
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 342)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
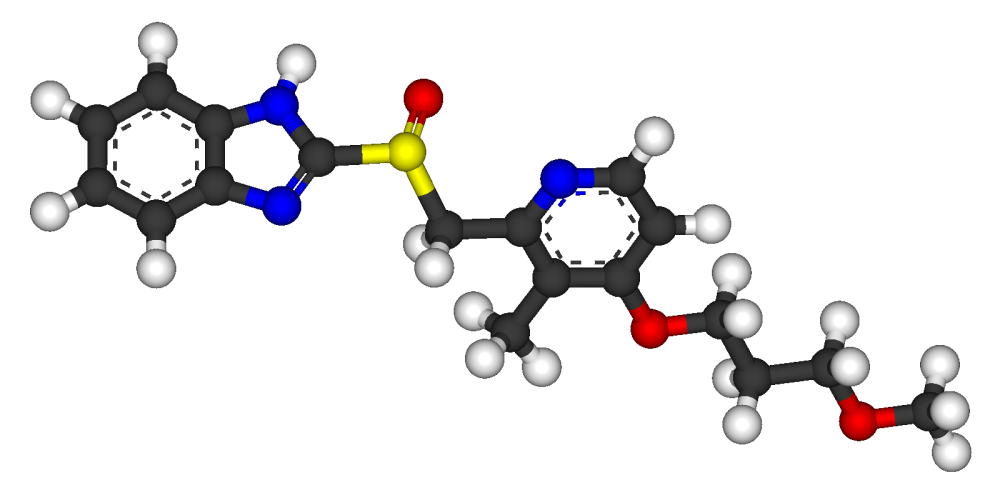
RABEPRAZOLE

Pariprazole sodium;Rabeprazole sodium;LY-307640;E-3810;Aciphex;Pariet
Rabeprazole /ˌræ.ˈbɛp.ræ.zɔːl/ is an antiulcer drug in the class of proton pump inhibitors. It was developed by Eisai Co. and is marketed by Janssen-Cilag as the sodium salt under the brand names AcipHex (/ˈæsɨfɛks/, referring to pH) in the US, Pariet in Europe, Brazil, Canada, Japan, Russia and Australia, Acigard, Cyra, Rabium, Esoon,Orporo, Parit, Rabemac, Rabiloz, Razo, Rabifast, Rablet and Rabsiv in India, and Zechin in Pakistan.
Rabeprazole, 2-[[[4-(3-Methoxypropoxy)-3-methyl-2-pyridinyl]methyl]sulfinyl]-1H-benzimidazole has the following structural formula

Rabeprazole belongs to a class of antisecretory compounds (substituted benzimidazole proton-pump inhibitors) that do not exhibit anticholinergic or histamine H2-receptor antagonist properties, but suppress gastric acid secretion by inhibiting the gastric H+, K+ATPase at the secretory surface of the gastric parietal cell. Because this enzyme is regarded as the acid (proton) pump within the parietal cell, rabeprazole has been characterized as a gastric proton-pump inhibitor. Rabeprazole blocks the final step of gastric acid secretion. So that it can effectively inhibit the secretion of an acid and is therefore effective in the therapy or prevention of human and animal peptic ulcer.
-
US 5045552 discloses the preparation of Rabeprazole sodium by known traditional procedures, such as dissolution of the product in a mixture of stoichiometric quantity of aqueous sodium hydroxide and ethanol, then removal of water azeotropically, thereafter drying the residue at low pressure and then crystallization of the residue with less polar solvent such as diethyl ether, tert-butyl methyl ether.
The U.S. Pat. No. 5,045,552 discloses the Rabeprazole and many other substituted benzimidazole-type compounds having anti-ulcer activity. This patent further discloses the process for preparation of Rabeprazole by oxidation of Rabeprazole sulfide using 85% m-chloroperbenzoic acid in a mixture of dichloromethane and diethyl ether followed by work up to get product as oil. The obtained oil is crystallized from a mixture of dichloromethane/ether. Optionally the oily crude is dissolved in aqueous solution of sodium hydroxide. The obtained solution is subjected to azeotropic distillation with ethanol to remove water and adding ether to get crystalline Rabeprazole base.

According to the prior art, Rabeprazole base is crystallized using dichloromethane/ether to obtain crystalline off white product. The HPLC purity is less than or equal to 99% and the isolation procedure involves azeotropic distillation of water, during which the product is exposed to high temperature and leads to certain impurities. Repeated crystallization is needed to remove impurities to get desired quality. Using large volumes of chlorinated solvents in the plant leads to environmental hazardous.
Japanese patent application JP2001039975 teaches that the product obtained by example 33 of U.S. Pat. No. 5,045,552 with a melting range of 140-141° C. corresponds to amorphous rabeprazole sodium
The U.S. Pat. No. 6,919,459 patent also discloses the process for the preparation of Rabeprazole by oxidation of Rabeprazole sulfide using m-Chloroperbenzoic acid (m-CPBA) in a suitable solvent. The reaction mass is subjected to repeated washings at different pH levels and isolate the product from aqueous layer.

Rabeprazole is not stable at acidic conditions and decomposes to form unknown impurities. To remove these impurities repeated crystallizations are required to get desire quality of the final product.
The WO2006/117802 PCT application discloses the process for the preparation of Rabeprazole sodium by oxidation of Rabeprazole sulfide with sodium hypo halite solution in water or a mixture of water and water miscible solvent medium using alkali metal hydroxide and catalyst. The reaction mass is saturated by inorganic saturating agents and the Rabeprazole sodium salt is extracted with water immiscible organic solvent. Organic solvent is distilled and the residue is dissolved in second organic solvent to get clear solution, which is precipitated by adding antisolvent.
The WO2006/120701 PCT application discloses process for manufacture of amorphous Rabeprazole sodium by the reaction of Rabeprazole base with aqueous sodium hydroxide. Ethanol is added to the obtained solution. Solvents are distilled from the solution to get thick mass. Organic solvent is added to the obtained residue to get clear solution, to which antisolvent is added to get amorphous Rabeprazole sodium.
The prior art methods cited above have many disadvantages, these methods involve more number of organic solvents and lack successive extractions and washings of the layers during work up procedure. It leads to many impurities that ultimately affect on purity and yield loss of final product.
The U.S. Pat. No. 6,180,652 and WO 2003101452 PCT application discloses the process for the preparation of amorphous rabeprazole sodium, which is obtained by lyophilization of an aqueous solution of rabeprazole sodium acetone complex and an aqueous NaOH solution of Rabeprazole respectively.

Lyophilization technique is not suitable for production at industrial scale and it needs more time cycle and involves the cost.
We observed that rabeprazole is rapidly degraded in chlorinated solvent like dichloromethane to form unknown impurities, due to impurities while distillation gummy material is formed. It leads to yellowish color in final product, finally it leads to yield loss in final product.
According to prior art methods,
-
- (a) Dichloromethane/ether is used for final crystallization gives off white product with HPLC purity less than or equal to 99% and
- (b) Rabeprazole sodium is isolated by using azeotropic distillation. It needs high temperature to remove water and the reaction mass is exposed to high temperature to form unknown impurities, to remove these impurities repeated crystallizations are required to get desire quality of the final product
US 6,313,303 discloses the preparation of sulfoxides by oxidizing thio ether with a peroxoborate salt in the presence of an acid anhydride or a metal catalyst; and the preparation of sulfoxides by oxidizing thio ether with an N- halosuccinimide, l,3-dihalo-5,5-dimethyl-hydantoin or dichloroisocyanuric acid salt in the presence of a base.
IN 192030 discloses the purification process of Rabeprazole, in which sulfone enriched Rabeprazole is treated with an amino alcohol e.g. ethanolamine in the presence of an organic solvent, further the reaction mixture washed with water to remove the sulfone impurities. US 7,439,367 (IN218648, 058/MUM/2003, 193/MUM/2003) discloses the preparation of Rabeprazole by oxidizing its corresponding sulfide compound, where aqueous hypohalite solution is used as an oxidizing agent. The said oxidation is carried out at a controlled temperature and pH. During said oxidation the pH of the reaction mixture is maintained in the range of 9 to 12. This process utilizes catalyst such as pyridine, di-isopropyl ethyl amine and N,N-dimethyl amino pyridine.
US 7,060,837 discloses the purification of lansoprazole using ammonia, ammonium hydroxide, diethylamine, triethylamine and methylamine in the presence of solvent. The said patent utilizes acid for the isolation of lanzoprazole in pure form.
US 2008/0161579 (IN190/MUM/2005) discloses a process for the preparation of Rabeprazole sodium comprising oxidation of Rabeprazole sulfide with sodium hypohalite in water or a mixture of water and water miscible solvent using alkali metal hydroxide and catalyst. It also discloses a process for the preparation of Rabeprazole sulfide.
WO 2008/045777 (1856/CHE/2006) discloses the preparation of
Rabeprazole by oxidizing the corresponding sulfide compound using about 0.8 to 1.25 equivalents of an oxidizing agent in the presence of less than or about 2.25 equivalents of a base where aqueous sodium hypohalite used as an oxidizing agent.
WO 2006/024890 discloses a process for the preparation of Rabeprazole in which the Rabeprazole obtained was treated with the triethylamine in hexane. The use of n-hexane in the final stage is not suitable for manufacturing point of view as it is difficult to remove residual hexane solvent. There are several disadvantages associated with such known processes; all the methods reported in these prior arts leads to the formation of many impurities which ultimately affects the purity of the final product.
US 5,045,552 patent discloses the preparation of Rabeprazole by oxidizing the Rabeprazole sulfide using m-chloroperbenzoic acid as shown in scheme-I. The crude Rabeprazole was dissolved in sodium hydroxide and the resulting solution was azeotropically distilled together with ethanol thrice to remove the water. Finally ether was added to get the crystals of Rabeprazole sodium
WO 03/101452 discloses a method for the preparation of Rabeprazole sodium comprising dissolving Rabeprazole base in aqueous sodium hydroxide and then subjecting to lyophilization.



Souda, S.; Ueda, N.; Miyazawa, S.; Tagami, K.; Nomoto, S.; Okita, M.; Shimomura, N.; Kaneko, T.; Fujimoto, M.; Murakami, M.; Oketani, K.; Fujisaki, H.; Shibata, H.; Wakabayashi, T. (Eisai Co., Ltd.); Pyridine derivs., pharmaceutical compsns. comprising the same, the use of the same for the manufacture of medicaments having therapeutic or preventative value, and a process for preparing the same. AU 8781138; EP 0268956; EP 0475456; EP 0654471; EP 0786461; JP 1989006270; JP 1993247035; JP 1995291967; US 5045552; US 5998445 .
Castaner, J.; Prous, J.; E-3810. Drugs Fut 1991, 16, 1, 19.
Sohda, S.; Tagami, K.; Chiku, S.; Synthesis of 14C-labelled sodium pariprazole (E3810). J Label Compd Radiopharm 1993, 33, 9, 849.

Rabeprazole as “CYRA” (Systopic Labs Pvt Ltd), “Elpizole” (Orchid Chemicals & Pharmaceuticals), Elpizole-20 (Orchid Chemicals & Pharmaceuticals), Rablet (Lupin), Acigard (3D), AcipHex, Rabeloc, Pariet, Rabider (Duta Formulations) Rabsiv 20 (Saharsh Biologicals) is supplied in:
- Tablet, enteric-coated; 10 mg
- Tablet, enteric-coated; 20 mg
- Pali-Schöll I, Jensen-Jarolim E (April 2011). “Anti-acid medication as a risk factor for food allergy”. Allergy 66 (4): 469–77. doi:10.1111/j.1398-9995.2010.02511.x. PMID 21121928.
HPLC METHOD
Rabeprazole with more impurities, particularly at 2.12 RRT (393 mass), 3.51 RRT (491 mass), 4.47 RRT (457 mass), 4.85 RRT (684 mass) and 4.54 RRT (893 mass). The mass (molecular or formula weight) number of the impurities were identified using LCMS. Particularly, the obtained product contains unknown impurities of higher molecular weight in the range of 0.1-1.0 % at relative retention time (RRT) of 2.12, 3.51, 4.47, 4.85, and 4.54 RRT as measured by high performance liquid chromatography (HPLC) method provided below.
The purity of the product obtained is determined by high performance liquid chromatography method under the conditions mentioned below.
Column: Prontosil Kromabond 100-5-C18 (250 x 4.6 mm), 5μ,
Mobile phase A: 1.36g KH2PO4 to 1 litre water, 0.5ml OfEt3N, Mobile phase B: Methanol: ACN (95:5),
Diluent: Mobile phase A and ACN (70:30),
Flow Rate: 1.0 mL/min,
Detection: UV at 280 nm,
Injection Volume: 20 μL, Run Time: 60 min.
Column oven temperature: 3O0C. Surprisingly the applicant identified a method in which, crude Rabeprazole was treated with diethylamine and optionally addition of TBAB (tetrabutylammmonium bromide) as catalyst, where the impurity level reduced. Though the reported amines like triethyl amine, ethanolamine, and ammonia are effectively used to minimize sulfone impurity, those are failed or unsatisfactory to remove the impurities at 2.12 RRT, 3.51 RRT, 4.47 RRT, 4.85 RRT and 4.54 RRT.
SPECTRAL DATA
EP 1869015 B1 FOR RABEPRAZOLE SODIUM
IR Spectra (KBr, cm-1): 3382, 2927, 1583, 1462, 1384, 1298, 1269, 1190, 1157, 1093, 1018, 745.
H NMR Spectra [200 M Hz, CD3OD] δ (ppm): 8.23 – 8.25 (1H, d, ArH); 7.57 – 7.62 (2H, m, ArH); 7.0 – 7.09 (2H, m, ArH); 6.87 – 6.90 (1H, d, ArH); 4.57 – 4.63 (2H, d, O=S-CH2-Ar); 4.0 – 4.1 (2H, t, -O-CH2-CH2-); 3.49 – 3.55 (2H, t, -CH2-O-CH3); 3.31 (3H, s, -OCH3); 2.1 (3H, s, Ar-CH3); 1.96 – 2.0 (2H, t, -CH2-CH2-CH2-).
MP
As per the process described and exemplified in the U. S. Patent No.
5,045,552, rabeprazole sodium is prepared by oxidizing 2-[[4-(3- methoxyporpoxy)-3-methylpyridine-2-yl]rnethylthio]-1 H-benzimidazole with m- chloroperbenzoic acid to afford the rabeprazole base which is further converted to its sodium salt by using 0.1 N aqueous solution of sodium hydroxide, followed by addition of ethanol. The water is removed by azeotropic distillation and the product is precipitated by using ether as solvent such as diethyl ether, tert-butyl methyl ether. The melting point of the disclosed rabeprazole sodium salt is 140- 1410C. The isolation process described in the U. S. Patent No. 5,045,552 has numerous disadvantages such as large volume of solvents is required for azeotropic removal of water during which the product is exposed to high temperature and leads to certain impurities. Based on these drawbacks the isolation process finds to be unsuitable for preparation of amorphous rabeprazole sodium at commercial scale operations.
Japanese patent application JP 2001039975 indicates that the product obtained by example 33 of the U. S. Patent No. 5,045,552 with a melting point of
140-1410C corresponds to amorphous rabeprazole sodium. In this application, the X-ray powder diffraction pattern of the amorphous rabeprazole sodium is shown.
The PCT patent publication No. WO 03/101452 discloses a method for the preparation of rabeprazole sodium comprising dissolving rabeprazole base in aqueous sodium hydroxide and then subjecting to lyophilization. U.S. Patent No. 6,180,652 B1 (the ‘652 patent) describes acetone complex of rabeprazole sodium, process for its production and characterizes it by powder X-ray diffraction, infra-red spectroscopy and 1H-NMR spectroscopy. The ‘652 patent further reports a process for preparation of amorphous rabeprazole sodium by lyophilizing (freeze-drying) an aqueous solution of rabeprazole sodium acetone complex.
However, lyophilization is a technique, which is not suitable for production at industrial scale because this process presents serious limitations on cost, time, equipment capability and environmental protection.
According to PCT patent publication No. WO 2004/085424A1 , amorphous rabeprazole sodium is obtained by heating the rabeprazole sodium acetone complex at elevated temperature, preferably between 100 and 1100C. It is well known that exposing rabeprazole-type compounds to high temperatures increases the risk of decomposition to form impurities and as such, heat treatment of rabeprazole sodium acetone complex into amorphous rabeprazole sodium is not adequate for the production of a rabeprazole which is suitable for pharmaceutical use.
PCT patent publication No. WO 2007/023393 A2 reports a process for preparation of amorphous rabeprazole sodium, the said process comprises: i) contacting rabeprazole sodium acetone complex with a first solvent system which includes a hydrocarbon solvent or an ether solvent or an alcohol solvent or mixtures thereof; ii) filtering the solid from the solvent system used in step i) or distilling the solvent system used in step i) under reduced or atmospheric pressure, to thereby obtain a residue; iii) contacting the wet solid or the residue of step ii) with a second solvent system which includes a hydrocarbon solvent or an ether solvent; and iv) filtering to obtain a wet solid from the solvent system used in step iii) to obtain a wet solid.
The methods for preparation of amorphous rabeprazole sodium as described in the patents U.S. Patent No. 6,180,652 B1 , PCT patent publication No. WO 2004/085424A1 and PCT patent publication No. WO 2007/023393 A2 involves lengthy process i.e., proceeds via rabeprazole sodium acetone complex intermediate and also the yields obtained in these processes are very low.
U.S. Patent Application No. US2004/0180935A1 teaches a process for production of amorphous rabeprazole sodium by dissolving rabeprazole acid in a mixture of sodium hydroxide and methanol at 25-350C, removing the solvent by evaporation and precipitating the product by adding petroleum ether.
PCT patent publication No. WO 2006/120701 A1 teaches a process for manufacture of amorphous rabeprazole sodium with mean particle diameter between 10 to 55 μm, the said process comprises, addition of rabeprazole to aqueous sodium hydroxide; addition of ethyl alcohol to the solution; distillation of solvents from the solution thus obtained till thick mass is obtained; addition of an organic solvent selected from ethyl acetate, dichloromethane, chloroform, butyl acetate, ethanol, isopropyl alcohol, methanol, tetrahydrofuran, to the residue to obtain a clear solution; addition of this clear solution to an anti-solvent includes diisopropyl ether, diethyl ether, methyl tert-butyl ether, under agitation and isolation of the product.
Since a solvent may play an important role in increasing the yield rate or in determination of physical properties of drug substance such as crystal form, purity, solubility, etc., even if such a solvent is known to be toxic, there may be many cases that the use thereof in the preparation of drug substance cannot be avoided in terms of risk benefits. In such cases, this guideline (ICH guidelines Q3C(R3)) decrees that a concentration of a residual solvent in drug substance should be not more than a specified value, which is toxicologically acceptable. The methods for preparation of amorphous rabeprazole sodium as described in the patents, U.S. Patent Application No. US2004/0180935A1 and PCT patent publication No. WO 2006/120701 A1 suffers with residual solvent problem and thereby commercially not viable. These methods utilize the solvents like diisopropyl ether and petroleum ether as precipitating solvents. These solvents are difficult to remove completely by practical manufacturing techniques. According to the ICH guidelines Q3C(R3), there is no adequate toxicological data for the solvents like diisopropyl ether and petroleum ether on which to base a PDE was found. However, a need still remains for an improved and commercially viable process of preparing pure amorphous rabeprazole sodium that would solve the aforesaid problems associated with processes described in the prior art, which will be suitable for largr-scale preparation, in terms of simplicity, chemical yield and purity of the product, and which would carry out with comparatively smaller volume of solvent
KAPVAY, CLONIDINE HYDROCHLORIDE, Patent expiry…13 th oct 2013


CLONIDINE

C9H9Cl2N3•HCl Mol. Wt. 266.56
Clonidine hydrochloride is an imidazoline derivative and exists as a mesomeric compound. The chemical name is 2-(2,6-dichlorophenylamino)-2-imidazoline hydrochloride. The following is the structural formula:
Clonidine hydrochloride is an odorless, bitter, white, crystalline substance soluble in water and alcohol.
KAPVAY
SHIONOGI INC
Drug Patent Expiration and Exclusivity
Drug Information
| Active Ingredient | Form | Dosage | Drug Type | Application | Product | |
|---|---|---|---|---|---|---|
| CLONIDINE HYDROCHLORIDE | TABLET, EXTENDED RELEASE; ORAL | 0.1MG | RX | 022331 | 003 | |
| CLONIDINE HYDROCHLORIDE | TABLET, EXTENDED RELEASE; ORAL | 0.2MG | RX | 022331 | 004 |
Patents
There are 1 patent(s) protecting SHIONOGI INC’s KAPVAY.
The last patent expires on 2013-10-13.
| Patent | Expiration | |
|---|---|---|
| US5869100 | Extended release clonidine formulation (tablet)
A method of providing a patient needing clonidine with an extended dosage of clonidine over a prolonged period of time. Such method involves administering to the patient an oral dosage unit comprising a homogenous mixture of a therapeutically effective amount of clonidine, about 30 to about 70 percent by weight of one or more cellulose ethers such as hydroxypropyl methylcellulose, and about 30 to about 70 percent by weight of an inert substance such as cornstarch. The oral dosage unit may be contained in a gelatin capsule or in the form of a tablet.
|
2013-10-13(expired) |
Exclusivity
Exclusivity is marketing rights granted by the FDA to the SHIONOGI INC.
Exclusivity ends on 2013-09-28.

loteprednol etabonate…Patent expiry this week……of October 20, 2013
loteprednol etabonate
82034-46-6 cas
Drug Patent Expiry for the week of October 20, 2013
Tradename….LOTEMAX
Applicant………Pharmos
SUSPENSION/DROPS; OPHTHALMIC, o.5%
Generic Name………loteprednol etabonate
Patent No.US 5,540,930
http://www.google.co.in/patents/US5540930
The invention provides novel compositions of matter containing water-insoluble steroid drugs suitable for therapeutic use. The invention provides stable aqueous suspensions of water-insoluble steroid drugs of particle sizes of ≦15 μm which remain in such a state so as to allow for immediate suspension, when desired, even after extended periods of settling.
| Date | Supplement No. | Action | Documents |
|---|---|---|---|
| 1998-03-09 | 000 | Approval |
| Publication number | US5540930 A |
| Publication type | Grant |
| Application number | US 08/142,743 |
| Publication date | 30 Jul 1996 |
| Filing date | 25 Oct 1993 |
| Priority date | 25 Oct 1993 |
| Fee status | Paid |
| Also published as | CA2174550A1, CA2174550C, DE69430635D1, DE69430635T2, EP0730443A1, EP0730443A4, EP0730443B1, US5747061, WO1995011669A1, Less «8 More » |
| Publication number | 08142743, 142743, US 5540930 A, US 5540930A, US-A-5540930, US5540930 A, US5540930A |
| Inventors | Doron I. Friedman, Yaacov J. Guy |
| Original Assignee | Pharmos Corporation |
| Company | |||
|---|---|---|---|
| LOTEMAX NDA (020583) | BAUSCH AND LOMB | LOTEPREDNOL ETABONATE | |
| ALREX NDA (020803) | BAUSCH AND LOMB | LOTEPREDNOL ETABONATE | |
| LOTEMAX NDA (020841) | PHARMOS | LOTEPREDNOL ETABONATE…expired | |
| ZYLET NDA (050804) | BAUSCH AND LOMB | LOTEPREDNOL ETABONATE; TOBRAMYCIN | |
| LOTEMAX NDA (200738) | BAUSCH AND LOMB | LOTEPREDNOL ETABONATE | |
| LOTEMAX NDA (202872) | BAUSCH AND LOMB | LOTEPREDNOL ETABONATE |
Loteprednol (as the ester loteprednol etabonate) is a corticosteroid used in optometry and ophthalmology. Marketed by Bausch and Lomb as Lotemax in the U.S., ocular applications for this drug include the treatment of inflammation of the eye due to allergies (according to the prescription information sheet), as well as chronic forms of keratitis (e.g.: adenoviral and Thygeson’s keratitis), vernal keratoconjunctivitis, pingueculitis, and episcleritis. The drug has little or no effect on intraocular pressure.

Druzgala, P.; Hochhaus, G.; Bodor, N.; J. Steroid Biochem. Mol. Biol. 1991, 38, 149.
http://dx.doi.org/10.1016/0960-0760(91)90120-T
- Steward, R; et al. (November 1998). “Double-masked, placebo-controlled evaluation of loteprednol etabonate 0.5% for postoperative inflammation”. J Cataract Surg 24: 1480–1489.
- Pavesio, CE; Decory, HH (2008). “Treatment of ocular inflammatory conditions with loteprednol etabonate”. Br J Ophthalmol 92 (4): 455–459. doi:10.1136/bjo.2007.132621. PMID 18245274.
GSK and Genmab seek FDA approval for ofatumumab combination therapy for CLL first-line treatment
GlaxoSmithKline (GSK) and Genmab have submitted a supplemental Biologics License Application (sBLA) to the US Food and Drug Administration (FDA) seeking the use of Arzerra (ofatumumab) in combination with an alkylator-based therapy in patients with chronic lymphocytic leukaemia (CLL) who have not received prior treatment.
READ AT
GSK and Genmab seek FDA approval for ofatumumab combination therapy for CLL first-line treatment
Iroko Pharmaceuticals Receives FDA Approval for ZORVOLEX™
![]()
Philadelphia, Pennsylvania, October 18, 2013 – Iroko Pharmaceuticals, LLC, a global specialty pharmaceutical company dedicated to advancing the science of analgesia, today announced that the U.S. Food and Drug Administration (FDA) has approved ZORVOLEX™ (diclofenac) capsules, a nonsteroidal anti-inflammatory drug (NSAID), for the treatment of mild to moderate acute pain in adults[i]. ZORVOLEX was approved at dosage strengths that are 20 percent lower than currently available diclofenac products. FDA approval of ZORVOLEX was supported by data from a Phase 3 multi-center, randomized study in which patients treated with ZORVOLEX reported significant pain relief compared with patients receiving placebo
Antifungal drugs-Antibiotics
Antifungal drugs-Antibiotics
by Parasuraman S, Senior Lecturer at AIMST University, Malaysia on Oct 20, 2013
Health Canada approves Levemir® FlexTouch® prefilled insulin pen for the treatment of type 1 and type 2 diabetes

MISSISSAUGA, ON, Oct. 18, 2013 /CNW/ – Novo Nordisk today announced that Health Canada has approved Levemir® FlexTouch®, a disposable prefilled insulin pen containing Levemir® (insulin detemir). Levemir® FlexTouch® has been designed to improve ease of use for insulin administration and to help decrease barriers to good treatment adherence for Canadians living with type 1 and type 2 diabetes mellitus (diabetes).
The approval was also announced at the 2013 Vascular Conference in Montreal, Quebec.
Health Canada approves Levemir® FlexTouch® prefilled insulin pen for the treatment of type 1 and type 2 diabetes
http://www.pharmalive.com/health-canada-approves-levemir-flextouch
Actelion wins crucial FDA approval for next-gen lung disease drug Opsumit

MACITENTAN
N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N’-propylsulfamide, CAS NO 441798-33-0
Late on Friday the FDA came through with an approval for Actelion’s pulmonary arterial hypertension (PAH) drug Opsumit (macitentan), its next-gen successor to the franchise drug Tracleer.
Read more: Actelion wins crucial FDA approval for next-gen lung disease drug Opsumit – FierceBiotech http://www.fiercebiotech.com/story/actelion-wins-crucial-fda-approval-next-gen-lung-disease-drug-opsumit/2013-10-18#ixzz2i7tDhpZT
Subscribe at FierceBiotech
Macitentan (Opsumit® )is a novel dual endothelin receptor antagonist that resulted from a tailored drug discovery process. Macitentan has a number of potentially key beneficial characteristics – i.e., increased in vivo preclinical efficacy vs. existing ERAs resulting from sustained receptor binding and tissue penetration properties. A clinical pharmacology program indicated a low propensity of macitentan for drug-drug interactions.
Macitentan is an investigational drug being studied for the treatment of pulmonary arterial hypertension. It acts as a dualendothelin receptor antagonist and is being developed by Actelion.[1] A Phase III clinical trial was successfully completed in 2012.[2]
on 22 October 2012 – Actelion (SIX: ATLN) announced that it has submitted a New Drug Application (NDA) to the US Food and Drug Administration (FDA) seeking approval for macitentan (Opsumit®) for the treatment of patients with pulmonary arterial hypertension
Actelion’s experimental lung drug macitentan prolonged overall survival by more than a third according to detailed study data, which the company hopes will convince investors it has a viable follow-up product to secure its commercial future.
Europe’s largest standalone biotech company wants the drug, which treats pulmonary arterial hypertension — a disease that causes high blood pressure in the arteries of the lungs — to replace blockbuster Tracleer.
Tracleer currently makes up 87 percent of sales but loses patent protection in 2015 and has also seen its market share eroded by Gilead’s Letairis.
Pharmacokinetics
Macitentan has an active metabolite, ACT-132577, which is an oxidative depropylation product. Both macitentan and ACT-132577 are mainly excreted in form of hydrolysis products via urine (about 2/3 of all metabolites) and faeces (1/3).[3]
Co-administration of ciclosporin has only a slight effect on the concentrations of macitentan and its active metabolite, whilerifampicin decreases the area under the curve (AUC) of the drug’s blood plasma concentration by 79%, and ketoconazoleapproximately doubles it. This corresponds to the finding that macitentan is mainly metabolised via the liver enzyme CYP3A4.[4]

- ^ Bolli, M. H.; Boss, C.; Binkert, C.; Buchmann, S.; Bur, D.; Hess, P.; Iglarz, M.; Meyer, S.; Rein, J.; Rey, M.; Treiber, A.; Clozel, M.; Fischli, W.; Weller, T. (2012). “The Discovery of N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N′-propylsulfamide (Macitentan), an Orally Active, Potent Dual Endothelin Receptor Antagonist”. Journal of Medicinal Chemistry 55 (17): 7849–7861. doi:10.1021/jm3009103. PMID 22862294. edit
- ^ “Macitentan”. Actelion. Retrieved 22 August 2012.
- ^ Bruderer, S.; Hopfgartner, G. R.; Seiberling, M.; Wank, J.; Sidharta, P. N.; Treiber, A.; Dingemanse, J. (2012). “Absorption, distribution, metabolism, and excretion of macitentan, a dual endothelin receptor antagonist, in humans”. Xenobiotica 42 (9): 901–910.doi:10.3109/00498254.2012.664665. PMID 22458347. edit
- ^ Bruderer, S.; Äänismaa, P. I.; Homery, M. C.; Häusler, S.; Landskroner, K.; Sidharta, P. N.; Treiber, A.; Dingemanse, J. (2011).“Effect of Cyclosporine and Rifampin on the Pharmacokinetics of Macitentan, a Tissue-Targeting Dual Endothelin Receptor Antagonist”. The AAPS Journal 14 (1): 68–78. doi:10.1208/s12248-011-9316-3. PMC 3282010. PMID 22189899. edit
External links
Actelion Ltd
Actelion Ltd is a biopharmaceutical company with its corporate headquarters in Allschwil/Basel, Switzerland. Actelion’s first drug Tracleer®, an orally available dual endothelin receptor antagonist, has been approved as a therapy for pulmonary arterial hypertension. Actelion markets Tracleer through its own subsidiaries in key markets worldwide, including the United States (based in South San Francisco), the European Union, Japan, Canada, Australia and Switzerland. Actelion, founded in late 1997, is a leading player in innovative science related to the endothelium – the single layer of cells separating every blood vessel from the blood stream. Actelion’s over 2,400 employees focus on the discovery, development and marketing of innovative drugs for significant unmet medical needs. Actelion shares are traded on the SIX Swiss Exchange (ticker symbol: ATLN) as part of the Swiss blue-chip index SMI (Swiss Market Index SMI®).
