beta cells of the pancreas to enhance glucose-stimulated insulin secretion, which, in turn, controls glucose concentrations in the blood stream (For reviews see: Diabetic Medicine 2007, 24(3), 223; Molecular and Cellular Endocrinology 2009, 297(1-2), 127; Experimental and Clinical Endocrinology & Diabetes 2001 , 109(Suppl. 2), S288).
The association between the incretin hormones GLP-1 and GIP and type 2 diabetes has been extensively explored. The majority of studies indicate that type 2 diabetes is associated with an acquired defect in GLP-1 secretion as well as GIP action (see Diabetes 2007, 56(8), 1951 and Current Diabetes Reports 2006, 6(3), 194). The use of exogenous GLP-1 for treatment of patients with type 2 diabetes is severely limited due to its rapid degradation by the protease DPP-4. Multiple modified peptides have been designed as GLP-1 mimetics that are DPP-4 resistant and show longer half-lives than endogenous GLP-1 . Agents with this profile that have been shown to be highly effective for treatment of type 2 diabetes include exenatide and liraglutide, however, these agents require injection. Oral agents that inhibit DPP-4, such as sitagliptin vildagliptin, and saxagliptin, elevate intact GLP-1 and modestly control circulating glucose levels (see Pharmacology & Therapeutics 2010, 125(2), 328; Diabetes Care 2007, 30(6), 1335; Expert Opinion on Emerging Drugs 2008, 13(4), 593). New oral medications that increase GLP-1 secretion would be desirable for treatment of type 2 diabetes.
Bile acids have been shown to enhance peptide secretion from the
gastrointestinal tract. Bile acids are released from the gallbladder into the small intestine after each meal to facilitate digestion of nutrients, in particular fat, lipids, and lipid-soluble vitamins. Bile acids also function as hormones that regulate cholesterol homeostasis, energy, and glucose homeostasis via nuclear receptors (FXR, PXR, CAR, VDR) and the G-protein coupled receptor TGR5 (for reviews see: Nature Drug Discovery 2008, 7, 672; Diabetes, Obesity and Metabolism 2008, 10, 1004). TGR5 is a member of the Rhodopsin-like subfamily of GPCRs (Class A) that is expressed in intestine, gall bladder, adipose tissue, liver, and select regions of the central nervous system. TGR5 is activated by multiple bile acids with lithocholic and deoxycholic acids as the most potent activators {Journal of Medicinal Chemistry 2008, 51(6), 1831 ). Both deoxycholic and lithocholic acids increase GLP-1 secretion from an enteroendocrine STC-1 cell line, in part through TGR5
{Biochemical and Biophysical Research Communications 2005, 329, 386). A synthetic TGR5 agonist INT-777 has been shown to increase intestinal GLP-1 secretion in vivo in mice {Cell Metabolism 2009, 10, 167). Bile salts have been shown to promote secretion of GLP-1 from colonic L cells in a vascularly perfused rat colon model {Journal of Endocrinology 1995, 145(3), 521 ) as well as GLP-1 , peptide YY (PYY), and neurotensin in a vascularly perfused rat ileum model {Endocrinology 1998, 139(9), 3780). In humans, infusion of deoxycholate into the sigmoid colon produces a rapid and marked dose responsive increase in plasma PYY and enteroglucagon concentrations (Gi/M993, 34(9), 1219). Agents that increase ileal and colonic bile acid or bile salt concentrations will increase gut peptide secretion including, but not limited to, GLP-1 and PYY.
Bile acids are synthesized from cholesterol in the liver then undergo conjugation of the carboxylic acid with the amine functionality of taurine and glycine. Conjugated bile acids are secreted into the gall bladder where accumulation occurs until a meal is consumed. Upon eating, the gall bladder contracts and empties its contents into the duodenum, where the conjugated bile acids facilitate absorption of cholesterol, fat, and fat-soluble vitamins in the proximal small intestine (For reviews see: Frontiers in Bioscience 2009, 74, 2584; Clinical Pharmacokinetics 2002,
41(10), 751 ; Journal of Pediatric Gastroenterology and Nutrition 2001 , 32, 407). Conjugated bile acids continue to flow through the small intestine until the distal ileum where 90% are reabsorbed into enterocytes via the apical sodium-dependent bile acid transporter (ASBT, also known as iBAT). The remaining 10% are deconjugated to bile acids by intestinal bacteria in the terminal ileum and colon of which 5% are then passively reabsorbed in the colon and the remaining 5% being excreted in feces. Bile acids that are reabsorbed by ASBT in the ileum are then transported into the portal vein for recirculation to the liver. This highly regulated process, called enterohepatic recirculation, is important for the body’s overall maintenance of the total bile acid pool as the amount of bile acid that is synthesized in the liver is equivalent to the amount of bile acids that are excreted in feces.
Pharmacological disruption of bile acid reabsorption with an inhibitor of ASBT leads to increased concentrations of bile acids in the colon and feces, a physiological consequence being increased conversion of hepatic cholesterol to bile acids to compensate for fecal loss of bile acids. Many pharmaceutical companies have pursued this mechanism as a strategy for lowering serum cholesterol in patients with dyslipidemia/hypercholesterolemia (For a review see: Current Medicinal Chemistry 2006, 73, 997). Importantly, ASBT-inhibitor mediated increase in colonic bile acid/salt concentration also will increase intestinal GLP-1 , PYY, GLP-2, and other gut peptide hormone secretion. Thus, inhibitors of ASBT could be useful for treatment of type 2 diabetes, type 1 diabetes, dyslipidemia, obesity, short bowel syndrome, Chronic Idiopathic Constipation, Irritable bowel syndrome (IBS), Crohn’s disease, and arthritis.
Certain 1 ,4-thiazepines are disclosed, for example in WO 94/18183 and WO 96/05188. These compounds are said to be useful as ileal bile acid reuptake inhibitors (ASBT).
Patent publication WO 201 1/137,135 dislcoses, among other compounds, the following compound. This patent publication also discloses methods of synthesis of the compound.

The preparation of the above compound is also disclosed in J. Med. Chem, Vol 56, pp5094-51 14 (2013).
PATENT
EXAMPLES
Patent publication WO 201 1/137,135 dislcoses general methods for preparing the compound. In addition, a detailed synthesis of the compound is disclosed in Example 26. J. Med. Chem, Vol 56, pp5094-51 14 (2013) also discloses a method for synthesising the compound.
The present invention discloses an improved synthesis of the compound.
The synthetic scheme of the present invention is depicted in Scheme 1 .
Treatment of 2-methoxyphenyl acetate with sulfur monochloride followed by ester hydrolysis and reduction with zinc gave rise to thiophenol (A). Epoxide ring opening of (+)-2-butyl-ethyloxirane with thiophenol (A) and subsequent treatment of tertiary alcohol (B) with chloroacetonitrile under acidic conditions gave chloroacetamide (C), which was then converted to intermediate (E) by cleavage of the chloroacetamide with thiourea followed by classical resolution with dibenzoyl-L-tartaric acid.
Benzoylation of intermediate (E) with triflic acid and benzoyl chloride afforded intermediate (H). Cyclization of intermediate (H) followed by oxidation of the sulfide to a sulphone, subseguent imine reduction and classical resolution with (+)-camphorsulfonic acid provided intermediate (G), which was then converted to intermediate (H). Intermediate (H) was converted to the target compound using the methods disclosed in Patent publication WO 201 1/137,135.
Scheme 1

Dibenzoyl-L-tataric acid


The present invention also discloses an alternative method for construction of the quaternary chiral center as depicted in Scheme 2. Reaction of intermediate (A) with (R)-2-ammonio-2-ethylhexyl sulfate (K) followed by formation of di-p-toluoyl-L-tartrate salt furnished intermediate (L).

The present invention also discloses an alternative synthesis of intermediate (H) as illustrated in Scheme 3. Acid catalyzed cyclization of intermediate (F) followed by triflation gave imine (M), which underwent asymmetric reduction with catalyst lr(COD)2BArF and ligand (N) to give intermediate (O). Oxidation of the sulfide in intermediate (O) gave sulphone intermediate (H).

The present invention differs from the synthesis disclosed in WO 201 1/137,135 and J. Med. Chem, Vol56, pp5094-51 14 (2013) in that intermediate (H) in the present invention is prepared via a new, shorter and more cost-efficient synthesis while the synthesis of the target compound from intermediate (H) remains unchanged.
Intermediate A: 3-Hydroxy-4-methoxythiophenol
![]()
A reaction vessel was charged with 2-methoxyphenyl acetate (60 g, 0.36 mol), zinc chloride (49.2 g, 0.36 mol) and DME (600 mL). The mixture was stirred and S2CI2 (53.6 g, 0.40 mol) was added. The mixture was stirred at ambient temperature for 2 h. Concentrated HCI (135.4 mL, 1 .63 mol) was diluted with water (60 mL) and added slowly to the rxn mixture, maintaining the temperature below 60 °C. The mixture was stirred at 60 °C for 2 h and then cooled to ambient
temperature. Zinc dust (56.7 g, 0.87 mol) was added in portions, maintaining the temperature below 60 °C. The mixture was stirred at 20-60 °C for 1 h and then concentrated under vacuum to -300 mL. MTBE (1 .2 L) and water (180 mL) were added and the mixture was stirred for 10 min. The layers were separated and the organic layer was washed twice with water (2x 240 mL). The layers were separated and the organic layer was concentrated under vacuum to give an oil. The oil was distilled at 1 10-1 15 °C/2 mbar to give the title compound (42 g, 75%) as colorless oil, which solidified on standing to afford the title compound as a white solid. M.P. 41 -42 °C. 1 H NMR (500 MHz, CDCI3)$ ppm 3.39 (s, 1 H), 3.88 (s, 3H), 5.65 (br. S, 1 H), 6.75 (d, J – 8.3 Hz, 1 H), 6.84 (ddd, J – 8.3, 2.2, 0.6 Hz, 1 H), 6.94 (d, J – 2.2 Hz).
Intermediate E: (R)-5-((2-amino-2-ethylhexyl)thio)-2-methoxyphenol, dibenzoyl-L-tartrate salt

A reaction vessel was charged with 3-hydroxy-4-methoxythiophenol (5.0 g, 25.2 mmol), (+)-2-butyl-2-ethyloxirane (3.56 g, 27.7 mmol) and EtOH (30 mL). The mixture was treated with a solution of NaOH (2.22 g, 55.5 mmol) in water (20 mL), heated to 40 °C and stirred at 40 °C for 5 h. The mixture was cooled to ambient temperature, treated with toluene (25 mL) and stirred for 10 min. The layers were separated and the organic layer was discarded. The aqueous layer was neutralized with 2N HCI (27.8 mL, 55.6 mmol) and extracted with toluene (100 mL). The organic layer was washed with water (25 mL), concentrated in vacuo to give an oil. The oil was treated with chloroacetonitrile (35.9 mL) and HOAc (4.3 mL) and cooled to 0 °C. H2SO4 (6.7 mL, 126 mmol, pre-diluted with 2.3 mL of water) was added at a rate maintaining the temperature below 10 °C. After stirred at 0 °C for 0.5 h, the reaction mixture was treated with 20% aqueous Na2CO3 solution to adjust the pH to
7-8 and then extracted with MTBE (70 ml_). The extract was washed with water (35 ml_) and concentrated in vacuo to give an oil. The oil was then dissolved in EOH (50 ml_) and treated with HOAc (10 ml_) and thiourea (2.30 g, 30.2 mmol). The mixture was heated at reflux overnight and then cooled to ambient temperature. The solids were filtered and washed with EtOH (10 ml_). The filtrate and the wash were combined and concentrated in vacuo, treated with MTBE (140 ml_) and washed successively with 10% aqueous Na2C03 and water. The mixture was concentrated in vacuo to give an oil. The oil was dissolved in MeCN (72 ml_), heated to -50 °C and then dibenzoyl-L-tartaric acid (9.0 g, 25.2 mmol) in acetonitrile (22 ml_) was added slowly. Seed crystals were added at -50 °C. The resultant slurry was stirred at 45-50 °C for 5 h, then cooled down to ambient temperature and stirred at ambient temperature overnight. The solids were filtered and washed with MeCN (2x 22 ml_). The wet cake was treated with MeCN (150 ml_) and heated to 50 °C. The slurry was stirred at 50 °C for 5 h, cooled over 1 h to ambient temperature and stirred at ambient temperature overnight. The solids were collected by filtration, washed with MeCN (2 x 20 ml_), dried under vacuum to give the title compound (5.5 g, 34% overall yield, 99.5% purity, 93.9% ee) as a white solid. 1 H NMR (500 MHz, DMSO-d6) δ ppm 0.78 (m, 6H), 1 .13 (m, 4H), 1 .51 (m, 2H), 1 .58 (q, J – 7.7 Hz, 2H), 3.08 (s, 2H), 3.75 (s, 3H), 5.66 (s, 2H), 6.88 (m, 2H), 6.93 (m, 1 H), 7.49 (m, 4H), 7.63 (m, 2H), 7.94 (m, 4H). EI-LCMS m/z 284 (M++1 of free base).
Intermediate F: (R)-(2-((2-amino-2-ethylhexyl)thio)-4-hydroxy-5-methoxyphenyl)(phenyl)methanone

A suspension of (R)-5-((2-amino-2-ethylhexyl)thio)-2-methoxyphenol, dibenzoyl-L-tartrate salt (29 g, 45.2 mmol) in DCM (435 mL) was treated with water (1 16 mL) and 10% aqueous Na2C03 solution (1 16 mL). The mixture was stirred at ambient temperature until all solids were dissolved (30 min). The layers were separated. The organic layer was washed with water (2 x 60 mL), concentrated under vacuum to give (R)-5-((2-amino-2-ethylhexyl)thio)-2-methoxyphenol (free base) as an off-white solid (13.0 g, quantitative). A vessel was charged with TfOH (4.68 ml, 52.9 mmol) and DCM (30 mL) and the mixture was cooled to 0 °C. 5 g (17.6 mmol) of (R)-5-((2-amino-2-ethylhexyl)thio)-2-methoxyphenol (free base) was dissolved in DCM (10 mL) and added at a rate maintaining the temperature below 10 °C. Benzoyl chloride (4.5 mL, 38.8 mmol) was added at a rate maintaining the temperature below 10 °C. The mixture was then heated to reflux and stirred at reflux for 48 h. The mixture was cooled to 30 °C. Water (20 mL) was added and the mixture was concentrated to remove DCM. EtOH (10 mL) was added. The mixture was heated to 40 ° C, treated with 50% aqueous NaOH solution (10 mL) and stirred at 55 °C. After 1 h, the mixture was cooled to ambient temperature and the pH was adjusted to 6-7 with cone. HCI. The mixture was concentrated in vacuo to remove EtOH. EtOAc (100 mL) was added. The mixture was stirred for 5 min and the layers were separated. The organic layer was washed successively with 10% aqueous Na2CO3 (25 mL) and water (25 mL) and then concentrated in vacuo. The resultant oil was treated with DCM (15 mL). The resultant thick slurry was further diluted with DCM (15 mL) followed by addition of Hexanes (60 mL). The slurry was stirred for 5 min, filtered, washed with DCM/hexanes (1 :2, 2 x 10 mL) and dried under vacuum to give the title compound (7.67 g, 80%) as a yellow solid. 1 NMR (500 MHz, DMSO-d6) δ ppm 0.70 (t, 7.1 Hz, 3 H), 0.81 (t, 7.1 Hz, 3H), 1 .04-1 .27 (m, 8H), 2.74 (s, 2H), 3.73 (s, 3H), 6.91 (s, 1 H), 7.01 (s, 1 H), 7.52 (dd, J – 7.8, 7.2 Hz, 2H), 7.63 (t, J = 7.2 Hz, 1 H), 7.67 (d, J = 7.8 Hz, 2H). EI-LCMS m/z 388 (M++1 ).
Intermediate G: (3R,5R)-3-butyl-3-ethyl-8-hydroxy-7-methoxy-5-phenyl-2,3,4,5-tetrahydrobenzo[f][1 ,4]thiazepine 1 ,1 -dioxide, (+)-camphorsulfonate salt

A vessel was charged with (R)-(2-((2-amino-2-ethylhexyl)thio)-4-hydroxy-5-methoxyphenyl)(phenyl)methanone (1 .4 g, 3.61 mmol), toluene (8.4 ml_) and citric acid (0.035 g, 0.181 mmol, 5 mol%). The mixture was heated to reflux overnight with a Dean-Stark trap to remove water. The mixture was concentrated under reduced pressure to remove solvents. Methanol (14.0 ml_) and oxone (2.22 g, 3.61 mmol, 1 .0 equiv) were added. The mixture was stirred at gentle reflux for 2 h. The mixture was cooled to ambient temperature, and filtered to remove solids. The filter cake was washed with small amount of Methanol. The filtrate was cooled to 5 °C, and treated with sodium borohydride (0.410 g, 10.84 mmol, 3.0 equiv.) in small portions. The mixture was stirred at 5 °C for 2 h and then concentrated to remove the majority of solvents. The mixture was quenched with Water (28.0 ml_) and extracted with EtOAc (28.0 ml_). The organic layer was washed with brine, and then concentrated to remove solvents. The residue was dissolved in MeCN (14.0 ml_) and concentrated again to remove solvents. The residue was dissolved in MeCN (7.00 ml_) and MTBE (7.00 ml_) at 40 °C, and treated with (+)-camphorsulfonic acid (0.839 g, 3.61 mmol, 1 .0 equiv.) at 40 °C for 30 min. The mixture was cooled to ambient temperature, stirred for 2 h, and filtered to collect solids. The filter cake was washed with MTBE/MeCN (2:1 , 3 ml_), and dried at 50 °C to give the title compound (0.75 g, 32% overall yield, 98.6 purity, 97% de, 99.7% ee) as white solids. 1 NMR (400 MHz, CDCI3) δ ppm 0.63 (s, 3H), 0.88 (t, J – 6.9 Hz, 3H), 0.97 (m, 6H), 1 .29-1 .39 (m, 5H), 1 .80-1 .97 (m, 6H), 2.08-2.10 (m, 1 H), 2.27 (d, J – 17.3 Hz, 1 H), 2.38-2.44 (m, 3H), 2.54 (b, 1 H), 2.91 (b, 1 H), 3.48 (d, J – 15.4 Hz, 1 H), 3.79 (s, 3H), 4.05 (d, J – 17.2 Hz, 1 H), 6.45 (s, 1 H), 6.56 (s, 1 H), 7.51 -7.56 (m, 4H), 7.68 (s, 1 H), 7.79 (b, 2H), 1 1 .46 (b, 1 H). EI-LCMS m/z 404 (M++1 of free base).
Intermediate H: (3R,5R)-3-butyl-3-ethyl-7-methoxy-1 ,1 -dioxido-5-phenyl-2, 3,4,5-tetrahydrobenzo[f][1 ,4]thiazepin-8-yl trifluoromethanesulfonate

Method 1 : A mixture of (3R,5R)-3-butyl-3-ethyl-8-hydroxy-7-methoxy-5-phenyl-2,3,4,5-tetrahydrobenzo[f][1 ,4]thiazepine 1 ,1 -dioxide, (+)-camphorsulfonate salt (0.5 g, 0.786 mmol), EtOAc (5.0 mL), and 10% of Na2C03 aqueuous solution (5 mL) was stirred for 15 min. The layers were separated and the aqueous layer was discarded. The organic layer was washed with dilute brine twice, concentrated to remove solvents. EtOAc (5.0 mL) was added and the mixture was concentrated to give a pale yellow solid free base. 1 ,4-Dioxane (5.0 mL) and pyridine (0.13 mL, 1 .57 mmol) were added. The mixture was cooled to 5-10 °C and triflic anhydride (0.199 mL, 1 .180 mmol) was added while maintaining the temperature below 15 °C. The mixture was stirred at ambient temperature until completion deemed by HPLC (1 h). Toluene (5 mL) and water (5 mL) were added. Layers were separated. The organic layer was washed with water, concentrated to remove solvents. Toluene (1 .0 mL) was added to dissolve the residue followed by Isooctane (4.0 mL). The mixture was stirred at rt overnight. The solids was filtered, washed with Isooctane (4.0 mL) to give the title compound (0.34 g, 81 %) as slightly yellow solids. 1 NMR (400 MHz, CDCI3) δ ppm 0.86 (t, J – 7.2 Hz, 3H), 0.94 (t, J – 7.6 Hz, 3H), 1 .12-1 .15 (m, 1 H), 1 .22-1 .36 (m, 3H), 1 .48-1 .60 (m, 2H), 1 .86-1 .93 (m, 2H), 2.22 (dt, J = 4.1 Hz, 12 Hz, 1 H), 3.10 (d, J – 14.8 Hz, 1 H), 3.49 (d, J – 14.8 Hz, 1 H), 3.64 (s, 3H), 6.1 1 (s, 1 H), 6.36 (s, 1 H), 7.38-7.48 (m, 5), 7.98 (s, 1 H).
Method 2: A mixture of (R)-3-butyl-3-ethyl-7-methoxy-5-phenyl-2,3-dihydrobenzo[f][1 ,4]thiazepin-8-yl trifluoromethanesulfonate (0.5 g, 0.997 mmol), ligand (N) (0.078 g, 0.1 10 mmol) and lr(COD)2BArF (0.127 g, 0.100 mmol) in DCM (10.0 mL) was purged with nitrogen three times, then hydrogen three times. The mixture was shaken in Parr shaker under 10 Bar of H2 for 24 h. The experiment described above was repeated with 1 .0 g (1 .994 mmol) input of (R)-3-butyl-3-ethyl-7-methoxy-5-phenyl-2,3-dihydrobenzo[f][1 ,4]thiazepin-8-yl
trifluoromethanesulfonate. The two batches of the reaction mixture were combined,
concentrated to remove solvents, and purified by silica gel chromatography
(hexanes:EtOAc =9:1 ) to give the sulfide (O) as slightly yellow oil (0.6 g, 40% yield, 99.7% purity). The oil (0.6 g, 1 .191 mmol) was dissolved in TFA (1 .836 mL, 23.83 mmol) and stirred at 40 °C. H202 (0.268 mL, 2.62 mmol) was added slowly over 30 min. The mixture was stirred at 40 °C for 2 h and then cooled to room temperature. Water (10 mL) and toluene (6.0 mL) were added. Layers were separated and the organic layer was washed successively with aqueous sodium carbonate solution and wate, and concentrated to dryness. Toluene (6.0 mL) was added and the mixture was concentrated to dryness. The residue was dissolved in toluene (2.4 mL) and isooctane (7.20 mL) was added. The mixture was heated to reflux and then cooled to room temperature. The mixture was stirred at room temperature for 30 min. The solid was filtered and washed with isooctane to give the title compound (0.48 g, 75%).
Intermediate L: (R)-5-((2-amino-2-ethylhexyl)thio)-2-methoxyphenol, di-p-toluoyl-L-tartrate salt

To a mixture of (R)-2-amino-2-ethylhexyl hydrogen sulfate (1 1 .1 g, 49.3 mmol) in water (23.1 mL) was added NaOH (5.91 g, 148 mmol). The mixture was stirred at reflux for 2 h. The mixture was cooled to room temperature and extracted with MTBE (30.8 mL). The extract was washed with brine (22 mL), concentrated under vacuum and treated with methanol (30.8 mL). The mixture was stirred under nitrogen and treated with 3-hydroxy-4-methoxythiophenol (7.70 g, 49.3 mmol). The mixture was stirred under nitrogen at room temperature for 1 h. The mixture was concentrated under vacuum, treated with acetonitrile (154 mL) and then heated to 45 °C. To the stirred mixture was added (2R,3R)-2,3-bis((4-methylbenzoyl)oxy)succinic acid (19.03 g, 49.3 mmol). The resultant slurry was
stirred at 45 °C. After 2 h, the slurry was cooled to room temperature and stirred for 5 h. The solids were filtered, washed twice with acetonitrile (30 mL) and dried to give the title compound (28.0 g, 85%) as white solids. 1 NMR (400 MHz, DMSO-d6) δ (ppm): 0.70-0.75 (m, 6H), 1 .17 (b, 4H), 1 .46-1 .55 (m, 4H), 2.30 (s, 6H), 3.71 (s, 3H), 5.58 (s, 2H), 6.84 (s, 2H), 6.89 (s, 1 H), 7.24 (d, J – 1 1 .6 Hz, 4H), 7.76 (d, J – 1 1 .6 Hz, 4H).
Intermediate M: (R)-3-butyl-3-ethyl-7-methoxy-5-phenyl-2,3
dihydrobenzo[f][1 ,4]thiazepin-8-yl trifluoromethanesulfonate

A flask was charged with (R)-(2-((2-amino-2-ethylhexyl)thio)-4-hydroxy-5-methoxyphenyl)(phenyl)methanone (3.5 g, 9.03 mmol), citric acid (0.434 g, 2.258 mmol), 1 ,4-Dioxane (17.50 mL) and Toluene (17.50 mL). The mixture was heated to reflux with a Dean-Stark trap to distill water azetropically. The mixture was refluxed for 20 h and then cooled to room temperature. EtOAc (35.0 mL) and water (35.0 mL) were added and layers were separated. The organic layer was washed with aqueous sodium carbonate solution and concentrated to remove solvents to give crude imine as brown oil. The oil was dissolved in EtOAc (35.0 mL) and cooled to 0-5 °C. To the mixture was added triethylamine (1 .888 mL, 13.55 mmol) followed by slow addition of Tf2O (1 .831 mL, 10.84 mmol) at 0-5 °C. The mixture was stirred at room temperature for 1 h. Water was added and layers were separated. The organic layer was washed with brine, dried over Na2SO4 and concentrated under vacuum. The crude triflate was purified by silica gel chromatography
(hexane:EtOAc =90:10) to give the title compound (3.4 g, 75%) as amber oil. 1 NMR (400 MHz, CDCI3) δ ppm 0.86 (t, J – 7.2 Hz, 3H), 0.92 (t, J – 7.9 Hz, 3H), 1 .19-1 .34 (m, 4H), 1 .47-1 .71 (m, 4H), 3.25 (s, 2H), 3.75 (s, 3H), 6.75 (s, 1 H), 7.35-7.43 (m, 3H), 7.48 (s, 1 H), 7.54 (d, J – 7.6 Hz, 2H).
PAPER
Journal of Medicinal Chemistry (2013), 56(12), 5094-5114.

The apical sodium-dependent bile acid transporter (ASBT) transports bile salts from the lumen of the gastrointestinal (GI) tract to the liver via the portal vein. Multiple pharmaceutical companies have exploited the physiological link between ASBT and hepatic cholesterol metabolism, which led to the clinical investigation of ASBT inhibitors as lipid-lowering agents. While modest lipid effects were demonstrated, the potential utility of ASBT inhibitors for treatment of type 2 diabetes has been relatively unexplored. We initiated a lead optimization effort that focused on the identification of a potent, nonabsorbable ASBT inhibitor starting from the first-generation inhibitor 264W94 (1). Extensive SAR studies culminated in the discovery of GSK2330672 (56) as a highly potent, nonabsorbable ASBT inhibitor which lowers glucose in an animal model of type 2 diabetes and shows excellent developability properties for evaluating the potential therapeutic utility of a nonabsorbable ASBT inhibitor for treatment of patients with type 2 diabetes.
//////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
PATENT
Example 26: 3-({[(3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-1 ,1 -dioxido-5-phenyl- 2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8-yl]methyl}amino)pentanedioic acid
Method 1 , Step 1 : To a solution of (3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-5- phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepine-8-carbaldehyde 1 ,1 -dioxide (683 mg, 1 .644 mmol) in 1 ,2-dichloroethane (20 mL) was added diethyl 3- aminopentanedioate (501 mg, 2.465 mmol) and acetic acid (0.188 mL, 3.29 mmol). The reaction mixture was stirred at room temperature for 1 hr then treated with NaHB(OAc)3 (697 mg, 3.29 mmol). The reaction mixture was then stirred at room temperature overnight and quenched with aqueous potassium carbonate solution. The mixture was extracted with DCM. The combined organic layers were washed with H2O, saturated brine, dried (Na2SO4), filtered, and concentrated under reduced pressure to give diethyl 3-({[(3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-1 ,1 -dioxido-5- phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8-yl]methyl}amino)pentanedioate (880 mg, 88%) as a light yellow oil: MS-LCMS m/z 603 (M+H)+.
Method 1 , Step 2: To a solution of diethyl 3-({[(3R,5R)-3-butyl-3-ethyl-7- (methyloxy)-l ,1 -dioxido-5-phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8- yl]methyl}amino)pentanedioate (880 mg, 1 .460 mmol) in a 1 :1 :1 mixture of
THF/MeOH/H2O (30 mL) was added lithium hydroxide (175 mg, 7.30 mmol). The reaction mixture was stirred at room temperature overnight then concentrated under reduced pressure. H2O and MeCN was added to dissolve the residue. The solution was acidified with acetic acid to pH 4-5, partially concentrated to remove MeCN under reduced pressure, and left to stand for 30 min. The white precipitate was collected by filtration and dried under reduced pressure at 50°C overnight to give the title compound (803 mg, 100%) as a white solid: 1 H NMR (MeOH-d4) δ ppm 8.05 (s, 1 H), 7.27 – 7.49 (m, 5H), 6.29 (s, 1 H), 6.06 (s, 1 H), 4.25 (s, 2H), 3.60 – 3.68 (m, 1 H), 3.58 (s, 3H), 3.47 (d, J = 14.8 Hz, 1 H), 3.09 (d, J = 14.8 Hz, 1 H), 2.52 – 2.73 (m, 4H), 2.12 – 2.27 (m, 1 H), 1 .69 – 1 .84 (m, 1 H), 1 .48 – 1 .63 (m, 1 H), 1 .05 – 1 .48 (m, 5H), 0.87 (t, J = 7.4 Hz, 3H), 0.78 (t, J = 7.0 Hz, 3H); ES-LCMS m/z 547 (M+H) Method 2: A solution of dimethyl 3-({[(3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-
1 ,1 -dioxido-5-phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8- yl]methyl}amino)pentanedioate (~ 600 g) in THF (2.5 L) and MeOH (1 .25 L) was cooled in an ice-bath and a solution of NaOH (206 g, 5.15 mol) in water (2.5 L) was added dropwise over 20 min (10-22°C reaction temperature). After stirring 20 min, the solution was concentrated (to remove THF/MeOH) and acidified to pH~4 with concentrated HCI. The precipitated product was aged with stirring, collected by filtration and air dried overnight. A second 600g batch of dimethyl 3-({[(3R,5R)-3- butyl-3-ethyl-7-(methyloxy)-1 ,1 -dioxido-5-phenyl-2,3,4,5-tetrahydro-1 ,4- benzothiazepin-8-yl]methyl}amino)pentanedioate was saponified in a similar fashion. The combined crude products (~2 mol theoretical) were suspended in CH3CN (8 L) and water (4 L) and the stirred mixture was heated to 65°C. A solution formed which was cooled to 10°C over 2 h while seeding a few times with an authentic sample of the desired crystalline product. The resulting slurry was stirred at 10°C for 2 h, and the solid was collected by filtration. The filter cake was washed with water and air-dried overnight. Further drying to constant weight in a vacuum oven at 55°C afforded crystalline 3-({[(3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-1 ,1 – dioxido-5-phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8- yl]methyl}amino)pentanedioic acid as a white solid (790 g).
Method 3: (3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-5-phenyl-2,3,4,5-tetrahydro- 1 ,4-benzothiazepine-8-carbaldehyde 1 ,1 -dioxide (1802 grams, 4.336 moles) and dimethyl 3-aminopentanedioate (1334 grams, 5.671 moles) were slurried in iPrOAc (13.83 kgs). A nitrogen atmosphere was applied to the reactor. To the slurry at 20°C was added glacial acetic acid (847 ml_, 14.810 moles), and the mixture was stirred until complete dissolution was observed. Solid sodium triacetoxyborohydride (1424 grams, 6.719 moles) was next added to the reaction over a period of 7 minutes. The reaction was held at 20°C for a total of 3 hours at which time LC analysis of a sample indicated complete consumption of the (3R,5R)-3-butyl-3-ethyl- 7-(methyloxy)-5-phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepine-8-carbaldehyde 1 ,1 – dioxide. Next, water (20.36 kgs) and brine (4.8 kgs) were added to the reactor. The contents of the reactor were stirred for 10 minutes and then settled for 10 minutes. The bottom, aqueous layer was then removed and sent to waste. A previously prepared, 10% (wt/wt) aqueous solution of sodium bicarbonate (22.5 L) was added to the reactor. The contents were stirred for 10 minutes and then settled for 10 minutes. The bottom, aqueous layer was then removed and sent to waste. To the reactor was added a second wash of 10% (wt/wt) aqueous, sodium bicarbonate
(22.5 L). The contents of the reactor were stirred for 10 minutes and settled for 10 minutes. The bottom, aqueous layer was then removed and sent to waste. The contents of the reactor were then reduced to an oil under vacuum distillation. To the oil was added THF (7.15 kgs) and MeOH (3.68 kgs). The contents of the reactor were heated to 55°C and agitated vigorously until complete dissolution was observed. The contents of the reactor were then cooled to 25°C whereupon a previously prepared aqueous solution of NaOH [6.75 kgs of water and 2.09 kgs of NaOH (50% wt wt solution)] was added with cooling being applied to the jacket. The contents of the reactor were kept below 42°C during the addition of the NaOH solution. The temperature was readjusted to 25°C after the NaOH addition, and the reaction was stirred for 75 minutes before HPLC analysis indicated the reaction was complete. Heptane (7.66 kgs) was added to the reactor, and the contents were stirred for 10 minutes and then allowed to settle for 10 minutes. The aqueous layer was collected in a clean nalgene carboy. The heptane layer was removed from the reactor and sent to waste. The aqueous solution was then returned to the reactor, and the reactor was prepared for vacuum distillation. Approximately 8.5 liters of distillate was collected during the vacuum distillation. The vacuum was released from the reactor, and the temperature of the contents was readjusted to 25°C. A 1 N HCI solution (30.76 kgs) was added to the reactor over a period of 40 minutes. The resulting slurry was stirred at 25°C for 10 hours then cooled to 5°C over a period of 2 hours. The slurry was held at 5°C for 4 hours before the product was collected in a filter crock by vacuum filtration. The filter cake was then washed with cold (5°C) water (6 kgs). The product cake was air dried in the filter crock under vacuum for approximately 72 hours. The product was then transferred to three drying trays and dried in a vacuum oven at 50°C for 79 hours. The temperature of the vacuum oven was then raised to 65°C for 85 additional hours. The product was off-loaded as a single batch to give 2568 grams (93.4% yield) of intermediate grade 3-({[(3R,5R)-3- butyl-3-ethyl-7-(methyloxy)-1 ,1 -dioxido-5-phenyl-2,3,4,5-tetrahydro-1 ,4- benzothiazepin-8-yl]methyl}amino)pentanedioic acid as an off-white solid.
Intermediate grade 3-({[(3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-1 ,1 -dioxido-5- phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8-yl]methyl}amino)pentanedioic acid was dissolved (4690 g) in a mixture of glacial acetic acid (8850 g) and purified water (4200 g) at 70°C. The resulting solution was transferred through a 5 micron polishing filter while maintaining the temperature above 30°C. The reactor and filter were rinsed through with a mixture of glacial acetic acid (980 g) and purified water (470 g). The solution temperature was adjusted to 50°C. Filtered purified water (4230 g) was added to the solution. The cloudy solution was then seeded with crystalline 3-({[(3 5R)-3-butyl-3-ethyl-7-(methyloxy)-1 ,1 -dioxido-5-phenyl-2,3 ,4,5- tetrahydro-1 ,4-benzothiazepin-8-yl]methyl}amino)pentanedioic acid (10 g). While maintaining the temperature at 50°C, filtered purified water was charged to the slurry at a controlled rate (1 1030 g over 130 minutes). Additional filtered purified water was then added to the slurry at a faster controlled rate (20740 g over 100 minutes). A final charge of filtered purified water (3780 g) was made to the slurry. The slurry was then cooled to 10°C at a linear rate over 135 minutes. The solids were filtered over sharkskin filter paper to remove the mother liquor. The cake was then rinsed with filtered ethyl acetate (17280 g) then the wash liquors were removed by filtration. The resulting wetcake was isolated into trays and dried under vacuum at 50°C for 23 hours. The temperature was then increased to 60°C and drying was continued for an additional 24 hours to afford crystalline 3-({[(3R,5R)-3-butyl-3-ethyl- 7-(methyloxy)-1 ,1 -dioxido-5-phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8- yl]methyl}amino)pentanedioic acid (3740 g, 79.7% yield) as a white solid.
To a slurry of this crystalline 3-({[(3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-1 ,1 – dioxido-5-phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8- yl]methyl}amino)pentanedioic acid (3660 g) and filtered purified water (3.6 L) was added filtered glacial acetic acid (7530 g). The temperature was increased to 60°C and full dissolution was observed. The temperature was reduced to 55°C, filtered, and treated with purified water (3.2 L). The solution was then seeded with crystalline 3-({[(3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-1 ,1 -dioxido-5-phenyl-2,3,4,5- tetrahydro-1 ,4-benzothiazepin-8-yl]methyl}amino)pentanedioic acid (18 g) to afford a slurry. Filtered purified water was charged to the slurry at a controlled rate (9 L over 140 minutes). Additional filtered purified water was then added to the slurry at a faster controlled rate (18 L over 190 minutes). The slurry was then cooled to
10°C at a linear rate over 225 minutes. The solids were filtered over sharkskin filter paper to remove the mother liquor. The cake was then rinsed with filtered purified water (18 L), and the wash liquors were removed by filtration. The resulting wetcake was isolated into trays and dried under vacuum at 60°C for 18.5 hours to afford a crystalline 3-({[(3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-1 ,1 -dioxido-5-phenyl- 2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8-yl]methyl}amino)pentanedioic acid (3330 g, 90.8% yield) as a white solid which was analyzed for crystallinity as summarized below.
Paper
Cowan, D. J.; Collins, J. L.; Mitchell, M. B.; Ray, J. A.; Sutton, P. W.; Sarjeant, A. A.; Boros, E. E.Enzymatic- and Iridium-Catalyzed Asymmetric Synthesis of a Benzothiazepinylphosphonate Bile Acid Transporter Inhibitor J. Org. Chem. 2013, 78 ( 24) 12726– 12734, DOI: 10.1021/jo402311e

A synthesis of the benzothiazepine phosphonic acid 3, employing both enzymatic and transition metal catalysis, is described. The quaternary chiral center of 3 was obtained by resolution of ethyl (2-ethyl)norleucinate (4) with porcine liver esterase (PLE) immobilized on Sepabeads. The resulting (R)-amino acid (5) was converted in two steps to aminosulfate 7, which was used for construction of the benzothiazepine ring. Benzophenone 15, prepared in four steps from trimethylhydroquinone 11, enabled sequential incorporation of phosphorus (Arbuzov chemistry) and sulfur (Pd(0)-catalyzed thiol coupling) leading to mercaptan intermediate 18. S-Alkylation of 18 with aminosulfate 7 followed by cyclodehydration afforded dihydrobenzothiazepine 20. Iridium-catalyzed asymmetric hydrogenation of 20 with the complex of [Ir(COD)2BArF] (26) and Taniaphos ligand P afforded the (3R,5R)-tetrahydrobenzothiazepine 30 following flash chromatography. Oxidation of 30 to sulfone 31 and phosphonate hydrolysis completed the synthesis of 3 in 12 steps and 13% overall yield.
Paper

Scheme 1. Current Route to Chiral Intermediate 4 in the Synthesis of GSK2330672
Development of an Enzymatic Process for the Production of (R)-2-Butyl-2-ethyloxirane
Gheorghe-Doru Roiban*† , Peter W. Sutton*‡, Rebecca Splain§, Christopher Morgan§, Andrew Fosberry∥, Katherine Honicker⊥, Paul Homes∥, Cyril Boudet#, Alison Dann#, Jiasheng Guo§, Kristin K. Brown∇, Leigh Anne F. Ihnken⊥, and Douglas Fuerst⊥
, Peter W. Sutton*‡, Rebecca Splain§, Christopher Morgan§, Andrew Fosberry∥, Katherine Honicker⊥, Paul Homes∥, Cyril Boudet#, Alison Dann#, Jiasheng Guo§, Kristin K. Brown∇, Leigh Anne F. Ihnken⊥, and Douglas Fuerst⊥†Synthetic Biochemistry, Advanced Manufacturing Technologies, ‡API Chemistry, ∥Protein and Cellular Sciences, GlaxoSmithKline, Medicines Research Centre, Gunnels Wood Road, Stevenage SG1 2NY, United Kingdom
§API Chemistry, ⊥Synthetic Biochemistry, Advanced Manufacturing Technologies, GlaxoSmithKline, 709 Swedeland Road, King of Prussia, Pennsylvania 19406, United States
# Biotechnology and Environmental Shared Service, Global Manufacturing and Supply, GlaxoSmithKline, Dominion Way, Worthing BN14 8PB, United Kingdom
∇ Molecular Design, Computational and Modeling Sciences, GlaxoSmithKline, 1250 S. Collegeville Road, Collegeville, Pennsylvania 19426, United States
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.7b00179
*E-mail: gheorghe.d.roiban@gsk.com., *E-mail: peter.sutton@uab.cat.

An epoxide resolution process was rapidly developed that allowed access to multigram scale quantities of (R)-2-butyl-2-ethyloxirane 2 at greater than 300 g/L reaction concentration using an easy-to-handle and store lyophilized powder of epoxide hydrolase from Agromyces mediolanus. The enzyme was successfully fermented on a 35 L scale and stability increased by downstream processing. Halohydrin dehalogenases also gave highly enantioselective resolution but were shown to favor hydrolysis of the (R)-2 epoxide, whereas epoxide hydrolase from Aspergillus nigerinstead provided (R)-7 via an unoptimized, enantioconvergent process.
REFERENCES
1: Nunez DJ, Yao X, Lin J, Walker A, Zuo P, Webster L, Krug-Gourley S, Zamek-Gliszczynski MJ, Gillmor DS, Johnson SL. Glucose and lipid effects of the ileal apical sodium-dependent bile acid transporter inhibitor GSK2330672: double-blind randomized trials with type 2 diabetes subjects taking metformin. Diabetes Obes Metab. 2016 Jul;18(7):654-62. doi: 10.1111/dom.12656. Epub 2016 Apr 21. PubMed PMID: 26939572.
2: Wu Y, Aquino CJ, Cowan DJ, Anderson DL, Ambroso JL, Bishop MJ, Boros EE, Chen L, Cunningham A, Dobbins RL, Feldman PL, Harston LT, Kaldor IW, Klein R, Liang X, McIntyre MS, Merrill CL, Patterson KM, Prescott JS, Ray JS, Roller SG, Yao X, Young A, Yuen J, Collins JL. Discovery of a highly potent, nonabsorbable apical sodium-dependent bile acid transporter inhibitor (GSK2330672) for treatment of type 2 diabetes. J Med Chem. 2013 Jun 27;56(12):5094-114. doi: 10.1021/jm400459m. Epub 2013 Jun 6. PubMed PMID: 23678871.
///////GSK 2330672, phase 2
CCCC[C@@]1(CS(=O)(=O)c2cc(c(cc2[C@H](N1)c3ccccc3)OC)CNC(CC(=O)O)CC(=O)O)CC
UPDATE
WO/2022/136335FORMS OF LINERIXIBAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022136335&_cid=P20-L54PZU-72661-1
Example lb – Preparation of Form I
Method 1
Form I may be prepared according to the procedures in WO2011/137135, Example 26.
Method 2
Form I of linerixibat was prepared according to the following procedure at a large scale (> 500 g). All charges were based on input linerixibat.
An intermediate grade linerixibat was dissolved in acetonitrile / Water (12 vols / 8 vols) at reflux (~76 °C). The solution was seeded at 70°C with Form I (2% w/w), cooled to 60 °C over 15 mins and aged at 60 °C for 2 hrs. Water (14 vols) was added over 8 hrs and aged for 1 hr. The suspension was cooled to 20 °C over 1 hr and aged for >30 mins. The slurry was filtered, washed with acetonitrile: water (6:11 v/v) (3.5 vols), then twice with water (2 vols) and blown down with nitrogen 8-18 hrs. Drying was carried out under vacuum at 40-50 °C without agitation until the Karl Fisher measurement (KF) was < 10% w/w. The batch was agitated at 4 rpm for 2 mins every 3 hrs until the KF <1 % w/w to provide Form I.
Form I can also be prepared by the above procedure without the step of seeding. Method 3
Form I of linerixibat was prepared according to the following procedure at a large scale (> 50 kg). A reactor (Reactor 1) was charged with 55.84 kg GSK2330672B (1.0 wt) intermediate grade (IG) followed by acetonitrile (12 vol) and purified water (8 vol). The mixture was heated to reflux (74-79°C), and held until complete dissolution is observed. The solution was then transferred to a reactor (Reactor 2) that had been pre-heated to 74-79°C via filter (0.22pm pipe-line filter). Reactor 1 was rinsed with acetonitrile (MeCN) (0.3 vol) and purified water (0.2 vol), and the solution in Reactor 1 was transferred to Reactor 2 via filter (0.22pm pipe-line filter). The contents of Reactor 2 were held until complete dissolution is observed. The solution in Reactor 2 was cooled to 69-72°C, then seeded with 2 w/w% (based on pure GSK2330672B input). The suspension was cooled to 58-62°C within 10-20 min. The suspension was held at 58-62°C for 2h. Purified water (14 vol) was added over 8 hrs. After the addition was complete, the slurry was held at 58-62°C for 60 min, then cooled to 18-25°C over 50-70 min. The slurry was stirred at 18-25°C for not less than 30 min, then the suspension filtered under vacuum. The reactor was rinsed with MeCN/water (6/11 v:v, 3.5 vol) and the rinse used to wash the cake. The cake was washed twice with water (2 vol). The cake was blown down with nitrogen and dried at 60°C under vacuum to give 46.35 kg GSK2330672B Form 1 solid.
Method 4
10.94g of GSK2330672B was charged and washed into a vessel using 131mL of acetonitrile (MeCN) and 88mL of water. The slurry was then heated to reflux. 4mL of 3:2v/v MeCN/water was charged followed by 5.5mL of 3:2v/v MeCN/water. The contents were cooled to 70 °C, then cooled to 60°C over 15 minutes and held stirring for 2 hours. 153 mL water was added over 8 hours, the contents were cooled to 20°C over 1 hour, and held stirring for 1 hour. The product was isolated, washed with 38mL of 6:11 MeCN/water, then washed twice with 22mL of water. After deliquoring, the product was dried at 45°C under vacuum, to give 9.35g (85.5%w/w) Form I GSK2330672B.

NEW DRUG APPROVALS
ONE TIME TO PAY SUBSCRIPTION
$10.00
GSK 2982772
CAS: 1622848-92-3 (free base), 1987858-31-0 (hydrate)
Chemical Formula: C20H19N5O3
Molecular Weight: 377.404
5-Benzyl-N-[(3S)-5-methyl-4-oxo-2,3,4,5-tetrahydro-1,5-benzoxazepin-3-yl]-4H-1,2,4-triazole-3-carboxamide
(S)-5-benzyl-N-(5-methyl-4-oxo-2,3,4,5-tetrahydrobenzo[b][l,4]oxazepin-3-yl)-4H-l,2,4- triazole-3-carboxamide
- 3-(Phenylmethyl)-N-[(3S)-2,3,4,5-tetrahydro-5-methyl-4-oxo-1,5-benzoxazepin-3-yl]-1H-1,2,4-triazole-5-carboxamide
- (S)-5-Benzyl-N-(5-methyl-4-oxo-2,3,4,5-tetrahydrobenzo[b][1,4]oxazepin-3-yl)-4H-1,2,4-triazole-3-carboxamide
GSK2982772 is a potent and selective receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate for the Treatment of Inflammatory Diseases. GSK2982772 is, currently in phase 2a clinical studies for psoriasis, rheumatoid arthritis, and ulcerative colitis. GSK2982772 potently binds to RIP1 with exquisite kinase specificity and has excellent activity in blocking many TNF-dependent cellular responses. RIP1 has emerged as an important upstream kinase that has been shown to regulate inflammation through both scaffolding and kinase specific functions.
GSK-2982772, an oral receptor-interacting protein-1 (RIP1) kinase inhibitor, is in phase II clinical development at GlaxoSmithKline for the treatment of active plaque-type psoriasis, moderate to severe rheumatoid arthritis, and active ulcerative colitis. A phase I trial was also completed for the treatment of inflammatory bowel disease using capsule and solution formulations.
- Originator GlaxoSmithKline
- Class Antipsoriatics
- Mechanism of Action Receptor-interacting protein serine-threonine kinase inhibitors
Highest Development Phases
- Phase II Plaque psoriasis; Rheumatoid arthritis; Ulcerative colitis
- Phase I Inflammatory bowel diseases
Most Recent Events
- 15 Dec 2016 Biomarkers information updated
- 01 Nov 2016 Phase-II clinical trials in Ulcerative colitis (Adjunctive treatment) in USA (PO) (NCT02903966)
- 01 Oct 2016 Phase-II clinical trials in Rheumatoid arthritis in Poland (PO) (NCT02858492)
PHASE 2 Psoriasis, plaque GSK
| Inflammatory Bowel Disease, Agents for Rheumatoid Arthritis, Treatment of Antipsoriatics |

Data Science and Informatics Leader | Innovation Advocate
GSK
University of North Carolina at Chapel Hill
He is a data scientist and innovator with experience in both early and late stages of drug development. his current role involves the late stage of drug product development. I’m leading a project to bring GSK’s large molecule process and analytical data onto our big data platform and develop new data analysis and modeling capabilities. Also, working within GSK’s Advanced Manufacturing Technology (AMT) initiative provides plenty of other opportunities to impact how we make medicines.
Previously as a computational chemist (i.e. a data scientist in drug discovery), he worked with scientists from many domains, including chemists, biologists, and other informaticians. he enjoys digging into all the computational aspects of life science research, and solving data challenges by exploiting adjacencies and connections – between diverse fields of knowledge, and the equally diverse scientists trained in them.
He has supported multiple drug discovery projects at GSK starting from target identification (“how should we modulate disease X?”) through to candidate selection and early clinical development (“let’s see if what we discovered can become a medicine”). Deriving insight by custom data integration is one of my specialties; recently he designed and implemented a platform for integrating data sets from multiple experiments that will be used by GSK screening scientists to find and combine hits.
A trained computer scientist and cheminformatician, he is an active member of the algorithms, data science and internal innovation communities at GSK, leading many of these efforts.
His Ph.D. work introduced new computational geometry techniques for structural bioinformatics and protein function prediction. I have touched on several other subject areas:
* data mining/machine learning (predictive modeling and graph mining),
* computer graphics and augmented reality (one of the pioneers of projection mapping)
* robotics (keen current interest and future aspiration)
Receptor-interacting protein- 1 (RIP1) kinase, originally referred to as RIP, is a TKL family serine/threonine protein kinase involved in innate immune signaling. RIPl kinase is a RHIM domain containing protein, with an N-terminal kinase domain and a C-terminal death domain ((2005) Trends Biochem. Sci. 30, 151-159). The death domain of RIPl mediates interaction with other death domain containing proteins including Fas and TNFR-1 ((1995) Cell 81 513-523), TRAIL-Rl and TRAIL-R2 ((1997) Immunity 7, 821-830) and TRADD ((1996) Immunity 4, 387-396), while the RHIM domain is crucial for binding other RHFM domain containing proteins such as TRIF ((2004) Nat Immunol. 5, 503-507), DAI ((2009) EMBO Rep. 10, 916-922) and RIP3 ((1999) J. Biol. Chem. 274, 16871-16875); (1999) Curr. Biol. 9, 539-542) and exerts many of its effects through these interactions. RIPl is a central regulator of cell signaling, and is involved in mediating both pro-survival and programmed cell death pathways which will be discussed below.
The role for RIPl in cell signaling has been assessed under various conditions
[including TLR3 ((2004) Nat Immunol. 5, 503-507), TLR4 ((2005) J. Biol. Chem. 280,
36560-36566), TRAIL ((2012) J .Virol. Epub, ahead of print), FAS ((2004) J. Biol. Chem. 279, 7925-7933)], but is best understood in the context of mediating signals downstream of the death receptor TNFRl ((2003) Cell 114, 181-190). Engagement of the TNFR by TNF leads to its oligomerization, and the recruitment of multiple proteins, including linear K63-linked polyubiquitinated RIPl ((2006) Mol. Cell 22, 245-257), TRAF2/5 ((2010) J. Mol. Biol. 396, 528-539), TRADD ((2008) Nat. Immunol. 9, 1037-1046) and cIAPs ((2008) Proc. Natl. Acad. Sci. USA. 105, 1 1778-11783), to the cytoplasmic tail of the receptor. This complex which is dependent on RIPl as a scaffolding protein (i.e. kinase
independent), termed complex I, provides a platform for pro-survival signaling through the activation of the NFKB and MAP kinases pathways ((2010) Sci. Signal. 115, re4).
Alternatively, binding of TNF to its receptor under conditions promoting the
deubiquitination of RIPl (by proteins such as A20 and CYLD or inhibition of the cIAPs) results in receptor internalization and the formation of complex II or DISC (death-inducing signaling complex) ((2011) Cell Death Dis. 2, e230). Formation of the DISC, which contains RIPl, TRADD, FADD and caspase 8, results in the activation of caspase 8 and the onset of programmed apoptotic cell death also in a RIPl kinase independent fashion ((2012) FEBS J 278, 877-887). Apoptosis is largely a quiescent form of cell death, and is involved in routine processes such as development and cellular homeostasis.
Under conditions where the DISC forms and RJP3 is expressed, but apoptosis is inhibited (such as FADD/caspase 8 deletion, caspase inhibition or viral infection), a third RIPl kinase-dependent possibility exists. RIP3 can now enter this complex, become phosphorylated by RIPl and initiate a caspase-independent programmed necrotic cell death through the activation of MLKL and PGAM5 ((2012) Cell 148, 213-227); ((2012) Cell 148, 228-243); ((2012) Proc. Natl. Acad. Sci. USA. 109, 5322-5327). As opposed to apoptosis, programmed necrosis (not to be confused with passive necrosis which is not programmed) results in the release of danger associated molecular patterns (DAMPs) from the cell.
These DAMPs are capable of providing a “danger signal” to surrounding cells and tissues, eliciting proinflammatory responses including inflammasome activation, cytokine production and cellular recruitment ((2008 Nat. Rev. Immunol 8, 279-289).
Dysregulation of RIPl kinase-mediated programmed cell death has been linked to various inflammatory diseases, as demonstrated by use of the RIP3 knockout mouse (where RIPl -mediated programmed necrosis is completely blocked) and by Necrostatin-1 (a tool inhibitor of RIPl kinase activity with poor oral bioavailability). The RIP3 knockout mouse has been shown to be protective in inflammatory bowel disease (including Ulcerative colitis and Crohn’s disease) ((2011) Nature 477, 330-334), Psoriasis ((2011) Immunity 35, 572-582), retinal-detachment-induced photoreceptor necrosis ((2010) PNAS 107, 21695-21700), retinitis pigmentosa ((2012) Proc. Natl. Acad. Sci., 109:36, 14598-14603), cerulein-induced acute pancreatits ((2009) Cell 137, 1100-1111) and Sepsis/systemic inflammatory response syndrome (SIRS) ((2011) Immunity 35, 908-918). Necrostatin-1 has been shown to be effective in alleviating ischemic brain injury ((2005) Nat. Chem. Biol. 1, 112-119), retinal ischemia/reperfusion injury ((2010) J. Neurosci. Res. 88, 1569-1576), Huntington’s disease ((2011) Cell Death Dis. 2 el 15), renal ischemia reperfusion injury ((2012) Kidney Int. 81, 751-761), cisplatin induced kidney injury ((2012) Ren. Fail. 34, 373-377) and traumatic brain injury ((2012) Neurochem. Res. 37, 1849-1858). Other diseases or disorders regulated at least in part by RIPl -dependent apoptosis, necrosis or cytokine production include hematological and solid organ malignancies ((2013) Genes
Dev. 27: 1640-1649), bacterial infections and viral infections ((2014) Cell Host & Microbe 15, 23-35) (including, but not limited to, tuberculosis and influenza ((2013) Cell 153, 1-14)) and Lysosomal storage diseases (particularly, Gaucher Disease, Nature Medicine Advance Online Publication, 19 January 2014, doi: 10.1038/nm.3449).
A potent, selective, small molecule inhibitor of RIP1 kinase activity would block RIP 1 -dependent cellular necrosis and thereby provide a therapeutic benefit in diseases or events associated with DAMPs, cell death, and/or inflammation.
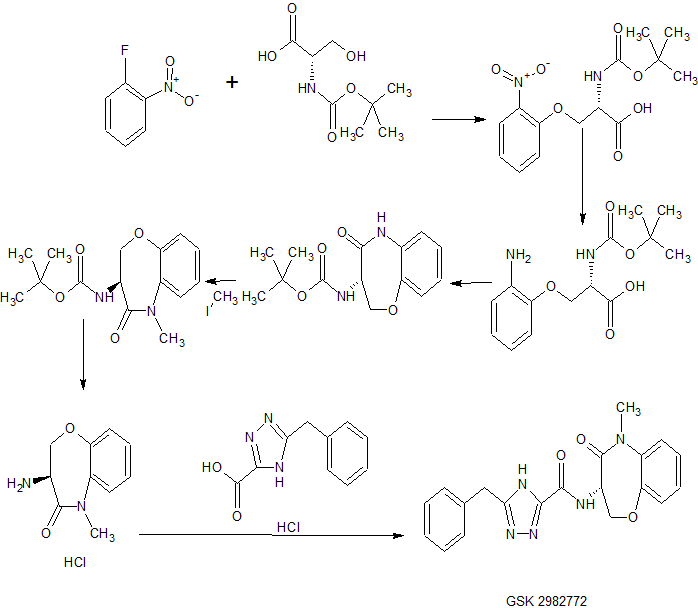
Patent

Example 12
Method H
(S)-5-benzyl-N-(5-methyl-4-oxo-2,3,4,5-tetrahydrobenzo[b][l,4]oxazepin-3-yl)-4H-l,2,4- triazole-3-carboxamide

A mixture of (S)-3-amino-5-methyl-2,3-dihydrobenzo[b][l,4]oxazepin-4(5H)-one, hydrochloride (4.00 g, 16.97 mmol), 5-benzyl-4H-l,2,4-triazole-3-carboxylic acid, hydrochloride (4.97 g, 18.66 mmol) and DIEA (10.37 mL, 59.4 mmol) in isopropanol (150 mL) was stirred vigorously for 10 minutes and then 2,4,6-tripropyl-l,3,5,2,4,6-trioxatriphosphinane 2,4,6-trioxide (T3P) (50% by wt. in EtOAc) (15.15 mL, 25.5 mmol) was added. The mixture was stirred at rt for 10 minutes and then quenched with water and concentrated to remove isopropanol. The resulting crude material is dissolved in EtOAc and washed with 1M HC1, satd. NaHC03 and brine. Organics were concentrated and purified by column chromatography (220 g silica column; 20-90% EtOAc/hexanes, 15 min.; 90%, 15 min.) to give the title compound as a light orange foam (5.37 g, 83%). 1H NMR (MeOH-d4) δ: 7.40 – 7.45 (m, 1H), 7.21 – 7.35 (m, 8H), 5.01 (dd, J = 11.6, 7.6 Hz, 1H), 4.60 (dd, J = 9.9, 7.6 Hz, 1H), 4.41 (dd, J = 11.4, 9.9 Hz, 1H), 4.17 (s, 2H), 3.41 (s, 3H); MS (m/z) 378.3 (M+H+).
Alternative Preparation:
To a solution of (S)-3-amino-5-methyl-2,3-dihydrobenzo[b][l,4]oxazepin-4(5H)-one hydrochloride (100 g, 437 mmol), 5-benzyl-4H-l,2,4-triazole-3-carboxylic acid hydrochloride (110 g, 459 mmol) in DCM (2.5 L) was added DIPEA (0.267 L, 1531 mmol) at 15 °C. The reaction mixture was stirred for 10 min. and 2,4,6-tripropyl-l, 3, 5,2,4,6-trioxatriphosphinane 2,4,6-trioxide >50 wt. % in ethyl acetate (0.390 L, 656 mmol) was slowly added at 15 °C. After stirring for 60 mins at RT the LCMS showed the reaction was complete, upon which time it was quenched with water, partitioned between DCM and washed with 0.5N HCl aq (2 L), saturated aqueous NaHC03 (2 L), brine (2 L) and water (2 L). The organic phase was separated and activated charcoal (100 g) and sodium sulfate
(200 g) were added. The dark solution was shaken for 1 h before filtering. The filtrate was then concentrated under reduced pressure to afford the product as a tan foam (120 g). The product was dried under a high vacuum at 50 °C for 16 h. 1H MR showed 4-5% wt of ethyl acetate present. The sample was dissolved in EtOH (650 ml) and stirred for 30 mins, after which the solvent was removed using a rotavapor (water-bath T=45 °C). The product was dried under high vacuum for 16 h at RT (118 g, 72% yield). The product was further dried under high vacuum at 50 °C for 5 h. 1H NMR showed <1% of EtOH and no ethyl acetate. 1H NMR (400 MHz, DMSO-i¾) δ ppm 4.12 (s, 2 H), 4.31 – 4.51 (m, 1 H), 4.60 (t, J=10.36 Hz, 1 H), 4.83 (dt, 7=11.31, 7.86 Hz, 1 H), 7.12 – 7.42 (m, 8 H), 7.42 – 7.65 (m, 1 H), 8.45 (br. s., 1 H), 14.41 (br. s., 1 H). MS (m/z) 378 (M + H+).
Crystallization:
(S)-5-Benzyl-N-(5-methyl-4-oxo-2,3,4,5-tetrahydrobenzo[b][l,4]oxazepin-3-yl)-4H-l,2,4-triazole-3-carboxamide (100 mg) was dissolved in 0.9 mL of toluene and 0.1 mL of methylcyclohexane at 60 °C, then stirred briskly at room temperature (20 °C) for 4 days. After 4 days, an off-white solid was recovered (76 mg, 76% recovery). The powder X-ray diffraction (PXRD) pattern of this material is shown in Figure 7 and the corresponding diffraction data is provided in Table 1.
The PXRD analysis was conducted using a PANanalytical X’Pert Pro
diffractometer equipped with a copper anode X-ray tube, programmable slits, and
X’Celerator detector fitted with a nickel filter. Generator tension and current were set to 45kV and 40mA respectively to generate the copper Ka radiation powder diffraction pattern over the range of 2 – 40°2Θ. The test specimen was lightly triturated using an agate mortar and pestle and the resulting fine powder was mounted onto a silicon background plate.
Table 1.

Paper
Discovery of a first-in-class receptor interacting protein 1 (RIP1) kinase specific clinical candidate (GSK2982772) for the treatment of inflammatory diseases
J Med Chem 2017, 60(4): 1247

http://pubs.acs.org/doi/pdf/10.1021/acs.jmedchem.6b01751
RIP1 regulates necroptosis and inflammation and may play an important role in contributing to a variety of human pathologies, including immune-mediated inflammatory diseases. Small-molecule inhibitors of RIP1 kinase that are suitable for advancement into the clinic have yet to be described. Herein, we report our lead optimization of a benzoxazepinone hit from a DNA-encoded library and the discovery and profile of clinical candidate GSK2982772 (compound 5), currently in phase 2a clinical studies for psoriasis, rheumatoid arthritis, and ulcerative colitis. Compound 5 potently binds to RIP1 with exquisite kinase specificity and has excellent activity in blocking many TNF-dependent cellular responses. Highlighting its potential as a novel anti-inflammatory agent, the inhibitor was also able to reduce spontaneous production of cytokines from human ulcerative colitis explants. The highly favorable physicochemical and ADMET properties of 5, combined with high potency, led to a predicted low oral dose in humans.
J. Med. Chem. 2017, 60, 1247−1261

(S)-5-Benzyl-N-(5-methyl-4-oxo-2,3,4,5-tetrahydrobenzo[b]- [1,4]oxazepin-3-yl)-4H-1,2,4-triazole-3-carboxamide (5).
EtOAc solvate. 1 H NMR (DMSO-d6) δ ppm 14.41 (br s, 1 H), 8.48 (br s, 1 H), 7.50 (dd, J = 7.7, 1.9 Hz, 1 H), 7.12−7.40 (m, 8 H), 4.83 (dt, J = 11.6, 7.9 Hz, 1 H), 4.60 (t, J = 10.7 Hz, 1 H), 4.41 (dd, J = 9.9, 7.8 Hz, 1 H), 4.12 (s, 2 H), 3.31 (s, 3 H). Anal. Calcd for C20H20N5O3·0.026EtOAc·0.4H2O C, 62.36; H, 5.17; N, 18.09. Found: C, 62.12; H, 5.05; N, 18.04.
Synthesis of (<it>S</it>)-3-amino-benzo[<it>b</it>][1,4]oxazepin-4-one via Mitsunobu and S<INF>N</INF>Ar reaction for a first-in-class RIP1 kinase inhibitor GSK2982772 in clinical trials
Tetrahedron Lett 2017, 58(23): 2306
Harris, P.A.
Identification of a first-in-class RIP1 kinase inhibitor in phase 2a clinical trials for immunoinflammatory diseases
ACS MEDI-EFMC Med Chem Front (June 25-28, Philadelphia) 2017, Abst
Harris, P.
Identification of a first-in-class RIP1 kinase inhibitor in phase 2a clinical trials for immuno-inflammatory diseases
253rd Am Chem Soc (ACS) Natl Meet (April 2-6, San Francisco) 2017, Abst MEDI 313
1H NMR AND 13C NMR PREDICT


////////////GSK 2982772, phase 2, Plaque psoriasis, Rheumatoid arthritis, Ulcerative colitis
CN3c4ccccc4OC[C@H](NC(=O)c2nnc(Cc1ccccc1)n2)C3=O
Debio-1452

Debio-1452, AFN 1252
AFN-1252; UNII-T3O718IKKM; API-1252; CAS 620175-39-5; CHEMBL1652621; (E)-N-methyl-N-((3-methylbenzofuran-2-yl)methyl)-3-(7-oxo-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl)acrylamide
- MFC22 H21 N3 O3
- 2-Propenamide, N-methyl-N-[(3-methyl-2-benzofuranyl)methyl]-3-(5,6,7,8-tetrahydro-7-oxo-1,8-naphthyridin-3-yl)-, (2E)-
- MW375.42
- Phase 2, clinical trials for the oral treatment of staphylococcal infections, including hospital and community-acquired MRSA and acute bacterial skin and skin structure infections
- Qualified Infectious Disease Product designation
GlaxoSmithKline plc INNOVATOR
![]()
Debiopharm SA,


Melioidosis, Enoyl ACP reductase Fabl inhibitor
Debio-1452, a novel class fatty acid biosynthesis (FAS) II pathway inhibitor, was studied in phase II clinical trials for the oral treatment of staphylococcal infections, including hospital and community-acquired MRSA and acute bacterial skin and skin structure infections. Debiopharm is developing oral and IV formulations of a prodrug of Debio-1452, Debio-1450.
Infections caused by or related to bacteria are a major cause of human illness worldwide. Unfortunately, the frequency of resistance to standard antibacterials has risen dramatically over the last decade, especially in relation to Staphylococcus aureus. For example, such resistant S. aureus includes MRSA, resistant to methicillin, vancomycin, linezolid and many other classes of antibiotics, or the newly discovered New Delhi metallo-beta-lactamase- 1 (NDM-1) type resistance that has shown to afford bacterial resistant to most known antibacterials, including penicillins, cephalosporins, carbapenems, quinolones and fluoroquinolones, macrolides, etc. Hence, there exists an urgent, unmet, medical need for new agents acting against bacterial targets..
In recent years, inhibitors of Fabl, a bacterial target involved in bacterial fatty acid synthesis, have been developed and many have been promising in regard to their potency and tolerability in humans, including a very promising Fabl inhibitor, (E)-N-methyl-N-((3-methylbenzofuran-2-yl)methyl)-3-(7-oxo-5,6,7,8-tetrahydro-l,8-naphthyridin-3-yl)acrylamide. This compound, however, has been found to be difficult or impracticable to formulate into acceptable oral and parenteral (e.g., intravenous or subcutaneous) formulations, and has marked insolubility, poor solution stability, and oral bioavailability. Much effort, over a decade or more, has been expended to design and synthesize an alternative compound that retains the significant inhibition of Fabl upon administration, but has improved physical and chemical characteristics that finally allow for practical oral and parenteral formulations. Up to now, no such compound has been identified that has adequate stability in the solid state, in aqueous solutions, together with excellent oral bioavailability that is necessary for oral and/or a parenteral administration, and is capable of being formulated into an oral and/or intravenous or intramuscular drug product using practical and commonly utilized methods of sterile formulation manufacture.
Debio-1452 is expected to have high potency against all drug-resistant phenotypes of staphylococci, including hospital and community-acquired MRSA.
Affinium obtained Debio-1452, also known as API-1252, through a licensing deal with GlaxoSmithKline. In 2014, Debiopharm acquired the product from Affinium.
In 2013, Qualified Infectious Disease Product designation was assigned to the compound for the treatment of acute bacterial skin and skin structure infections (ABSSSI).

SYNTHESIS

Heck coupling of 6-bromo-3,4-dihydro-1,8-naphthyridin-2-one with t-butyl acrylate in the presence of Pd(OAc)2, DIEA and P(o-tol)3 in propionitrile/DMF or acetonitrile/DMF affords naphthyridinyl-acrylate,
Whose t-butyl ester group is then cleaved using TFA in CH2Cl2 to furnish, after treatment with HCl in dioxane, 3-(7-oxo-6,8-dihydro-5H-1,8-naphthyridin-3-yl)acrylic acid hydrochloride
SEE BELOW………
Finally, coupling of acid with N-methyl-N-(3-methylbenzofuran-2-ylmethyl)amine using EDC, HOBt and DIEA in DMF provides the target AFN-1252

Preparation of N-methyl-N-(3-methylbenzofuran-2-ylmethyl)amine :

Chlorination of 3-methylbenzofuran-2-carboxylic acid with (COCl)2 and catalytic DMF, followed by condensation with CH3NH2 in CH2Cl2 yields the corresponding benzofuran-2-carboxamide,
Which is then reduced with LiAlH4 in THF to furnish N-methyl-N-(3-methylbenzofuran-2-ylmethyl)amine.
CONTD……..

Reduction of 2-aminonicotinic acid with LiAlH4 in THF gives (2-amino-3-pyridinyl)methanol ,
which upon bromination with Br2 in AcOH yields (2-amino-5-bromo-3-pyridinyl)methanol hydrobromide.
Substitution of alcohol with aqueous HBr at reflux provides the corresponding bromide,
which undergoes cyclocondensation with dimethyl malonate in the presence of NaH in DMF/THF to furnish methyl 6-bromo-2-oxo-1,2,3,4-tetrahydro-1,8-naphthyridine-3-carboxylate.
Hydrolysis of ester with NaOH in refluxing MeOH, followed by decarboxylation in refluxing HCl leads to 6-bromo-3,4-dihydro-1,8-naphthyridin-2-one
PATENT
US-20170088822

Aurigene Discovery Technologies Ltd

Novel co-crystalline polymorphic form of a binary enoyl-acyl carrier protein reductase (FabI) and FabI inhibitor ie AFN-1252. The FabI was isolated from Burkholderia pseudomallei (Bpm). The co-crystal is useful for identifying an inhibitor of FabI, which is useful for treating BpmFabI associated disease ie melioidosis. Appears to be the first patenting to be seen from Aurigene Discovery Technologies or its parent Dr Reddy’s that focuses on BpmFabI crystal; however, see WO2015071780, claiming alkylidine substituted heterocyclyl derivatives as FabI inhibitors, useful for treating bacterial infections. Aurigene was investigating FabI inhibitors, for treating infectious diseases, including bacterial infections such as MRSA infection, but its development had been presumed to have been discontinued since December 2015; however, publication of this application would suggest otherwise.
WO2015071780
PATENTS
US 20060142265
http://www.google.co.in/patents/US20060142265
PATENT
////////////Debio-1452, AFN 1252,AFN-1252, UNII-T3O718IKKM, API-1252, 620175-39-5, PRECLINICAL, Phase 2, Qualified Infectious Disease Product designation
CC1=C(OC2=CC=CC=C12)CN(C)C(=O)C=CC3=CC4=C(NC(=O)CC4)N=C3
Tradipitant, традипитант , تراديبيتانت , 曲地匹坦 ,

Tradipitant
VLY-686, LY686017
традипитант
تراديبيتانت [Arabic]
曲地匹坦 [Chinese]
- Molecular Formula C28H16ClF6N5O
- Average mass 587.903 Da
622370-35-8 CAS
Methanone, [2-[1-[[3,5-bis(trifluoromethyl)phenyl]methyl]-5-(4-pyridinyl)-1H-1,2,3-triazol-4-yl]-3-pyridinyl](2-chlorophenyl)-
(2-(1-(3,5-bis(trifluoromethyl)benzyl)-5-(pyridin-4-yl)-1H-1,2,3-triazol-4-yl)pyridin-3-yl)(2-chlorophenyl)methanone
[2-[1-[[3,5-bis(trifluoromethyl)phenyl]methyl]-5-(4-pyridinyl)-1H-1,2,3-triazol-4-yl]-3-pyridinyl](2-chlorophenyl)methanone
PHASE 2, Gastroparesis; Pruritus
FDA 2025, APPROVALS 2025, 12/30/2025, To treat vomiting associated with motion
pyridine-containing NK-1 receptor antagonist ie tradipitant, useful for treating anxiety, pruritus and alcoholism.
Vanda Pharmaceuticals, under license from Eli Lilly, was developing tradipitant, a NK1 antagonist, for treating anxiety disorder, pruritus and alcohol dependence. The company was also investigating the drug for treating gastroparesis. In February 2017, tradipitant was reported to be in phase 2 clinical development for treating anxiety and pruritus.
- Originator Eli Lilly
- Developer Eli Lilly; National Institute on Alcohol Abuse and Alcoholism; Vanda Pharmaceuticals
- Class Antipruritics; Anxiolytics; Chlorobenzenes; Pyridines; Small molecules; Triazoles
- Mechanism of Action Neurokinin 1 receptor antagonists; Substance P inhibitors
Highest Development Phases
- Phase II Gastroparesis; Pruritus
- Discontinued Alcoholism; Social phobia
- The drug had been in phase II clinical trials at Lilly and the National Institute on Alcohol Abuse and Alcoholism for the treatment of alcoholism; however, no recent development has been reported for this research.
- A phase II clinical trial for the treatment of social phobia has been completed by Lilly.
PATENT WO 2003091226
SYNTHESIS

Condensation of 2-chloropyridine with thiophenol in the presence of K2CO3 in DMF at 110ºC yields sulfide intermediate,
which is then oxidized by means of NaOCl in AcOH to give 2-(benzenesulfonyl)pyridine.
This is treated with (iPr)2NH and n-BuLi in THF at -60 to -70°C and subsequently couples with 2-chlorobenzaldehyde in THF at -60 to -70°C to furnish (2-(phenylsulfonyl)pyridin-3-yl)-(2-chlorophenyl)methanone.
Ketone couples with the enolate of 4-acetylpyridine (formed by treating 4-acetylpyridine (VII) with t-BuOK in DMSO) in the presence of LiOH in DMSO and subsequently is treated with PhCOOH in iPrOAc to give rise to pyridine benzoate derivative.
This finally couples with 1-azidomethyl-3,5-bistrifluoromethylbenzene (obtained by treating 3,5-bis(trifluoromethyl)benzylchloride with NaN3 ini DMSO) in the presence of K2CO3 in t-BuOH to afford the title compound Tradipitant.
Tradipitant (VLY-686 or LY686017) is an experimental drug that is a neurokinin 1 antagonist. It works by blocking substance P, a small signaling molecule. Originally, this compound was owned by Eli Lilly and named LY686017. VLY-686 was purchased by Vanda Pharmaceuticals from Eli Lilly and Company in 2012.[1] Vanda Pharmaceuticals is a U.S. pharmaceutical company that as of November 2015 only has 3 drugs in their product pipeline: tasimelteon, VLY-686, and iloperidone.[2]
Tachykinins are a family of peptides that are widely distributed in both the central and peripheral nervous systems. These peptides exert a number of biological effects through actions at tachykinin receptors. To date, three such receptors have been characterized, including the NK-1 , NK-2, and NK-3 subtypes of tachykinin receptor.
The role of the NK-1 receptor subtype in numerous disorders of the central nervous system and the periphery has been thoroughly demonstrated in the art. For instance, NK-1 receptors are believed to play a role in depression, anxiety, and central regulation of various autonomic, as well as cardiovascular and respiratory functions. NK- 1 receptors in the spinal cord are believed to play a role in pain transmission, especially the pain associated with migraine and arthritis. In the periphery, NK-1 receptor activation has been implicated in numerous disorders, including various inflammatory disorders, asthma, and disorders of the gastrointestinal and genitourinary tract.
There is an increasingly wide recognition that selective NK-1 receptor antagonists would prove useful in the treatment of many diseases of the central nervous system and the periphery. While many of these disorders are being treated by new medicines, there are still many shortcomings associated with existing treatments. For example, the newest class of anti-depressants, selective serotonin reuptake inhibitors (SSRIs), are increasingly prescribed for the treatment of depression; however, SSRIs have numerous side effects, including nausea, insomnia, anxiety, and sexual dysfunction. This could significantly affect patient compliance rate. As another example, current treatments for chemotherapy- induced nausea and emesis, such as the 5-HT3receptor antagonists, are ineffective in managing delayed emesis. The development of NK-1 receptor antagonists will therefore greatly enhance the ability to treat such disorders more effectively. Thus, the present invention provides a class of potent, non-peptide NK-1 receptor antagonists, compositions comprising these compounds, and methods of using the compounds.
Indications
Pruritus
It is being investigated by Vanda Pharmaceuticals for chronic pruritus (itchiness) in atopic dermatitis. In March 2015, Vanda announced positive results from a Phase II proof of concept study.[3] A proof of concept study is done in early stage clinical trials after there have been promising preclinical results. It provides preliminary evidence that the drug is active in humans and has some efficacy.[4]
Alcoholism
VLY-686 reduced alcohol craving in recently detoxified alcoholic patients as measured by the Alcohol Urge Questionnaire.[5] In a placebo controlled clinical trial of recently detoxified alcoholic patients, VLY-686 significantly reduced alcohol craving as measured by the Alcohol Urge Questionnaire. It also reduced the cortisol increase seen after a stress test compared to placebo. The dose given was 50 mg per day.
Social anxiety disorder
In a 12-week randomized trial of LY68017 in 189 patients with social anxiety disorder, 50 mg of LY68017 did not provide any statistically significant improvement over placebo.[6]
PATENT
WO03091226,
https://www.google.com/patents/WO2003091226A1?cl=en
PATENT
The compound {2-[l-(3,5-bis-trifluoromethyl-benzyl)-5-pyridin-4-yl-lH-[l,2,3]triazol-4-yl]- pyridin-3-yl}-(2-chlorophenyl)-methanone, depicted below as the compound of Formula I, was first described in PCT published application WO2003/091226.

(I)
Because the compound of Formula I is an antagonist of the NK-I subtype of tachykinin receptor, it is useful for the treatment of disorders associated with an excess of tachykinins. Such disorders include depression, including major depressive disorder; anxiety, including generalized anxiety disorder, panic disorder, obsessive compulsive disorder, and social phobia or social anxiety disorder; schizophrenia and other psychotic disorders, including bipolar disorder; neurodegenerative disorders such as dementia, including senile dementia of the Alzheimer’s type or Alzheimer’s disease; disorders of bladder function such as bladder detrusor hyper-reflexia and incontinence, including urge incontinence; emesis, including chemotherapy-induced nausea and acute or delayed emesis; pain or nociception; disorders associated with blood pressure, such as hypertension; disorders of blood flow caused by vasodilation and vasospastic diseases, such as angina, migraine, and Reynaud’s disease; hot flushes; acute and chronic obstructive airway diseases such as adult respiratory distress syndrome, bronchopneumonia, bronchospasm, chronic bronchitis, drivercough, and asthma; inflammatory diseases such as inflammatory bowel disease; gastrointestinal disorders or diseases associated with the neuronal control of viscera such as ulcerative colitis, Crohn’s disease, functional dyspepsia, and irritable bowel syndrome (including constipation-predominant, diarrhea- -?-
predominant, and mixed irritable bowel syndrome); and cutaneous diseases such as contact dermatitis, atopic dermatitis, urticaria, and other eczematoid dermatitis.
In PCT published application, WO2005/042515, novel crystalline forms of the compound of Formula I, identified as Form IV and Form V, are identified. Also described in WO2005/042515 is a process for preparation of the compound of Formula I, comprising reacting (2-chlorophenyl)-[2-(2- hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone or a phosphate salt thereof with l-azidomethyl-3,5- bistrifluoromethylbenzene in the presence of a suitable base and a solvent. Use of this procedure results in several shortcomings for synthesis on a commercial scale. For example, use of the solvent DMSO, with (2- chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone phosphate, requires a complex work-up that has a propensity to emulsify. This process also requires extraction with CH2CI2, the use of which is discouraged due to its potential as an occupational carcinogen, as well as the use of MgSC>4 and acid-washed carbon, which can generate large volumes of waste on a commercial scale. Conducting the reaction with (2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone in isopropyl alcohol, as also described in WO2005/042515, is also undesirable due to the need to incorporate a free base step. Furthermore, variable levels of residual l-azidomethyl-3,5-bistrifluoromethylbenzene, a known mutagen, are obtained from use of the procedures described in WO2005/042515.
An improved process for preparing the compound of Formula I would control the level of 1- azidomethyl-3,5-bistrifluoromethylbenzene impurity, and improve the yield. We have discovered that use of the novel salt, (2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone benzoate, as well as use of tert-butanol as the reaction solvent, improves reaction times and final yield, and decreases impurities in the final product. In addition, a novel process for the preparation of (2-chlorophenyl)- [2-(2- hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone benzoate, in which a pre-formed enolate of 4-acetyl pyridine is added to (2-phenylsulfonyl-pyridine-3-yl)-(2-chlorophenyl)methanone, results in an overall improved yield and improved purity, and is useful on a commercial scale.
EXAMPLES
Example 1 {2-[l-(3,5-bistrifluoromethylbenzyl)-5-pyridin-4-yl-lH-[l,2,3]triazol-4-yl]-pyridin-3-yl}-(2-chlorophenyl)- methanone (Form IV)

Suspend (2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl] methanone benzoate (204.7 g; 1.04 equiv; 445 mmoles) in t-butanol (614 mL) and treat the slurry with potassium carbonate (124.2 g; 898.6 mmoles). Heat to 7O0C with mechanical stirring for 1 hour. Add l-azidomethyl-3,5- bistrifluoromethylbenzene (115.6 g; 1.00 equiv; 429.4 mmoles) in a single portion, then heat the mixture to reflux. A circulating bath is used to maintain a condenser temperature of 3O0C. After 18 hours at reflux, HPLC reveals that the reaction is complete (<2% l-azidomethyl-3,5-bistrifluoromethylbenzene remaining). The mixture is cooled to 7O0C, isopropanol (818 mL) is added, then the mixture is stirred at 7O0C for 1 hour. The mixture is filtered, and the waste filter cake is rinsed with isopropanol (409 mL). The combined filtrate and washes are transferred to a reactor, and the mechanically stirred contents are heated to 7O0C. To the dark purple solution, water (1.84 L) is added slowly over 35 minutes. The solution is cooled to 6O0C, then stirred for 1 hour, during which time a thin precipitate forms. The mixture is slowly cooled to RT, then the solid is filtered, washed with 1 : 1 isopropanol/water (614 mL), subsequently washed with isopropanol (410 mL), then dried in vacuo at 450C to produce 200.3 g of crude {2-[l-(3,5- bistrifluoromethylbenzyl)-5-pyridin-4-yl-lH-[l,2,3]triazol-4-yl]-pyridin-3-yl}-(2-chlorophenyl)-methanone as a white solid. Crude {2-[l-(3,5-bistrifluoromethylbenzyl)-5-pyridin-4-yl-lH-[l,2,3]triazol-4-yl]-pyridin- 3-yl}-(2-chlorophenyl)-methanone (200.3 g) and isopropyl acetate (600 mL) are charged to a 5L 3-neck jacketed flask, then the contents heated to 750C. After dissolution is achieved, the vessel contents are cooled to 550C, then the solution polish filtered through a 5 micron filter, and the filter rinsed with a volume of isopropyl acetate (200 mL). After the polish filtration operation is complete, the filtrates are combined, and the vessel contents are adjusted to 5O0C. After stirring for at least 15 minutes at 5O0C, 0.21 grams of {2-[l-(3,5-bistrifluoromethylbenzyl)-5-pyridin-4-yl-lH-[l,2,3]triazol-4-yl]-pyridin-3-yl}-(2- chlorophenyl)-methanone Form IV seed (d90 = 40 microns) is added, and the mixture stirred at 5O0C for at least 2 h. Heptanes (1.90 L) are then added over at least 2 h. After the heptanes addition is completed, the slurry is stirred for an hour at 5O0C, cooled to 230C at a rate less then 2O0C per hour, then aged at 230C for an hour prior to isolation. The mixture is then filtered in portions through the bottom outlet valve in the reactor into a 600 mL filter. The resulting wetcake is washed portionwise with a solution containing heptanes (420 mL) and isopropyl acetate (180 mL), which is passed directly through the 5L crystallization vessel. The wetcake is blown dry for 5 minutes with nitrogen, then transferred to a 500 mL plastic bottle. The product is dried at 5O0C for 4 h. to produce 190.3g of pure {2-[l-(3,5- bistrifluoromethylbenzyl)-5-pyridin-4-yl-lH-[l,2,3]triazol-4-yl]-pyridin-3-yl}-(2-chlorophenyl)- methanone, Form IV in 75.0% yield with 100% purity, as determined by HPLC analysis. Particle size is reduced via pin or jet mill. 1H NMR (400 MHz, CDCl3): 5.46 (s, 2H); 7.19 (m, 5H); 7.36 (dd, IH, J = 4.9, 7.8); 7.45 (s, 2H); 7.59 (m, IH); 7.83 (s, IH); 7.93 (dd, IH, J = 1.5, 7.8); 8.56 (dd, IH, J= 1.5, 4.9); 8.70 (d, 2H, J= 5.9).
Preparation 1-A (2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone benzoate Charge powdered KOfBu (221.1 g, 1.93 moles, 1.40 eq.) to Reactor A, then charge DMSO (2 L) at
250C over 10 min. The KOfBu/DMSO solution is stirred for 30 min at 230C, then a solution of 4-acetyl pyridine (92 mL, 2.07 moles, 1.50 eq) in DMSO (250 mL) is prepared in reactor B. The contents of reactor B are added to Reactor A over 10 minutes, then the Reactor A enolate solution is stirred at 230C for Ih. In a separate 12-L flask (Reactor C), solid LiOH (84.26 g, 3.45 moles, 2.0 eq) is poured into a mixture of (2- phenylsulfonyl-pyridin-3-yl)-(2-chlorophenyl)methanone (500.0 g, 1.34 moles, 1.0 eq) and DMSO (2L), with stirring, at 230C. The enolate solution in reactor A is then added to Reactor C over a period of at least 15 minutes, and the red suspension warmed to 4O0C. The reaction is stirred for 3h, after which time HPLC analysis reveals less than 2% (2-phenylsulfonyl-pyridin-3-yl)-(2-chlorophenyl)methanone. Toluene (2.5 L) is charged, and the reactor temperature cooled to 3O0C. The mixture is quenched by addition of glacial acetic acid (316 mL, 5.52 moles, 4.0 eq), followed by 10 % NaCl (2.5 L). The biphasic mixture is transferred to a 22-L bottom-outlet Morton flask, and the aqueous layer is removed. The aqueous layer is then extracted with toluene (750 mL). The combined organic layers are washed with 10 % NaCl (750 mL), then concentrated to 4 volumes and transferred to a 12-L Morton flask and rinsed with isopropyl acetate (4 vol, 2 L). The opaque amber solution is warmed to 75 degrees to 750C over 40 min. Benzoic acid (171. Ig, 1.34 moles, 1.0 eq) is dissolved in hot isopropyl acetate (1.5 L), and charged to the crude free base solution over at least 30 min. The crude solution containing benzoate salt is stirred for 0.5 h at 750C then cooled to 23 0C. When solids are first observed, the cooling is stopped and the mixture is aged for an hour at the temperature at which crystals are first observed. Alternatively, if seed crystal is available, the mixture may be seeded with (2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone benzoate (2.25g) at 750C, followed by stirring for 0.5 h at 750C, then cooling to 230C over at least 1.5 h. The mixture is then cooled to <5 0C, then filtered through paper on a 24cm single-plate filter. The filtercake is then rinsed with cold z‘-PrOAc (750 mL) to produce granular crystals of bright orange-red color. The wet solid is dried at 550C to produce 527.3 g (83% yield) with 99.9% purity. (2-chlorophenyl)-[2-(2-hydroxy-2- pyridin-4-yl-vinyl)pyridin-3-yl]methanone benzoate. Anal. Calcd. for C26Hi9N2ClO4: C, 68.05; H, 4.17; N, 7.13. Found: C, 67.89; H, 4.15; N 6.05. HRMS: calcd for C19H13ClN2O2, 336.0666; found 336.0673.
The synthesis of(2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone benzoate proceeds optimally when the potassium enolate of 4-acetyl pyridine is pre-formed using KOfBu in DMSO. Pre-formation of the enolate allows the SNAR (nucleophilic aromatic substitution) reaction to be performed between room temperature and 4O0C, which minimizes the amount of degradation. Under these conditions, the SNAR is highly regioselective, resulting in a ratio of approximately 95:5 preferential C – acylation. In all cases, less polar solvents such as THF or toluene, or co-solvents of these solvents mixed with DMSO, results in a substantial increase of acylation at the oxygen in the SNAR, and leads to a lower yield of product. This is a substantial improvement over the procedures described in WO2005/042515 for synthesis of the free base or the phosphate salt, in which the SNAR is performed at 60-700C, resulting in a substantial increase in chemical impurity. Using the conditions described in WO2005/042515, when scaled to 2kg, results in maximum yields of 55%, with sub-optimal potency. In comparison, the improved conditions described herein can be run reproducibly from 0.4 to 2kg scale to give yields of 77-83%, with >99% purity. In addition, the reaction can be held overnight at 4O0C with minimal degradation, whereas holding the reaction for 1 h past completion at 60-70°C results in substantial aromatized impurity. The reaction may also be performed using sodium tert-amylate as the base, in combination with an aprotic solvent, such as DMSO or DMF.
The title compound exists as a mixture of tautomers and geometric isomers. It is understood that each of these forms is encompassed within the scope of the invention.

Preparation 1-B
(2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone toluate The procedure described in Preparation 1-A is followed, with the following exception. Solid toluic acid (1.0 eq) is added to the crude free base solution at 550C, then the solution cooled to 45 0C. The solution is stirred for one hour at 45 0C, then slowly cooled to 23 0C. When solids are first observed, the cooling is stopped and the mixture is aged for an hour at the temperature at which crystals are first observed. Alternatively, if seed crystal is available, the mixture may be seeded, aged for 3 h at 450C , then cooled to O0C over 4 h. The isolation slurry is filtered, and the wetcake washed with MeOH (3 volumes). The wetcake is dried at 5O0C to provide 14.0 g (76.4%) of (2-chlorophenyl)-[2-(2-hydroxy-2-pyridin-4-yl- vinyl)pyridin-3-yl]methanone toluate as a light red powder.
As with the benzoate salt, the toluate salt can also exist as a mixture of tautomers and geometric isomers, each of which is encompassed within the scope of the invention. (2-chlorophenyl)-[2-(2-hydroxy- 2-pyridin-4-yl-vinyl)pyridin-3-yl]methanone toluate . 13C NMR (125 MHz,DMS0-d6) δ 194.5, 167.8, 167.4, 155.5, 150.7 (2C), 147.4, 144.0, 143.4, 142.7, 138.6, 133.0, 130.8, 130.7, 130.5, 129.8(2C), 129.5(2C), 128.5, 128.0, 127.9, 119.9 (2C), 118.6, 92.6, 21.5.
Preparation 1-C
(2-phenylsulfonyl-pyridin-3-yl)-(2-chlorophenyl)methanone
A solution of 1.3 eq of diisopropylamine (based on 2-benzenesulfonyl pyridine) in 5 volumes of THF in a mechanically stirred 3 -necked flask is cooled to -70 to -75 0C. To this solution is added 1.05 eq of w-butyllithium (1.6M in hexanes) at such a rate as to maintain the temperature below -6O0C. The light yellow solution is stirred at -60 to -70 0C for 30 minutes. Once the temperature has cooled back down to – 60 to -650C, 1.0 eq of 2-benzene-sulfonyl pyridine, as a solution in 3 volumes of THF, is added at the fastest rate that will maintain the reaction temperature under -6O0C. A yellow suspension forms during the addition that becomes yellow-orange upon longer stirring. This mixture is stirred for 3 hours at -60 to – 750C, and then 1.06 eq of 2-chlorobenzaldehyde, as a solution in 1 volume of THF, is added dropwise at a sufficient rate to keep the temperature under -55 0C. The suspension gradually turns orange-red, thins out, and then becomes a clear red solution. The reaction mixture is allowed to stir at -60 to -7O0C for 1 hour, 3N aqueous HCl (7 volumes) is added over 20-30 minutes, and the temperature is allowed to exotherm to 0-100C. The color largely disappears, leaving a biphasic yellow solution. The solution is warmed to at least 1O0C, the layers are separated, and the aqueous layer is back-extracted with 10 volumes of ethyl acetate. The combined organic layers are washed with 10 volumes of saturated sodium bicarbonate solution and concentrated to about 2 volumes. Ethyl acetate (10 volumes) is added, and the solution is once again concentrated to 2 volumes. The thick solution is allowed to stand overnight and is taken to the next step with no purification of the crude alcohol intermediate. The crude alcohol intermediate is transferred to a 3 -necked flask with enough ethyl acetate to make the total solution about 10 volumes. The yellow solution is treated with 3.2 volumes of 10% aqueous (w/w) potassium bromide, followed by 0.07 eq of 2,2,6,6-Tetramethylpiperidine-N-oxide (TEMPO). The orange mixture is cooled to 0-50C and treated with a solution of 1.25 eq of sodium bicarbonate in 12% w/w sodium hypochlorite (9 volumes) and 5 volumes of water over 30-60 minutes while allowing the temperature to exotherm to a maximum of 2O0C. The mixture turns dark brown during the addition, but becomes yellow, and a thick precipitate forms. The biphasic light yellow mixture is allowed to stir at ambient temperature for 1-3 hours, at which time the reaction is generally completed. The biphasic mixture is cooled to 0-50C and stirred for 3 hours at that temperature. The solid is filtered off, washed with 4 volumes of cold ethyl acetate, followed by 4 volumes of water, and dried in vacuo at 450C to constant weight. Typical yield is 80-83% with a purity of greater than 98%. 1H NMR (600 MHz, CDCl3-^) δ ppm 7.38 (td, ./=7.52, 1.28 Hz, 1 H) 7.47 (dd, ./=7.80, 1.30 Hz, 1 H) 7.51 (td, ./=7.79, 1.60 Hz, 1 H) 7.51 (t, ./=7.89 Hz, 2 H) 7.50 – 7.54 (m, J=7.75, 4.63 Hz, 1 H) 7.60 (t, J=7.43 Hz, 1 H) 7.73 (dd, J=7.75, 1.60 Hz, 1 H) 7.81 (dd, J=7.79, 1.56 Hz, 1 H) 8.00 (dd, ./=8.44, 1.10 Hz, 2 H) 8.76 (dd, ./=4.63, 1.61 Hz, 1 H).
Preparation 1-D 1 -azidomethyl-3,5-bistrifluoromethyl-benzene
Sodium azide (74.3 g, 1.14 mol) is suspended in water (125 mL), then DMSO (625 mL) is added. After stirring for 30 minutes, a solution consisting of 3,5-Bis(trifluoromethyl)benzyl chloride (255.3 g, 0.97 moles) and DMSO (500 mL) is added over 30 minutes. (The 3,5-Bis(trifluoromethyl)benzyl chloride is heated to 350C to liquefy prior to dispensing (MP = 30-320C)). The benzyl chloride feed vessel is rinsed with DMSO (50 mL) into the sodium azide solution, the mixture is heated to 4O0C, and then maintained for an hour at 4O0C, then cooled to 230C.
In Process Analysis: A drop of the reaction mixture is dissolved in d6-DMSO and the relative intensities of the methylene signals are integrated (NMR verified as a 0.35% limit test for 3,5- Bis(trifluoromethyl)benzyl Chloride). Work-up: After the mixture reaches 230C , it is diluted with heptanes (1500 mL), then water (1000 mL) is added, and the mixture exotherms to 350C against a jacket setpoint of 230C. The aqueous layer is removed (-2200 mL), then the organic layer (approximately 1700 mL) is washed with water (2 X 750 mL). The combined aqueous layers (-3700 mL) are analyzed and discarded.
The solvent is then partially removed via vacuum distillation with a jacket set point of 850C, pot temperature of 60-650C and distillate head temperature of 50-550C to produce 485g (94.5% yield) of 51 Wt% solution title compound as a clear liquid. Heptanes can be either further removed by vacuum distillation or wiped film evaporation technology. 1H NMR (400 MHz, CDCl3): 4.58 (s, 2H); 7.81 (s, 2H); 7.90 (s, IH).
Preparation 1-E 2-benzene-sulfonyl pyridine Charge 2-chloropyridine (75 mL, 790 mmol), thiophenol (90 mL, 852 mmol), and DMF (450 mL) to a 2L flask. Add K2CO3 (134.6 g, 962 mmol), then heat to HO0C and stir for 18 hours. Filter the mixture, then rinse the waste cake with DMF (195 mL). The combined crude sulfide solution and rinses are transferred to a 5-L flask, and the waste filtercake is discarded. Glacial acetic acid (57 mL, 995 mmol) is added to the filtrate, then the solution is heated to 4O0C, and 13 wt % NaOCl solution (850 mL, 1.7 mol) is added over 2 hours. After the reaction is complete, water (150 mL) is added, then the pH of the mixture adjusted to 9 with 20 % (w/v) NaOH solution (250 mL). The resulting slurry is cooled to <5 0C, stirred for 1.5 h, then filtered, and the cake washed with water (3 x 200 mL). The product wetcake is dried in a 550C vacuum oven to provide 2-benzene-sulfonyl pyridine (149 g, 676 mmol) in 86 % yield: 1H NMR (500 MHz, CDCl3) δ 8.66 (d, J = 5.5 Hz, IH), 8.19 (d, J = 1.1 Hz, IH), 8.05 (m, 2H), 7.92 (ddd, J= 9.3, 7.7, 1.6 Hz, IH), 7.60 (m, IH), 7.54 (m, 2H), 7.44 (m, IH); IR (KBr) 788, 984, 1124, 1166, 1306, 1424, 1446, 1575, 3085 cm“1; MS (TOF) mlz 220.0439 (220.0427 calcd for C11H10NO2S, MH); Anal, calcd for C11H9NO2S: C, 60.26; H, 4.14; N, 6.39; S, 14.62. Found: C, 60.40; H, 4.02; N, 6.40; S, 14.76.
As noted above, use of the improved process of the present invention results in an improved habit of the crystalline Form IV compound of Formula I. The improved habit reduces surface area of the crystal, improves the filtration, and washing, and improves the efficiency of azide mutagen rejection. These improvements are described in greater detail below.
In patent application WO2005/042515, the polish filtration is carried out in 7 volumes (L/kg) of isopropanol near its boiling point (65-83 0C), a process that is difficult and hazardous to execute in commercial manufacturing because of the high risk of crystallization on the filter and/or vessel transfer lines due to supersaturation. In the preferred crystallization solvent, isopropyl acetate, the polish filtration is conducted in four volumes of isopropyl acetate at temperatures from 45 to 55 0C. This temperature range is 35 to 45 0C lower than the boiling point of isopropyl acetate, which provides a key safety advantage.
PATENT
PATENT
WO 2017031215
EXAMPLES
Example 1: Preparation of Compound (I) via Negishi Coupling Route
Example 1 provides a scheme including preparations 1A-1D, described below, for the synthesis of the compound of Formula (I) and intermediates used in the route. An overview of the scheme is as follows:

80 on ma s ale
Example 1A: Preparation of Compound (I)

Zinc dust (200 mg, 3.06 mmol) combined with 2.0 mL of dimethylformamide was treated with 0.010 mL of 1,2-dibromoethane and heated to 65°C for 3 minutes. The mixture was cooled to ambient temperature and treated with 0.010 mL of trimethylsilyl chloride. After 5 minutes, 1.26 mL of 1M zinc chloride in diethyl ether was added to the mixture followed by Compound (Ila) (600 mg, 1.20 mmol). The mixture was heated to 65°C and further treated with 0.020 mL each of 1,2-dibromoethane and trimethylsilyl chloride. After 2.5 hours, via HPLC chromatogram, the reaction showed some formation of the zincate and was allowed to stir at ambient temperature for 16 hours. At this time
tetrakis(triphenylphosphine)palladium(0) (70 mg, 0.06 mmol), Compound (Ilia) (357 mg, 1.20 mmol) were added to the reaction and the mixture heated to 65°C. HPLC analysis showed the formation of Compound (I) in the reaction.
IB: Preparation of Comp

To a solution of Compound (IV) (8.00 g, 18 mmol) in 40 mL of 1,2-dichloroethane was added a solution of iodine monochloride (10.7 g, 65.9 mmol) in 40 mL of 1,2-dichloroethane resulting in a slurry. The slurry was heated to 75°C for 4 hours then cooled to ambient temperature. The solids were collected by filtration, washed with heptane, then combined with 90 mL of ethyl acetate and 80 mL of saturated sodium thiosulfate solution. The organic phase was washed with saturated sodium chloride solution and dried with sodium sulfate. The mixture was concentrated to yield 7.80 g (87%) of Compound (Ila) as a yellow solid. The product could be further purified by silica gel chromatography. Thus 2.0 g of yellow solid was dissolved in dichloromethane and charged onto a silica gel column. The product was eluted using tert-butyl methyl ether to provide 1.87 g (93% recovery) of Compound (Ila) as a white powder. Analytical data: Iodine monochloride complex: ¾ NMR (500 MHz, DMSO-de) δ 8.80 (2 H), 8.05 (1 H), 7.77 (2 H), 7.59 (2 H), 5.86 (2 H).
Uncomplexed: ¾ NMR (500 MHz, DMSO-de) δ 8.71 (2 H), 8.03 (1 H), 7.74 (2 H), 7.44 (2 H), 5.86 (2 H).
It was observed that the iodination proceeded smoothly as a suspension in 1,2-dichloroethane with IC1 (4.0 equiv) at 75°C. An ICl-Compound (Ila) complex was initially isolated by filtration. Compound (Ila) was then obtained in approximately 85% yield by treatment of the ICl-Compound (Ila) complex with sodium thiosulfate. This protocol provided a viable means of isolation of Compound (Ila) without the use of DMF.
Example 1C: Preparation of silyl substituted triazole (Compound IV)

A mixture of Compound (V) (8.07 g, 30.0 mmol) and Compound (VI) (5.12 g, 29.2 mmol) was heated to 100°C for 18 hours. To the mixture was added 40 mL of heptane and the reaction was allowed to cool with rapid stirring. After 1 hour the solids were collected by filtration and washed with heptane then dried to 9.30 g (72%) of Compound (IV) as a tan solid. Analytical data: ¾ NMR (500 MHz, DMSO-de) δ 8.66 (2 H), 8.04 (1 H), 7.67 (2 H), 7.32 (2 H), 5.72 (2 H), 0.08 (9 H).
It was further found that combining Compound (V) and Compound (VI) (neat) and heating at 95 – 105°C afforded a 92: 8 mixture of regioisomers as shown below:

Crystallization of the mixture from heptane afforded Compound (IV) in 62-72% yield, thus obviating the need for chromatography to isolate Compound (IV).
Example ID: Preparation of starting material Compound (VI)

Zinc bromide (502 g, 2.23 mole) was added in approximately 100 g portions to 2.0 L of tetrahydrofuran cooled to between 0 and 10°C. To this cooled solution was added 4-bromopyridine hydrochloride (200 g, 1.02 mol), triphenylphosphine (54 g, 0.206 mol), and palladium (II) chloride (9.00 g, 0.0508 mol). Triethylamine (813 g, 8.03 mol) was then added at a rate to maintain the reaction temperature at less than 10°C, and finally
trimethylsilylacetylene (202 g, 2.05 mol) was added. The mixture was heated to 60°C for 4.5 hours. The reaction was cooled to -5°C and combined with 2.0 L of hexanes and treated with 2 L of 7.4 M NH4OH. Some solids were formed and were removed as much as possible with the aqueous phase. The organic phase was again washed with 2.0 L of 7.4 M NH4OH, followed by 2 washes with 500 mL of water, neutralized with 1.7 L of 3 M hydrochloric acid, dried with sodium sulfate, and concentrate to a thick slurry. The slurry was combined with 1.0 L of hexanes to give a precipitate. The precipitate was removed by filtration and the filtrate was concentrated to 209 g of dark oil. The product was purified by distillation (0.2 torr, 68°C) to give 172 g (96%) of Compound (VI) as colorless oil. Analytical data: ¾ NMR (500 MHz, DMDO-de) δ 8.57 (2 H), 7.40 (2 H), 0.23 (9 H).
EXAMPLE 2 – Preparation of Compound (Ilia)
Example 2 provides a morpholine amide route for the synthesis of Compound (Ilia). In this approach, morpholine amide (Compound VII) was prepared from 2-chlorobenzoyl chloride (Preparation 2A). Metallation of 2-bromopyridine with LDA (1.09 equiv.) in THF at -70°C followed by addition of (Compound VII) afforded Compound (Ilia) in 37% yield after crystallization from IP A/heptane (Preparation 2B). This sequence provides a direct route to Compound (Ilia), and a means to isolate Compound (Ilia) without the use of
chromatography. Compound (Ilia) may then be used to form Compound (I) as shown in Example 1A above (Preparation 2C).
Preparation 2A: Preparation of Compound (VII)

Toluene (1.5 L) was added to Compound (IX) (150 g, 0.86 mol) and cooled to 10°C. Morpholine (82 mL, 0.94 mol) was added to the clear solution over 10 minutes. The resulting white slurry was stirred for 20 minutes then pyridine (92 mL, 1.2 mol) was added dropwise over 20 minutes. The cloudy white mixture was stirred in a cold bath for 1 hour. Water (600 mL) was added in a single portion and the cold bath removed. The mixture was stirred for 20 minutes and the layers are separated. The organic layer was washed with a mixture of 1 N HC1 and water (2: 1, 500 mL:250 mL). The pH of the aqueous layer was ~ 2. The organic layer was washed with a mixture of saturated NaHCCb and water (1 : 1, 100 mL: 100 mL). The pH of the aqueous layer was ~ 9. The layers were separated. The organic layer was concentrated in vacuo to an oil. The oil was dissolved in IPA (70 mL) and heated at 60°C for 30 min. The clear solution was allowed to cool to 30°C, then heptane (700 mL, 4.7 v) was added dropwise. The resulting slurry was stirred at RT for 2 hours then cooled to 0°C for 1 hour. The slurry was filtered at RT, washed with heptane then dried under vacuum at 30°C overnight. Compound (VII) (156.2 g, 81%) was obtained as a white solid. Analytical data: ¾ NMR (500 MHz, CDCh) δ 7.42-7.40 (m, 1 H), 7.35-7.29 (m, 3 H), 3.91-3.87 (m, 1 H), 3.80-3.76 (m, 3 H), 3.71 (ddd, J= 11.5, 6.8, 3.3 Hz, 1 H), 3.60 (ddd, J = 11.2, 6.4, 3.4 Hz, 1 H), 3.28 (ddd, J= 13.4, 6.3, 3.2 Hz, 1 H), 3.22 (ddd, J= 13.7, 6.8, 3.3 Hz, 1 H); LRMS (ES+) calcd for CnHi3F6ClN02 (M+H)+ 226.1, found 225.9 m/z.
Preparation 2B: Preparation of Compound (Ilia)

THF (75 mL) was added to diisopropyl amine (4.9 mL, 34.8 mmol) and cooled to a
temperature of -70°C under N2 atmosphere. 2.5 M w-BuLi in hexanes (13.9 mL, 34.8 mmol) was added in a single portion (a 30-40°C exotherm) to the clear solution and cooled back to -70°C. Compound (VIII) (5.0 g, 31.6 mmol) was added neat to the LDA solution (a 2 to 5°C exotherm) followed by a THF (10 mL) rinse, keeping T< -65°C. This clear yellow solution was stirred at -70°C for 15 min. Compound (VII) (7.1 g, 31.6 mmol) in THF (30 mL) was added keeping T< -65°C. The resulting clear orange solution was stirred at -70°C for 3 hours. MeOH (3 mL) was added to quench reaction mixture and the cold bath was removed. 5 N HC1 (25 mL) was added to the reaction solution. MTBE (25 mL) was added, and the layers were separated. The organic layer was washed with water (25 mL X 2). The organic layer was dried over MgS04 and filtered. The organic layer was concentrated in vacuo to an orange oil. The oil was dissolved in IPA (15 mL, 3 vol) at ambient temperature. Heptane (25 mL) was added dropwise and the resulting slurry was stirred at RT for 1 hour. The slurry was cooled to 0°C for 1 hour and filtered. The filter cake was rinsed with chilled heptane (20 mL) and dried under vacuum at 30°C overnight. Compound (Ilia) (4.25 g, 45%) was obtained as a yellow solid.
Several reactions were run at different temperatures and with different addition rates of Compound (VII). If the reaction temperature was maintained below -65°C and Compound (VII) was added in <5 min, it was found that the reaction worked well. If the temperature was increased and/or the addition time of Compound (VII) was increased, then yields suffered, and the work-up was complicated by emulsions.
Preparation 2C: Preparation of Compound (I)
Compound (Ilia) may then reacted with Compound (Ila) to produce Compound (I) as shown in Preparation 1A.
EXAMPLE 3
Example 3 describes a new route for the synthesis of an intermediate free base, which may be used to form Compound (I) as described further below.
Example 3A: Preparation of starting material (Compound X) from 2-Chloronicotinonitrile

A mixture of NaH (40.0 g, 1 mol, 60% dispersion in mineral oil) and 2-chloronicotinonitrile (69.3 g, 500 mmol) in THF (1 L) was heated to reflux. A solution of 4-acetylpyridine (60.6 g, 500 mmol) in THF (400 mL) was added over a period of 40 min. The resulting dark brown mixture was stirred at reflux for ~ 2 h. The heating mantle was then removed, and AcOH (58 mL, 1 mol) was added. EtOAc (1 L) and H2O (1 L) were then added, and the layers were separated. The organic layer was concentrated to afford an oily solid. CH3CN (500 mL) was added, and the mixture was stirred for 30 min. H2O (1 L) was then added. The mixture was stirred for 1 h then filtered. The solid was rinsed with 2: 1
CH3CN-H2O (900 mL) and hexanes (400 mL) then dried under vacuum at 45°C overnight to afford 61.4 g (55% yield) of Compound (X) as yellow solid. Compound (X) exists as an approximate 95:5 enol-ketone mixture in CDCI3. Analytical data for enol: IR (CHCI3): 3024, 2973, 2229, 1631, 1597, 1579, 1550, 1497; ¾ NMR (500 MHz, CDCI3) δ 8.69 (dd, J= 4.4,
1.7 Hz, 2H), 8.55 (dd, J = 5.2, 1.8 Hz, 1H), 7.97 (dd, J= 7.9, 1.8 Hz, 1H), 7.70 (dd, J= 4.6, 1.5 Hz, 2H, 7.17 (dd, J = 7.8, 5.0 Hz, 1H), 6.59 (s, 1H); LRMS (ES+) calcd for C13H10N3O (M+H)+ 224.1, found 224.0 m/z.
Preparation 3B: Preparation of Compound (XI)
Preparation 3B(1):

(X) (XI)
Compound (XI) may be prepared using Compound (X).
Preparation 3B(2):
Alternatively, the following procedure for the conversion of nitrile into an acid which may also yield compound (XI). A mixture of Compound (X) (1 eq) and NaOH (1.5 eq) in 1 : 1 fhO-EtOH (3.5 mL/g of Compound (X)) was heated at 65°C overnight. The reaction mixture was cooled to RT then added to CH2C12 (12.5 mL/g of Compound (X)) and H20 (12.5 mL/g of Compound (X)). Cone. HC1 (2.5 mL/g of Compound (X)) was then added, and the layers were separated. The aqueous layer was extracted with CH2CI2 (10 mL/g of Compound (X)). The combined organic extracts were washed with H2O (12.5 ml/g of Compound (X)), dried (MgS04), filtered and concentrated to afford Compound (XI).
Preparation 3C
Compound Compound (XI) may then be converted into a Stage C intermediate free base, with observed 87% conversion in Grignard reaction as shown above. A complete synthesis route for Com ound (I) starting from compound Compound (XI) is depicted below.

Detailed experimental procedures for the synthesis of benzoate salt and final step are given in
International Patent Application Publication WO 2008/079600 Al .
References
- “Company Overview of Eli Lilly & Co., Worldwide License to Develop and Commercialize VLY-686”. Bloomberg Business. Retrieved 16 November 2015.
- [1]
- “Vanda Pharmaceuticals Announces Tradipitant Phase II Proof of Concept Study Results for Chronic Pruritus in Atopic Dermatitis”. PR Newswire. Retrieved 16 November 2015.
- Schmidt, B (2006). “Proof of principle studies”. Epilepsy Res. 68 (1): 48–52. doi:10.1016/j.eplepsyres.2005.09.019. PMID 16377153.
- George, DT; Gilman, J; Hersh, J; et al. (2008). “Neurokinin 1 receptor antagonism as a possible therapy for alcoholism.”. Science. 6: 1536–1539. doi:10.2147/SAR.S70350. PMC 4567173. PMID 26379454.
- Tauscher, J; Kielbasa, W; Iyengar, S; et al. (2010). “Development of the 2nd generation neurokinin-1 receptor antagonist LY686017 for social anxiety disorder”. European Neuropsychopharmacology. 20 (2): 80–87. doi:10.1016/j.euroneuro.2009.10.005. PMID 20018493.
George, D.T.; Gilman, J.; Hersh, J.; Thorsell, A.; Herion, D.; Geyer, C.; Peng, X.; Kielbasa, W.; Rawlings, R.; Brandt, J.E.; Gehlert, D.R.; Tauscher, J.T.; Hunt, S.P.; Hommer, D.; Heilig, M. Neurokinin 1 receptor antagonism as a possible therapy for alcoholism, Science 2008, 319(5869): 1536
Gackenheimer, S.L.; Gehlert, D.R.In vitro and in vivo autoradiography of the NK-1 antagonist (3H)-LY686017 in guinea pig brain39th Annu Meet Soc Neurosci (October 17-21, Chicago) 2009, Abst 418.16
Tonnoscj, K.; Zopey, R.; Labus, J.S.; Naliboff, B.D.; Mayer, E.A.
The effect of chronic neurokinin-1 receptor antagonism on sympathetic nervous system activity in irritable bowel syndrome (IBS) Dig Dis Week (DDW) (May 30-June 4, Chicago) 2009, Abst T1261
Kopach, M.E.; Kobierski, M.E.; Coffey, D.S.; et al.
Process development and pilot-plant synthesis of (2-chlorophenyl)[2-(phenylsulfonyl)pyridin-3-yl]methanone
Org Process Res Dev 2010, 14(5): 1229
| Patent ID | Patent Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2016060250 | NOVEL INTERMEDIATE AND PROCESS USEFUL IN THE PREPARATION OF -(2-CHLOROPHENYL)-METHANONE | 2015-11-10 | 2016-03-03 |
| US2015320866 | PHARMACEUTICAL COMPOSITION COMPRISING ANTIEMETIC COMPOUNDS AND POLYORTHOESTER | 2013-12-13 | 2015-11-12 |
| US2014206877 | NOVEL INTERMEDIATE AND PROCESS USEFUL IN THE PREPARATION OF -(2-CHLOROPHENYL)-METHANONE | 2014-03-27 | 2014-07-24 |
| US2012225904 | New 7-Phenyl-[1, 2, 4]triazolo[4, 3-a]Pyridin-3(2H)-One Derivatives | 2010-11-09 | 2012-09-06 |
| US2010056795 | NOVEL INTERMEDIATE AND PROCESS USEFUL IN THE PREPARATION OF -(2-CHLOROPHENYL)-METHANONE | 2010-03-04 | |
| US7381826 | Crystalline forms of {2-[1-(3, 5-bis-trifluoromethyl-benzyl)-5-pyridin-4-yl-1H-[1, 2, 3]triazol-4-yl]-pyridin-3-yl}-(2-chlorophenyl)-methanone | 2007-04-05 | 2008-06-03 |
| US7320994 | Triazole derivatives as tachykinin receptor antagonists | 2005-10-27 | 2008-01-22 |
 |
|
| Legal status | |
|---|---|
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| Chemical and physical data | |
| Formula | C28H16ClF6N5O |
| Molar mass | 587.90 g/mol |
| 3D model (Jmol) | |
Tradipitant
Tradipitant is being evaluated in a Phase II study in treatment resistant pruritus in atopic dermatitis.
Tradipitant is an NK-1 receptor antagonist licensed from Eli Lilly in 2012. Tradipitant has demonstrated proof-of-concept in alcohol dependence in a study published by the NIH1. In that study tradipitant was shown to reduce alcohol cravings and voluntary alcohol consumption among patients with alcohol dependence. NK-1R antagonists have been evaluated in a number of indications including chemotherapy-induced nausea and vomiting (CINV), post-operative nausea and vomiting (PONV), alcohol dependence, anxiety, depression, and pruritus.
The NK-1R is expressed throughout different tissues of the body, with major activity found in neuronal tissue. Substance P (SP) and NK-1R interactions in neuronal tissue regulate neurogenic inflammation locally and the pain perception pathway through the central nervous system. Other tissues, including endothelial cells and immune cells, have also exhibited SP and NK-1R activity2. The activation of NK-1R by the natural ligand SP is involved in numerous physiological processes, including the perception of pain, behavioral stressors, cravings, and the processes of nausea and vomiting1,2,3. An inappropriate over-expression of SP either in nervous tissue or peripherally could result in pathological conditions such as substance dependence, anxiety, nausea/vomiting, and pruritus1,2,3,4. An NK-1R antagonist may possess the ability to reduce this over-stimulation of the NK-1R, and as a result address the underlying pathophysiology of the symptoms in these conditions.
References
- George DT, Gilman J, Hersh J, Thorsell A, Herion D, Geyer C, Peng X, Keilbasa W, Rawlings R, Brandt JE, Gehlert DR, Tauscher JT, Hunt SP, Hommer D, Heilig M. Neurokinin 1 receptor antagonism as a possible therapy for alcoholism. Science. 2008; 319(5869):1536-9
- Almeida TA, Rojo J, Nieto PM, Pinto FM, Hernandez M, et al. Tachykinins and tachykinin receptors: structure and activity relationships. Current Medicinal Chemistry. 2004;11:2045-2081.
- Hargreaves R, Ferreira JC, Hughes D, Brands J, Hale J, Mattson B, Mill S. Development of aprepitant, the first neurokinin-1 receptor antagonist for the prevention of chemotherapy-induced nausea and vomiting. Annals of the New York Academy of Sciences. 2011; 1222:40-48.
- Stander S, Weisshaar E, Luger A. Neurophysiological and neurochemical basis of modern pruritus treatment. Experimental Dermatology. 2007;17:161-69.
///////////////////tradipitant, PHASE 2, VLY-686, LY686017, традипитант , تراديبيتانت , 曲地匹坦 , VANDA, ELI LILLY, Gastroparesis Pruritus, FDA 2025, APPROVALS 2025, vomiting associated with motion