
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 227)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
2-aminooctahydrocyclopentalene-3a-carboxamides as potent CCR2 antagonists

((2R,3aR,6aR)-2-((3R,4R)-3-Methoxytetrahydro-2H-pyran-4-ylamino)octahydropentalen-3a-yl)(3-(trifluoromethyl)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)methanone
((2R,3aR,6aR)-2-((3R,4R)-3-Methoxytetrahydro-2H-pyran-4-ylamino)octahydropentalen-3a-yl)(3-(trifluoromethyl)-7,8-dihydro-1,6-naphthyridin-6(5H)-yl)methanone Semisuccinate
D-erythro-Pentitol, 1,5-anhydro-3,4-dideoxy-3-[[(2R,3aR,6aR)-3a-[[7,8-dihydro-3-(trifluoromethyl)-1,6-naphthyridin-6(5H)-yl]carbonyl]octahydro-2-pentalenyl]amino]-2-O-methyl-, rel–
cas 1416178-01-2
Abbott Laboratories, Abbott Laboratories Trading (Shanghai) Company, Ltd.
Antagonists of the CC-chemokine receptor 2 (CCR-2) have been vigorously pursued by a number of pharmaceutical companies as a target for drug discovery, in that compounds could have the potential for use in the acute and chronic conditions of inflammatory and autoimmune diseases associated with infiltration of monocytes, macrophages, lymphocytes, dendritic cells, NK cells, eosinophils, basophils, natural killer (NK cells), and memory T-cells. A compound of interest that was discovered in the Janssen laboratories that met the initial criteria set out during the in vitro screening phase of drug discovery was bicyclic 1
Chemokines are chemotactic cytokines that are released by a wide variety of cells to attract leukocytes, as illustrated by macrophages, T cells, B cells, eosinophils, basophils, and neutrophils to and from sites of inflammation or within specific compartments, as illustrated by lymph nodes (reviewed in Schall, Cytokine 1991 ; 3: 165- 183; Schall, et al., Curr. Opin. Immunol. 1994; 6:865- 873; and Murphy, Rev. Immun. 1994; 12:593-633). In addition to stimulating chemotaxis, other changes can be selectively induced by chemokines in responsive cells, including changes in cell shape, transient rises in the concentration of intracellular free calcium ions ([Ca2+]), granule exocytosis, integrin upregulation, formation of bioactive lipids (e.g., leukotrienes), and respiratory burst, associated with leukocyte activation. Thus, the chemokines are early modulators of inflammatory response, effecting inflammatory mediator release, chemotaxis and extravasation to sites of infection or inflammation.
There are four classes of chemokines, CXC (a), CC (β), C (γ), and CX3C (δ), depending on whether the first two cysteines are separated by a single amino acid (C-X- C), are adjacent (C-C), have a missing cysteine pair (C), or are separated by three amino acids (CX3C). The a-chemokines, such as interleukin-8 (IL-8), melanoma growth stimulatory activity protein (MGSA), and stromal cell derived factor 1 (SDF-1) are chemotactic primarily for neutrophils and lymphocytes, whereas β-chemokines, such as RANTES, ΜΙΡ-Ια, ΜΙΡ- Ι β, monocyte chemotactic protein- 1 (MCP-1), MCP-2, MCP-3, and eotaxin are chemotactic for macrophages, T-cells, eosinophils and basophils (Deng, et al., Naturel996; 381 :661-666). The C chemokine lymphotactin shows specificity for lymphocytes (Kelner, et al., Science 1994; 266: 1395-1399) while the CX3C chemokine fractalkine shows specificity for lymphocytes and monocytes (Bazan, et al., Nature 1997; 385:640-644).
Chemokines bind specific cell-surface receptors belonging to the family of G- protein-coupled seven-transmembrane-domain proteins (reviewed in Horuk, Trends
Pharm. Sci. 1994; 15: 159-165) termed “chemokine receptors.” On binding their cognate ligands, chemokine receptors transduce an intracellular signal through the associated heterotrimeric G protein, resulting in a rapid increase in intracellular calcium concentration. There are at least twelve human chemokine receptors that bind or respond to β-chemokines with the following characteristic pattern: CCR1 (or “CKR-1 ” or “CC- CKR-1 “) ΜΙΡ-Ια, ΜΙΡ-Ι β, MCP-3, RANTES (Ben-Barruch, et al., J. Biol. Chem. 1995; 270:22123-22128; Neote, et al., Cell 1993; 72:415425); CCR2A and CCR2B (or “CKR- 2A’7″CKR-2A” or “CC-CKR-2A”/”CC-CKR2A”) MCP-1, MCP-2, MCP-3, MCP-4; CCR3 (or “CKR-3” or “CC-CKR-3”) eotaxin, RANTES, MCP; (Ponath, et al., J. Exp. Med. 1996; 183:2437-2448); CCR4 (or “CKR-4” or “CC-CKR-4”) TARC, MDC (Imai, et al., J. Biol. Chem. 1998; 273: 1764- 1768); CCR5 (or “CKR-5” or “CC-CKR-5”) MIP- la, RANTES, MIP-Ι β; (Sanson, et al., Biochemistry 1996; 35:3362-3367); CCR6 MIP-3a (Greaves, et al., J. Exp. Med. 1997; 186:837-844); CCR7 ΜΙΡ-3β and 6Ckine (Campbell, et al., J. Cell. Biol. 1998; 141 : 1053- 1059); CCR8 I- 309, HHV8 vMIP-I, HHV-8 vMIP-II, MCV vMCC-I (Dairaghi, et al., J. Biol. Chem. 1999; 274:21569-21574); CCR9 TECK (Zaballos, et al., J. Immunol. 1999; 162:5671-5675), D6 MIP-1 beta, RANTES, and MCP-3 (Nibbs, et al., J. Biol. Chem. 1997; 272:32078-32083), and the Duffy blood-group antigen RANTES, MCP-1 (Chaudhun, et al., J. Biol. Chem. 1994; 269:7835-7838).
Chemokine receptors, such as CCR1, CCR2, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CXCR1, CXCR2, CXCR3, CXCR4, CXCR5, CX3CR1, and XCRl have been implicated as being important mediators of inflammatory and immunoregulatory disorders and diseases, including asthma and allergic diseases, as well as autoimmune pathologies such as rheumatoid arthritis and atherosclerosis.
The CCR2 chemokine receptor is expressed primarily in monocytes and activated T lymphocytes, and its functional activity can be measured by cytosolic calcium elevation or chemotaxis. CCR2 exists in two isoforms, CCR2A and CCR2B. These two isoforms are alternatively spliced variants of a single MCP- 1 receptor gene and differ only in the carboxyl-terminal tails. The chromosomal location of the CCR2 gene is localized to
3p21. The CC chemokines, MCP-1, MCP-2, MCP-3, and MCP-4, have been identified as the ligands that are selective and of high affinity to the CCR2 receptor.
The highly selective expression of CCR2 makes it an ideal target for intervention to interrupt inappropriate monocyte and T cell trafficking. The clinical indications for such intervention are in inflammatory diseases and T-cell mediated autoimmune diseases such as multiple sclerosis, rheumatoid arthritis, asthma, allergy, chronic obstructive pulmonary disease, atherosclerosis, restinosis, type I and type II diabetes, metabolic syndrome, and pain. Ectopic expression of MCP-1 and CCR2 in certain tumors indicates that selective modulation (such as antagonism or inhibition) of CCR2 can have value in tumor immunotherapy, particularly attenuation of metastasis.
The native peptide ligand of CCR2 is monocyte chemoattractant protein- 1 (MCP- 1 or CCL2) containing two adjacent disulfied bonds. Ample evidence exists for the role of the CCR2/MCP-1 system in preclinical animal models of pain (White F.A., Jung F., and Miller R.J., Proc. Natl. Acad. Sci. USA 2007; 51 :20151). Although CCR2 and MCP- 1 have limited expression levels in the CNS tissues under normal conditions, significant upregulation of CCR2 and MCP- 1 has been observed following a neuropathic injury in tissue relevant to pain, including neurons and glia in the spinal cord, rostroventromedial medulla (RVM) and DRG (Wang H., Zou S., Wei F., Dubner R., and Ren K., Soc for Neurosci Poster 2009; 72.3). MCP- 1 has been shown to increase the excitability of neurons acutely dissociated from the DRG tissue (Sun J.H., Yang B., Donnelly D.F., Ma C, and LaMotte R.H., J Neurophysiol. 2006; 96:2189). In addition, direct injection of MCP- 1 in the spinal cord induces thermal hyperalgesia and mechanical allodynia (Dansereau et al. Neurochem. 2008; 106:7), and the MCP- 1 induced pronociception can be blocked by a CCR2 antagonist, INCB3344. Similarly, the hyperalgesia induced by MCP-1 injection in the RVM is reversed by another CCR2 antagonist, RS I 02895 (Wang H., Zou S., Wei F., Dubner R. and Ren K., Soc for Neurosci Poster 2009; 72.3). In addition, CCR2 knock out mice exhibit significantly reduced mechanical allydonia following nerve injury and reduced nocifensive behavior in the second phase of the formalin model, whereas they exhibit normal sensitivity to acute pain stimulation in the hot plate model (Abbadie C, Lindia J.A., Cumiskey A.M., Peterson L.B., Mudgett J.S., Bayne E.K., DeMartino J.A., Maclntyre D.E., and Forrest M.J., Proc Natl Aca Sci USA 2003; 100:7947). Treatment with AZ889 (Serrano A., Pare M., Mcintosh F., Elmes S.J.R. Martino G., Jomphe C, Lessard E., Lembo P.M.C., Vaillancourt F., Perkins M.N., and Cao C.Q., Mol. Pain 2010; 6:90), a CCR2 antagonist, abolished CCL2-evoked neuronal excitation, confirming that this activity is CCR2 -mediated. Neuronal and non-neuronal cells in the spinal cord were also excited by CCL2 applications indicating an important role of spinal CCR2 in neuropathic pain. In vivo spinal intrathecal injection of AZ889 produced dose-dependent analgesia in chronic constriction injury rats (Serrano A., Pare M., Mcintosh F., Elmes S.J.R., Martino G., Jomphe C, Lessard E., Lembo P.M.C., Vaillancourt F., Perkins M.N., and Cao C.Q., Mol. Pain 2010; 6:90). Additionally, application of AZ889 to the exposed spinal cord inhibited evoked neuronal activity and confirmed that CCR2-mediated analgesia involved predominantly the spinal cord. In view of the clinical importance of CCR2, the identification of compounds that modulate CCR2 function represents an attractive avenue into the development of new therapeutic agents that can be used to treat diseases such as inflammatory, autoimmune disease, cancer, and pain, that are associated with chemokine receptor expression or activity. Such compounds are provided herein.
……………………………………………………….

51 cas 1421063-65-1
succinate cas 1421065-31-7
52 cas 1416178-01-2
…………………………………..
http://www.google.com/patents/WO2013010453A1?cl=en
Example 74
1 -phenyl-4-[(2S,3aR,6aR)-3a- {[3-(trifluoromethyl)-7,8-dihydro- 1 ,6-naphthyridin-6(5H)- yl]carbonyl}octahydropentalen-2-yl]piperazin-2-one The title compound was obtained from the procedure of Example 63. Ή NMR
(400 MHz, CDCI3) δ ppm 8.70 (s, 1 H), 7.65 (s, 1 H), 7.35 (m, 2 H), 7.20 (m, 3 H), 4.60 – 4.90 (m, 2 H), 3.85 (m, 2H), 3.65 (m, 2H), 3.30 – 3.45 (m, 3H), 3.09 (m, 2H), 2.90 (m, 1H), 2.75 (m, 1H), 2.60 (m, 1H), 2.40 (m, 1H), 2.15 (m, 1H), 1.92 (m, 1H), 1.45 – 1.82 (m, 6H), 1.25 (m, 1H); MS (ESI) nVz 513 (M+H)+.

Example 75 C
l,5-anhydro-2,3-dideoxy-4-0-methyl-3-{[(2S,3aR,6aR)-3a-{[3-(trifluoromethyl)-7,8- dihydro- 1 ,6-naphthyridin-6(5H)-yl]carbonyl} octahydropentalen-2-yl]amino} -L-threo- pentitol
The title compound was prepared according to method A of Example 79G, substituting Example 75B for Example 79F, and isolated as the major isomer. Ή NMR (400 MHz, CDC13) δ 8.68 (s, 1H), 7.66 (s, 1H), 4.75 (m, 2H), 4.10 (m, 1H), 3.78 – 3.95 (m, 3H), 3.52 (m, 2H), 3.40 (s, 3H), 3.30 (m, 2H), 3.02 – 3.16 (m, 3H), 2.96 (m, 1H), 2.00 – 2.17 (m, 3H), 1.55 – 1.95 (m, 8H), 1.30 (m, 2H); MS (ESI) m/z 468 (M+H)+.

Example 76
l,5-anhydro-2,3-dideoxy-4-0-methyl-3-{[(2S,3aR,6aR)-3a-{[3-(trifluoromethyl)-7,8- dihydro- l,6-naphthyridin-6(5H)-yl]carbonyl}octahydropentalen-2-yl]amino}-D-erythro- pentitol
The title compound was prepared and purified according to the method A described in Example 79G, and was isolated as the major product. Ή NMR (400 MHz, CDC13) δ 8.70 (s, 1H), 7.70 (s, 1H), 4.70 – 4.90 (m, 2H), 4.10 (m, 1H), 3.80 – 3.95 (m, 3H), 3.35 – 3.50 (m, 2H), 3.42 (s, 3H), 3.30 (m, 2H), 3.15 (m, 2H), 3.05 (m, 1H), 2.75 (m, lH), 2.35 (m, lH), 2.15 (m, 1H), 1.50 – 2.00 (m, 9H), 0.90-1.10 (m, 2H). MS (ESI) m/z 468 (M+H)+.

Example 78
l,5-anhydro-2,3-dideoxy-4-0-methyl-3-{methyl[(2R,3aR,6aR)-3a-{[3-(trifluoromethyl)- 7,8-dihydro-l,6-naphthyridin-6(5H)-yl]carbonyl}octahydropentalen-2-yl]amino}-D- erythro-pentitol
To the solution of Example 79G (10 mg, 0.03 mmol) in dioxane (1 mL) was added formic acid (0.6 mL) and aqueous formalin solution (37%, 0.6 mL ). The mixture was heated at 80 °C overnight under nitrogen. The reaction mixture was concentrated in vaccum and was purified by HPLC to afford title compound as white solid (8.0 mg). Ή NMR (400 MHz, CD3OD) δ 8.70 (s, 1H), 8.06 (s, 1H), 4.75 – 4.90 (m, 2H), 4.30 (m, 1H), 3.90 – 4.10 (m, 4H), 3.50 – 3.72 (m, 4H), 3.45 (s, 3H), 3.15 (m, 2H), 2.78 (s, 3H), 2.60 (m, 1H), 1.85 – 2.20 (m, 9H), 1.80 (m, 1H), 1.58 (m, 1H), 1.42 (m, 1H); MS (ESI) m/z 482 (M+H)+.
To a solution of Example 78 (400 mg, 0.83 mmol) in MeOH (20 mL) was added succinic acid (98 mg 0.83 mmol) then the mixture was heated to 80 °C for 4 hours, then the solvent was concentrated under in vacuum to give the corresponding succinate salt (480 mg, 0.81 mmol, 96%). ‘H NMR (400 MHz, CD3OD) δ 8.70 (s, 1H), 8.05 (s, 1H), 4.85 (m, 2H), 4.25 (m, 1H), 4.05 (m, 2H), 3.95 (m, 2H), 3.70 (s, 1H), 3.50 (m, 2H), 3.42 (s, 3H), 3.33 (m, 1H), 3.12 (m, 2H), 2.70 (s, 3H), 2.55 (m, 1H), 2.50 (s, 4H), 2.15 (m, 3H), 1.70 – 2.02 (m, 7H), 1.55 (m, 1H), 1.40 (m, 1H).
ABS

4,4-dimethoxytetrahydro-2H-pyran
To the solution of dihydro-2H-pyran-4(3H)-one (50 g, 0.5 mol) in MeOH (500 mL) was added TiCl4 (1 g, 5 mmol) and Et3N (500 mg, 5 mmol), and the mixture stirred at room temperature for 10 hours, followed by the addition of Et3N (2 g, 20 mmol). The mixture was concentrated to 100 mL, diluted by methyl tert-butyl ether (1.5 L) and washed with ¾0 (500 mL), and brine. The organic layer was dried over Na2S04, filtered, and concentrated to yield Example 79A (60 g, 84% yield). Example 79B
4,4-dimethoxytetrahydro-2H-pyran compound with 4-methoxy-3,6-dihydro-2H-pyran To the solution of Example 79A (60 g, 0.42 mol) in dichloromethane (800 mL) at -78 °C was added TiCl4 (84 g, 0.42 mol). The mixture was stirred at -78 °C for 1 hour, followed by the addition of pyridine (66 g, 0.84 mol) and KOH (47 g, 0.84 mol). The mixture was stirred at -78 °C for additional 0.5 hour then warmed to room temperature and stirred overnight. The mixture was filtered. The filtrate was washed with water (1 L), dried over MgS04, filtered, and concentrated to yield Example 79B (52 g, 100%).
Example 79C
4,4-dimethoxytetrahydro-2H-pyran-3-ol
To the solution of Example 79B (2 g, 17.5 mmol) in MeOH (50 mL) was added meta-chloroperoxy benzoic acid (6 g, 35 mmol) in MeOH (6 mL) at 0 – 6 °C via addition funnel. After addition, the mixture was stirred at 0 °C for 4 hours. Upon reaction completion, the mixture was concentrated to yield white solid, which was then dissolved in dichloromethane (40 mL). Calcium hydroxide (14.8 g, 200 mmol) was added to the solution, and the solution was stirred for an additional 2 hours. The mixture was filtered and the filtrate was concentrated to yield crude title compound (2 g, 66.7%), which was used into next step without further purification.
Example 79D 3-methoxydihydro-2H-pyran-4(3H)-one
To the mixture of NaH (60%, 5.04 g, 12.6 mmol) in THF (200 mL) was added Example 79C (20 g, 12.6 mmol) in THF (150 mL). The mixture was stirred at 0 °C for 0.5 hour, followed by addition of iodomethane (200 g, 15.5 mmol), and was stirred overnight. HC1 (12 M, 12 mL) was added to the mixture, stirred at room temperature for additional 1.5 hours and concentrated. The residue was purified by column
chromatography to Example 79D (20 g, 100%).
Example 79E
(3S,4S)-3-methoxy-N-((S)-l-phenylethyl)tetrahydro-2H-pyran-4-amine To the solution of Example 79D (20 g, 153.8 mmol) and (S)- 1 -phenylethanamine
(18.6 g, 153.8 mmol) in dichloromethane (200 mL) was added Ti(i-OPr)4 (87.7 g, 310 mmol) and diisopropylethyl amine (40 g, 310 mmoL). The mixture was then stirred at room temperature for 18 hours, following by addition of sodium triacetoxyborohydride (65.1 g, 310 mmol), and MeOH (15 mL). The mixture was stirred for additional 4 hours, then poured into saturated NaHC(¾ solution, stirred for 1.5 hours and filtered. The filtrate was extracted with dichloromethane (2 x 200 mL) and concentrated. The residue was purified by preparative HPLC followed by chiral SFC separation to yield title compound (10 g, 27.7%).
Example 79F
(3S,4S)-3-methoxytetrahydro-2H-pyran-4-amine
To the solution of Example 79E (5.8 g, 21.3 mmol) in EtOH (100 mL) was added Pd/C (6 g), and the mixture was then submitted to hydrogenolysis at 50 °C (¾, 50 Psi) for 48 hours. The mixture was filtered and the filtrate was concentrated to yield 1.5 g (55%) of Example 79F which was used into next step directly without further purification.
Example 79G
l,5-anhydro-2,3-dideoxy-4-0-methyl-3- {[(2R,3aR,6aR)-3a-{[3-(trifluoromethyl)-7,8- dihydro- l,6-naphthyridin-6(5H)-yl]carbonyl}octahydropentalen-2-yl]amino}-D-erythro- pentitol
To the mixture of Example 1H (4.8 g, 13.6 mmol), Example 79F (1.5 g, 0.87 mmol) in dichloroethane (120 mL) was added Ti(i-OPr)4 (974 mg, 3.48 mmol), N,N- diisopropylethyl amine (1.2 g, 10 mmol). The mixture was then stirred overnight followed by addition of NaBH4 (132 mg, 3.48 mmol) and MeOH (5 mL). The mixture was stirred for another 12 hours before it was poured into saturated NaHC(¾, stirred at room temperature for 2 hours, filtered, and the filtrate was extracted by dichloromethane (3 x 200 mL). The organic layer was concentrated and the residue was purified by preparative HPLC to afford a mixture of two diastereomers. This mixture was purified by chiral SFC to yield Exmaple 76 as the first eluent, as well as the title compound (600 mg) as the second eluent. Ή NMR (400 MHz, CDC13) δ 8.70 (s, 1H), 7.70 (s, 1H), 4.68 – 4.85 (m, 2H), 4.22 (m, 1H), 4.05 (m, 1H), 3.70 – 3.90 (m, 3H), 3.55 (m, 2H), 3.35 – 3.45 (m, 2H), 3.40 (s, 3H), 3.30 (m, 1H), 3.10 (m, 2H), 2.45 (m, 1H), 2.30 (m, 1H), 2.00 – 2.20 (m, 3H), 1.55 – 1.95 (m, 6H), 1.30 (m, 2H); MS (ESI) m/z 468 (M+H)+.
The succinate salt of the above compound was prepared as follows: A mixture of free base of Example 79G (600 mg, 1.25 mmol), succinic acid (152 mg, 1.25 mmol) in EtOH (50 mL) was heated at 65 °C for 2 hours and then concentrated. The residue was washed with Et20 (25 mL) to yield white solid (610 mg, 86.4%). Ή NMR (400 MHz, CD3OD) δ 8.70 (s, 1H), 8.05 (s, 1H), 4.85 (m, 2H), 4.22 (d, J= 14.8 Hz, 1H), 3.95 (m, 3H), 3.70 (m, 1H), 3.30 – 3.53 (m, 4H), 3.38 (s, 3H), 3.12 (m, 2H), 2.55 (m, 1H), 2.52 (s, 4H), 2.15 (m, 1H), 1.70 – 2.05 (m, 9H), 1.55 (m, 1H), 1.38 (m, 1H).
Method B
Example 79H
( 1 R,4S)-methyl 4-aminocyclopent-2-enecarboxylate To a cooled mixture of (lR,4S)-2-azabicyclo[2.2.1]hept-5-en-3-one (13 g, 1 19 mmol) in MeOH (150 mL) was added SOCI2 (20 mL) dropwise to keep the temperature of the reaction under 15 °C. Upon completion of addition, the mixture was stirred at 5 °C for 3 hours. The solvent was removed under reduced pressure to yield liquid, which was dried under high vacuum to give Example 79H as white solid (23 g).
Example 791
(lR,4S)-methyl 4-(2,5 -dimethyl- lH-pyrrol-l-yl)cyclopent-2-enecarboxylate To a mixture of Example 79H (23 g, 163 mmol) in MeOH (100 ml) was added diisopropylethyl amine (23 g, 179 mmol) and acetyl acetone (20 g, 170 mmol), then the mixture was stirred at room temperature for 16 hours. The solvent was removed under reduced pressure, and the crude product was purified by column chromatography (S1O2, petroleum ethenEtOAc = 20: 1) to give Example 791 as yellow oil (20 g).
Example 79J (lR,4S)-methyl 1 -(3-bromopropyl)-4-(2,5-dimethyl- lH-pyrrol- 1 -yl)cyclopent-2- enecarboxylate
To a solution of Example 791 (16.5 g, 74.4 mmol) in THF (200 ml) was added dropwise lithium hexamethyl bis(trimethylsilyl)amide (1 M, 1 19 mL) at -50 °C, stirredd for 1 hour, allowed to warm to -20 °C, followed by the dropwise addition of 1,3- dibromopropane (150 g, 744 mmol) over 1 hour. The reaction mixture was stirred at -20 °C for 1 hour, quenched with aqueous NH4C1 solution (6%, 600 mL), and extracted with ethyl acetate. The organic layer was washed with NH4CI solution, brine, dried over Na2S04, filtered, and concentrated. The residued was purified by silica gel column chromatography (petroleum ethenEtOAc = 80: 1) to give Example 79J (16 g).
Example 79K
(2R,3aR,6aR)-methyl 2-(2,5-dimethyl- lH-pyrrol- 1 -yl)octahydropentalene-3a-carboxylate To a solution of compound 79 J (16 g, 47 mmol) and azobisisobutyronitrile (1.6 g, 10 mmol) in toluene (1.8 L) was added a solution of tributyl tin hydride (32 mL) in toluene (200 mL) at 1 10 °C over 1 hour. After refluxing for 3 hours, the reaction mixture was quenched by saturated aqueous KF (200 mL), and extracted with ethyl acetate. The organic layer was washed with brine, dried over Na2S04;filtered, and concentrated. The residue was purified by column chromatography (S1O2, petroleum ethenEtOAc = 50: 1) to give compound Example 79K (8 g) as white solid.
Example 79L
(2R,3aR,6aR)-2-(2,5-dimethyl-lH-pyrrol-l-yl)octahydropentalene-3a-carboxylic acid To a solution of Example 79K (5.3 g, 20.3 mmol) in MeOH (33 mL) and water (15 mL) was added a aqueous solution of NaOH (3.2 g, 80 mmol in 4 mL water) and the mixture was heated at 65 °C for 16 hours. The mixture was cooled to room temperature, adjusted the pH to about 4 with HC1 solution (4 N) and filtered to collect Example 79L (4.5 g) as yellow solid and used in next step without purification.
Example 79M
((2R,3aR,6aR)-2-(2,5-dimethyl- lH-pyrrol- 1 -yl)octahydropentalen-3a-yl)(3- (trifluoromethyl)-7,8-dihydro-l,6-naphthyridin-6(5H)-yl)methanone To a solution of compound Example 79L (5 g, 20.2 mmol) in dichloromethane (50 mL) was added hydroxybenzotriazole (4.2 g, 30.9 mmol), l-ethyl-3-(3- dimethylaminopropyl) carbodiimide hydrochloride (5.8 g, 30.4 mmol), and Et3N (6.0 g, 59.4 mmol), and the mixture was stirred at room temperature for 16 hours. The reaction mixture was suspended in water and extracted with dichloromethane (3 x 300 mL). The combined organic layer was washed with brine, dried over Na2S04, filtered, and concentrated to result in the title compound (8 g), which was used in the next step without purification.
Example 79N
((2R,3aR,6aR)-2-aminooctahydropentalen-3a-yl)(3-(trifluoromethyl)-7,8-dihydro- l,6- naphthyridin-6(5H)-yl)methanone
To a solution of Example 79M (8.0 g, 18.6 mmol) in MeOH (100 mL) was added hydroxylamine hydrochloride (8.0 g, 1 15.9 mmol), 50% hydroxylamine hydrate (12 mL) and ¾0 (40 mL). The mixture was heated at reflux for 13 hours and cooled to room temperature. The mixture was treated with NaOH (10 N) to adjust the pH to about 1 1, and extracted with dichloromethane (3 x 300 mL). The combined organic layer was washed with brine, dried over MgS04, filtered, and concentrated. A solution of HCl in EtOAc (30 mL) was added to the residue with stirring at room temperature for 1 hour. The solvent was removed and the HCl salt of Example 79N (6.5 g) was used for next step without purification.
Example 790
tert-butyl(3,6-dihydro-2H-pyran-4-yloxy)dimethylsilane To a mixture of the tetrahydro-4H-pyran-4-one (38.9 g, 0.38 mol) and Et3N (76.8 g, 0.76 mol) in dichloromethane (800 mL) was added trimethyl trifluoromethanesulfonate (105.5 g, 0.399 mol) dropwise over 3 hours. After addition, the reaction was allowed to warm to room temperature and stirred overnight. Water was added and the resulting solution was extracted with dichloromethane (2 x 500 mL). The combined organic phase was washed with water (2 x 500 mL) and brine (2 x 200 mL), dried over Na2S04, filtered, and concentrated to give Example 790 (78 g, 85%) as an oil.
Example 79P
sodium (3R)-3,4-dihydroxytetrahydro-2H-pyran-4-sulfonate To a solution of (DHQD^PHAL (hydroquinidein 1 ,4-phthalazinediyl diether) (3.06 g, 3.93 mol), K20s04 (723 mg, 1.96 mol) and N-methylmorpholine-N-oxide (58.4 g, 0.432 mol) in acetone/H20 (700 mL, 10/1) at 0 °C was added slowly a solution of Example 790 (84 g, 0.393 mol) in acetone (100 mL) for 5 hours. The resulting solution was stirred at 10-20 °C overnight. A freshly prepared solution of Na2S20s (44.8 g, 0.236 mol) in water (315 mL) was added followed by acetic acid (67.3 mL). After stirring for 16 hours at room temperature, the solid was filtered and washed with isopropanol (400 mL) and dried to provide Example 79P (60 g, 73%) as a white solid.
Example 79Q
(R)-4,4-dimethoxytetrahydro-2H-pyran-3-ol To a solution of Example 79P (60 g, 0.294 mol) and HC(OCH3)3 (69.3 g, 0.647 mol) in MeOH (500 mL) at 50 °C was added HCl/MeOH (68 mL, 5-6 N) slowly over 30 minutes. Then the slurry was cooled to 5 °C and 50% of NaOH (100 mL in water) was added over 1 hour. The solid was filtered and the filtrate was concentrated. The resulting solution was washed with toluene for several times and then concentrated to give Example 79Q (38 g, yield: 88%) as an oil.
Example 79R
(R)-3-methoxydihydro-2H-pyran-4(3H)-one To a solution of Example 79Q (9.5 g, 64.68 mol) in THF (300 mL) was added sodium tert-butoxide (9.3 g, 97.02 mmol) at ice bath. Then dimethyl sulfate (13.4 g, 106 mmol) was added over 20 minutes, maintaining an interal temp below 36 °C. After addition, the reaction mixture was stirred for 4 hours at room temperature. Water (200 mL) was added followed by addition of 2N HC1 (100 mL). The apparent pH is below 1. After 16 hours of reaction, NaHC(¾ (20 g) was added and the mixture was extracted with EtOAc (4 x 300 mL), dried over Na2S04, filtered, and concentrated to give Example 79R (6 g, yield: 71 %) as an oil.
Example 79G
l,5-anhydro-2,3-dideoxy-4-0-methyl-3- {[(2R,3aR,6aR)-3a-{[3-(trifluoromethyl)-7,8- dihydro- l,6-naphthyridin-6(5H)-yl]carbonyl}octahydropentalen-2-yl]amino}-D-erythro- pentitol
A solution of Example 79N (3.2 g, 9.0 mmol) in isopropyl acetate (80 mL) was cooled with ice bath and tributylamine (3.3 g, 20.7 mmol) was added dropwise, followed by the addition of isopropyl alcohol (1.6 ml, 20.7 mmol). Sodium triacetoxyborohydride (4.4 g, 20.7 mmol) was added. After 1 hour at room temperature, a solution of Exampole 79R (1.75 g, 13 mmol) in isopropyl acetate (10 mL) was added to the mixture at 1 °C. Then the mixture was stirred at room temperature for 15 hours and partitioned between satureated aqueous NaHC(¾ (80 mL), water (50 mL) and EtOAc (400 mL). The aqueous phase was further extracted with EtOAc (200 mL). The combined organic phase was washed with saturated.aqeous NaHC03 solution, dried with Na2S04, filtered, concentrated. The residue was purified by column chromatography (S1O2,
dichloromethane:MeOH = 20: 1) to give Example 79G as a mixture of diastereomers, which was then further purified by chiral SFC to yield title compound as the first eluent and white solid upon concentration, as well as Example 80 as the second eluent.

…………………………………………………………
Bioorganic & Medicinal Chemistry Letters, 23(1), 351-354; 2013
http://www.sciencedirect.com/science/article/pii/S0960894X12013601

Regents and conditions: (a) (Boc)2O, DMAP, THF, 58 °C; (b) AcOCH2C(
……………………………
http://pubs.acs.org/doi/full/10.1021/op500265z

………………………………..

YM758 Monophosphate, A Novel If Channel Inhibitor


| N-[2-[(3R)-3-[(3,4-Dihydro-6,7-dimethoxy-2(1H)-isoquinolinyl)carbonyl]-1-piperidinyl]ethyl]-4-fluorobenzamide Phosphate; (R)-(-)-N-[2-[3-[(6,7-Dimethoxy-1,2,3,4 -tetrahydroisoquinolin-2-yl)carbonyl]piperidino]ethyl]-4-fluorobenzamide monophosphate; YM 758; |
| CAS Number: 312752-86-6 |
U.S. Patent No. 6,573,279, incorporated herein by reference, describes isoquinoline compounds with 1 channel blocker activity and their use in treating a variety of cardiovascular diseases. U.S. Patent Application Publication Nos. 20060084807 and
20070129357, each of which is incorporated herein by reference, describe methods for making those isoquinoline compounds as well as crystals of certain fluorobenzamide derivatives of them. U.S. Patent Publication No. 20090247572, incorporated herein by reference, relates to the use of one of these isoquinoline fluorobenzamide derivatives, (-)-N- {2-[(i?)-3-(6,7-dimethoxy-l ,2,3,4-tetrahydroisoquinoline-2-carbonyl)piperidino]ethyl}-4- fluorobenzamide monophosphate (referred to in that patent publication as “compound A” and “chemical formulation I” and referred to herein as “YM758”), for treating atrial fibrillation.
To date, however, these isoquinoline compounds in general and YM758 in particular have not been developed as cardiovascular drugs. Thus, there remains a need for methods of using these compounds, alone and in combination with other cardiovascular drugs, to treat cardiovascular disease, as well as a need for pharmaceutical formulations and unit dose forms useful in such methods. This invention meets those needs.
Provided herein are fast-acting (immediate release) and modified (sustained) release oral formulations as well as intravenous formulations of YM758. The present invention also provides unit dose forms of these formulations. The present invention also provides methods for using these formulations and unit dose forms alone and in combination with other drugs for the treatment of cardiovascular disease, including but not limited to stable angina, atrial fibrillation, and heart failure. In these methods, the pharmaceutical formulations and unit dose forms of the invention may be dosed alone or in combination with other drugs, including but not limited to drugs such as beta-blockers, anti-arrhythmia drugs, calcium channel blockers, sodium channel blockers, potassium channel blockers, adenosine, and digitalis. The invention also provides formulations and unit dose forms of YM758 and another drug selected from the group of drugs including beta-blockers, anti-arrhythmia drugs, calcium channel blockers, sodium channel blockers, potassium channel blockers, adenosine, and digitalis. The single agent and combination pharmaceutical formulations and unit dose forms of the invention include capsule, tablet, and solution formulations and unit dose forms that provide either immediate or sustained release. The pharmaceutical formulations in solution forms are, in various embodiments, suitable for intravenous, subcutaneous, intraperitoneal, and intramuscular administration.
Thus, in one aspect, the present invention provides an oral formulation comprising or consisting essentially of YM758 and optionally an excipient. As used herein, the excipient is suitable for administration to human patients with various cardiovascular diseases and includes, without limitation, one or more of the following: an additive, an anti- foaming agent, a binder, a chemical stabilizer, a coloring agent, a diluent, a disintegrating agent, an emulsifying agent, a filler, a flavoring agent, a glidant, a lubricant, a pH modifier, a plasticizer, a solubilizer, a swelling enhancer, a spheronization aid, a solubility enhancer, and a suspending agent. In some embodiments, the formulation is provided in a unit dose form, which may be, for example, a tablet or capsule. In various embodiments, the unit dose forms contain from about 5 mg to about 80 mg of YM758. In some embodiments, the unit dose forms contain from about 5 mg to about 50 mg of YM758. In other embodiments, the unit dose form contains from about 10 mg to about 40 mg of YM758. In one embodiment, the unit dose form contains about 25 mg of YM758.
In another aspect, the present invention provides formulations comprising or consisting essentially of YM758 and optionally an excipient that are suitable for intravenous, subcutaneous, intraperitoneal, and intramuscular administration. As used herein, the excipient is suitable for administration to human cardiovascular disease patients and includes, without limitation, one or more of the following: an additive, an anti-foaming agent, a chemical stabilizer, a diluent, an emulsifying agent, a pH modifier, a buffering agent, an osmolality modifier, a salt, a solubilizer, a solubility enhancer, and a suspending agent. In some embodiments, the formulation is a solution formulation. In one embodiment, the solution formulation is provided in a unit dose form, which may be, for example, in a vial, an ampoule or an intravenous bag. In various embodiments, the unit dose forms contain from about 5 mg to about 80 mg of YM758. In some embodiments, the unit dose forms contain from about 5 mg to about 50 mg of YM758. In another embodiment, the unit dose form contains from about 10 mg to about 40 mg of YM758. In one embodiment, the unit dose form contains about 25 mg of YM758. [0008] In various embodiments, the oral formulation is an immediate release formulation, including, and unit dose forms of this formulation include, without limitation, a gelatin capsule comprising a YM758 formulation.
NMR: δ 1.25-1.50 (1H, m), 1.53-1.76 (3H, m), 2.15-2.80 (6H, m), 2.95-3.10 (3H m), 3.40-3.50 (2H, m), 3.60-3.75 (8H, m), 4.50 (1H, q), 4.63 (1H, q), 6.73 (1H, s), 6.78, 6.85 (1H, s in combination), 7.29 (2H, t), 7.91-7.95 (2H, m), 8.60 (1H, br).
FAB-MS m/z: 470 (M++1).


A novel, practical, and efficient synthesis of (−)-N-{2-[(R)-3-(6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline-2-carbonyl)piperidino]ethyl}-4-fluorobenzamide monophosphate (YM758 monophosphate, (R)-1·H3PO4 (Figure 1) is described. The target molecule (R)-1 has a potent If current channel inhibitor. Medicinal chemistry synthetic routes were very long and suffered from extensive use of chlorinated solvents and silica-gel column chromatography. A number of steps in the medicinal chemistry route were also unattractive for large-scale synthesis due to some reasons for example the use of unstable intermediates.
An important objective of a new synthetic route was avoidance of such a use of unstable intermediate, and it was achieved by the discovery of an important 4,5-dihydrooxazole intermediate 19 and ring-opening N-alkylation of chiral amine with 19 under acidic condition. The new procedure does not require any purification by column chromatography for all steps.
The overall yield was significantly improved from 14% or 34% to 49% compared to that of the medicinal synthetic routes. This highly efficient process was successfully demonstrated at a pilot-scale operation, yielding 36.5 kg of (R)-1·H3PO4.
(−)-N-{2-[(R)-3-(6,7-Dimethoxy-1,2,3,4-tetrahydroisoquinoline-2-carbonyl)piperidino]ethyl}-4-fluorobenzamide Monophosphate (YM758 Monophosphate, (R)-1·H3PO4)
Cefditoren pivoxil


Cefditoren pivoxil
ME-1207, Spectracef, Meiact
117467-28-4
- (-)-(6R,7R)-2,2-dimethylpropionyloxymethyl 7-((Z)-2-(2-aminothiazol-4-yl)-2-methoxyiminoacetamido)-3-((Z)-2-(4-methylthiazol-5-yl)ethenyl)-8-oxo-5-thia-1-azabicyclo(4.2.0)oct-2-ene-2-carboxylate
- 7-(2-(2-aminothiazol-4-yl)-2-(methoxyimino)acetamido)-3-(2-(4-methylthiazol-5-yl)ethenyl)cephem-4-carboxylic acid pivaloyloxymethyl ester
- CDTR-PI
- cefditoren pivoxil
- ME 1207
- ME-1207
- Spectracef
Novel crystalline form of cefditoren pivoxil (first disclosed in EP175610). Represents Yungjin Pharma’s first interest in this API, which was developed and launched by Meiji Seika and previous licensee TAP Pharmaceuticals, and now marketed by Merus Labs, for treating chronic bronchitis and community acquired pneumonia caused by bacterial infections
Cefditoren is a third-generation cephalosporin antibiotic for oral use. It is commonly marketed under the trade name Spectracef by Vansen Pharma Inc.
Cefditoren is also marketed under the name Meiact by Meiji Seika Pharma Co., Ltd.[1]
Spectrum of bacterial susceptibility
Cefditoren has a broad spectrum of activity and has been used to treat bacterial infections of the skin and respiratory tract, including bronchitis, pneumonia, and tonsillitis. The following represents MIC susceptibility data for a few medically significant microorganisms.
- Haemophilus influenzae: ≥0.063 – 0.25 μg/ml
- Staphylcoccus aureus: 0.25 – >128 μg/ml (includes MRSA)
- Streptococcus pyogenes: ≤0.004 – 2 μg/ml[2]
Cefditoren is a broad-spectrum antibiotic against Gram-negative and Gram-positive bacteria, but does not have antibacterial activity against Pseudomonas aeruginosa.[3]
Clinical use
Indications
Cefditoren is used to treat uncomplicated skin and skin structure infections, community-acquired pneumonia, acute bacterial exacerbation of chronic bronchitis, pharyngitis, and tonsillitis.
Formulations
Cefditoren is available as 200- and 400-mg tablets. It can be formulated as the prodrug cefditoren pivoxil.

Chemical structure of cefditoren pivoxil

References
- Meiact Full Description
- http://www.toku-e.com/Assets/MIC/Cefditoren%20sodium.pdf
- “Disease relevance of Cefditoren”. Retrieved June 24, 2014.
- Chem Pharm Bull 1992,39(9),2433
- J Antibiot 1990,43(8),1047
Synthesis Reference
Kiyoshi Yasui, Masahiro Onodera, Masamichi Sukegawa, Tatsuo Watanabe, Yuichi Yamamoto, Yasushi Murai, Katsuharu Iinuma, “Crystalline substance of cefditoren pivoxyl and the production of the same.” U.S. Patent US6294669, issued March, 1986.
| Patent | Submitted | Granted |
|---|---|---|
| Therapy for Treating Resistant Bacterial Infections [US2009275552] | 2009-11-05 | |
| Process for the preparation of thiazole intermediate [US6833459] | 2003-10-30 | 2004-12-21 |
| Nanoparticulate and Controlled Release Compositions Comprising Cefditoren [US8119163] | 2008-11-13 | 2012-02-21 |
External links
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (7R)-7-((Z)-2-(2-Aminothiazol-4-yl)-2-(methoxyimino)acetamido)-3-((Z)-2-(4-methylthiazol-5-yl)vinyl)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid | |
| Clinical data | |
| Trade names | Spectracef |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a605003 |
| Legal status |
?
|
| Identifiers | |
| CAS number | 104145-95-1 |
| ATC code | J01DD16 |
| PubChem | CID 9870843 |
| DrugBank | DB01066 |
| ChemSpider | 8046534 |
| UNII | 81QS09V3YW |
| Chemical data | |
| Formula | C19H18N6O5S3 |
| Mol. mass | 506.58 g/mol |
WO-2014189308, Yungjin Pharmaceutical Co Ltd
I did not run away from a NaCN Exotherm

DELTAMETHRIN
DID NOT RUN AWAY FROM NACN ie sodium cyanide EXOTHERM
ALMOST VIRTUAL ACCIDENT AT ISAGRO.RPG LIFESCIENCES (SEARLE) PANOLI GUJARAT INDIA 1999-2000
DELTAMETHRIN PROJECT, 1999-2000 Panoli Gujarat India
ww were trying to add acid chloride into an aldehyde at zero degrees cent using PTC conditions and one of ingredient was sodium cyanide, cooling was done by brine
We Did not run away when instead of adding acid chloride in 2 hrs the operator added it on 10 min…………..I waited at the reactor and controlled an exotherm in plant by switching off brine supply to other reactors,
The reaction got controlled at 59 deg cent and luckily was ok…………the exotherm was fearful. This extreme tension is experienced in a lifetime when everyone runs away and no one to help, FEARFUL, In this case the ‘I’ worked not the ‘we’
Despite all odds God saves us
 MAKE IN INDIA
MAKE IN INDIA
http://makeinindia.com/
http://makeinindia.com/sector/pharmaceuticals/

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
web link

New Drug Approvals, ALL ABOUT DRUGS, WORLD DRUG TRACKER
MEDICINAL CHEM INTERNATIONAL, DRUG SYN INTERNATIONAL
SCALEUP OF DRUGS, ALL FOR DRUGS ON WEB,
MY CHINA, VIETNAM AND JAPAN BLOGS
ICELAND, RUSSIA, ARAB
BOBRDOBR, BLAND ICELAND, 100zakladok, adfty
GROUPS
you can post articles and will be administered by me on the google group which is very popular across the world
OPD GROUPSPACES, SCOOP OCI, organic-process-development GOOGLE, TVINX, MENDELEY WDT, SCIPEOPLE OPD, EPERNICUS OPD, SYNTHETIC ORGANIC CHEMISTRYLinkedIn group,DIIGO OPD, LINKEDIN OPD, WDT LINKEDIN, WDTI ZING




I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my familyJanssen seeks FDA approval for Yondelis (Trabectedin) drug to treat advanced STS

ET-743, Yondelis (trabectedin)
Trabectedin, Ecteinascidin 743, NSC-684766, ET-743, Yondelis, ID0YZQ2TCP
cas 114899-77-3
(-)-(1’R,6R,6aR,7R,13S,14S,16R)-5-Acetoxy-6′,8,14-trihydroxy-7′,9-dimethoxy-4,10,23-trimethyl-1′,2′,3′,4′,6a,7,12,13,14,16-decahydro-6H-spiro[6,16-(epithiopropanoxymethano)-7,13-epimino-1,3-dioxolo[7,8]isoquino[3,2-b][3]benzazocine-20,1′-isoquinolin]-19-one
Janssen seeks FDA approval for Yondelis drug to treat advanced STS
Janssen Research & Development is seeking approval from US Food and Drug Administration (FDA) for its Yondelis (trabectedin) to treat patients with advanced soft tissue sarcoma (STS).
http://www.pharmaceutical-technology.com/news/newsjanssen-yondelis-sts-4451060?WT.mc_id=DN_News
Trabectedin, also referred as ET-743 during its development, is a marine derived antitumoral agent discovered in the Carribean tunicate _Ecteinascidia turbinata_ and now produced synthetically. Trabectedin has a unique mechanism of action. It binds to the minor groove of DNA interfering with cell division and genetic transcription processes and DNA repair machinery.It is approved for use in Europe, Russia and South Korea for the treatment of advanced soft tissue sarcoma. It is also undergoing clinical trials for the treatment of breast, prostate, and paediatric sarcomas. The European Commission and the U.S. Food and Drug Administration (FDA) have granted orphan drug status to trabectedin for soft tissue sarcomas and ovarian cancer.
Trabectedin (also known as ecteinascidin 743 or ET-743) is an anti-tumor drug. It is sold by Zeltia and Johnson and Johnson under the brand name Yondelis. It is approved for use in Europe, Russia and South Korea for the treatment of advanced soft tissue sarcoma. It is also undergoing clinical trials for the treatment of breast, prostate, and paediatric sarcomas. The European Commission and the U.S. Food and Drug Administration (FDA) have granted orphan drug status to trabectedin for soft tissue sarcomas and ovarian cancer.
Discovery and development
The ecteinascidins (herein abbreviated ETs) are exceedingly potent antitumor agents isolated from the marine tunicate Ecteinascidia turbinata. Several ecteinascidins have been reported previously in the patent and scientific literature. See, for example U.S. Pat. No. 5,089,273, which describes novel compounds of matter extracted from the tropical marine invertebrate Ecteinascidia turbinata, and designated therein as ecteinascidins 729, 743, 745, 759A, 759B and 770. These compounds are useful as antibacterial and/or antitumor agents in mammals. U.S. Pat. No. 5,478,932 describes other novel ecteinascidins isolated from the Caribbean tunicate Ecteinascidia turbinata, which provide in vivo antitumor activity against P388 lymphoma, B16 melanoma, M5076 ovarian sarcoma, Lewis lung carcinoma, and the LX- I human lung and MX- 1 human mammary carcinoma xenografts.
One of the ETs, ecteinascidin 743 (ET-743), is a tetrahydroisoquinoline alkaloid with considerable in vitro and in vivo antitumor activity in murine and human tumors, and potent antineoplastic activity against a variety of human tumor xenografts grown in athymic mice, including melanoma, ovarian and breast carcinoma.
ET-743 is a natural compound with the following structure:

ET-743 is also known with the generic name trabectedin and the trademark Yondelis®, and it is currently approved in Europe for the treatment of soft tissue sarcoma. The clinical development of trabectedin continues in phase 11/ III clinical trials in breast, ovarian and prostate cancer. A clinical development program of ET-743 in cancer patients was started with phase I studies investigating 1- hour, 3-hour, 24-hour, and 72-hour intravenous infusion schedules and a 1 hour daily x 5 (dx5) schedule. Promising responses were observed in patients with sarcoma, breast and ovarian carcinoma.
Therefore this new drug is currently under intense investigation in several phase 11/ III clinical trials in cancer patients with a variety of neoplastic diseases. Further information regarding the dosage, schedules, and administration of ET-743 for the treatment of cancer in the human body, either given alone or in combination is provided in WO 00/69441 , WO 02/36135, WO 03/39571 , WO 2004/ 105761 , WO 2005/039584, WO 2005/049031 , WO 2005/049030, WO 2005/049029, WO 2006/046080, WO 2006/005602, and PCT/US07/98727, which are incorporated by reference herein in their entirety.
A review of ET-743, its chemistry, mechanism of action and preclinical and clinical development can be found in Kesteren, Ch.
Van et al., Anti-Cancer Drugs, 2003, 14 (7), 487-502: “ET-743 (trabectedin, ET-743): the development of an anticancer agent of marine origin”, and references therein.
During the past 30 years medical oncologists have focused to optimise the outcome of cancer patients and it is just now that the new technologies available are allowing to investigate polymorphisms, gene expression levels and gene mutations aimed to predict the impact of a given therapy in different groups of cancer patients to tailor chemotherapy. Representative examples include the relationship between the Thymidylate Synthase (TS) mRNA expression and the response and the survival with antifolates, beta tubulin III mRNA levels and response to tubulin interacting agents, PTEN gene methylation and resistance to CPT- I l and, STAT3 over expression and resistance to Epidermal Growth Factor (EGF) interacting agents.
A molecular observation of potential clinical impact relates to the paradoxical relation between the efficiency of the Nucleotide Excision Repair (NER) pathway and the cytotoxicity of ET-743. In fact, tumour cells that are efficient in this DNA repair pathway appear to be more sensitive to ET-743. This evidence is in contrast with the pattern noted with platin based therapeutic regimens which are highly dependent on the lack of activity of this repair pathway (ie. an increase in ERCCl expression has been associated to clinical resistance to platinum-based anti-cancer therapy).
There are evidences on the key role of NER pathways on the cytotoxicity of ET-743 in cell lines. ET-743 binds to G residues in the minor groove of DNA forming adducts that distort the DNA helix structure and they are recognised by NER mechanisms (Pourquier, P. et al., 2001 , Proceedings of the American Association for Cancer Research Annual Meeting, Vol. 42, pp. 556. 92nd Annual Meeting of the American Association for Cancer Research. New Orleans, LA, USA. March 24-28, 2001. ISSN: 0197-016X). Takebayasi et al. (Nature Medicine, 2001 , 7(8), 961-966) have proposed that the presence of these DNA adducts in transcribed genes, blocks the Transcription Coupled NER (TC-NER) system by stalling the cleavage intermediates and producing lethal Single Strand Breaks (SSBs). It is known from Grazziotin et al (Proc.Natl.Acad.Sic.USA, 104: 13062- 13067) that the DNA adducts formed by exposure to ET-743 are transformed into double strand DNA breaks.
The fact that NER mediates ET-743 ‘s cytotoxicity has also been found in the yeast Saccharomyces cerevisae by Grazziotin et al. (Biochemical Pharmacology, 2005, 70, 59-69) and in the yeast Schizosaccharomyces pombe by Herrero et al. (Cancer Res. 2006, 66(16), 8155-8162).
In addition, Bueren et al. (Proceedings AACR Annual Meeting 2007, Abstract no. 1965) have been shown that ET-743 induces double-strand breaks in the DNA in early S phase that are detected and repaired by the Homologous Recombination Repair (HRR) pathway. In addition, Erba et al (Eur. J. Cancer, 2001 , 37(1), 97- 105) and Bueren et al (Proceedings AACR Annual Meeting 2007, Abstract no. 1965) have shown that inactivation/ mutations of genes related to the Double Strand Break detection such as DNA-PK, ATM and ATR and of genes related to Homologous Recombination Repair pathway, such as Fanconi Anemia genes, BRCAl , BRCA2 and RAD51 make cells more sensitive to trabectedin. Such unique finding is the opposite to the pattern with conventional DNA interacting agents, like in the case of microtubule poisons such as taxanes and vinorelbine.
Finally, pharmacogenomic studies prior have demonstrated that increased expression of the NER genes ERCCl and XPD in the tumor tissue does not impact the outcome of patients treated with
ET-743. However, the low expression of BRCAl in the tumor tissue is correlated with a better outcome in cancer patents treated with
ET-743. Further information can be found in WO 2006/005602, which is incorporated by reference herein in its entirety.
Three rare, autosomal recessive inherited human disorders are associated with impaired NER activity: xeroderma pigmentosum (XP), Cockayne Syndrome (CS), and trichothiodystrophy (Bootsma et al. The Genetic Basis of Human Cancer. McGraw-Hill, 1998, 245- 274). XP patients exhibit extreme sensitivity to sunlight, resulting in a high incidence of skin cancers (Kraemer et al. Arch. Dermatol. 123, 241-250, and Arch. Dermatol. 130, 1018- 1021). About 20% of XP patients also develop neurologic abnormalities in addition to their skin problems. These clinical findings are associated with cellular defects, including hypersensitivity to killing and mutagenic effects of UV, and inability of XP cells to repair UV-induced DNA damage (van Steeg et al. MoI. Med. Today, 1999, 5, 86-94).
Seven different NER genes, which correct seven distinct genetic XP complementation groups (XPA-XPG), have been identified (Bootsma et al. The Genetic Basis of Human Cancer. McGraw-Hill, 1998, 245-274). The human gene responsible for XP group G was identified as ERCC5 (Mudgett et al. Genomics, 1990, 8, 623-633; O’Donovan et al. Nature, 1993, 363, 185- 188; and Nouspikel et al. Hum. MoI. Genet. 1994, 3, 963-967). The XPG gene codes for a structure-specific endonuclease that cleaves damaged DNA ~5 nt 3′ to the site of the lesion and is also required non-enzymatically for subsequent 5’ incision by the XPF/ ERCCl heterodimer during the NER process (Aboussekhra et al. Cell, 1995, 80, 859-868; Mu et al. J. Biol. Chem. 1996, 271 , 8285-8294; and Wakasugi et al. J. Biol. Chem. 1997, 272, 16030- 16034). There is also evidence suggesting that XPG is also involved in transcription-coupled repair of oxidative DNA lesions (Le Page et al. Cell, 101 , 159- 171).
Takebayashi et al. (Cancer Lett., 2001 , 174: 1 15- 125) have observed an increase in heterozygosity loss and microsatellite instability in a substantial percentage of samples of ovarian, lung and colon carcinoma. Le Moirvan et al, (Int.J. Cancer, 2006,1 19: 1732- 1735) have described the presence of polymorphisms in the XPG gene in sarcoma patients. It is also known from Takebayashi et al. (Proceedings of the American Association forCancer Research Annual Meeting, March, 2001 , Vol. 42, pp. 813.92nd Annual Meeting of the American Association for Cancer
Research. New Orleans, LA, USA. March 24-28, 2001) that cells deficient in the NER system are resistant to treatment with ET-743 (Zewail-Foote, M. et al., 2001 , Chemistry and Biology, 8: 1033- 1049 and Damia, G. et al., 2001 , Symposium AACR NCI EORTC) and that the antiproliferative effects of ET-743 require a functional XPG gene.
Since cancer is a leading cause of death in animals and humans, several efforts have been and are still being undertaken in order to obtain an antitumor therapy active and safe to be administered to patients suffering from a cancer. Accordingly, there is a need for providing additional antitumor therapies that are useful in the treatment of cancer.
Trabectedin is a tetrahydroisoquinoline, a novel marine-derived antitumor agent isolated from the colonial tunicate Ecteinascidia turbinate. The drug binds to the minor groove of the DNA, bending the DNA towards the major groove, blocking the activation of genes in a unique way via several pathways, including selective inhibition of the expression of key genes (including oncogenes) involved in cell growth and drug resistance, inhibition of genetic repair pathways and inhibition of cell cycle progression leading to p53-independent programmed cell death.
In July 2003, the European Committee of Proprietary Medicinal Products (CPMP) recommended against granting marketing authorization to trabectedin for soft tissue sarcoma. PharmaMar appealed the decision in September 2003. Later that year, the CPMP rejected the company’s appeal. In 2006, the company filed another regulatory application for this indication and, finally, in 2007, a positive opinion was received in the E.U. for the treatment of metastatic soft tissue sarcoma. First commercialization of the product in the E.U. took place in October 2007 in the U.K. and Germany.
The compound is also available in several other countries. In 2008, the compound was filed for approval in the U.S. and the E.U. for the treatment of relapsed advanced ovarian cancer in combination with liposomal doxorubicin, and in 2009 approval was received in both countries. Trabectedin is available in several European countries, including the U.K. and Germany. Also in 2009 the drug candidate was approved in Philippines for the ovarian cancer indication.
The compound had been in phase II development by Johnson & Johnson for the treatment of prostate cancer; however, no recent development has been reported for this research. PharmaMar is evaluating the compound in phase II trials for the treatment of breast cancer. Additional early clinical trials are ongoing at the National Cancer Institute (NCI) to evaluate trabectedin for potential use in the treatment of advanced, persistent or recurrent uterine leiomyosarcomas and solid tumors.
In 2011, a regulatory application that had been filed in the U.S. seeking approval for the treatment of relapsed advanced disease in combination with liposomal doxorubicin was withdrawn by the company based on the FDA’s recommendation that an additional phase III study be conducted to obtain approval. In 2014, Janssen Research & Development, LLC submitted an NDA for trabectedin to the FDA for the treatment of patients with advanced soft tissue sarcoma (STS), including liposarcoma and leiomyosarcoma subtypes, who have received prior chemotherapy including an anthracycline.
Trabectedin was developed by PharmaMar, a subsidiary of Zeltia. The drug was being codeveloped and comarketed in partnership with Ortho Biotech, a subsidiary of Johnson & Johnson pursuant to an agreement signed in 2001. However, in 2008 the license agreement between the two companies was terminated.
The compound was granted orphan drug designation for the treatment of soft tissue sarcoma and for the treatment of ovarian cancer by the FDA and the EMEA. In 2011, orphan drug designation was granted in Japan for the treatment of malignant soft tissue tumor accompanied with chromosomal translocation. In 2009, the product was licensed to Taiho by PharmaMar in Japan for the treatment of cancer.
During the 1950s and 1960s, the National Cancer Institute carried out a wide ranging program of screening plant and marine organism material. As part of that program extract from the sea squirt Ecteinascidia turbinata was found to have anticancer activity in 1969.[1] Separation and characterisation of the active molecules had to wait many years for the development of sufficiently sensitive techniques, and the structure of one of them, Ecteinascidin 743, was determined by KL Rinehart at the University of Illinois in 1984.[2] Rinehart had collected his sea squirts by scuba diving in the reefs of the West Indies.[3]
Recently, the biosynthetic pathway responsible for producing the drug, has been determined to come from Candidatus Endoecteinascidia frumentensis, a microbial symbiont of the tunicate.[4] The Spanish company PharmaMar licensed the compound from the University of Illinois before 1994 and attempted to farm the sea squirt with limited success.[3]
Yields from the sea squirt are extremely low – it takes 1 tonne of animals to isolate 1 gram of trabectedin – and about 5 grams were believed to be needed for a clinical trial[5] so Rinehart asked the Harvard chemist E. J. Corey to search for a synthetic method of preparation. His group developed such a method and published it in 1996.[6] This was later followed by a simpler and more tractable method which was patented by Harvard and subsequently licensed to PharmaMar.[3] The current supply is based on a semisynthetic process developed by PharmaMar starting from Safracin B, an antibiotic obtained by fermentation of the bacterium Pseudomonas fluorescens.[7] PharmaMar have entered into an agreement with Johnson and Johnson to market the compound outside Europe.
Trabectedin was first dosed in humans in 1996.In 2007, the EMEA gave authorisation for the marketing of trabectedin, under the trade name Yondelis, for the treatment of patients with advanced soft tissue sarcoma, after failure of anthracyclines and ifosfamide, or who are unsuited to receive these agents. The agency’s evaluating committee, the CHMP observed that trabectedin had not been evaluated in an adequately designed and analyzed randomized trial against current best care, and that the clinical efficacy data was mainly based on patients with liposarcoma and leiomyosarcoma. However the pivotal study did show a significant difference between two different trabectedin treatment regimens, and due to the rarity of the disease the CHMP considered that marketing authorisation could be granted under exceptional circumstances.[8] As part of the approval PharmaMar agreed to conduct a further trial to identify whether any specific chromosomal translocations could be used to predict responsiveness to trabectedin.[9] Trabectedin is also approved in South Korea[10] and Russia.
In 2008 the submission was announced of a registration dossier to the European Medicines Agency (EMEA) and the FDA for Yondelis when administered in combination with pegylated liposomal doxorubicin (Doxil, Caelyx) for the treatment of women with relapsed ovarian cancer. In 2011, Johnson&Johnson voluntarily withdrew the submission in the United States following a request by the FDA for an additional Phase III study to be done in support of the submission.[11]
Trabectedin is also in phase II trials for prostate, breast and paediatric cancers.[12]
Structure

Trabectedin is composed of 3 tetrahydroisoquinoline moieties, 8 rings including one 10-membered heteocyclic ring containing a cysteine residue, and 7 chiral centers.
Biosynthesis
The biosynthesis of Trabectedin in Candidatus Endoecteinascidia frumentensis starts with a fatty acid loading onto the acyl-ligase domain of the EtuA3 module. A cysteine and glycine are then loaded as canonical NRPS amino acids. A tyrosine residue is modified by the enzymes EtuH, EtuM1, and EtuM2 to add a hydroxyl at the meta position of the phenol, and adding two methyl groups at the para-hydroxyl and the meta carbon position. This modified tyrosine reacts with the original substrate via a Pictet-Spangler reaction, where the amine group is converted to an imine by deprotonation, then attacks the free aldehyde to form a carbocation that is quenched by electrons from the methyl-phenol ring. This is done in the EtuA2 T-domain. This reaction is done a second time to yeid a dimer of modified tyrosine residues that have been further cyclized via Pictet-spangler reaction, yielding a bicyclic ring moiety. The EtuO and EtuF3 enzymes continue to post-translationally modify the molecule, adding several functional groups and making a sulfide bridge between the original cysteine residue and the beta-carbon of the first tyrosine to form ET-583, ET-597, ET-596, and ET-594 which have been previously isolated.[4] A third o-methylated tyrosine is added and cyclized via Pictet-Spangler to yield the final product.[4]

Proposed biosynthetic scheme for the biosynthesis of Trabecteden (ET-743)
Synthesis
The total synthesis by E.J. Corey used this proposed biosynthesis to guide their synthetic strategy. The synthesis uses such reactions as the Mannich reaction, Pictet-Spengler reaction, the Curtius rearrangement, and chiral rhodium-based diphosphine–catalyzed enantioselective hydrogenation. A separate synthetic process also involved the Ugi reaction to assist in the formation of the pentacyclic core. This reaction was unprecedented for using such a one pot multi-component reaction in the synthesis of such a complex molecule.

Org Lett 2000,2(7),993
The previously reported synthesis of 139221 (scheme 13922101a) has been investigated in order to find a more efficient, reproducible and economical route to work in the mutikilogram scale. Herein it is reported a new process which is simpler and proceeds with an overall yield of 54% (the original process, 35%). The condensation of intermediate aminolactone (I) (scheme 13922101a, intermediate (VII)) with acid (XLII) (the acid derived from scheme 13922101a, intermediate ester (IX)) by means of 2-chloro-1,3-dimethylimidazolidinium hexafluorophosphate (CIP), and 1-hydroxy-7-azabenzotriazole (HOAt) in THF/dichloromethane gives the coupling product (XLIII), which is allylated with allyl bromide (XLIV) and Cs2CO3 in DMF yielding the allyl ether (XLV). The reduction of the lactone group of (XLV) with LiAlH2(OEt)2 in ethyl ether affords the lactol (XLVI), which is desilylated with KF in methanol to provide the phenolic compound (XLVII). The opening of the lactol ring of (XLVII) with simultaneous cyclization by means of Tf-OH in water/trifluoroethanol gives the hexacyclic intermediate (XLVIII), which is finally reductocondensed with KCN by means of LiAlH2(OEt)2 in THF to furnish the previously reported pentacyclic intermediate (XI) (scheme 13922101a, intermediate (XI)).
……………………………………………

Reaction of cyanosafracin B (I) with Boc2O in ethanol gives the amino-protected compound (II), which is treated with methoxymethyl bromide (MOM-Br), DIEA and DMAP in acetonitrile yielding the O-protected compound (III). The demethylation of (III) with NaOH in methanol affords the hydroxyquinone (IV), which is reduced with H2 over Pd/C and cyclized with bromochloromethane and Cs2CO3 in hot DMF to provide compound (V). Reaction of (V) with allyl bromide (VI) and Cs2CO3 in DMF gives the allyl ether (VII), which first is treated with TFA, phenyl isothiocyanate and HCl to yield the primary amine (VIII) and then protected at the free NH2 group with Troc-Cl and pyridine, to afford the amino protected compound (IX).Org Lett 2000,2(16),2545
……………………………….

Reaction of (IX) with MOM-Br and DIEA as before affords the ether (X), which is treated with Zn/HOAc in order to regenerate the primary amino group giving (XI). The reaction of (XI) with NaNO2 and HOAc eliminates the NH2 group, affording the primary alcohol (XII), which is esterified with the protected (S)-cysteine (XIII) by means of EDC and DMAP in dichloromethane furnishing the cysteine ester (XIV). Reaction of (XIV) with Bu3SnH and PdCl2(PPh3)2, followed by oxidation with (PhSeO)2O in dichloromethane gives the hydroxyketone (XV), which is cyclized with Tf2O and Ac2O yielding the heptacyclic compound (XVI). Elimination of the MOM protecting group with TMSCl and NaI in CH3CN/CH2Cl2 affords the phenolic compound (XVII).
…………………….

Intermediate (XVII) by a treatment with Zn and HOAc eliminates the Troc protecting group, giving the primary amine (XVIII). This compound by treatment with 4-formyl-1-methylpyridinium iodide (NMPC), DBU and oxalic acid in order to convert the nitrile group into an alcohol, provides compund (XIX), which is finally cyclized with 2-(3-hydroxy-4-methoxyphenyl)ethylamine (XX) by means of SiO2 / EtOH, followed treatment with and AgNO3 in acetonitrile/water.
……………………….
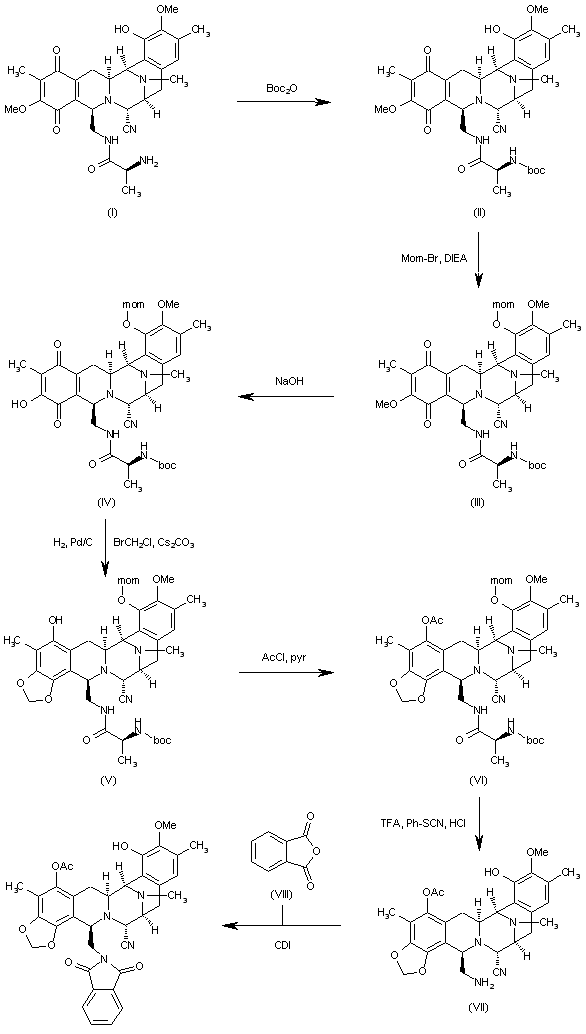
The reaction of cyanosafracin B (I) with Boc2O in ethanol gives the amino protected compound (II), which is treated with Mom-Br, DIEA and DMAP in acetonitrile yielding the O-protected compound (III). The demethylation of (III) with NaOH in methanol affords the hydroxyquinone (IV), which is reduced with H2 over Pd/C and cyclized with bromochloromethane and Cs2CO3 in hot DMF providing the methylenedioxy compound (V). The reaction of (V) with acetyl chloride and pyridine in dichloromethane gives the acetate (VI), which is treated with TFA, phenyl isothiocyanate and HCl yielding the primary amine (VII). Finally, this compound is treated with phthalic anhydride (VIII) and CDI in dichloromethane to afford the target phthalimide (phthalascidin Pt-650)
………………………………
http://pubs.acs.org/doi/abs/10.1021/ol0056729

Enantioselective Total Synthesis of Ecteinascidin 743
Ecteinascidins are a group of marine alkaloid having antineoplasticity which is isolated from the extracted products from the marine tunicate habitat of the Caribbean sea by a very small amount. Arming the ecteinascidins, Et 743 has a very strong antineoplastic activity, studies to put it into practical use as a carcinostatic agent are limited, and the phase II clinical tests are now being carried out in ten countries in Europe and America. It is known that Et 743 has an effect of depressing the proliferation of cancer cells by 10 to 100 times more potent than (IC50=0.1-1 nM) Toxol, Camptotesin, Adriamycin or Mitomycin which are currently used carcinostatic agents.
From the background mentioned above, various studies for synthesis were carried out; however, the complete synthesis was only reported by Prof. E. J. Corey of Harvard University in the U.S.A. (J. Am. Chem. Soc. 1996, 118, 9202-9203, reference document A).
In the process of the total synthesis disclosed in Document A (refer to page 9202), the main feature of the process is that Et 743 is synthesized from the analogous compound to the compound represented by general formula 1 of the present invention via intermediates 4 and 8. That is, according to said process, the C4 site of ring B (regarding the location of rings, and the sites of atoms comprising the 6 membered ring, refer to general formula 1), which composes a 6 membered ring, is formed from the intermediate 4 at the first step. Since the atom C4 composing the ring B of the 6-membered ring H, which lacks reactivity, is bonded, it becomes necessary to perform an oxidation reaction at the C4 site on the B ring. This oxidation reaction is not effective and is carried out under harsh conditions; therefore production on an industrial scale is difficult, and also the yield is not good. Further, since the atom N12 site of the synthesized intermediate is substituted by an alkyl group which lacks reactivity, in this case substituted by a methyl group, it is not suited to the synthesis of various compounds. Although total synthesis was reported, the supplying source of Et 743 still depends on the natural sample whose supply is very scarce. Therefore, the establishment of the method for a large scale production of Et 743 is desired and requires accomplishing an effective synthesizing process.
Since ET 743 is known as a medicine having high antineoplasticity, and phthalascidin induced from the intermediate product at the synthesis of Et 743 displays the same activity to ET 743, the establishment of an effective and mild method for synthesis of ET 743 and analogous compounds thereof is strongly desired.
Therefore, the subject of the present invention is to accomplish the effective method for total synthesis of Et 743, and further, to provide not only Et 743 but also analogous compounds.
To dissolve the subject, the present invention uses retrosynthetic analysis for easy synthesis. It will be possible to form a B ring by a ring forming reaction at the ortho position of phenol, which binds an A ring to inner molecular aldehyde in a compound generated by the 4-8 reaction. Further, the present invention contemplates that the generated compound by the 4-8 reaction can be synthesized based on the polycondensation reaction of general formula 4, and general formula 5 via a compound of general formula 3. Then the total synthesis of Et 743, which is the aimed compound, can be accomplished by way of the compounds represented by general formulae 5, 4, 3, 2 and 1 and the specific structure of general formulae 1 and 2. This synthetic route provides for the analogous compounds of Et 743.





Mechanism of action
The biological mechanism of action is believed to involve the production of superoxide near the DNA strand, resulting in DNA backbone cleavage and cell apoptosis. The actual mechanism is not yet known, but is believed to proceed from reduction of molecular oxygen into superoxide via an unusual auto-redox reaction on a hydroxyquinone moiety of the compound following. There is also some speculation the compound becomes ‘activated’ into its reactive oxazolidine form.

Schematic of the unique and complex mode of action of trabectedin. The antitumor effects of trabectedin are due to multiple mechanisms involving DNA binding in the minor groove, interactions with DNA repair mechanisms, modulation of transcription regulation, and induction of microenvironment changes.
References
- Lichter et al. Worthen LW, ed. “Food-drugs from the sea. Proc: Aug 20–23, 1972.” 173. Marine Tech Soc. pp. 117–127.
- Rinehart KL (January 2000). “Antitumor compounds from tunicates”. Med Res Rev 20 (1): 1–27. doi:10.1002/(SICI)1098-1128(200001)20:1<1::AID-MED1>3.0.CO;2-A. PMID 10608919.
- “Potent cancer drugs made — Sea squirts provide recipe”.
- Rath CM et al (November 2011). “Meta-omic characterization of the marine invertebrate microbial consortium that produces the chemotherapeutic natural product ET-743”. ACS Chemical Biology 6 (11): 1244–56. doi:10.1021/cb200244t. PMC 3220770. PMID 21875091.
- “New Scientist”.
- E. J. Corey, David Y. Gin, and Robert S. Kania (1996). “Enantioselective Total Synthesis of Ecteinascidin 743”. J. Am. Chem. Soc. 118 (38): 9202–9203. doi:10.1021/ja962480t.
- C. Cuevas et al. (2000). “Synthesis of ecteinascidin ET-743 and phthalascidin PT-650 from cyanosafracin”. B. Org. Lett. 2: 2545–2548.
- “CHMP evaluation”.
- “PharmaMar website”.
- S.Korea approves Zeltia cancer drug Yondelis, Reuters.com, May 8, 2008
- Grogan, Kevin (3 May 2011). “J&J pulls submission for Zeltia’s Yondelis”. PharmaTimes Magazine (London, England). Online PharmaTimes. Archived from the original on 7 May 2011. Retrieved 7 May 2011.
- “PharmaMar website”.
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (1′R,6R,6aR,7R,13S,14S,16R)-6′,8,14-trihydroxy-7′,9-dimethoxy-4,10,23-trimethyl-19-oxo-3′,4′,6,7,12,13,14,16-octahydrospiro[6,16-(epithiopropano-oxymethano)-7,13-imino-6aH-1,3-dioxolo[7,8]isoquino[3,2-b][3]benzazocine-20,1′(2′H)-isoquinolin]-5-yl acetate | |
| Clinical data | |
| AHFS/Drugs.com | International Drug Names |
| Licence data | EMA:Link |
| Legal status | |
| Routes | Intravenous |
| Pharmacokinetic data | |
| Bioavailability | Not applicable (IV only) |
| Protein binding | 94 to 98% |
| Metabolism | Hepatic (mostly CYP3A4-mediated) |
| Half-life | 180 hours (mean) |
| Excretion | Mostly fecal |
| Identifiers | |
| CAS number | 114899-77-3 |
| ATC code | L01CX01 |
| PubChem | CID 108150 |
| IUPHAR ligand | 2774 |
| DrugBank | DB05109 |
| ChemSpider | 16736970 |
| UNII | ID0YZQ2TCP |
| Chemical data | |
| Formula | C39H43N3O11S |
| Mol. mass | 761.84 g/mol |
……..
1 Corey, “Enantioselective Total Synthesis of Ecteinascidin 743“, J. Am. Chem. Soc. 1996, vol. 118, 9202-9203.
| 2 | * | Endo, “Synthetic Study on Ecteinascidin 743 Starting From D-Glucose“, Synlett 1999, No. 7, 1103-1105. |
| 3 | * | Endo, “Total Synthesis of Ecteinascidin 743“, J. Am. Chem. Soc. 2002, vol. 124, 6552-6554. |
| 4 | * | Hinterding, “Synthesis and In Vitro Evaluation of the Ras Farnesyltransferase Inhibitor Pepticinnamin E“, Angew. Chem. Int. Ed. 1998, 37, No. 9 1236-1239. |
| 5 | * | Tohma, “Synthesis of Optically Active alpha-Arylglycines: Stereoselective Mannich-Type Reaction with a New Chiral Template“, Synlett 2001, No. 7, 1179-1181.Hamprecht, D.W.; Berge, J.M.; Copley, R.C.B.; Eggleston, D.S.; Houge-Frydrych, C.S.V.; Jarvest, R.L.; Mensah, L.M.; O’Hanlon, P.J.; Pope, A.J.; Rittenhouse, S. Derivatives of the natural product SB-219383 and synthetic analogues: Potent inhibitors of bacterial tyrosyl tRNA synthetase 16th Int Symp Med Chem (September 18-22, Bologna) 2000, Abst PA-155Cuevas, C.; Perez, M.; Martin, M.J.; et al. Synthesis of ecteinascidin ET-743 and phathalascidin Pt-650 from cyanosafracin B Org Lett 2000, 2(16): 2545
|
| Patent | Submitted | Granted |
|---|---|---|
| Assay for identifying biological targets of polynucleotide-binding compounds [US2008096201] | 2008-04-24 | |
| Compounds of the saframycin-ecteinascidin series, uses, and synthesis thereof [US6936714] | 2004-07-01 | 2005-08-30 |
| Method For Total Synthesis Of Ecteinascidins And Intermediate Compounds Thereof [US7807833] | 2009-08-06 | 2010-10-05 |
| Method For Total Synthesis Of Ecteinascidins And Intermediate Compounds Thereof [US7820838] | 2009-02-05 | 2010-10-26 |
| Assay for identifying biological targets of polynucleotide-binding compounds [US7183054] | 2004-12-09 | 2007-02-27 |
Synthesis of Ibuprofen Using Silica-Supported Preyssler Nanoparticles as an Eco-Friendly, Inexpensive, and Efficient Catalyst,
 |
|
|
Synthesis of Ibuprofen Using Silica-Supported Preyssler Nanoparticles as an Eco-Friendly, Inexpensive, and Efficient Catalyst,
Organic Chemistry International
Volume 2014 (2014), Article ID 906801, 6 pages
http://dx.doi.org/10.1155/2014/906801
http://www.hindawi.com/journals/oci/2014/906801/
Ali Gharib,1,2 Nader Noroozi Pesyan,3 Leila Vojdani Fard,4 and Mina Roshani1
1Department of Chemistry, Islamic Azad University, Mashhad, Iran
2Agricultural Researches and Services Center, Mashhad, Iran
3Department of Chemistry, Faculty of Science, Urmia University, Urmia 57159, Iran
4Education Organization of Razavi Khorasan, Education Ministry, Mashhad, Iran
Received 5 January 2014; Revised 15 February 2014; Accepted 31 March 2014; Published 6 May 2014
Academic Editor: Jonathan White
Copyright © 2014 Ali Gharib et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
This paper describes an alternative and simple procedure for the synthesis of Ibuprofen using Silica-Supported Preyssler Nanoparticles (H14[NaP5W30O110]/SiO2) (SPNPs), as an eco-friendly, inexpensive, and efficient catalyst. High yields, simplicity of operation, and easy work-up procedure are some advantages of this protocol. Silica-Supported Preyssler Nanoparticles (H14[NaP5W30O110]/SiO2) (SPNPs) offer the advantages of a higher hydrolytic and thermal stability. The salient features of Preyssler’s anion are availability, nontoxicity and reusability. We believe this methodology can find usefulness in organic synthesis.
Synthesis of Ibuprofen (6)
To a solution of ethyl-2-(4-isobutylphenyl) propanoate (1 g, 4.27 mmol) in 6 mL of CH3OH a solution of KOH was added (479 mg, 8.55 mmol) in 5 mL of H2O. The resultant solution was stirred at room temperature for 4 h. Methanol was removed under reduced pressure and the resulting solution was extracted with ethyl acetate and the organic extracts were washed with H2O, dried over anhydrous Na2SO4, and concentrated under reduced pressure to give compound 6.
M.P (°C) 130-133,
IR (KBr, cm−1): 3100, 2920, 2870, 1716, 1408, 1419, 1321, 1230, 1184, 935, 779, 668, 583. 1H NMR (400 MHz, CDCl3) 7.15 (d, J = 8.1 Hz, 2H), 7.02 (d, J = 8.1 Hz, 2H), 3.64 (q, J = 7.2 Hz, 1H), 2.37 (d, J = 7.1 Hz, 2H), 1.75 (m, 1H), 1.43 (d, J = 7.1 Hz, 3H), 0.82 (d, J = 6.6 Hz, 6H).
13C NMR (100 MHz, CDCl3): 22.81, 22.82, 29.07, 42.64, 44.50, 128.80, 128.93, 128.95, 132.22, 140.23, 181.26. Anal. Calcd. for C13H18O2: C, 75.69; H, 8.80%. Found: C, 75.61; H, 8.70%.
HRMS (EI) Calcd. for C26H25FN4O6 [M]+, 206.1600, Found 206.1009.
KEBUZONE…….An antirheumatic agent.

Kebuzone (or ketophenylbutazone) is a non-steroidal anti-inflammatory drug.
Structural formula

4-(3-oxobutyl)-1,2-diphenylpyrazolidine-3,5-dione
- BRN 0308507
- Chebutan
- Chepirol
- Chetazolidin
- Chetil
- Copirene
- EINECS 212-715-7
- Hichillos
- Kebuzone
- Kebuzonum
- Kebuzonum [INN-Latin]
- Keobutane-jade
- Ketason
- Ketazone
- Ketophenylbutazone
- Ketophenylbutazonum
- KPB
- Pecnon
- Quebuzona
- Quebuzona [INN-Spanish]
- Recheton
- UNII-4VD83UL6Y6
Anti-inflammatory agents that are non-steroidal in nature. In addition to anti-inflammatory actions, they have analgesic, antipyretic, and platelet-inhibitory actions.They act by blocking the synthesis of prostaglandins by inhibiting cyclooxygenase, which converts arachidonic acid to cyclic endoperoxides, precursors of prostaglandins. Inhibition of prostaglandin synthesis accounts for their analgesic, antipyretic, and platelet-inhibitory actions; other mechanisms may contribute to their anti-inflammatory effects.
UV – range
 |
||||
| Conditions : Concentration – 1 mg / 100 ml | ||||
| The solvent designation graphics | Methanol |
Water |
0.1М HCl |
0.1M NaOH |
|---|---|---|---|---|
| Maximum absorption | 244 nm | – | 237 nm | 262 nm |
 |
448 | – | 404 | 617 |
| e | 14440 | – | 13020 | 19890 |
IR – spectrum
| Wavelength (μm) |
|---|
 |
| Wave number (cm -1 ) |
Reference
-
UV and IR Spectra. H.-W. Dibbern, R.M. Muller, E. Wirbitzki, 2002 ECV
-
NIST/EPA/NIH Mass Spectral Library 2008
-
Handbook of Organic Compounds. NIR, IR, Raman, and UV-Vis Spectra Featuring Polymers and Surfactants, Jr., Jerry Workman. Academic Press, 2000.
-
Handbook of ultraviolet and visible absorption spectra of organic compounds, K. Hirayama. Plenum Press Data Division, 1967.
Brief background information
| Salt | ATC | Formula | MM | CAS |
|---|---|---|---|---|
| – | M01AA06 | C 19 H 18 N 2 O 3 | 322.36 g / mol | 853-34-9 |
| 4-(3-oxobutyl)-1,2-di(phenyl)pyrazolidine-3,5-dione | |
| Clinical data | |
|---|---|
| Legal status |
?
|
| Identifiers | |
| CAS number | 853-34-9 |
| ATC code | M01AA06 |
| PubChem | CID 3824 |
| ChemSpider | 3692 |
| UNII | 4VD83UL6Y6 |
| KEGG | D01567 |
| ChEBI | CHEBI:31749 |
| Chemical data | |
| Formula | C19H18N2O3 |
| Mol. mass | 322.35782 g/mol |
Application
-
anti-inflammatory
-
antirheumatic
-
Synthesis pathway
| Synthesis of a) |
|---|
   |
| Synthesis b) |
 |
Trade names
| Country | Trade name | Manufacturer |
|---|---|---|
| Germany | Kebuzon | Steiner |
| France | Ketazon | Beytout |
| Italy | Chetopir | Sarm |
| Ukraine | no | no |
Formulations
-
ampoules of 1 g / 5 ml;
-
250 mg capsule
Reference
-
Synthesis of a)
-
Denss, R. et al .: Helv. Chim. Acta (HCACAV) 40, 402 (1957).
-
material:
-
Kühn, M .: J. Prakt. Chem. (JPCEAO) 156 (II), 103 (1940).
-
-
-
Synthesis b)
-
AT 198 263 (Synfarma; appl. 1955).
-
References: Prepn: Deuss et al., US 2910481 (1959 to Geigy).
Review of pharmacology: Horakova et al.,Pharmacotherapeutica 1950-1959, 335-350 (1963), C.A. 60, 6072g (1964).
Metabolism: Nemecek et al., Arzneim.-Forsch. 16,1339 (1966); Queisnerova, Nemecek, Cesk. Farm. 20, 55 (1971), C.A. 75, 47077u (1971).
Herrenknecht, Christine; Guernet-Nivaud, Elisabeth; Lafont, Olivier; Guernet, Michel; Gueutin, Claire
Canadian Journal of Chemistry, 1988 , v. 66, pg. 1199 – 1202
Cizmarik; Lycka
Pharmazie, 1988 , v. 43, 11 pg. 794 – 795
Gueutin-Pelinard, Claire; Nivaud, Elisabeth; Boucly, Patrick; Guernet, Michel
Canadian Journal of Chemistry, 1981 , v. 59, pg. 759 – 762
Denss et al.
Helvetica Chimica Acta, 1957 , v. 40, pg. 402,406
Patent: CS124279 , 1965 ;Chem.Abstr., 1968 , v. 69, 52134r
SPOFA; United Pharmaceutical Work Patent: FR1500627 , 1965 ;Chem.Abstr., 1968 , v. 69, 96715k
Nippon Shinyaju Co., Ltd. Patent: US5811547 A1, 1998 ;
Fisnerova,L. et al. Collection of Czechoslovak Chemical Communications, 1974 , v. 39, pg. 624 – 633
EU approves Lilly diabetes drug Trulicity, dulaglutide
Regulators in Europe have given the green light to Eli Lilly’s Trulicity, its once-weekly glucagon-like peptide-1 receptor agonist for type 2 diabetes.
Read more at: http://www.pharmatimes.com/Article/14-11-25/EU_approves_Lilly_diabetes_drug_Trulicity.aspx
Dulaglutide is a glucagon-like peptide 1 receptor agonist (GLP-1 agonist) for the treatment of type 2 diabetes that can be used once weekly.[1][2]GLP-1 is a hormone that is involved in the normalization of level of glucose in blood (glycemia). The FDA approved dulaglutide for use in the United States in September 2014.[3] The drug is manufactured by Eli Lilly under the brand name Trulicity.[3]

Mechanism of action
Dulaglutide binding to glucagon-like peptide 1 receptor, slows gastric emptying and increases insulin secretion by beta cells in the pancreas. Simultaneously the compound reduces the elevated glucagon secretion by alpha cells of the pancreas, which is known to be inappropriate in the diabetic patient. GLP-1 is normally secreted by L cells of the gastrointestinal mucosa in response to a meal.[4]
Medical uses[
The compound is indicated for adults with type 2 diabetes mellitus as an adjunct to diet and exercise to improve glycemic control. Dulaglutide is not indicated in the treatment of subjects with type 1 diabetes mellitus or patients with diabetic ketoacidosis. Dulaglutide can be used either stand-alone or in combination with other medicines for type 2 diabetes, in particular metformin, sulfonylureas, thiazolidinediones, and insulin taken concomitantly with meals.[5]
Side effects
The most common side effects include gastrointestinal disorders, such as dyspepsia, decreased appetite, nausea, vomiting, abdominal pain, diarrhea.[6] Some patients may experience serious adverse reactions: acute pancreatitis (symptoms include persistent severe abdominal pain, sometimes radiating to the back and accompanied by vomiting),hypoglycemia, renal impairment (which may sometimes require hemodialysis). The risk of hypoglycemia is increased if the drug is used in combination with sulfonylureas orinsulin.[7][8]
Contraindications
The compound is contraindicated in subjects with hypersensitivity to active principle or any of the product’s components. As a precautionary measure patients with a personal or family history of medullary thyroid carcinoma or affected by multiple endocrine neoplasia syndrome type 2 should not take dulaglutide, because for now it is unclear whether the compound can increase the risk of these cancers.[9]
![]()
References
- JCourtney Aavang Tibble, Tricia Santos Cavaiola, Robert R Henry (2013). “Longer Acting GLP-1 Receptor Agonists and the Potential for Improved Cardiovascular Outcomes: A Review of Current Literature”. Expert Rev Endocrinol Metab 8 (3): 247–259.doi:10.1586/eem.13.20.
- “Lilly’s Once-Weekly Dulaglutide Shows Non-Inferiority to Liraglutide in Head-to-Head Phase III Trial for Type 2 Diabetes”. Eli Lilly. Feb 25, 2014.
- “FDA approves Trulicity to treat type 2 diabetes” (Press release). FDA. Sep 18, 2014.
- Nadkarni P, Chepurny OG, Holz GG (2014). “Regulation of glucose homeostasis by GLP-1”. Prog Mol Biol Transl Sci 121: 23–65. doi:10.1016/B978-0-12-800101-1.00002-8.PMC 4159612. PMID 24373234. Retrieved 2014-09-29.
- Terauchi Y, Satoi Y, Takeuchi M, Imaoka T (July 2014). “Monotherapy with the once weekly GLP-1 receptor agonist dulaglutide for 12 weeks in Japanese patients with type 2 diabetes: dose-dependent effects on glycaemic control in a randomised, double-blind, placebo-controlled study”. Endocr. J. PMID 25029955. Retrieved 2014-09-29.
- Nauck M, Weinstock RS, Umpierrez GE, Guerci B, Skrivanek Z, Milicevic Z (August 2014). “Efficacy and safety of dulaglutide versus sitagliptin after 52 weeks in type 2 diabetes in a randomized controlled trial (AWARD-5)”. Diabetes Care 37 (8): 2149–58.doi:10.2337/dc13-2761. PMID 24742660.
- Amblee A (April 2014). “Dulaglutide for the treatment of type 2 diabetes”. Drugs Today50 (4): 277–89. doi:10.1358/dot.2014.50.4.2132740. PMID 24918645.
- Monami M, Dicembrini I, Nardini C, Fiordelli I, Mannucci E (February 2014). “Glucagon-like peptide-1 receptor agonists and pancreatitis: a meta-analysis of randomized clinical trials”. Diabetes Res. Clin. Pract. 103 (2): 269–75. doi:10.1016/j.diabres.2014.01.010.PMID 24485345.
- Samson SL, Garber A (April 2013). “GLP-1R agonist therapy for diabetes: benefits and potential risks”. Curr Opin Endocrinol Diabetes Obes 20 (2): 87–97.doi:10.1097/MED.0b013e32835edb32. PMID 23403741. Retrieved 2014-09-30.
| Identifiers | |
|---|---|
| CAS number | 923950-08-7 |
| ATC code | None |
| Chemical data | |
| Formula | C2646H4044N704O836S18 |
| Mol. mass | 59669.81 g/mol |
NETOGLITAZONE

NETOGLITAZONE, isaglitazone
- 5-((6-((2-fluorophenyl)methoxy)-2-naphthalenyl)methyl)-2,4-thiazolidinedione
- MCC 555
- MCC-555
- netoglitazone
- RWJ-241947
Netoglitazone (MCC-555) is a hypoglycemic agent.
Synthesis

US 5594016
http://www.google.co.in/patents/US5594016
Reaction of aldehyde (III) with 2-fluorobenzyl alcohol (VIII) by means of triphenylphosphine and diethyl azodicarboxylate (DEAD) in THF furnishes 6-(2-fluorobenzyloxy)naphthalene-2-carbaldehyde (IX) , which is then reduced with NaBH4 in ethanol/THF to give the naphthalenemethanol derivative (X). Halogenation of (X) by means of iodide, triphenylphosphine and imidazole in THF yields the naphthylmethyl iodide derivative (XI), which is finally condensed with thiazolidine-2,4-dione (IV) by means of HMPA and butyl lithium in THF.
Ueno, H.; Oe, T.; Suehiro, I.; Nakamura, F. (Mitsubishi Chemical Corp.); Naphthalene derivs.. EP 0604983; JP 1994247945; US 5594016 .
http://www.google.co.in/patents/EP0604983B1?cl=en
References
Sorbera, L.A.; Castañer, J.; Del Fresno, M.; Silvestre, J. (2002). “Netoglitazone”. Drugs of the Future 27 (2): 132.doi:10.1358/dof.2002.027.02.657482.
 |
|
| Systematic (IUPAC) name | |
|---|---|
| 5-[(6-[(2-fluorophenyl)methoxy]naphthalen-2-yl)methyl]-1,3-thiazolidine-2,4-dione | |
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS number | 161600-01-7 |
| ATC code | ? |
| PubChem | CID 204109 |
| UNII | QOV2JZ647A |
| KEGG | D05150 |
| Chemical data | |
| Formula | C21H16FNO3S |
| Mol. mass | 381.420 g/mol |
| Pharmaceutical composition comprising a glitazone and a 4-oxobutanoic acid, and the use thereof for treating diabetes [US2005085489] | 2005-04-21 | |
| Compositions of a cyclooxygenase-2 selective inhibitor and a peroxisome proliferator activated receptor agonist for the treatment of ischemic mediated central nervous system disorders [US2005107387] | 2005-05-19 | |
| Pharmaceutical composition comprising an ACAT inhibitor and an insulin resistance reducing agent [US2005119314] | 2005-06-02 | |
| Medical devices to treat or inhibit restenosis [US2005149174] | 2005-07-07 | |
| Medicinal compositions containing diuretic and insulin resistance-improving agent [US2005288339] | 2005-12-29 | |
| Crystals of 5-[{6-(2-fluorobenzyl)oxy-2-naphthyl}methyl]-2,4-thiazolidinedione [US2006149075] | 2006-07-06 | |
| Concomitant drug as therapeutic agent for inflammatory bowel disease [US2006177444] | 2006-08-10 | |
| Combination of FBPase inhibitors and insulin sensitizers for the treatment of diabetes [US2004167178] | 2004-08-26 | |
| Crystals of 5-[{6-(2-fluorobenzyl)oxy-2-naphthyl}methyl]-2,4-thiazolidinedione [US2003158241] | 2003-08-21 | |
| Pharmacological method for treatment of neuropathic pain [US2007249561] | 2007-10-25 |
| Patent | Submitted | Granted |
|---|---|---|
| Medicinal composition containing diabetes remedy [US7943584] | 2008-02-14 | 2011-05-17 |
| Medicinal compositions containing diuretic and insulin resistance-improving agent [US7199139] | 2004-03-18 | 2007-04-03 |
| Crystals of 5-[{6-(2-fluorobenzyl)oxy-2-naphthyl}methyl]-2,4-thiazolidinedione [US6541493] | 2003-04-01 | |
| Combination of FBPase inhibitors and insulin sensitizers for the treatment of diabetes [US6756360] | 2004-06-29 | |
| Roflumilast for the Treatment of Diabetes Mellitus [US8017633] | 2008-09-04 | 2011-09-13 |
| Combination of FBPase Inhibitors and Insulin Sensitizers for the Treatment of Diabetes [US2008004226] | 2008-01-03 | |
| Pharmaceutical Composition Comprising Ppar Regulator [US2008153882] | 2008-06-26 | |
| Pharmaceutical combination comprising vitamin k [US2009137614] | 2009-05-28 | |
| Pharmaceutical Composition Containing PPARgamma Agonist [US2009137626] | 2009-05-28 | |
| Pharmaceutical agent comprising insulin resistance improving agent [US2009124626] | 2009-05-14 | |
| ROFLUMILAST FOR THE TREATMENT OF DIABETES MELLITUS [US2011269750] | 2011-11-03 | |
| Combination treatment for diabetes mellitus [US2010179131] | 2010-07-15 | |
| Therapeutic agent for diabetes containing insulin resistance improving agent [US2007049515] | 2007-03-01 | |
| PHARMACEUTICAL COMBINATION COMPRISING VITAMIN K [US2011028499] | 2011-02-03 | |
| RESPIRATORY DISEASE TREATMENT [US8236786] | 2011-03-03 | 2012-08-07 |


NAVEGLITAZAR (LY519818)
NAVEGLITAZAR
2(S)-Methoxy-3-[4-[3-(4-phenoxyphenoxy)propoxy]phenyl]propionic acid
476436-68-7
C25 H26 O6, 422.4703
- CCRIS 9448
- LY 519818
- LY 9818
- LY519818
- LY9818
- Naveglitazar
- UNII-Y995M7GM0G
http://clinicaltrials.gov/search/intervention=NAVEGLITAZAR
Naveglitazar, a peroxisome proliferator-activated receptor (PPAR) modulator, had been in phase II clinical trials for the once-daily oral treatment of type 2 diabetes, however, no recent development for this indication has been reported. The compound was originally discovered through an ongoing research collaboration between Lilly and Ligand, but, in 2006, Lilly discontinued the development program.
Naveglitazar [LY519818; benzenepropanoic acid, alpha-methoxy-4-[3-(4-phenoxyphenoxy)propoxy], (alpha-S)-] is a nonthiozolidinedione peroxisome proliferator-activated receptor alpha-gamma dual, gamma-dominant agonist that has shown glucose-lowering potential in animal models and in the clinic.
Studies have been conducted to characterize the disposition, metabolism, and excretion of naveglitazar in mice, rats, and monkeys after oral and/or i.v. bolus administration.
………………………………
Martín JA, Brooks DA, Prieto L, González R, Torrado A, Rojo I, López de Uralde B, Lamas C, Ferritto R, Dolores Martín-Ortega M, Agejas J, Parra F, Rizzo JR, Rhodes GA, Robey RL, Alt CA, Wendel SR, Zhang TY, Reifel-Miller A, Montrose-Rafizadeh C, Brozinick JT, Hawkins E, Misener EA, Briere DA, Ardecky R, Fraser JD, Warshawsky AM.
Bioorg Med Chem Lett. 2005 Jan 3;15(1):51-5.
………………………………………..
http://www.google.im/patents/US20050020684?cl=un
EXAMPLE 153
′2-Methoxy-3-{3-[3-(4-phenoxy-phenoxy)-propoxy]-phenyl}-propionic acid

The title compound was prepared from 3-(3-Hydroxy-phenyl)-2-methoxy-propionic acid methyl ester from Example 152, Step D with 4-(3-bromopropoxy)1-phenoxybenzene in a manner analogous as in Example 152, Step E. MS (ES) for C25H26O6[M+NH4]+: 440.2, [M+Na]+: 445.2. 1H-NMR (CDCl3, 200.15 MHz): 7.33-7.17 (m, 3H), 7.07-6.78 (m, 10H), 4.15 (dt, 4H, J=1.9, 6.2), 4.03 (dd, 1H, J=7.3, 4.3), 3.40 (s, 3H), 3.13 (dd, 1H, J=14.2, 4.6), 2.98 (dd, 1H, J=14.0, 7.5), 2.25 (qui, 2H, J=5.9)ppm.
