
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 323)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
RUXOLITINIB…FOR THE TREATMENT OF INT OR HIGH-RISK MYELOFIBROSIS


Ruxolitinib
(3R)-3-cyclopentyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propanenitrile, cas no 941678-49-5
(R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile
-
1H-Pyrazole-1-propanenitrile, beta-cyclopentyl-4-(7H-pyrrolo(2,3-d)pyrimidin-4-yl)-,(betaR)-
Formula: C17H18N6
Molecular Weight: 306.37

Phosphate salt
(R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile phosphate
INCB 018424
- INCB 018424
- INCB018424
- Ruxolitinib
- UNII-82S8X8XX8H
CAS No.: 1092939-17-7
M.Wt: 404.36
Formula: C17H21N6O4P
Ruxolitinib phosphate
LAUNCHED 2011, INCYTE FOR MYELOFIBROSIS, NDA202192, 2011-11-16
CLINICAL TRIALS.http://clinicaltrials.gov/search/intervention=INCB018424+OR+ruxolitinib
HPLC, MS, NMR…http://www.medkoo.com/Product-Data/Ruxolitinib/Ruxolitinib-QC-LC20130225.pdf
http://file.selleckchem.com/downloads/nmr/S137803-INCB018424-HNMR-Selleck.pdf
http://file.selleckchem.com/downloads/hplc/S137803-INCB018424-HPLC-Selleck.pdf
Ruxolitinib phosphate is a kinase inhibitor with the chemical name (R)-3-(4-(7H-pyrrolo[2,3d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile phosphate and a molecular weight of 404.36.
Ruxolitinib is a janus-associated kinase inhibitor indicated to treat bone marrow cancer, specifically intermediate or high-risk myelofibrosis. FDA approved on November 16, 2011.
INCB018424 is the first potent, selective, JAK1/2 inhibitor to enter the clinic with IC50 of 3.3 nM/2.8 nM, >130-fold selectivity for JAK1/2 versus JAK3
Ruxolitinib phosphate has the following structural formula:
|
Ruxolitinib phosphate is a white to off-white to light pink powder and is soluble in aqueous buffers across a pH range of 1 to 8.
Jakafi (ruxolitinib) Tablets are for oral administration. Each tablet contains ruxolitinib phosphate equivalent to 5 mg, 10 mg, 15 mg, 20 mg and 25 mg of ruxolitinib free base together with microcrystalline cellulose, lactose monohydrate, magnesium stearate, colloidal silicon dioxide, sodium starch glycolate, povidone and hydroxypropyl cellulose
.NCI: /Ruxolitinib phosphate/ The phosphate salt form of ruxolitinib, an orally bioavailable Janus-associated kinase (JAK) inhibitor with potential antineoplastic and immunomodulating activities. Ruxolitinib specifically binds to and inhibits protein tyrosine kinases JAK 1 and 2, which may lead to a reduction in inflammation and an inhibition of cellular proliferation. The JAK-STAT (signal transducer and activator of transcription) pathway plays a key role in the signaling of many cytokines and growth factors and is involved in cellular proliferation, growth, hematopoiesis, and the immune response; JAK kinases may be upregulated in inflammatory diseases, myeloproliferative disorders, and various malignancies. (NCI Thesaurus)
patent expiry
US pat 7598257 exp 24/12/27
US pat 8415362 exp 24/12/27
NCE.Nov 16, 2016
Discovered by Incyte, ruxolitinib phosphate is an inhibitor of Janus-associated kinase 2 (JAK2), a protein involved in signal transduction. This orally available compound was approved and launched in the U.S. in 2011 for the treatment of patients with intermediate or high-risk myelofibrosis (MF), including primary MF, post-polycythemia vera MP and post-essential thrombocythemia MF. A regulatory application in the E.U. was filed in 2011 and a positive opinion was assigned in April 2012. Final E.U. approval was obtained in August 2012. In November 2012, the product was launched in the U.K. for the treatment of disease-related splenomegaly or symptoms in primary myelofibrosis or myelofibrosis due to polycythemia vera or essential thrombocythemia. In 2012, the product has been filed for approval in Japan for the treatment of myelofibrosis.
Phase II clinical trials are also ongoing for the treatment of multiple myeloma, leukemia, pancreas cancer, thrombocytopenia and for the treatment of relapsed or refractory diffuse large B-cell or peripheral T-cell non-Hodgkin lymphoma following donor stem cell transplant. In Japan, the product is under development in phase III trials for the treatment of polycythemia vera and in phase II trials for the treatment of myelofibrosis. No recent development has been reported for clinical trials for the treatment of rheumatoid arthritis (RA), for the treatment of psoriasis or for the treatment of metastatic prostate cancer. Columbia University is evaluating the compound in preclinical studies for the treatment of alopecia areata. The University of Pennsylvania is conducting phase II clinical trials for the treatment of breast cancer.
In 2008 and 2009, the compound was assigned orphan drug designation in the U.S. and the E.U., respectively, for the treatment of myelofibrosis. This designation was assigned in Japan for this indication in 2011. Additional orphan drug designation was assigned by the FDA in 2010 for the treatment of polycythemia vera and for the treatment of essential thrombocythemia. In 2013, an orphan drug designation was assigned in U.S. for the treatment of pancreatic cancer. In 2009, fast track designation was assigned to ruxolitinib phosphate in the U.S .for the treatment of myelofibrosis.
Ruxolitinib (trade names Jakafi and Jakavi, by Incyte Pharmaceuticals and Novartis) is a drug for the treatment of intermediate or high-risk myelofibrosis, a type of bone marrow cancer.It is also being investigated for the treatment of other types of cancer (such as lymphomas and pancreatic cancer), for polycythemia vera, and for plaque psoriasis.
The phase III Controlled Myelofibrosis Study with Oral JAK Inhibitor-I (COMFORT-I) and COMFORT-II trials showed significant benefits by reducing spleen size, relieving debilitating symptoms, and improving overall survival.
INCYTE developed in cooperation with companies and NOVARTIS jak2 selective inhibitor Ruxolitinib(INCB-018424) – has been approved by the FDA successfully listed). (Safety and Efficacy of INCB018424, a JAK1 and JAK2 Inhibitor, in Myelofibrosis. Srdan Verstovsek, MD, Ph.D., Hagop Kantarjian, MD, Ruben A. Mesa. MD, et al. N Engl J Med 2010; 363: 1117-1127).
The presently disclosed a series of patent applications JAK inhibitors, including WO2001042246, WO200200066K WO2009054941 and WO2011013785, etc.

Mechanism of action
Ruxolitinib is a Janus kinase inhibitor with selectivity for subtypes 1 and 2 of this enzyme.
Side effects
Immunologic side effects have included herpes zoster (1.9%) and case reports of opportunistic infections.[10] Metabolic side effects have included weight gain (7.1%). Laboratory abnormalities have included alanine transaminase (ALT) abnormalities (25.2%), aspartate transaminase (AST) abnormalities (17.4%), and elevated cholesterol levels (16.8%).
Legal status
In November 2011, ruxolitinib was approved by the USFDA for the treatment of intermediate or high-risk myelofibrosis based on results of the COMFORT-I and COMFORT-II Trials.
Some analysts believe this to be a potential blockbuster drug.[3] As of the end of March 2012, and according to an Incyte spokesman, approximately 1000 physicians had prescribed the drug in the United States, out of a total 6500 hematologists and oncologists nationwide.

The US Food and Drug Administration had approved Incyte’s Jakafi (ruxolitinib) to treat patients with the bone marrow disease myelofibrosis (MF). Jakafi is the first and only drug granted license specifically for the treatment of the rare blood cancer.
Jakafi approved by FDA to treat rare bone marrow disease
Posted By Edward Su On November 17th, 2011
MF is a rare, potentially life-threatening blood cancer with limited treatment methods. Patients with the bone marrow disoder, characterized by bone marrow failure, enlarged spleen (splenomegaly), suffer from the symptoms of fatigue, night sweats and pruritus, poor quality of life, weight loss and shortened survival. The US drug firm Incyte estimates the disease affects about 16,000-18,500 people in the USA. Currently, the disease is treated with chemotherapy or bone marrow transplant.
Incyte’s Jakafi, the first drug to reach market from the Wilmington-based drug company, was approved by the FDA as a twice-a-day pill for the treatment of patients with intermediate or high-risk myelofibrosis (MF), including primary MF, post-polycythemia vera MF and post-essential thrombocythemia MF. The US regulators reviewed Jakafi under its priority review program for important new therapies.
The approval of Jakafi was based on the results from two clinical studies involved 528 patients with the disease. Patients in the Jakafi treatment arm experienced a significant reduction in the size of their spleen as well as a 50 percent decrease in symptoms, including pain, discomfort and night sweats.
Jakafi, generically known as ruxolitinib, works by blocking JAK1 and JAK2 enzymes associated with the disease. The company has co-developed the drug with Novartis as part of their collaboration signed in 2009. The Swiss drug firm has the rights to market Jakafi in other countries.
“The availability of Jakafi is a significant medical advancement for people living with myelofibrosis, a debilitating disease,” said Paul A. Friedman, M.D., President and Chief Executive Officer of Incyte. “This milestone marks a tremendous achievement for Incyte because a scientific discovery from our research laboratories has become the first JAK inhibitor to reach the market and provide a clinical benefit to patients.”
Richard Pazdur, director of the Office of Hematology and Oncology Drug Products in the FDA’s Center for Drug Evaluation and Research, said that Jakafi “represents another example of an increasing trend in oncology where a detailed scientific understanding of the mechanisms of a disease allows a drug to be directed toward specific molecular pathways”.
Incyte says Jakafi will be available next week, and the drug will cost $7,000 per month, or $84,000 for a year’s supply for insured patients. The company plans to provide Jakafi free to uninsured patients and will offer co-pay assistance to patients with financial need.
……………
NMR free base
http://www.google.com/patents/US8410265
For (R)-13 (free base): 1H NMR (DMSO-d6, 400 MHz) δ ppm 12.1 (bs, 1H), 8.80 (d, 1H, J=0.42 Hz), 8.67 (s, 1H), 8.37 (s, 1H), 7.59 (dd, 1H, J=2.34, 3.51 Hz), 6.98 (dd, 1H, J=1.40, 3.44 Hz), 4.53 (td, 1H, J=19.5, 4.63 Hz), 3.26 (dd, 1H, J=9.77, 17.2 Hz), 3.18 (dd, 1H, J=4.32, 17.3 Hz), 2.40 (m, 1H), 1.79 (m, 1H), 1.65 to 1.13 (m, 7H); C17H18N6 (MW, 306.37) LCMS (EI) m/e 307 (M++H).
http://www.google.com/patents/US8410265
phosphate
For (R)-14 (phosphate): mp. 197.6° C.; 1H NMR (DMSO-d6, 500 MHz) δ ppm 12.10 (s, 1H), 8.78 (s, 1H), 8.68 (s, 1H), 8.36 (s 1H), 7.58 (dd, 1H, J=1.9, 3.5 Hz), 6.97 (d, 1H, J=3.6 Hz), 4.52 (td, 1H, J=3.9, 9.7 Hz), 3.25 (dd, 1H, J=9.8, 17.2 Hz), 3.16 (dd, 1H, J=4.0, 17.0 Hz), 2.41, (m, 1H), 1.79 (m, 1H), 1.59 (m, 1H), 1.51 (m, 2H), 1.42 (m, 1H), 1.29 (m, 2H), 1.18 (m, 1H); 13C NMR (DMSO-d6, 125 MHz) δ ppm 152.1, 150.8, 149.8, 139.2, 131.0, 126.8, 120.4, 118.1, 112.8, 99.8, 62.5, 44.3, 29.1, 29.0, 24.9, 24.3, 22.5; C17H18N6(MW, 306.37 for free base) LCMS (EI) m/e 307 (M++H, base peak), 329.1 (M++Na).
………………….
SYNTHESIS
http://www.google.com/patents/US8410265



………………………
SYNTHESIS
(R)-Methyl 3-cyclopentyl-3-(4-(7-((2-(trimethylsilypethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)propanoate ((R)-22). A solution of (E)-methyl 3-cyclopentyl-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)acrylate (21, 815 mg) in tetrahydrofuran (THF, 8.0 mL) in a pressure glass tube was treated with the catalyst [Rh(COD)(−)-DuanPhos](BF4) (4.6 mg) under nitrogen before the reaction mixture was pressurized with hydrogen gas to 50 bar pressure. The reaction mixture was stirred at 35° C. under this hydrogen pressure for 22 h. When HPLC analysis showed that the substrate was almost completely consumed, the reaction mixture was cooled down to room temperature. The enantiomeric excess of the reaction mixture was determined to be 94.7% ee (97.35% of the second peak, (R)-22; 2.65% of the first peak, (S)-22) by chiral HPLC analysis. The reaction mixture was then filtered through a thin silica gel pad and the pad was washed with tetrahydrofuran (THF, 5 mL). The filtrate was then concentrated under reduced pressure to dryness. The resultant foamy solid (778 mg) was analyzed by chiral HPLC analysis and result showed a 94.7% of enantiomeric excess favoring the second peak (97.35% of the second peak, (R)-22; 2.65% of the first peak, (S)-22).

(3R)-3-Cyclopentyl-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)propanoic acid ((R)-23). To a stirred solution of (3R)-methyl 3-cyclopentyl-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)propanoate ((R)-22, 2.47 g, 5.26 mmol) in THF (30 mL) at room temperature was added a solution of lithium hydroxide monohydrate (LiOH—H2O, 265 mg, 6.31 mmol, 1.2 equiv) in water (15 mL). The reaction mixture was stirred at room temperature for 3 h. When LCMS showed the reaction was complete, the reaction mixture was then acidified with 1 N aqueous HCl solution to pH 5 before it was extracted with EtOAc (2×25 mL). The combined organic layers were washed with brine, dried over magnesium sulfate (MgSO4), filtered and concentrated under reduced pressure to afford (3R)-3-cyclopentyl-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)propanoic acid ((R)-23, 2.40 g, 2.40 g theoretical, 100% yield) as a colorless oil, which solidified upon standing at room temperature in vacuo. For (R)-23: 1H NMR (CDCl3, 300 MHz) δ ppm 8.95 (s, 1H), 8.95 (bs, 1H), 8.36 (s, 1H), 7.57 (d, 1H, J=3.7 Hz), 6.99 (d, 1H, J=3.7 Hz), 5.74 (s, 2H), 4.65 (dt, 1H, J=3.1, 10.3 Hz), 3.58 (t, 2H, J=8.2 Hz), 3.24 (dd, 1H, J=16.5, 10.3 Hz), 3.04 (dd, 1H, J=16.2, 3.1 Hz), 2.59 (m, 1H), 2.00 (m, 1H), 1.77-1.24 (m, 7H), 0.97 (t, 2H, J=8.2 Hz), 0.00 (s, 9H); C23H33N5O3Si (MW, 455.63), LCMS (EI) m/e 456.1 (M++H).
(3R)-3-Cyclopentyl-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)propanamide ((R)-24). To a stirred solution of (3R)-3-cyclopentyl-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)propanoic acid ((R)-23, 20 mg, 0.044 mmol) in DMF (1 mL) at room temperature was added N,N-carbonyldiimidazole (CDI, 21 mg, 0.13 mmol, 3.0 equiv). The reaction mixture was then stirred at room temperature and TLC was used to follow the reaction for formation of acyl imidazole (consumption of acid to a higher Rf spot with 30% EtOAc/hexane). When TLC showed that the acyl imidazole transformation was complete, ammonia gas was then bubbled through the stirred solution for 30 min to afford the amide (followed by LCMS). The excess amount of ammonia gas was evaporated by bubbling nitrogen vigorously through the solution. The crude product, (3R)-3-cyclopentyl-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)propanamide ((R)-24), in DMF was used directly to the following reaction to convert amide ((R)-24) into the corresponding nitrile ((R)-20).
(3R)-cyclopentyl-3-{4-[7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl]pyrazol-1-yl}propionitrile ((R)-20). Method A. To a stirred solution of (3R)-3-cyclopentyl-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)propanamide ((R)-24, 20 mg, 0.044 mmol) in DMF (1 mL) at 0° C. was added methylene chloride (1 mL) and triethylamine (0.12 mL, 0.88 mmol, 20.0 equiv), followed by trichloroacetyl chloride (0.052 ml, 0.462 mmol, 10.5 equiv). The resulting reaction mixture was stirred at 0° C. for 1 h. When LCMS showed the reaction was complete, the reaction mixture was quenched with saturated sodium bicarbonate solution (NaHCO3, 5 mL) before being extracted with EtOAc (2×10 mL). The combined organic layers were washed with brine, dried over magnesium sulfate (MgSO4), filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography with 0-75% EtOAc/hexane gradient elution to give (3R)-cyclopentyl-3-{4-[7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl]pyrazol-1-yl}propionitrile ((R)-20, 10 mg, 19 mg theoretical, 53% yield). For (R)-20: 1H NMR (DMSO-d6, 400 MHz) δ ppm 8.83 (s, 1H), 8.75 (s, 1H), 8.39 (s, 1H), 7.77 (d, 1H, J=3.7 Hz), 7.09 (d, 1H, J=3.7 Hz), 5.63 (s, 2H), 4.53 (td, 1H, J=19.4, 4.0 Hz), 3.51 (t, 2H, J=8.1 Hz), 3.23 (dq, 2H, J=9.3, 4.3 Hz), 2.41 (m, 1H), 1.79 (m, 1H), 1.66-1.13 (m, 7H), 0.81 (t, 2H, J=8.2 Hz), 0.124 (s, 9H); C23H32N6OSi (MW, 436.63), LCMS (EI) m/e 437 (M−+H) and 459 (M++Na).
(3R)-Cyclopentyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propionitrile ((R)-13, free base). Method B. To a solution of (3R)-cyclopentyl-3-{4-[7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl]pyrazol-1-yl}propionitrile ((R)-20, 463 g, 1.06 mol, 98.6% ee) in acetonitrile (4.5 L) was added water (400 mL) followed immediately by lithium tetrafluoroborate (LiBF4, 987.9 g, 10.5 mol, 10.0 equiv) at room temperature. The reaction temperature was observed to decrease from ambient to 12° C. upon addition of the water and then increase to 33° C. during the addition of lithium tetrafluoroborate (LiBF4). The resulting reaction mixture was heated to reflux (about 80° C.) for overnight. An aliquot was quenched into ethyl acetate/water and checked by LCMS and TLC (95:5 ethyl acetate/methanol, v/v). When LCMS and TLC analyses showed both the hydroxyl methyl intermediate ((R)-25) and fully de-protected material ((R)-13, free base) produced but no starting material ((R)-20) left, the reaction mixture was cooled gradually to <5° C. before a 20% aqueous solution of ammonium hydroxide (NH4OH, 450 mL) was added gradually to adjust the pH of the reaction mixture to 9 (checked with pH strips). The cold bath was removed and the reaction mixture was gradually warmed to room temperature and stirred at room temperature for overnight. An aliquot was quenched into ethyl acetate/water and checked by LCMS and TLC (95:5 ethyl acetate/methanol, v/v) to confirm complete de-protection. When LCMS and TLC showed the reaction was deemed complete, the reaction mixture was filtered and the solids were washed with acetonitrile (1 L). The combined filtrates were then concentrated under reduce pressure, and the residue was partitioned between ethyl acetate (EtOAc, 6 L) and half-saturated brine (3 L). The two layers were separated and the aqueous layer was extracted with ethyl acetate (2 L). The combined organic layers were washed with half-saturated sodium bicarbonate (NaHCO3, 3 L) and brine (3 L), dried over sodium sulfate (Na2SO4), and concentrated under reduced pressure to give the crude product as an orange oil. The crude material was then purified by flash column chromatography (SiO2, 40 to 100% ethyl acetate/heptane gradient elution) to afford (3R)-cyclopentyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propionitrile ((R)-13, free base, 273 g, 324.9 g theoretical, 84% yield) as a white foam. This material was checked by 19F NMR to ensure no lithium tetrafluoroborate (LiBF4) remained and by chiral HPLC (Chiralcel OD, 90:10 hexane/ethanol) to confirm enantiomeric purity and was used without further purification to prepare the corresponding phosphate salt. For (R)-13 (free base): 1H NMR (DMSO-d6, 400 MHz) δ ppm 12.1 (bs, 1H), 8.80 (d, 1H, J=0.42 Hz), 8.67 (s, 1H), 8.37 (s, 1H), 7.59 (dd, 1H, J=2.34, 3.51 Hz), 6.98 (dd, 1H, J=1.40, 3.44 Hz), 4.53 (td, 1H, J=19.5, 4.63 Hz), 3.26 (dd, 1H, J=9.77, 17.2 Hz), 3.18 (dd, 1H, J=4.32, 17.3 Hz), 2.40 (m, 1H), 1.79 (m, 1H), 1.65 to 1.13 (m, 7H); C17H18N6(MW, 306.37) LCMS (EI) m/e 307 (M++H).

3-Cyclopentylacrylonitrile (8). A solution of diethyl cyanomethylphosphonate (7, 742.5 g, 4.2 mol, 1.1 equiv) in dry THF (5.75 L) was stirred under nitrogen on an ice-water-methanol bath and a solution of 1 M potassium tert-butoxide in THF (4 L, 4.0 mol, 1.05 equiv) was added at such a rate as to keep the temperature below 0° C. After addition of 1 M potassium tert-butoxide in THF was complete, the stirring was continued on the cold bath for 1 h and a solution of cyclopentanecarbaldehyde (6, 374 g, 3.81 mol) in dry THF (290 mL) was added at such a rate as to maintain the temperature below 0° C. The cold bath was removed, and the reaction mixture was gradually warmed to room temperature and stirred at room temperature for overnight. When the reaction was deemed complete, the reaction mixture was partitioned between methyl tent-butyl ether (MTBE, 14 L), water (10 L) and brine (6 L). The two layers were separated, and the combined organic phase was washed with brine (6 L). The aqueous phase was extracted with MTBE (10 L) and washed with brine (6 L). The combined organic extracts were concentrated under reduced pressure and the residue was distilled (65-78° C./6 torr) to afford 3-cyclopentylacrylonitrile (8, 437.8 g, 461.7 g theoretical, 94.8% yield) as a colorless oil, which was found to be a mixture of E- and Z-isomer. For 8: 1H NMR (DMSO-d6, 400 MHz, for Z-isomer) δ ppm 6.58 (t, 1H, J=10.6 Hz), 5.55 (dd, 1H, J=10.8, 0.59 Hz), 2.85 (m, 1H), 1.9-1.46 (m, 6H), 1.34 (m, 2H) and (for E-isomer) δ ppm 6.83 (q, 1H, J=8.3 Hz), 5.66 (dd, 1H, J=16.5, 1.4 Hz), 2.60 (m, 1H), 1.9-1.46 (m, 6H), 1.34 (m, 2H); 13C NMR (DMSO-d6, 100 MHz, for Z-isomer) δ ppm 159.8, 116.6, 97.7, 42.3, 32.3, 25.1 and (for E-isomer) δ ppm 160.4, 118.1, 97.9, 43.2, 31.5, 24.8; C8H11N (MW, 121.18), GCMS (EI) m/e 120 (M+−H).
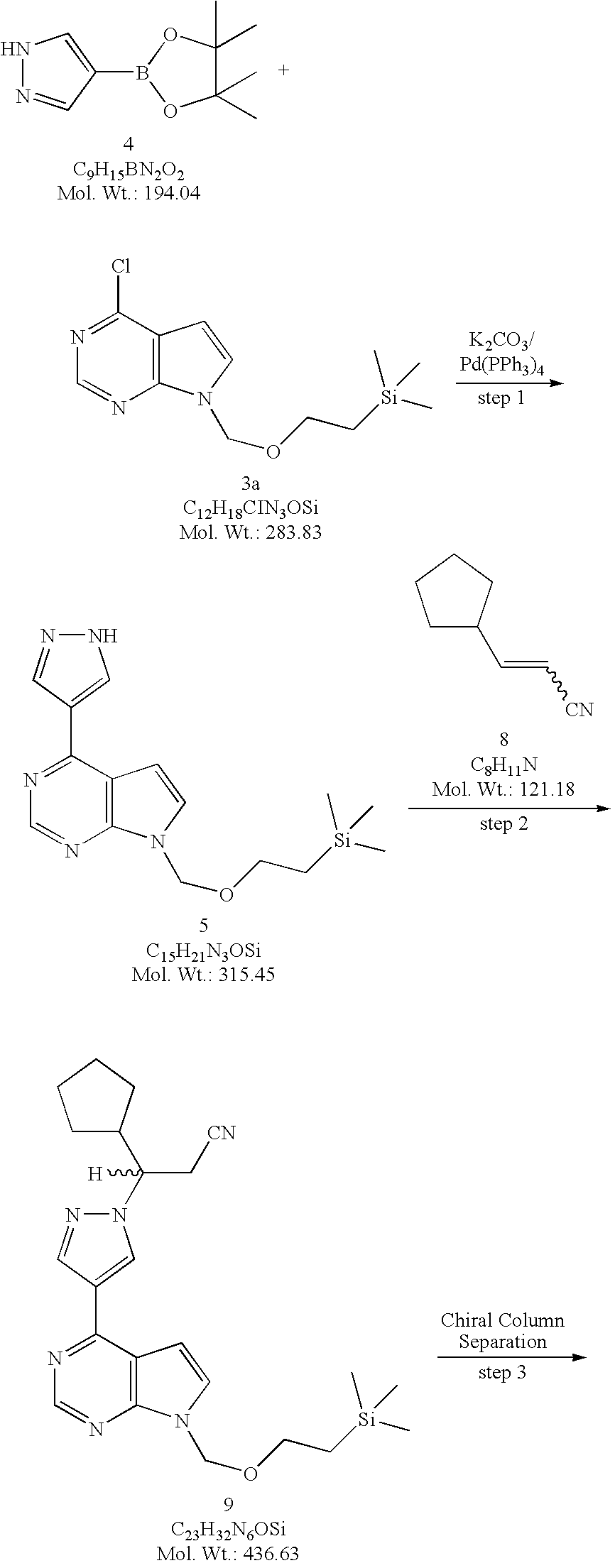

4-(1H-Pyrazol-4-yl)-7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidine (5). Method A. To a flask equipped with a reflux condenser, a nitrogen inlet, mechanical stirrer, and a thermowell was added 4-chloro-7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidine (3a, 817 g, 2.88 mol) and dioxane (8 L). To this solution was added 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (4, 728 g, 3.75 mol, 1.30 equiv) followed by a solution of potassium carbonate (K2CO3, 1196 g, 8.67 mol, 3.0 equiv) in water (4 L). The solution was degassed by passing a stream of nitrogen through the solution for 15 minutes before being treated with tetrakis(triphenylphosphine)palladium(0) (167 g, 0.145 mol, 0.05 equiv) and the resulting reaction mixture was heated at reflux (about 90° C.) for 2 hours. When the reaction was deemed complete by TLC (1:1 heptane/ethyl acetate) and LCMS, the reaction mixture was cooled to room temperature, diluted with ethyl acetate (24 L) and water (4 L). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (4 L). The combined organic layers were washed with water (2×2 L), brine (2 L), dried over sodium sulfate (Na2SO4), and concentrated under reduced pressure. The residue was suspended in toluene (4 L) and the solvent was removed under reduced pressure. The residue was finally triturated with methyl tert-butyl ether (MTBE, 3 L) and the solids were collected by filtration and washed with MTBE (1 L) to afford 4-(1H-pyrazol-4-yl)-7-(2-trimethylsilanyl-ethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidine (5, 581.4 g, 908.5 g theoretical, 64% yield) as white crystalline solids. For 5: 1H NMR (DMSO-d6, 400 MHz) δ ppm 13.41 (bs, 1H), 8.74 (s, 1H), 8.67 (bs, 1H), 8.35 (bs, 1H), 7.72 (d, 1H, J=3.7 Hz), 7.10 (d, 1H, J=3.7 Hz), 5.61 (s, 2H), 3.51 (t, 2H, J=8.2 Hz), 0.81 (t, 2H, J=8.2 Hz), 0.13 (s, 9H); C15H21N5OSi (MW, 315.45), LCMS (EI) m/e 316 (M++H).
Racemic 3-cyclopentyl-3-{4-[7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl]pyrazol-1-yl}propionitrile (9, racemic SEM-protected compound). Method A. 3-Cyclopentylacrylonitrile (8, 273.5 g, 2.257 mol, 1.20 equiv) and DBU (28 mL, 0.187 mol, 0.10 equiv) was added to a suspension of 4-(1H-pyrazol-4-yl)-7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidine (5, 591.8 g, 1.876 mol) in acetonitrile (4.7 L) at room temperature. The resulting reaction mixture was heated to 50-60° C. for 17 hours (a clear solution developed midway through heating) then to 70-80° C. for 8 hours. When LCMS analysis showed the reaction was deemed complete, the reaction mixture was cooled to room temperature. The cooled solution was then concentrated under reduced pressure to give the crude product (9) as a thick amber oil. The crude product was dissolved in dichloromethane (DCM) and absorbed onto silica gel then dry-loaded onto a silica column (3 Kg) packed in 33% EtOAc/heptanes. The column was eluted with 33% EtOAc/heptanes (21 L), 50% EtOAc/heptanes (28 L), 60% EtOAc/heptanes (12 L) and 75% EtOAc/heptanes (8 L). The fractions containing the desired product (9) were combined and concentrated under reduced pressure to generate a yellow oil, which was transferred to a 3 L flask with EtOAc. The solvent was removed under reduced pressure and the residual EtOAc by co-evaporating with heptanes. The residue was further dried under high vacuum for overnight to afford racemic 3-cyclopentyl-3-{4-[7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl]pyrazol-1-yl}propionitrile (9, racemic SEM-protected compound, 800 g, 819.1 g theoretical, 97.7% yield) as an extremely viscous yellow oil. For 9: 1H NMR (DMSO-d6, 400 MHz) δ ppm 8.83 (s, 1H), 8.75 (s, 1H), 8.39 (s, 1H), 7.77 (d, 1H, J=3.7 Hz), 7.09 (d, 1H, J=3.7 Hz), 5.63 (s, 2H), 4.53 (td, 1H, J=19.4, 4.0 Hz), 3.51 (t, 2H, J=8.1 Hz), 3.23 (dq, 2H, J=9.3, 4.3 Hz), 2.41 (m, 1H), 1.79 (m, 1H), 1.66-1.13 (m, 7H), 0.81 (t, 2H, J=8.2 Hz), 0.124 (s, 9H); C23H32N6OSi (MW, 436.63), LCMS (EI) m/e 437 (M++H) and 459 (M++Na).
(3R)-Cyclopentyl-3-{4-[7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo [2,3-d]pyrimidin-4-yl]pyrazol-1-yl}propionitrile ((R)-10) and (3S)-Cyclopentyl-3-{4-[7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl]pyrazol-1-yl}propionitrile ((S)-10) A slurry of 1.5 Kg of 20-micron Chiralcel® OD chiral stationary phase (CSP) made by Daicel in 3.0 L of isopropanol (IPA) was packed into a PROCHROM Dynamic Axial Compression Column LC110-1 (11 cm ID×25 cm L; Column Void Vol.: approximate 1.5 L) under 150 bar of packing pressure. The packed column was then installed on a Novasep Hipersep HPLC unit. The column and the Hipersep unit were flushed with methanol (17 L) followed by the mobile phase made of a mixture of isopropanol and hexane (2:8 by volume, 17 L). The feed solution was then prepared by dissolving 3-cyclopentyl-3-{4-[7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl]pyrazol-1-yl}propionitrile (9, racemic SEM-protected compound, 2795 g, 6.4 mol) in the mobile phase to a concentration of 80 g/L. The feed solution was then sequentially injected into the preparative chiral column for separation. Each injection was 120 ml in volume. The chiral column was eluted with the mobile phase at a flow rate of 570 mL/min at room temperature. The column elution was monitored by UV at a wavelength of 330 nm. Under these conditions a baseline separation of the two enantiomers was achieved. The retention times were 16.4 minutes (Peak 1, the undesired (S)-enantiomer (S)-10) and 21.0 minutes (Peak 2, the desired (R)-enantiomer (R)-10), respectively. The cycle time for each injection was 11 minutes and a total of 317 injections were performed for this separation process. Fractions for Peak 1 (the undesired (S)-enantiomer, (S)-10) and Peak 2 (the desired (R)-enantiomer, (R)-10) were collected separately from each injection. The collected fractions collected were continuously concentrated in the 1-square feet and 2-square feet ROTOTHERM evaporator, respectively, at 40° C. under reduced pressure (40-120 bar). The residue from each evaporator was further dried under high vacuum to constant weight to afford (3R)-cyclopentyl-3-{4-[7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl]pyrazol-1-yl}propionitrile ((R)-10, 1307 g, 1397.5 g theoretical, 93.5%) from Peak 2 as a light yellow oil and (3S)-cyclopentyl-3-{4-[7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl]pyrazol-1-yl}propionitrile ((S)-10, 1418 g, 1397.5 g theoretical, 101.5%) from Peak 1 as an yellow oil.
(3R)-Cyclopentyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propionitrile ((R)-12, free base). Method A. To a solution of (3R)-cyclopentyl-3-{4-[7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl]pyrazol-1-yl}propionitrile ((R)-10, 463 g, 1.06 mol, 98.6% ee) in acetonitrile (4.5 L) was added water (400 mL) followed immediately by lithium tetrafluoroborate (LiBF4, 987.9 g, 10.5 mol, 10.0 equiv) at room temperature. The reaction temperature was observed to decrease from ambient to 12° C. upon addition of the water and then increase to 33° C. during the addition of lithium tetrafluoroborate (LiBF4). The resulting reaction mixture was heated to reflux (about 80° C.) for overnight. An aliquot was quenched into ethyl acetate/water and checked by LCMS and TLC (95:5 ethyl acetate/methanol, v/v). When LCMS and TLC analyses showed both the hydroxyl methyl intermediate ((R)-11) and fully de-protected material ((R)-12, free base) produced but no starting material ((R)-10) left, the reaction mixture was cooled gradually to <5° C. before a 20% aqueous solution of ammonium hydroxide (NH4OH, 450 mL) was added gradually to adjust the pH of the reaction mixture to 9 (checked with pH strips). The cold bath was removed and the reaction mixture was gradually warmed to room temperature and stirred at room temperature for overnight. An aliquot was quenched into ethyl acetate/water and checked by LCMS and TLC (95:5 ethyl acetate/methanol, v/v) to confirm complete de-protection. When LCMS and TLC showed the reaction was deemed complete, the reaction mixture was filtered and the solids were washed with acetonitrile (1 L). The combined filtrates were then concentrated under reduce pressure, and the residue was partitioned between ethyl acetate (6 L) and half-saturated brine (3 L). The two layers were separated and the aqueous layer was extracted with ethyl acetate (2 L). The combined organic layers were washed with half-saturated sodium bicarbonate (NaHCO3, 3 L) and brine (3 L), dried over sodium sulfate (Na2SO4), and concentrated under reduced pressure to give the crude product as an orange oil. The crude material was then purified by flash column chromatography (SiO2, 40 to 100% ethyl acetate/heptane gradient elution) to afford (3R)-cyclopentyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propionitrile ((R)-12, free base, 273 g, 324.9 g theoretical, 84% yield) as a white foam. This material was checked by 19F NMR to ensure no lithium tetrafluoroborate (LiBF4) remained, and by chiral HPLC (Chiralcel® OD-H, 90:10 hexane/ethanol) to confirm enantiomeric purity (98.7% ee), and was used without further purification to prepare the corresponding phosphate salt. For (R)-12 (free base): 1H NMR (DMSO-d6, 400 MHz) δ ppm 12.1 (bs, 1H), 8.80 (d, 1H, J=0.42 Hz), 8.67 (s, 1H), 8.37 (s, 1H), 7.59 (dd, 1H, J=2.34, 3.51 Hz), 6.98 (dd, 1H, J=1.40, 3.44 Hz), 4.53 (td, 1H, J=19.5, 4.63 Hz), 3.26 (dd, 1H, J=9.77, 17.2 Hz), 3.18 (dd, 1H, J=4.32, 17.3 Hz), 2.40 (m, 1H), 1.79 (m, 1H), 1.65 to 1.13 (m, 7H); C17H18N6(MW, 306.37) LCMS (EI) m/e 307 (M++H).

(R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile (R)-10. A solution of (R)-3-cyclopentyl-3-(4-(7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)propanenitrile ((R)-10, 75.0 g, 0.172 mol, 98.8% ee) in acetonitrile (600 mL) was cooled to 0-5° C. To the cooled solution was added boron trifluoride diethyl etherate (54.4 mL, 0.429 mol) over 10 minutes while maintaining the internal reaction temperature below 5° C. Following the addition, the cold bath was removed and the reaction mixture was allowed to warm to room temperature. When HPLC analysis indicated that the level of (R)-10 was below 1%, the initial phase of the deprotection reaction was considered complete. The reaction was then cooled to 0-5° C., followed by the slow addition of water (155 mL). Following the water addition, the cold bath was removed and the resulting reaction mixture was allowed to warm to 13-17° C., and stirred for an additional 2-3 hours. The resulting reaction mixture was cooled again to 0-5° C. To the cooled reaction mixture was added slowly a solution of ammonia in water [prepared by mixing aqueous 28% ammonia solution (104.5 mL) and water (210.5 mL)] while maintaining the internal reaction temperature at below 5° C. After the aqueous ammonia solution was added, the cold bath was removed and the reaction was allowed to warm to room temperature. The hydrolysis was deemed complete when the level of the hydroxylmethyl intermediate was below 1% by HPLC analysis.
The resulting reaction mixture was diluted with ethyl acetate (315 mL) and washed with 20% brine (315 mL). The aqueous fraction was back extracted with ethyl acetate (315 mL). The organic fractions were combined and concentrated under vacuum with a bath temperature of 40° C. to a volume of 380 mL. The concentrated residue was diluted with ethyl acetate (600 mL) and washed with 1M NaHCO3 (2×345 mL) and 20% brine (345 mL). The aqueous washes were combined and back extracted with ethyl acetate (345 mL). The organic fractions were combined and polish filtered into a clean 2L round bottom flask. The organic fraction was washed with warm water (50° C., 2×450 mL) and then treated with activated charcoal at 65° C. with stirring for 1.5 hours. The slurry was filtered through a celite bed. The filtrate was concentrated under vacuum with a bath temperature of 40° C. The resulting syrup was placed under high vacuum to provide (R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile [(R)-12, 54.2g, 103% yield] as a light yellow foam. This material was checked by 19F NMR to ensure that the product was not contaminated by any fluorinated impurities. The chemical purity of the isolated free base was 96.3%. The chiral purity of the free base was 98.8% by chiral HPLC (chiralcel OD, 90:10 hexane/ethanol). The free base was used without further purification to prepare the phosphate salt. 1H NMR (DMSO-d6, 400 MHz) δ 12.11(bs, 1H), 8.79(d, 1H, J=0.43 Hz), 8.67(s, 1H), 8.37(s, 1H), 7.59(q, 1H, J=2.3 Hz), 6.98(q, 1H, J=1.6 Hz), 4.53(td, 1H, J=19.2, 4.1 Hz), 3.22(dq, 2H, J=9.8, 4.3 Hz), 2.40(m, 1H), 1.79(m, 1H), 1.65-1.13(m, 7H). C17H16N6 (MW, 306.37), LCMS (EI) m/e 307 (M++H).
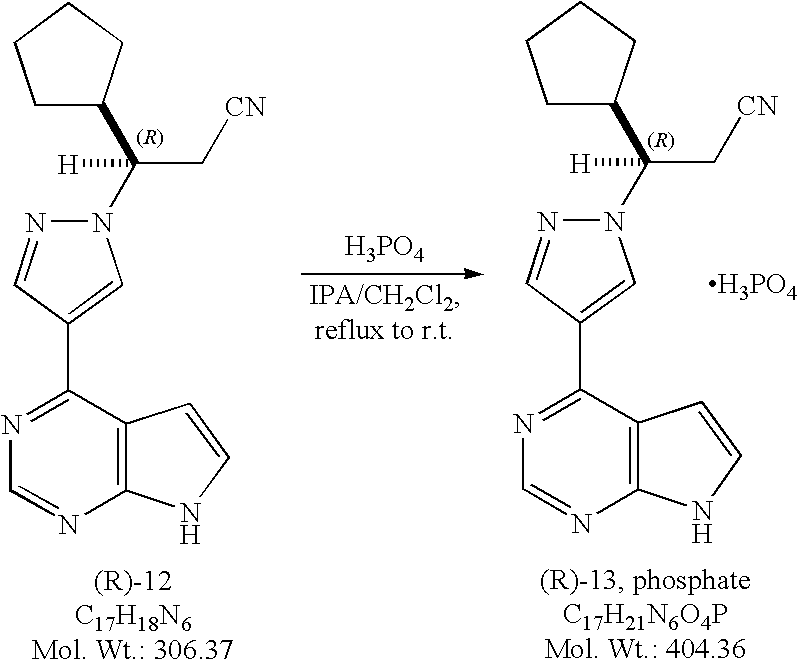
(3R)-Cyclopentyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propionitrile phosphate salt ((R)-13, phosphate).
Method A. To a solution of (3R)-cyclopentyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propionitrile ((R)-12, free base, 572 g, 1.87 mol) in isopropanol (IPA, 8 L) at 60-65° C. was added a solution of phosphoric acid (186.2 g, 1.9 mol, 1.10 equiv) in isopropanol (1.6 L). No exotherm was observed while adding a solution of phosphoric acid, and a precipitate was formed almost immediately. The resulting mixture was then heated at 76° C. for 1.5 hours, then cooled gradually to ambient temperature and stirred at room temperature for overnight. The mixture was filtered and the solids were washed with a mixture of heptanes and isopropanol (1/1, v/v, 3 L) before being transferred back to the original flask and stirred in heptanes (8 L) for one hour. The solids were collected by filtration, washed with heptanes (1 L), and dried in a convection oven in vacuum at 40° C. to a constant weight to afford (3R)-cyclopentyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]propionitrile phosphate salt ((R)-13, phosphate, 634.2 g , 755 g theoretical, 84% yield) as white to off-white crystalline solids. For (R)-13, phosphate: mp. 197.6° C.; 1H NMR (DMSO-d6, 500 MHz) δ ppm 12.10 (s, 1H), 8.78 (s, 1H), 8.68 (s, 1H), 8.36 (s 1H), 7.58 (dd, 1H, J=1.9, 3.5 Hz), 6.97 (d, 1H, J=3.6 Hz), 4.52 (td, 1H, J=3.9, 9.7 Hz), 3.25 (dd, 1H, J=9.8, 17.2 Hz), 3.16 (dd, 1H, J=4.0, 17.0 Hz), 2.41, (m, 1H), 1.79 (m, 1H), 1.59 (m, 1H), 1.51 (m, 2H), 1.42 (m, 1H), 1.29 (m, 2H), 1.18 (m, 1H); 13C NMR (DMSO-d6, 125 MHz) δ ppm 152.1, 150.8, 149.8, 139.2, 131.0, 126.8, 120.4, 118.1, 112.8, 99.8, 62.5, 44.3, 29.1, 29.0, 24.9, 24.3, 22.5; C17H18N6(MW, 306.37 for free base) LCMS (EI) m/e 307 (M++H, base peak), 329.1 (M++Na).
Method B. To a solution of (R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H -pyrazol-1-yl)-3-cyclopentylpropanenitrile ((R)-12, 54.2 g, 177 mol) in dichloromethane (782 mL) and 2-propanol (104 mL) at reflux was added a solution of phosphoric acid (19.9 g, 0.173 mol, 1.15 equiv) in 2-propanol (34.0 mL) over a period of 47 minutes. Following the acid addition, the resulting mixture was heated to reflux for an additional 1 hour. The mixture was gradually cooled to ambient temperature and stirred for 3 hours. The solids were collected by filtration and washed with dichloromethane (390 mL), followed by n-heptane (390 mL). The solids were partially dried under vacuum at room temperature and then under vacuum at 62° C. to afford (R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile phosphate (60.1 g, 84% yield) as white to off-white crystalline solids. Analysis by chiral HPLC (chiralcel OD, 90:10 hexane/ethanol) gave the enantiopurity as 99.2% ee.
1H NMR (DMSO-d6, 400 MHz) δ 12.11(bs, 1H), 8.79(d, 1H, J=0.59 Hz), 8.67(s, 1H), 8.36(s, 1H), 7.59(q, 1H, J=2.3 Hz), 6.98(q, 1H, J=1.6 Hz), 4.53(td, 1H, J=19.6, 4.4 Hz), 3.22(dq, 2H, J=9.6, 4.3 Hz), 2.40(m, 1H), 1.79(m, 1H), 1.65-1.13(m, 7H). C17H21N6O4P (MW, 404.36), LCMS (EI) m/e 307 (M++H) and m/e 329 (M++Na).

(R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile phosphate.
Into a 1L round bottom flask, equipped with stir bar, distillation head, addition funnel and heating mantle, were charged methanol (520 mL) and (R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile phosphate ((R)-13, phosphate, 40.0 grams, 98.92 mmol). The slurry was heated to 55° C. to generate a slightly pink solution. The solution was cooled to 50° C. and filtered into a 2 L flask equipped with an overhead stirrer, distillation head, addition funnel and heating mantle. The 1 L round bottom flask and the filter funnel were rinsed with additional methanol (104.0 mL). The filtrate solution was heated to reflux to distill methanol (281 mL) over 1 hour under atmospheric pressure. Isopropyl alcohol (IPA) (320 mL) was charged slowly via the addition funnel over 80 minutes while maintaining the internal temperature approximately at 65° C. Precipitation of the phosphate salt was observed during IPA addition. After the addition of IPA was complete, n-heptane (175 mL) was added slowly at the same temperature. Distillation was continued under atmospheric pressure. Additional n-heptane (825 mL) was added at approximately the same rate as the distillation rate while maintaining the internal temperature at approximately 65° C. The distillation was complete when the volume of the distillate reached 742 mL (excluding the volume of 281 mL of methanol from the previous distillation). The distillation took approximately 1 hour. The vapor temperature during the distillation was in the range of 54-64° C. and the internal temperature was 67° C. at the end of the distillation. The mixture was slowly cooled to room temperature and stirred for an additional 3 hours. The solids were collected by filtration. The wet cake was washed with 16.7% (v/v) of isopropyl alcohol in n-heptane (384.0 mL), followed by n-heptane (280.0 mL), and dried under vacuum at 55° C. to provide 36.1 grams of the desired product as white solids in 90% yield. The chemical purity is 99.79% by HPLC analysis. The chiral purity is 99.8% by chiral HPLC analysis.
1H NMR (499.7 MHz, DMSO-d6) δ (ppm): 12.21 (s, 1H), 10.71 (s, 3H), 8.80 (s, 1H), 8.72 (s, 1H), 8.40 (s, 1H), 7.60 (d, J=3.5 Hz, 1H), 7.00 (d, J=3.5 Hz, 1H), 4.51 (td, J=9.75, 4.0 Hz, 1H), 3.25 (dd, J=17.3, 9.75 Hz, 1H), 3.14 (dd, J=17.0, 4.0 Hz, 1H), 2.43-2.35 (m, 1H), 1.79-1.73 (m, 1H), 1.58-1.42 (m, 3H), 1.41-1.33 (m, 1H), 1.30-1.23 (m, 2H), 1.19-1.12 (m, 1H);
13C NMR (125.7 MHz, DMSO-d6) δ (ppm): 152.8, 151.2, 150.3, 140.0, 131.8, 127.7, 120.8, 118.8, 113.5, 100.7, 63.3, 45.0, 29.8, 25.6, 25.0, 23.2;
LCMS m/z: calculated for C17H18N6 (M+H)+:=307.2. Found (M+H)+: 307.0.
……………………….
(JAK1, JAK2) inhibitor, developed by the Incyte Corporation, trade name Jakafi.
Ruxolitinib synthetic route as shown below. 4 – bromo-pyrazole ( 1 ) with ethyl vinyl ether ( 2 ) to protect, and then with a Grignard reagent to a halogen – exchanged with isopropyl magnesiumpinacol ester ( 3 ) quenching to obtain 4 . Compound 5 is obtained consisting of hydrogen is protected 6 , and then with a boronic acid ester 4 Suzuki coupling occurs under acidic conditions after removal of the protecting group pyrazolyl 7 , 7 and α, β-unsaturated aldehyde 8 chiral catalyst 9 of under the catalysis of asymmetric Michael addition to give ( R ) -10 (90% EE). ( R) -10 , after reaction with ammonia to obtain an imine oxidation with iodine nitrile 11 , respectively, with different conditions for the final removal of the protecting group to afford Ruxolitinib.
…………………………………
Bioorganic and Medicinal Chemistry Letters, 2013 , vol. 23, # 9 p. 2793 – 2800
http://www.sciencedirect.com/science/article/pii/S0960894X13001868

Structures of tofacitinib (1a) and ruxolitinib (1b).
………………………….
Organic Letters, 2009 , vol. 11, 9 p. 1999 – 2002
http://pubs.acs.org/doi/abs/10.1021/ol900350k

An enantioselective synthesis of INCB018424 via organocatalytic asymmetric aza-Michael addition of pyrazoles (16 or 20) to (E)-3-cyclopentylacrylaldehyde (23) using diarylprolinol silyl ether as the catalyst was developed. Michael adducts (R)-24 and (R)-27 were isolated in good yield and high ee and were readily converted to INCB018424
http://pubs.acs.org/doi/suppl/10.1021/ol900350k/suppl_file/ol900350k_si_001.pdf
COMPD 1 IS RUXOLITINIB
(3R)-Cyclopentyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol
-1-yl]propionitrile (1, INCB018424).
. Method A. To a solution of (3R)-cyclopentyl-3-
{4-[7-(2-trimethylsilanylethoxymethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-yl]pyrazol-1-
yl}proprionitrile ((R)-25, INCB032306, 463 g, 1.06 mol, 98.6% ee) in acetonitrile (4.5 L)
was added water (400 mL) followed immediately by lithium tetrafluoroborate (LiBF4,
987.9 g, 10.5 mol, 10.0 equiv) at room temperature. The resulting reaction mixture was
heated to reflux for overnight. An aliquot was quenched into ethyl acetate/water and
checked by LCMS and TLC (95:5 ethyl acetate/methanol, v/v). When LCMS and TLC
analyses indicated that both the hydroxyl methyl intermediate (R)-26, INCB021499) and
the fully deprotected product (1, INCB018424) SEE LINKhttp://pubs.acs.org/doi/suppl/10.1021/ol900350k/suppl_file/ol900350k_si_001.pdf
:1H NMR
(DMSO-d6, 400 MHz) δ ppm 12.1 (bs, 1H), 8.80 (d, 1H, J = 0.4 Hz), 8.67 (s, 1H), 8.37
(s, 1H), 7.59 (dd, 1H, J = 2.3, 3.5 Hz), 6.98 (dd, 1H, J = 1.4, 3.4 Hz), 4.53 (td, 1H, J =
19.5, 4.6 Hz), 3.26 (dd, 1H, J = 9.8, 17.2 Hz), 3.18 (dd, 1H, J = 4.3, 17.3 Hz), 2.40 (m, S-12
1H), 1.79 (m, 1H), 1.65 to 1.13 (m, 7H);
13C NMR (DMSO-d6, 100MHz) δ ppm 152.1,
151.0, 149.9, 139.3, 131.0, 126.8, 120.6, 118.2, 112.8, 99.8, 62.5, 44.3, 29.1, 25.0, 24.3,
22.5; IR (KBr) 3197, 3118, 2956, 2865, 1731, 1581, 1448, 1344, 1251 cm-1;
HRMS (CI)
m/z calculated for C17H19N6 (M + H)+
307.1671, found 307.1665
REFERENCES
- Jakafi (ruxolitinib) dosing, indications, interactions, adverse effects, and more”. Medscape Reference. WebMD. Retrieved 16 February 2014.
- Mesa, Ruben A.; Yasothan, Uma; Kirkpatrick, Peter (2012). “Ruxolitinib”. Nature Reviews Drug Discovery 11 (2): 103–4.doi:10.1038/nrd3652. PMID 22293561.
- Harrison, C; Mesa, R; Ross, D; Mead, A; Keohane, C; Gotlib, J; Verstovsek, S (2013). “Practical management of patients with myelofibrosis receiving ruxolitinib”. Expert Review of Hematology 6 (5): 511–23. doi:10.1586/17474086.2013.827413. PMID 24083419.
- Natoli, Cori Anne (May 5, 2012), “Incyte looks to ride on drug’s success”, The News Journal, retrieved May 6, 2012
- Harrison, C.; Kiladjian, J. J.; Al-Ali, H. K.; Gisslinger, H.; Waltzman, R.; Stalbovskaya, V.; McQuitty, M.; Hunter, D. S.; Levy, R.; Knoops, L.; Cervantes, F.; Vannucchi, A. M.; Barbui, T.; Barosi, G. (2012). “JAK Inhibition with Ruxolitinib versus Best Available Therapy for Myelofibrosis”. New England Journal of Medicine 366 (9): 787–798.doi:10.1056/NEJMoa1110556. PMID 22375970. edit
- Verstovsek, S.; Mesa, R. A.; Gotlib, J.; Levy, R. S.; Gupta, V.; Dipersio, J. F.; Catalano, J. V.; Deininger, M.; Miller, C.; Silver, R. T.; Talpaz, M.; Winton, E. F.; Harvey Jr, J. H.; Arcasoy, M. O.; Hexner, E.; Lyons, R. M.; Paquette, R.; Raza, A.; Vaddi, K.; Erickson-Viitanen, S.; Koumenis, I. L.; Sun, W.; Sandor, V.; Kantarjian, H. M. (2012). “A Double-Blind, Placebo-Controlled Trial of Ruxolitinib for Myelofibrosis”. New England Journal of Medicine 366 (9): 799–807. doi:10.1056/NEJMoa1110557.PMID 22375971. edit
- Tefferi, A. (2012). “Challenges Facing JAK Inhibitor Therapy for Myeloproliferative Neoplasms”. New England Journal of Medicine 366(9): 844–846. doi:10.1056/NEJMe1115119. PMID 22375977. edit
- ASCO Annual Meeting 2011: JAK Inhibitor Ruxolitinib Demonstrates Significant Clinical Benefit in Myelofibrosis
- Mesa, RA (2010). “Ruxolitinib, a selective JAK1 and JAK2 inhibitor for the treatment of myeloproliferative neoplasms and psoriasis”. IDrugs : the investigational drugs journal 13 (6): 394–403.PMID 20506062. edit
- Pardanani, A.; Tefferi, A. (2011). “Targeting myeloproliferative neoplasms with JAK inhibitors”. Current Opinion in Hematology 18 (2): 1. doi:10.1097/MOH.0b013e3283439964. PMID 21245760. edit
- Wysham, NG; Allada G, Sullivan DR (2013). Chest 143 (5): 1478–9.PMID 23648912.
- “FDA Approves Incyte’s Jakafi(TM) (ruxolitinib) for Patients with Myelofibrosis” (Press release). Incyte. Retrieved 2012-01-02.
- Harrison, C.; Kiladjian, J.-J.; Al-Ali, H. K.; Gisslinger, H.; Waltzman, R.;Stalbovskaya, V.; McQuitty, M.; Hunter, D. S.; Levy, R.; Knoops, L.;Cervantes, F.; Vannucchi, A. M.; Barbui, T.; Barosi, G. N. Eng. J. Med. 2012,366, 787.Zhou, J.; Liu, P.; Lin, Q.; Metcalf, B. W.; Meloni, D.; Pan, Y.; Xia, M.; Li, M.; Yue,T.-Y.; Rodgers, J. D.; Wang, H. WO 2010083283 A2, 2010.Rodgers, J. D.; Shepard, S.; Maduskuie, T. P.; Wang, H.; Falahatpisheh, N.;Rafalski, M.; Arvanitis, A. G.; Storace, L.; Jalluri, R. K.; Fridman, J. S.; Vaddi, K.U.S. 20070135461 A1, 2007.Lin, Q.; Meloni, D.; Pan, Y.; Xia, M.; Rodgers, J.; Shepard, S.; Li, M.; Galya, L.;Metcalf, B.; Yue, T.-Y.; Liu, P.; Zhou, J. Org. Lett. 1999, 2009, 11.http://www.google.com/patents/US8410265
| US7598257 * | Dec 12, 2006 | Oct 6, 2009 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as janus kinase inhibitors |
| US20090233903 * | Mar 10, 2009 | Sep 17, 2009 | Incyte Corporation | Azetidine and cyclobutane derivatives as jak inhibitors |
| WO2007070514A1 * | Dec 12, 2006 | Jun 21, 2007 | Incyte Corp | Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as janus kinase inhibitors |
| WO2007117494A1 * | Apr 5, 2007 | Oct 18, 2007 | Vertex Pharma | Deazapurines useful as inhibitors of janus kinases |
| US8309718 | Nov 13, 2008 | Nov 13, 2012 | Incyte Corporation | 4-pyrazolyl-N-arylpyrimidin-2-amines and 4-pyrazolyl-N-heteroarylpyrimidin-2-amines as janus kinase inhibitors |
| US8410265 | Jan 14, 2010 | Apr 2, 2013 | Incyte Corporation | Processes for preparing JAK inhibitors and related intermediate compounds |
| US8415362 | Jun 12, 2008 | Apr 9, 2013 | Incyte Corporation | Pyrazolyl substituted pyrrolo[2,3-b]pyrimidines as Janus kinase inhibitors |
| US8486902 | Oct 8, 2010 | Jul 16, 2013 | Incyte Corporation | Hydroxyl, keto, and glucuronide derivatives of 3-(4-(7H-pyrrolo[2,3-d] pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US8530485 | Mar 30, 2011 | Sep 10, 2013 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as Janus kinase inhibitors |
| US8541425 | Aug 27, 2009 | Sep 24, 2013 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as Janus kinase inhibitors |
| US8604043 | May 21, 2010 | Dec 10, 2013 | Incyte Corporation | 3-[4-(7H-pyrrolo[2,3-D]pyrimidin-4-yl)-1H-pyrazol-1-yl]octane- or heptane-nitrile as jak inhibitors |
FDA Accepts Eliquis sNDA
FDA Accepts For Review ELIQUIS® (apixaban) Supplemental New Drug Application for the Treatment of Deep Vein Thrombosis (DVT) and Pulmonary Embolism (PE), and for the Reduction in the Risk of Recurrent DVT and PE
PRINCETON, N.J. & NEW YORK, December 19, 2013–(BUSINESS WIRE)–Bristol-Myers Squibb Company (NYSE:BMY) and Pfizer Inc. (NYSE:PFE) today announced that the U.S. Food and Drug Administration (FDA) has accepted for review a Supplemental New Drug Application (sNDA) for ELIQUIS® (apixaban) for the treatment of deep vein thrombosis (DVT) and pulmonary embolism (PE), and for the reduction in the risk of recurrent DVT and PE. The Prescription Drug User Fee Act (PDUFA) goal date for a decision by the FDA is August 25, 2014.
MARAVIROC

MARAVIROC
UK-427857; Selzentry; Celsentri
376348-65-1 CAS NO
Maraviroc M.Wt: 513.67
Maraviroc Formula: C29H41F2N5O
Maraviroc (brand-named Selzentry, or Celsentri outside the U.S.) is an antiretroviral drug in the CCR5 receptor antagonist class used in the treatment of HIV infection. It is also classed as an entry inhibitor. It also appeared to reduce graft-versus-host disease in patients treated withallogeneic bone marrow transplantation for leukemia, in a phase 1/2 study.[3][4]
Maraviroc is an entry inhibitor. Specifically, maraviroc is a negative allosteric modulator of the CCR5 receptor, which is found on the surface of certain human cells. The chemokine receptor CCR5 is an essential co-receptor for most HIV strains and necessary for the entry process of the virus into the host cell. The drug binds to CCR5, thereby blocking the HIV protein gp120 from associating with the receptor. HIV is then unable to enter humanmacrophages and T-cells.[5] Because HIV can also use other coreceptors, such as CXCR4, an HIV tropism test such as a trofile assay must be performed to determine if the drug will be effective.[6]
Development and approval
Maraviroc, originally designated UK-427857, was developed by the drug company Pfizer in its UK labs located in Sandwich. On April 24, 2007 the U.S. Food and Drug Administration advisory panel reviewing maraviroc’s New Drug Application unanimously recommended approval for the new drug,[7] and the drug received full FDA approval on August 6, 2007 for use in treatment experienced patients.[8]
On September 24, 2007, Pfizer announced that the European Commission approved maraviroc. Industry experts forecast annual maraviroc sales of $500 million by 2011.[9]
SELZENTRY (maraviroc) is a selective, slowly reversible, small moleculeantagonist of the interaction between human CCR5 and HIV-1 gp120. Blocking this interaction prevents CCR5-tropic HIV-1 entry into cells.
SELZENTRY is available as film-coated tablets for oral administration containing either 150 or 300 mg of maraviroc and the following inactive ingredients: dibasic calcium phosphate (anhydrous), magnesium stearate, microcrystalline cellulose, and sodium starch glycolate. The film coat (Opadry® II Blue [85G20583]) contains FD&C blue #2 aluminum lake, soya lecithin, polyethylene glycol (macrogol 3350), polyvinyl alcohol, talc, and titanium dioxide.
Maraviroc is chemically described as 4,4-difluoro-#-((15′)-3-[exo-3-(3-isopropyl-5- methyl-4#-1,2,4-triazol-4-yl)-8-azabicyclo[3.2.1]oct-8-yl]-1- phenylpropyl} cycl ohexanecarboxamide.
The molecular formula is C29H41F2N5O and the structural formula is:
 |
Maraviroc is a white to pale-colored powder with a molecular weight of 513.67. It is highly soluble across the physiological pH range (pH 1.0 to 7.5).
Two randomized, placebo-controlled clinical trials, known as MOTIVATE 1 & 2, compared 209 patients receiving optimized therapy plus a placebo to 426 patients receiving optimized therapy plus 150 mg maraviroc once daily and 414 patients receiving optimized therapy plus 150 mg maraviroc twice daily. At 48 weeks, 55% of participants receiving maraviroc once daily and 60% of participants receiving the drug twice daily achieved a viral load of less than 400 copies/mL compared with 26% of those taking placebo; about 44% of the once-daily and 45% of the twice-daily maraviroc group had a viral load of less than 50 copies/mL compared with about 23% of those who received placebo. In addition, those who received the entry inhibitor had a mean increase in CD4 cells of 110 cells/µL in the once-daily group, 106 cells/µL in the twice-daily group, and 56 cells/µL in the placebo group.[10][11][12]
The MOTIVATE trials showed no clinically relevant differences in safety between the maraviroc and placebo groups. However, researchers question the long-term safety of blocking CCR5, a receptor whose function in the healthy individual is not fully understood.[10]
- Abel S, Russell D, Whitlock LA, Ridgway CE, Nedderman AN, Walker DK (April 2008). “Assessment of the absorption, metabolism and absolute bioavailability of maraviroc in healthy male subjects”. British Journal of Clinical Pharmacology 65 (Suppl 1): 60–7. doi:10.1111/j.1365-2125.2008.03137.x. PMC 2311408. PMID 18333867.
- Abel S, Back DJ, Vourvahis M (2009). “Maraviroc: pharmacokinetics and drug interactions”. Antiviral Therapy 14 (5): 607–18. PMID 19704163.
- http://www.uphs.upenn.edu/news/News_Releases/2012/07/hiv/
- Blocade of lymphocyte chemotaxis in visceral graft-versus-host disease, Ran Reshef et al., New England Journal of Medicine, 367:135 (July 12, 2012)
- Levy JA (January 2009). “HIV pathogenesis: 25 years of progress and persistent challenges”. AIDS 23 (2): 147–60. doi:10.1097/QAD.0b013e3283217f9f. PMID 19098484.
- Biswas P, Tambussi G, Lazzarin A (May 2007). “Access denied? The status of co-receptor inhibition to counter HIV entry”. Expert Opinion on Pharmacotherapy 8 (7): 923–33.doi:10.1517/14656566.8.7.923. PMID 17472538.
- Gay News From 365Gay.com
- Krauskopf, Lewis (August 6, 2007). “Pfizer wins U.S. approval for new HIV drug”. Reuters. Retrieved 2007-08-06.
- Reuters, Europe gives final approval to Pfizer HIV drug
- ^ Jump up to:a b Stephenson J (April 2007). “Researchers buoyed by novel HIV drugs: will expand drug arsenal against resistant virus”. JAMA 297 (14): 1535–6. doi:10.1001/jama.297.14.1535.PMID 17426263.
- Emmelkamp JM, Rockstroh JK (October 2007). “CCR5 antagonists: comparison of efficacy, side effects, pharmacokinetics and interactions–review of the literature”. European Journal of Medical Research 12 (9): 409–17. PMID 17933722.
- “Maraviroc reduces viral load in naive patients at 48 weeks”. AIDS Patient Care and STDs 21 (9): 703–4. September 2007. PMID 17941136.
- BBC News story: Drug ‘stops HIV’s entry to cells’
- Maraviroc data at aidsmap
- Maraviroc early access program
- New HIV Drug Recommended for Approval
- maraviroc at the US National Library of Medicine Medical Subject Headings (MeSH)
…………………
Maraviroc and its pharmaceutically acceptable salt or solvate thereof were disclosed in U.S. Patent No. 6,667,314 (herein after refer to ‘314 patent). Maraviroc is chemically, N- {(lS)-3-[3-(3-isopropyl-5-methyl-4H-l,2,4-triazol-4-yl)-exo-8-azabicyclo- [3.2.1]oct-8-yl]-l- phenylpropyl}-4,4-difluorocyclohexanecarboxamide and has the structural formula:

Maraviroc as modulators of the chemokine receptor CCR5 and thus useful in the treatment of retroviral diseases caused by viruses that utilize CCR5 to enter cells. In particular maraviroc has been disclosed as being a useful therapeutic in the treatment of HIV, a retroviral infection genetically related to HIV, AIDS, or an inflammatory disease.
Solvent medium and mode of crystallization play very important role in obtaining a crystalline form over the other
Maraviroc or its salts can exist in different polymorphic forms, which may differ from each other in terms of stability, physical properties, spectral data and methods of preparation.
According to the ‘314 patent, maraviroc can be prepared by reacting a solution of (l S)-3-[3-(3-isopropyl-5-methyl-4H-l,2,4-triazol-4-yl)-exo-8-azabicyclo[3.2.1]oct-8-yl]-l- phenyl- 1 -propanamine in methylene chloride and saturated sodium carbonate with a solution of 4,4-difluorocyclohexanecarbonyl chloride in toluene, and isolating to obtain maraviroc.
Crystalline polymorph form A and form B of maraviroc were disclosed in U.S. patent no. 7,576,097.
An overview of the key routes to the best selling 5-membered ring heterocyclic pharmaceuticals
 , Ian R. Baxendale, Steven V. Ley and Nikzad Nikbin Corresponding author email
, Ian R. Baxendale, Steven V. Ley and Nikzad Nikbin Corresponding author emailA novel and very promising HIV treatment is Pfizer’s maraviroc (286, Celsentri). HIV uses a member of the G-protein coupled receptor family called CCR-5 as an anchor to attach itself to white blood cells such as T-cells and macrophages followed by viral fusion and entry into white blood cells. Maraviroc blocks this pathway by acting as an antagonist for the CCR-5 receptor hence disrupting HIV life cycle. The structural features of this molecule are a geminal difluorocyclohexyl carboxamide which is linked to a β-aminoacid, and a tropinone-type unit bound to a 1,2,4-triazole ring. Relatively simple and straightforward chemical transformations are used to assemble the main fragments of maraviroc such as amide bond formation and reductive amination (Scheme 57) [86]. The triazole ring incorporation is achieved at an early stage by N-acylation of the tropinone fragment 287 with 2-methylpropanoyl chloride (288). The resulting amide 289 is then converted to the corresponding imidoyl chloride 290 using phosphorous pentachloride in dichloromethane (which proved to be superior to phosphoryl chloride) followed by condensation with acetic hydrazide (291). It was found that the dryness of the acetic hydrazide was crucial in order to minimise the hydrolysis of the starting amide 289.
![[1860-5397-7-57-i57]](https://i0.wp.com/beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i57.png)

TET LETT 46, 2005, PG5005
FDA Breakthrough Therapy Designation: 28 And Counting
On December 20, GlaxoSmithKline (GSK) and Medicines for Malaria Venture (MMV), announce that the FDA grants a Breakthrough Therapy Designation (BTD) for Tafenoquine, an investigational medicine for the treatment and relapse prevention of Plasmodium vivax malaria. This is the 28th BTD that is announced by a sponsor company – GSK and MMV. This is the 3rd FDA BTD that GSK receives in 2013:
Row Num | Drug Name | Sponsor Company | Indication |
1 | Drisapersen | GlaxoSmithKline | Duchenne Muscular
Dystrophy |
2 | Ofatumumab | Genmab/GSK | Chronic Lymphocytic
Leukemia (CLL) |
3 | Tafenoquine | GSK/Medicines for Malaria Venture | Plasmodium Vivax Malaria |
Plasmodium vivax malaria is a “neglected” tropical disease and a major cause of uncomplicated malaria. It is prevalent in Latin America, Southeast Asia, and the horn of Africa, where the majority of the approximately 70-390 million annual clinical cases exist. Plasmodium vivax malaria of all the…
View original post 164 more words
Ondansetron

 ondansetron
ondansetron
Ondansetron hydrochloride dihydrate, cas 99614-01-4, GG-032, SN-307, GR-C505/75,

Ondansetron hydrochloride dihydrate is a serotonin-3 (5-HT3) receptor antagonist. J Org Chem1980, 45, (15): 2938 Heterocycles1997, 45, (10): 2041 EP 0595111 WO 0172716, Ondansetron (INN) (/ɒnˈdænsɛtrɒn/; developed and first marketed by GlaxoSmithKline as Zofran) is a serotonin 5-HT3 receptor antagonist used mainly as an antiemetic (to treat nausea and vomiting), often following chemotherapy. It affects both peripheral and central nerves. Ondansetron reduces the activity of the vagus nerve, which deactivates the vomiting center in the medulla oblongata, and also blocks serotonin receptors in thechemoreceptor trigger zone. It has little effect on vomiting caused by motion sickness, and does not have any effect on dopamine receptors ormuscarinic receptors. Although an effective anti-emetic agent, the high cost of brand-name ondansetron initially limited its use to controlling postoperative nausea and vomiting (PONV) and chemotherapy-induced nausea and vomiting (CINV). The active ingredient in ZOFRAN Tablets and ZOFRAN Oral Solution is ondansetron hydrochloride (HC1) as the dihydrate, the racemic form of ondansetron and a selective blocking agent of the serotonin 5-HT3 receptor type. Chemically it is (±) 1, 2, 3, 9-tetrahydro-9-methyl-3-[(2-methyl-lH-imidazol-l-yl)methyl]-4H-carbazol-4-one, monohydrochloride, dihydrate. It has the following structural formula:
 |
The empirical formula is C18H19N3O•HCl•2H2O, representing a molecular weight of 365.9. Ondansetron HC1 dihydrate is a white to off-white powder that is soluble in water and normal saline. The active ingredient in ZOFRAN ODT Orally Disintegrating Tablets is ondansetron base, the racemic form of ondansetron, and a selective blocking agent of the serotonin 5-HT3 receptor type. Chemically it is (+) 1, 2, 3, 9-tetrahydro-9-methyl-3-[(2-methyl-lH-imidazol-l-yl)methyl]-4H-carbazol-4-one. It has the following structural formula:
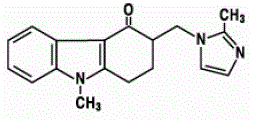 |
The empirical formula is C18H19N3O representing a molecular weight of 293.4. The 5-HT3 receptor antagonists are the primary drugs used to treat and prevent chemotherapy-induced nausea and vomiting (CINV). A common use case is to give them intravenously about 30 minutes before commencement of a chemotherapy treatment.
Ondansetron is used off-label to treat morning sickness and hyperemesis gravidarum of pregnancy. A cohort study of over 600,000 pregnancies in Denmark found that ondansetron administration during pregnancy is not associated with a significantly increased risk of spontaneous abortion,stillbirth, major birth defect, preterm birth, low birth weight, or small for gestational age. However, in practice, ondansetron is typically used after trials of other drugs have failed.
Ondansetron is one of several anti-emetic agents used during the vomiting phase of cyclic vomiting syndrome. Trials in emergency department (ED) settings support the use of ondansetron to reduce vomiting associated with gastroenteritis and dehydration.A retrospective review found that it was used commonly for this purpose, being administered in over 58% of cases. Its use reduced hospital admissions, but was also associated with higher rates of return visits to the ED. Furthermore, patients who had initially received ondansetron were more likely to be admitted on the return visit than patients who had not received the drug. However, this effect may simply be due to the agent being used more frequently in patients who present with more severe illness. Its use was not found to mask serious diagnoses.
.JPG)
A vial of ondansetron for intravenous injection
Ondansetron was developed around 1984 by scientists working at Glaxo’s laboratories in London. It is in both the imidazole and carbazole families of heterocyclic compounds. After several attempts the company successfully filed for U.S. patent protection for the drug in 1986 and was granted in June 1988 while a use patent was granted in June 1988. A divisional use patent was granted on November 26, 1996. Ondansetron was granted FDA approval as Zofran in January 1991. Glaxo did pediatric research on Zofran’s uses, and gained a patent extension as a result, extending U.S. exclusivity until December 24, 2006. The FDA subsequently approved the first generic versions in December 2006, with marketing approval granted to Teva Pharmaceuticals USA and SICOR Pharmaceuticals.
Ondansetron is marketed by GlaxoSmithKline (GSK) under the trade name Zofran. Other manufacturers include Pfizer Injectables (Ondanzetron), Opsonin Pharma Bangladesh (Anset), Strativa Pharmaceuticals (Zuplenz), Indswift Ltd. (Ondisolv), Cipla Ltd. (Emeset), Gedeon Richter Ltd. (Emetron), Korea United Pharmaceuticals (Emodan), Zentiva a.s. (Ondemet), Strides Arcolab (Setronax), Emistat (Unimed and Unihealth Bangladesh Ltd.)Glenmark Generics Ltd. (India) (Ondansetron) and Novell Pharmaceutical Laboratories (Ondavell). On May 29, 2006, Baxter Healthcare received tentative approval to market its own label of Ondansetron Injection, USP, 8 mg/50 mL and 32 mg/50 mL iso-osmotic sodium chloride solution, beginning upon expiration of GSK’s patent later that year.
In 1997, ondansetron was the subject of a meta-analysis case-study published in the British Medical Journal. Researchers examined 84 trials, with 11,980 patients receiving ondansetron, published between 1991 and September 1996. Intravenous ondansetron 4 mg versus placebo was investigated in 16 reports and three further reports which had been duplicated six times. The number needed to treat (NNT) to prevent vomiting within 24 hours was 9.5, with 95% confidence interval 6.9 to 15, in the 16 non-duplicated reports. In the three duplicated reports, the NNT was significantly lower at 3.9 (3.3 to 4.8) with P<0.00001. When all 25 reports were combined the apparent number needed to treat improved to 4.9 (4.4 to 5.6). Inclusion of duplicate reports led to a 23% overestimation of ondansetron’s antiemetic efficacy. In addition, the authors found that the covert duplication of reports on ondansetron was not easy to detect, because of lack of cross-referencing between papers, and that reports containing duplicate findings were cited in eight reviews of the drug. Their analysis was a subject of an editorial in the Journal of the American Medical Association in 1999. ………………………
patents
AU 8538097; BE 0901576; CH 664152; ES 8609309; ES 8708224; ES 8801247; FR 2561244; GB 2153821; JP 1985214784
……………..
(±) l,2,3,9-Tetrahydro-9-methyl-3-[2-methyl-lh-imidazol-l-yl)methyl]-4h- carbazol-4-one having the molecular structure

is a selective 5-HT3 receptor antagonist. It is known by the generic nameondansetron. Ondansetron reduces nausea in patients undergoing chemotherapy. Grunberg, S.M.; Hesketh, P.J. “Control of Chemotherapy-Induced emesis” N. Engl. J. Med. 1993, 329, 1790-96. Ondansetron is indicated for prevention of nausea and vomiting associated with some cancer chemotherapy, radiotherapy and postoperative nausea and/or vomiting.
Several chemical processes are known from the literature for the synthesis ofondansetron. GB-Pat. 2 153 821 and 2 192 885 describe syntheses starting from carbazolone derivative, and EP-Pat. 595 111 as well as a Hungarian patent application ( P 00-01287 ) give detailed information about some different chemical procedures.
Ondansetron is currently available as an anti-emetic agent, particularly in cancer chemotherapy, and in some other uses such as anti-depressive, anti- migraine and anti-psychotic. It is commonly used in the alleviation of cognitive disorders as in Alzheimer disease, in treatment of rhinitis, psychiatric disorders and for increased vigilance and for control of dependence on narcotics.
U.S. Patent No. 4,695,578, assigned to the Glaxo Group Limited, describes a process of preparing ondansetron and uses thereof. However, ondansetronprepared according to said process contains impurities and by-products such as l,2,3,9-tetrahydro-9-methyl-3-methylene-4H-carbazol-4-one.
The hydrochloride salt of ondansetron is generally safe for oral administration to a patient without causing irritation or other adverse effect. The hydrochloride salt is marketed in tablet form and in oral solution form under the brand name Zofran®. The tablet’s active ingredient is a dihydrate of ondansetronhydrochloride containing two molecules of bound water in ondansetronhydrochloride’ s crystal lattice. The present invention relates to the solid state physical properties of ondansetron hydrochloride. These properties can be influenced by controlling the conditions under which the hydrochloride salt is obtained in solid form. Solid state physical properties include, for example, the flowability of the milled solid. Flowability affects the ease with which the material is handled during processing into a pharmaceutical product. When particles of the powdered compound do not flow past each other easily, a formulation specialist must take that fact into account in developing a tablet or capsule formulation, which may necessitate the use of glidants such as colloidal silicon dioxide, talc, starch or tribasic calcium phosphate.
These important physical characteristics are influenced by the conformation and orientation of molecules in the unit cell, which defines a particular polymorphic form of a substance. Llacer and coworkers have postulated that different spectroscopic characteristics of samples of ondansetron free base prepared differently could be attributable to two different configurations about the methylene bridge between the 1, 2, 3, 9-tetrahydrocarbazol-4-one ring and the imidazole ring. Llacer, J.M.; Gallardo, V.; Parera, A. Ruiz, M.A. InternJ.Pharm., 177, 1999, 221-229.
(±)1,2,3,9-Tetrahydro-9-methyl-3-[2-methyl-1h-imidazol-1-yl)methyl]-4h-carbazol-4-one having the molecular structure

is a selective 5-HT3 receptor antagonist. It is known by the generic nameondansetron. Ondansetron reduces nausea in patients undergoing chemotherapy. Grunberg, S. M.; Hesketh, P. J. “Control of Chemotherapy-Induced emesis” N. Engl. J. Med. 1993, 329, 1790-96. Ondansetron is indicated for prevention of nausea and vomiting associated with some cancer chemotherapy, radiotherapy and postoperative nausea and/or vomiting.
The hydrochloride salt of ondansetron is generally safe for oral administration to a patient without causing irritation or other adverse effect. The hydrochloride salt is marketed in tablet form and in oral solution form under the brand name Zofran®. The tablet’s active ingredient is a dihydrate of ondansetronhydrochloride containing two molecules of bound water in ondansetronhydrochloride’s crystal lattice.
The present invention relates to the solid state physical properties ofondansetron hydrochloride. These properties can be influenced by controlling the conditions under which the hydrochloride salt is obtained in solid form. Solid state physical properties include, for example, the flowability of the milled solid. Flowability affects the ease with which the material is handled during processing into a pharmaceutical product. When particles of the powdered compound do not flow past each other easily, a formulation specialist must take that fact into account in developing a tablet or capsule formulation, which may necessitate the use of glidants such as colloidal silicon dioxide, talc, starch or tribasic calcium phosphate.
Another important solid state property of a pharmaceutical compound is its rate of dissolution in aqueous fluid. The rate of dissolution of an active ingredient in a patient’s stomach fluid can have therapeutic consequences since it imposes an upper limit on the rate at which an orally-administered active ingredient can reach the patient’s bloodstream. The rate of dissolution is also a consideration in formulating syrups, elixirs and other liquid medicaments. The solid state form of a compound may also affect its behavior on compaction and its storage stability.
These important physical characteristics are influenced by the conformation and orientation of molecules in the unit cell, which defines a particular polymorphic form of a substance. Llacer and coworkers have postulated that different spectroscopic characteristics of samples ofondansetron free base prepared differently could be attributable to two different configurations about the methylene bridge between the 1,2,3,9-tetrahydrocarbazol-4-one ring and the imidazole ring. Llacer, J. M.; Gallardo, V.; Parera, A. Ruiz, M. A. Intern.J.Pharm., 177, 1999, 221-229.
A crystalline polymorphic form of a compound may exhibit different thermal behavior from amorphous material or another polymorphic form. Thermal behavior is measured in the laboratory by such techniques as capillary melting point, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) and can be used to distinguish some polymorphic forms from others. A particular polymorphic form may also give rise to distinct spectroscopic properties that may be detectable by powder X-ray crystallography, solid state 13C NMR spectrometry and infrared spectrometry. There is a wide variety of techniques that have the potential of producing different crystalline forms of a compound. Examples include crystallization, crystal digestion, sublimation and thermal treatment.
U.S. Pat. No. 4,695,578, Example 1a, discloses a preparation ofondansetron by alkylation of 2-methylimidazole with 2,3,4,9 tetrahydro-N,N,N,9-tetramethyl-4-oxo-1H-carbazole-3-methanaminium iodide. In this example,ondansetron was isolated as its hydrochloride salt by suspending the reaction product in a mixture of absolute ethanol and ethanolic HCl, warming the suspension, filtering to remove impurities and precipitating the hydrochloride salt with dry ether.
In Example 10 of the ‘578 patent, ondansetron free base was converted into a hydrochloride salt dihydrate by dissolving the free base in a mixture of isopropanol and water and treating it with concentrated hydrochloric acid. After filtration at elevated temperature, ondansetron was driven out of solution by adding additional isopropanol and cooling. The dihydrate was obtained as a white crystalline solid by recrystallizing it from a 6:10 mixture of water and isopropanol. Ondansetron hydrochloride dihydrate obtained by following Example 10 of the ‘578 patent is denominated Form A in this disclosure. Powdered samples of Form A produce a powder X-ray diffraction pattern essentially the same as the pattern shown in FIG. 1.
U.S. Pat. No. 5,344,658 describes ondansetron having a particular particle size distribution and the use of such ondansetron in a pharmaceutical composition. The particle size of ondansetron hydrochloride dihydrate obtained by crystallization from a solvent is reduced by desolvating them, e.g. by heating, and then exposing the desolvated crystals to a humid atmosphere. A collection of crystals obtained by this particle size reduction process is said to consist exclusively of crystals of less than 250 micron size and to contain 80% or more crystals of less than 63 microns. Crytals size was determined by air jet seive analysis.
According to the ‘658 patent, ondansetron hydrochloride dehydrate having the same particle size distribution as the rehydrated ondansetron hydrochloride also is provided as part of that invention. Since only one process for dehydratingondansetron hydrochloride is described in the ‘658 patent, a dehydrate is evidently the intermediate compound that is rehydrated in the particle size reduction process.
U.S. Pat. Nos. 4,695,578 and 5,344,658 are incorporated herein by reference.
U.S. Pat. No. 4,695,578 (‘578 patent) discloses a process for preparingondansetron hydrochloride dihydrate having a large particle size (e.g., less than about 60% of the particles are smaller than 250 μm). The ‘578 patent process involves the step of cooling a solution of ondansetron hydrochloride, isopropanol, and water, optionally followed by an additional step of recrystallizing from a mixture of water and isopropanol.
U.S. Pat. No. 5,722,720 (the ‘720 patent) discloses a non-conventional technique for reducing particle size. In particular, the ‘720 patent discloses a multistep process in which ondansetron hydrochloride dihydrate is first dried at elevated temperature and reduced or atmospheric pressure, and is then cooled to ambient temperature. The process requires the heating step to be performed until the ondansetron hydrochloride dihydrate is desolvated, and requires the cooling step to be performed until the ondansetron hydrochloride is rehydrated to form ondansetron hydrochloride dihydrate.
The ‘720 patent process has several disadvantages. First, the ‘720 patent process requires a prolonged time period (i.e., 16-24 hours) for the drying/desolvating step, plus an additional prolonged time period for the cooling/rehydrating step. Second, the ‘720 patent process requires vigorous and carefully controlled drying conditions. For example, when the drying step is performed at 48-52° C., a reduced pressure of 100-200 torr is required. When the drying step is performed at ambient pressure, an elevated temperature of 1 00° C. is required.
An overview of the key routes to the best selling 5-membered ring heterocyclic pharmaceuticals
, Ian R. Baxendale, Steven V. Ley and Nikzad Nikbin Corresponding author emailA completely different strategy was used in the synthesis of the serotonin 5-HT3 receptor antagonist ondansetron (119, Zofran). In this synthesis a palladium-catalysed intramolecular Heck-reaction was used to build the tricyclic indole core in a short and concise sequence (Scheme 26) [35,36].
![[1860-5397-7-57-i26]](https://i0.wp.com/beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i26.png)
Alternatively, a direct Fischer indole synthesis between phenylmethyl hydrazine and a cyclic 1,3-dione derivative could be utilised to prepare the desired fully substituted tricyclic core of ondansetron (Scheme 27) [37].
![[1860-5397-7-57-i27]](https://i0.wp.com/beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i27.png)
- 35………..Godfrey, N.; Coates, I. H.; Bell, J. A.; Humber, D. C.; Ewan, G. B. Process for Preparing N-Heterocyclic Compounds. U.S. Patent 4,957,609, Sept 18, 1990.
- 36…………Iida, H.; Yuasa, Y.; Kibayashi, C. J. Org. Chem. 1980, 45, 2938–2942. doi:10.1021/jo01303a003
- Oxford, A. W.; Eldred, C. D.; Coates, I. H.; Bell, J. A.; Humber, D. C.; Ewan, G. B. Process for Preparing Tetrahydrocarbazolones. U.S. Patent 4,739,072, April 19, 1988.
OLMESARTAN

Mol. mass 558.585 g/mol
Olmesartan medoxomil (trade names: Benicar in the US, Olmetec in EU, Canada and Japan, WinBP, Golme in India, Erastapex in Egypt) is an angiotensin II receptor antagonist used to treat high blood pressure.
Olmesartan is indicated for the treatment of hypertension. It may be used alone or in combination with other antihypertensive agents.[1] The U.S. Food and Drug Administration (FDA) has determined that the benefits of Benicar continue to outweigh its potential risks when used for the treatment of patients with high blood pressure according to the drug label.[2]
Angiotensin-II receptor antagonists should be used with caution in renal artery stenosis. Monitoring of plasma-potassium concentration is advised, particularly in the elderly and in patients with renal impairment; lower initial doses may be appropriate in these patients. Angiotensin-II receptor antagonists should be used with caution in aortic or mitral valve stenosis and in hypertrophic cardiomyopathy. Those with primary aldosteronism, and Afro-Caribbean patients (particularly those with left ventricular hypertrophy), may not benefit from an angiotensin-II receptor antagonist.
Structure
The olmesartan molecule includes one tetrazole group (a 5-member heterocyclic ring of four nitrogen and one carbon atom) and one imidazole group (a 5-membered planar heterocyclic aromatic ring of two nitrogen and three carbon atoms, classified as an alkaloid).
Olmesartan as the starting material can be easily produced according to the method described in Japanese Examined Patent Application (Kokoku) No. Hei 7-121918 (Japanese Patent No. 2082519 ; US Patent No. 5616599 ) or the like
Olmesartan is a prodrug that works by blocking the binding of angiotensin II to the AT1 receptors in vascular muscle; it is therefore independent of angiotensin II synthesis pathways, unlike ACE inhibitors. By blocking the binding rather than the synthesis of angiotensin II, olmesartan inhibits the negative regulatory feedback on renin secretion. As a result of this blockage, olmesartan reduces vasoconstriction and the secretion of aldosterone. This lowers blood pressure by producing vasodilation, and decreasing peripheral resistance.
The usual recommended starting dose of olmesartan is 20 mg once daily. The dose may be increased to 40 mg after two weeks of therapy, if further reduction in blood pressure is desirable. Doses above 40 mg do not appear to have greater effect, and twice-daily dosing offers no advantage over the same total dose given once daily.[1] No adjustment of dosage is typically necessary for advanced age, renal impairment, or hepatic dysfunction. For patients with possible depletion of intravascular volume (e.g., patients treated with diuretics), olmesartan should be initiated with caution; consideration should be given to use of a lower starting dose in such cases.[1] If blood pressure is not controlled by Benicar alone, a diuretic may be added. Benicar may be administered with other antihypertensive agents. Benicar may be administered with or without food.[1]

Olmesartan and Sevikar HCT is marketed worldwide by Daiichi Sankyo, in India by Abbott Healthcare Pvt. Ltd. under the trade name WinBP, by Zydus Cadila under the trade name Olmy, by Ranbaxy Laboratories Ltd. under the trade name Olvance, and in Canada by Schering-Plough as Olmetec. Benicar HCT is the brand name of a medication containing olmesartan medoxomil in combination with hydrochlorothiazide, a thiazide diuretic. Three dosage combinations are available: 20 mg or 40 mg of olmesartan medoxomil combined with 12.5 mg of hydrochlorothiazide, or 40 mg of olmesartan medoxomil combined with 25 mg of hydrochlorothiazide. Benitec H, another medication containing olmesartan medoxomil and hydrochlorothiazide, is marketed by GlaxoSmithKline in India. In Poland as Olesartan Medoxomil by TEVA, Olimestra and Co-Olimestra (with HCTZ) by Miklich Lab., Elestar (with amlodipine) and Elestar HCT (with amlodipine, HCTZ) by Menarini, Sevikar HCT (with amoldipine, HCTZ) by Aiichi Sankyo.
Research
Two clinical studies (MORE [6] and OLIVUS [7])[8] report that Benicar reduced arterial plaque during therapy for high-blood pressure (hypertension).
- RxList Inc. (5 July 2007). “Benicar (olmesartan medoxomil)”. RxList Inc. Retrieved 22 July 2010.
- “FDA Alert: Benicar (olmesartan): Ongoing Safety Review”. Drugs.com. Retrieved 2013-06-27.
- Angiotensin II receptor blocker induced fetopathy: 7 cases. Hünseler C, Paneitz A, Friedrich D, Lindner U, Oberthuer A, Körber F, Schmitt K, Welzing L, Müller A, Herkenrath P, Hoppe B, Gortner L, Roth B, Kattner E, Schaible T. Klin Padiatr. 2011 Jan;223(1):10-4. Epub 2011 Jan 26.
- “BENICAR Prescribing Information”. Retrieved 2011-01-20.
- Rubio-Tapia, Alberto; Herman, Margot L.; Ludvigsson, Jonas F.; Kelly, Darlene G.; Mangan, Thomas F.; Wu, Tsung-Teh; Murray, Joseph A. (NaN undefined NaN). “Severe Spruelike Enteropathy Associated With Olmesartan”. Mayo Clinic Proceedings 87 (8): 732–738. doi:10.1016/j.mayocp.2012.06.003.
- as referenced in http://www.medicalnewstoday.com/releases/91285.php “Olmetec(R) Is First Angiotensin Receptor Blocker (ARB) To Suggest Atherosclerosis Regression (In Hypertensives With Cardiovascular Risk), UK”
- Cardiovascular Research Foundation (2008, October 16). Drug May Reduce Coronary Artery Plaque. ScienceDaily. Retrieved January 5, 2013, from http://www.sciencedaily.com /releases/2008/10/081012121318.htm
- (Review) R Preston Mason, Cardiovascular Division, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, and Elucida Research, Beverly, MA, USA. Vascular Health and Risk Management, Dovepress, Published Date June 2011 Volume 2011:7 Pages 405 – 416. Optimal therapeutic strategy for treating patients with hypertension and atherosclerosis: focus on olmesartan medoxomil. Retrieved January 5, 2013, from http://www.dovepress.com/optimal-therapeutic-strategy-for-treating-patients-with-hypertension-a-peer-reviewed-article-VHRM
4-Isopropenyl-2-propyl-1-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]imidazole-5-carboxylic acid [olmesartan dehydrate, compound 34b described in J. Med. Chem., 39, 323-338 (1996)]
- Daiichi-Sankyo Benicar page
- Benicar HCT from RXlist.com
- Mayo Clinic Proceedings vol.87 Issue 8, pages 732-738

-
Olmesartan medoxomil is known by two names,
- (a)(5-Methyl-2-oxo-1,3-dioxolen-4-yl)methyl 4-(1-hydroxy-1-methylethyl)-2-propyl-1-[4-(2(tetrazole-5yl)phenyl]phenyl]methylimidazole-5-carboxylate
- (b)4-(1-Hydroxy-1-methylethyl)-2-propyl-1-[[2′-(1H-tetrazol-5-yl)[1,1′-biphenyl]-4-yl]methyl]-1H-imidazol-5-carboxylic acid (5-methyl-2-oxo-1,-3 dioxol-4-yl)methyl ester, and has a CAS No. [144689-63-4].
-
The structural formula is represented below:
 Olmesartan medoxomil, which is an angiotensin II receptor antagonist, is useful as an active ingredient in medicaments for treatment and prophylaxis of hypertension (for example, Patent documents 1 to 5 or Non-patent document 1 and 2). Techniques for producing high-purity olmesartan medoxomil are necessary for use of olmesartan medoxomil as a medicament.
Olmesartan medoxomil, which is an angiotensin II receptor antagonist, is useful as an active ingredient in medicaments for treatment and prophylaxis of hypertension (for example, Patent documents 1 to 5 or Non-patent document 1 and 2). Techniques for producing high-purity olmesartan medoxomil are necessary for use of olmesartan medoxomil as a medicament. -
Olmesartan medoxomil is produced from olmesartan by the steps described below, but there is the problem that olmesartan which is a starting material, olmesartan medoxomil dehydrate which is a by-product, or the like, is present as an impurity.


-
- Patent document 1: Japanese Examined Patent Application (Kokoku) No. Hei 7-121918 (Japanese Patent No. 2082519 )
- Patent document 2: US Patent No. 5616599
- Patent document 3: International Patent Publication No. WO2006/029056
- Patent document 4: International Patent Publication No. WO2006/029057
- Patent document 5: International Patent Publication No. WO2006/073519
-
- Non-patent document 1: J. Med. Chem., 39, 323-338 (1996)
- Non-patent document 2: Annu. Rep. Sankyo Res. Lab. (Sankyo Kenkyusho Nempo) 55, 1-91 (2003)
-
The prior art synthetic methods involve coupling reaction between the substituted imidazole and substituted biphenyl methyl bromide. J. Med. Chem. 1996 vol. 39 No.1, page 323-38 describes the synthesis of Olmesartan medoxomil as follow. [Scheme-1 ]

-
US 5616599 describes a process for the preparation of olmesartan medoxomil as follows.
-
[0006]4-(1-hydroxyl-1-methylethyl)-2-propyl imidazole-5carboxylic acid is reacted with 5-methyl-2-oxo-1, 3-dioxolene-4-yl)methyl chloride using N,N-diisopropylethyl amine as base in N,N-dimethyl acetamide at 60°C to give (5-methyl-2-oxo-1,3-dioxolen-4-yl)methyl4-(1-hydroxy-1-methylethyl)-2-propyl imidazole-5-carboxylate. The resulting product is coupled with N-(triphenylmethyl)-5-[4′-(bromomethyl)biphenyl-2-yl]tetrazole [herein referred to as TTBB] at 60°C in N, N-dimethyl acetamide using potassium carbonate as base to give protected olmesartan medoxomil. The protected olmesartan medoxomil is deprotected using 75 % acetic acid to give olmesartan medoxomil.
-
This process involves column chromatographic purification of intermediates which is not desirable on commercial scale operation.
-
US 5616599 describes another process for the preparation of olmesartan Medoxomil which involves addition of methyl Magnesium chloride on diethyl 2-propylimidazole-4, 5-dicarboxylate in tetrahydrofuran at -30 to -20°C to give ethyl-4-(1-hydroxy-1-methylethyl)-2-propylimidazole -5-carboxylate, which is coupled with TTBB using sodium hydride as base in N, N-dimethylformamide at 60°C to give ethyl-4-(1-hydroxy-1-methylethyl)2-propyl-1-[[2′-[2-(triphenylmethyl)-2H-tetrazol-. 5yl]biphenyl-4-yl]methyl]imidazole-5-carboxylate. The product thus formed is hydrolyzed using lithium hydroxide monohydrate as base in 1,4-dioxane at 5-10°C to give lithium salt of 4-(1-hydroxy-1-methylethyl)-2-propyl-1-[[2′-[2-(triphenylmethyl)-2H-tetrazol-5yl]biphenyl-4-yl]methyl]imidazole-5-carboxylic acid, which is then coupled with 5-methyl-2-oxo-(1,3-dioxolene-4-yl)methyl chloride using K2CO3 as base in N,N-dimethylacetamide at 50°C to give trityl protected olmesartan medoxomil which on deprotection using 75% acetic acid gives Olmesartan Medoxomil.
-
During the condensation of ethyl-4-(1-hydroxy-1-methylethyl)-2-propylimidazole -5-carboxylate, with TTBB using sodium hydride as base in N, N-dimethylformamide, various impurities are formed, and isolation of the product involves extractive workup.



 , Ian R. Baxendale, Steven V. Ley and Nikzad Nikbin Corresponding author email
, Ian R. Baxendale, Steven V. Ley and Nikzad Nikbin Corresponding author emailThe structurally related imidazole core of olmesartan is formed in a different fashion (Scheme 36). Condensation between diaminomaleonitrile and trimethyl orthobutyrate furnishes the trisubstituted imidazole 181 in high yield [53,54]. Acid-mediated nitrile hydrolysis followed by esterification results in the corresponding diester unit 182. Treatment of 182 with four equivalents of methylmagnesium chloride in a mixture of diethyl ether and dichloromethane selectively provides tertiary alcohol 183. In subsequent steps this imidazole is alkylated with the tetrazole containing biphenyl appendage, followed by ester hydrolysis and alkylation of the resulting carboxylate with 4-(chloromethyl)-5-methyl-2-oxo-1,3-dioxole to yield olmesartan (Scheme 36).
![[1860-5397-7-57-i36]](https://i0.wp.com/beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i36.png)
- 53…….Yanagisawa, H.; Fujimoto, K.; Amemiya, Y.; Shimoji, Y.; Kanazaki, T.; Koike, H.; Sada, T. Angiotensin II Antagonist 1-Biphenylmethylimidazole Compounds and their Therapeutic Use. U.S. Patent 5,616,599, April 1, 1997.
Return to citation in text: [1] - 54……….Yanagisawa, H.; Amemiya, Y.; Kanazaki, T.; Shimoji, Y.; Fujimoto, K.; Kitahara, Y.; Sada, T.; Mizuno, M.; Ikeda, M.; Miyamoto, S.; Furukawa, Y.; Koike, H. J. Med. Chem. 1996, 39, 323–338. doi:10.1021/jm950450f
Return to citation in text: [1]
Reacting ethyl-4-(1-hydroxy-1-methylethyl)-2-propylimidazole-5-carboxylate with N-(Triphenylmethyl)-5-[4′-(bromomethyl)biphenyl-2- yl]tetrazole in an organic solvent in presence of a base and a phase transfer catalyst in non-aqueous system to give after workup, ethyl-4-(1-hydroxy-1-methylethyl)-2-propyl-1-[[2′-[2-(triphenylmethyl)-2H-tetrazol-5yl]biphenyl-4-yl]methyl]imidazole-5-carboxylate, which is further processed, by following improved reaction conditions in three steps to provide substantially pure [HPLC purity 99.3 to 99.7 %] olmesartan medoxomil.
-
To a 3M solution of MeMgCl(55.86 g, 0.74 mol) in tetrahydrofuran was added a solution of diethyl 2-propyl imidazole- 4,5-dicarboxylate (50 g,0.19 mol) in tetrahydrofuran (200 ml) at -10 to 0°C under N2 atmosphere. The mixture was stirred at -5 to 0°C for 10 minutes. Reaction mass was quenched into 400 ml 25 % ammonium chloride solution followed by extraction with ethyl acetate (300 ml). The organic phase was separated, washed with brine, dried over Na2SO4, and concentrated in vacuo to give a syrup, which was crystallized using diisopropyl ether.
Yield: 85-90 %,
Purity by HPLC: 88-93 %.
1H-NMR (CDCl3) δ: 7.8-8.1 (s, 1H), 5.8(s, 1H)., 4.35(q, 2H), 2.68(t, 2H), 1.78(m, 2H), 1.61(s, 6H), 1.36(t, 3H), 0.96(t, 3H).
Example-2Preparation of Ethyl-4-(1-hydroxy-1-methylethyl)-2-propyl-1-[[2′-[2-(triphenylaiethyl)-2H-tetrazol-5yl]biphenyl-4-yl]methyl]imidazole
- -5-
carboxylate
-
Mixture of Ethyl-4-(1-hydroxy-1-methylethyl)-2-propylimidazole -5-carboxylate (41 g, 0.17 mol), potassium carbonate (47g, 0.34 mol) and tetrabutylammonium bromide (4.9 g, 0.01 mol) in acetone was stirred at room temperature for 1hr. Then TTBB (93% Purity, 92.89g, 0.15 mol) was charged, refluxed for 14hrs. Potassium salts were filtered off from the reaction mass and the filtrate was charcoalised for 1hr. It was filtered over celite bed and the filtrate was distilled off completely to get a semi solid mass. 250 ml of Methanol was added to the residue and stirred for 2-3 hrs to give a solid product, which was filtered and washed with chilled methanol and dried.
Yield: 80-85%,
Purity by HPLC: 85-90%.
1H-NMR (CDCl3) δ: 7.8-8.1 (m, 1H), 6.7-7.61 (m, 22H), 5.78 (bs, 1H), 5.38(s, 2H), 4.12 (q, 2H), 2.52 (t, 2H), 1.64(s, 6H), 1.5-1.8(m, 2H), 1.08(t, 3H), 0.88(t, 3H).
Example-3Preparation of lithium salt of 4-(1-hydroxy-1-methylethyl) 2-propyl-1-[[2′-[2-(triphenylmethyl)-2H-tetrazol-5yl] biphenyl-4-yl] methyl] imidazole -5-carboxylic acid
-
To a solution of Ethyl-4-(1-hydroxy-1-methylethyl) 2-propyl- 1-[[2′-[2-(triphenylmethyl)-2H-tetrazol-5yl]biphenyl-4-yl]methyl]imidazole-5-carboxylate (105 g, 0.14 mol) in tetrahydrofuran , was added LiOH.H2O (7.8 g, 0.18 mol) solution below 10°C. The reaction mixture was stirred at room temperature for 15 hours. Reaction mass was concentrated under vacuum at 35°C to 1/4 th of its volume. 300 ml of ethyl acetate and NaCl (130 g) were added to the residue under stirring. The organic phase was separated, dried over sodium sulphate and concentrated under vacuum to get the product. The crude product was taken as such to the next stage.
Example-4Preparation of trityl protected olmesartan medoxomil
-
To the solution of lithium salt of 4-(1-hydroxy-1-methylethyl) 2-propyl- 1-[[2′-[2-(triphenylmethyl)-2H-tetrazol-5yl] biphenyl-4-yl] methyl] imidazole -5-carboxylic acid , (97 g, 0.13 mol) in N, N-dimethyl acetamide(200 ml) was added triethylamine(12.7 g, 0.12 mol), stirred at room temperature for 0.5 hours. 5-methyl-2-oxo-(1,3-dioxolene-4-yl)methyl chloride (85% purity, 37.3 g, 0.25 mol) was added below 10°C. The mixture was stirred at 50-55°C for 4 hours, checked TLC. Dichloromethane (400 ml) and chilled water (500 ml) were added under stirring. The organic phase was separated, given brine wash (50 ml), dried over sodium sulphate and concentrated under vacuum to get a residue. To the residue methanol was added, stirred for 1hr, cooled to 5-10°, filtered and washed with chilled methanol and dried.
Yield: 75-80%,
Purity by HPLC: 96-98%.
1H-NMR (CDCl3) δ: 7.87(d, 1H), 6.90-7.52(m, 20H), 6.68(d, 2H), 5.61(s, 1H), 5.3(s, 2H), 4.7(s, 2H), 2.54(t, 2H), 1.97(s, 3H), 1.6-1.75(m, 2H), 1.62(s, 6H), 0.87(t, 3H).
Example-5Preparation of olmesartan medoxomil
-
To the suspension of trityl protected olmesartan medoxomil (50g, 0.06 mol) in 250 ml 75% acetic acid was stirred at 50-55°C for 1.5hrs and cooled to 5-10°C. The by-product trityl alcohol was filtered off and washed with 75% acetic acid. The filtrate was concentrated under vacuum to get syrup, which was crystallized using isopropyl alcohol.
Yield: 85-88%,
Purity by HPLC: 95-98%.
The material was further purified with ethyl methyl ketone. It was filtered and washed with ethyl methyl ketone and dried to give substantially pure olmesartan medoxomil.
Yield: 70-75%,
Purity by HPLC: 99.3-99.7%.
1H-NMR (CDCl3) δ: 7.81(dd, 1H), 7.43-7.6(m, 3H), 7.09d, 2H), 6.79(d, 2H), 5.41(s, 1H),4.95(s, 1H), 2.56(t, 3H), 2.17(s, 3H), 1.58-1.69(m, 2H), 1.58(s, 6H), 0.92(t,3H).
Ipragliflozin

Ipragliflozin
ASP-1941 , 1(S)-[3-(1-benzothien-2-ylmethyl)-4-fluorophenyl]-1-deoxy-beta-D-glucopyranose L-proline cocrystal

Kotobuki (Originator)
| (1S)-1,5-Anhydro-1-C-[3-[(1-benzothiophen-2-yl)methyl]-4-fluorophenyl]-D-glucitol |
| Molecular Formula | C21H21FO5S | |
| Molecular Weight | 404.45 | |
| CAS Registry Number | 761423-87-4 |
Ipragliflozin (formerly ASP1941) has been filed in Japan on the back of phase III trials which showed that it could provide significant reductions in glycated haemoglobin levels (HbA1c) levels – a marker of glucose control over time – compared to placebo
According to Astellas’ latest R&D pipeline update in February 2013, Astellas is developing ipragliflozin only in Japan. The same document in August 2012 indicated it was also carrying out phase II studies with the drug in the US and Japan.
Astellas Pharma Inc.: Submits Application for Marketing Approval of
Ipragliflozin (ASP1941), SGLT2 Inhibitor for Treatment of
Type 2 Diabetes, in Japan
TOKYO, March 13, 2013 – Astellas Pharma Inc. (“Astellas”; Tokyo:4503; President and CEO:
Yoshihiko Hatanaka) announced today that it has submitted a market authorization application for aSGLT2 inhibitoripragliflozin (generic name; development code: ASP1941) to the Ministry of Health, Labour and Welfare in Japan seeking an approval forthe indication of type 2 diabetes.
Ipragliflozin is a selective SGLT2 (sodium-glucose co-transporter 2)inhibitor discovered through research collaboration with Kotobuki Pharmaceutical Co., Ltd. SGLTs are membrane proteins that
exist on the cell surface and transfer glucose into cells. SGLT2 is a subtype of the sodium-glucose co-transporters and plays a key role in the reuptake of glucose in the proximal tubule of the kidneys.
Ipragliflozin reduces blood glucose levels by inhibiting the reuptake of glucose.
In the Phase III pivotal study in monotherapy for type 2 diabetesin Japan, ipragliflozin
demonstrated significant decreases of HbA1c, an index of glycemic control, in change from baseline compared to placebo. Based on the safety resultsin this study, ipragliflozin was safe and well tolerated. Patients with type 2 diabetes generally need combination therapy, so it is important
for a novel oral hypoglycemic agent to be safe to use with existing diabetes therapies. In this regard, Astellas has conducted six Phase III studies to investigate the safety and efficacy of ipragliflozin
used in combination with other hypoglycemic agentsfor a long term period. In these Phase IIIstudies, effectiveness and favorable safety of ipragliflozin was confirmed even in combination with
other hypoglycemic agents.
Astellas expects to provide an additional therapeutic option and further contribute to the treatment of type 2 diabetes by introducing ipragliflozin, an oral hypoglycemic agent with a novel mechanism
of action, into the Japanese market.
About Type 2 Diabetes
Diabetes (medically known as diabetes mellitus) is a disorder in which the body has difficulty regulating its blood glucose (sugar) level. There are two major types of diabetes: type 1 and type 2.
Type 2 diabetes (formerly called non-insulin-dependent diabetes mellitus or adult-onset diabetes) is a disorder that is characterized by high blood glucose in the context of insulin resistance and relative insulin deficiency. Patients are instructed to increase exercise and diet restrictions, but most
require treatment with an anti-diabetic agent to control blood glucose.
structure:

The compound and methods of its synthesis are described in WO 2004/080990, WO 2005/012326 and WO 2007/114475 for example.
The gluconolactone method: In 1988 and 1989 a general method was reported to prepare C-arylglucosides from tetra-6>-benzyl protected gluconolactone, which is an oxidized derivative of glucose (see J. Org. Chem. 1988, 53, 752-753 and J. Org. Chem. 1989, 54, 610- 612). The method comprises: 1) addition of an aryllithium derivative to the hydroxy-protected gluconolactone to form a hemiketal (a.k.ci., a lactol), and 2) reduction of the resultant hemiketal with triethylsilane in the presence of boron trifluoride etherate. Disadvantages of this classical, but very commonly applied method for β-C-arylglucoside synthesis include:
1) poor “redox economy” (see J. Am. Chem. Soc. 2008, 130, 17938-17954 and Anderson, N. G. Practical Process Research & Development, 1st Ed.; Academic Press, 2000 (ISBN- 10: 0120594757); pg 38)— that is, the oxidation state of the carbon atom at CI, with respect to glucose, is oxidized in the gluconolactone and then following the arylation step is reduced to provide the requisite oxidation state of the final product. 2) due to a lack of stereospecificity, the desired β-C-arylglucoside is formed along with the undesired a-C-arylglucoside stereoisomer (this has been partially addressed by the use of hindered trialkylsilane reducing agents (see Tetrahedron: Asymmetry 2003, 14, 3243-3247) or by conversion of the hemiketal to a methyl ketal prior to reduction (see J. Org. Chem. 2007, 72, 9746-9749 and U.S. Patent 7,375,213)).
Oxidation Reduction

Glucose Gluconoloctone Hemiketal a-anomer β-anomer
R = protecting group
The metalated glucal method: U.S. Patent 7,847,074 discloses preparation of SGLT2 inhibitors that involves the coupling of a hydroxy-protected glucal that is metalated at CI with an aryl halide in the presence of a transition metal catalyst. Following the coupling step, the requisite formal addition of water to the C-arylglucal double bond to provide the desired C-aryl glucoside is effected using i) hydroboration and oxidation, or ii) epoxidation and reduction, or iii) dihydroxylation and reduction. In each case, the metalated glucal method represents poor redox economy because oxidation and reduction reactions must be conducted to establish the requisite oxidation states of the individual CI and C2 carbon atoms.
U.S. Pat. Appl. 2005/0233988 discloses the utilization of a Suzuki reaction between a CI -boronic acid or boronic ester substituted hydroxy-protected glucal and an aryl halide in the presence of a palladium catalyst. The resulting 1- C-arylglucal is then formally hydrated to provide the desired 1- C-aryl glucoside skeleton by use of a reduction step followed by an oxidation step. The synthesis of the boronic acid and its subsequent Suzuki reaction, reduction and oxidation, together, comprise a relatively long synthetic approach to C-arylglucosides and exhibits poor redox economy. Moreover, the coupling catalyst comprises palladium which is toxic and therefore should be controlled to very low levels in the drug substance.

R = protecting group; R’ = H or alkyl
The glucal epoxide method: U.S. Patent 7,847,074 discloses a method that utilizes an organometallic (derived from the requisite aglycone moiety) addition to an electrophilic epoxide located at C1-C2 of a hydroxy-protected glucose ring to furnish intermediates useful for SGLT2 inhibitor synthesis. The epoxide intermediate is prepared by the oxidation of a hydroxy- protected glucal and is not particularly stable. In Tetrahedron 2002, 58, 1997-2009 it was taught that organometallic additions to a tri-6>-benzyl protected glucal-derived epoxide can provide either the a-C-arylglucoside, mixtures of the a- and β-C-arylglucoside or the β-C-arylglucoside by selection of the appropriate counterion of the carbanionic aryl nucleophile (i.e., the
organometallic reagent). For example, carbanionic aryl groups countered with copper (i.e., cuprate reagents) or zinc (i.e., organozinc reagents) ions provide the β-C-arylglucoside, magnesium ions provide the a- and β-C-arylglucosides, and aluminum (i.e., organoaluminum reagents) ions provide the a-C-arylglucoside.

or Zn
The glycosyl leaving group substitution method: U.S. Patent 7,847,074, also disclosed a method comprising the substitution of a leaving group located at CI of a hydroxy-protected glucosyl species, such as a glycosyl halide, with a metalated aryl compound to prepare SGLT2 inhibitors. U.S. Pat. Appl. 2011/0087017 disclosed a similar method to prepare the SGLT2 inhibitor canagliflozin and preferably diarylzinc complexes are used as nucleophiles along with tetra- >-pivaloyl protected glucosylbromide.

Glucose Glucosyl bromide β-anomer
Methodology for alkynylation of 1,6-anhydroglycosides reported in Helv. Chim. Acta. 1995, 78, 242-264 describes the preparation of l,4-dideoxy-l,4-diethynyl^-D-glucopyranoses (a. La., glucopyranosyl acetylenes), that are useful for preparing but-l,3-diyne-l,4-diyl linked polysaccharides, by the ethynylating opening (alkynylation) of partially protected 4-deoxy-4-C- ethynyl-l,6-anhydroglucopyranoses. The synthesis of β-C-arylglucosides, such as could be useful as precursors for SLGT2 inhibitors, was not disclosed. The ethynylation reaction was reported to proceed with retention of configuration at the anomeric center and was rationalized (see Helv. Chim. Acta 2002, 85, 2235-2257) by the C3-hydroxyl of the 1,6- anhydroglucopyranose being deprotonated to form a C3-0-aluminium species, that coordinated with the C6-oxygen allowing delivery of the ethyne group to the β-face of the an oxycarbenium cation derivative of the glucopyranose. Three molar equivalents of the ethynylaluminium reagent was used per 1 molar equivalent of the 1,6-anhydroglucopyranose. The
ethynylaluminium reagent was prepared by the reaction of equimolar (i.e., 1:1) amounts of aluminum chloride and an ethynyllithium reagent that itself was formed by the reaction of an acetylene compound with butyllithium. This retentive ethynylating opening method was also applied (see Helv. Chim. Acta. 1998, 81, 2157-2189) to 2,4-di-<9-triethylsilyl- 1,6- anhydroglucopyranose to provide l-deoxy-l-C-ethynyl- -D-glucopyranose. In this example, 4 molar equivalents of the ethynylaluminium reagent was used per 1 molar equivalent of the 1,6- anhydroglucopyranose. The ethynylaluminium regent was prepared by the reaction of equimolar (i.e., 1: 1) amounts of aluminum chloride and an ethynyl lithium reagent that itself was formed by reaction of an acetylene compound with butyllithium.
It can be seen from the peer-reviewed and patent literature that the conventional methods that can be used to provide C-arylglucosides possess several disadvantages. These include (1) a lack of stereoselectivity during formation of the desired anomer of the C- arylglucoside, (2) poor redox economy due to oxidation and reduction reaction steps being required to change the oxidation state of CI or of CI and C2 of the carbohydrate moiety, (3) some relatively long synthetic routes, (4) the use of toxic metals such as palladium, and/or (5) atom uneconomic protection of four free hydroxyl groups. With regard to the issue of redox economy, superfluous oxidation and reduction reactions that are inherently required to allow introduction of the aryl group into the carbohydrate moiety of the previously mention synthetic methods and the subsequent synthetic steps to establish the required oxidation state, besides adding synthetic steps to the process, are particular undesirable for manufacturing processes because reductants can be difficult and dangerous to operate on large scales due to their flammability or ability to produce flammable hydrogen gas during the reaction or during workup, and because oxidants are often corrosive and require specialized handling operations (see Anderson, N. G. Practical Process Research & Development, 1st Ed.; Academic Press, 2000 (ISBN-10: 0120594757); pg 38 for discussions on this issue).
-
The C-glycoside derivative represented by the formula (1) and its salt [hereinafter, they are referred to as “compound (1)” or “compound of formula (1)” in some cases] is known to be useful for treatment and prevention of diabetes such as insulin-dependent diabetes (type 1 diabetes), non-insulin-dependent diabetes (type 2 diabetes) and the like and various diabetes-related diseases including insulin-resistant diseases and obesity (Patent Literature 1).
-


-
The method for producing the C-glycoside derivative represented by the formula (1), described in the Patent Literature 1 is understood to be represented by the below-shown reaction formula (I), by referring to the Examples and Reference Examples, described in the Patent Literature 1. Roughly explaining, it is a method which comprises reacting [1-benzothien-2-yl(5-bromo-2-fluorophenyl)methoxy]tert-butyl)dimethylsilane (synthesized in accordance with Reference Example 37 of the Literature) in a manner shown in Example 65 of the Literature, to obtain (1S)-1,5-anhydro-1-[3-(1-benzothien-2-ylmethyl)-4-fluorophenyl]-2,3,4,6-tetra-O-benzyl-D-glucitol and then reacting the obtained compound in accordance with Example 100 of the Literature to synthesize intended (1S)-1,5-anhydro-1-C-[3-(1-benzothiophene-2-ylmethyl)-4-fluorophenyl]-D-glucitol.
-


-
However, the method for producing the C-glycoside derivative of the formula (1), disclosed in the Patent Literature 1 is not industrially satisfactory in yield and cost, as is seen in later-shown Reference Example 1 of the present Description.
-
For example, as described later, the method includes a step of low product yield (for example, a step of about 50% or lower yield) and the overall yield of the C-glycoside derivative (final product) represented by the formula (1) from the compound (8) (starting raw material) is below 7%; therefore, the method has problems in yield and cost from the standpoint of medicine production and has not been satisfactory industrially. In addition, the method includes an operation of purification by column chromatography which uses chloroform as part of purification solvents; use of such a solvent poses a problem in environmental protection and there are various restrictions in industrial application of such an operation; thus, the method has problems in providing an effective medicine.
-
Also, an improved method of conducting an addition reaction with trimethylsilyl carbohydrate instead of benzyl carbohydrate and then conducting deprotection for acetylation, is known for a compound which has a structure different from that of the compound of the formula (1) but has a structure common to that of the compound of the formula (1) (Patent Literature 2). It is described in the Patent Literature 2 that the improved method enhances the overall yield to 6.2% from 1.4%. Even in the improved method, however, the yield is low at 6.2% which is far from satisfaction in industrial production.
- Patent Literature 1: WO 2004/080990 Pamphlet
- Patent Literature 2: WO 2006/006496 Pamphlet





http://www.google.com/patents/EP2105442A1
-
Into a tetrahydrofuran (20 ml) solution of benzo[b]thiophene (5.0 g) was dropwise added a n-hexane solution (25 ml) of n-butyl lithium (1.58 M) at -78°C in an argon atmosphere, followed by stirring at -78°C for 10 minutes. Into this solution was dropwise added a tetrahydrofuran (80 ml) solution of 5-bromo-2-fluorobenzaldehyde (8.0 g), followed by stirring at -78°C for 2.5 hours. The temperature of the reaction mixture was elevated to room temperature. Water was added thereto, followed by extraction with ethyl acetate. The organic layer was washed with a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, filtered, and concentrated. The residue was purified by silica gel column chromatography (n-hexane/ethyl acetate) to obtain 1-benzothien-2-yl(5-bromo-2-fluorophenyl)methanol (10.5 g, yield: 83.6%).
1H-NMR (CDCl3): δ
2.74 (1H, d), 6.35 (1H, d), 6.93 (1H, dd), 7.14 (1H, s), 7.27-7.38 (2H, m), 7.39 (1H, m), 7.68 (1H, dd), 7.74 (2H, m)
- First step: synthesis of 1-benzothien-2-yl(5-bromo-2-fluorophenyl)methanol
Second step: synthesis of [1-benzothien-2-yl(5-bromo-2-fluorophenyl)methoxy](tert-butyl)dimethylsilane
-
To a dimethylformamide (20 ml) solution of 1-benzothien-2-yl(5-bromo-2-fluorophenyl)methanol (5.0 g) were added imidazole (1.3 g), a catalytic amount of 4-(dimethylamino)pyridine and tert-butyldimethylchlorosilane (2.7 g), followed by stirring at room temperature for 7 hours. To the reaction mixture was added a saturated aqueous ammonium chloride solution, followed by extraction with ethyl acetate. The organic layer was washed with a saturated aqueous ammonium chloride solution and a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, filtered and concentrated. The residue was purified by silica gel column chromatography (n-hexane/ethyl acetate) to obtain [1-benzothien-2-yl(5-bromo-2-fluorophenyl)methoxy](tert-butyl)dimethylsilane (5.22 g, yield: 78.0%).
MS: 451 (M+)
1H-NMR (CDCl3): δ
0.05 (3H, s), 0.11 (3H, s), 0.95 (9H, s), 6.34 (1H, s), 6.91 (1H, t), 7.08 (1H, d), 7.23-7.38 (2H, m), 7.64-7.68 (1H, m), 7.75-7.28 (2H, m)
Third step: Synthesis of 1-C-[3-(1-benzothien-2-yl{[tert-butyl-(dimethyl)silyloxy}methyl)4-fluorophenyl]-2,3,4,6-tetra-O-benzyl-D-glucopyranose
-
Into a tetrahydrofuran (15 ml) solution of [1-benzothien-2-yl(5-bromo-2-fluorophenyl)methoxy](tert-butyl)dimethylsilane (1.5 g) was dropwise added a n-hexane solution (2.2 ml) of n-butyl lithium (1.58 M) in an argon atmosphere at -78°C, followed by stirring at -78°C for 30 minutes. Into the solution was dropwise added a tetrahydrofuran (20 ml) solution of 2,3,4,6-tetra-O-benzyl-D-glucono-1,5-lactone (1.9 g), followed by stirring at -78°C for 15 minutes and then at 0°C for 1.5 hours. To the reaction mixture was added a saturated aqueous ammonium chloride solution, followed by extraction with ethyl acetate. The organic layer was washed with a saturated aqueous ammonium chloride solution and a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, filtered and concentrated. The residue was purified by silica gel column chromatography (n-hexane/chloroform/acetone) to obtain 1-C-[3-(1-benzothien-2-yl{[tert-butyl-(dimethyl)silyloxy}methyl)-4-fluorophenyl]-2,3,4,6-tetra-O-benzyl-D-glucopyranose (1.52 g, yield: 50.2%). MS: 933 (M+Na)
Fourth step: Synthesis of 1-C-{3-[1-benzothien-2-yl(hydroxy)methyl]-4-fluorophenyl}-2,3,4,6-tetra-O-benzyl-D-glucopyranose
-
To a tetrahydrofuran (15 ml) solution of 1-C-[3-(1-benzothien-2-yl{[tert-butyl-(dimethyl)silyloxy}methyl)-4-fluorophenyl]-2,3,4,6-tetra-O-benzyl-D-glucopyranose (1.52 g) was added a tetrahydrofuran solution (2.0 ml) of tetrabutylammonium fluoride (1.0 M), followed by stirring at room temperature for 1 hour. The reaction mixture was concentrated per se. The residue was purified by silica gel column chromatography (n-hexane/ethyl acetate) to obtain 1-C-{3-[1-benzothien-2-yl(hydroxy)methyl]-4-fluorophenyl}-2,3,4,6-tetra-O-benzyl-D-glucopyranose (0.99 g, yield: 74.7%). MS: 819 (M+Na), 779 (M+H-H2O)
Fifth step: Synthesis of (1S)-1,5-anhydro-1-[3-(1-benzothien-2-ylmethyl)-4-fluorophenyl]-2,3,4,6-tetra-O-benzyl-D-glucitol
-
To an acetonitrile (5.0 ml) solution of 1-C-{3-[1-benzothien-2-yl(hydroxy)methyl]-4-fluorophenyl}-2,3,4,6-tetra-O-benzyl-D-glucopyranose (500 mg) were added triethylsilane (175 mg) and boron trifluoride-diethyl ether complex (196 mg) in an argon atmosphere at -20°C, followed by stirring at -20°C for 5 hours. To the reaction mixture was added a saturated aqueous sodium bicarbonate solution, followed by extraction with chloroform. The organic layer was washed with a saturated aqueous sodium bicarbonate solution and a saturated aqueous sodium chloride solution, dried over anhydrous magnesium sulfate, filtered and concentrated. The residue was purified by silica gel column chromatography (n-hexane/ethyl acetate) to obtain (1S)-1,5-anhydro-1-[3-(1-benzothien-2-ylmethyl)-4-fluorophenyl]-2,3,4,6-tetra-O-benzyl-D-glucitol (150 mg, yield: 30.2%) MS: 787 (M+Na)
1H-NMR (CDCl3): δ
3.42-3.48 (1H, m), 3.55-3.58 (1H, m), 3.72-3.78 (4H, m), 3.83 (1H, d), 4.14-4.30 (3H, m), 4.39 (1H, d), 4.51-4.67 (4H, m), 4.83-4.94 (2H, m), 6.86-6.90 (1H, m), 6.98 (1H, brs), 7.06-7.37 (24H, m), 7.57-7.60 (1H, m), 7.66-7.69 (1H, m)
Sixth step: Synthesis of (1S)-1,5-anhydro-1-C-[3-(1-benzothiophene-2-ylmethyl)-4-fluorophenyl]-D-glucitol
-
To a dichloromethane (10 ml) solution of (1S)-1,5-anhydro-1-[3-(1-benzothien-2-ylmethyl)-4-fluorophenyl]-2,3,4,6-tetra-O-benzyl-D-glucitol (137 mg) were added pentamethylbenzene (382 mg) and a n-heptane solution (0.75 ml) of boron trichloride (1.0 M) in an argon atmosphere at -78°C, followed by stirring at -78°C for 3 hours. Methanol was added to the reaction mixture, the temperature of the resulting mixture was elevated to room temperature, and the mixture was concentrated per se. The residue was purified by silica gel column chromatography (chloroform/methanol) to obtain (1S)-1,5-anhydro-1-C-[3-(1-benzothiophene-2-ylmethyl)-4-fluorohenyl]-D-glucitol OR IPRAGLIFLOZIN (63 mg, yield: 87.8%).
1H-NMR (CD3OD): δ
3.29-3.48 (4H, m), 3.68 (1H, dd), 3.87 (1H, dd), 4.11 (1H, d), 4.20-4.29 (2H, m), 7.03 (1H, s), 7.08 (1H, dd), 7.19-7.29 (2H, m), 7.35 (1H, m), 7.42 (1H, dd), 7.64 (1H, d), 7.72 (1H, d)
(1S)-1,5-anhydro-1-C-[3-(1-benzothiophene-2-ylmethyl)-4-fluorohenyl]-D-glucitol OR IPRAGLIFLOZIN
