
Home » Phase2 drugs (Page 13)
Category Archives: Phase2 drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
BMS 911543

BMS 911543
N,N-dicyclopropyl-4-((1,5-dimethyl-1H-pyrazol-3-yl)amino)-6-ethyl-1-methyl-1,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridine-7-carboxamide
cas 1271022-90-2
Chemical Formula: C23H28N8O
Exact Mass: 432.23861
UNII-7N03P021J8;
N,N-dicyclopropyl-4-((1,5-dimethyl-1H-pyrazol-3-yl)amino)-6-ethyl-1-methyl-1,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridine-7-carboxamide
Bristol-Myers Squibb Company innovator
BMS-911543 is an orally available small molecule targeting a subset of Janus-associated kinase (JAK) with potential antineoplastic activity. JAK2 inhibitor BMS-911543 selectively inhibits JAK2, thereby preventing the JAK/STAT (signal transducer and activator of transcription) signaling cascade, including activation of STAT3. This may lead to an induction of tumor cell apoptosis and a decrease in cellular proliferation. JAK2, often upregulated or mutated in a variety of cancer cells, mediates STAT3 activation and plays a key role in tumor cell proliferation and survival.

The JAK2 selective compound BMS911543 (WO2011028864) is in phase II clinical trials for the treatment of m elofibrosis. BMS91 1543 is shown below.

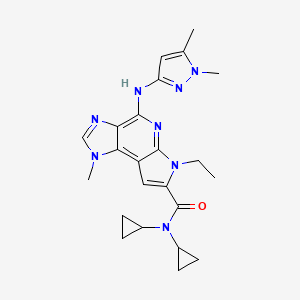
PAPER
ACS Medicinal Chemistry Letters (2015), 6(8), 850-855
Discovery of a Highly Selective JAK2 Inhibitor, BMS-911543, for the Treatment of Myeloproliferative Neoplasms

JAK2 kinase inhibitors are a promising new class of agents for the treatment of myeloproliferative neoplasms and have potential for the treatment of other diseases possessing a deregulated JAK2-STAT pathway. X-ray structure and ADME guided refinement of C-4 heterocycles to address metabolic liability present in dialkylthiazole 1 led to the discovery of a clinical candidate, BMS-911543 (11), with excellent kinome selectivity, in vivo PD activity, and safety profile
MS (ESI) m/z 434.3 (M+H). 1H NMR (CDCl3) δ: 7.96 (s, 1H), 7.65 (s, 1H), 6.83 (s, 1H), 4.67 (q, J = 7.1 Hz, 2H), 4.01 (s, 3H), 3.82 (s, 3H), 2.77 – 2.84 (m, 2H), 2.43 (s, 3H), 1.48 (t, J = 7.2 Hz, 3H), 0.79 – 0.86 (m, 4H), 0.71 – 0.77 (m, 4H).
PAPER
Journal of Organic Chemistry (2015), 80(12), 6001-601
Click to access jo5b00572_si_001.pdf
Ni-Catalyzed C–H Functionalization in the Formation of a Complex Heterocycle: Synthesis of the Potent JAK2 Inhibitor BMS-911543

BMS-911543 is a complex pyrrolopyridine investigated as a potential treatment for myeloproliferative disorders. The development of a short and efficient synthesis of this molecule is described. During the course of our studies, a Ni-mediated C–N bond formation was invented, which enabled the rapid construction of the highly substituted 2-aminopyridine core. The synthesis of this complex, nitrogen-rich heterocycle was accomplished in only eight steps starting from readily available materials.
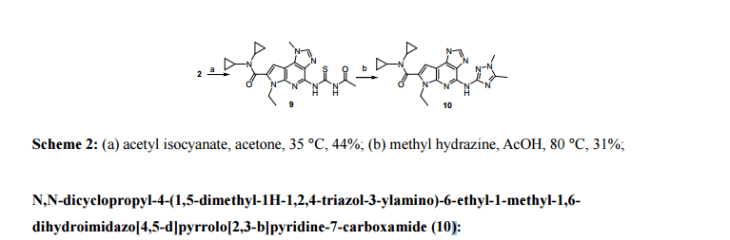
N,N-Dicyclopropyl-4-((1,5-dimethyl-1H-pyrazol-3-yl)amino)-6-ethyl-1-methyl-1,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridine-7-carboxamide, 1
PATENT
WO 2015031562
These Schemes are illustrative and are not meant to limit the possible techniques one skilled in the art may use to manufacture compounds disclosed herein.
As shown below in Scheme 1, the general preparation of compound 7 is described. Trichloroacetyl pyrrole (Compound 1) is reacted with a halogenating agent to give the C4-bromo pyrrole (Compound 2). Alcoho lysis occurs in the presence of an alcohol and base to generate ester (Compound 3), which can be selectively nitrated through contact with an appropriate nitrating agent (defined as a species that generates N02 ), yielding C5-nitro pyrrole (Compound 4). Compound 4 can be isolated as its free form, or optionally as a salt with an appropriate base. Ethylation with an appropriate alkylating agent generates the N-ethyl pyrrole (Compound 5), which in the presence of an imidazole, base, palladium and an appropriate phosphine ligand, will undergo a coupling process to form Compound 6. Reduction of the nitro-group of Compound 6 in the presence of hydrogen, a metal catalyst and optionally a base will produce Compound 7.
Scheme 1

As shown below in Scheme 2, the preparation of Compound 13 is described. Trichloroacetyl pyrrole is treated with NBS in acetonitrile to produce Compound 8. Treatment with sodium ethoxide in EtOH yields the ethyl ester Compound 9. This may be treated with a range of nitrating systems, in this example, NaNC /SCVPy, to generate nitro-pyrrole Compound 10, which can be isolated directly or as a salt form with an appropriate base, preferably dibenzylamine. Ethylation with ethyl iodide generates Compound 11 which may be isolated, or optionally telescoped directly into the arylation with Compound 32. Arylation proceeds in the presence of palladium, Xantphos, potassium pivylate and Hunig’s base to generate Compound 12. Hydrogenation presence of Pt/C followed by cyclization with NaOEt yields Compound 13.
Scheme 2

Another process of the invention is disclosed in Scheme 3 shown below. Compound 14 is prepared from Compound 3 in the presence of an alkylating agent. Treatment with a suitable diboron reagent produces Compound 15, which can then be coupled with a suitably functionalized imidazole derivative to yield Compound 16. Amino lysis with a suitable nitrogen donor produces Compound 17, which can cyclize under appropriate conditions to produce Compound 7.
Scheme 3
Step 3 Step 4 Step 5

As shown below in Scheme 4, ethylation of Compound 9 with ethyl iodide produces Compound 18. This may be directly reacted with dipinacol-diboron in the presence of Pd(OAc)2 and tricyclohexylphosphin hexafluorophosphate and
tetramethylammonium acetate to yield Compound 19. Subsequent coupling with 5-Br-imidazole derivative yields Compound 20. Treatment with hydroxylamine hydrochloride in the presence of triethylamine yields the Compound 21. Subsequent cyclization with Piv20 in the presence of PRICAT™ and hydrogen yields Compound 13.
Scheme 4
77% isolated over 2-steps%

18
Step 5 Pd(OAc)2
PPh3
78%

As shown below in Scheme 5, Compound 23 may be converted to Compound 26 by two pathways. In one option, Compound 23 can be treated with palladium, ligand and a mild base to prepare Compound 25. Reaction of Compound 25 with a metal hydroxide produces Compound 26.
Alternately, Compound 23 can be treated with palladium and ligand in the presence of a soluble hydroxide base, followed by treatment with the metal counter-ion to prepare Compound 26 directly. Once Compound 26 is formed, it can be coupled to Compound 27 to form compound I.


A solution of Compound 1 in acetonitrile (1238.0 kg, 264.9 kg after correction) was charged into a 5000 L glass-lined reactor at a temperature of 20-30 °C. The mixture was added with stirring over about 2 h and then cooled to 0 °C. NBS (221.8 kg) was charged into the mixture at intervals of 20-30 min at 0-20 °C. The mixture was cooled to 0-5 °C and reacted until the content of Compound 8was < 1.0%. Additional NBS (4.0 kg) was charged into the mixture at 0-20 °C. The mixture was reacted over 3 h until the content of Compound 8 was < 1.0%. Purified water (2650.0 kg) was added over about 1.5 – 2.5 h at 0-20 °C. The mixture was cooled to 0-5 °C and then stirred for about 1 h for crystallization. The mixture was filtered and the filter cake was rinsed with water.
Example 2

While maintaining the temperature at 20-30 °C, anhydrous ethanol (950.0 kg) was charged into a 3000 L glass-lined reactor followed by Compound 8 (342.7 kg). The mixture was cooled to 0-5 °C over about 2 h. Sodium alcoholate solution in ethanol (21%, 36.4 kg) was added dropwise over about 1-1.5 h at 0-5 °C. The reaction mixture was then heated to about 25-30 °C and tested until the content of Compounds 8/9 was < 1.0%. The reaction mixture was concentrated at a temperature < 50 °C until about 1.3-1.4 volume of Compound 8 was left. The concentrated mixture was cooled at 25-30 °C. The mixture was quenched into cooled water (3427.0 kg) over about 2 h. After addition, the mixture was stirred at 0-5 °C over about 2 h for crystallization. The mixture was filtered and the filter cake was rinsed. The solid was dried at 30-40 °C over 40-45 h to afford 234.3 kg of Compound 9 , 99.9% purity and 91.3% yield.
Example 3

9 10
A mixture of NaN03, NaHS04, and Na2S04 in CH3CN is wet-milled to constant particle size of -50 micron. To the slurry of inorganic salts is added S03 -pyridine and Compound 9. The reaction mixture is agitated at 25 °C until 90-95% conversion is achieved. The reaction is quenched with aqueous sodium hydroxide and the spent inorganic salts are removed by filtration. The filtrate is passed through a carbon pad and distilled under constant volume distillation and diluted with water to a target 15
volumes/kg of Compound 9 and a target ratio 1.0:2.0 vol/vol MeCN to water. The resulting solids are deliquored, washed, and dried to afford Compound 10.
Example 4

Toluene (10 L/Kg)
65 °C
Compound 10 (1.0 eq) and TBABr (1.0 eq) were added to a biphasic mixture of toluene (8 L/kg 10) and potassium carbonate (1.5 eq) in water (5 L/kg 10). The batch temperature was held at 25 °C. The resulting triphasic slurry was heated to 60-65 °C and diethylsulfate (1.5 eq, in a solution of toluene 2 L/kg 10) was slowly added over ~ 1 h. The reaction was aged until less than 1 RAP of Compound 10 (10:11) remained. The resulting homogeneous biphasic mixture was cooled to 20 °C and the lean aq. phase was removed. The rich organic phase was washed with water (2×7 L/kg 10) and concentrated to 6 mL/g 10. The concentrated stream was dried via azeotropic, constant volume distillation with toluene until the water content of the stream was <0.1 wt %. The resulting stream was telescoped into the subsequent direct arylation reaction.
Example 5

11 28 12
To the toluene stream of Compound 11, with potassium pivalate (1.5 equiv.) was charged, followed by DIPEA (3 eq.), Compound 28 (3 eq.) and Pd(Xantphos)Cl2 (0.04 eq.). The vessel was evacuated to < 200 torr and backfilled with nitrogen (3 X) followed by heating to 95 °C until residual Compound 11 was less than 1 RAP (11: 12). The reaction mixture was cooled to 25 °C and diluted with ethyl acetate (15 mL/g vs input pyrrole) and aq. N-acetylcysteine (0.2 eq., 5 wt % solution, 1.8 mL/g vs. input pyrrole) and heated to 50 °C for 1 h. The biphasic mixture was cooled to 25 °C. The lower aqueous layer was removed. The ethyl acetate stream was washed with water (2×7 mL/g vs. input pyrrole). The rich organic phase was polish filtered followed by a vessel/polish filter rinse with ethyl acetate (2 mL/g vs. input pyrrole). The rich organic stream was concentrated to 4 mL/g vs. input pyrrole via vacuum distillation, while maintaining the batch temperature above 50 °C. If spontaneous nucleation did not occur, Compound 12 seeds (1 wt %) were charged, followed by aging for 30 min at temperature. MTBE (5 mL/g vs. 11) was charged to the slurry over 1 hour while maintaining the batch temperature above 40 °C, followed by aging at 40 °C for 1 h. The slurry was cooled to 0 °C over 6 h and aged at 0°C for 6 h. The slurry was filtered and washed with
EtO Ac : Toluene : MTBE (1.5: 1.0: 1.5, 2 mL/g vs. input 11 ). The wet cake was dried (50 °C, 100 torr) until LOD was < 1 wt %.
Example 6

Compound 12 (1 eq., limiting reagent (LR)) is dissolved in THF/NMP (20 Vol wrt LR, 9/1 ratio) and submitted to hydrogenation using 10 wt% (wrt LR) Pt/C (5 wt%) at 25 to 40° C for 5-10 h. The reaction containing the corresponding amine is filtered. The rich organic stream is concentrated to Compound 12 Vol (wrt LR) and subjected to 0.1 eq of 21 wt% NaOEt/EtOH for 5 h at 20-25 °C, upon which Compound 13 forms. The stream is cooled to 0-10 °C, and water (5L/Kg, wrt to LR) is added and then filtered to isolate Compound 13. The product is dried at 50 °C under vacuum.
Example 7

in toluene solution
9
18
Compound 18 was prepared by treating the pyrrole with ethyl iodide and pulverized potassium carbonate in DMF at 25-30°C under inert atmosphere. After the reaction was completed, the batch mass was cooled to 15°C to 20°C and quenched by slow addition of water then MTBE. The MTBE layer was separated and washed with water. The MTBE layer was distilled to 4 Vol and solvent swapped with toluene. The toluene stream was then taken into the next step.
Example 8

18 19
Tetra-methyl ammonium acetate in toluene slurry was heated to 75-80°C to get a clear solution. The mass was cooled to below 30°C and pyrrole in toluene and bis (pinacolato) diborane were added. The reactor was inerted by nitrogen purging then the reaction was heated to 75-80°C. A freshly prepared catalyst/ligand complex (0.0 leq of palladium acetate, 0.025eq of tricyclohexyl phosphino hexafluoroborate and 0.2eq of tetra methyl ammonium acetate in toluene) was charged under nitrogen atmosphere at RT and stirred for 2h. The mass was then stirred at 75-80°C under nitrogen atmosphere. After the reaction was completed, the mixture was cooled below 30°C and quenched with aq. sodium bisulphate solution. The organic layer was polish filtered through a Celite bed and the filtrate was washed with water. The solvent swapped to ethanol until the toluene content became less than 0.5 %. The solution was cooled to 0-5°C and water was added for crystallization. The product was then isolated by filtration.
Example 9

Compound 20 was prepared by treating Compound 19 with Compound 34 in the presence of palladium acetate, triphenyl phosphine and potassium carbonate in dimethyl acetamide with the water mixture as the solvent. Dimethyl acetamide, water, potassium carbonate and the two starting materials were charged into the reactor. The mixture was made inert with nitrogen for 30 min and then charged with freshly prepared catalyst mixture (palladium acetate, triphenyl phosphine and potassium carbonate in dimethyl acetamide). The temperature was raised to 78-83 °C then the mass was stirred at this temperature. After the reaction was completed, the reaction mass was cooled to ambient temperature and purified water was added slowly into the mass for product
crystallization. The mass was stirred for a period of 3 h and filtered. The wet cake was washed with purified water and dried in VTD at 50-55 °C under vacuum.
Example 10

Compound 21 was prepared by treating Compound 20 with hydroxylamine hydrochloride and triethyl amine using ethanol as the solvent. Compound 20 was added into ethanol (15 Vol) and the reaction mass was heated to 38-40 °C. Hydroxylamine hydrochloride was charged and stirred for 10 min, then triethyl amine was added slowly at 38-40 °C over a period of lh. The above mass was stirred at 38-40 °C until Compound 20 becomes less than 5.0%, typically in about 15 h. After the reaction was completed, the above reaction mass was cooled to ambient temperature (below 30 °C) and filtered. The wet cake was washed with purified water (4 Vol) and dried under vacuum in VTD at 55-60 °C.
Example 11

Initially Compound 21 was treated with pivalic anhydride using toluene and acetic acid mixture as solvent under inert atmosphere until Compound 21 becomes less than 3.0% with respect to Compound 21, typically in about 30 min. PRICAT Nickel was then added under nitrogen atmosphere. The reaction mass was inerted with nitrogen for three cycle times and then degassed with hydrogen gas for three cycle times. Following this, 3.0 kg/cm2 hydrogen pressure was applied to the reaction mass which was stirred for about 12h. After the reaction was completed, the reaction mixture was filtered through a sparkler filter. The filtrate was distilled and the solvent exchanged with toluene until the ratio of acetic acid & toluene reaches 1 :20. At this time, n-Heptane was charged and cooled to 15°C. Then the product was filtered and the wet cake was dried in VTD at 50-55°C under vacuum.

Compound 30 was prepared by the coupling of Compound 22 with Compound 29, 3 -bromo- 1,5 -dimethyl- lH-pyrazole in the presence of
Tris(dibenzylideneacetone)dipalladium chloroform adduct, t-Brettphos and potassium phosphate in tert-amyl alcohol at 98-103 °C under inert atmosphere. After completion of the reaction (typical level of Int.9 -5% & typical reaction hrs 20 h), the mass was cooled to ambient temperature and t-amyl alcohol (4 Vol) and 20 Vol of water were charged into the reaction mass. The reaction mass was stirred for 15 min. and then phase split. The organic layer was diluted with 10 Vol of MTBE and product was extracted with 20 Vol of 1M methane sulphonic acid. The MSA stream was treated with 15 wt % charcoal to reduce the residual palladium numbers. The filtrate was cooled to below 20 °C and the pH was adjusted to 1.7-1.9 using IN NaOH for product crystallization and then iltered. The wet cake was washed with purified water (3 x 5 Vol), followed by methanol (5 Vol). The cake was vacuum dried for 3 h. then the wet cake and dimethyl sulfoxide (20 Vol) were charged into a reactor. The mass was heated to 120-125 °C to get clear solution then the mass was cooled to ambient temperature and stirred for 2 h, then filtered. The wet cake was washed with methanol (3x 4.0 Vol) and vacuum dried for 2 h. The wet cake was dried in VTD at below 55°C under vacuum.
Example 13

Compound 30 , ethanol (16.5 Vol), water and aq sodium hydroxide solution were charged into a reactor then the mass was heated to 70-75 °C and stirred until Compound 30 becomes less than 1.0%. After the reaction was completed, the mass was diluted with ethanol for complete product precipitation at 65-75 °C. Then the mass was cooled to 50 °C for a period of lh and stirred for lh at 50 °C. The mass was further cooled to 20 °C and stirred for lh at 20 °C and then filtered. The wet cake was washed with 5 Vol of 15% aqueous ethanolic solution followed by THF. The wet cake was dried under vacuum at 70-75 °C till LOD comes to less than 5.0 %, typically in about 40 h.
Example 14

In a vessel 36.5 mmol (-42.6 mL) of Compound 29 solution in 2-methyl-2-butanol was combined with 30.7g (65.1 mmol) tetrabutylammonium hydroxide (55 wt% in water), 8.01g (27.0 mmol) Compound 13 , and 10 mL 2-methyl-2-butanol. The mixture was heated at 70 °C until hydrolysis of Compound 13 was complete (full dissolution, <15 min). The solution was cooled to 60 °C and 1.12g (2.22 mmol) of tBuBippyPhos followed by 384 mg (1.028 mmol) allylpalladium chloride dimer (L:Pd = 1 :1) was added. The mixture was heated to 80 °C and was aged at this temperature for 20h before cooling to 22 °C.
Water was added and the mixture concentrated, a constant volume distillation was then performed to swap to ethanol (40-55 °C, 150 mbar). The resulting solution was passed through a 5 micron filter to remove any particulates. The solution was heated to 55 °C and 8.10 mL (40.52 mmol, 1.5 equiv) 5N NaOH (aq) was added dropwise over a 3 h period. Crystals of Compound 31 began to form, and after aging for an additional lh, the mixture was cooled to 20 °C over 3 h. After an additional 6h of aging, crystals were collected on a frit and the cake was washed with 40 mL of 90: 10 ethanol: water, followed by 48 mL acetone. After drying at 80 °C in a vacu-oven for 16 h, Compound 31 was collected as an off-white solid (8.89g, 85%).
Example 15

Compound 31 was added into dichloromethane (20 Vol) and cooled to 15-20 °C. The reaction mass was charged with DMC in DCM solution (1.4 eq of DMC in 5.0 Vol of DCM). The mixture was stirred until Compound 31 becomes less than 2.0% with respect to the corresponding acid chloride, typically in about lh. After completion of the reaction, Compound 27 (1.4 eq) and N,N-diisopropylethyleneamine (3.0 eq) were charged and the mixture was stirred. After completion of the reaction, the mass was quenched with 12 Vol of water then the layers were separated. The organic layer was washed with water and filtered through a celite bed. The filtrate was concentrated to ~6.0 vol and then the mass was cooled to 35 °C. To the resulting solution was added THF, followed by seeds of product, then stirred for 3 h. The solvent was swapped with THF until
dichloromethane becomes less than 2 wt% (wrt THF). The mass was cooled to -5 to 0 °C over a period of 2 h and stirred for 2 h. The reaction mass was then filtered under a nitrogen atmosphere. The material was slurried with pre-cooled THF (2*2 Vol) and filtered. The wet cake was dried in VTD at 60 °C under vacuum till LOD becomes < 1%, typically in about 20 h.
Example 16

DC , RT
I
To a slurry of Compound 31 (15.00 g, 40.0 mmol) in dichloromethane (300 ml) was added diphenylphosphinic chloride (12.29 g, 51.9 mmol). The mixture was stirred at room temperature for 2 h and Ν,Ν-diisopropylethylamine ( 16.53 g, 127.9 mmol) was then added and stirred for another 30 min. Compound 27 (6.94 g, 51.9 mmol) and 4-dimethylaminopyridine (0.49 g, 4.0 mmol) were subsequently added and stirred for 16 h until the reaction was completed. The reaction mixture was treated with N-acetyl-L-cysteine (3.26 g, 20.0 mmol) and citric acid (10.10 g, 48.0 mmol) in deionized water (180 ml) for 2 h. After phase split, the dichloromethane phase was washed once with 0.42 N NaOH solution (180 ml) and washed twice with deionized water (180 ml each). The final dichloromethane phase was concentrated (to 90 ml) and acetone (30 ml) was added. The solution was cooled to 35 °C and N-2 form seed of Compound 1 ( 150 mg ) was added and aged for 1 h. The resulting slurry was solvent-swapped to acetone (DCM < 10% v/v), and cooled to 0 °C. The solid was filtered and washed with cold acetone and dried to afford 14.69 g (85%) of Compound I (HPLC AP 99.8) as off-white crystals.
Patent
WO 2011028864
http://www.google.com/patents/WO2011028864A1?cl=en
Compounds of general formula I in which the R group is thiazole (as in Ial) and R1 and R2 groups are CF3 or alkyl or cycloalkyl or combine to form a saturated carbocyclic or heterocyclic ring or where R2 group is COORb could be prepared using the general method depicted in Scheme 1. Dichloro intermediate II (prepared using procedure reported in WO200612237) could be combined with a 2,4-dimethoxybenzyl and the resulting secondary amine is capped with suitable protective group (Boc) (III). The second chlorine atom could be converted into the
corresponding amine (IV) through the benzophenone imine intermediate. The amino compound could be halogenated to intermediate V. V could be subjected to transition metal mediated indole ring formation and the resulting indole nitrogen is capped with ethyl iodide to afford VI. Ester hydrolysis followed by amide bond formation and cleavage of protective groups with acid treatment would yield amine VII. Amine VII could be converted into thiourea VIII by first coupling with benzoyl isothiocyanate followed treatment with aqueous base. Formation of thiazole could be achieved by condensation with an a-bromoketone derivative (R^HBrCOR2).


a) 2,4-dimethoxybenzylamine, heat; b) NaHMDS, Boc20; c) (Ph)2=NH; d) HCl; e) NIS; f) Pd2(dba)3, ethyl pyruvate; g) Etl, Cs2C03; h) NaOH (aq); i) dicyclopropylamine HCl, HATU, DIPEA; j) TFA; k) Benzoyl isothiocyanate;
1) NaOH (aq); m) I^CHBrCOR1
Scheme 1
Compounds of general formula Ia2 in which the R1 group is CONRaRa could be made using Scheme 2. Thiourea intermediate (VIII) could be combined with Et02CCHBrCOR1 to afford the thiazole ester (IX). The ester could be hydrolyzed and the acid could be coupled with amine to afford thiazole amide derivative (la)

a) Et02CCHBrCOR1; b) NaOH (aq); c) HNRaRa, HATU, DIPEA
Scheme 2
Similarly, compounds of general formula Ia3 in which the R1 group is CONRaRa could be prepared using the general protocol depicted in Scheme 3.

a) R2CHBrCOC02Me; b) NaOH (aq); c) HNRaRa, HATU, DIPEA
Scheme 3
Compounds of general formula la in which R1 is halogen (CI, Br or I) could be prepared by condensing an a,a’-dihaloketone as depicted in Scheme 4.
a) R2COCH(Hal)2
Scheme 4
Alternatively, thiourea derivative VIII could be converted to room temperature into C-5 un-substituted thiazole XI and then directly halogenated using electrophilic halogen source or through metallation followed by quenching with an electrophilic halogenating agent (Scheme 5).

a) BrCH2COR2; b) Selectfluor or NCS or NBS or NIS or tBuLi followed Selectfluor or NBS or NCS
Scheme 5
Compounds of general formula Ia5 in which R1 is S02Rb could be synthesized using the general synthetic approach shown in Scheme 6


a) Br2-acetic acid; b) EtOH, heat
Scheme 6
Compounds with general formula la in which R1 and R2 combine to form an aromatic or heteroaromatic ring could be prepared using Scheme 7.

X = hal, -S02Me
a) Pd(0) catalyst, NaOtBu, phosphine ligand, heat
Scheme 7
Alternatively, these compounds could be made by first coupling aniline or heteroaniline (XVI) with the isothiocyanate (XV) followed by oxidative cyclization (Scheme 8).

a) 1, 1 ‘-Thiocarbonyldi-2( 1 H)-pyridone; b) NaH; c) NIS
Scheme 8
Compounds of general formula Ibl could be prepared using the general synthetic approach depicted in Scheme 9. Aniline VII could be combined with γ-dithiomethylketone compound XVII, (prepared using the procedure reported at room temperature in Synlett, p 2331 (2008)) under basic condition to afford XVIII.
Stepwise condensation of the Boc-protected hydrazine derivative would give the required pyrazole Ibl.

a) NaH, THF; b) R1N(Boc)NH2, AcOH, 35-40°C; c) HCO2H or TFA, 60°C
Scheme 9
Compounds of general formula Ibl or Ifl and If could also be prepared by coupling C-4 halo derivative (XIX) with an appropriately substituted 2-aminopyrazole derivative (XX) using a transition metal catalyzed reaction (Scheme 10).

a) isoamyl nitrite, CH2I2 or isoamyl nitrite, CH2Br2; b) Pd2(dba)3, Xanphos, Cs2C03
Scheme 10
Compounds of general formula Ib2 in which R2 group is CONRaRa could be synthesized using Scheme 11. Aniline VII could be combined with γ-dithiomethylketone derivative XXII, (prepared using the procedure from
Tetrahedron, p 2631 (2003)) to afford intermediate XXIII. Stepwise condensation of Boc-protected hydrazine derivative would give the required pyrazole aldehyde XXIV. Aldehyde could be oxidized using oxone or sodium hypochlorite to furnish carboxylic acid XXV. Coupling of acid XXV with amine would give pyrazole amide Ib2.

a) NaH, THF, heat; b) R1N(Boc)NH2, AcOH; c) TFA; d) oxone or sodium hypochlorite; e) HNRaRa, HATU, DIPEA
Scheme 11
Compounds of general formula Icl could be prepared using the general protocol as shown in Scheme 12. Aniline VII could be coupled with chloroacetyl chloride and the resulting amide could be treated with thioamide (R2CS H2) to furnish thiazole Icl .

a) chloroacetyl chloride, base; b) R2CSNH2
Scheme 12
00120] Compounds of general formula ldl could be made as per Scheme 13. Previously described isothiocyanate derivative XV could be combined with amidine XXV under dehydrating reaction conditions to give 1,2,4-thiadiazole (ldl).

Scheme 13
Compounds of general formula lei could be prepared using a synthetic approach as shown in Scheme 14. Isothiocyanate XV could be combined with azide XXVI in the presence of phosphine to yield 1,3-oxazole Iel .

Scheme 14
Compounds of general formula lgl could be prepared using a synthetic approach as shown in Scheme 15. Amine VII could be combined with acyl isothiocyanate XXVII. The acylthioureaido could be condensed with hydrazine derivative to yield the 1,2,4-triazol derivative lgl.

igi
Scheme 15
without a methyl

Preparation of 7V,7V-dicyclopropyl-6-ethyl-l-methyl-4-(5-m ethyl- lH-pyrazol-3- ylamino)-l,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridine-7-carboxamide
[00437] Prepared using similar protocol as for example 72 from hydrazine.
[00438] MS (ESI) m/z 419.3 (M+H)
[00439] 1H NMR (400 MHz, CHLOROFORM-d) δ ppm 8.70 (br s, 1 H), 7.91 (br s, 1 H), 6.87 (s, 1 H), 6.09 (br s, 1 H), 4.64 (q, 2 H, J= 7.03 Hz), 4.08 (s, 3 H), 2.74 -2.95 (m, 2 H), 2.41 (s, 3 H), 1.51 (t, 3 H, J= 7.15 Hz), 0.81 – 0.95 (m, 4 H), 0.70 -0.81 (m, 4 H)
with an ethyl

7V,iV-dicyclopropyl-6-ethyl-4-(l-ethyl-5-methyl-lH-pyrazol-3-ylamino)-l-methyl- 1,6-dihydroimidazo [4,5-d] pyrrolo [2,3-b] pyridine-7-carboxamide
74A Preparation of fe/t-butyl l,3-dioxoisoindolin-2-yl(ethyl)carbamate

Diisopropyl azodicarboxylate (2.92 mL, 15.00 mmol) was added in one portion to a solution of tert-butyl l,3-dioxoisoindolin-2-ylcarbamate (2.62 g, 10 mmol, prepared following the procedure described by Nicolas Brosse et al. in Eur. J. Org. Chem. 4757-4764, 2003), triphenylphosphine (3.93 g, 15.00 mmol) and ethanol (0.691 g, 15.00 mmol) in THF (20 mL) at 0 °C and the reaction solution was stirred at room temperature for lh (monitored by TLC until completion). Solvent was evaporated and the residue was purified by flash chromatography on silica gel using an automated ISCO system (80 g column, eluting with 5-35% ethyl acetate / hexanes) to provide tert-butyl l,3-dioxoisoindolin-2-yl(ethyl)carbamate (2.6 g, 90 % yield) as a white solid which was used as it in the next step
74B Preparation of fe/t-butyl l-ethylhydrazinecarboxylate
Boc
H2N-N
\
Methylhydrazine (1.415 niL, 26.9 mmol) was added to a solution oi tert-butyl l,3-dioxoisoindolin-2-yl(ethyl)carbamate (example 74A, 5.2 g, 17.91 mmol) in THF (40 mL) at 0 °C and the reaction mixture was stirred at room temperature overnight. A white precipitate formed and was filtered off through a pad of Celite, The filtrate was concentrated in vacuo. The residue was dissolved in ethyl acetate (50 ml) and extracted with IN HC1 (3×30 ml), the acid layer was washed with ethyl acetate (50 ml) and basified to pH 10 by addition of 20% NaOH. The basic solution was then extracted with ethyl acetate (3×50 ml) and the combined organic layers were washed with brine, dried over magnesium sulfate, filtered and concentrated in vacuo to give tert-butyl 1 -ethylhydrazinecarboxylate (2.5 g, 87 % yield) as colorless oil.
XH NMR (400 MHz, CDC13) δ: 3.90 (br. s., 2H), 3.35 (q, J = 7.0 Hz, 2H), 1.42 (s, 9H), 1.07 (t, J = 7.0 Hz, 3H)
74 Preparation of N.N-dicyclopropyl-6-ethyl-4-(l-ethyl-5-methyl-lH-pyrazol-3-ylamino)-l-methyl-l ,6-dihydroimidazor4,5-d1pyrrolor2,3-b1pyridine-7-carboxamide
A mixture of (Z)-N,N-dicyclopropyl-6-ethyl- 1 -methyl-4-( 1 -(methylthio)-3-oxobut-l-enylamino)-l,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridine-7-carboxamide (example 74B, 70 mg, 0.155 mmol) and tert-butyl 1-ethylhydrazinecarboxylate (49.6 mg, 0.309 mmol) in acetic acid (1 mL) wan stirred at 35 °C for 4 h (monitored by LC/MS until no starting material left). Formic acid (1 mL) was added and the reaction mixture stirred at 60 °C for 6 h. The solvent was evaporated and the crude product was purified by flash chromatography on silica gel using an automated ISCO system (12 g column, eluting with 2-10% methanol / dichloromethane). The material was further purified by preparative HPLC to afford N,N-dicyclopropyl-6-ethyl-4-( 1 -ethyl-5-methyl- lH-pyrazol-3-ylamino)- 1 -methyl- 1 ,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridine-7-carboxamide (38 mg, 53.4 % yield) as an off-white solid.
MS (ESI) m/z 447.3 (Μ+Η).
XH NMR (500 MHz, CDC13) δ: 8.08 (s, 1H), 7.61 (s, 1H), 6.93 (s, 1H),
6.84 (s, 1H), 4.66 (q, J = 7.1 Hz, 2H), 4.02 (q, J = 7.2 Hz, 2H), 3.98 (s, 3H), 2.79 – 2.85 (m, 2H), 2.34 (s, 3H), 1.49 (t, J = 7.1 Hz, 3H), 1.41 (t, J = 7.2 Hz, 3H), 0.82 -0.87 (m, 4H), 0.72 – 0.78 (m, 4H).
Patent
JAK2 INHIBITORS AND THEIR USE FOR THE TREATMENT OF MYELOPROLIFERATIVE DISEASES AND CANCER [US8202881]2011-03-102012-06-19
JAK2 inhibitors and their use for the treatment of myeloproliferative diseases and cancer [US8673933]2012-04-302014-03-18
: Purandare AV, McDevitt TM, Wan H, You D, Penhallow B, Han X, Vuppugalla R, Zhang Y, Ruepp SU, Trainor GL, Lombardo L, Pedicord D, Gottardis MM, Ross-Macdonald P, de Silva H, Hosbach J, Emanuel SL, Blat Y, Fitzpatrick E, Taylor TL, McIntyre KW, Michaud E, Mulligan C, Lee FY, Woolfson A, Lasho TL, Pardanani A, Tefferi A, Lorenzi MV. Characterization of BMS-911543, a functionally selective small-molecule inhibitor of JAK2. Leukemia. 2012 Feb;26(2):280-8. doi: 10.1038/leu.2011.292. Epub 2011 Oct 21. PubMed PMID: 22015772.
Characterization of BMS-911543, a functionally selective small-molecule inhibitor of JAK2http://www.nature.com/leu/journal/vaop/ncurrent/full/leu2011292a.html
GRAPHS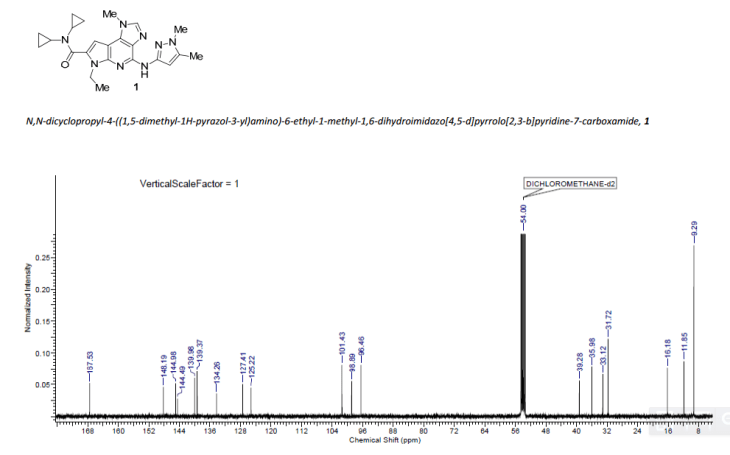
Click to access jo5b00572_si_001.pdf
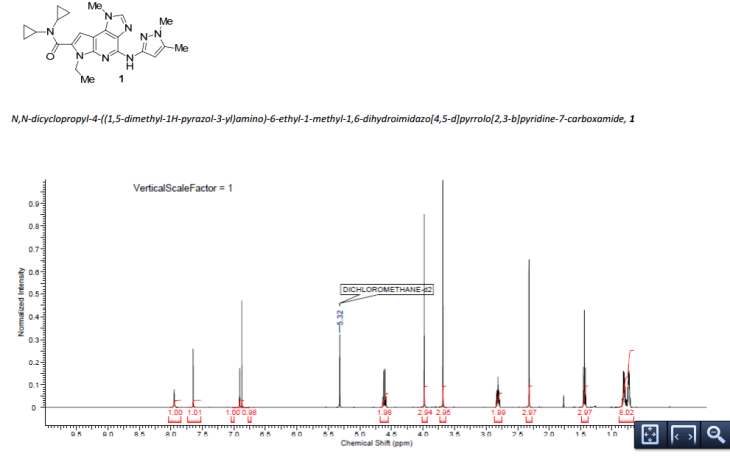
//////BMS 911543, phase 2, bms,
AT 9283

AT9283, AT 9283
N-cyclopropyl-N’-[3-[6-(4-morpholinylmethyl)-1H-benzimidazol-2-yl]-1H-pyrazol-4-yl]urea
1-cyclopropyl-3-[(3Z)-3-[5-(morpholin-4-ylmethyl)benzimidazol-2-ylidene]-1,2-dihydropyrazol-4-yl]urea
| 896466-04-9 | |
| Molecular Weight | 381.43 |
| Molecular Formula | C19H23N7O2 |
CAS
896466-04-9, 896466-57-2 ((±)-Lactic acid), 896466-61-8 (HCl), 896466-55-0 (methanesulfonate)
MolFormulaC22H29N7O5
MolWeight471.5096
CAS 896466-76-5 L LACTATE
(2S)-2-Hydroxypropanoic acid compd. with N-cyclopropyl-N’-[3-[6-(4-morpholinylmethyl)-1H-benzimidazol-2-yl]-1H-pyrazol-4-yl]urea
Astex Therapeutics Ltd, INNOVATOR

AT-9283 is a potent AuroraA/AuroraB and multi-kinase inhibitor. AT-9283 has shown to inhibit growth and survival of multiple solid tumor cell lines and is efficacious in mouse xenograft models.
AT 9283 is a substance being studied in the treatment of some types of cancer. It is small molecule a multi-targeted c-ABL, JAK2, Aurora A and B inhibition with 4, 1.2, 1.1 ad approximate 3 nM for Bcr-Abl (T3151), Jak2 and Jak3 aurora A and B, respectively. It blocks enzymes (Aurora kinases) involved in cell division and may kill cancer cells
WO2006070195 to Astex Therapeuitcs discloses pyrazole compounds of the general structure shown below as kinase inhibitors.

The compound AT9283 is in phase II clinical trials for treating advanced or metastatic solid tumors or Non-Hodgkin’s Lymphoma. AT9283 is shown below.



a Reagents and conditions:
(a) SOCl2, THF, DMF; (b) morpholine, THF, Et3N; ………FORMATION OOF ACID CHLORIDE AND COUPLING WITH MORPHOLINE
(c) NaBH4, BF3.OEt2, THF; …………..KETO TO CH2
(d) 10% Pd-C, H2, EtOH; TWO NITRO GPS TO TWO AMINO , REDN
(e) EDC, HOBt, DMF; (f) AcOH, reflux;COUPLING WITH 4-Nitro-lH-pyrazole-3-carboxylic acid
(g) 10%Pd-C, H2, DMF; NITRO GP TO AMINO
(h) standard amide and urea coupling methods
WO2006070195
https://www.google.co.in/patents/WO2006070195A1?cl=en
Stage 10: Synthesis of l-cvclopropyl-3-[3-(5-morpholin-4-ylmethyl-lH- beiizoimidazol-2-ylV 1 H-pyrazol-4-yli -urea.

To a mixture of 7-morpholin-4-ylmethyl-2,4-dihydro- 1 ,2,4,5a, 10- pentaaza- cyclopenta[a]fluoren-5-one (10.7 g, 32.9 mmol) in NMP (65 mL) was added cyclopropylamine (6.9 mL, 99 mmol). The mixture was heated at 100 0C for 5 h. LC/MS analysis indicated -75% conversion to product, therefore a further portion of cyclopropylamine (2.3 mL, 33 mmol) was added, the mixture heated at 100 0C for 4 h and then cooled to ambient. The mixture was diluted with water (100 mL) and extracted with EtOAc (100 niL). The organic portion was washed with sat. aq. NH4Cl (2 x 50 mL) and brine (50 rnL) and then the aqueous portions re-extracted with EtOAc (3 x 100 mL). The combined organic portions were dried over MgSO4 and reduced in vacuo to give l-cycloρropyl-3-[3-(5-morpholin-4-ylmethyl-lH- benzoimidazol-2-yl)-lH-pyrazol-4-yl]-urea as an orange glassy solid (9.10 g).
Stage 11: Synthesis of l-cvclopropyl-S-P-fS-morpholin^-ylmethyl-lH- benzoimidazol-2-yl)-lH-pyrazol-4-yll-urea, L-lactate salt

To a solution of l-cyclopropyl-3-[3-(5-morpholin-4-ylmethyl-lH-benzoimidazol-2- yl)-lH-pyrazol-4-yl]-urea (9.10 g, 24 mmol) in EtOAc-iPrOH (1 :1, 90 mL) was added L-lactic acid (2.25 g, 25 mmol). The mixture was stirred at ambient temperature for 24 h then reduced in vacuo. The residue was given consecutive slurries using toluene (100 mL) and Et2O (100 mL) and the resultant solid collected and dried (8.04 g).
This solid was purified by recrystallisation from boiling iPrOH (200 mL) to give after drying l-cyclopropyl-3-[3-(5-morpholin-4-ylmethyl-lH-benzoimidazol-2-yl)- lH-pyrazol-4-yl]-urea, L-lactate salt (5.7 g) as a beige solid.
EXAMPLE 66
Stage 1: Preparation of (3,4-dinitrophenyl)-morpholin-4-yl-methanone

3,4-Dinitrobenzoic acid (1.000Kg, 4.71mol, l.Owt), tetiuhydrofuran (10.00L5 lO.Ovol), and dimethylformamide (0.010L, O.Olvol) were charged to a flask under nitrogen. Thionyl chloride (0.450L, 6.16mol, 0.45vol) was added at 20 to 3O0C and the reaction mixture was heated to 65 to 7O0C. Reaction completion was determined by 1H NMR analysis (d6-DMSO), typically in 3 hours. The reaction mixture was cooled to 0 to 50C and triethylamine (1.25L, 8.97mol, 1.25vol) was added at 0 to 100C. Morpholine (0.62L, 7.07mol, 0.62vol) was charged to the reaction mixture at 0 to 1O0C and the slurry was stirred for 30 minutes at 0 to 1O0C. Reaction completion was determined by H NMR analysis (d6-DMSO). The reaction mixture was warmed to 15 to 2O0C and water (4.00L, 4.0vol) was added. This mixture was then charged to a 4OL flange flask containing water (21.0OL, 21.0vol) at 15 to 250C to precipitate the product. The flask contents were cooled to and aged at 0 to 50C for 1 hour and the solids were collected by filtration. The filter-cake was washed with water (4x 5.00L, 4x 5.0vol) and the pH of the final wash was found to be pH 7. The wet filter-cake was analysed by H NMR for the presence of triethylamine hydrochloride. The filter-cake was dried at 40 to 450C under vacuum until the water content by KF <0.2%w/w, to yield (3,4-dinitrophenyl)-morpholin-4-yl-methanone (1.286Kg, 97.0%, KF 0.069%w/w) as a yellow solid.
Stage 2: Preparation of 4-(3,4-dinitro-benzyl)-morpholine

C11H11N3O6 C11H13N3O5
FW:281.22 FW:267.24
(3,4-DinitiOphenyl)-morpholin-4-yl-methanone (0.750Kg, 2.67mol, l.Owt) and tetrahydrofuran (7.50L, lO.Ovol) were charged to a flask under nitrogen and cooled to 0 to 50C. Borontrifluoride etherate (0.713L, 5.63mol, 0.95vol) was added at 0 to 50C and the suspension was stirred at this temperature for 15 to 30 minutes. Sodium borohydride (0.212Kg, 5.60mol, 0.282wt) was added in 6 equal portions over 90 to 120 minutes. (A delayed exotherm was noted 10 to 15 minutes after addition of the first portion. Once this had started and the reaction mixture had been re-cooled, further portions were added at 10 to 15 minute intervals, allowing the reaction to cool between additions). The reaction mixture was stirred at 0 to 50C for 30 minutes. Reaction completion was determined by 1H NMR analysis (d6-DMSO). Methanol (6.30L, 8.4vol) was added drop wise at 0 to 1O0C to quench the reaction mixture (rapid gas evolution, some foaming). The quenched reaction mixture was stirred at 0 to 1O0C for 25 to 35 minutes then warmed to and stirred at 20 to 3O0C (exotherm, gas/ether evolution on dissolution of solid) until gas evolution had slowed. The mixture was heated to and stirred at 65 to 7O0C for 1 hour. The mixture was cooled to 30 to 4O0C and concentrated under vacuum at 40 to 450C to give crude 4-(3,4-dinitro-benzyl)-morpholine (0.702Kg, 98.4%) as a yellow/orange solid.
4-(3,4-Dinitro-benzyl)-niorpholme (2.815kg, 10.53mol, l.Owt) and methanol (12.00L, 4.3vol) were charged to a flask under nitrogen and heated to 65 to 7O0C. The temperature was maintained until complete dissolution. The mixture was then cooled to and aged at 0 to 50C for 1 hour. The solids were isolated by filtration. The filter-cake was washed with methanol (2x 1.50L, 2x 0.5vol) and dried under vacuum at 35 to 45°C to give 4-(3,4-dinitro-benzyl)-morpholine (2.353Kg, 83.5% based on input Stage 2, 82.5% overall yield based on total input Stage 1 material,) as a yellow solid.
Stage 3: Preparation of 4-morpholin-4-yl-methyl-benzene-L2-diamine

C11H13N3O5 C11H17N3O
FW:267.24 FW:207.27
4-(3,4-Dinitro-benzyl)-morρholine (0.800Kg, 2.99mol, l.Owt), and ethanol (11.20L, 14.0vol) were charged to a suitable flask and stirred at 15 to 250C and a vacuum / nitrogen purge cycle was performed three times. 10% Palladium on carbon (10%Pd/C, 50%wet paste, 0.040Kg, 0.05wt wet weight) was slurried in ethanol (0.80L, l.Ovol) and added to the reaction. The mixture was cooled to 10 to 2O0C and a vacuum / nitrogen purge cycle was performed three times. A vacuum / hydrogen purge cycle was performed three times and the reaction was stirred under a hydrogen atmosphere at 10 to 2O0C. Reaction completion was determined by 1H NMR analysis (d6-DMSO), typically 14 to 20 hours. A vacuum / nitrogen purge cycle was performed three times and the reaction mixture was filtered through glass microfibre paper under nitrogen. The filter-cake was washed with ethanol (3x 0.80L, 3x l.Ovol) and the combined filtrate and washes were concentrated to dryness under vacuum at 35 to 450C to give 4-morpholin-4-yl-methyl-benzene-l,2- diamine (0.61 IKg 98.6%) as a brown solid.
Stage 4: Preparation of 4-nitiO-lH-pyrazole-3-carboxγlic acid methyl ester

C4H3N3O4 C5H5N3O4
FW: 157.09 FW: 171.11
4-Nitro-lH-pyrazole-3-carboxylic acid (1.00kg, 6.37mol, l.Owt) and methanol (8.00L, 8.0vol) were charged to a flange flask equipped with a mechanical stirrer, condenser and thermometer. The suspension was cooled to 0 to 5°C under nitrogen and thionyl chloride (0.52L, 7.12mol, 0.52vol) was added at this temperature. The mixture was warmed to 15 to 25°C over 16 to 24 hours. Reaction completion was determined by 1H NMR analysis (d6-DMSO). The mixture was concentrated under vacuum at 35 to 45°C. Toluene (2.00L, 2.0vol) was charged to the residue and removed under vacuum at 35 to 450C. The azeotrope was repeated twice using toluene (2.00L, 2.0vol) to give 4-nitro-lH-pyrazole-3-carboxylic acid methyl ester (1.071Kg, 98.3%) as an off white solid.
Stage 5: Preparation of 4-amino-lH-pyrazole-3-carboxylic acid methyl ester. O2Me

C5H 5N3O4 C5H7N3O2 FW: 171.11 FW: 141.13
A suspension of 4-nitro-lH-pyrazole-3-carboxylic acid methyl ester (1.084Kg, 6.33mol, l.Owt) and ethanol (10.84L, lO.Ovol) was heated to and maintained at 30 to 35°C until complete dissolution occurred. 10% Palladium on carbon (10% Pd/C wet paste, 0.152Kg, 0.14wt) was charged to a separate flask under nitrogen and a vacuum / nitrogen purge cycle was performed three times. The solution of 4-nitro- lH-pyrazole-3-carboxylic acid methyl ester in ethanol was charged to the catalyst and a vacuum / nitrogen purge cycle was performed three times. A vacuum / hydrogen purge cycle was performed three times and the reaction was placed under an atmosphere of hydrogen. The reaction mixture was stirred at 28 to 30°C until deemed complete by 1H NMR analysis (d6-DMSO). The mixture was filtered under nitrogen and concentrated under vacuum at 35 to 450C to give 4-amino-lH- pyrazole-3-carboxylic acid methyl ester (0.883Kg, 98.9%) as a purple solid.
Stage 6: Preparation of 4-fert-butoxycarbonylamino-lH-pyrazole-3-carboxylic acid

C5H7N3O2 C9H13N3O4
FW: 141.13 FW:227.22
4-Amino-lH-pyrazole-3-carboxylic acid methyl ester (1.024Kg, 7.16mol, l.Owt) and dioxane (10.24L, lO.Ovol) were charged to a flange flask equipped with a mechanical stirrer, condenser and thermometer. 2M aq. Sodium hydroxide solution (4.36L, 8.72mol, 4.26vol) was charged at 15 to 250C and the mixture was heated to 45 to 550C. The temperature was maintained at 45 to 550C until reaction completion, as determined by 1H NMR analysis (d6-DMSO). Di-te/Y-butyl dicarbonate (Boc anhydride, 1.667Kg, 7.64mol, 1.628wt) was added at 45 to 55°C and the mixture was stirred for 55 to 65 minutes. 1H NMR IPC analysis (d6-DMSO) indicated the presence of 9% unreacted intermediate. Additional di-fert-butyl dicarbonate (Boc anhydride, 0.141Kg, 0.64mol, 0.14wt) was added at 55°C and the mixture was stirred for 55 to 65 minutes. Reaction completion was determined by 1H NMR analysis (d6-DMSO). The dioxane was removed under vacuum at 35 to 450C and water (17.60L, 20.0vol) was added to the residue. The pH was adjusted to pH 2 with 2M aq. hydrochloric acid (4.30L, 4.20vol) and the mixture was filtered. The filter-cake was slurried with water (10.00L3 9.7vol) for 20 to 30 minutes and the mixture was filtered. The filter-cake was washed with heptanes (4.10L, 4.0vol) and pulled dry on the pad for 16 to 20 hours. The solid was azeodried with toluene (5x 4.00L, 5x 4.6vol) then dried under vacuum at 35 to 45°C to give 4-tert- butoxycarbonylamino-lH-pyrazole-3-carboxylic acid (1.389Kg, 85.4%) as a purple solid.
Stage 7: Preparation of [3-(2-amino-4-moipholin-4-ylmetliyl-phenylcarbamoviy lH-pyrazol-4-yl]-carbamic acid tert-butyl ester

C9H13N3O4 C11H17N3O C20H28N6O4
FW: 227.22 FW: 207.27 FW: 416.48
+ regioisomer
4-førf-Butoxycarbonylamino-lH-pyrazole-3-carboxylic acid (0.750Kg, 3.30 mol, l.Owt), 4-morpholin-4yl-methyl-benzene-l,2-diamine (0.752Kg, 3.63mol, l.Owt) and N,N’-dimethylformamide (11.25L, 15.0vol) were charged under nitrogen to a flange flask equipped with a mechanical stirrer and thermometer. 1- Hydroxybenzotriazole (HOBT, 0.540Kg, 3.96mol, 0.72wt) was added at 15 to 250C. N-(3-Dimethylaminopropyl)-N’-ethylcarbodiimide (EDC, 0.759Kg, 3.96mol, 1.01 wt) was added at 15 to 250C and the mixture was stirred at this temperature for 16 to 24 hours. Reaction completion was determined by 1H NMR analysis. The reaction mixture was concentrated under vacuum at 35 to 45°C. The residue was partitioned between ethyl acetate (7.50L, lO.Ovol) and sat. aq. sodium hydrogen carbonate solution (8.03L, 10.7vol) and the layers were separated. The organic phase was washed with brine (3.75L, 5.0vol), dried over magnesium sulfate (1.00Kg, 1.33wt) and filtered. The filter-cake was washed with ethyl acetate (1.50L, 2.0vol). The combined filtrate and wash were concentrated under vacuum at 35 to 450C to give [3-(2-amino-4-morpholin-4-ylmethyl-phenylcarbamoyl)-lH-pyrazol- 4-yl]-carbamic acid tert-butyl ester (1.217Kg, 88.6%) as a dark brown solid.
Stage 8 : Preparation of 3 -f 5-morpholin-4-ylmethyl- 1 H-benzoimidazol-2-ylV 1 H- pyrazol-4-ylamme

C15H19N6O

FW: 298.35
As a mixture of two regioisomers
[3-(2-Amino-4-morpholin-4-ylmethyl-phenylcarbamoyl)-lH-pyrazol-4-yl]- carbamic acid tert-butyl ester (1.350Kg, 3.24 mol, l.Owt) and ethanol (6.75L, 5.0vol) were charged to a flange flask equipped with a mechanical stirrer, condenser and thermometer. Cone. aq. hydrochloric acid (1.10L, 13.2 mol, 0.80vol) was added at 15 to 3O0C under nitrogen and the contents were then heated to 70 to 😯0C and maintained at this temperature for 16 to 24 hours. A second portion of hydrochloric acid (0.1 IL, 1.32 mol, O.OSOvol) was added at 70 to 😯0C and the reaction was heated for a further 4 hours. Reaction completion was determined by HPLC analysis. The reaction mixture was cooled to 10 to 200C and potassium carbonate (1.355Kg, 9.08mol, l.Owt) was charged portionwise at this temperature. The suspension was stirred until gas evolution ceased and was then filtered. The filter-cake was washed with ethanol (1.35L, l.Ovol) and the filtrates retained. The filter-cake was slurried with ethanol (4.00L, 3.0vol) at 15 to 250C for 20 to 40 minutes and the mixture was filtered. The filter-cake was washed with ethanol (1.35L3 1.Ovol) and the total combined filtrates were concentrated under vacuum at 35 to 450C. Ethanol (4.00L, 3. Ovol) was charged to the residue and removed under vacuum at 35 to 450C. Tetrahydrofuran (5.90L, 4.4vol) was added to the residue and stirred for 10 to 20 minutes at 15 to 25°C. The resulting solution was filtered, the filter-cake was washed with tetrahydrofuran (1.35L, l.Ovol) and the combined filtrates were concentrated under vacuum at 35 to 450C. Tetrahydrofuran (5.40L, 4. Ovol) was charged to the concentrate and removed under vacuum at 35 to 450C. Tetrahydrofuran (5.40L, 4. Ovol) was charged to the concentrate and removed under vacuum at 35 to 45°C to give the desired product, 3-(5-morpholin-4-ylmethyl-lH- benzoimidazol-2-yl)-lH-pyrazol-4-ylamine (0.924Kg, 95.5%, 82.84% by HPLC area) as a purple foam.
Stage 9: Preparation of 7-morpholin-4-ylmethyl-2,4-dihydro- 1,2,4,5a ,10-pentaaza- cyclopentaFal fluoren-5 -one

C15H18N6O C16H16N6O2 FW: 298.35 FW: 324.34
As a mixture of two regioisomers
3-(5-Morpholin-4-ylmethyl-lH-benzoimidazol-2-yl)-lH-pyrazol-4-ylamine (0.993Kg, 3.33 mol, l.Owt) and tetrahydrofuran (14.0L, 15.0vol) were charged to a flange flask equipped with a mechanical stirrer, condenser and thermometer. The contents were stirred under nitrogen at 15 to 25°C and l,l ‘-carbonyldiimidazole (0.596Kg, 3.67 mol, O.όOwt) was added. The contents were then heated to 60 to 700C and stirred at this temperature for 16 to 24 hours. Reaction completion was determined by TLC analysis. The mixture was cooled to 15 to 200C and filtered. The filter-cake was washed with tetrahydrofuran (4.00L, 4. Ovol) and pulled dry for 15 to 30 minutes. The solid was dried under vacuum at 35 to 450C to yield 7- morpholin-4-ylmethyl-2,4-dihydro- 1 ,2,4,5a, 10-pentaaza-cyclopenta[a]fluoren-5- one (0.810Kg, 75.0%th, 92.19% by HPLC area) as a purple solid. Stage 10: Preparation of l-cvclopropyl-3-[3-(5-morpholin-4-ylmethyl-lH- benzoimidazol-2-vD- 1 H-pyrazol-4-yll -urea

C16H16N6O2 C19H23N7O2
FW: 324.34 FW: 381.44
As a mixture of two regioisomers
7-Morpholin-4-ylmethyl-254-dihydro-l,2,4,5a,10-pentaaza-cyclopenta[a]fluoren-5- one (0.797Kg, 2.46mol, l.Owt) and l-methyl-2-pyrrolidinone (2.40L, 3.0vol) were charged to a flange flask equipped with a mechanical stirrer, condenser and thermometer. Cyclopropylamine (0.279Kg, 4.88mol, 0.35 lwt) was added at 15 to 30°C under nitrogen. The contents were heated to 95 to 105°C and stirred at this temperature for 16 to 24 hours. Reaction completion was determined by 1H NMR analysis. The reaction mixture was cooled to 10 to 200C and ethyl acetate (8.00L, lO.Ovol) and sat. aq. sodium chloride (2.50L, 3.0vol) were charged, the mixture was stirred for 2 to 5 minutes and the layers separated. The organic phase was stirred with sat. aq. sodium chloride (5.00L, ό.Ovol) for 25 to 35 minutes, the mixture filtered and the filter-cake washed with ethyl acetate (0.40L, 0.5vol). The filter-cake was retained and the filtrates were transferred to a separating funnel and the layers separated. The procedure was repeated a further 3 times and the retained solids were combined with the organic phase and the mixture concentrated to dryness under vacuum at 35 to 450C. The concentrate was dissolved in propan-2-ol (8.00L, lO.Ovol) at 45 to 55°C and activated carbon (0.080Kg5 O.lwt) was charged. The mixture was stirred at 45 to 550C for 30 to 40 minutes and then hot filtered at 45 to 55°C. The filter-cake was washed with propan-2-ol (0.40L, 0.5vol). Activated carbon (0.080L, O.lwt) was charged to the combined filtrates and wash and the mixture stirred at 45 to 550C for 30 to 40 minutes. The mixture was hot filtered at 45 to 550C and the filter-cake washed with propan-2-ol (0.40L, 0.5vol). The filtrates and wash were concentrated under vacuum at 35 to 450C. Ethyl acetate (8.00, lO.Ovol) and water (2.20L, 3.0vol) were charged to the concentrate at 25 to 350C and the mixture stirred for 1 to 2 minutes. The layers were separated and the organic phase was concentrated under vacuum at 35 to 45°C. Ethyl acetate (4.00L, 5.0vol) was charged to the residue and concentrated under vacuum at 35 to 450C. Ethyl acetate (4.00L, 5.0vol) was charged to the residue and the mixture was stirred for 2 to 20 hours at 15 to 250C. The mixture was cooled to and aged at 0 to 5°C for 90 to 120 minutes and then filtered. The filter-cake was washed with ethyl acetate (0.80L, l.Ovol) and pulled dry for 15 to 30 minutes. The solid was dried under vacuum at 35 to 450C to yield l-cyclopropyl-3-[3-(5-morpholin-4-ylmethyl-lH- benzoimidazol-2-yl)-lH-pyrazol-4-yl]-urea (0.533Kg, 56.8%, 93.20% by HPLC area) as a brown solid.
Several batches of Stage 9 product were processed in this way and the details of the quantities of starting material and product for each batch are set out in Table IA.
Table IA – Yields from urea formation step – Stage 10

Stage 11 : Preparation of l-cyclopiOpyl-3-r3-(5-moipholin-4-ylmethyl-lH- benzoimidazol-2-yls)-lH-pyrazol-4-yll-urea £-lactic acid salt L-Lactic acid


acid
C19H23N7O2 C22H29N7O5
FW: 381.44 FW: 471.52 l-Cyclopropyl-3-[3-(5-morpholin-4-ylmethyl-lH-benzoimidazol-2-yl)-lH-ρyrazol- 4-yl]-urea (1.859Kg, 4.872mol, l.Owt), propan-2-ol (9.00L5 5.0vol) and ethyl acetate (8.0OL, 4.5vol) were charged to a flange flask equipped with a mechanical stirrer and thermometer. The contents were stirred under nitrogen and L-lactic acid (0.504Kg, 5.59mol, 0.269wt) was added at 15 to 25°C followed by a line rinse of ethyl acetate (0.90L, 0.5vol). The mixture was stirred at 15 to 25°C for 120 to 140 minutes. The solid was isolated by filtration, the filter-cake washed with ethyl acetate (2x 2.00L, 2x l.Ovol) and pulled dry for 20 to 40 minutes. The filter-cake was dissolved in ethanol (33.00L, 17.7vol) at 75 to 850C, cooled to 65 to 700C and the solution clarified through glass microfibre paper. The filtrates were cooled to and aged at 15 to 250C for 2 to 3 hours. The crystallised solid was isolated by filtration, the filter-cake washed with ethanol (2x 1.00L, 2x 0.5vol) and pulled dry for at least 30 minutes. The solid was dried under vacuum at 35 to 45°C to yield 1- cyclopropyl-3 – [3-(5 -morpholin-4-ylmethyl- 1 H-benzoimidazol-2-yl)- 1 H-pyrazol-4- yl]-urea l-lactic acid salt (1.386Kg, 58.7%th, 99.47% by HPLC area,) as a dark pink uniform solid.
The infra-red spectrum of the lactate salt (KBr disc method) included characteristic peaks at 3229, 2972 and 1660 cm“1.
Without wishing to be bound by any theory, it is believed that the infra red peaks can be assigned to structural components of the salt as follow:
Peak: Due to:
3229 cm“1 N-H
2972 cm“1 aliphatic C-H
1660 cm“1 urea C=O EXAMPLE 67
Synthesis of Crystalline Free Base And Crystalline Salt Forms Of l-Cyclopropyl-3-
[3-(5-Morpholin-4-ylmethyl-lH-Benzoimidazol-2-vπ-lH-Pyrazol-4-yll-Urea
A. Preparation of l-Cvclopropyl-3-[3-f5-Moφholm-4-ylmethyl-lH- Benzoimidazol-2-yl)-lH-Pyrazol-4-yll-Urea free base
A sample of crude l-cyclopropyl-3-[3-(5-morpholin-4-ylmethyl-lH-benzoimidazol- 2-yl)-lH-pyrazol-4-yl]-urea free base was prepared as outlined in Example 60 and initially purified by column chromatography on silica gel, eluting with EtOAc- MeOH (98:2 – 80:20). A sample of the free base obtained was then recrystallised from hot methanol to give crystalline material of l-cyclopropyl-3-[3-(5-morpholin- 4-ylmethyl- 1 H-benzoimidazol-2-yl)- 1 H-pyrazol-4-yl] -urea free base.
B. Preparation of l-Cyclopropyl-S-rS-fS-Morpholin^-ylmethyl-lH-Benzoimidazol- 2-yl)-lH-Pyrazol-4-yl]-Urea free base dihydrate
A sample of crude l-cyclopropyl-3-[3-(5-moφholm-4-ylmethyl-lH-benzoimidazol- 2-yl)-l H-pyrazol-4-yl] -urea free base was dissolved in THF and then concentrated in vacuo to a minimum volume (~4 volumes). To the solution was added water dropwise (2 – 4 volumes) until the solution became turbid. A small amount of THF was added to re-establish solution clarity and the mixture left to stand overnight to give a crystalline material which was air-dried to give l-cyclopropyl-3-[3-(5- morpholin-4-ylmethyl- 1 H-benzoimidazol-2-yl)- 1 H-pyrazol-4-yl] -urea free base dihydrate.
C. Preparation of l-Cyclopl^pyl-3-[3-(5-Morpholm-4-ylmethyl-lH-Benzoimidazol- 2-ylVlH-Pyrazol-4-yl]-Urea hydrochloride salt
A sample of crude l-cyclopropyl-3-[3-(5-moφholin-4-ylmethyl-lH-benzoimidazol- 2-yl)-l H-pyrazol-4-yl] -urea free base was dissolved in the minimum amount of MeOH and then diluted with EtOAc. To the solution at 0 °C was slowly added 1.1 equivalents of HCl (4M solution in dioxane). Following addition, solid precipitated from solution which was collected by filtration. To the solid was added MeOH and the mixture reduced in vacuo. To remove traces of residual MeOH the residue was evaporated from water and then dried at 60 0C/ 0.1 mbar to give the hydrochloride salt.
D. Preparation of l-Cyclopropyl-3-[3-(‘5-Morpholm-4-ylmethyl-lH- Benzoimidazol-2-yiyiH-Pyrazol-4-yl1-Urea ethanesulfonate salt
To a solution of l-cyclopropyl-3-[3-(5-morpholin-4-ylmethyl-lH-benzoimidazol-2- yl)-lH-pyrazol-4-yl]-urea free base in MeOH-EtOAc was added 1 equivalent of ethanesulfonic acid. The mixture was stirred at ambient temperature and then reduced in vacuo. The residue was taken up in MeOH and to the solution was added Et2O. Mixture left to stand for 72 h and the solid formed collected by filtration and dried to give l-cyclopropyl-3-[3-(5-morpholin-4-ylmethyl-lH- benzoimidazol-2-yl)-lH-pyrazol-4-yl]-urea ethanesulfonate salt.
E. Preparation of l-Cvclopropyl-3-[3-(‘5-Morpholm-4-ylmethyl-lH-Benzoimidazol- 2-yl)-lH-Pyrazol-4-yl]-Urea methanesulfonate salt
To a solution of l-cyclopropyl-3-[3-(5-morpholin-4-ylmethyl-lH-benzoimidazol-2- yl)-lH-pyrazol-4-yl]-urea free base (394 mg) in MeOH-EtOAc was added 1 equivalent of methanesulfonic acid (67 μl). A solid was formed which was collected by filtration, washing with EtOAc. The solid was dissolved in the minimum amount of hot MeOH, allowed to cool and then triturated with Et2O. The solid was left to stand for 72 h and then collected by filtration, washing with MeOH, to give l-cyclopropyl-3-[3-(5-morpholin-4-ylmethyl-lH-benzoimidazol-2- yl)-lH-pyrazol-4-yl]-urea methanesulfonate salt.
EXAMPLE 68
Characterisation of l-Cvclopropyl-3-[3-(5-Morpholin-4-ylmethyl-lH-
Benzoimidazol-2-yl)-lH-Pyrazol-4-yll-Urea Free Base and Salts
Various forms of l-cyclopropyl-3-[3-(5-morpholin-4-ylmethyl-lH-benzoimidazol- 2-yl)-lH-pyrazol-4-yl]-urea were characterised. The forms selected for characterisation were identified from studies which primarily investigated extent of polymorphism and salt stability. The salts selected for further characterisation were the L-lactate salt, Free base dihydrate, Esylate salt, Free base and Hydrochloride salt.

Paper
Fragment-Based Discovery of the Pyrazol-4-yl Urea (AT9283), a Multitargeted Kinase Inhibitor with Potent Aurora Kinase Activity†
†
Coordinates of the protein complexes with compounds 5, 7, 9, 10, and 16 have been deposited in the Protein Data Bank under accession codes 2w1d, 2w1f, 2w1c, 2w1e, 2w1g (Aurora A), 2w1h (CDK2), and 2w1i (JAK2).
, * To whom correspondence should be addressed. Phone: +44 (0)1223 226209. Fax: +44 (0)1223 226201. E-mail: s.howard@astex-therapeutics.com.
Abstract

Here, we describe the identification of a clinical candidate via structure-based optimization of a ligand efficient pyrazole-benzimidazole fragment. Aurora kinases play a key role in the regulation of mitosis and in recent years have become attractive targets for the treatment of cancer. X-ray crystallographic structures were generated using a novel soakable form of Aurora A and were used to drive the optimization toward potent (IC50 ≈ 3 nM) dual Aurora A/Aurora B inhibitors. These compounds inhibited growth and survival of HCT116 cells and produced the polyploid cellular phenotype typically associated with Aurora B kinase inhibition. Optimization of cellular activity and physicochemical properties ultimately led to the identification of compound16 (AT9283). In addition to Aurora A and Aurora B, compound 16 was also found to inhibit a number of other kinases including JAK2 and Abl (T315I). This compound demonstrated in vivo efficacy in mouse xenograft models and is currently under evaluation in phase I clinical trials.
1-Cyclopropyl-3-[3-(5-morpholin-4-ylmethyl-1H-benzoimidazol-2-yl)-1H-pyrazol-4-yl]urea (16), Hydrochloride Salt
///////////
C1CC1NC(=O)NC2=CNNC2=C3N=C4C=CC(=CC4=N3)CN5CCOCC5
VAL-083

VAL-083
(1R,2S)-1-((R)-oxiran-2-yl)-2-((S)-oxiran-2-yl)ethane-1,2-diol
Galactitol, 1,2:5,6-dianhydro-
- 1,2:5,6-Dianhydrodulcitol
- 1,2:5,6-Dianhydrogalactitol
- 1,2:5,6-Diepoxydulcitol
Dianhydrodulcitol; Dianhydrogalactitol; VAL083; VAL 083, Dulcitol diepoxide, NSC 132313
CAS 23261-20-3
MF C6H10O4, MW 146.14
VAL-083 is a bi-functional alkylating agent; inhibit U251 and SF188 cell growth in monolayer better than TMZ and caused apoptosis
VAL-083 is a bi-functional alkylating agent, with potential antineoplastic activity. Upon administration, VAL-083 crosses the blood brain barrier (BBB) and appears to be selective for tumor cells. This agent alkylates and crosslinks DNA which ultimately leads to a reduction in cancer cell proliferation. In addition, VAL-083 does not show cross-resistance to other conventional chemotherapeutic agents and has a long half-life in the brain. Check for active clinical trials or closed clinical trials using this agent
Currently, VAL-083 is approved in China to treat chronic myelogenous leukemia and lung cancer, while the drug has also secured orphan drug designation in Europe and the US to treat malignant gliomas.
LAUNCHED CHINA FOR Cancer, lung
Del Mar Pharmaceuticals Inc……..Glioblastoma…………..PHASE2
DelMar and MD Anderson to accelerate development of anti-cancer drug VAL-083
DelMar Pharmaceuticals has collaborated with the University of Texas MD Anderson Cancer Center (MD Anderson) to speed up the clinical development of its VAL-083 anti-cancer drug.

VAL-083 is a BI-Functional alkylating agent; INHIBIT U251 and SF188 Cell Growth in monolayer Better than TMZ and Caused apoptosis. IC50 Value : 5 uM (INHIBIT U251, SF188, T98G Cell Growth in monolayer after 72h) [1]. in vitro :.. VAL-083 INHIBITED U251 and SF188 Cell Growth in monolayer and as neurospheres Better than TMZ and Caused apoptosis after 72 hr Formation Assay In the colony, VAL-083 (5 uM) SF188 Growth suppressed by about 95% are T98G cells classically TMZ-resistant and express MGMT, but VAL-083 inhibited their growth in monolayer after 72 hr in a dose-dependent manner (IC50, 5 uM). VAL-083 also inhibited the growth of CSCs (BT74, GBM4, and GBM8) . by 80-100% in neurosphere self-Renewal assays Conversely, there was minimal normal Effect on Human Neural stem cells [1]. in Vivo : Clinical Trial : Safety Study of VAL-083 in Patients With Recurrent Malignant glioma or Secondary Progressive Brain Tumor. Phase 1 / Phase 2

VAL-083 has demonstrated activity in cyclophosphamide, BCNU and phenylanine mustard resistant cell lines and no evidence of cross-resistance has been encountered in published clinical studies. Based on the presumed alkylating functionality of VAL-083, published literature suggests that DNA repair mechanisms associated with Temodar and nitrosourea resistance, such as 06-methylguanine methyltransferace (MGMT), may not confer resistance to VAL-083. VAL-083 readily crosses the blood brain barrier where it maintains a long half-life in comparison to the plasma. Published preclinical and clinical research demonstrates that VAL-083 is selective for brain tumor tissue. VAL-083 has been assessed in multiple studies as chemotherapy in the treatment of newly diagnosed and recurrent brain tumors. In published clinical studies, VAL-083 has previously been shown to have a statistically significant impact on median survival in high grade gliomas when combined with radiation vs. radiation alone. The main dose-limiting toxicity related to the administration of VAL-083 in previous clinical studies was myelosuppression

Glioblastoma is the most common form of primary brain cancer
DelMar Pharmaceuticals has collaborated with the University of Texas MD Anderson Cancer Center (MD Anderson) to speed up the clinical development of its VAL-083 anti-cancer drug.
VAL-083 is a small-molecule chemotherapeutic designed to treat glioblastoma multiforme (GBM), the most common and deadly cancer that starts within the brain.
Under the deal, MD Anderson will begin a new Phase II clinical trial with VAL-083 in patients with GBM at first recurrence / progression, prior to Avastin (bevacizumab) exposure.
During the trial, eligible patients will have recurrent GBM characterised by a high expression of MGMT, the DNA repair enzyme implicated in drug-resistance, and poor patient outcomes following current front-line chemotherapy.
The company noted that MGMT promoter methylation status will be used as a validated biomarker for enrollment and tumours must exhibit an unmethylated MGMT promoter for patients to be eligible for the trial.
DelMar chairman and CEO Jeffrey Bacha said: “The progress we continue to make with our research shows that VAL-083 may offer advantages over currently available chemotherapies in a number of tumour types.
“This collaboration will allow us to leverage world-class clinical and research expertise and a large patient population from MD Anderson as we extend and accelerate our clinical focus to include GBM patients, following first recurrence of their disease.
“We believe that VAL-083’s unique cytotoxic mechanism offers promise for GBM patients across the continuum of care as a potential superior alternative to currently available cytotoxic chemotherapies, especially for patients whose tumours exhibit a high-expression of MGMT.”
The deal will see DelMar work with the scientists and clinicians at MD Anderson to accelerate its research in order to transform the treatment of patients whose cancers fail or are unlikely to respond to existing treatments.
In more than 40 clinical trials, VAL-083 showed clinical activity against several cancers including lung, brain, cervical, ovarian tumours and leukemia both as a single-agent and in combination with other treatments.
PATENT
WO 2012024368
https://www.google.com/patents/WO2012024368A3?cl=en
Dianhydrogalactitol (DAG or dianhydrodulcitol) can be synthesized from dulcitol which can be produced from natural sources (such as Maytenus confertiflora) or commercial sources.The structure of DAG is given below as Formula (I).

One method for the preparation of dulcitol from Maytenus confertiflora is as follows: (1) The Maytenus confertiflora plant is soaked in diluted ethanol (50-80%) for about 24 hours, and the soaking solution is collected. (2) The soaking step is repeated, and all soaking solutions are combined. (3) The solvent is removed by heating under reduced pressure. (4) The concentrated solution is allowed to settle overnight and the clear supernatant is collected. (5) Chloroform is used to extract the supernatant. The chloroform is then removed under heat and reduced pressure. (6) The residue is then dissolved in hot methanol and cooled to allow crystallization. (7) The collected crystals of dulcitol are filtered and dried under reduced pressure. The purified material is dulcitol, contained in the original Maytenus confertiflora plant at a concentration of about 0.1% (1/1000).
DAG can be prepared by two general synthetic routes as described below:
Route 1 :
Dulcitol DAG
Route 2. Dulcitol

In Route 1 , “Ts” represents the tosyl group, or p-toluenesulfonyl group. PATENT
However, the intermediate of Route 1, 1,6-ditosy)dulcitol, was prepared with low yield (~36%), and the synthesis of 1,6-ditosyldulcitol was poorly reproducible. Therefore, the second route process was developed, involving two major steps: (1) preparation of dibromodulcitol from dulcitol; and (2) preparation of dianhydrodulcitol from dibromodulcitol.
Dibromodulcitol is prepared from dulcitol as follows: (1) With an aqueous HBr solution of approximately 45% HBr concentration, increase the HBr concentration to about 70% by reacting phosphorus with bromine in concentrated HBr in an autoclave. Cool the solution to 0° C. The reaction is:
2P+3Br2→2PBr3+H20→HBr†+H3P04. (2) Add the dulcitol to the concentrated HBr solution and reflux at 80° C to complete the reaction. (3) Cool the solution and pour the mixture onto ice water. Dibromodulcitol is purified through recrystallization.
The results for the preparation of dibromodulcitol (DBD) are shown in Table 1, below.
TABLE 1

For the preparation of DAG from DBD, DBD was poorly dissolved in methanol and ethanol at 40° C (different from what was described in United States PATENT
Patent No. 3,993,781 to Horvath nee Lengyel et al., incorporated herein by this reference). At refluxing, DBD was dissolved but TLC showed that new impurities formed that were difficult to remove from DBD.
The DBD was reacted with potassium carbonate to convert the DBD to dianhydrogalactitol.
The results are shown in Table 2, below.
TABLE 2

In the scale-up development, it was found the crude yield dropped significantly. It is unclear if DAG could be azeotropic with BuOH. It was confirmed that t-BuOH is essential to the reaction. Using MeOH as solvent would result in many impurities as shown spots on TLC. However, an improved purification method was developed by using a slurry with ethyl ether, which could provide DAG with good purity. This was developed after a number of failed attempts at recrystallization of DAG.

Bromination of dulcitol with HBr at 80°C gives dibromodulcitol , which upon epoxidation in the presence of K2CO3 in t-BuOH or NaOH in H2O or in the presence of ion exchange resin Varion AD (OH) (4) affords the target dianhydrogalactitol .
PATENT



SCHEME 5



PATENT
CN 103923039
http://www.google.com/patents/CN103923039A?cl=en
The resulting Dulcitol 9g and 18ml mass percent concentration of 65% hydrobromic acid at 78 ° C under reflux for 8 hours to give 1,6-dibromo dulcitol, and the product is poured into ice crystals washed anhydrous tert-butyl alcohol, and dried to give 1,6-dibromo dulcitol crystal, then 10.0gl, 6- dibromo dulcitol sample is dissolved in t-butanol, adding solid to liquid 2 % obtained through refining process 1,6_ dibromo dulcitol seed stirred and cooled to 0 ° C, allowed to stand for seven days to give 1,6_ dibromo dulcitol crystal, anhydrous t-butanol, dried to give 1,6-dibromo dulcitol. 5g of the resulting 1,6_ dibromo Euonymus dissolved in 50ml tert-butanol containing 5g of potassium carbonate, the elimination reaction, at 80 ° C under reflux time was 2 hours, the resulting product was dissolved in t-butanol, Join I% stock solution to the water quality of 1,2,4,5_ two Dulcitol including through a purification step to get less than 1% of 1,2,5,6_ two to water Dulcitol seeded stirring, cooling to 0 ° C, allowed to stand for I-day, two to go get 1,2,5,6_ water Dulcitol crystals washed anhydrous tert-butyl alcohol, and dried to give 1,2,5,6 two to crystalline water Dulcitol and lyophilized to give two to water Dulcitol lyophilized powder, containing I, 2,4,5- two to water Dulcitol less than 0.3%.
PATENT
PATENT
-
DAG can be prepared by two general synthetic routes as described below:
-

-
In Route 1, “Ts” represents the tosyl group, or p-toluenesulfonyl group.
-
However, the intermediate of Route 1, 1,6-ditosyldulcitol, was prepared with low yield (˜36%), and the synthesis of 1,6-ditosyldulcitol was poorly reproducible. Therefore, the second route process was developed, involving two major steps: (1) preparation of dibromodulcitol from dulcitol; and (2) preparation of dianhydrodulcitol from dibromodulcitol.
-
Dibromodulcitol is prepared from dulcitol as follows: (1) With an aqueous HBr solution of approximately 45% HBr concentration, increase the HBr concentration to about 70% by reacting phosphorus with bromine in concentrated HBr in an autoclave. Cool the solution to 0° C. The reaction is: 2P+3Br2→2PBr3+H2O→HBr↑+H3PO4. (2) Add the dulcitol to the concentrated HBr solution and reflux at 80° C. to complete the reaction. (3) Cool the solution and pour the mixture onto ice water. Dibromodulcitol is purified through recrystallization.

PATENT
US 20150329511
PAPER
Antibacterial and Anti-Quorum Sensing Molecular Composition Derived from Quercus cortex (Oak bark) Extract
Journal of the American Chemical Society, 1991 , vol. 113, 7 pg. 2786 – 2787

REFERENCES
Currently, VAL-083 is approved in China to treat chronic myelogenous leukemia and lung cancer, while the drug has also secured orphan drug designation in Europe and the US to treat malignant gliomas.
[1]. Fotovati A, Hu KJ, Wakimoto H, VAL-083, A NOVEL N7 ALKYLATING AGENT, SURPASSES TEMOZOLOMIDE ACTIVITY AND INHIBITS CANCER STEM CELLS, PROVIDING A NEW POTENTIAL TREATMENT OPTION FOR GLIOBLASTOMA MULTIFORME. Neuro-oncology, 2012, 14, AbsET-37, Suppl. 6
1: Szende B, Jeney A, Institoris L. The diverse modification of N-butyl-N-(4-hydroxybutyl) nitrosamine induced carcinogenesis in urinary bladder by dibromodulcitol and dianhydrodulcitol. Acta Morphol Hung. 1992;40(1-4):187-93. PubMed PMID: 1365762.
2: Anderlik P, Szeri I, Bános Z. Bacterial translocation in dianhydrodulcitol-treated mice. Acta Microbiol Hung. 1988;35(1):49-54. PubMed PMID: 3293340.
3: Huang ZG. [Clinical observation of 15 cases of chronic myelogenous leukemia treated with 1,2,5,6-dianhydrodulcitol]. Zhonghua Nei Ke Za Zhi. 1982 Jun;21(6):356-8. Chinese. PubMed PMID: 6957285.
4: Anderlik P, Szeri I, Bános Z, Wessely M, Radnai B. Higher resistance of germfree mice to dianhydrodulcitol, a lymphotropic cytostatic agent. Acta Microbiol Acad Sci Hung. 1982;29(1):33-40. PubMed PMID: 6211912.
5: Bános Z, Szeri I, Anderlik P. Effect of Bordetella pertussis vaccine on the course of lymphocytic choriomeningitis (LCM) virus infection in suckling mice pretreated with dianhydrodulcitol (DAD). Acta Microbiol Acad Sci Hung. 1979;26(2):121-5. PubMed PMID: 539467.
6: Bános Z, Szeri I, Anderlik P. Dianhydrodulcitol treatment of lymphocytic choriomeningitis virus infection in suckling mice. Acta Microbiol Acad Sci Hung. 1979;26(1):29-34. PubMed PMID: 484266.
7: Gerö-Ferencz E, Tóth K, Somfai-Relle S, Gál F. Effect of dianhydrodulcitol (DAD) on the primary immune response of normal and tumor bearing rats. Oncology. 1977;34(4):150-2. PubMed PMID: 335301.
8: Kopper L, Lapis K, Institóris L. Incorporation of 3H-dibromodulcitol and 3H-dianhydrodulcitol into ascites tumor cells. Autoradiographic study. Neoplasma. 1976;23(1):47-52. PubMed PMID: 1272473.
9: Bános S, Szeri I, Anderlik P. Combined phytohaemagglutinin and dianhydrodulcitol treatment of lymphocytic choriomeningitis virus infection in mice. Acta Microbiol Acad Sci Hung. 1975;22(3):237-40. PubMed PMID: 1155228.
Carbohydrate Research, 1982 , vol. 108, p. 173 – 180
Deryabin, Dmitry G.; Tolmacheva, Anna A.
Molecules, 2015 , vol. 20, 9 pg. 17093 – 17108
Gati; Somfai-Relle
Arzneimittel-Forschung/Drug Research, 1982 , vol. 32, 2 pg. 149 – 151
| WO2013128285A2 * | Feb 26, 2013 | Sep 6, 2013 | Del Mar Pharmaceuticals | Improved analytical methods for analyzing and determining impurities in dianhydrogalactitol |
| WO2013128285A3 * | Feb 26, 2013 | Dec 27, 2013 | Del Mar Pharmaceuticals | Improved analytical methods for analyzing and determining impurities in dianhydrogalactitol |
| US9029164 | Nov 18, 2013 | May 12, 2015 | Del Mar Pharmaceuticals | Analytical methods for analyzing and determining impurities in dianhydrogalactitol |
| US3470179 * | Jun 14, 1966 | Sep 30, 1969 | Sandoz Ag | 4-substituted-3,4-dihydroquinazolines |
| US20020032230 * | May 21, 2001 | Mar 14, 2002 | Dr. Reddy’s Laboratories Ltd. | Novel compounds having antiinflamatory activity: process for their preparation and pharmaceutical compositions containing them |
| US20020037328 * | May 31, 2001 | Mar 28, 2002 | Brown Dennis M. | Hexitol compositions and uses thereof |
| CN101045542A * | Apr 6, 2007 | Oct 3, 2007 | 中国科学院过程工程研究所 | Method for preparing water softening aluminium stone of sodium aluminate solution carbonation resolving |
| CN101654270A * | Sep 10, 2009 | Feb 24, 2010 | 沈阳工业大学 | Method for eliminating periodic thinning of granularity of seed product |
| CN101775413A * | Mar 23, 2010 | Jul 14, 2010 | 禹城绿健生物技术有限公司 | Technique for producing xylitol and dulcitol simultaneously |
| CN103270035A * | Aug 17, 2011 | Aug 28, 2013 | 德玛医药 | Method of synthesis of substituted hexitols such as dianhydrogalactitol |
/////////////////
C1C(O1)C(C(C2CO2)O)O
O[C@H]([C@H]1OC1)[C@@H](O)[C@H]2CO2
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Facebook 

 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
GS 9883, Bictegravir an HIV-1 integrase inhibitor
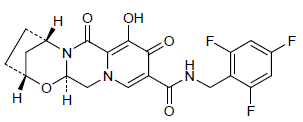

GS 9883, bictegravir
CAS 1611493-60-7
PHASE 3
HIV-1 integrase inhibitor
(2R,5S,13aR)-8-hydroxy-7,9-dioxo-N-[(2,4,6-trifluorophenyl)methyl]-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide
2,5-Methanopyrido(1′,2′:4,5)pyrazino(2,1-b)(1,3)oxazepine-10-carboxamide, 2,3,4,5,7,9,13,13a-octahydro-8-hydroxy-7,9-dioxo-N-((2,4,6-trifluorophenyl)methyl)-, (2R,5S,13aR)-
(2R,5S,13aR)-8-hydroxy-7,9-dioxo-N-(2,4,6-trifluorobenzyl)-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide
(2 ,5S,13aI )-8-hydroxy-7,9-dioxo-N-(2,4,6-trifluoroheoctahydro-2,5-methanopyrido[ 1 ‘,2’:4,5]pyrazino[2, 1 -b][ 1 ,3]oxazepine- 10-carboxamide
MF C21H18F3N3O5,
| MW | 449.37993 g/mol |
|---|
UNII-8GB79LOJ07; 8GB79LOJ07

BICTEGRAVIR
- 16 Nov 2015 Phase-III clinical trials in HIV-1 infections (Combination therapy, Treatment-naive) in USA (PO) (Gilead Pipeline, November 2015)
- 01 Jul 2015 Gilead Sciences completes a phase I trial in HIV-1 infections in USA and New Zealand (NCT02400307)
- 01 Apr 2015 Phase-I clinical trials in HIV-1 infections (In volunteers) in New Zealand (PO) (NCT02400307)
UPDATE Biktarvy (bictegravir/emtricitabine/tenofovir alafenamide); Gilead; For the treatment of HIV-1 infection in adults, Approved February 2018
Human immunodeficiency virus infection and related diseases are a major public health problem worldwide. Human immunodeficiency virus type 1 (HIV-1) encodes three enzymes which are required for viral replication: reverse transcriptase, protease, and integrase. Although drugs targeting reverse transcriptase and protease are in wide use and have shown effectiveness, particularly when employed in combination, toxicity and development of resistant strains have limited their usefulness (Palella, et al. N. Engl. J Med. (1998) 338:853-860; Richman, D. D. Nature (2001) 410:995-1001). Accordingly, there is a need for new agents that inhibit the replication of HIV and that minimize PXR activation when co-administered with other drugs.
Certain polycyclic carbamoylpyridone compounds have been found to have antiviral activity, as disclosed in PCT/US2013/076367. Accordingly, there is a need for synthetic routes for such compounds.
SYNTHESIS
WO 2014100323

PATENTS
xample 42
Preparation of Compound 42
(2 ,5S,13aI )-8-hydroxy-7,9-dioxo-N-(2,4,6-trifluorohe
octahydro-2,5-methanopyrido[ 1 ‘,2’:4,5]pyrazino[2, 1 -b][ 1 ,3]oxazepine- 10-carboxamide

42

Step 1
l-(2,2-dimethoxyethyl)-5-methoxy-6-(methoxycarbonyl)-4-oxo-l ,4-dihydropyridine-3-carboxylic acid (3.15 g, 10 mmol) in acetonitrile (36 mL) and acetic acid (4 mL) was treated with methanesuffhnic acid (0.195 mL, 3 mmol) and placed in a 75 deg C bath. The reaction mixture was stirred for 7 h, cooled and stored at -10 °C for 3 days and reheated to 75 °C for an additional 2 h. This material was cooled and carried on crude to the next step.
Step 2
Crude reaction mixture from step 1 (20 mL, 4.9 mmol) was transferred to a flask containing (lR,3S)-3-aminocyclopentanol (0.809 g, 8 mmol). The mixture was diluted with acetonitrile (16.8 mL), treated with potassium carbonate (0.553 g, 4 mmol) and heated to 85 °C. After 2 h, the reaction mixture was cooled to ambient temperature and stirred overnight. 0.2M HQ (50 mL) was added, and the clear yellow solution was extracted with dichloromethane (2×150 mL). The combined organic layers were dried over sodium sulfate, filtered and concentrated to 1.49 g of a light orange solid. Recrystallization from dichloimethane:hexanes afforded the desired intermediate 42 A: LC S-ESI (m/z): [M+H]+ calculated for Ci5Hi7N206: 321.1 1 ; found: 321.3.
Step 3
Intermediate 42-A (0.225 g, 0.702 mmol) and (2,4,6-trifluorophenyl)methanamine (0.125 g, 0.773 mmol) were suspended in acetonitrile (4 mL) and treated with N,N-diisopropylethylamine (DIPEA) (0.183 mmol, 1.05 mmol). To this suspension was added (dimethyiammo)- V,A/-dimethyi(3H-[l ,2,3]triazolo[4,5-&]pyridm~3-yiox.y)methammimum hexafluorophosphate (HATU, 0.294 g, 0.774 mmol). After 1.5 hours, the crude reaction mixture was taken on to the next step. LfJMS-ESlT (m/z): [M+H calculated for (\ ,l l.,, i \\:0< : 464.14; found: 464.2.
Step 4
To the crude reaction mixture of the previous step was added MgBr2
(0.258 g, 1.40 mmol). The reaction mixture was stirred at 50 °C for 10 minutes, acidified with 10% aqueous HC1, and extract twice with dichloromethane. The combined organic phases were dried over MgS04, filtered, concentrated, and purified by silica gel chromatography (EtOH/dichlormethane) followed by HPLC (ACN H2O with 0.1 % TFA modifier) to afford compound 42: 1H~ M (400 MHz, DMSO-</6) δ 12.43 (s, 1H), 10.34 (t, J = 5.7 Hz, IH), 8.42 (s, 1H), 7.19 (t, J = 8.7 Hz, 2H), 5.43 (dd, ./’ 9.5, 4.1 Hz, I H), 5.08 (s, i l l ). 4.66 (dd, ./ 12.9, 4.0 Hz, IH), 4.59 (s, 1 1 1 ). 4.56 4.45 (m, 2H), 4.01 (dd, J = 12.7, 9.7 Hz, IH), 1.93 (s, 4H), 1.83 (d, J —— 12.0 Hz, I H),
1.56 (dt, J = 12.0, 3.4 Hz, I H). LCMS-ESI+ (m/z): [M+H]+ calculated for { · Ί ί ] ΝΓ :Χ.¾ϋ : 450.13; found: 450.2.
PATENT
WO2015177537
PATENT
WO2015196116
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015196116&redirectedID=true
PATENT
WO2015196137
PATENT
http://www.google.com/patents/US20140221356
Example 42 Preparation of Compound 42 (2R,5S,13aR)-8-hydroxy-7,9-dioxo-N-(2,4,6-trifluorobenzyl)-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide

Step 1
-
1-(2,2-dimethoxyethyl)-5-methoxy-6-(methoxycarbonyl)-4-oxo-1,4-dihydropyridine-3-carboxylic acid (3.15 g, 10 mmol) in acetonitrile (36 mL) and acetic acid (4 mL) was treated with methanesulfonic acid (0.195 mL, 3 mmol) and placed in a 75 deg C. bath. The reaction mixture was stirred for 7 h, cooled and stored at −10° C. for 3 days and reheated to 75° C. for an additional 2 h. This material was cooled and carried on crude to the next step.
Step 2
-
Crude reaction mixture from step 1 (20 mL, 4.9 mmol) was transferred to a flask containing (1R,3S)-3-aminocyclopentanol (0.809 g, 8 mmol). The mixture was diluted with acetonitrile (16.8 mL), treated with potassium carbonate (0.553 g, 4 mmol) and heated to 85° C. After 2 h, the reaction mixture was cooled to ambient temperature and stirred overnight. 0.2M HCl (50 mL) was added, and the clear yellow solution was extracted with dichloromethane (2×150 mL). The combined organic layers were dried over sodium sulfate, filtered and concentrated to 1.49 g of a light orange solid. Recrystallization from dichlormethane:hexanes afforded the desired intermediate 42A: LCMS-ESI+ (m/z): [M+H]+ calculated for C15H17N2O6: 321.11; found: 321.3.
Step 3
-
Intermediate 42-A (0.225 g, 0.702 mmol) and (2,4,6-trifluorophenyl)methanamine (0.125 g, 0.773 mmol) were suspended in acetonitrile (4 mL) and treated with N,N-diisopropylethylamine (DIPEA) (0.183 mmol, 1.05 mmol). To this suspension was added (dimethylamino)-N,N-dimethyl(3H-[1,2,3]triazolo[4,5-b]pyridin-3-yloxy)methaniminium hexafluorophosphate (HATU, 0.294 g, 0.774 mmol). After 1.5 hours, the crude reaction mixture was taken on to the next step. LCMS-ESI+ (m/z): [M+H]+ calculated for C22H21F3N3O5: 464.14; found: 464.2.
Step 4
-
To the crude reaction mixture of the previous step was added MgBr2 (0.258 g, 1.40 mmol). The reaction mixture was stirred at 50° C. for 10 minutes, acidified with 10% aqueous HCl, and extract twice with dichloromethane. The combined organic phases were dried over MgSO4, filtered, concentrated, and purified by silica gel chromatography (EtOH/dichlormethane) followed by HPLC (ACN/H2O with 0.1% TFA modifier) to afford compound 42: 1H-NMR (400 MHz, DMSO-d6) δ 12.43 (s, 1H), 10.34 (t, J=5.7 Hz, 1H), 8.42 (s, 1H), 7.19 (t, J=8.7 Hz, 2H), 5.43 (dd, J=9.5, 4.1 Hz, 1H), 5.08 (s, 1H), 4.66 (dd, J=12.9, 4.0 Hz, 1H), 4.59 (s, 1H), 4.56-4.45 (m, 2H), 4.01 (dd, J=12.7, 9.7 Hz, 1H), 1.93 (s, 4H), 1.83 (d, J=12.0 Hz, 1H), 1.56 (dt, J=12.0, 3.4 Hz, 1H). LCMS-ESI+ (m/z): [M+H]+ calculated for C21H19F3N3O5: 450.13; found: 450.2.
PATENT
General Scheme I:

General Scheme II:



General Scheme II
General Scheme III:


General Scheme III
General Scheme IV:


G-1
General Scheme V:

II
EXAMPLES
In order for this invention to be more fully understood, the following examples are set forth. These examples are for the purpose of illustrating embodiments, and are not to be construed as limiting the scope of this disclosure in any way. The reactants used in the examples below may be obtained either as described herein, or if not described herein, are themselves either commercially available or may be prepared from commercially available materials by methods known in the art.
In one embodiment, a multi-step synthetic method for preparing a compound of Formula I is provided, as set forth below. In certain embodiments, each of the individual steps of the Schemes set forth below is provided. Examples and any combination of two or more successive steps of the below Examples are provided.
A. Acylation and amidation of Meldrum ‘s acid to form C-la:

[0520] In a reaction vessel, Meldrum’s acid (101 g, 1.0 equivalent) and 4-dimethylaminopyridine (1.8 g, 0.2 equivalents) were combined with acetonitrile (300 mL). The resulting solution was treated with methoxyacetic acid (6.2 mL, 1.2 equivalents). Triethylamine (19.4 mL, 2.0 equivalents) was added slowly to the resulting solution, followed by pivaloyl chloride (9.4 mL, 1.1 equivalents). The reaction was then heated to about 45 to about 50 °C and aged until consumption of Meldrum’s acid was deemed complete.
A separate reaction vessel was charged with acetonitrile (50 mL) and J-la (13.4 g, 1.2 equivalents). The resulting solution was treated with trifluoroacetic acid (8.0 mL, 1.5 equivalents), and then this acidic solution was added to the acylation reaction in progress at about 45 to about 50 °C.
The reaction was allowed to age for at least 18 hours at about 45 to about 50 °C, after which time the solvent was removed under reduced pressure. The crude residue was dissolved in ethyl acetate (150 mL), and the organic layer was washed with water. The combined aqueous layers were extracted with ethyl acetate. The combined organic layers were washed with saturated sodium bicarbonate solution, and the combined bicarbonate washes were back extracted with ethyl acetate. The combined organic layers were dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The resulting crude material was purified twice via silica gel chromatography to yield C-la.
lH NMR (400 MHz, CDC13): δ 7.12 (br, 1H), 6.66 (app t, J= 8.1 Hz, 2H), 4.50 (app d, J= 5.7 Hz, 2H), 4.08 (s, 2H), 3.44 (s, 2H), 3.40 (s, 3H). 13C NMR (100 MHz, CDC13): δ 203.96, 164.90, 162.37 (ddd, J= 250.0, 15.7, 15.7 Hz), 161.71 (ddd, J = 250.3, 14.9, 10.9 Hz), 110.05 (ddd, J= 19.7, 19.7, 4.7 Hz), 100.42 (m), 77.58, 59.41, 45.71, 31.17 (t, J= 3.5 Hz). LCMS, Calculated: 275.23, Found: 275.97 (M).
I l l
B. Alkylation of C-la to form E-la:

A solution of C-la (248 mg, 1.0 equivalent) and 2-methyl tetrahydrofuran (1.3 niL) was treated with N,N-dimethylformamide dimethylacetal (0.1 mL, 1.1 equivalent) and stirred at room temperature overnight (~14 hours). The reaction was treated with aminoacetaldehyde dimethyl acetal (0.1 mL, 1.0 equivalents), and was allowed to age for about 2 hours, and then was quenched via the addition of 2 Ν HC1
(1.5 mL).
The reaction was diluted via the addition of ethyl acetate, and phases were separated. The aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with brine, dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The crude residue was purified via silica gel chromatography to yield E-la.
1H NMR (400 MHz, CDC13): δ 10.85 (s, 1H), 9.86 (s, 1H), 8.02 (d, J= 13.1 Hz, 1H), 6.65 (dd, J= 8.7, 7.7 Hz, 2H), 4.53 (d, J= 3.9 Hz, 2H), 4.40 (t, J= 5.1 Hz, 1H), 4.18 (s, 2H), 3.42 (s, 6H), 3.39 (m, 2H), 3.37 (s, 3H). 13C MR (100 MHz, CDC13): δ 193.30, 169.15, 162.10 (ddd, J= 248.9, 15.5, 15.5 Hz), 161.7 (ddd, J =
250.0, 14.9, 1 1.1 Hz), 161.66, 1 11.08 (ddd J= 19.9, 19.9, 4.7 Hz) 103.12, 100.29 (ddd, J= 28.1, 17.7, 2.3 Hz), 76.30, 58.83, 54.98, 53.53, 51.57, 29.89 (t, J= 3.3 Hz). LCMS, Calculated: 390.36, Found: 390.92 (M).
c. Cyclization of E-la to form F-la:

E-1a F-1a
] E-la (0.2 g, 1.0 equivalent), dimethyl oxalate (0.1 g, 2.5 equivalents) and methanol (1.5 mL) were combined and cooled to about 0 to about 5 °C. Sodium methoxide (0.2 mL, 30% solution in methanol, 1.75 equivalents) was introduced to the reaction slowly while keeping the internal temperature of the reaction below about 10 °C throughout the addition. After the addition was completed the reaction was heated to about 40 to about 50 °C for at least 18 hours.
After this time had elapsed, the reaction was diluted with 2 N HC1 (1.5 mL) and ethyl acetate (2 mL). The phases were separated, and the aqueous phase was extracted with ethyl acetate. The combined organic layers were washed with brine, dried over magnesium sulfate, filtered, and solvent was removed under reduced pressure. The resulting crude oil was purified via silica gel chromatography to afford F-la.
lR NMR (400 MHz, CDC13): δ 10.28 (t, J= 5.5 Hz, 1H), 8.38 (s, 1H), 6.66 – 6.53 (m, 2H), 4.58 (d, J= 5.6 Hz, 2H), 4.43 (t, J= 4.7 Hz, 1H), 4.00 (d, J= 4.7 Hz, 2H), 3.92 (s, 3H), 3.88 (s, 3H), 3.32 (s, 6H). 13C NMR (100 MHz, CDC13): δ 173.08, 163.81, 162.17, 162.14 (ddd, J= 249.2, 15.6, 15.6 Hz), 161.72 (ddd, J= 250.5, 15.0, 10.9 Hz), 149.37, 144.64, 134.98, 119.21, 1 10.53 (ddd, J= 19.8, 4.7, 4.7 Hz), 102.70, 100.22 (m), 60.68, 56.75, 55.61, 53.35, 30.64. LCMS, Calculated: 458.39, Found: 459.15 (M+H).
D. Alkylation and cyclization of C-la to form F-la:
1 . DMFDMA

C-1a NaOMe, MeOH, 40 °C F-1a
To a reaction vessel were added C-la (245 mg, 1.0 equivalent) and N,N-dimethylformamide dimethylacetal (0.5 mL, 4.3 equivalent). The reaction mixture was agitated for approximately 30 minutes. The reaction was then treated with 2-methyl tetrahydrofuran (2.0 mL) and aminoacetaldehyde dimethyl acetal (0.1 mL, 1.0 equivalent). The reaction was allowed to age for several hours and then solvent was removed under reduced pressure.
The resulting material was dissolved in methanol and dimethyl oxalate was added (0.3 g, 2.5 equivalents). The reaction mixture was cooled to about 0 to about 5 °C, and then sodium methoxide (0.4 mL, 30% solution in methanol, 1.75 equivalents) was introduced to the reaction slowly. After the addition was completed the reaction was heated to about 40 to about 50 °C.
After this time had elapsed, the reaction was cooled to room temperature and quenched via the addition of 2 Ν HC1 (1.5 mL). The reaction was then diluted with ethyl acetate, and the resulting phases were separated. The aqueous layer was extracted with ethyl acetate. The combined organic layers were dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The crude residue was purified via silica gel chromatography to yield F-la.
lR NMR (400 MHz, CDC13): δ 10.28 (t, J= 5.5 Hz, 1H), 8.38 (s, 1H), 6.66 – 6.53 (m, 2H), 4.58 (d, J= 5.6 Hz, 2H), 4.43 (t, J= 4.7 Hz, 1H), 4.00 (d, J= 4.7 Hz, 2H), 3.92 (s, 3H), 3.88 (s, 3H), 3.32 (s, 6H). 13C NMR (100 MHz, CDC13): δ 173.08, 163.81, 162.17, 162.14 (ddd, J= 249.2, 15.6, 15.6 Hz), 161.72 (ddd, J= 250.5, 15.0, 10.9 Hz), 149.37, 144.64, 134.98, 119.21, 1 10.53 (ddd, J= 19.8, 4.7, 4.7 Hz), 102.70, 100.22 (m), 60.68, 56.75, 55.61, 53.35, 30.64. LCMS, Calculated: 458.39, Found: 459.15 (M+H).
E. Condensation of F-la with N-la to form G-la:

K2C03, MeCN, 75 °C
To a reaction vessel were added F-la (202 mg, 1.0 equivalent) and acetonitrile (1.4 mL). The resulting solution was treated with glacial acetic acid (0.2 mL, 6.0 equivalents) and methane sulfonic acid (0.01 mL, 0.3 equivalents). The reaction was then heated to about 70 to about 75 °C.
After 3 hours, a solid mixture of N-la (0.128g, 1.5 equivalents) and potassium carbonate (0.2 g, 2.7 equivalents) was introduced to the reaction at about 70 to about 75 °C. After the addition was completed, the reaction was allowed to progress for at least about 1 hour.
After this time had elapsed, water (1.4 mL) and dichloromethane (1.4 mL) were introduced to the reaction. The phases were separated, and the aqueous layer was extracted with dichloromethane. The combined organic layers were dried over magnesium sulfate, then were filtered and concentrated under reduced pressure. The resulting crude material was purified via silica gel chromatography to obtain G-la.
lR NMR (400 MHz, CDC13): δ 10.23 (t, J= 5.5 Hz, 1H), 8.39 (s, 1H), 6.60 (t, J= 8.1 Hz, 2H), 5.29 (dd, J= 9.5, 3.7 Hz, 2H), 4.57 (d, J= 5.4 Hz, 3H), 4.33 (dd, J = 12.8, 3.8 Hz, 1H), 4.02 – 3.87 (m, 1H), 3.94 (s, 3H), 2.06 – 1.88 (m, 4H), 1.78 (dd, J = 17.2, 7.5 Hz, 1H), 1.55 – 1.46 (m, 1H). 13C MR (100 MHz, CDC13): δ 174.53, 163.75, 162.33 (dd, J= 249.4, 15.7, 15.7 Hz), 161.86 (ddd, J= 250.4, 14.9, 10.9 Hz), 154.18, 154.15, 142.44, 129.75, 1 18.88, 1 10.58 (ddd, J= 19.8, 4.7, 4.7 Hz), 100.42 (m), 77.64, 74.40, 61.23, 54.79, 51.13, 38.31, 30.73, 29.55, 28.04. LCMS, Calculated: 463.14, Found: 464.15 (M+H).
Γ. Deprotection of G-la to form a compound of Formula la:
G-la (14 g) was suspended in acetonitrile (150 mL) and dichloromethane (150 mL). MgBr2 (12 g) was added. The reaction was heated to 40 to 50 °C for approximately 10 min before being cooled to room temperature. The reaction was poured into 0.5M HC1 (140 mL) and the layers separated. The organic layer was washed with water (70 mL), and the organic layer was then concentrated. The crude product was purified by silica gel chromatography (100% dichloromethane up to 6% ethanol/dichloromethane) to afford la.
REFERENCES
| Patent | Submitted | Granted |
|---|---|---|
| POLYCYCLIC-CARBAMOYLPYRIDONE COMPOUNDS AND THEIR PHARMACEUTICAL USE [US2014221356] | 2013-12-19 | 2014-08-07 |
| US9216996 | Dec 19, 2013 | Dec 22, 2015 | Gilead Sciences, Inc. | Substituted 2,3,4,5,7,9,13,13a-octahydropyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepines and methods for treating viral infections |
see full gravir series at…………..http://medcheminternational.blogspot.in/p/ravir-series.html
//////////
C1CC2CC1N3C(O2)CN4C=C(C(=O)C(=C4C3=O)O)C(=O)NCC5=C(C=C(C=C5F)F)F
OR
c1c(cc(c(c1F)CNC(=O)c2cn3c(c(c2=O)O)C(=O)N4[C@H]5CC[C@H](C5)O[C@@H]4C3)F)F
![]()

BICTEGRAVIR, NEW PATENT, WO 2018005328, CONCERT PHARMA
WO2018005328) DEUTERATED BICTEGRAVIR
CONCERT PHARMACEUTICALS, INC.
TUNG, Roger, D.; (US)

Concert CEO Roger Tung
Novel deuterated forms of bictegravir is claimed. Gilead Sciences is developing the integrase inhibitor bictegravir as an oral tablet for the treatment of HIV-1 infection.
This invention relates to deuterated forms of bictegravir, and pharmaceutically acceptable salts thereof. In one aspect, the invention provides a compound of Formula (I) or a pharmaceutically acceptable salt thereof, wherein each of Y1, Y2, Y3, Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, Y8, Y9, Y10a, Y10b, Y11a, and Y11b is independently hydrogen or deuterium; provided that if each Y1, Y2, Y3, Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, Y8, Y9, Y10a, Y10b, and Y11 is hydrogen, then Y11b is deuterium.

Many current medicines suffer from poor absorption, distribution, metabolism and/or excretion (ADME) properties that prevent their wider use or limit their use in certain indications. Poor ADME properties are also a major reason for the failure of drug candidates in clinical trials. While formulation technologies and prodrug strategies can be employed in some cases to improve certain ADME properties, these approaches often fail to address the underlying ADME problems that exist for many drugs and drug candidates. One such problem is rapid metabolism that causes a number of drugs, which otherwise would be highly effective in treating a disease, to be cleared too rapidly from the body. A possible solution to rapid drug clearance is frequent or high dosing to attain a sufficiently high plasma level of drug. This, however, introduces a number of potential treatment problems such as poor patient compliance with the dosing regimen, side effects that become more acute with higher doses, and increased cost of treatment. A rapidly metabolized drug may also expose patients to undesirable toxic or reactive metabolites.
[3] Another ADME limitation that affects many medicines is the formation of toxic or biologically reactive metabolites. As a result, some patients receiving the drug may experience toxicities, or the safe dosing of such drugs may be limited such that patients receive a suboptimal amount of the active agent. In certain cases, modifying dosing intervals or formulation approaches can help to reduce clinical adverse effects, but often the formation of such undesirable metabolites is intrinsic to the metabolism of the compound.
[4] In some select cases, a metabolic inhibitor will be co-administered with a drug that is cleared too rapidly. Such is the case with the protease inhibitor class of drugs that are used to treat HIV infection. The FDA recommends that these drugs be co-dosed with ritonavir, an inhibitor of cytochrome P450 enzyme 3A4 (CYP3A4), the enzyme typically responsible for their metabolism (see Kempf, D.J. et al., Antimicrobial agents and chemotherapy, 1997, 41(3): 654-60). Ritonavir, however, causes adverse effects and adds to the pill burden for HIV patients who must already take a combination of different drugs. Similarly, the
CYP2D6 inhibitor quinidine has been added to dextromethorphan for the purpose of reducing rapid CYP2D6 metabolism of dextromethorphan in a treatment of pseudobulbar affect. Quinidine, however, has unwanted side effects that greatly limit its use in potential combination therapy (see Wang, L et al., Clinical Pharmacology and Therapeutics, 1994, 56(6 Pt 1): 659-67; and FDA label for quinidine at http://www.accessdata.fda.gov).
[5] In general, combining drugs with cytochrome P450 inhibitors is not a satisfactory strategy for decreasing drug clearance. The inhibition of a CYP enzyme’s activity can affect the metabolism and clearance of other drugs metabolized by that same enzyme. CYP inhibition can cause other drugs to accumulate in the body to toxic levels.
[6] A potentially attractive strategy for improving a drug’s metabolic properties is deuterium modification. In this approach, one attempts to slow the CYP-mediated metabolism of a drug or to reduce the formation of undesirable metabolites by replacing one or more hydrogen atoms with deuterium atoms. Deuterium is a safe, stable, non-radioactive isotope of hydrogen. Compared to hydrogen, deuterium forms stronger bonds with carbon. In select cases, the increased bond strength imparted by deuterium can positively impact the ADME properties of a drug, creating the potential for improved drug efficacy, safety, and/or tolerability. At the same time, because the size and shape of deuterium are essentially identical to those of hydrogen, replacement of hydrogen by deuterium would not be expected to affect the biochemical potency and selectivity of the drug as compared to the original chemical entity that contains only hydrogen.
[7] Over the past 35 years, the effects of deuterium substitution on the rate of metabolism have been reported for a very small percentage of approved drugs (see, e.g., Blake, MI et al, J Pharm Sci, 1975, 64:367-91; Foster, AB, Adv Drug Res 1985, 14:1-40 (“Foster”); Kushner, DJ et al, Can J Physiol Pharmacol 1999, 79-88; Fisher, MB et al, Curr Opin Drug Discov Devel, 2006, 9:101-09 (“Fisher”)). The results have been variable and unpredictable. For some compounds deuteration caused decreased metabolic clearance in vivo. For others, there was no change in metabolism. Still others demonstrated increased metabolic clearance. The variability in deuterium effects has also led experts to question or dismiss deuterium modification as a viable drug design strategy for inhibiting adverse metabolism (see Foster at p.35 and Fisher at p.101).
[8] The effects of deuterium modification on a drug’s metabolic properties are not predictable even when deuterium atoms are incorporated at known sites of metabolism. Only by actually preparing and testing a deuterated drug can one determine if and how the rate of metabolism will differ from that of its non-deuterated counterpart. See, for example, Fukuto et al. (J. Med. Chem.1991, 34, 2871-76). Many drugs have multiple sites where metabolism is possible. The site(s) where deuterium substitution is required and the extent of deuteration necessary to see an effect on metabolism, if any, will be different for each drug.
Exemplary Synthesis
[72] Deuterium-modified analogs of bictegravir can be synthesized by means known in the art of organic chemistry. For instance, using methods described in US Patent No.9,216,996 (Haolun J. et al., assigned to Gilead Sciences, Inc. and incorporated herein by reference), using deuterium-containing reagents provides the desired deuterated analogs.
[73] Such methods can be carried out utilizing corresponding deuterated and optionally, other isotope-containing reagents and/or intermediates to synthesize the compounds delineated herein, or invoking standard synthetic protocols known in the art for introducing isotopic atoms to a chemical structure.
[74] A convenient method for synthesizing compounds of Formula I is depicted in the Schemes below.
 [75] A generic scheme for the synthesis of compounds of Formula I is shown in Scheme 1 above. In a manner analogous to the procedure described in Wang, H. et al. Org. Lett.2015, 17, 564-567, aldol condensation of compound 1 with appropriately deuterated compound 2 affords enamine 3. Enamine 3 is then reacted with primary amine 4 to afford enamine 5, which then undergoes cyclization with dimethyl oxalate followed by ester hydrolysis to provide carboxylic acid 7.
[75] A generic scheme for the synthesis of compounds of Formula I is shown in Scheme 1 above. In a manner analogous to the procedure described in Wang, H. et al. Org. Lett.2015, 17, 564-567, aldol condensation of compound 1 with appropriately deuterated compound 2 affords enamine 3. Enamine 3 is then reacted with primary amine 4 to afford enamine 5, which then undergoes cyclization with dimethyl oxalate followed by ester hydrolysis to provide carboxylic acid 7.
[76] In a manner analogous to the procedure described in US 9,216,996, acetal deprotection of carboxylic acid 7 followed by cyclization with appropriately deuterated aminocyclopentanol 9 provides carboxylic acid intermediate 10. Amide coupling with appropriately deuterated benzylamine 11 followed by deprotection of the methyl ether ultimately affords a compound of Formula I in eight overall steps from compound 1.
[77] Use of appropriately deuterated reagents allows deuterium incorporation at the Y1, Y2, Y3, Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, Y8, Y9, Y10a, Y10b, Y11a, and Y11bpositions of a compound of Formula I or any appropriate intermediate herein, e.g., about 90%, about 95%, about 97%, about 98%, or about 99% deuterium incorporation at any Y1, Y2, Y3, Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, Y8, Y9, Y10a, Y10b, Y11a, and/or Y11b.
[78] Appropriately deuterated intermediates 2a and 2b, for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents as exemplified in Scheme 2 below.
S h 2 S th i f C d 2 d 2b

[79] Synthesis of compound 2a (wherein Y3=H) by acetal formation of N,N-dimethylformamide (DMF) with dimethylsulfate has been described in Mesnard, D. et. al. J. Organomet. Chem.1989, 373, 1-10. Replacing DMF with N,N-dimethylformamide-d1 (98-99 atom % D; commercially available from Cambridge Isotope Laboratories) in this reaction would thereby provide compound 2b (wherein Y3=D).
[80] Use of appropriately deuterated reagents allows deuterium incorporation at the Y3 position of a compound of Formula I or any appropriate intermediate herein, e.g., about 90%, about 95%, about 97%, about 98%, or about 99% deuterium incorporation at Y3.
[81] Appropriately deuterated intermediates 4a-4d, for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents as exemplified in Scheme 3 below.

[82] As described in Malik, M. S. et. al. Org. Prep. Proc. Int.1991, 26, 764-766, acetaldehyde is converted to alkylhalide 14a via reaction with chlorine gas and subsequent acetal protection with CaCl2 in methanol. As described in CN 103739506, reaction of 14a with aqueous ammonia and then sodium hydroxide provides primary amine 4a (wherein Y9=Y10a=Y10b=H). Replacing acetaldehyde with acetaldehyde-d1, acetaldehyde-2,2,2-d3, or acetaldehyde-d4 (all commercially available from CDN Isotopes with 98-99 atom % D) in the sequence then provides access to compounds 4b (Y9=D, Y10a=Y10b=H), 4c (Y9=H,
Y10a=Y10b=D) and 4d (Y9=Y10a=Y10b=D) respectively (Schemes 3b-d).
[83] Use of appropriately deuterated reagents allows deuterium incorporation at the Y9, Y10a, and Y10b positions of a compound of Formula I or any appropriate intermediate herein, e.g., about 90%, about 95%, about 97%, about 98%, or about 99% deuterium incorporation at any Y9, Y10a, and/or Y10b.
[84] Appropriately deuterated intermediates 9a-9d, for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents as exemplified in Scheme 4 below.
 [85] Following the procedures described by Gurjar, M. et. al. Heterocycles, 2009, 77, 909-925, meso-diacetate 16a is prepared in 2 steps from cyclopentadiene. Desymmetrization of 16a is then achieved enzymatically by treatment with Lipase as described in Specklin, S. et. al. Tet. Lett.201455, 6987-6991, providing 17a which is subsequently converted to aminocyclopentanol 9a (wherein Y4a=Y4b=Y5a=Y5b=Y6=Y7a=Y7b=Y8=H) via a 3 step sequence as reported in WO 2015195656.
[85] Following the procedures described by Gurjar, M. et. al. Heterocycles, 2009, 77, 909-925, meso-diacetate 16a is prepared in 2 steps from cyclopentadiene. Desymmetrization of 16a is then achieved enzymatically by treatment with Lipase as described in Specklin, S. et. al. Tet. Lett.201455, 6987-6991, providing 17a which is subsequently converted to aminocyclopentanol 9a (wherein Y4a=Y4b=Y5a=Y5b=Y6=Y7a=Y7b=Y8=H) via a 3 step sequence as reported in WO 2015195656.
[86] As depicted in Scheme 4b, aminocyclopentanol 9b (Y4a=Y4b=Y5a=Y5b=Y6=Y7a=Y7b= Y8=D) is obtained through an analogous synthetic sequence using cyclopentadiene-d6 and performing the penultimate hydrogenation with D2 in place of H2. Cyclopentadiene-d6 is prepared according to the procedure described in Cangoenuel, A. et. al. Inorg. Chem.2013, 52, 11859-11866.
[87] Alternatively, as shown in Scheme 4c, the meso-diol obtained in Scheme 4a is oxidized to the diketone following the procedure reported by Rasmusson, G.H. et. al. Org. Syn.1962, 42, 36-38. Subsequent mono-reduction with sodium borodeuteride and CeCl3 then affords the D1-alcohol in analogy to the method described in WO 2001044254 for the all-protio analog using sodium borohydride. Reduction of the remaining ketone using similar conditions provides the meso-D2-diol in analogy to the method reported in Specklin, S. et. al. Tet. Lett.2014, 55, 6987-6991 for the all protio analog using sodium borohydride. The meso-D2-diol is then converted to 9c (Y4a=Y4b=Y5a=Y5b=Y7a=Y7b=H, Y6=Y8=D) following the same procedures outlined in Scheme 4a.
[88] Likewise, the meso-diol obtained in Scheme 4b may be converted to 9d
(Y4a=Y4b=Y5a=Y5b=Y7a=Y7b=D, Y6=Y8=H) in an analogous manner as depicted in Scheme 4d. The use of deuterated solvents such as D2O or MeOD may be considered to reduce the risk of D to H exchange for ketone containing intermediates.
[89] Use of appropriately deuterated reagents allows deuterium incorporation at the Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, and Y8 positions of a compound of Formula I or any appropriate intermediate herein, e.g., about 90%, about 95%, about 97%, about 98%, or about 99% deuterium incorporation at any Y4a, Y4b, Y5a, Y5b, Y6, Y7a, Y7b, and/or Y8.
[90] Appropriately deuterated intermediates 11a-11d, for use in the preparation of compounds of Formula I according to Scheme 1, may be prepared from corresponding deuterated reagents exemplified in Scheme 5 below.
Scheme 5. Synthesis of Benzylamines 11a-11d

//////////////////
AN 2898

AN2898
(5-(3,4-dicyanophenoxy)-1-hydroxy -1,3-dihydro-2,1-benzoxaborole)
1,2-Benzenedicarbonitrile, 4-((1,3-dihydro-1-hydroxy-2,1-benzoxaborol-5-yl)oxy)-,
AN-2898
cas: 906673-33-4
UNII: 6O60L94RMB,
MW 276.0581, MF C15 H9 B N2 O3
A PDE4 inhibitor potentially for the treatment of fungal infection.
AN-2898, a novel topical anti-inflammatory compound that inhibits phosphodiesterase 4 and 7 enzyme activit
PHASE 2 FUNGAL INFECTION, Anacor Pharmaceuticals for the treatment of atopic dermatitis
![]()
| Anacor Pharmaceuticals Inc. | |
| Description | Boron-containing small molecule phosphodiesterase-4 (PDE-4) inhibitor that reduces the production of tumor necrosis factor (TNF) alpha, IL-12 and IL-23 |
| Molecular Target | Phosphodiesterase-4 (PDE-4) |
| Mechanism of Action | Phosphodiesterase-4 (PDE-4) inhibitor |
| Therapeutic Modality | Small molecule |
AN2898 (5-(3,4-dicyanophenoxy)-1-hydroxy -1,3-dihydro-2,1-benzoxaborole) is a broad spectrum anti-inflammatory compound currently in development for the topical treatment of plaque and atopic psoriasis.
AN2898 inhibited phosphodiesterase 4 (PDE4) enzyme activity (IC50 0.060 μM) and the release of multiple cytokines including TNF-α (IC50 0.16 μM) from peripheral blood mononuclear cells (hPBMCs) stimulated by lipopolysaccharide (LPS) or phytohemag- glutinin.
Further, AN2898 was also found to inhibit IL-23 release (IC50 1.0 μM) from THP-1 cells stimulated by LPS and IFN-γ. Investigation of the structure-activity relation-ship around this compound was reported to identify a more potent dual TNF-α/IL-23 inhibitor
( ref………. Akama T, Antunes J, Freund Y, Kimura R, Dong C, Sanders V, et al. Structure-activity studies of novel oxaborole dual inhibitors of PDE4 and IL-23 release. 69th Annu Meet Soc Invest Dermatol (May 6-9, Montreal) 2009 Abst 282. ).
PATENT
WO 2007095638
https://www.google.co.in/patents/WO2007095638A2?cl=en
PATENT
WO 2006089067
http://www.google.co.in/patents/WO2006089067A2?cl=en
US 7582621
http://www.google.co.in/patents/US7582621
WO 2009111676
http://www.google.im/patents/WO2009111676A2?cl=en
WO 2007078340
http://www.google.im/patents/WO2007078340A2?cl=en
US 20070286822
http://www.google.com/patents/US20070286822
REFERENCES
1 Structure-activity studies led to the discovery of AN2898 in development for topical treatment of psoriasis and atopic dermatitis, J Am Acad Dermatol 2009, 60(3, Suppl. 1): Abst P1317
2 FEBS Letters (2012), 586(19), 3410-3414
See all Bboroles at………http://apisynthesisint.blogspot.in/p/borole-compds.html
/////////AN2898, AN 2898, ANACOR, BOROLE
B1(c2ccc(cc2CO1)Oc3ccc(c(c3)C#N)C#N)O
Ataciguat

Ataciguat HMR-1766
Hoechst Marion Roussel De Gmbh
5-Chloro-2-[[(5-chloro-2-thienyl)sulfonyl]amino]-N-[4-(4-morpholinylsulfonyl)phenyl]benzamide
C21H19Cl2N3O6S3
UNII-QP166M390Q;
576.49306 g/mol
A guanylate cyclase activator potentially for the treatment of aortic valve stenosis.
![]()
CAS No. 254877-67-3
- Originator sanofi-aventis
- Developer Mayo Clinic; National Center for Advancing Translational Sciences; Sanofi; sanofi-aventis
- Class Anthranilic acids; Benzamides; Cardiovascular therapies; Chlorobenzenes; Morpholines; Small molecules; Sulfonamides; Thiophenes
- Mechanism of Action Guanylate cyclase stimulants
- 30 Jun 2015 Mayo Clinic plans a phase II trial for Aortic valve stenosis in USA (NCT02481258)
- 29 Jan 2014 Phase-I clinical trials in Aortic valve stenosis in USA (PO)
- 01 Jan 2010 Discontinued – Phase-II for Peripheral arterial occlusive disorders in Austria, Canada, France, Germany, Italy, Poland, Portugal, Russia, South Africa and USA (PO) prior to 2010
SYNTHESIS

The Intermediates hown above is used in next step shown below

Paper
Organic Letters (2013), 15(7), 1638-1641
http://pubs.acs.org/doi/abs/10.1021/ol400411v
http://pubs.acs.org/doi/suppl/10.1021/ol400411v/suppl_file/ol400411v_si_001.pdf

The Ru(II)-catalyzed intermolecular o-C–H amidation of arenes in N-benzoylated sulfoximine with sulfonyl azides is demonstrated. The reaction proceeds with broad substrate scope and tolerates various functional groups. Base hydrolysis of the amidation product provides the anthranilic acid derivatives and methylphenyl sulfoximine (MPS) directing group. This method is successfully employed for the synthesis of HMR 1766.
PATENT
WO 2009043495
http://www.google.com/patents/WO2009043495A1?cl=en
Patent
http://www.google.com/patents/WO2008124505A2?cl=en
HMR-1766 (ataciguat sodium, see patent publication WO2000002851)

PATENT
http://www.google.com/patents/WO2000002851A1?cl=en
| Patent | Submitted | Granted |
|---|---|---|
| TRA COMBINATION THERAPIES [US2007238674] | 2007-10-11 | |
| sGC STIMULATORS OR sGC ACTIVATORS ALONE AND IN COMBINATION WITH PDE5 INHBITORS FOR THE TREATMENT OF CYSTIC FIBROSIS [US2013035340] | 2011-02-03 | 2013-02-07 |
| SOLUBLE GUANYLATE CYCLASE (SGC) MODULATORS FOR TREATMENT OF LIPID RELATED DISORDERS [US2013123354] | 2013-01-08 | 2013-05-16 |
| Novel combination [US2005059660] | 2004-07-29 | 2005-03-17 |
| SGC STIMULATORS OF SGC ACTIVATORS IN COMBINATION WITH PDE5 INHBITORS FOR THE TREATMENT OF ERECTILE DYSFUNCTION [US2014288079] | 2014-03-18 | 2014-09-25 |
| Patent | Submitted | Granted |
|---|---|---|
| novel use of activators and stimulators of soluble guanylate cyclase for the prevention or treatment of renal disorders [US2010016305] | 2010-01-21 | |
| HETEROARYL-SUBSTITUTED PIPERIDINES [US8119663] | 2009-12-10 | 2012-02-21 |
| Use of soluble guanylate cyclase activators for the treatment of Raynaud’s Phenomenon [US2009215769] | 2009-08-27 | |
| Use of Activators of Soluble Guanylate Cyclase for Promoting Wound Healing [US2009221573] | 2009-09-03 | |
| Use of Suluble Guanylate Cyclase Acitvators for Treating Acute and Chronic Lung Diseases [US2009286781] | 2009-11-19 | |
| Use of Activators of Soluble Guanylate Cyclase for Treating Reperfusion Damage [US2009298822] | 2009-12-03 | |
| HETEROCYCLIC DERIVATIVE AND USE THEREOF [US2011028493] | 2011-02-03 | |
| SUBSTITUTED PIPERIDINES [US8202862] | 2010-12-02 | 2012-06-19 |
| METHODS AND COMPOSITIONS FOR TREATING CARDIAC DYSFUNCTIONS [US2009022729] | 2009-01-22 | |
| sGC STIMULATORS [US2014323448] | 2014-04-29 | 2014-10-30 |



/////////
C1COCCN1S(=O)(=O)C2=CC=C(C=C2)NC(=O)C3=C(C=CC(=C3)Cl)NS(=O)(=O)C4=CC=C(S4)Cl
Umbralisib, TGR-1202, a Phosphoinositide-3 kinase delta inhibitor, Rhizen Pharmaceuticals S.A./TG Therapeutics

| Molecular Formula: | C31H24F3N5O3 |
|---|---|
| Molecular Weight: | 571.54917 g/mol |
RP-5307
TGR-1202
TGR-1202 PTSA
FU8XW5V3FS (UNII code)
RP-5264 (free base)
A PI3K inhibitor potentially for treatment of chronic lymphocytic leukemia, leukemia,lymphoma,B-cell
TGR‐1202, a next generation PI3K-δ delta inhibitor. TGR-1202 (RP-5264) is a highly specific, orally available, PI3K delta inhibitor, targeting the delta isoform with nanomolar potency and several fold selectivity over the alpha, beta, and gamma isoforms of PI3K.
TG Therapeutics, under license from Rhizen Pharmaceuticals, is developing TGR-1202 (structure shown; formerly RP-5264), a lead from a program of PI3K delta inhibitors, for the potential oral treatment of hematological cancers including Hodgkin lymphoma, non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), B-cell lymphoma and mantle cell lymphoma (MCL)
Incozen Therapeutics Pvt Ltd
TG Therapeutics
TGR-1202 potential to perform as the best PI3K inhibitor in its class and the possible superiority of TG-1101 over Rituxan®.
| Rhizen Pharmaceuticals S.A. | |
| Description | Phosphoinositide 3-kinase (PI3K) delta inhibitor |
Leukemia, chronic lymphocytic PHASE 3, TG Therapeutics
Orphan Drug
Umbralisib is a novel phosphatidylinositol 3-kinase delta (PI3Kdelta) inhibitor under development at TG Therapeutics in phase III clinical trials, in combination with ublituximab, for the treatment of chronic lymphocytic leukemia (CLL) and for the treatment of diffuse large B-cell lymphoma (DLBCL). The company refers to the combination regimen of ublituximab and TGR-1202 as TG-1303. The drug is also in phase II clinical development for the oral treatment of hematologic malignancies, as a single agent or in combination therapy. Phase I clinical trials are ongoing in patients with select relapsed or refractory solid tumors, such as adenocarcinoma of the pancreas, adenocarcinoma of the colon, rectum, gastric and GE junction cancer, and GI Stromal Tumor (GIST).
In 2016, orphan drug designation was assigned to the compound in the U.S. for the treatment of CLL. In 2017, additional orphan drug designation was granted in the U.S. for the treatment of CLL and DLBCL, in combination with ublituximab.
Originated by Rhizen Pharmaceuticals, the product was jointly developed by Rhizen Pharmaceuticals and TG Therapeutics since 2012. In 2014, exclusive global development and commercialization rights (excluding India) were licensed to TG Therapeutics.
CLINICAL TRIALS……….https://clinicaltrials.gov/search/intervention=TGR-1202
B-cell lymphoma; Chronic lymphocytic leukemia; Hematological neoplasm; Hodgkins disease; Mantle cell lymphoma; Non-Hodgkin lymphoma
Phosphoinositide-3 kinase delta inhibitor
SYNTHESIS


Rhizen Pharmaceuticals Announces Out-licensing Agreement for TGR-1202, a Novel Next Generation PI3K-delta Inhibitor
Rhizen to receive upfront payment of $8.0 million — Rhizen to retain global manufacturing and supply rights — Rhizen to retain development and commercialization for India
Rhizen to retain development and commercialization for India
| Source: Rhizen Pharmaceuticals SA
La Chaux-de-Fonds, Switzerland, Sept. 23, 2014 (GLOBE NEWSWIRE) — Rhizen Pharmaceuticals S.A. today announced an out-licensing agreement for TGR-1202, a novel next generation PI3K-delta inhibitor. TG Therapeutics exercised its option for early conversion to a licensing agreement from a 50:50 joint venture partnership.
In exchange for this licensing agreement, TG Therapeutics will pay Rhizen an upfront payment of $8.0 million ($4.0 million in cash and $4.0 million in TG Therapeutics common stock). In addition to the upfront payment, Rhizen will be eligible to receive regulatory filing, approval and sales based milestones in the aggregate of approximately $240 million, and tiered royalties based on net sales.
Swaroop Vakkalanka, Ph.D. and President of Rhizen stated, “We are extremely happy and take pride in discovering a novel, next generation, once-daily PI3K-delta inhibitor under active development led by TG Therapeutics. We are encouraged by the progress of TRG-1202 to date, and the speed at which TG Therapeutics is developing the asset in various hematological malignancies. We look forward to the day this novel drug reaches cancer patients in need of new and safe therapies.”
About Rhizen Pharmaceuticals S.A.:
Rhizen Pharmaceuticals is an innovative, clinical-stage biopharmaceutical company focused on the discovery and development of novel therapeutics for the treatment of cancer, immune and metabolic disorders. Since its establishment in 2008, Rhizen has created a diverse pipeline of proprietary drug candidates targeting several cancers and immune associated cellular pathways. Rhizen is headquartered in La-Chaux-de-Fonds, Switzerland. For additional information, please visit Rhizen’s website, www.rhizen.com.

TGR-1202.with Idelalisib and IPI-145 (left to right) for comparison.
IPI 145
PATENTS
WO 2011055215
http://www.google.com/patents/WO2011055215A2?cl=en

PATENT
WO 2015181728
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015181728
TGR-1202, chemically known as (S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-3-(3-fluorophenyl)-4H-chromen-4-one, has the following chemical structure:

Example 1: Preparation of the PTSA Salt of TGR-1202 (Form A)

7100 g of TGR-1202 was charged in a reactor containing 56.8 litres of acetone and stirred at ambient temperature. 4680 g of p-toluene sulphonic acid was added and the reaction mixture was heated at a temperature of 60-65° C for about 6 hours. The solvent was removed by distillation under reduced pressure to obtain a wet residue. The wet residue was degassed and allowed to cool to < 20° C. Approximately 142 litres of diethyl ether was then added and the resulting mixture was stirred overnight, then filtered to obtain a solid mass which was washed with diethyl ether and dried in vacuo to yield a solid mass. The solid mass was re-suspended in diethyl ether, stirred for 6 hours, and then filtered to yield a solid mass which was subsequently dissolved in 56.8 litres of acetone, filtered through a HiFlow bed, and concentrated under reduced pressure. The resulting residue mass was stirred with water overnight, then filtered and vacuum dried to yield 6600 g of the PTSA salt of TGR-1202. HPLC: 99.21% and chiral purity of 99.64:0.36 (S:R).
Example 2: Preparation of the PTSA Salt of TGR-1202 (Form B)

1000 g of TGR-1202 was charged in a reactor containing 8 litres of acetone and stirred at ambient temperature. 666 g of p-toluene sulphonic acid was then added and the reaction mixture was heated at a temperature of 60-65 °C for about 6 hours. The solvent was removed by distillation under reduced pressure to obtain a wet residue. The wet residue was degassed and allowed to cool to < 20° C. Approximately 20 litres of diethyl ether was added and the resulting mixture was stirred overnight, then filtered to obtain a solid mass which was washed with diethyl ether and dried in vacuo to yield a solid mass which was then vacuum dried to yield 1150 g of the PTSA salt of TGR-1202. HPLC: 99.33% and chiral purity: 99.61:0.39 (S:R).
Table 1 lists the XRPD pattern peaks and relative peak intensities for the products of Examples 1 and 2.
TABLE 1

The tablet composition comprising a PTSA salt of TGR-1202 prepared according to Example 2 exhibited a Cmax about 2.5 fold and an area under the curve (AUC) about 1.9 fold greater than that of the tablet composition comprising a PTSA salt of TGR-1202 prepared according to Example 1. The results are provided in Table 8 below.
TABLE 8

PATENT
WO 2014071125
http://www.google.com/patents/WO2014071125A1?cl=en
formula (A) that is a ΡΒΚδ selective inhibitor,

(A)
Synthesis of Compound of Formula A
Unless otherwise stated, purification implies column chromatography using silica gel as the stationary phase and a mixture of petroleum ether (boiling at 60-80°C) and ethyl acetate or dichloromethane and methanol of suitable polarity as the mobile phases. The term “RT” refers to ambient temperature (25-28°C).
Intermediate 1 : 2-( l-bromoethyl)-6-fluoro-3-f3-fluorophenyl)-4H-chromen-4-one
Step-1 [l-(5-Fluoro-2-hydroxyphenyl)-2-(3-fluorophenyl)ethanone]: 3- Fluorophenylacetic acid (7.33 g, 47.56 mmoles) was dissolved in 25 ml dichloromethane. To this mixture, oxalylchloride (7.54 g, 59.46 mmoles) and DMF (3 drops) were added at 0°C and stirred for 30 min. The solvent was evaporated and dissolved in 25 ml dichloromethane. To this mixture, 4-fluoroanisole (5.00 g, 39.64 mmoles) was added and cooled to 0°C. At 0°C A1C13 (7.95 g, 59.46 mmoles) was added and the reaction mixture was warmed to RT and stirred for 12 hours. The reaction mixture was quenched by the addition of 2N HC1, extracted with ethyl acetate, dried over sodium sulphate and concentrated. The crude product was purified by column chromatography with ethyl acetate :petroleum ether to afford the title compound as colorless solid (4.5 g, 45% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 11.34 (s, 1H), 7.75 (dd, J=9.4, 3.1 Hz, 1H), 7.42 (m, 2H), 7.12 (m, 3H), 7.05 (dd, J=9.0, 4.5 Hz, 1H), 4.47 (s, 2H).
Step-2 [2-Ethyl-6-fiuoro-3-(3-fluorophenyl)-4H-chromen-4-one]: l-(5-Fluoro-2- hydroxyphenyl)-2-(3-fluorophenyl)ethanone obtained from Step-1 (3.00 g, 12.08 mmoles) was placed in a round bottom flask and to this triethylamine (25 ml) and propionic anhydride (4.92 g, 37.82 mmoles) were added, and the mixture was refluxed for 24 hours. After cooling to RT, the reaction mixture was acidified by the addition of IN HC1 solution, extracted with ethyl acetate, washed with sodium bicarbonate solution, dried with sodium sulphate and concentrated. The crude product was purified by column chromatography with ethyl acetate :petroleum ether to afford the title compound as off-yellow solid (1.80 g, 52% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 7.80 (m, 1H), 7.76 (m, 2H), 7.51 (dd, J=8.0, 6.4 Hz), 7.22 (m, 1H), 7.18 (m, 2H), 2.56 (q, J=7.6 Hz, 2H), 1.20 (t, J=7.6 Hz, 3H).
Step-3: To a solution of 2-Ethyl-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one obtained from Step-2 (1.80 g, 6.28 mmoles) in carbon tetrachloride (20 ml), N- bromosuccinimide (1.11 g, 6.28 mmoles) was added and heated to 80°C. Azobisisobutyronitrile (10 mg) was added to the reaction mixture at 80°C. After 12 hours, the reaction mixture was cooled to RT, diluted with dichloromethane and washed with water. The organic layer was dried over sodium sulphate and concentrated under reduced pressure to afford the crude title compound as yellow solid (1.25 g, 55% yield). 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 7.91 (dd, J=9.2, 4.3 Hz, 1H), 7.81 (dt, j=8.2, 2.8 Hz, 1H), 7.74 (dd, J=8.3, 3.1 Hz, 1H), 7.57 (m, 1H), 7.32 (dt, J=8.5, 2.4 Hz, 1H), 7.19 (m, 2H), 5.00 (q, J=6.8 Hz, 1H), 1.97 (d, J=6.8 Hz, 3H).
Intermediate 2: 6-fluoro-3-f3-fluorophenyl)-2-fl-hvdroxyethyl)-4H-chromen-4-one

To a solution of Intermediate 1 (15.0 g, 40.84 mmol) in DMSO (150 ml), n-butanol (7.5 ml) was added and heated to 120°C for 3 hours. The reaction mixture was cooled to RT, quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (7.90 g, 64%). 1H-NMR (δ ppm, CDC13, 400 MHz): 7.85 (dd, J = 8.1, 3 Hz, 1H), 7.54 (dd, J = 9.2, 4.2 Hz, 1H), 7.47-7.37 (m, 2H), 7.15-6.98 (m, 3H), 4.74 (quintet, J= 6.8 Hz, 1H), 2.23 (d, J = 7.4 Hz, 1H), 1.54 (d, J = 6.6 Hz, 3H).
Intermediate 3 : 2-acetyl-6-fluoro-3-( 3-fluorophenyl)-4H-chromen-4-one

DMSO (5.60 ml, 79.14 mmol) was added to dichloromethane (40 ml), and cooled to – 78°C, followed by oxalyl chloride (3.40 ml, 39.57 mmol). After 10 min., intermediate 2 (6.00 g, 19.78 mmol) in dichloromethane (54 ml) was added dropwise and stirred for 20 min.
Triethylamine (12 ml) was added and stirred for 1 hour. The reaction mixture was quenched with water and extracted with dichloromethane. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (4.2 g, 71%) which was used as such in the next step.
Intermediate 4: fS)-6-fluoro-3-f3-fluorophenyl)-2-fl-hvdroxyethyl)-4H-chromen-4-one

To intermediate 3 (2.00 g, 6.66 mmol), R-Alpine borane (0.5 M in THF, 20 ml) was added and heated to 60°C for 20 hours. The reaction mixture quenched with 2N HC1, and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (1.51 g, 75%).
Enantiomeric excess: 94.2%, enriched in the fast eluting isomer (retention time: 8.78 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 5: fR)-l-f6-fluoro-3-f3-fluorophenyl)-4-oxo-4H-chromen-2-yl)ethyl 4- chlorobenzoate

To a solution of intermediate 4 (1.45 g, 4.78 mmol) in THF (15 ml), 4-chlorobenzoic acid (0.748 g, 4.78 mmol) and triphenylphosphine (1.88 g, 7.17 mmol) were added and heated to 45°C followed by diisopropylazodicarboxylate (1.4 ml, 7.17 mmol). After 1 hour, the reaction mixture was concentrated and the residue was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as an off-white solid (1.81 g, 86%) which was used without purification in the next step. Intermediate 6: fR)-6-fluoro-3-f3-fluorophenyl)-2-fl-hvdroxyethyl)-4H-chromen-4-one

Method A
Intermediate 5 (1.75 g, 3.96 mmol) in methanol (17 ml) was cooled to 10°C, potassium carbonate (0.273 g, 1.98 mmol) was added and stirred for 30 min. The reaction mixture was concentrated, acidified with 2N HCl solution, extracted with ethyl acetate, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow solid (1.05 g, 87% yield). Enantiomeric excess: 93.6%>, enriched in the late eluting isomer (retention time: 11.12 min.) as determined by HPLC on a chiralpak AD-H column.
Method B
Step-1 [(R)-2-(l-(benzyloxy)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one]: To l-(5-fluoro-2-hydroxyphenyl)-2-(3-fluorophenyl)ethanone (11.00 g, 44.31 mmol) in dichloromethane, HATU (33.7 g, 88.63 mmol) and R-(+)2-benzyloxypropionic acid (9.58 g, 53.17 mmol) were added and stirred for 10 min. Triethylamine (66.7 ml, 0.47 mol) was added dropwise and stirred at RT for 24 hours. The reaction mixture was quenched with water, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate:
petroleum ether to afford the title compound as a yellow solid (10.5 g, 60%> yield). 1H-NMR (δ ppm, CDCls, 400 MHz): 7.85 (dd, J = 8.1,3 Hz, 1H), 7.58 (dd, J = 9.1, 4.1 Hz, 1H), 7.47-7.39 (m, 1H), 7.39-7.34 (m, 1H), 7.28-7.20 (m, 3H), 7.20-7.14 (m, 2H), 7.16-7.07 (m, 1H), 6.99-6.89 (m, 2H), 4.50-4.31 (m, 3H), 1.56 (d, J = 6.4 Hz, 3H).
Step-2: (R)-2-(l-(benzyloxy)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one obtained in Step-1 (10.5 g, 26.69 mmol) in dichloromethane (110 ml) was cooled to 0°C, aluminium chloride (5.35 g, 40.03 mmol) was added portionwise and stirred at RT for 6 hours. The reaction mixture was quenched with 2N HCl solution, extracted with dichloromethane, dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford intermediate 6 a yellow solid (6.1 g, 76% yield). Enantiomeric excess: 97.7%, enriched in the late eluting isomer (retention time: 11.12 min.) as determined by HPLC on a chiralpak AD-H column.
Intermediate 7: 4-bromo-2-fluoro-l-isopropoxybenzene

To a solution of 4-bromo-3-fluorophenol (10 g, 52.35 mmol) in THF (100ml), isopropyl alcohol (4.8 ml, 62.62 mmol) and triphenylphosphine (20.6 g, 78.52 mmol) were added and heated to 45°C followed by diisopropylazodicarboxylate (15.4 ml, 78.52 mmol). The mixture was refluxed for 1 hour, concentrated and the residue was purified by column
chromatography with ethyl acetate: petroleum ether to afford the title compound as a colorless liquid (13.1 g, 99% yield), which was used without purification in the next step.
Intermediate 8: 2-f3-fluoro-4-isopropoxyphenyl)-4,4,5.,5-tetramethyl-l,3i2-dioxaborolane

Potassium acetate (10.52 g, 107.2 mmol) and bis(pinacolato)diboron (15 g, 58.96 mmol) were added to a solution of intermediate 7 (10.52 g, 107.2 mmol) in dioxane (125 ml), and the solution was degassed for 30 min. [l, -Bis(diphenylphosphino)ferrocene]dichloro palladium(II) CH2CI2 (4.4 g, 5.36 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12 hours, the reaction mixture was filtered through celite and concentrated. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow oil (13.9g, 99%) which was used without purification in the next step.
Intermediate 9: 3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-dlpyrimidin-4-amine

To a solution of 3-iodo-lH-pyrazolo[3,4-d]pyrimidin-4-amine (11.0 g, 42.14 mmol) in DMF (110 ml), ethanol (55 ml) and water (55 ml), intermediate 8 (23.4 g, 84.28 mmol) and sodium carbonate (13.3 g, 126.42 mmol) were added and degassed for 30 min.
Tetrakis(triphenylphosphine)palladium(0) (2.4 g, 2.10 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12 hours, the reaction mixture was filtered through celite, concentrated and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was triturated with diethyl ether, filtered and dried under vacuum to afford the title compound as light brown solid (3.2 g, 26% yield) which is used as such for the next step.
(RS)- 2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
To a solution of intermediate 9 (0.080 g, 0.293 mmol) in DMF (2 ml), potassium carbonate (0.081 g, 0.587 mmol) was added and stirred at RT for 10 min. To this mixture intermediate 1 (0.215 g, 0.587 mmol) was added and stirred for 12 hours. The reaction mixture was diluted with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with methanol: dichloromethane to afford the title compound as a pale yellow solid (0.045 g). MP: 175-177°C. 1H-NMR (δ ppm, DMSO-D6, 400 MHz): δ 8.20 (s, 1H), 7.85 (dd, J = 81, 3.0 Hz, 1H), 7.48-7.33 (m, 5H), 7.14 (t, J= 8.3 Hz, 1H), 7.02 (m, 2H), 6.90 (m, 1H), 6.10 (q, J = 7.1 Hz, 1H), 5.42 (s, 2H), 4.64 (quintet, J = 6.0 Hz, 1H), 1.99 (d, J = 7.1 Hz, 3H), 1.42 (d, J= 6.1 Hz, 6H).
fS)-2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (“S-isomer”)
To a solution of intermediate 9 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 6 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.15 ml, 0.749 mmol) was added heated to 45°C. After 2 hours, the reaction mixture was quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as an off-white solid (0.049 g, 20 % yield). MP: 139-142°C. Mass: 571.7 (M+). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 10.64 min.). fR)-2-fl-f4-amino-3-f3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3.,4-(ilpyrimi(iin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-ehromen-4-one
To a solution of intermediate 8 (0.284 g, 0.989 mmol) in THF (5.0 ml), intermediate 4 (0.250 g, 0.824 mmol) and tris(4-methoxy)phenylphosphine (0.435 g, 1.23 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.25 ml, 1.23 mmol) was added stirred at RT. After 12 hours, the reaction mixture was quenched with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate :
petroleum ether to afford the title compound as an off-white solid (0.105 g, 22 % yield). MP: 145-148°C. Mass: 571.7 (M+). Enantiomeric excess: 95.4% as determined by HPLC on a chiralpak AD-H column, enriched in the late eluting isomer (retention time = 14.83 min.).
PATENT
WO 2014006572
http://www.google.com/patents/WO2014006572A1?cl=en
 B1 IS DESIRED
B1 IS DESIRED
(S)-2- (l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l-yl)ethyl)-6- fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (compound-B l)
Intermediate 11
[119] Intermediate 11: 4-bromo-2-fluoro-l-isopropoxybenzene:To a solution of 4-bromo-2- fluorophenol (lOg, 52.35 mmol) in THF (100ml), isopropyl alcohol (4.8ml, 62.62 mmol) and triphenylphosphine (20.6g, 78.52 mmol) were added and heated to 45 C followed by diisopropylazodicarboxylate (15.4ml, 78 52 mmol). The mixture was refluxed for lh, concentrated and the residue was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a colourless liquid (13. lg, 99%) which was used without purification in the next step. Intermediate 12
[120] Intermediate 12: 2-(3-fluoro-4-isopropoxyphenyl)-4,4,5,5-tetramethyl- 1,3,2- dioxaborolane: Potassium acetate (10.52 g, 107.2 mmol) and bis(pinacolato)diboron (15g, 58.96 mmol) were added to a solution of intermediate 11 (10.52 g, 107.2 mmol) in dioxane (125 ml), and the solution was degassed for 30 min. [1,1 ‘- Bis(diphenylphosphino)ferrocene]dichloro palladium(II).CH2Cl2 (4.4g, 5.36 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12h the reaction mixture was filtered through celite and concentrated. The crude product was purified by column chromatography with ethyl acetate: petroleum ether to afford the title compound as a yellow oil (13.9g, 99%) which was used without purification in the next step.
Intermediate 13
[121] Intermediate 13: 3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-4- amine: To a solution of 3-iodo-lH-pyrazolo[3,4-d]pyrimidin-4-amine (11.0 g, 42.14 mmol) in DMF 110 ml), ethanol (55 ml) and water (55 ml), intermediate 12 (23.4 g, 84.28 mmol) and sodium carbonate (13.3 g, 126.42 mmol) were added and degassed for 30 min. Tetrakis(triphenylphosphine)palladium(0) (2.4 g, 2.10 mmol) was added under nitrogen atmosphere and heated to 80°C. After 12h, the reaction mixture was filtered though celite, concentrated and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was triturated with diethyl ether, filtered and dried under vacuum to afford the title compound as light brown solid (3.2 g, 26% yield) which is used as such for the next step.
Example Bl
(S)-2-(l-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-lH-pyrazolo[3,4-d]pyrimidin-l- yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
[127] To a solution of intermediate 13 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 5 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate ( 0.15 ml, 0.749 mmol) was added heated to 45°C. After 2h, the reaction mixture was quenched with with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate : petroleum ether to afford the title compound as an off-white solid (0.049 g, 20 %). MP: 139- 142°C. Mass : 571.7 (M H-NMR (δ ppm, CDC13, 400 MHz): 8.24 (s, 1H), 7.85 (dd, J = 8.2,3.1 Hz, 1H), 7.50-7.29 (m, 5H), 7.14 (t, J = 8.4 Hz, 1H), 7.02 (m, 2H), 6.92 (d, J = 8.4 Hz, 1H), 6.11 (q, J = 7.1 Hz, 1H), 5.40 (s, 2H), 4.66 (quintet, J = 6.1 Hz, 1H), 2.00 (d, J = 7.1Hz, 3H), 1.42 (d, J = 6.1 Hz, 6H). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time = 10.64min.).
PATENT
US 2014/0011819 describe the synthesis of TGR-1202 (Example B l)
http://www.google.co.in/patents/US20140011819
Example B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one
-
To a solution of intermediate 13 (0.134 g, 0.494 mmol) in THF (2.0 ml), intermediate 5 (0.150 g, 0.494 mmol) and triphenylphosphine (0.194 g, 0.741 mml) were added and stirred at RT for 5 min. Diisopropylazodicarboxylate (0.15 ml, 0.749 mmol) was added heated to 45° C. After 2 h, the reaction mixture was quenched with with water and extracted with ethyl acetate. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The crude product was purified by column chromatography with ethyl acetate:petroleum ether to afford the title compound as an off-white solid (0.049 g, 20%). MP: 139-142° C. Mass: 571.7 (M+).1H-NMR (δ ppm, CDCl3, 400 MHz): 8.24 (s, 1H), 7.85 (dd, J=8.2, 3.1 Hz, 1H), 7.50-7.29 (m, 5H), 7.14 (t, J=8.4 Hz, 1H), 7.02 (m, 2H), 6.92 (d, J=8.4 Hz, 1H), 6.11 (q, J=7.1 Hz, 1H), 5.40 (s, 2H), 4.66 (quintet, J=6.1 Hz, 1H), 2.00 (d, J=7.1 Hz, 3H), 1.42 (d, J=6.1 Hz, 6H). Enantiomeric excess: 89.8% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=10.64 min)
4-Methylbenzenesulfonate Salt of Compound B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one 4-methylbenzenesulfonate
-

-
(S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one 4-methylbenzenesulfonate: To (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (22.7 g, 39.69 mmol) in isopropanol (600 ml), p-toluenesulphonic acid (8.30 g, 43.66 mmol) was added and refluxed for 1 h. The reaction mixture was concentrated, co-distilled with petroleum ether and dried. To the residue water (300 ml) was added and stirred for 30 min. The solid was filtered, washed with petroleum ether and dried under vacuum to afford the title compound as off-white solid (28.2 g, 95%). MP: 138-141° C. 1H-NMR (δ ppm, CDCl3, 400 MHz): 8.11 (s, 1H), 7.85 (dd, J=8.0, 3.0 Hz, 1H), 7.80 (d, J=8.2 Hz, 2H), 7.51 (dd, J=9.3, 4.3 Hz, 1H), 7.45 (dd, J=7.5, 3.1 Hz, 1H), 7.42-7.31 (m, 3H), 7.29 (m, 2H), 7.22 (d, J=8.0 Hz, 2H), 7.16 (t, J=8.3 Hz, 1H), 7.08 (dt, J=8.5, 2.5 Hz, 1H), 6.97 (br s, 1H), 6.88 (br s, 1H), 6.11 (q, J=7.2 Hz, 1H), 4.67 (quintet, J=6.0 Hz, 1H), 2.36 (s, 3H), 2.03 (d, J=7.1 Hz, 3H), 1.43 (d, J=6.0 Hz, 6H). Mass: 572.4 (M++1-PTSA). Enantiomeric excess: 93.4% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=12.35 min.)

Sulphate Salt of Compound B1 (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one sulfate
-

-
(S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one sulphate: To (S)-2-(1-(4-amino-3-(3-fluoro-4-isopropoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)ethyl)-6-fluoro-3-(3-fluorophenyl)-4H-chromen-4-one (15.0 g, 26.24 mmol) in isopropanol (600 ml) was cooled to 0° C. To this Sulphuric acid (2.83 g, 28.86 mmol) was added and stirred at room temperature for 24 h. The reaction mass was filtered and washed with petroleum ether and dried under vacuum. To the solid, water (150 ml) was added and stirred for 30 min. The solid was filtered, washed with petroleum ether and dried under vacuum to afford the title compound as off-white solid (13.5 g, 76%). MP: 125-127° C. 1H-NMR (δ ppm, CDCl3, 400 MHz): 8.11 (s, 1H), 7.85 (dd, J=8.0, 3.0 Hz, 1H), 7.51 (dd, J=9.2, 4.2 Hz, 1H), 7.45-7.31 (m, 3H), 7.29 (m, 1H), 7.15 (t, J=8.3 Hz, 1H), 7.08 (dt, J=8.5, 2.4 Hz, 1H), 6.96 (br s, 1H), 6.88 (br s, 1H), 6.09 (q, J=7.1 Hz, 1H), 4.676 (quintet, J=6.1 Hz, 1H), 2.01 (d, J=7.1 Hz, 3H), 1.42 (d, J=6.1 Hz, 6H). Mass: 572.2 (M++1-H2SO4). Enantiomeric excess: 89.6% as determined by HPLC on a chiralpak AD-H column, enriched in the fast eluting isomer (retention time=12.08 min.)
-
Various other acid addition salts of compound B1 were prepared as provided in Table 1.
-
TABLE 1 Melting Point Acid Method of preparation (° C.) Hydro- Compound B1 (1 eq.) dissolved in THF, 130-132 chloric excess HCl/Et2O was added, the clear acid solution obtained was evaporated completely. The residue obtained was washed with water. p- Compound B1 (1 eq.) dissolved in 138-141° C. Toluene- isopropyl alcohol (IPA), refluxed for sulfonic 30 min., acid (1.1 eq.) in IPA was added, acid the clear solution obtained was evaporated completely. The residue obtained was washed with water. Benzene- Compound B1 (1 eq.) dissolved in IPA, 170-172 sulphonic refluxed for 30 min., acid(1.1 eq.) in IPA acid was added, the clear solution not obtained, the residue was evaporated completely and was washed with water. Maleic Compound B1 (1 eq.) dissolved in IPA, 107-109 acid refluxed for 30 min., acid (1.1 eq.) in IPA was added, the clear solution not obtained, the residue was evaporated completely and was washed with water. Camphor Compound B1 (1 eq.) dissolved in IPA, 120-121 sulfonic refluxed for 30 min., acid (1.1 eq.) in IPA acid was added, the clear solution not obtained, the residue was evaporated completely and was washed with water. Sulphuric Compound B1 (1 eq.) dissolved in IPA, 125-127 acid refluxed for 30 min., acid(1.1 eq.) in IPA was added, the clear solution obtained was evaporated completely. The residue obtained was washed with water.

REFERENCES
WO 2014/006572 and U.S. Patent Publication No. 2014/0011819,
http://www.tgtherapeutics.com/O’ConnorTGR202Single%20AgentEHA&Lugano2015.pdf
-
TGR-1202: Phase I/II started 09/28/2015
Week in Review, Clinical StatusRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) … -
Ublituximab: Phase I/II started 09/28/2015
Week in Review, Clinical StatusLFB S.A., Les Ulis, France TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: Ublituximab (TGTX-1101, TG-1101, LFB-R603) Business: Cancer Molecular target: CD20 Description: Glycoengineered mAb against CD20 … -
The Daily Extra, Company NewsTG Therapeutics Inc. (NASDAQ:TGTX) rose $2.65 (23%) to $14.37 after the company said it received an SPA from FDA for the Phase III UNITY-CLL trial of ublituximab (TG-1101) in combination with TGR-1202 to treat chronic …
-
Targets & Mechanisms: The battle for IRAK 04/23/2015
Nimbus, Aurigene and TG Therapeutics are chasing IRAK4 inhibitors for cancerBC Innovations, Targets & MechanismsNow that Nimbus has put IRAK4 on the map for B cell lymphoma, several companies are closing in with their own inhibitors, and they’re all on track for IND-enabling studies this year. -
TGR-1202: Additional Phase I/II data 01/26/2015
Week in Review, Clinical ResultsRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) … -
Ublituximab: Additional Phase I/II data 01/26/2015
Week in Review, Clinical ResultsLFB S.A., Les Ulis, France TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Ildong Pharmaceutical Co. Ltd. (KSE:000230), Seoul, South Korea Product: Ublituximab (TGTX-1101, TG-1101, LFB-R603) Business: Cancer … -
TGR-1202: Phase I started 12/15/2014
Week in Review, Clinical StatusRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Product: TGR-1202 (formerly RP5264) Business: Cancer Molecular target: Phosphoinositide 3-kinase (PI3K) … -
Rhizen, TG Therapeutics deal 12/08/2014
Week in Review, DealsRhizen Pharmaceuticals S.A., La Chaux-de-Fonds, Switzerland TG Therapeutics Inc. (NASDAQ:TGTX), New York, N.Y. Business: Cancer TG Therapeutics exercised an option under a 2012 deal to license exclusive, worldwide …
| Patent | Submitted | Granted |
|---|---|---|
| NOVEL SELECTIVE PI3K DELTA INHIBITORS [US2014011819] | 2013-07-02 | 2014-01-09 |
| Treatment Of Cancers Using PI3 Kinase Isoform Modulators [US2014377258] | 2014-05-30 | 2014-12-25 |
////////Umbralisib
CC(C)OC1=C(C=C(C=C1)C2=NN(C3=C2C(=NC=N3)N)C(C)C4=C(C(=O)C5=C(O4)C=CC(=C5)F)C6=CC(=CC=C6)F)F
DRL 17822 from Reddy US Therapeutics/Dr Reddy’s



CAS 920493-71-6 and CAS 898911-09-6



DRL 17822
MW 603.6045, MFC30 H31 F6 N7
| Molecular Formula: | C30H31F6N7 |
|---|---|
| Molecular Weight: | 603.604459 g/mol |
Cas 898911-09-6, 1454689-50-9
3-([[3,5-Bis(trifluoromethyl)benzyl](2-methyl-2H-tetrazol-5-yl)amino]methyl)-N,N-bis(cyclopropylmethyl)-8-methylquinolin-2-amine
3-Quinolinemethanamine, 2-[bis(cyclopropylmethyl)amino]-N-[[3,5-bis(trifluoromethyl)phenyl]methyl]-8-methyl-N-(2-methyl-2H-tetrazol-5-yl)-
3-(((3,5-bis(trifluoromethyl)benzyl)(2- methyl-2H-tetrazol-5-yl)amino)methyl)-N,N-bis(cyclopropylmethyl)-8- methylquinolin-2-amine
(3-{ [3,5-bis-trifluoromethyl-benzyl)-(2-methyl-2H-tetrazoIe-5-yl)- amino]-methyl}-8-methyl-quinolin-2-yl)-bis-cyclopropylmethyl-amine
Reddy US Therapeutics (Innovator)




Treatment of Atherosclerosis Therapy Lipoprotein Disorders,
CETP inhibitor (dyslipidemia/atherosclerosis/cardiovascular diseases), Dr Reddy’s
Selective inhibitor of cholesteryl ester transfer protein (CETP)
- 30 Jun 2012Dr Reddy’s Laboratories completes a phase II trial in Hypercholesterolaemia in Italy, Poland and Ukraine (NCT01388816)
- 09 Mar 2012Dr Reddy’s Laboratories completes enrolment in its phase II trial for Hypercholesterolaemia in Italy, Poland, and Ukraine (NCT01388816)
- 02 Sep 2011Phase-II clinical trials in Hypercholesterolaemia in Ukraine (PO)
CLINICAL TRIALS…..Type II Hyperlipidemia PHASE 2…………https://clinicaltrials.gov/ct2/show/NCT01388816
Cardiovascular disease is a leading cause of death worldwide. Among cardiovascular disorders, coronary heart disease (CHD) caused by atherosclerosis is the most common cause of morbidity and mortality. Prevention, stabilization and regression of atherosclerotic plaques may have a major impact on reducing the risk of acute coronary events.
LDL-C lowering agents, primarily the statins, are the current mainstay in the pharmacologic management of dyslipidemia. However even with stain use, residual CHD risk from dyslipidemia remains. Epidemiologic and observational studies have shown that HDL-C is also a strong independent predictor of CHD, suggesting that raising HDL-C levels might afford clinical benefit in the reduction of cardiovascular risk.
Presently only niacin is approved by the FDA for HDL-C elevation and can raise HDL-C levels by 20-30%. However its use can be limited by a high incidence of flushing and, less commonly, by elevation of blood glucose and potential hepatic toxicity.
Cholesteryl ester transfer protein (CETP) inhibitors are being explored for their ability to elevate HDL-C. A small molecule CETP inhibitor, torcetrapib, has been demonstrated to elevate HDL-C by 60-100%. However, a large clinical trial (ILLUMINATE) where it increased HDL-C by a mean of 72% compared to baseline was halted as it failed to show benefit. Post-hoc analysis of this study implicated an off-target increase in blood pressure as potentially counteracting any anti-atherosclerotic benefits. Post-hoc subgroup analysis showed that patients in the highest HDL-C quartile had a 57% reduction in the risk of cardiovascular events.
Increased blood pressure appears to be specifically related to torcetrapib as two other small molecule CETP inhibitors, anacetrapib and dalcetrapib, have not shown this in clinical trials and have been well tolerated. DRL-17822 has also not shown elevation of blood pressure in either animals or in normal volunteers.
This study will investigate the efficacy and tolerability of DRL-17822 as dyslipidemia monotherapy in patients with Type II hyperlipidemia.
Hyperlipidemia or an elevation in serum lipids is associated with an increase incidence of cardiovascular disease and atherosclerosis. Primary hyperlipidemia is a term used to describe a defect in lipoprotein metabolism. The lipoproteins commonly affected are low density lipoprotein (LDL) cholesterol, which transports mainly cholesterol, and very low density lipoprotein-cholesterol (VLDL-cholesterol), which transports mainly triglycerides (TG). Most subjects with hyperlipidemia have a defect in LDL metabolism, characterized by raised cholesterol, LDL-C levels, with or without raised triglyceride levels; such subjects are termed hypercholesterolemic (Fredrickson Type II). Familial hypercholesterolemia (FH) is caused by any one of a number of genetically-determined defects in the LDL receptor, which is important for the entry of cholesterol into cells. The condition is characterized by a reduced number of functional LDL receptors, and is therefore associated with raised serum LDL-C levels due to an increase in LDL.

It is reasonably known in the art that the likelihood of cardiovascular disease can be decreased, if the serum lipids, and in particular LDL-C, can be reduced. It is further known that the progression of atherosclerosis can be retarded or the regression of atherosclerosis can be induced if serum lipids can be lowered. In such cases, individuals diagnosed with hyperlipidemia or hypercholesteremia should consider lipid-lowering therapy to retard the progression or induce the regression of atherosclerosis for purposes of reducing their risk of cardiovascular disease, and in particular coronary artery disease.
Cholesteryl ester-transfer protein (CETP) is an important player in metabolism of lipoproteins, such as, for example, a high density lipoprotein (HDL). CETP is a 70 kDa plasma glycoprotein that is physically associated with HDL particles. It facilitates the transport of cholesteryl ester from HDL to apolipoprotein B-containing lipoproteins. This transfer is accompanied by transfer of triglycerides in the opposite direction. Thus, a decrease in CETP activity can result in an increase in the level of HDL cholesterol and a decrease in the level of very low density lipoprotein (VLDL) and low density lipoprotein (LDL). CETP can therefore simultaneously affect the concentrations of pro-atherogenic (for example, LDL) and anti-atherogenic (for example, HDL) lipoproteins.
Several CETP inhibitors are currently in various clinical phases of development for treating various aforementioned disorders. In spite of having various advantages, CETP inhibitors are proven to be difficult to formulate for oral administration. CETP inhibitors are of a highly lipophilic nature and have extremely low solubility in water. Due to their poor solubility, bioavailability of conventional oral compositions is very poor. The lipophilic nature of CETP inhibitors not only leads to low solubility but also tends to poor wettability, further reducing their tendency to be absorbed from the gastrointestinal tract. In addition to the low solubility, CETP inhibitors also tend to have significant, “food effect”, where a significant difference in rate and amount of drug absorption is observed when the drug is administered with or without a meal. This “food effect”, often complicates the dosing regimen and may require high dosing to achieve the desired therapeutic effect, resulting in potentially unwanted side effects.
Several attempts have been made to improve the solubility of CETP inhibitors, but have generally ended up with limited success. At the outset, most methods aimed at enhancing aqueous concentration and bioavailability of low-solubility drugs only offer moderate improvements. References describing improving the dissolution of poorly soluble drugs include: U.S. Patent Nos. 5,456,923, 5,993,858, 6,057,289, 6,096,338, 6,267,985, 6,280,770, 6,436,430, 6,451,339, 6,531,139, 6,555,558, 6,638,522, 6,962,931 and 7,374,779.
PATENT
WO 2014128564
https://www.google.co.in/patents/WO2014128564A2?cl=en
WO-2014076568
http://www.google.com/patents/WO2014076568A2?cl=en
EXAMPLES
In the following Examples 1-17, various compositions in accordance with the present application were prepared comprising 3-(((3,5-bis(trifluoromethyl)benzyl)(2- methyl-2H-tetrazol-5-yl)amino)methyl)-N,N-bis(cyclopropylmethyl)-8- methylquinolin-2-amine as the CETP inhibitor.:
EXAMPLE 1 :

1. 3-(((3,5-bis(trifluoromethyl)benzyl)(2-methyl-2H-tetrazol-5-yl)amino)methyl)- N,N-bis(cyclopropylmethyl)-8-methylquinolin-2-amineand hydroxypropyl methyl cellulose acetate succinate were mixed together in given solvent mixture to form clear solution.
2. To the solution of step I, Polyoxyl 35 castor oil and talc were added to form a homogenous suspension.
3. The suspension of step 2 was sprayed over inert sugar spheres and dried.
4. The drug layered spheres of step 3 were coated with dispersion made from given seal layer ingredients.
5. The coated spheres of step 4 were formulated further as capsule dosage form.
PATENT
WO 2013046045
https://www.google.co.in/patents/WO2013046045A1?cl=en
PATENT
WO 2013024358
PATENT
WO 2007075194
https://www.google.co.in/patents/WO2007075194A1?cl=en
Syntheis construction





Example 1
Synthesis of (3-{[3,5-bis trifluoromethyl-benzyl )-(2-cyclopropyImethyI-2H- tetrazole -5-yl)-amino]-methyl-}-8-methyI-quinolme-2-yl)-bis- cyclopropylmethyl-amine Step (i): Synthesis of 2~chloro-8-methyl-quinoline-3-carbaldehyde
DMF (1.22 g, 16.7 mmol) was taken in a flask equipped with a drying tube and POCl3 (7.32 g, 46.7 mmol) was added dropwise with stirring at 0° C. To this solution, TV-o-Tolyl acetamide (1.00 g, 6.7 mmol) was added and the solution was refluxed for 6 h at 90° C. The excess POCl3 was distilled off, water was added to the residue and this was stirred at room temperature for 10 min. The solid was filtered and dried under vacuum..This crude compound was purified over silica gel (100-200 mesh) using 6% ethyl acetate and petroleum ether to give the product as a yellowish solid (yield: 78%). 1H NMR (CDCl3, 200 MHz): δ 10.5 (s, IH)5 8.71 (s, IH), 7.83- 7.79 (m, IH), 7.74- 7.70 (m, IH), 7.56-7.49 (m, IH), 2.79 (s, 3H); m/z (EI-MS): 206 (M+, 100%). Step (ϋ): Synthesis of 2-(bis(cyclopropylmethyl)amino)-8-methylquinoline-3- carbaldehyde:
2-Chloro-8-methyl-quinoline-3-carbaldehyde (.115 g, 0.559 mmol), and potassium carbonate (0.231 g, 1.67 mmol) were put in a 25 mL two necked RB flask. To this, 3 mL of DMF was added followed by dropwise addition of bis- cyclopropylmethyl amine (0.083 g, 0.67 mmol). The reaction mixture was refluxed for 2 h and was cooled to RT. It was then poured on crushed ice (10 mL) and extracted with EtOAc (3 x 10 mL). The organic layer was washed with brine and dried over sodium sulphate. The solvent was evaporated under vacuum to give a yellow colored oil (0.081 g, 50%).
1H NMR (CDCl3, 400 MHz): δ 10.5 (s, IH), 8.71 (s, IH), 7.83- 7.79 (m, IH),
7.74-7.70 (m, IH), 7.56-7.49 (m, IH), 3.55-3.47 (m, 4H), 2.79 (s, 3H), 1.73-1.72
(m, 2H), 1.70-1.46 (m, 4H), 1.20-1.11 (m, 4H); m/z (ES-MS ): 295 (M+H-I5
100%); IR (neat, cm“1): 3385, 2948, 1691.
Step (iii): Synthesis of 3-((3,5-bis(trifluoromethyl)benzylamino)methyl)-N,N- bis(cyclopropylmethyl)-8-methylquinolin-2-amine
2-(Bis(cyclopropylmethyl)amino)-8-methylquinoline-3-carbaldehyde (0.081 g, 0.39 mmol), 3,5-bis-trifluoromethylbenzylamine (0.096 g, 0.39 mmol) and acetic acid (0.047 g, 0.78 mmol) were put in a 25 mL RB flask. To this, 2 rnL of methanol was added and stirred at RT for 15 min. Sodium cyanoborohydri.de (0.075 g, 0.77 mmol) was added portionwise and stirring was continued at RT for another 1 h. Methanol was removed from the reaction mixture under vacuum, water was added to this crude and was extracted with ethyl acetate (3 x 50 mL). The organic layer was washed with saturated NaHCO3 solution, brine and dried over sodium sulphate. The solvent was evaporated and the crude residue was purified by column chromatography over silica gel (100-200 mesh) eluting with 4% ethyl acetate in petroleum ether to give the title amine (0.142 g, yield: 99%). 1R NMR (CDCl3, 400 MHz): δ 7.89-7.86 (m, IH), 7.80 (m, IH), 7.75-7.74 (m, IH), 7.60-7.40 (m, 3H), 7.30-7.26 (m,lH), 4.12 (s, 2H), 3.88 (s, 2H), 3.24-3.22 (m, 4H), 2.72 (s, 3H), 0.99-0.92 (m, 2H), 0.44-0.35 (m, 4H), 0.11-0.05 (m, 4H); m/z (EI-MS ): 522 (M++l, 100%); IR (neat, cm“1): 3357, 2929, 2851.
Step (iv): Synthesis of N-(3,5-bis(trifluoromethyl)benzyl)-N-((2- (bis(cyclopropylmethyl)amino)-8-methylqumolin-3-yl)methyl)cyanamide
To a solution of 3-((3,5~bis(trifluoromethyl)benzylamino)methyl)-N,N- bis(cyclopropylmethyl)-8-methylquinolin-2-amine (0.176 g , 0.33 mmol ), obtained in step (iii) , in MeOH (4 mL) under N2 atmosphere was added sodium bicarbonate (0.056 g, 0.67 mmol ) followed by the addition of cyanogen bromide (0.063 g, 0.60 mmol). The reaction mixture was stirred at RT for 2 h. The solvent was removed under vacuum to give the crude residue which was dissolved in water, extracted with ethyl acetate and dried over sodium sulphate. The solvent was evaporated and concentrated in vacuo to afford N-(3,5-bis(trifluoromethyl)benzyl)- N-((2-(bis(cyclopropylmethyl)amino)-8-methylquinolin-3-yl)methyl)cyanamide (0.118 g, 64%).
1H NMR (CDCl3, 400 MHz ): δ 8.07 (s, IH) , 7.82 (s, IH), 7.70 (s, 2H), 7.56-7.55 (m, IH), 7.50-7.49 (m, IH), 4.49 (s, 2H), 4.23 (s, 2H), 3.17 -3.15 (m, 4H), 2.71 (s, 3H), 0.097-0.085 (m, 2H), 0.405-0.401 (m, 4H), 0.385-0.381 (m, 4H); m/z (ES- MS): 547 (M++l, 100%); IR(KBr ,Cm“1 ) : 2273, 1280.
Step (v): Synthesis of (3-{[(3,5-bistrifluoromethyl-benzyl)-(2H-tetrazol-5-yl)- amino]-methyl}-8-methyl-quinolin-2-yl)-bis-cyclopropylmethyl-amine
7V-(3,5-Bis(tiifluoromethyl)benzyl)-N-((2-(bis(cyclopropylmethyl)amino)- 8-methylqumolin-3-yl)methyl)cyanamide (0.118 g, 0.216 mmol), sodium azide (0.70 g 1.08 mmol) and ammonium chloride (.058 g, 1.08 mmol) were put in a RB flask under N2atmosphere. To this reaction mixture, DMF (2 mL) was added and was refluxed for 1 h. The reaction mixture was cooled to RT and ice was added to this and extracted with ethylacetate (3×10 mL). The combined organic layer was washed with brine, dried over sodium sulphate and then concentrated under vacuum to afford of (3-{[(3,5-bistrifluoromethyl-benzyl)-(2H-tetrazol-5-yl)- amino]-methyl}-8-methyl-quinolin-2-yl)-bis-cyclopropylmethyl-amine as a yellow solid (0.125 g, 99%).
1H NMR (CDCl3, 400 MHz ): δ 7.99 (s, IH) , 7.79 -7.74 (m, 4H ), 7.41-7.40 (m,
IH ), 7.33-7.31 (m, IH), 4.99 (s, 2H), 4.80 (s, 2H), 3.68 (s, 4H), 2.16 (s, IH) 1.56-
1.06 (m, HH); m/z (ES-MS): 578 (M++l, 100%); IR (KBr , cm“1) 3680 , 2922 ,
1660 , 1616.
METHYLATION SHOULD GIVE THE PRODUCT
Scheme 1


PATENT
WO 2006073973
http://www.google.co.in/patents/WO2006073973A2?cl=en
Example 47
Synthesis of [2-(bis-cycIopropylmethyI-amino)-8-methyl-quinolin-3-ylmethyI]-(3,5- bis-trifluoromethyl-benzyl)-carbamic acid methyl ester
Step (i): Synthesis of bis-cyclopropylmethyl-amine
(i) a. Synthesis of cyclopropanecarboxylic acid cyclopropylmethyl-amide:

Cyclopropyl carboxylic acid (1.0 g, 11.63 mmol) was added to a 50 mL two neck round bottom flask, along with DCM (25 mL). This mixture was cooled to 0° C, EDCI (4.15 g, 13.95 mmol) was added portionwise to the mixture with stirring under nitrogen atmosphere, and the temperature was maintained for 0.5 h. After this time, hydroxybenzotriazole (1.88 g, 13.95 mmol) was added to the 0° C mixture which was stirred for 10 min, then triethylamine (1.7 g, 11.63 mmol) was added, and stirring of the mixture was continued at the same temperature for another 0.5 h. Then, cyclopropylmethylamine (0.825 g, 11.63 mmol) was added, and the reaction was allowed to reach RT, and stirring was continued overnight. The solvent was then removed in vacuo, and the crude residue was purified by passing through a column over 60-120 silica gel, eluting with dichloromethane, to afford the title compound (1.6 g), yield: 87%. 1H NMR (CDCl3, 200 MHz): d 5.75 (br s, NH, D2O exchangeable), 3.17-3.16 (m, 2H), 1.00-0.80 (m, 4H), 0.77-0.67 (m, 2H), 0.56-0.43 (m, 2H), 0.24-0.16 (m, 2H) m/z (CI-MS): 139 (M+, 100%) (i) b. Synthesis of bis-cyclopropylmethyl-amine

To a suspension of lithium aluminum hydride (1.3 g, 9.35mmol) in 10 mL dry ether, a solution of N-cyclopentenoyl-ethylamine (1.7 g, 13.3 mmol) in dry ether (10 mL) was added under a nitrogen atmosphere. This reaction was stirred at RT for 8 h and the reaction mixture was then quenched with saturated sodium sulfate solution, filtered, and the precipitate was washed with diethyl ether. The filtrate was concentrated to afford the title amine (0.8 g), yield: 69%.
1H NMR (CDCl3, 200 MHz): d 5.75 (br s, NH, D2O exchangeable), 3.16-3.09 (m, 2H), 2.50-2.4 (m, 2H), 0.56-0.43 (m, 4H), 0.24-0.21 (m, 3H), 0.21-0.13 (m, 3H) m/z (ES-MS): 139 (M^+14, 100%)
Step (ii): Synthesis of [2-(bis-cyclopropylmethyl-amino)-8-methyl-quinolin-3-ylmethyl]- (S^-bis-trifluoromethyl-benzy^-carbamicacid methyl ester

The title compound was synthesized by using the same procedure as in Example 35, except using o-tolyl acetanilide in step (i) instead of acetanilide and bis- cyclopropylmethyl amine in step (iii), which yielded the desired product as a light yellow, viscous liquid (0.05 g), yield:40%, of purity 98.8% (HPLC: Symmetry Shield RP8, [0.01M KH2PO4: CH3CN], 217 nM, Rt12.719 min).
1H NMR (CDCl3, 400 MHz): d 7.7 (s, IH), 7.68-7.44 (m, 3H), 7.27-7.24 (m, 2H), 4.78- 4.65 (m, 2H), 4.47-4.4 (m, 2H), 3.8 (s, 3H), 3.16-3.14 (d, J=7Hz, 2H), 2.7 (s, 3H), 1.55 (s, 3H), 1.01-0.9(m, IH), 0.38-0.34 (m, 4H), 0.07-0.05 (m, 4H); m/z (CI-MS): 579 (M+, 100%)
Example 57
Synthesis of (3-{ [3,5-bis-trifluoromethyl-benzyl)-(2-methyl-2H-tetrazoIe-5-yl)- amino]-methyl}-8-methyl-quinolin-2-yl)-bis-cyclopropylmethyl-amine
The title compound was prepared as an oil by following the same synthetic procedures as in Example 52, except using {3-[(3,5-bis-trifluoromethyl-benzylamino)- methyl]-8-methyl-quinolin-2-yl}-bis-cyclopropylmethyl-amine in step (i) instead of {3- [(3,5-bis-trifluoromethyl-benzylamino)-methyl]-quinolin-2-yl}-cyclopentylmethyl-ethyl- amine (0.07 g), yield: 52%.
Purity: 95.53% (HPLC: Symmetry Shield RP8, [0.01M KH2PO4: CH3CN], 217 nM, Rt 9.538 min).
IR (neat, cm4) 3079, 2925, 1582;
1H NMR (CDCl3, 400 MHz): d 7.82 (s, IH), 7.69-7.67 (m, 2H), 7.44-7.41 (m, IH), 7.23- 7.2 (m, 3H), 4.91 (s, 2H), 4.65 (s, 2H), 4.21 (s, 3H), 3.29 -3.19 (m, 4H)5 2.71 (s, 3H), 1.01-1.00 (m, 2H), 0.99-0.83 (m, 2H), 0.39-0.34 (m, 3H), 0.08-0.07 (m, 3H). m/z (ES-MS): 604 (M++!, 100%)
Dr. Reddy’s announces start of Phase II study with the CETP inhibitor, DRL-17822 in dyslipidemia patients
Hyderabad, India, September 02, 2011: Dr Reddy’s Laboratories (NYSE: RDY) announced the initiation of dosing with DRL-17822 in patients with diagnosis of type II dyslipidaemia. DRL-17822, is a selective, orally bioavailable inhibitor of cholesteryl ester transfer protein (CETP), for the treatment and/or prevention of dyslipidaemia, atherosclerosis and associated cardiovascular disease.
The current study is being conducted under a CTA in a number of countries in Europe. The objective of the study is to evaluate the efficacy and safety of DRL-17822 in patients with Type-II dyslipidemia. This is a randomized, double blind, placebo controlled, parallel group study in 160 subjects. The primary outcome measure is to assess the elevation in HDL cholesterol and reduction in LDL cholesterol from baseline to end of treatment compared to placebo. Three doses (50, 150 & 300 mg) of DRL-17822 given once daily for 4 weeks will be evaluated during this study.
Three human Phase I studies with DRL-17822 had already been conducted in Europe, where DRL-17822 was shown to be safe and well tolerated. In these studies, the proof of mechanism had been demonstrated by dose-dependent inhibition of plasma CETP activity as well as by significant increase in HDL cholesterol & decrease in LDL cholesterol levels.
Cardiovascular disease is a leading cause of death among men and women worldwide. Among cardiovascular disorders, coronary heart disease (CHD), caused by atherosclerosis is the most common cause of morbidity and mortality. Stabilization and/or regression of atherosclerotic plaques may have a major impact on reducing the risk of acute coronary events. Low-density lipoprotein cholesterol lowering agents, primarily the statins, are the current mainstay in the pharmacological management of dyslipidaemia. However, significant residual cardiovascular risk remains despite use of statins.
Epidemiological and observational studies demonstrate that reduced high density lipoprotein cholesterol levels are a strong, independent predictor of CHD, suggesting that raising HDL cholesterol levels might afford clinical benefit in the reduction of cardiovascular risk. One approach to raise HDL level has been inhibition of CETP activity. Currently it is believed that, raising HDL cholesterol and lowering LDL cholesterol through CETP inhibition would lead to a significant benefit in terms of CHD risk reduction.
Dr. K. Anji Reddy, Founder Chairman, Dr. Reddy’s Laboratories added, “We are committed to delivering products of differentiated value in this area of high global clinical unmet need. We are excited to continue to advance our CETP program and look forward to the data from our Phase II study. This class of therapy could transform the treatment of CHD and DRL 17822 is in a position to be one of the front-running products in the class”.
Disclaimer
This press release includes forward-looking statements, as defined in the U.S. Private Securities Litigation Reform Act of 1995. We have based these forward-looking statements on our current expectations and projections about future events. Such statements involve known and unknown risks, uncertainties and other factors that may cause actual results to differ materially. Such factors include, but are not limited to, changes in local and global economic conditions, our ability to successfully implement our strategy, the market acceptance of and demand for our products, our growth and expansion, technological change and our exposure to market risks. By their nature, these expectations and projections are only estimates and could be materially different from actual results in the future.
About Dr. Reddy’s
Dr. Reddy’s Laboratories Ltd. (NYSE: RDY) is an integrated global pharmaceutical company, committed to providing affordable and innovative medicines for healthier lives. Through its three businesses – Pharmaceutical Services and Active Ingredients, Global Generics and Proprietary Products – Dr. Reddy’s offers a portfolio of products and services including APIs, custom pharmaceutical services, generics, biosimilars, differentiated formulations and NCEs. Therapeutic focus is on gastro-intestinal, cardiovascular, diabetology, oncology, pain management, anti-infective and pediatrics. Major markets include India, USA, Russia and CIS, Germany, UK, Venezuela, S. Africa, Romania, and New Zealand. For more information, log on to: www.drreddys.com
For more information please contact:
Investors and Financial Analysts:
Kedar Upadhye at kedaru@drreddys.com / +91-40-66834297
Raghavender R at raghavenderr@drreddys.com / +91-40-49002135
Milan Kalawadia (USA) at mkalawadia@drreddys.com / +1 908-203-4931
Media:
S Rajan at rajans@drreddys.com / +91-40- 49002445
| WO2005011634A1 * | Jul 21, 2004 | Feb 10, 2005 | William John Curatolo | Dosage forms providing controlled release of cholesteryl ester transfer protein inhibitors and immediate release of hmg-coa reductase inhibitors |
| WO2006073973A2 * | Dec 28, 2005 | Jul 13, 2006 | Reddy Us Therapeutics Inc | Novel benzylamine derivatives as cetp inhibitors |
| WO2006129167A1 * | May 22, 2006 | Dec 7, 2006 | Pfizer Prod Inc | PHARMACEUTICAL COMPOSITIONS OF CHOLESTERYL ESTER TRANSFER PROTEIN INHIBITORS AND HMG-CoA REDUCTASE INHIBITORS |
| WO2007128568A1 * | May 8, 2007 | Nov 15, 2007 | Novartis Ag | Bicyclic derivatives as cetp inhibitors |
| EP0298666A2 * | Jul 1, 1988 | Jan 11, 1989 | American Home Products Corporation | Spray dried ibuprofen compositions |
| EP1741424A2 * | Jul 27, 1998 | Jan 10, 2007 | Pfizer Products Inc. | Solid pharmaceutical dispersions with enhanced bioavailabilty |
| US5456923 | Dec 23, 1993 | Oct 10, 1995 | Nippon Shinyaku Company, Limited | Method of manufacturing solid dispersion |
| US5474989 | Mar 1, 1994 | Dec 12, 1995 | Kurita Water Industries, Ltd. | Drug composition |
| US5985326 | May 30, 1996 | Nov 16, 1999 | Icos Corporation | Method of producing a solid dispersion of a poorly water soluble drug |
| US6350786 | Sep 7, 1999 | Feb 26, 2002 | Hoffmann-La Roche Inc. | Stable complexes of poorly soluble compounds in ionic polymers |
| US6548555 | Jan 31, 2000 | Apr 15, 2003 | Pfizer Inc | Basic drug compositions with enhanced bioavailability |
| US6638522 | Dec 10, 1999 | Oct 28, 2003 | Pharmasolutions, Inc. | Microemulsion concentrate composition of cyclosporin |
| US6730679 | Mar 20, 1997 | May 4, 2004 | Smithkline Beecham Corporation | Pharmaceutical formulations |
| US7008640 | Jul 16, 2001 | Mar 7, 2006 | Yamanouchi Pharmaceutical Co., Ltd. | Pharmaceutical composition for oral use with improved absorption |
| US7034013 | Mar 19, 2002 | Apr 25, 2006 | Cydex, Inc. | Formulations containing propofol and a sulfoalkyl ether cyclodextrin |
| US7037528 | Jun 5, 2001 | May 2, 2006 | Baxter International Inc. | Microprecipitation method for preparing submicron suspensions |
| US7078057 | Dec 19, 2000 | Jul 18, 2006 | Kerkhof Nicholas J | Process for producing nanometer particles by fluid bed spray-drying |
| US7081255 | Aug 14, 2002 | Jul 25, 2006 | Janssen Pharmaceutica, N.V. | Antifungal compositions with improved bioavailability |
| US8030359 | Feb 9, 2007 | Oct 4, 2011 | Merck Sharp & Dohme Corp. | Polymer formulations of CETP inhibitors |
| US20060178514 | Dec 28, 2005 | Aug 10, 2006 | Anima Baruah | Novel benzylamine derivatives as CETP inhibitors |
| Reference | ||
|---|---|---|
| 1 | * | EINFAL T ET AL: “Methods of amorphization and investigation of the amorphous state“, ACTA PHARMACEUTICA 20130901 CROATIAN PHARMACEUTICAL SOCIETY DEU, vol. 63, no. 3, 1 September 2013 (2013-09-01), pages 305-334, XP002721717, ISSN: 1330-0075 |
| 2 | * | KAI TOSHIYA ET AL: “Oral absorption improvement of poorly soluble drug using solid dispersion technique“, CHEMICAL AND PHARMACEUTICAL BULLETIN (TOKYO), vol. 44, no. 3, 1996, pages 568-571, XP002721716, ISSN: 0009-2363 |
| 3 | * | KIM TAE-WAN ET AL: “Characterization of dual layered pellets for sustained release of poorly water-soluble drug“, CHEMICAL & PHARMACEUTICAL BULLETIN (TOKYO), vol. 55, no. 7, July 2007 (2007-07), pages 975-979, XP002721715, ISSN: 0009-2363 |
| 4 | * | KIM TAE-WAN ET AL: “Modified release of coated sugar spheres using drug-containing polymeric dispersions.“, ARCHIVES OF PHARMACAL RESEARCH JAN 2007, vol. 30, no. 1, January 2007 (2007-01), pages 124-130, XP002721714, ISSN: 0253-6269 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014128564A2 * | Feb 21, 2014 | Aug 28, 2014 | Dr. Reddy’s Laboratories Ltd. | Pharmaceutical compositions of cetp inhibitors |
| Patent | Submitted | Granted |
|---|---|---|
| Novel benzylamine derivatives and their utility as cholesterol ester-transfer protein inhibitors [US2007015758] | 2007-01-18 | |
| Novel benzylamine derivatives as CETP inhibitors [US2006178514] | 2006-08-10 |
| Publication Number | Publication Date | IPCR Assignee/Applicant | Structure hits | Tools | |
|---|---|---|---|---|---|
|
1.
EP-2919765-A2 |
2015-09-23 |
EN
|
|
||
|
2.
US-20150216866-A1 |
2015-08-06 |
|
|||
|
3.
US-9040558-B2 |
2015-05-26 |
|
|||
|
4.
WO-2014128564-A2 |
2014-08-28 |
EN
|
|
||
|
5.
WO-2014076568-A2 |
2014-05-22 |
EN
|
|
||
|
6.
US-20140134235-A1 |
2014-05-15 |
|
|||
|
7.
US-20070015758-A1 |
2007-01-18 |
|
SEE
http://circ.ahajournals.org/cgi/content/meeting_abstract/122/21_MeetingAbstracts/A13981
////////
Cn1nc(nn1)N(Cc2cc5cccc(C)c5nc2N(CC3CC3)CC4CC4)Cc6cc(cc(c6)C(F)(F)F)C(F)(F)F
CC1=CC=CC2=CC(=C(N=C12)N(CC3CC3)CC4CC4)CN(CC5=CC(=CC(=C5)C(F)(F)F)C(F)(F)F)C6=NN(N=N6)C
Pfizer’s PF 04937319 glucokinase activators for the treatment of Type 2 diabetes

PF 04937319
N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide
MW 432.43
CLINICAL TRIALS
A trial to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of single doses of PF-04937319 in subjects with type 2 diabetes mellitus (NCT01044537)
SYNTHESIS

Glucokinase is a key regulator of glucose homeostasis and small molecule activators of this enzyme represent a promising opportunity for the treatment of Type 2 diabetes. Several glucokinase activators have advanced to clinical studies and demonstrated promising efficacy; however, many of these early candidates also revealed hypoglycemia as a key risk. In an effort to mitigate this hypoglycemia risk while maintaining the promising efficacy of this mechanism, we have investigated a series of substituted 2-methylbenzofurans as “partial activators” of the glucokinase enzyme leading to the identification of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide as an early development candidate.

Diabetes is a major public health concern because of its increasing prevalence and associated health risks. The disease is characterized by metabolic defects in the production and utilization of carbohydrates which result in the failure to maintain appropriate blood glucose levels. Two major forms of diabetes are recognized. Type I diabetes, or insulin-dependent diabetes mellitus (IDDM), is the result of an absolute deficiency of insulin. Type Il diabetes, or non-insulin dependent diabetes mellitus (NIDDM), often occurs with normal, or even elevated levels of insulin and appears to be the result of the inability of tissues and cells to respond appropriately to insulin. Aggressive control of NIDDM with medication is essential; otherwise it can progress into IDDM. As blood glucose increases, it is transported into pancreatic beta cells via a glucose transporter. Intracellular mammalian glucokinase (GK) senses the rise in glucose and activates cellular glycolysis, i.e. the conversion of glucose to glucose-6-phosphate, and subsequent insulin release. Glucokinase is found principally in pancreatic β-cells and liver parenchymal cells. Because transfer of glucose from the blood into muscle and fatty tissue is insulin dependent, diabetics lack the ability to utilize glucose adequately which leads to undesired accumulation of blood glucose (hyperglycemia). Chronic hyperglycemia leads to decreases in insulin secretion and contributes to increased insulin resistance. Glucokinase also acts as a sensor in hepatic parenchymal cells which induces glycogen synthesis, thus preventing the release of glucose into the blood. The GK processes are thus critical for the maintenance of whole body glucose homeostasis.
It is expected that an agent that activates cellular GK will facilitate glucose-dependent secretion from pancreatic beta cells, correct postprandial hyperglycemia, increase hepatic glucose utilization and potentially inhibit hepatic glucose release. Consequently, a GK activator may provide therapeutic treatment for NIDDM and associated complications, inter alia, hyperglycemia, dyslipidemia, insulin resistance syndrome, hyperinsulinemia, hypertension, and obesity. Several drugs in five major categories, each acting by different mechanisms, are available for treating hyperglycemia and subsequently, NIDDM (Moller, D. E., “New drug targets for Type 2 diabetes and the metabolic syndrome” Nature 414; 821 -827, (2001 )): (A) Insulin secretogogues, including sulphonyl-ureas (e.g., glipizide, glimepiride, glyburide) and meglitinides (e.g., nateglidine and repaglinide) enhance secretion of insulin by acting on the pancreatic beta-cells. While this therapy can decrease blood glucose level, it has limited efficacy and tolerability, causes weight gain and often induces hypoglycemia. (B) Biguanides (e.g., metformin) are thought to act primarily by decreasing hepatic glucose production. Biguanides often cause gastrointestinal disturbances and lactic acidosis, further limiting their use. (C) Inhibitors of alpha-glucosidase (e.g., acarbose) decrease intestinal glucose absorption. These agents often cause gastrointestinal disturbances. (D) Thiazolidinediones (e.g., pioglitazone, rosiglitazone) act on a specific receptor (peroxisome proliferator-activated receptor-gamma) in the liver, muscle and fat tissues. They regulate lipid metabolism subsequently enhancing the response of these tissues to the actions of insulin. Frequent use of these drugs may lead to weight gain and may induce edema and anemia. (E) Insulin is used in more severe cases, either alone or in combination with the above agents. Ideally, an effective new treatment for NIDDM would meet the following criteria: (a) it would not have significant side effects including induction of hypoglycemia; (b) it would not cause weight gain; (c) it would at least partially replace insulin by acting via mechanism(s) that are independent from the actions of insulin; (d) it would desirably be metabolically stable to allow less frequent usage; and (e) it would be usable in combination with tolerable amounts of any of the categories of drugs listed herein.
Substituted heteroaryls, particularly pyridones, have been implicated in mediating GK and may play a significant role in the treatment of NIDDM. For example, U.S. Patent publication No. 2006/0058353 and PCT publication No’s. WO2007/043638, WO2007/043638, and WO2007/117995 recite certain heterocyclic derivatives with utility for the treatment of diabetes. Although investigations are on-going, there still exists a need for a more effective and safe therapeutic treatment for diabetes, particularly NIDDM.
Designing glucokinase activators with reduced hypoglycemia risk: discovery of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide as a clinical candidate for the treatment of type 2 diabetes mellitus
E-mail: jeffrey.a.pfefferkorn@pfizer.com
Tel: +860 686 3421
DOI: 10.1039/C1MD00116G
http://pubs.rsc.org/en/content/articlelanding/2011/md/c1md00116g/unauth#!divAbstract
http://www.rsc.org/suppdata/md/c1/c1md00116g/c1md00116g.pdf
N,N-Dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)carbamoyl)-benzofuran-4- yloxy)pyrimidine-2-carboxamide (28). To a solution of the 5-methyl-2-aminopyrazine (38.9 g, 356 mmol) in dimethoxyethane (315 mL) in a 3-neck flask equipped with overhead stirring and a condenser at 0 o C was added Me2AlCl (1 M solution in hexanes) (715 mL). The mixture was warmed to room temperature and stirred for 1.5 h. In a separate flask, 26 (52.6 g, 142.5 mmol) was dissolved in dimethoxyethane (210 mL). This mixture was then added to the amine mixture. A gum precipitated and upon scratching the flask it dissipated into a solid. The reaction was refluxed for 3.5 h. Aq. Rochelle’s salt (5 L) and 2-MeTHF (2 L) was added to the mixture and this was allowed to stir with overhead stirring for 14 h, after which time, a yellow solid precipitated. The solid was collected by filtration, washing with 2-MeTHF. The resulting solid was dried in a vacuum oven overnight to afford the desired material (50.0g) in 81% yield.
1 H NMR (400MHz, CDCl3) δ 9.54 (d, J = 1.56 Hz, 1H), 8.50 (s, 2H), 8.37 (s, 1H), 8.14 (d, J = 0.78 Hz, 1H), 7.88 – 7.92 (m, 1H), 7.52 (d, J = 1.37 Hz, 1H), 6.28 (t, J = 0.98 Hz, 1H), 3.14 (s, 3H), 2.98 (s, 3H), 2.55 (s, 3H), 2.49 (d, J = 1.17 Hz, 3H);
MS(ES+ ): m/z 433.4 (M+1), MS(ES- ): m/z 431.3 (M-1).
PAPER

http://pubs.rsc.org/en/content/articlelanding/2013/md/c2md20317k#!divAbstract
PAPER
Bioorganic & Medicinal Chemistry Letters (2013), 23(16), 4571-4578
http://www.sciencedirect.com/science/article/pii/S0960894X13007452

Figure 1.
Glucokinase activators 1 and 2.
PATENT
WO 2010103437
https://www.google.co.in/patents/WO2010103437A1?cl=en
Scheme I outlines the general procedures one could use to provide compounds of the present invention having Formula (I).


Preparations of Starting Materials and Key Intermediates
Preparation of Intermediate (E)-3-(ethoxycarbonyl)-4-(5-methylfuran-2-yl)but- 3-enoic acid (I- 1a):

(Ma) To a vigorously stirred solution of 5-methyl-2-furaldehyde (264 ml_, 2650 mmol) and diethyl succinate (840 ml_, 5050 mmol) in ethanol (1.820 L) at room temperature was added sodium ethoxide (0.93 L of a 21 weight % solution in ethanol) in one portion. The reaction mixture was then heated at reflux for 13 hours. After cooling to room temperature, the mixture was concentrated in vacuo (all batches were combined at this point). The resulting residue was partitioned between ethyl acetate (1 L) and hydrochloric acid (1 L of a 2M aqueous solution). After separation, the aqueous layer was extracted with ethyl acetate (2 x 1 L). The combined organic extracts were then extracted with sodium hydrogen carbonate (2 x 1 L of a saturated aqueous solution). These aqueous extracts were combined and adjusted to pH 2 with hydrochloric acid (2M aqueous solution) then extracted with ethyl acetate (2 x 1 L). These organic extracts were combined and concentrated in vacuo to give desired (E)-3-(ethoxycarbonyl)-4-(5-methylfuran-2-yl)but-3-enoic acid (J1 Ia: 34.34 g, 5%). The original organic extract was extracted with sodium hydroxide (2 L of a 2M aqueous solution). This aqueous extract was adjusted to pH 2 with hydrochloric acid (2M aqueous solution) then extracted with ethyl acetate (2 x 1 L). These organic extracts were combined and concentrated in vacuo to give additional desired materials (395.2 gram, 63%) as red liquid. 1H NMR (CDCI3, 300 MHz) δ ppm 7.48 (s, 1 H), 6.57 (d, 1 H), 6.09 (d, 1 H), 4.24 (q, 2H), 3.87 (s, 2H), 2.32 (s, 3H), 1.31 (t, 3H).
Preparation of Intermediate ethyl 4-acetoxy-2-methylbenzofuran-6- carboxylate (1-1 b):

(M b) To a vigorously stirred solution of (E)-3-(ethoxycarbonyl)-4-(5- methylfuran-2-yl)but-3-enoic acid (1-1 a: 326.6 g, 1 .371 mol) in acetic anhydride (1 .77 L, 18.72 mol) at room temperature was added sodium acetate (193 g, 2350 mmol) in one portion. The reaction mixture was then heated at reflux for 2.5 hours. After cooling to room temperature, the mixture was concentrated in vacuo (all batches were combined at this point). The resulting residue was suspended in dichloromethane (1 .5 L) and filtered, washing the solids with dichloromethane (3 x 500 ml_). The combined filtrate and washings were then washed with sodium hydrogencarbonate (2 x 1 L of a saturated aqueous solution) and brine (2 L), then concentrated in vacuo to give desired ethyl 4-acetoxy-2-methylbenzofuran-6-carboxylate (H b: 549.03 g, quantitative). 1H NMR (CDCI3, 300 MHz) δ ppm 8.00-7.99 (m, 1 H), 7.64 (d, 1 H), 6.32-6.32 (m, 1 H), 4.38 (q, 2H), 2.47 (d, 3H), 2.37 (s, 3H), 1 .39 (t, 3H).
Preparation of Intermediate ethyl 4-hydroxy-2-methylbenzofuran-6- carboxylate (1- 1 c):

(He) To a stirred solution of ethyl 4-acetoxy-2-methylbenzofuran-6- carboxylate (Hb: 549.03 g, 1 .37 mol) in ethanol (4.00 L) at room temperature was added potassium carbonate (266 g, 1 .92 mol) in one portion. The reaction mixture was then heated at 600C for 3 hours. Potassium carbonate (100 g, 0.720 mol) was then added in one portion and the reaction mixture was heated at 600C for a further 3 hours. After cooling to room temperature the mixture was diluted with dichloromethane (2 L) and the suspension filtered, washing the solids with dichloromethane (2 x 1 L) (all batches were combined at this point). The combined filtrate and washings were then washed with citric acid (2.5 L of a 1 M aqueous solution), then concentrated in vacuo and the resulting residue purified by dry flash chromatography (hexane then 2:1 hexane:ethyl acetate). All fractions containing the desired product were combined and concentrated in vacuo. The resulting residue, which solidified on standing, was slurried with cold toluene and filtered. The solids were then stirred with hot toluene and decolourising charcoal for 1 hour, followed by filtration of the hot mixture through a pad of celite. The filtrate was allowed to cool and the resulting precipitate isolated by filtration to give desired ethyl 4-hydroxy-2- methylbenzofuran-6-carboxylate (1-1 c: 360 g, 90%) as orange powder.
1H NMR (CDCI3, 300 MHz) δ ppm 7.73-7.73 (m, 1 H), 7.45 (d, 1 H), 6.51 -6.50 (m, 1 H), 5.85 (s, 1 H), 4.39 (q, 2H), 2.48 (d, 3H), 1.40 (t, 3H). LCMS (liquid chromatography mass spectrometry): m/z 221.06 (96.39 % purity).
Preparation of SM-25-bromo-N,N-dimethylpyrimidine-2-carboxamide (SM-
£1:

(SM-2) Oxalyl chloride (47.4g, 369mmol) was added to a suspension of 5-
Bromo-pyrimidine-2-carboxylic acid (5Og, 250mmol) in dichloromethane (821 ml) at room temperature followed by 1 -2 drop of dimethylformamide. The reaction mixture was stirred under nitrogen for 2 hours LCMS in methanol indicated the presence of the methyl ester and some acid. Dimethylformamide (0.2ml) was added to the reaction mixture. The acid dissolved after 30 minutess. LCMS showed corresponding methyl ester and no starting material peak was observed. The solvent was removed and dried in vacuo to afford the crude 5-Bromo-pyrimidine-2-carbonyl chloride (55g, 100%). The 5-Bromo-pyrimidine-2-carbonyl chloride (55g, 250mmol) was dissolved in tetrahydrofuran (828ml) and dimethyl-amine (2M solution in tetrahydrofuran) (373ml, 745mmol) was added portionwise at room temperature. The reaction was stirred at room temperature under nitrogen for 16 hours, after which time, LCMS indicated completion. The mixture was diluted with ethyl acetate (500ml) and washed with H2O (500ml). The water layer was further extracted with CH2CI2 (5x500ml), all organics combined, and dried over magnesium sulfate. The filtrate was concentrated in vacuo and then suspended in methyl-/-butylether (650ml). The solution was then heated to reflux. The hot solution was allowed to cool overnight to afford pink crystals. The crystals were filtered and washed with cold methyl-t-butylether (100ml) the solid was dried in a vacuum oven at 550C for 12 hourrs to afford the title compound 5-bromo-N,N-dimethylpyhmidine-2-carboxamide (SM-2: 44g, 77%) as a pink solid.
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 2.94 (s, 3 H) 3.13 (s, 3 H) 8.85 (s, 2 H) m/z (M+1 ) = 232.
Preparation of Intermediate Ethyl 4-(2-(dimethylcarbamoyl)Dyrimidin-5- yloxy)-2-methylbenzofuran-6-carboxylate (l-2a):

A mixture of Cs2CO3 (62.1 g, 191 mmol), 5-bromo-N,N- dimethylpyrimidine-2-carboxamide (SM-2: 24g, 104mmol) and ethyl 4- hydroxy-2-methylbenzofuran-6-carboxylate (1-1 c: 2Og, 91 mmol); 1 ,10- phenanthroline (1.64g, 9.07mmol) and copper iodide (864mg, 4.54mmol) in dimethylformamide (200ml) was purged with N2 gas and then heated to 90°C using a mechanical stirrer. The heterogeneous reaction mixture was stirred at this temperature for 18 hours. HPLC indicated near completion. The reaction mixture was cooled to 350C and diluted with ethyl acetate (300ml). The mixture was filtered to remove any cesium carbonate. The filtrate was then partitioned between water (500ml) and ethyl acetate (500ml); however, no separation was observed. Concentrated HCL (20ml) was added to the mixture. When the aqueous phase was about pH1 , the phases separated. The organics were separated and the aqueous layer reextracted with ethyl acetate (2x500ml). All organics were combined and back extracted with water (200ml) and brine (500ml). The organics were separated and treated with activated charcoal (10g) and magnesium sulfate. The mixture was allowed to stir for 10 minutes and then filtered through a plug of celite to afford a crude yellow solution. The filter cake was washed with ethyl acetate (100 ml_). The organics were concentrated in vacuo to afford a crude solid this was dried under high vacuum for 4 days. The dry crude solid was triturated using methanol (80 ml_). The solids were dispersed into a fine light orange crystalline powder with a red liquor. The solids were isolated by filtration and rinsed with methanol (20 ml_). The solid was dried in the vacuum oven at 550C for 12 hours to afford ethyl 4-(2- (dimethylcarbamoyl)pyrimidin-5-yloxy)-2-methylbenzofuran-6-carboxylate (J1 2a) as a yellow solid (18.2g, 54%)
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.41 (t, J=7.12 Hz, 3 H) 2.50 (d, J=0.98 Hz, 3 H) 3.00 (s, 3 H) 3.17 (s, 3 H) 4.41 (d, J=7.22 Hz, 2 H) 6.29 (s, 1 H) 7.62 (d, J=1.17 Hz, 1 H) 8.06 (s, 1 H) 8.50 (s, 2 H). m/z (M+1 ) = 370.5
Preparation of Starting material 5-bromo-N-ethyl-N-methylpyrimidine-2- carboxamide (SM-3):

(SM-3) Oxalyl chloride (1 .45g, 1 1 .1 mmol) was added to a suspension of 5-
Bromo-pyrimidine-2-carboxylic acid (1 .5g, 7.4mmol) in dichloromethane (50ml) at room temperature followed by 1 -2 drop of dimethylformamide. The reaction mixture was stirred under nitrogen for 2 hours LCMS in methanol indicated the presence of the methyl ester and some acid. Dimethylformamide (0.2ml) was added to the reaction mixture and all of the acid dissolved after 30 minutes. LCMS showed corresponding methyl ester and no starting material peak was observed. The solvent was removed and dried in vacuo to afford the crude 5-Bromo-pyrimidine-2-carbonyl chloride (1 -6g). 5-Bromo-pyrinnidine-2-carbonyl chloride (1600mg, 7.225mnnol) was dissolved in dichloromethane (25ml) and triethylamine (4.03ml, 28.9mmol) was added followed by ethyl-methyl-amine (0.68 mL, 7.92 mmol). The reaction was stirred at room temperature under nitrogen for 16 ours, after which time, LCMS indicated completion. The mixture was diluted with dichloromethane (50ml) and washed with water (50ml) followed by 10% citric acid (50ml) and brine (50ml). The organic layer was separated and dried over MgSO4, the residue was filtered and the solvent was removed in vacuo to afford the title compound 5-bromo-N-ethyl-N-methylpyrimidine-2- carboxamide (SM-3): (1.4g, 79.4%) as a brown oil.
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.08 – 1.31 (m, 3 H) 2.99 (d, J=79.05 Hz, 3 H) 3.19 (q, J=7.22 Hz, 1 H) 3.59 (q, J=7.22 Hz, 1 H) 8.84 (d, J=3.12 Hz, 2 H)
Example 2
Preparation of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2- yl)carbamoyl)-benzofuran-4-yloxy)Dyrimidine-2-carboxamide (2):

(2)
To a solution of the 5-methyl-2-aminopyrazine (38.9 g, 356 mmol) in dimethylether (315 ml_) in a 3-neck flask equipped with overhead stirring and a condensor at O0C was added Me2AICI (1 M solution in hexanes) (715 ml_). The mixture was warmed at room temperature and stirred for 1.5 hours. In a separate flask, ethyl 4-(2-(dimethylcarbamoyl)pyrimidin-5-yloxy)-2- methylbenzofuran-6-carboxylate (l-2a: 52.6g, 142.5mmol) was dissolved in dimethylether (210 ml_). This mixture was then added to the complexed amine. A gum precipitated upon scratching the flask and dissipated into a solid. The resultant reaction was refluxed for 3.5 hours HPLC indicated 93% complete. Five liters of Rochelles salt made up in water and 2 liters of 2- methyltetrahydrofuran was added to the mixture. The reaction mixture was then poured into the biphasic system. The mixture was allowed to stir with overhead stirring for 14 hours, after which time, a yellow solid precipitated. The solid was collected through filteration. The solid retained was washed with 2-methyltetrahydrofuran. The resultant solid was dried in vacuo oven overnight to afford the title compound N,N-dimethyl-5-(2-methyl-6-((5- methylpyrazin-2-yl)carbamoyl)benzofuran-4-yloxy)pyhmidine-2-carboxamide (2): (49.98g, 81 %)
1H NMR (400 MHz, CHLOROFORM-d) d ppm 2.49 (d, J=1 .17 Hz, 3H) 2.55 (s, 3H) 2.98 (s, 3 H) 3.14 (s, 3 H) 6.28 (t, J=0.98 Hz, 1 H) 7.52 (d, J=1 .37 Hz, 1 H) 7.88 – 7.92 (m, 1 H) 8.14 (d, J=0.78 Hz, 1 H) 8.37 (s, 1 H) 8.50 (s, 2 H) 9.54 (d, J=1 .56 Hz, 1 H).
m/z (M+1 ) = 433.4, m/z (M-1 )= 431 .5
REFERENCES
Beebe, D.A.; Ross, T.T.; Rolph, T.P.; Pfefferkorn, J.A.; Esler, W.P.
The glucokinase activator PF-04937319 improves glycemic control in combination with exercise without causing hypoglycemia in diabetic rats
74th Annu Meet Sci Sess Am Diabetes Assoc (ADA) (June 13-17, San Francisco) 2014, Abst 1113-P
Amin, N.B.; Aggarwal, N.; Pall, D.; Paragh, G.; Denney, W.S.; Le, V.; Riggs, M.; Calle, R.A.
Two dose-ranging studies with PF-04937319, a systemic partial activator of glucokinase, as add-on therapy to metformin in adults with type 2 diabetes
Diabetes Obes Metab 2015, 17(8): 751
Study to compare single dose of three modified release formulations of PF-04937319 with immediate release material-sparing-tablet (IR MST) formulation previously studied in adults with type 2 diabetes mellitus (NCT02206607)
OTHERS

///////////Pfizer , PF 04937319, glucokinase activators, Type 2 diabetes
WCK 2349 in phase II trials by Wockhardt
 . CH3SO3H
. CH3SO3H
WCK 2349: A novel fluoroquinolone (FQ) prodrug-13 week oral (PO) safety profile in cynomolgus monkeys
47th Intersci Conf Antimicrob Agents Chemother (ICAAC) (September 17-20, Chicago) 2007, Abst F1-2133a
8-{4-[2(S)-Amino-propionyloxy] piperidine-l-yl}-9-fluoro-5 (S)-methyl-ό, 7-dihydro-l- oxo-lH, 5H-benzo[i,j]quinolizine-2-carboxylic acid of structural Formula I can be used to treat bacterial Gram-positive, Gram-negative and anaerobic infections; especially infections caused by resistant Gram-positive organism and Gram-negative organism, mycobacterial infections and emerging nosocomial pathogen infections.
Formula I
U.S. Patent Nos. 6,750,224 and 7,247,642 describes optically pure S-(-)-benzoquinolizine carboxylic acids, their derivatives, salts, pseudopolymorphs, polymorphs and hydrates thereof, their processes of preparation and their pharmaceutical compositions.
PATENT
WO 2007102061
http://www.google.co.in/patents/WO2007102061A2?cl=en

Scheme 1
Experimental:
(S)-9-Fluoro-6,7-dihydro-8-(4-hydroxypiperidin-l-yl)-5-methyl-l-oxo-lH,5H-benzo[ij] quinolizine-2-carboxylic acid was prepared as per procedure described in Chem. Pharm. Bull. 1996, 44(4), 642-645.
Example-l
Preparation of (2’S,5S)-9-fluoro-6,7-dihydro-8-(4-(N-tert-butoxycarbonyI-L-aIaninyl- oxy)-piperidin-l-yl)-5-methyl-l-oxo-lH,5H-benzo[i,j]quinolizine-2-carboxylic acid:
Method-1 : To a mixture of N-tert-butoxycarbonyl-L-alanine (473 g) in dichloromethane (2 L), dicyclohexylcarbodiimide (515 g) dissolved in dichloromethane (2 L) was charged at -10 to 0 0C to provide a turbid suspension. To the turbid suspension, 300 g of (S)-9-fluoro-6,7- dihydro-8-(4-hydroxy-piperidin- 1 -yl)-5-methyl- 1-oxo- lH,5H-benzo[i,j]quinolizine-2- carboxylic acid was added followed by 4-N,N-dimethylamino pyridine (58 g) and the reaction mixture was stirred at -10 to 5 °C temperature over a period of 2 h. Suspension was filtered and solid was washed with 500 ml of dichloromethane. The filtrate was washed with water. Filtrate was dried over anhydrous sodium sulfate. Dried organic layer was then concentrated to its half volume where upon solid was precipitated. The solid was filtered and washed with 300 ml of dichloromethane. Clear organic filtrate was concentrated to dryness to provided an oily mass. Oily mass was triturated with diethyl ether (4 L) to provide white solid. The solid was filtered under suction and washed with diethyl ether (1 L) to provide title compound in 415 g (94%) quantity.
Method-2: To a mixture of triethylamine (98.0 ml) and N-tert-butoxycarbonyl-L-alanine (110 g) in tetrahydrofuran (1050 ml) and N,N-dimethyl formamide (350 ml) mixture, was added 2,4,6-trichlorobenzoyl chloride (100 ml). The resultant mixture was stirred at a temperature -5 to 0 °C for 5 h. To the > reaction mixture 4-N,N-dimethylamino pyridine (24g) and (S)-9-fluoro-6,7-dihydro-8-(4-hydroxy-piperidin-l-yl)-5-methyl-l-oxo-lH,5H- benzo[i,j]quinolizine-2-carboxylic acid (70 g) was added. The reaction mixture was stirred for additional 7 h at -5 to 0 0C temperature. The suspension was filtered at room temperature and the filtrate was extracted with ethyl acetate after addition of water. The evaporation of organic layer under reduced pressure provided a sticky solid, which upon triturating with diethyl ether provided a white solid in 85 g quantity.
Method-3: To a solution N-tert-butoxycarbonyl-L-alanine (7.9 g) in tetrahydrofuran (75 ml) and N,N-dimethyl formamide (25 ml) mixture at -10 to 0°C was added methanesulfonyl chloride (2.42 ml) dropwise. To the above solution triethylamine (8.7 ml) was added dropwise over 5 min. the reaction was stirred for 1.5 h maintaining the temperature between at -10 to 0 0C. To the reaction mixture (S)-9-fluoro-6,7-dihydro-8-(4-hydroxy-piperidin-l- yl)-5-methyl-l-oxo-lH,5H-benzo[ij]quinolizine-2-carboxylic acid (5.01 g) and 4-N5N- dimethylamino pyridine (1.70 g) was added. The reaction mixture was stirred for additional 1 h at -5 to 0 °C temperature. The suspension was filtered at room temperature and the filtrate was diluted with water (300 ml) and extracted with ethyl acetate (150 ml x 2). The evaporation of organic layer under reduced pressure provided a sticky solid, which upon triturating with diethyl ether provided a white solid in 6.38 g (86%) quantity.
Example-2
Preparation of (2’S, 5S)-9-fluoro-6,7-dihydro-8-(4-L-alaninyl-oxy-piperidin-l-yl)-5-methyl- l-oxo-lH,5H-benzo[i,j]quinolizine-2-carboxylic acid methanesulfonic acid salt:
To a mixture of (2’S, 5S)-9-fluoro-6,7-dihydro-8-(4-N-tert-butoxycarbonyl-L-alaninyloxy- piperidin-l-yl)-5-methyl-l-oxo-lH,5H-benzo[i,j]quinolizine-2-carboxylic acid (415 g) in acetone (4.5 L) was charged methanesulfonic acid (66 ml). Reaction mixture was stirred at 65-67 °C temperature for overnight. The suspension was filtered at 40-45 0C. Solid was washed with acetone (1.5 L) followed by diethyl ether (1.5 L). Off white solid was dried under 40 to 45 mm vacuum at 55-60 °C temperature over the period of 3-4 h. Title compound was obtained as a free flowing off white material 383.0 g (93%).
For MF: C23H30FN3O8S, MS (ES+) m/z 432 (obtained as free base for MF: C22H26FN3O5);
M.P. 278.50 0C by DSC

PATENT

Patent
PATENT
The tablets may optionally be coated with film forming agents and/or pharmaceutically acceptable excipients. Particularly suitable for use are commercially available coating compositions comprising film-forming polymers marketed under various trade names, such as Opadry® and Eudragit®. The coating layers over the tablet may be applied as solution/dispersion of coating ingredients using conventional techniques known in the art.
The present invention is further illustrated by the following examples which are provided merely to be exemplary of the invention and do not limit the scope of the invention. Certain modifications and equivalents will be apparent to those skilled in the art and are intended to be included within the scope of the present invention.
Example 1 :
Table 1 provides the composition of batches of the present invention.
Table 1

Procedure: The compound of Formula I or pharmaceutically acceptable salts, esters or products thereof, lactose and croscannellose sodium were sifted and dry mixed in a rapid mixer granulator. The above mass was granulated by spraying aqueous solution of povidone. The granules were dried in a fluidized bed drier, sifted and oversize granules were milled in a Quadra mill. The resultant granules were mixed with talc, croscarmellose sodium, microcrystalline cellulose and sodium stearyl fumarate in a double cone blender. The lubricated granules were compressed into tablets using suitable tooling. Tablets were coated with aqueous dispersion of opadry.
Table 2 provides the dissolution data for the compound of formula I or pharmaceutically acceptable salts, esters or products thereof tablets prepared as per the formula given in Table 1. For determination of drug release rate, USP Type 2 Apparatus (rpm 50) was used wherein 0.1 N hydrochloric acid (900 ml) was used as a medium. Table 2: Dissolution data



NEW DELHI: Drug maker WockhardtBSE -1.83 % today said that two of its anti-infective drugs
have received Qualified Infectious Disease Product (QIDP) status from the US
health regulator.Two drugs – WCK 771 and WCK 2349 – have received QIDP
status, which allows fast-track review of the drug application by the US Food and Drug Administration (USFDA),
Wockhardt said in a statement.
http://economictimes.indiatimes.com/articleshow/41359481.cms?utm_source=contentofinterest&utm_medium=text&utm_campaign=cppst
RN: 306748-89-0
-
C19-H21-F-N2-O4.C6-H14-N4-O2
- MW: 534.5855
-
L-Arginine, mono((5S)-9-fluoro-6,7-dihydro-8-(4-hydroxy-1-piperidinyl)-5-methyl-1-oxo-1H,5H-benzo(ij)quinolizine-2-carboxylate)

J Med Chem 2005, 48(16): 5232
| WO1991012815A1 * | Feb 25, 1991 | Sep 5, 1991 | Squibb Bristol Myers Co | COMPOSITIONS AND METHODS FOR TREATING INFECTIONS CAUSED BY ORGANISMS SENSITIVE TO β-LACTAM ANTIBIOTICS |
| WO2000068229A2 * | May 8, 2000 | Nov 16, 2000 | S K Agarwal | (s)-benzoquinolizine carboxylic acids and their use as antibacterial agents |
| WO2001085095A2 * | May 3, 2001 | Nov 15, 2001 | Shiv Kumar Agarwal | Chiral fluoroquinolizinone arginine salt forms |
| WO2002009758A2 * | Jul 31, 2001 | Feb 7, 2002 | Satish B Bhawsar | Inhibitors of cellular efflux pumps of microbes |
| EP2062582A1 * | Aug 14, 2007 | May 27, 2009 | Tianjin Hemey Bio-Tech Co., Ltd. | The antibiotics composition comprising beta-lactam antibiotics and buffers |
| US4524073 * | Jul 20, 1983 | Jun 18, 1985 | Beecham Group P.1.C. | β-Lactam compounds |
| US6465428 * | Aug 25, 2000 | Oct 15, 2002 | Aventis Pharma S.A. | Pharmaceutical combinations based on dalfopristine and quinupristine, and on cefepime |
| US20040254381 * | Aug 15, 2003 | Dec 16, 2004 | Day Richard A. | Antibiotic compositions and methods of using the same |
| US20050148571 * | Nov 29, 2002 | Jul 7, 2005 | Nancy Niconovich | Method of treating bacterial infections using gemifloxacin or a salt thereof and a betha-Lactam antibiotic |
| US20090148512 * | Apr 17, 2008 | Jun 11, 2009 | Lannett Co Inc | Novel uses of chloramphenicol and analogous thereof |
| US20090232744 * | Feb 26, 2009 | Sep 17, 2009 | Pari Pharma Gmbh | Macrolide compositions having improved taste and stability |
| WO2002009758A2 * | 31 Jul 2001 | 7 Feb 2002 | Satish B Bhawsar | Inhibitors of cellular efflux pumps of microbes |
| US6750224 | 17 Aug 2000 | 15 Jun 2004 | Wockhardt Limited | Antibacterial optically pure benzoquinolizine carboxylic acids, processes, compositions and methods of treatment |


Mr Habil Khorakiwala, Chairman, Wockhardt Ltd.

///////////keywords USFDA, Qualified Infectious Disease Product status, Wockhardt, drugs, WCK 2349, QIDP
ORGANIC SPECTROSCOPY