


| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT01706783 | A Trial Investigating the Safety, Tolerability, Availability and Distribution in the Body of Once-weekly Long-acting Growth Hormone (Somapacitan) Compared to Once Daily Norditropin NordiFlex® in Adults With Growth Hormone Deficiency | Phase 1 | Completed | 2018-05-25 |
| NCT01973244 | A Trial Investigating Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of a Single Dose of Long-acting Growth Hormone (Somapacitan) Compared to Daily Dosing of Norditropin® SimpleXx® in Children With Growth Hormone Deficiency | Phase 1 | Completed | 2018-05-25 |
| NCT02962440 | A Trial Investigating the Absorption, Metabolism and Excretion of Somapacitan After Single Dosing in Healthy Male Subjects | Phase 1 | Completed | 2017-06-07 |
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 71)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Clascoterone
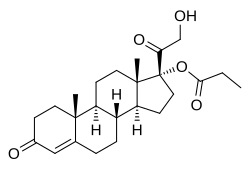
Clascoterone
(1R,3aS,3bR,9aR,9bS,11aS)-1-(2-hydroxyacetyl)-9a,11a-dimethyl-7-oxo-1H,2H,3H,3aH,3bH,4H,5H,7H,8H,9H,9aH,9bH,10H,11H,11aH-cyclopenta[a]phenanthren-1-yl propanoate
| Formula |
C24H34O5
|
|---|---|
| CAS |
19608-29-8
|
| Mol weight |
402.5238
|
FDA APPROVED, 2020/8/26, Winlevi
|
クラスコステロン;
|
Anti-acne, Androgen receptor antagonist
Clascoterone, sold under the brand name Winlevi, is an antiandrogen medication which is used topically in the treatment of acne.[1][2][3] It is also under development for the treatment of androgen-dependent scalp hair loss.[2] The medication is used as a cream by application to the skin, for instance the face and scalp.[3]
Clascoterone is an antiandrogen, or antagonist of the androgen receptor (AR), the biological target of androgens such as testosterone and dihydrotestosterone.[4][5] It shows no systemic absorption when applied to skin.[3]
The medication, developed by Cassiopea and Intrepid Therapeutics,[2] was approved by the US Food and Drug Administration (FDA) for acne in August 2020.[6][7]
Medical uses
Clascoterone is indicated for the topical treatment of acne vulgaris in females and males age 12 years and older.[1][8] It is applied to the affected skin area in a dose of 1 mg cream (or 10 mg clascoterone) twice per day, once in the morning and once in the evening.[1] The medication should not be used ophthalmically, orally, or vaginally.[1]
Available forms
Clascoterone is available in the form of a 1% (10 mg/g) cream for topical use.[1]
Contraindications
Clascoterone has no contraindications.[1]
Side effects
The incidences of local skin reactions with clascoterone were similar to placebo in two large phase 3 randomized controlled trials.[1][9] Suppression of the hypothalamic–pituitary–adrenal axis (HPA axis) may occur during clascoterone therapy in some individuals due to its cortexolone metabolite.[1][8] HPA axis suppression as measured by the cosyntropin stimulation test was observed to occur in 3 of 42 (7%) of adolescents and adults using clascoterone for acne.[1][8] HPA axis function returned to normal within 4 weeks following discontinuation of clascoterone.[1][8] Hyperkalemia (elevated potassium levels) occurred in 5% of clascoterone-treated individuals and 4% of placebo-treated individuals.[1]
Pharmacology
Pharmacodynamics
Clascoterone is an steroidal antiandrogen, or antagonist of the androgen receptor (AR), the biological target of androgens such as testosterone and dihydrotestosterone (DHT).[1][4][5] In a bioassay, the topical potency of the medication was greater than that of progesterone, flutamide, and finasteride and was equivalent to that of cyproterone acetate.[10] Likewise, it is significantly more efficacious as an antiandrogen than other AR antagonists such as enzalutamide and spironolactone in scalp dermal papilla cells and sebocytes in vitro.[5]\
Pharmacokinetics
Steady-state levels of clascoterone occur within 5 days of twice daily administration.[1] At a dosage of 6 g clascoterone cream applied twice daily, maximal circulating levels of clascoterone were 4.5 ± 2.9 ng/mL, area-under-the-curve levels over the dosing interval were 37.1 ± 22.3 h*ng/mL, and average circulating levels of clascoterone were 3.1 ± 1.9 ng/mL.[1] In rodents, clascoterone has been found to possess strong local antiandrogenic activity, but negligible systemic antiandrogenic activity when administered via subcutaneous injection.[10] Along these lines, the medication is not progonadotropic in animals.[10]
The plasma protein binding of clascoterone is 84 to 89% regardless of concentration.[1]
Clascoterone is rapidly hydrolyzed into cortexolone (11-deoxycortisol) and this compound is a possible primary metabolite of clascoterone based on in-vitro studies in human liver cells.[1][8] During treatment with clascoterone, cortexolone levels were detectable and generally below or near the low limit of quantification (0.5 ng/mL).[1] Clascoterone may also produce other metabolites, including conjugates.[1]
The elimination of clascoterone has not been fully characterized in humans.[1]
Chemistry
Clascoterone, also known as cortexolone 17α-propionate or 11-deoxycortisol 17α-propionate, as well as 17α,21-dihydroxyprogesterone 17α-propionate or 17α,21-dihydroxypregn-4-en-3,20-dione 17α-propionate, is a synthetic pregnane steroid and a derivative of progesterone and 11-deoxycortisol (cortexolone).[11] It is specifically the C17α propionate ester of 11-deoxycortisol.[10]
An analogue of clascoterone is 9,11-dehydrocortexolone 17α-butyrate (CB-03-04).[12]
History
C17α esters of 11-deoxycortisol were unexpectedly found to possess antiandrogenic activity.[10] Clascoterone, also known as cortexolone 17α-propionate, was selected for development based on its optimal drug profile.[10] The medication was approved by the US Food and Drug Administration (FDA) for the treatment of acne in August 2020.[6]
Two large phase 3 randomized controlled trials evaluated the effectiveness of clascoterone for the treatment of acne over a period of 12 weeks.[1][8][9] Clascoterone decreased acne symptoms by about 8 to 18% more than placebo.[1][9] The defined treatment success endpoint was achieved in about 18 to 20% of individuals with clascoterone relative to about 7 to 9% of individuals with placebo.[1][8][9] The comparative effectiveness of clascoterone between males and females was not described.[1][9]
A small pilot randomized controlled trial in 2011, found that clascoterone cream decreased acne symptoms to a similar or significantly greater extent than tretinoin 0.05% cream.[8][13] No active comparator was used in the phase III clinical trials of clascoterone for acne.[8] Hence, it’s unclear how clascoterone compares to other therapies used in the treatment of acne.[8]
The FDA approved clascoterone based on evidence from two clinical trials (Trial 1/NCT02608450 and Trial 2/NCT02608476) of 1440 participants 9 to 58 years of age with acne vulgaris.[14] The trials were conducted at 99 sites in the United States, Poland, Romania, Bulgaria, Ukraine, Georgia, and Serbia.[14]
Participants applied clascoterone or vehicle (placebo) cream twice daily for 12 weeks.[14] Neither the participants nor the health care providers knew which treatment was being given until after the trial was completed.[14] The benefit of clascoterone in comparison to placebo was assessed after 12 weeks of treatment using the Investigator’s Global Assessment (IGA) score that measures the severity of disease (on a scale from 0 to 4) and a decrease in the number of acne lesions.[14]
Society and culture
Names
Clascoterone is the generic name of the drug and its INN and USAN.[11][15]
Research
Clascoterone has been suggested as a possible treatment for hidradenitis suppurativa (acne inversa), an androgen-dependent skin condition.[16]
………………………………………………………………………….
PATENT
CN 112028956
https://patents.google.com/patent/CN112028956A/en

Example 1
Preparation of intermediate I
Wherein R is DMTr
Dissolving the compound 11-deoxycortisol (1.04g, 3.0mmol, 1eq.) in 10mL of anhydrous pyridine, dissolving dried DMTrCl (1.2-1.5eq) in 5mL of anhydrous dichloromethane, dropwise adding a dichloromethane solution of DMTrCl into the reactant solution at room temperature, and reacting for 4 hours at room temperature; the reaction was quenched with methanol and the solvent was evaporated to dryness with an oil pump to give intermediate I in 85% yield (the next reaction was carried out without work-up, the solvent environment and catalyst were similar to the reaction of this step).
1 H NMR (600MHz, CDCl 3 ) (ppm) 7.25-7.31 (m, 5H, H-DMTr), 7.15-7.18 (m, 4H, H-DMTr), 6.81-6.84 (m, 4H, H-DMTr), 5.73(1H,s,H-4),4.65(1H,dd,J=19.8,4.8Hz,H-21),4.30(1H,dd,J=19.8,4.8Hz,H-21),3.80(6H ,s),2.71(s,1H,17-OH),2.66-2.71(m,1H,H-16β),2.27-2.45(m,4H),1.19(3H,s,H-19),0.96- 1.87(m, 14H), 0.72(s, 3H, H-18).
MS + 303(DMTr protecting group fragment), 649[M + H] +
Melting point: 95-97 deg.C
Example 2:
preparation of intermediate II
Wherein R is DMTr
Under the protection of nitrogen, dissolving the intermediate product I (1eq.) in 5mL of anhydrous dichloromethane, adding DMAP (0.1eq.) into the solution, dropwise adding triethylamine (1.2eq.) and propionic anhydride or propionyl chloride (1.2eq. ), reacting at 40 ℃ for 12 hours after dropwise adding, and evaporating the solvent to obtain an intermediate product II.
Or under the protection of nitrogen, dissolving the intermediate product I (1eq.) in 5mL of anhydrous pyridine, adding DMAP (0.1eq.) into the solution, dropwise adding triethylamine (1.2eq.) and propionic anhydride or propionyl chloride (1.2eq .), reacting at 80 ℃ for 4 hours after dropwise adding, and evaporating the solvent to obtain an intermediate product II. (the reaction in the step can be directly carried out for the next step of removing DMTr protecting group to obtain the reaction after solvent evaporation without strict purification post-treatment)
1 H NMR (600MHz, CDCl 3 ) (ppm) 7.26-7.32 (m, 5H, H-DMTr), 7.14-7.18 (m, 4H, H-DMTr), 6.81-6.84 (m, 4H, H-DMTr), 5.72(1H,s,H-4),4.65(1H,dd,J=19.8,4.8Hz,H-21),4.30(1H,dd,J=19.8,4.8Hz,H-21),3.81(6H ,s),2.66-2.71(m,1H,H-16β),2.35(m,2H,-CH 2 CH 3 ),2.27-2.45(m,4H),1.19(3H,s,H-19), 1.15 (t, 3H, J=7.8Hz, -CH 2 CH 3 ), 0.96-1.87 (m, 14H), 0.72 (s, 3H, H-18);
MS + :303(DMTr protecting group fragment), 727[ M + Na [)] + ,768[M+Na+CH 3 CN] + .
Example 3:
preparation of target Compound 1 (21-hydroxy-17- (1-oxopropoxy) pregn-4-ene-3, 20-dione)
Dissolving the concentrated intermediate product II in an ethyl acetate solution, slowly dropwise adding 0.5M hydrochloric acid solution or 2% trifluoroacetic acid-ethyl acetate solution at 0 ℃, reacting for 5 minutes at 0 ℃, removing DMTr protective groups, adding 5% sodium bicarbonate aqueous solution at 0 ℃, stirring, neutralizing acid in a reaction system, washing an ethyl acetate organic layer twice by using 5% sodium bicarbonate aqueous solution, removing acid and other water-soluble impurities in the ethyl acetate organic layer, drying the ethyl acetate organic layer by anhydrous sodium sulfate, evaporating to remove part of ethyl acetate solvent, adding petroleum ether into the remaining small amount of ethyl acetate solution, and recrystallizing in a system with 10 times of solvent amount of ethyl acetate-petroleum ether (5:1) to obtain a target product with high purity of 90%. The total yield from 11-deoxycortisol is up to 70%. The final product was free of isomerized by-products by HPLC and was not found by LCMS.
1 H NMR (600 MHz, CDCl 3 ) (ppm): 5.75 (s, 1H, H-4), 4.28 (d, 1H, J=18.0 Hz, H-21), 4.23 (d, 1H, J=18.0 Hz, H-21), 3.05(s, 1H, 21-OH), 2.81-2.86(m, 1H, H-16β), 2.34-2.46(m, 3H), 2.35(m, 2H, -CH 2 CH 3 ) ,2.28-2.33(m,1H),2.03-2.07(m,1H),1.86-1.94(m,2H),1.67-1.77(m,3H),1.55-1.64(m,3H),1.35-1.46( m, 3H), 1.19(s, 3H, H-19), 1.15(t, 3H, J=7.8Hz, -CH 2 CH 3 ), 1.08-1.11(m, 1H), 1.00-1.05(m, 1H ),0.69(s,3H,H-18);
MS + : 403[M+H] + , 444[M+H+CH 3 CN] +
Melting point: 128-130 ℃.
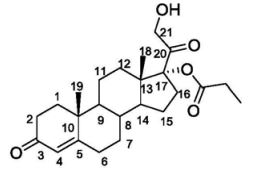
PATENT
WO 2009019138,
EXAMPLES Example 1
Alcoho lysis with CCL of cortexolone 17α, 21-dipropionate
Add butanol (0.4g, 5.45 mmoles) and CCL (17.4g, 3.86 U/mg, FLUKA) to a solution of cortexolone- 17α,21-dipropionate (0.5g, 1.09 mmoles) in toluene (50ml). Maintain the mixture under stirring, at 30 0C, following the progress of the reaction in TLC (Toluene/ethyl acetate 6/4) until the initial material is dissolved (24h). Remove the enzyme by means of filtration using a Celite layer. Recover the cortexolone 17α-propionate (0.437, 99%) after evaporation under low pressure. Through crystallisation, from diisopropyl ether you obtain a product with a purity >99% in HPLC.
1 H-NMR (500MHz, CDCl3) relevant signals δ (ppm) 5.78 (br s, 1 H, H-4), 4.32 (dd, 1 H, H-21), 4.25 (dd, IH, H-21), 1.22 (s, 3H, CH3-19), 1.17 (t, 3H, CH3), 0.72 (s, 3H5 CH3-18). P.f. 114 0C Example 2
According to the method described in example 1 prepare cortexolone- 17α- butanoate.
1H-NMR relevant signals δ (ppm) 5.78 (br s, IH, H-4), 4.32 (dd, IH, H-21), 4.26 (dd, IH, H-21), 1.23 (s, 3H, CH3-19), 0.97 (t, 3H, CH3), 0.73 (s, 3H, CH3-18). P.F. 134-136 0C
Example 3
According to the method described in the example prepare cortexolone- 17α- valerate.
1H-NMR relevant signals δ (ppm) 5.77 (br s, IH, H-4), 4.32 (dd, IH, H-21), 4.26
(dd, IH, H-21), 1.22 (s, 3H, CH3-19), 0.95 (t, 3H, CH3), 0.72 (s, 3H, CH3-18). P.f.
114 0C (diisopropyl ether).
Example 4
According to the method described in the example prepare 9,11-dehydro- cortexolone- 17α-butanoate.
1 H-NMR relevant signals δ (ppm) 5.77 (br s, IH, H-4), 5.54 (m, IH, H-9), 4.29
(dd, IH, H-21), 4.24 (dd, IH, H-21), 1.32 (s, 3H, CH3-19), 0.94(t, 3H, CH3), 0.68
(s, 3H, CH3– 18). P.f. 135-136 0C (acetone/hexane).
Example 5
Alcoho lysis with CALB of cortexolone- 17α, 21-dipropionate
Dissolve cortexolone, 17α, 2-dipropionate (0.5g, 1 .09 mmoles) in acetonitrile
(40ml), add CALB (2.3g, 2.5 U/mg Fluka) and octanol (0.875ml). Leave the mixture under stirring, at 30 0C, for 76 hrs. Remove the enzyme by means of filtration using a paper filter. Once the solvents evaporate, recover a solid
(0.4758) which upon analysis 1H-NMR shall appear made up of cortexolone- 17α- propionate at 91%.
Example 6
Crystallisation
Add the solvent (t-butylmethylether or diisopropylether) to the sample according to the ratios indicated in Table 3. Heat the mixture to the boiling temperature of the solvent, under stirring, until the sample dissolves completely. Cool to room temperature and leave it at this temperature, under stirring, for 6 hours. Filter using a buchner funnel and maintain the solid obtained, under low pressure, at a room temperature for 15 hours and then, at 400C, for 5 hours.
Example 7
Precipitation Disslove the sample in the suitable solvent (dichloromethane, acetone, ethyl acetate or ethanol) according to the ratios indicated in table 3 and then add the solvent, hexane or water, according to the ratios indicated in table 3, maintaining the mixture, under stirring, at room temperature. Recover the precipitate by filtration using a buchner funnel and desiccate as in example 6. Example 8.
Obtaining a pharmaceutical form containing the medication in a defined crystalline form.
Prepare a fluid cream containing 2 % cetylic alcohol, 16% glyceryl monostearate, 10% vaseline oil, 13 % propylene glycol, 10% poly ethylengly col with low polymerization 1.5% polysorbate 80 and 47.5 % purified water. Add 1 g of cortexolone 17α-propionate of crystalline form III to 100 g of this cream and subject the mixture to homogenisation by means of a turbine agitator until you obtain homogeneity. You obtain a cream containing a fraction of an active ingredient dissolved in the formulation vehicle and a non-dissolved fraction of an active ingredient, present as a crystal of crystalline form III. This preparation is suitable for use as a formulation vehicle for skin penetration tests on Franz cells, where a coefficient of penetration in the range of 0.04 to 0.03 cm/h is observed on the preparation. Example 9.
Obtaining the pharmaceutical form containing the medication in solvate form IV for replacing the solvent during the galenic formulation procedure Dissolve lOOg of cortexolone 17α-propionate of crystalline form III in 2500 g of propylene glycol under stirring at room temperature. Separately prepare, by using a turbo emulsifϊer raising the temperature up to about 700C, an emulsion with 250 g of Cetylic alcohol, 1500 g of glyceryl monostearate, 1000 g of liquid paraffin, 5 g of mixed tocopherols, 100 g of polysorbate 80 and 4650 g of water. After cooling the emulsion up to about 300C, add – under stirring and under negative pressure – the cortexolone 17α-propionate solution in propylene glycol. Maintain the emulsioned cream under stirring until you obtain homogeneity, making sure the temperature remains low by means the circulation of a coolant. The cream contains a dispersed crystalline fraction, made up of an active ingredient in solvate crystalline form IV, formed due to the precipitation of the active ingredient itself from the glycolic solution which contained it when the latter was added to the predominantly aqueous formulation. The DRX spectra of the crystalline form present in the cream are indicated in Fig. 30.
PAPER
Tetrahedron Letters, 49(31), 4610-4612; 2008
Abstract
Several 17α-monoesters of cortexolone and its Δ9-derivative are endowed with antiandrogenic activity. Their synthesis can be accomplished by means of a lipase-catalyzed chemoselective alcoholysis of the corresponding 17α,21-diesters.
Graphical abstract

1H NMR (500 MHz, CDCl3): selected data δ 5.78 (br s, 1H, H-4), 4.32 (dd, 1H, H-21, J18.3 and 4.9 Hz), 4.25 (dd, 1H, H-21, J18.3 and 4.9 Hz), 1.22 (s, 3H, CH3-19), 1.17 (t, 3H, CH3, J7.6 Hz), 0.72 (s, 3H, CH3-18) MP 133 °C (t-butylmethylether)
…………………………………………………………………..
PATENT
https://patents.google.com/patent/EP2503005B1/en
-
Cortexolone derivatives in which the hydroxyl group at position C-17α is esterified with short chain aliphatic or aromatic acids and the derivatives of the corresponding 9,11-dehydro derivative, are known to have an antiandrogenic effect.
- [0002]
EP 1421099 describes cortexolone 17α-propionate and 9,11-dehydro-cortexolone-17-α-butanoate regarding a high antiandrogenic biological activity demonstrated both “in vitro” and “in vivo” on the animal.
- [0003]
US3530038 discloses the preparation of a crystalline form of cortexolone-17α-propionate having a melting point of 126-129 °C and an IR spectrum with bands at (cm-1): 3500, 1732, 1713, 1655 and 1617.
- [0004]
A method for obtaining the above mentioned derivatives is described by Gardi et al. (Gazz. Chim. It. 63, 43 1,1963) and in the United States patent US3152154 providing for the transformation of cortexolone, or transformation of 9,11-dehydrocortexolone, in the intermediate orthoester using orthoesters available in the market as a mixture of aprotic solvents such as cyclohexane and DMF, in presence of acid catalysis (ex. PTSA.H20). The intermediate orthoester thus obtained can be used as is or upon purification by suspension in a solvent capable of solubilising impurities, preferably in alcohols. The subsequent hydrolysis in a hydroalcoholic solution, buffered to pH 4-5 preferably in acetate buffer, provides the desired monoester.
- [0005]
Such synthesis is indicated in the diagram 1 below

- [0006]
However, the monoesters thus obtained were, in the reaction conditions, unstable and, consequently hard to manipulate and isolate (R. Gardi et al Tetrahedron Letters, 448, 1961). The instability is above all due to the secondary reaction of migration of the esterifying acyl group from position 17 to position 21.
- [0007]
It is thus known that in order to obtain the above mentioned monoesters with a chemical purity in such a manner to be able to proceed to the biological tests, it is necessary to use, at the end of the synthesis, a purification process which is generally performed by means of column chromatography.
- [0008]
Furthermore, US3152154 describes how the hydrolysis of the diester in a basic environment is not convenient due to the formation of a mixture of 17α,21-diol, of 17- and 21 -monoesters, alongside the initial non-reacted product.
- [0009]
Now, it has been surprisingly discovered that an alcoholysis reaction using a lipase from Candida as a biocatalyst can be usefully applied during the preparation of 17α monoesters of cortexolone, or its 9,11-dehydroderivatives.
- [0010]
As a matter of fact, it has been discovered that such enzymatic alcoholysis of the 17,21-diester of the cortexolone, or of its derivative 9,11-dehydro, selectively occurs in position 21 moving to the corresponding monoester in position 17, as shown in diagram 2 below:

- [0011]
The chemoselectivity of the special enzymatic reaction in alcoholysis conditions, according to the present invention, opens new perspectives for preparation, at industrial level with higher yields, of 17α-monoesters with respect to the methods already indicated in literature.
- [0012]
The diesters serving as a substrate for the reaction of the invention can be prepared according to the prior art, for example following the one described in B.Turner, (Journal of American Chemical Society, 75, 3489, 1953) which provides for the esterification of corticosteroids with a linear carboxylic acid in presence of its anhydride and PTSA monohydrate.
EXAMPLES
-
- Example 1
Alcoholysis with CCL of cortexolone 17α, 21-dipropionate
-
-
- [0055]
Add butanol (0.4g, 5.45 mmoles) and CCL (17.4g, 3.86 U/mg, FLUKA) to a solution of cortexolone-17α,21-dipropionate (0.5g, 1.09 mmoles) in toluene (50ml). Maintain the mixture under stirring, at 30 °C, following the progress of the reaction in TLC (Toluene/ethyl acetate 6/4) until the initial material is dissolved (24h). Remove the enzyme by means of filtration using a Celite layer. Recover the cortexolone 17α-propionate (0.437, 99%) after evaporation under low pressure. Through crystallisation, from diisopropyl ether you obtain a product with a purity >99% in HPLC.
- [0056]
1H-NMR (500MHz, CDCl3) relevant signals δ (ppm) 5.78 (br s, 1 H, H-4), 4.32 (dd, 1 H, H-21), 4.25 (dd, 1H, H-21), 1.22 (s, 3H, CH3-19), 1.17 (t, 3H, CH3), 0.72 (s, 3H, CH3-18). P.f. 114 °C
- [0055]
-
Example 2 (comparative)
-
-
- [0057]
According to the method described in example 1 prepare cortexolone-17α-butanoate.
- [0058]
1H-NMR relevant signals δ (ppm) 5.78 (br s, 1H, H-4), 4.32 (dd, 1H, H-21), 4.26 (dd, 1H, H-21), 1.23 (s, 3H, CH3-19), 0.97 (t, 3H, CH3), 0.73 (s, 3H. CH3-18). P.F. 134-136 °C
- [0057]
-
Example 3 (comparative)
According to the method described in the example prepare cortexolone-17α-valerate.
-
-
- [0059]
1H-NMR relevant signals δ (ppm) 5.77 (br s, 1H, H-4), 4.32 (dd, 1H, H-21), 4.26 (dd, 1H, H-21), 1.22 (s, 3H, CH3-19), 0.95 (t, 3H, CH3), 0.72 (s, 3H, CH3-18). P.f. 114 °C (diisopropyl ether).
- [0059]
-
Example 4 (comparative)
According to the method described in the example prepare 9, 11-dehydro-cortexolone-17α-butanoate.
-
-
- [0060]
1H-NMR relevant signals δ (ppm) 5.77 (br s, 1H, H-4), 5.54 (m, 1H, H-9), 4.29 (dd, 1H, H-21), 4.24 (dd, 1H, H-21), 1.32 (s, 3H, CH3-19), 0.94(t, 3H, CH3), 0.68 (s, 3H, CH3-18). P.f. 135-136 °C (acetone/hexane).
- [0060]
-
Example 5
Alcoholysis with CALB of cartexolone-17α, 21-dipropionate
-
-
- [0061]
Dissolve cortexolone, 17α, 2-dipropionate (0.5g, 1.09 mmoles) in acetonitrile (40ml), add CALB (2.3g, 2.5 U/mg Fluka) and octanol (0.875ml). Leave the mixture under stirring, at 30 °C, for 76 hrs. Remove the enzyme by means of filtration using a paper filter. Once the solvents evaporate, recover a solid (0.4758) which upon analysis 1H-NMR shall appear made up of cortexolone-17α-propionate at 91%.
- [0061]
-
Example 6
Crystallisation
-
-
- [0062]
Add the solvent (t-butylmethylether or diisopropylether) to the sample according to the ratios indicated in Table 3. Heat the mixture to the boiling temperature of the solvent, under stirring, until the sample dissolves completely. Cool to room temperature and leave it at this temperature, under stirring, for 6 hours. Filter using a buchner funnel and maintain the solid obtained, under low pressure, at a room temperature for 15 hours and then, at 40°C, for 5 hours.
- [0062]
-
Example 7 (comparative)
Precipitation
-
-
- [0063]
Disslove the sample in the suitable solvent (dichloromethane, acetone, ethyl acetate or ethanol) according to the ratios indicated in table 3 and then add the solvent, hexane or water, according to the ratios indicated in table 3, maintaining the mixture, under stirring, at room temperature. Recover the precipitate by filtration using a buchner funnel and desiccate as in example 6.
- [0063]
-
Example 8.
Obtaining a pharmaceutical form containing the medication in a defined crystalline form.
- [0064]
Prepare a fluid cream containing 2 % cetylic alcohol, 16% glyceryl monostearate, 10% vaseline oil, 13 % propylene glycol, 10% polyethylenglycol with low polymerization 1.5% polysorbate 80 and 47.5 % purified water. Add 1 g of cortexolone 17α-propionate of crystalline form III to 100 g of this cream and subject the mixture to homogenisation by means of a turbine agitator until you obtain homogeneity. You obtain a cream containing a fraction of an active ingredient dissolved in the formulation vehicle and a non-dissolved fraction of an active ingredient, present as a crystal of crystalline form III. This preparation is suitable for use as a formulation vehicle for skin penetration tests on Franz cells, where a coefficient of penetration in the range of 0.04 to 0.03 cm/h is observed on the preparation.
References
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u v w “Winlevi (clascoterone) cream, for topical use”(PDF). Cassiopea. Retrieved 9 September 2020.
- ^ Jump up to:a b c http://adisinsight.springer.com/drugs/800026561
- ^ Jump up to:a b c Kircik LH (July 2019). “What’s new in the management of acne vulgaris”. Cutis. 104(1): 48–52. PMID 31487336.
- ^ Jump up to:a b Rosette C, Rosette N, Mazzetti A, Moro L, Gerloni M (February 2019). “Cortexolone 17α-Propionate (Clascoterone) is an Androgen Receptor Antagonist in Dermal Papilla Cells In Vitro”. J Drugs Dermatol. 18 (2): 197–201. PMID 30811143.
- ^ Jump up to:a b c Rosette C, Agan FJ, Mazzetti A, Moro L, Gerloni M (May 2019). “Cortexolone 17α-propionate (Clascoterone) Is a Novel Androgen Receptor Antagonist that Inhibits Production of Lipids and Inflammatory Cytokines from Sebocytes In Vitro”. J Drugs Dermatol. 18 (5): 412–418. PMID 31141847.
- ^ Jump up to:a b “Cassiopea Receives FDA Approval for Winlevi (clascoterone cream 1%), First-in-Class Topical Acne Treatment Targeting the Androgen Receptor”. Cassiopea (Press release). Retrieved 2020-08-30.
- ^ “Winlevi: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 9 September 2020.
- ^ Jump up to:a b c d e f g h i j Barbieri, John S. (2020). “A New Class of Topical Acne Treatment Addressing the Hormonal Pathogenesis of Acne”. JAMA Dermatology. 156 (6): 619–620. doi:10.1001/jamadermatol.2020.0464. ISSN 2168-6068. PMID 32320045.
- ^ Jump up to:a b c d e Hebert A, Thiboutot D, Stein Gold L, Cartwright M, Gerloni M, Fragasso E, Mazzetti A (April 2020). “Efficacy and Safety of Topical Clascoterone Cream, 1%, for Treatment in Patients With Facial Acne: Two Phase 3 Randomized Clinical Trials”. JAMA Dermatol. 156 (6): 621–630. doi:10.1001/jamadermatol.2020.0465. PMC 7177662. PMID 32320027.
- ^ Jump up to:a b c d e f Celasco G, Moro L, Bozzella R, Ferraboschi P, Bartorelli L, Quattrocchi C, Nicoletti F (2004). “Biological profile of cortexolone 17alpha-propionate (CB-03-01), a new topical and peripherally selective androgen antagonist”. Arzneimittelforschung. 54 (12): 881–6. doi:10.1055/s-0031-1297043. PMID 15646372.
- ^ Jump up to:a b https://chem.nlm.nih.gov/chemidplus/rn/19608-29-8
- ^ Celasco G, Moroa L, Bozzella R, Ferraboschi P, Bartorelli L, Di Marco R, Quattrocchi C, Nicoletti F (2005). “Pharmacological profile of 9,11-dehydrocortexolone 17alpha-butyrate (CB-03-04), a new androgen antagonist with antigonadotropic activity”. Arzneimittelforschung. 55 (10): 581–7. doi:10.1055/s-0031-1296908. PMID 16294504.
- ^ Trifu V, Tiplica GS, Naumescu E, Zalupca L, Moro L, Celasco G (2011). “Cortexolone 17α-propionate 1% cream, a new potent antiandrogen for topical treatment of acne vulgaris. A pilot randomized, double-blind comparative study vs. placebo and tretinoin 0·05% cream”. Br. J. Dermatol. 165 (1): 177–83. doi:10.1111/j.1365-2133.2011.10332.x. PMID 21428978. S2CID 38404925.
- ^ Jump up to:a b c d e “Drug Trial Snapshot: Winlevi”. U.S. Food and Drug Administration (FDA). 26 August 2020. Retrieved 10 September 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 82”. WHO Drug Information. 33 (3): 106. hdl:10665/330879.
- ^ Der Sarkissian SA, Sun HY, Sebaratnam DF (August 2020). “Cortexolone 17 α-proprionate for hidradenitis suppurativa”. Dermatol Ther: e14142. doi:10.1111/dth.14142. PMID 32761708.
External links
- “Clascoterone”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02608450 for “A Study to Evaluate the Safety and Efficacy of CB-03-01 Cream, 1% in Subjects With Facial Acne Vulgaris (25)” at ClinicalTrials.gov
- Clinical trial number NCT02608476 for “A Study to Evaluate the Safety and Efficacy of CB-03-01 Cream, 1% in Subjects With Facial Acne Vulgaris (26)” at ClinicalTrials.gov
 |
|
| Clinical data | |
|---|---|
| Trade names | Winlevi |
| Other names | CB-03-01; Breezula; 11-Deoxycortisol 17α-propionate; 17α-(Propionyloxy)- deoxycorticosterone; 21-Hydroxy-3,20-dioxopregn-4-en-17-yl propionate |
| License data |
|
| Routes of administration |
Topical (cream) |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.210.810 |
| Chemical and physical data | |
| Formula | C24H34O5 |
| Molar mass | 402.531 g·mol−1 |
| 3D model (JSmol) | |
/////////Clascoterone, クラスコステロン , FDA 2020, 2020 APPROVALS, ANTI ACNE
[H][C@@]12CC[C@](OC(=O)CC)(C(=O)CO)[C@@]1(C)CC[C@@]1([H])[C@@]2([H])CCC2=CC(=O)CC[C@]12C
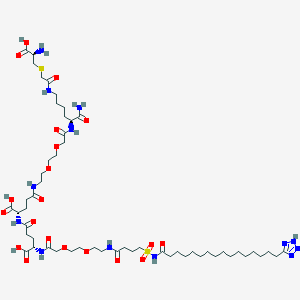
Somapacitan, ソマパシタン;
FPTIPLSRLF DNAMLRAHRL HQLAFDTYQE FEEAYIPKEQ KYSFLQNPQT SLCFSESIPT
PSNREETQQK SNLELLRISL LLIQSWLEPV QFLRSVFANS CVYGASDSNV YDLLKDLEEG
IQTLMGRLED GSPRTGQIFK QTYSKFDTNS HNDDALLKNY GLLYCFRKDM DKVETFLRIV
QCRSVEGSCG F
(Disulfide bridge: 53-165, 182-189)
Somapacitan
FDA APPROVED, 2020/8/28, SOGROYA
Growth hormone (GH) receptor agonist
CAS: 1338578-34-9
(2S)-5-[2-[2-[2-[[(2S)-1-amino-6-[[2-[(2R)-2-amino-2-carboxyethyl]sulfanylacetyl]amino]-1-oxohexan-2-yl]amino]-2-oxoethoxy]ethoxy]ethylamino]-2-[[(4S)-4-carboxy-4-[[2-[2-[2-[4-[16-(2H-tetrazol-5-yl)hexadecanoylsulfamoyl]butanoylamino]ethoxy]ethoxy]acetyl]amino]butanoyl]amino]-5-oxopentanoic acid
| Formula |
C1038H1609N273O319S9
|
|---|---|
| Mol weight |
23305.1048
|
| JAP | ソマパシタン; |
Treatment of growth hormone dificiency
albumin-binding growth hormone
UNII-F1 component VTUYEWRWJTWXPQ-IWWWZYECSA-N
Somapacitan, also known as NNC0195-0092,3 is a growth hormone analog indicated to treat adults with growth hormone deficiency.2,6 This human growth hormone analog differs by the creation of an albumin binding site, and prolonging the effect so that it requires weekly dosing rather than daily.5
Somapacitan was granted FDA approval on 28 August 2020.7
Somapacitan, sold under the brand name Sogroya, is a growth hormone medication.[2] Somapacitan is a human growth hormone analog.[1] Somapacitan-beco is produced in Escherichia coli by recombinant DNA technology.[1]
The most common side effects include: back pain, joint paint, indigestion, a sleep disorder, dizziness, tonsillitis, swelling in the arms or lower legs, vomiting, adrenal insufficiency, hypertension, increase in blood creatine phosphokinase (a type of enzyme), weight increase, and anemia.[2]
It was approved for medical use in the United States in August 2020.[2][3][4]
Somapacitan (Sogroya) is the first human growth hormone (hGH) therapy that adults only take once a week by injection under the skin; other FDA-approved hGH formulations for adults with growth hormone deficiency must be administered daily.[2]
Medical uses
Somapacitan is indicated for replacement of endogenous growth hormone in adults with growth hormone deficiency.[2]
Contraindications
Somapacitan should not be used in people with active malignancy, any stage of diabetic eye disease in which high blood sugar levels cause damage to blood vessels in the retina, acute critical illness, or those with acute respiratory failure, because of the increased risk of mortality with use of pharmacologic doses of somapacitan in critically ill individuals without growth hormone deficiency.[2]
History
Somapacitan was evaluated in a randomized, double-blind, placebo-controlled trial in 300 particpants with growth hormone deficiency who had never received growth hormone treatment or had stopped treatment with other growth hormone formulations at least three months before the study.[2] Particpants were randomly assigned to receive injections of weekly somapacitan, weekly placebo (inactive treatment), or daily somatropin, an FDA-approved growth hormone.[2] The effectiveness of somapacitan was determined by the percentage change of truncal fat, the fat that is accumulated in the trunk or central area of the body that is regulated by growth hormone and can be associated with serious medical issues.[2]
At the end of the 34-week treatment period, truncal fat decreased by 1.06%, on average, among particpants taking weekly somapacitan while it increased among particpants taking the placebo by 0.47%.[2] In the daily somatropin group, truncal fat decreased by 2.23%.[2] Particpants in the weekly somapacitan and daily somatropin groups had similar improvements in other clinical endpoints.[2]
It was approved for medical use in the United States in August 2020.[2][4] The U.S. Food and Drug Administration (FDA) granted the approval of Sogroya to Novo Nordisk, Inc.[2][4]
References
- ^ Jump up to:a b c d “Sogroya (somapacitan-beco) injection, for subcutaneous use” (PDF). Retrieved 1 September 2020.
- ^ Jump up to:a b c d e f g h i j k l m n o “FDA approves weekly therapy for adult growth hormone deficiency”. U.S. Food and Drug Administration (FDA) (Press release). 1 September 2020. Retrieved 1 September 2020. This article incorporates text from this source, which is in the public domain.
- ^ “FDA approves once-weekly Sogroya for the treatment of adult growth hormone deficiency”. Novo Nordisk (Press release). 28 August 2020. Retrieved 1 September 2020.
- ^ Jump up to:a b c “Sogroya: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 2 September 2020.
External links
- “Somapacitan”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Sogroya |
| Other names | somapacitan-beco, NNC0195-0092 |
| License data |
|
| Routes of administration |
Subcutaneous[1] |
| Drug class | Human growth hormone analog |
| ATC code |
|
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C1038H1609N273O319S9 |
| Molar mass | 23305.42 g·mol−1 |
ClinicalTrials.gov
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT02616562 | Investigating Efficacy and Safety of Once-weekly NNC0195-0092 (Somapacitan) Treatment Compared to Daily Growth Hormone Treatment (Norditropin® FlexPro®) in Growth Hormone Treatment naïve Pre-pubertal Children With Growth Hormone Deficiency | Phase 2 | Recruiting | 2020-03-25 |
| NCT03075644 | A Trial to Evaluate the Safety of Once Weekly Dosing of Somapacitan (NNC0195-0092) and Daily Norditropin® FlexPro® for 52 Weeks in Previously Human Growth Hormone Treated Japanese Adults With Growth Hormone Deficiency | Phase 3 | Completed | 2019-10-18 |
| NCT03905850 | A Study to Compare the Uptake Into the Blood of Two Strengths of Somapacitan After Injection Under the Skin in Healthy Subjects | Phase 1 | Completed | 2019-08-06 |
| NCT03212131 | Investigation of Pharmacokinetics, Pharmacodynamics, Safety and Tolerability of Multiple Doses of Somapacitan in Subjects With Mild and Moderate Degrees of Hepatic Impairment Compared to Subjects With Normal Hepatic Function. | Phase 1 | Completed | 2019-05-24 |
| NCT01514500 | First Human Dose Trial of NNC0195-0092 (Somapacitan) in Healthy Subjects | Phase 1 | Completed | 2018-05-25 |
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT03811535 | A Research Study in Children With a Low Level of Hormone to Grow. Treatment is Somapacitan Once a Week Compared to Norditropin® Once a Day | Phase 3 | Recruiting | 2020-09-03 |
| NCT03878446 | A Research Study in Children Born Small and Who Stayed Small. Treatment is Somapacitan Once a Week Compared to Norditropin® Once a Day | Phase 2 | Recruiting | 2020-08-27 |
| NCT02382939 | A Trial to Compare the Safety of Once Weekly Dosing of Somapacitan With Daily Norditropin® FlexPro® for 26 Weeks in Previously Human Growth Hormone Treated Adults With Growth Hormone Deficiency | Phase 3 | Completed | 2020-07-09 |
| NCT02229851 | Trial to Compare the Efficacy and Safety of NNC0195-0092 (Somapacitan) With Placebo and Norditropin® FlexPro® (Somatropin) in Adults With Growth Hormone Deficiency. | Phase 3 | Completed | 2020-07-07 |
| NCT03186495 | Investigation of Pharmacokinetics, Pharmacodynamics, Safety and Tolerability of Multiple Doses of Somapacitan in Subjects With Various Degrees of Impaired Renal Function Compared to Subjects With Normal Renal Function | Phase 1 | Completed | 2020-04-17 |
EU Clinical Trials Register
| EudraCT | Title | Phase | Status | Date |
|---|---|---|---|---|
| 2018-000232-10 | A dose-finding trial evaluating the effect and safety of once-weekly treatment of somapacitan compared to daily Norditropin® in children with short stature born small for gestational age with no catch-up growth by 2 years of age or older | Phase 2 | Ongoing, Prematurely Ended | 2019-05-15 |
| 2015-000531-32 | A randomised, multinational, active-controlled,(open-labelled), dose finding, (double-blinded), parallel group trial investigating efficacy and safety of once-weekly NNC0195-0092 treatment compared to daily growth hormone treatment (Norditropin® FlexPro®) in growth hormone treatment naïve pre-pubertal children with growth hormone deficiency | Phase 2 | Ongoing, Completed | 2015-12-10 |
| 2014-000290-39 | A multicentre, multinational, randomised, open-labelled, parallel-group, active-controlled trial to compare the safety of once weekly dosing of NNC0195-0092 with daily Norditropin® FlexPro® for 26 weeks in previously human growth hormone treated adults with growth hormone deficiency | Phase 3 | Completed | 2014-11-07 |
| 2013-002892-16 | A multicentre, multinational, randomised, parallel-group, placebo-controlled (double blind) and active-controlled (open) trial to compare the efficacy and safety of once weekly dosing of NNC0195-0092 with once weekly dosing of placebo and daily Norditropin® FlexPro® in adults with growth hormone deficiency for 35 weeks, followed by a 53-week open-label extension period | Phase 3 | Completed | 2014-10-07 |
| 2018-000231-27 | A trial comparing the effect and safety of once weekly dosing of somapacitan with daily Norditropin® in children with growth hormone deficiency | Phase 3 | Ongoing |
EU Clinical Trials Register
| EudraCT | Title | Phase | Status | Date |
|---|---|---|---|---|
| 2013-000013-20 | A randomised, open-labelled, active-controlled, multinational, dose-escalation trial investigating safety, tolerability, pharmacokinetics and pharmacodynamics of a single dose of long-acting growth hormone (NNC0195-0092) compared to daily dosing of Norditropin® SimpleXx® in children with growth hormone deficiency | Phase 1 | Ongoing, Completed | 2013-12-09 |
///////////Somapacitan, PEPTIDE.2020 APPROVALS, FDA 2020, ソマパシタン, NN8640
C(CCCCCCCC1=NNN=N1)CCCCCCCC(=O)NS(=O)(=O)CCCC(=O)NCCOCCOCC(=O)NC(CCC(=O)NC(CCC(=O)NCCOCCOCC(=O)NC(CCCCNC(=O)CSCC(C(=O)O)N)C(=O)N)C(=O)O)C(=O)O
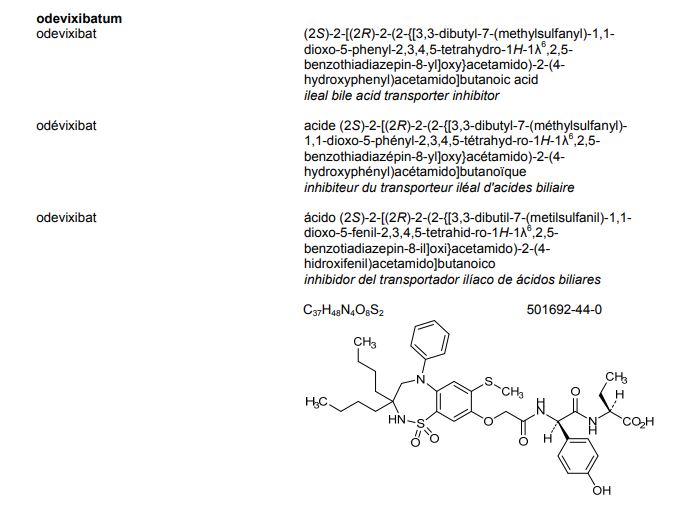
Odevixibat

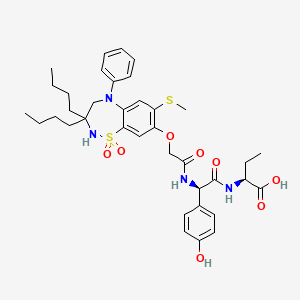
Odevixibat
A-4250, AR-H 064974
CAS 501692-44-0
BUTANOIC ACID, 2-(((2R)-2-((2-((3,3-DIBUTYL-2,3,4,5-TETRAHYDRO-7-(METHYLTHIO)-1,1-DIOXIDO-5-PHENYL-1,2,5-BENZOTHIADIAZEPIN-8-YL)OXY)ACETYL)AMINO)-2-(4-HYDROXYPHENYL)ACETYL)AMINO)-, (2S)-
(2S)-2-[[(2R)-2-[[2-[(3,3-dibutyl-7-methylsulfanyl-1,1-dioxo-5-phenyl-2,4-dihydro-1λ6,2,5-benzothiadiazepin-8-yl)oxy]acetyl]amino]-2-(4-hydroxyphenyl)acetyl]amino]butanoic acid
| Molecular Formula | C37H48N4O8S2 |
| Molecular Weight | 740.929 |
-
-
-
- UPDATE 7/20/2021FDA APPROVED, To treat pruritus,
-
- New Drug Application (NDA): 215498
Company: ALBIREO PHARMA INC
-
- AZD8294WHO 10706AR-H064974HY-109120CS-0078340D11716US9694018, 5Originator Albireo AB
- Developer Albireo AB; Albireo Pharma
- ClassAcetamides; Butyric acids; Hepatoprotectants; Small molecules; Sulfones; Thiazepines
- Mechanism of Action Sodium-bile acid cotransporter inhibitors
- Orphan Drug Status Yes – Primary biliary cirrhosis; Biliary atresia; Intrahepatic cholestasis; Alagille syndrome
- New Molecular Entity Yes
- Phase III Biliary atresia; Intrahepatic cholestasis
- Phase II Alagille syndrome; Cholestasis; Primary biliary cirrhosis
- No development reported Non-alcoholic steatohepatitis
- 22 Jul 2020 Albireo initiates an expanded-access programme for Intrahepatic cholestasis in USA, Canada, Australia and Europe
- 14 Jul 2020 Phase-III clinical trials in Biliary atresia (In infants, In neonates) in Belgium (PO) after July 2020 (EudraCT2019-003807-37)
- 14 Jul 2020 Phase-III clinical trials in Biliary atresia (In infants, In neonates) in Germany, France, United Kingdom, Hungary (PO) (EudraCT2019-003807-37)
UPDATE Bylvay, FDA APPROVED2021/7/20 AND EMA 2021/7/16
Odevixibat, sold under the trade name Bylvay, is a medication for the treatment of progressive familial intrahepatic cholestasis (PFIC).[1]
The most common side effects include diarrhea, abdominal pain, hemorrhagic diarrhea, soft feces, and hepatomegaly (enlarged liver).[1]
Odevixibat is a reversible, potent, selective inhibitor of the ileal bile acid transporter (IBAT).[1][2]
In May 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) recommended granting a marketing authorization in the European Union for odevixibat for the treatment of PFIC in people aged six months or older.[1][3]
A-4250 (odevixibat) is a selective inhibitor of the ileal bile acid transporter (IBAT) that acts locally in the gut. Ileum absorbs glyco-and taurine-conjugated forms of the bile salts. IBAT is the first step in absorption at the brush-border membrane. A-4250 works by decreasing the re-absorption of bile acids from the small intestine to the liver, whichreduces the toxic levels of bile acids during the progression of the disease. It exhibits therapeutic intervention by checking the transport of bile acids. Studies show that A-4250 has the potential to decrease the damage in the liver cells and the development of fibrosis/cirrhosis of the liver known to occur in progressive familial intrahepatic cholestasis. A-4250 is a designated orphan drug in the USA for October 2012. A-4250 is a designated orphan drug in the EU for October 2016. A-4250 was awarded PRIME status for PFIC by EMA in October 2016. A-4250 is in phase II clinical trials by Albireo for the treatment of primary biliary cirrhosis (PBC) and cholestatic pruritus. In an open label Phase 2 study in children with cholestatic liver disease and pruritus, odevixibat showed reductions in serum bile acids and pruritus in most patients and exhibited a favorable overall tolerability profile.
![]()
Odevixibat is a highly potent, non-systemic ileal bile acid transport inhibitor (IBATi) that has has minimal systemic exposure and acts locally in the small intestine. Albireo is developing odevixibat to treat rare pediatric cholestatic liver diseases, including progressive familial intrahepatic cholestasis, biliary atresia and Alagille syndrome.
With normal function, approximately 95 percent of bile acids released from the liver into the bile ducts to aid in liver function are recirculated to the liver via the IBAT in a process called enterohepatic circulation. In people with cholestatic liver diseases, the bile flow is interrupted, resulting in elevated levels of toxic bile acids accumulating in the liver and serum. Accordingly, a product capable of inhibiting the IBAT could lead to a reduction in bile acids returning to the liver and may represent a promising approach for treating cholestatic liver diseases.
The randomized, double-blind, placebo-controlled, global multicenter PEDFIC 1 Phase 3 clinical trial of odevixibat in 62 patients, ages 6 months to 15.9 years, with PFIC type 1 or type 2 met its two primary endpoints demonstrating that odevixibat reduced serum bile acids (sBAs) (p=0.003) and improved pruritus (p=0.004), and was well tolerated with a low single digit diarrhea rate. These topline data substantiate the potential for odevixibat to be first drug for PFIC patients. The Company intends to complete regulatory filings in the EU and U.S. no later than early 2021, in anticipation of regulatory approval, issuance of a rare pediatric disease priority review voucher and launch in the second half of 2021.
Odevixibat is being evaluated in the ongoing PEDFIC 2 open-label trial (NCT03659916) designed to assess long-term safety and durability of response in a cohort of patients rolled over from PEDFIC 1 and a second cohort of PFIC patients who are not eligible for PEDFIC 1.
Odevixibat is also currently being evaluated in a second Phase 3 clinical trial, BOLD (NCT04336722), in patients with biliary atresia. BOLD, the largest prospective intervention trial ever conducted in biliary atresia, is a double-blind, randomized, placebo-controlled trial which will enroll approximately 200 patients at up to 75 sites globally to evaluate the efficacy and safety of odevixibat in children with biliary atresia who have undergone a Kasai procedure before age three months. The company also anticipates initiating a pivotal trial of odevixibat for Alagille syndrome by the end of 2020.
For more information about the PEDFIC 2 or BOLD studies, please visit ClinicalTrials.gov or contact medinfo@albireopharma.com.
The odevixibat PFIC program, or elements of it, have received fast track, rare pediatric disease and orphan drug designations in the United States. In addition, the FDA has granted orphan drug designation to odevixibat for the treatment of Alagille syndrome, biliary atresia and primary biliary cholangitis. The EMA has granted odevixibat orphan designation, as well as access to the PRIority MEdicines (PRIME) scheme for the treatment of PFIC. Its Paediatric Committee has agreed to Albireo’s odevixibat Pediatric Investigation Plan for PFIC. EMA has also granted orphan designation to odevixibat for the treatment of biliary atresia, Alagille syndrome and primary biliary cholangitis.

PATENT
https://patents.google.com/patent/US9694018B1/en
Example 5
1,1-Dioxo-3,3-dibutyl-5-phenyl-7-methylthio-8-(N—{(R)-α-[N—((S)-1-carboxypropyl) carbamoyl]-4-hydroxybenzyl}carbamoylmethoxy)-2,3,4,5-tetrahydro-1,2,5-benzothiadiazepine, Mw. 740.94.
This compound is prepared as described in Example 29 of WO3022286.
PATENT
https://patents.google.com/patent/WO2003022286A1/sv
Example 29
1,1-Dioxo-3,3-dibutyl-5-phenyl-7-methylthio-8-(N-((R)-α-[N-((S)- 1-carboxypropyl) carbamoyl]-4-hydroxybenzyl}carbamoylmethoxy)-2,3,4,5-tetrahydro-1,2,5-benzothiadiazepine
A solution of 1,1-dioxo-3,3-dibutyl-5-phenyl-7-methylthio-8-[N-((R)-α-carboxy-4-hydroxybenzyl)carbamoylmethoxy]-2,3,4,5-tetrahydro-1,2,5-benzothiadiazepine (Example 18; 0.075 g, 0.114 mmol), butanoic acid, 2-amino-, 1,1-dimethylethyl ester, hydrochloride, (2S)-(0.031 g, 0.160 mmol) and Ν-methylmorpholine (0.050 ml, 0.457 mmol) in DMF (4 ml) was stirred at RT for 10 min, after which TBTU (0.048 g, 0.149 mmol) was added. After 1h, the conversion to the ester was complete. M/z: 797.4. The solution was diluted with toluene and then concentrated. The residue was dissolved in a mixture of DCM (5 ml) and TFA (2 ml) and the mixture was stirred for 7h. The solvent was removed under reduced pressure. The residue was purified by preparative HPLC using a gradient of 20-60% MeCΝ in 0.1M ammonium acetate buffer as eluent. The title compound was obtained in 0.056 g (66 %) as a white solid. ΝMR (400 MHz, DMSO-d6): 0.70 (3H, t), 0.70-0.80 (6H, m), 0.85-1.75 (14H, m), 2.10 (3H, s), 3.80 (2H, brs), 4.00-4.15 (1H, m), 4.65 (1H, d(AB)), 4.70 (1H, d(AB)), 5.50 (1H, d), 6.60 (1H, s), 6.65-7.40 (11H, m), 8.35 (1H, d), 8.50 (1H, d) 9.40 (1H, brs).
PATENT
https://patents.google.com/patent/US20140323412A1/en
PATENT
https://patents.google.com/patent/WO2013063526A1/e
PATENT
https://patents.google.com/patent/WO2019245448A1/en
The compound l,l-dioxo-3,3-dibutyl-5-phenyl-7-methylthio-8-(A/-{(R)-a-[A/-((S)-l-carboxypropyl) carbamoyl]-4-hydroxybenzyl}carbamoylmethoxy)-2,3,4,5-tetrahydro-l,2,5-benzothiadiazepine (odevixibat; also known as A4250) is disclosed in WO 03/022286. The structure of odevixibat is shown below.

As an inhibitor of the ileal bile acid transporter (IBAT) mechanism, odevixibat inhibits the natural reabsorption of bile acids from the ileum into the hepatic portal circulation. Bile acids that are not reabsorbed from the ileum are instead excreted into the faeces. The overall removal of bile acids from the enterohepatic circulation leads to a decrease in the level of bile acids in serum and the liver. Odevixibat, or a pharmaceutically acceptable salt thereof, is therefore useful in the treatment or prevention of diseases such as dyslipidemia, constipation, diabetes and liver diseases, and especially liver diseases that are associated with elevated bile acid levels.
According to the experimental section of WO 03/022286, the last step in the preparation of odevixibat involves the hydrolysis of a tert-butyl ester under acidic conditions. The crude compound was obtained by evaporation of the solvent under reduced pressure followed by purification of the residue by preparative HPLC (Example 29). No crystalline material was identified.
Amorphous materials may contain high levels of residual solvents, which is highly undesirable for materials that should be used as pharmaceuticals. Also, because of their lower chemical and physical stability, as compared with crystalline material, amorphous materials may display faster
decomposition and may spontaneously form crystals with a variable degree of crystallinity. This may result in unreproducible solubility rates and difficulties in storing and handling the material. In pharmaceutical preparations, the active pharmaceutical ingredient (API) is for that reason preferably used in a highly crystalline state. Thus, there is a need for crystal modifications of odevixibat having improved properties with respect to stability, bulk handling and solubility. In particular, it is an object of the present invention to provide a stable crystal modification of odevixibat that does not contain high levels of residual solvents, that has improved chemical stability and can be obtained in high levels of crystallinity.
Example 1
Preparation of crystal modification 1
Absolute alcohol (100.42 kg) and crude odevixibat (18.16 kg) were charged to a 250-L GLR with stirring under nitrogen atmosphere. Purified water (12.71 kg) was added and the reaction mass was stirred under nitrogen atmosphere at 25 ± 5 °C for 15 minutes. Stirring was continued at 25 ± 5 °C for 3 to 60 minutes, until a clear solution had formed. The solution was filtered through a 5.0 m SS cartridge filter, followed by a 0.2 m PP cartridge filter and then transferred to a clean reactor.
Purified water (63.56 kg) was added slowly over a period of 2 to 3 hours at 25 ± 5 °C, and the solution was seeded with crystal modification 1 of odevixibat. The solution was stirred at 25 ± 5 °C for 12 hours. During this time, the solution turned turbid. The precipitated solids were filtered through centrifuge and the material was spin dried for 30 minutes. The material was thereafter vacuum dried in a Nutsche filter for 12 hours. The material was then dried in a vacuum tray drier at 25 ± 5 °C under vacuum (550 mm Hg) for 10 hours and then at 30 ± 5 °C under vacuum (550 mm Hg) for 16 hours. The material was isolated as an off-white crystalline solid. The isolated crystalline material was milled and stored in LDPE bags.
An overhydrated sample was analyzed with XRPD and the diffractogram is shown in Figure 2.
Another sample was dried at 50 °C in vacuum and thereafter analysed with XRPD. The diffractogram of the dried sample is shown in Figure 1.
The diffractograms for the drying of the sample are shown in Figures 3 and 4 for 2Q ranges 5 – 13 ° and 18 – 25 °, respectively (overhydrated sample at the bottom and dry sample at the top).
References
- ^ Jump up to:a b c d “First treatment for rare liver disease”. European Medicines Agency (EMA) (Press release). 21 May 2021. Retrieved 21 May 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Odevixibat”. Albireo Pharma. Retrieved 21 May 2021.
- ^ “Bylvay: Pending EC decision”. European Medicines Agency (EMA). 19 May 2021. Retrieved 21 May 2021.
External links
- “Odevixibat”. Drug Information Portal. U.S. National Library of Medicine.
ClinicalTrials.gov
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT04336722 | Efficacy and Safety of Odevixibat in Children With Biliary Atresia Who Have Undergone a Kasai HPE (BOLD) | Phase 3 | Recruiting | 2020-09-02 |
| NCT04483531 | Odevixibat for the Treatment of Progressive Familial Intrahepatic Cholestasis | Available | 2020-08-25 | |
| NCT03566238 | This Study Will Investigate the Efficacy and Safety of A4250 in Children With PFIC 1 or 2 | Phase 3 | Active, not recruiting | 2020-03-05 |
| NCT03659916 | Long Term Safety & Efficacy Study Evaluating The Effect of A4250 in Children With PFIC | Phase 3 | Recruiting | 2020-01-21 |
| NCT03608319 | Study of A4250 in Healthy Volunteers Under Fasting, Fed and Sprinkled Conditions | Phase 1 | Completed | 2018-09-19 |
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT02630875 | A4250, an IBAT Inhibitor in Pediatric Cholestasis | Phase 2 | Completed | 2018-03-29 |
| NCT02360852 | IBAT Inhibitor A4250 for Cholestatic Pruritus | Phase 2 | Terminated | 2017-02-23 |
| NCT02963077 | A Safety and Pharmakokinetic Study of A4250 Alone or in Combination With A3384 | Phase 1 | Completed | 2016-11-16 |
EU Clinical Trials Register
| EudraCT | Title | Phase | Status | Date |
|---|---|---|---|---|
| 2019-003807-37 | A Double-Blind, Randomized, Placebo-Controlled Study to Evaluate the Efficacy and Safety of Odevixibat (A4250) in Children with Biliary Atresia Who Have Undergone a Kasai Hepatoportoenterostomy (BOLD) | Phase 3 | Ongoing | 2020-07-29 |
| 2015-001157-32 | An Exploratory Phase II Study to demonstrate the Safety and Efficacy of A4250 | Phase 2 | Completed | 2015-05-13 |
| 2014-004070-42 | An Exploratory, Phase IIa Cross-Over Study to Demonstrate the Efficacy | Phase 2 | Ongoing | 2014-12-09 |
| 2017-002325-38 | An Open-label Extension Study to Evaluate Long-term Efficacy and Safety of A4250 in Children with Progressive Familial Intrahepatic Cholestasis Types 1 and 2 (PEDFIC 2) | Phase 3 | Ongoing | |
| 2017-002338-21 | A Double-Blind, Randomized, Placebo-Controlled, Phase 3 Study to Demonstrate Efficacy and Safety of A4250 in Children with Progressive Familial Intrahepatic Cholestasis Types 1 and 2 (PEDFIC 1) | Phase 3 | Ongoing, Completed |
.
 |
|
| Clinical data | |
|---|---|
| Trade names | Bylvay |
| Routes of administration |
By mouth |
| ATC code |
|
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C37H48N4O8S2 |
| Molar mass | 740.93 g·mol−1 |
| 3D model (JSmol) | |
////////////odevixibat, Orphan Drug Status, phase 3, Albireo, A-4250, A 4250, AR-H 064974
CCCCC1(CN(C2=CC(=C(C=C2S(=O)(=O)N1)OCC(=O)NC(C3=CC=C(C=C3)O)C(=O)NC(CC)C(=O)O)SC)C4=CC=CC=C4)CCCC
| publicationnumber | |||||||
| US-2020046635-A1 | |||||||
| US-2020046636-A1 | US-2020046757-A1 | US-2020046758-A1 | US-2020002299-A1 | WO-2019245448-A1 | WO-2019245449-A1 | US-2019046451-A1 | US-2019070217-A1 |
| US-10441605-B2 | |||||||
| US-2017224720-A1 | |||||||
| US-2017224721-A1 | |||||||
| US-2018264029-A1 | |||||||
| US-2018360869-A1 | |||||||
| US-2018360870-A1 | |||||||
| US-2018360871-A1 | |||||||
| WO-2017133517-A1 | |||||||
| US-2017143738-A1 | |||||||
| US-2017143783-A1 | |||||||
| EP-2968230-A2 | |||||||
| EP-2968262-A1 | |||||||
| US-2014271734-A1 | |||||||
| US-2014275090-A1 | |||||||
| WO-2014144485-A1 | |||||||
| WO-2014144485-A9 | |||||||
| WO-2014144650-A2 | |||||||
| EP-2770990-A1 | |||||||
| EP-2771003-A1 | |||||||
| EP-2771003-B1 | |||||||
| EP-3266457-A1 | |||||||
| EP-3278796-A1 | |||||||
| US-10512657-B2 | |||||||
| US-2013108573-A1 | |||||||
| US-2013109671-A1 | |||||||
| US-2013338093-A1 | |||||||
| US-2014243281-A1 | |||||||
| US-2014323412-A1 | |||||||
| US-2016310518-A1 | |||||||
| US-2017368085-A1 | |||||||
| US-2019169217-A1 | |||||||
| US-2020069715-A1 | |||||||
| WO-2013063512-A1 | |||||||
| WO-2013063526-A1 | |||||||
| EP-2739286-A2 | |||||||
| WO-2013020108-A2 | |||||||
| EP-2637646-B1 | |||||||
| EP-2637668-B1 | |||||||
| EP-3023102-A1 | |||||||
| EP-3023102-B1 | |||||||
| EP-3400944-A1 | |||||||
| US-10000528-B2 | |||||||
| US-10011633-B2 | |||||||
| US-10093697-B2 | |||||||
| US-10221212-B2 | |||||||
| US-2012114588-A1 | |||||||
| US-2013225511-A1 | |||||||
| US-2013236541-A1 | |||||||
| US-2015031636-A1 | |||||||
| US-2015031637-A1 | |||||||
| US-2016193277-A1 | |||||||
| US-2016194353-A1 | |||||||
| US-2017182059-A1 | |||||||
| US-2017182115-A1 | |||||||
| US-2018022776-A1 | |||||||
| US-2018030088-A1 | |||||||
| US-2018030089-A1 | |||||||
| US-2018162904-A1 | |||||||
| US-2018362577-A1 | |||||||
| US-9688720-B2 | |||||||
| US-9694018-B1 | |||||||
| US-10555950-B2 | |||||||
| US-2016220577-A1 | |||||||
| US-9339480-B2 | |||||||
| WO-2008039829-A2 | |||||||
| US-2009069285-A1 | |||||||
| US-7842684-B2 | |||||||
| WO-2007051995-A2 | |||||||
| EP-1896408-A1 | |||||||
| EP-1896409-A1 | |||||||
| EP-1896457-A1 | |||||||
| US-2010048529-A1 | |||||||
| US-2010048530-A1 | |||||||
| US-2010137273-A1 | |||||||
| US-2010152156-A1 | |||||||
| US-2010168039-A1 | |||||||
| US-2010168075-A1 | |||||||
| US-7893048-B2 | |||||||
| US-7906502-B2 | |||||||
| WO-2006137792-A1 | |||||||
| WO-2006137794-A1 | |||||||
| WO-2006137795-A1 | |||||||
| US-2010216759-A1 | |||||||
| US-7863265-B2 | |||||||
| US-2008194494-A1 | |||||||
| US-2009186834-A1 | |||||||
| WO-2006102674-A2 | |||||||
| US-2009005321-A1 | |||||||
| EP-1831151-A1 | |||||||
| US-2008114064-A1 | |||||||
| WO-2006065214-A1 | |||||||
| EP-1699759-A1 | |||||||
| US-2007142304-A1 | |||||||
| US-2008064676-A1 | |||||||
| US-2010099657-A2 | |||||||
| US-7871998-B2 | |||||||
| WO-2005061452-A1 | |||||||
| EP-1638922-A1 | |||||||
| EP-1638926-A1 | |||||||
| EP-1638930-A1 | |||||||
| EP-1675820-A2 | |||||||
| EP-1676833-A1 | |||||||
| US-2005148656-A1 | |||||||
| US-2006142389-A1 | |||||||
| US-2006178432-A1 | |||||||
| US-2006194879-A1 | |||||||
| US-2006258866-A1 | |||||||
| US-2007099928-A1 | |||||||
| US-2007099997-A1 | |||||||
| US-2007244198-A1 | |||||||
| US-7309720-B2 | |||||||
| WO-2004110984-A1 | |||||||
| WO-2004113270-A2 | |||||||
| WO-2004113276-A1 | |||||||
| WO-2004113283-A1 | |||||||
| EP-1610770-A1 | |||||||
| EP-1610770-B1 | |||||||
| EP-1894564-A2 | |||||||
| US-2006199797-A1 | |||||||
| US-7514421-B2 | |||||||
| WO-2004089350-A1 | |||||||
| EP-1572626-A1 | |||||||
| US-2005131068-A1 | |||||||
| WO-2004056748-A1 | |||||||
| EP-1539120-A1 | |||||||
| US-2006083790-A1 | |||||||
| WO-2004006899-A1 | |||||||
| EP-1521742-A1 | |||||||
| US-2005239766-A1 | |||||||
| US-7470678-B2 | |||||||
| WO-2004005247-A1 | |||||||
| EP-1517679-A1 | |||||||
| EP-1517679-B1 | |||||||
| EP-1517883-A1 | |||||||
| EP-1517883-B1 | |||||||
| EP-1517883-B8 | |||||||
| US-2005222261-A1 | |||||||
| US-2005256198-A1 | |||||||
| US-2005267149-A1 | |||||||
| US-7351858-B2 | |||||||
| US-7355069-B2 | |||||||
| US-7521461-B2 | |||||||
| WO-2004000294-A1 | |||||||
| WO-2004000790-A1 | |||||||
| EP-1478368-A1 | |||||||
| US-2005124557-A1 | |||||||
| WO-03061663-A1 | |||||||
| EP-1458672-A1 | |||||||
| EP-1458672-B1 | |||||||
| EP-1458673-A1 | |||||||
| EP-1458673-B1 | |||||||
| EP-1458677-A1 | |||||||
| EP-1458677-B1 | |||||||
| US-2005113362-A1 | |||||||
| US-2005171204-A1 | |||||||
| US-2005215630-A1 | |||||||
| US-2005282822-A1 | |||||||
| US-7256307-B2 | |||||||
| US-7276539-B2 | |||||||
| US-7488844-B2 | |||||||
| US-7514471-B2 | |||||||
| WO-03051821-A1 | |||||||
| WO-03051822-A1 | |||||||
| WO-03051826-A1 | |||||||
| EP-1427423-B1 | |||||||
| EP-1427423-B9 | |||||||
| US-2005038009-A1 | |||||||
| US-7132416-B2 |
Copper Cu 64 dotatate, 銅(Cu64)ドータテート;
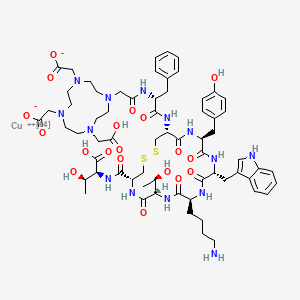

Copper Cu 64 dotatate
銅(Cu64)ドータテート;
UNII-N3858377KC
N3858377KC
Copper 64-DOTA-tate
Copper Cu-64 dotatate
Copper dotatate Cu-64
Diagnostic (neuroendocrine tumors), Radioactive agent
| Formula |
C65H86CuN14O19S2. 2H
|
|---|---|
| CAS: |
1426155-87-4
|
| Mol weight |
1497.1526
|
FDA APPROVED 2020. 2020/9/3. Detectnet
2-[4-[2-[[(2R)-1-[[(4R,7S,10S,13R,16S,19R)-10-(4-aminobutyl)-4-[[(1S,2R)-1-carboxy-2-hydroxypropyl]carbamoyl]-7-[(1R)-1-hydroxyethyl]-16-[(4-hydroxyphenyl)methyl]-13-(1H-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-1,2-dithia-5,8,11,14,17-pentazacycloicos-19-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-2-oxoethyl]-10-(carboxylatomethyl)-7-(carboxymethyl)-1,4,7,10-tetrazacyclododec-1-yl]acetate;copper-64(2+)
Copper Cu 64 dotatate, sold under the brand name Detectnet, is a radioactive diagnostic agent indicated for use with positron emission tomography (PET) for localization of somatostatin receptor positive neuroendocrine tumors (NETs) in adults.[1]
Common side effects include nausea, vomiting and flushing.[2]
It was approved for medical use in the United States in September 2020.[1][2]
History
The U.S. Food and Drug Administration (FDA) approved copper Cu 64 dotatate based on data from two trials that evaluated 175 adults.[3]
Trial 1 evaluated adults, some of whom had known or suspected NETs and some of whom were healthy volunteers.[3] The trial was conducted at one site in the United States (Houston, TX).[3] Both groups received copper Cu 64 dotatate and underwent PET scan imaging.[3] Trial 2 data came from the literature-reported trial of 112 adults, all of whom had history of NETs and underwent PET scan imaging with copper Cu 64 dotatate.[3] The trial was conducted at one site in Denmark.[3] In both trials, copper Cu 64 dotatate images were compared to either biopsy results or other images taken by different techniques to detect the sites of a tumor.[3] The images were read as either positive or negative for presence of NETs by three independent image readers who did not know participant clinical information.[3]
PATENT
https://patents.google.com/patent/WO2013029616A1/en
PATENT
https://patents.google.com/patent/US20140341807
-
Known imaging techniques with tremendous importance in medical diagnostics are positron emission tomography (PET), computed tomography (CT), magnetic resonance imaging (MRI), single photon computed tomography (SPECT) and ultrasound (US). Although today’s imaging technologies are well developed they rely mostly on non-specific, macroscopic, physical, physiological, or metabolic changes that differentiate pathological from normal tissue.
- [0003]
Targeting molecular imaging (MI) has the potential to reach a new dimension in medical diagnostics. The term “targeting” is related to the selective and highly specific binding of a natural or synthetic ligand (binder) to a molecule of interest (molecular target) in vitro or in vivo.
- [0004]
MI is a rapidly emerging biomedical research discipline that may be defined as the visual representation, characterization and quantification of biological processes at the cellular and sub-cellular levels within intact living organisms. It is a novel multidisciplinary field, in which the images produced reflect cellular and molecular pathways and in vivo mechanism of disease present within the context of physiologically authentic environments rather than identify molecular events responsible for disease.
- [0005]
Several different contrast-enhancing agents are known today and their unspecific or non-targeting forms are already in clinical routine. Some examples listed below are reported in literature.
- [0006]
For example, Gd-complexes could be used as contrast agents for MRI according to “Contrast Agents I” by W. Krause (Springer Verlag 2002, page one and following pages). Furthermore, superparamagnetic particles are another example of contrast-enhancing units, which could also be used as contrast agents for MRI (Textbook of Contrast Media, Superparamagnetic Oxides, Dawson, Cosgrove and Grainger Isis Medical Media Ltd, 1999, page 373 and following pages). As described in Contrast Agent II by W. Krause (Springer Verlag 2002, page 73 and following pages), gas-filled microbubbles could be used in a similar way as contrast agents for ultrasound. Moreover “Contrast Agents II” by W. Krause (Springer Verlag, 2002, page 151 and following pages) reports the use of iodinated liposomes or fatty acids as contrast agents for X-Ray imaging.
- [0007]
Contrast-enhancing agents that can be used in functional imaging are mainly developed for PET and SPECT.
- [0008]
The application of radiolabelled bioactive peptides for diagnostic imaging is gaining importance in nuclear medicine. Biologically active molecules which selectively interact with specific cell types are useful for the delivery of radioactivity to target tissues. For example, radiolabelled peptides have significant potential for the delivery of radionuclides to tumours, infarcts, and infected tissues for diagnostic imaging and radiotherapy.
- [0009]
DOTA (1,4,7,10-tetrakis(carboxymethyl)-1,4,7,10tetraazacyclododecane) and its derivatives constitute an important class of chelators for biomedical applications as they accommodate very stably a variety of di- and trivalent metal ions. An emerging area is the use of chelator conjugated bioactive peptides for labeling with radiometals in different fields of diagnostic and therapeutic nuclear oncology.
- [0010]
There have been several reports in recent years on targeted radiotherapy with radiolabeled somatostatin analogs.
- [0011]
US2007/0025910A1 discloses radiolabled somatostatin analogs primarily based on the ligand DOTA-TOC. The radionucleotide can be (64)Copper and the somatostatin analog may be octreotide, lanreotide, depreotide, vapreotide or derivatives thereof. The compounds of US2007/0025910A1 are useful in radionucleotide therapy of tumours.
- [0012]
US2007/0025910A1 does not disclose (64)Cu-DOTA-TATE. DOTA-TATE and DOTA-TOC differ clearly in affinity for the 5 known somatostatin receptors (SST1-SST2). Accordingly, the DOTA-TATE has a 10-fold higher affinity for the SST2 receptor, the receptor expressed to the highest degree on neuroendocrine tumors. Also the relative affinity for the other receptor subtypes are different. Furthermore, since 177Lu-DOTATATE is used for radionuclide therapy, only 64Cu-DOTATATE and not 64Cu-DOTATOC can be used to predict effect of such treatment by a prior PET scan.
- [0013]
There exists a need for further peptide-based compounds having utility for diagnostic imaging techniques, such as PET.
-
- EXAMPLE
- [0033]
Preparation of “Cu-Dotatate-DOTA-TATE
- [0034]
64Cu was produced using a GE PETtrace cyclotron equipped with a beamline. The 64Cu was produced via the 64Ni (p,n) 64Cu reaction using a solid target system consisting of a water cooled target mounted on the beamline. The target consisted of 64Ni metal (enriched to >99%) electroplated on a silver disc backing. For this specific type of production a proton beam with the energy of 16 MeV and a beam current of 20 uA was used. After irradiation the target was transferred to the laboratory for further chemical processing in which the 64Cu was isolated using ion exchange chromatography. Final evaporation from aq. HCl yielded 2-6 GBq of 64Cu as 64CuCl2 (specific activity 300-3000 TBq/mmol; RNP >99%). The labeling of 64Cu to DOTA-TATE was performed by adding a sterile solution of DOTA-TATE (0.3 mg) and Gentisic acid (25 mg) in aq Sodium acetate (1 ml; 0.4M, pH 5.0) to a dry vial containing 64CuCl2 (˜1 GBq). Gentisic acid was added as a scavenger to reduce the effect of radiolysis. The mixture was left at ambient temperature for 10 minutes and then diluted with sterile water (1 ml). Finally, the mixture was passed through a 0.22 μm sterile filter (Millex GP, Millipore). Radiochemical purity was determined by RP-HPLC and the amount of unlabeled 64Cu2+ was determined by thin-layer chromatography. All chemicals were purchased from Sigma-Aldrich unless specified otherwise. DOTA-Tyr3-Octreotate (DOTA-TATE) was purchased from Bachem (Torrance, Calif.). Nickel-64 was purchased in +99% purity from Campro Scientific Gmbh. All solutions were made using Ultra pure water (<0.07 μSimens/cm). Reversed-phase high pressure liquid chromatography was performed on a Waters Alliance 2795 Separations module equipped with at Waters 2489 UV/Visible detector and a Caroll Ramsey model 105 S-1 radioactivity detector—RP-HPLC column was Luna C18, HST, 50×2 mm, 2.5 μm, Phenomenex. The mobile phase was 5% aq. acetonitrile (0.1% TFA) and 95% aq. acetonitrile (0.1% TFA).
- [0035]
Thin layer chromatography was performed with a Raytest MiniGita Star TLC-scanner equipped with a Beta-detector. The eluent was 50% aq methanol and the TLC-plate was a Silica60 on Al foil (Fluka). Ion exchange chromatography was performed on a Dowex 1×8 resin (Chloride-form, 200-400 mesh).
References
- ^ Jump up to:a b “FDA approval letter” (PDF). 3 September 2020. Retrieved 5 September 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “RadioMedix and Curium Announce FDA Approval of Detectnet (copper Cu 64 dotatate injection) in the U.S.” (Press release). Curium. 8 September 2020. Retrieved 9 September 2020 – via GlobeNewswire.
- ^ Jump up to:a b c d e f g h “Drug Trials Snapshots: Detectnet”. U.S. Food and Drug Administration (FDA). 3 September 2020. Retrieved 10 September 2020. This article incorporates text from this source, which is in the public domain.
External links
- “Copper dotatate Cu-64”. Drug Information Portal. U.S. National Library of Medicine.
- “copper Cu 64 dotatate injection safety data sheet” (PDF). Curium US LLC. 15 March 2020.



The FDA has approved copper Cu 64 dotatate injection (Detectnet) for the localization of somatostatin receptor–positive neuroendocrine tumors (NETs), according to an announcement from RadioMedix Inc. and Curium Pharma.1
The positron emission tomography (PET) diagnostic agent is anticipated to launch immediately, according to Curium. Doses will be accessible through several nuclear pharmacies or through the nuclear medicine company.
“Detectnet brings an exciting advancement in the diagnosis of NETs for healthcare providers, patients, and their caregivers,” Ebrahim Delpassand MD, CEO of RadioMedix, stated in a press release. “The phase 3 results demonstrate the clinical sensitivity and specificity of Detectnet which will provide a great aid to clinicians in developing an accurate treatment approach for their [patients with] NETs.”
Copper Cu 64 dotatate adheres to somatostatin receptors with highest affinity for subtype 2 receptors (SSTR2). Specifically, the agent binds to somatostatin receptor–expressing cells, including malignant neuroendocrine cells; these cells overexpress SSTR2. The agent is a positron-producing radionuclide that possesses an emission yield that permits PET imaging.
“Perhaps most exciting is that the 12.7-hour half-life allows Detectnet to be produced centrally and shipped to sites throughout the United States,” added Delpassand. “This will help alleviate shortages or delays that have been experienced with other somatostatin analogue PET agents.”
Two single-center, open-label studies confirmed the efficacy of the diagnostic agent, according to Curium.2 In Study 1, investigators conducted a prospective analysis of 63 patients, which included 42 patients with known or suspected NETs according to histology, conventional imaging, or clinical evaluations, and 21 healthy volunteers. The majority of the participants, or 88% (n = 37) had a history of NETs at the time that they underwent imaging. Just under half of patients (44%; n = 28) were men and the majority were white (86%). Moreover, patients had a mean age of 54 years.
Images produced by the PET agent were interpreted to be either positive or negative for NET via 3 independent readers who had been blinded to the clinical data and other imaging information. Moreover, the results from the diagnostic agent were compared with a composite reference standard that was comprised of 1 oncologist’s blinded evaluation of patient diagnosis based on available histopathology results, reports of conventional imaging that had been done within 8 weeks before the PET imaging, as well as clinical and laboratory findings, which involved chromogranin A and serotonin levels.
Additionally, the percentage of patients who tested positive for disease via composite reference as well as through PET imaging was used to quantify positive percent agreement. Conversely, the percentage of participants who did not have disease per composite reference and who were determined to be negative for disease per PET imaging was used to quantify negative percent agreement.
Results showed that the percent reader agreement for positive detection in 62 scans was 91% (95% CI, 75-98) and negative detection was 97% (95% CI, 80-99). For reader 2, these percentages were 91% (95% CI, 75-98) and 80% (95% CI, 61-92), respectively, for 63 scans. Lastly, the percent reader agreement for reader 3 in 63 scans was 91% (95% CI, 75-98) positive and 90% (95% CI, 72-97) negative.
Study 2 was a retrospective analysis in which investigators examined published findings collected from 112 patients; 63 patients were male, while 43 were female. The mean age of patients included in the analysis was 62 years. All patients had a known history of NETs. Results demonstrated similar performance with the PET imaging agent.
In both safety and efficacy trials, a total of 71 patients were given a single dose of the diagnostic agent; the majority of these patients had known or suspected NETs and 21 were healthy volunteers. Adverse reactions such as nausea, vomiting, and flushing were reported at a rate of less than 2%. In all clinical experience that has been published, a total of 126 patients with a known history of NETs were given a single dose of the PET diagnostic agent. A total of 4 patients experienced nausea immediately after administration.
“Curium is excited to bring the first commercially available Cu 64 diagnostic agent to the US market,” Dan Brague, CEO of Curium, North America, added in the release. “Our unique production capabilities and distribution network allow us to deliver to any nuclear pharmacy, hospital, or imaging center its full dosing requirements first thing in the morning, to provide scheduling flexibility to the institution and its patients. We look forward to joining with healthcare providers and our nuclear pharmacy partners to bring this highly efficacious agent to the market.”
References
1. RadioMedix and Curium announce FDA approval of Detectnet (copper Cu 64 dotatate injection) in the US. News release. RadioMedix Inc and Curium. September 8, 2020. Accessed September 9, 2020. https://bit.ly/3m6iC0q.
2. Detectnet. Prescribing information. Curium Pharma; 2020. Accessed September 9, 2020. https://bit.ly/32eZxS3.
///////////////Copper Cu 64 dotatate, 銅(Cu64)ドータテート , FDA 2020, 2020 APPROVALS, Diagnostic, neuroendocrine tumors, Radioactive agent,
CC(C1C(=O)NC(CSSCC(C(=O)NC(C(=O)NC(C(=O)NC(C(=O)N1)CCCCN)CC2=CNC3=CC=CC=C32)CC4=CC=C(C=C4)O)NC(=O)C(CC5=CC=CC=C5)NC(=O)CN6CCN(CCN(CCN(CC6)CC(=O)[O-])CC(=O)[O-])CC(=O)O)C(=O)NC(C(C)O)C(=O)O)O.[Cu+2]
CILOFEXOR
CILOFEXOR
| 586.8 g/mol |
1418274-28-8
GS-9674, Cilofexor (GS(c)\9674)
UNII-YUN2306954
YUN2306954
2-[3-[2-chloro-4-[[5-cyclopropyl-3-(2,6-dichlorophenyl)-1,2-oxazol-4-yl]methoxy]phenyl]-3-hydroxyazetidin-1-yl]pyridine-4-carboxylic acid
Cilofexor is under investigation in clinical trial NCT02943447 (Safety, Tolerability, and Efficacy of Cilofexor in Adults With Primary Biliary Cholangitis Without Cirrhosis).
Cilofexor (GS-9674) is a potent, selective and orally active nonsteroidal FXR agonist with an EC50 of 43 nM. Cilofexor has anti-inflammatory and antifibrotic effects. Cilofexor has the potential for primary sclerosing cholangitis (PSC) and nonalcoholic steatohepatitis (NASH) research.
Gilead , following a drug acquisition from Phenex , is developing cilofexor tromethamine (formerly GS-9674), the lead from a program of farnesoid X receptor (FXR; bile acid receptor) agonists, for the potential oral treatment of non-alcoholic steatohepatitis (NASH), primary biliary cholangitis/cirrhosis (PBC) and primary sclerosing cholangitis. In March 2019, a phase III trial was initiated for PSC; at that time, the trial was expected to complete in August 2022.

PATENT
Product case WO2013007387 , expiry EU in 2032 and in the US in 2034.
https://patents.google.com/patent/WO2013007387A1/en
PATENT
WO2020150136 claiming 2,6-dichloro-4-fluorophenyl compounds.
PATENT
Novel crystalline forms of cilofexor as FXR agonists useful for treating nonalcoholic steatohepatitis. Gilead , following a drug acquisition from Phenex , is developing cilofexor tromethamine (formerly GS-9674), the lead from a program of farnesoid X receptor (FXR; bile acid receptor) agonists, for the potential oral treatment of non-alcoholic steatohepatitis (NASH), primary biliary cholangitis/cirrhosis (PBC) and primary sclerosing cholangitis. In March 2019, a phase III trial was initiated for PSC; at that time, the trial was expected to complete in August 2022. Family members of the cilofexor product case WO2013007387 , expire in the EU in 2032 and in the US in 2034.
solid forms of compounds that bind to the NR1H4 receptor (FXR) and act as agonists or modulators of FXR. The disclosure further relates to the use of the solid forms of such compounds for the treatment and/or prophylaxis of diseases and/or conditions through binding of said nuclear receptor by said compounds.
[0004] Compounds that bind to the NR1H4 receptor (FXR) can act as agonists or modulators of FXR. FXR agonists are useful for the treatment and/or prophylaxis of diseases and conditions through binding of the NR1H4 receptor. One such FXR agonist is the compound of Formula I:
I.
[0005] Although numerous FXR agonists are known, what is desired in the art are physically stable forms of the compound of Formula I, or pharmaceutically acceptable salt thereof, with desired properties such as good physical and chemical stability, good aqueous solubility and good bioavailability. For example, pharmaceutical compositions are desired that address
challenges of stability, variable pharmacodynamics responses, drug-drug interactions, pH effect, food effects, and oral bioavailability.
[0006] Accordingly, there is a need for stable forms of the compound of Formula I with suitable chemical and physical stability for the formulation, therapeutic use, manufacturing, and storage of the compound.
[0007] Moreover, it is desirable to develop a solid form of Formula I that may be useful in the synthesis of Formula I. A solid form, such as a crystalline form of a compound of Formula I may be an intermediate to the synthesis of Formula F A solid form may have properties such as bioavailability, stability, purity, and/or manufacturability at certain conditions that may be suitable for medical or pharmaceutical uses.
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||
| Description | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 & Target |
EC50: 43 nM (FXR)[1] |
||||||||||||||||||||||||||||||||||||||||||||||||||
| In Vivo |
Cilofexor (GS-9674; 30 mg/kg; oral gavage; once daily; for 10 weeks; male Wistar rats) treatment significantly increases Fgf15 expression in the ileum and decreased Cyp7a1 in the liver in nonalcoholic steatohepatitis (NASH) rats. Liver fibrosis and hepatic collagen expression are significantly reduced. Cilofexor also significantly reduces hepatic stellate cell (HSC) activation and significantly decreases portal pressure, without affecting systemic hemodynamics[3].
|
||||||||||||||||||||||||||||||||||||||||||||||||||
| Clinical Trial |
|
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2019142814 | Novel FXR (NR1H4) binding and activity modulating compounds | 2019-01-15 | |
| US2019055273 | ACYCLIC ANTIVIRALS | 2017-03-09 | |
| US10220027 | FXR (NR1H4) binding and activity modulating compounds | 2017-10-13 | |
| US10071108 | Methods and pharmaceutical compositions for the treatment of hepatitis b virus infection | 2018-02-19 | |
| US2018000768 | INTESTINAL FXR AGONISM ENHANCES GLP-1 SIGNALING TO RESTORE PANCREATIC BETA CELL FUNCTIONS | 2017-09-06 |
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9820979 | NOVEL FXR (NR1H4) BINDING AND ACTIVITY MODULATING COMPOUNDS | 2016-12-05 | |
| US9539244 | NOVEL FXR (NR1H4) BINDING AND ACTIVITY MODULATING COMPOUNDS | 2015-08-12 | 2015-12-03 |
| US9895380 | METHODS AND PHARMACEUTICAL COMPOSITIONS FOR THE TREATMENT OF HEPATITIS B VIRUS INFECTION | 2014-09-10 | 2016-08-04 |
| US2017355693 | FXR (NR1H4) MODULATING COMPOUNDS | 2017-06-12 | |
| US2016376279 | FXR AGONISTS AND METHODS FOR MAKING AND USING | 2016-09-12 |
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9139539 | NOVEL FXR (NR1H4) BINDING AND ACTIVITY MODULATING COMPOUNDS | 2012-07-12 | 2014-08-07 |
| US2018133203 | METHODS OF TREATING LIVER DISEASE | 2017-10-27 |
ClinicalTrials.gov
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT03890120 | Safety, Tolerability, and Efficacy of Cilofexor in Non-Cirrhotic Adults With Primary Sclerosing Cholangitis | Phase 3 | Recruiting | 2020-08-31 |
| NCT02781584 | Safety, Tolerability, and Efficacy of Selonsertib, Firsocostat, and Cilofexor in Adults With Nonalcoholic Steatohepatitis (NASH) | Phase 2 | Recruiting | 2020-08-13 |
| NCT03987074 | Safety, Tolerability, and Efficacy of Monotherapy and Combination Regimens in Adults With Nonalcoholic Steatohepatitis (NASH) | Phase 2 | Completed | 2020-07-29 |
| NCT02943460 | Safety, Tolerability, and Efficacy of Cilofexor in Adults With Primary Sclerosing Cholangitis Without Cirrhosis | Phase 2 | Completed | 2020-06-09 |
| NCT02943447 | Safety, Tolerability, and Efficacy of Cilofexor in Adults With Primary Biliary Cholangitis Without Cirrhosis | Phase 2 | Completed | 2020-02-11 |
ClinicalTrials.gov
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT03449446 | Safety and Efficacy of Selonsertib, Firsocostat, Cilofexor, and Combinations in Participants With Bridging Fibrosis or Compensated Cirrhosis Due to Nonalcoholic Steatohepatitis (NASH) | Phase 2 | Completed | 2019-12-24 |
| NCT02854605 | Evaluating the Safety, Tolerability, and Efficacy of GS-9674 in Participants With Nonalcoholic Steatohepatitis (NASH) | Phase 2 | Completed | 2019-01-29 |
| NCT02808312 | Pharmacokinetics and Pharmacodynamics of GS-9674 in Adults With Normal and Impaired Hepatic Function | Phase 1 | Completed | 2018-10-30 |
| NCT02654002 | Study in Healthy Volunteers to Evaluate the Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of GS-9674, and the Effect of Food on GS-9674 Pharmacokinetics and Pharmacodynamics | Phase 1 | Completed | 2016-07-27 |
EU Clinical Trials Register
| EudraCT | Title | Phase | Status | Date |
|---|---|---|---|---|
| 2019-000204-14 | A Phase 3, Randomized, Double-Blind, Placebo-Controlled Study Evaluating the Safety, Tolerability, and Efficacy of Cilofexor in Non-Cirrhotic Subjects with Primary Sclerosing Cholangitis | Phase 3 | Restarted, Ongoing | 2019-09-11 |
| 2016-002496-10 | A Phase 2, Randomized, Double-Blind, Placebo-Controlled Study Evaluating the Safety, Tolerability, and Efficacy of GS-9674 in Subjects with Nonalcoholic Steatohepatitis (NASH) | Phase 2 | Completed | 2017-02-21 |
| 2016-002443-42 | A Phase 2, Randomized, Double-Blind, Placebo Controlled Study Evaluating the Safety, Tolerability, and Efficacy of GS-9674 in Subjects with Primary Biliary Cholangitis Without Cirrhosis | Phase 2 | Completed | 2017-01-09 |
| 2016-002442-23 | A Phase 2, Randomized, Double-Blind, Placebo Controlled Study Evaluating the Safety, Tolerability, and Efficacy of GS-9674 in Subjects with Primary Sclerosing Cholangitis Without Cirrhosis | Phase 2 | Completed | 2017-01-09 |
///////////CILOFEXOR, Cilofexor (GS(c)\9674), GS-9674, phase 3
C1CC1C2=C(C(=NO2)C3=C(C=CC=C3Cl)Cl)COC4=CC(=C(C=C4)C5(CN(C5)C6=NC=CC(=C6)C(=O)O)O)Cl
LAZUVAPAGON

LAZUVAPAGON
KRPN-118
CAS 2379889-71-9
Chemical Formula: C27H32N4O3
Molecular Weight: 460.58
(4S)-N-[(2S)-1-hydroxypropan-2-yl]-methyl-1-[2-methyl-4-(3- methyl-1H-pyrazol-1-yl)benzoyl]-2,3,4,5-tetrahydro-1H-1-benzazepine-4-carboxamide
Vasopressin V2 receptor agonist
Kyorin Pharmaceutical under license from Sanwa Kagaku Kenkyusho , is developing SK-1404 ([14C]-SK-1404, presumed to be lazuvapagon), for the iv treatment of nocturia, and as an oral formulation, as KRPN-118
PATENT
WO2020171055
PATENT
WO2014104209
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014104209
PATENT
WO-2020171073
Process for preparing benzazepine derivatives, particularly lazuvapagon a V2 receptor agonist, and their intermediates, useful for treating diabetes insipidus, hemophilia and overactive bladder.
[Fifth Step] to [Sixth Step]
[Chemical
 Formula 33] [In the formula, R 1 and R 2 have the same meanings as those in the first step, and * represents an asymmetric center. ]
Formula 33] [In the formula, R 1 and R 2 have the same meanings as those in the first step, and * represents an asymmetric center. ]
[Chemical
[0074]
In the fifth step and the sixth step, the reaction can be performed according to a conventional method.
In the fifth step, compound (IX) is treated with a base (eg, sodium hydroxide, potassium hydroxide, etc.) in a suitable solvent (eg, alcohol solvent such as methanol, ethanol, etc., water), usually at room temperature to an organic solvent. A carboxylic acid compound of the compound (X) can be obtained by reacting at a temperature of the boiling point of the solvent for 30 minutes to 1 day. Next, in the sixth step, the obtained carboxylic acid compound is subjected to amidation with L-alaninol to obtain the compound (V). For the amidation, a method using a condensing agent, a method of reacting L-alaninol with a mixed acid anhydride or acid chloride of carboxylic acid, and the like can be used. In the method using a condensing agent, for example, the carboxylic acid compound and L-alaninol are condensed in a suitable organic solvent (chloroform, dimethylformamide, etc.) in the presence of a base (eg, diisopropylethylamine, triethylamine, etc.) (for example, 1 , 3-dicyclohexylcarbodiimide (DCC), 1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide (EDC), etc.) alone or in combination with 1-hydroxybenztriazole (HOBt). (V) can be obtained. Further, in the method using a mixed acid anhydride, for example, a carboxylic acid derivative in an appropriate organic solvent (eg, dichloromethane, toluene, etc.) in the presence of a base (eg, pyridine, triethylamine, etc.), an acid chloride (eg, pivaloyl chloride, Tosyl chloride, etc.) or an acid derivative (eg, ethyl chloroformate, isobutyl chloroformate, etc.), and the resulting mixed acid anhydride is reacted with L-alaninol usually at 0° C. to room temperature to give compound (V). Can be obtained. Further, in the method of passing through an acid chloride, for example, an acid chloride is obtained by using a chlorinating agent (eg, thionyl chloride, oxalyl chloride, etc.) in a suitable organic solvent (eg, toluene, xylene, etc.) Acid chloride in the presence of a base (eg sodium carbonate, triethylamine etc.) in a suitable organic solvent (eg ethyl acetate, toluene etc.) with L-alaninol,
In the fifth step, compound (IX) is treated with a base (eg, sodium hydroxide, potassium hydroxide, etc.) in a suitable solvent (eg, alcohol solvent such as methanol, ethanol, etc., water), usually at room temperature to an organic solvent. A carboxylic acid compound of the compound (X) can be obtained by reacting at a temperature of the boiling point of the solvent for 30 minutes to 1 day. Next, in the sixth step, the obtained carboxylic acid compound is subjected to amidation with L-alaninol to obtain the compound (V). For the amidation, a method using a condensing agent, a method of reacting L-alaninol with a mixed acid anhydride or acid chloride of carboxylic acid, and the like can be used. In the method using a condensing agent, for example, the carboxylic acid compound and L-alaninol are condensed in a suitable organic solvent (chloroform, dimethylformamide, etc.) in the presence of a base (eg, diisopropylethylamine, triethylamine, etc.) (for example, 1 , 3-dicyclohexylcarbodiimide (DCC), 1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide (EDC), etc.) alone or in combination with 1-hydroxybenztriazole (HOBt). (V) can be obtained. Further, in the method using a mixed acid anhydride, for example, a carboxylic acid derivative in an appropriate organic solvent (eg, dichloromethane, toluene, etc.) in the presence of a base (eg, pyridine, triethylamine, etc.), an acid chloride (eg, pivaloyl chloride, Tosyl chloride, etc.) or an acid derivative (eg, ethyl chloroformate, isobutyl chloroformate, etc.), and the resulting mixed acid anhydride is reacted with L-alaninol usually at 0° C. to room temperature to give compound (V). Can be obtained. Further, in the method of passing through an acid chloride, for example, an acid chloride is obtained by using a chlorinating agent (eg, thionyl chloride, oxalyl chloride, etc.) in a suitable organic solvent (eg, toluene, xylene, etc.) Acid chloride in the presence of a base (eg sodium carbonate, triethylamine etc.) in a suitable organic solvent (eg ethyl acetate, toluene etc.) with L-alaninol,
[0075]
Compound (V) can also exist as a solvate. The solvate of compound (V) can be obtained by a conventional method for producing a solvate. Specifically, it can be obtained by mixing the compound (V) with a solvent while heating if necessary, and then cooling and crystallizing the mixture while stirring or standing. It is desirable that the cooling be carried out while adjusting the cooling rate if necessary in consideration of the influence on the quality of crystal, grain size and the like. For example, cooling at a cooling rate of 20 to 1° C./hour is preferable, and cooling at a cooling rate of 10 to 3° C./hour is more preferable. As the organic solvent used in these methods, alcohol solvents such as methanol, ethanol, propanol, isopropanol, normal propanol, and tertiary butanol are preferable. The amount of the organic solvent used is preferably 3 to 20 times by weight, more preferably 5 to 10 times by weight, of the compound (V).
PATENT
WO-2020171055
The present inventors have investigated the method described in Patent Document 1 by using N-[(S)-1-hydroxypropan-2-yl]-4-methyl-1-[2-methyl-4-(3-methyl-1H). -Pyrazol-1-yl)benzoyl]-2,3,4,5-tetrahydro-1H-benzo[b]azepine-4-carboxamide chiral compound was prepared and analyzed. As a result, the compound was amorphous (amorphous). Solid). Amorphous is known to be a thermodynamically non-equilibrium metastable state and generally has high solubility and dissolution rate, but is low in stability and is often unfavorable in terms of drug development. Therefore, an object of the present invention is to increase the applicability as a drug substance to (S)-N-[(S)-1-hydroxypropan-2-yl]-4 represented by the formula (I). -Methyl-1-[2-methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl]-2,3,4,5-tetrahydro-1H-benzo[b]azepine-4-carboxamide It is to provide an alcohol solvate or a crystal thereof.
[Chemical 1]
[Reference Example 1] Compound (I) (amorphous)
Compound (I) was produced by the following method.
[Chemical 5]

Compound (I) was produced by the following method.
[Chemical 5]
[0046]
(First Step)
1-(2-Methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl)-5-oxo-2,3,4,5-tetrahydro-1H-benzo[b] Azepine-4-carboxylic acid ethyl ester was treated with methyl bromide in the presence of (R,R)-3,5-bistrifluoromethylphenyl-NAS bromide, cesium carbonate and cesium fluoride in a mixed solvent of benzene bromide and water. By carrying out methylation using (R)-4-methyl-1-(2-methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl)-5-oxo-2,3,4 ,5-Tetrahydro-1H-benzo[b]azepine-4-carboxylic acid ethyl ester was obtained.
1-(2-Methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl)-5-oxo-2,3,4,5-tetrahydro-1H-benzo[b] Azepine-4-carboxylic acid ethyl ester was treated with methyl bromide in the presence of (R,R)-3,5-bistrifluoromethylphenyl-NAS bromide, cesium carbonate and cesium fluoride in a mixed solvent of benzene bromide and water. By carrying out methylation using (R)-4-methyl-1-(2-methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl)-5-oxo-2,3,4 ,5-Tetrahydro-1H-benzo[b]azepine-4-carboxylic acid ethyl ester was obtained.
[0047]
(Second Step)
(R)-4-Methyl-1-(2-methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl)-5-oxo-2,3,4,5- Reduction of the ketone portion of tetrahydro-1H-benzo[b]azepine-4-carboxylic acid ethyl ester with a borane-ammonia complex prepared from sodium borohydride and ammonium sulfate in a toluene solvent gave (4R)-5. -Hydroxy-4-methyl-1-(2-methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl)-2,3,4,5-tetrahydro-1H-benzo[b]azepine- 4-Carboxylic acid ethyl ester was obtained.
(R)-4-Methyl-1-(2-methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl)-5-oxo-2,3,4,5- Reduction of the ketone portion of tetrahydro-1H-benzo[b]azepine-4-carboxylic acid ethyl ester with a borane-ammonia complex prepared from sodium borohydride and ammonium sulfate in a toluene solvent gave (4R)-5. -Hydroxy-4-methyl-1-(2-methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl)-2,3,4,5-tetrahydro-1H-benzo[b]azepine- 4-Carboxylic acid ethyl ester was obtained.
[0048]
(Third Step)
(4R)-5-hydroxy-4-methyl-1-(2-methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl)-2,3,4,5- By chlorinating the hydroxyl group of tetrahydro-1H-benzo[b]azepine-4-carboxylic acid ethyl ester with phosphorus oxychloride in the presence of pyridine in a toluene solvent, (4S)-5-chloro-4-methyl-1 -(2-Methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl)-2,3,4,5-tetrahydro-1H-benzo[b]azepine-4-carboxylic acid ethyl ester was obtained. It was
(4R)-5-hydroxy-4-methyl-1-(2-methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl)-2,3,4,5- By chlorinating the hydroxyl group of tetrahydro-1H-benzo[b]azepine-4-carboxylic acid ethyl ester with phosphorus oxychloride in the presence of pyridine in a toluene solvent, (4S)-5-chloro-4-methyl-1 -(2-Methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl)-2,3,4,5-tetrahydro-1H-benzo[b]azepine-4-carboxylic acid ethyl ester was obtained. It was
[0049]
(Step 4)
(4S)-5-chloro-4-methyl-1-(2-methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl)-2,3,4,5- By stirring tetrahydro-1H-benzo[b]azepine-4-carboxylic acid ethyl ester in a methanol solvent in the presence of 10% palladium-carbon under slightly pressurized conditions of hydrogen gas, (S)-4-methyl- 1-(2-methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl)-2,3,4,5-tetrahydro-1H-benzo[b]azepine-4-carboxylic acid ethyl ester Obtained.
(4S)-5-chloro-4-methyl-1-(2-methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl)-2,3,4,5- By stirring tetrahydro-1H-benzo[b]azepine-4-carboxylic acid ethyl ester in a methanol solvent in the presence of 10% palladium-carbon under slightly pressurized conditions of hydrogen gas, (S)-4-methyl- 1-(2-methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl)-2,3,4,5-tetrahydro-1H-benzo[b]azepine-4-carboxylic acid ethyl ester Obtained.
[0050]
(Fifth Step)
(S)-4-Methyl-1-(2-methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl)-2,3,4,5-tetrahydro-1H- Benzo[b]azepine-4-carboxylic acid ethyl ester is hydrolyzed with 30% sodium hydroxide in a solvent of water and methanol to give (S)-4-methyl-1-(2-methyl-4-( 3-Methyl-1H-pyrazol-1-yl)benzoyl)-2,3,4,5-tetrahydro-1H-benzo[b]azepine-4-carboxylic acid was obtained.
(S)-4-Methyl-1-(2-methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl)-2,3,4,5-tetrahydro-1H- Benzo[b]azepine-4-carboxylic acid ethyl ester is hydrolyzed with 30% sodium hydroxide in a solvent of water and methanol to give (S)-4-methyl-1-(2-methyl-4-( 3-Methyl-1H-pyrazol-1-yl)benzoyl)-2,3,4,5-tetrahydro-1H-benzo[b]azepine-4-carboxylic acid was obtained.
[0051]
(Sixth Step)
(S)-4-Methyl-1-(2-methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl)-2,3,4,5-tetrahydro-1H- Benzo[b]azepine-4-carboxylic acid was converted to an acid chloride form using thionyl chloride in a toluene solvent. This acid chloride and L-alaninol are reacted in a mixed solvent of ethyl acetate and water in the presence of sodium carbonate to give (S)-N-((S)-1-hydroxypropan-2-yl)-4-methyl. -1-(2-methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl)-2,3,4,5-tetrahydro-1H-benzo[b]azepine-4-carboxamide (compound ( I)) was obtained.
(S)-4-Methyl-1-(2-methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl)-2,3,4,5-tetrahydro-1H- Benzo[b]azepine-4-carboxylic acid was converted to an acid chloride form using thionyl chloride in a toluene solvent. This acid chloride and L-alaninol are reacted in a mixed solvent of ethyl acetate and water in the presence of sodium carbonate to give (S)-N-((S)-1-hydroxypropan-2-yl)-4-methyl. -1-(2-methyl-4-(3-methyl-1H-pyrazol-1-yl)benzoyl)-2,3,4,5-tetrahydro-1H-benzo[b]azepine-4-carboxamide (compound ( I)) was obtained.
[0052]
FIG. 7 shows the powder X-ray diffraction spectrum of the compound (I) obtained in the first to sixth steps. No clear peak was observed in the X-ray diffraction pattern, and the compound (I) of Reference Example 1 was found to be amorphous.
[0053]
[Example 1] Isopropanol solvate
of compound (I) To 5.0 g of amorphous compound (I) of Reference Example 1, 65 mL of isopropanol was added, and the mixture was stirred at room temperature for 30 minutes. After the precipitated suspension was dissolved by heating, it was allowed to cool to room temperature and stirred overnight at 5°C. The suspension was filtered, washed with chilled isopropanol and dried at 40° C. overnight to give 4.9 g of a white solid.
of compound (I) To 5.0 g of amorphous compound (I) of Reference Example 1, 65 mL of isopropanol was added, and the mixture was stirred at room temperature for 30 minutes. After the precipitated suspension was dissolved by heating, it was allowed to cool to room temperature and stirred overnight at 5°C. The suspension was filtered, washed with chilled isopropanol and dried at 40° C. overnight to give 4.9 g of a white solid.
[0054]
When the obtained compound was analyzed by a thermogravimetric apparatus, the content of isopropanol was 8.2% with respect to the compound (I), and the molar ratio was 0.7 times the amount with respect to the compound (I). It was
[0055]
The powder X-ray diffraction spectrum and the infrared absorption spectrum of the compound obtained in Example 1 are shown in FIG. 1 and FIG. 2, respectively. The characteristic peaks shown in Table 1 were shown as the diffraction angle (2θ) or as the interplanar spacing d. The obtained compound was crystalline.
[0056]
[table 1]

FIG. 2 shows an infrared absorption spectrum of the compound obtained in Example 1.
/////////////LAZUVAPAGON, KRPN-118
CC1=NN(C=C1)C2=CC(=C(C=C2)C(=O)N3CCC(CC4=CC=CC=C43)(C)C(=O)NC(C)CO)C
MOLINDONE, молиндон موليندون 吗茚酮


MOLINDONE
C16H24N2O2,, 276.374
SPN 810, SPN 801M, AFX 2201
cas 15622-65-8 hcl
Molindone is used for the management of the manifestations of psychotic disorders.
молиндон
موليندون
吗茚酮
(±)-Molindone
2376
3-Ethyl-2-methyl-5-(4-morpholinylmethyl)-1,5,6,7-tetrahydro-4H-indol-4-one [ACD/IUPAC Name]
3-Ethyl-2-methyl-5-(morpholin-4-ylmethyl)-1,5,6,7-tetrahydro-4H-indol-4-one
4H-Indol-4-one, 3-ethyl-1,5,6,7-tetrahydro-2-methyl-5-(4-morpholinylmethyl)-
7416-34-4 [RN]
RT3Y3QMF8N
UNII:RT3Y3QMF8N
Supernus Pharmaceuticals , under license from Afecta Pharmaceuticals , is developing molindone hydrochloride (SPN-810; SPN-801M; AFX-2201; presumed to be Zalvari), as a capsule formulation, for the potential oral treatment of conduct disorder in patients with attention deficit hyperactivity disorder. In 3Q15, the company initiated two phase III trials (CHIME 1 and CHIME 2) for compulsive aggression in ADHD. In November 2019, the trial was expected to complete in June 2020.
Molindone, sold under the brand name Moban, is an antipsychotic which is used in the United States in the treatment of schizophrenia.[1][2] It works by blocking the effects of dopamine in the brain, leading to diminished symptoms of psychosis. It is rapidly absorbed when taken orally.
It is sometimes described as a typical antipsychotic,[3] and sometimes described as an atypical antipsychotic.[4]
Molindone was discontinued by its original supplier, Endo Pharmaceuticals, on January 13, 2010.[5]
Availability and Marketing in the USA
After having been produced and subsequently discontinued by Core Pharma in 2015-2017, Molindone is available again from Epic Pharma effective December, 2018.[6]
Adverse effects
The side effect profile of molindone is similar to that of other typical antipsychotics. Unlike most antipsychotics, however, molindone use is associated with weight loss.[4][7]
Chemistry
Synthesis

Molindone synthesis: SCHOEN KARL, J PACHTER IRWIN; BE 670798 (1965 to Endo Lab).
Condensation of oximinoketone 2 (from nitrosation of 3-pentanone), with cyclohexane-1,3-dione (1) in the presence of zinc and acetic acid leads directly to the partly reduced indole derivative 6. The transformation may be rationalized by assuming as the first step, reduction of 2 to the corresponding α-aminoketone. Conjugate addition of the amine to 1 followed by elimination of hydroxide (as water) would give ene-aminoketone 3. This enamine may be assumed to be in tautomeric equilibrium with imine 4. Aldol condensation of the side chain carbonyl group with the doubly activated ring methylene group would then result in cyclization to pyrrole 5; simple tautomeric transformation would then give the observed product. Mannich reaction of 6 with formaldehyde and morpholine gives the tranquilizer molindone (7).
US-20200262788
Process for preparing molindone and its intermediates useful for treating schizophrenia..
|
Molindone is chemically known as 4H-Indol-4-one, 3-ethyl-1,5,6,7-tetrahydro-2-methyl-5-(4-morpholinylmethyl) and represented by formula I. Molindone is indicated for management of schizophrenia and is under clinical trial for alternate therapies.
|
EXAMPLES
Example 1: Preparation of methyl 2-chloro-2-ethyl-3-oxobutanoate
Example 2: Preparation of 3-chloropentan-2-one
Example 3: Preparation of 3-chloropentan-2-one
Example 4: Preparation of 2-(2-oxopentan-3-yl)cyclohexane-1,3-dione (4)
Example 5: Preparation of 2-methyl-3-ethyl-4-oxo-4,5,6,7-tetrahydroindole (5)
Example 6: Preparation of 2-methyl-3-ethyl-4-oxo-4,5,6,7-tetrahydroindole (5)
Example 7: Preparation of Molindone Hydrochloride
References
- ^ “molindone”. F.A. Davis Company.
- ^ “Molindone”.
- ^ Aparasu RR, Jano E, Johnson ML, Chen H (October 2008). “Hospitalization risk associated with typical and atypical antipsychotic use in community-dwelling elderly patients”. Am J Geriatr Pharmacother. 6 (4): 198–204. doi:10.1016/j.amjopharm.2008.10.003. PMID 19028375.
- ^ Jump up to:a b Bagnall A, Fenton M, Kleijnen J, Lewis R (2007). Bagnall A (ed.). “Molindone for schizophrenia and severe mental illness”. Cochrane Database Syst Rev (1): CD002083. doi:10.1002/14651858.CD002083.pub2. PMID 17253473.
- ^ https://www.fda.gov/Drugs/DrugSafety/DrugShortages/ucm050794.htm
- ^ “NEWS”. http://www.epic-pharma.com. Retrieved 2018-12-12.
- ^ Allison DB, Mentore JL, Heo M, et al. (1999). “Antipsychotic-induced weight gain: a comprehensive research synthesis”. Am J Psychiatry. 156 (11): 1686–96. doi:10.1176/ajp.156.11.1686 (inactive 2020-01-22). PMID 10553730. Free full text
 |
|
| Clinical data | |
|---|---|
| Pronunciation | /moʊˈlɪndoʊn/ moh-LIN-dohn |
| Trade names | Moban |
| AHFS/Drugs.com | Consumer Drug Information |
| MedlinePlus | a682238 |
| Pregnancy category |
|
| Routes of administration |
By mouth (tablets) |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Metabolism | Hepatic |
| Elimination half-life | 1.5 hours |
| Excretion | Minor, renal and fecal |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.254.109 |
| Chemical and physical data | |
| Formula | C16H24N2O2 |
| Molar mass | 276.380 g·mol−1 |
| 3D model (JSmol) | |
//////////MOLINDONE, SPN 810, SPN 801M, AFX 2201, молиндон, موليندون , 吗茚酮 ,
TILDACERFONT
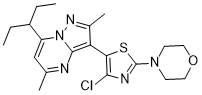
TILDACERFONT
| Synonyms: |
Tildacerfont 1014983-00-6 3-(4-Chloro-2-morpholin-4-yl-thiazol-5-yl)-7-(1-ethyl-propyl)-2,5-dimethyl-pyrazolo[1,5-a]pyrimidine 7-(1-ethyl-propyl)-3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-2,5-dimethyl-pyrazolo[1,5-a]pyrimidine |
|---|---|
| MW/ MF | 420 g/mol/ C20H26ClN5OS |
- Originator Spruce Biosciences
- Class2 ring heterocyclic compounds; Morpholines; Pyrazoles; Pyrimidines; Small molecules; Thiazoles
- Mechanism of Action Corticotropin receptor antagonists
- Orphan Drug Status Yes – Congenital adrenal hyperplasia
- New Molecular Entity Yes
- Phase II Congenital adrenal hyperplasia
- 09 Jul 2020 Spruce Biosciences initiates a phase II trial in Congenital adrenal hyperplasia in USA (PO) (NCT04457336)
- 24 Sep 2019 Spruce Biosciences completes a phase II trial in Congenital adrenal hyperplasia in USA (NCT03687242)
- 19 Sep 2019 Updated safety and efficacy data from a phase II trial in Congenital adrenal hyperplasia release by Spruce Biosciences
Deuterated pyrazolo[1,5-a]pyrimidine derivatives, particularly tildacerfont (SPR-001), useful as CRF antagonists for treating congenital adrenal hyperplasia. Spruce Bioscience is developing tildacerfont under license from Lilly as an oral capsule formulation for the treatment of congenital adrenal hyperplasia; in July 2017, a phase II trial for CAH was initiated.
Corticotropin releasing factor (CRF) is a 41 amino acid peptide that is the primary physiological regulator of proopiomelanocortin (POMC) derived peptide secretion from the anterior pituitary gland. In addition to its endocrine role at the pituitary gland, immunohistochemical localization of CRF has demonstrated that the hormone has a broad extrahypothalamic distribution in the central nervous system and produces a wide spectrum of autonomic, electrophysiological and behavioral effects consistent with a neurotransmitter or neuromodulator role in the brain. There is also evidence that CRF plays a significant role in integrating the response in the immune system to physiological, psychological, and immunological stressors.
PATENT
Product case, WO2008036579 ,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2008036579
Example 16
3-(4-Chloro-2-morpholin-4-yl-thiazol-5-yl)-7-(l-ethyl-propyl)-2,5-dimethyl- pyrazolo [ 1 ,5 -α]pyrimidine
Under a nitrogen atmosphere dissolve 3-(4-bromo-2-morpholin-4-yl-thiazol-5-yl)-7-(l-ethyl-propyl)-2,5-dimethyl-pyrazolo[l,5-α]pyrimidine (116 mg, 0.25 mmol) in THF (1.5 mL) and chill to -78 0C. Add n-butyl lithium (0.1 mL. 2.5 M in hexane, 0.25 mmol) and stir at -78 0C for 30 min. Add N-chlorosuccinimide (33.4 mg, 0.25 mmol) and stir for another 30 min, slowly warming to room temperature. After stirring overnight, quench the reaction by adding a solution of saturated ammonia chloride and extract with ethyl acetate. Wash the organic layer with brine, dry over sodium sulfate, filter, and concentrate to a residue. Purify the crude material by flash chromatography, eluting with hexanes:dichloromethane: ethyl acetate (5:5:2) to provide the title compound (54 mg). MS (APCI) m/z (35Cl) 420.6 (M+l)+; 1H NMR (400 MHz, CDCl3): 6.44 (s, IH), 3.79 (t, 4H, J=4.8 Hz), 3.63-3.56 (m, IH), 3.47 (t, 4H, J=4.8 Hz), 2.55 (s, 3H), 2.45 (s, 3H), 1.88-1.75 (m, 4H), 0.87 (t, 6H, J=7.5 Hz).
Alternate Preparation from Preparation 6:
Combine 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-α]pyrimidine, (9 g,
26.2 mmol) and 4-chloro-2-morpholino-thiazole (7.5 g, 36.7 mmol) in
dimethylformamide (90 mL) previously degassed with nitrogen. Add cesium carbonate (17.8 g, 55 mmol), copper iodide (250 mg, 1.31 mmol), triphenylphosphine (550 mg, 2.09 mmol) and palladium acetate (117 mg, 0.52 mmol). Heat the mixture to 125 0C for 16 h and then cool to 22 0C. Add water (900 mL) and extract with methyl-?-butyl ether (3 x 200 mL). Combine the organic portions and evaporate the solvent. Purify by silica gel chromatography eluting with hexanes/ethyl acetate (4/1) to afford the title compound (6.4 g, 62%). ES/MS m/z (35Cl) 420 (M+l)+.
Example 16a
3-(4-Chloro-2-morpholin-4-yl-thiazol-5-yl)-7-(l-ethyl-propyl)-2,5-dimethyl- pyrazolo[l,5-α]pyrimidine, hydrochloride
Dissolve 3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-7-(l-ethyl-propyl)-2,5-dimethyl-pyrazolo[l,5-α]pyrimidine (1.40 g, 3.33 mmol) in acetone (10 mL) at 50 0C and cool to room temperature. Add hydrogen chloride (2 M in diethyl ether, 2.0 mL, 4.0 mmol) and stir well in a sonicator. Concentrate the solution a little and add a minimal amount of diethyl ether to crystallize the HCl salt. Cool the mixture in a refrigerator overnight. Add additional hydrogen chloride (2 M in diethyl ether, 2.0 mL, 4.0 mmol) and cool in a refrigerator. Filter the crystalline material and dry to obtain the title compound (1.15 g, 75%). ES/MS m/z (35Cl) 420 (M+l)+; 1H NMR(CDCO): 9.18 (br, IH), 6.86 (s, IH), 3.72 ( m, 4H), 3.49(m, IH), 3.39 (m, 4H), 2.48 (s, 3H), 2.38(s, 3H), 1.79 (m, 4H), 0.79 (m, 6H).
PATENT
US-20200255436
PATENT
WO2019210266
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019210266
claiming the use of CRF-1 antagonists (eg tildacerfont).
PATENT
WO 2010039678
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2010039678
EXAMPLES
Example 1 : 7-(l-ethyl-propyl)-3-(‘2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolori ,5-alpyrimidine nthroline
Charge 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine (1.03 g, 3.00 mmoles), K3PO4 (1.95 g, 9.00 mmoles), 2,4-dichlorothiazole (0.58 g, 3.75 mmoles), 1,10 phenanthroline (0.05 g, 0.30 mmoles) and anhydrous DMAC (5 mL) to a round bottom flask equipped with a magnetic stir bar, thermal couple and N2 inlet. Degas the yellow heterogeneous reaction mixture with N2 (gas) for 30 min. and then add CuI (0.06 g, 0.30 mmoles) in one portion followed by additional 30 min. degassing with N2 (gas). Stir the reaction mixture at 120 0C for about 6 hr. Cool the reaction mixture to room temperature overnight, add toluene (10 mL) and stir for 1 hr. Purify the mixture through silica gel eluting with toluene (10ml). Extract with 1 M HCl (10 mL), water (10 mL), brine (10 mL) and concentrate under reduced pressure to give a yellow solid. Recrystallize the solid from methanol (5ml) to yield the title compound as a yellow crystalline solid. (0.78 g, 70% yield, >99% pure by LC) MS(ES) = 369 (M+ 1). 1H NMR (CDCl3)= 6.5 (IH, s); 3.6 (IH, m); 2.6 (3H, s); 2.5 (3H, s); 1.9 (4H, m); 0.9 (6H, t).
Example 2: 7-(l-ethyl-propyl)-3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-2,5-dimethyl-pyrazolol“! ,5-aipyrimidine
Charge 7-(l-ethyl-propyl)-3-(2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine (0.37 g, 1.00 mmoles), K2CO3 (0.28 g, 2.00 mmoles) and anhydrous morpholine (3 mL) to a round bottom flask equipped with a magnetic stir bar and N2 inlet. Stir the yellow mixture at 100 0C for about 4 hr., during which time the reaction becomes homogeneous. Cool the reaction mixture to room temperature, add H2O (10 mL) and stir the heterogeneous reaction mixture overnight at room temperature. Collected the yellow solid by filtration, wash with H2O and allowed to air dry overnight to give the crude title compound (391mg). Recrystallize from isopropyl alcohol (3 mL) to yield the title compound as a light yellow crystalline solid (380 mg, 90.6% yield, >99% by LC). MS(ES) = 420 (M+l). 1H NMR (CDCl3)= 6.45 (IH, s); 3.81 (m, 4H); 3.62 (IH, m); 3.50 (m, 4H); 2.6 (3H, s); 2.45 (3 H, s); 1.85 (4H, m); 0.9 (6H, t).
Example 3 :
The reactions of Example 1 are run with various other catalysts, ligands, bases and solvents, which are found to have the following effects on yield of 7-(l-ethyl-propyl)-3-(2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine. (See Tables 1 – 4).
Table 1 : Evaluation of different li ands
(Reactions are carried out in parallel reactors with 1.2 mmol 2,4-dichlorothiazole, 1 mmol 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine, 0.5 mmol CuI, 0.5 mmol ligand and 2.1 mmol Cs2CO3 in 4 mL DMAC. The reactions are degassed under N2 for 30 min. and then heated at between 80 and
1000C overnight under N2. Percent product is measured as the percent of total area under the HPLC curve for the product peak. Longer reaction times are shown in parenthesis) Table 2: Evaluation of various solvents
(Reactions are carried out in parallel reactors with 1.2 mmol 2,4-dichlorothiazole 1 mmol 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine, 0.25 mmol CuI, 0.25 mmol 1,10-phenanthroline and 2.1 mmol Cs2CO3 in 3 mL specified solvent. The reactions are degassed under N2 for 30 minutes and then heated at 1000C overnight under N2. Percent product is measured as the percent of total area under the HPLC curve for the product peak.)
Table 3 : Evaluation of different copper sources
(Reactions are carried out in in parallel reactors with 1 mmol 2,4-dichlorothiazole 1 mmol 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine, 0.05 mmol CuX, 0.01 mmol 1,10-phenanthroline and 3 equivalents K3PO4 in 3 mL DMAC. The reactions are degassed under N2 for 30 minutes and then heated at 1000C overnight under N2. Percent product is measured as the percent of total area under the HPLC curve for the product peak.)
Table 4: Evaluation of various inorganic bases
(Reactions are carried out in in parallel reactors with 1 mmol 2,4-dichlorothiazole 1 mmol 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine, 0.1 mmol CuI, 0.1 mmol 1,10-phenanthroline and 2.1 mmol base and degassed for 30 minutes prior to the addition of 3 mL DMAC. The reactions are degassed under N2 for 10 minutes and then heated at 1000C overnight under N2. Percent product is measured as the percent of total area under the HPLC curve for the product peak.)
Example 4. Use of morpholine both as a reactant and base in 2-MeTHF as solvent.
solvent
7-(l-ethyl-propyl)-3-(2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-ajpyrimidine (15.2 g, 41.16 mmoles) is charged into a 250 mL 3-necked round bottomed flask, followed by addition of 2-MeTHF (61 mL, 4.0 volumes), the yellowish brown slurry is stirred at about 20 0C for 5 min. Then morpholine (19 g, 218.18 mmoles) is added over 2-5 minutes. Contents are heated to reflux and maintained at reflux for 12 hr. The slurry is cooled to 25 0C, followed by addition of 2-MeTHF (53 mL, 3.5 volumes) and water ( 38 mL 2.5 volumes). The reaction mixture is warmed to 40 0C, where upon a homogenous solution with two distinct layers formed. The layers are separated, the organic layer is filtered and concentrated to ~3 volumes at atmospheric pressure. Four volumes 2-propanol (61 mL) are added. The solution is concentrated to ~3 volumes followed by addition of 4 volumes 2-propanol (61 mL), re-concentrated to ~3 volumes, followed by addition of another 6 volumes 2-propanol (91 mL), and refluxed for 15 min. The clear solution is gradually cooled to 75 0C, seeded with 0.45 g 7-(l-ethyl-propyl)-3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine slurried in 2 mL 2-propanol, rinsed with an additional 2 mL 2-propanol and transferred to a crystallization flask. The slurry is cooled to between 0-5 0C, maintained for 1 hr, filtered and the product rinsed with 2-propanol (30 mL, 2 volumes). The solid is dried at 60 0C in a vacuum oven to afford 16.92 g 7-(l-ethyl-propyl)-3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine. Purity of product by HPLC assay is 100.00 %. XRPD and DSC data of product is consistant with reference sample. MS(ES) = 420 (M+ 1).
Example 5. Use of morpholine as both reactant and base in 2-propanol as solvent.
7-(l-ethyl-propyl)-3-(2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-ajpyrimidine (11.64 mmoles) is charged into a 100 mL 3 -necked round bottomed flask followed by addition of 2-propanol ( 16 mL, 3.72 volumes). The yellowish brown slurry is stirred at about 20 0C for 5 min. Then morpholine (3.3 g, 37.84 mmoles) is added over 2-5 minutes. Contents are refluxed for 6 hr. The slurry is cooled to 25 0C. 2-Propanol ( 32 mL, 7.44 volumes) and water ( 8.6 mL, 2.0 volumes) are added and the mixture warmed to 70-75 0C, filtered and concentrated to ~ 9 volumes at atmospheric pressure. The clear solution is gradually cooled to 55 0C, seeded with 0.06 g of crystalline 7-(l-ethyl-propyl)-3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine slurried in 0.5 mL 2-propanol, rinsed with additional 0.5 mL 2-propanol and added to crystallization flask. The slurry is cooled to 0-5 0C, maintained for 1 hr., filtered and the product rinsed with 2-propanol ( 9 mL, 2.1 volumes). Suctioned dried under vacuum at 60 0C to afford 4.6 g of dry 7-(l-ethyl-propyl)-3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine (88.8 % yield, purity by HPLC assay is 99.88 % ). MS(ES) = 420 (M+ 1).
Example 6: 7-(l-ethyl-propyl)-3-(2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolori ,5-alpyrimidine
7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine (10 g, 29.17 mmoles), 2, 4-dichlorothiazole (5.2 g , 33.76 mmoles), cesium carbonate(19.9g, 61.07 mmoles) and 1,10-phenanthroline (1 g, 5.5 mmoles) are charged into a 250 mL 3-necked round bottomed flask, followed by 2-MeTHF (36 mL, 3.6 volumes). The reaction mixture is degassed with nitrogen and then evacuated. Cuprous chloride (0.57 g, 5.7 mmoles), DMAC (10 mL, 1 volume) and 2-MeTHF (4 mL, 0.4 volumes) are added in succession. The reaction mixture is degassed with nitrogen and then evacuated. The contents are refluxed for 20 hr. The reaction mixture is cooled to -70 0C and 2-MeTHF (100 mL, 10 volumes) is added. The contents are filtered at ~70 0C and the residual cake is washed with 2-MeTHF (80 mL, 8 volumes) at about 65-72°C. The filtrate is transferred into a separatory funnel and extracted with water. The organic layer is separated and washed with dilute HCl. The resulting organic layer is treated with Darco G60, filtered hot (600C). The filtrate is concentrated at atmospheric pressure to -2.8 volumes. 25 mL 2-propanol is added, followed by re-concentration to -2.8 volumes. An additional 25 mL 2-propanol is added, followed again by re-concentration to -2.8 volumes. Finally, 48 mL 2-propanol is added. The contents are cooled to -7 0C, maintained at -7 0C for 1 hr., filtered and rinsed with 20 mL chilled 2-propanol. Product is suction dried and then vacuum dried at 60 0C to afford 9.41 g 7-(l-ethyl-propyl)-3-(2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine (purity of product by HPLC assay is 95.88 %). MS(ES) = 369 (M+ 1).
Example 7. Synthesis of 7-(l-ethyl-propyl)-3-(2, 4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolori,5-a1pyrimidine using 1,4-Dioxane solvent and CuCl catalyst
Add dioxane (9.06X), Cs2CO3 (2.00X), 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine (1.0 equivalent), 2,4-dichlorothiazole (0.54 equivalent) to a reactor under N2. Purge the reactor with N2 three times, degas with N2 for 0.5-1 hr., and then add 1,10-phenanthroline (0.3 eq) and CuCl (0.3eq) under N2 , degassing with N2 for 0.5-1 hr. Heat the reactor to 1000C -1100C under N2 . Stir the mixture for 22-24 hr. at 100 0C -1100C. Cool to 10~20°C and add water (10V) and CH3OH (5V), stir the mixture for 1-1.5 hr. at 10~20°C. Filter the suspension, resuspend the wet cake in water, stirr for 1-1.5 hr. at 10~20°C, and filter the suspension again. Charge the wet cake to n-heptane (16V) and EtOAc (2V) under N2. Heat the reactor to 40 °C~500C under N2.
Active carbon (0. IX) is added at 40 °C~500C. The reactor is heated to 55°C~650C under N2 and stirred at 55 °C~650C for 1-1.5 hr. The suspension is filtered at 40~55°C through diatomite (0.4 X). The cake is washed with n-heptane (2.5V). The filtrate is transferred to another reactor. EtOAc (10V) is added and the the organic layer washed with 2 N HCl (10V) three times, followed by washing two times with water (10X, 10V). The organic layer is concentrated to 3-4V below 500C. The mixture is heated to 80-90 0C. The mixture is stirred at this temperature for 40-60 min. The mixture is cooled to 0~5°C, stirred for 1-1.5 hr. at 0~5°C and filtered. The cake is washed with n-heptane (IV) and vacuum dried at 45-500C for 8-10 hr. The crude product is dissolved in 2-propanol (7.5V) under N2, and re-crystallized with 2-propanol. The cake is dried in a vacuum oven at 45°C~50°C for 10-12 hr. (55-80% yield). 1H NMR56.537 (s, IH) 3.591-3.659 (m, IH, J=6.8Hz), 2.593 (s, 3H), 2.512 (s, 3H), 1.793-1.921(m, 4H), 0.885-0.903 (m, 6H).
REFERENCES
1: Zorrilla EP, Logrip ML, Koob GF. Corticotropin releasing factor: a key role in the neurobiology of addiction. Front Neuroendocrinol. 2014 Apr;35(2):234-44. doi: 10.1016/j.yfrne.2014.01.001. Epub 2014 Jan 20. Review. PubMed PMID: 24456850; PubMed Central PMCID: PMC4213066.
/////////////tildacerfont, SPR 001, Orphan Drug Status, Congenital adrenal hyperplasia, SPRUCE BIOSCIENCES, PHASE 2
CCC(CC)C1=CC(=NC2=C(C(=NN12)C)C3=C(N=C(S3)N4CCOCC4)Cl)C
SULCARDINE SULPHATE
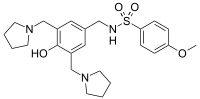
sulcardine, HBI-3000
B 87823
- Molecular FormulaC24H33N3O4S
- Average mass459.602 Da
N-[[4-hydroxy-3,5-bis(pyrrolidin-1-ylmethyl)phenyl]methyl]-4-methoxybenzenesulfonamide
Benzenesulfonamide, N-[[4-hydroxy-3,5-bis(1-pyrrolidinylmethyl)phenyl]methyl]-4-methoxy-
N-[4-Hydroxy-3,5-bis(1-pyrrolidinylmethyl)benzyl]-4-methoxybenzenesulfonamide
343935-60-4 [RN]
heart arrhythmia
.gif)
CAS No. : 343935-61-5 (Sulcardine sulfate)
| Synonyms: | B-87823; HBI-3000; B87823; HBI3000; B 87823; HBI 3000;N-(4-hydroxy-3,5-bis(pyrrolidin-1-ylmethyl)benzyl)-4-methoxybenzenesulfonamide sulfate |
| Molecular Formula: | C24H35N3O8S2 |
| Molecular Weight: | 557.67 |
- Originator Jiangsu Furui Pharmaceuticals; Shanghai Institute of Materia Medica
- Developer HUYA Bioscience International; Jiangsu Furui Pharmaceuticals
- Class Antiarrhythmics; Small molecules
- Mechanism of ActionIon channel antagonists
- Phase I Atrial fibrillation
- No development reported Arrhythmias
- 13 Mar 2020 Chemical structure information added
- 28 Feb 2020 No recent reports of development identified for preclinical development in Arrhythmias in USA (IV)
- 16 Dec 2019 Adverse events data from a phase I trial in Atrial fibrillation (In volunteers) presented at the American Heart Association Scientific Sessions 2019 (AHA-2019)
HUYA Bioscience , under license from Shanghai Institute of Materia Medica (SIMM), is developing sulcardine (HBI-3000, oral, i.v, heart arrhythmia), a myocardial ion channel inhibitory compound, for the treatment of arrhythmia; In September 2016, the drug was still in phase II development, as of August 2020, the company website states that a phase II trial was pending in China.
HBI-3000 (sulcardine sulfate) is an experimental drug candidate that is currently in phase II of human clinical trials as an antiarrhythmic agent.[1][needs update] Clinical investigation will test the safety and efficacy of HBI-3000 as a treatment for both atrial and ventricular arrhythmias.[2]
The molecular problem
Anti-arrhythmic medication is taken to treat irregular beating of the heart. This irregular beating results from a deregulation of the initiation or propagation of the electrical stimulus of the heart. The most common chronic arrhythmia is atrial fibrillation.[3] There is an increased incidence of atrial fibrillation in the elderly and some examples of complications include heart failure exacerbation, hypotension and thrombembolic events.[3]
Most anti-arrhythmic medications exert their effects by decreasing the permeability of potassium ion channels (IKr) in heart cells. These potassium channel blockers delay ventricular repolarization and prolong action potential duration (APD; the prolongation of the electrical stimulus within heart cells). These changes can lower heart rate, eliminate atrial fibrillation, and ultimately sudden cardiac death.[4][5]
Mechanism of action in ventricular myocytes
Ventricular myocytes are heart muscle cells found in the lower chambers of the heart. Heart rate is dependent on the movement of an electrical stimulus through the individual heart cells. This is mediated by the opening of ion channels on cell surfaces. HBI-3000 exerts its effects on the heart by inhibiting multiple ion channels (INa-F, INa-L, ICa-L and IKr), but predominantly the INa-L ion channel . By decreasing the ion permeability of these channels, HBI-3000 slightly prolongs APD (due to IKr); however, unlike pure IKr channel blockers, it is self-limited (due to the decreased permeability of INa-L and ICa-L). This is similar to the medications ranolazine and amiodarone.[5] HBI-3000 suppresses early afterdepolarizations (EADs; a change in the normal net flow of ions during repolarization), does not produce any electrical abnormalities, and displays minimally pronounced prolongation of APD during a slow heart rate (i.e. stimulated at a slower frequency). Pronounced prolongation of APD during a slow heart rate can lead to proarrythmias. Overall, HBI-3000 seems to have a low proarrhythmic risk. The effect of HBI-3000 on contractility and cardiac conduction requires further investigation.[5]
Studies
Animal model
In a canine model, the intravenous injection of HBI-3000 demonstrated to be an effective anti-arrhythmic and anti-fribrillatory agent.[6]
Cellular isolation
The administration of HBI-3000 to isolated heart muscle cells demonstrated the potential to improve arrhythmias while having low proarrhythmic risk.[5]
Human studies
Jiangsu Furui Pharmaceuticals Co., Ltd is currently recruiting participants in their study.[1][
PAPER
http://www.simm.cas.cn/wyp/wyp_lw/201804/W020180420480084769998.pdf

N-[3,5-bis(1-pyrrolidylmethyl)-4-hydroxybenzyl]-4-methoxybenzenesulfamide (sulcardine, 6f) and the sulfate (sulcardine sulfate) (1) To a suspension of 4-hydroxybenzylamine (133 g, 1.08 mol) in DMF (500 mL) was added dropwise 4-methoxybenzensul-fonyl chloride (206 g, 1.00 mol) in DMF (320 mL) over a period of 30 min at 0–10 °C with stirring, followed by the addition of triethylamine (158 mL, 1.12 mol) over 30 min at the same temperature. The stirring was continued for an additional 1.5 h at room temperature. The reaction mixture was poured into ice-water (5 L). After stirring for 10 min, the suspension was allowed to stand for 2 h. The solid was filtered, washed with water (300 mL×3), and dried in a desiccator over anhydrous calcium chloride, yielding N-(4-hydroxybenzyl)-4-methoxybenzenesulfamide (11) (248 g, 85%) as a white solid, mp 160–162 °C. The authentic sample was obtained by recrystallization from ethyl acetate, mp 161–162 °C. 1 H NMR (CD3OD) δ 3.70 (s, 3H), 3.76 (s, 2H), 6.48 (d, J=8.4 Hz, 2H), 6.82(d, J=8.4 Hz, 2H), 6.86 (d, J=8.7 Hz, 2H), 7.56 (d, J=8.7 Hz, 2H). EIMS (m/z): 293 (M+ ), 254, 195, 185, 171, 155, 149, 122 (100), 107, 99, 77, 65. Anal. (C14H15NO4S) C, H, N.
(2) A mixture of 11 (230 g, 0.78 mmol), pyrrolidine (200 mL, 2.44 mol) and 36% aqueous formaldehyde (250 mL, 3.30 mol) in ethanol (800 mL) was stirred under reflux for 8 h. The reaction mixture was concentrated under vacuum to dryness. The resulting oil residue was dissolved in chloroform (350 mL), and the solution was washed with water (300 mL×3). Under stirring, the organic layer was mixed with water (300 mL), and then concentrated hydrochloric acid (approximately 165 mL) was added portionwise at 0-10 °C to adjust the pH of the aqueous phase to ~2. The aqueous phase was washed with chloroform (200 mL) and then mixed with additional chloroform (300 mL). Under stirring, the two-phase mixture was treated portionwise with 25%–28% aqueous ammonia (~300 mL) to adjust the pH of the aqueous phase to 9–10. The organic layer was separated, and the aqueous layer was further extracted with chloroform (200 mL×2). The combined organic layer was dried over anhydrous sodium sulfate and concentrated under vacuum to dryness. The oily residue was treated with acetone (45 mL) and isopropyl ether (290 mL), and the mixture was heated under reflux until the suspension became a solution. The solution was cooled to room temperature, seeded with an authentic sample, and allowed to stand at 0°C overnight. The solid was filtered and dried under vacuum, yielding product 6f (290 g, 81%) as a yellowish solid, mp 96–98 °C. The authentic sample was obtained by preparative TLC or column chromatography (silica gel; CHCl3:MeOH:25% NH4OH=92:7:1). The compound could be recrystallized from ethanol-water, mp 101–102 °C. 1 H NMR (CDCl3) δ 1.77–1.86 (m, 8H), 2.53–2.63 (m, 8H), 3.68 (s, 4H), 3.86 (s, 3H), 3.97 (s, 2H), 6.86 (s, 2H), 6.95 (d, J=8.7 Hz, 2H), 7.78 (d, J=8.6 Hz 2H). EIMS (m/z): 459 (M+ ), 390, 388, 202, 171, 148, 107, 84, 70 (100). Anal. (C24H33N3O4S) C, H, N.
(3) Under stirring, the Mannich base 6f (150.5 g, 0.327 mol) was mixed with 2 mol/L H2SO4 (172 mL, 0.344 mol), and the mixture was heated at 80 °C until the solid dissolved. The solution was cooled to room temperature, seeded with an authentic sample, and the sulfate of 6f was formed as crystals. To the stirred mixture was added anhydrous ethanol (520 mL), and the mixture was allowed to stand at 0°C for 24 h. The solid was filtered, washed with ethanol, and recrystallized with 80% ethanol (250 mL). The sulfate was dried over concentrated sulfuric acid in a desiccator, giving the sulfate of 6f (143 g, 71%) as a trihydrate, mp 125–140°C. 1 H NMR (D2O) δ 2.00–2.13 (m, 4H), 2.14–2.25 (m, 4H), 3.12–3.22 (m, 4H), 3.45– 3.55 (m, 4H), 3.90 (s, 3H), 4.20 (s, 2H), 4.33 (s, 4H), 7.06 (d, J=8.7 Hz, 2H), 7.28 (s, 2H), 7.66 (d, J=8.9 Hz, 2H). 13C NMR (D2O) δ 24.7, 47.6, 55.7, 56.1, 58.1, 116.6, 122.5, 131.3, 132.3, 133.3, 136.0, 155.8, 164.8. EIMS (m/z): 459, 390, 388, 202, 171, 148, 107, 84, 70 (100). Anal. (C24H33N3O4S∙H2SO4∙3H2O) C, H, N, S.
PATENT
Preparation of sulcardine sulfate salt has been reported in U.S. Patent No. 6,605,635.
https://patents.google.com/patent/US6605635
Synthesis and antiarrhythmic activities of changrolin (1) have been reported (Liangquan Li, et al., Scientia Sinica, 1979, 7, 723; Weizhou Chen, et al., Acta Pharmaceutica Sinica, 1979, 14, 710). Thereafter, investigations of the chemical structural modifications and the physiological activities have successively been carried out by domestic and foreign scientists (Cunji Sun, et al., Acta Pharmaceutica Sinica, 1981, 16, 564; 1986, 21, 692; Mulan Lin, et al., ibid., 1982, 17, 212; D. M. Stout, et al. J. Med. Chem., 1983, 26, 808; 1984, 27, 1347; 1985, 28, 295; 1989, 32, 1910; R. J. Chorvat, et al., ibid., 1993, 36, 2494).

Changrolin is an effective antiarrhythmic agent. Ventricular premature beats disappear 2-3 days after oral administration of changrolin to patients suffering from arrhythmia; I.v. injection or instillaton may result in significant reduction or even disappearence of ventricular premature beats and ventricular tachycardia. However, oral administration of changrolin for a period of over one month may cause a reversible pigmentation on the skin of patients, which gradually retrogresses after ceasing the administration. This pigmentation is associated to the subcutaneous oxidation of certain structural moieties in changrolin molecule or to its instability in solution.
EXAMPLE 1N-[3,5-bis(1-Piperidinomethyl)-4-hydroxy]phenyl-1-naphthalenesulfonamide (B-87836)
(1) To a solution of 4-aminophenol (4.5 g) in dioxane (20 ml) was added dropwise a solution of 1-naphthalenesulfonyl chloride (4.4 g) in dioxane (20 ml). The mixture was further stirred at room temperatue for 4.5 hours and poured into water. The precipitate was collected by filtration, recrystallized from ethanol and decolored with activated carbon to give N-(ρ-hydroxyphenyl)-1-naphthalenesulfonamide (4.2 g), mp 195-196° C.
(2) A mixture of N-(ρ-hydroxyphenyl)-1-naphthalenesulfonamide (2.0 g), 37% aqueous formaldehyde (4.5 g) and piperidine (5.6 g) in ethanol (100 ml) was heated to reflux for 50 hours. The ethanol was removed by evaporation in vacuo and chloroform was added to the residue. The organic layer was washed with water then dried over anhydrous Na2SO4. Then the chloroform was removed in vacuo and the residue was triturated in water to give a solid, which was then recrystallized from ethanol to give the titled product (1.4 g), mp 197-198° C.
1HNMR(CDCl3): 1.30-1.50(m, 12H), 2.10-2.21(m, 8H), 3.28(s, 4H), 6.45(s, 2H), 7.24-8.04(m, 6H), 8.56(m, 1H). Elemental analysis (C28H35N3O3S ): Calcd. (%): C, 68.12; H, 7.15; N, 8.51. Found (%): C, 67.96; H, 7.16; N, 8.56.
PATENT
WO-2020159959
Novel crystalline forms of acid salts of sulcardine useful for treating arrhythmia and atrial fibrillation.
4-Methoxy-N-(3,5-bis-(l-pyrrolidinylmethyl)-4-hydroxybenzyl)benzene sulfonamide (or N-(4-hydroxy-3,5-bis(pyrrolidin-l-ylmethyl)benzyl)-4-methoxybenzenesulfonamide), also known as sulcardine, and its salts, such as sulcardine sulfate, constitute a group of compounds with potent anti -arrhythmic activity. Sulcardine is a multi-ion channel blocker that specifically inhibits iNa-Peak, iNa-Late, Ica,L, and Ixrwith similar in vitro potencies (and Ito and IKUT to a lesser degree) in human atrial cardiomyocytes and represents what may be the sole example of a substituted sulfonamide class of anti-arrhythmic. Sulcardine salts can be used as an intravenous injectable or as oral doses for the treatment of arrhythmias, including supraventricular tachyarrhythmia, premature ventricular contractions, ventricular tachycardia, ventricular fibrillation, and atrial fibrillation. See, e.g ., U.S. Patent Nos. 8,541,464 and 8,637,566. Preparation of sulcardine sulfate salt has been reported in U.S. Patent No. 6,605,635.
[0004] In addition, the evidence to date suggests that one advantage of sulcardine and its salts is that they lack significant pro-arrhythmic activity, as demonstrated in rigorous preclinical safety models, including a post-MI sudden-death conscious canine model and the validated rabbit ventricular wedge model. Additionally, it has been shown that they do not significantly increase defibrillation threshold, nor increase defibrillation failure risk in a post-MI canine model as was seen with flecainide. On the basis of these data, sulcardine and salts, with their very low apparent pro-arrhythmic potential, could potentially be used to treat acute and recurrent atrial fibrillation in the presence of organic heart disease, prolonged QR syndrome, and ventricular arrhythmias, including premature ventricular contractions (PVCs), ventricular tachycardia (VT), and ventricular fibrillation (VF), in either acute- or chronic-administration settings owing to their ability to be formulated into intravenous and oral dosing formulations.
Sulcardine has a chemical name of 4-methoxy-N-(3,5-bis-(l-pyrrolidinylmethyl)- 4-hydroxybenzyl)benzene sulfonamide (or N-(4-hydroxy-3,5-bis(pyrrolidin-l-ylmethyl)benzyl)-4-methoxybenzenesulfonamide), and has the following structure:
[0062] Sulcardine sulfate has the following structure:
[0063] Sulcardine sulfate can exist in a hydrated form. One such form is a trihydrate.
HPLC analysis was performed on a Dionex Ultimate 3000 instrument with the following parameters:
Column: Phenomenex Luna C18, 150×4.6mm, 5pm
Column Temperature: 30°C
Mobile Phase A: 0.2% Phosphoric Acid
Mobile Phase B: Methanol
Diluent: 50:50 MeOH:H20
Runtime: 12 minutes
Flow Rate: l.OmL/min
Injection Volume: 5pL
Detection: 237 nm
Gradient:
EXAMPLE 2 – PREPARATION OF FREE BASE AND SCREENING
[00348] Sulcardine sulfate trihydrate was dissolved in ethyl acetate (16 vol.) and saturated sodium bicarbonate solution (16 vol.). The biphasic solution was transferred to a separating funnel and the layers separated. The organic layer was dried over sodium sulfate and then the solvent was removed by rotary evaporation and the resulting oil dried under vacuum at ambient temperature for ca. 3 hr. FIG. 4 is an XRPD pattern of the resulted amorphous sulcardine free base. In all cases, the initial screening work detailed below was performed on 10 mg of sulcardine free base. All XRPD diffractograms were compared with sulcardine sulfate trihydrate, sulcardine free base and relevant counterions and found to be distinct.
Patent
WO2020123824
claiming treatment of atrial fibrillation (AF) by intravenously administering sulcardine sulfate .
PATENT
References
- ^ Jump up to:a b Jiangsu Furui Pharmaceuticals (November 5, 2010). “Efficacy and safety of sulcardine sulfate tablets in patients with premature ventricular contractions”. ClinicalTrials.gov. U.S. National Library of Medicine. Retrieved 2019-12-20.
- ^ “HUYA Bioscience Int’l announces clinical trial milestones in China for promising new anti-arrhythmic compound; Data supports desirable safety profile” (Press release). San Francisco, California: HUYA Bioscience International. Retrieved 2019-12-20.
- ^ Jump up to:a b Mashal, Abdallah; Katz, Amos; Shvartzman, Pesach (2011). “Atrial fibrillation: A primary care cross-sectional study”. Israel Medical Association Journal. 13 (11): 666–671. PMID 22279699.
- ^ Farkas, András; Leprán, István; Papp, Julius Gy. (1998). “Comparison of the antiarrhythmic and the proarrhythmic effect of almokalant in anaesthetised rabbits”. European Journal of Pharmacology. 346 (2–3): 245–253. doi:10.1016/S0014-2999(98)00067-3. PMID 9652366.
- ^ Jump up to:a b c d Guo, Donglin; Liu, Que; Liu, Tengxian; Elliott, Gary; Gingras, Mireille; Kowey, Peter R.; Yan, Gan-Xin (2011). “Electrophysiological properties of HBI-3000: A new antiarrhythmic agent with multiple-channel blocking properties in human ventricular myocytes”. Journal of Cardiovascular Pharmacology. 57 (1): 79–85. doi:10.1097/FJC.0b013e3181ffe8b3. PMID 20980921.
- ^ Lee, Julia Y.; Gingras, Mireille; Lucchesi, Benedict R. (2010). “HBI-3000 prevents sudden cardiac death in a conscious canine model”. Heart Rhythm. 7 (11): 1712. doi:10.1016/j.hrthm.2010.09.028.
 |
|
| Names | |
|---|---|
| IUPAC name
N-({4-Hydroxy-3,5-bis[(pyrrolidin-1-yl)methyl]phenyl}methyl)-4-methoxybenzene-1-sulfonamide
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChemSpider | |
|
PubChem CID
|
|
| UNII | |
| Properties | |
| C24H33N3O4S | |
| Molar mass | 459.61 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
| Infobox references | |
////////////////sulcardine sulfate, phase 2, china, HBI 3000, atrial fibrillation, B 87823,
COC1=CC=C(C=C1)S(=O)(=O)NCC2=CC(=C(C(=C2)CN3CCCC3)O)CN4CCCC4
Diquafosol
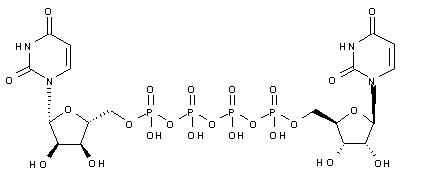
Diquafosol
- Molecular FormulaC18H26N4O23P4
- Average mass790.307 Da
{Oxybis[(hydroxyphosphoryl)oxy]}bis[hydrogéno(phosphonate)] de bis{[(2R,3S,4R,5R)-5-(2,4-dioxo-3,4-dihydro-1(2H)-pyrimidinyl)-3,4-dihydroxytétrahydro-2-furanyl]méthyle}
59985-21-6 [RN]
7828VC80FJ
8326
Bis{[(2R,3S,4R,5R)-5-(2,4-dioxo-3,4-dihydro-1(2H)-pyrimidinyl)-3,4-dihydroxytetrahydro-2-furanyl]methyl} {oxybis[(hydroxyphosphoryl)oxy]}bis[hydrogen (phosphonate)]
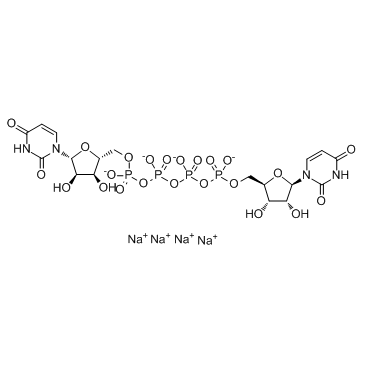
Diquafosol tetrasodium
- Molecular FormulaC18H22N4Na4O23P4
- Average mass878.234 Da
1) uridine, 5′-(pentahydrogen tetraphosphate), P”’->5′-ester with uridine, tetrasodium salt
211427-08-6[RN]
Diquafosol tetrasodium[USAN]
uridine(5′)tetraphospho(5′)uridine tetrasodium salt
Diquafosol tetrasodium (USAN)
INS365
P1,P4-Diuridine 5′-tetraphosphate tetrasodium salt
Prolacria
U2P4
UNII:X8T9SBH9LL
INS-365; DE-089; KPY-998
Title: Diquafosol
CAS Registry Number: 59985-21-6
CAS Name: Uridine 5¢-(pentahydrogen tetraphosphate) P¢¢¢®5¢-ester with uridine
Additional Names:P1,P4-diuridine 5¢-tetraphosphate; UP4U
Molecular Formula: C18H26N4O23P4
Molecular Weight: 790.31
Percent Composition: C 27.36%, H 3.32%, N 7.09%, O 46.56%, P 15.68%
Literature References: Uridine nucleotide analog. P2Y2 purinoceptor agonist; stimulates mucin secretion from goblet cells. Prepn: M. J. Stutts, III et al.,WO9640059 (1996 to Univ. North Carolina at Chapel Hill); and receptor activity: W. Pendergast et al.,Bioorg. Med. Chem. Lett.11, 157 (2001). Ocular pharmacology: T. Fujihara et al.,J. Ocul. Pharmacol. Ther.18, 363 (2002). Review of development and therapeutic potential: J. Fischbarg, Curr. Opin. Invest. Drugs4, 1377-1383 (2003); K. K. Nichols et al.,Expert Opin. Invest. Drugs13, 47-54 (2004). Clinical trial in dry eye disease: J. Tauber et al., Cornea23, 784 (2004).
Derivative Type: Tetrasodium salt
CAS Registry Number: 211427-08-6
Manufacturers’ Codes: INS-365
Molecular Formula: C18H22N4Na4O23P4
Molecular Weight: 878.23
Percent Composition: C 24.62%, H 2.52%, N 6.38%, Na 10.47%, O 41.90%, P 14.11%
Therap-Cat: In treatment of dry eye disease.
Keywords: Purinoceptor P2Y Agonist.
- Company:
- Santen (Originator)
- Sales:
- $80 Million (Y2015);

$71.7 Million (Y2014);
$79.3 Million (Y2013);
$67.1 Million (Y2012);
$36 Million (Y2011); - ATC Code:
- S01
Approved Countries or Area 2010-04-16, JAPAN
Diquafosol tetrasodium was approved by Pharmaceuticals Medical Devices Agency of Japan (PMDA) on April 16, 2010. It was developed and marketed as Diquas® by Santen Pharmaceutical Corporation in Japan.
Diquafosol tetrasodium is a P2Y2 purinoceptor receptor agonist. It is indicated for improve dry eye symptoms by promoting secretion of mucin and water, thereby bringing the tear film closer to a normal state. No serious ocular or systemic adverse drug reactions were found during the clinical trials. Dry eye begins with symptoms of ocular discomfort such as burning, stinging or a foreign body sensation.
Diquas® is available as solution for ophthalmic use, containing 3% of Diquafosol tetrasodium. The recommended dose is 1 drop at a time, 6 times a day.
Index:
Diquafosol (tradename Diquas) is a pharmaceutical drug for the treatment of dry eye disease. It was approved for use in Japan in 2010.[1] It is formulated as a 3% ophthalmic solution of the tetrasodium salt.
Its mechanism of action involves agonism of the P2Y2 purinogenic receptor.[2]
SYN
INS-365 can also been obtained by the following ways: 4) Dimerization of uridine-5′-monophosphate tributyl-ammonium salt (I) with bis(tributylammonium) pyrophosphate (II) by means of CDI, followed by purification by semipreparative ion璭xchange chromatography. 5) Dimerization of uridine-5′-monophosphate tributyl-ammonium salt (I) with pyrophosphoryl chloride (III) in pyridine, followed by chromatographic purification as before. 6) Condensation of uridine (IV) with POCl3 and bis(tributylammonium) pyrophosphate (II) by means of tributylamine in trimethyl phosphate, followed by chromatographic purification as before. 7) Dimerization of uridine-5′-diphosphate tributylammonium salt (V) by means of CDI in DMF, followed by purification over Dowex 50Wx4 Na+. 8) Condensation of uridine-5′-triphosphate tributylammonium salt (VI) with uridine-5′-monophosphate tributyl-ammonium salt (I) by means of DCC in DMF, followed by chromatographic purification as before. 9) Reaction of uridine-5′-monophosphate tributylammonium salt (I) with CDI in DMF, followed by condensation with uridine-5′-triphosphate (VI) and chromatographic purification as before.
CLIP
Route 1


Reference:1. WO9905155A2.
Route 2


Reference:1. WO1999005155.
Route 3


Reference:1. WO1999005155.
Route 4


Reference:1. WO2014103704.
SYN
Practical and Efficient Approach to the Preparation of Diquafosol Tetrasodium
-
- Pengfei Xu
Publication Date:June 30, 2020

https://pubs.acs.org/doi/suppl/10.1021/acs.oprd.0c00209/suppl_file/op0c00209_si_001.pdf
https://pubs.acs.org/doi/10.1021/acs.oprd.0c00209
A scalable and practical route to synthesize the P2Y2 receptor agonist diquafosol tetrasodium has been described. Diquafosol tetrasodium was obtained via a four-step process starting from commercially available 5′-uridylic acid disodium salt. The whole procedure gives the target product in a 45% overall yield with high purity (>99%). Key steps in this process including isolation of impurities and the target product by using anion-exchange resin are discussed in detail. The optimized process has been successfully demonstrated on a large scale to support the development of diquafosol tetrasodium in China.




References
- ^ “Santen and Inspire Announce Approval of DIQUAS for Dry Eye Treatment in Japan”. April 16, 2010.
- ^ Pendergast, W; Yerxa, BR; Douglass Jg, 3rd; Shaver, SR; Dougherty, RW; Redick, CC; Sims, IF; Rideout, JL (2001). “Synthesis and P2Y receptor activity of a series of uridine dinucleoside 5′-polyphosphates”. Bioorganic & Medicinal Chemistry Letters. 11 (2): 157–60. doi:10.1016/S0960-894X(00)00612-0. PMID 11206448.
| Names | |
|---|---|
| IUPAC name
[[[[(2R,3S,4R,5R)-5-(2,4-Dioxopyrimidin-1-yl)-3,4-dihydroxy-tetrahydrofuran-2-yl]methoxy-hydroxy-phosphoryl]oxy-hydroxy-phosphoryl]oxy-hydroxy-phosphoryl] [(2R,3S,4R,5R)-5-(2,4-dioxopyrimidin-1-yl)-3,4-dihydroxy-tetrahydrofuran-2-yl]methyl hydrogen phosphate
|
|
| Other names
P1,P4-Bis(5′-uridyl) tetraphosphate; INS-365; Diquafosol tetrasodium
|
|
| Identifiers | |
|
|
|
3D model (JSmol)
|
|
| ChEMBL | |
| ChemSpider | |
|
PubChem CID
|
|
| UNII | |
|
CompTox Dashboard (EPA)
|
|
| Properties | |
| C18H26N4O23P4 | |
| Molar mass | 790.306 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
| Infobox references | |
///////////// INS 365, Diquafosol, INS-365, DE 089, KPY 998, JAPAN 16
| 59985-21-6 (Diquafosol ); 211427-08-6 (Diquafosol Tetrasodium); |
