
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 263)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
The US FDA has issued a Warning Letter to Tianjin Zhongan Pharmaceutical Co. Ltd. in Tianjin, China.
![]()
FDA issues Warning Letter for API Facility
http://www.gmp-compliance.org/enews_4367_FDA%20issues%20Warning%20Letter%20for%20API%20Facility_8509,S-WKS_n.html
The US FDA has issued a Warning Letter to Tianjin Zhongan Pharmaceutical Co. Ltd. in Tianjin, China. The company produces APIs and failed to establish adequate GMP procedures at the facility. Read more about the FDA Warning Letter.
FDA issues Warning Letter for API Facility
The US FDA has issued a Warning Letter to Tianjin Zhongan Pharmaceutical Co. Ltd. in Tianjin, China. The company produces APIs and failed to establish adequate GMP procedures at the facility.
For quite some time India was in the center of attention and very little was heard about GMP problems in China (see also RAPS article). This is a bit surprising because a number of non-compliant facilities have been detected in the past. Also the facilities which caused the Heparin Scandal were located in China. The last enforcement action from FDA which became public referred to Import Alerts for the manufacturer Zhejiang Jiuzhou Pharmaceutical and for Zhejiang Zonebanne in China.
The new Warning Letter for Tianjin Zhongan Pharmaceutical lists a number of different non compliance findings. These findings refer to equipment cleaning (risk of cross contamination), not adequate Change Control procedures and failure to adequately review and investigate product deviations. However, in difference to the Warning Letters sent to Indian manufacturers recently data integrity issues have not been detected.
Interestingly the API manufacturer Tianjin Zhongan Pharmaceutical is not listed in EudraGMDP the inspection database in the EU. No entry for GMP Certificates or GMP Non-Compliance Report are available.
Source: FDA Warning Letter to Tianjin Zhongan Pharmaceutical

Tianjin Zhong’an Pharmaceutical Company Ltd. locates in the southwest of Tianjin. Set up in 1988, the company underwent operation mechanism reform in 2002 and acquired the present name.
The company covers an area of 82,000 square meters, with a construction area of 31,600 square meters and an afforested area of 13,300 square meters. It has over 600 staff members, more than 50 of whom have been conferred intermediate or advanced titles of technical post. The company produces 10 major chemical bulk pharmaceuticals, including Caffeine, Theophylline, Aminophylline, Metronidazole, Metronidazole Benzoate, Nifedipine, Secnidazole and Xanthinol Nicotinate. Its annual production capacity amounts to 4000 ton, with a sales volume of approximately RMB 250 million. Two leading products in the company are Caffeine and Metronidazole.
Zhong’an Pharmaceutical enjoys a self-management power over import and export., 90% of its products are aimed at international market, with 60% of which sold directly to world-renowned pharmaceutical and beverage enterprises. It has established a sales network which covers more than 30 countries and regions, including some European and American countries, Hong Kong, Taiwan and Macao, countries in Southeast and Midwest Asia, and Russia.
Zhong’an Pharmaceutical has set up a full set of quality assurance system, and owns a central laboratory with advanced analyse instruments that has got the title of Export Enterprise Lab and approved by Tianjin entry-exit inspection and quarantine bureau. All workshops of the company are dedicated to avoid the issue of cross pollution. Every stage of production strict complies with the cGMP, which led to the products has been enjoying a high reputation both domestic and overseas market.
Since 2000, the company has obtained GMP and ISO9001certificates, and conferred the title of Tianjin High-new Tech Enterprise and Municipal-level Key Technical Center. Both of leading products Metronidazole and Caffeine have got COS issued by EDQM and DMF register number from US FDA. Also Caffeine has got “Foreign Manufacturer Validation Certificate” from Japan Health Ministry and KOF-K and Halal certificates.
US Orphan Drug Market Outlook 2018 ……….download available

US Orphan Drug Market Outlook 2018
Academia.edu
US Orphan Drug Pipeline Insight by Phase & Indication 5.1 Research 5.2 Preclinical 5.3 Phase I 5.4 Phase I/II 5.5 Phase II 5.6 Phase II/III 5.7 Phase III …

http://www.academia.edu/7453102/US_Orphan_Drug_Market_Outlook_2018 …………… download at this site
![]()
Market Overview
In the largest market for orphan drugs, USA, there was a shortage of adequate therapies for treating many rare diseases. These therapies were not developed as companies did not expect these drugs to be highly profitable. Hence there was a lack of interest and thus investment on the part of pharma companies in the USA. Therefore, the FDA introduced incentives for developing such drugs. This step taken by the FDA was successful in creating a thriving market for orphan drugs. It was in the USA first that a special law exclusively for governing orphan drugs was framed in the form of the Orphan Drug Act of 1983. This led to an increase in the popularity of orphan drugs. The FDA also has been continuously increasing its efforts to support this market by providing significant financial and non-financial incentives to the pharmaceutical companies to attract them. This has been one of the major drivers of growth for the US orphan drugs market.
Figure 3-1: US Orphan Drug Market (US$ Billion), 2012-2018
2012201320142015201620172018
Source: KuicK Research
see my profile
http://ictmumbai.academia.edu/AnthonyMelvinCrastoPhD

Study Finds Shu Gan Liang Xue Herbal Formula Has Breast Cancer Anti Tumor Effect
There are a host of herbal formulas that show anti tumor properties for various cancers in clinical studies. Chinese researchers publishing in the Journal of Ethnopharmacology recently conducted a study looking at the effect of Shu Gan Liang Xue Formula on breast cancer tumors – particularly estrogen receptor positive breast cancer line ZR-75-1. Researchers investigated these anti-tumor functions in vitro and in vivo.
Shu Gan Liang Xue is a traditional Chinese herbal formula, it is comprised of the following herbs:
- Chai Hu
- Bai Shao
- Wu Wei Zi
- Dan Pi
- Bai Wei
- Zi Cao
Researchers understand that estrogen is a driver behind breast cancer. Two other substances, aromatase and steroid sulfatase are enzymes which contribute to estrogen synthesis. The researchers found that Shu Gan Liang Xue inhibits aromatase and steriod sulfatase which decreases their expression and their effect of estrogen synthesis. They found this both in vitro and in vivo.
In addition to…
View original post 87 more words
Too-clean homes may encourage child allergies, asthma
http://www.live5news.com/story/25711359/too-clean-homes-may-encourage-child-allergies-asthma-study
(HealthDay News) — Cleanliness may be next to godliness, but a home that’s too clean can leave a newborn child vulnerable to allergies and asthma later in life, a new study reports.
Infants are much less likely to suffer from allergies or wheezing if they are exposed to household bacteria and allergens from rodents, roaches and cats during their first year of life, the study found.
The results stunned researchers, who had been following up on earlier studies that found an increased risk of asthma among inner-city dwellers exposed to high levels of roach, mouse and pet droppings and allergens.
“What we found was somewhat surprising and somewhat contradictory to our original predictions,” said study co-author Dr. Robert Wood, chief of the Division of Allergy and Immunology at the Johns Hopkins Children’s Center in Baltimore. “It turned out to be completely opposite — the more of those three allergens…
View original post 719 more words
Odor Code for Food Based on a Few Volatile Substances

The actual flavor of a food is experienced through our sense of smell rather than with our tongue. However, of the large number of volatile compounds in foods, only about 230 are involved in the scent, as reported by German scientists in the journal Angewandte Chemie. The different smells derive from characteristic combinations of three to forty of these odorants.
The Discovery of MK-4256, a Potent SSTR3 Antagonist as a Potential Treatment of Type 2 Diabetes

somatostatin receptor antagonist
C27 H23 F N8 O
494.5229
3(R)-[4-(4-Fluorophenyl)-1H-imidazol-2-yl]-1(R)-(5-methyl-1,2,4-oxadiazol-3-yl)-1-(1-methyl-1H-pyrazol-4-yl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole
3(R)-[4-(4-Fluorophenyl)-1H-imidazol-2-yl]-1(R)-(5-methyl-1,2,4-oxadiazol-3-yl)-1-(1-methyl-1H-pyrazol-4-yl)-2,3,4,9-tetrahydro-1H-beta-carboline
1H-Pyrido[3,4-b]indole, 3-[5-(4-fluorophenyl)-1H-imidazol-2-yl]-2,3,4,9-tetrahydro-1-(5-methyl-1,2,4-oxadiazol-3-yl)-1-(1-methyl-1H-pyrazol-4-yl)-, (1R,3R)-
3-((1R,3R)-3-(4-(4-fluorophenyl)-1H-imidazol-2-yl)-1-(1-methyl-1H-pyrazol-4-yl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-1-yl)-5-methyl-1,2,4-oxadiazole
Merck & Co. (Originator)
Somatostatin srif1C (sst3) Antagonists
The Discovery of MK-4256, a Potent SSTR3 Antagonist as a Potential Treatment of Type 2 Diabetes
(ACS Medicinal Chemistry Letters) Thursday May 10th 2012
Author(s): Shuwen He, Zhixiong Ye, Quang Truong, Shrenik Shah, Wu Du, Liangqin Guo, Peter H. Dobbelaar, Zhong Lai, Jian Liu,Tianying Jian, Hongbo Qi, Raman K. Bakshi, Qingmei Hong, James Dellureficio, Alexander Pasternak, Zhe Feng, Reynalda deJesus, Lihu Yang, Mikhail Reibarkh, Scott A. Bradley, Mark A. Holmes, Richard G. Ball, Rebecca T. Ruck, Mark A. Huffman,Frederick Wong, Koppara Samuel, Vijay B. Reddy, Stan Mitelman, Sharon X. Tong, Gary G. Chicchi, Kwei-Lan Tsao, Dorina Trusca, Margaret Wu, Qing Shao, Maria E. Trujillo, George J. Eiermann, Cai Li, Bei B. Zhang, Andrew D. Howard, Yun-Ping Zhou,Ravi P. Nargund, William K. Hagmann,
DOI:10.1021/ml300063m
GO TO: [Article]
http://pubs.acs.org/doi/suppl/10.1021/ml300063m/suppl_file/ml300063m_si_001.pdf
The fast eluting diastereomer(52 mg, 10%) was 3-((1R,3R)-3-(4-(4-fluorophenyl)-1H-imidazol-2-yl)-1-(1-methyl-1H-pyrazol-4-yl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-1-yl)-5-methyl-1,2,4-oxadiazole(8, MK-4256).[α]D= +24.2, c=10 mg/mL in MeOH. LC-MS: m/z 495.3 (M+ H)+.
……………………………….
By:Ruck, RT (Ruck, Rebecca T.)[ 1 ] ; Huffman, MA (Huffman, Mark A.)[ 1 ] ; Stewart, GW (Stewart, Gavin W.)[ 2 ] ; Cleator, E (Cleator, Ed)[ 2 ] ; Kandur, WV (Kandur, Wynne V.)[ 1 ] ; Kim, MM (Kim, Mary M.)[ 1 ] ; Zhao, DL (Zhao, Dalian)[ 1 ]
ORGANIC PROCESS RESEARCH & DEVELOPMENT
Volume:16Issue:8Pages:1329-1337
DOI:10.1021/op300128c
Reprint Address: Ruck, RT (reprint author)
| Merck & Co Inc, Dept Proc Chem, Merck Res Labs, Rahway, NJ 07065 USA. |
Addresses:
| [ 1 ] Merck & Co Inc, Dept Proc Chem, Merck Res Labs, Rahway, NJ 07065 USA | |
| [ 2 ] Merck Sharp & Dohme Res Labs, Dept Proc Chem, Hoddesdon EN11 9BU, Herts, England |
http://pubs.acs.org/doi/abs/10.1021/op300128c
http://pubs.acs.org/doi/suppl/10.1021/op300128c/suppl_file/op300128c_si_001.pdf

Route development and demonstration on multikilogram scale for the first GMP delivery of MK-4256 are described. Key aspects of the convergent route include a regioselective green iodination, one-pot oxadiazole synthesis, and an efficient ketone Pictet–Spengler reaction with diastereomeric upgrade via crystallization to afford 6 kg of API. A recycle procedure augmented the yield of desired diastereomer in the Pictet–Spengler reaction from a mixture of diastereomers heavily enriched in the undesired diastereomer.
Residual metals were <10 ppm. Chiral method: Chiralcel OD-H, 250 mm × 4.6 mm, 40 °C, 1 mL/min, 260 nm, 30 min run time, 20% (1:1 IPA/MeOH) in heptane +0.1% TEA isocratic: rt (1): 7.61 min, rt (enantiomer-1): 14.45 min. By HPLC assay, final product was 99.60 LCAP 1, 0.17 LCAP 22, 0.24 LCAP enantiomer-22, enantiomer-1 was undetectable.
……………………………….
http://www.google.com/patents/WO2009011836A1?cl=en
WO 2009011836
Several methods for preparing the compounds of this invention are illustrated in the following Schemes and Examples. Starting materials are either commercially available or made by known procedures in the literature or as illustrated. The present invention further provides processes for the preparation of compounds of structural formula I as defined above, hi some cases the order of carrying out the foregoing reaction schemes may be varied to facilitate the reaction or to avoid unwanted reaction products. The following examples are provided for the purpose of illustration only and are not to be construed as limitations on the disclosed invention. All temperatures are degrees Celsius unless otherwise noted. The assignment of stereochemistry at the stereogenic carbon center indicated by an ** in Structure G of Scheme 3 from the Pictet-Spengler cyclization reaction to elaborate the β-carboline nucleus was determined using the aid of nuclear Overhauser effect (NOE) NMR spectroscopy. For a thorough discussion of the theory and application of NOE NMR spectroscopy, reference is made to Ernst, R.R.; Bodenhausen, B.; Wokaun, A., “Principles of Nuclear Magnetic Resonances in One or Two Dimensions”, Oxford University Press, 1992; Neuhaus, D.; Williamson, M. P., “The Nuclear Overhauser Effect in Structural and Conformational Analysis, 2nd Edition”, in “Methods in Stereochemical Analysis”, Marchand, A. P. (series editor), John A. Wiley and Sons, New York 2000.
SCHEME l

In Scheme 1 , substituted indoles A are treated with dimethylamine and paraformaldehyde in a Mannich reaction to form 3-(dimethylamino)methyl-indole B. Reaction of B with nitro ester C affords the 3-(indol-3-yl)-2-nitro-propionic acid, ethyl ester D which is reduced to tryptophan derivative E. Acylation of the amine in E and hydrolysis of the ester F affords the appropriately protected tryptophan derivative G. Separation of the isomers of F or G by chiral column chromatography yields the individual enantiomers.
SCHEME 2

In Scheme 2, substituted indole A is reacted with L-serine in the presence of acetic anhydride and acetic acid to form tryptophan B. Hydrolysis of the amide followed by amine protection affords the desired substituted tryptophan intermediate D.
SCHEME 3

In Scheme 3, substituted tryptophan derivative A is reacted with α-bromo-ketone B to afford ester C. Reaction with ammonium acetate effects cyclization to form substituted imidazole D. Removal of the N-Boc protecting group with acid yields indole imidazole E which is reacted with aldehydes or ketones F in a Pictet-Spengler cyclization to afford the desired product G.
EXAMPLE 21

(3i?Vr4-(4-Fluorophenvn-lH-imidazol-2-yll-l-r5-methyl-1.2.4-oxadiazol-3-vn-l-π-methyl-lH- pyrazol-4-yl)-23,4,9-tetrahydro-lH-β-carboline
(IR)-I -[4-(4-Fluorophenyl)- 1 H-imidazol-2-yl] -2-( 1 H-indol-3 -yl) ethanamine hydrochloride (370 mg, 1.037 mmol) [prepared by treatment of tert-butyl (lR)-2-(l H-indol-3 -yl)- l-(4-(4-fluorophenyl)-l H-imidazol-2-yl)- 1-ethylcarbamate with hydrochloric acid] was treated with pyridine (4 mL) followed by reaction with l-methyl-pyrazol-4-yl 5-methyl-l,2,4-triazol-3-yl ketone (Intermediate 22) (219 mg, 1.141 mmol). The reaction was heated under N2 (oil bath 7O0C) for 48 h followed by additional heating (oil bath 850C) for 3 d. The reaction mixture was concentrated and azeotroped with toluene. The residue was purified with preparative TLC eluting with 10% MeOH in CH2Cl2 to give (3i?)-[4-(4-fluorophenyl)-lH-imidazol-2-yl]-l-(5- methyl-1 ,2,4-oxadiazol-3-yl)-l-(l-methyl-pyrazol-4-yl)-2,3,4,9-tetrahydro-lH-β-carboline as a mixture of diastereoisomers which were separated by chiral ΗPLC. The isomers were characterized by an analytical chiral AD column eluting with 20% IPA in heptane. (3i?)-[4-(4- Fluorophenyl)- 1 H-imidazol-2-yl] – 1 -(5 -methyl- 1 ,2,4-oxadiazol-3 -yl)-( 1 R)-( 1 -methyl-pyrazol-4- yl)-2,3,4,9-tetrahydro-lH-β-carboline (faster eluting isomer: retention time 18.13 min): 1H NMR (500 MHz, MeOH-(I4): δ 7.74 (m, 2H), 7.65 (s, IH), 7.52 (m, 2H), 7.37 (m, 2H), 7.13 (m, 3H), 7.04 (s, IH), 4.47 (dd, IH), 3.87 (s, 3H), 3.24 (dd, IH), 3.16 (dd, IH), 2.63 (s, 3H). LC-MS: m/z 495.3 (M + H)+ (2.56 min).
(3i?)-[4-(4-Fluorophenyl)-lH-imidazol-2-yl]-l-(5-methyl-l,2,4-oxadiazol-3-yl)-(lS)-(l-methyl- pyrazol-4-yl)-2,3,4,9-tetrahydro-l//-β-carboline (slower eluting isomer: retention time 24.62 min): 1H NMR (500 MHz, MeOH-Cl4): δ 7.73 (m, 2H), 7.54 (d, IH), 7.48 (s, IH), 7.43 (s, IH),
7.40 (d, IH), 7.36 ( brs, IH), 7.13 (m, 3H), 7.06 (t, IH), 4.40 (dd, IH), 3.84 (s, 3H), 3.26 (dd, IH), 3.16 (dd, IH), 2.63 (s, 3H). LC-MS: m/z 495.3 (M + H)+ (2.61 min).
The relative stereochemistry of the two diastereoisomers was determined by nuclear Overhauser effect (nθe) NMR spectroscopy. The slower eluting diastereisoomer afforded an nOe signal between the C-3 and C-5 hydrogens on the C-I pyrazole and the C-3 hydrogen on the β-carboline and the faster eluting product did not. Therefore, the diastereoisomer that eluted first from the preparative chiral HPLC purification was assigned as the c/s-isomer (imidazole and pyrazole are cis) and the slower eluting isomer as the trørøs-isomer.
…………………..
Dobbelaar, P. H.; Du, W.; Guo, L.; Hagmann, W. K.; He, S.; Jian, T.; Liu, J.; Nargund, R. P.; Pasternak, A.; Shah, S. K.; Truong, Q. T.; Ye, Z.; Dellureficio, J.; Bakshi, R.WO/2009/011836 A1, 2009.
Drugs Fut 2012, 37(5): 379
The discovery of MK-4256, a potent SSTR3 antagonist as a potential treatment of type 2 diabetes
ACS Med Chem Lett 2012, 3(6): 484
Route development and multikilogram GMP delivery of a somatostatin receptor antagonist
Org Process Res Dev 2012, 16(8): 1329
Addressing cardiovascular issues of SSTR3 antagonists in K-4256 structural class
247th ACS Natl Meet (March 16-20, Dallas) 2014, Abst MEDI 213
Discovery of MK-4256, a subtype selective SSTR antagonist as a potential treatment of type-2 diabetes
243rd ACS Natl Meet (March 25-29, San Diego) 2012, Abst MEDI 186
………………………………
| US6586445 * | Jun 8, 1999 | Jul 1, 2003 | Société de Conseils de Recherches et d’Applications Scientifiques, S.A.S. | Racemic mixtures of 1,2,3,4-tetra hydro-1-(4-methoxyphenyl)-3 -(4-phenyl-1H-imidazol-2-yl)-9H- pyrido(3,4-b)indole, which bind to somatostatin receptors and block sodium channel modulators; antidiabetic, antiinflammatory agents; diarrhea |
| US6864253 * | Oct 1, 2002 | Mar 8, 2005 | Orth-Mcneil Pharmaceutical, Inc. | Heterocyclic amines such as 1-(3,4-methylenedioxyphenyl)-2-(5 -(3,4-dimethoxyphenyl)pyrimidin-2-yl)- 2,3,4,9-tetrahydro-1H-beta-carboline, used as enzyme inhibitors for prophylaxix of sexual disorders |
| US6933303 * | Oct 18, 2002 | Aug 23, 2005 | Transtech Pharma, Inc. | Antidiabetic agents |
| WO2010083136A1 * | Jan 12, 2010 | Jul 22, 2010 | Merck Sharp & Dohme Corp. | Oxadiazole beta carboline derivatives as antidiabetic compounds |
| WO2011012661A1 | Jul 28, 2010 | Feb 3, 2011 | Novartis Ag | Pyridine and pyrazine derivatives as protein kinase modulators |
| WO2011028455A1 | Aug 23, 2010 | Mar 10, 2011 | Merck Sharp & Dohme Corp. | Aminotetrahydropyrans as dipeptidyl peptidase-iv inhibitors for the treatment or prevention of diabetes |
| WO2011088025A1 | Jan 11, 2011 | Jul 21, 2011 | Merck Sharp & Dohme Corp. | Oxadiazole beta carboline derivatives as antidiabetic compounds |
| WO2012101062A1 | Jan 20, 2012 | Aug 2, 2012 | Novartis Ag | Substituted bi-heteroaryl compounds as cdk9 inhibitors and their uses |
| WO2012164071A1 | Jun 1, 2012 | Dec 6, 2012 | Intervet International B.V. | Imidazole derivatives |
| WO2013068328A1 | Nov 6, 2012 | May 16, 2013 | Intervet International B.V. | Bicyclo [2.2.2] octan-1-ylcarboxylic acid compounds as dgat-1 inhibitors |
| WO2013068439A1 | Nov 8, 2012 | May 16, 2013 | Intervet International B.V. | 4-amino-5-oxo-7,8-dihydropyrimido[5, 4 -f] [1, 4] oxazepine compounds as dgat1 inhibitors |
| EP2676959A1 | Nov 11, 2009 | Dec 25, 2013 | Merck Sharp & Dohme Corporation | Combination drugs comprising aminotetrahydropyrans as Dipeptidyl Peptidase-IV Inhibitors for the Treatment or Prevention of Diabetes |
| EP2676960A1 | Nov 11, 2009 | Dec 25, 2013 | Merck Sharp & Dohme Corp. | Combination drugs comprising aminotetrahydropyrans as Dipeptidyl Peptidase-IV Inhibitors for the Treatment or Prevention of Diabetes |
| EP2676961A1 | Nov 11, 2009 | Dec 25, 2013 | Merck Sharp & Dohme Corporation | Combination drugs comprising aminotetrahydropyrans as Dipeptidyl Peptidase-IV Inhibitors for the Treatment or Prevention of Diabetes |
| US20120264777 * | Jan 11, 2011 | Oct 18, 2012 | Merck Sharp & Dohme Corp. | Oxadiazole beta carboline derivatives as antidiabetic compounds |
Memantine

Memantine
1-amino-3,5-dimethyl adamantane
REVIEW BY ARK…..http://learnabout.arkpatentintelligence.com/drug-in-focus-memantine…… GREAT REVIEW BY

Please visit www.arkpatentintelligence.com to find out more about Pipeline Patent Intelligence and how it can assist your generic drug development
Visit THEIR product page OR
Request a call to be contacted by an Ark Patent Intelligence staff member

Memantine is an orally active NMDA (N-methyl-D-aspartate) receptor antagonist which works by blocking the NMDA receptors in the brain. It blocks the excessive activity of glutamate, but still allows the normal activation of these receptors that occurs when the brain forms a memory. Therefore it improves the brain functioning in Alzheimer’s disease, and may also block the glutamate activity that could cause further damage to the brain cells.
Memantine hydrochloride is commercially available in the market in products sold under the trademark NAMENDA. It is available for oral administration as capsule shaped film-coated tablets containing 5 mg and 10 mg of memantine hydrochloride. U.S. Patent No. 3,391 ,142 discloses memantine and its related compounds, and their pharmaceutically acceptable salts.
| SynthesisReference | DrugSyn.orgUS3391142 |
|---|
……………………………….
| Country | Patent Number | Approved | Expires (estimated) |
|---|---|---|---|
| Canada | 2426492 | 2006-10-03 | 2023-05-08 |
| United States | 5061703 | 1995-04-11 | 2015-04-11 |
| Memantine is an amantadine derivative with low to moderate-affinity for NMDA receptors. It is a noncompetitive NMDA receptor antagonist that binds preferentially to NMDA receptor-operated cation channels. It blocks the effects of excessive levels of glutamate that may lead to neuronal dysfunction. It is under investigation for the treatment of Alzheimer’s disease, but there has been no clinical support for the prevention or slowing of disease progression. |

OLD CLIP
Forest Announces U.S. Availability of New Once-Daily NAMENDA XR
– Treatment for moderate to severe Alzheimer’s Disease is now available to patients in a convenient extended release formulation –
NYSE:FRX.NEW YORK–(BUSINESS WIRE)–Forest Laboratories, Inc. announced today that NAMENDA XR(TM) (memantine hydrochloride) once-daily formulation is now available in pharmacies throughout the United States. NAMENDA XR is approved by the U.S. Food and Drug Administration (FDA) for the treatment of moderate to severe dementia of the Alzheimer’s type.
http://www.pharmalive.com/once-daily-namenda-xr-launched-in-us


Memantine is the first in a novel class of Alzheimer’s disease medications acting on theglutamatergic system by blocking NMDA-type glutamate receptors. It was first synthesized by Eli Lilly and Company in 1968. Memantine is marketed under the brandsAxura and Akatinol by Merz, Namenda by Forest, Ebixa and Abixa by Lundbeck andMemox by Unipharm. Memantine has been shown to have a modest effect in moderate-to-severe Alzheimer’s disease and in dementia with Lewy bodies. Despite years of research, there is little evidence of effect in mild Alzheimer’s disease. 1-4


Medical use
Memantine is approved by the U.S. F.D.A and the European Medicines Agency for treatment of moderate-to-severe Alzheimer’s disease,[5] and has now received a limited recommendation by the UK’s National Institute for Clinical Excellence for patients who fail other treatment options.[6] Within the new guidance memantine is recommended as an option for managing Alzheimer’s disease for people with: moderate Alzheimer’s disease who are intolerant of or have a contraindication to AChE (acetylcholinesterase) inhibitors or those with severe Alzheimer’s disease.
Memantine has been associated with a moderate decrease in clinical deterioration[7] with only a small positive effect on cognition, mood, behavior, and the ability to perform daily activities in moderate to severe Alzheimer’s disease.[8] There does not appear to be any benefit in mild disease.[4]
Adverse effects
Memantine is, in general, well-tolerated.[8] Common adverse drug reactions (≥1% of patients) include confusion, dizziness, drowsiness, headache, insomnia, agitation, and/or hallucinations. Less common adverse effects include vomiting, anxiety, hypertonia, cystitis, and increased libido.[7][9] It has been reported to induce reversible neurological impairment in multiple sclerosis patients, which led to the halt of an ongoing clinical trial.[10][11] Though exceedingly rare, extrapyramidal side effects (such as dystonic reactions, etc.) may occur, in particular, in the younger population.[citation needed]
A recent study demonstrates therapeutically-relevant doses of memantine in the mouse can lead to disruption of cognitive flexibility.[12]

Biochemistry
The drug belongs to a class of drugs called NMDA receptor antagonists, which reduce certain types of brain activity by binding to NMDA receptors on brain cells and blocking the activity of the neurotransmitter glutamate. At normal levels, glutamate aids in memory and learning, but if levels are too high, glutamate appears to overstimulate nerve cells, killing them through excitotoxicity.
Pharmacology
Glutamatergic (NMDA receptor)
A dysfunction of glutamatergic neurotransmission, manifested as neuronal excitotoxicity, is hypothesized to be involved in the etiology of Alzheimer’s disease. Targeting the glutamatergic system, specifically NMDA receptors, offers a novel approach to treatment in view of the limited efficacy of existing drugs targeting the cholinergic system.[13]
Memantine is a low-affinity voltage-dependent uncompetitive antagonist at glutamatergic NMDA receptors.[14][15][16][17][18] By binding to the NMDA receptor with a higher affinity than Mg2+ ions, memantine is able to inhibit the prolonged influx of Ca2+ ions, particularly from extrasynaptic receptors, which forms the basis of neuronal excitotoxicity. The low affinity, uncompetitive nature, and rapid off-rate kinetics of memantine at the level of the NMDA receptor-channel, however, preserves the function of the receptor at synapses, as it can still be activated by physiological release of glutamate following depolarization of the presynaptic neuron.[19][20][21][22][15][16][23][24][25] The interaction of memantine with NMDA receptors plays a major role in the symptomatic improvement that the drug produces in Alzheimer’s disease. Moreover, there is no evidence as yet that the ability of memantine to protect against NMDA receptor-mediated excitotoxicity has a disease-modifying effect in Alzheimer’s, although this has been suggested in animal models.[22]
Serotonergic (5-HT3 receptor)
Memantine acts as a non-competitive antagonist at the 5-HT3 receptor, with a potency similar to that for the NMDA receptor.[26] The clinical significance of this serotonergic activity in the treatment of Alzheimer’s disease is unknown.
Cholinergic (nicotinic acetylcholine receptor)
Memantine acts as a non-competitive antagonist at different neuronal nicotinic acetylcholine receptors (nAChRs) at potencies possibly similar to the NMDA and 5-HT3 receptors, but this is difficult to ascertain with accuracy because of the rapid desensitization of nAChR responses in these experiments. It can be noted that memantine is an antagonist at alpha-7 nAChR, which may contribute to initial worsening of cognitive function during early memantine treatment. Alpha-7 nAChR upregulates quickly in response to antagonism, which could explain the cognitive-enhancing effects of chronic memantine treatment.[24][27][28] It has been shown that the number of nicotinic receptors in the brain are reduced in Alzheimer’s disease, even in the absence of a general decrease in the number of neurons, and nicotinic receptor agonists are viewed as interesting targets for anti-Alzheimer drugs.[29] Consequently, this may also suggest that administration of nicotine itself may act against the effects of Alzheimer’s disease. In fact a recent small double blind placebo controlled randomized trial published in the Journal Neurology demonstrated a significant benefit from use of 15 mg nicotine patches in non-smoking elderly patients with mild cognitive impairment. The small sample sizes suggest that the effect size is substantial due to the limited statistical power inherent in a small N trial. (see : http://www.ncbi.nlm.nih.gov/pubmed/22232050)
Dopaminergic (D2 receptor)
Memantine acts as an agonist at the dopamine D2 receptor.[30]
Availability
The hydrochloride (Memantine HCl) is white water soluble powder available as capsule-shaped film-coated tablets or oral solution. The tablets are available as 5 mg, 10 mg or 20 mg of memantine hydrochloride. The oral solution contains 2 mg of memantine hydrochloride per ml.
History
Memantine was first synthesized and patented by Eli Lilly and Company in 1968 (as cited in the Merck Index), and then developed by Merz in collaboration with Neurobiological Technologies, Inc. and Children’s Hospital, Boston/Harvard Medical School, and then licensed to Forest for the U.S. and Lundbeck for selected European and international markets.
Sales of the drug reached $1.8 billion for 2010.
Research
Memantine is also being tested for generalized anxiety disorder, epilepsy, opioid dependence, systemic lupus erythematosus, depression, bipolar disorder,[31] obsessive compulsive disorder, Tourette Syndrome, problem gambling, attention-deficit hyperactivity disorder (ADHD),[32] glaucoma, tinnitus, neuropathic pain including Complex Regional Pain Syndrome,[33] pervasive developmental disorders, HIV associated dementia,[34] nystagmus,[35] multiple sclerosis,[10] autism,[36] migraine,[37] amyotrophic lateral sclerosis,[38] Down syndrome[39] and for protection of cognitive function during whole brain radiation
Memantine (1-amino-3,5-dimethyl adamantane, disclosed, e.g., in U.S. Patents No. 4,122,193; 4,273,774; 5,061 ,703) is a systemically-active uncompetitive NMDA receptor antagonist having moderate affinity for the receptor and strong voltage dependency and rapid blocking/unblocking kinetics. Memantine has been shown to be useful in alleviation of various progressive neurodegenerative disorders such as dementia in patients with moderate to severe Alzheimer’s disease, Parkinson’s disease, and spasticity (see, e.g., U. S. Patents No. 5,061 ,703; 5,614,560, and 6,034,134; Parsons et al., Neuropharmacology 1999 Jun; 38(6):735-67; Mόbius, ADAD, 1999,13:S172-178; Danysz et al., Neurotox. Res., 2000, 2:85-97; Winblad and Poritis, Int. J. Geriatr. Psychiatry, 1999, 14:135-146; Danysz et al., Cυrr. Pharm. Des., 2002, 8:835-843; Jirgensons et. al., Eur. J. Med. Chem., 2000, 35: 555-565). Memantine has also been suggested to be useful in the treatment of AIDS dementia (U.S. Patent No. 5,506,231), neuropathic pain (U.S. Patent No. 5,334,618), epilepsy, glaucoma, hepatic encephalopathy, multiple sclerosis, stroke, tardive dyskinesia (Parsons et al., 1999, supra), autism, Attention-Deficit/Hyperactivity Disorder (ADHD) and other autistic spectrum disorders (US Published Application No. 2006/0079582). Memantine is currently approved in Europe and the United States for the treatment of Alzheimer’s disease.
[0003] US Patent No. 3,391 ,142 discloses a process for the synthesis of adamantylamines, including memantine hydrochloride, involving treatment of 1-bromo- 3,5-dimethyladamantane with acetonitrile and concentrated sulfuric acid to yield the corresponding 1-acetamido-3,5-dimethyladamantane which is hydrolyzed with sodium hydroxide to yield 1-amino-3,5-dimethyladamantane which is converted to memantine hydrochloride via treatment with hydrochloric acid.

[0004] US Patent No. 4,122,193 discloses a process for the synthesis of i-amino-3,5- dialkyl adamantane derivatives, including memantine hydrochloride, which involves treatment of a 1-halo-3,5-dialkyl adamantane derivatives with a urea followed by treatment with hydrochloric acid.

R,, R2 = lower alkyl
[0005] US Patent No. 5,061 ,703 discloses a process for the synthesis of aminoadamantanes, including memantine hydrochloride, which involves halogenation and/or alkylation of the adamantane ring followed by introduction of the amino group via treatment of the halogenated derivative with formamide and subsequent hydrolysis.
HCONH2 HCI



R = H, alkyl X = Cl, Br [0006] Czech Republic Patent No. 288445 discloses a process for the synthesis of 1- amino-3,5-dimethyladannantane hydrochloride wherein 1-chloro-3,5- dimethyladamantane is reacted with formamide followed by treatment of the formamide intermediate with aqueous hydrochloric acid to yield 1-amino-3,5-dimethyladamantane hydrochloride.
HCONH2


[0007] Czech Republic Patent No. 282398 discloses a process for the synthesis of 1- amino-3,5-dimethyladamantane hydrochloride which involves treatment of 1-acetamido- 3,5-dimethyl adamantane with a base (such as postassium hydroxide) in a solvent such as methanol, ethanol, or 2-propanol.

[0008] Chinese Published Patent Publication No. CN 1566075 discloses a process for preparing 1-aminoadamantane derivatives, including memantine hydrochloride, wherein a halogenated adamantane compound (which may have substituents, including alkyl groups, in the 3 and 5 positions) is reacted with formamide or a substituted formamide, followed by deformylation under acidic conditions to yield a 1-aminoadamantane derivative.

X = Cl, Br, I R1, R2, R3 = H, alkyl, etc.
[0009] US Patent No. 5,599,998 discloses a process for the synthesis of 1- aminoadamantane derivatives, including memantine, which involves treatment of a 1- halo adamantane derivative with lithium metal to yield the lithiated intermediate which is treated with an aminating agent (such as NH2CI) under sonication conditions.
NH,CI


X = Cl, Br, I R3, R5, R7, R8 = H, F, Me
[0010] US Published Application No. 2006/025885 discloses a process for the synthesis of 1-aminoadamantane derivatives, including memantine hydrochloride, wherein a halogenated adamantane compound (which may have alkyl substituents, in the 3 and 5 positions of the adamantane ring) is reacted with acetonitrile in the presence of glacial acetic acid and concentrated sulfuric acid, followed by hydrolysis in the presence of an alkaline earth metal in a solvent such as 1-methoxy-2-propanol to yield the 1- aminoadamantane derivative which may then be converted to an acid addition salt via treatment with the appropriate acid (e.g., hydrochloric acid).

X = = F, Cl, Br, I
R1, R2 = = H, C.^alkyl
[0011] International Publication No. WO 2006/076562 discloses a process for the synthesis of memantine hydrochloride, wherein 1-halo-3,5-dimethyladamantane is reacted with acetonitrile in the presence of phosphoric acid to yield N-acetyl-1-amino- 3,5-dimethyladamantane, which may be converted to memantine (for example, via basic hydrolysis) which may then be treated with hydrochloric acid to yield memantine hydrochloride.

X = F, Cl, Br, I
[0012] International Publication No. WO 2006/122238 discloses processes for preparing memantine or an acid addition salt of memantine, which involve either reaction of 1- bromo-3,5-dimethyladamantane with formamide to form N-formyl-1-amino-3,5- dimethyladamantane or reaction of 1-hydroxy-3,5-dimethyladamantane with a hydrogen halide to obtain 1-halo-3,5-dimethyl adamantane which is then reacted with formamide to yield N-formyl-1-amino-3,5-dimethyladamantane. The N-formyl-1-amino-3,5- dimethyladamantane intermediate is deformylated under acidic conditions to yield memantine hydrochloride.

[0013] International Publication No. WO 2005/062724 discloses a process for the synthesis of 1-aminoadamantane derivatives, including memantine, which involves bromination of a compound of formula Ma, followed by hydrolysis to yield a compound of formula Md, which is treated with acetonitrile in the presence of an acid (e.g., sulfuric acid) to yield the acetamido intermediate of formula Hc. The compound of formula Mc is then hydrolyzed in the presence of acid or base to yield the aminoadamantane derivative of formula I.

R1, R2 = C1-talkyl R3 = H, C,^alkyl
[0014] International Publication No. WO 2007/101536 discloses a process for the synthesis of 1-formamido-3,5-dimethyladmantane which involves treatment of 1 ,3- dimethyladamantane with formamide in a concentrated acid, as well as a process for the conversion of 1-formamido-3,5-dimethyladmantane to memantine hydrochloride via hydrolysis with hydrochloric acid. add


[0015] Pharmaceutical active ingredients must meet strict regulatory requirements with respect to purity. Typically, products meeting such purity requirements may obtained by purifying a crude active pharmaceutical ingredient using standard purification techniques.
[0016] Memantine produced by processes known in the art may contain trace impurities including 1-amino-3,5,7-trimethyladamantane. As such trace impurities are closely related to memantine, isolating memantine from crude preparations using standard purification techniques is difficult.
[0017] Thus, a need exists to develop a process for producing memantine which is substantially free of impurities, such as 1-amino-3,5,7-trimethyladamantane.
………………………………………..
http://www.google.com/patents/EP2389351A1?cl=en

DETAILED DESCRIPTION OF THE INVENTION
[0030] A process for the preparation of 1 ,3-dimethyladamantane is shown in Scheme 1. [0031] Acenaphthene (1) is hydrogenated over a catalyst such as Raney Nickel at elevated temperature and pressure to yield perhydroacenaphthene (2). Perhydroacenaphthene is treated with a Lewis acid such as AICI3 and/or AIBr3 in the presence or absence of HCI to yield 1 ,3-dimethyladamantane which may be further purified (e.g., via fractional distillation) to provide 1 ,3-dimethyladamantane (4) containing 0.05% or less of the impurity 1,3,5-trimethyladamantane.
hydrogenation raney nickel

temp./ press.

AICL

Scheme 1 – Preparation of 1,3-dimethyladamantane
[0032] 1 ,3-dimethyladamantane containing 0.05% or less of the impurity 1 ,3,5- trimethyladamantane may be converted to memantine, or a pharmaceutically acceptable salt thereof (e.g., memantine hydrochloride), which is substantially free of the impurity 1-amino-3,5,7-trimethyladamantane, according to Scheme 2.
[0033] 1 ,3-dimethyladamantane (4), which contains 0.05% or less of the impurity 1 ,3,5- trimethyladamantane, may be treated with a halogenating agent (e.g., bromine, chlorine, or t-butylchloride) to yield 1-halo-3,5-dimethyladamantane derivative 5. Derivative 5 may be treated with formamide to yield 1-formamido-3,5- dimethyladamantane derivative 6. Alternatively, 1 ,3-dimethyladamantane, which contains 0.05% or less of the impurity 1 ,3,5-trimethyladamantane, may be treated with formamide in the presence of concentrated acid to yield 1-formamido-3,5- dimethyladamantane derivative 6. Derivative 6 may be hydrolyzed under basis or acidic conditions to provide memantine, which is substantially free of the impurity 1-amino- 3,5,7-trimethyladamantane, or a pharmaceutically acceptable salt thereof.
[0034] Derivative 5 may be also treated with acetonitrile in the presence of acid (e.g., sulfuric acid, phosphoric acid, nitric acid, a combination of acetic acid and sulfuric acid, or mixtures thereof) to yield 1-acetamido-3,5-dimethyladamantane derivative 7. Derivative 7 may be hydrolyzed to provide memantine, which is substantially free of the impurity 1-amino-3,5,7-trimethyladamantane, which may be converted to a pharmaceutically acceptable salt via treatment with a pharmaceutically acceptable acid.

Scheme 2 – Synthesis of memantine [0035] As used herein, the term halogen refers to fluorine, chlorine, bromine, and iodine.
[0036] As used herein, the term “substantially free of the impurity 1 ,3,5- trimethyladamantane” used in conjunction with 1 ,3-dimethyladamantane includes 1 ,3- dimethyladamantane which contains 0.05% or less of the impurity 1 ,3,5- trimethyladamantane.
[0037] As used herein, the term “substantially free of the impurity 1-amino-3,5,7- trimethyladamantane” used in conjunction with memantine (or a pharmaceutically acceptable salt thereof, e.g., memantine hydrochloride) includes memantine (or a pharmaceutically acceptable salt thereof, e.g., memantine hydrochloride) which contains 0.02% or less of the impurity 1-amino-3,5,7-trimethyladamantane.
Example 1
Analytical method for determining impurities in 1 ,3-dimethyladamantane
[0059] Analysis is performed on DANI 86.10 HT gas chromatograph equipped with CTC 200S automatic sampler. Millennium 3.20 software is applied for acquisition and processing of the data. All further calculations are made by MS Excel 97 software, statistics are calculated according to “Validierung in Chromotographie” Novia GmbH. 08.02.1996, Frankfurt am Main.
[0060] Gas chromatographic analysis to determine the impurity profile of 1 ,3- dimethyladamantane is performed on an SE-52 capillary column (length: 25 ; ID: 0.32 mm; df = 0.25 μm). An flame ionization detector (FID) is applied for detection. The injector temperature is 2500C and the detector temperature is also 2500C. The carrier gas is nitrogen.
[0061] Both calibration standards and analytical samples are prepared by dissolution of the components in hexane. 1-Hydroxyadamantane is applied as internal standard. All impurities are calibrated against the 1 ,3-dimethyladamantane peak.
[0062] Results for a high purity sample of 1 ,3-dimethyladamantane are shown in Table 1 below and in Figure 1.
Table 1 – Impurity profile for high purity 1 ,3-dimethyladamantane

Example 2
Preparation of 1, 3-dimethyladamantane
[0063] Acenaphthene is washed with bentonite in toluene, followed by a second wash with cyclohexene. The acenaphthene is then hydrogenated over Raney Nickel at 150 bar at a temperature of 140 to 180 0C. The catalyst is filtered off and the crude perhydroacenaphthene (240 kg) is treated with AICI3 (45 kg) and HCI (100 ml_) at 80 to 90 0C for 4 h. The reaction mixture is then heated to 120 0C for 8 h. An additional 100 ml_ of hydrochloric acid and an additional 5 kg of AICI3 are added and the reaction mixture is heated to 80 to 900C for 4 h. The crude 1 , 3-dimethyladamantane is purified via fractional distillation on a DN 300 column with oriented Sulzer type packing providing a minium of 60 theoretical plates, with the temperature profile being dependent on column pressure. The critical point is estimated on the basis of trend and is confirmed by analytical control. 1 ,3-Dimethyladamantane containing 0.05% or less of the impurity 1 ,3,5, trimethyladamantane is obtained in about 75% yield.
Example 3
Preparation of 1-bromo-3,5-dimethyladamantane
[0064] 1 , 3-dimethyladamantane containing 0.05% or less of the impurity 1 ,3,5, trimethyladamantane, as prepared in Example 2, is treated with bromine (3 equivalents) and heated to reflux for 16 h. The reaction mixture is cooled to about 15 0C and quenched with sodium bisulphite in methylene chloride. The aqueous layer is removed, and the organic layer is washed with water. The organic layer is concentrated in vacuo to yield 1-bromo-3,5-dimethyladamantane as an oil.
Example 4
Preparation of 1-amino-3,5-dimethyladamantane hydrochloride
[0065] 1-bromo-3,5-dimethyladamantane as prepared in Example 3, is treated with an excess of formamide and heated to 120 0C for 3 to 5 h. The reaction mixture is cooled and diluted with methylene chloride. This mixture is washed 4 times with a 30% sodium hydroxide solution. The organic layer is concentrated via distillation followed by addition of water. The distillation is continued to remove the organic layer, and the mixture is then cooled to below 800C. The N-formyl-1-amino-3,5-dimethyladamantane intermediate, which is optionally isolated, is then hydrolyzed by addition of a 37% hydrochloric acid solution. The reaction mixture is heated to reflux for about 3 h, and the reaction mixture is then cooled to 5 0C to yield crude 1-amino-3,5- dimethyladamantane hydrochloride which is isolated by centrifugation and washed with water followed by ethyl acetate. The crude 1-amino-3,5-dimethyladamantane hydrochloride is then reprecipitated to yield the title compound.
[0066] The purity of memantine hydrochloride prepared according to Examples 3-4 is shown in Table 2.
Table 2

Example 5
Preparation of 1-formamido-3,5-dimethyladamantane
[0067] 1,3-dimethyladamantane containing 0.05% or less of the impurity 1 ,3,5, trimethyladamantane, as prepared in Example 2, is treated with nitric acid followed by sulfuric acid at 0 0C. The reaction is stirred over night at 0 0C. The reaction mixture is poured onto 100 ml_ formamide (at 0 0C) in a round bottom flask which is equipped with a drying tube. The reaction is stirred at 0 0C for 30 min and then at room temperature for 90 min. Dichloromethane and water are then added. The organic phase is removed and washed with water and a 2 % NaHCO3-solution, dried over Na2SO4 and concentrated in vacuo. The resulting oil is purified via column chromatography to yield the title compound as a solid. Example 6
Preparation of 1-amino-3,5-dimethyladamantane
[0068] 1-formamido-3,5-dimethyladamantane, as prepared in Example 5, is hydrolyzed by addition of a 37% hydrochloric acid solution. The reaction mixture is heated to reflux for about 3 h, and the reaction mixture is then cooled to 5 0C to yield crude 1-amino-3,5- dimethyladamantane hydrochloride which is filtered and washed with water followed by ethyl acetate. The crude 1-amino-3,5-dimethyladamantane hydrochloride is then reprecipitated to yield the title compound.
Example 7
Relationship between content of the 1-amino-3,5, 7-trimethyladamantane (TMM) impurity in memantine and the 1 ,3,5-thmethyladamantane impurity in 1,3- dimethyladamantane
[0069] Memantine is synthesized according to Examples 5-6 starting from 1 ,3- Dimethyladamantane (1 ,3-DMA) spiked with different levels of the alkyl adamantane impurity 1,3,5-trimethyladamantane (TMA). The level of the corresponding aminoalkyl adamantane impurity 1-amino-3,5,7-trimethyladamantane (TMM) in the final memantine product is determined. The results (shown in Table 3) demonstrate that the amount of the 1-amino-3,5,7-trimethyladamantane (TMM) impurity in the memantine final product is dependent on the amount of 1 ,3,5-trimethyladamantane (TMA) impurity present in the 1,3-dimethyladamantane starting material.
Table 3

* * * * *
…………………………
http://www.google.com/patents/WO2006122238A1?cl=en
Memantine has the chemical name 1-amino-3,5-dimethyl adamantane (hereinafter referred to by the officially adopted name “memantine”), and can be represented by the structural Formula 1.

Formula I
Memantine is an orally active NMDA (N-methyl-D-aspartate) receptor antagonist which works by blocking the NMDA receptors in the brain. It blocks the excessive activity of glutamate, but still allows the normal activation of these receptors that occurs when the brain forms a memory. Therefore it improves the brain functioning in Alzheimer’s disease, and may also block the glutamate activity that could cause further damage to the brain cells.
Memantine hydrochloride is commercially available in the market in products sold under the trademark NAMENDA. It is available for oral administration as capsule shaped film-coated tablets containing 5 mg and 10 mg of memantine hydrochloride. U.S. Patent No. 3,391 ,142 discloses memantine and its related compounds, and their pharmaceutically acceptable salts. This patent also describes a process for the preparation of memantine as depicted in Scheme 1.
International Application Publication No. WO 2005/062724 A2 describes an alternate process for the preparation of memantine as depicted in Scheme 2, and U.S. Patent No. 5,061 ,703 discloses a process for the preparation of derivatives of memantine.
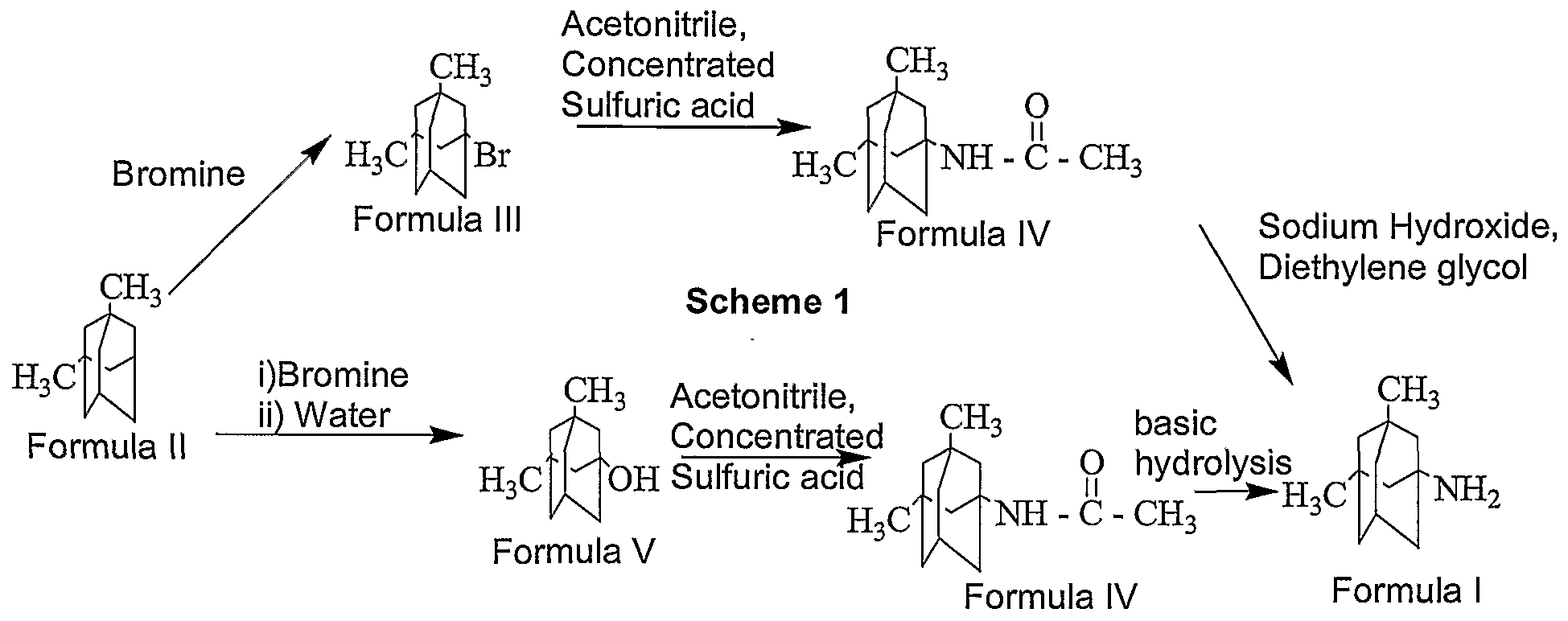
Scheme 2
A process with a reduced number of stages, and which does not require isolation of unstable or hazardous intermediates, will be helpful. Also a process which is easily scalable and is industrially feasible will be helpful.
PREPARATION OF 1-BROMO-3.5-DIMETHYL ADAMANTANE (FORMULA III)
8.0 liters of bromine was taken into a reactor and 5.1 kg of 1 ,3-dimethyl adamantane was added into the reactor slowly at 28° C in 2.5 hours. The reaction mass was maintained at 28° C for 24 hours. Reaction completion was checked using a gas chromatographic technique. After the reaction was completed, the bromine was distilled off from the reaction mass at a temperature of below 40° C. After the completion of distillation, 13.4 kg of the title compound in the form of a residue was recovered from the reactor.
Purity by GC: 98.35%,
1 ,3-dimethyl adamantane: 0.53%.
1-hydroxy-3,5-di methyl adamantane: 0.66%.
EXAMPLE 2
PREPARATION OF 1-N-FORMYL-3,5-DIMETHYL ADAMANTANE (FORMULA Vl)
64 liters of formamide was taken into a reactor and 2.55 kg of the 1-bromo- 3,5-dimethyl adamantane of Formula III obtained above was added to it. The reaction mass was heated to a temperature of 157° C and maintained for 14 hours. Reaction completion was checked using gas chromatography. After the reaction is completed, the reaction mass was cooled to 28° C. The reaction mass was then further cooled to 4° C and maintained for 4.25 hours. 37.5 liters of chilled water was added to the reaction mass slowly below 5° C. Then 37.5 liters of dichloromethane was added to the reaction mass. The temperature of the reaction mass was raised to 25° C. The reaction mass was filtered over a celite bed and the bed was washed with 12.5 liters of dichloromethane. The filtrate was taken into another reactor and stirred at 25° C for 10 minutes. The layers were separated and the aqueous layer was extracted with 15 liters of dichloromethane in 2 equal lots. The combined dichloromethane layer was washed with 51 liters of 10% sodium bicarbonate solution in 2 equal lots. The dichloromethane layer was dried over sodium sulphate and distilled under vacuum at a temperature of 40° C to yield 2.3 kg of the title compound in the form of a residue.
Purity by GC: 76.4 %,
1 ,3-dimethyl adamantane: 0.11 %, 1-hydroxy-3,5-dimethyl adamantane: 21.28%.
EXAMPLE 3
PREPARATION OF MEMANTINE HYDROCHLORIDE (FORMULA VIII): 21 liters of 36% aqueous hydrochloric acid was taken into a reactor and 2.1 kg of 1-N-formyl-3,5-dimethyl adamantane of Formula Vl obtained above was added to it. The reaction mass was heated to a temperature of 104° C and maintained for 6.5 hours. Reaction completion was checked using gas chromatographic technique. After the reaction was completed, the reaction mass was cooled to 4° C and maintained for 3 hours. The reaction mass was filtered in a centrifuge and the filtered solid was washed with 2.1 liters of chilled water. The wet cake was taken into another reactor and 8.5 liters of acetone was added to it. The reaction mass was cooled to 4° C and maintained for 3.5 hours. The reaction mass was then filtered and the filtered cake was washed with 2.1 liters of chilled acetone (chilled to a temperature of 5° C). The wet compound was dried at 73° C for 7 hours to yield 1.05 kg of the title compound. The dried compound was then milled in a micronizer (Manufacturer: Microtech Engineering company, Model: M- 50).
Purity by GC: 99.9%, 1-hydroxy-3,5-dimethyl adamantane: Less than 0.004%,
1-bromo-3,5-dimethyl adamantane: 0.01 %, 1-formamido-3,5-dimethyl adamantane: 0.03%. Residual Solvents: methanol = 231 ppm, dichloromethane = 249 ppm, cyclohexane = 43 ppm, toluene = 122 ppm. Particle size distribution: Before milling: Di0 = 3.2 μm; D50 = 18.4 μm; D90
= 96.6 μm. After milling: D10 = 1.7 μm; D50 = 9.6 μm; Dg0 = 48.3 μm.
Bulk Density: Before milling: Before tapping: 0.20 g/ml ; After tapping: 0.38 g/ml. After milling: Before tapping: 0.20 g/ml; After tapping: 0.39 g/ml. EXAMPLE 4
PREPARATION OF I-CHLORO-S.δ-DIMETHYL ADAMANTANE (FORMULA VII) 15 g of 1-hydroxy-3,5-dimethyl adamantane was taken into a round bottom flask and 300 ml of 36% aqueous hydrochloric acid was added to it. The reaction mass was stirred at 28° C for 21 hours. The reaction mass was kept for layer separation. The upper layer was separated to yield 13.0 g of the title compound in the form of crude. Purity by GC: 99.88%.
EXAMPLE 5
PREPARATION OF 1-N-FORMYL-3.5-DIMETHYL ADAMANTANE (FORMULA Vl)
10 g of 1-chloro-3,5-dimethyl adamantane of Formula VII prepared above was taken into a round bottom flask and 250 ml of formamide was added to it. The reaction mass was heated to 152° C and maintained at that temperature for 10 hours. The reaction mass was then cooled to 10 ° C, and 150 ml of water chilled to a temperature of 5° C was added to it slowly. Then 150 ml of dichloromethane was added to it and the temperature of the reaction mass was brought up to 28° C. The reaction mass was filtered through a celite bed and the celite bed was washed with 50 ml of dichloromethane. The filtrate was transferred into a separating funnel and the organic layer was separated. The aqueous layer was extracted with 150 ml of dichloromethane in two equal lots. The combined organic layer was washed with 200 ml of 10% sodium bicarbonate solution in two equal lots. The organic layer was dried over sodium sulphate and distilled under vacuum at 38° C to yield 9.6 g of the title compound. Purity by GC: 77.35%. EXAMPLE 6
PREPARATION OF 1-AMINO-3.5-DIMETHYL ADAMANTANE HYDROCHLORIDE (FORMULA VIII) 9.0 g of 1-N-formyl-3,5-dimethyl adamantane of Formula Vl and 90 ml of
36% aqueous hydrochloric acid were taken into a round bottom flask and heated to 102° C. The reaction mass was maintained at 102° C for 6 hours. The reaction mass was then allowed to cool to 2° C and maintained for 2 hours. The reaction mass was then filtered and the solid was washed with 20 ml of water chilled to a temperature of 5° C. The wet compound was taken into another round bottom flask and 54 ml of acetone was added to it. The reaction mass was stirred at 28° C for one hour, and then the reaction mass was cooled to 2° C. The reaction mass was maintained at 2° C for 2 hours. The reaction mass was then filtered and the solid was washed with 18 ml of acetone. The solid was suction dried under vacuum of 400 mm Hg and then dried in a vacuum oven at a temperature of 60° C and vacuum of 400 mm Hg for 6 hours to yield 5.0 g of the title compound. Purity by GC: 99.59%
EXAMPLE 7
PREPARATION OF 1-N-FORMYL-3.5-DIMETHYL ADAMANTANE (FORMULA VI)
13 liters of bromine was taken into a reactor and 8.3 kg of 1,3-dimethyl adamantane was added to it at 30° C. The reaction mass was maintained at 30° C for 24 hours. Reaction completion was checked using a gas chromatographic technique. 200 liters of formamide was taken in another reactor and the reaction mass from the previous reactor was added to it. The previous reactor was rinsed with 8 liters of formamide and added to the reaction mass. The temperature of the reaction mass was raised to 155° C. The reaction mass was maintained at 155° C for 8 hours. Reaction completion was checked using gas chromatography. After the reaction was completed, the reaction mass was cooled to 4° C and maintained for 1.5 hours. 125 liters of chilled water was added to the reaction mass at 4° C. 125 liters of dichloromethane was added to the reaction mass and the temperature was raised to 25° C. The reaction mass was then filtered over a celite bed and the bed was washed with 42 liters of dichloromethane. The filtrate was allowed to settle and the organic layer was separated. The aqueous layer was extracted with 124.5 liters of dichloromethane in two equal lots. The combined dichloromethane layer was washed with 166 liters of 10% aqueous solution of sodium bicarbonate in two equal lots. The dichloromethane layer was dried over sodium sulfate and distilled atmospherically to dryness at 40° C to yield 18.2 kg of the title compound.
Purity by GC: 96.29% 1 ,3-dimethyl adamantane: less than 0.004%.
1-hydroxy-3,5-dimethyl adamantane: 2.81 %.
1-bromo-3,5-dimethyl adamantane: 0.05%.
EXAMPLE 8
PREPARATION OF MEMANTINE HYDROCHLORIDE (FORMULA VIII)
24 liters of water was taken into a reactor and 24 liters of 36% aqueous hydrochloric acid was added to it. 10 kg of 1-N-formyl-3,5-dimethyl adamantane of Formula Vl was added to the reaction mass. The reaction mass was heated to a temperature of 102° C. The reaction mass was maintained at 102° C for 24 hours. Reaction completion was checked using gas chromatographic technique. After the reaction was completed the reaction mass was cooled to 5° C. The reaction mass was maintained at 5° C for 2 hours and 30 minutes and then s olids were separated using a centrifuge (Manufacturer: Nima Engineering Pvt. Ltd. Model: 14″ x 7″ – LAD CANT). The solid was washed with 10 liters of chilled water. The wet solid was then taken into another reactor and 20 liters of isopropanol was added to it. The reaction mass was stirred at 28° C for 45 minutes. Then the reaction mass was cooled to 5° C and maintained for 2.5 hours. The reaction mass was then filtered and the filtered solid was washed with 10 liters of chilled isopropanol. The wet cake was dried in an oven at 92° C for 11 hours to yield 4.6 kg of the title compound.
Purity by GC: 99.98%
1-chloro-3,5-dimethyl adamantane: less than 0.005%. 1-hydroxy-3,5-dimethyl adamantane: 0.003%.
1-N-formyl-3,5-dimethyl adamantane: 0.01%.
Residual Solvents: methanol = 33 ppm, dichloromethane = 14 ppm, acetone= 2 ppm, isopropanol = 383 ppm. Fig. 2 shows the X-ray powder diffraction pattern for the product, obtained using Cu Ka- 1 radiation (1.541 A wavelength).
EXAMPLE 9
Gas Chromatography method for memantine hydrochloride and impurities.
Chromatographic conditions:
Column: Agilent Ultra 2, or equivalent (crosslinked 5% phenylmethylsiloxane).
Length: 50 meters.
ID: 0.32 mm. Film thickness: 0.52 microns.
Column Temperature: Initial temperature: 50° C, Hold time-1: 0 minutes, Heating rate-1 : 5° C; Final temperature: 145° C, Hold time-2: 0 minutes, Heating rate-2:
10° C; Final temperature: 250° C, Final hold time: 20 minutes, Injection port temperature: 220° C, Detector temperature: 300° C. Carrier gas: Helium
Flow rate: 4.0 ml/minute
Injection mode (Split): 1 : 50
Injection volume: 1 μl.
Solvent: Hexane.
PEAK LOCATIONS:

Gas Chromatography method for residual organic solvents.
Chromatographic Conditions:
Column: Alltech AT-624 capillary, or equivalent. Length: 30 meters.
ID: 0.53 mm.
Film thickness: 3.0 μm of 6% cyanopropylphenyl-94% methylpolysiloxane.
Injector temperature: 140° C.
Detector temperature: 260° C. Mode of injection [split]: 1 :5
Carrier gas: Helium
Flow rate: 4.0 ml/minute.
Sample: 1 μl.
Diluent: N,N-dimethylacetamide. Temperature: column temperature is programmed according to the following steps: it is held at 40° C for 8 minutes then increased to 165° C at a rate of 10° C per minute and held at 165° C for 5 minutes. Again increased to 250° C at a rate of 35° C and held at 250° C for 20 minutes.
…………………………..
| WO2008062472A2 * | Oct 22, 2007 | May 29, 2008 | Cadila Healthcare Ltd | Process for the preparation of memantine |
| WO2009010806A1 * | Jul 18, 2008 | Jan 22, 2009 | Generics Uk Ltd | Assay methods for memantine |
| WO2010015415A1 * | Aug 7, 2009 | Feb 11, 2010 | Merz Pharma Gmbh & Co. Kgaa | Process for manufacturing adamantane derivatives with high yield |
| WO2010069555A1 | Dec 16, 2009 | Jun 24, 2010 | Merz Pharma Gmbh & Co. Kgaa | Method for producing memantine |
| WO2010083996A1 * | Jan 20, 2010 | Jul 29, 2010 | Merz Pharma Gmbh & Co. Kgaa | A process for preparing memantine |
| EP2555616A1 * | Apr 8, 2010 | Feb 13, 2013 | Hetero Research Foundation | Process for the preparation of memantine hydrochloride |
| WO2006122238A1 * | May 11, 2006 | Nov 16, 2006 | Mukunda Reddy Jambula | Process for preparing memantine |
| WO2007101536A1 * | Feb 20, 2007 | Sep 13, 2007 | Univ Giessen Justus Liebig | Method for producing 1-formamido-3,5-dimethyladamantane |
| WO2007126886A1 * | Mar 27, 2007 | Nov 8, 2007 | Teva Pharm Fine Chemicals Srl | Process for preparing memantine hydrochloride substantially free of impurities |
| EP1908748A1 * | Oct 5, 2006 | Apr 9, 2008 | Krka | Process for the preparation of memantine and its hydrochloric acid salt form |
References
- Reisberg B, Doody R, Stöffler A, Schmitt F, Ferris S, Möbius HJ; Memantine Study Group. (2003) Memantine in moderater-to-severe Alzheimer’s disease. New Engl. J. Med. 348(14) 1333-41
- Aarsland, D; Ballard, C; Walker, Z; Bostrom, F; Alves, G; Kossakowski, K; Leroi, I; Pozo-Rodriguez, F; Minthon, L; Londos, E (July 2009). “Memantine in patients with Parkinson’s disease dementia or dementia with Lewy bodies: a double-blind, placebo-controlled, multicentre trial.”. Lancet neurology 8 (7): 613–8. doi:10.1016/S1474-4422(09)70146-2. PMID 19520613.
- Johansson, C; Ballard, C; Hansson, O; Palmqvist, S; Minthon, L; Aarsland, D; Londos, E (February 2011). “Efficacy of memantine in PDD and DLB: an extension study including washout and open-label treatment.”. International journal of geriatric psychiatry 26 (2): 206–13. doi:10.1002/gps.2516. PMID 20665553.
- Schneider, LS; Dagerman, KS, Higgins, JP, McShane, R (August 2011). “Lack of evidence for the efficacy of memantine in mild Alzheimer disease.”. Archives of neurology 68 (8): 991–8. doi:10.1001/archneurol.2011.69. PMID 21482915.
- Mount C, Downton C (July 2006). “Alzheimer disease: progress or profit?”. Nat Med. 12 (7): 780–4. doi:10.1038/nm0706-780. PMID 16829947.
- NICE technology appraisal January 18, 2011 Azheimer’s disease – donepezil, galantamine, rivastigmine and memantine (review): final appraisal determination
- Rossi S, editor. Australian Medicines Handbook 2006. Adelaide: Australian Medicines Handbook; 2006.
- Areosa SA, Sherriff F, McShane R (2005). “Memantine for dementia”. In Areosa Sastre, Almudena. Cochrane Database Syst Rev (3): CD003154. doi:10.1002/14651858.CD003154.pub4. PMID 16034889.
- Joint Formulary Committee (2004). British National Formulary (47th ed.). London: BMA and the Royal Pharmaceutical Society of Great Britain. ISBN 0-85369-584-9.
- Villoslada P, Arrondo G, Sepulcre J, Alegre M, Artieda J (December 2008). “Memantine induces reversible neurologic impairment in patients with MS”. Neurology 72 (19): 1630–3. doi:10.1212/01.wnl.0000342388.73185.80. PMID 19092106.
- Green AJ (February 2009). “Understanding pseudo. The symptoms are real, the cause is unclear”. Neurology 72 (19): 1626–7. doi:10.1212/01.wnl.0000345879.39454.68. PMID 19246422.
- Bechara J. Saab, Ruxandra M. Luca, Wing B. Yuen, Adam M. P. Saab, John C. Roder “Memantine Affects Cognitive Flexibility in the Morris Water Maze”, Journal of Alzheimer’s Disease Volume 27 Issue 3 (December 2011).
- Cacabelos R, Takeda M, Winblad B (January 1999). “The glutamatergic system and neurodegeneration in dementia: preventive strategies in Alzheimer’s disease”. Int J Geriatr Psychiatry 14 (1): 3–47. doi:10.1002/(SICI)1099-1166(199901)14:1<3::AID-GPS897>3.0.CO;2-7. PMID 10029935.
- Kornhuber J, Bormann J, Retz W, Hübers M, Riederer P (1989). “Memantine displaces [3H]MK-801 at therapeutic concentrations in postmortem human frontal cortex”. Eur.J.Pharmacol 166: 589–590. doi:10.1016/0014-2999(89)90384-1. PMID 2680528.
- Chen HSV, Pellegrini JW, Aggarwal SK, Lei SZ, Warach S, Jensen FE, Lipton SA (1 November 1992). “Open-channel block of N-methyl-D-aspartate (NMDA) responses by memantine: therapeutic advantage against NMDA receptor-mediated neurotoxicity”. J. Neurosci. 12 (11): 4427–36. PMID 1432103.
- Chen HSV, Lipton SA (15 February 1997). “Mechanism of memantine block of NMDA-activated channels in rat retinal ganglion cells: uncompetitive antagonism”. J. Physiol. (Lond.) 499 (Pt 1): 27–46. PMC 1159335. PMID 9061638.
- Rogawski, MA; Wenk GL (2003). “The neuropharmacological basis for the use of memantine in the treatment of Alzheimer’s disease”. CNS Drug Rev 9 (3): 275–308. doi:10.1111/j.1527-3458.2003.tb00254.x. PMID 14530799.
- Robinson, DM; Keating GM (2006). “Memantine: a review of its use in Alzheimer’s disease”. Drugs 66 (11): 1515–34. doi:10.2165/00003495-200666110-00015. PMID 16906789.
- Xia P, Chen HSV, Zhang D, Lipton SA (2010). “Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses”. J. Neurosci 30 (33): 11246–11250. doi:10.1523/JNEUROSCI.2488-10.2010. PMC 2932667. PMID 20720132.
- Kornhuber J, Weller M (1997). “Psychotogenicity and NMDA receptor antagonism: implications for neuroprotective pharmacotherapy”. Biol. Psychiatry 41 (2): 135–144. doi:10.1016/S0006-3223(96)00047-9. PMID 9018383.
- Rogawski, MA (2000). “Low affinity channel blocking (uncompetitive) NMDA receptor antagonists as therapeutic agents—toward an understanding of their favorable tolerability”. Amino Acids 19 (1): 133–49. doi:10.1007/s007260070042. PMID 11026482.
- Parsons CG, Stöffler A, Danysz W (November 2007). “Memantine: a NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system — too little activation is bad, too much is even worse”. Neuropharmacology 53 (6): 699–723. doi:10.1016/j.neuropharm.2007.07.013. PMID 17904591.
- Lipton SA (February 2006). “Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond”. Nature Reviews Drug Discovery 5 (2): 160–70. doi:10.1038/nrd1958. PMID 16424917.
- Chen HS, Lipton SA (June 2006). “The chemical biology of clinically tolerated NMDA receptor antagonists”. J Neurochem. 97 (6): 1611–26. doi:10.1111/j.1471-4159.2006.03991.x. PMID 16805772.
- Lipton SA (October 2007). “Pathologically activated therapeutics for neuroprotection”. Nature Reviews Neuroscience 8 (10): 803–8. doi:10.1038/nrn2229. PMID 17882256.
- Rammes G, Rupprecht R, Ferrari U, Zieglgänsberger W, Parsons CG (June 2001). “The N-methyl-D-aspartate receptor channel blockers memantine, MRZ 2/579 and other amino-alkyl-cyclohexanes antagonise 5-HT(3) receptor currents in cultured HEK-293 and N1E-115 cell systems in a non-competitive manner”. Neurosci Lett. 306 (1–2): 81–4. doi:10.1016/S0304-3940(01)01872-9. PMID 11403963.
- Buisson B, Bertrand D (1 March 1998). “Open-channel blockers at the human alpha4beta2 neuronal nicotinic acetylcholine receptor”. Mol Pharmacol. 53 (3): 555–63. PMID 9495824.
- Aracava Y, Pereira EF, Maelicke A, Albuquerque EX (March 2005). “Memantine blocks alpha7* nicotinic acetylcholine receptors more potently than n-methyl-D-aspartate receptors in rat hippocampal neurons”. J Pharmacol Exp Ther. 312 (3): 1195–205. doi:10.1124/jpet.104.077172. PMID 15522999.
- Gotti C, Clementi F (December 2004). “Neuronal nicotinic receptors: from structure to pathology”. Prog Neurobiol. 74 (6): 363–96. doi:10.1016/j.pneurobio.2004.09.006. PMID 15649582.
- Seeman P, Caruso C, Lasaga M (February 2008). “Memantine agonist action at dopamine D2High receptors”. Synapse 62 (2): 149–53. doi:10.1002/syn.20472. PMID 18000814.
- http://www.psycheducation.org/depression/meds/memantine.htm
- Open-Label Pilot Study of Namenda in Adult Subjects With ADHD and ADHD NOS [1]
- Dan Ziegler. “New drugs to prevent or treat diabetic polyneuropathy” (pdf). Retrieved 2008-01-07.[dead link]
- Schifitto G, Navia BA, Yiannoutsos CT, et al. (September 2007). “Memantine and HIV-associated cognitive impairment: a neuropsychological and proton magnetic resonance spectroscopy study”. AIDS 21 (14): 1877–86. doi:10.1097/QAD.0b013e32813384e8. PMID 17721095.
- Corbett J (September 2007). “Memantine/Gabapentin for the treatment of congenital nystagmus”. Curr Neurol Neurosci Rep 7 (5): 395–6. doi:10.1007/s11910-007-0061-z. PMID 17764629.
- Aman, Michael (Interviewee) (2010-07-29). Drug Used in Alzheimer’s Tested In Kids With Autism. Ohio: Ohio State University Medical Center.
- Borghol, Amne; Kirkwood A, Hawawini F (May 2010). “Memantine for the Treatment of Migraine”. US Pharm 35 (5): 28–35.
- Wang, R.; Zhang, D. (2005). “Memantine prolongs survival in an amyotrophic lateral sclerosis mouse model”. European Journal of Neuroscience 22 (9): 2376–2380. doi:10.1111/j.1460-9568.2005.04431.x. PMID 16262676.
- Costa, ACS; Boada R, Hutaff-Lee C, Schrader A, Weitzenkamp D, Benke TA, Goldson EJ (July 17, 2012). “Antagonism of NMDA receptors as a potential treatment for Down syndrome: a pilot randomized controlled trial”. Translational Psychiatry 2 (e141). doi:10.1038/tp.2012.66.
|
6-4-1981
|
Salts of dihalo-2-quinoxaline carboxylic acids, their preparation and pharmaceutical formulations containing them.
|
|
|
2-25-1981
|
Salts of dihalo-3,4-dihydro-3-oxo-2-quinoxaline carboxylic acids and hindered amines
|
|
|
10-25-1978
|
Drugs or medicines for influencing the central nervous system
|
|
12-16-2011
|
NOVEL THERAPEUTIC COMPOUNDS
|
|
|
12-16-2011
|
PROCESS FOR THE PREPARATION OF 1-BROMO-3,5-DIMETHYL ADAMANTANE
|
|
|
11-18-2011
|
PROCESS FOR PREPARING MEMANTINE
|
|
|
11-2-2011
|
CARNITINE CONJUGATES OF ADAMANTANAMINES DERIVATIVES AS DUAL PRODRUGS FOR VARIOUS USES
|
|
|
10-28-2011
|
PROCESS FOR MANUFACTURING ADAMANTANE DERVATIVES WITH HIGH YIELD
|
|
|
9-30-2011
|
Immediate release formulations of 1-aminocyclohexane compounds, memantine and neramexane
|
|
|
8-19-2011
|
Compositions, Methods, and Kits for Treating Influenza Viral Infections
|
|
|
7-13-2011
|
Carnitine Conjugates of Adamantanamines and Neramexane Derivatives as Dual Prodrugs for Various Uses
|
|
|
5-6-2011
|
SALTS OF MEMANTINE AND COX-INHIBITORS AND THEIR CRYSTAL FORM IN THE TREATMENT OF PAIN
|
|
|
5-4-2011
|
FLUORO-CONTAINING DERIVATIVES OF HYDROGENATED PYRIDO[4,3-B]INDOLES WITH NEUROPROTECTIVE AND COGNITION ENHANCING PROPERTIES, PROCESS FOR PREPARING, AND USE
|
Further reading
- Lipton SA (2005). “The molecular basis of memantine action in Alzheimer’s disease and other neurologic disorders: low-affinity, uncompetitive antagonism”. Current Alzheimer research 2 (2): 155–65. doi:10.2174/1567205053585846. PMID 15974913.
External links
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| 3,5-dimethyltricyclo[3.3.1.13,7]decan-1amine or 3,5-dimethyladamantan-1-amine |
|
| Clinical data | |
| Trade names | Namenda |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a604006 |
| Licence data | EMA:Link, US FDA:link |
| Pregnancy cat. | B2 (AU) B (US) |
| Legal status | Prescription Only (S4) (AU) POM (UK) ℞-only (US) |
| Routes | Oral |
| Pharmacokinetic data | |
| Bioavailability | ~100% |
| Metabolism | Hepatic (<10%) |
| Half-life | 60–100 hours |
| Excretion | Renal |
| Identifiers | |
| CAS number | 19982-08-2 |
| ATC code | N06DX01 |
| PubChem | CID 4054 |
| DrugBank | DB01043 |
| ChemSpider | 3914 |
| UNII | W8O17SJF3T |
| KEGG | D08174 |
| ChEMBL | CHEMBL807 |
| Chemical data | |
| Formula | C12H21N |
| Mol. mass | 179.3 g/mol |
| |
|
High Cholesterol: An Obstacle to Pregnancy

High Cholesterol: An Obstacle to Pregnancy
Elevated concentrations of cholesterol in the blood circulation are associated to a reduced fecundity
Read more
http://www.chemistryviews.org/details/news/6210791/High_Cholesterol_An_Obstacle_to_Pregnancy.html
