
Home » APPROVALS 2025 (Page 2)
Category Archives: APPROVALS 2025
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Brensocatib
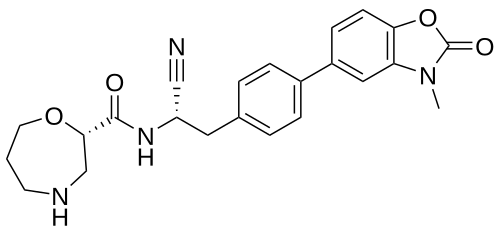

Brensocatib
WeightAverage: 420.469
Monoisotopic: 420.179755269
Chemical FormulaC23H24N4O4
- AZD7986
- CAS 1802148-05-5
- INS1007
- AZD 7986
- WHO 11097
(2S)-N-[(1S)-1-cyano-2-[4-(3-methyl-2-oxo-1,3-benzoxazol-5-yl)phenyl]ethyl]-1,4-oxazepane-2-carboxamide
- 1,4-Oxazepine-2-carboxamide, N-((1S)-1-cyano-2-(4-(2,3-dihydro-3-methyl-2-oxo-5-benzoxazolyl)phenyl)ethyl)hexahydro-, (2S)-
- (2S)-N-((1S)-1-cyano-2-(4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl)ethyl)-1,4-oxazepane-2-carboxamide
- (2S)-N-{(1S)-1-Cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}-1,4-oxazepane-2-carboxamide
- Brensocatib [USAN]
- (S)-N-((S)-1-cyano-2-(4-(3-methyl-2-oxo-2,3-dihydrobenzo[d]oxazol-5-yl)phenyl)ethyl)-1,4-oxazepane-2-carboxamide
- UNII-25CG88L0BB
- (2S)-N-[(1S)-1-cyano-2-[4-(3-methyl-2-oxo-1,3-benzoxazol-5-yl)phenyl]ethyl]-1,4-oxazepane-2-carboxamide
FDA 8/12/2025. Brinsupri, To treat non-cystic fibrosis bronchiectasis
Brensocatib is an investigational new drug that is being evaluated to treat bronchiectasis.[1] It is a dipeptidyl-peptidase I (also known as cathepsin C) inhibitor.[2]
A phase 3 clinical trial, known as the ASPEN trial, was conducted to evaluate the safety and efficacy of brensocatib in patients with non-cystic fibrosis bronchiectasis.[3] Brensocatib tablets (Brinsupri) by Insmed Inc. was approved by the FDA in August 2025 after it received breakthrough therapy designation and was reviewed on a priority timeline.
Brensocatib is an orally bioavailable, small molecule, reversible inhibitor of dipeptidyl peptidase 1 (DPP1), with potential anti-inflammatory activity. Upon oral administration, brensocatib reversibly binds to and inhibits the activity of DPP1, thereby inhibiting the activation of neutrophil serine proteases (NSPs), including neutrophil elastase (NE), during neutrophil maturation. This inhibits the activity of NSPs, and may prevent lung inflammation and injury and improve lung function associated with NSPs-induced respiratory diseases. NSPs, serine proteases released by neutrophils during inflammation, is upregulated in a number of respiratory diseases.
SYN
J. Med. Chem. 2016, 59, 9457–9472, DOI: 10.1021/acs.jmedchem.6b01127
https://www.thieme-connect.com/products/ejournals/pdf/10.1055/s-0040-1719365.pdf
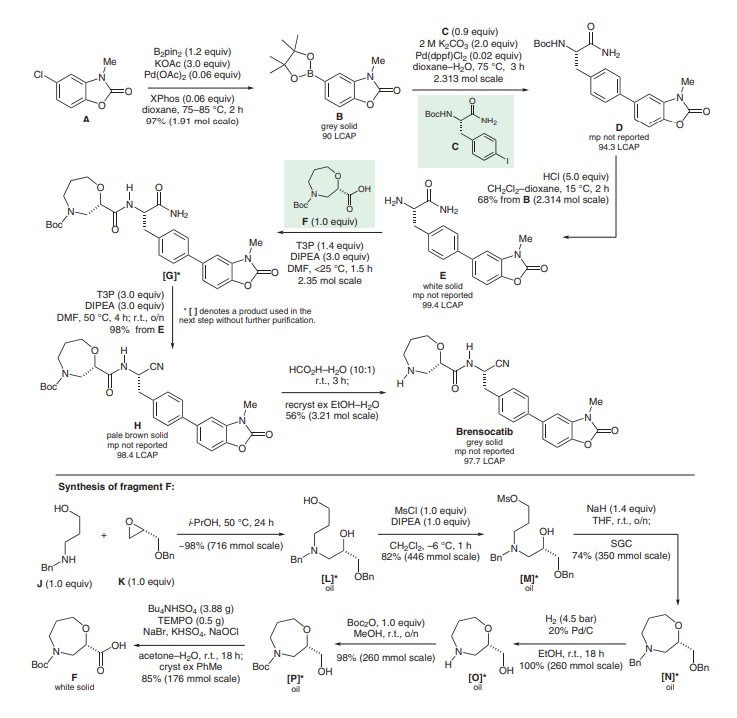
SYN
Brensocatib is now a clinical candidate to impair proteasedriven tissue degradation in COVID-19 (B. Korkmaz,
A. Lesner, S. Marchand-Adam, C. Moss, D. E. Jenne
J. Med. Chem. 2020, 63, 13258).
PAT
https://patents.google.com/patent/US9522894B2/en
A compound according to claim 1
which is (2S)—N-{(1S)-1-cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}-1,4-oxazepane-2-carboxamide

EXAMPLESExample 1(2S)—N-[(1S)-1-Cyano-2-(4′-cyanobiphenyl-4-yl)ethyl]-1,4-oxazepane-2-carboxamide

i) tert-Butyl(2S)-2-{[(1S)-1-cyano-2-(4′-cyanobiphenyl-4-yl)ethyl]carbamoyl}-1,4-oxazepane-4-carboxylate
2-Pyridinol-1-oxide (0.155 g, 1.4 mmol), TEA (0.36 g, 3.6 mmol) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (0.268 g, 1.4 mmol) were added to a solution of (2S)-4-(tert-butoxycarbonyl)-1,4-oxazepane-2-carboxylic acid (Intermediate 3, 0.294 g, 1.2 mmol) in DCM (15 mL). After 20 min
4′-[(2S)-2-amino-2-cyanoethyl]biphenyl-4-carbonitrile (Intermediate 1, 0.296 g, 1.2 mmol) was added and the mixture was stirred for 3 h and allowed to stand at rt for 18 h. The mixture was heated at 40° C. for 4 h before water (15 mL) was added. After 10 min the DCM was dried (phase separating cartridge) and evaporated under reduced pressure. The resultant yellow oil was purified by silica gel column chromatography to give the subtitled compound (0.29 g, 52%). Used without further purification in the next step.ii) (2S)—N-[(1S)-1-Cyano-2-(4′-cyanobiphenyl-4-yl)ethyl]-1,4-oxazepane-2-carboxamide
Prepared according to procedure in Method A step ii) using tert-butyl(2S)-2-{[(1S)-1-cyano-2-(4′-cyanobiphenyl-4-yl)ethyl]carbamoyl}-1,4-oxazepane-4-carboxylate to afford the title compound as a white solid (60 mg, 28%).
1H NMR (400 MHz, CDCl3): δ 7.77-7.65 (m, 4H), 7.62-7.57 (m, 2H), 7.40 (d, 2H), 7.11 (d, 1H), 5.18-5.11 (m, 1H), 4.19-4.14 (m, 1H), 4.06-3.96 (m, 2H), 3.75-3.69 (m, 1H), 3.56-3.48 (m, 2H), 3.18-3.05 (m, 3H), 2.95-2.90 (m, 1H), 2.70 (ddd, 1H) (1 exchangeable proton not observed).
LCMS (10 cm_ESCI_Formic_MeCN) tR 2.57 (min) m/z 375 (MH+).Example 2(2S)—N-{(1S)-1-Cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}-1,4-oxazepane-2-carboxamide

i) tert-Butyl(2S)-2-({(1S)-1-cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}carbamoyl)-1,4-oxazepane-4-carboxylate

N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (468 mg, 2.44 mmol) and 2-pyridinol 1-oxide (271 mg, 2.44 mmol) were added to a solution of (2S)-4-(tert-butoxycarbonyl)-1,4-oxazepane-2-carboxylic acid (Intermediate 3, 490 mg, 2.0 mmol) in DCM (15 mL). The reaction was stirred at rt for 30 min before the addition of (2S)-2-amino-3-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]propanenitrile (Intermediate 2, 586 mg, 2.0 mmol) and DiPEA (1.79 mL, 10 mmol). The reaction was stirred at rt for 18 h before transferring to a separating funnel. The mixture was washed with 2 M hydrochloric acid, saturated sodium hydrogen carbonate solution and brine. The organic extract was run through a hydrophobic frit/phase separator and concentrated under reduced pressure. The crude material was purified by silica gel column chromatography eluting with 0-60% EtOAc in iso-hexane to afford the subtitled compound as an oil (457 mg, 44%). 1H NMR (400 MHz, CDCl3): δ 7.63-7.52 (m, 2H), 7.38 (d, 2H), 7.36-7.24 (m, 2H), 7.35-6.98 (m, 2H), 5.18 (t, 1H), 4.22-3.97 (m, 2H), 3.76-3.67 (m, 0.5H), 4.10-2.94 (m, 4.5H), 3.35-3.26 (m, 1H), 3.24-3.04 (m, 3H), 2.06-1.82 (m, 2H), 1.47 (s, 10H).ii) (2S)—N-{(1S)-1-Cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}-1,4-oxazepane-2-carboxamide
tert-Butyl(2S)-2-({(1S)-1-cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}carbamoyl)-1,4-oxazepane-4-carboxylate (457 mg, 0.85 mmol) was dissolved in formic acid (3 mL) and heated at 50° C. for 10 min on a pre-heated stirrer hotplate. After this time the reaction was concentrated under reduced pressure, dissolved in DCM and washed with saturated sodium hydrogen carbonate solution. The organic extract was run through a hydrophobic frit/phase separator and concentrated under reduced pressure. The resultant foam was purified by silica gel column chromatography eluting with 0-5% methanolic ammonia (7 N) in DCM to afford the title compound as solid material (230 mg, 64%).
1H NMR (400 MHz, CDCl3): δ 7.59-7.51 (m, 2H), 7.39 (dd, 2H), 7.33-7.23 (m, 3H), 7.14 (d, 1H), 5.23-5.12 (m, 1H), 4.12-4.06 (m, 1H), 4.05-3.95 (m, 1H), 3.81-3.71 (m, 1H), 3.46 (s, 3H), 3.34-3.26 (m, 1H), 3.19-3.00 (m, 3H), 2.99-2.82 (m, 2H), 1.92-1.77 (m, 2H) (one exchangeable proton not observed).
LCMS (10 cm_ESCI_Formic_MeCN) tR 2.48 (min) m/z 375 (MH+).Example 2Alternative Synthesis(2S)—N-{(1S)-1-Cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}-1,4-oxazepane-2-carboxamidei) 5-Chloro-1,3-benzoxazol-2(3H)-one

To a solution of 2-amino-4-chlorophenol (400 g, 2.79 mol) in 2-MeTHF (6 L) was added CDI (497 g, 3.07 mol) under N2 (exotherm 11.0° C.-22.0° C.). The reaction mixture was heated at reflux for 1 h. The mixture was cooled to rt, washed with 2 M HCl(aq) (6 L), 8% NaHCO3(aq) (6 L) and brine (3 L). The organic layer was dried over MgSO4, filtered and evaporated. This gave the product as a pale brown solid (456.1 g, 97% yield, LC purity >99%).
1H NMR (270 MHz, DMSO-d6): δ 12.0-11.5 (br s, 1H), 7.31 (d, 1H), 7.12 (m, 2H).
LCMS (5 cm_ESCI, aq. formic acid_methanol) tR 3.87 (min) m/z 169.8 (MH+).ii) 5-Chloro-3-methyl-1,3-benzoxazol-2(3H)-one

To a solution of 5-chloro-1,3-benzoxazol-2(3H)-one (stage i) (1111.8 g, 6.56 mol) in DMF (4.12 L) was added Cs2CO3 (2136.4 g, 6.56 mol) maintaining the temperature between 0-5° C. MeI (450 ml, 7.21 mol) was then added slowly maintaining the temperature between 0-5° C. The reaction mixture was allowed to warm-up to rt and stirred overnight. The mixture was cooled to 0-5° C. and H2O (4.12 L) was added slowly. The reaction mixture was then warmed to rt and stirred for 15 min. The solids were filtered off and washed with water (4×980 ml). The filter cake was dried under vacuum at 55° C. overnight (1149.9 g, 96% yield, LC purity >99%, H2O: (Karl Fischer) 0.1%).
1H NMR (270 MHz, DMSO-d6): δ 7.45 (d, 1H), 7.35 (d, 1H), 7.15 (dd, 1H), 3.35 (s, 3H). LCMS (5 cm_ESCI_aq. formic acid_methanol) tR 4.13 (min) m/z 183.8 (M+).iii) 3-Methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3-benzoxazol-2(3H)-one

A solution of 5-chloro-3-methyl-1,3-benzoxazol-2(3H)-one (stage ii)) (350 g, 1.91 mol), B2pin2 (581.0 g, 2.29 mol) and KOAc (561.3 g, 5.72 mol) was vacuum degassed and purged with N2 (×3). Pd(OAc)2 (12.9 g, 57.2 mmol) and XPhos (54.6 g, 114 mmol) were added and the mixture was vacuum degassed and purged with N2 (×3). The mixture was heated to 75° C. A large exotherm was observed at ˜70° C. which warmed-up the mixture to reflux (100° C.). The reaction mixture was stirred for 1 h with no heating. HPLC analysis indicated 2.5% of the starting material remaining therefore the mixture was heated at 85° C. for 1 h. At this stage, no further change was observed. Additional portions of B2pin2 (14.6 g, 57.2 mmol), KOAc (5.7 g, 57.2 mmol), Pd(OAc)2 (12.9 g, 57.2 mmol) and XPhos (27.3 g, 57.2 mmol) were added and the mixture was stirred for 1 h at 75° C. HPLC analysis showed no starting material remaining. The mixture was cooled to rt, filtered through a pad of Celite (501 g) and the cake was washed with EtOAc (2240 ml). The filtrate was combined with two other batches prepared in the same way (2×350 g) and evaporated. This gave 1865.1 g of the product as a grey solid (97% yield, 90.0% pure by LC, 82±2% pure by 1H NMR (DMSO-d6) assay vs TCNB).
1H NMR (270 MHz, DMSO-d6): δ 7.40-7.50 (m, 2H), 7.30 (d, 1H), 3.40 (s, 3H), 1.30 (s, 12H).
LCMS (5 cm_ESCI_aq. formic acid_methanol_) tR 4.91 (min) m/z 276.1 (MH+).iv) Nα-(tert-Butoxycarbonyl)-4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)-L-phenylalaninamide

To a suspension of 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3-benzoxazol-2(3H)-one (stage iii)) (859 g, 700 g active, 2.544 mol) and tert-butyl (S)-1-carbamoyl-2-(4-iodophenypethylcarbamate (prepared according to the procedure in WO 2009/074829 p. 47), (903 g, 2.313 mol) in dioxane (4.1 L) was added 2 M K2CO3 (2.3 L). The suspension was vacuum degassed and purged with N2 (×3). Pd(dppf)Cl2.DCM (28.33 g, 0.0347 mol) was added and the reaction mixture was heated at 75° C. for 3 h. The mixture was cooled to rt and diluted with water (6.4 L). The suspension was stirred at rt overnight; the solid was filtered off and washed with water (3×1 L). The product was dried at 45° C. for 3 days (1269.1 g, yield 133%—by 1H NMR contains pinacol related impurity and dioxane, LC 94.3% pure, H2O: (Karl Fischer) 3.35%).
1H NMR (270 MHz, DMSO-d6): δ 7.62-7.34 (m, 7H), 7.04 (brs, 2H), 6.86 (d, 1H) 4.12 (m, 1H), 3.40 (s, 3H), 3.00 (dd, 1H), 2.78 (dd, 1H), 1.30 (s, 9H).
LCMS (5 cm_ESI_Water_MeCN) tR 4.51 (min) m/z 312 (MH+).v) 4-(3-Methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)-L-phenylalaninamide

To a very thick suspension of Nα-(tert-butoxycarbonyl)-4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)-L-phenylalaninamide (stage iv)) (1269 g, active 952 g assumed 100% conversion at stage iv), 2.3138 mol) in DCM (2.1 L) under N2 was added dropwise 4.1 M HCl in dioxane (2.7 L, 11.06 mol) over 1 h maintaining the temperature at 15° C. (suspension became more mobile after addition of approx. 0.5 L of 4.1 M HCl dioxane). After 2 h, the mixture was diluted with water (5.6 L) and stirred for 30 min at rt. The mixture was then filtered through a pad of Celite (500 g) to remove undissolved material—very slow filtration; the Celite was checked for product by LC. The pad was washed with water (400 ml). The layers DCM/dioxane-water were separated. The aqueous layer was cooled to ˜5° C. and 35% NH3 (aq) (700 ml) was added slowly to achieve pH=9-10. The suspension was stirred overnight then the product was filtered off and washed with water (3×400 ml). The product was dried at 45° C. in vacuo for 2 days (off white solid, 489.4 g, 68% yield over two stages, 99.4% pure by LC, >99% EP, 98±2% pure by 1H NMR assay vs TCNB in DMSO, H2O: (Karl Fischer) 0.92%).
1H NMR (270 MHz, DMSO-d6): δ 7.59-7.30 (m, 7H), 6.98 (brs, 1H), 3.36 (m, 4H), 2.95 (dd, 1H), 2.67 (dd, 1H) 1.86 (brs, 2H).
LCMS (5 cm_ESI_Water_MeCN) tR 2.76 (min) m/z 312 (MH+).vi) tert-Butyl(2S)-2-({(1S)-1-cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}carbamoyl)-1,4-oxazepane-4-carboxylate

To a solution of 4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)-L-phenylalaninamide (stage v)) (756 g, active 733 g, 2.354 mol) and (2S)-4-(tert-butoxycarbonyl)-1,4-oxazepane-2-carboxylic acid (577 g, 2.354) (Intermediate 3) in DMF (3 L) was added DiPEA (1230 ml, 7.062 mol) under N2. T3P in DMF (50% w/w, 1924 ml, 3.296 mol) was added dropwise over 1.5 h maintaining the temperature<25° C. After 30 min, LC completion check indicated completion of the coupling reaction. DiPEA (1230 ml, 7.062 mol) was then added and the reaction mixture was heated to 50° C. T3P in DMF (50% w/w, 3986 ml, 6.827 mol) was added portionwise over 1 h (no exotherm observed). The reaction mixture was stirred at 50° C. for 4 h and then at rt overnight. The mixture was cooled to 10° C., diluted with 2-MeTHF (4 L) and water (5.6 L, exothermic). The layers were separated and the aqueous layer was extracted with 2-MeTHF (2×4 L). The combined organic extracts were dried over MgSO4, filtered and concentrated under reduced pressure. This delivered the product as a pale brown solid in 98% yield (1242 g (active 1205 g), corrected yield 98%, LC purity 98.4%, 1H NMR assay vs TCNB 97±2%, main impurities by 1H NMR: 2-MeTHF 1.9%, DMF 0.6%).vii) (2S)—N-{(1S)-1-Cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}-1,4-oxazepane-2-carboxamide
A solution of tert-butyl(2S)-2-({(1S)-1-cyano-2-[4-(3-methyl-2-oxo-2,3-dihydro-1,3-benzoxazol-5-yl)phenyl]ethyl}carbamoyl)-1,4-oxazepane-4-carboxylate (stage vi)) (1776 g, active 1671 g, 3.210 mol) in formic acid/water (4.2 L/440 ml) was stirred on a buchi at 35-37° C. under reduced pressure (300-500 mbar). After 3 h, LCMS completion check indicated 93.95% of the product and 0.5% of the starting material. The mixture was concentrated (4 h) to give an oily residue. The residue was dissolved in water (4.4 L) and washed with TBME (2.2 L). The aqueous layer was vigorously stirred and treated with NH3(aq) (1.8 L) at <25° C. to achieve pH=9-10. The mixture was stirred at rt for 3 h. The solid was filtered off and washed with water (3×1 L). The filter cake was dried at 45° C. overnight. This gave the product as a pale brown solid (1498 g, active 1333 g, LC 91.5%, 1H NMR assay vs TCNB 89±2%, H2O: (Karl Fischer) 4.63%).
The crude product was re-crystallised from EtOH/H2O in two batches (2×747 g).
Batch A: The crude product (747 g) was dissolved in EtOH (8 L) at reflux under N2. Water (1.6 L) was added slowly. The mixture was hot filtered (65° C.) to remove black particles (filtrate temperature
50° C.) and then stirred at 40° C. overnight. The suspension was cooled to 10° C. over 4 h and held at that temperature for 3 h. The product was filtered off and washed with EtOH/H2O (8:2, 3×500 ml) then water (3×500 ml). The filter cake was dried at 45° C. overnight (473 g, 97.7% pure by LC, Pd level 71.4 ppm).
Batch B gave 436 g of the product (95.8% pure by LC, Pd level 65.8 ppm).
The liquors from both batches were combined and concentrated to ˜8 L. The liquors were left overnight at rt. The solids were filtered off and washed with EtOH/H2O (8:2, 3×400 ml) then water (3×400 ml). The product was dried at 45° C. overnight. This gave additional 88 g of the product (LC purity 95.0%).
The products (LC purity of the blend 95.69%) were re-crystallised from EtOH/H2O in two batches (Batch C: 520 g, Batch D: 520 g).
Batch C: The crude product (520 g) was dissolved in EtOH (6.24 L) at reflux under N2. Water (1248 ml) was added slowly. The mixture was allowed to cool down to 40° C. (3 h), seeded with 0.5 g of the title compound and stirred at 40° C. for 10 h. The mixture was then cooled to 26° C. over 7 h. The resulting suspension was cooled to 10° C. and stirred at that temperature for 6 h. The product was filtered off, washed with EtOH/water (8:2, 3×500 ml) and water (3×500 ml). The filter cake was dried at 45° C. for 2 d. The product was obtained as a grey solid (418 g, yield ˜56%, LCMS purity 97.5%, chiral LC
100%, 1H NMR (DMSO-d6) assay vs TCNB
100±2%).
Batch D: 418 g, yield 56%, LCMS purity 97.5%, chiral LC
100%, 1H NMR (DMSO-d6) assay vs TCNB
100±2%
The product was blended with the material from an intermediate scale reaction performed in the same way and re-analysed (968 g, LC purity 98.04%, chiral LC
100%, 1H NMR assay vs TCNB 99±2%, 0.35% EtOH by 1H NMR, H2O: (Karl Fischer) 4.58%, Pd 57.6 ppm, XRPD (X-ray powder diffraction) Form A.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
References
- “Brensocatib – Insmed”. AdisInsight. Springer Nature Switzerland AG.
- Chalmers JD, Usansky H, Rubino CM, Teper A, Fernandez C, Zou J, et al. (October 2022). “Pharmacokinetic/Pharmacodynamic Evaluation of the Dipeptidyl Peptidase 1 Inhibitor Brensocatib for Non-cystic Fibrosis Bronchiectasis”. Clinical Pharmacokinetics. 61 (10): 1457–1469. doi:10.1007/s40262-022-01147-w. PMC 9553789. PMID 35976570.
- Chalmers JD, Burgel PR, Daley CL, De Soyza A, Haworth CS, Mauger D, et al. (April 2025). “Phase 3 Trial of the DPP-1 Inhibitor Brensocatib in Bronchiectasis”. The New England Journal of Medicine. 392 (16): 1569–1581. doi:10.1056/NEJMoa2411664. PMID 40267423.
| Clinical data | |
|---|---|
| Other names | AZD7986; INS1007 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1802148-05-5 |
| PubChem CID | 118253852 |
| IUPHAR/BPS | 9412 |
| DrugBank | DB15638 |
| ChemSpider | 67896269 |
| UNII | 25CG88L0BB |
| KEGG | D12120 |
| ChEMBL | ChEMBL3900409 |
| Chemical and physical data | |
| Formula | C23H24N4O4 |
| Molar mass | 420.469 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
////////Brensocatib, APPROVALS 2025, FDA 2025, Brinsupri, non-cystic fibrosis, AZD7986, 1802148-05-5, INS1007, AZD 7986, WHO 11097
Dordaviprone
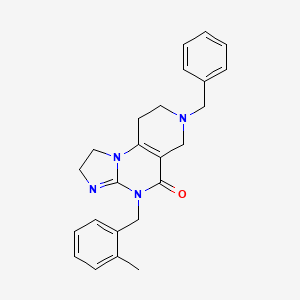
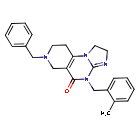
Dordaviprone
WeightAverage: 386.499
Monoisotopic: 386.210661473
Chemical FormulaC24H26N4O
- TIC10
- CAS 1616632-77-9
- Dordaviprone
- ONC201
- ONC 201
- 9U35A31JAI
- NSC-350625
11-benzyl-7-[(2-methylphenyl)methyl]-2,5,7,11-tetrazatricyclo[7.4.0.02,6]trideca-1(9),5-dien-8-one
Product Ingredients
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Dordaviprone dihydrochloride | 53VG71J90J | 1638178-82-1 | Not applicable |
- 11-benzyl-7-(o-tolylmethyl)-2,5,7,11-tetrazatricyclo[7.4.0.02,6]trideca-1(9),5-dien-8-one
- 11-benzyl-7-[(2-methylphenyl)methyl]-2,5,7,11-tetrazatricyclo[7.4.0.0(2,6)]trideca-1(9),5-dien-8-one
- 7-Benzyl-4-(2-methylbenzyl)-2,4,6,7,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one
- 7-benzyl-4-[(2-methylphenyl)methyl]-2,4,6,7,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one
FDA 8/6/2025, Modeyso, To treat diffuse midline glioma harboring an H3 K27M mutation with progressive disease following prior therapy
Dordaviprone, sold under the brand name Modeyso is an anti-cancer medication used for the treatment of diffuse midline glioma (a type of brain tumor).[1][2] Dordaviprone is a protease activator of the mitochondrial caseinolytic protease P.[1] It is dopamine receptor D2 antagonist and an allosteric activator of the mitochondrial caseinolytic protease P.[3]
Dordaviprone was approved for medical use in the United States in August 2025.[2] It is the first approval of a systemic therapy for H3 K27M-mutant diffuse midline glioma by the US Food and Drug Administration.[2]
Dordaviprone is an organic heterotricyclic compound that is 2,4,6,7,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(1H)-one substituted by 2-methylbenzyl and benzyl groups at positions 4 and 7, respectively. It is a selective antagonist of the dopamine receptor D2 and an allosteric agonist of mitochondrial protease caseinolytic protease P. It has a role as an antineoplastic agent, a dopamine receptor D2 antagonist and an apoptosis inducer. It is a member of toluenes, a member of benzenes and an organic heterotricyclic compound.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- https://pp.jazzpharma.com/pi/modeyso.en.USPI.pdf [bare URL PDF]
- “FDA grants accelerated approval to dordaviprone for diffuse midline glioma”. U.S. Food and Drug Administration (FDA). 6 August 2025. Retrieved 7 August 2025.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - Prabhu VV, Morrow S, Rahman Kawakibi A, Zhou L, Ralff M, Ray J, et al. (December 2020). “ONC201 and imipridones: Anti-cancer compounds with clinical efficacy”. Neoplasia. 22 (12). New York, N.Y.: 725–744. doi:10.1016/j.neo.2020.09.005. PMC 7588802. PMID 33142238.
- “Jazz Pharmaceuticals Announces U.S. FDA Approval of Modeyso (dordaviprone) as the First and Only Treatment for Recurrent H3 K27M-mutant Diffuse Midline Glioma” (Press release). Jazz Pharmaceuticals. 6 August 2025. Retrieved 10 August 2025 – via PR Newswire.
- World Health Organization (2023). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 89”. WHO Drug Information. 37 (1). hdl:10665/366661.
External links
- Clinical trial number NCT02525692 for “Oral ONC201 in Adult Recurrent Glioblastoma” at ClinicalTrials.gov
- Clinical trial number NCT03295396 for “ONC201 in Adults With Recurrent H3 K27M-mutant Glioma” at ClinicalTrials.gov
- Clinical trial number NCT03416530 for “ONC201 in Pediatric H3 K27M Gliomas” at ClinicalTrials.gov
- Clinical trial number NCT05392374 for “Expanded Access Use of ONC201 in a Patient With Diffuse Intrinsic Pontine Gliomas” at ClinicalTrials.gov
- Clinical trial number NCT03134131 for “Expanded Access to ONC201 for Patients With H3 K27M-mutant and/or Midline High Grade Gliomas” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Modeyso |
| Other names | ONC201, ONC-201 |
| AHFS/Drugs.com | Modeyso |
| License data | US DailyMed: Dordaviprone |
| Routes of administration | By mouth |
| Drug class | Protease activator |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1616632-77-9as HCl: 1638178-82-1 |
| PubChem CID | 73777259 |
| DrugBank | DB14844as HCl: DBSALT003291 |
| ChemSpider | 30904994 |
| UNII | 9U35A31JAIas HCl: 53VG71J90J |
| KEGG | D12733as HCl: D12734 |
| ChEBI | CHEBI:232328 |
| ChEMBL | ChEMBL4297310 |
| Chemical and physical data | |
| Formula | C24H26N4O |
| Molar mass | 386.499 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////Dordaviprone, Modeyso, FDA 2025, APPROVALS 2025, TIC10, 1616632-77-9, Dordaviprone, ONC201, ONC 201, 9U35A31JAI, NSC 350625
Zongertinib
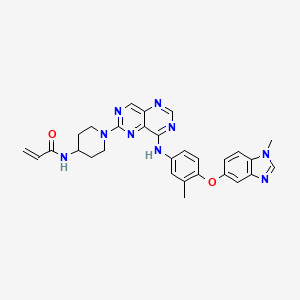
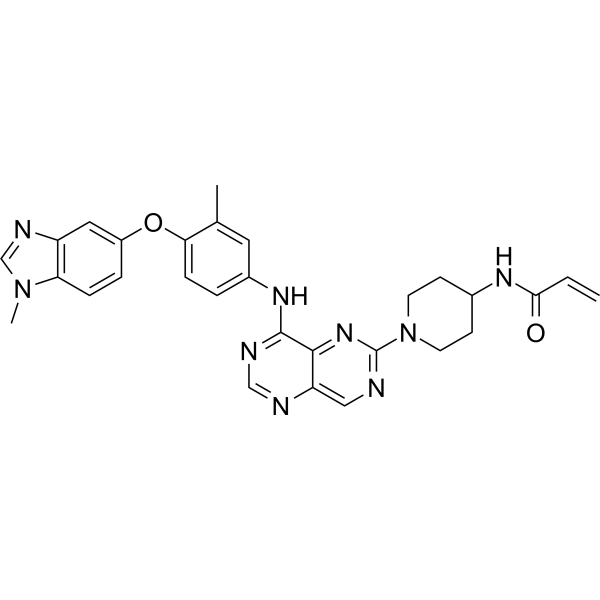
Zongertinib
CAS No. : 2728667-27-2,
BI-1810631, BI1810631
| Molecular Weight | 535.60 |
|---|---|
| Formula | C29H29N9O2 |
FDA 8/8/2025, Hernexeos, To treat adults with unresectable or metastatic non-squamous non-small cell lung cancer whose tumors have HER2 tyrosine kinase domain activating mutations, as detected by an FDA-approved test, and who have received prior systemic therapy
- N-(1-(8-((3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)amino)pyrimido[5,4-d]pyrimidin-2-yl)piperidin-4-yl)acrylamide
- N-(1-(8-((3-methyl-4-((1-methyl-1H-benzo(d)imidazol-5-yl)oxy)phenyl)amino)pyrimido(5,4-d)pyrimidin-2-yl)piperidin-4-yl)acrylamide
- 884-819-6
Zongertinib is an orally bioavailable inhibitor of the receptor tyrosine kinase human epidermal growth factor receptor 2 (HER2; ErbB2; HER-2), with potential antineoplastic activity. Upon oral administration, zongertinib covalently binds to and inhibits the activity of both wild-type and HER2 mutants, including HER2 mutants with exon 20 insertion (ex20ins) mutations. This prevents HER2-mediated signaling and may lead to cell death in HER2-expressing tumor cells. HER2, a receptor tyrosine kinase overexpressed on a variety of tumor cell types, plays an important role in tumor cell proliferation and tumor vascularization.
REF
Synthesis of zongertinib (N-(1-(8-((3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-
548 yl)oxy)phenyl)amino)pyrimido[5,4-d]pyrimidin-2-yl)piperidin-4-yl)acrylamide)
Methods
Synthesis of Zongertinib (N-(1-(8-((3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)amino)pyrimido[5,4-d]pyrimidin-2-yl)piperidin-4-yl)acrylamide)
An overview of the synthetic routes to zongertinib and BI-3999 is shown in Supplementary Fig. S1, and graphical NMR spectra are shown in Supplementary Fig. S2.
3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)aniline (500 mg, 1.97 mmol) and 8-chloro-2-(methylthio)pyrimido[5,4-d]pyrimidine hydrochloride (492 mg, 1.97 mmol) were suspended in isopropanol, and the resulting reaction mixture stirred at 50°C for 3 hours, at which time high-performance liquid chromatography–mass spectrometry (HPLC-MS) indicated full conversion. The reaction mixture was concentrated under reduced pressure, and the crude product was redissolved in dichloromethane and washed with aqueous NaHCO3. The organic layer was dried over Na2SO4 and concentrated, and the resulting crude product was purified by column chromatography (SiO2, gradient of 0%–15% methanol in dichloromethane) to afford the product (840 mg).
N-(3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)-6-(methylthio)pyrimido[5,4-d]pyrimidin-4-amine (860 mg, 90%, 1.80 mmol) was suspended in dichloromethane (30 mL), and the resulting mixture was cooled to 0°C to 5°C. mCPBA (3-chloroperbenzoic acid, 444 mg, 77%, 1.98 mmol) was added portionwise over 1 hour, and the resulting reaction mixture was stirred at room temperature overnight, at which time HPLC-MS indicated full conversion. The reaction mixture was diluted with dichloromethane and washed with aqueous NaHCO3. The organic layer was dried over Na2SO4 and concentrated, and the resulting crude product which was used directly in the next step (767 mg, crude).
N-(3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)-6-(methylsulfinyl)pyrimido[5,4-d]pyrimidin-4-amine (5.42 g, 80%, 9.73 mmol) was dissolved in N,N-dimethyl formamide (DMF, 50 mL) and diisopropylethylamine (2.8 mL, 16 mmol). 4-Boc-amino-1-piperidine (2.39 g, 11.9 mmol) was added, and the reaction was stirred at 60°C overnight. Then, the reaction mixture was concentrated, and the crude product was used directly in the next step (5.66 g, crude).
Tert-butyl (1-(8-((3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)amino)pyrimido[5,4-d]pyrimidin-2-yl)piperidin-4-yl)carbamate (5.66 g, 9.73 mmol) was dissolved in dichloromethane (100 mL) and methanol (30 mL). Four mol/L HCl in dioxane (11 mL, 44 mmol) was added, and the resulting reaction mixture was heated to 45°C for 7 hours. HPLC-MS indicated some remaining starting material; therefore, the reaction mixture was stirred at room temperature overnight. Four mol/L HCl in dioxane (1 mL, 0.40 mmol) was added, and the reaction mixture was reheated to 45°C for 4 hours, at which time HPLC-MS indicated full conversion. The reaction mixture was concentrated, and the resulting crude product was purified by column chromatography (SiO2, gradient of 0%–20% methanol in dichloromethane) to afford the product (4.5 g, 70% purity).
1-[8-({3-methyl-4-[(1-methyl-1H-1,3-benzodiazol-5-yl)oxy]phenyl}amino)-[1,3]diazino[5,4-d]pyrimidin-2-yl]piperidin-4-amine (4.5 g, 70%, 6.9 mmol) was suspended in dichloromethane (150 mL) and triethyl amine (4 mL, 28 mmol), and dimethylaminopyridine (115 mg, 0.941 mmol) was added. Then, acroyloyl anhydride (1.36 g, 95%, 10.3 mmol) was added, and the resulting reaction mixture was stirred at room temperature for 1 hour, at which time HPLC-MS indicated full conversion. The reaction mixture was diluted with dichloromethane (50 mL) and washed with aqueous NaHCO3 and brine. The organic layer was dried over Na2SO4 and concentrated, and the resulting crude product was purified by column chromatography (SiO2, gradient of 0%–20% methanol in dichloromethane) to afford the product (2.49 g).
1H NMR (DMSO-d6, 500 MHz) δ 9.58 (s, 1H), 9.08 (s, 1H), 8.39 (s, 1H), 8.19 (s, 1H), 8.10 (d, 1H, J = 7.6 Hz), 7.84 (d, 1H, J = 2.2 Hz), 7.77 (dd, 1H, J = 8.8 Hz, J = 2.2 Hz), 7.57 (d, 1H, J = 8.8 Hz), 7.09 (d, 1H, J = 2.2 Hz), 7.00 (dd, 1H, J = 2.2, 8.5 Hz), 6.89 (d, 1H, J = 8.8 Hz), 6.20 (dd, 1H, J = 10.1, 17.0 Hz), 6.10 (dd, 1H, J = 2.2, 17.0 Hz), 5.6 (dd, 1H, J = 2.2, 9.8 Hz), 4.86 (m, 2H), 3.99 (m, 1H), 3.84 (s, 3H), 3.25 (m, 2H), 2.26 (s, 3H), 1.92 (m, 2H), and 1.43 (m, 2H).
Synthesis of BI-3999 (N-(1-(8-((3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)amino)pyrimido[5,4-d]pyrimidin-2-yl)piperidin-4-yl)acetamide)
6-(4-aminopiperidin-1-yl)-N-(3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)pyrimido[5,4-d]pyrimidin-4-amine (100 mg, 208 mmol) and 4-dimethylaminopyridine (2.5 mg, 0.02 mmol) were suspended in 5 mL dichloromethane. Acetic anhydride (25 μL, 0.23 mmol) was added, and the resulting reaction mixture was stirred at room temperature for one hour. Then, the reaction mixture was diluted with dichloromethane and washed with aqueous NaHCO3 and brine. Then, the layers were separated, and the organic layer was dried over MgSO4 and concentrated. The crude product was purified by column chromatography (SiO2, gradient of 0%–10% methanol in dichloromethane) to afford the product (75 mg).
1H NMR (DMSO-d6, 400 MHz) δ 9.58 (s, 1H), 9.07 (s, 1H), 8.39 (s, 1H), 8.17 (s, 1H), 7.88 (d, 1H, J = 7.9 Hz), 7.84 (d, 1H, J = 2.5 Hz), 7.77 (dd, 1H, J = 2.7, 8.7 Hz), 7.57 (d, 1H, J = 8.9 Hz), 7.09 (d, 1H, J = 2.3 Hz), 7.00 (dd, 1H, J = 2.3, 8.6 Hz), 6.89 (d, 1H, J = 8.6 Hz), 4.85 (m, 2H), 3.90 (m, 1H), 3.84 (s, 3H), 3.23 (m, 2H), 2.26 (s, 3H), 1.88 (m, 2H), 1.82 (s, 3H), and 1.38 (m, 2H).
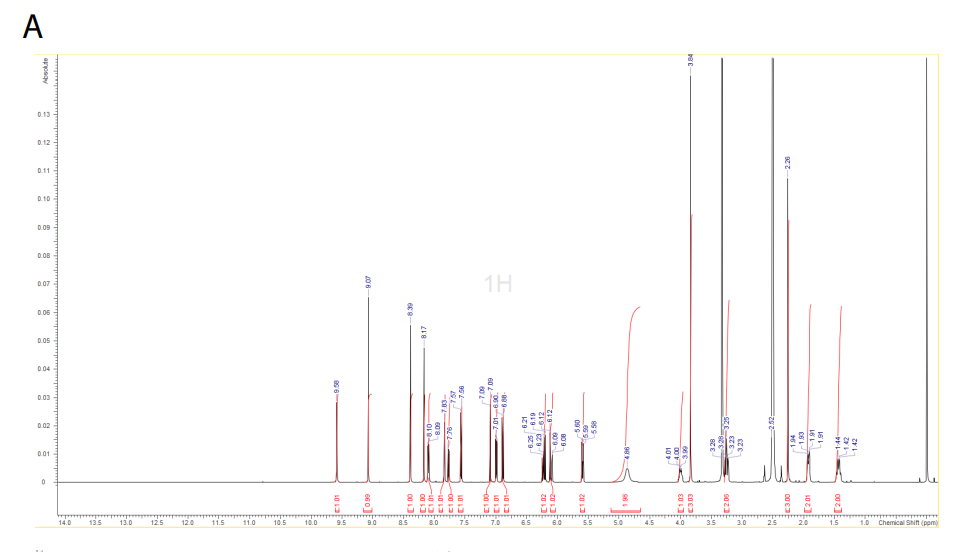
A) 1H NMR spectrum of zongertinib
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021213800&_cid=P10-ME52KD-62836-1
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
- [1]. WHO Drug Informat ion – World Health Organization (WHO).[2]. Wilding Birgit, et al. Synthesis of diazino-pyrimidines as anticancer agents: World Intellectual Property Organization, WO2021213800. 2021-10-28.[3]. Li S, et al. Emerging Targeted Therapies in Advanced Non-Small-Cell Lung Cancer. Cancers (Basel). 2023 May 24;15(11):2899. [Content Brief]
////////////Zongertinib, Hernexeos, APPROVALS 2025, FDA 2025, lung cancer, BI-1810631, BI1810631, DRH7R67UVL
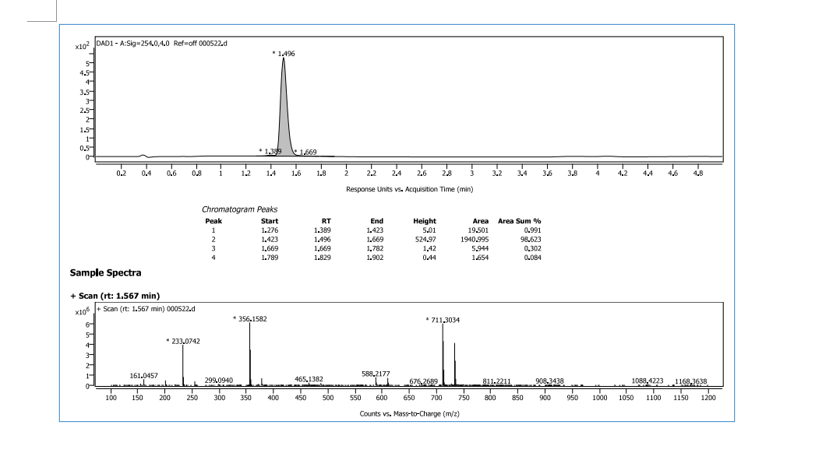
Ropotrectinib
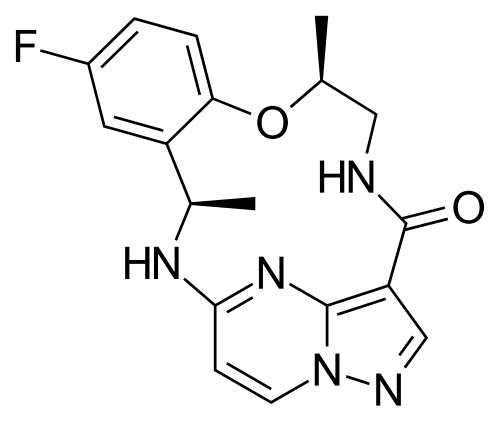
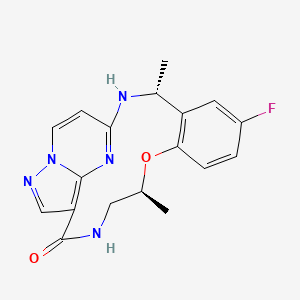
Ropotrectinib
- CAS 1802220-02-5
- TPX-0005
- Augtyro
- 08O3FQ4UNP
WeightAverage: 355.373
Monoisotopic: 355.144453003
Chemical FormulaC18H18FN5O2
- repotrectinibum
- (3R,11S)-6-fluoro-3,11-dimethyl-10-oxa-2,13,17,18,21-pentazatetracyclo[13.5.2.04,9.018,22]docosa-1(21),4(9),5,7,15(22),16,19-heptaen-14-one
- 1,15-Etheno-1H-pyrazolo(4,3-F)(1,4,8,10)benzoxatriazacyclotridecin-4(5H)-one, 11-fluoro-6,7,13,14-tetrahydro-7,13-dimethyl-, (7S,13R)-
- 1,15-Etheno-1H-pyrazolo(4,3-f)(1,4,8,10)benzoxatriazacyclotridecin-4(5H)-one, 11-fluoro-2,6,7,13-tetrahydro-7,13-dimethyl-, (14Z)-
- (1Z)-6-Fluoro-3,11-dimethyl-10-oxa-2,13,17,18,21-pentaazatetracyclo(13.5.2.04,9.018,22)docosa-1,4,6,8,15,19,21-heptaen-14-one
- (3R,11S)-6-fluoro-3,11-dimethyl-10-oxa-2,13,17,18,21-pentaazatetracyclo[13.5.2.0,.0,]docosa-1(21),4(9),5,7,15(22),16,19-heptaen-14-one
- (7S,13R)-11-fluoro-7,13-dimethyl-6,7,13,14- tetrahydro-1,15-ethenopyrazolo[4,3- f][1,4,8,10]benzoxatriazacyclotridecin-4(5H)- one
- (3R,6S,)-45-FLUORO-3,6-DIMETHYL-5-OXA-2,8-DIAZA-1(5,3)-PYRAZOLO(1,5-A)PYRIMIDINA-4(1,2)-BENZENANONAPHAN-9-ONE
- (7S,13R)-11-Fluoro-7,13-Dimethyl-6,7,13,14-Tetrahydro-1,15-Ethenopyrazolo[4,3-F][1,4,8,10]Benzoxatriazacyclotridecin-4(5H)-One
- 1,15-ETHENO-1H-PYRAZOLO(4,3-F)(1,4,8,10)BENZOXATRIAZACYCLOTRIDECIN-4(5H)-ONE, 11-FLUORO-6,7,13,14-TETRAHYDRO-7,13-DIMETHYL-, (7S,13R)-
Repotrectinib, sold under the brand name Augtyro, is an anti-cancer medication used for the treatment of non-small cell lung cancer.[2][5] It is taken by mouth.[2] Repotrectinib is an inhibitor of proto-oncogene tyrosine-protein kinase ROS1 (ROS1) and of the tropomyosin receptor tyrosine kinases (TRKs) TRKA, TRKB, and TRKC.[2]
The most common adverse reactions include dizziness, dysgeusia, peripheral neuropathy, constipation, dyspnea, ataxia, fatigue, cognitive disorders, and muscular weakness.[5]
Repotrectinib was approved for medical use in the United States in November 2023,[5][6] and in the European Union in January 2025.[3][4] CHINA 2024
SYN
https://chemistry-europe.onlinelibrary.wiley.com/doi/abs/10.1002/slct.202405153
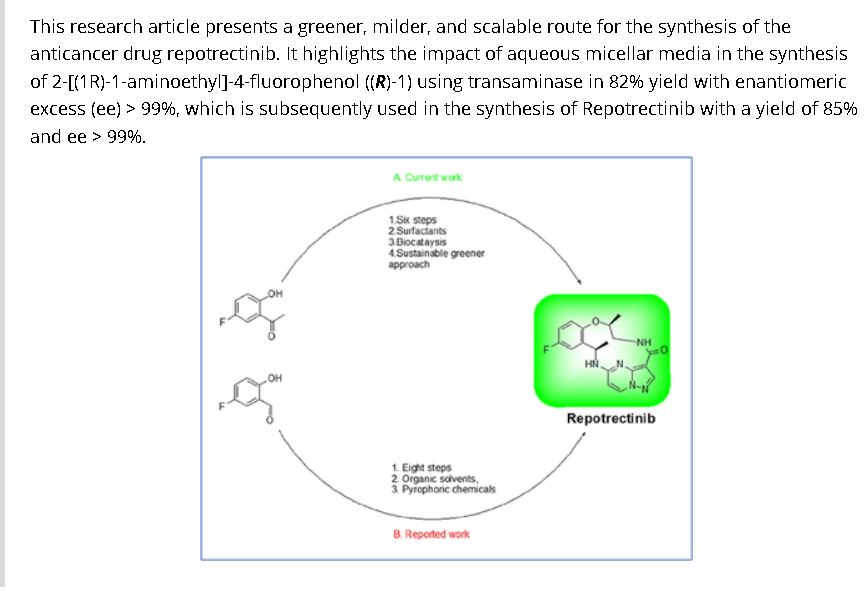
Synthesis of Repotrectinib
To a stirred solution of 5-{[(1R)-1-(2-{[(2S)-1-aminopropan-2-yl]oxy}-5-fluorophenyl)ethyl]amino}pyrazolo[1,5-a]pyrimidine-3-carboxylic acid 15 (0.25 g, 0.000611 mol, 1.0 eq.) in DMF (4.0 mL, 16V) was slowly added to solution of DIPEA (0.6 mL, 0.00488 mol, 8.0 eq.) in DCM (1.8 mL, 7V) at 0-5 °C. Then FDPP (0.25 g, 0.000672 mol, 1.1 eq.) was added at 0-5 °C. The reaction mixture was allowed to stirr for 1-2h at 25-30 °C. The reaction was monitored by TLC for disappearance of starting material. Then the resulting reaction mixture was diluted with ethyl acetate (50 mL), washed with water (20 mL) and brine solution (20 mL). The separated organic layer was dried over sodium sulphate and concentrated under reduced pressure at 45 °C. The obtained crude product was purified by silica gel (60-120 mesh) column chromatography to get repotrectinib asawhite solid (0.18 g, 85%).
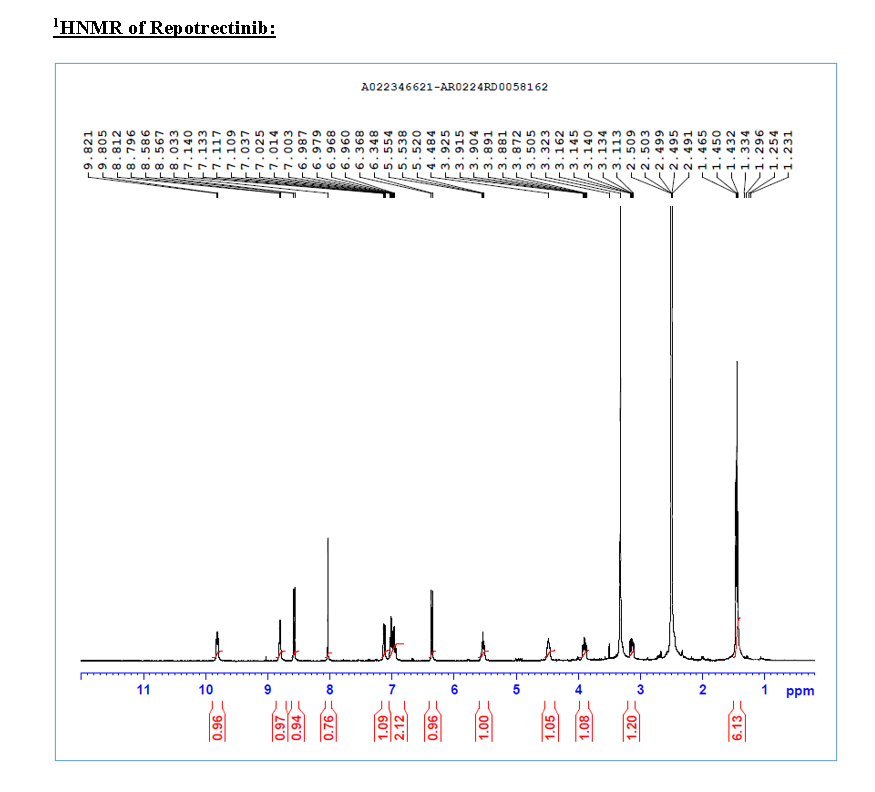
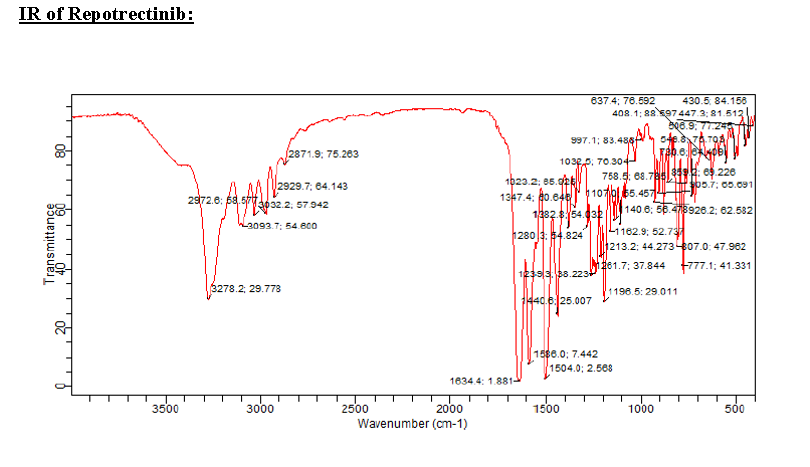

HRMS
SYN
https://pubs.acs.org/doi/10.1021/acs.oprd.3c00152
REF
https://pubs.acs.org/doi/10.1021/acs.oprd.4c00061
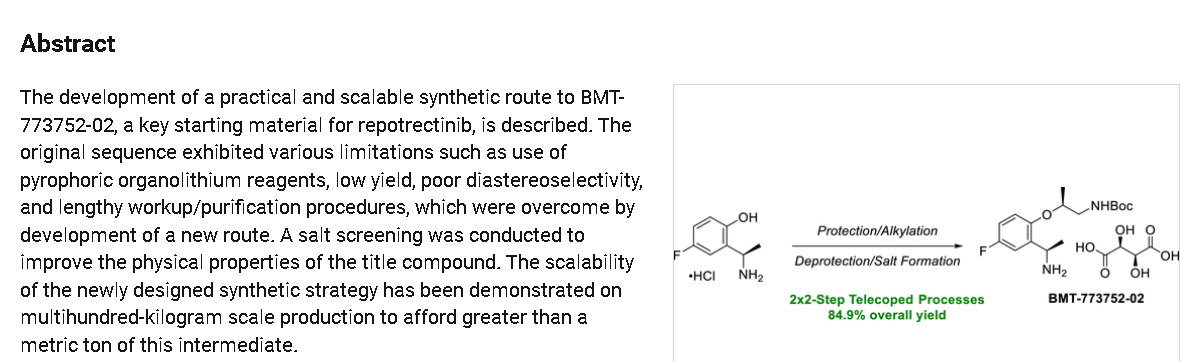
REF
US20180194777
https://patentscope.wipo.int/search/en/detail.jsf?docId=US222923082&_cid=P11-ME283N-03701-1
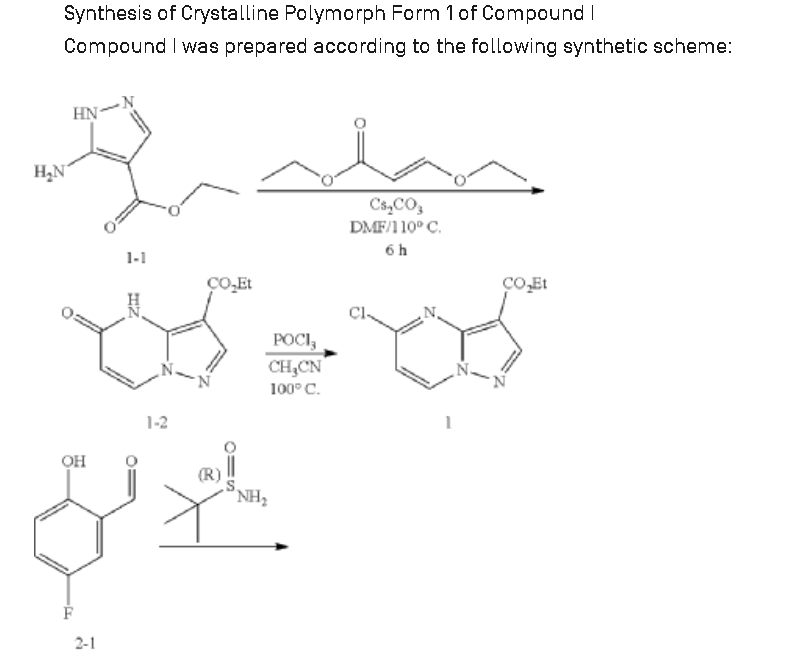
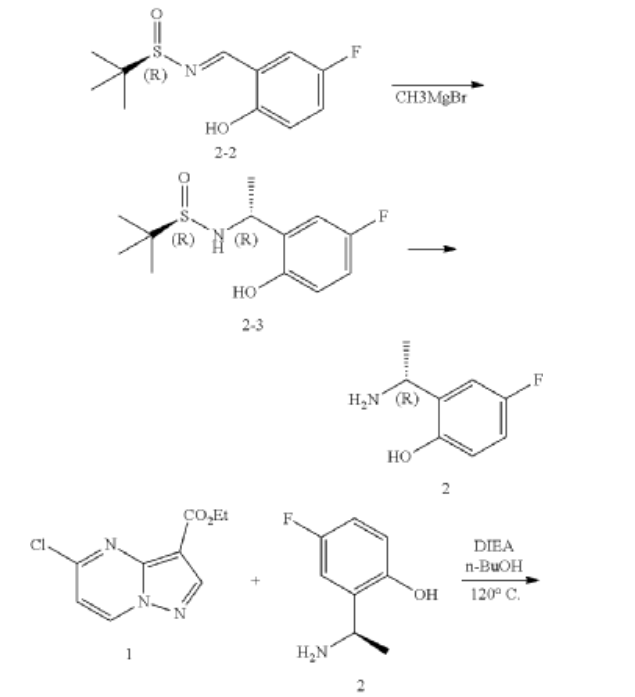
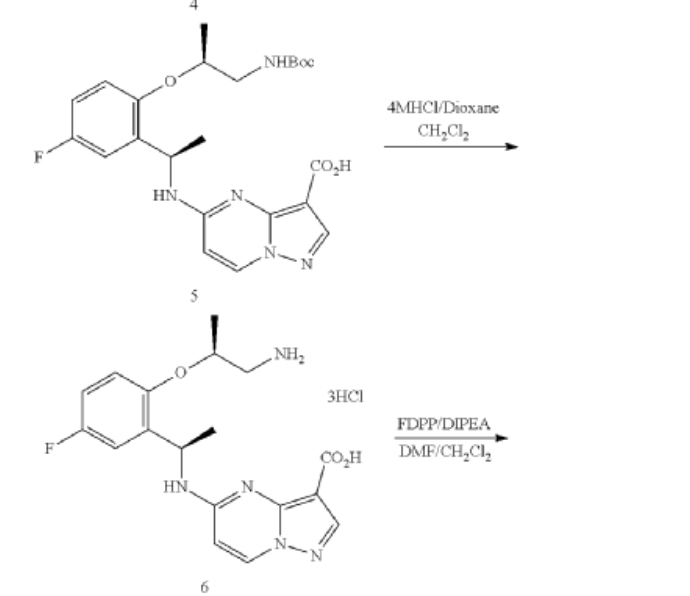
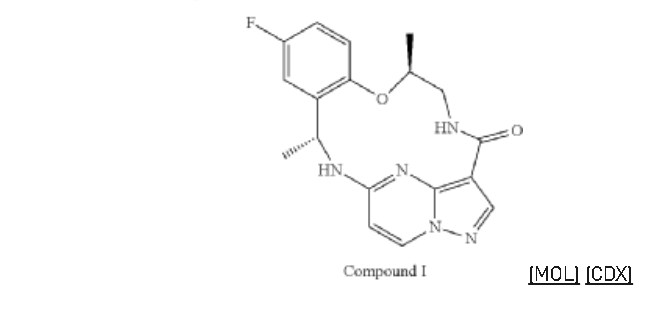
Example 1: Preparation of 5-chloropyrazolo[1,5-a]pyrimidine-3-carboxylate (1)
Step 1: Preparation of ethyl 5-oxo-4H-pyrazolo[1,5-a]pyrimidine-3-carboxylate (1-2)
Step 2: Preparation of 5-chloropyrazolo[1,5-a]pyrimidine-3-carboxylate (1)
PATENT
https://patents.google.com/patent/US10246466B2/en
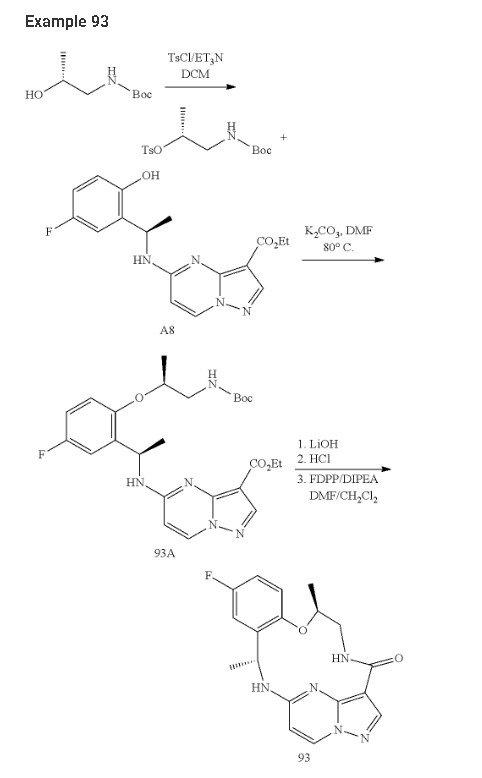
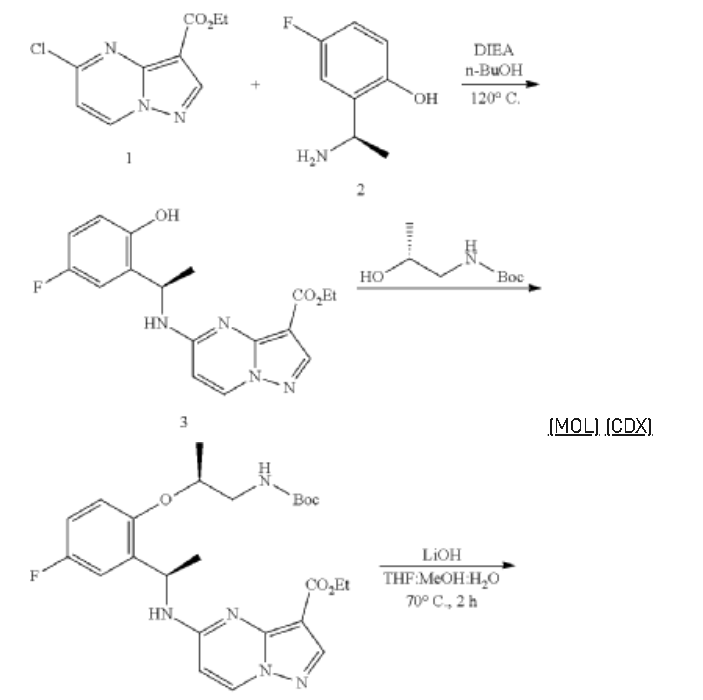
Step 1. To a solution of tert-butyl (R)-(2-hydroxypropyl)carbamate (1.00 g, 5.71 mmol) and tosyl chloride (1.14 g, 6.00 mmol) in DCM (29 mL) was added triethylamine (1.44 g, 14.28 mmol and the mixture was stirred at room temp for 48 hour. The reaction solution was concentrated under reduced pressure and the residue was purified with flash chromatography (ISCO system, silica (40 g), 0-20% ethyl acetate in hexane) to provide (R)-1-((tert-butoxycarbonyl)amino)propan-2-yl 4-methylbenzenesulfonate (1.12 g, 3.40 mmol, 59.54% yield).
Step 2. To a solution of A8 (100.00 mg, 0.290 mmol) and (R)-1-((tert-butoxycarbonyl)amino)propan-2-yl 4-methylbenzenesulfonate (143.50 mg, 0.436 mmol) in DMF (1.45 mL) was added K2CO3 (200.7 mg, 1.45 mmol) and heated at 80° C. with stirring for 16 hour. The reaction was cooled to ambient temperature and diluted with DCM (3 mL), filtered through a syringe filter, and concentrated under reduced pressure. Flash chromatography (ISCO system, silica (12 g), 0-60% ethyl acetate in hexane) provided 93A (32.90 mg, 0.0656 mmol, 22.59% yield).
Step 3. To a solution of 93A (32.90 mg, 0.0656 mmol) in MeOH (3 mL) and THF (2 mL) was added LiOH aqueous solution (2M, 2 mL) at ambient temperature. The reaction solution was heated at 70° C. for 2 hours The reaction flask was cooled to ambient temperature, diluted with water and methanol, and then quenched with HCl aqueous solution (2 M, 2 mL) to pH<5. The mixture was extracted with DCM (3×5 mL), dried with Na2SO4, concentrated under reduced and dried on high vacuum overnight. To a solution of the acid product in DCM (4 mL) was added 4 M HCl in 1,4-dioxane (2.0 mL). The mixture was stirred at room temperature for 3 hours, and then concentrated under reduced pressure and dried on high vacuum. To a solution of the de-Boc product and FDPP (27.62 mg, 0.0719 mmol) in DMF (1.6 mL) was added Hunig’s base (42.23 mg, 0.327 mmol) at room temperature. The mixture was stirred for 2.5 hours, and then quenched the reaction with 2 M Na2CO3 solution (2 mL). The mixture was stirred for 15 min then extracted with DCM (4×10 mL). The combined extracts were dried with Na2SO4 and concentrated under reduced pressure. The residue was purified with flash chromatography (ISCO system, silica (12 g), 0-10% methanol in dichloromethane) to provide 93 (10.1 mg, 0.0284 mmol, 43.49% yield for three steps).
PATENT
SYN
European Journal of Medicinal Chemistry 265 (2024) 116124
Repotrectinib (Augtyro) Repotrectinib, developed by Turning Point Therapeutics, Inc., was granted FDA approval on November 15, 2023. It is indicated to treat locally advanced or metastatic ROS proto-oncogene 1, receptor tyrosine kinase (ROS1)-positive non-small cell lung cancer (NSCLC). Repotrectinib is a highly effective inhibitor of ROS1 (ICtyrosine receptor kinase (TRK) (IC5050= 0.07 nM) and
=0.83/0.05/0.1 nM for TRKA/B/C) [87]. After undergoing currently approved targeted therapies, patients with tumors containing ROS1 and neurotrophic tyrosine kinase receptor (NTRK) gene fusions frequently acquire resistance mutations [88,89]. These mutations restrict the ability of drugs to bind to their
targets, ultimately resulting in the advancement of tumors. Repotrectinib, a novel tyrosine kinase inhibitor (TKI), is the pioneering drug developed to specifically target ROS1 or NTRK-positive metastatic
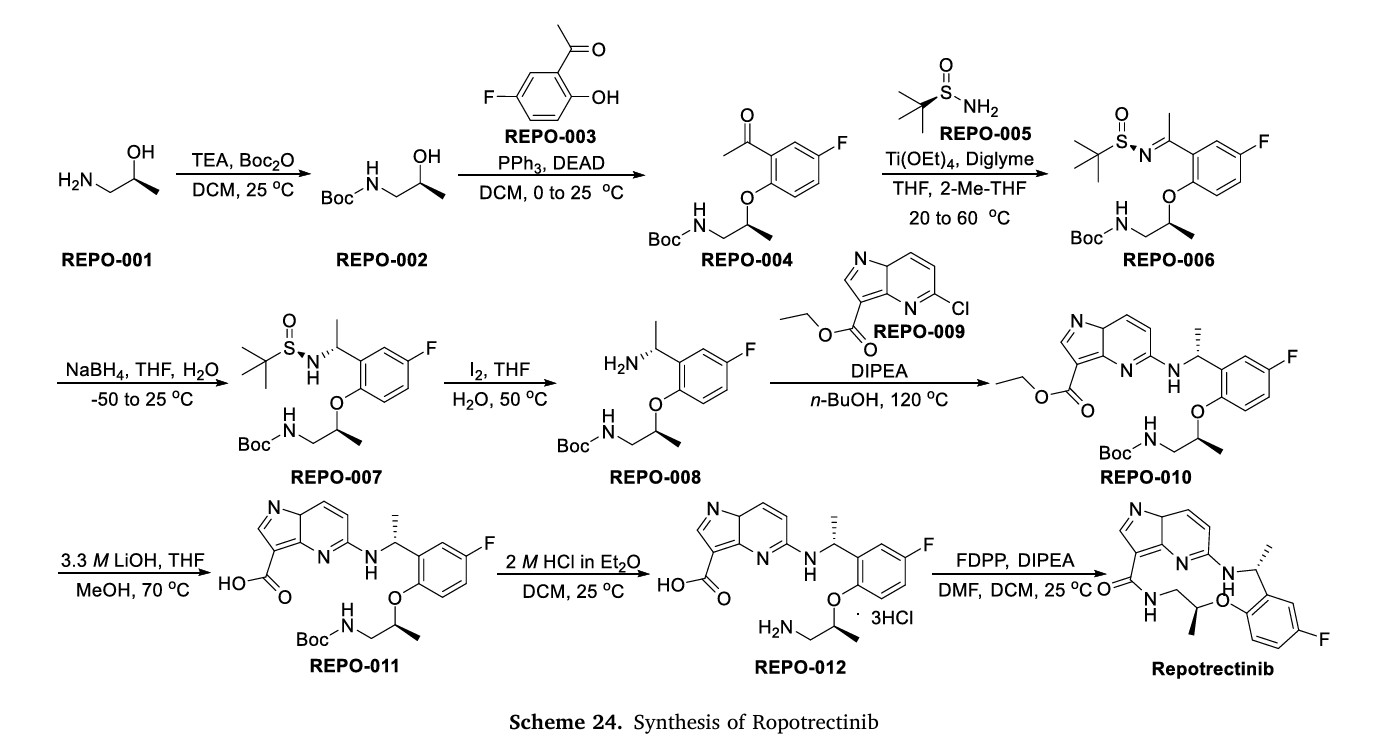
NSCLC and effectively combat the primary factors contributing to disease advancement [90].Preparation of Repotrectinib is described as Scheme 24 [91].Protecting the amino group of REPO-001 with Boc group in the presence of Kgave REPO-002, followed by intermolecular dehydration with
1-(5-fluoro-2-hydroxyphenyl)ethan-1-one (REPO-003) to give the ester REPO-004. REPO-004 was reacted with chiral auxiliary REPO-005 to give REPO-006, which was reduced by NaBH4
to obtain REPO-007. Then REPO-008 was obtained by removing the chiral auxiliary under iodine conditions. Substitution of REPO-008 with REPO-009 gave REPO-010, which was further hydrolyzed under alkaline conditions to obtain REPO-011. Salt formation of REPO-011 with hydrochloric acid
yielded REPO-012, which underwent intramolecular condensation to obtain the product Repotrectinib.
[87] D. Zhai, W. Deng, Z. Huang, E. Rogers, J.J. Cui, The novel, rationally-designed,
ALK/SRC inhibitor TPX-0005 overcomes multiple acquired resistance
mechanisms to current ALK inhibitors, Cancer Res. 76 (2016) 2132.
[88] C. Keddy, P. Shinde, K. Jones, S. Kaech, R. Somwar, U. Shinde, M.A. Davare,
Resistance profile and structural modeling of next-generation ROS1 tyrosine
kinase inhibitors, Mol. Cancer Therapeut. 21 (2022) 336–346.
[89] E. Cocco, M. Scaltriti, A. Drilon, NTRK fusion-positive cancers and TRK inhibitor
therapy, Nat. Rev. Clin. Oncol. 15 (2018) 731–747.
[90] A. Drilon, S.I. Ou, B.C. Cho, D.W. Kim, J. Lee, J.J. Lin, V.W. Zhu, M.J. Ahn, D.
R. Camidge, J. Nguyen, D. Zhai, W. Deng, Z. Huang, E. Rogers, J. Liu, J. Whitten,
J.K. Lim, S. Stopatschinskaja, D.M. Hyman, R.C. Doebele, J.J. Cui, A.T. Shaw,
Repotrectinib (TPX-0005) is a next-generation ROS1/TRK/ALK inhibitor that
potently inhibits ROS1/TRK/ALK solvent-front mutations, Cancer Discov. 8
(2018) 1227–1236.
[91] J.J. Cui, E.W. Rogers, Gialir Macrocyclic Polymorph, 2018. US20180194777A1.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Syn
European Journal of Medicinal Chemistry 291 (2025) 117643
Repotrectinib, developed by Bristol-Myers Squibb and marketed under the brand name Augtyro, is an oral tyrosine kinase inhibitor (TKI) targeting ROS1 and TRK oncogenic drivers. In 2024, NMPA condition
ally approved Repotrectinib for adult patients with ROS1-positive locally advanced or metastatic NSCLC [15]. Repotrectinib exerts its antitumor activity by inhibiting ROS1 and TRK kinases, thereby disrupting the downstream signaling pathways that facilitate tumor cell proliferation and survival [16]. This argeted mechanism is particularly effective against tumors that harbor ROS1 or NTRK gene fusions. The clinical efficacy of Repotrectinib has been through validated the Phase 1/2 TRIDENT-1 trial (NCT03093116) [17]. In the study cohort, treat ment-naïve patients harboring ROS1-positive NSCLC exhibited an overall response rate (ORR) of 79 %, characterized by a median duration of response (DOR) reaching 34.1 months. Conversely, among those who had previously received ROS1 TKI therapy, the ORR was documented at 38 %, accompanied by a median DOR of 14.8 months. With respect to safety profiles, the adverse event spectrum commonly encompassed dizziness, dysgeusia, peripheral neuropathy, constipation, dyspnea, fatigue, ataxia, cognitive impairment, muscular weakness, and nausea
[18,19]. These side effects are generally manageable, but patients should be monitored for potential severe adverse events.
The synthetic route of Repotrectinib, shown in Scheme 4, begins with condensation reaction between Repo-001 and Repo-002 to afford Repo-003, which is chlorinated to yield Repo-004 [20]. This intermediate undergoes nucleophilic substitution with Repo-005 to form Repo-006,
followed by second nucleophilic substitution with Repo-007 to produce Repo-008. Ester hydrolysis of Repo-008 affords Repo-009, which undergoes acid-mediated deprotection to generate Repo-010. Final
intramolecular amidation of Repo-010 delivers Repotrectinib. In parallel, Repo-011 and Repo-012 undergo condensation to form imine Repo-013, which undergoes Grignard addition to afford Repo-014.
Acidification of Repo-014 then yields Repo-005. Concurrently, Repo-015 undergoes nucleophilic substitution to generate Repo-007.
[15] S. Dhillon, Repotrectinib: first approval, Drugs 84 (2024) 239–246.
[16] T. Rais, A. Shakeel, L. Naseem, N. Nasser, M. Aamir, Repotrectinib: a promising
new therapy for advanced nonsmall cell lung cancer, Ann Med Surg (Lond) 86
(2024) 7265–7269.
[17] A. Drilon, S.I. Ou, B.C. Cho, D.W. Kim, J. Lee, J.J. Lin, V.W. Zhu, M.J. Ahn, D.
R. Camidge, J. Nguyen, D. Zhai, W. Deng, Z. Huang, E. Rogers, J. Liu, J. Whitten, J.
K. Lim, S. Stopatschinskaja, D.M. Hyman, R.C. Doebele, J.J. Cui, A.T. Shaw,
Repotrectinib (TPX-0005) is a next-generation ROS1/TRK/ALK inhibitor that
potently inhibits ROS1/TRK/ALK solvent-front mutations, Cancer Discov. 8 (2018)
1227–1236.
[18] Repotrectinib, Drugs and Lactation Database (Lactmed®), National Institute of
Child Health and Human Development, Bethesda (MD), 2006.
[19] H. Zhong, J. Lu, M. Wang, B. Han, Real-world studies of crizotinib in patients with
ROS1-positive non-small-cell lung cancer: experience from China, J Comp Eff Res
14 (2024) e240043.
[20] J.J. Cui, E.W. Rogers, Preparation of
Fluorodimethyltetrahydroethenopyrazolobenzoxatriazacyclotridecinone
Derivatives for Use as Antitumor Agents, 2017. US20180194777A1.
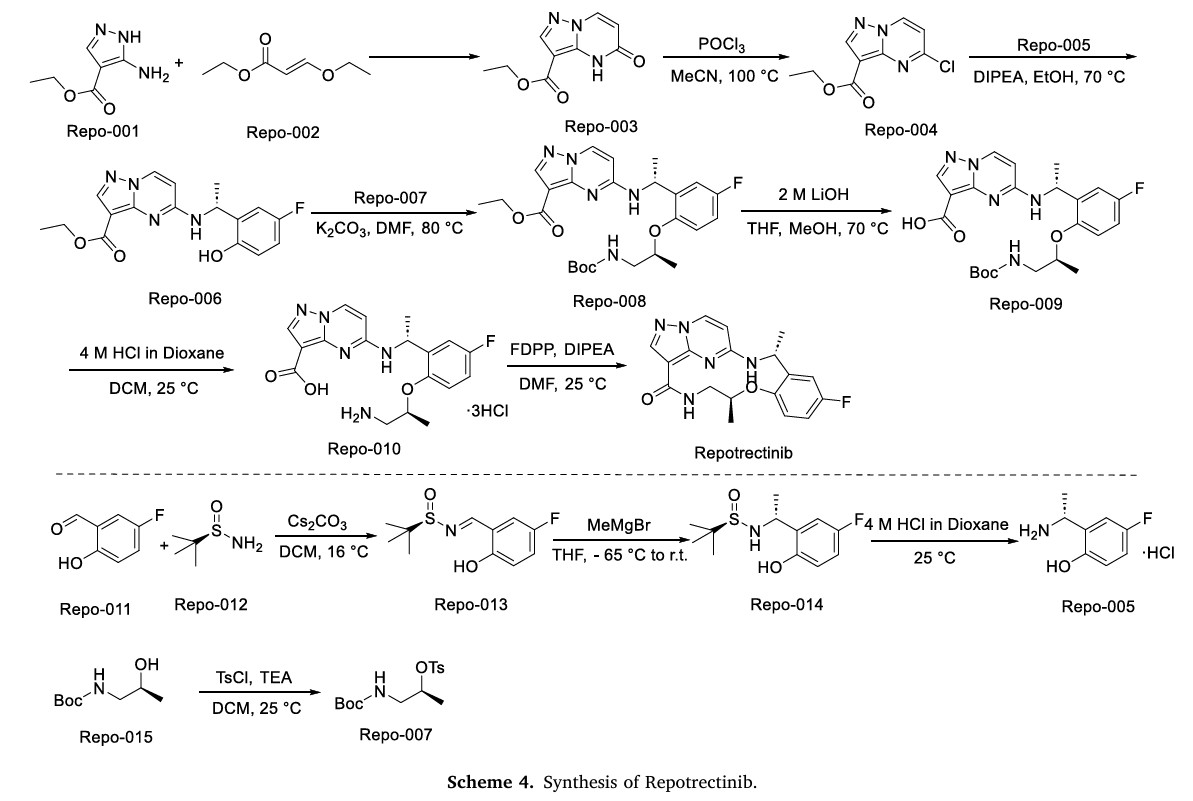
Repotrectinib is indicated for the treatment of adults with locally advanced or metastatic ROS1-positive non-small cell lung cancer.[2][5]
In June 2024, the US Food and Drug Administration (FDA) expanded the indication to include the treatment of people twelve years of age and older with solid tumors that have a neurotrophic tyrosine receptor kinase (NTRK) gene fusion, are locally advanced or metastatic or where surgical resection is likely to result in severe morbidity, and that have progressed following treatment or have no satisfactory alternative therapy.[7][8]
References
- “Register of Innovative Drugs”. Health Canada. 3 November 2006. Retrieved 23 May 2025.
- “Augtyro- repotrectinib capsule”. DailyMed. 15 November 2023. Archived from the original on 12 December 2023. Retrieved 12 December 2023.
- “Augtyro EPAR”. European Medicines Agency (EMA). 14 November 2024. Retrieved 16 November 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- “Augtyro PI”. Union Register of medicinal products. 14 January 2025. Retrieved 16 January 2025.
- “FDA approves repotrectinib for ROS1-positive non-small cell lung cancer”. U.S. Food and Drug Administration (FDA). 15 November 2023. Archived from the original on 16 November 2023. Retrieved 17 November 2023. This article incorporates text from this source, which is in the public domain.
- “U.S. Food and Drug Administration Approves Augtyro (repotrectinib), a Next-Generation Tyrosine Kinase Inhibitor (TKI), for the Treatment of Locally Advanced or Metastatic ROS1-Positive Non-Small Cell Lung Cancer (NSCLC)” (Press release). Bristol Myers Squibb. 16 November 2023. Archived from the original on 16 November 2023. Retrieved 17 November 2023 – via Business Wire.
- “FDA grants accelerated approval to repotrectinib for adult and pediatric participants with neurotrophic tyrosine receptor kinase gene fusion-positive solid tumors”. U.S. Food and Drug Administration. 13 June 2024. Archived from the original on 13 June 2024. Retrieved 13 June 2024. This article incorporates text from this source, which is in the public domain.
- “Cancer Accelerated Approvals”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 6 December 2024.
- Turning Point Therapeutics, Inc. (5 February 2024). A Phase 1/2, Open-Label, Multi-Center, First-in-Human Study of the Safety, Tolerability, Pharmacokinetics, and Anti-Tumor Activity of TPX-0005 in Patients With Advanced Solid Tumors Harboring ALK, ROS1, or NTRK1-3 Rearrangements (TRIDENT-1) (Report). clinicaltrials.gov. Archived from the original on 18 June 2024. Retrieved 18 June 2024.
- “Meeting highlights from the Committee for Medicinal Products for Human Use (CHMP) 11-14 November 2024”. European Medicines Agency (EMA). 15 November 2024. Retrieved 16 November 2024.
Further reading
- Drilon A, Ou SI, Cho BC, Kim DW, Lee J, Lin JJ, et al. (October 2018). “Repotrectinib (TPX-0005) Is a Next-Generation ROS1/TRK/ALK Inhibitor That Potently Inhibits ROS1/TRK/ALK Solvent- Front Mutations”. Cancer Discovery. 8 (10): 1227–1236. doi:10.1158/2159-8290.CD-18-0484. PMID 30093503.
External links
- “Repotrectinib (Code C133821)”. NCI Thesaurus. 25 September 2023. Retrieved 17 November 2023.
| Clinical data | |
|---|---|
| Trade names | Augtyro |
| Other names | TPX-0005 |
| AHFS/Drugs.com | Augtyro |
| License data | US DailyMed: Repotrectinib |
| Routes of administration | By mouth |
| Drug class | Tyrosine kinase inhibitor |
| ATC code | L01EX28 (WHO) |
| Legal status | |
| Legal status | CA: ℞-only[1]US: ℞-only[2]EU: Rx-only[3][4] |
| Identifiers | |
| CAS Number | 1802220-02-5 |
| PubChem CID | 135565923 |
| DrugBank | DB16826 |
| ChemSpider | 64853849 |
| UNII | 08O3FQ4UNP |
| KEGG | D11454 |
| ChEBI | CHEBI:229220 |
| ChEMBL | ChEMBL4298138 |
| PDB ligand | 7GI (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C18H18FN5O2 |
| Molar mass | 355.373 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
- (3R,6S,)-45-FLUORO-3,6-DIMETHYL-5-OXA-2,8-DIAZA-1(5,3)-PYRAZOLO(1,5-A)PYRIMIDINA-4(1,2)-BENZENANONAPHAN-9-ONE
- (7S,13R)-11-Fluoro-7,13-Dimethyl-6,7,13,14-Tetrahydro-1,15-Ethenopyrazolo[4,3-F][1,4,8,10]Benzoxatriazacyclotridecin-4(5H)-One
- 1,15-ETHENO-1H-PYRAZOLO(4,3-F)(1,4,8,10)BENZOXATRIAZACYCLOTRIDECIN-4(5H)-ONE, 11-FLUORO-6,7,13,14-TETRAHYDRO-7,13-DIMETHYL-, (7S,13R)-
////////Ropotrectinib, FDA 2023, APPROVALS 2023, Turning Point , EU 2025, APPROVALS 2025, EMA 2025, Augtyro, TPX 0005, CHINA 2024, APPROVALS 2024
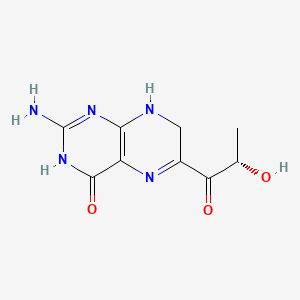
Sepiapterin
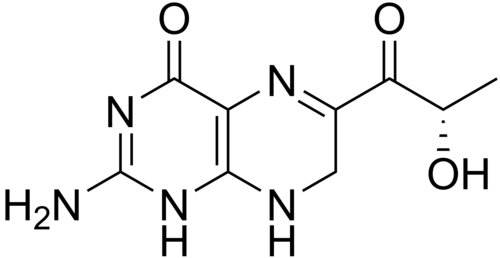
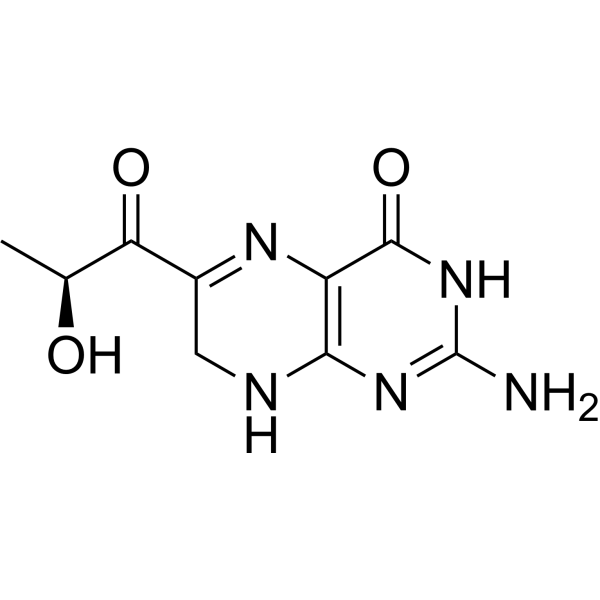
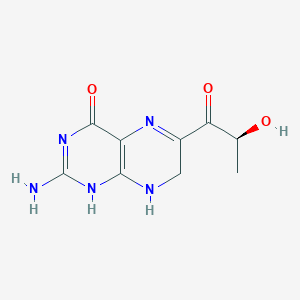
Sepiapterin
- Sepiapterine
- CNSA-001
- CJQ26KO7HP
| Molecular Weight | 237.22 |
|---|---|
| Formula | C9H11N5O3 |
2-amino-6-[(2S)-2-hydroxypropanoyl]-7,8-dihydro-3H-pteridin-4-one
(S)-2-Amino-6-(2-hydroxypropanoyl)-7,8-dihydropteridin-4(3H)-one
- 1-(2-amino-7,8-dihydro-4-hydroxy-6-pteridinyl)-2-hydroxy-1-Propanone
- 2-amino-6-[(2S)-2-hydroxypropanoyl]-7,8-dihydropteridin-4(1H)-one
- 2-amino-7,8-dihydro-6-[(2S)-2-hydroxy-1-oxopropyl]-4(1H)Pteridinone
- S(-)-2-Amino-7,8-dihydro-6-(2-hydroxy-1-oxopropyl)-4(1H)-pteridione
- S-(-)-2-Amino-7,8-dihydro-6-(2-hydroxy-1-oxopropyl)-4(1H)-pteridinone
- 2-AMINO-7,8-DIHYDRO-6-((2S)-2-HYDROXY-1-OXOPROPYL)-4(3H)-PTERIDINONE
- 4(1H)-Pteridinone, 2-amino-7,8-dihydro-6-(2-hydroxy-1-oxopropyl)-, (S)-
7/28/2025 fda approved, Sephience, To treat hyperphenylalaninemia in patients with sepiapterin-responsive phenylketonuria, in conjunction with a phenylalanine-restricted diet
Sepiapterin, sold under the brand name Sephience, is a medication used for the treatment of hyperphenylalaninemia.[2][3] Sepiapterin is a phenylalanine hydroxylase activator.[1]
The most common side effects are upper respiratory tract infection, headache, diarrhea, abdominal pain, hyperphenylalaninemia and discoloration of feces.[2]
Syn
https://patents.google.com/patent/WO2013168693A1/en
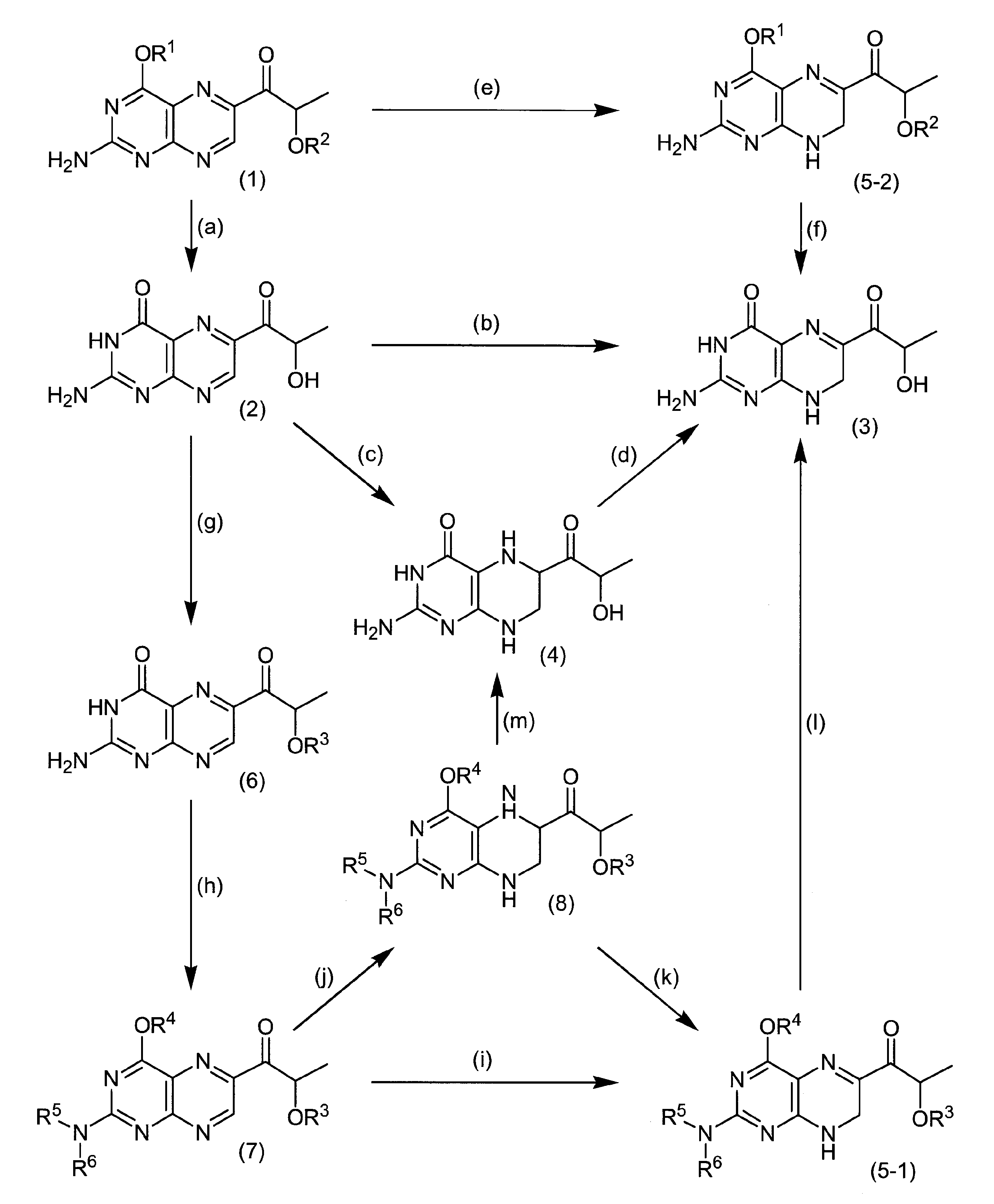
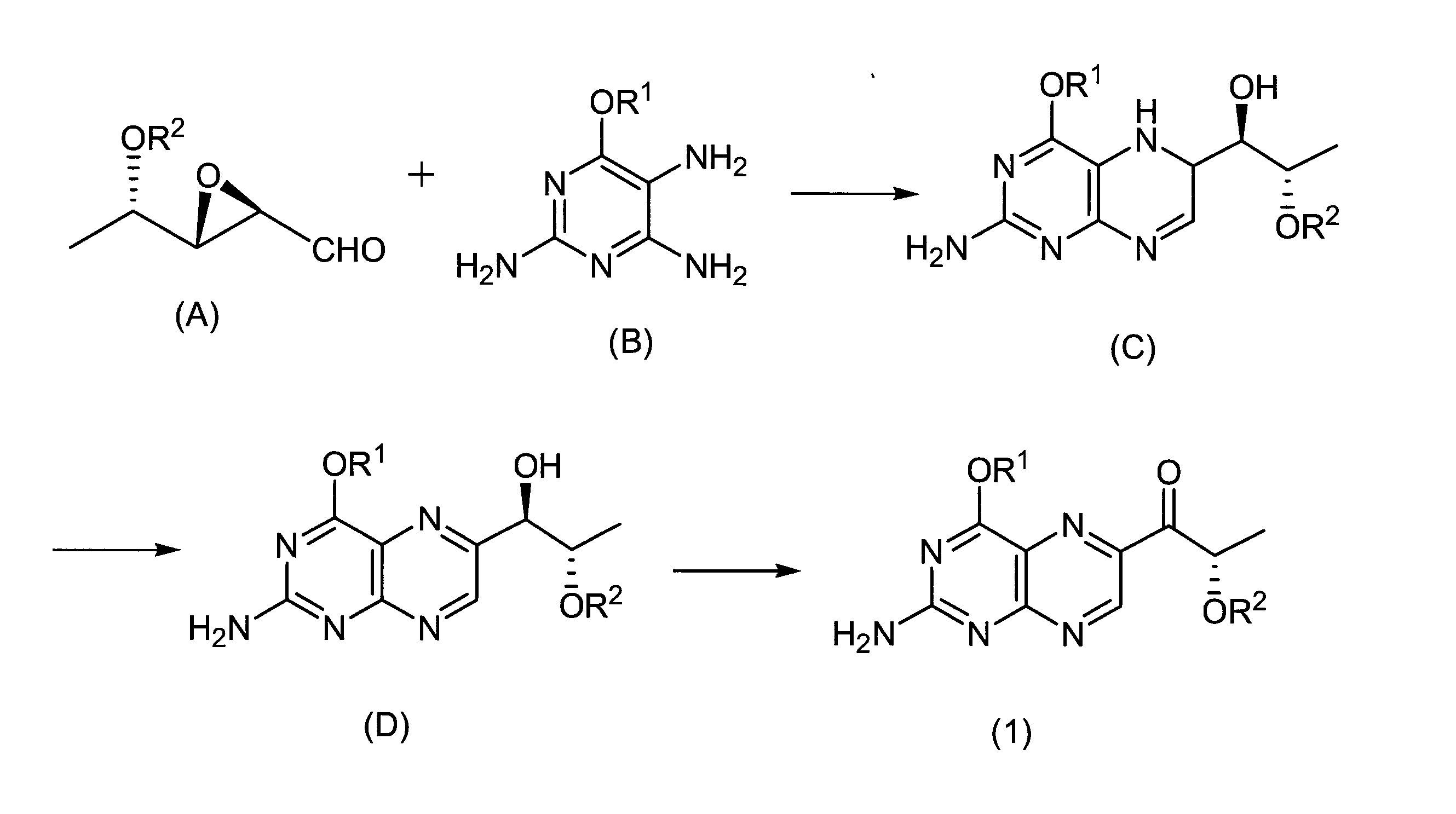
Sepiapterin is synthesized by a method of reacting 7,8-dihydropterin and α-keto-β-hydroxybutyric acid in the presence of zinc chloride (Non-patent Document 1), and a method of oxidizing BH4 in air for 6 days. (Non-Patent Document 2) is known.
As a method for synthesizing lactoylpterin, it is known that it can be obtained by oxidizing sepiapterin (Non-patent Documents 3 and 4).International Publication No. 2011/132435
However, the method described in Non-Patent Document 1 produces only a trace amount of sepiapterin and cannot be a stable supply method. Further, in the method of Non-Patent Document 2, very expensive BH4 is used as a raw material, and this cannot be a method that can be industrially stably supplied. Further, the method of Non-Patent Document 2 has a problem that the reaction time is long and many by-products such as biopterin in which BH4 is oxidized and deoxysepiapterin from which the β-position hydroxyl group of the side chain is eliminated are also generated. . In addition, the methods for synthesizing lactoylpterin of Non-Patent Documents 3 and 4 use sepiapterin, which is difficult to obtain industrially, as a raw material, and the yield is low, which cannot be a stable supply method.
Accordingly, an object of the present invention is to provide a novel production method capable of stably supplying sepiapterin, lactoylpterin and tetrahydrolactoylpterin, which have recently been found to be useful as pharmaceuticals.
Therefore, the present inventor has studied a method for synthesizing sepiapterin, lactoylpterin, and tetrahydrolactoylpterin using available raw materials. As a starting material, the compound of the following formula (1) or the compound of formula (7) is used. As a result, it was found that sepiapterin, lactoylpterin and tetrahydrolactoylpterin can be obtained in good yield, and these compounds can be stably supplied as a medicine for the first time, thereby completing the present invention.
Example 1
Synthesis of S-lactoylpterin (2)

1- (2-Amino-4-cyclohexyloxypteridin-6-yl) -2S-methoxymethoxypropan-1-one (compound (1)) 24.7 g (68.2 mmol) in methanol 50 mL, 3 mol / L hydrochloric acid 250 mL And stirred at 50 ° C. for 3 hours. The reaction solution was adjusted to pH = 7 with an aqueous sodium hydroxide solution, collected by filtration, and dried under reduced pressure to obtain 15.1 g (64.2 mmol, 94% yield) of S-lactoylpterin.
(S-lactoylpterin: (2))
1 H NMR (DMSO-d 6 ): δ / ppm = 1.32 (d, 3H, J = 6.8Hz), 5.16 (br, 1H), 5.32 (q, 1H, J = 6.8Hz), 9.09 (s, 1H )
Example 2 (Synthesis of 1- (2-amino-4-cyclohexyloxypteridin-6-yl) -2S-hydroxypropan-1-one)

1- (2-Amino-4-cyclohexyloxypteridin-6-yl) -2St-butyldimethylsilanoxypropan-1-one (compound (1)) (4.0 g, 9.27 mmol) was added to THF 40 mL, 70% 6.92 g (18.5 mmol) of tetrabutylammonium fluoride was added and stirred at 10 ° C. or lower for 2 hours. Water was added to the reaction mixture, and the mixture was extracted with chloroform. The organic layer was dehydrated and concentrated under reduced pressure. The crude product was purified by flash chromatography to give 2.09 g (6.59 mmol, 71% yield) of 1- (2-amino-4-cyclohexyloxypteridin-6-yl) -2S-hydroxypropan-1-one. Got.
1 H NMR (DMSO-d 6 ): δ / ppm = 1.38 (d, 3H, J = 6.6Hz), 1.37-1.79 (m, 8H), 1.98-1.99 (m, 2H), 5.20 (d, 1H, J = 6.3Hz), 5.34 (dq, 1H, J = 6.6Hz), 5.29-5.37 (m, 1H), 7.68 (br, 1H), 7.82 (br, 1H), 9.22 (s, 1H)
Example 3
Synthesis of S-lactoylpterin hydrochloride

To 500 mg (2.13 mmol) of S-lactoylpterin were added 1.25 mL of 6 mol / L hydrochloric acid and 10 mL of ethanol, and the mixture was stirred for 30 minutes. The crystals were collected by filtration and dried under reduced pressure, and 465 mg of S-lactoylpterin hydrochloride (1. 71 mmol, yield 80%).
(S-lactoylpterin hydrochloride)
1 H NMR (DMSO-d 6 ): δ / ppm = 1.34 (d, 3H, J = 6.9Hz), 3.91 (br, 3H), 5.34 (q, 1H, J = 6.9Hz), 9.12 (s, 1H )
Example 4 Synthesis of 2-amino-6- (2S-hydroxypropionyl) -7,8-dihydro-3H-pteridin-4-one (S-sepiapterin)

To 500 mg (2.13 mol) of S-lactoylpterin were added 125 mL of methanol, 2.08 mL (14.9 mmol) of triethylamine, 250 mg of 8.4% Pd / C (Ph 2 S) (containing 50% water), and an external temperature of 40 ° C. The hydrogenation reaction was carried out for 3 hours. After completion of the reaction, the reaction solution was stirred in air at room temperature for 1 hour, and then the catalyst was filtered off from the reaction solution and concentrated under reduced pressure. The crude product was separated and purified by flash chromatography and 296 mg (1.25 mmol) of S-sepiapterin. Yield 59%).
(S-sepiapterin: (3))
1 H NMR (DMSO-d 6 ): δ / ppm = 1.21 (d, 3H, J = 6.6Hz), 4.11 (s, 2H), 4.89 (d, 1H, J = 6.6Hz), 5, 10 (quin ., 1H, J = 6.6Hz), 6.81 (br-s, 2H), 7.51 (s, 1H), 10.26 (s, 1H)
Example 5
To 20 mg (0.085 mmol) of S-lactoylpterin were added 2 mL of saturated aqueous sodium hydrogen carbonate and 76 mg (0.44 mmol) of sodium dithionite, and the mixture was stirred at room temperature for 2 hours to give S-sepiapterin as a mixture.
Example 6
A reaction was carried out in the same manner as in Example 5 except that 20% (0.085 mmol) of S-lactoylpterin was used and the saturated aqueous sodium bicarbonate solution was changed to an aqueous sodium borate solution to give S-sepiapterin as a mixture.
Example 7
Synthesis of S-sepiapterin hydrochloride

To 620 mg (2.61 mmol) of S-sepiapterin were added 2.5 mL of 6 mol / L hydrochloric acid and 5.0 mL of ethanol, and the mixture was stirred at 0 ° C. for 30 minutes. The crystals were collected by filtration and dried under reduced pressure to obtain 650 mg (2.38 mmol, yield 91%) of S-sepiapterin hydrochloride.
(S-sepiapterin hydrochloride)
1 H NMR (DMSO-d 6 ): δ / ppm = 1.22 (d, 3H, J = 6.9Hz), 4.14 (s, 2H), 4.89 (d, 1H, J = 6.6Hz), 5.11 (q, 1H , J = 6.9Hz), 7.40 (br-s, 4H), 7.80 (br-s, 1H)
Example 8 (Synthesis of 2-amino-6- (2S-hydroxypropionyl) -5,6,7,8-tetrahydro-3H-pteridin-4-one (S-tetrahydrolactoylpterin) dihydrochloride)

Methanol 50 mL, 6 mol / L hydrochloric acid 5 mL, and borane pyridine complex 593 mg (6.38 mmol) were added to 1.00 g (4.25 mmol) of S-lactoylpterin, and the mixture was stirred at an external temperature of 0 ° C. for 1 hour. After completion of the reaction, 5 mL of acetone was added, concentrated under reduced pressure, azeotropically dehydrated with ethanol, ethanol was added, the crystals were filtered and dried under reduced pressure, and a mixture 1 of S-tetrahydrolactoylpterin dihydrochloride (4a) and (4b) 1 .12 g (3.59 mmol, 85% yield) was obtained.
(6S—S-tetrahydrolactoylpterin dihydrochloride: (4a))
1 H NMR (DMSO-d 6 ): δ / ppm = 1.24 (d, 3H, J = 6.9Hz), 3.45 (dd, 1H, J = 7.2, 13.5Hz), 3.87 (dd, 1H, J = 3.3, 13.5Hz), 4.34 (q, 1H, J = 6.9Hz), 4.53 (dd, 1H, J = 3.3, 7.2Hz), 7.03 (br-s, 4H), 7.67 (br-s, 1H)
(6R-S-tetrahydrolactoylpterin dihydrochloride: (4b))
1 H NMR (DMSO-d 6 ): δ / ppm = 1.24 (d, 3H, J = 6.9Hz), 3.45 (dd, 1H, J = 6.9, 13.5Hz), 3.91 (dd, 1H, J = 3.3, 13.5Hz), 4.31 (q, 1H, J = 6.6Hz), 4.55 (dd, 1H, J = 3.3, 6.9Hz), 7.12 (br-s, 3H), 7.71 (br-s, 2H)
Example 9
To 3.00 g (12.8 mmol) of S-lactoylpterin was added 150 mL of methanol, 15 mL of 6 mol / L hydrochloric acid, and 1.78 g (19.1 mmol) of borane pyridine complex, and the mixture was stirred at an external temperature of 0 ° C. for 1 hour. After completion of the reaction, 45 mL of concentrated hydrochloric acid was added, and the mixture was stirred overnight at the same temperature. The crystals were collected by filtration and dried under reduced pressure, and 1.63 g (5.2 mmol, yield) of 6S-S-tetrahydrolactoylpterin dihydrochloride (4a) 41%). The filtrate was concentrated under reduced pressure, azeotropically dehydrated with ethanol, ethanol was added, the crystals were collected by filtration and dried under reduced pressure, and 1.38 g (4.4 mmol) of 6R-S-tetrahydrolactoylpterin dihydrochloride (4b) was collected. Yield 35%). It was confirmed that the obtained compound was consistent with the spectrum data described in Example 8.
Example 10
Methanol 5 mL, 6 mol / L hydrochloric acid 0.5 mL, and borane pyridine complex 59 mg (0.64 mmol) were added to 100 mg (0.43 mmol) of S-lactoylpterin, and the mixture was stirred overnight at an external temperature of 0 ° C. The precipitated crystals were collected by filtration and dried under reduced pressure to obtain 46 mg (0.15 mmol, yield 35%) of 6S—S-tetrahydrolactoylpterin dihydrochloride (4a). It was confirmed that the obtained compound was consistent with the spectrum data described in Example 8.
Example 11
To 200 mg (0.85 mol) of S-lactoylpterin, 50 mL of methanol, 0.62 mL (5.95 mmol) of diethylamine and 100 mg of 8.4% Pd / C (Ph 2 S) (containing 50% water) were added, and the external temperature was 40 ° C. The hydrogenation reaction was carried out for 2.5 hours. After completion of the reaction, concentrated hydrochloric acid is added, the catalyst is filtered off, concentrated under reduced pressure, azeotropically dehydrated with ethanol, ethanol is added, the crystals are filtered and dried under reduced pressure, and S-tetrahydrolactoylpterin dihydrochloride (4a) and 122 mg (0.39 mmol, 46% yield) of a mixture of (4b) was obtained. It was confirmed that the obtained compound was consistent with the spectrum data described in Example 8.
Example 12 (Synthesis of 2-amino-6- (2S-hydroxypropionyl) -5,6,7,8-tetrahydro-3H-pteridin-4-one (S-tetrahydrolactoylpterin) ditoluenesulfonate)

To 100 mg (0.43 mmol) of S-lactoylpterin was added 5 mL of methanol, 0.5 mL of water, 566 mg (2.98 mmol) of p-toluenesulfonic acid monohydrate, and 59 mg (0.64 mmol) of borane pyridine complex. Stir at 0 ° C. for 1 hour. After completion of the reaction, 0.5 mL of acetone was added and concentrated under reduced pressure. After azeotropic dehydration with ethanol, acetone was added, the crystals were collected by filtration and dried under reduced pressure, and S-tetrahydrolactoylpterin ditoluenesulfonate 158 mg (0.27 mmol, Yield 63%) was obtained.
(S-tetrahydrolactoylpterin ditoluenesulfonate)
1 H NMR (DMSO-d 6 ): δ / ppm = 1.25 (d, 3H, J = 7.2Hz), 2.29 (S, 6H), 3.35 (dd, 1H, J = 7.5, 13.5Hz), 3.84 (dd , 1H, J = 3.0, 13.5Hz), 4.35 (q, 1H, J = 6.9Hz), 4.49 (dd, 1H, J = 3.0, 7.5Hz), 6.72 (br-s, 2H), 7.13 (d, 4H, J = 8.1Hz), 7.49 (d, 4H, J = 8.1Hz), 7.62 (br-s, 1H), 10.66 (br-s, 1H)
1 H NMR (DMSO-d 6 ): δ / ppm = 1.25 (d, 3H, J = 7.2Hz), 2.29 (S, 6H), 3.33 (dd, 1H, J = 7.5, 13.5Hz), 3.84 (dd , 1H, J = 3.0, 13.5Hz), 4.32 (q, 1H, J = 6.9Hz), 4.49 (dd, 1H, J = 3.0, 7.5Hz), 6.72 (br-s, 2H), 7.13 (d, 4H, J = 8.1Hz), 7.49 (d, 4H, J = 8.1Hz), 7.62 (br-s, 1H), 10.66 (br-s, 1H)
Example 13 Synthesis of 2-amino-6- (2S-hydroxypropionyl) -7,8-dihydro-3H-pteridin-4-one (S-sepiapterin)

6 mL of water and 6 mL of ethanol were added to 1.00 g (3.20 mmol) of S-tetrahydrolactoylpterin dihydrochloride, and 363 mg (3.20 mmol) of 30% aqueous hydrogen peroxide was added at an external temperature of −10 ° C. at the same temperature. Stir for 2 hours. A sodium sulfite aqueous solution was added to the reaction solution, and the crystals were collected by filtration and dried under reduced pressure to obtain 676 mg (2.85 mmol, yield 89%) of S-sepiapterin. It was confirmed that the obtained compound was consistent with the spectral data described in Example 4.
Example 14
46 mg of S-sepiapterin was prepared in the same manner as in Example 13 except that 100 mg (0.32 mmol) of S-tetrahydrolactoylpterin dihydrochloride was changed to 68 mg (0.32 mmol) of 36% peracetic acid with 30% hydrogen peroxide. 0.19 mmol, 61% yield). It was confirmed that the obtained compound was consistent with the spectral data described in Example 4.
Example 15
S-Sepia was prepared in the same manner as in Example 13 except that 100 mg (0.32 mmol) of S-tetrahydrolactoylpterin dihydrochloride was changed to 85 mg of m-CPBA (content 65%, 0.32 mmol) with 30% hydrogen peroxide. 35 mg (0.15 mmol, 46% yield) of pterin was obtained. It was confirmed that the obtained compound was consistent with the spectral data described in Example 4.
Example 16
20 mL of methanol and 0.89 mL (6.40 mmol) of triethylamine were added to 200 mg (0.64 mmol) of S-tetrahydrolactoylpterin dihydrochloride, and the mixture was stirred at room temperature for 1 hour in air. The reaction mixture was concentrated under reduced pressure, water was added, the crystals were collected by filtration and dried under reduced pressure to obtain 105 mg (0.44 mmol, yield 69%) of S-sepiapterin. It was confirmed that the obtained compound was consistent with the spectral data described in Example 4.
Example 17
Add 20 mL of methanol to 200 mg (0.64 mmol) of S-tetrahydrolactoylpterin dihydrochloride, neutralize with 0.16 mL (1.28 mmol) of 8 mol / L aqueous sodium hydroxide solution, and stir in air at room temperature for 1 hour did. The reaction mixture was concentrated under reduced pressure, water was added, the crystals were collected by filtration and dried under reduced pressure to obtain 87 mg (0.37 mmol, yield 58%) of S-sepiapterin. It was confirmed that the obtained compound was consistent with the spectral data described in Example 4.
Example 18 (Synthesis of 1- (2-amino-4-cyclohexyloxy-7,8-dihydropteridin-6-yl) -2S-methoxymethoxypropan-1-one)

1- (2-Amino-4-cyclohexyloxypteridin-6-yl) -2S-methoxymethoxypropan-1-one (1.00 g, 2.77 mmol), ethyl acetate 60 mL, 10% Pd—C 500 mg, potassium carbonate 3 .82 g (27.6 mmol) was added, and the hydrogenation reaction was performed at an external temperature of 50 ° C. for 3 hours. After the catalyst was filtered off, the reaction solution was concentrated under reduced pressure. The crude product was separated and purified by flash chromatography to obtain 257 mg (0.71 mmol) of 1- (2-amino-4-cyclohexyloxy-7,8-dihydropteridin-6-yl) -2S-methoxymethoxypropan-1-one. Yield 26%).
1 H NMR (CDCl 3 ): δ / ppm = 1.33-1.47 (m, 3H), 1.44 (d, 3H, J = 6.9Hz), 1.54-1.63 (m, 3H), 1.79 (m, 2H), 1.91 (m, 2H), 3.37 (s, 3H), 4.36 (d, 1H, J = 15.6), 4.43 (d, 1H, J = 15.6), 4.71 (d, 1H, J = 6.6Hz), 4.74 (d , 1H, J = 6.6Hz), 4.90 (br-s, 2H), 5.00 (br-s, 1H), 5.05-5.11 (m, 1H), 5.34 (q, 1H, J = 6.9Hz)
Example 19 (Synthesis of 1- (2-amino-4-cyclohexyloxy-7,8-dihydropteridin-6-yl) -2S-methoxyethoxymethoxypropan-1-one)

100 mg (0.56 mmol) of ascorbic acid was weighed and 2 mL of water was added. 1- (2-amino-4-cyclohexyloxypteridin-6-yl) -2S-methoxyethoxymethoxypropane- dissolved in 2 mL of methanol after neutralizing the pH of the solution with 1 mol / L aqueous sodium hydroxide solution 20 mg (0.054 mmol) of 1-one was added. To this, 80 mg (0.46 mmol) of Na 2 S 2 O 4 was added and stirred at room temperature for 1 hour. Water was added to the reaction solution, and the mixture was extracted with ethyl acetate. After dehydrating the organic phase, the solvent was concentrated under reduced pressure. Separation and purification by silica gel column chromatography gave 4.4 mg (0.011 mmol) of 1- (2-amino-4-cyclohexyloxy-7,8-dihydropteridin-6-yl) -2S-methoxyethoxymethoxypropan-1-one. Yield 20%).
1 H NMR (CDCl 3 ): δ / ppm = 1.13 (m, 1H), 1.44 (d, 3H, J = 6.8 Hz), 1.63 (m, 1H), 1.80 (m, 2H), 1.93 (m, 2H ), 2.06 (m, 2H), 3.37 (s, 3H), 3.52 (m, 2H), 3.70 (t, J = 4.6 Hz, 2H), 4.40 (m, 2H), 4.81 (m, 2H), 5.11 (tt, J = 3.9, 8.5 Hz, 1H), 5.35 (q, J = 6.8 Hz, 1H)
Example 20
Synthesis of S-sepiapterin (3)

To 10 mg of 1- (2-amino-4-cyclohexyloxy-7,8-dihydropteridin-6-yl) -2S-methoxymethoxypropan-1-one (compound (5-2)) was added 0.1 mL of concentrated hydrochloric acid. , Warmed up. The reaction solution was diluted with water, neutralized to pH 6-7 with an aqueous sodium hydroxide solution, and the precipitated crystals were filtered off. The filtrate was concentrated under reduced pressure to obtain S-sepiapterin as a mixture. The resulting compound was consistent with the spectral data described in Example 4.
Example 21
Synthesis of S-sepiapterin (3)

4.0 mg (9.8 μmol) of 1- (2-amino-4-cyclohexyloxy-7,8-dihydropteridin-6-yl) -2S-methoxyethoxymethoxypropan-1-one in 2 mL of methanol, ascorbic acid 3 0.02 mg was added, 2 mL of 3 mol / L hydrochloric acid was added thereto, and the mixture was stirred at 50 ° C. for 6 hours while shielding light. The solution was adjusted to pH 7 with 28% aqueous ammonia, washed with ethyl acetate, and purified by Florisil column chromatography to obtain 2.0 mg (8.4 μmol, yield 86%) of S-sepiapterin. As a result of HPLC measurement, the retention time and the UV waveform of the peak coincided with the standard S-sepiapterin.
Example 22 (Synthesis of 2-amino-6- [2S- (tert-butyldimethylsilanyloxy) -propionyl] -3H-pteridin-4-one (6))

To 3.00 g (12.8 mmol) of S-lactoylpterin were added 30 mL of DMF, 2.61 g (38.3 mmol) of imidazole and 3.84 g (25.5 mmol) of TBSCl, and the mixture was stirred for 1 hour under ice cooling. Water was added to the reaction mixture, and the crystals were collected by filtration and dried under reduced pressure to give 3.91 g of 2-amino-6- [2S- (tert-butyldimethylsilanyloxy) -propionyl] -3H-pteridin-4-one (6). (11.2 mmol, 88% yield) was obtained.
1 H NMR (DMSO-d 6 ): δ / ppm = 0.01 (s, 3H), 0.06 (s, 3H), 0.83 (s, 9H), 1.36 (d, 3H, J = 6.9Hz), 5.55 (q , 1H, J = 6.9Hz), 9.10 (s, 1H), 11.73 (br-s, 1H)
Example 23 (Synthesis of 2-amino-6- [2S- (triisopropylsilanyl) -propionyl] -3H-pteridin-4-one (6))
2-amino-6- [2S- (triisopropylsilanyl) -propionyl] -3H- was prepared in the same manner as in Example 22 except that TBSCl was changed to TIPSCl from 300 mg (1.28 mmol) of S-lactoylpterin. Pteridin-4-one (6) (339 mg, 0.87 mmol, yield 68%) was obtained.
1 H NMR (DMSO-d 6 ): δ / ppm = 0.89-1.15 (m, 21H), 1.40 (d, 3H, J = 6.9Hz), 5.71 (q, 1H, J = 6.9Hz), 9.13 (s , 1H), 11.74 (br-s, 1H)
Example 24 (Synthesis of 2-amino-6- [2S- (tert-butyldiphenylsilanyl) -propionyl] -3H-pteridin-4-one (6))

2-amino-6- [2S- (triisopropylsilanyl) -propionyl] -3H- was prepared in the same manner as in Example 22 except that TBSCl was changed to TBDPSCl from 300 mg (1.28 mmol) of S-lactoylpterin. 498 mg (1.05 mmol, yield 82%) of pteridin-4-one (6) was obtained.
1 H NMR (DMSO-d 6 ): δ / ppm = 1.03 (s, 9H), 1.38 (d, 3H, J = 6.9Hz), 5.71 (q, 1H, J = 6.9Hz), 7.23-7.33 (m , 3H), 7.37-7.45 (m, 3H), 7.50-7.59 (m, 2H), 7.61-7.71 (m, 2H), 8.96 (s, 1H), 11.67 (br-s, 1H)
Example 25 (1- [4-tert-butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) aminopteridin-6-yl] -2S-tert-butyldimethylsilanyloxypropan-1-one Synthesis of (7))

2-amino-6- [2S- (tert-butyldimethylsilanyloxy) -propionyl] -3H-pteridin-4-one 1.50 g (4.29 mmol) and 75 mL of ethyl acetate, di-tert-butyl dicarbonate 4 .68 g (21.4 mmol) and N, N-dimethylaminopyridine 52 mg (0.43 mmol) were added, and the mixture was heated to reflux for 1 hour. The reaction solution is washed with water, and the organic layer is dehydrated and concentrated under reduced pressure to give 2- (N, N-di-tert-butylcarbonyl) -amino-6- [2S- (tert-butyldimethylsilanyloxy) -propionyl]- 2.18 g (3.35 mmol, yield 78%) of 3H-pteridin-4-one (7) was obtained.
1 H NMR (DMSO-d 6 ): δ / ppm = 0.01 (s, 3H), 0.08 (s, 3H), 0.75 (s, 9H), 1.40 (d, 3H, J = 6.6Hz), 1.48 (s , 18H), 1.71 (s, 9H), 5.59 (q, 1H, J = 6.6Hz), 9.53 (s, 1H)
Example 26 (1- [4-tert-butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) aminopteridin-6-yl] -2S-tert-butoxycarbonyloxypropan-1-one (7 )

To 1.00 g (4.25 mmol) of S-lactoylpterin was added 50 mL of THF, 4.64 g (21.3 mmol) of di-tert-butyl dicarbonate, and 30 mg (0.25 mmol) of N, N-dimethylaminopyridine. Heated to reflux for hours. The reaction solution was concentrated under reduced pressure, and the crude product was separated and purified by flash chromatography, and 1- [4-tert-butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) aminopteridin-6-yl] There was obtained 0.30 g (0.47 mmol, yield 11%) of -2S-tert-butoxycarbonyloxypropan-1-one (7).
1 H NMR (CDCl 3 ): δ / ppm = 1.26 (s, 9H), 1.27 (d, 3H, J = 7.2Hz), 1.45 (s, 18H), 1.71 (s, 9H), 6.11 (q, 1H , J = 7.2Hz), 6.73 (s, 1H)
Example 27 (Synthesis of 1- [4-cyclohexyloxy-2- (N, N-di-tert-butoxycarbonyl) aminopteridin-6-yl] -2S-methoxymethoxypropan-1-one)

1- (2-Amino-4-cyclohexyloxypteridin-6-yl) -2S-methoxymethoxypropan-1-one (1.00 g, 2.76 mmol) was added with 20 mL of THF and 1.27 g of di-tert-butyl dicarbonate ( 5.82 mmol) and 3.4 mg (0.03 mmol) of N, N-dimethylaminopyridine were added, and the mixture was heated to reflux for 1 hour. The reaction solution was concentrated under reduced pressure to give 1- [4-cyclohexyloxy-2- (N, N-di-tert-butylcarbonyl) aminopteridin-6-yl] -2S-methoxymethoxypropan-1-one (7) 1 Obtained .55 g (2.76 mmol, 100% yield).
1 H NMR (DMSO-d 6 ): δ / ppm = 1.45-1.88 (m, 8H), 1.53 (s, 18H), 1.58 (d, 3H, J = 6.9Hz), 2.10-2.14 (m, 2H) , 3.38 (s, 3H), 4.78 (d, 1H, J = 6.9Hz), 4.84 (d, 1H, J = 6.9Hz), 5.36-5.45 (m, 1H), 5.55 (q, 1H, J = 6.9 Hz), 9.65 (s, 1H)
Example 28 (1- [4-tert-butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) amino-7,8-dihydropteridin-6-yl] -2S-tert-butyldimethylsilanyl Synthesis of oxypropan-1-one)

1- [4-tert-Butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) aminopteridin-6-yl] -2S-tert-butyldimethylsilanyloxypropan-1-one 1.31 g ( 2.02 mmol) was added with 130 mL of ethyl acetate, 655 mg of 10% Pd—C and 2.78 g (20.1 mmol) of potassium carbonate, and the hydrogenation reaction was carried out for 1 hour at an external temperature of 50 ° C. under normal pressure (H 2 balloon). . After the catalyst was filtered off, the reaction solution was stirred in air at room temperature overnight, and the reaction solution was concentrated under reduced pressure. The crude product was separated and purified by flash chromatography, and 1- [4-tert-butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) amino-7,8-dihydropteridin-6-yl]- 684 mg (1.05 mmol, 66% yield) of 2S-tert-butyldimethylsilanyloxypropan-1-one was obtained.
1 H NMR (DMSO-d 6 ): δ / ppm = 0.01 (s, 3H), 0.07 (s, 3H), 0.82 (s, 9H), 1.24 (d, 3H, J = 6.6Hz), 1.42 (s , 18H), 1.53 (s, 9H), 4.23 (d, 1H, J = 16.5Hz), 4.32 (d, 1H, J = 16.5Hz), 5.39 (q, 1H, J = 6.6Hz), 7.92 (s , 1H)
Example 29 (Synthesis of 2-amino-6S- (2S-hydroxypropionyl) -5,6,7,8-tetrahydro-3H-pteridin-4-one (6S-S-tetrahydrolactoylpterin) dihydrochloride)

1- [4-tert-butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) aminopteridin-6-yl] -2S-tert-butyldimethylsilanyloxypropan-1-one 4.92 g ( 7.57 mmol) was added 250 mL of ethyl acetate, 2.46 g of 10% Pd—C, and 10.5 g (76.0 mmol) of K 2 CO 3 , and the hydrogenation reaction was performed at an external temperature of 50 ° C. under normal pressure (H 2 balloon). It went for 1 hour. After the catalyst was filtered off, the reaction solution was concentrated under reduced pressure, 49 mL of concentrated hydrochloric acid was added, and the mixture was concentrated under reduced pressure. Ethanol was added to the concentrate, and the crystals were collected by filtration and dried under reduced pressure to obtain 1.79 g (5.73 mmol, yield 76%) of 6S—S-tetrahydrolactoylpterin dihydrochloride (4a). The compound obtained agreed with the spectral data described in Example 8.
Example 30
1- [4-tert-Butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) aminopteridin-6-yl] -2S-tert-butyldimethylsilanyloxypropan-1-one 500 mg (0. 164 mg (0.53 mmol, 68% yield) of 6S—S-tetrahydrolactoylpterin dihydrochloride (4a) was obtained in the same manner as in Example 29 except that the amount of 10% Pd / C was changed to 100 mg from 77 mmol). Obtained. It was confirmed that the obtained compound was consistent with the spectrum data described in Example 8.
Example 31 (1- [4-tert-butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) amino-5,6,7,8-tetrahydropteridin-6-yl] -2S-tert- Synthesis of butyldimethylsilanyloxypropan-1-one)

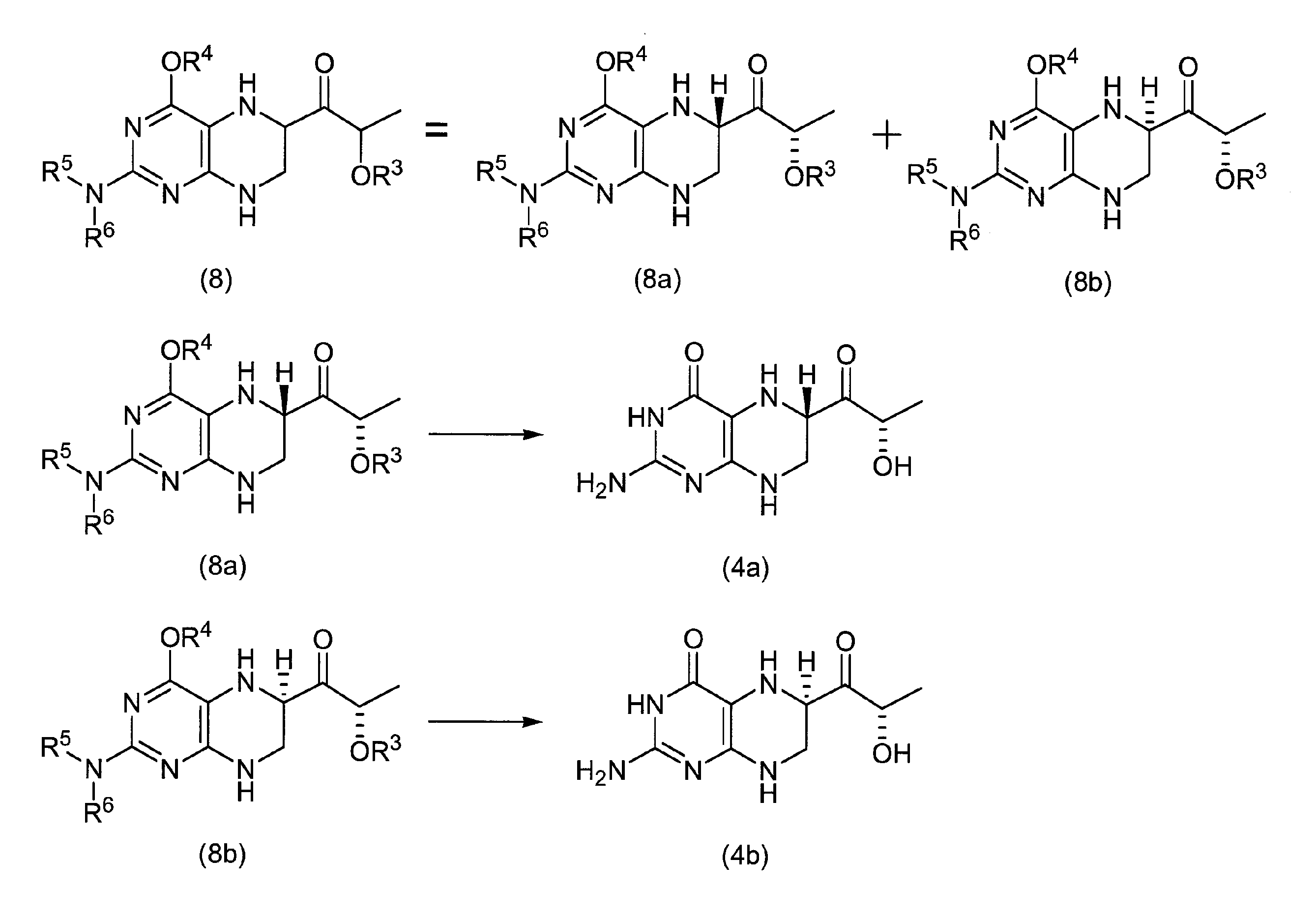
1- [4-tert-butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) aminopteridin-6-yl] -2S-tert-butyldimethylsilanyloxypropan-1-one 100 mg (0. 15 mmol), 10 mL of ethyl acetate, 20 mg of 10% Pd—C and 156 mg (1.54 mmol) of triethylamine were added, and the hydrogenation reaction was carried out for 1 hour at an external temperature of 50 ° C. under normal pressure (H 2 balloon). After the catalyst was filtered off, the reaction solution was concentrated under reduced pressure, and the crude product was separated and purified by flash chromatography to give 1- [4-tert-butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) amino. -5,6,7,8-tetrahydropteridin-6S-yl] -2S-tert-butyldimethylsilanyloxypropan-1-one (8a) 30 mg (0.045 mmol, 30% yield) and 1- [4 -Tert-butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) amino-5,6,7,8-tetrahydropteridin-6R-yl] -2S-tert-butyldimethylsilanyloxypropane-1 -30 mg (0.045 mmol, 30% yield) of ONE (8b) was obtained.
(8a)
1 H NMR (DMSO-d 6 ): δ / ppm = 0.08 (s, 3H), 0.09 (s, 3H), 0.89 (s, 9H), 1.21 (d, 3H, J = 6.6Hz), 1.37 (s , 18H), 1.49 (s, 9H), 3.56-3.67 (m, 2H), 4.39 (m, 1H), 4.42 (q, 1H, J = 6.6Hz), 4.79 (s, 1H), 7.00 (s, 1H)
(8b)
1 H NMR (DMSO-d 6 ): δ / ppm = 0.08 (s, 3H), 0.09 (s, 3H), 0.89 (s, 9H), 1.23 (d, 3H, J = 6.6Hz), 1.37 (s , 18H), 1.49 (s, 9H), 3.40-3.53 (m, 2H), 4.35 (m, 1H), 4.44 (q, 1H, J = 6.6Hz), 4.93 (s, 1H), 7.09 (s, 1H)
Example 32 (1- [4-cyclohexyloxy-2- (N, N-di-tert-butoxycarbonyl) amino-5,6,7,8-tetrahydropteridin-6-yl] -2S-methoxymethoxypropane- Synthesis of 1-one)

From Example 1 from 200 mg (0.36 mmol) of 1- [4-cyclohexyloxy-2- (N, N-di-tert-butoxycarbonyl) aminopteridin-6-yl] -2S-methoxymethoxypropan-1-one In a similar manner, 1- [4-cyclohexyloxy-2- (N, N-di-tert-butoxycarbonyl) amino-5,6,7,8-tetrahydropteridin-6-yl] -2S-methoxymethoxypropane- 76 mg (0.13 mmol, yield = 38%) of 1-one was obtained.
1 H NMR (DMSO-d 6 ): δ / ppm = 1.21 (d, 3H, J = 6.9Hz), 1.32-1.37 (m, 3H), 1.38 (s, 18H), 1.43-1.51 (m, 3H) , 1.73 (m, 2H), 1.89-1.91 (m, 2H), 3.27 (s, 3H), 3.51-3.56 (m, 2H), 4.33-4.35 (m, 1H), 4.41 (q, 1H, J = 6.9Hz), 4.59 (d, 1H, J = 6.9Hz), 4.67 (d, 1H, J = 6.9Hz), 4.86-4.89 (m, 1H), 4.95 (d, 1H, J = 2.7Hz), 7.08 (s, 1H)
1 H NMR (DMSO-d 6 ): δ / ppm = 1.24 (d, 3H, J = 6.9Hz), 1.32-1.37 (m, 3H), 1.38 (s, 18H), 1.43-1.51 (m, 3H) , 1.73 (m, 2H), 1.89-1.91 (m, 2H), 3.32 (s, 3H), 3.51-3.56 (m, 2H), 4.33-4.35 (m, 1H), 4.39 (q, 1H, J = 6.9Hz), 4.59 (d, 1H, J = 6.9Hz), 4.67 (d, 1H, J = 6.9Hz), 4.86-4.89 (m, 1H), 5.01 (d, 1H, J = 2.4Hz), 7.08 (s, 1H)
Example 33 Synthesis of 2-amino-6- (2S-hydroxypropionyl) -7,8-dihydro-3H-pteridin-4-one (S-sepiapterin)

1- [4-tert-Butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) amino-7,8-dihydropteridin-6-yl] -2S-tert-butyldimethylsilanyloxypropane-1 -To 300 mg (0.46 mmol) of ON was added 3 mL of acetonitrile and 6 mL of 2 mol / L hydrochloric acid, and the mixture was stirred at an external temperature of 40 ° C for 3 hours. The reaction solution was adjusted to pH = 7 with an aqueous sodium hydroxide solution, and the crystals were collected by filtration and dried under reduced pressure to obtain 96 mg (0.40 mmol, yield 88%) of S-sepiapterin. It was confirmed that the obtained compound was consistent with the spectral data described in Example 4.
Example 34 (Synthesis of 2-amino-6R- (2S-hydroxypropionyl) -5,6,7,8-tetrahydro-3H-pteridin-4-one (6R-S-tetrahydrolactoylpterin) dihydrochloride)

1- [4-tert-Butoxycarbonyl-2- (N, N-di-tert-butoxycarbonyl) amino-5,6,7,8-tetrahydropteridin-6R-yl] -2S-tert-butyldimethylsilanyl To 393 mg (0.60 mmol) of oxypropan-1-one (8b) was added 10 mL of concentrated hydrochloric acid, and the mixture was concentrated under reduced pressure. Ethanol was added to the concentrate, and the crystals were collected by filtration and dried under reduced pressure to obtain 106 mg (0.34 mmol, yield 56%) of 6R—S-tetrahydrolactoylpterin dihydrochloride (4b). The compound obtained agreed with the spectral data described in Example 8.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Medical uses
Sepiapterin is indicated for the treatment of hyperphenylalaninemia in people with phenylketonuria.[1][2]
Side effects
The most common side effects are upper respiratory tract infection, headache, diarrhea, abdominal pain, hyperphenylalaninemia and discoloration of feces.[2]
Society and culture
Legal status
In April 2025, the Committee for Medicinal Products for Human Use of the European Medicines Agency adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Sephience, intended for the treatment of hyperphenylalaninemia in adults and children with phenylketonuria.[2] The applicant for this medicinal product is PTC Therapeutics International Limited.[2] Sepiapterin was authorized for medical use in the European Union in June 2025.[2][3]
Sepiapterin was approved for medical use in the United States in July 2025.[1]
Research
Deficiency of tetrahydrobiopterin can cause toxic buildup of phenylalanine (phenylketonuria) as well as deficiencies of dopamine, norepinephrine, and epinephrine, leading to dystonia and other neurological illnesses. This has led to clinical study of sepiapterin in humans to treat tetrahydrobiopterin deficiency.[4]
Since atherosclerosis and other circulatory diseases associated with diabetes are also associated with tetrahydrobiopterin deficiency, animal studies of the value of sepiaterin in these vascular diseases have been done. These studies show that relaxation of the blood vessels studied was impaired after animals were given sepiapterin, even though their levels of tetrahydrobiopterin were replenished.[5]
References
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/219666s000lbl.pdf
- “Sephience EPAR”. European Medicines Agency (EMA). 25 April 2025. Retrieved 2 May 2025. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- “Sephience Product information”. Union Register of medicinal products. 25 June 2025. Retrieved 27 June 2025.
- Smith N, Longo N, Levert K, Hyland K, Blau N (April 2019). “Phase I clinical evaluation of CNSA-001 (sepiapterin), a novel pharmacological treatment for phenylketonuria and tetrahydrobiopterin deficiencies, in healthy volunteers”. Molecular Genetics and Metabolism. 126 (4): 406–412. doi:10.1016/j.ymgme.2019.02.001. ISSN 1096-7192. PMID 30922814. S2CID 85564348.
- Vasquez-Vivar J, Duquiane D, Whitsett J, Kalyanaraman B, Rajagopalan S (October 2002). “Altered Tetrahydrobiopterin Metabolism in Atherosclerosis”. Arteriosclerosis, Thrombosis, and Vascular Biology. 22 (10): 1655–1661. doi:10.1161/01.ATV.0000029122.79665.D9. PMID 12377745.
| Names | |
|---|---|
| IUPAC name2-amino-6-[(2S)-2-hydroxypropanoyl]-7,8-dihydro-1H-pteridin-4-one | |
| Other namesSephience | |
| Identifiers | |
| CAS Number | 17094-01-8 |
| 3D model (JSmol) | Interactive image |
| ChEMBL | ChEMBL1255653 |
| ChemSpider | 58746 |
| KEGG | C00835 |
| PubChem CID | 65253 |
| UNII | CJQ26KO7HP |
| InChI | |
| SMILES | |
| Properties | |
| Chemical formula | C9H11N5O3 |
| Molar mass | 237.22 g/mol |
| Pharmacology | |
| ATC code | None |
| Routes of administration | By mouth |
| Legal status | US: ℞-only[1]EU: Rx-only[2][3] |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
- [1]. Pannirselvam M, et al. Chronic oral supplementation with sepiapterin prevents endothelial dysfunction and oxidative stress in small mesenteric arteries from diabetic (db/db) mice. Br J Pharmacol. 2003;140(4):701‐706. [Content Brief][2]. Cho YR, et al. Sepiapterin inhibits cell proliferation and migration of ovarian cancer cells via down-regulation of p70S6K-dependent VEGFR-2 expression. Oncol Rep. 2011;26(4):861‐867. [Content Brief]
//////////Sepiapterin, approvals 2025, fda 2025, Sephience, Sepiapterine, CNSA 001, CJQ26KO7HP, PTC 923, WHO 11848,
Sebetralstat
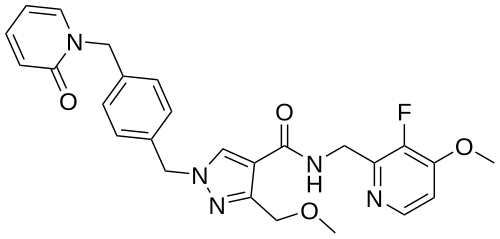
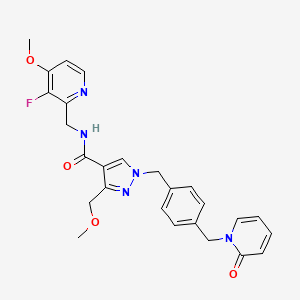
Sebetralstat, KVD 900
CAS 1933514-13-6
491.5 g/mol
FDA 7/3/2025, Ekterly, To treat acute attacks of hereditary angioedema
N-[(3-fluoro-4-methoxypyridin-2-yl)methyl]-3-(methoxymethyl)-1-[[4-[(2-oxopyridin-1-yl)methyl]phenyl]methyl]pyrazole-4-carboxamide
Sebetralstat, sold under the brand name Ekterly, is a medication used for the treatment of hereditary angioedema.[1] Sebetralstat is a plasma kallikrein inhibitor.[1]
Sebetralstat was approved for medical use in the United States in July 2025.[1][2]
SYN
https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c00921
aReagents and conditions: (a) 2-Hydroxypyridine (1.2 equiv), K2CO3 (3.0 equiv), acetone, 50 °C, 18 h, 78%; (b) methanesulfonyl chloride (1.3 equiv), Et3N, (1.4 equiv), dichloromethane, rt, 18h, 93%; (c) methyl 3-(methoxymethyl)-1H-pyrazole-4-carboxylate (0.83 equiv), K2CO3 (2.0 equiv), DMF, 60 °C, 18 h, 54%; (d) NaOH (3.0 equiv), THF-MeOH-H2O, rt, 18 h, 34%; (e) 22a (1.0 equiv), C-(3-fluoro-4-methoxy-pyridin-2-yl)-methylamine (1.0 equiv), HATU (1.1 equiv), Et3N (6.0 equiv), dichloromethane, rt, 4 h, 64%.
Synthesis of Sebetralstat
1-(4-Hydroxymethyl-benzyl)-1H-pyridin-2-one (19)
4-(Chloromethyl)benzyl alcohol 18 (5.0 g, 31.9 mmol) was added to a solution of potassium carbonate (13.2 g, 96 mmol) and 2-hydroxypyridine (3.6 g, 38.3 mmol) in acetone (250 mL). The reaction mixture was heated at 50 °C for 18 h and then concentrated in vacuo. The residue was partitioned between dichloromethane (300 mL) and water (300 mL). The organic layer was separated, and the aqueous layer was extracted with dichloromethane (2 × 300 mL). The combined organic layers were washed with brine (300 mL), dried over magnesium sulfate, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica (0–10% methanol in dichloromethane) to afford 19 (5.4 g, 25.1 mmol, 78% yield) as a white solid. MS (ESI) m/z 216.0 (M + H)+. 1H NMR (400 MHz, DMSO-d6) δ 7.76 (dd, J = 6.8, 2.1 Hz, 1H), 7.41 (ddd, J = 9.0, 6.6, 2.1 Hz, 1H), 7.34–7.21 (m, 4H), 6.41 (dd, J = 9.1, 1.3 Hz, 1H), 6.22 (td, J = 6.7, 1.4 Hz, 1H), 5.15 (t, J = 5.7 Hz, 1H), 5.07 (s, 2H), 4.46 (d, J = 5.7 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 161.4, 141.9, 140.0, 139.0, 135.7, 127.5, 126.6, 119.8, 105.4, 62.6, 50.8.
1-(4-Chloromethyl-benzyl)-1H-pyridin-2-one (20)
A reaction flask containing 1-(4-hydroxymethyl-benzyl)-1H-pyridin-2-one (19) (8.45 g, 39.3 mmol), dry dichloromethane (80 mL), and triethylamine (7.66 mL, 55.0 mmol) was cooled in an ice–water bath. Methanesulfonyl chloride (3.95 mL, 51.0 mmol) was added to the reaction at 0 °C, and ice–water bath cooling continued. After 15 min, the ice–water bath was removed and stirring continued at room temperature overnight. The reaction mixture was partitioned between dichloromethane (100 mL) and saturated aqueous ammonium chloride solution (100 mL). The aqueous layer was extracted with further dichloromethane (2 × 50 mL), and the combined organic layers were washed with brine (50 mL), dried over sodium sulfate, filtered, and concentrated to afford 20 (8.65 g, 36.6 mmol, 93% yield) as a pale yellow solid. MS (ESI) m/z 234.1 (M + H)+. 1H NMR (400 MHz, DMSO-d6) δ 7.79 (ddd, J = 6.8, 2.1, 0.7 Hz, 1H), 7.49–7.39 (m, 1H), 7.40 (d, J = 7.8 Hz, 2H), 7.28 (d, J = 8.4 Hz, 2H), 6.42 (ddd, J = 9.2, 1.3, 0.7 Hz, 1H), 6.24 (td, J = 6.7, 1.4 Hz, 1H), 5.09 (s, 2H), 4.73 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 161.4, 140.1, 139.1, 137.6, 136.9, 129.0, 127.9, 119.9, 105.5, 50.8, 45.8.
Methyl 3-(Methoxymethyl)-1-(4-((2-oxopyridin-1(2H)-yl)methyl)benzyl)-1H-pyrazole-4-carboxylate (21a) and Methyl 5-(Methoxymethyl)-1-(4-((2-oxopyridin-1(2H)-yl)methyl)benzyl)-1H-pyrazole-4-carboxylate (21b)
Methyl 3-(methoxymethyl)-1H-pyrazole-4-carboxylate (2.11 g, 11.77 mmol; CAS No. 318496-66-1) was added to a solution of potassium carbonate (3.25 g, 23.54 mmol) and 1-(4-chloromethyl-benzyl)-1H-pyridin-2-one 20 (3.30 g, 14.12 mmol) in N,N-dimethylformamide (5 mL) and heated at 70 °C for 3 h. The reaction mixture was diluted with ethyl acetate (50 mL) and washed with brine (2 × 100 mL), and the organic layer was dried over magnesium sulfate, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography (120 g column, 0–100% (10% ethanol in ethyl acetate) in isohexanes to afford two regioisomers: 21a (2.03 g, 5.47 mmol, 47% yield) as an off-white solid and 21b (350 mg, 0.92 mmol, 8% yield). 21a MS (ESI) m/z 368.1 (M + H)+. 1H NMR (400 MHz, DMSO-d6) δ 8.42 (s, 1H), 7.76 (dd, J = 6.8, 2.2 Hz, 1H), 7.41 (ddd, J = 8.9, 6.5, 2.1 Hz, 1H), 7.25 (d, J = 1.2 Hz, 4H), 6.40 (dt, J = 9.1, 1.0 Hz, 1H), 6.22 (td, J = 6.7, 1.4 Hz, 1H), 5.30 (s, 2H), 5.07 (s, 2H), 4.49 (s, 2H), 3.72 (s, 3H), 3.23 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 163.2, 161.8, 150.5, 140.6, 139.6, 137.6, 136.3, 135.6, 128.5, 128.4, 120.3, 111.8, 106.0, 66.0, 58.0, 55.1, 51.5, 51.2. 21b MS (ESI) m/z 368.1 (M + H)+. 1H NMR (400 MHz, DMSO-d6) δ 7.88 (s, 1H), 7.76 (dd, J = 6.8, 2.1 Hz, 1H), 7.41 (ddd, J = 8.9, 6.6, 2.1 Hz, 1H), 7.28–7.21 (m, 2H), 7.17 (d, J = 8.2 Hz, 2H), 6.43–6.36 (m, 1H), 6.22 (td, J = 6.7, 1.4 Hz, 1H), 5.35 (s, 2H), 5.06 (s, 2H), 4.78 (s, 2H), 3.75 (s, 3H), 3.25 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 163.4, 161.8, 142.4, 140.9, 140.5, 139.6, 137.4, 136.2, 128.3, 120.3, 112.8, 106.0, 61.7, 58.2, 53.0, 51.7, 51.2.
3-(Methoxymethyl)-1-(4-((2-oxopyridin-1(2H)-yl)methyl)benzyl)-1H-pyrazole-4-carboxylic acid (22a)
To methyl 3-(methoxymethyl)-1-(4-((2-oxopyridin-1(2H)-yl)methyl)benzyl)-1H-pyrazole-4-carboxylate 21a (3.77 g, 10.26 mmol) in tetrahydrofuran (5 mL) and methanol (5 mL) was added 2 M aqueous sodium hydroxide solution (15.39 mL, 30.80 mmol), and the reaction mixture was stirred at room temperature overnight. The reaction was acidified with 1 M aqueous HCl solution (50 mL) and extracted with ethyl acetate (50 mL). The organic layer was washed with brine (50 mL), dried over magnesium sulfate, filtered, and concentrated in vacuo to afford 22a (1.22 g, 3.45 mmol, 34% yield) as a white solid. MS (ESI) m/z 354.2 (M + H)+. 1H NMR (400 MHz, DMSO-d6) δ 12.32 (s, 1H), 8.32 (s, 1H), 7.76 (ddd, J = 6.8, 2.1, 0.7 Hz, 1H), 7.41 (ddd, J = 8.9, 6.6, 2.1 Hz, 1H), 7.30–7.20 (m, 4H), 6.40 (ddd, J = 9.1, 1.4, 0.7 Hz, 1H), 6.22 (td, J = 6.7, 1.4 Hz, 1H), 5.29 (s, 2H), 5.07 (s, 2H), 4.50 (s, 2H), 3.22 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 164.3, 161.8, 150.5, 140.6, 139.6, 137.6, 136.4, 135.6, 128.5, 128.4, 120.3, 113.0, 106.0, 66.0, 58.0, 55.1, 51.2.
3-Methoxymethyl-1-[4-(2-oxo-2H-pyridin-1-ylmethyl)-benzyl]-1H-pyrazole-4-carboxylic Acid (3-Fluoro-4-methoxy-pyridin-2-ylmethyl)-amide (14w)
3-(Methoxymethyl)-1-(4-((2-oxopyridin-1(2H)-yl)methyl)benzyl)-1H-pyrazole-4-carboxylic acid 22a (75 mg, 0.212 mmol), C-(3-fluoro-4-methoxy-pyridin-2-yl)-methylamine (49 mg, 0.212 mmol; CAS No. 1256812-75-5), and HATU (89 mg, 0.233 mmol) were suspended in anhydrous dichloromethane (3 mL) to which triethylamine (177 μL, 1.270 mmol) was added, sonicated, and then left to stir at room temperature for 4 h. The solvent was removed under reduced pressure, and the resulting residue was quenched with saturated aqueous ammonium chloride solution (5 mL). An off-white solid resulted, which was sonicated, filtered under reduced pressure, washed with water, and dried in a vacuum oven at 40 °C overnight. The residue was purified by chromatography eluting with 1% NH3 in MeOH/dichloromethane to afford 14w as a white solid (67 mg, 64% yield). MS (ESI) m/z 492.0 (M + H)+. 1H NMR (400 MHz, DMSO-d6) δ: 8.42 (t, J = 5.4 Hz, 1H), 8.29–8.21 (m, 2H), 7.75 (ddd, J = 0.7, 2.1, 6.8 Hz, 1H), 7.41 (ddd, J = 2.1, 6.6, 8.9 Hz,1H), 7.28–7.17 (m, 5H), 6.39 (ddd, J = 0.7, 1.4, 9.2 Hz, 1H), 6.22 (td, J = 1.4, 6.7 Hz, 1H), 5.28 (s, 2H), 5.07 (s, 2H), 4.57–4.46 (m, 4H), 3.92 (s, 3H), 3.25 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 161.9, 161.3, 153.0 (JC–F = 8.7 Hz), 147.5, 146.8 (JC–F = 253.5 Hz), 146.0 (JC–F = 7.2 Hz), 145.2 (JC–F = 11.6 Hz), 140.1, 139.1, 137.1, 136.0, 133.2, 128.1, 127.9, 119.9, 116.3, 108.7, 105.5, 66.3, 57.5, 56.4, 54.6, 50.7, 38.3.
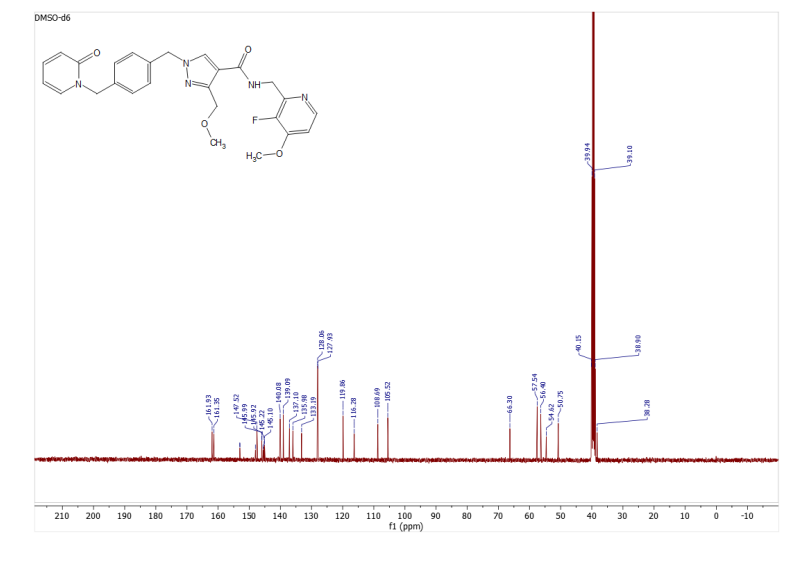
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US232820883&_cid=P22-MCYEU3-92408-1
Example 41
3-Fluoro-4-methoxy-pyridine-2-carbonitrile
(3-Fluoro-4-methoxy-pyridin-2-ylmethyl)-carbamic acid tert-butyl ester
C-(3-Fluoro-4-methoxy-pyridin-2-yl)-methylamine hydrochloride salt
3-Methoxymethyl-1-[4-(2-oxo-2H-pyridin-1-ylmethyl)-benzyl]-1H-pyrazole-4-carboxylic acid (3-fluoro-4-methoxy-pyridin-2-ylmethyl)-amide
Medical uses
Sebetralstat is indicated for the treatment of acute attacks of hereditary angioedema.[1]
Pharmacology
Sebetralstat is a plasma kallikrein inhibitor that contains the unusual 2-pyridone heterocycle.[3]
Society and culture
Legal status
Sebetralstat was approved for medical use in the United States in July 2025.[1] The US Food and Drug Administration granted the application for sebetralstat orphan drug designation.[4]
Names
Sebetralstat is the international nonproprietary name.[5]
Sebetralstat is sold under the brand name Ekterly.[1]
References
- ^ Jump up to:a b c d e f g “Ekterly- sebetralstat tablet”. DailyMed. 7 July 2025. Retrieved 9 July 2025.
- ^ “KalVista Pharmaceuticals Announces FDA Approval of Ekterly (sebetralstat), First and Only Oral On-demand Treatment for Hereditary Angioedema” (Press release). Kalvista. 7 July 2025. Retrieved 9 July 2025 – via Business Wire.
- ^ Davie RL, Edwards HJ, Evans DM, Hodgson ST, Stocks MJ, Smith AJ, et al. (October 2022). “Sebetralstat (KVD900): A Potent and Selective Small Molecule Plasma Kallikrein Inhibitor Featuring a Novel P1 Group as a Potential Oral On-Demand Treatment for Hereditary Angioedema”. Journal of Medicinal Chemistry. 65 (20): 13629–13644. doi:10.1021/acs.jmedchem.2c00921. PMC 9620001. PMID 36251573.
- ^ “Sebetralstat Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). Retrieved 9 July 2025.
- ^ World Health Organization (2022). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 87”. WHO Drug Information. 36 (1). hdl:10665/352794.
External links
- “Sebetralstat (Code – C184930)”. EVS Explore.
- Clinical trial number NCT05259917 for “A Phase III, Crossover Trial Evaluating the Efficacy and Safety of KVD900 (Sebetralstat) for On-Demand Treatment of Angioedema Attacks in Adolescent and Adult Patients With Hereditary Angioedema (HAE)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Ekterly |
| Other names | KVD-900, KVD900 |
| License data | US DailyMed: Sebetralstat |
| Routes of administration | By mouth |
| ATC code | B06AC08 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1933514-13-6 |
| PubChem CID | 121365142 |
| IUPHAR/BPS | 11947 |
| DrugBank | DB18305 |
| ChemSpider | 115006749 |
| UNII | O5ZD2TU2B7 |
| KEGG | D12396 |
| ChEMBL | ChEMBL5095248 |
| Chemical and physical data | |
| Formula | C26H26FN5O4 |
| Molar mass | 491.523 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
//////////Sebetralstat, FDA 2025, APPROVALS 2025, Ekterly, angioedema, KVD 900, O5ZD2TU2B7
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
Sunvozertinib
Sunvozertinib
CAS 2370013-12-8
DZD9008, 584.1 g/mol, C29H35ClFN7O3, A-1801, L1Q2K5JYO8
N-[5-[[4-[5-chloro-4-fluoro-2-(2-hydroxypropan-2-yl)anilino]pyrimidin-2-yl]amino]-2-[(3R)-3-(dimethylamino)pyrrolidin-1-yl]-4-methoxyphenyl]prop-2-enamide
FDA Zegfrovy, 7/2/2025
To treat locally advanced or metastatic non-small cell lung cancer with epidermal growth factor receptor exon 20 insertion mutations, as detected by an FDA-approved test, with disease progression on or after platinum-based chemotherapy |
Sunvozertinib (DZD9008) is a potent ErbBs (EGFR, Her2, especially mutant forms) and BTK inhibitor. Sunvozertinib shows IC50s of 20.4, 20.4, 1.1, 7.5, and 80.4 nM for EGFR exon 20 NPH insertion, EGFR exon 20 ASV insertion, EGFR L858R and T790M mutations, and Her2 Exon20 YVMA, and EGFR WT A431, respectively (patent WO2019149164A1, example 52).
Sunvozertinib, sold under the brand name 舒沃哲, among others is an anti-cancer medication used for the treatment of non-small-cell lung cancer.[2][3] It is an epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor.[2][4]
Sunvozertinib was approved for medical use in the United States in July 2025.[1]
Medical uses
In the US, sunvozertinib is indicated for the treatment of adults with locally advanced or metastatic non-small cell lung cancer with epidermal growth factor receptor exon 20 insertion mutations, as detected by an FDA-approved test, whose disease has progressed on or after platinum-based chemotherapy.[1]
Side effects
The US FDA prescribing information for sunvozertinib includes warnings and precautions for interstitial lung disease/pneumonitis, gastrointestinal adverse reactions, dermatologic adverse reactions, ocular toxicity, and embryo-fetal toxicity.[1]
History
Sunvozertinib is being developed by Dizal Pharmaceutical.[5] In China, it was conditionally approved in 2023 for the treatment of NSCLC and full approval is contingent on results of phase 3 clinical trials.[6] In the United States, it has been designated by the Food and Drug Administration as a breakthrough therapy for patients with locally advanced or metastatic NSCLCs with an EGFR exon 20 insertion mutation.[7]
Efficacy was evaluated in WU-KONG1B (NCT03974022), a multinational, open-label, dose randomization trial.[1] Eligible participants had locally advanced or metastatic non-small cell lung cancer with epidermal growth factor receptor exon 20 insertion mutations with disease progression on or after platinum-based chemotherapy.[1] The primary efficacy population was in 85 participants who received sunvozertinib 200 mg orally once daily with food until disease progression or intolerable toxicity.[1]
The US Food and Drug Administration granted the application for sunvozertinib priority review and breakthrough therapy designations.[1]
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019149164&_cid=P10-MCWSEU-52423-1
[1286]
(R) -N- (5- (4- (5-chloro-4-fluoro-2- (2-hydroxypropan-2-yl) phenylamino) pyrimidin-2-ylamino) -2- (3- (dimethylamino) pyrrolidin-1-yl) -4-methoxyphenyl) acrylamide
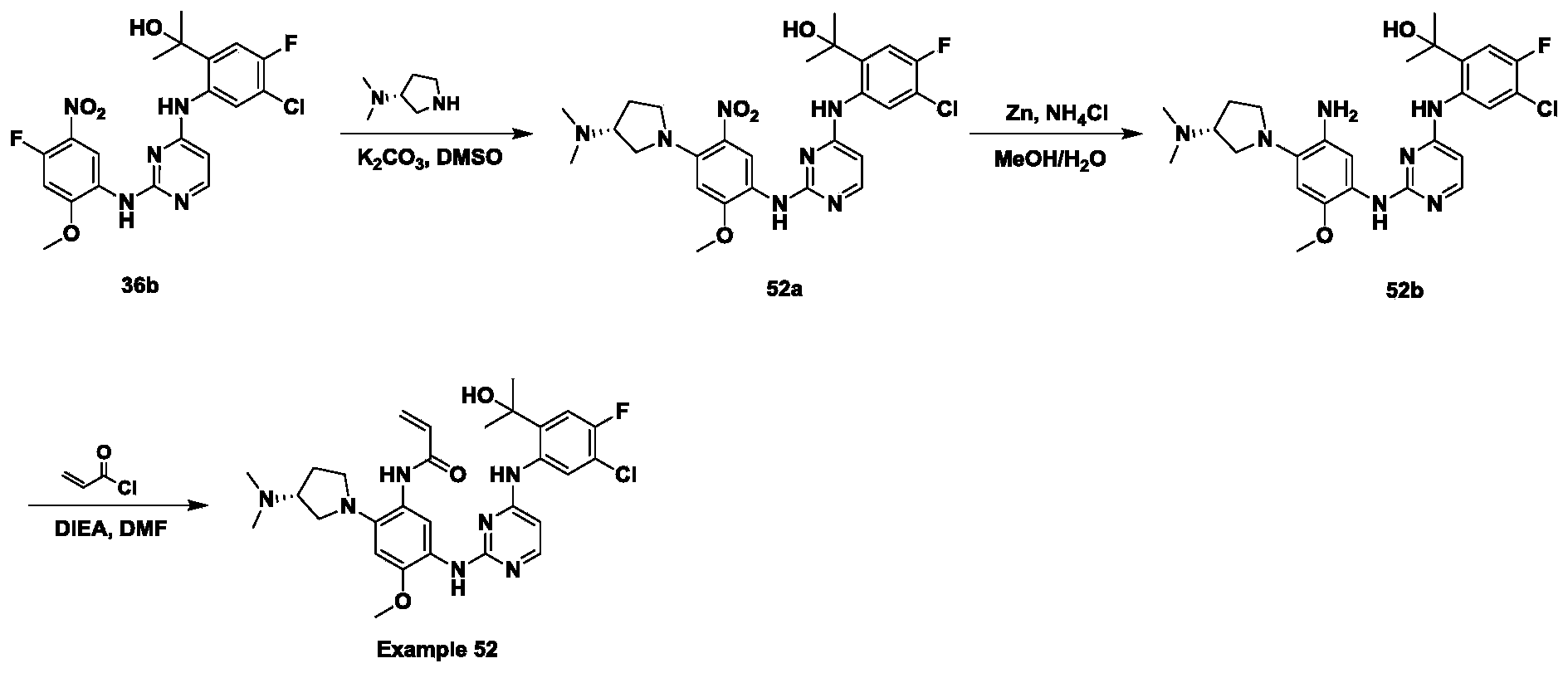
Procedure for the preparation of compound 52a:
[1289]
To a solution of compound 36b (200 mg, 0.429 mmol) and K 2CO 3(119 mg, 0.858 mmol) in DMSO (3 mL) was added (R) -N, N-dimethylpyrrolidin-3-amine (59 mg, 0.515 mmol) . The reaction mixture was stired at 22-30℃ for 4 h, then 50℃ for 1 h while color changed from brown to deep orange. The reaction mixture was added drop wise into H 2O (40 mL) under ice water bath. The precipitated solid was collected by filtration and washed with H 2O (15 mL × 3) , the filter cake was dissolved with CH 2Cl 2(20 mL) , dried over Na 2SO 4and concentrated in vacuum to give compound 52a (210 mg, 87.4%yield) as an orange solid.
[1290]
LCMS: R t=0.677 min in 5-95AB_220&254. lcm chromatography (MERCK RP18 2.5-2mm) , MS (ESI) m/z=559.9 [M+H] +.
[1291]
1H NMR (400MHz, Methanol-d 4) δ 8.42 (s, 1H) , 7.99 (d, J=7.6 Hz, 1H) , 7.96 (d, J=5.6 Hz, 1H) , 7.24 (d, J=10.8 Hz, 1H) , 6.50 (s, 1H) , 6.15 (d, J=5.6 Hz, 1H) , 3.96 (s, 3H) , 3.54 (dt, J=6.4, 10.4 Hz, 1H) , 3.36 (t, J=9.2 Hz, 1H) , 3.28 -3.24 (m, 1H) , 3.09 (dd, J=6.8, 10.0 Hz, 1H) , 2.93 -2.81 (m, 1H) , 2.33 (s, 6H) , 2.30 -2.25 (m, 1H) , 1.97 -1.82 (m, 1H) , 1.59 (d, J=3.6 Hz, 6H) .
[1292]
Procedure for the preparation of compound 52b:
[1293]
To a solution of compound 52a (210 mg, 0.375 mmol) in 5 mL MeOH/H 2O=5/1 (v/v) was added Zn (147 mg, 2.25 mmol) and NH 4Cl (120 mg, 2.25 mmol) . The resulting mixture was heated at 90℃ for 2 h while color changed from orange to brown. The reaction mixture was filtered, and the filtrate was concentrated in vacuum to give the crude residue, which was dissolved with CH 2Cl 2(20 mL) , washed with water (15 mL×3) , then dried over Na 2SO 4and concentrated in vacuum to give compound 52b (125 mg, 63%yield) as a brown solid.
[1294]
LCMS: R t=0.629 min in 5-95AB_220&254. lcm chromatography (MERCK RP18 2.5-2mm) , MS (ESI) m/z=530.1 [M+H] +.
[1295]
1H NMR (400MHz, CDCl 3) δ 8.85 (s, 1H) , 8.15 (d, J=7.2 Hz, 1H) , 8.03 (d, J=6.0 Hz, 1H) , 7.87 (s, 1H) , 7.43 (s, 1H) , 7.08 (d, J=10.4 Hz, 1H) , 6.66 (s, 1H) , 6.05 (d, J=5.6 Hz, 1H) , 3.81 (s, 3H) , 3.20 -3.11 (m, 2H) , 3.06 -2.97 (m, 2H) , 2.91 -2.83 (m, 1H) , 2.28 (s, 6H) , 2.17 -2.09 (m, 1H) , 1.92 -1.81 (m, 1H) , 1.65 (s, 6H) .
[1296]
Procedure for the preparation of Example 52:
[1297]
To a solution of compound 52b (125 mg, 0.236 mmol) and DIEA (46 mg, 0.354 mmol) in DMF (1.5 mL) was added acryloyl chloride (21 mg, 0.236 mmol) in ice water bath. The resulting mixture was stirred at 5-10℃ for 15 min. The reaction was quenched by H 2O (0.1 mL) and then filtered, the filtrate was purified by pre-HPLC directly (Column: Xtimate C18 150*25mm*5um; Condition: 35-65%B (A: 0.04%NH 3·H 2O+10mM NH 4HCO 3, B: CH 3CN) ; Flow Rate: 30 ml/min) and then lyophilized to give Example 52 (14.5 mg, 10.5%yield) as an off-white solid.
[1298]
LCMS: R t=2.028 min in 10-80CD_3min_220&254. lcm chromatography (XBrige Shield RP18 2.1*50mm, 5um) , MS (ESI) m/z=584.3 [M+H] +.
[1299]
1H NMR (400MHz, CDCl 3) δ 9.62 (s, 1H) , 9.43 (br s, 1H) , 8.59 (br s, 1H) , 8.08 (d, J=5.2 Hz, 1H) , 7.53 (d, J=6.8 Hz, 1H) , 7.47 (br s, 1H) , 7.14 (d, J=10.8 Hz, 1H) , 6.75 (s, 1H) , 6.41 -6.31 (m, 3H) , 5.77 (t, J=5.4 Hz, 1H) , 3.86 (s, 3H) , 3.15 -3.02 (m, 4H) , 2.97 -2.84 (m, 1H) , 2.30 (s, 6H) , 2.21 -2.14 (m, 1H) , 1.99 -1.94 (m, 1H) , 1.72 (s, 6H) .
PATENT
PAPER
https://www.mdpi.com/1420-3049/29/7/1448
Sunvozertinib, a novel TKI manufactured by Dizal Pharmaceuticals, represents an advancement arising from the need to overcome resistance mechanisms and thereby replaces previous generations of EGFR inhibitors. As marketed under the proprietary name DZD9008, this therapeutic entity embodies an innovative modality aimed at addressing NSCLC characterized by distinct mutations within the EGFR gene [24]. The pharmacological efficacy of sunvozertinib is predicated upon its specific capacity to inhibit EGFR with Exon 20 mutations, alongside targeting human epidermal growth factor receptor 2 (HER2) with Exon 20 insertions. This targeted inhibition is of significance due to the diminished responsiveness of cancer cells with these mutations to earlier generations of EGFR inhibitors. By competitively obstructing the ATP-binding site of these mutated tyrosine kinases, sunvozertinib effectively hinders the proliferative signaling pathways, exhibiting potent anti-tumor activity (ClinicalTrials.gov Identifier: NCT03974022). Preclinical models have demonstrated sunvozertinib’s capacity to inhibit tumor growth, especially in NSCLC models harboring Exon 20 insertions. Moreover, its substantiated ability to traverse the blood–brain barrier has provided favorable support for its prospective efficacy in managing central nervous system metastases. In clinical settings, sunvozertinib has shown promising efficacy, with ongoing trials further assessing its utility as a targeted intervention for individuals presenting specific EGFR mutations. The toxicity profile has been comparable to other EGFR inhibitors, with manageable side effects that do not significantly diminish its therapeutic value [25].
The preparation of sunvozertinib is shown in Scheme 1 [26]. SUNV-001 and SUNV-002 engage in nucleophilic substitution reactions under alkaline conditions, yielding the formation of SUNV-003 through a subsequent nucleophilic substitution process involving SUNV-004, ultimately leading to the generation of SUNV-005. SUNV-005 and SUNV-006 consecutively engage in nucleophilic substitution reactions, culminating in the formation of SUNV-007. The nitro moiety present in SUNV-007 undergoes a reduction process to yield an amino functional group, through the utilization of hydrogen gas as the reducing agent and platinum carbon as the catalytic mediator, ultimately affording the formation of SUNV-008. The amino moiety present in SUNV-008 and the acyl chloride functionality of SUNV-009 engage in a condensation reaction, leading to the formation of the amide compound SUNV-010. SUNV-010 is subjected to an elimination reaction in alkaline environments, leading to the formation of sunvozertinib.
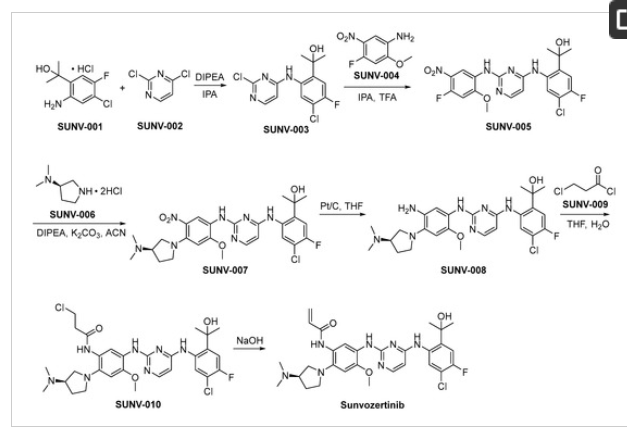
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- ^ Jump up to:a b c d e f g h “FDA grants accelerated approval to sunvozertinib for metastatic non-small cell lung cancer with EGFR exon 20 insertion mutations”. U.S. Food and Drug Administration (FDA). 2 July 2025. Retrieved 7 July 2025. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b Wang M, Yang JC, Mitchell PL, Fang J, Camidge DR, Nian W, et al. (2022). “Sunvozertinib, a Selective EGFR Inhibitor for Previously Treated Non–Small Cell Lung Cancer with EGFR Exon 20 Insertion Mutations”. Cancer Discovery. 12 (7): 1676–1689. doi:10.1158/2159-8290.CD-21-1615. PMC 9262839. PMID 35404393.
- ^ Wang M, Fan Y, Sun M, Wang Y, Zhao Y, Jin B, et al. (2024). “Sunvozertinib for patients in China with platinum-pretreated locally advanced or metastatic non-small-cell lung cancer and EGFR exon 20 insertion mutation (WU-KONG6): Single-arm, open-label, multicentre, phase 2 trial”. The Lancet Respiratory Medicine. 12 (3): 217–224. doi:10.1016/S2213-2600(23)00379-X. PMID 38101437.
- ^ Hidetoshi Hayashi (2024). “Sunvozertinib: the next candidate of TKI for NSCLC”. The Lancet Respiratory Medicine. 12 (3): 185–186. doi:10.1016/S2213-2600(23)00419-8. PMID 38101435.
- ^ “ASH: With high tumor response, AstraZeneca spinout Dizal explores FDA path and US partner for PTCL drug”. Fierce Biotech. 11 December 2023.
- ^ Dhillon S (2023). “Sunvozertinib: First Approval”. Drugs. 83 (17): 1629–1634. doi:10.1007/s40265-023-01959-5. PMID 37962831.
- ^ “FDA Grants Breakthrough Therapy Designation to Sunvozertinib in EGFR Exon20+ NSCLC”. targetedonc.com. 9 April 2024.
External links
- Clinical trial number NCT03974022 for “Assessing an Oral EGFR Inhibitor, Sunvozertinib in Patients Who Have Advanced Non-small Cell Lung Cancer with EGFR or HER2 Mutation (WU-KONG1)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | 舒沃哲, Zegfrovy |
| Routes of administration | Oral |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1]Rx in China |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2370013-12-8 |
| PubChem CID | 139377809 |
| DrugBank | DB18925 |
| UNII | L1Q2K5JYO8 |
| Chemical and physical data | |
| Formula | C29H35ClFN7O3 |
| Molar mass | 584.09 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
//////////Sunvozertinib, DZD9008, DZD 9008, FDA 2025, APPROVALS 2025, A-1801, A 1801, L1Q2K5JYO8
PALTUSOTINE



PALTUSOTINE
CAS 2172870-89-0
- CRN00808
- F2IBD1GMD3
WeightAverage: 456.497
Monoisotopic: 456.17616767
Chemical FormulaC27H22F2N4O
3-[4-(4-Amino-1-piperidinyl)-3-(3,5-difluorophenyl)-6-quinolinyl]-2-hydroxybenzonitrile
fda 2025, approvals 2025, To treat acromegaly in adults who had an inadequate response to surgery and/or for whom surgery is not an option
- OriginatorCrinetics Pharmaceuticals
- ClassAmines; Antineoplastics; Antisecretories; Fluorobenzenes; Nitriles; Piperidines; Quinolines; Small molecules
- Mechanism of ActionSomatostatin receptor 2 agonists
- Orphan Drug Status – Acromegaly
- PreregistrationAcromegaly
- Phase IIMalignant carcinoid syndrome
- 08 May 2025Crinetics Pharmaceuticals expects potential EMA decision for paltusotine in Acromegaly, in the first half of 2026
- 08 May 2025FDA assigns PDUFA action date of 25/09/2025 for paltusotine for acromegaly
- 08 May 2025Crinetics Pharamceuticals plans the phase III CAREFNDR trial for Malignant carcinoid syndrome (PO), in the second quarter of 2025
Paltusotine is a selective somatostatin receptor type 2 (SST2) agonist in development by Crinetics Pharmaceuticals for the treatment of acromegaly and certain neuroendocrine tumors. It is a small molecule delivered orally.[1][2][3][4]
SCHEME
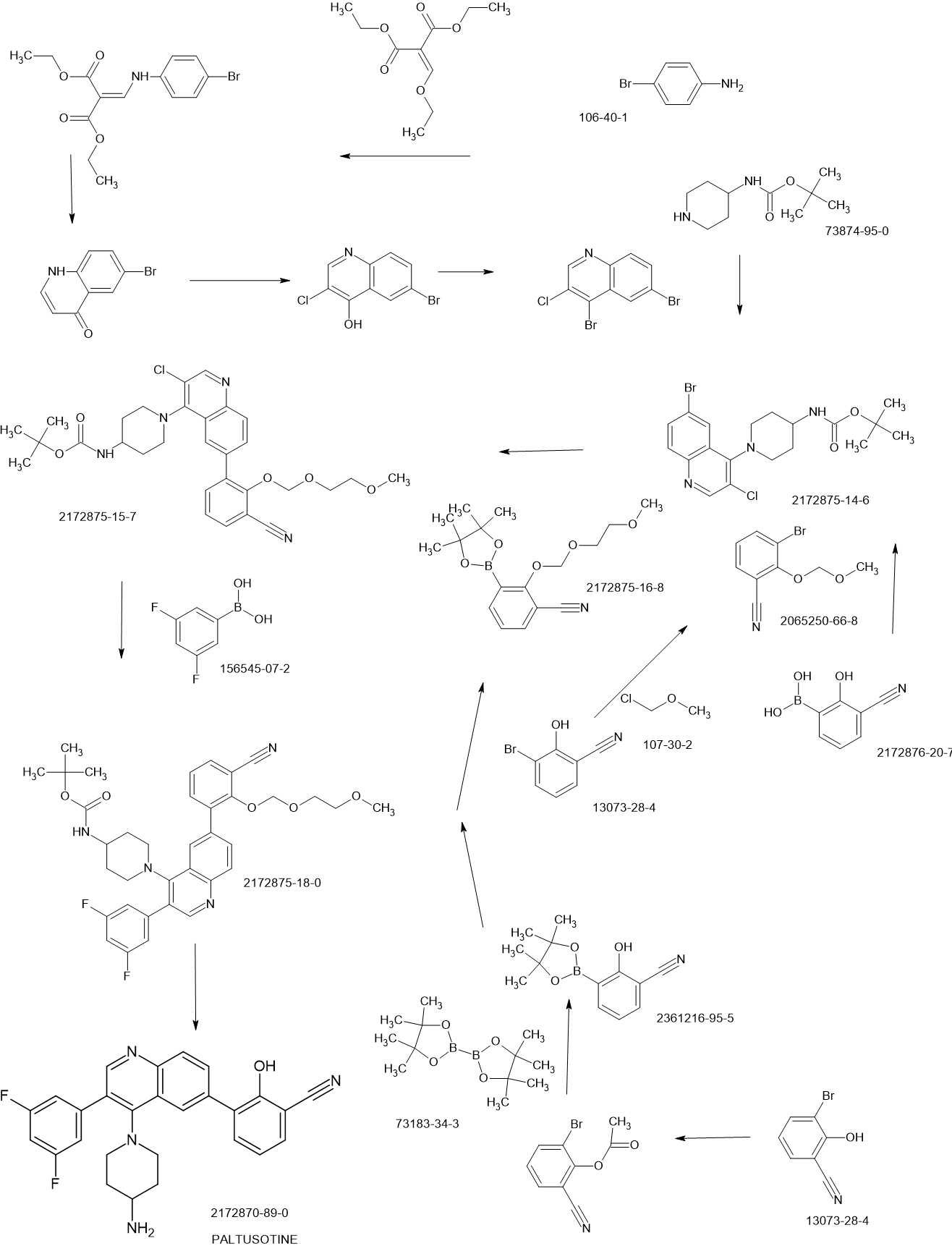

PAPER
https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00431
Discovery of Paltusotine (CRN00808), a Potent, Selective, and Orally Bioavailable Non-peptide SST2 Agonist
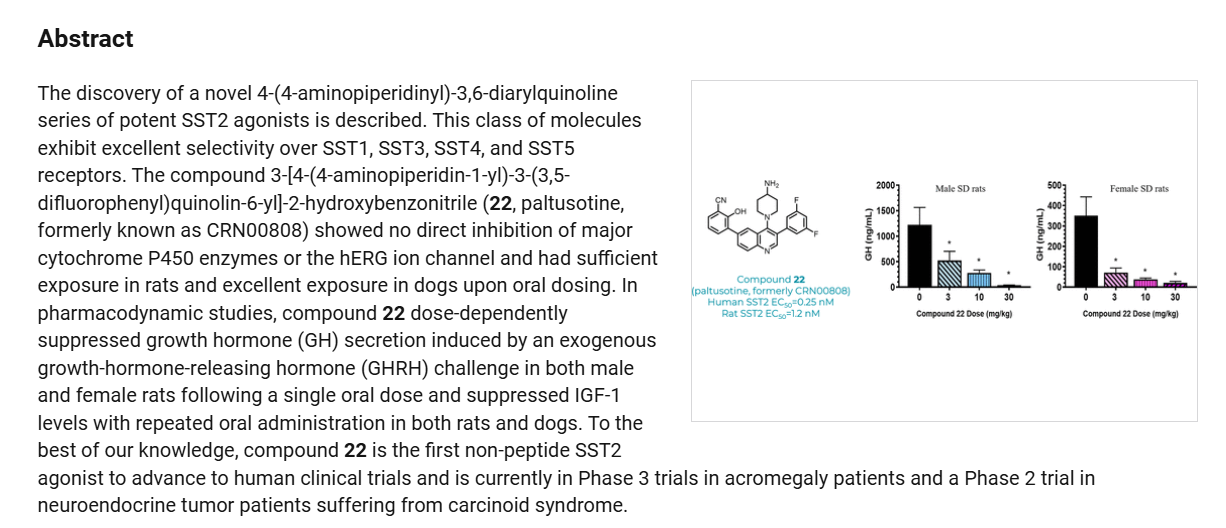

Step 2-1, preparation of [1-(6-bromo-3-chloro-quinolin-4-yl)-piperidin-4-yl]-carbamic acid tertbutyl ester: To a DMSO solution of 6-bromo-3,4-dichloroquinoline (950 mg, 1 Eq, 3.43 mmol)
was added tert-butyl piperidin-4-ylcarbamate (841 mg, 98% Wt, 1.2 Eq, 4.12 mmol) and DIPEA
(1.19 g, 1.60 mL, 3 Eq, 10.3 mmol). The resulting mixture was heated at 60 °C for overnight.
The reaction crude was quenched with water, extracted with EtOAc, washed with brine,
concentrated and purified by silica gel chromatography to afford tert-butyl (1-(6-bromo-3-
chloroquinolin-4-yl)piperidin-4-yl)carbamate (0.95 g, 2.2 mmol, 63 %) as an off-white solid. 1H
NMR (500 MHz, CDCl3) δ 8.66 (s, 1H), 8.25 (d, J=5 Hz, 1H), 7.94 (d, J=10 Hz, 1H), 7.74 (d,
J=10 Hz, 1H), 4.61 (s, 1H), 3.76 (s, 1H), 3.51 (m, 2H), 3.37 (m, 2H), 2.13-2.15 (m, 2H), 1.73-
1.65 (m, 2H), 1.48 (s, 9H). MS [M+H]
+= 442.0.
Step 4-2, preparation of 1-{3-chloro-6-[3-cyano-2-(2-methoxy-ethoxymethoxy)-phenyl]-
quinolin-4-yl}-piperidin-4-yl)-carbamic acid tert-butyl ester: To a THF (5.0 mL) solution of [1-
(6-bromo-3-chloro-quinolin-4-yl)-piperidin-4-yl]-carbamic acid tert-butyl ester (1.0 mmol, 440
mg) and 2-(2-methoxy-ethoxymethoxy)-3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-
benzonitrile (1.4 eq., 1.4 mmol, 460 mg) was added PdCl2dppf (0.1 eq., 0.1 mmol, 75 mg) and
KOAc (3.0 eq., 3.0 mmol, 300 mg). N2 was bubbled through the reaction solution for 5 min and
0.5 mL water was added. The resulting mixture was heated at 80 °C for 1 h. LCMS analysis
showed about 50% of the starting material has been converted to the desired product. Additional
2-(2-methoxy-ethoxymethoxy)-3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzonitrile
(1.4 eq., 1.4 mmol, 460 mg), PdCl2dppf (0.1 eq., 0.1 mmol, 75 mg) and KOAc (3.0 eq., 3.0
mmol, 300 mg) were added and the resulting solution was heated at 80 °C for another 2 h. The
reaction solution was combined with silica gel and concentrated. The residue obtained was
purified by silica gel chromatography eluting with ethyl acetate/hexane (0~50%) to give 0.512 g
of the desired product as white solid. MS [M+H]
+= 567.6.
Step 4-3, preparation of {1-[6-[3-cyano-2-(2-methoxy-ethoxymethoxy)-phenyl]-3-(3,5-difluorophenyl)-quinolin-4-yl]-piperidin-4-yl}-carbamic acid tert-butyl ester: To a dioxane (5 mL)
solution of (1-{3-chloro-6-[3-cyano-2-(2-methoxy-ethoxymethoxy)-phenyl]-quinolin-4-yl}-
piperidin-4-yl)-carbamic acid tert-butyl ester (0.5 mmol, 283 mg) was added Pd(amphos)Cl2 (0.1
eq., 0.05 mmol, 37 mg), 3, 5-difluorophenyl boronic acid (3.0 eq., 1.5 mmol, 250 mg) and
K2CO3 (4.0 eq., 2.0 mmol, 276 mg). N2 was bubbled through the reaction solution for 5 min and
0.5 mL water was added. The resulting mixture was heated at 95 °C for 0.5 h and LCMS analysis
showed that starting material was completely consumed. The reaction solution was concentrated
with silica gel and purified by silica gel chromatography eluting with ethyl acetate/hexane
(0~50%) to give 0.170 g of the desired product as white solid. MS (M+H)+= 645.6.
Step 4-4, preparation of 3-[4-(4-amino-piperidin-1-yl)-3-(3,5-difluoro-phenyl)-quinolin-6-yl]-2-hydroxybenzonitrile: to the dichloromethane (5.0 mL) solution of {1-[6-[3-cyano-2-(2-methoxyethoxymethoxy)-phenyl]-3-(3,5-difluoro-phenyl)-quinolin-4-yl]-piperidin-4-yl}-carbamic acid
tert-butyl ester (0.264 mmol, 170 mg) was added trifluroroacetic acid (2.0 mL) and the resulting
mixture was stirred at ambient temperature for 2 h. The reaction solution was concentrated and
purified by C18 reversed phase chromatography eluting with MeCN/water (0~40%). Pure
fractions were combined, neutralized with saturated NaHCO3, extracted with ethyl acetate and
dried with MgSO4. The organic solution was concentrated with HCl in ether (2.0 M) to give the
final compound as HCl salt (68 mg, 0.138 mmol, 52%).
1H NMR (500 MHz, DMSO-d6) δ 10.77
(br s, 1H), 8.78 (s, 1H), 8.29-8.15 (m, 5H), 7.79 (dd, J=20 Hz, 5 Hz, 2H), 7.41 (m, 1H), 7.26-
7.19 (m, 3H), 3.59 (t, J=12 Hz, 2H), 3.31 (m, 1H), 3.00 (t, J=12 Hz, 2H), 2.05-1.99 (m, 2H),
1.76-1.74 (m, 2H). MS [M+H]
+= 457.5. 13C NMR (DMSO-d6) δ 30.2, 47.4, 50.8, 102.4, 103.2,
113.4, 117.2, 121.4, 124.6, 130.7, 133.1, 134.6, 136.0, 141.7, 156.6, 161.2, 163.2. LCMS purity
98% (254&220 nM). HRMS m/z [M+H]+ Calcd for C27H23F2N4O 457.1834; found 457.1833.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US235548187&_cid=P20-MCSHXW-73235-1
PATENTS
WO2021011641
WO2018013676
References
- ^ Madan, Ajay; Markison, Stacy; Betz, Stephen F.; Krasner, Alan; Luo, Rosa; Jochelson, Theresa; Lickliter, Jason; Struthers, R. Scott (April 2022). “Paltusotine, a novel oral once-daily nonpeptide SST2 receptor agonist, suppresses GH and IGF-1 in healthy volunteers”. Pituitary. 25 (2): 328–339. doi:10.1007/s11102-021-01201-z. PMC 8894159. PMID 35000098.
- ^ Zhao, Jian; Wang, Shimiao; Markison, Stacy; Kim, Sun Hee; Han, Sangdon; Chen, Mi; Kusnetzow, Ana Karin; Rico-Bautista, Elizabeth; Johns, Michael; Luo, Rosa; Struthers, R. Scott; Madan, Ajay; Zhu, Yunfei; Betz, Stephen F. (12 January 2023). “Discovery of Paltusotine (CRN00808), a Potent, Selective, and Orally Bioavailable Non-peptide SST2 Agonist”. ACS Medicinal Chemistry Letters. 14 (1): 66–74. doi:10.1021/acsmedchemlett.2c00431. PMC 9841592. PMID 36655128.
- ^ Gadelha, Monica R; Gordon, Murray B; Doknic, Mirjana; Mezősi, Emese; Tóth, Miklós; Randeva, Harpal; Marmon, Tonya; Jochelson, Theresa; Luo, Rosa; Monahan, Michael; Madan, Ajay; Ferrara-Cook, Christine; Struthers, R Scott; Krasner, Alan (13 April 2023). “ACROBAT Edge: Safety and Efficacy of Switching Injected SRLs to Oral Paltusotine in Patients With Acromegaly”. The Journal of Clinical Endocrinology & Metabolism. 108 (5): e148 – e159. doi:10.1210/clinem/dgac643. PMC 10099171. PMID 36353760. S2CID 253445337.
- ^ Zhao, Jie; Fu, Hong; Yu, Jingjing; Hong, Weiqi; Tian, Xiaowen; Qi, Jieyu; Sun, Suyue; Zhao, Chang; Wu, Chao; Xu, Zheng; Cheng, Lin; Chai, Renjie; Yan, Wei; Wei, Xiawei; Shao, Zhenhua (21 February 2023). “Prospect of acromegaly therapy: molecular mechanism of clinical drugs octreotide and paltusotine”. Nature Communications. 14 (1): 962. Bibcode:2023NatCo..14..962Z. doi:10.1038/s41467-023-36673-z. ISSN 2041-1723. PMC 9944328. PMID 36810324.
| Legal status | |
|---|---|
| Legal status | Investigational |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2172870-89-0 |
| PubChem CID | 134168328 |
| ChemSpider | 81367268 |
| UNII | F2IBD1GMD3 |
| Chemical and physical data | |
| Formula | C27H22F2N4O |
| Molar mass | 456.497 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
////////PALTUSOTINE, ORPHAN DRUG, Acromegaly, CRN 00808, F2IBD1GMD3, fda 2025, approvals 2025
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

NEW DRUG APPROVALS
ONE TIME
$10.00
Taletrectinib
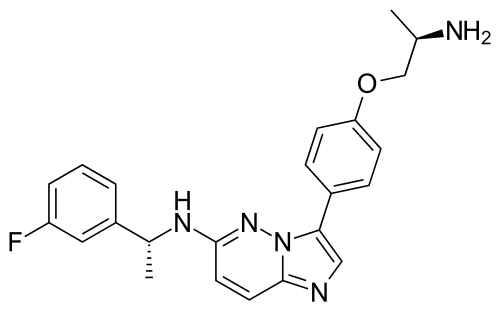
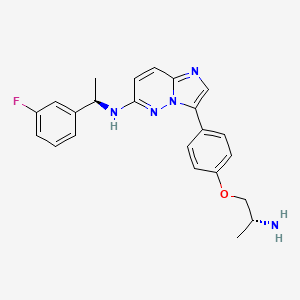
Taletrectinib
CAS 1505514-27-1
as salt: 1505515-69-4, Taletrectinib adipate
FDA 6/11/2025, Ibtrozi, To treat locally advanced or metastatic ROS1-positive non-small cell lung cancer ALSO CHINA 2024 APPROVED |
405.5 g/mol, C23H24FN5O, UNII-W4141180YD
3-[4-[(2R)-2-aminopropoxy]phenyl]-N-[(1R)-1-(3-fluorophenyl)ethyl]imidazo[1,2-b]pyridazin-6-amine
Taletrectinib adipate
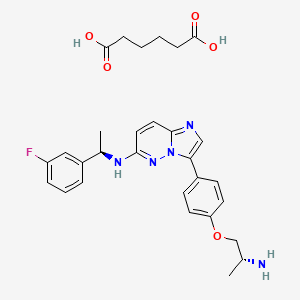
WeightAverage: 551.619
Monoisotopic: 551.254397378
Chemical FormulaC29H34FN5O5
DS-6051B, CAS 1505515-69-4,
6KLL51GNBG, 3-{4-[(2R)-2-aminopropoxy]phenyl}-N-[(1R)-1-(3-fluorophenyl)ethyl]imidazo[1,2-b]pyridazin-6-amine; hexanedioic acid
Taletrectinib, sold under the brand name Ibtrozi, is an anti-cancer medication used for the treatment of non-small cell lung cancer.[1][2] It is used as the salt, taletrectinib adipate.[1] Taletrectinib is a kinase inhibitor.[1] It is taken by mouth.[1]
Taletrectinib was approved for medical use in the United States in June 2025.[3]
SYN
US20200062765
https://patentscope.wipo.int/search/en/detail.jsf?docId=US289038418&_cid=P12-MCIHV1-02369-1
Example 1
tert-Butyl [(2R)-1-(4-bromophenoxy)propan-2-yl]carbamate (1)
Example 2
6-Fluoroimidazo[1,2-b]pyridazine methanesulfonate (2)
Example 3
tert-Butyl {(2R)-1-[4-(6-fluoroimidazo[1,2-b]pyridazin-3-yl)phenoxy]propan-2-yl}carbamate (3)
Example 4
tert-Butyl {(2R)-1-[4-(6-{[(1R)-1-(3-fluorophenyl)ethyl]amino}imidazo[1,2-b]pyridazin-3-yl)phenoxy]propan-2-yl}carbamate hydrochloride (4)
Example 5
3-{4-[(2R)-2-Aminopropoxy]phenyl}-N-[(1R)-1-(3-fluorophenyl)ethylimidazo[1,2-b]pyridazin-6-amine dihydrochloride (5)
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023272701&_cid=P12-MCIHPU-95869-1
The NMR data for the crystalline form A of Compound 1 adipate are as follows: 1H NMR (500 MHz, DMSO) δ 1.13-1.14 (d, J=5.0 Hz, 3H) , 1.47-1.48 (d, J=5.0 Hz, 7H) , 2.15-2.18 (t, J=5.0 Hz, J=10.0 Hz, 4H) , 3.25-3.29 (m, 1H) , 3.79-3.83 (m, 2H) , 4.80-4.85 (m, 1H) , 6.76-6.77 (d, J=5.0 Hz, 1H) , 6.92-6.94 (d, J=10.0 Hz, 2H) , 7.01-7.05 (t, J=10.0 Hz, 1H) , 7.23-7.28 (m, 2H) , 7.37-7.42 (m, 1H) , 7.64-7.65 (d, J=5.0 Hz, 1H) , 7.72-7.76 (t, J=10.0 Hz, 4H) .
[0148]
The IR data for the crystalline form A of Compound 1 adipate are as follows: IR (cm -1) : 1701, 1628, 1612, 1586, 1463, 1333, 1246, 1110, 829, 821.
Example 5: Preparation and Characterization of Crystalline Form A of Compound 1 Free Base
[0212]
Compound 1 HCl (75.5 g) (e.g., obtained by using the method described in Example 5 of U.S. Application Publication No. 2020/0062765) was dissolved in ethanol (604 mL) at 50℃. Sodium hydroxide (68.1 g) was added to the above solution. The mixture was cooled to 1℃ in 1.5 hours and stirred for 18.5 hours. The mixture was then filtered, and the solid thus obtained was washed with a cooled mixture of ethanol (151 mL) and water (151 mL) and dried. The solid thus obtained was confirmed to be the crystalline form A of Compound 1 free base.
[0213]
The NMR data for the crystalline form A of Compound 1 free base are as follows: 1H NMR (500 MHz, DMSO) δ 1.09-1.10 (d, J=5.0 Hz, 3H) , 1.48-1.49 (d, J=5.0 Hz, 3H) , 3.16-3.20 (m, 1H) , 3.75-3.79 (m, 2H) , 4.82-4.86 (m, 1H) , 6.76-6.78 (d, J=10.0 Hz, 1H) , 6.92-6.94 (m, 2H) , 7.01-7.05 (m, 1H) , 7.23-7.28 (m, 2H) , 7.37-7.42 (m, 1H) , 7.62-7.63 (d, J=5.0 Hz, 1H) , 7.72-7.75 (m, 4H) .
[0214]
The IR data for the crystalline form A of Compound 1 free base are as follows: IR (cm -1) : 3350, 3247, 3055, 2961, 2923, 2864, 1611, 1586, 1349, 829, 819.
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Taletrectinib is an oral, next-generation ROS1 TKI developed by Nuvation Bio Inc. for the treatment of ROS1-positive NSCLC. In 2024, the NMPA approved taletrectinib for adult patients with locally advanced or metastatic ROS1-positive NSCLC, regardless of prior ROS1TKI treatment [47]. Under an exclusive license agreement, Innovent Biologics will commercialize taletrectinib in China under the brand
name DOVBLERON®. Taletrectinib exerts its pharmacological action through the mechanism of selectively impeding the ROS1 receptor tyrosine kinase, which effectively disrupts the signaling cascades which are responsible for facilitating the growth and survival of cancer cells in ROS1-positive NSCLC. This inhibition of the ROS1 receptor tyrosine kinase is a key event in the drug’s mode of action, as it specifically targets the molecular processes that drive the progression of the disease in ROS1-positive NSCLC cases [48]. The NMPA granted approval founded on the data sourced from the crucial Phase 2 TRUST – I study. This study substantiated that patients administered with taletrectinib achieved sustained responses and extended PFS. Regarding safety, taletrectinib boasted a generally good tolerability. It presented an advantageous safety profile and favorable tolerability characteristics, as evidenced by the low incidences of dose reduction and treatment discontinuation triggered by adverse effects. [49]. Overall, taletrectinib represents a promising therapeutic option for patients with advanced ROS1-positive NSCLC, offering efficacy in both TKI-naïve and TKI-pretreated populations, including those with CNS metastases [50–52].
The synthesis of Taletrectinib, illustrated in Scheme 12, commences with Mitsunobu coupling of Tale-001 and Tale-002 to afford Tale-003, which then undergoes Suzuki coupling with Tale-004 constructing
Tale-005 [53]. Sequential acidolysis/deprotection of Tale-005 ultimately delivers Taletrectinib
[47] M. P´ erol, N. Yang, C.M. Choi, Y. Ohe, S. Sugawara, N. Yanagitani, G. Liu, F.G.M.
D. Braud, J. Nieva, M. Nagasaka, 1373P efficacy and safety of taletrectinib in
patients (pts) with ROS1+ non-small cell lung cancer (NSCLC): interim analysis of
global TRUST-II study, Ann. Oncol. 34 (2023) S788–S789.
[48] G. Harada, F.C. Santini, C. Wilhelm, A. Drilon, NTRK fusions in lung cancer: from
biology to therapy, Lung Cancer 161 (2021) 108–113.
[49] W. Li, A. Xiong, N. Yang, H. Fan, Q. Yu, Y. Zhao, Y. Wang, X. Meng, J. Wu, Z. Wang,
Y. Liu, X. Wang, X. Qin, K. Lu, W. Zhuang, Y. Ren, X. Zhang, B. Yan, C.M. Lovly,
C. Zhou, Efficacy and safety of taletrectinib in Chinese patients with ROS1+ non-
small cell lung cancer: the phase II TRUST-I study, J. Clin. Oncol. 42 (2024)
2660–2670.
[50] M. Nagasaka, D. Brazel, S.I. Ou, Taletrectinib for the treatment of ROS-1 positive
non-small cell lung cancer: a drug evaluation of phase I and II data, Expert Opin
Investig Drugs 33 (2024) 79–84.
[51] S. Waliany, J.J. Lin, Taletrectinib: TRUST in the continued evolution of treatments
for ROS1 fusion-positive lung cancer, J. Clin. Oncol. 42 (2024) 2622–2627.
[52] M. Nagasaka, Y. Ohe, C. Zhou, C.M. Choi, N. Yang, G. Liu, E. Felip, M. P´ erol,
B. Besse, J. Nieva, L. Raez, N.A. Pennell, A. Dimou, F. Marinis, F. Ciardiello,
T. Seto, Z. Hu, M. Pan, W. Wang, S. Li, S.I. Ou, TRUST-II: a global phase II study of
taletrectinib in ROS1-positive non-small-cell lung cancer and other solid tumors,
Future Oncol. 19 (2023) 123–135.
[53] Y. Takeda, K. Yoshikawa, Y. Kagoshima, Y. Yamamoto, R. Tanaka, Y. Tominaga,
M. Kiga, Y. Hamada, Preparation of imidazo[1,2-b]pyridazine Derivatives as
Potent Inhibitors of ROS1 Kinase and NTRK Kinase, 2013. WO2013183578A1.

Medical uses
Taletrectinib is indicated for the treatment of adults with locally advanced or metastatic ROS1-positive non-small cell lung cancer.[1][2]
Adverse effects
The FDA prescribing information for taletrectinib includes warnings and precautions for hepatotoxicity, interstitial lung disease/pneumonitis, QTc interval prolongation, hyperuricemia, myalgia with creatine phosphokinase elevation, skeletal fractures, and embryo-fetal toxicity.[1][3]
History
The efficacy of taletrectinib to treat ROS1-positive non-small cell lung cancer was evaluated in participants with locally advanced or metastatic, ROS1-positive non-small cell lung cancer enrolled in two multi-center, single-arm, open-label clinical trials, TRUST-I (NCT04395677) and TRUST-II (NCT04919811).[3] The efficacy population included 157 participants (103 in TRUST-I; 54 in TRUST-II) who were naïve to treatment with a ROS1 tyrosine kinase inhibitor (TKI) and 113 participants (66 in TRUST-I; 47 in TRUST-II) who had received one prior ROS1 tyrosine kinase inhibitor.[3] Participants may have received prior chemotherapy for advanced disease.[3] The US Food and Drug Administration (FDA) granted the application for taletrectinib priority review, breakthrough therapy, and orphan drug designations.[3]
Society and culture
Legal status
Taletrectinib was approved for medical use in the United States in June 2025.[3][4]
Names
Taletrectinib is the international nonproprietary name.[5]
Taletrectinib is sold under the brand name Ibtrozi.[3][4]
References
- ^ Jump up to:a b c d e f g “Prescribing Information for NDA 219713, Supplement 000” (PDF). Drugs@FDA. U.S. Food and Drug Administration. April 2025. Retrieved 14 June 2025.
- ^ Jump up to:a b Khan I, Sahar A, Numra S, Saha N, Nidhi, Parveen R (April 2025). “Efficacy and safety of taletrectinib for treatment of ROS1 positive non-small cell lung cancer: A systematic review”. Expert Opinion on Pharmacotherapy. 26 (6): 765–772. doi:10.1080/14656566.2025.2487150. PMID 40170301.
- ^ Jump up to:a b c d e f g h “FDA approves taletrectinib for ROS1-positive non-small cell lung cancer”. U.S. Food and Drug Administration (FDA). 11 June 2025. Retrieved 13 June 2025. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “U.S. Food and Drug Administration Approves Nuvation Bio’s Ibtrozi (taletrectinib), a Next-Generation Oral Treatment for Advanced ROS1-Positive Non-Small Cell Lung Cancer”. Nuvation Bio (Press release). 12 June 2025. Retrieved 13 June 2025.
- ^ World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 85”. WHO Drug Information. 35 (1). hdl:10665/340684.
External links
- Clinical trial number NCT04395677 for “A Study of AB-106 in Subjects With Advanced NSCLC Harboring ROS1 Fusion Gene” at ClinicalTrials.gov
- Clinical trial number NCT04919811 for “Taletrectinib Phase 2 Global Study in ROS1 Positive NSCLC (TRUST-II)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Ibtrozi |
| License data | US DailyMed: Taletrectinib |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 1505514-27-1as salt: 1505515-69-4 |
| PubChem CID | 72202474as salt: 72694302 |
| DrugBank | DB18711 |
| ChemSpider | 114934673as salt: 88297530 |
| UNII | W4141180YDas salt: 6KLL51GNBG |
| KEGG | D12363as salt: D12364 |
| ChEMBL | ChEMBL4650989as salt: ChEMBL4650361 |
| Chemical and physical data | |
| Formula | C23H24FN5O |
| Molar mass | 405.477 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
/////////Taletrectinib, FDA 2025, APPROVALS 2025, Ibtrozi, CANCER, AB-106, DS-6051a, UNII-W4141180YD, DS 6051B, APPROVALS 2024, CHINA 2024, Nuvation Bio Inc
Acoltremon
Acoltremon
CAS 68489-09-8
WeightAverage: 289.419
Monoisotopic: 289.204179113
Chemical FormulaC18H27NO2
FDA 2025, 5/28/2025, To treat the signs and symptoms of dry eye disease
Tryptyr |
WS 12
(1R,2S,5R)-N-(4-methoxyphenyl)-5-methyl-2-(propan-2-yl)cyclohexane-1-carboxamide
Fema No. 4681
N-(4-methoxyphenyl)-p-menthanecarboxamide
- OriginatorInstituto de Neurociencias de Alicante
- DeveloperAlcon; AVX Pharma
- ClassCyclohexanes; Ethers; Eye disorder therapies; Small molecules
- Mechanism of ActionTRPM8 protein stimulants
- RegisteredDry eyes
- 30 May 2025Alcon plans to launch Acoltremon for Dry eyes in USA in the third quarter of 2025
- 28 May 2025Registered for Dry eyes in USA (Ophthalmic) – First global approval
- 05 May 2025FDA assigns PDUFA action date of 30/05/2025 for Acoltremon for Dry eyes
Acoltremon sold under the brand name Tryptyr, is a medication used for the treatment of dry eye syndrome.[1]
PATENT
US 217370
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023114986&_fid=RU437402572
https://patentscope.wipo.int/search/en/detail.jsf?docId=US193167995&_cid=P11-MCE7BB-27500-1
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012032209&_fid=US193167995
Medical uses
Acoltremon was approved for medical use in the United States in May 2025, for the treatment of signs and symptoms associated with dry eye disease.[2]
Pharmacology
Acoltremon acts as a potent and selective activator (opener) of the TRPM8 calcium channel, which is responsible for the sensation of coldness produced by menthol.[3] It is slightly less potent as a TRPM8 activator compared to icilin, but is a much more selective TRPM8 ligand when compared to menthol.[4]
Society and culture
Legal status
Acoltremon was approved for medical use in the United States in May 2025.[5]
References
- ^ Jump up to:a b https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/217370s000lbl.pdf
- ^ “Novel Drug Approvals for 2025”. U.S. Food and Drug Administration (FDA). 29 May 2025. Archived from the original on 3 March 2025. Retrieved 29 May 2025.
- ^ Ma S, Gisselmann G, Vogt-Eisele AK, Doerner JF, Hatt H (October 2008). “Menthol derivative WS-12 selectively activates transient receptor potential melastatin-8 (TRPM8) ion channels”. Pakistan Journal of Pharmaceutical Sciences. 21 (4): 370–378. PMID 18930858.
- ^ Kühn FJ, Kühn C, Lückhoff A (February 2009). “Inhibition of TRPM8 by icilin distinct from desensitization induced by menthol and menthol derivatives”. The Journal of Biological Chemistry. 284 (7): 4102–4111. doi:10.1074/jbc.M806651200. PMID 19095656.
- ^ “Alcon Announces FDA Approval of Tryptyr (acoltremon ophthalmic solution) 0.003% for the Treatment of the Signs and Symptoms of Dry Eye Disease” (Press release). Alcon. 28 May 2025. Archived from the original on 29 May 2025. Retrieved 29 May 2025 – via Business Wire.
External links
- Clinical trial number NCT05285644 for “Study Evaluating the Safety and Efficacy of AR-15512 (COMET-2)” at ClinicalTrials.gov
- Clinical trial number NCT05360966 for “Study Evaluating the Safety and Efficacy of AR-15512 (COMET-3)” at ClinicalTrials.gov
| molecular structure | |
| 3D representation | |
| Clinical data | |
|---|---|
| Trade names | Tryptyr |
| Other names | AVX-012, WS-12 |
| License data | US DailyMed: Acoltremon |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 68489-09-8 |
| PubChem CID | 11266244 |
| DrugBank | DB19202 |
| ChemSpider | 9441255 |
| UNII | 1L7BVT4Z4Z |
| KEGG | D13125 |
| ChEMBL | ChEMBL2441929 |
| CompTox Dashboard (EPA) | DTXSID10460636 |
| Chemical and physical data | |
| Formula | C18H27NO2 |
| Molar mass | 289.419 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
- [1]. Beck B, et al. Prospects for prostate cancer imaging and therapy using high-affinity TRPM8 activators. Cell Calcium. 2007 Mar;41(3):285-94. [Content Brief][2]. Ma S, et al. Menthol derivative WS-12 selectively activates transient receptor potential melastatin-8 (TRPM8) ion channels. Pak J Pharm Sci. 2008 Oct;21(4):370-8. [Content Brief]
///////Acoltremon, FDA 2025, APPROVALS 2025, WS-12, WS 12, Fema No. 4681, Tryptyr, 1L7BVT4Z4Z, AR-15512

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}