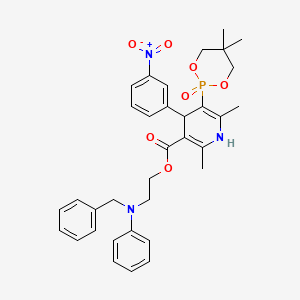
| FDA Orange Book Patents: 1 of 1 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 9549999 |
| Expiration | Mar 10, 2030 |
| Applicant | GE HEALTHCARE |
| Drug Application |
|
from FDA Orange Book
WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 94)
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
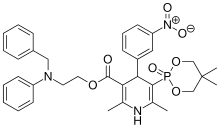
Efonidipine
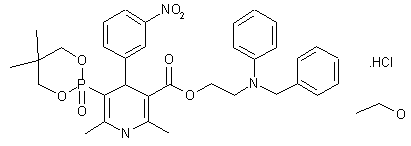
| Molecular Formula: | C36H45ClN3O8P |
|---|---|
| Molecular Weight: | 714.193 g/mol |
LD50:> 5 g/kg (R, p.o.)
Efonidipine Hydrochloride Ethanolate Bulk & Tablets 10 mg/20mg/40mg, 
Efonidipine (INN) is a dihydropyridine calcium channel blocker marketed by Shionogi & Co. of Japan. It was launched in 1995, under the brand name Landel (ランデル). The drug blocks both T-type and L-type calcium channels.[1] Drug Controller General of India (DCGI) granted approval to M/s. Zuventus pharma Ltd for marketing efonidipine under brand name Efnocar in India .[2]
Efonidipine is a dual Calcium Channel Blocker (L & T-type). It has a unique chemical structure. The phosphonate moiety (Figure 1) at the C5 position of the dihydropyridine ring is considered to be important for the characteristic pharmacological profile of the drug. (figure-1)

Figure-1:Efonidipine: Chemical Structure
Efonidipine, a new generation dihydropyridine (DHP) calcium channel blocker, inhibits both L-type and T-type calcium channels.[1]
Peak plasma concentration is achieved in about 1.5 to 3.67 hours after administration. Half life is approximately 4 hours. The pharmacokinetic parameters of Efonidipine are depicted in Table-1.
Table 1: PK Parameters in Adult Healthy Male Subjects
| Variable | Efonidipine | |
| Mean | Range | |
| Cmax(ng/ml) | 36.25 | 9.66-66.91 |
| Tmax (hour) | 2.59 | 1.50-3.67 |
| T1/2 (hour) | 4.18 | 2.15-6.85 |
*Data on file
Efonidipine has a slow onset and a long duration of action. This unique characteristic of Efonidipine is because of the following reasons:[13]
Efonidipine is primarily metabolized in the liver. The important metabolites are N-dephenylated Efonidipine (DPH), deaminated Efonidipine (AL) and N-debenzylated Efonidipine (DBZ). DBZ and DPH exhibit activity as calcium antagonists. The vasodilating properties of DBZ and DPH were about two-thirds and one-third respectively than that of the parent compound. Results suggest that the majority of the pharmacological effect after oral dosing of Efonidipine hydrochloride in man is due to unchanged compound and its metabolites make a small contribution to the pharmacological effect.[14]
Biliary route is the main pathway of excretion. No significant amount of unchanged drug was excreted in urine. In the urine collected for 24 h after an oral dosing, 1.1 % of the dose was excreted as deaminated Efonidipine, and 0.5% as a pyridine analogue of deaminated Efonidipine.
The common side effects are hot flushes, facial flushing and headache. In addition, elevation in serum total cholesterol, ALT (SGPT), AST (SGOT) and BUN may occur. Frequent urination, pedal edema, increased triglycerides occurs in less than 0.1%.[17]
One common adverse effect of the L-type Ca2+ channel blockers like Amlodipine is vasodilatory Pedal edema. Combined L-/T-type Ca2+ channel blockers, such as Efonidipine, display antihypertensive efficacy similar to their predecessors (Amlodipine) with much less propensity of pedal edema formation. Efonidipine equalizes the hydrostatic pressure across the capillary bed through equal arteriolar and venular dilatation, thus reducing vasodilatory edema. These incremental microcirculatory benefits of efonidipine over the conventional L-type Ca2+ channel blockers (Amlodipine) are likely attributed to their additional T-type Ca2+ channel blocking properties and the increased presence of T-type Ca2+channels in the microvasculature (e.g. arterioles, capillaries, venules etc).[18]
Among the CCBs, Efonidipine (<0.1%)[17] has lowest incidence of pedal edema compared to amlodipine ( 5-16%)[19], cilnidipine (5%)[20], benidipine (5%)[21] and azelnidipine (15.5%).[22]
The drug should be started at low dose (20 mg/day) in elderly. Patient should be carefully observed for development of hypo-tension. Dose may be halved if there is intolerance to the 20 mg/day dosage regimen.
The drug should not be administered to pregnant women and women suspected of being pregnant. Administration to lactating women should be avoided unless benefit significantly surpasses the risk to the child. Mothers on Efonidipine treatment should avoid breast feeding.
Safety of Efonidipine in low birth weight infants, newborns, infants and children has not been established.
Efonidipine is unique among clinically available CCBs. Its antihypertensive efficacy is superior or at par with other CCBs. But, in terms of pleiotropic effects leading to enhanced cerebral, cardiac and renal protection, Efonidipine scores over the other CCBs.
1. Better renoprotection by:
2. Preferred in angina with hypertension due to negative chronotropic action[26]
3. Better control of reflex tachycardia[3]
4. Reduces cardiac remodelling, arterial stiffness and prevents atherogenesis[27]
5. More useful in patients with diabetes & nephropathy[28]
6. Better protection against cardiac hypertrophy by significant reduction in LVMI[29]
7. Less adverse effects compared to Amlodipine[30]
8. Reduces endothelial dysfunction and oxidative stress(anti-oxidant property)[10]
1. Strong negative chronotropic effect (less tachycardia) compared to Cilnidipine[3]
2. Significant improvement in exercise tolerance.[31]Better choice in hypertensive patients with angina.
3. Better BP control by marked urinary Na+ excretion[32]
4. Better renoprotection by:
5. Better choice in diabetic hypertensives[36]
6. Prevents cardiac remodelling by suppression of aldosterone secretion[5]
7. Superior anti-oxidant activity[10]
8. Less adverse effects compared to Cilnidipine[30]
L & T-type CCBs have invoked a lot of interest in the management of hypertension because of their unique pharmacological profile. Several novel agents have been developed including Azelnidipine, Barnidipine, Benidipine, Efonidipine, Manidipine and Nilvadipine. Among all the agents, Efonidipine has emerged as the best among its peers. The advantages of Efonidipine over Benidipine are summarized below.
1. More selective blockade of T-type calcium channels [37][38]
2. More balanced renal arteriolar dilatation than benidipine[37][38]
3. Superior anti-proteinuric effect [15]
4. Greater reduction of serum aldosterone [39]
5. Renoprotection by reducing plasma renin unlike Benidipine [39]
6. Greater negative chronotropic effect
7. Efonidipine has anti-platelet activity[12]
8. Efonidipine reduces Insulin Resistance [40]
9. Significantly lower incidence of pedal edema & constipation compared to Benidipine
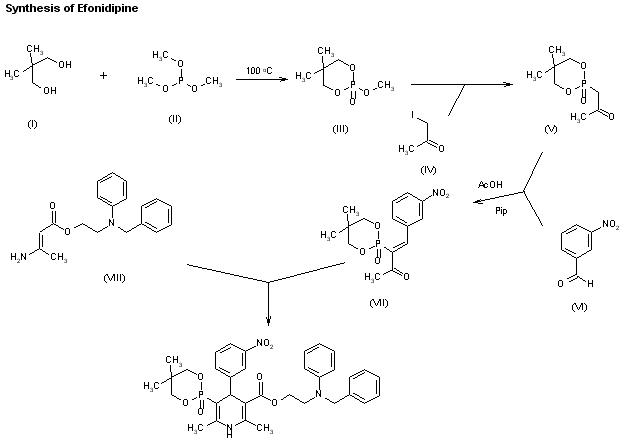
A new synthesis for (4S)-efonidipine has been described: The reaction of 5,5-dimethyl-2-(2-oxopropyl)-1,3,2-dioxaphosphorinan-2-one (I) with dimorpholino(3-nitrophenyl)methane (II) by means of trifluoroacetic acid in hot toluene gives 5,5-dimethyl-2-[1-acetyl-2-(3-nitrophenyl)vinyl]-1,3,2-dioxaphosphorina n-2-one (III), which is cyclized with 3-aminocrotonic acid 2(S)-methoxy-2-phenylethyl ester (IV) in refluxing toluene; the recrystallization of the resulting product affords 5-(5,5-dimethyl-2-oxo-1,3,2-dioxaphosphorinan-2-yl)-2,6-dimethyl-4(S)-(3-nitrophenyl)-1,4-dihydropyridine-3-carboxylic acid 2(S)-methoxy-2-phenylethyl ester (V). The protection of the NH group of (V) with chloromethyl methyl ether and NaH in THF yields the N-methoxymethyl derivative (VI), which is transesterified with 2-(N-benzyl-N-methylamino)ethanol (VII) and NaH in DMSO, giving the protected final product (VIII). Finally, this compound is deprotected with HCl in ethanol.
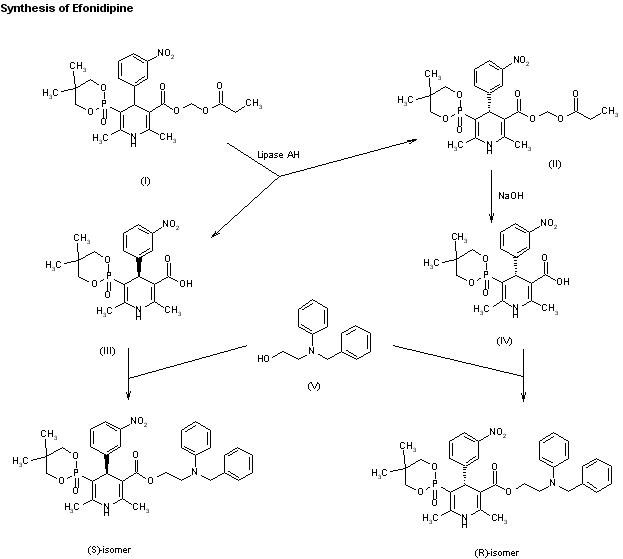
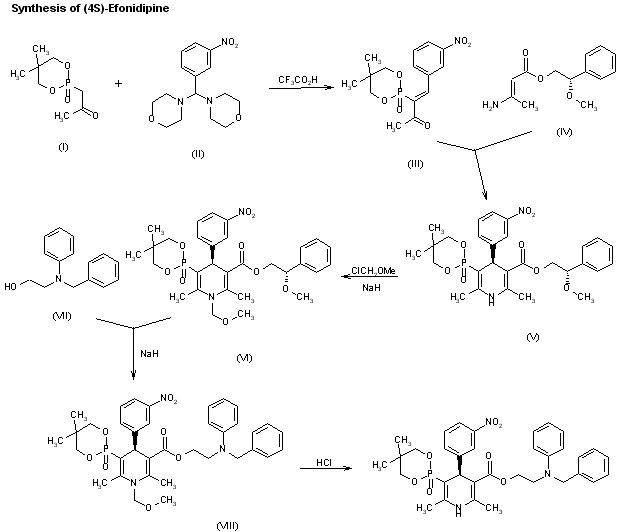
An enantioselective synthesis of efonidipine has been described: The enantioselective hydrolysis of 5-(5,5-dimethyl-2-oxo-1,3,2-dioxaphosphorinan-2-yl)-2,6-dimethyl-4-(3-n itrophenyl)-1,4-dihydropyridine-3-carboxylic acid propionyloxymethyl ester (I) with lipase AH in 2,5-dimethyltetrahydrofuran saturated with water gives the corresponding free acid of the (S)-isomer (III), while the propionyloxymethyl ester of the (R)-isomer (II) remains undisturbed. After chromatographic separation, the (R)-ester (II) is hydrolyzed with NaOH in methanol to the (R)-acid (IV). Finally, both enantiomerically pure acids (III) and (IV) are separately esterified with 2-(N-benzyl-N-phenylamino)ethanol in the usual way
CLIP
PAPER
Synthesis of 1,4-dihydropyridine-5-phosphonates and their calcium antagonistic and antihypertensive activities: Novel calcium-antagonist 2-[benzyl(phenyl)amino]ethyl 5-(5,5-dimethyl-2-oxo-1,3,2-dioxaphosphorinan-2-yl)-1,4-dihydro-2, 6-dimethyl-4-(3-nitrophenyl)-3-pyridinecarboxylate hydrochloride ethanol (NZ-105) and its crystal structure
Chem Pharm Bull 1992, 40(9): 2362
PATENT
IN 201501586
http://ipindiaservices.gov.in/PatentSearch/PatentSearch/ViewPDF
|title= (help)|title= (help)|title= (help) |
|
| Clinical data | |
|---|---|
| Trade names | Landel (ランデル) |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration |
Oral |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C34H38N3O7P |
| Molar mass | 631.65 g/mol |
| 3D model (JSmol) | |
///////////Efonidipine, エホニジピン, IND 2017, Landel , NZ 105, Efonidipine Hydrochloride Ethanolate
CC1=C(C(C(=C(N1)C)P2(=O)OCC(CO2)(C)C)C3=CC(=CC=C3)[N+](=O)[O-])C(=O)OCCN(CC4=CC=CC=C4)C5=CC=CC=C5
Chemists, engineers, scientists, lend us your ears… Carbon capture, utilisation, and storage (CCUS) is among the largest challenges on the horizon and we need your help. In this perspective, we focus on identifying the critical research needs to make CCUS a reality, with an emphasis on how the principles of green chemistry (GC) and green engineering can be used to help address this challenge. We identify areas where GC principles can readily improve the energy or atom efficiency of processes or reduce the environmental impact. Conversely, we also identify dilemmas where the…
View original post 115 more words


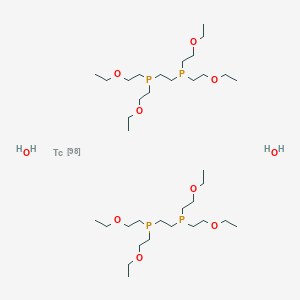
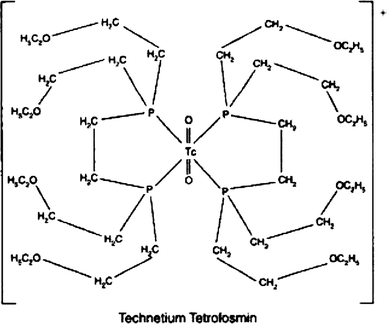
Technetium (99mTc) tetrofosmin, 99mTc-Tetrofosmin
テトロホスミンテクネチウム (99mTc)
| Formula | C36H80O10P4Tc |
|---|---|
| Molar mass | 895.813 g/mol |
| CAS Number |
|
|---|
UNII42FOP1YX93
2-[bis(2-ethoxyethyl)phosphanyl]ethyl-bis(2-ethoxyethyl)phosphane;technetium-98;dihydrate
Technetium Tc 99m tetrofosmin; Technetium Tc-99m tetrofosmin; TECHNETIUM TC-99M TETROFOSMIN KIT; Tc-99m tetrofosmin; Technetium-99 tetrofosmin; Technetium (99mTc) tetrofosmin
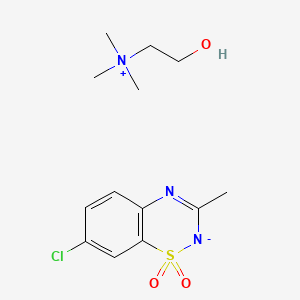
Technetium Tc-99m Tetrofosmin is a radiopharmaceutical consisting of tetrofosmin, composed of two bidentate diphosphine ligands chelating the metastable radioisotope technetium Tc-99 (99mTc), with potential imaging activity upon SPECT (single photon emission computed tomography). Upon administration, technetium Tc 99m tetrofosmin is preferentially taken up by, and accumulates in, myocardial cells. Upon imaging, myocardial cells can be visualized and changes in ischemia and/or perfusion can be detected.
Technetium Tc-99m tetrofosmin is a drug used in nuclear myocardial perfusion imaging. The radioisotope, technetium-99m, is chelated by two 1,2-bis[di-(2-ethoxyethyl)phosphino]ethane ligands which belong to the group of diphosphines and which are referred to as tetrofosmin. It is a lipophilic technetium phosphine dioxo cation that was formulated into a freeze-dried kit which yields an injection.[A31592] Technetium Tc-99m tetrofosmin was developed by GE Healthcare and FDA approved on February 9, 1996.
Technetium Tc-99m tetrofosmin is a drug used in nuclear myocardial perfusion imaging. The radioisotope, technetium-99m, is chelated by two 1,2-bis[di-(2-ethoxyethyl)phosphino]ethane ligands which belong to the group of diphosphines and which are referred to as tetrofosmin. It is a lipophilic technetium phosphine dioxo cation that was formulated into a freeze-dried kit which yields an injection.[1] Technetium Tc-99m tetrofosmin was developed by GE Healthcare and FDA approved on February 9, 1996.
Technetium (99mTc) tetrofosmin is a drug used in nuclear medicine cardiac imaging. It is sold under the brand name Myoview (GE Healthcare). The radioisotope, technetium-99m, is chelated by two 1,2-bis[di-(2-ethoxyethyl)phosphino]ethane ligands which belong to the group of diphosphines and which are referred to as tetrofosmin.[1][2]
Tc-99m tetrofosmin is rapidly taken up by myocardial tissue and reaches its maximum level in approximately 5 minutes. About 66% of the total injected dose is excreted within 48 hours after injection (40% urine, 26% feces). Tc-99m tetrofosmin is indicated for use in scintigraphic imaging of the myocardium under stress and rest conditions. It is used to determine areas of reversible ischemia and infarcted tissue in the heart. It is also indicated to detect changes in perfusion induced by pharmacologic stress (adenosine, lexiscan, dobutamine or persantine) in patients with coronary artery disease. Its third indication is to assess left ventricular function (ejection fraction) in patients thought to have heart disease. No contraindications are known for use of Tc-99m tetrofosmin, but care should be taken to constantly monitor the cardiac function in patients with known or suspected coronary artery disease. Patients should be encouraged to void their bladders as soon as the images are gathered, and as often as possible after the tests to decrease their radiation doses, since the majority of elimination is renal. The recommended dose of Tc-99m tetrofosmin is between 5 and 33 millicuries (185-1221 megabecquerels). For a two-dose stress/rest dosing, the typical dose is normally a 10 mCi dose, followed one to four hours later by a dose of 30 mCi. Imaging normally begins 15 minutes following injection.[3]

Amersham (formerly Nycomed Amersham , now GE Healthcare ) has developed and launched 99mTc-tetrofosmin (Myoview) as an injectable nuclear imaging agent for ischemic heart disease in several major territories and for use in detecting breast tumors
Technetium (99mTc) tetrofosmin is a drug used in nuclear medicine cardiac imaging. It is sold under the brand name Myoview (GE Healthcare). The radioisotope, technetium-99m, is chelated by two 1, 2-bis-[bis-(2-ethoxyethyl)phosphino] ethane ligands, which belong to the group of diphosphines and which are referred to as tetrofosmin and has the structural Formula 1 :
Formula 1
99mTc -based radiopharmaceuticals are commonly used in diagnostic nuclear medicine, especially for in vivo imaging (e.g. via immunoscintigraphy or radiolabeling). Usually cold kits are manufactured in advance in accordance with strict requirements of Good Manufacturing Practice (GMP) Guidelines, containing the chemical ingredients (e.g. 99mTc -coordinating ligands, preservatives) in lyophilized form. The radioactive isotope 99mTc (ti/2 = 6h) is added to those kits shortly before application to the patient via intravenous or subcutaneous injection.
Tc-99m tetrofosmin is rapidly taken up by myocardial tissue and reaches its maximum level in approximately 5 minutes. About 66% of the total injected dose is excreted within 48 hours after injection (40% urine, 26% feces). Tc-99m tetrofosmin is indicated for use in scintigraphic imaging of the myocardium under stress and rest conditions. It is used to determine areas of reversible ischemia and infarcted tissue in the heart. It is also indicated to detect changes in perfusion induced by pharmacologic stress (adenosine, lexiscan, dobutamine or persantine) in patients with coronary artery disease. Its third indication is to assess left ventricular function (ejection fraction) in patients thought to have heart disease. No contraindications are known for use of Tc-99m tetrofosmin, but care should be taken to constantly monitor the cardiac function in patients with known or suspected coronary artery disease. Patients should be encouraged to void their bladders as soon as the images are gathered, and as often as possible after the tests to decrease their radiation doses, since the majority of elimination is renal. The recommended dose of Tc-99m tetrofosmin is between 5 and 33 millicuries (185-1221 megabecquerels). For a two-dose stress/rest dosing, the typical dose is normally a 10 mCi dose, followed one to four hours later by a dose of 30 mCi. Imaging normally begins 15 minutes following injection.
99mTc -Tetrofosmin is also described to be useful for tumor diagnostics, in particular of breast cancer and parathyroid gland cancer, and for multidrug resistance (MDR) research.
US5045302 discloses 99mTc-coordinating diphosphine ligands (L), wherein one preferred example thereof is the ether functionalized diphosphine ligand l,2-bis[bis(2-ethoxy- ethyl)phosphino]ethane according to Formula 1, called tetrofosmin (“P53”), that forms a dimeric cationic technetium (V) dioxo phosphine complex, [TCO2L2] with 99mTc, useful as myocardial imaging agent. Example 1 of said patent described the process for preparing tetrofosmin by reacting ethyl vinyl ether, bis(diphosphino)ethane in the presence of a-azo-isobutyronitrile (AIBN) in a fischer pressure-bottle equipped with a teflon stirring bar followed by removal of volatile materials and non-distillable material obtained, as per below mentioned Scheme 1.
Scheme 1
Formula 2 Formula 3 Formula 1
CN 1184225 C discloses tetrofosmin salts containing chloride or bromide or aryl sulfonates as negatively charged counter ions, which can be used for the preparation of a 99mTc- Tetrofosmin radiopharmaceutical composition. According to this patent tetrofosmin hydrochloride is a viscous liquid. Own experiments of the inventors of the present invention revealed that the halide salts of tetrofosmin are hygroscopic oils, which are complicated to handle, e.g. when weighed. The oily and hygrospcopic
properties of tetrofosmin hydrochloride hampers its use in pharmaceutical preparations. Attempts to synthesize the subsalicylate salt of tetrofosmin failed because the starting material sulfosalicylic acid was not soluble in ether in the concentration specified in the patent (3.4 g in 15 ml).
WO2006/064175A1 discloses tetrofosmin was converted to tetrofosmin subsalicylate by reaction with 2.3 to 2.5 molar equivalents of 5-sulfosalicyclic acid at room temperature in ethanol, followed by recrystallisation from ethanol/ether.
WO2015/114002A1 relates to tetrafluoroborate salt of tetrafosmin and its process for the preparation thereof. Further this application also discloses one-vial and two vial kit formulation with tetrafluoroborate salt of tetrafosmin.
The article Proceedings of the International Symposium, 7th, Dresden, Germany, June 18-22, 2000 by Amersham Pharmacia Biotech UK Limited titled “The synthesis of [14C]tetrofosmin, a compound vital to the development of Myoview, Synthesis and Applications of Isotopically Labelled Compounds” disclosed a process for the preparation of tetrofosmin as per below mentioned Scheme 2:
Scheme 2
Formula 1A Formula 7
The starting material was bis(2- ethoxyethyl)benzylphosphine of Formula 4 . This was prepared from benzyl phosphonate, PhCH2P(0)(OEt)2 by reduction with lithium aluminium hydride to give the intermediate benzylphosphine, PhCH2PH2, followed by a photolysis reaction in the presence of ethyl vinyl ether to give compound of Formula 4. The compound of Formula 4 in acetonitrile was treated with dibromo[U-14C]ethane to give compound of Formula 6, further it was treated with excess of 30% aqueous sodium hydroxide in ethanol. The mixture was stirred at room temperature for 24 hours. The solvent was removed and the residue was treated with excess concentrated hydrochloric acid at 0°C. Aqueous work up gave compound of Formula 7. Then compound of Formula 7 in dry benzene was treated with hexachlorodisilane and hydrolysed with excess 30% aqueous sodium hydroxide at 0°C. Aqueous work up followed by flash column chromatography on silica gave [bisphosphinoethane- 1,2-14C]tetrofosmin of formula 1A.
The article Polyhedron (1995), 14(8), 1057-65, titled “Synthesis and characterization of Group 10 metal complexes with a new trifunctional ether phosphine. The X-ray crystal structures of bis[bis(2-ethoxyethyl)benzylphosphine]dichloronickel(II) and bis[bis(2-ethoxyethyl)benzylphosphine]chlorophenylnickel(II)” disclosed the process for the preparation of bis(2-ethoxyethyl)benzylphosphine as per below mentioned Scheme 3:
Scheme 3
Formula 8 Formula 9 Formula 4
The compound bis(2-ethoxyethyl)benzylphosphine of Formula 4 was prepared by first reduction of diethylbenzylphosphonate of Formula 8 using lithium aluminium hydride to obtain benzyl phosphine of Formula 9 followed by radical catalysed coupling reaction with ethyl vinyl ether carried out by using UV photolysis.
Tetrofosmin is extremely sensitive to atmospheric oxygen, which makes synthesis of the substance, as well as manufacturing and handling of the kit complicated as the substance has constantly to be handled in an oxygen free atmosphere.
High purity and stability under dry and controlled conditions are pivotal requirements for chemical compounds used as active ingredients in pharmaceuticals.
The processes disclosed in prior art for the preparation of compound of Formula 4 involves that coupling reaction of benzyl phosphine of Formula 9 with ethyl vinyl ether carried out by using photolytic conditions. Such technology is expensive as it requires separate instruments including isolated facility (to avoid the UV radiation exposure etc.), also it is not suitable for commercial scale production.
PATENT
WO-2018162964
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018162964&tab=PCTDESCRIPTION&maxRec=1000
Example 1
Preparation of benzyl phosphine:
A mixture of lithium aluminium hydride (25 g) in methyl tertiary butyl ether (MTBE) (800 ml) was cooled to 0 to 5°C and added a solution of diethylbenzylphosphonate in methyl tertiary butyl ether (100 g in 200ml). The temperature of reaction mixture was raised to 25 to 30 °C and stirred for 14 to 16 hour. After completion of the reaction, the reaction mixture was cooled to 0 to 5°C and 6N hydrochloric acid was added slowly. Further raised the temperature of reaction mixture to 25 to 30 °C and stirred for 30-45 minutes. The layers were separated, the aqueous layer was extracted with MTBE (250ml) and the combined organic layer was washed with deoxygenated water. The organic layer was dried over sodium sulfate and concentrated to obtain the title compound as non-distillable liquid.
Example 2
Preparation of benzylbis(2-ethoxyethyl)phosphane:
To a mixture of benzyl phosphine (obtained from example 1) and vinyl ethyl ether (250 ml) in pressure RB flask was added a-azo-isobutyronitrile (AIBN) (1.5g). The resulting reaction mixture was maintained at 80 to 90°C for 14 to 16 hours. The mixture was cooled to 20 to 30°C and AIBN (0.5g) added, then continued to heat the reaction mixture at 80 to 90°C for 6 to 7 hours. After completion of the reaction, the reaction mixture was allowed to cool to room temperature and distilled under vacuum to obtain title compound as an oil (107 g).
Example 3
Preparation of Ethane- 1,2-diylbis (benzylbis(2-ethoxyethyl) phosphonium) bromide:
To a mixture of benzylbis(2-ethoxyethyl)phosphane 107.g) in acetonitrile (100ml) in pressure bottle was added 1, 2-dibromoethane (30.5 g). The reaction mixture was maintained at 80 to 90°C for 20 to 25 hours. After completion of the reaction, the reaction mass was cooled to room temperature and stirred for 45 to 60 minutes to obtain the solid. To the solid obtained was added methyl tertiary butyl ether (MTBE) (500ml) and stirred at room temperature for 2 to 3 hour. The reaction mass was filtered, washed with MTBE and suck dried. Further the filtered solid was heated in acetone (400ml) at 50 to 55°C for 2 to 3 hour. Then cooled the reaction mixture to room temperature, stirred, filtered and washed with acetone to obtain the title compound as white solid. (85g)
Example 4
Preparation of Ethane- 1, 2-diylbis (bis (2-ethoxy ethyl) phosphine oxide):
To a mixture of Ethane- 1,2-diylbis (benzylbis(2-ethoxyethyl) phosphonium) bromide (80g) in ethanol (480 ml) was added an aq. solution of sodium hydroxide ( 48g in 160 ml water) at room temperature. The reaction mass was maintained at 25 to 35°C for 10 to 12 hour. After completion of the reaction, the reaction mass was cone, under vacuum to obtained the residue. The residue was dissolved in deoxygenated water (400 ml) and washed with MTBE (400 ml x 2). The layers were separated, the aqueous layer was cooled to 10 to 20°C and 6N hydrochloric acid (200 ml) was added slowly. Then extracted the aqueous layer with dichloromethane (2000 ml), washed the organic layer with deoxygenated water (160 ml), dried the organic layer using sodium sulfate, filtered, and distilled under vacuum to obtain the residue. Further MTBE (160 ml x 2) was added to the residue and continued distillation under vacuum, degassed to obtain the solid. To the obtained solid, MTBE (400 ml) was added and heated at 45 to 50°C for 1-2 hour, further slowly cooled the reaction mass to 25 to 30°C, filtered the solid product. Again MTBE (400 ml) was added to the solid product and heated at 45 to 50°C for 1-2 hour, further slowly cooled the reaction mass to 25 to 30°C, filtered, washed with MTBE and dried under vacuum to obtain the title compound as white solid (32g).
Example 5
Preparation of tetrofosmin free base:
To a mixture of ethane- 1, 2-diylbis (bis (2-ethoxyethyl) phosphine oxide (18g) in toluene (180ml) in pressure RB flask argon/nitrogen gas was purged for 5 minute and hexachlorodisilane (30g) was added. The reaction mixture was heated to 80 to 90°C, stirred for 10 to 12 hour, further slowly cooled to -5 to 0°C and slowly added 30% aqueous sodium hydroxide solution (45g sodium hydroxide in 150 ml deoxygenated water) the temperature of reaction mixture was raised to 25 to 30°C and stirred for 1 to 2 hour. The layers were separated and the aq. layer was extracted with Toluene (180 ml). The combined organic layer was washed with deoxygenated water (180 ml). Further dried the organic layer using sodium sulfate, distilled under vacuum to obtain the residue of tetrofosmin free base (15.5g).
Example 6
Preparation of tetrofosmin disulfosalicylate salt:
To the residue of tetrofosmin free base (15.5g) was added an aq. solution of 5-sulfosalicylic acid dihydrate (21.6g in 75ml deoxygenated water) and stirred at 25 to 30°C for 25 to 30 minutes. Further heated the reaction mass to 55 to 60°C, stirred for 15 to 30 minute, slowly cooled the reaction mass to 10 to 15°C and stirred for 1-2 hour. Filtered, washed with chilled deoxygenated water, and dried under vacuum to obtain the title compound as white solid. (30g).
Example 7
Preparation of Form J of tetrofosmin disulfosalicylate salt:
An aq. solution of 5-sulfosalicylic acid dihydrate (21.6g in 75ml deoxygenated water) was added slowly into tetrofosmin free base (15.5g) and stirred at room temperature for 30 to 40 minutes. The temperature of reaction mixture was further raised to 50 to 60°C, stirred for 20 to 30 minute, cooled the reaction mass to 10 to 15°C and stirred for 1-2 hour. Filtered, washed with chilled deoxygenated water, and dried under vacuum to obtain the title compound.
PATENT
EP337654 ,
PATENT
US9549999
| FDA Orange Book Patents: 1 of 1 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 9549999 |
| Expiration | Mar 10, 2030 |
| Applicant | GE HEALTHCARE |
| Drug Application |
|
 |
|
| Clinical data | |
|---|---|
| Routes of administration |
Intravenous |
| ATC code | |
| Pharmacokinetic data | |
| Bioavailability | N/A |
| Identifiers | |
| CAS Number |
|
| Chemical and physical data | |
| Formula | C36H80O10P4Tc |
| Molar mass | 895.813 g/mol |
Myoview Prescribing Information Page
//////////99mTc-Tetrofosmin, Technetium (99mTc) tetrofosmin, テトロホスミンテクネチウム (99mTc)
CCOCCP(CCOCC)CCP(CCOCC)CCOCC.CCOCCP(CCOCC)CCP(CCOCC)CCOCC.O.O.[Tc]

Tasimelteon
N-([(1R,2R)-2-(2,3-Dihydro-1-benzofuran-4-yl)cyclopropyl]methyl)propanamide,
609799-22-6 cas, BMS-214778; VEC-162, ATC:N05CH03
| Hetlioz | Vanda Pharmaceuticals, 2014 |
Approved fda 2014
EMA
Tasimelteon is a white to off-white crystalline powder, it is non hygroscopic, soluble in water across relevant pH values and freely soluble in alcohols, cyclohexane, and acetonitrile. Conducted in vivo studies demonstrate that tasimelteon is highly permeable substance. Photostability testing and testing on stress conditions demonstrated that the active substance degrades in light.
Tasimelteon exhibits stereoisomerism due to the presence of two chiral centres. Active substance is manufactured as a single, trans-1R,2R isomer. Enantiomeric purity is controlled routinely during manufacture of active substance intermediates by chiral HPLC/specific optical rotation and additionally controlled in the active substance. Stability data indicates tasimelteon is isomerically stable.
Polymorphism has been observed in polymorphic screening studies for tasimelteon and two forms have been identified. The thermodynamically more stable form has been chosen for development and the manufacturing process consistently yields active substance of single, desired polymorphic form. It was demonstrated that milling of the active substance does not affect polymorphic form. Polymorphism is additionally controlled in active substance release and shelf-life specifications using X-ray powder diffraction analysis.
Tasimelteon is synthesized in nine main steps using linear synthesis and using commercially available well-defined starting materials with acceptable specifications. Three intermediates are isolated for control of active substance quality including stereochemical control. The active substance is isolated by slow recrystallisation or precipitation of tasimelteon from an ethanol/water mixture which ensures the formation of desired polymorphic form. Up to two additional, optional recrystallisations may be performed for unmilled tasimelteon to ensure that milled tasimelteon active substance is of high purity. Seed crystals complying with active substance specifications can be used optionally. Active substance is jet milled (micronised) to reduce and control particle size, which is critical in finished product performance with regards to content uniformity and dissolution…….http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/003870/WC500190309.pdf
launched in 2014 in the U.S. by Vanda Pharmaceuticals for the treatment of non-24-hour sleep-wake disorder in totally blind subjects. In 2015, the European Committee for Medicinal Products of the European Medicines Agency granted approval for the same indication. In 2010 and 2011, orphan drug designations were assigned for the treatment of non-24 hour sleep/wake disorder in blind individuals without light perception in the U.S. and the E.U., respectively.
Tasimelteon (trade name Hetlioz) is a drug approved by the U.S. Food and Drug Administration (FDA)[2] in January 2014 for the treatment of non-24-hour sleep–wake disorder (also called Non-24, N24 and N24HSWD).[3] In June 2014, the European Medicines Agency accepted an EU filing application for tasimelteon[4] and in July 2015, the drug was approved in Europe for the treatment of non-24-hour sleep-wake rhythm disorder in totally blind adults,[5] but not in the rarer case of non-24 in sighted people.
Tasimelteon is a selective agonist for the melatonin receptors MT1 and MT2, similar to other members of the melatonin receptor agonistclass of which ramelteon (2005) and agomelatine (2009) were the first approved.[6] As a treatment for N24HSWD, as with melatonin or other melatonin derivatives, the patient may experience improved sleep timing while taking the drug. Reversion to baseline sleep performance occurs within a month of discontinuation.[7]
![]()
Tasimelteon (previously known as BMS-214,778) was developed for the treatment of insomnia and other sleep disorders. A phase II trial on circadian rhythm sleep disorders was concluded in March 2005.[8] A phase III insomnia trial was conducted in 2006.[9] A second phase III trial on insomnia, this time concerning primary insomnia, was completed in June 2008.[10] In 2010, the FDA granted orphan drug status to tasimelteon, then regarded as an investigational medication, for use in totally blind adults with N24HSWD.[11] (Through mechanisms such as easing the approval process and extending exclusivity periods, orphan drug status encourages development of drugs for rare conditions that otherwise might lack sufficient commercial incentive.)
On completion of Phase III trials, interpretations of the clinical trials by the research team concluded that the drug may have therapeutic potential for transient insomnia in circadian rhythm sleep disorders.[12] A year-long (2011–2012) study at Harvard tested the use of tasimelteon in blind subjects with non-24-hour sleep-wake disorder. The drug has not been tested in children nor in any non-blind people.
In May 2013 Vanda Pharmaceuticals submitted a New Drug Application to the Food and Drug Administration for tasimelteon for the treatment of non-24-hour sleep–wake disorder in totally blind people. It was approved by the FDA on January 31, 2014 under the brand name Hetlioz.[3] In the opinion of Public Citizen, an advocacy group, the FDA erroneously allowed it to be labelled without stating that it is only approved for use by totally blind people.[13] However, FDA updated its press release on Oct. 2, 2014 to clarify the approved use of Hetlioz, which includes both sighted and blind individuals. The update did not change the drug labeling (prescribing information).[14]
Experiments with rodents revealed fertility impairments, an increase in certain cancers, and serious adverse events during pregnancy at dosages in excess of what is considered the “human dose”.[15][16]
As expected, advisors to the US Food and Drug Administration have recommended approval of Vanda Pharmaceuticals’ tasimelteon, to be sold as Hetlioz, for the treatment of non-24-hour disorder in the totally blind.http://www.pharmatimes.com/Article/13-11-14/FDA_panel_backs_Vanda_body_clock_drug_for_blind.aspx
The master body clock controls the timing of many aspects of physiology, behavior and metabolism that show daily rhythms, including the sleep-wake cycles, body temperature, alertness and performance, metabolic rhythms and certain hormones which exhibit circadian variation. Outputs from the
suprachiasmatic nucleus (SCN) control many endocrine rhythms including those of melatonin secretion by the pineal gland as well as the control of Cortisol secretion via effects on the hypothalamus, the pituitary and the adrenal glands. This master body clock, located in the SCN, spontaneously generates rhythms of approximately 24.5 hours. These non-24-hour rhythms are synchronized each day to the 24-hour day-night cycle by light, the primary environmental time cue which is detected by specialized cells in the retina and transmitted to the SCN via the retino-hypothalamic tract. Inability to detect this light signal, as occurs in most totally blind individuals, leads to the inability of the master body clock to be reset daily and maintain entrainment to a 24-hour day.
Non-24-Hour Disorder, Non-24, also referred to as Non-24-Hour Sleep-Wake Disorder, (N24HSWD) or Non-24-Hour Disorder, is an orphan indication affecting approximately 65,000 to 95,000 people in the U.S. and 140,000 in Europe. Non- 24 occurs when individuals, primarily blind with no light perception, are unable to synchronize their endogenous circadian pacemaker to the 24-hour light/dark cycle. Without light as a synchronizer, and because the period of the internal clock is typically a little longer than 24 hours, individuals with Non-24 experience their circadian drive to initiate sleep drifting later and later each day. Individuals with Non-24 have abnormal night sleep patterns, accompanied by difficulty staying awake during the day. Non-24 leads to significant impairment, with chronic effects impacting the social and occupational functioning of these individuals.
In addition to problems sleeping at the desired time, individuals with Non-24 experience excessive daytime sleepiness that often results in daytime napping.
The severity of nighttime sleep complaints and/or daytime sleepiness complaints varies depending on where in the cycle the individual’s body clock is with respect to their social, work, or sleep schedule. The “free running” of the clock results in approximately a 1-4 month repeating cycle, the circadian cycle, where the circadian drive to initiate sleep continually shifts a little each day (about 15 minutes on average) until the cycle repeats itself. Initially, when the circadian cycle becomes desynchronous with the 24h day-night cycle, individuals with Non-24 have difficulty initiating sleep. As time progresses, the internal circadian rhythms of these individuals becomes 180 degrees out of synchrony with the 24h day-night cycle, which gradually makes sleeping at night virtually impossible, and leads to extreme sleepiness during daytime hours.
Eventually, the individual’s sleep-wake cycle becomes aligned with the night, and “free-running” individuals are able to sleep well during a conventional or socially acceptable time. However, the alignment between the internal circadian rhythm and the 24-hour day-night cycle is only temporary.
In addition to cyclical nighttime sleep and daytime sleepiness problems, this condition can cause deleterious daily shifts in body temperature and hormone secretion, may cause metabolic disruption and is sometimes associated with depressive symptoms and mood disorders.
It is estimated that 50-75% of totally blind people in the United States (approximately 65,000 to 95,000) have Non-24. This condition can also affect sighted people. However, cases are rarely reported in this population, and the true rate of Non-24 in the general population is not known.
The ultimate treatment goal for individuals with Non-24 is to entrain or synchronize their circadian rhythms into an appropriate phase relationship with the 24-hour day so that they will have increased sleepiness during the night and increased wakefulness during the daytime. Tasimelteon
Tasimelteon is a circadian regulator which binds specifically to two high affinity melatonin receptors, Mella (MT1R) and Mellb (MT2R). These receptors are found in high density in the suprachiasmatic nucleus of the brain (SCN), which is responsible for synchronizing our sleep/wake cycle. Tasimelteon has been shown to improve sleep parameters in prior clinical studies, which simulated a desynchronization of the circadian clock. Tasimelteon has so far been studied in hundreds of individuals and has shown a good tolerability profile.
Tasimelteon has the chemical name: tr ns-N-[[2-(2,3-dihydrobenzofuran- 4-yl)cycloprop-lyl] methyl] propanamide, has the structure of Formula I:

Formula I
and is disclosed in US 5856529 and in US 20090105333, both of which are incorporated herein by reference as though fully set forth.
Tasimelteon is a white to off-white powder with a melting point of about 78°C (DSC) and is very soluble or freely soluble in 95% ethanol, methanol, acetonitrile, ethyl acetate, isopropanol, polyethylene glycols (PEG-300 and PEG- 400), and only slightly soluble in water. The native pH of a saturated solution of tasimelteon in water is 8.5 and its aqueous solubility is practically unaffected by pH. Tasimelteon has 2-4 times greater affinity for MT2R relative to MTIR. It’s affinity (¾) for MTIR is 0.3 to 0.4 and for MT2R, 0.1 to 0.2. Tasimelteon is useful in the practice of this invention because it is a melatonin agonist that has been demonstrated, among other activities, to entrain patients suffering from Non-24.
Metabolites of tasimelteon include, for example, those described in “Preclinical Pharmacokinetics and Metabolism of BMS-214778, a Novel
Melatonin Receptor Agonist” by Vachharajani et al., J. Pharmaceutical Sci., 92(4):760-772, which is hereby incorporated herein by reference. The active metabolites of tasimelteon can also be used in the method of this invention, as can pharmaceutically acceptable salts of tasimelteon or of its active metabolites. For example, in addition to metabolites of Formula II and III, above, metabolites of tasimelteon also include the monohydroxylated analogs M13 of Formula IV, M12 of Formula V, and M14 of Formula VI.
Formula IV

Formula V
MO

Formula VI
Thus, it is apparent that this invention contemplates entrainment of patients suffering free running circadian rhythm to a 24 hour circadian rhythm by administration of a circadian rhythm regulator (i.e., circadian rhythm modifier) capable of phase advancing and/or entraining circadian rhythms, such as a melatonin agonist like tasimelteon or an active metabolite oftasimelteon or a pharmaceutically acceptable salt thereof. Other MT1R and MT2R agonists, i.e., melatonin agonists, can have similar effects on the master body clock. So, for example, this invention further contemplates the use of melatonin agonists such as but not limited to melatonin, N-[l-(2,3-dihydrobenzofuran-4- yl)pyrrolidin-3-yl]-N-ethylurea and structurally related compounds as disclosed in US 6,211,225, LY-156735 ((R)-N-(2-(6-chloro-5-methoxy-lH-indol- 3yl) propyl) acetamide) (disclosed in U.S. Patent No. 4,997,845), agomelatine (N- [2-(7-methoxy-l-naphthyl)ethyl]acetamide) (disclosed in U.S. Patent No.
5,225,442), ramelteon ((S)-N-[2-(l,6,7,8-tetrahydro-2H-indeno- [5,4-b] furan-8- yl)ethyl]propionamide), 2-phenylmelatonin, 8-M-PDOT, 2-iodomelatonin, and 6- chloromelatonin.
Additional melatonin agonists include, without limitation, those listed in U.S. Patent Application Publication No. 20050164987, which is incorporated herein by reference, specifically: TAK-375 (see Kato, K. et al. Int. J.
Neuropsychopharmacol. 2000, 3 (Suppl. 1): Abst P.03.130; see also abstracts P.03.125 and P.03.127), CGP 52608 (l-(3-allyl-4-oxothiazolidine-2-ylidene)-4- met- hylthiosemicarbazone) (See Missbach et al., J. Biol. Chem. 1996, 271, 13515-22), GR196429 (N-[2-[2,3,7,8-tetrahydro-lH-fur-o(2,3-g)indol-l- yl] ethyl] acetamide) (see Beresford et al., J. Pharmacol. Exp. Ther. 1998, 285, 1239-1245), S20242 (N-[2-(7-methoxy napth-l-yl) ethyl] propionamide) (see Depres-Brummer et al., Eur. J. Pharmacol. 1998, 347, 57-66), S-23478 (see Neuropharmacology July 2000), S24268 (see Naunyn Schmiedebergs Arch. June 2003), S25150 (see Naunyn Schmiedebergs Arch. June 2003), GW-290569, luzindole (2-benzyl-N-acetyltryptamine) (see U.S. Patent No. 5,093,352), GR135531 (5-methoxycarbonylamino-N-acetyltrypt- amine) (see U.S. Patent Application Publication No. 20010047016), Melatonin Research Compound A, Melatonin Agonist A (see IMSWorld R&D Focus August 2002), Melatonin
Analogue B (see Pharmaprojects August 1998), Melatonin Agonist C (see Chem. Pharm. Bull. (Tokyo) January 2002), Melatonin Agonist D (see J. Pineal Research November 2000), Melatonin Agonist E (see Chem. Pharm. Bull. (Tokyo) Febrary 2002), Melatonin Agonist F (see Reprod. Nutr. Dev. May 1999), Melatonin Agonist G (see J. Med. Chem. October 1993), Melatonin Agonist H (see Famaco March 2000), Melatonin Agonist I (see J. Med. Chem. March 2000), Melatonin Analog J (see Bioorg. Med. Chem. Lett. March 2003), Melatonin Analog K (see MedAd News September 2001), Melatonin Analog L, AH-001 (2-acetamido-8- methoxytetralin) (see U.S. Patent No. 5,151,446), GG-012 (4-methoxy-2- (methylene propylamide)indan) (see Drijfhout et al., Eur. J. Pharmacol. 1999, 382, 157-66), Enol-3-IPA, ML-23 (N-2,4-dinitrophenyl-5-methoxy-tryptamine ) (see U.S. Patent No. 4,880,826), SL-18.1616, IP-100-9 (US 5580878), Sleep Inducing Peptide A, AH-017 (see U.S. Patent No. 5,151,446), AH-002 (8-methoxy- 2-propionamido-tetralin) (see U.S. Patent No. 5,151,446), and IP-101.
Metabolites, prodrugs, stereoisomers, polymorphs, hydrates, solvates, and salts of the above compounds that are directly or indirectly active can, of course, also be used in the practice of this invention.
Melatonin agonists with a MT1R and MT2R binding profile similar to that of tasimelteon, which has 2 to 4 time greater specificity for MT2R, are preferred.
Tasimelteon can be synthesized by procedures known in the art. The preparation of a 4-vinyl-2,3-dihydrobenzofuran cyclopropyl intermediate can be carried out as described in US7754902, which is incorporated herein by reference as though fully set forth.
Pro-drugs, e.g., esters, and pharmaceutically acceptable salts can be prepared by exercise of routine skill in the art.
In patients suffering a Non-24, the melatonin and Cortisol circadian rhythms and the natural day/night cycle become desynchronized. For example, in patients suffering from a free-running circadian rhythm, melatonin and Cortisol acrophases occur more than 24 hours, e.g., >24.1 hours, prior to each previous day’s melatonin and Cortisol acrophase, respectively, resulting in desynchronization for days, weeks, or even months, depending upon the length of a patient’s circadian rhythm, before the melatonin, Cortisol, and day /night cycles are again temporarily synchronized.
Chronic misalignment of Cortisol has been associated with metabolic, cardiac, cognitive, neurologic, neoplastic, and hormonal disorders. Such disorders include, e.g., obesity, depression, neurological impairments.


INTRODUCTION
Tasimelteon has the chemical name: trans-N-[[2-(2,3-dihydrobenzofuran-4-yl)cycloprop-1yl]methyl]propanamide, has the structure of Formula I:

and is disclosed in U.S. Pat. No. 5,856,529 and in US 20090105333, both of which are incorporated herein by reference as though fully set forth.
Tasimelteon is a white to off-white powder with a melting point of about 78° C. (DSC) and is very soluble or freely soluble in 95% ethanol, methanol, acetonitrile, ethyl acetate, isopropanol, polyethylene glycols (PEG-300 and PEG-400), and only slightly soluble in water. The native pH of a saturated solution of tasimelteon in water is 8.5 and its aqueous solubility is practically unaffected by pH. Tasimelteon has 2-4 times greater affinity for MT2R relative to MT1R. It’s affinity (Ki) for MT1R is 0.3 to 0.4 and for MT2R, 0.1 to 0.2. Tasimelteon is useful in the practice of this invention because it is a melatonin agonist that has been demonstrated, among other activities, to entrain patients suffering from Non-24.
SYNTHESIS
(1R-trans)-N-[[2 – (2,3-dihydro-4 benzofuranyl) cyclopropyl] methyl] propanamide PATENT: BRISTOL-MYERS SQUIBB PRIORITY DATE: 1996 HYPNOTIC


PREPARATION OF XV
XXIV D-camphorsulfonic acid IS REACTED WITH THIONYL CHLORIDE TO GIVE
…………XXV (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonyl chloride
TREATED WITH
XXVI ammonium hydroxide
TO GIVE
XXVII (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonamide
TREATED WITH AMBERLYST15
….XXVIII (3aS, 6R) -4,5,6,7-tetrahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
TREATED WITH LAH, ie double bond is reduced to get
…..XV (3aS, 6R, 7aR)-hexahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide

Intermediate
I 3-hydroxybenzoic acid methyl ester
II 3-bromo-1-propene
III 3 – (2-propenyloxy) benzoic acid methyl ester
IV 3-hydroxy-2-(2-propenyl) benzoic acid methyl ester
V 2,3-dihydro-4-hydroxy-2-benzofurancarboxylic acid methyl ester
VI benzofuran-4-carboxylic acid methyl ester
VII benzofuran-4-carboxylic acid
VIII 2,3-dihydro-4-benzofurancarboxylic acid
IX 2,3-dihydro-4-benzofuranmethanol
X 2,3-dihydro-4-benzofurancarboxaldehyde
XI Propanedioic acid
XII (E) -3 – (2,3-dihydro-4-benzofuranyl) propenoic acid
XIII thionyl chloride
XIV (E) -3 – (2,3-dihydro-4-benzofuranyl) propenoyl chloride
XV (3aS, 6R, 7aR)-hexahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
XVI (3aS,6R,7aR)-1-[(E)-3-(2,3-dihydro-4-benzofuranyl)-1-oxo-2-propenyl]hexahydro-8,8-dimethyl-3H-3a,6-methano-2,1-benzisothiazole-2,2-dioxide
XVII (3aS,6R,7aR)-1-[[(1R,2R)-2-(2,3-dihydro-4-benzofuranyl)cyclopropyl]carbonyl]hexahydro-8,8-dimethyl-3H-3a,6-methano-2,1-benzisothiazole-2,2-dioxide
XVIII [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanemethanol
XIX [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanecarboxaldehyde
XX hydroxylamine hydrochloride
XXI [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanecarbaldehyde oxime
XXII [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanemethanamine
XXIII propanoyl chloride
XXIV D-camphorsulfonic acid
XXV (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonyl chloride
XXVI ammonium hydroxide
XXVII (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonamide
XXVIII (3aS, 6R) -4,5,6,7-tetrahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
Bibliography
– Patents: Benzofuran and dihydrobenzofuran melatonergic agents: US5856529 (1999)
Priority: US19960032689P, 10 Dec. 1996 (Bristol-Myers Squibb Company, U.S.)
– Preparation III (quinazolines): US2004044015 (2004) Priority: EP20000402845, 13 Oct. 2000
– Preparation of VII (aminoalkylindols): Structure-Activity Relationships of Novel Cannabinoid Mimetics Eissenstat et al, J.. Med. Chem. 1995, 38, 3094-3105
– Preparation XXVIII: Towson et al. Organic Syntheses, Coll. Vol. 8, p.104 (1993) Vol. 69, p.158 (1990)
– Preparation XV: Weismiller et al. Organic Syntheses, Coll. Vol. 8, p.110 (1993) Vol. 69, p.154 (1990).
– G. Birznieks et al. Melatonin agonist VEC-162 Improves sleep onset and maintenance in a model of transient insomnia. Sleep 2007, 30, 0773 Abstract.
-. Rajaratnam SM et al, The melatonin agonist VEC-162 Phase time immediately advances the human circadian system, Sleep 2006, 29, 0159 Abstract.
-. AK Singh et al, Evolution of a manufacturing route for a highly potent drug candidate, 229th ACS Natl Meet, March 13-17, 2005, San Diego, Abstract MEDI 576.
– Vachharajani NN et al, Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist, J Pharm Sci. 2003 Apr; 92 (4) :760-72.
. – JW Scott et al, Catalytic Asymmetric Synthesis of a melotonin antagonist; synthesis and process optimization. 223rd ACS Natl Meet, April 7-11, Orlando, 2002, Abstract ORGN 186.
SYNTHESIS CONSTRUCTION AS IN PATENT
GENERAL SCHEMES
Reaction Scheme 1

The syntheses of the 4-aryl-propenoic acid derivatives, 2 and 3, are shown in Reaction Scheme 1. The starting aldehydes, 1 , can be prepared by methods well known to those skilled in the art. Condensation of malonic acid with the aldehydes, 1, in solvents such as pyridine with catalysts such as piperidine or pyrrolidine, gives the 4-aryl- propenoic acid, 2. Subsequent conversion of the acid to the acid chloride using reagents such as thionyl chloride, phosphoryl chloride, or the like, followed by reaction with N,0-dimethyl hydroxylamine gives the amide intermediate 3 in good yields. Alternatively, aldehyde 1 can be converted directly to amide 3 using reagents such as diethyl (N-methoxy- N-methyl-carbamoylmethyl)phosphonate with a strong base such as sodium hydride.
Reaction Scheme 2

The conversion of the amide intermediate 3 to the racemic, trans- cyclopropane carboxaldehyde intermediate, 4, is shown in Reaction Scheme 2. Intermediate 3 was allowed to react with cyclopropanating reagents such as trimethylsulfoxonium iodide and sodium hydride in solvents such as DMF, THF, or the like. Subsequent reduction using reagents such as LAH in solvents such as THF, ethyl ether, or the like, gives the racemic, trans-cyclopropane carboxaldehyde intermediates, 4.
Reaction Scheme 3

Racemic cyclopropane intermediate 5 (R = halogen) can be prepared from intermediate 2 as shown in Reaction Scheme 3. Intermediate 2 was converted to the corresponding allylic alcohol by treatment with reducing agents such as sodium borohydride plus iodine in solvents such as THF. Subsequent acylation using reagents such as acetic anhydride in pyridine or acetyl chloride gave the allylic acetate which was allowed to react with cyclopropanating reagents such as sodium chloro-difluoroacetate in diglyme to provide the racemic, trans- cyclopropane acetate intermediates, 5. Reaction Scheme 4

The conversion of the acid 2 to the chiral cyclopropane carboxaldehyde intermediate, (-)-(trans)-4, is shown in Reaction Scheme 4. Intermediate 2 is condensed with (-)-2,10-camphorsultam under standard conditions, and then cyclopropanated in the presence of catalysts such as palladium acetate using diazomethane generated from reagents such as 1-methyl-3-nitro-1-nitrosoguanidine. Subsequent reduction using reagents such as LAH in solvents such as THF, followed by oxidation of the alcohol intermediates using reagents such as DMSO/oxalyl chloride, or PCC, gives the cyclopropane carboxaldehyde intermediate, (-)-(trans)-4, in good yields. The enantiomer, (+)-(trans)-4, can also be obtained employing a similar procedure using (+)-2,10- camphorsultam in place of (-)-2,10-camphorsultam.
When it is desired to prepare compounds of Formula I wherein m = 2, the alcohol intermediate may be activated in the conventional manner such as with mesyl chloride and treated with sodium cyanide followed by reduction of the nitrile group with a reducing agent such as LAH to produce the amine intermediate 6.
Reaction Scheme 5


Reaction Scheme 5 shows the conversion of intermediates 4 and 5 to the amine intermediate, 7, and the subsequent conversion of 6. or 7 to compounds of Formula I. The carboxaldehyde intermediate, 4, is condensed with hydroxylamine and then reduced with reagents such as LAH to give the amine intermediate, 7. The acetate intermediate 5 is hydrolyzed with potassium hydroxide to the alcohol, converted to the mesylate with methane sulfonyl chloride and triethyl amine in CH2CI2and then converted to the azide by treatment with sodium azide in solvents such as DMF. Subsequent reduction of the azide group with a reducing agent such as LAH produced the amine intermediate 7. Further reaction of 6 or 7 with acylating reagents gives compounds of Formula I. Suitable acylating agents include carboxylic acid halides, anhydrides, acyl imidazoles, alkyl isocyanates, alkyl isothiocyanates, and carboxylic acids in the presence of condensing agents, such as carbonyl imidazole, carbodiimides, and the like. Reaction Scheme 6

Reaction Scheme 6 shows the alkylation of secondary amides of Formula I (R2 = H) to give tertiary amides of Formula I (R2 = alkyl). The secondary amide is reacted with a base such as sodium hydride, potassium tert-butoxide, or the like, and then reacted with an alkylating reagent such as alkyl halides, alkyl sulfonate esters, or the like to produce tertiary amides of Formula I.
Reaction Scheme 7

Reaction Scheme 7 shows the halogenation of compounds of Formula I. The carboxamides, i (Q1 = Q2 = H), are reacted with excess amounts of halogenating agents such as iodine, N-bromosuccinimide, or the like to give the dihalo-compounds of Formula I (Q1 = Q2 = halogen). Alternatively, a stoichiometric amount of these halogenating agents can be used to give the monohalo-compounds of Formula I (Q1 = H, Q2 = halogen; or Q1 = halogen, Q2 = H). In both cases, additives such as lead IV tetraacetate can be used to facilitate the reaction. Biological Activity of the Compounds
The compounds of the invention are melatonergic agents. They have been found to bind human melatonergic receptors expressed in a stable cell line with good affinity. Further, the compounds are agonists as determined by their ability, like melatonin, to block the forskolin- stimulated accumulation of cAMP in certain cells. Due to these properties, the compounds and compositions of the invention should be useful as sedatives, chronobiotic agents, anxiolytics, antipsychotics, analgesics, and the like. Specifically, these agents should find use in the treatment of stress, sleep disorders, seasonal depression, appetite regulation, shifts in circadian cycles, melancholia, benign prostatic hyperplasia and related conditions
EXPERIMENTAL PROCEDURES
SEE ORIGINAL PATENT FOR CORECTIONS
Preparation 1
Benzofuran-4-carboxaldehyde
Step 1 : N-Methoxy-N-methyl-benzofuran-4-carboxamide
A mixture of benzofuran-4-carboxylic acid [Eissenstat, et al.. J. Medicinal Chemistry, 38 (16) 3094-3105 (1995)] (2.8 g, 17.4 mmol) and thionyl chloride (25 mL) was heated to reflux for 2 h and then concentrated in vacuo. The solid residue was dissolved in ethyl acetate (50 mL) and a solution of N,O-dimethylhydroxylamine hydrochloride (2.8 g) in saturated NaHC03(60 mL) was added with stirring. After stirring for 1.5 h, the ethyl acetate layer was separated. The aqueous layer was extracted with ethyl acetate. The ethyl acetate extracts were combined, washed with saturated NaHCO3 and concentrated in vacuo to give an oil (3.2 g, 95.4%).
Step 2: Benzofuran-4-carboxaldehyde
A solution of N-methoxy-N-methyl-benzofuran-4-carboxamide (3.2 g, 16.6 mmol) in THF (100 mL) was cooled to -45°C and then LAH (0.7 g, 18.7 mmol) was added. The mixture was stirred for 15 min, allowed to warm to -5°C, and then recooled to -45°C. Saturated KHS04 (25 mL) was added with vigorous stirring, and the mixture was allowed to warm to room temperature. The precipitate was filtered and washed with acetone. The filtrate was concentrated in vacuo to give an oil (2.3 g, 94%). Preparation 2
2,3-Dihydrobenzofuran-4-carboxaldehyde
Step 1 : 2,3-Dihydrobenzofuran-4-carboxylic acid
Benzofuran-4-carboxylic acid (10.0 g, 61 .7 mmol) was hydrogenated (60 psi) in acetic acid (100 mL) over 10% Pd/C (2 g) for 12 hr. The mixture was filtered and the filtrate was diluted with water (500 mL) to give 2,3- dihydrobenzofuran-4-carboxylic acid as a white powder (8.4 g, 83%). A sample was recrystallized from isopropanol to give fine white needles (mp: 185.5-187.5°C).
Step 2: (2,3-Dihydrobenzofuran-4-yl)methanol
A solution of 2,3-dihydrobenzofuran-4-carboxylic acid (10 g, 61 mmol) in THF (100 mL) was stirred as LAH (4.64 g, 122 mmol) was slowly added. The mixture was heated to reflux for 30 min. The mixture was cooled and quenched cautiously with ethyl acetate and then with 1 N HCI (150 mL). The mixture was then made acidic with 12 N HCI until all the inorganic precipitate dissolved. The organic layer was separated, and the inorganic layer was extracted twice with ethyl acetate. The organic layers were combined, washed twice with brine, and then concentrated in vacuo. This oil was Kϋgelrohr distilled to a clear oil that crystallized upon cooling (8.53 g, 87.6%).
Step 3: 2.3-Dihydrobenzofuran-4-carboxaldehyde
DMSO (8.10 mL, 1 14 mmol) was added at -78°C to a stirred solution of oxalyl chloride in CH2CI2 (40 mL of a 2M solution). A solution of (2,3- dihydrobenzofuran-4-yl)methanol (8.53 g, 56.9 mmol) in CH2CI2 (35 mL) was added dropwise, and the solution stirred at -78°C for 30 min. Triethyl amine (33 mL, 228 mmol) was added cautiously to quench the reaction. The resulting suspension was stirred at room temperature for 30 min and diluted with CH2CI2 (100 mL). The organic layer was washed three times with water, and twice with brine, and then concentrated in vacuo to an oil (8.42 g, 100%) that was used without purification.
Preparation 16
(±)-(trans)-2-(2,3-Dihyd robenzofuran-4-yl)cyclopropane- carboxaldehyde
Step 1 : (±Htrans)-N-Methoxy-N-methyl-2-(2.3-dihydrobenzofuran-4- yhcyclopropanecarboxamide
Trimethylsulfoxonium iodide (9.9 g, 45 mmol) was added in small portions to a suspension of sodium hydride (1 .8 g, 45 mmol) in DMF (120 mL). After the foaming had subsided (10 min), a solution of (trans)- N-methoxy-N-methyl-3-(2,3-dihydrobenzofuran-4-yl)propenamide (3.5 g, 15 mmol) in DMF (60 mL) was added dropwise, with the temperature maintained between 35-40°C. The mixture was stirred for 3 h at room temperature. Saturated NH4CI (50 mL) was added dropwise and the mixture was extracted three times with ethyl acetate. The organic extracts were combined, washed with H2O and brine, dried over K2CO3, and concentrated in vacuo to give a white wax (3.7 g, 100%).
Step 2: (±)-(trans)- 2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane- carboxaldehyde
A solution of (±)-(trans)-N-methoxy-N-methyl-2-(2,3-dihydrobenzofuran- 4-yl)cyclopropanecarboxamide (3.7 g, 15 mmol) in THF (10 mL) was added dropwise to a rapidly stirred suspension of LAH (683 mg, 18 mmol) in THF (50 mL) at -45°C, maintaining the temperature below -40°C throughout. The cooling bath was removed, the reaction was allowed to warm to 5°C, and then the reaction was immediately recooled to -45°C. Potassium hydrogen sulfate (3.4 g, 25.5 mmol) in H20 (50 mL) was cautiously added dropwise, the temperature maintained below – 30°C throughout. The cooling bath was removed and the suspension was stirred at room temperature for 30 min. The mixture was filtered through Celite and the filter cake was washed with ether. The combined filtrates were then washed with cold 1 N HCI, 1 N NaOH, and brine. The filtrates were dried over MgSO4, and concentrated in vacuo to give a clear oil (2.6 g, 99%).
Preparation 18
(-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane-carboxaldehyde
Step 1 : (-Htrans)-N-[3-(2.3-Dihvdrobenzofuran-4-yl)-propenoyll-2.10- camphorsultam
To a solution of (-)-2,10-camphorsultam (8.15 g, 37.9 mmol) in 50 mL toluene at 0°C was added sodium hydride (1.67 g, 41.7 mmol). After stirring for 0.33 h at 0°C and 0.5 h at 20°C and recooling to 0°C, a solution of 3-(2,3-dihydrobenzofuran-4-yl)-2-propenoyl chloride
(37.9 mmol), prepared in situ from the corresponding acid and thionyl chloride (75 mL), in toluene (50 mL), was added dropwise. After stirring for 18 h at 20°C, the mixture was diluted with ethyl acetate and washed with water, 1 N HCI, and 1 N NaOH. The organic solution was dried and concentrated in vacuo to give 15.8 g of crude product. Recrystallization form ethanol-methanol (600 mL, 1 :1) gave the product (13.5 g, 92%, mp 199.5-200°C).
Step 2: (-)-N-[[(trans)-2-(2,3-Dihydrobenzofuran-4-yl)-cyclopropylj- carbonylj-2, 10-camphorsultam
1 -Methyl-3-nitro-1 -nitrosoguanidine (23.88g 163 mmol) was added in portions to a mixture of 10 N sodium hydroxide (60 mL) and ether (200 mL) at 0°C. The mixture was shaken vigorously for 0.25 h and the ether layer carefully decanted into a solution of (-)-N-[3-(2,3-dihydrobenzofuran-4-yl)-2-propenoyl]-2,10-camphorsultam (9.67 g, 25 mmol) and palladium acetate (35 mg) in methylene chloride (200 mL). After stirring for 18 h, acetic acid (5 mL) was added to the reaction and the mixture stirred for 0.5 h. The mixture was washed with 1 N HCI, 1 N NaOH and brine. The solution was dried, concentrated in vacuo and the residue crystallized twice from ethanol to give the product (6.67 g, 66.5%, mp 157-159°C).
Step 3: (-)-(trans)-2-(2,3-Dihydrobenzofuran-4-yl)cyclopropane- methanol
A solution of (-)-N-[(trans)-2-(2,3-dihydrobenzofuran-4-yl)cyclo-propanecarbonylj-2,10-camphorsultam (4.3 g, 10.7 mmol) in THF (50 mL) was added dropwise to a mixture of LAH (0.81 g, 21.4 mmol) in THF (50 mL) at -45°C. The mixture was stirred for 2 hr while it warmed to 10°C. The mixture was recooled to -40°C and hydrolyzed by the addition of saturated KHS0 (20 mL). The mixture was stirred at room temperature for 30 minutes and filtered. The precipitate was washed twice with acetone. The combined filtrate and acetone washes were concentrated in vacuo. The gummy residue was dissolved in ether, washed with 1 N NaOH and 1 N HCI, and then dried in vacuo to give the product (2.0 g, 98.4%).
Step 4: (-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane- carboxaldehyde DMSO (1.6 g, 21 mmol) was added to oxalyl chloride in CH2CI2(7.4 mL of 2 M solution, 14.8 mmole) at -78°C. The (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)-cyclopropylmethanol (2.0 g, 10.5 mmol) in CH2CI2(15 mL) was added. The mixture was stirred for 20 min and then triethylamine (4.24 g, 42 mmol) was added. The mixture was warmed to room temperature and stirred for 30 min. The mixture was diluted with CH2CI2 and washed with water, 1 N HCI, and then 1 N NaOH. The organic layer was dried and concentrated iι> vacuo to give the aldehyde product (1.98 g, 100%).
Preparation 24
(-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane-methanamine A mixture of (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)cyclopropane-carboxaldehyde (1.98 g, 10.5 mmol), hydroxylamine hydrochloride (2.29 g, 33 mmol), and 30% NaOH (3.5 mL, 35 mmol), in 5:1
ethanol/water (50 mL) was heated on a steam bath for 2 h. The solution was concentrated in vacuo. and the residue mixed with water. The mixture was extracted with CH2CI2. The organic extracts were dried and concentrated in vacuo to give a solid which NMR analysis showed to be a mixture of the cis and trans oximes. This material was dissolved in THF (20 mL) and added to solution of alane in THF [prepared from LAH (1.14 g, 30 mmol) and H2S04 (1.47 g, 15 mmol) at 0°Cj. The reaction was stirred for 18 h, and quenched successively with water (1.15 mL), 15% NaOH (1.15 mL), and then water (3.45 mL). The mixture was filtered and the filtrate was concentrated in vacuo. The residue was mixed with ether and washed with water and then 1 N HCI. The acid washes were made basic and extracted with CH2CI . The extracts were dried and concentrated in vacuo to give the amine product (1.4 g, 70.5%). The amine was converted to the fumarate salt in ethanol (mp: 197-198°C).
Anal. Calc’d for C12H15NO • C4H404: C, 62.94; H, 6.27; N, 4.59.
Found: C, 62.87; H, 6.31 ; N, 4.52.
FINAL PRODUCT TASIMELTEON
Example 2
(-)-(trans)-N-[[2-(2,3-Dihydrobenzofuran-4-yl)cycloprop-1-yl]methyl]propanamide
This compound was prepared similar to the above procedure using propionyl chloride and (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)- cyclopropanemethanamine to give an oil that solidified upon standing to an off-white solid (61 %, mp: 71-72°C). IR (NaCI Film): 3298, 1645, 1548, 1459, 1235 cm“1.
Mo5 : -17.3°
Anal. Calc’d for C15H19N02: C, 73.44; H, 7.87; N, 5.71 . Found: C, 73.28; H, 7.68; N, 5.58

Tasimelteon (Hetlioz)Tasimelteon, which is marketed by Vanda Pharmaceuticals as Hetlioz and developed in partnership with Bristol-Myers Squibb,is a drug that was approved by the US FDA in January 2014 for the treatment of non-24-hour sleep–wake disorder (also called Non-24, N24 and N24HSWD).234 Tasimelteon is a melatonin MT1
and MT2 receptor agonist; because it exhibits a greater affinity to the MT2 receptor than MT1, is also known as Dual Melatonin
Receptor Agonist.234 Two randomized controlled trials (phases II
and III) demonstrated that tasimelteon improved sleep latency
and maintenance of sleep with a shift in circadian rhythms, and
therefore has the potential to treat patients with transient insomnia
associated with circadian rhythm sleep disorders.235 Preclinical
studies showed that the drug has similar phase-shifting properties
to melatonin, but with less vasoconstrictive effects.236 The most
likely scale preparation of the drug, much of which has been published
in the chemical literature, is described below in Scheme 44.
Activation of commercial bis-ethanol 250 with 2.5 equivalents
of the Vilsmeier salt 251 followed by treatment with base resulted
an intramolecular cyclization reaction with the proximal phenol
and concomitant elimination of the remaining imidate to deliver
the vinylated dihydrobenzofuran 252 in 76% yield.237 Interestingly,
this reaction could be performed on multi-kilogram scale, required
no chromatographic purification, and generated environmentallyfriendly
DMF and HCl as byproducts.237 Sharpless asymmetric
dihydroxylation of olefin 252 delivered diol 253 in 86% yield and
impressive enantioselectivity (>99% ee). This diol was then activated
with trimethylsilyl chloride and then treated with base to generate epoxide 254.238 Next, a modified Horner–Wadsworth–
Emmons reaction involving triethylphosphonoacetate (TEPA, 255)
was employed to convert epoxide 254 to cyclopropane 256.239
The reaction presumably proceeds through removal of the acidic
TEPA proton followed by nucleophilic attack at the terminal epoxide
carbon. The resulting alkoxide undergoes an intramolecular
phosphoryl transfer reaction resulting in an enolate, which then attacked the newly formed phosphonate ester in an SN2 fashion
resulting in the trans-cyclopropane ester, which was ultimately
saponified and re-acidified to furnish cyclopropane acid 256.239
Conversion of this acid to the corresponding primary amide preceded
carbonyl reduction with sodium borohydride. The resulting
amine was acylated with propionyl chloride to furnish tasimelteon
(XXXI) as the final product in 86% yield across the four-step
sequence.
PATENTS
| US2010261786 | 10-15-2010 | PREDICTION OF SLEEP PARAMETER AND RESPONSE TO SLEEP-INDUCING COMPOUND BASED ON PER3 VNTR GENOTYPE |
| US2009209638 | 8-21-2009 | TREATMENT FOR DEPRESSIVE DISORDERS |
| US6060506 | 5-10-2000 | Benzopyran derivatives as melatonergic agents |
| US5981571 | 11-10-1999 | Benzodioxa alkylene ethers as melatonergic agents |
| WO9825606 | 6-19-1998 | BENZODIOXOLE, BENZOFURAN, DIHYDROBENZOFURAN, AND BENZODIOXANE MELATONERGIC AGENTS |
| WO2007137244A1 * | May 22, 2007 | Nov 29, 2007 | Gunther Birznieks | Melatonin agonist treatment |
| US4880826 | Jun 25, 1987 | Nov 14, 1989 | Nava Zisapel | Melatonin antagonist |
| US4997845 | May 10, 1990 | Mar 5, 1991 | Eli Lilly And Company | β-alkylmelatonins as ovulation inhibitors |
| US5093352 | May 16, 1990 | Mar 3, 1992 | Whitby Research, Inc. | Antidepressant agents |
| US5151446 | Mar 28, 1991 | Sep 29, 1992 | Northwestern University | Substituted 2-amidotetralins as melatonin agonists and antagonists |
| US5225442 | Jan 3, 1992 | Jul 6, 1993 | Adir Et Compagnie | Compounds having a naphthalene structure |
| US5580878 | Jun 7, 1995 | Dec 3, 1996 | Interneuron Pharmaceuticals, Inc. | Substituted tryptamines phenalkylamines and related compounds |
| US5856529 | Dec 9, 1997 | Jan 5, 1999 | Bristol-Myers Squibb Company | Benzofuran and dihydrobenzofuran melatonergic agents |
| US6211225 | Jun 6, 2000 | Apr 3, 2001 | Bristol-Meyers Squibb | Heterocyclic aminopyrrolidine derivatives as melatonergic agents |
| US7754902 | May 18, 2006 | Jul 13, 2010 | Vanda Pharmaceuticals, Inc. | Ruthenium(II) catalysts for use in stereoselective cyclopropanations |
| US20010047016 | Apr 12, 2001 | Nov 29, 2001 | Gregory Oxenkrug | Method for treating depression |
| US20050164987 | Dec 22, 2004 | Jul 28, 2005 | Barberich Timothy J. | Melatonin combination therapy for improving sleep quality |
| US20090105333 | May 22, 2007 | Apr 23, 2009 | Gunther Birznieks | Melatonin agonist treatment |
extra info
|
 |
This preparation is referenced from:
a US 5 856 529 (Bristol-Myers Squibb; 5.1.1999; appl. 9.12.1997; USA-prior. 10.12.1996).
In animal studies, administration of tasimelteon during pregnancy resulted in developmental toxicity (embryofetal mortality, neurobehavioral impairment, and decreased growth and development in offspring) at doses greater than those used clinically.
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Hetlioz |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Oral |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | not determined in humans[1] |
| Protein binding | 89–90% |
| Metabolism | extensive hepatic, primarily CYP1A2 and CYP3A4-mediated |
| Elimination half-life | 0.9–1.7 h / 0.8–5.9 h (terminal) |
| Excretion | 80% in urine, 4% in feces |
| Identifiers | |
| CAS Number | |
| PubChemCID | |
| IUPHAR/BPS | |
| ChemSpider | |
| UNII | |
| ChEBI | |
| ECHA InfoCard | 100.114.889 |
| Chemical and physical data | |
| Formula | C15H19NO2 |
| Molar mass | 245.32 g/mol |
| 3D model (JSmol) | |

DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
Tasimelteon has two stereogenic centers. Besides the medically used trans-1 R , 2 R isomer (in the picture above left), there are thus three further stereoisomers that do not arise in the synthesis.

Tasimelteon is a white to off-white crystalline non-hygroscopic substance, soluble in water at physiologically relevant pH levels and readily soluble in alcohols, cyclohexane and acetonitrile. The compound occurs in two crystal forms. It is an anhydrate melting at 74 ° C and a hemihydrate . [4] The hemihydrate is from about 35 ° C the water of hydration and converts thereby in the anhydrate form to. [4] The anhydrate crystallizes in a monoclinic lattice with the space group P 2 1 , and the hemihydrate crystallizes in a tetragonal lattice with the space group P 4 3 21 2. [4]
4 Kaihang Liu, Zhou Xinbo, Zhejing Xu, Bai Hongzhen, Jianrong Zhu Jianming Gu, Guping Tang, Liu Xingang, Hu Xiurong: anhydrate and hemihydrate of Tasimelteon: Synthesis, structure, and pharmacokinetic study in J. Pharm. Biomed. Anal. 151 (2018) 235-243, doi : 10.1016 / j.jpba.2017.12.035 .

| Molecular Formula: | C26H35NO6S |
|---|---|
| Molecular Weight: | 489.627 g/mol |
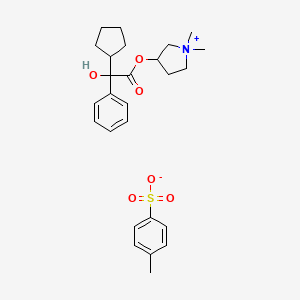
(1,1-dimethylpyrrolidin-1-ium-3-yl) 2-cyclopentyl-2-hydroxy-2-phenylacetate;4-methylbenzenesulfonate
CAS 873295-46-6 , C19 H28 N O3 . C7 H7 O3 S, Pyrrolidinium, 3-[(2-cyclopentyl-2-hydroxy-2-phenylacetyl)oxy]-1,1-dimethyl-, 4-methylbenzenesulfonate (1:1)
Glycopyrronium tosylate monohydrate
Molecular Formula, C19-H28-N-O3.C7-H8-O3-S.H2-O, Molecular Weight, 508.6522
https://chem.nlm.nih.gov/chemidplus/structure/1624259-25-1?maxscale=30&width=300&height=300
CAS 1624259-25-1, C19 H28 N O3 . C7 H7 O3 S . H2 O, Pyrrolidinium, 3-[(2-cyclopentyl-2-hydroxy-2-phenylacetyl)oxy]-1,1-dimethyl-, 4-methylbenzenesulfonate, hydrate (1:1:1)
Dermira (Originator)
DRM-04
DRM-04B
In 2018, the product was approved in the U.S. for the treatment of primary axillary hyperhidrosis in adult and pediatric patients 9 years of age and older.
In 2016, Maruho signed an exclusive license agreement with Dermina for product development and marketing in Japan for the treatment of axillary hyperhidrosis.
PATENT
https://patents.google.com/patent/US8558008B2/en
PATENT
https://patents.google.com/patent/US20130211101A1/en
Example 7 Preparation of Glycopyrrolate Tosylate
Example 8 Preparation of Glycopyrrolate Tosylate Form D
Example 9 Single Crystal Preparation of Form D
Example 10 Preparation of Dehydrated Form D
Example 11 Preparation of Form C Glycopyrrolate Tosylate
Example 12 Preparation of Form C Glycopyrrolate Tosylate
Example 13 Preparation of Form C Glycopyrrolate Tosylate
Example 14 Amorphous Glycopyrrolate Tosylate
In a dark room, silver tosylate (3.5 g) was dissolved in water (~ 100 mL) by sonication. The solution was heated to approximately 40°C. and additional water was added (-15 mL). An equimolar amount of glycopyrrolate bromide (5 g) (mixture of R,S and S,R diastereomers) was added and imme diately resulted in a yellow precipitate. The slurry was stirred at approximately 40°C. overnight, and then slowly cooled while stirring to ambient temperature. At ambient tempera ture, the solids were vacuum filtered and the wet cake was washed three times with approximately 10 mL of water. The mother liquor was collected and filtered two times through a 0.2 pm nylon filter with glass microfiber (GMF). A clear solution was observed after filtration and was lyophilized at approximately -50°C. After 6 days, a mixture of white, needle-like and slightly sticky, glassy solids was observed. Toluene (-20 mL) was added, and the slurry was briefly sonicated and then stirred at ambient temperature. Additional toluene (-80 mL) was added for easier stirring, and the mix ture was allowed to stand at ambient conditions for 1 day. Solids of glycopyrrolate tosylate were collected by vacuum filtration and vacuum drying at ambient temperature for 1 day. Glycopyrrolate Tosylate.
PAtent
https://patents.google.com/patent/CN103159659A/en

glycopyrrolate (I)
Methyl ethyl ketone (20mL) IOOmL three-necked flask was added 8 (4.6g, 15mmol) was, at (Γ5 ° C was added dropwise dibromomethane (2.9g, 30mmol) in butanone (5 mL) was added dropwise completed, continued The reaction was stirred for 15min, and a white solid precipitated, was allowed to stand 36h at room temperature, filtered off with suction, the filter cake was sufficiently dried to give crude ketone was recrystallized twice to give a white powdery crystals I (3.9g, 66%) mp 191~193 ° C chromatographic purity 99.8% [HPLC method, mobile phase: lmol / L triethylamine acetate – acetonitrile – water (1: 150: 49); detection wavelength: 230nm, a measurement of the area normalization method] .MS m / z: 318 ( m-BrO 1HNMR (CD3OD) δ:! 1.33~1.38 (m, 2H), 1.55~1.70 (m, 6H), 2.11~2.21 (m, 1H), 2.67~2.80 (m, 1H), 3.02 (m, 1H), 3.06 (s, 3H), 3.23 (s, 3H), 3.59~3.71 (m, 3H), 3.90 (dd, /=13.8,1H), 5.47 (m, 1H), 7.27 (t, 1H) , 7.35 (t, 2H), 7.62 (dd, 2H) .13C bandit R (DMSO) δ: 27.0, 27.4, 28.0, 31.3, 47.8, 53.8, 54.3, 66.0, 71.3, 74.6, 81.1, 126.9,128.7,129.3 , 143.2 17 5.00
Patent
https://patents.google.com/patent/WO2016204998A1/en

PAPER
https://link.springer.com/article/10.1007/s41981-018-0015-4
Journal of Flow Chemistry, pp 1–8| Cite as
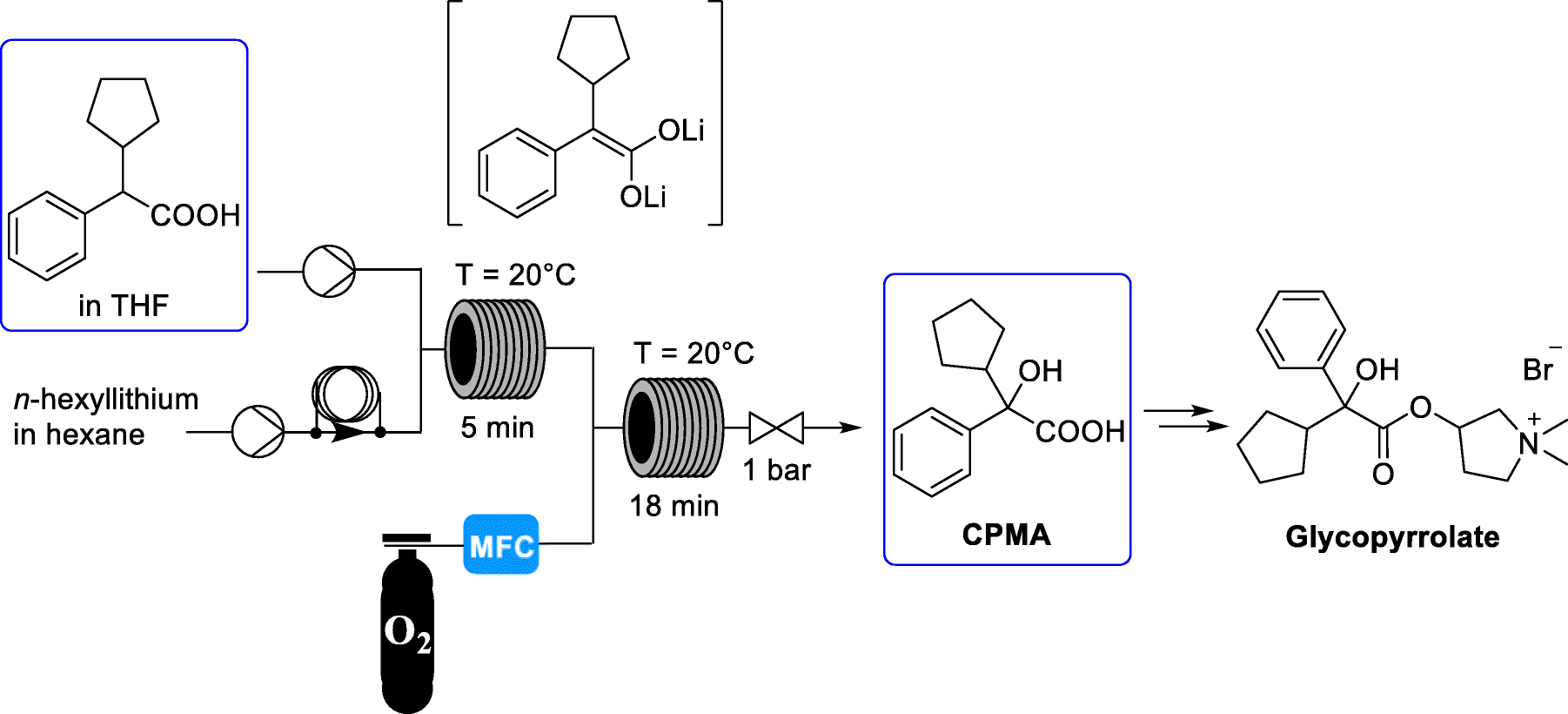
The medicinal properties of glycopyrronium bromide (glycopyrrolate, 4) were first identified in the late 1950s [1]. Glycopyrrolate is an antagonist of muscarinic cholinergic receptors and is used for the treatment of drooling or excessive salivation (sialorrhea) [2], excess sweating (hyperhidrosis) [3], and overactive bladder and for presurgery treatment. In addition, it has recently been introduced as an effective bronchodilator for the treatment of chronic obstructive pulmonary disease (COPD) for asthma patients [4]. Glycopyrrolate displays few side effects because it does not pass through the blood brain barrier. Cyclopentyl mandelic acid (CPMA, 1), or its corresponding ester derivatives, are key intermediates in the synthetic routes to 4. CPMA (1) reacts with 1-methyl-pyrrolidin-3-ol (2) to form tertiary amine 3. N-Methylation of 3 by methyl bromide gives quaternary ammonium salt glycopyrrolate 4 as a racemate (Scheme 1) [5].
Scheme 1
Synthesis of glycopyrrolate 4 from CPMA (1)
CPMA (1) is a synthetically challenging intermediate to prepare (Scheme 2). Routes A to D are most likely to be the commercially applied methods because these procedures are described in patents [5]. The published descriptions for the yields of 1 range from 28 to 56% for routes A to D. Ethyl phenylglyoxylate is reacted with cyclopentyl magnesium bromide to form an ester which is then hydrolyzed (route A) [6]. Phenylglyoxylic acid can be reacted in a similar manner with cyclopentyl magnesium bromide to directly form 1 (route B) [7]. Alternatively, the inverse addition of phenyl-Grignard reagent to cyclopentyl glyoxylic acid ester is reported (route C) [8]. Cyclopentyl glyoxylic acid ester can also be reacted with cyclopentadienyl magnesium bromide which is followed by an additional hydrogenation step with Pd/C and H2 to afford 1 (route D) [9, 10].
Scheme 2
Existing synthetic pathways to CPMA (1)
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9440056 | DEVICE AND METHOD FOR DISPENSING A DRUG |
2015-09-29
|
2016-03-31
|
| US2016058735 | METHODS OF TREATING HYPERHIDROSIS |
2015-08-27
|
2016-03-03
|
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2017157088 | GLYCOPYRROLATE SALTS |
2017-02-21
|
|
| US9610278 | Glycopyrrolate Salts |
2016-01-07
|
2016-04-28
|
| US2016052879 | PROCESS FOR PRODUCTION OF GLYCOPYRRONIUM TOSYLATE |
2015-08-19
|
2016-02-25
|
| US9006461 | CRYSTALLINE GLYCOPYRROLATE TOSYLATE |
2013-09-11
|
2014-08-28
|
| US2016243345 | DEVICE AND METHOD FOR DISPENSING A DRUG |
2016-05-04
|
2016-08-25
|
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2016354315 | DOSAGE FORMS AND USE THEREOF |
2016-06-03
|
|
| US9259414 | Glycopyrrolate Salts |
2015-03-10
|
2015-07-16
|
| US9006462 | Glycopyrrolate Salts |
2014-08-29
|
2014-12-18
|
| US8558008 | Crystalline glycopyrrolate tosylate |
2013-02-28
|
2013-10-15
|
| US8859610 | Crystalline glycopyrrolate tosylate |
2013-09-11
|
2014-10-14
|
///////////Glycopyrrolate Tosylate, DRM-04 , DRM-04B , FDA 2018, Qbrexza
CC1=CC=C(C=C1)S(=O)(=O)[O-].C[N+]1(CCC(C1)OC(=O)C(C2CCCC2)(C3=CC=CC=C3)O)C
Diazoxide choline,
RN: 1098065-76-9
UNII: 2U8NRZ7P8L
Diazoxide choline; UNII-2U8NRZ7P8L; 2U8NRZ7P8L; YLLWQNAEYILHLV-UHFFFAOYSA-N
| Molecular Formula: | C13H20ClN3O3S |
|---|---|
| Molecular Weight: | 333.831 g/mol |
Ethanaminium, 2-hydroxy-N,N,N-trimethyl-, compd. with 7-chloro-3-methyl-2H-1,2,4-benzothiadiazine dioxide (1:1)
7-chloro-3-methyl-1$l^{6},2,4-benzothiadiazin-2-ide 1,1-dioxide;2-hydroxyethyl(trimethyl)azanium
 Diazoxide
Diazoxide
CAS: 364-98-7 FREE FORM
2H-1,2,4-Benzothiadiazine, 7-chloro-3-methyl-, 1,1-dioxide
Diazoxide (INN; brand name Proglycem[1]) is a potassium channel activator, which causes local relaxation in smooth muscle by increasing membrane permeability to potassium ions. This switches off voltage-gated calcium ion channels, preventing calcium flux across the sarcolemma and activation of the contractile apparatus.
In the United States, this agent is only available in the oral form and is typically given in hospital settings.[2]
Diazoxide is used as a vasodilator in the treatment of acute hypertension or malignant hypertension.[3]
Diazoxide also inhibits the secretion of insulin by opening ATP-sensitive potassium channel of beta cells of the pancreas, thus it is used to counter hypoglycemia in disease states such as insulinoma (a tumor producing insulin)[4] or congenital hyperinsulinism.
Diazoxide acts as a positive allosteric modulator of the AMPA and kainate receptors, suggesting potential application as a cognitive enhancer.[5]
The Food and Drug Administration published a Safety Announcement in July 2015 highlighting the potential for development of pulmonary hypertension in newborns and infants treated with this drug.[2]Diazoxide interferes with insulin release through its action on potassium channels.[6] Diazoxide is one of the most potent openers of the K+ ATP channels present on the insulin producing beta cells of the pancreas. Opening these channels leads to hyperpolarization of cell membrane, a decrease in calcium influx, and a subsequently reduced release of insulin.[7] This mechanism of action is the mirror opposite of that of sulfonylureas, a class of medications used to increase insulin release in Type 2 Diabetics. Therefore, this medicine is not given to non-insulin dependent diabetic patients.
SYN
Medicinal Chemistry Research, 12(9), 457-470; 2004

PATENT
WO 2009006483
https://patents.google.com/patent/WO2009006483A1/enIt
PATENT
US 20120238554
PATENT
WO 2013130411
able 16. Characterization of Forms A and B of Diazoxide Choline Salt In
Screening Study
Experiment Form A Form B
*Maj or peaks (2-Θ):
Form A (9.8, 10.5, 14.9, 17.8, 17.9, 18.5, 19.5, 22.1, 22.6, 26.2, 29.6, 31.2);
Form B (8.9, 10.3, 12.0, 18.3, 20.6, 24.1, 24.5, 26.3, 27.1, 28.9).
** Unique FTIR (ATR) absorbances (cm 1):
Form A (2926, 2654, 1592, 1449, 1248);
Form B (3256, 2174, 2890, 1605, 1463, 1235).
6.1.5.1. Solubility Screen in organic solvents.
[00725] Diazoxide choline, prepared in MEK using choline hydroxide as 50 wt % solution in water (see above) displayed some solubility in the following solvents:
acetonitrile, acetone, ethanol, IPA, MEK, DMF, and methanol. These solvents were chosen due to differences in functionality, polarity, and boiling points and their ability to dissolve diazoxide. Other solvents which showed poor ability to dissolve salts were used as antisolvents and in slurry experiments where some solubility was observed: dioxane, MTBE, EtOAc, IP Ac, THF, water, cyclohexane, heptane, CH2C12, and toluene.
[00726] Solvents for crystallizations during screening were chosen based on the solubility screen summarized in Table 17. Crystallizations of diazoxide choline from all conditions afforded a total of two forms, A and B. Forms A and B were found to be anhydrous polymorphs of diazoxide choline. Form B was observed to be generated from most solvents used. It was difficult to isolate pure Form A on large scales (>50 mg) as conditions observed to produce Form A on a smaller scale (approximately 50 mg or less) were found to result in Form B or mixtures of both forms on larger scales. Based on room-temperature slurry experiments, anhydrous Form B was found to be the most thermodynamically stable form in this study. Form A readily converted to Form B in all slurry solvents utilized.
Table 17. Solubility Screen for Diazoxide Choline Salt
Solvent Cmpd Solvent Cone. Temp. Soluble
(mg) (mL) (mg/niL) (°C)
CH2CI2 1.3 5.00 0.26 55 Partially
Toluene 1.4 5.00 0.28 55 No
.1.5.2. Single-Solvent Crystallizations
[00727] Fast cooling procedure: Diazoxide (approximately 20 mg) was weighed out into vials and enough solvent (starting with 0.25 mL) was added until the material completely dissolved at elevated temperature. After hot filtration the vials were placed in a refrigerator (4 °C) for 16 hours. After the cooling-process the samples were observed for precipitates which were isolated by filtration. Vials not demonstrating precipitates were evaporated down to dryness using a gentle stream of nitrogen. All solids were dried in vacuo at ambient temperature and 30 in. Hg.
[00728] Slow cooling procedure: Diazoxide (approximately 30 mg of choline salt) was weighed out into vials and enough solvent was added until the material went into solution at elevated temperature. After hot filtration the vials were then slowly cooled to room temperature at the rate of 20 °C/h and stirred at room temperature for 1-2 hours. All solids were dried in vacuo at ambient temperature and 30 in. Hg.
[00729] Based on the initial solubility study, seven solvents were selected for the fast-cooling crystallization: acetonitrile, acetone, ethanol, IPA, MEK, DMF, and methanol. Table 18 shows a list of the solvents that were used and the amount of solvent needed to dissolve the material. After the cooling-process precipitates were noticed in samples # 2, 3, 5, and 6, the solids were isolated by filtration. The other samples (# 1, 4, and 7) were evaporated down to dryness using a gentle stream of nitrogen. The diazoxide choline salts were found to be consistent with Form A by XRPD analysis for all solids with the exception of sample #2 (consistent with the freeform) and sample #5 (consistent with Form B with preferred orientation observed).
Table 18. Single- Solvent Crystallization of Diazoxide Choline Salt Using Fast- Cooling Procedure
[00730] In accordance with the data obtained from fast-cooling experiments, four solvents which showed precipitation of solids were chosen for the slow-cooling experiments: MeOH, EtOH, MeCN, and IPA (Table 19). All obtained analyzable solids of the choline salt were found to be consistent with Form B by XRPD with the exception of Entry #1 which was consistent with diazoxide freeform and Entry #2 which was not analyzable. Mother liquor of Entry #2 was concentrated to dryness and the residual solids were analyzed by XRPD and found to be Form B material. As a result of obtaining freeform material from the single- solvent crystallizations in methanol, three more alcohols were tested for the single- solvent crystallizations using fast- and slow-cooling procedures. Tables 20 and 21 provide a list of the solvents that were used and the amount of solvent needed to dissolve the material. XRPD patterns of the fast-cooling procedure showed freeform of diazoxide from isobutanol, Form B from isoamyl alcohol, and Form A from tert-amyl alcohol compared to the slow-cooling procedure, which afforded Form B material from all three solvents.
Table 19. Single-Solvent Crystallization of Diazoxide Choline Salt Using Slow- Cooling Procedure
Table 20. Single- Solvent Crystallization of Diazoxide Choline Salt Using Fast- Cooling Procedure
Table 21. Single-Solvent Crystallization of Diazoxide Choline Salt Using Slow- Cooling Procedure
[00731] The results of the choline salt single- solvent fast- and slow-cooling crystallizations (see Tables 19 to 21) indicated that Form A was more likely to be isolated with fast-cooling profiles and Form B with slow-cooling profiles.
6.1.5.3. Binary Solvent Crystallizations
[00732] Binary- solvent crystallizations of the choline salt were performed using four primary solvents (MeOH, EtOH, IPA, and MeCN) and nine cosolvents (MTBE, EtOAc, IPAc, THF, c-hexane, heptane, toluene, CH2CI2, and dioxane) with a fast-cooling profile (supra). XRPD patterns showed that Form B was obtained from mixtures of MeOH with MTBE, EtOAc, IPAc, toluene, and dioxane. As shown in Table 22, Form A was obtained from mixtures of MeOH with THF and with CH2CI2 after evaporating the solvent to dryness. The mixtures of MeOH with cyclohexane and heptane provided the freeform of diazoxide. All solids obtained from fast-cooling procedures with EtOH, IPA, and MeCN as primary solvents provided Form B material.
Table 22. Binary-Solvent Crystallizations of Choline Salt of Diazoxide Using Fast- Cooling Procedure and MeOH as a Primary Solvent
* Solids were dissolved at 62 °C.
** Freeform of diazoxide.
[00733] Binary- solvent recrystallizations of the choline salt with the slow-cooling procedure were performed using two primary solvents (IPA and MeCN) and nine cosolvents (MTBE, EtOAc, IPAc, THF, c-hexane, heptane, toluene, CH2C12, and dioxane). All solids obtained from a slow-cooling procedure with IPA and MeCN as primary solvents provided Form B material based on XRPD analysis. The results of
binary- solvent crystallizations indicated that Form B was the most thermodynamic ally stable form of diazoxide choline.
6.1.5.4. Binary Solvent Crystallizations Using Water as a Cosolvent
[00734] In an attempt to investigate the formation of hydrates of the choline salt, experiments was performed using fast- and slow-cooling procedures and water as a cosolvent.
[00735] The fast cooling procedure (supra) was used with the exception of using different primary solvents which were miscible with water: acetone, acetonitrile, DMF, IPA, i-BuOH, i-AmOH, and t-AmOH. Water was utilized in these crystallizations as a cosolvent. All solids obtained from the fast-cooling procedure with water as the cosolvent provided diazoxide freeform material by XRPD analysis.
[00736] To compare the results obtained from the fast-cooling procedure a set of experiments was performed using a slow-cooling procedure and water as a cosolvent. All obtained solids were analyzed by XRPD and afforded patterns consistent with diazoxide freeform. Without wishing to be bound by theory, these results suggest that the conditions used for crystallization caused dissociation of the choline salt. A small amount of a second crop was obtained in each sample, but only two samples were analyzable by XRPD and indicated that the samples were freeform material. All mother liquors were evaporated to dryness and the residual solids were also analyzed by XRPD to afford patterns consistent with Form B of the choline salt.
6.1.5.5. Metastable Zone Width Estimation
[00737] Form B: To produce a robust process, an understanding of the solubility profiles of the various solid forms under consideration is required. From a practical standpoint, this involves the measurement of the metastable zone width (MSZW) of pure forms, whereby the saturation and supersaturation curves of the different forms are generated over a well defined concentration and temperature range. This knowledge can then be used to design a crystallization protocol that should ideally favor a selective crystal growth of the desired form.
[00738] Form B of diazoxide choline salt showed moderate solubility in a solvent mixture made of MeCN/MeOH/MtBE (10: 1: 12, volume ratios). The wide width of the metastable zone as shown in Table 23 gives many seeding options. During the MSZW measurement, aliquots from the crystallizing material were withdrawn and analyzed by XRPD to ensure that no form conversion occurred during the experiment. Indeed, the material remained unchanged during the test.
Table 23. Meta-Stable Zone Width For Form B Diazoxide Choline Salt in
MeCN/MeOH/MtBE (10:1:12) (v/v).
[00739] Form A: The metastable zone width for Form could not be estimated because this polymorphic form converted during the experiment to Form B.
6.1.5.6. Crystallization of Form A of Diazoxide Choline Salt
[00740] The choline salt of diazoxide (160.3 mg) was dissolved in 1 mL of IPA at 55 °C which was then passed through a Millipore 0.45 μΜ filter into a clean vial. This vial was placed in freezer a -20 °C overnight. Solids were not noticed and the flask was scratched with a micro- spatula. The vial was placed back in the freezer and nucleation was noticed after ten minutes. The solids were collected by vacuum filtration and washed with 1 mL of MtBE. The solids were dried in vacuo at 40 °C and 30 in. Hg to afford 70 mg (43.6% recovery) of Form A as determined by XRPD.
6.1.5.7. 500-mg Scale Crystallization of Form B of Diazoxide Choline Salt
[00741] The choline salt of diazoxide (524.3 mg) was dissolved in 3 mL of IPA at 78 °C and this solution was then cooled to 55 °C for the addition of MtBE. The MtBE (4 mL) was added until nucleation was observed. After nucleation the batch was allowed to cool to room temperature at a rate of 20 °C /h. The solids were collected by vacuum filtration and washed with 1 mL of MtBE. The solids were dried in vacuo at 40 °C and 30 in. of Hg to afford 426.7 mg (81.3% recovery) of Form B as determined by XRPD.
6.1.5.8. 2-g Scale Crystallization of Form B of Diazoxide Choline Salt
[00742] The choline salt of diazoxide (2.0015 g) was dissolved in 5.5 mL of IPA at 78 °C to afford a clear solution. This solution was passed through a Millipore Millex FH 0.45 μΜ filter. This solution was then cooled to 55 °C. MtBE was added in 1 mL portions, with a two minute interval between portions. Nucleation was noted after the second addition of MtBE. This suspension was allowed to cool to room temperature at a rate of 20 °C /h and stirred at this temperature for 16 hours. The solids were collected by vacuum filtration and washed with 1 mL of MtBE. The solids were dried in vacuo at 40 °C and 30 in. of Hg to afford 1.6091 g (80.4% recovery) of Form B as determined by XRPD.
6.1.5.9. Detection of Form Impurities
[00743] Mixtures of diazoxide choline Forms A and B were prepared by adding a minor amount of Form A to Form B. Samples were lightly ground by hands with a mortar and pestle for approximately one minute. Samples were then analyzed by XRPD analysis. XRPD analysis was found to be suitable for detecting 5% of Form A in Form B.
 |
|
| Clinical data | |
|---|---|
| Trade names | Proglycem |
| AHFS/Drugs.com | Monograph |
| Pregnancy category |
|
| Routes of administration |
Oral, intravenous |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Protein binding | 90% |
| Metabolism | Hepatic oxidation and sulfate conjugation |
| Elimination half-life | 21-45 hours |
| Excretion | Renal |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.006.063 |
| Chemical and physical data | |
| Formula | C8H7ClN2O2S |
| Molar mass | 230.672 g/mol |
| 3D model (JSmol) | |
//////////////Diazoxide choline
CC1=NC2=C(C=C(C=C2)Cl)S(=O)(=O)[N-]1.C[N+](C)(C)CCO
CC1=NC2=C(C=C(C=C2)Cl)S(=O)(=O)[N-]1.C[N+](C)(C)CCO


Zoledronic acid
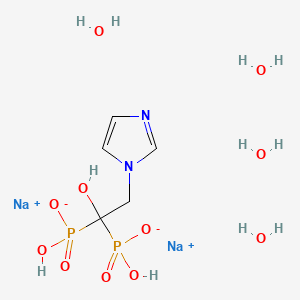
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Zoledronate disodium | 7D7GS1SA24 | 165800-07-7 | IEJZOPBVBXAOBH-UHFFFAOYSA-L |
| Zoledronate trisodium | ARL915IH66 | 165800-08-8 | Not applicable |
| Zoledronic acid hemipentahydrate | 1K9U67HDID | Not Available | AZZILOGHCMYHQY-UHFFFAOYSA-N |
| Zoledronic acid monohydrate | 6XC1PAD3KF | 165800-06-6 | FUXFIVRTGHOMSO-UHFFFAOYSA-N |
Zoledronate (zoledronic acid, marketed by Novartis under the trade names Zometa and Reclast) is a bisphosphonate. Zometa is used to prevent skeletal fractures in patients with cancers such as multiple myeloma and prostate cancer. It can also be used to treat hypercalcemia of malignancy and can be helpful for treating pain from bone metastases.
An annual dose of Zoledronate may also prevent recurring fractures in patients with a previous hip fracture.
Zoledronate is a single 5 mg infusion for the treatment of Paget’s disease of bone. In 2007, the FDA also approved Reclast for the treatment of postmenopausal osteoporosis.
Zoledronic acid, also known as zoledronate, is a medication used to treat a number of bone diseases.[1] This include osteoporosis, high blood calcium due to cancer, bone breakdown due to cancer, and Paget’s disease of bone.[1] It is given by injection into a vein.[1]
Common side effects include fever, joint pain, high blood pressure, diarrhea, and feeling tired.[1] Serious side effects may include kidney problems, low blood calcium, and osteonecrosis of the jaw.[1] Use during pregnancy may result in harm to the baby.[1] It is in the bisphosphonate family of medications.[1] It works by blocking the activity of osteoclast cells and thus decreases the breakdown of bone.[1]
Zoledronic acid was approved for medical use in the United States in 2001.[1] It is on the World Health Organization’s List of Essential Medicines, the most effective and safe medicines needed in a health system.[3] The wholesale cost in the developing world is between 5.73 USD and 26.80 USD per vial.[4] In the United Kingdom, as of 2015, a dose costs the NHS about 220 pounds.[5]
Zoledronic acid is used to prevent skeletalfractures in patients with cancers such as multiple myeloma and prostate cancer, as well as for treating osteoporosis.[6] It can also be used to treat hypercalcemia of malignancy and can be helpful for treating pain from bone metastases.[7]
It can be given at home rather than in hospital. Such use has shown safety and quality-of-life benefits in people with breast cancer and bone metastases.[8]
Zoledronic acid may be given as a 5 mg infusion once per year for treatment of osteoporosis in men and post-menopausal women at increased risk of fracture.[9]
In 2007, the U.S. Food and Drug Administration (FDA) also approved it for the treatment of postmenopausal osteoporosis.[10][11]
A single 5 mg dose of zoledronic acid is used for the treatment of Paget’s disease.[medical citation needed][12]
Side effects can include fatigue, anemia, muscle aches, fever, and/or swelling in the feet or legs. Flu-like symptoms are common after the first infusion, although not subsequent infusions, and are thought to occur because of its potential to activate human γδ T cells(gamma/delta T cells).
There is a risk of severe renal impairment. Appropriate hydration is important prior to administration, as is adequate calcium and vitamin D intake prior to Aclasta therapy in patients with preexisting hypocalcaemia, and for ten days following Aclasta in patients with Paget’s disease of the bone. Monitoring for other mineral metabolism disorders and the avoidance of invasive dental procedures for those who develop osteonecrosis of the jaw is recommended.[14]
Zoledronate is rapidly processed via the kidneys; consequently its administration is not recommended for patients with reduced renal function or kidney disease.[15] Some cases of acute renal failure either requiring dialysis or having a fatal outcome following Reclast use have been reported to the U.S. Food and Drug Administration (FDA).[16] This assessment was confirmed by the European Medicines Agency (EMA), whose Committee for Medicinal Products for Human Use (CHMP) specified new contraindications for the medication on 15 December 2011, which include hypocalcaemia and severe renal impairment with a creatinine clearance of less than 35 ml/min.[17]
A rare complication that has been recently observed in cancer patients being treated with bisphosphonates is osteonecrosis of the jaw. This has mainly been seen in patients with multiple myeloma treated with zoledronate who have had dental extractions.[18]
Atypical fractures : After approving the drug on 8 July 2009, the European Medicines Agency conducted a class review of all bisphosphonates, including Zoledronate, after several cases of atypical fractures were reported.[19] In 2008, the EMA’s Pharmacovigilance Working Party (PhVWP) noted that alendronic acid was associated with an increased risk of atypical fracture of the femur that developed with low or no trauma. In April 2010, the PhVWP noted that further data from both the published literature and post-marketing reports were now available which suggested that atypical stress fractures of the femur may be a class effect. The European Medicines Agency then reviewed all case reports of stress fractures in patients treated with bisphosphonates, relevant data from the published literature, and data provided by the companies which market bisphosphonates. The Agency recommended that doctors who prescribe bisphosphonate-containing medicines should be aware that atypical fractures may occur rarely in the femur, especially after long-term use, and that doctors who are prescribing these medicines for the prevention or treatment of osteoporosis should regularly review the need for continued treatment, especially after five or more years of use.[19]
Zoledronic acid slows down bone resorption, allowing the bone-forming cells time to rebuild normal bone and allowing bone remodeling.[20]
Zoledronic acid has been found to have a direct antitumor effect and to synergistically augment the effects of other antitumor agents in osteosarcoma cells.[21]
Zoledronate has shown significant benefits versus placebo over three years, with a reduced number of vertebral fractures and improved markers of bone density.[22][11] An annual dose of zoledronic acid may also prevent recurring fractures in patients with a previous hip fracture.[9]
Zoledronate also attenuates accumulation of DNA damage in mesenchymal stem cells and protects their function.[23] Given this characteristic, its potential to affect conditions arising from stem-cell dysfunction makes it a promising medicine for a range of age-related diseases[24]
An increase in disease-free survival (DFS) was found in the ABCSG-12 trial, in which 1,803 premenopausal women with endocrine-responsive early breast cancer received anastrozole with zoledronic acid.[25] A retrospective analysis of the AZURE trial data revealed a DFS survival advantage, particularly where estrogen had been reduced.[26]
In a meta-analysis of trials where upfront zoledronic acid was given to prevent aromatase inhibitor-associated bone loss, active cancer recurrence appeared to be reduced.[27]
As of 2010 “The results of clinical studies of adjuvant treatment on early-stage hormone-receptor-positive breast-cancer patients under hormonal treatment – especially with the bisphosphonate zoledronic acid – caused excitement because they demonstrated an additive effect on decreasing disease relapses at bone or other sites. A number of clinical and in vitro and in vivo preclinical studies, which are either ongoing or have just ended, are investigating the mechanism of action and antitumoral activity of bisphosphonates.”[28]
A 2010 review concluded that “adding zoledronic acid 4 mg intravenously every 6 months to endocrine therapy in premenopausal women with hormone receptor-positive early breast cancer … is cost-effective from a US health care system perspective”.[29]
PAPER
J Med Chem 2002,45(17),3721
https://pubs.acs.org/doi/10.1021/jm020819i

Bisphosphonates (BPs) are pyrophosphate analogues in which the oxygen in P−O−P has been replaced by a carbon, resulting in a metabolically stable P−C−P structure. Pamidronate (1b, Novartis), a second-generation BP, was the starting point for extensive SAR studies. Small changes of the structure of pamidronate lead to marked improvements of the inhibition of osteoclastic resorption potency. Alendronate (1c, MSD), with an extra methylene group in the N-alkyl chain, and olpadronate (1h, Gador), the N,N-dimethyl analogue, are about 10 times more potent than pamidronate. Extending one of the N-methyl groups of olpadronate to a pentyl substituent leads to ibandronate (1k, Roche, Boehringer-Mannheim), which is the most potent close analogue of pamidronate. Even slightly better antiresorptive potency is achieved with derivatives having a phenyl group linked via a short aliphatic tether of three to four atoms to nitrogen, the second substituent being preferentially a methyl group (e.g., 4g, 4j, 5d, or 5r). The most potent BPs are found in the series containing a heteroaromatic moiety (with at least one nitrogen atom), which is linked via a single methylene group to the geminal bisphosphonate unit. Zoledronic acid (6i), the most potent derivative, has an ED50 of 0.07 mg/kg in the TPTX in vivo assay after sc administration. It not only shows by far the highest therapeutic ratio when comparing resorption inhibition with undesired inhibition of bone mineralization but also exhibits superior renal tolerability. Zoledronic acid (6i) has thus been selected for clinical development under the registered trade name Zometa. The results of the clinical trials indicate that low doses are both efficacious and safe for the treatment of tumor-induced hypercalcemia, Paget’s disease of bone, osteolytic metastases, and postmenopausal osteoporosis.
SYN 1
AU 8781453; EP 0275821; JP 1988150291; US 4939130
Zoledronate sodium can be prepared by reaction of 2-(1-imidazolyl)acetic acid hydrochloride (I) with PCl3, with optional presence of phosphoric acid, in refluxing chlorobenzene, followed by hydrolysis with refluxing 9N hydrochloric acid and final formation of the sodium salt by treatment with aqueous NaOH.
SYN
PAPER
https://www.sciencedirect.com/science/article/pii/S0969804311006385

Clip
https://link.springer.com/article/10.1007/s11094-015-1205-0
A One-Pot and Efficient Synthesis of Zoledronic Acid Starting from Tert-butyl Imidazol-1-yl Acetate
A one-pot synthesis of zoledronic acid in high yield is described. The procedure involves a non-aqueous ester cleavage of the tert-butyl imidazol-1-yl acetate under dry conditions in the presence of methanesulfonic acid as solubilizer and chlorobenzene as solvent to afford in situthe corresponding imidazolium methanesulfonate salt which yields zoledronic acid upon reaction with phosphoric acid and phosphorus oxychloride. A possible chemical mechanism for the synthesis of this acid is described.

Paper
https://www.beilstein-journals.org/bjoc/articles/4/42


To a solution of imidazole (10.0 g, 0.15 mol) in ethyl acetate (160 mL) was added powdered K2CO3 (29.0 g, 0.21 mol) followed by tert-butyl chloroacetate (25.7 mL, 0.18 mol) at room temperature and the mixture was refluxed for 10.0 h. After completion of the reaction as indicated by TLC (10% MeOH/CHCl3, I2 active), the reaction mass was quenched with cold water (80 mL) and the ethyl acetate layer was separated. The aqueous layer was extracted with ethyl acetate (2 × 80 mL) and the combined ethyl acetate layers were washed with brine, dried with anhydrous sodium sulfate and then concentrated under vacuum. The resulting solid was stirred with hexane (50 mL) at RT, filtered and washed with hexane (2 × 20 mL) to afford the title compound as an off-white solid (20.0 g, 75%). mp: 111.3–113.2 °C (Lit [10]: 111–113 °C). IR (cm−1): 3458, 3132, 3115, 2999, 2981, 2884, 1740, 1508, 1380, 1288, 1236, 1154, 1079, 908, 855, 819, 745, 662, 583; 1H NMR (300 MHz, CDCl3) δ 1.47 (s, 9H), 4.58 (s, 2H), 6.94 (s, 1H), 7.09 (s, 1H), 7.49 (s, 1H); 13C NMR (75 MHz, CDCl3) δ 27.7, 48.6, 82.9, 119.8, 129.2, 137.7, 166.3; MS (m/z) 183.0 [M+1, 100%], 127.0.
To a solution of imidazol-1-yl-acetic acid tert-butyl ester (2) (10.0 g, 0.05 mol) in dichloromethane (100 mL) was added titanium tetrachloride (8.0 mL, 0.07 mol) dropwise slowly at −15 to −10 °C over 1 h and the mixture was stirred at −5 to 0 °C for 2 h. Isopropyl alcohol (25 mL) was added at 0 to −10 °C over 0.5 h and the reaction mass was stirred at room temperature for 0.5 h. Additional isopropyl alcohol (125 mL) was added dropwise at room temperature over 0.5 h and the mixture was stirred for 1 h. Dichloromethane was distilled out under a low vacuum and the resulting crystalline solid precipitated was filtered to afford the title compound as an off-white crystalline solid (7.4 g, 83%). mp 200.3–202.3 °C; IR (cm−1): 3175, 3125, 3064, 2945, 2869, 2524, 2510, 1732, 1581, 1547, 1403, 1223, 1193, 1081, 780, 650; 1H NMR (300 MHz, D2O + 3-(trimethylsilyl)propionic acid sodium salt) δ 5.1 (s, 3H, -CH2– + HCl), 7.5 (br s, 2H), 8.7 (s, 1H); 13C NMR (75 MHz, D2O + 3-(trimethylsilyl)propionic acid sodium salt) 52.7, 122.4, 125.9, 138.8, 172.8; MS (m/z) 127.0 [M+1, 100%]; HCl-content: found 21.8% (along with 3.25% moisture), calcd 22.43% for C5H6N2O2·HCl.
To a suspension of imidazol-1-yl-acetic acid hydrochloride (6) (7.0 g, 0.043 mol) and phosphorous acid (9.5 g, 0.116 mol) in chlorobenzene (50 mL) was added phosphorous oxychloride (9.6 ml, 0.103 mol) at 80–85 °C over a period of 2 h then heated to 90–95 °C for 2.5 h. The reaction mass was cooled to 60–65 °C and water (100 mL) was added at the same temperature. The aqueous layer was separated, collected and refluxed for 18 h. It was then cooled to room temperature and diluted with methanol (140 mL). The mixture was cooled to 0–5 °C and stirred for 3 h. The precipitated solid was filtered, washed with cold water followed by methanol and then dried under vacuum at 60 °C for 12 h to afford the title compound (6.6 g, 57% yield) as a white solid; mp 237–239 °C (lit [1] 239 °C with decomposition).
PATENT
https://patents.google.com/patent/CN104610357A/en
Sodium Zoledronic (Zoledronate sodium, I), chemical name [1-yl light -2- (lH- imidazol-1-yl) ethylidene] bisphosphonic acid monosodium salt monohydrate, is by the Novartis (Novartis) developed imidazole heterocyclic bisphosphonates, belongs to the third generation of bisphosphonates bisphosphonate drugs, in October 2000, first marketed in Canada. Subsequently approved in the European Union, the United States more than 80 countries or regions, trade name Zometa, for the treatment of hypercalcemia of malignancy (HCM) and multiple myeloma and bone metastases of solid tumors. The drug is effective in treating cancer caused by HCM, advanced bone metastases and Paget’s disease, reduce the incidence of skeletal related events, relieve symptoms and improve quality of life, is also expected to be used to treat osteoporosis. Compared with other similar drugs, high efficacy, dosage, ease of administration, better security, etc., is currently the only FDA-approved for metastatic bone tumor effective bisphosphonate drugs.Currently bisphosphonate drugs in our country is still in the initial stages of clinical applications, but in recent years has made rapid progress, broad market prospect.
[0003] The prior art synthesis reaction conditions zoledronate sodium harsh, toxicity and use methanol, chloroform and chlorobenzene, easily exceeding the amount of residual organic solvents, low yield, low product purity, contamination environment, does not meet the medical criteria, is not conducive to industrial production. Environmental pollution has attracted increasing attention around the world today, the development of new green efficient synthesis of a pharmaceutical drug synthesis is an important issue facing the Institute. In recent years, room temperature ionic liquids as a reaction medium is environmentally friendly, has been widely used in a variety of organic synthesis reactions. Compared with traditional organic solvents, ionic liquids have very low vapor pressure, non-flammable, good thermal stability, both as a reaction medium underway catalysis, can be recycled and many other advantages.

Example 1 of zoledronic alendronate

(1) Synthesis of imidazol-1-yl acetate were added successively imidazole (13.62g, 0.2mol) and [bmim] BF4 (IOOmL) a three-necked flask, heated with stirring warmed to 60 ° C, incubated under reflux was slowly added dropwise chlorination ethyl acetate (24. 51g, 0. 2mol), dropwise addition time is about 2h, dropwise, with stirring maintained at reflux for 16 h, the reaction monitored by TLC showed no starting material end point, completion of the reaction, cooled to room temperature, to give imidazole -1 – ethyl crude, about 24g, crude without purification, was used directly in the next reaction.
[0014] (2) Synthesis of imidazol-1-yl acetate hydrochloride A solution of 24g 1-yl imidazole prepared above was added crude ethyl necked flask, concentrated hydrochloric acid (34 mL), exotherm to 85 ° C, warmed to reflux heating was continued, the reaction was stirred at reflux for 10H, the reaction was completed, the solvent was evaporated under reduced pressure, 20ml of absolute ethanol was added to the residue, vigorously stirred for 2h, filtered off with suction, the filter cake finally at 80 ° C blast pressure and dried to give a white solid imidazol-1-yl acetate hydrochloride about 25. 65g, 79.4% overall yield.
[0015] (3) Synthesis of zoledronic acid monohydrate were added imidazol-1-yl acetate hydrochloride (17. 26g, 0. 137mol) a three-necked flask, in an ionic liquid with stirring – n-butyl-3- methylimidazolium tetrafluoroborate [bmim] BF4 (40mL) and concentration of 85% phosphoric acid solution (16mL), heating to 60 ° C was added dropwise phosphorus trichloride (30mL) , about 4h dropwise, reaction was continued under reflux for 4h at 65 ° C, the reaction was complete, cooled to 40 ° C, filtered off with suction, the filter cake was added to a molar concentration in 80mL 9mol / L hydrochloric acid, heated with stirring state the reaction was refluxed for 6h, the reaction was completed, filtered hot, the filter cake was added to a molar concentration in 80mL 9mol / L hydrochloric acid, the above-described operation is repeated to continue the combined filtrate was evaporated to dryness under reduced pressure to give a yellow oily residue was slowly added to the residue volume ratio of 1: 1 acetone – ethanol mixture 240 mL, was stirred, and the precipitated solid was 15min, filtered off with suction, the filter cake was recrystallized in 30mL of deionized water, suction filtered to give a white solid that is zoledronic acid monohydrate , about 35. 8g, yield 90.1%, determined by HPLC, purity> 98.5%.
After [0016] (4) Synthesis of zoledronic sodium phosphinate obtained above azole zoledronic acid monohydrate (46.4g, 0. 16mol) washed with water (450 mL of) was dissolved, was added sodium hydroxide (5. 6g, 0. IOmol), were refluxed for 30min, cooling and crystallization, filtration, to obtain a crude product zoledronate sodium, crude mother liquor was concentrated and then half with distilled water (410 mL), isopropanol (60 mL), heated to dissolve the combined, activated carbon bleaching, charcoal filtered off, cooling and crystallization, filtration, washed with water, dried at 40-60 ° C to about crystallization water containing one to give zoledronate sodium (42. 4g, 85%), total yield of more than 60% by HPLC assay, purity 99. 8%, mp239 ° C. IR: 711011 ^ 671 (^ 1 is the stretching vibration peak of the PC, 1643〇 ^ 1 = 0 (: stretching vibration peak, 3011〇 ^ 1 = (: – stretching vibration peak 11 ^ 1 is CN 1406〇 the stretching vibration, 1643CHT1 is C = N stretching vibration peak of 3447 (3485 ^^ (^ 1 is the stretching vibration peak of OH, 1459CHT1 symmetrical bending vibration of CH, 2830CHT1 stretching vibration of CH, 1324CHT1 is P = O the stretching vibration, 1094CHT1 stretching vibration peak of .1HNMR PO (400MHz, D20), S: 8.68 (lH, s), 7.48 (lH, s), 7.34 (lH, s), 4.67 (2H, t).
Effects [0017] Example 2 was added dropwise phosphorus trichloride fixed time on the yield other conditions remain unchanged, only the changes of phosphorus trichloride dropwise addition, dropping zoledronic Table 1 Effect of Sodium yield Experimental results show that excessive phosphorus trichloride was added dropwise, and instantly generate a large amount of gas, the reaction is very intense, the liquid splashing, a rapid rise in temperature, resulting in the low yield, if slowly added dropwise, the reaction rate is too slow, consumption too long, and therefore is the best 4h dropping time.

Zoledronic fixed effect of sodium yield other conditions remain unchanged, imidazol-1-yl acetic acid hydrochloride with phosphorus trichloride and phosphoric acid condensation reaction temperature is zoledronic acid monohydrate embodiment the reaction temperature Example 3 Effect yield (Table 2). The results show that, with increasing temperature, increasing the yield, but at higher temperatures to reflux, shows a decreasing trend in yield, due to decomposition sake phosphorus trichloride, resulting in reduction reaction. Further, when the temperature is too high, solvent evaporation and solvent leakage losses will increase, the reflux temperature is low, the reaction rate is slow, the reaction is insufficient, therefore the yield is low, and therefore the optimum reaction temperature is about 65 ° C.

Example 4

Effect of the ionic liquid frequency reuse sodium zoledronic yield of the reaction medium can be recovered and reused important concern is “green chemistry” used in the present embodiment examines the sodium ionic liquid used in the synthesis of zoledronic repeated use, the experiment results shown in Table 3. Seen from Table 3, the ionic liquid after 5 subsequent to use, product yield began to decrease, the ionic liquid may be recovered and reused effectively, and repeated five times using good performance, and therefore is an ionic liquid in this reaction green solvents may be recycled.

Example 5 different ionic liquids zoledronic same impact conditions were examined yield sodium 1-butyl-3-methylimidazolium tetrafluoroborate ionic liquids ([bmim] BF4), N- ethyl pyridinium tetrafluoroborate ([EPy] BF4), l- butyl-3-methylimidazolium hexafluorophosphate ([bmim] PF6), 1- hydroxyethyl-2,3-dimethyl imidazolium chloride (LOH), 1- propyl-3-carbonitrile methylimidazolium chloride (the LCN) and 1-carboxyethyl-3-methyl imidazolium chloride (LOOH) Effects of sodium zoledronic yield the results are shown in Table 4, the test results show little effect on the synthesis of ionic liquids yield.

Clip
Mar 5, 2013 –
Dr. Reddy’s Laboratories announced today that it has launched Zoledronic Acid Injection (4 mg/5 mL), a bioequivalent generic version of Zometa® (zoledronic acid) 4 mg/5 mL Injection in the US market on March 4, 2013, following the approval by the United States Food & Drug Administration (USFDA) of Dr. Reddy’s ANDA for Zoledronic Acid Injection (4 mg/5 mL).
Dr. Reddy’s Zoledronic Acid Injection 4 mg/5mL is available in a single use vial of concentrate.
Zoledronic acid (INN) or zoledronate (marketed by Novartis under the trade names Zometa, Zomera, Aclasta and Reclast) is a bisphosphonate. Zometa is used to prevent skeletal fractures in patients with cancers such as multiple myeloma and prostate cancer, as well as for treating osteoporosis.It can also be used to treat hypercalcemia of malignancy and can be helpful for treating pain from bone metastases.
An annual dose of zoledronic acid may also prevent recurring fractures in patients with a previous hip fracture.
Reclast is a single 5 mg infusion for the treatment of Paget’s disease of bone. In 2007, the U.S. Food and Drug Administration (FDA) also approved Reclast for the treatment of postmenopausal osteoporosis.

Dr. Reddy’s Laboratories Ltd. (NYSE: RDY) is an integrated global pharmaceutical company, committed to providing affordable and innovative medicines for healthier lives. Through its three businesses – Pharmaceutical Services and Active Ingredients, Global Generics and Proprietary Products – Dr. Reddy’s offers a portfolio of products and services including APIs, custom pharmaceutical services, generics, biosimilars, differentiated formulations and NCEs. Therapeutic focus is on gastro-intestinal, cardiovascular, diabetology, oncology, pain management, anti-infective and pediatrics. Major markets include India, USA, Russia and CIS, Germany, UK, Venezuela, S. Africa, Romania, and New Zealand. For more information, log on to: http://www.drreddys.com
Zometa® is a registered trademark of Novartis AG
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Reclast, Zometa, others[2] |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a605023 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
Intravenous |
| Drug class | Bisphosphonate[1] |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Protein binding | 22% |
| Metabolism | Nil |
| Elimination half-life | 146 hours |
| Excretion | Kidney (partial) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| PDB ligand | |
| Chemical and physical data | |
| Formula | C5H10N2O7P2 |
| Molar mass | 272.09 g/mol |
| 3D model (JSmol) | |
Judith Aronhime, Revital Lifshitz-Liron, “Zoledronic acid crystal forms, zoledronate sodium salt crystal forms, amorphous zoledronate sodium salt, and processes for their preparation.” U.S. Patent US20050054616, issued March 10, 2005., US20050054616
/////////////////disodium zoledronate tetrahydrate, zoledronic acid, ZOMETA, CGP-42446, CGP-42446A
OC(CN1C=CN=C1)(P(O)(O)=O)P(O)(O)=O

Arteolane Maleate
C26H40N2O8
Molecular Weight: 508.612
CAS 959520-73-1
664338-39-0 (free base) 959520-73-1 (maleate) 959520-79-7 (acetate) 664338-40-3 (tosylate) 959520-82-2 (tartrate) 959520-83-3 (citrate)
N-(2-amino-2-methylpropyl)-2-((1R,3R,4”S,5R,5’s,7R)-dispiro[adamantane-2,3′-[1,2,4]trioxolane-5′,1”-cyclohexan]-4”-yl)acetamide maleate
Dispiro[cyclohexane-1,3′-[1,2,4]trioxolane-5′,2”-tricyclo[3.3.1.13,7]decane]-4-acetamide, N-(2-amino-2-methylpropyl)-, cis-, (2Z)-2-butenedioate (1:1)
APPROVED 4.11.2017 CDSCO
Arteolane Maleate and Piperaquine phosphate Dispersible tablets (37.5 mg +187.5 mg

cas 664338-39-0
Arterolane
Arterolane, also known as OZ277 or RBx 11160, is a substance that was tested for antimalarial activity[1] by Ranbaxy Laboratories.[2] It was discovered by US and European scientists who were coordinated by the Medicines for Malaria Venture (MMV).[3] Its molecular structure is uncommon for pharmacological compounds in that it has both a ozonide (trioxolane) group and an adamantanesubstituent.[4]
Initial results were disappointing, and in 2007 MMV withdrew support, after having invested $20M in the research;[5] Ranbaxy said at the time that it intended to continue developing the drug combination on its own.[2] Ranbaxy started a Phase II clinical trial of arterolane, in combination with piperaquine in 2009 that published in 2015.[6][7]
In 2012, Ranbaxy obtained approval to market the arterolane/piperaquine combination drug in India, under the brand name Synriam,[5]and in 2014 received approval to market it in Nigeria, Uganda, Senegal, Cameroon, Guinea, Kenya and Ivory Coast; it had already received approval in Uganda.[8]

Ranbaxy launched India’s first new drug, SynriamTM, treating Plasmodium falciparummalaria in adults. The drug provides quick relief from most malaria-related symptoms, including fever, and has a high cure rate of over 95 %.
Just one tablet per day is required, for three days, instead of two to four tablets, twice daily, for three or more days with other medicines. The drug is independent of dietary restrictions for fatty foods or milk.
Ranbaxy developed Synriam as a fixed-dose combination of arterolane maleate and piperaquine phosphate, where arterolane is the new chemical entity (NCE) that was developed as an alternative to artemisinin. It is the first recently developed antimalarial not based on artemisinin, one of the most effective treatments for malaria, which has shown problems with resistance in recent years. Arterolane was discovered by a collaborative drug discovery project funded by the Medicines for Malaria Venture. Since SynriamTM has a synthetic source, unlike artemisinin-based drugs, production can be scaled up whenever required and a consistent supply can be maintained at a low cost.

The new drug, has been approved by the Drug Controller General of India (DCGI) for marketing in India and conforms to the recommendations of the World Health Organization (WHO) for using combination therapy in malaria. Ranbaxy is also working to make it available in African, Asian and South American markets where Malaria is rampant. SynriamTM trials are ongoing for Plasmodium vivax malaria and a paediatric formulation.
Derek Lowe of the famous In the Pipeline blog had written about arterolane in 2009. At the time it was in Phase III trial, which I assumed were the trials that Ranbaxy was conducting. But it turned out that arterolane was developed by a collaboration between researchers in the US, the UK, Switzerland and Australia who were funded by the World Health Organization and Medicines for Malaria Venture (a Swiss non-profit). They published this work in Nature in 2004 and further SAR (Structure Activity Relationship) studies in J Med Chem in 2010. So Ranbaxy did not develop the drug from scratch? But the press release quotes Arun Sawhney, CEO and Managing Director of Ranbaxy which misleads people to think so: “It is indeed gratifying to see that Ranbaxy’s scientists have been able to gift our great nation its first new drug, to treat malaria, a disease endemic to our part of the world. This is a historic day for science and technology in India as well as for the pharmaceutical industry in the country. Today, India joins the elite and exclusive club of nations of the world that have demonstrated the capability of developing a new drug”. So Ranbaxy mixes a known active compound (piperaquine) with a new compound that someone else found to be active (arterolane) and claims that they developed a new drug? In an interview in LiveMint, Sawhney says, “Ranbaxy spent around $30 million on Synriam and the contribution from DST [India’s Department of Science & Technology] was Rs.5 crore. The drug went through several phases of development since the project began in 2003. We did not look at this as a commercial development. Instead, this is a CSR [Corporate Social Responsibility] venture for us.” That’s a give away because developing a new drug from scratch has to cost more than $30 million + Rs.50 million.
![]() now taken over by sun
now taken over by sun
SynriamTM
|
|
Description SynriamTM is a fixed dose combination of two antimalarial active ingredients arterolane maleate and piperaquine phosphate.
Arterolane maleate is a synthetic trioxolane compound. The chemical name of arterolane maleate is cis-adamantane-2-spiro-3’-8’-[[[(2’-amino-2’ methylpropyl) amino] carbonyl] methyl] 1’,2’,4’-trioxaspiro [4.5] decane hydrogen maleate. The molecular formula is C26H40N2O8 and molecular weight is 508.61. The structural formula is as follows:

| MALARIA |
| Malaria is one of the most prevalent and deadly parasitic diseases in the world. Up to 289 million cases of malaria may have occurred in 2010, causing between 660,000 and 1.25 million deaths, mainly in Africa and mostly of children younger than 5 years. |
| (WHO: http://www.who.int/malaria/publications/world_malaria_report_2012/en/index.html; Fidock, D. A. Eliminating Malaria. Science 2013, 340, 1531-1533.) |
|

Malaria, the most common parasitic disease of humans, remains a major health and economic burden in most tropical countries. Large areas of Central and South America, Hispaniola (Haiti and the Dominican Republic), Africa, the Middle East, the Indian subcontinent, Southeast Asia, and Oceania are considered as malaria-risk areas. It leads to a heavy toll of illness and death, especially amongst children and pregnant women.
According to the World Health Organization, it is estimated that the disease infects about 400 million people each year, and around two to three million people die from malaria every year. There are four kinds of malaria parasites that infect human: Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale and Plasmodium malariae.
Malaria spreads from one person to another by the bite of mosquito, Anopheles gambiae, which serves as vector. When a mosquito sucks the blood of human, sporozoites are transfused into the human body together with saliva of the mosquito. The sporozoites enter into the hepatocytes, reproduce asexually and finally enter into the blood stream. The parasites continue to multiply inside the red blood cells, until they burst and release large number of merozoites. This process continues, destroying a significant number of blood cells and causing the characteristic paroxysm (“chills and fever”) associated with the disease. In the red blood cells, some of the merozoites become male or female gametocytes. These gametocytes are ingested by the mosquito when it feeds on blood. The gametocytes fuse in the vector’s gut; sporozoites are produced and are migrated to the vector’s salivary glands.
The clinical symptoms of malaria are generally associated with the bursting of red blood cells causing an intense fever associated with chills that can leave the infected individual exhausted and bedridden. More severe symptoms associated with repeat infections and/or infection by Plasmodium falciparum include anaemia, severe headaches, convulsions, delirium and, in some instances, death.
Quinine, an antimalarial compound that is extracted from the bark of cinchona tree, is one of the oldest and most effective drugs in existence. Chloroquine and mefloquine are the synthetic analogs of quinine developed in 1940’s, which due to their effectiveness, ease of manufacture, and general lack of side effects, became the drugs of choice. The downside to quinine and its derivatives is that they are short-acting and have bitter taste. Further, they fail to prevent disease relapses and are also associated with side effects commonly known as “Chinchonism syndrome” characterized by nausea, vomiting, dizziness, vertigo and deafness. However, in recent years, with the emergence of drug- resistant strains of parasite and insecticide-resistant strains of vector, the treatment and/or control of malaria is becoming difficult with these conventional drugs.
Malarial treatment further progressed with the discovery of Artemisinin
(qinghaosu), a naturally occurring endoperoxide sesquiterpene lactone isolated from the plant Artemisia annua (Meshnick et al., Microbiol. Rev. 1996, 60, p. 301-315; Vroman et al., Curr. Pharm. Design, 1999, 5, p. 101-138; Dhingra et al., 2000, 66, p. 279-300), and a number of its precursors, metabolites and semi-synthetic derivatives which have shown to possess antimalarial properties. The antimalarial action of artemisinin is due to its reaction with iron in free heme molecules of the malaria parasite, with the generation of free radicals leading to cellular destruction. This initiated a substantial effort to elucidate its molecular mechanism of action (Jefford, dv. Drug Res. 1997, 29, p. 271-325; Cumming et al., Adv. Pharmacol. 1997, 37, p. 254-297) and to identify novel antimalarial peroxides (Dong and Vennerstrom, Expert Opin. Ther. Patents 2001, 1 1, p. 1753-1760).
Although the clinically useful artemisinin derivatives are rapid acting and potent antimalarial drugs, they have several disadvantages including recrudescence,
neurotoxicity, (Wesche et al., Antimicrob. Agents. Chemother. 1994, 38, p. 1813-1819) and metabolic instability (White, Trans. R. Soc. Trop. Med. Hyg., 1994, 88, p. 41-43). A fair number of these compounds are quite active in vitro, but most suffer from low oral activity (White, Trans. R. Soc. Trop. Med. Hyg., 1994, 88, p. 41-43 and van Agtmael et al., Trends Pharmacol. Sci., 1999, 20, p. 199-205). Further all these artemisinin derivatives are conventionally obtained from plant source and are therefore expensive. As the cultivation of the plant material is dependent on many factors including the weather conditions, the supply source thus becomes finite and there are chances of varying yield and potency. This leads to quality inconsistencies and supply constraints. As malaria is more prevalent in developing countries, a switch to cheaper and effective medicine is highly desirable.
Thus there exists a need in the art to identify new peroxide antimalarial agents, especially those which are not dependent on plant source and can be easily synthesized, are devoid of neurotoxicity, and which possess improved solubility, stability and pharmacokinetic properties.
Following that, many synthetic antimalarial 1 ,2,4-trioxanes (Jefford, Adv. Drug Res. 1997, 29, p. 271-325; Cumming et al., Adv. Pharmacol. 1997, 37, p. 254-297), 1,2,4,5-tetraoxanes (Vennerstrom et al., J. Med. Chem., 2000, 43, p. 2753-2758), and other endoperoxides have been prepared. Various patents/applications disclose means and method for treating malaria using Spiro or dispiro 1,2,4-trioxolanes for example, U.S.
Patent Application No. 2004/0186168 and U.S. Patent Nos. 6,486, 199 and 6,825,230. The present invention relates to solid dosage forms of the various spiro or dispiro 1 ,2,4- trioxolanes antimalarial compounds disclosed in these patents/applications and are incorporated herein by reference.
Active compounds representing various Spiro and dispiro 1 ,2,4-trioxolane derivatives possess excellent potency, efficacy against Plasmodium parasites, and a lower degree of neurotoxicity, in addition to their structural simplicity and ease of synthesis. Furthermore, these compounds have half-lives which are believed to permit short-term treatment regimens comparing favorably to other artemisinin-like drugs. In general, the therapeutic dose of trioxolane derivative may range between about 0.1-1000 mg/kg/day, in particular between about 1-100 mg/kg/day. The foregoing dose may be administered as a single dose or may be divided into multiple doses. For malaria prevention, a typical dosing schedule could be, for example, 2.0-1000 mg/kg weekly beginning 1-2 weeks prior to malaria exposure, continued up to 1-2 weeks post-exposure.
Monotherapy with artemisinin (natural or synthetic) class of drugs might cure the patients within 3 days, however perceiving the potential threat of the malarial parasite developing resistance towards otherwise very potent artemisinin class of drugs, WHO had strictly called for an immediate halt to the provision of single-drug artemisinin malaria pills. Combination therapy in case of malaria retards the development of resistance, improve efficacy by lowering recrudescence rate, provides synergistic effect, and increase exposure of the parasite to the drugs.
Artemsinin based combinations are available in the market for a long time.
Artemether-lumafentrine (Co-artem®) was the first fixed dose antimalarial combination containing an artemisinin derivative and has been known since 1999. This combination has passed extensive safety and efficacy trials and has been approved by more than 70 regulatory agencies. Co-artem® is recommended by WHO as the first line treatment for uncomplicated malaria.
Other artemisinin based combinations include artesunate and amodiaquine (Coarsucam®), and dihydroartemisin and piperaquine (Eurartesim®). Unfortunately, all the available artemisinin based combinations have complicated dosage regimens making it difficult and inconvenient for a patient to comply completely with the total prescribed duration. For example, the dosage regimen of Co-artem® for an adult having body weight of more than 35 kg includes 6 doses over three days. The first dose comprises four tablets initially, the second dose comprises four tablets after eight hours, the third to sixth doses comprise four tablets twice for another two days; making it a total of 24 tablets. The dosage regimen of Coarsucam® for an adult having body weight of more than 36 kg or age above 14 years includes three doses over three days; each dose comprises two tablets; making it a total of six tablets. The dosage regimen of Eurartesim® for an adult having body weight between 36 kg – 75 kg includes 3 doses over three days, each dose comprises of three tablets, making it a total of nine tablets.
It is evident that the available artemisinin-based combinations have a high pill burden on patients as they need to consume too many tablets. As noted above, this may increase the possibility of missing a few doses, and, consequently, could result in reduced efficacy due to non-compliance and may even lead to development of resistance for the drug. Therefore, there is an urgent and unmet need for anti-malarial combinations with a simplified daily dosing regimen that reduces the pill burden and would increase patient compliance.
Apart from simplifying the regimen, there are certain limitations for formulators developing formulations with trioxolones, the first being their susceptibility to degradation in presence of moisture that results in reduced shelf lives. Another is their bitter taste, which can result in poor compliance of the regimen or selection of another, possibly less effective, therapeutic agent.
……………………..
http://www.google.st/patents/US6906205

……………………
http://www.google.st/patents/WO2013008218A1?cl=en
structural Formula II.

Formula II
Active compound includes one or more of the various spiro and dispiro trioxolane derivatives disclosed in U.S. Application No. 2004/0186168 and U.S. Patent Nos.
6,486,199 and 6,825,230, which are incorporated herein by reference. These trioxolanes are relatively sterically hindered on at least one side of the trioxolane heterocycle which provides better in vivo activity, especially with respect to oral administration. Particularly, spiro and dispiro 1,2,4-trioxolanes derivatives possess excellent potency and efficacy against Plasmodium parasites, and a lower degree of neurotoxicity.
The term “Active compound I” herein means cis-adamantane-2-spiro-3′-8′-[[[(2′- amino-2′-methylpropyl)amino]carbonyl]-methyl]- 1 ‘,2′,4’-trioxaspiro[4.5]decane hydrogen maleate. The Active compound I may be present in an amount of from about 5% to about 25%, w/w based on the total dosage form.

………………
http://www.google.st/patents/WO2007138435A2?cl=en
A synthetic procedure for preparing compounds of Formula I, salts of the free base c«-adamantane-2-spiro-3′-8′-[[[(2′-amino-2′-methyl propyl) amino] carbonyl] methyl]- 1 ‘, 2′, 4’-trioxaspiro [4.5] decane has been disclosed in U.S. 6,906,205.

The process for the preparation of compounds of Formula I wherein a compound of Formula II (wherein R is lower alkyl) is reacted with a compound of Formula III (wherein R is lower alkyl) to obtain compound of Formula IV;


Formula Formula IV
followed by hydrolysis of the compounds of Formula IV to give a compound of Formula V;

Formula V followed by the reaction of the compound of Formula V with an activating agent, for example, methyl chloroformate, ethyl chloroformate, propyl chloro formate, n-butyl chloro formate, isobutyl chloroformate or pivaloyl chloride leads to the formation of mixed anhydride, which is reacted in situ reaction with 1 ,2-diamino-2-methyl propane to give a compound of Formula VI; and

Formula Vl reacting the compound of Formula VI with an acid of Formula HX (wherein X can be the same as defined earlier) to give compounds of Formula I.
Example 1 : Preparation of O-methyl-2-adamantanone oxime
To a solution of 2-adamantanone (50 g, 0.3328 mol, 1 equiv.) in methanol (0.25 lit), sodium hydroxide solution (15 g, 0.3761mol, 1.13 equiv, in 50 mL water) was added followed by methoxylamine hydrochloride (37.5 g x 81.59% Purity= 30.596 g, 0.366 mol, 1.1 equiv) at room temperature under stirring. The reaction mixture was stirred at room temperature for 1 to 2 h. The reaction was monitored by HPLC. The reaction mixture was concentrated at 40- 45°C under vacuum to get a thick residue. Water (250 mL) was added at room temperature and the reaction mixture was stirred for half an hour. The white solid was filtered, washed with water (50 mL), and dried at 40 to 45°C under reduced pressure. O-methyl 2- adamantanone oxime (57 g, 95 % yield) was obtained as a white solid.
(M++l) 180, 1HNMR (400 MHz, CDCl3 ): δ 1.98 – 1.79 (m, 12H), 2.53 (s, IH), 3.46 ( s, IH), 3.81 (s, 3H).
Example 2: Preparation of 4-(methoxycarbonvmethvPcvclohexanone A high pressure autoclave was charged with a mixture of methyl (4- hydroxyphenyl)acetate (50 g, 0.30 mol), palladium ( 5g) (10 %) on carbon (50 % wet) and O- xylene (250 mL). The reaction mixture was stirred under 110 to 115 psi of hydrogen pressure for 7 to 8 h at 1400C. The reaction was monitored by HPLC. The reaction mixture was then cooled to room temperature, and the catalyst was filtered off. Filtrate was concentrated under reduced pressure to get 4-(methoxycarbonylmethyl)cyclohexanone as light yellow to colorless oily liquid (48.7 g, 97.4 %).
(M++!) 171, ‘ HNMR (400 MHz, CDCl 3): δ 1.48 – 1.51 ( m, 2H), 2.1 1-2.07 (m, 2H), 2.4- 2.23 (m, 7H), 3.7 (s, 3H).
Example 3: Preparation of methyl (Is, 4s)-dispiro [cyclohexane-l, 3′-f 1,2,4] trioxolane-5′, 2″-tricvclor3.3.1.13–71decan1-4-ylacetate
A solution of O-methyl-2-adamantanone oxime (example 1) (11.06 g, 61.7 mmol, 1.5 equiv.) and 4-(methoxycarbonymethyl)cyclohexanone (example 2) (7.0 g, 41.1 mmol, 1 equiv.) in cyclohexane ( 200ml) and dichloromethane (40 mL) was treated with ozone (ozone was produced with an OREC ozone generator [0.6 L/min. O2, 60 V] passed through an empty gas washing bottle that was cooled to -780C). The solvent was removed after the reaction was complete. After removal of solvents, the crude product was purified by crystallization from 80% aqueous ethanol (200 mL) to afford the title compound as a colorless solid. Yield: 10.83 g, 78%, mp: 96-980C; 1HNMR (500 Hz3CDCl3): δ 1.20-1.33 (m, 2H), 1.61-2.09 (m, 5 21H), 2.22 (d, J = 6.8Hz, 2H), 3.67(s,3H).
Example 4: Preparation of (Is, 4s)-dispiro [cyclohexane-1, 3′-[l,2,4] trioxolane-5′, 2″- tricvclo [3.3.1.13‘7] decanl-4-ylacetic acid
Sodium hydroxide (3.86 g, 96.57 mmol, 3 equiv.) in water (80 mL) was added to a solution of methyl (\s, 4s)-dispiro [cyclohexane-1, 3′-[l,2,4] trioxolane-5′, 2″-tricyclo
10 [3.3.1.I3‘7] decan]-4-ylacetate (example 3) (10.83 g, 32.19 mmol, 1 equiv.) in 95% ethanol (150 mL). The mixture was stirred at 500C for about 4 h, cooled to O0C, and treated with IM hydrochloric acid (129ml, 4 equiv). The precipitate was collected by filtration, washed with 50 % aqueous ethanol (150 mL) and dried in vacuum at 40 0C to give the title compound as colorless solid. Yield: 9.952 g, 96%, mp: 146-1480C ( 95% ethanol), 1HNMR (500 Hz,
15 CDCl3): δ 1.19-1.41 (m,2H), 1.60-2.05 (m,21H), 2.27 (d, J=6.8 Hz,2H).
Example 5: Preparation of c?s-adamantane-2-spiro-3′-8′-[[[(2′-amino-2′-methyl propyl) amino] carbonyl] methyl]-! ‘, T , 4’-trioxaspiro [4.5] decane
Method A:
(Is, 4s)-dispiro[cyclohexane- 1 ,3 ‘-[ 1 ,2,4]trioxolane-5 ‘,2 ‘ ‘-tricyclo[3.3.1.13‘7]decan]-4-
.0 ylacetic acid (example 4) (5 g ,15.5mmol, 1 equiv) was mixed with triethylamine (2.5 g , 24.8 mmol, 1.6 equiv) in 100ml of dichloromethane. The reaction mixture was cooled to – 1O0C to 00C. Ethyl chloro formate (1.68 g, 17 mmol, 1.0 equiv) in 15 mL dichloromethane was charged to the above reaction mixture at – 100C to 00C. The reaction mixture was stirred at the same temperature for 10 to 30 minutes. The resulting mixed anhydride reaction mixture
15 was added dropwise to a previously prepared solution of l,2-diamino-2-methylpropane (1.64 g, 18.6 mmol, 1.2 equiv), in 100 mL dichloromethane at -100C to O0C. The temperature of reaction mixture was raised to room temperature. The reaction mixture was stirred at the same temperature till the reaction was complete. Reaction monitoring was done by thin layer chromatography using 5 to 10% methanol in dichloromethane. The reaction was complete
>0 within 2 h. Nitrogen atmosphere was maintained throughout the reaction. Water (50 mL) was charged, organic layer was separated and washed with 10% sodium bicarbonate solution (50 mL) and water (50 mL) at room temperature. The organic layer was dried over sodium sulphate and the solvent was removed at 25 to 4O0C under reduced pressure. Hexane (50ml) was added to obtain residue under stirring at room temperature. The mixture was filtered and washed with 5 mL of chilled hexane. The solid was dried under reduced pressure at room 5 temperature.
Yield: 5.2 g (85.4 %), (M++l) 393, 1HNMR (400 MHz, DMSO-J6 ): δ 0.929 ( s, 6H), 1.105 – 1.079 (m, 2H), 1.887-1.641 (m, 21H), 2.030-2.017 (d, 2H), 2.928 (d, 2H).
Method B:
(Is, 4s)-dispiro [cyclohexane-1, 3′-[l,2,4] trioxolane-5′, 2″-tricyclo [3.3.1.I3‘7]
10 decan]-4-ylacetic acid (example 4) (10 g, 31mmol, 1 equiv) was treated with isobutyl chloroformate (4.5 g, 33mmol, 1.1 equiv) in presence of organic base like triethyl amine (5 g, 49.6mmol, 1.6 equiv) at 00C to 7°C in 250ml of dichloromethane. The solution was stirred at O0C to 7°C for aboutlO to 30 minutes. To the above reaction mixture, previously prepared solution of l,2-diamino-2-methylpropane (3.27 g, 37 mmol, 1.2 equiv), in 50 mL of
15 dichloromethane was added at O0C to 7°C in one lot. The temperature of reaction mixture was raised to room temperature. The reaction mixture was stirred at the room temperature till reaction was over. Reaction monitoring was done by thin layer chromatography using 5 to 10% methanol in dichloromethane. Reaction was complete within 2 h. The reaction nitrogen atmosphere was maintained throughout the reaction. Water (250 mL) was charged, organic
20 layer was separated and washed with 10% sodium bicarbonate solution (200 mL) and water (100 mL) at room temperature and the solvent was removed at 25 to 4O0C under reduced pressure. Hexane (100ml) was added to the residue, under stirring, at room temperature. The mixture was filtered and washed with chilled hexane (10 mL). The resultant solid was dried under reduced pressure at room temperature. Yield: 10.63 g (87%), (M++l) 393, 1HNMR
>5 (400 MHz, DMSO-J6 ) :δ 0.928 ( s, 6H), 1.102 – 1.074 (m, 2H), 1.859-1.616 (m, 21H), 2.031- 2.013 (d, 2H), 2.94-2.925 (d, 2H). Method C:
(\s, 4s)-dispiro[cyclohexane-l,3′-[l,2,4]trioxolane-5′,2″-tricyclo[3.3.1.13>7]decan]-4- ylacetic acid (example 4) (5 g, 15.5mmol, 1 equiv) was treated with pivaloyl chloride (1.87 g, 15.5 mmol, 1 equiv) and triethylamine (2.5gm, 24.8mmol, 1.6 equiv) at -15°C to -100C in dichloromethane (125 mL). The solution was stirred at -150C to -100C for aboutlO to 30 minutes. It resulted in the formation of mixed anydride. To the above reaction mixture, previously prepared solution of 1 ,2-diamino-2-methylpropane (1.64 g, 18.6 mmol, 1.2 equiv) in 25 mL dichloromethane was added at -15°C to -100C. The temperature of reaction mixture was raised to room temperature. The reaction mixture was stirred at the room temperature till reaction was over. Reaction monitoring was done by thin layer chromatography using 5 to 10% methanol in dichloromethane. The reaction was complete within 2 h. Nitrogen atmosphere was maintained throughout the reaction. Water (125 mL) was charged, organic layer was separated and washed with 50 mL of 10% sodium bicarbonate solution and 125 mL of water, respectively at room temperature. Finally solvent was removed at 25 to 4O0C under reduced pressure. 50 mL of 5% Ethyl acetate – hexane solvent mixture was added to the residue under stirring at room temperature. The mixture was filtered and washed with 5 mL of chilled hexane. Solid was dried under reduced pressure at room temperature. Yield: 5.03 g (83 %), (M++l) 393, 1JINMR (400 MHz, OMSO-d6 ):δ 0.93 ( s, 6H), 1.113 – 1.069 (m, 2H), 1.861-1.644 (m, 21H), 2.033-2.015 (d, 2H), 2.948-2.933 (d, 2H).
Example 6: Preparation of c/s-adamantane-2-spiro-3′ -8 ‘-πT(2′-amino-2’ -methyl propyl) amino! carbonyl] methyli-l ‘, 2\ 4′-U-JoXaSpJrQ [4.51 decane maleate To a solution of c/s-adamantane-2-spiro-3′-8′-[[[(2′-amino-2’-methyl propyl) amino] carbonyl] methyl]-! ‘, 2′, 4’-trioxaspiro [4.5] decane (example 5) (60 g, 0.153 moles) in ethanol (150 mL) was added a solution of maleic acid (17.3 g, 0.15 moles, 0.98 equiv. in ethanol 90 mL) and the reaction mixture was stirred for about 1 h. To this clear solution, n- heptane (720 mL) was added at room temperature in 1 h and the reaction mixture was stirred for 3 h. It was then cooled to 0 to 100C and filtered. The cake was washed with n-heptane (60 mL) and dried under vacuum at 40-450C.
Yield: 67 g, 77.4%, mp: 1490C (decomp), (M++l) 393.5, 1HNMR (300 MHz, DMSO-^ ): δ 1.05-1.11 (2H,m), 1.18 (6H,s), 1.64-1.89 (21H,m), 2.07(2H,d), 3.21 (2H,d), 6.06 (2H,d), 7.797 (2H, bs), 8.07 (IH, t).



References
|
5-27-2011
|
PROCESS FOR THE PREPARATION OF DISPIRO 1,2,4-TRIOXOLANE ANTIMALARIALS (OZ277)
|
|
|
2-13-2009
|
STABLE DOSAGE FORMS OF SPIRO AND DISPIRO 1,2,4-TRIOXOLANE ANTIMALARIALS
|
|
|
6-15-2005
|
Spiro and dispiro 1,2,4-trioxolane antimalarials
|
|
|
11-31-2004
|
Spiro and dispiro 1,2,4-trixolane antimalarials
|
ANTIMALARIALS



http://www.rsc.org/chemistryworld/2013/03/new-antimalarial-drug-class-resistance-elq-300-quinolone

http://www.nature.com/nrd/journal/v9/n11/full/nrd3301.html
Structure of NITD609; the 1R,3Sconfiguration is fundamental for its antimalarial activity
1: Valecha N, Savargaonkar D, Srivastava B, Rao BH, Tripathi SK, Gogtay N, Kochar SK, Kumar NB, Rajadhyaksha GC, Lakhani JD, Solanki BB, Jalali RK, Arora S, Roy A, Saha N, Iyer SS, Sharma P, Anvikar AR. Comparison of the safety and efficacy of fixed-dose combination of arterolane maleate and piperaquine phosphate with chloroquine in acute, uncomplicated Plasmodium vivax malaria: a phase III, multicentric, open-label study. Malar J. 2016 Jan 27;15:42. doi: 10.1186/s12936-016-1084-1. PubMed PMID: 26818020; PubMed Central PMCID: PMC4728808.
2: Saha N, Moehrle JJ, Zutshi A, Sharma P, Kaur P, Iyer SS. Safety, tolerability and pharmacokinetic profile of single and multiple oral doses of arterolane (RBx11160) maleate in healthy subjects. J Clin Pharmacol. 2014 Apr;54(4):386-93. doi: 10.1002/jcph.232. PubMed PMID: 24242999.
3: Toure OA, Rulisa S, Anvikar AR, Rao BS, Mishra P, Jalali RK, Arora S, Roy A, Saha N, Iyer SS, Sharma P, Valecha N. Efficacy and safety of fixed dose combination of arterolane maleate and piperaquine phosphate dispersible tablets in paediatric patients with acute uncomplicated Plasmodium falciparum malaria: a phase II, multicentric, open-label study. Malar J. 2015 Nov 25;14:469. doi: 10.1186/s12936-015-0982-y. PubMed PMID: 26608469; PubMed Central PMCID: PMC4660726.
4: Toure OA, Valecha N, Tshefu AK, Thompson R, Krudsood S, Gaye O, Rao BH, Sagara I, Bose TK, Mohanty S, Rao BS, Anvikar AR, Mwapasa V, Noedl H, Arora S, Roy A, Iyer SS, Sharma P, Saha N, Jalali RK; AM–PQP Study Team.. A Phase 3, Double-Blind, Randomized Study of Arterolane Maleate-Piperaquine Phosphate vs Artemether-Lumefantrine for Falciparum Malaria in Adolescent and Adult Patients in Asia and Africa. Clin Infect Dis. 2016 Apr 15;62(8):964-71. doi: 10.1093/cid/ciw029. PubMed PMID: 26908796; PubMed Central PMCID: PMC4803108.
5: Fontaine SD, Spangler B, Gut J, Lauterwasser EM, Rosenthal PJ, Renslo AR. Drug delivery to the malaria parasite using an arterolane-like scaffold. ChemMedChem. 2015 Jan;10(1):47-51. doi: 10.1002/cmdc.201402362. PubMed PMID: 25314098; PubMed Central PMCID: PMC4420023.
6: Gupta A, Singh Y, Srinivas KS, Jain G, Sreekumar VB, Semwal VP. Development and validation of a headspace gas chromatographic method for the determination of residual solvents in arterolane (RBx11160) maleate bulk drug. J Pharm Bioallied Sci. 2010 Jan;2(1):32-7. doi: 10.4103/0975-7406.62706. PubMed PMID: 21814428; PubMed Central PMCID: PMC3146089.
7: Gautam A, Ahmed T, Sharma P, Varshney B, Kothari M, Saha N, Roy A, Moehrle JJ, Paliwal J. Pharmacokinetics and pharmacodynamics of arterolane maleate following multiple oral doses in adult patients with P. falciparum malaria. J Clin Pharmacol. 2011 Nov;51(11):1519-28. doi: 10.1177/0091270010385578. PubMed PMID: 21148048.
8: Patil C, Katare S, Baig M, Doifode S. Fixed dose combination of arterolane and piperaquine: a newer prospect in antimalarial therapy. Ann Med Health Sci Res. 2014 Jul;4(4):466-71. doi: 10.4103/2141-9248.139270. Review. PubMed PMID: 25221689; PubMed Central PMCID: PMC4160665.
9: Valecha N, Looareesuwan S, Martensson A, Abdulla SM, Krudsood S, Tangpukdee N, Mohanty S, Mishra SK, Tyagi PK, Sharma SK, Moehrle J, Gautam A, Roy A, Paliwal JK, Kothari M, Saha N, Dash AP, Björkman A. Arterolane, a new synthetic trioxolane for treatment of uncomplicated Plasmodium falciparum malaria: a phase II, multicenter, randomized, dose-finding clinical trial. Clin Infect Dis. 2010 Sep 15;51(6):684-91. doi: 10.1086/655831. PubMed PMID: 20687837.
10: Lanteri CA, Chaorattanakawee S, Lon C, Saunders DL, Rutvisuttinunt W, Yingyuen K, Bathurst I, Ding XC, Tyner SD. Ex vivo activity of endoperoxide antimalarials, including artemisone and arterolane, against multidrug-resistant Plasmodium falciparum isolates from Cambodia. Antimicrob Agents Chemother. 2014 Oct;58(10):5831-40. doi: 10.1128/AAC.02462-14. PubMed PMID: 25049252; PubMed Central PMCID: PMC4187925.
11: Valecha N, Krudsood S, Tangpukdee N, Mohanty S, Sharma SK, Tyagi PK, Anvikar A, Mohanty R, Rao BS, Jha AC, Shahi B, Singh JP, Roy A, Kaur P, Kothari M, Mehta S, Gautam A, Paliwal JK, Arora S, Saha N. Arterolane maleate plus piperaquine phosphate for treatment of uncomplicated Plasmodium falciparum malaria: a comparative, multicenter, randomized clinical trial. Clin Infect Dis. 2012 Sep;55(5):663-71. doi: 10.1093/cid/cis475. PubMed PMID: 22586253.
12: Dong Y, Wittlin S, Sriraghavan K, Chollet J, Charman SA, Charman WN, Scheurer C, Urwyler H, Santo Tomas J, Snyder C, Creek DJ, Morizzi J, Koltun M, Matile H, Wang X, Padmanilayam M, Tang Y, Dorn A, Brun R, Vennerstrom JL. The structure-activity relationship of the antimalarial ozonide arterolane (OZ277). J Med Chem. 2010 Jan 14;53(1):481-91. doi: 10.1021/jm901473s. PubMed PMID: 19924861.
13: Jourdan J, Matile H, Reift E, Biehlmaier O, Dong Y, Wang X, Mäser P, Vennerstrom JL, Wittlin S. Monoclonal Antibodies That Recognize the Alkylation Signature of Antimalarial Ozonides OZ277 (Arterolane) and OZ439 (Artefenomel). ACS Infect Dis. 2016 Jan 8;2(1):54-61. PubMed PMID: 26819968; PubMed Central PMCID: PMC4718528.
14: Fügi MA, Wittlin S, Dong Y, Vennerstrom JL. Probing the antimalarial mechanism of artemisinin and OZ277 (arterolane) with nonperoxidic isosteres and nitroxyl radicals. Antimicrob Agents Chemother. 2010 Mar;54(3):1042-6. doi: 10.1128/AAC.01305-09. PubMed PMID: 20028825; PubMed Central PMCID: PMC2825978.
15: Mossallam SF, Amer EI, El-Faham MH. Efficacy of Synriam™, a new antimalarial combination of OZ277 and piperaquine, against different developmental stages of Schistosoma mansoni. Acta Trop. 2015 Mar;143:36-46. doi: 10.1016/j.actatropica.2014.12.005. PubMed PMID: 25530543.
16: Longo M, Zanoncelli S, Brughera M, Colombo P, Wittlin S, Vennerstrom JL, Moehrle J, Craft JC. Comparative embryotoxicity of different antimalarial peroxides: in vitro study using the rat whole embryo culture model (WEC). Reprod Toxicol. 2010 Dec;30(4):583-90. doi: 10.1016/j.reprotox.2010.07.011. PubMed PMID: 20708075.
17: Abiodun OO, Brun R, Wittlin S. In vitro interaction of artemisinin derivatives or the fully synthetic peroxidic anti-malarial OZ277 with thapsigargin in Plasmodium falciparum strains. Malar J. 2013 Jan 31;12:43. doi: 10.1186/1475-2875-12-43. PubMed PMID: 23368889; PubMed Central PMCID: PMC3566918.
18: White NJ. Can new treatment developments combat resistance in malaria? Expert Opin Pharmacother. 2016 Jul;17(10):1303-7. doi: 10.1080/14656566.2016.1187134. PubMed PMID: 27191998.
19: Wang X, Dong Y, Wittlin S, Charman SA, Chiu FC, Chollet J, Katneni K, Mannila J, Morizzi J, Ryan E, Scheurer C, Steuten J, Santo Tomas J, Snyder C, Vennerstrom JL. Comparative antimalarial activities and ADME profiles of ozonides (1,2,4-trioxolanes) OZ277, OZ439, and their 1,2-dioxolane, 1,2,4-trioxane, and 1,2,4,5-tetraoxane isosteres. J Med Chem. 2013 Mar 28;56(6):2547-55. doi: 10.1021/jm400004u. PubMed PMID: 23489135.
20: Marfurt J, Chalfein F, Prayoga P, Wabiser F, Wirjanata G, Sebayang B, Piera KA, Wittlin S, Haynes RK, Möhrle JJ, Anstey NM, Kenangalem E, Price RN. Comparative ex vivo activity of novel endoperoxides in multidrug-resistant plasmodium falciparum and P. vivax. Antimicrob Agents Chemother. 2012 Oct;56(10):5258-63. doi: 10.1128/AAC.00283-12. PubMed PMID: 22850522; PubMed Central PMCID: PMC3457353.
 |
|
| Clinical data | |
|---|---|
| Routes of administration |
Oral |
| ATC code |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| Chemical and physical data | |
| Formula | C22H36N2O4 |
| Molar mass | 392.531 g/mol |
| 3D model (JSmol) | |
////////////Arteolane Maleate, OZ-277, RBx-11160, OZ 277, RBx 11160, OZ277, RBx11160, IND 2017
O=C(NCC(C)(N)C)C[C@H](CC1)CC[C@@]21OC3(OO2)[C@H]4C[C@H]5C[C@@H]3C[C@@H](C4)C5.O=C(O)/C=C\C(O)=O
Bepotastine Besilate
ベポタスチンベシル酸塩
| 10 mg Tablets | For the treatment of allergic rhinitis | 27.03.2017 CDSCO
APPROVED |
USFDA
NDA 22-288 Bepotastine Besilate 1.5% Ophthalmic Solution ISTA Pharmaceuticals, Inc.
https://www.accessdata.fda.gov/drugsatfda_docs/nda/2009/02228s000_ChemR.pdf

Drug Substance Bepotastine besilate is manufactured by Ube Industries and the information for the NDA is submitted through DMF #19966. Bepotastine besilate is a white crystalline powder with no odor and bitter taste. It is very soluble in but sparingly soluble in . It is stable when exposed to light, and optically active. The S-isomer is the active drug and is controlled as an impurity through synthesis. The distribution coefficient in 1-octanol is higher than in aqueous buffer in the pH 5-9 range. There are 10 potential impurities but only one impurity is above 0.1%. Two potential genotoxic impurities are controlled below . Residual is controlled below Bepotastine besilate is stable under long term storage conditions for (25ºC/60% RH) over 5 years
Bepotastine besilate was originally developed as an oral tablet dosage form and got approval in Japan in 2000 for allergic rhinitis. It is a non-sedating anti-allergic drug. The proposed NDA is an ophthalmic solution indicated for allergic conjunctivitis. Bepotastine besilate ophthalmic solution 1.5% is a sterile solution. It is an aqueous solution to be administered as drops at or near physiological pH range of tears. The formulation contains sodium chloride, monobasic sodium phosphate as dihydrate, benzalkonium chloride, sodium hydroxide and purified water; typically these components are used for , preservative action, pH adjustment,
INTRO
Bepotastine is a non-sedating, selective antagonist of the histamine 1 (H1) receptor. Bepotastine was approved in Japan for use in the treatment of allergic rhinitis and uriticaria/puritus in July 2000 and January 2002, respectively, and is marketed by Tanabe Seiyaku Co., Ltd. under the brand name Talion. It is available in oral and opthalmic dosage forms in Japan. The opthalmic solution is FDA approved since Sept 8, 2009 and is under the brand name Bepreve.
Tae Hee Ha, Chang Hee Park, Won Jeoung Kim, Soohwa Cho, Han Kyong Kim, Kwee Hyun Suh, “PROCESS FOR PREPARING BEPOTASTINE AND INTERMEDIATES USED THEREIN.” U.S. Patent US20100168433, issued July 01, 2010., US20100168433
BEPREVE® (bepotastine besilate ophthalmic solution) 1.5% is a sterile, topically administered drug for ophthalmic use. Each mL of BEPREVE contains 15 mg bepotastine besilate.
Bepotastine besilate is designated chemically as (+) -4-[[(S)-p-chloro-alpha -2pyridylbenzyl] oxy]-1-piperidine butyric acid monobenzenesulfonate. The chemical structure for bepotastine besilate is:
 |
Bepotastine besilate is a white or pale yellowish crystalline powder. The molecular weight of bepotastine besilate is 547.06 daltons. BEPREVE ophthalmic solution is supplied as a sterile, aqueous 1.5% solution, with a pH of 6.8.
The osmolality of BEPREVE (bepotastine besilate ophthalmic solution) 1.5% is approximately 290 mOsm/kg.
ベポタスチンベシル酸塩 JP17
Bepotastine Besilate

C21H25ClN2O3▪C6H6O3S : 547.07
[190786-44-8]
Bepotastine (Talion, Bepreve) is a 2nd generation antihistamine.[1] It was approved in Japan for use in the treatment of allergic rhinitisand urticaria/pruritus in July 2000 and January 2002, respectively. It is currently marketed in the United States under the brand-name Bepreve, by ISTA Pharmaceuticals.
Bepotastine besilate is a second-generation antihistamine that was launched in a tablet formulation under a collaboration between Tanabe Seiyaku and Ube in 2000 and in 2002 for the treatment of allergic rhinitis including sneeze, mucus discharge and solidified mucus, and for the treatment of urticaria, respectively. An orally disintegrating tablet was made available in Japan in 2006, while a dry syrup formulation for the treatment of allergic rhinitis was studied in clinical trials at Tanabe Seiyaku for the treatment of allergic rhinitis
Originally developed at Ube, bepotastine besilate was later licensed to Tanabe Seiyaku as part of a collaboration agreement. In 2010, rights were licensed to Dong-A and Mitsubishi Tanabe Pharma in Korea for the treatment of eye disorders.
Bepotastine is available as an ophthalmic solution and oral tablet. It is a direct H1-receptor antagonist that inhibits the release of histamine from mast cells.[2] The ophthalmic formulation has shown minimal systemic absorption, between 1 and 1.5% in healthy adults.[3] Common side effects are eye irritation, headache, unpleasant taste, and nasopharyngitis.[3] The main route of elimination is urinary excretion, 75-90% excreted unchanged.[3]
It is marketed in Japan by Tanabe Seiyaku under the brand name Talion. Talion was co-developed by Tanabe Seiyaku and Ube Industries, the latter of which discovered bepotastine. In 2001, Tanabe Seiyaku granted Senju, now owned by Allergan, exclusive worldwide rights, with the exception of certain Asian countries, to develop, manufacture and market bepotastine for ophthalmic use. Senju, in turn, has granted the United States rights for the ophthalmic preparation to ISTA Pharmaceuticals.
In 2011, ISTA pharmaceuticals experienced a 2.4% increase in net revenues from 2010, which was driven by the sales of Bepreve. Their net revenue for 2011 was $160.3 million.[4] ISTA Pharmaceuticals was acquired by Bausch & Lomb in March 2012 for $500 million.[5] Bausch & Lomb hold the patent for bepotastine besilate (https://www.accessdata.fda.gov/scripts/cder/ob/docs/temptn.cfm. On November 26, 2014, Bausch & Lomb sue Micro Labs USA for patent infringement.[6] Bausch & Lomb was recently bought out by Valeant Pharmaceuticals in May 2013 for $8.57 billion, Valeant’s largest acquisition to date, causing the company’s stock to rise 25% when the deal was announced.[7]
A Phase III clinical trial was carried out in 2010 to evaluate the effectiveness of bepotastine besilate ophthalmic solutions 1.0% and 1.5%.[8] These solutions were compared to a placebo and evaluated for their ability to reduce ocular itchiness. The study was carried out with 130 individuals and evaluated after 15 minutes, 8 hours, or 16 hours. There was a reduction in itchiness at all-time points for both ophthalmic solutions. The study concluded that bepotastine besilate ophthalmic formulations reduced ocular itchiness for at least 8 hours after dosing compared to placebo. Phase I and II trials were carried out in Japan.
Studies have been performed in animals and bepotastine besilate was not found to be teratogenic in rats during fetal development, even at 3,300 times more that typical use in humans.[3] Evidence of infertility was seen in rats at 33,000 times the typical ocular does in humans.[3] The safety and efficacy has not been established in patients under 2 years of age and has been extrapolated from adults for patients under 10 years of age.[3]
SYN
EP 0335586; JP 1989242574; JP 1990025465; JP 1993294929; US 4929618
The reaction of 4-[1-(4-chlorophenyl)-1-(2-pyridyl)methoxy]piperidine (I) with ethyl 4-bromobutyrate (II) by means of K2CO3 in refluxing acetone gives the corresponding condensation product (III), which is then hydrolyzed with NaOH in ethanol/water yielding compound (IV).
SYN 2
JP 1998237070; JP 2000198784; WO 9829409
A new synthesis of betotastine has been developed: The racemic 4-[1-(4-chlorophenyl)-1-(2-pyridyl)methoxy]piperidine (I) is submitted to optical resolution with N-acyl amino acids such as N-acetyl-L-phenylalanine (preferred), N-acetyl-L-leucine, N-(benzyloxycarbonyl)-L-phenylalanine, N-(benzyloxycarbonyl)-L-valine, N-(benzyloxycarbonyl)-L-threonine, N-(benzyloxycarbonyl)-L-serine or with (2R,3R)-3-(5-chloro-2-nitrophenylsulfanyl)-2-hydroxy-3-(4-methoxyphenyl)propionic acid (preferred) or (2R,3R)-2-hydroxy-3-(4-methoxyphenyl)-3-(2-nitrophenylsulfanyl)propionic acid as chiral intermediates, yielding the (S)-isomer (II). The condensation of (II) with ethyl 4-bromobutyrate (III) by means of a base such as Na2CO3, NaHCO3, K2CO3 or KHCO3 gives the expected 4-(1-piperidinyl)butyric acid ester (IV), which is finally hydrolyzed with NaOH or KOH in aqueous ethanol or methanol.
SYN 3
A new synthesis of betotastine has been developed: The racemic 4-[1-(4-chlorophenyl)-1-(2-pyridyl)methoxy]piperidine (I) is submitted to optical resolution with N-acyl amino acids such as N-acetyl-L-phenylalanine (preferred), N-acetyl-L-leucine, N-(benzyloxycarbonyl)-L-phenylalanine, N-(benzyloxycarbonyl)-L-valine, N-(benzyloxycarbonyl)-L-threonine, N-(benzyloxycarbonyl)-L-serine or with (2R,3R)-3-(5-chloro-2-nitrophenylsulfanyl)-2-hydroxy-3-(4-methoxyphenyl)propionic acid (preferred) or (2R,3R)-2-hydroxy-3-(4-methoxyphenyl)-3-(2-nitrophenylsulfanyl)propionic acid as chiral intermediates, yielding the (S)-isomer (II). The condensation of (II) with ethyl 4-bromobutyrate (III) by means of a base such as Na2CO3, NaHCO3, K2CO3 or KHCO3 gives the expected 4-(1-piperidinyl)butyric acid ester (IV), which is finally hydrolyzed with NaOH or KOH in aqueous ethanol or methanol.
CLIP
A Novel Synthetic Method for Bepotastine, a Histamine H1 Receptor …
file:///C:/Users/91200291/Downloads/B130241_549.pdf


Scheme 1. Synthesis of bepotastine l-menthyl ester N-benzyloxycarbonyl-L-aspartic acid complex (3), bepotastine besilate (4) and bepotastine calcium (5). Reagents and conditions; i) 4-bromobutanoic acid l-menthyl ester, K2CO3, acetone, reflux, 7 h, 95-99%; ii) N-benzyloxycarbonyl-L-aspartic acid (NCbzLAA), ethyl acetate, rt, 12 h, 71-73%; iii) Ethyl acetate/H2O, NaHCO3, 97-99%; iv) EtOH:H2O = 1:1, NaOH, rt, 12 h, 3.0 N-HCl Neutralization, 92- 95%; v) AcOH, reflux, 12 h, racemization 97-100%; vi) Bezensulfonic acid, acetonitrile, rt, 12 h, 64-67%; vii) NaOH, H2O, CaCl2, rt, 12 h, 86-89%.
Synthesis of (S)-Bepotastine Besilate (4). Bepotastine (50 g, 0.13 mol) was dissolved in 500 mL of acetonitrile, and benzenesulfonic acid monohydrate (20 g, 0.11 mol) was added to the reaction mixture. Bepotastine besilate (0.5 g, 1.28 mmol) was seeded in the reaction mixture and stirred at rt for 12 h. The solid precipitate was filtered and dried. The product was obtained 38 g (yield: 64%, optical purity: 99.5% ee) as a pale white crystalline powder. Melting point: 161- 163 o C. Water: 0.2% (Karl-Fischer water determination). MS: m/z 389.1 [M+H]; 1 H-NMR (300 MHz, DMSO-d6) δ 9.2 (br s, 1H), 8.5 (d, J = 4.1 Hz, 1H), 7.8 (t, J = 7.7 Hz, 1H), 7.6 (m, 3H), 7.4 (m, 4H), 7.3 (m, 4H), 5.7 (s, 1H), 3.7 (br s, 2H), 3.3 (br s, 3H), 3.1 (br s, 2H), 2.3 (t, J = 14.1 Hz, 2H), 2.2 (m, 1H), 2.0 (m, 1H), 1.8 (m, 3H), 1.7 (m, 1H); IR (KBr, cm−1 ): 3422, 2996, 2909, 2735, 2690, 2628, 1719, 1592, 1572, 1488, 1470, 1436, 1411, 1320, 1274, 1221, 1160, 1123, 1066, 1031, 1014, 996, 849, 830, 771, 759, 727, 693, 612, 564
| FDA Orange Book Patents: 1 of 3 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 8784789 |
| Expiration | Sep 5, 2024 |
| Applicant | BAUSCH AND LOMB INC |
| Drug Application | N022288 (Prescription Drug: BEPREVE. Ingredients: BEPOTASTINE BESILATE) |
| FDA Orange Book Patents: 2 of 3 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 6780877 |
| Expiration | Sep 19, 2019 |
| Applicant | BAUSCH AND LOMB INC |
| Drug Application | N022288 (Prescription Drug: BEPREVE. Ingredients: BEPOTASTINE BESILATE) |
| FDA Orange Book Patents: 3 of 3 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 8877168 |
| Expiration | Jul 30, 2023 |
| Applicant | BAUSCH AND LOMB INC |
| Drug Application | N022288 (Prescription Drug: BEPREVE. Ingredients: BEPOTASTINE BESILATE) |
 |
|
| Clinical data | |
|---|---|
| Trade names | Bepreve |
| AHFS/Drugs.com | International Drug Names |
| MedlinePlus | a610012 |
| Pregnancy category |
|
| Routes of administration |
Oral, topical (eye drops) |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | High (oral) Minimal (topical) |
| Protein binding | ~55% |
| Excretion | Renal (75–90%) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C21H25ClN2O3 |
| Molar mass | 388.88 g/mol |
| 3D model (JSmol) | |
|url= value (help).////////////Bepotastine Besilate, ベポタスチンベシル酸塩 ,Talion , tau284, TAU-284DS, TAU-284, DA-5206
HL-151 , SNJ-1773
C1CN(CCC1OC(C2=CC=C(C=C2)Cl)C3=CC=CC=N3)CCCC(=O)O.C1=CC=C(C=C1)S(=O)(=O)O

Fipronil
APPROVED CDSCO INDIA 25.06.2018
Fipronil 50mg/134mg/268mg/402 mg spot on solution for cats and dogs , For treatment of flea and tick infestation in cats and dogs (for veterinary use only)
Fipronil is a broad-spectrum insecticide that belongs to the phenylpyrazole chemical family. Fipronil disrupts the insect central nervous system by blocking GABA-gated chloride channels and glutamate-gated chloride (GluCl) channels. This causes hyperexcitation of contaminated insects’ nerves and muscles. Fipronil’s specificity towards insects is believed to be due to its greater affinity to the GABA receptor in insects relative to mammals and its effect on GluCl channels, which do not exist in mammals.[1]
Because of its effectiveness on a large number of pests, fipronil is used as the active ingredient in flea control products for pets and home roach traps as well as field pest control for corn, golf courses, and commercial turf. Its widespread use makes its specific effects the subject of considerable attention. This includes ongoing observations on possible off-target harm to humans or ecosystems as well as the monitoring of resistance development.[2]
Fipronil is or has been used in:
Fipronil is classed as a WHO Class II moderately hazardous pesticide, and has a rat acute oral LD50 of 97 mg/kg.
It has moderate acute toxicity by the oral and inhalation routes in rats. Dermal absorption in rats is less than 1% after 24 h and toxicity is considered to be low. It has been found to be very toxic to rabbits.
The photodegradate MB46513 or desulfinylfipronil, appears to have a higher acute toxicity to mammals than fipronil itself by a factor of about 10.[9]
Symptoms of acute toxicity via ingestion includes sweating, nausea, vomiting, headache, abdominal pain, dizziness, agitation, weakness, and tonic-clonic seizures. Clinical signs of exposure to fipronil are generally reversible and resolve spontaneously. As of 2011, no data were available regarding the chronic effects of fipronil on humans. The U.S. EPA has classified fipronil as a group C (possible human) carcinogen based on an increase in thyroid follicular cell tumors in both sexes of the rat. However, as of 2011, no human data is available regarding the carcinogenic effects of fipronil.[10]
Two Frontline TopSpot products were determined by the New York State Department of Environmental Conservation to pose no significant exposure risks to workers applying the product. However, concerns were raised about human exposure to Frontline spray treatment in 1996, leading to a denial of registration for the spray product. Commercial pet groomers and veterinarians were considered to be at risk from chronic exposure via inhalation and dermal absorption during the application of the spray, assuming they may have to treat up to 20 large dogs per day.[3] Fipronil is not volatile, so the likelihood of humans being exposed to this compound in the air is low.[10]
In contrast to neonicotinoids such as acetamiprid, clothianidin, imidacloprid, and thiamethoxam, which are absorbed through the skin to some extent, fipronil is not absorbed substantially through the skin.[11]
Fipronil may be quantitated in plasma by gas chromatography-mass spectrometry or liquid chromatography-mass spectrometry to confirm a diagnosis of poisoning in hospitalised patients or to provide evidence in a medicolegal death investigation.[12]
Fipronil is highly toxic for crustaceans, insects and zooplankton,[13] as well as bees, termites, rabbits, the fringe-toed lizard, and certain groups of gallinaceous birds. It appears to reduce the longevity and fecundity of female braconid parasitoids. It is also highly toxic to many fish, though its toxicity varies with species. Conversely, the substance is relatively innocuous to passerines, wildfowl, and earthworms.
Its half-life in soil is four months to one year, but much less on soil surface because it is more sensitive to light (photolysis) than water (hydrolysis).[14]
Few studies of effects on wildlife have been conducted, but studies of the nontarget impact from emergency applications of fipronil as barrier sprays for locust control in Madagascar showed adverse impacts of fipronil on termites, which appear to be very severe and long-lived. Also, adverse effects were indicated in the short term on several other invertebrate groups, one species of lizard (Trachylepis elegans), and several species of birds (including the Madagascar bee-eater).
Nontarget effects on some insects (predatory and detritivorous beetles, some parasitic wasps and bees) were also found in field trials of fipronil for desert locust control in Mauritania, and very low doses (0.6-2.0 g a.i./ha) used against grasshoppers in Niger caused impacts on nontarget insects comparable to those found with other insecticides used in grasshopper control. The implications of this for other wildlife and ecology of the habitat remain unknown, but appear unlikely to be severe.[3] Unfortunately, this lack of severity was not observed in bee species in South America. Fipronil is also used in Brazil and studies on the stingless bee Scaptotrigona postica have shown adverse reactions to the pesticide, including seizures, paralysis, and death with a lethal dose of .54 ng a.i./bee and a lethal concentration of .24 ng a.i./μl diet. These values are highly toxic in Scaptotrigona postica and bees in general.[15] Toxic baiting with fipronil has been shown to be effective in locally eliminating German wasps. All colonies within foraging range were completely eliminated within one week.[16][17][5]
In May 2003, the French Directorate-General of Food at the Ministry of Agriculture determined that a case of mass bee mortality observed in southern France was related to acute fipronil toxicity. Toxicity was linked to defective seed treatment, which generated dust. In February 2003, the ministry decided to temporarily suspend the sale of BASF crop protection products containing fipronil in France.[18] The seed treatment involved has since been banned.[citation needed] Fipronil was used in a broad spraying to control locusts in Madagascar in a program that began in 1997.[19]
Notable results from wildlife studies include:
Fipronil is one of the main chemical causes blamed for the spread of colony collapse disorder among bees. It has been found by the Minutes-Association for Technical Coordination Fund in France that even at very low nonlethal doses for bees, the pesticide still impairs their ability to locate their hive, resulting in large numbers of forager bees lost with every pollen-finding expedition.[22] A synergistic toxic effect of fipronil with the fungal pathogen Nosema ceranae was recently reported[23]. The functional basis for this toxic effect is now understood: the synergy between fipronil and the pathogenic fungus induces changes in male physiology leading to infertility[24] A 2013 report by the European Food Safety Authorityidentified fipronil as “a high acute risk to honeybees when used as a seed treatment for maize and on July 16, 2013 the EU voted to ban the use of fipronil on corn and sunflowers within the EU. The ban took effect at the end of 2013.”[25][26]
Fipronil acts by binding to allosteric sites of GABAA receptors and GluCl receptors (of insects) as an antagonist (a form of noncompetitive inhibition). This prevents the opening of chloride ion channels normally encouraged by GABA, reducing the chloride ions’ ability to lower a neuron’s membrane potential. This results in an overabundance of neurons reaching action potential and likewise CNS toxicity via overstimulation.[27][28][29][30]
In animals and humans, fipronil poisoning is characterized by vomiting, agitation, and seizures, and can usually be managed through supportive care and early treatment of seizures, generally with benzodiazepine use.[31][32]
Fipronil was discovered and developed by Rhône-Poulenc between 1985 and 1987, and placed on the market in 1993 under the B2 U.S. Patent 5,232,940 B2. Between 1987 and 1996, fipronil was evaluated on more than 250 insect pests on 60 crops worldwide, and crop protection accounted for about 39% of total fipronil production in 1997. Since 2003, BASF holds the patent rights for producing and selling fipronil-based products in many countries.
The 2017 Fipronil eggs contamination is an incident in Europe and South Korea involving the spread of insecticide contaminated eggs and egg products. Chicken eggs were found to contain Fipronil and distributed to 15 European Union countries, Switzerland, and Hong Kong.[33][34] Approximately 700,000 eggs are thought to have reached shelves in the UK alone.[35] Eggs at 44 farms in Taiwan were also found with excessive Fipronil levels.[36]
SYN


SYN 2

SYN 3

SYN 4

PATENT
http://www.allindianpatents.com/patents/271132-a-process-for-the-synthesis-of-fipronil
5-Amino-l-(2,6-dichloro-4-trifluoromethylphenyl)-3-cyano-4-trifluoromethyl
sulfinyl pyrazole or 5-Amino-[2,6-dichloro-4-(trif]uoromethyl)phenyl]-4-[-(1 (R,S)-trifluoromethyl)sulfinyl]-1H-pyrazole-3-carbonitrile also known as Fipronil is a novel pesticide characterized by high efficiency, low toxicity and especially low residue.
There are various routes to synthesize Fipronil by oxidation of thiopyrazole with various other oxidizing agents in suitable solvents. Oxidation of sulfides is a very useful route for the preparation of sulfoxides. Literature is replete with the conversion of sulfides to sulfoxides and/or sulfones. However, most of the existing methods use expensive, toxic or rare oxidizing reagents, which are difficult to prepare, are very expensive and cannot be used on commercial scale. Many of these processes suffer from poor selectivity.
WO01/30760 describes oxidation of 5-amino-l-(2,6-dichloro-4-trifluoromethylphenyl)-3-cyano-4-trifluoromethylthio-pyrazole with trifluoro-acetic acid and hydrogen peroxide in the presence of boric acid. The quantity
of trifluoroacetic acid used is 14.5 molar equivalents. The patent also
discloses the preparation of 5-amino-1-(2,6-dichloro-4-trifluoromethyl
phenyl)-3-cyano-4-trifluoromethylthio-pyrazole from 5-amino-1-(2,6-
dichloro-4-trifluoromethyl phenyl)-3-cyano pyrazole-4-yl disulphide.
European Patent publication No.295117 describes the preparation of 5-amino-l-(2,6-dichloro-4-trifluoromethylphenyl)-3-cyano-4-trifluoromethylsulphinyl pyrazole starting from 2,6-Dichloro-4-trifluoromethylaniline to give an intermediate 5-amino-l-(2,6-dichloro-4-trifluoromethylphenyl)-3-cyano-4-trifluoromethylthiopyrazole which is oxidized with meta-chloroperbenzoic acid in chloroform to give desired product.
Oxidizing agents such as perbenzoic acids do not provide effective and regioselective oxidation of electron deficient sulfides such as trifluoromethylsulphides which are less readily oxidized than other sulfides. Trifluoroacetic acid and trichloroacetic acid are found to be very efficient and regioselective oxidation medium for oxidation of 5-amino-l-(2,6-dichloro-4-trifluoromethylphenyl)-3-cyano-4-trifluoromethylthio-pyrazole in presence of hydrogen peroxide. Trichloroacetic acid can not be used alone due to higher melting point. Trifluoroacetic acid on the other hand is very regioselective with respect to conversion and low by-products formation. However, it is expensive, water miscible, corrosive to metal as well as glass, comparatively lower boiling and it’s recovery (in anhydrous form) is complex in nature.
W000/35851/2000 talks about synthesis of 2,6-Dichloro-4-trifluoromethylaniline starting from 3,4,5-trichloro-benzotrifluoride in the presence of alkaline fluorides like lithium fluoride and ammonia in the
presence of N-methylpyrrolidone at 250°C to give 97% conversion and 87% selectivity. The main drawback of the above process is the synthesis of 3,4,5-trichlorobenzotrifluoride in high yield and purity. Chlorination of p-chlorobenzotrifluoride gives a mixture of 3,4,5-trichlorobenzotrifluoride in 72% GLC conversions, 3,4-dichloro and tetrachlorobenzotrifluoride. The process to get pure 3,4,5-isomer from this mixture by fractionation followed by crystallization is very tedious. Moreover in-spite of using very pure intermediates, substantial amount of an undesired isomer (3-amino-4,5-dichlorobenzotrifluoride) is also obtained.
Another approach to generate 3,4,5-trichlorobenzotrifluoride with high yield and purity is to perform denitrochlorination of 4-chloro-3,5-dinitrobenzotrifluoride in the presence of a catalyst as described in GB Patent 2154581A. Even though the process produces 3,4,5-trichlorobenzotrifluoide in high yield and purity, the reaction conditions are too drastic to be employed for an industrial process.
The known commercial processes for the manufacture of Fipronil uses corrosive and expensive chemical such as trifluoroaceticacid, hydrogen peroxide and m-chloroperbenzoicacid Trifluoroacetic acid is expensive and generally not used in large quantities, as well as of m-chloroperbenzoic acid is difficult to handle at commercial scale due to its un-stability and detonating effect. Also the raw material used such as 2,6-Dichloro-4-trifluoromethylaniline are not easily available or made. The overall process for the Fipronil as disclosed above is found to be unsatisfactory in one respect or the other.
Thus, there is felt a need for preparing Fipronil from easily available raw materials in a simple and economical manner at an industrial level, with high yields and purity.
PATENT
https://patents.google.com/patent/US20130030190A1/en
Example 19 Purification of Fipronil
CN 101250158 [2008 to Hunan Res Inst of of chemical Ind.]
WO2005/44806 A1, ; Page/Page column 7-8; 12 ;
WO2009/77853 A1, ; Page/Page column 28-29 ;
US 5,618,945 [1995, to Rhone-Poulenc]
CN 102060774
IN 178903 [1997, to Rallis India Ltd.]
WO 2009/077,853 [2009 to Vetoquinol SA ]
BG 109983 [2008 to BASF Agro B V]
US 8,507,693 [2013, to Gharda]
US 5,618,945 [1995, to Rhone-Poulenc]
WO 2007/122,440 [ 2007 to Gharda Chemicals Ltd.]
FR 2,925,493 [2009 to to Vetoquinol SA ]
CN 1176078 [ 2002 to Jiangsu Prov Inst of Pesticide]
EP 0,374,061 [ 1990 to Rhone Poulenc Agrochimie]
US 5,232,940 [1993, to May and Baker]
PAPER
Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), , # 24 p. 3371 – 3376
Synthesis 2008, 11, 1682-1684
Synthesis 2007, 22, 3507-3511
Tetrahedron Letters, , 2007, 48(48), 8518-8520
Tetrahedron Letters 2008 ,49. 3463-3465
 |
|
 |
|
| Names | |
|---|---|
| IUPAC name
(RS)-5-Amino-1-[2,6-dichloro-4-(trifluoromethyl)phenyl]-4-(trifluoromethylsulfinyl)pyrazole-3-carbonitrile
|
|
| Other names
Fipronil
Fluocyanobenpyrazole Termidor |
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChEBI | |
| ChEMBL | |
| ChemSpider | |
| ECHA InfoCard | 100.102.312 |
| KEGG | |
|
PubChem CID
|
|
| UNII | |
| Properties | |
| C12H4Cl2F6N4OS | |
| Molar mass | 437.14 g·mol−1 |
| Density | 1.477-1.626 g/cm3 |
| Melting point | 200.5 °C (392.9 °F; 473.6 K) |
| Pharmacology | |
| QP53AX15 (WHO) | |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
/////////////Fipronil, INDIA 2018, フィプロニル , HSDB 7051, RM 1601, veterinary, ind 2018
C1=C(C=C(C(=C1Cl)N2C(=C(C(=N2)C#N)S(=O)C(F)(F)F)N)Cl)C(F)(F)F
“ DRUG APPROVALS INTERNATIONAL” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent