
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 195)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
GSK1904529A, GSK 4529

GSK1904529A, GSK 4529
GSK1904529A is a selective inhibitor of IGF1R with IC50 of 27 nM.
| 851.96 | |
| Formula | C44H47F2N9O5S |
| CAS Number | 1089283-49-7 |
N-(2,6-difluorophenyl)-5-[3-[2-[5-ethyl-2-methoxy-4-[4-(4-methylsulfonylpiperazin-1-yl)piperidin-1-yl]anilino]pyrimidin-4-yl]imidazo[1,2-a]pyridin-2-yl]-2-methoxybenzamide,
N-(2,6-Difluorophenyl)-5-[3-[2-[[5-ethyl-2-(methyloxy)-4-[4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl]phenyl]amino]-4-pyrimidinyl]imidazo[1,2-a]pyridin-2-yl]-2-(methyloxy)benzamide
NMR……http://www.abmole.com/download/gsk1904529a-hnmr.pdf

GSK1904529A, selectively inhibits IGF-IR and IR with IC50s of 27 and 25 nmol/L, respectively. It is a promising candidate for therapeutic use in solid and hematologic cancers. IC50s for GSK1904529A in tumor cell lines ranged from 35 nmol/L to >30 umol/L. The tumor histologic types showing the greatest sensitivity to this compound were Ewing’s sarcoma and multiple myeloma, where IC50s in three of five Ewing’s sarcoma cell lines were <100 nmol/L and IC50s in five of eight multiple myeloma cell lines were <200 nmol/L.
GSK1904529A is a small-molecule inhibitor of the insulin-like growth factor-I receptor (IGF-IR) with IC50 value of 27 nM 1.
GSK1904529A is a reversible and ATP-competitive inhibitor with Ki value of 1.6 nM. In NIH-3T3/LISN cells, GSK1904529A potently inhibited phosphorylation of IGF-IR with IC50 value of 22 nM. It also demonstrated to be a selective inhibitor since it showed poor inhibitory activity against 45 other serine/threonine and tyrosine kinases. When treated with whole-cell extracts, GSK1904529A significantly inhibited the ligand-induced phosphorylation of IGF-IR and decreased phosphorylation of downstream signaling including AKT, IRS-1 and ERK at concentrations > 0.01μM. GSK1904529A suppressed cell proliferation in a variety of tumor cells. The IC50 values for NCI-H929, TC-71, SK-N-MC, COLO 205, MCF7 and PREC are 81, 35, 43, 124, 137 and 68 nM, respectively. In COLO 205, MCF-7, and NCI-H929 cells, GSK1904529A treatment resulted in cell accumulation in G1 and decrease in S and G2-M phases. Moreover, in NIH-3T3/LISN xenograft model, once daily administration of GSK1904529A at 30 mg/kg inhibited 56% of tumor growt

…………..
Intermediates
 ,
,  ,
,  ,
, 
 ,
,

 ,
, ![]() ,
, ![]()
 ,
, 
 u can construct your synthesis
u can construct your synthesis
http://www.google.com/patents/US20080300242
Intermediate Example 2 5-[3-(2-chloro-4-pyrimidinyl)imidazo[1,2-a]pyridin-2-yl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide
Step A: Methyl 3-formyl-4-hydroxybenzoate
Methyl 4-hydroxybenzoate (3.00 g, 19.7 mmol) and magnesium chloride (2.81 g, 29.5 mmol) were stirred in 100 mL of acetonitrile. TEA (10.3 mL, 73.9 mmol) was added via syringe. Paraformaldehyde (12.0 g, 133 mmol) was added in a single portion and the reaction was heated to reflux. The reaction was stirred at reflux for 24 hours and cooled to rt. The reaction was quenched by the addition of approximately 100 mL of 1N HCl and poured into EtOAc. The layers were separated, and the organic layer was washed with brine. The combined aqueous layers were extracted with EtOAc. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography. The clean fractions (by TLC) were concentrated in vacuo to afford 2.06 g (58%) of the desired product. 1H NMR (400 MHz, DMSO-d6): δ 11.54 (s, 1H), 10.27 (s, 1H), 8.21 (d, J=2.4 Hz, 1H), 8.03 (dd, J=8.8, 2.4 Hz, 1H), 7.07 (d, J=8.8 Hz, 1H), 3.79 (s, 3H).
Step B: methyl 3-formyl-4-(methyloxy)benzoate
Methyl 3-formyl-4-hydroxybenzoate (2.06 g, 11.4 mmol) and K2CO3 (2.36 g, 17.1 mmol) were stirred in 50 mL of DMF. Methyl iodide (1.42 mL, 22.8 mmol) was added via syringe, and the reaction was stirred for 6 hours at rt. The reaction was poured into H2O and diethyl ether, and the layers were separated. The organic layer was washed with brine, and the combined aqueous layers were extracted with diethyl ether. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo to afford 2.24 g of crude desired product. 1H NMR (400 MHz, DMSO-d6): δ 10.33 (s, 1H), 8.23 (d, J=2.2 Hz, 1H), 8.20 (dd, J=8.8, 2.2 Hz, 1H), 7.36 (d, J=8.8 Hz, 1H), 3.99 (s, 3H), 3.83 (s, 3H).
Step C: 2-(methyloxy)-5-[(methyloxy)carbonyl]benzoic acid
Crude methyl 3-formyl-4-(methyloxy)benzoate from the previous step was dissolved in 40 mL of dioxane with stirring. Sulfamic acid (5.87 g, 60.5 mmol) in 20 mL of H2O was added to the stirring solution. Sodium chlorite (1.68 g, 80% by weight, 18.6 mmol) in 20 mL of H2O was added dropwise via addition funnel. The reaction was stirred for 40 min and poured into EtOAc and H2O. The layers were separated, and the organic layer was washed with brine. The combined aqueous layers were extracted with EtOAc, and the combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The solid was transferred to an Erlenmeyer flask with the aid of 30-40 mL of DCM. Approximately 50 mL of hexanes was added. Air was blown over the solution to allow most of the DCM to evaporate. Diethyl ether was added (20-30 mL), and the suspension was filtered. The solid was washed with hexanes, collected, and dried to afford 1.96 g (82% over 2 steps) of the desired compound. 1H NMR (400 MHz, DMSO-d6): δ 12.92 (brs, 1H), 8.22 (d, J=2.2 Hz, 1H), 8.07 (dd, J=8.8, 2.2 Hz, 1H), 7.24 (d, J=8.8 Hz, 1H), 3.88 (s, 3H), 3.82 (s, 3H).
Step D: methyl 3-{[(2,6-difluorophenyl)amino]carbonyl}-4-(methyloxy)benzoate
2-(Methyloxy)-5-[(methyloxy)carbonyl]benzoic acid (1.96 g, 9.33 mmol) was suspended in 60 mL of DCM with stirring. DMF (0.036 mL, 0.46 mmol) was added via syringe. Oxalyl chloride (7.0 mL, 2.0M in dichloromethane, 14 mmol) was added dropwise via addition funnel. The addition funnel was rinsed with 10 mL of DCM. The reaction was stirred for 2 hours and concentrated in vacuo. The resultant solid was further dried under high vacuum pressure. The solid was dissolved in 60 mL of DCM with stirring. Pyridine (3.8 mL, 47 mmol), (4-dimethylamino)pyridine (0.0570 g, 0.467 mmol), and 2,6-difluoroaniline (3.0 mL, 28 mmol) were added to the solution. The reaction was stirred for 18 hours and poured into 1N HCl. The layers were separated, and the aqueous layer was washed once with DCM and once with diethyl ether. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography. The clean fractions (by TLC) were concentrated in vacuo to afford 1.56 g (52%) of the desired product. 1H NMR (400 MHz, DMSO-d6): δ 9.81 (s, 1H), 8.31 (d, J=2.0 Hz, 1H), 8.10 (dd, J=8.8, 2.0 Hz, 1H), 7.38 (m, 1H), 7.31 (d, J=88 Hz, 1H), 7.22-7.13 (m, 2H), 3.97 (s, 3H), 3.82 (s, 3H).
Step E: 5-[(2-Chloro-4-pyrimidinyl)acetyl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide and 5-[(E)-2-(2-chloro-4-pyrimidinyl)-1-hydroxyethenyl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide
Methyl 3-{[(2,6-difluorophenyl)amino]carbonyl}-4-(methyloxy)benzoate (1.56 g, 4.86 mmol) was dissolved in 50 mL of THF with stirring and cooled to 0° C. Lithium bis(trimethylsilyl)amide (14.6 mL, 1.0M in THF, 14.6 mmol) was added slowly via syringe. 2-Chloro-4-methylpyrimidine (0.750 g, 5.83 mmol) was dissolved in 10 mL of THF and added dropwise via addition funnel. The addition funnel was rinsed with 10 mL of THF. The reaction was stirred at 0° C. for 1 hour and quenched with saturated ammonium chloride solution. The mixture was poured into H2O and EtOAc, and the layers were separated. The organic layer was washed with brine, and the combined aqueous layers were extracted with EtOAc. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography. The clean fractions (by TLC) were concentrated in vacuo to afford 1.26 g (62%) of the desired product. The proton NMR is a mixture of the keto and enol tautomers (˜2:1). 1H NMR (400 MHz, DMSO-d6): δ 13.58 (s, 1H, enol), 9.83 (s, 1H, keto), 9.82 (s, 1H, enol), 8.72 (m, 1H, keto), 8.54 (m, 1H, enol), 8.34 (s, 1H, keto), 8.22 (m, 1H, both), 8.06 (m, 1H, enol), 7.56 (m, 1H, keto), 7.42-7.31 (m, 2H, both+1H, enol), 7.22-7.14 (m, 2H, both), 6.55 (s, 1H, enol), 4.66 (s, 2H, keto), 4.00 (s, 3H, keto), 3.97 (s, 3H, enol).
Step F: 5-[3-(2-chloro-4-pyrimidinyl)imidazo[1,2-a]pyridin-2-yl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide
A tautomeric mixture of 5-[(2-Chloro-4-pyrimidinyl)acetyl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide and 5-[(E)-2-(2-chloro-4-pyrimidinyl)-1-hydroxyethenyl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide (1.26 g, 3.02 mmol) was dissolved in 60 mL of DCM with stirring. NBS (0.538 g, 3.02 mmol) was added in a single portion. The reaction was stirred for 20 minutes and concentrated in vacuo. The residue was dissolved in 60 mL of dioxane with stirring, and 2-aminopyridine (0.853 g, 9.06 mmol) was added in a single portion. The reaction was heated at 60° C. with an oil bath for 24 hours and cooled to rt. The reaction was stirred at rt for an additional 40 hours. The reaction was poured into half-saturated NaHCO3 solution and EtOAc, and the layers were separated. The organic layer was washed with brine, and the combined aqueous layers were extracted twice with EtOAc. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography. Impure fractions were concentrated and further purified by flash chromatography. The combined clean fractions (by TLC) from both runs were combined and concentrated in vacuo to afford 1.07 g (72%) of the desired product. 1H NMR (400 MHz, DMSO-d6): δ 9.80 (s, 1H), 9.40 (d, J=7.0 Hz, 1H), 8.57 (d, J=5.1 Hz, 1H), 8.10 (d, J=1.5 Hz, 1H), 7.84-7.77 (m, 2H), 7.57 (m, 1H), 7.39 (m, 1H), 7.33-7.26 (m, 2H), 7.24-7.14 (m, 3H), 3.99 (s, 3H).
Step A: 1,1-dimethylethyl 4-(methylsulfonyl)-1-piperazinecarboxylate
To 1,1-dimethylethyl 1-piperazinecarboxylate (568 g, 3.05 mol) in DCM (4 L) was added TEA (617 g, 6.10 mol). After stirring for 10 min at 0° C., methanesulfonyl chloride (384 g, 3.35 mol) was added via addition funnel. The mixture was stirred at rt overnight. The mixture was poured into H2O (1 L) and extracted with DCM (1 L). The organic layer was separated, washed with H2O (1 L), dried (Na2SO4), and rotovapped down to provide the title compound of step A (720 g, 2.72 mol, 90%) which was used without further purification. 1H NMR (400 MHz, CDCl3) δ 1.44 (s, 9H), 2.76 (s, 3H), 3.11-3.17 (m, 4H), 3.50-3.53 (m, 4H).
Step B: 1-(methylsulfonyl)piperazine hydrochloride
To 1,1-dimethylethyl 4-(methylsulfonyl)-1-piperazinecarboxylate (360 g, 1.36 mol) in MeOH (1 L) was added HCl (6 M in MeOH, 2 L) dropwise. The mixture was stirred at rt for 1 h. About 1 L of MeOH was rotovapped off. The resultant precipitate was filtered, washed with MeOH, and dried on high vacuum to provide the title compound of Step B (A combination of 2 batches, 570 g) which was used without further purification. 1H NMR (400 MHz, D2O) δ 2.95 (s, 3H), 3.27-3.29 (m, 4H), 3.42-3.46 (m, 4H).
Step C: 1-(methylsulfonyl)-4-(4-piperidinyl)piperazine dihydrochloride
To 1-(methylsulfonyl)piperazine hydrochloride (150 g, 632 mmol) in DCE (3.5 L) was added TEA (192 g, 1.90 mol). The mixture was stirred at rt for 1 h and then acetic acid (94.8 g, 1.58 mol) and 1,1-dimethylethyl 4-oxo-1-piperidinecarboxylate (251 g, 1.26 mol) was added. After stirring another h, the reaction was cooled with an ice water bath and NaBH(OAc)3 (294 g, 1.39 mol) was added in four portions. The mixture was stirred overnight at rt. The reaction mixture was neutralized with saturated Na2CO3 to pH 8-9. The organic phase was washed with brine and H2O, dried (Na2SO4), and rotovapped down to provide the crude Boc-protected amine (A combination of 3 batches, 720 g). This amount was split into 2 batches and used without further purification. To 1,1-dimethylethyl 4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinecarboxylate (360 g, 1.04 mol) in MeOH (1 L) was added HCl (6 M in MeOH, 2 L). The mixture was stirred at rt for 30 min. About 1 L of MeOH was rotovapped off. The resultant precipitate was filtered, washed with MeOH, and dried on high vacuum to provide the title compound of Step C (A combination of 2 batches, 600 g, 1.87 mol, 89% over 2 steps). 1H NMR (400 MHz, D2O) δ 1.87-1.91 (m, 2H), 2.33-2.36 (m, 2H), 2.97 (s, 3H), 2.99-3.05 (m, 2H), 3.45-3.59 (m, 11H).
Step A: 1-{1-[2-ethyl-5-(methyloxy)-4-nitrophenyl]-4-piperidinyl}-4-(methylsulfonyl)piperazine
A mixture of 1-ethyl-2-fluoro-4-(methyloxy)-5-nitrobenzene (Example 187, step C) (0.93 g, 4.67 mmol), 1-(methylsulfonyl)-4-(4-piperidinyl)piperazine (Example 204, step C) (1.16 g, 4.67 mmol) and K2CO3 (0.774 g, 5.60 mmol) in DMSO (20 mL) was heated at 90° C. for 48 h. The reaction had not progressed sufficiently so the reaction was then heated at 120° C. for an additional 4 h. The reaction was cooled to rt, poured into H2O and extracted with DCM. Some saturated brine solution was added and the resultant was exhaustively extracted with DCM. The combined organics were washed with H2O then dried over MgSO4. The resultant solution was concentrated onto silica and purified by flash chromatography to afford 1-{1-[2-ethyl-5-(methyloxy)-4-nitrophenyl]-4-piperidinyl}-4-(methylsulfonyl)piperazine (1.12 g, 56%). 1H NMR (400 MHz, DMSO-d6) δ ppm 7.73-7.80 (m, 1H), 6.75 (s, 1H), 3.91 (s, 3H), 3.23-3.30 (m, 1H), 3.05-3.19 (m, 3H), 2.87 (s, 2H), 2.70-2.84 (m, 2H), 2.53-2.67 (m, 5H), 1.77-1.94 (m, 2H), 1.48-1.67 (m, 2H), 1.19 (t, J=7.42 Hz, 3H).
Step B: 5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}aniline
A mixture of 1-{1-[2-ethyl-5-(methyloxy)-4-nitrophenyl]-4-piperidinyl}-4-(methylsulfonyl)piperazine (1.12 g, 2.63 mmol) and sulfided platinum on carbon (0.410 g, 0.105 mmol) in EtOAc (40 mL) was sealed in a round bottom flask with a rubber septum. The reaction mixture was purged with N2 gas and then a balloon of H2 gas was connected and the vessel was flushed with the H2 gas. The reaction was stirred at rt for 2 d. TLC analysis showed the complete consumption of the starting nitro compound so the reaction mixture was filtered through celite to remove the catalyst. The filtrate was concentrated onto silica gel and purified by flash chromatography to afford 5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}aniline (0.479 g, 46%).
1H NMR (400 MHz, DMSO-d6) δ ppm 6.60 (s, 1H), 6.46 (s, 1H), 4.35 (br. s., 2H), 3.71 (s, 3H), 3.03-3.16 (m, 4H), 2.81-2.93 (m, 5H), 2.56-2.68 (m, 6H), 2.29-2.42 (m, 1H), 1.72-1.89 (m, 2H), 1.44-1.62 (m, 2H), 1.09 (t, J=7.51 Hz, 3H).
Example 237 N-(2,6-difluorophenyl)-5-(3-{2-[(5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}phenyl)amino]-4-pyrimidinyl}imidazo[1,2-a]pyridin-2-yl)-2-(methyloxy)benzamide
A mixture of 5-[3-(2-chloro-4-pyrimidinyl)imidazo[1,2-a]pyridin-2-yl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide (Intermediate Example 2) (0.60 g, 1.22 mmol), 5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}aniline (Example 206, Step B) (0.48 g, 1.22 mmol) and HCl (4N,1,4-Dioxane, 0.61 mL, 2.44 mmol) in trifluoroethanol (15 mL) was heated at 170° C. for 40 min in the microwave. The reaction mixture was concentrated onto silica gel and purified by flash column chromatography. Recrystallization from DCM and EtOH afforded the title compound N-(2,6-difluorophenyl)-5-(3-{2-[(5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}phenyl)amino]-4-pyrimidinyl}imidazo[1,2-a]pyridin-2-yl)-2-(methyloxy)benzamide (0.61 g, 56%).
1H NMR (400 MHz, DMSO-d6)
δ ppm 9.80 (s, 1H), 9.36 (br. s., 1H), 8.50 (s, 1H), 8.26 (d, J=5.22 Hz, 1H), 8.12 (d, J=2.11 Hz, 1H), 7.80 (dd, J=8.80, 2.02 Hz, 1H), 7.71 (d, J=9.07 Hz, 1H), 7.53 (s, 1H), 7.36-7.50 (m, 2H), 7.30 (d, J=8.80 Hz, 1H), 7.14-7.25 (m, 2H), 6.91-7.00 (m, 1H), 6.83 (s, 1H), 6.58 (d, J=5.22 Hz, 1H), 4.00 (s, 3H), 3.80 (s, 3H), 3.08-3.15 (m, 4H), 3.00-3.07 (m, 2H), 2.88 (s, 3H), 2.67-2.76 (m, 2H), 2.61-2.66 (m, 4H), 2.56 (q, J=7.51 Hz, 2H), 2.38-2.46 (m, 1H), 1.80-1.91 (m, 2H), 1.50-1.68 (m, 2H), 1.11 (t, J=7.51 Hz, 3H).
MS (M+H, ES+) 852.
Separately, the Title Compound was Prepared in the Following Manner:
A mixture of 5-[3-(2-chloro-4-pyrimidinyl)imidazo[1,2-a]pyridin-2-yl]-N-(2,6-difluorophenyl)-2-(methyloxy)benzamide (Intermediate Example 2) (23.0 g, 46.8 mmol), 5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}aniline (Example 206, Step B) (18.6 g, 46.8 mmol) and HCl (4N,1,4-Dioxane, 23.4 mL, 93.6 mmol) in trifluoroethanol (200 mL) was heated in a sealed vessel at 85° C. for 48 h. After cooling to rt, the reaction mixture was treated with an excess of 7N NH3 in MeOH and then subjected to filtration. The filtrate was concentrated onto silica gel and purified by flash chromatography. The chromatographed product was dissolved in DCM and treated with an excess of diethyl ether. The resultant bright yellow precipitate was collected by filtration and then recrystallized from DCM and EtOH to afford the title compound N-(2,6-difluorophenyl)-5-(3-{2-[(5-ethyl-2-(methyloxy)-4-{4-[4-(methylsulfonyl)-1-piperazinyl]-1-piperidinyl}phenyl)amino]-4-pyrimidinyl}imidazo[1,2-a]pyridin-2-yl)-2-(methyloxy)benzamide (28.2 g, 67%).
……………..
Discovery and optimization of imidazo[1,2-a]pyridine inhibitors of insulin-like growth factor-1 receptor (IGF-1R)
Bioorg Med Chem Lett 2009, 19(3): 1004……http://www.sciencedirect.com/science/article/pii/S0960894X08014376


Scheme 1.
Reagents and conditions: (a) (ClCO)2, DMF, CH2Cl2; (b) 2,6-difluoroaniline, pyridine, CH2Cl2 (84%, 2 steps); (c) LiN(SiMe3)2, THF (83%); (d) NBS, CH2Cl2, then 2-aminopyridine, dioxane, 60 °C (77%); (e) HCl or p-TSA·H2O, trifluoroethanol or isopropanol, 80–100 °C or 140–180 °C (μw) (50–90%).
References
Antitumor activity of GSK1904529A, a small-molecule inhibitor of the insulin-like growth factor-I receptor tyrosine kinase.
Sabbatini et al. Clin Cancer Res. 2009 May 1;15(9):3058-67. PMID: 19383820.
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Facebook 

 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
 LIONEL MY SON
LIONEL MY SON
/////////////GSK1904529A, IGF1R, GSK 4529, preclinical
GSK 1059615
GSK1059615; 958852-01-2; GSK-1059615; UNII-07YMO87363;
- GSK 615
(5Z)-5-[(4-pyridin-4-ylquinolin-6-yl)methylidene]-1,3-thiazolidine-2,4-dione
5-[[4-(4-Pyridinyl)-6-quinolinyl]methylene]-2,4-thiazolidenedione
| C18H11N3O2S | |
| Molecular Weight: | 333.36384 |
|---|
CAS 958852-01-2
nmr……..http://file.selleckchem.com/downloads/nmr/S136001-GSK1059615-NMR-Selleck.pdf
GSK1059615 is a potent, ATP-competitive inhibitor of PI 3-kinase alpha (PI3Kα) with IC50 of 2 nM. Phosphatidylinositol-3 kinases (PI3K) are critical for malignant cellular processes including growth, proliferation, and survival. GSK1059615 is also a novel inhibitor of PI3Kβ, PI3Kδ, PI3Kγ and mTOR with IC50 of 0.6 nM, 2 nM, 5 nM and 12 nM, respectively. GSK1059615 (25 mg/kg) effectively inhibits tumor growth in xenograft mice models of BT474 or HCC1954 breast cancer cells and attenuates MAPK signaling.
GSK1059615 is a phosphoinositide 3-kinase (PI3K) inhibitor with potential antineoplastic activity. PI3K inhibitor GSK1059615 inhibits PI3K in the PI3K/AKT kinase signaling pathway, which may trigger the translocation of cytosolic Bax to the mitochondrial outer membrane and an increase in mitochondrial membrane permeability, followed by apoptosis. Bax is a member of the proapoptotic Bcl-2 family of proteins. PIK3, an enzyme often overexpressed in cancer cells, plays a crucial role in tumor cell regulation and survival.


Patent
http://www.google.com/patents/US20090306074

http://www.google.com/patents/US20090306074

Example 1 (5Z)-5-{[4-(4-pyridinyl)-6-quinolinyl]methylidene}-1,3-thiazolidine-2,4-dione

a) 4-chloro-6-ethenylquinoline
A mixture of 6-bromo-4-chloroquinoline (6.52 g, 26.88 mmol; see J. Med. Chem., 21, 268 (1978)), tributyl(vinyl)tin (8.95 g, 28.22 mmol), and tetrakistriphenylphosphine palladium (0) (0.62 g, 0.54 mmol) in 1,4-dioxane (150 mL) was refluxed for 2.0 h, cooled to room temperature, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (0-4% MeOH:CH2Cl2) to give the title compound (5.1 g) as a pale yellow solid. MS (ES)+m/e 190 [M+H]+. This material was used directly in the next step.
b) 4-chloro-6-quinolinecarbaldehyde
A mixture of 4-chloro-6-ethenylquinoline (5.1 g, 26.88 mmol), 2,6-lutidine (5.76 g, 53.75 mmol), sodium (meta) periodate (22.99 g, 107.51 mmol), and osmium tetroxide (5.48 g of a 2.5% solution in tert-butanol, 0.538 mmol) in 1,4-dioxane:H2O (350 mL of 3:1 mixture) was stirred for 3.5 h at room temperature and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (CH2Cl2) to give the title compound (4.26 g, 83% for 2 steps) as a pale yellow solid. MS (ES)+ m/e 192 [M+H]+.
c) 4-(4-pyridinyl)-6-quinolinecarbaldehyde
A mixture of 4-chloro-6-quinolinecarbaldehyde (3.24 g, 16.92 mmol), 4-pyridylboronic acid (3.12 g, 25.38 mmol), tetrakistriphenylphosphine palladium (0) (0.978 g, 0.846 mmol), and 2M aqueous K2CO3 (7.02 g, 50.76 mmol, 25.4 mls of 2M solution) in DMF (100 mL) was heated at 100° C. for 3.0 h and cooled to room temperature. The mixture was filtered through celite and the celite was washed with EtOAc. The filtrate was transferred to a separatory funnel, washed with water and saturated NaCl, dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (5% MeOH:CH2Cl2) to give the title compound (2.03 g, 51%) as a tan solid. MS (ES)+ m/e 235 [M+H]+.
d) (5Z)-5-{[4-(4-pyridinyl)-6-quinolinyl]methylidene}-1,3-thiazolidine-2,4-dione
A mixture of 4-(4-pyridinyl)-6-quinolinecarbaldehyde (0.108 g, 0.463 mmol), 2,4-thiazolidinedione (0.0417 g, 0.356 mmol), piperidine (0.0303 g, 0.356 mmol), and acetic acid (0.0214 g, 0.356 mmol) in EtOH (5 mL) was heated at 150° C. for 30 minutes in a microwave oven. The reaction was cooled to room temperature and the resulting precipitate was filtered and dried in a Buchner funnel to give the title compound (0.0594 g, 50%) as a tan solid. MS (ES)+ m/e 334 [M+H]+. 1H NMR (400 MHz, DMSO-d6) □ ppm 9.08 (d, J=4.42 Hz, 1H) 8.80-8.88 (m, 2H) 8.25 (d, J=8.72 Hz, 1H) 8.00-8.07 (m, 2H) 7.98 (s, 1H) 7.65-7.68 (m, 2H) 7.63 (d, J=4.42 Hz, 1H).
……………..
http://www.google.com/patents/WO2007136940A2?cl=en
Schemes/Experimentals
Scheme I:

Conditions: a) Tributyl(vinyl)tin, Pd(PPh3)4, dioxane, reflux; b) OsO4, NaIO4, 2,6- lutidine, f-BuOH, dioxane, H2O, rt; c) heteroaryl (R) boronic acid, Pd(PPh3)4, 2 M K2CO3, DMF, 10O 0C; d) 2,4-thiazolidinedione, piperidine, AcOH, EtOH, μwave, 150 0C.
Examples:
Example 1 : (5Z)-5-ff4-(4-pyridinyl)-6-quinolinvnmethylidene}-1 ,3-thiazolidine-
2,4-dione

a) 4-chloro-6-ethenylquinoline
A mixture of 6-bromo-4-chloroquinoline (6.52 g, 26.88 mmol; see J. Med. Chem., 21_, 268 (1978) ), tributyl(vinyl)tin (8.95 g, 28.22 mmol), and tetrakistriphenylphosphine palladium (0) (0.62 g, 0.54 mmol) in 1 ,4-dioxane (150 ml.) was refluxed for 2.0 h, cooled to room temperature, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (0-4% MeOH:CH2CI2) to give the title compound (5.1 g) as a pale yellow solid. MS(ES)+ m/e 190 [M+H]+. This material was used directly in the next step.
b) 4-chloro-6-quinolinecarbaldehyde
A mixture of 4-chloro-6-ethenylquinoline (5.1 g, 26.88 mmol), 2,6-lutidine (5.76 g, 53.75 mmol), sodium (meta) periodate (22.99 g, 107.51 mmol), and osmium tetroxide (5.48 g of a 2.5% solution in tert-butanol, 0.538 mmol) in 1 ,4- dioxane:H2O (350 ml. of 3:1 mixture) was stirred for 3.5 h at room temperature and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (CH2CI2) to give the title compound (4.26 g, 83% for 2 steps) as a pale yellow solid. MS(ES)+ m/e 192 [M+H]+.
c) 4-(4-pyridinyl)-6-quinolinecarbaldehyde
A mixture of 4-chloro-6-quinolinecarbaldehyde (3.24 g, 16.92 mmol), 4- pyridylboronic acid (3.12 g, 25.38 mmol), tetrakistriphenylphosphine palladium (0) (0.978 g, 0.846 mmol), and 2M aqueous K2CO3 (7.02 g, 50.76 mmol, 25.4 mis of 2M solution) in DMF (100 ml.) was heated at 1000C for 3.0 h and cooled to room temperature. The mixture was filtered through celite and the celite was washed with EtOAc. The filtrate was transferred to a separatory funnel , washed with water and saturated NaCI, dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (5% MeOHiCH2CI2) to give the title compound (2.03 g, 51%) as a tan solid. MS(ES)+ m/e 235 [M+H]+.
d) (5Z)-5-{[4-(4-pyridinyl)-6-quinolinyl]methylidene}-1 ,3-thiazolidine-2,4-dione
A mixture of 4-(4-pyridinyl)-6-quinolinecarbaldehyde (0.108 g, 0.463 mmol), 2,4-thiazolidinedione (0.0417 g, 0.356 mmol), piperidine (0.0303 g, 0.356 mmol), and acetic acid (0.0214 g, 0.356 mmol) in EtOH (5 ml.) was heated at 15O0C for 30 minutes in a microwave oven. The reaction was cooled to room temperature and the resulting precipitate was filtered and dried in a Buchner funnel to give the title compound (0.0594 g, 50%) as a tan solid. MS(ES)+ m/e 334 [M+H]+. 1 H NMR (400 MHz, DMSO-d6) D ppm 9.08 (d, J=4.42 Hz, 1 H) 8.80 – 8.88 (m, 2 H) 8.25 (d, J=8.72 Hz, 1 H) 8.00 – 8.07 (m, 2 H) 7.98 (s, 1 H) 7.65 – 7.68 (m, 2 H) 7.63 (d, J=4.42 Hz, 1 H).
| Patent | Submitted | Granted |
|---|---|---|
| THIAZOLIDINEDIONE DERIVATIVES AS PI3 KINASE INHIBITORS [US2008255115] | 2008-10-16 | |
| THIAZOLIDINEDIONE DERIVATIVES AS P13 KINASE INHIBITORS [US2009306074] | 2009-12-10 | |
| Role of PI3K p110 delta Signaling in Retroviral Infection and Replication [US2011135655] | 2011-06-09 | |
| PI3 KINASE INHIBITORS AND USES THEREOF [US2011230476] | 2011-09-22 |
Identification of druggable targets for radiation mitigation using a small interfering RNA screening assay.
Zellefrow CD,et al. Radiat Res. 2012 Sep;178(3);150-9. PMID: 22747550.
Saadia et al (2009) Phosphatidylinositol-3-kinase as a therapeutic target in melanoma. Clin.Cancer Res. 15 3029. PMID: 19383818.
Knight et al (2010) Discovery of GSK2126458, a highly potent inhibitor of PI3K and the mammalian target of rapamycin. ACS Med.Chem.Lett. 1 39.
////////GSK 1059615, GSK 615
Ombitasvir

Ombitasvir; ABT-267; ABT 267; UNII-2302768XJ8; 1258226-87-7;
| C50H67N7O8 | |
| Molecular Weight: | 894.10908 g/mol |
|---|
Anti-Viral Compounds [US2010317568]
Dimethyl (2S,2′S)-1,1′-((2S,2′S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-Butylphenyl)pyrrolidine-2,5-diyl)bis(4,1-phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-2,1-diyl)dicarbamate
methyl N-[(2S)-1-[(2S)-2-[[4-[(2S,5S)-1-(4-tert-butylphenyl)-5-[4-[[(2S)-1-[(2S)-2-(methoxycarbonylamino)-3-methylbutanoyl]pyrrolidine-2-carbonyl]amino]phenyl]pyrrolidin-2-yl]phenyl]carbamoyl]pyrrolidin-1-yl]-3-methyl-1-oxobutan-2-yl]carbamate

Ombitasvir is an antiviral drug for the treatment of hepatitis C virus (HCV) infection. In the United States, it is approved by theFood and Drug Administration for use in combination with paritaprevir, ritonavir and dasabuvir in the product Viekira Pak for the treatment of HCV genotype 1,[1][2] and with paritaprevir and ritonavir in the product Technivie for the treatment of HCV genotype 4.[3][4]
Ombitasvir is in phase II clinical development at AbbVie for the treatment of chronic hepatitis C infection in combination with ABT-450/ritonavir and, in combination with peginterferon alpha-2a/ribavirin (pegIFN/RBV) in treatment naïve Hepatitis C virus (HCV) genotype 1 infected patients.
Ombitasvir is part of a fixed-dose formulation with ABT-450/ritonavir that is approved in the U.S. and the E.U.
Ombitasvir acts by inhibiting the HCV protein NS5A.[5]
In 2013, breakthrough therapy designation was assigned in the U.S. for the treatment of genotype 1 hepatitis C in combination with ABT-450, ritonavir and ABT-333, with and without ribavirin.


DeGoey, DA, Discovery of ABT-267, a Pan-genotypic Inhibitor of HCV NS5A, J. Med. Chem., 2014, 57 (5), pp 2047-2057
http://pubs.acs.org/doi/full/10.1021/jm401398x
http://pubs.acs.org/doi/suppl/10.1021/jm401398x/suppl_file/jm401398x_si_001.pdf

We describe here N-phenylpyrrolidine-based inhibitors of HCV NS5A with excellent potency, metabolic stability, and pharmacokinetics. Compounds with 2S,5S stereochemistry at the pyrrolidine ring provided improved genotype 1 (GT1) potency compared to the 2R,5Ranalogues. Furthermore, the attachment of substituents at the 4-position of the central N-phenyl group resulted in compounds with improved potency. Substitution with tert-butyl, as in compound 38 (ABT-267), provided compounds with low-picomolar EC50 values and superior pharmacokinetics. It was discovered that compound 38 was a pan-genotypic HCV inhibitor, with an EC50 range of 1.7–19.3 pM against GT1a, -1b, -2a, -2b, -3a, -4a, and -5a and 366 pM against GT6a. Compound 38 decreased HCV RNA up to 3.10 log10 IU/mL during 3-day monotherapy in treatment-naive HCV GT1-infected subjects and is currently in phase 3 clinical trials in combination with an NS3 protease inhibitor with ritonavir (r) (ABT-450/r) and an NS5B non-nucleoside polymerase inhibitor (ABT-333), with and without ribavirin.
Dimethyl (2S,2′S)-1,1′-((2S,2′S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-Butylphenyl)pyrrolidine-2,5-diyl)bis(4,1-phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-2,1-diyl)dicarbamate (38)…desired
and
Dimethyl (2S,2′S)-1,1′-((2S,2′S)-2,2′-(4,4′-((2R,5R)-1-(4-tert-Butylphenyl)pyrrolidine-2,5-diyl)bis(4,1-phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2,1-diyl))bis(3-methyl-1-oxobutane-2,1-diyl)dicarbamate (39)…….undesired
………..
PATENT
WO 2011156578
dimethyl (2S,2,S)-l,l ‘-((2S,2’S)-2,2′-(4,4’-((2S,5S)-l-(4-fert-butylphenyl)pyrrolidine- 2,5-diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3- methyl- l-oxobutane-2,l-diyl)dicarbamate

hereinafter Compound IA),..http://www.google.com/patents/WO2011156578A1?cl=en
……………………………..
PATENT
US 20100317568
https://www.google.co.in/patents/US20100317568
Example 34
Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate and
Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2R,5R)-1-(4-ter/’-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 – oxobutane-2, 1 -diyl)dicarbamate

Example 34A l-(4-fer?-butylphenyl)-2,5-bis(4-nitrophenyl)pyrrolidine The product from Example 1C (3.67 g, 7.51 mmol) and 4-tert-butylaniline (11.86 ml, 75 mmol) in DMF (40 ml) was stirred under nitrogen at 50 °C for 4 h. The resulting mixture was diluted into ethyl acetate, treated with IM HCl, stirred for 10 minutes and filtered to remove solids. The filtrate organic layer was washed twice with brine, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (5% to 30%) to give a solid. The solid was triturated in a minimal volume of 1 :9 ethyl acetate/hexane to give a light yellow solid as a mixture of trans and cis isomers (1.21 g, 36%).
Example 34B 4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)dianiline and 4,4′-((2R,5R)-1-(4-fert- butylphenyl)pyrrolidine-2,5-diyl)dianiline To a solution of the product from Example 34A (1.1 g, 2.47 mmol) in ethanol (20 ml) and
THF (20 ml) was added PtC>2 (0.22 g, 0.97 mmol) in a 50 ml pressure bottle and stirred under 30 psi hydrogen at room temperature for 1 h. The mixture was filtered through a nylon membrane and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (20% to 60%). The title compound eluted as the first of 2 stereoisomers (trans isomer, 0.51 g, 54%).
Example 34C
(2S,2’S)-tert-Butyl 2,2′-(4,4′-((2S,5S)-1-(4-fer/’-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)dipyrrolidine- 1 -carboxylate and (2S,2’S)-tert-Butyl 2,2′- (4,4′-((2R,5R)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)dipyrrolidine-1-carboxylate To a mixture of the product from Example 34B (250 mg, 0.648 mmol), (S)-1-(tert- butoxycarbonyl)pyrrolidine-2-carboxylic acid (307 mg, 1.427 mmol) and HATU (542 mg, 1.427 mmol) in DMSO (10 ml) was added Hunig’s base (0.453 ml, 2.59 mmol). The reaction mixture was stirred at room temperature for 1 h. The mixture was partitioned with ethyl acetate and water. The organic layer was washed with brine, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (10% to 50%) to give the title compound (500 mg, 99%).
Example 34D
(2S,2’S)-N,N’-(4,4′-((2S,5S)-1-(4-ter/’-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))dipyrrolidine-2-carboxamide and (2S,2’S)-N,N’-(4,4′-((2R,5R)-1-(4-tert- butylphenyl)pyrrolidine-2,5-diyl)bis(4,l-phenylene))dipyrrolidine-2-carboxamide To the product from Example 34C (498 mg, 0.638 mmol) in dichloromethane (4 ml) was added TFA (6 ml). The reaction mixture was stirred at room temperature for 1 h and concentrated in vacuo. The residue was partitioned between 3: 1 CHCl3dsopropyl alcohol and saturated aq. NaHCO3. The aqueous layer was extracted by 3: 1 CHCl3:isopropyl alcohol again. The combined organic layers were dried over
filtered and concentrated to give the title compound (345 mg, 93%).
Example 34E Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-fert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate and
Dimethyl (2S,2’S)-1, r-((2S,2’S)-2,2′-(4,4′-((2R,5R)-1-(4-fert-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 – oxobutane-2, 1 -diyl)dicarbamate
The product from Example 34D (29.0 mg, 0.050 mmol), (S)-2-(methoxycarbonylamino)-3- methylbutanoic acid (19.27 mg, 0.110 mmol), EDAC (21.09 mg, 0.110 mmol), HOBT (16.85 mg,
0.110 mmol) and N-methylmorpholine (0.027 ml, 0.250 mmol) were combined in DMF (2 ml). The reaction mixture was stirred at room temperature for 3 h. The mixture was partitioned with ethyl acetate and water. The organic layer was washed with brine twice, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (50% to 80%) to give a solid. The solid was triturated with ethyl acetate/hexane to give the title compound (13 mg, 29%). 1H NMR (400 MHz, DMSO-D6) δ ppm 0.85 – 0.95 (m, 12 H) 1.11 (s, 9 H) 1.59 – 1.65 (m, 2 H) 1.79 – 2.04 (m, 8 H) 2.10 – 2.18 (m, 2 H) 2.41-2.46 (m, 2H) 3.52 (s, 6 H)
3.57 – 3.67 (m, 2 H) 3.76 – 3.86 (m, 2 H) 4.00 (t, J=7.56 Hz, 2 H) 4.39 – 4.46 (m, 2 H) 5.15 (d, J=7.00
Hz, 2 H) 6.17 (d, J=7.70 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=7.37 Hz, 4 H) 7.30 (d, J=8.20
Hz, 2 H) 7.50 (d, J=8.24 Hz, 4 H) 9.98 (s, 2 H); (ESI+) m/z 895 (M+H)+. The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
Example 35
Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate
 ………………desired
………………desiredThe product from Example 34E was purified by chiral chromatography on a Chiralpak AD-H semi-prep column eluting with a 2:1 mixture of hexane:(2: l isopropyl alcohol: EtOH). The title compound was the first of the 2 diastereomers to elute. 1H NMR (400 MHz, DMSO-D6) δ ppm 0.88 (d, J=6.61 Hz, 6 H) 0.93 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.42 Hz, 2 H) 1.80 – 2.04 (m, 8 H) 2.09 – 2.19 (m, 2 H) 2.44 – 2.47 (m, 2 H) 3.52 (s, 6 H) 3.59 – 3.66 (m, 2 H) 3.77 – 3.84 (m, 2 H) 4.02 (t, J=8.40 Hz, 2 H) 4.42 (dd, J=7.86, 4.83 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.67 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.50 (d, J=8.35 Hz, 4 H) 9.98 (s, 2 H). The title compound showed an EC50 value of less than about 0.1 nM in HCV Ib- Conl replicon assays in the presence of 5% FBS.
Example 36 Dimethyl (2S,2’S)-1, r-((2S,2’S)-2,2′-(4,4′-((2R,5R)-1-(4-fert-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 – oxobutane-2, 1 -diyl)dicarbamate
 …….undesired
…….undesiredThe product from Example 34E was purified by chiral chromatography on a Chiralpak AD-H semi-prep column eluting with a 2:1 mixture of hexane:(2: l isopropyl alcohol: EtOH). The title compound was the second of 2 diastereomers to elute. 1H NMR (400 MHz, DMSO-D6) δ ppm 0.87
(d, J=6.51 Hz, 6 H) 0.92 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.53 Hz, 2 H) 1.82 – 2.04 (m, 8
H) 2.09-2.18 (m, 2 H) 2.41 – 2.47 (m, 2 H) 3.52 (s, 6 H) 3.58 – 3.67 (m, 2 H) 3.75 – 3.84 (m, 2 H) 4.02
(t, J=7.26 Hz, 2 H) 4.43 (dd, J=7.92, 4.88 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.78 Hz, 2 H) 6.94 (d, J=8.67 Hz, 2 H) 7.12 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.49 (d, J=8.46 Hz, 4 H)
9.98 (s, 2 H). The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
Example 37 Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-fert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate
 ……………desired
……………desiredExample 37A (S)-2,5-dioxopyrrolidin-1-yl 2-(methoxycarbonylamino)-3-methylbutanoate To a mixture of (S)-2-(methoxycarbonylamino)-3-methylbutanoic acid (19.66 g, 112 mmol) and N-hydroxysuccinimide (13.29g, 116 mmol) was added ethyl acetate (250 ml), and the mixture was cooled to 0-5 °C. Diisopropylcarbodiimide (13.88 g, 110 mmol) was added and the reaction mixture was stirred at 0-5 °C for about 1 hour. The reaction mixture was warmed to room temperature. The solids (diisopropylurea by-product) were filtered and rinsed with ethyl acetate. The filtrate was concentrated in vacuo to an oil. Isopropyl alcohol (200 ml) was added to the oil and the mixture was heated to about 50 °C to obtain a homogeneous solution. Upon cooling, crystalline solids formed. The solids were filtered and washed with isopropyl alcohol (3 x 20 ml) and dried to give the title compound as a white solid (23.2 g, 77% yield).
Example 37B
(S)- 1 -((S)-2-(methoxycarbonylamino)-3-methylbutanoyl)pyrrolidine-2-carboxylic acid To a mixture of L-proline (4.44g, 38.6 mmol), water (20 ml), acetonitrile (20 ml) and DIEA (9.5 g, 73.5 mmol) was added a solution of the product from Example 37A (1Og, 36.7 mmol) in acetonitrile (20 inL) over 10 minutes. The reaction mixture was stirred overnight at room temperature. The solution was concentrated under vacuum to remove the acetonitrile. To the resulting clear water solution was added 6N HCl (9 ml) until pH ~ 2 .The solution was transferred to a separatory funnel and 25% NaCl (10 ml) was added and the mixture was extracted with ethyl acetate (75 ml), and then again with ethyl acetate (6 x 20 ml), and the combined extracts were washed with 25% NaCl (2 x 10ml). The solvent was evaporated to give a thick oil. Heptane was added and the solvent was evaporated to give a foam, which was dried under high vacuum. Diethyl ether was added and the solvent was evaporated to give a foam, which was dried under high vacuum to give the title compound (10.67g) as a white solid.
The compound of Example 37B can also be prepreared according to the following procedure: To a flask was charged L- valine (35 g, 299 mmol), IN sodium hydroxide solution (526 ml,
526 mmol) and sodium carbonate (17.42 g, 164 mmol). The mixture was stirred for 15 min to dissolve solids and then cooled to 15 °C. Methyl chloroformate (29.6 g, 314 mmol) was added slowly to the reaction mixture. The mixture was then stirred at rt for 30 min. The mixture was cooled to 15 °C and pH adjusted to -5.0 with concentrated HCl solution. 100 inL of 2-methytetrahydrofuran (2- MeTHF) was added and the adjustment of pH continued until the pH reached ~ 2.0. 150 mL of 2- MeTHF was added and the mixture was stirred for 15 min. Layers were separated and the aqueous layer extracted with 100 mL of 2-MeTHF. The combined organic layer was dried over anhyd Na2SC^ and filtered, and Na2SC^ cake was washed with 50 mL of 2-MeTHF. The product solution was concentrated to ~ 100 mL, chased with 120 mL of IPAc twice. 250 mL of heptanes was charged slowly and then the volume of the mixture was concentrated to 300 mL. The mixture was heated to 45 °C and 160 mL of heptanes charged. The mixture was cooled to rt in 2h, stirred for 30 min, filtered and washed with 2-MeTHF/heptanes mixture (1:7, 80 inL). The wetcake was dried at 55 °C for 24 h to give 47.1 g of Moc-L- VaI-OH product as a white solid (90%).
Moc-L- VaI-OH (15O g, 856 mmol), HOBt hydrate (138 g, 899 mmol) and DMF (1500 ml) were charged to a flask. The mixture was stirred for 15 min to give a clear solution. EDC hydrochloride (172 g, 899 mmol) was charged and mixed for 20 min. The mixture was cooled to 13
°C and (L)-proline benzyl ester hydrochloride (207 g, 856 mmol) charged. Triethylamine (109 g,
1079 mmol) was then charged in 30 min. The resulting suspension was mixed at rt for 1.5 h. The reaction mixture was cooled to 15 °C and 1500 mL of 6.7% NaHCO3 charged in 1.5 h, followed by the addition of 1200 mL of water over 60 min. The mixture was stirred at rt for 30 min, filtered and washed with water/DMF mixture (1 :2, 250 mL) and then with water (1500 mL). The wetcake was dried at 55 °C for 24 h to give 282 g of product as a white solid (90%).
The resulting solids (40 g) and 5% Pd/ Alumina were charged to a Parr reactor followed by THF (160 mL). The reactor was sealed and purged with nitrogen (6 x 20 psig) followed by a hydrogen purge (6 x 30 psig). The reactor was pressurized to 30 psig with hydrogen and agitated at room temperature for approximately 15 hours. The resulting slurry was filtered through a GF/F filter and concentrated to approximately 135 g solution. Heptane was added (120 mL), and the solution was stirred until solids formed. After an addition 2 – 3 hours additional heptane was added drop-wise (240 mL), the slurry was stirred for approximately 1 hour, then filtered. The solids were dried to afford the title compound.
Example 37C
(lR,4R)-1,4-bis(4-nitrophenyl)butane-1,4-diyl dimethanesulfonate
The product from Example 32 (5.01 g, 13.39 mmol) was combined with 2- methyltetrahydrofuran (70 mL) and cooled to -5 °C, and N,N-diisopropylethylamine (6.81 g, 52.7 mmol) was added over 30 seconds. Separately, a solution of methanesulfonic anhydride (6.01 g, 34.5 mmol) in 2-methyltetrahydrofuran (30 mL) was prepared and added to the diol slurry over 3 min., maintaining the internal temperature between -15 °C and -25 °C. After mixing for 5 min at -15 °C, the cooling bath was removed and the reaction was allowed to warm slowly to 23 °C and mixed for 30 minutes. After reaction completion, the crude slurry was carried immediately into the next step.
Example 37D
(2S,5S)-1-(4-tert-butylphenyl)-2,5-bis(4-nitrophenyl)pyrrolidine
To the crude product solution from Example 37C (7.35 g, 13.39 mmol) was added 4-tert- butylaniline (13.4 g, 90 mmol) at 23 °C over 1 minute. The reaction was heated to 65 °C for 2 h. After completion, the reaction mixture was cooled to 23 °C and diluted with 2-methyltetrahydrofuran (100 mL) and 1 M HCl (150 mL). After partitioning the phases, the organic phase was treated with 1 M HCl (140 mL), 2-methyltetrahydrofuran (50 mL), and 25 wt% aq. NaCl (100 mL), and the phases were partitioned. The organic phase was washed with 25 wt% aq. NaCl (50 mL), dried over MgSO/t, filtered, and concentrated in vacuo to approximately 20 mL. Heptane (30 mL) and additional 2- methyltetrahydrofuran were added in order to induce crystallization. The slurry was concentrated further, and additional heptane (40 mL) was slowly added and the slurry was filtered, washing with 2- methyltetrahydrofuran:heptane (1:4, 20 mL). The solids were suspended in MeOH (46 mL) for 3 h, filtered, and the wet solid was washed with additional MeOH (18 mL). The solid was dried at 45 °C in a vacuum oven for 16 h to provide the title compound (3.08 g, 51% 2-step yield).
Example 37E
4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)dianiline
To a 160 ml Parr stirrer hydrogenation vessel was added the product from Example 37D (2 g, 4.49 mmol), followed by 60 ml of THF, and Raney Nickel Grace 2800 (1 g, 50 wt% (dry basis)) under a stream of nitrogen. The reactor was assembled and purged with nitrogen (8 x 20 psig) followed by purging with hydrogen (8 x 30 psig). The reactor was then pressurized to 30 psig with hydrogen and agitation (700 rpm) began and continued for a total of 16 h at room temperature. The slurry was filtered by vacuum filtration using a GF/F Whatman glass fiber filter. Evaporation of the filtrate to afford a slurry followed by the addition heptane and filtration gave the crude title compound, which was dried and used directly in the next step.
Example 37F dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4, l- phenylene)bis(azanediyl)bis(oxomethylene))bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diy 1) die arb amate To a solution of the product from Example 37E (1.64 g, 4.25 mmol) in DMF (20 ml), the product from Example 37B (2.89 g, 10.63 mmol), and HATU (4.04 g, 10.63 mmol) in DMF (15OmL) was added triethylamine (1.07 g, 10.63 mmol), and the solution was stirred at room temperature for 90 min. To the reaction mixture was poured 20 mL of water, and the white precipitate obtained was filtered, and the solid was washed with water (3×5 mL). The solid was blow dried for Ih. The crude material was loaded on a silica gel column and eluted with a gradient starting with ethyl acetate/ heptane (3/7), and ending with pure ethyl acetate. The desired fractions were combined and solvent distilled off to give a very light yellow solid, which was dried at 45 °C in a vacuum oven with nitrogen purge for 15 h to give the title compound (2.3 g, 61% yield). 1H NMR (400 MHz, DMSO- D6) δ ppm 0.88 (d, J=6.61 Hz, 6 H) 0.93 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.42 Hz, 2 H) 1.80 – 2.04 (m, 8 H) 2.09 – 2.19 (m, 2 H) 2.44 – 2.47 (m, 2 H) 3.52 (s, 6 H) 3.59 – 3.66 (m, 2 H) 3.77 – 3.84 (m, 2 H) 4.02 (t, J=8.40 Hz, 2 H) 4.42 (dd, J=7.86, 4.83 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.67 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.50 (d, J=8.35 Hz, 4 H) 9.98 (s, 2 H).
Alternately, the product from example 37E (11.7 g, 85 wt%, 25.8 mmol) and the product from example 37B (15.45 g, 56.7 mmol) are suspended in EtOAc (117 mL), diisopropylethylamine (18.67 g, 144 mmol) is added and the solution is cooled to 0 °C. In a separate flask, 1-propanephosphonic acid cyclic anhydride (T3P®) (46.0 g, 50 wt% in EtOAc, 72.2 mmol) was dissolved in EtOAc (58.5 mL), and charged to an addition funnel. The T3P solution is added to the reaction mixture drop-wise over 3-4 h and stirred until the reaction is complete. The reaction is warmed to room temperature,and washed with IM HCl/7.5 wt% NaCl (100 mL), then washed with 5% NaHCO3 (100 mL), then washed with 5% NaCl solution (100 mL). The solution was concentrated to approximately 60 mL, EtOH (300 mL) was added, and the solution was concentrated to 84 g solution.
A portion of the EtOH solution of product (29 g) was heated to 40 °C, and added 134 g 40 w% EtOH in H2O. A slurry of seeds in 58 wt/wt% EtOH/H2O was added, allowed to stir at 40 °C for several hours, then cooled to 0 °C. The slurry is then filtered, and washed with 58wt/wt% EtOH/H2O. The product is dried at 40 – 60 °C under vacuum, and then rehydrated by placing a tray of water in the vacuum oven to give the title compound. The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
……………..
PATENT
http://www.google.com/patents/EP2337781A2?cl=en
Example 34
Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate and
Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2R,5R)-1-(4-ter/’-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 – oxobutane-2, 1 -diyl)dicarbamate

Example 34A l-(4-fer?-butylphenyl)-2,5-bis(4-nitrophenyl)pyrrolidine The product from Example 1C (3.67 g, 7.51 mmol) and 4-tert-butylaniline (11.86 ml, 75 mmol) in DMF (40 ml) was stirred under nitrogen at 50 °C for 4 h. The resulting mixture was diluted into ethyl acetate, treated with IM HCl, stirred for 10 minutes and filtered to remove solids. The filtrate organic layer was washed twice with brine, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (5% to 30%) to give a solid. The solid was triturated in a minimal volume of 1 :9 ethyl acetate/hexane to give a light yellow solid as a mixture of trans and cis isomers (1.21 g, 36%).
Example 34B 4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)dianiline and 4,4′-((2R,5R)-1-(4-fert- butylphenyl)pyrrolidine-2,5-diyl)dianiline To a solution of the product from Example 34A (1.1 g, 2.47 mmol) in ethanol (20 ml) and
THF (20 ml) was added PtC>2 (0.22 g, 0.97 mmol) in a 50 ml pressure bottle and stirred under 30 psi hydrogen at room temperature for 1 h. The mixture was filtered through a nylon membrane and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (20% to 60%). The title compound eluted as the first of 2 stereoisomers (trans isomer, 0.51 g, 54%).
Example 34C
(2S,2’S)-tert-Butyl 2,2′-(4,4′-((2S,5S)-1-(4-fer/’-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)dipyrrolidine- 1 -carboxylate and (2S,2’S)-tert-Butyl 2,2′- (4,4′-((2R,5R)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)dipyrrolidine-1-carboxylate To a mixture of the product from Example 34B (250 mg, 0.648 mmol), (S)-1-(tert- butoxycarbonyl)pyrrolidine-2-carboxylic acid (307 mg, 1.427 mmol) and HATU (542 mg, 1.427 mmol) in DMSO (10 ml) was added Hunig’s base (0.453 ml, 2.59 mmol). The reaction mixture was stirred at room temperature for 1 h. The mixture was partitioned with ethyl acetate and water. The organic layer was washed with brine, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (10% to 50%) to give the title compound (500 mg, 99%).
Example 34D
(2S,2’S)-N,N’-(4,4′-((2S,5S)-1-(4-ter/’-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))dipyrrolidine-2-carboxamide and (2S,2’S)-N,N’-(4,4′-((2R,5R)-1-(4-tert- butylphenyl)pyrrolidine-2,5-diyl)bis(4,l-phenylene))dipyrrolidine-2-carboxamide To the product from Example 34C (498 mg, 0.638 mmol) in dichloromethane (4 ml) was added TFA (6 ml). The reaction mixture was stirred at room temperature for 1 h and concentrated in vacuo. The residue was partitioned between 3: 1 CHCl3dsopropyl alcohol and saturated aq. NaHCO3. The aqueous layer was extracted by 3: 1 CHCl3:isopropyl alcohol again. The combined organic layers were dried over
filtered and concentrated to give the title compound (345 mg, 93%).
Example 34E Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-fert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate and
Dimethyl (2S,2’S)-1, r-((2S,2’S)-2,2′-(4,4′-((2R,5R)-1-(4-fert-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 – oxobutane-2, 1 -diyl)dicarbamate
The product from Example 34D (29.0 mg, 0.050 mmol), (S)-2-(methoxycarbonylamino)-3- methylbutanoic acid (19.27 mg, 0.110 mmol), EDAC (21.09 mg, 0.110 mmol), HOBT (16.85 mg,
0.110 mmol) and N-methylmorpholine (0.027 ml, 0.250 mmol) were combined in DMF (2 ml). The reaction mixture was stirred at room temperature for 3 h. The mixture was partitioned with ethyl acetate and water. The organic layer was washed with brine twice, dried with sodium sulfate, filtered and evaporated. The residue was purified by chromatography on silica gel eluting with ethyl acetate in hexane (50% to 80%) to give a solid. The solid was triturated with ethyl acetate/hexane to give the title compound (13 mg, 29%). 1H NMR (400 MHz, DMSO-D6) δ ppm 0.85 – 0.95 (m, 12 H) 1.11 (s, 9 H) 1.59 – 1.65 (m, 2 H) 1.79 – 2.04 (m, 8 H) 2.10 – 2.18 (m, 2 H) 2.41-2.46 (m, 2H) 3.52 (s, 6 H)
3.57 – 3.67 (m, 2 H) 3.76 – 3.86 (m, 2 H) 4.00 (t, J=7.56 Hz, 2 H) 4.39 – 4.46 (m, 2 H) 5.15 (d, J=7.00
Hz, 2 H) 6.17 (d, J=7.70 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=7.37 Hz, 4 H) 7.30 (d, J=8.20
Hz, 2 H) 7.50 (d, J=8.24 Hz, 4 H) 9.98 (s, 2 H); (ESI+) m/z 895 (M+H)+. The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
Example 35
Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate
 ………….desired
………….desiredThe product from Example 34E was purified by chiral chromatography on a Chiralpak AD-H semi-prep column eluting with a 2:1 mixture of hexane:(2: l isopropyl alcohol: EtOH). The title compound was the first of the 2 diastereomers to elute. 1H NMR (400 MHz, DMSO-D6) δ ppm 0.88 (d, J=6.61 Hz, 6 H) 0.93 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.42 Hz, 2 H) 1.80 – 2.04 (m, 8 H) 2.09 – 2.19 (m, 2 H) 2.44 – 2.47 (m, 2 H) 3.52 (s, 6 H) 3.59 – 3.66 (m, 2 H) 3.77 – 3.84 (m, 2 H) 4.02 (t, J=8.40 Hz, 2 H) 4.42 (dd, J=7.86, 4.83 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.67 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.50 (d, J=8.35 Hz, 4 H) 9.98 (s, 2 H). The title compound showed an EC50 value of less than about 0.1 nM in HCV Ib- Conl replicon assays in the presence of 5% FBS.
Example 36 Dimethyl (2S,2’S)-1, r-((2S,2’S)-2,2′-(4,4′-((2R,5R)-1-(4-fert-butylphenyl)pyrrolidine-2,5- diyl)bis(4, 1 -phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 – oxobutane-2, 1 -diyl)dicarbamate
 ……….undesired
……….undesiredThe product from Example 34E was purified by chiral chromatography on a Chiralpak AD-H semi-prep column eluting with a 2:1 mixture of hexane:(2: l isopropyl alcohol: EtOH). The title compound was the second of 2 diastereomers to elute. 1H NMR (400 MHz, DMSO-D6) δ ppm 0.87
(d, J=6.51 Hz, 6 H) 0.92 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.53 Hz, 2 H) 1.82 – 2.04 (m, 8
H) 2.09-2.18 (m, 2 H) 2.41 – 2.47 (m, 2 H) 3.52 (s, 6 H) 3.58 – 3.67 (m, 2 H) 3.75 – 3.84 (m, 2 H) 4.02
(t, J=7.26 Hz, 2 H) 4.43 (dd, J=7.92, 4.88 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.78 Hz, 2 H) 6.94 (d, J=8.67 Hz, 2 H) 7.12 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.49 (d, J=8.46 Hz, 4 H)
9.98 (s, 2 H). The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
Example 37 Dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-fert-butylphenyl)pyrrolidine-2,5-diyl)bis(4,l- phenylene))bis(azanediyl)bis(oxomethylene)bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diyl)dicarbamate
 ………………desired
………………desiredExample 37A (S)-2,5-dioxopyrrolidin-1-yl 2-(methoxycarbonylamino)-3-methylbutanoate To a mixture of (S)-2-(methoxycarbonylamino)-3-methylbutanoic acid (19.66 g, 112 mmol) and N-hydroxysuccinimide (13.29g, 116 mmol) was added ethyl acetate (250 ml), and the mixture was cooled to 0-5 °C. Diisopropylcarbodiimide (13.88 g, 110 mmol) was added and the reaction mixture was stirred at 0-5 °C for about 1 hour. The reaction mixture was warmed to room temperature. The solids (diisopropylurea by-product) were filtered and rinsed with ethyl acetate. The filtrate was concentrated in vacuo to an oil. Isopropyl alcohol (200 ml) was added to the oil and the mixture was heated to about 50 °C to obtain a homogeneous solution. Upon cooling, crystalline solids formed. The solids were filtered and washed with isopropyl alcohol (3 x 20 ml) and dried to give the title compound as a white solid (23.2 g, 77% yield).
Example 37B
(S)- 1 -((S)-2-(methoxycarbonylamino)-3-methylbutanoyl)pyrrolidine-2-carboxylic acid To a mixture of L-proline (4.44g, 38.6 mmol), water (20 ml), acetonitrile (20 ml) and DIEA (9.5 g, 73.5 mmol) was added a solution of the product from Example 37A (1Og, 36.7 mmol) in acetonitrile (20 inL) over 10 minutes. The reaction mixture was stirred overnight at room temperature. The solution was concentrated under vacuum to remove the acetonitrile. To the resulting clear water solution was added 6N HCl (9 ml) until pH ~ 2 .The solution was transferred to a separatory funnel and 25% NaCl (10 ml) was added and the mixture was extracted with ethyl acetate (75 ml), and then again with ethyl acetate (6 x 20 ml), and the combined extracts were washed with 25% NaCl (2 x 10ml). The solvent was evaporated to give a thick oil. Heptane was added and the solvent was evaporated to give a foam, which was dried under high vacuum. Diethyl ether was added and the solvent was evaporated to give a foam, which was dried under high vacuum to give the title compound (10.67g) as a white solid.
The compound of Example 37B can also be prepreared according to the following procedure: To a flask was charged L- valine (35 g, 299 mmol), IN sodium hydroxide solution (526 ml,
526 mmol) and sodium carbonate (17.42 g, 164 mmol). The mixture was stirred for 15 min to dissolve solids and then cooled to 15 °C. Methyl chloroformate (29.6 g, 314 mmol) was added slowly to the reaction mixture. The mixture was then stirred at rt for 30 min. The mixture was cooled to 15 °C and pH adjusted to -5.0 with concentrated HCl solution. 100 inL of 2-methytetrahydrofuran (2- MeTHF) was added and the adjustment of pH continued until the pH reached ~ 2.0. 150 mL of 2- MeTHF was added and the mixture was stirred for 15 min. Layers were separated and the aqueous layer extracted with 100 mL of 2-MeTHF. The combined organic layer was dried over anhyd Na2SC^ and filtered, and Na2SC^ cake was washed with 50 mL of 2-MeTHF. The product solution was concentrated to ~ 100 mL, chased with 120 mL of IPAc twice. 250 mL of heptanes was charged slowly and then the volume of the mixture was concentrated to 300 mL. The mixture was heated to 45 °C and 160 mL of heptanes charged. The mixture was cooled to rt in 2h, stirred for 30 min, filtered and washed with 2-MeTHF/heptanes mixture (1:7, 80 inL). The wetcake was dried at 55 °C for 24 h to give 47.1 g of Moc-L- VaI-OH product as a white solid (90%).
Moc-L- VaI-OH (15O g, 856 mmol), HOBt hydrate (138 g, 899 mmol) and DMF (1500 ml) were charged to a flask. The mixture was stirred for 15 min to give a clear solution. EDC hydrochloride (172 g, 899 mmol) was charged and mixed for 20 min. The mixture was cooled to 13
°C and (L)-proline benzyl ester hydrochloride (207 g, 856 mmol) charged. Triethylamine (109 g,
1079 mmol) was then charged in 30 min. The resulting suspension was mixed at rt for 1.5 h. The reaction mixture was cooled to 15 °C and 1500 mL of 6.7% NaHCO3 charged in 1.5 h, followed by the addition of 1200 mL of water over 60 min. The mixture was stirred at rt for 30 min, filtered and washed with water/DMF mixture (1 :2, 250 mL) and then with water (1500 mL). The wetcake was dried at 55 °C for 24 h to give 282 g of product as a white solid (90%).
The resulting solids (40 g) and 5% Pd/ Alumina were charged to a Parr reactor followed by THF (160 mL). The reactor was sealed and purged with nitrogen (6 x 20 psig) followed by a hydrogen purge (6 x 30 psig). The reactor was pressurized to 30 psig with hydrogen and agitated at room temperature for approximately 15 hours. The resulting slurry was filtered through a GF/F filter and concentrated to approximately 135 g solution. Heptane was added (120 mL), and the solution was stirred until solids formed. After an addition 2 – 3 hours additional heptane was added drop-wise (240 mL), the slurry was stirred for approximately 1 hour, then filtered. The solids were dried to afford the title compound.
Example 37C
(lR,4R)-1,4-bis(4-nitrophenyl)butane-1,4-diyl dimethanesulfonate
The product from Example 32 (5.01 g, 13.39 mmol) was combined with 2- methyltetrahydrofuran (70 mL) and cooled to -5 °C, and N,N-diisopropylethylamine (6.81 g, 52.7 mmol) was added over 30 seconds. Separately, a solution of methanesulfonic anhydride (6.01 g, 34.5 mmol) in 2-methyltetrahydrofuran (30 mL) was prepared and added to the diol slurry over 3 min., maintaining the internal temperature between -15 °C and -25 °C. After mixing for 5 min at -15 °C, the cooling bath was removed and the reaction was allowed to warm slowly to 23 °C and mixed for 30 minutes. After reaction completion, the crude slurry was carried immediately into the next step.
Example 37D
(2S,5S)-1-(4-tert-butylphenyl)-2,5-bis(4-nitrophenyl)pyrrolidine
To the crude product solution from Example 37C (7.35 g, 13.39 mmol) was added 4-tert- butylaniline (13.4 g, 90 mmol) at 23 °C over 1 minute. The reaction was heated to 65 °C for 2 h. After completion, the reaction mixture was cooled to 23 °C and diluted with 2-methyltetrahydrofuran (100 mL) and 1 M HCl (150 mL). After partitioning the phases, the organic phase was treated with 1 M HCl (140 mL), 2-methyltetrahydrofuran (50 mL), and 25 wt% aq. NaCl (100 mL), and the phases were partitioned. The organic phase was washed with 25 wt% aq. NaCl (50 mL), dried over MgSO/t, filtered, and concentrated in vacuo to approximately 20 mL. Heptane (30 mL) and additional 2- methyltetrahydrofuran were added in order to induce crystallization. The slurry was concentrated further, and additional heptane (40 mL) was slowly added and the slurry was filtered, washing with 2- methyltetrahydrofuran:heptane (1:4, 20 mL). The solids were suspended in MeOH (46 mL) for 3 h, filtered, and the wet solid was washed with additional MeOH (18 mL). The solid was dried at 45 °C in a vacuum oven for 16 h to provide the title compound (3.08 g, 51% 2-step yield).
Example 37E
4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)dianiline
To a 160 ml Parr stirrer hydrogenation vessel was added the product from Example 37D (2 g, 4.49 mmol), followed by 60 ml of THF, and Raney Nickel Grace 2800 (1 g, 50 wt% (dry basis)) under a stream of nitrogen. The reactor was assembled and purged with nitrogen (8 x 20 psig) followed by purging with hydrogen (8 x 30 psig). The reactor was then pressurized to 30 psig with hydrogen and agitation (700 rpm) began and continued for a total of 16 h at room temperature. The slurry was filtered by vacuum filtration using a GF/F Whatman glass fiber filter. Evaporation of the filtrate to afford a slurry followed by the addition heptane and filtration gave the crude title compound, which was dried and used directly in the next step.
Example 37F dimethyl (2S,2’S)-l,r-((2S,2’S)-2,2′-(4,4′-((2S,5S)-1-(4-tert-butylphenyl)pyrrolidine-2,5-diyl)bis(4, l- phenylene)bis(azanediyl)bis(oxomethylene))bis(pyrrolidine-2, 1 -diyl))bis(3-methyl- 1 -oxobutane-2, 1 – diy 1) die arb amate To a solution of the product from Example 37E (1.64 g, 4.25 mmol) in DMF (20 ml), the product from Example 37B (2.89 g, 10.63 mmol), and HATU (4.04 g, 10.63 mmol) in DMF (15OmL) was added triethylamine (1.07 g, 10.63 mmol), and the solution was stirred at room temperature for 90 min. To the reaction mixture was poured 20 mL of water, and the white precipitate obtained was filtered, and the solid was washed with water (3×5 mL). The solid was blow dried for Ih. The crude material was loaded on a silica gel column and eluted with a gradient starting with ethyl acetate/ heptane (3/7), and ending with pure ethyl acetate. The desired fractions were combined and solvent distilled off to give a very light yellow solid, which was dried at 45 °C in a vacuum oven with nitrogen purge for 15 h to give the title compound (2.3 g, 61% yield). 1H NMR (400 MHz, DMSO- D6) δ ppm 0.88 (d, J=6.61 Hz, 6 H) 0.93 (d, J=6.72 Hz, 6 H) 1.11 (s, 9 H) 1.63 (d, J=5.42 Hz, 2 H) 1.80 – 2.04 (m, 8 H) 2.09 – 2.19 (m, 2 H) 2.44 – 2.47 (m, 2 H) 3.52 (s, 6 H) 3.59 – 3.66 (m, 2 H) 3.77 – 3.84 (m, 2 H) 4.02 (t, J=8.40 Hz, 2 H) 4.42 (dd, J=7.86, 4.83 Hz, 2 H) 5.14 (d, J=6.18 Hz, 2 H) 6.17 (d, J=8.67 Hz, 2 H) 6.94 (d, J=8.78 Hz, 2 H) 7.13 (d, J=8.46 Hz, 4 H) 7.31 (d, J=8.35 Hz, 2 H) 7.50 (d, J=8.35 Hz, 4 H) 9.98 (s, 2 H).
Alternately, the product from example 37E (11.7 g, 85 wt%, 25.8 mmol) and the product from example 37B (15.45 g, 56.7 mmol) are suspended in EtOAc (117 mL), diisopropylethylamine (18.67 g, 144 mmol) is added and the solution is cooled to 0 °C. In a separate flask, 1-propanephosphonic acid cyclic anhydride (T3P®) (46.0 g, 50 wt% in EtOAc, 72.2 mmol) was dissolved in EtOAc (58.5 mL), and charged to an addition funnel. The T3P solution is added to the reaction mixture drop-wise over 3-4 h and stirred until the reaction is complete. The reaction is warmed to room temperature,and washed with IM HCl/7.5 wt% NaCl (100 mL), then washed with 5% NaHCO3 (100 mL), then washed with 5% NaCl solution (100 mL). The solution was concentrated to approximately 60 mL, EtOH (300 mL) was added, and the solution was concentrated to 84 g solution.
A portion of the EtOH solution of product (29 g) was heated to 40 °C, and added 134 g 40 w% EtOH in H2O. A slurry of seeds in 58 wt/wt% EtOH/H2O was added, allowed to stir at 40 °C for several hours, then cooled to 0 °C. The slurry is then filtered, and washed with 58wt/wt% EtOH/H2O. The product is dried at 40 – 60 °C under vacuum, and then rehydrated by placing a tray of water in the vacuum oven to give the title compound. The title compound showed an EC50 value of less than about 0.1 nM in HCV lb-Conl replicon assays in the presence of 5% FBS.
Intermediates
Example 32
( 1 R,4R)- 1 ,4-bis(4-mtrophenyl)butane- 1 ,4-diol
To (S)-(-)-α,α-diphenyl-2-pyrrohdinemethanol (2 71 g, 10 70 mmol) was added THF (80 mL) at 23 °C The very thin suspension was treated with t11methyl borate (1 44 g, 13 86 mmol) over 30 seconds, and the resulting solution was mixed at 23 °C for 1 h The solution was cooled to 16-19 °C, and N,N-diethylanilme borane (21 45 g, 132 mmol) was added dropwise via syringe over 3-5 mm (caution vigorous H2 evolution), while the internal temperature was maintained at 16-19 °C After 15 mm, the H2 evolution had ceased To a separate vessel was added the product from Example IA (22 04 g, 95 wt%, 63 8 mmol), followed by THF (80 mL), to form an orange slurry After cooling the slurry to 11 °C, the borane solution was transferred via cannula into the dione slurry over 3-5 min During this period, the internal temperature of the slurry rose to 16 °C After the addition was complete, the reaction was maintained at 20-27 °C for an additional 2 5 h After reaction completion, the mixture was cooled to 5 °C and methanol (16 7 g, 521 mmol) was added dropwise over 5-10 mm, maintaining an internal temperature <20 °C (note vigorous H2 evolution) After the exotherm had ceased (ca 10 mm), the temperature was adjusted to 23 °C, and the reaction was mixed until complete dissolution of the solids had occurred Ethyl acetate (300 mL) and 1 M HCl (120 mL) were added, and the phases were partitioned The organic phase was then washed successively with 1 M HCl (2 x 120 mL), H2O (65 mL), and 10% aq NaCl (65 mL) The orgamcs were dried over MgSO4, filtered, and concentrated in vacuo Crystallization of the product occurred during the concentration The slurry was warmed to 50 °C, and heptane (250 inL) was added over 15 min. The slurry was then allowed to mix at 23 °C for 30 min and filtered. The wet cake was washed with 3: 1 heptane:ethyl acetate (75 mL), and the orange, crystalline solids were dried at 45 °C for 24 h to provide the title compound (15.35 g, 99.3% ee, 61% yield), which was contaminated with 11% of the meso isomer (vs. dl isomer).
References
- “VIEKIRA PAK™ (ombitasvir, paritaprevir and ritonavir tablets; dasabuvir tablets), for Oral Use. Full Prescribing Information”(PDF). AbbVie Inc., North Chicago, IL 60064. Retrieved 30 July 2015.
- “FDA approves Viekira Pak to treat hepatitis C”. Food and Drug Administration. December 19, 2014.
- “TECHNIVIE™ (ombitasvir, paritaprevir and ritonavir) Tablets, for Oral Use. Full Prescribing Information” (PDF). AbbVie Inc., North Chicago, IL 60064. Retrieved 28 July 2015.
- “FDA approves Technivie for treatment of chronic hepatitis C genotype 4”. Food and Drug Administration. July 24, 2015.
- Jordan J. Feld, Kris V. Kowdley, Eoin Coakley, Samuel Sigal, David R. Nelson, Darrell Crawford, Ola Weiland, Humberto Aguilar, Junyuan Xiong, Tami Pilot-Matias, Barbara DaSilva-Tillmann, Lois Larsen, Thomas Podsadecki, and Barry Bernstein (2014). “Treatment of HCV with ABT-450/r–Ombitasvir and Dasabuvir with Ribavirin”. N Engl J Med 370: 1594–1603.doi:10.1056/NEJMoa1315722.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
Dimethyl ({(2S,5S)-1-[4-(2-methyl-2-propanyl)phenyl]-2,5-pyrrolidinediyl}bis{4,1-phenylenecarbamoyl(2S)-2,1-pyrrolidinediyl[(2S)-3-methyl-1-oxo-1,2-butanediyl]})biscarbamate
|
|
| Clinical data | |
| Trade names | Viekira Pak (with ombitasvir, paritaprevir, ritonavir and dasabuvir), Technivie (with ombitasvir, paritaprevir, and ritonavir) |
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | not determined |
| Protein binding | ~99.9% |
| Metabolism | amide hydrolysis followed by oxidation |
| Onset of action | ~4 to 5 hours |
| Biological half-life | 21 to 25 hours |
| Excretion | mostly with feces (90.2%) |
| Identifiers | |
| CAS Registry Number | 1258226-87-7 |
| PubChem | CID: 54767916 |
| ChemSpider | 31136214 |
| ChEBI | CHEBI:85183 |
| Synonyms | ABT-267 |
| Chemical data | |
| Formula | C50H67N7O8 |
| Molecular mass | 894.11 g/mol |
rx list
VIEKIRA PAK is ombitasvir, paritaprevir, ritonavir fixed dose combination tablets copackaged with dasabuvir tablets.
Ombitasvir, paritaprevir, ritonavir fixed dose combination tablet includes ahepatitis C virus NS5A inhibitor (ombitasvir), a hepatitis C virus NS3/4Aprotease inhibitor (paritaprevir), and a CYP3A inhibitor (ritonavir) that inhibits CYP3A mediated metabolism of paritaprevir, thereby providing increased plasma concentration of paritaprevir. Dasabuvir is a hepatitis C virus nonnucleoside NS5B palm polymerase inhibitor, which is supplied as separate tablets in the copackage. Both tablets are for oral administration.
Ombitasvir
The chemical name of ombitasvir is Dimethyl ([(2S,5S)-1-(4-tert-butylphenyl) pyrrolidine-2,5diyl]bis{benzene-4,1-diylcarbamoyl(2S)pyrrolidine-2,1-diyl[(2S)-3-methyl-1-oxobutane-1,2diyl]})biscarbamate hydrate. The molecular formula is C50H67N7O8•4.5H2O (hydrate) and the molecular weight for the drug substance is 975.20 (hydrate). The drug substance is white to light yellow to light pink powder, and is practically insoluble in aqueous buffers but is soluble in ethanol. Ombitasvir has the following molecular structure:
![]()
Paritaprevir
The chemical name of paritaprevir is (2R,6S,12Z,13aS,14aR,16aS)-N-(cyclopropylsulfonyl)-6{[(5-methylpyrazin-2-yl)carbonyl]amino}-5,16-dioxo-2-(phenanthridin-6-yloxy)1,2,3,6,7,8,9,10,11,13a,14,15,16,16a-tetradecahydrocyclopropa[e]pyrrolo[1,2-a][1,4] diazacyclopentadecine-14a(5H)-carboxamide dihydrate. The molecular formula is C40H43N7O7S•2H2O (dihydrate) and the molecular weight for the drug substance is 801.91 (dihydrate). The drug substance is white to off-white powder with very low water solubility. Paritaprevir has the following molecular structure:
 |
Ritonavir
The chemical name of ritonavir is [5S-(5R*,8R*,10R*,11R*)]10-Hydroxy-2-methyl-5-(1methyethyl)-1-[2-(1-methylethyl)-4-thiazolyl]-3,6-dioxo-8,11-bis(phenylmethyl)-2,4,7,12tetraazatridecan-13-oic acid,5-thiazolylmethyl ester. The molecular formula is C37H48N6O5S2 and the molecular weight for the drug substance is 720.95. The drug substance is white to off white to light tan powder practically insoluble in water and freely soluble in methanol and ethanol. Ritonavir has the following molecular structure:
![]()
Ombitasvir, Paritaprevir, Ritonavir Fixed-Dose Combination Tablets
Ombitasvir, paritaprevir, and ritonavir film-coated tablets are co-formulated immediate release tablets. The tablet contains copovidone, K value 28,vitamin E polyethylene glycol succinate, propylene glycol monolaurate Type I, sorbitan monolaurate, colloidal silicon dioxide/colloidal anhydrous silica, sodium stearyl fumarate, polyvinyl alcohol, polyethylene glycol 3350/macrogol 3350, talc, titanium dioxide, and iron oxide red. The strength for the tablet is 12.5 mg ombitasvir, 75 mg paritaprevir, 50 mg ritonavir.
Dasabuvir
The chemical name of dasabuvir is Sodium 3-(3-tert-butyl-4-methoxy-5-{6[(methylsulfonyl)amino]naphthalene-2-yl}phenyl)-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-ide hydrate (1:1:1). The molecular formula is C26H26N3O5S•Na•H2O (salt, hydrate) and the molecular weight of the drug substance is 533.57 (salt, hydrate). The drug substance is white to pale yellow to pink powder, slightly soluble in water and very slightly soluble in methanol and isopropyl alcohol. Dasabuvir has the following molecular structure:
 |
Dasabuvir is formulated as a 250 mg film-coated, immediate release tablet containing microcrystalline cellulose (D50-100 um), microcrystalline cellulose (D50-50 um), lactose monohydrate, copovidone, croscarmellose sodium, colloidal silicon dioxide/anhydrous colloidal silica, magnesium stearate, polyvinyl alcohol, titanium dioxide, polyethylene glycol 3350/macrogol 3350, talc, and iron oxide yellow, iron oxide red and iron oxide black. Each tablet contains 270.3 mg dasabuvir sodium monohydrate equivalent to 250 mg dasabuvir.
//////////fda 2014, Ombitasvir, orphan drug, Abbvie Inc.
IPI 926, Saridegib, Patidegib


IPI 926, Saridegib, Patidegib
C29H48N2O3S
Exact Mass: 504.33856
1037210-93-7
-
Patidegib hydrochloride
-
Saridegib hydrochloride
-
-
C29-H48-N2-O3-S.Cl-H
- 541.2361
-
Methanesulfonamide, N-((2S,3R,3’R,3aS,4’aR,6S,6’aR,6’bS,7aR,12’aS,12’bS)-2′,3′,3a,4,4′,4’a,5,5′,6,6′,6’a,6’b,7,7′,7a,8′,10′,12′,12’a,12’b-eicosahydro-3,6,11′,12’b-tetramethylspiro(furo(3,2-b)pyridine-2(3H),9′(1’H)-naphth(2,1-a)azulen)-3′-yl)-, hydrochloride (1:1)
CAS 1169829-40-6 HCL
Saridegib also known as IPI-926 is an experimental drug candidate undergoing clinical trials for the treatment of various types of cancer, including hard to treat hematologic malignancies such as myelofibrosis and ligand-dependant tumors such as chondrosarcoma.[1] IPI-926 exhibits its pharmacological effect by inhibition of the G protein-coupled receptor smoothened, a component of the hedgehog signaling pathway.[2]
Chemically, it is a semi-synthetic derivative of the alkaloid cyclopamine. The process begins with cyclopamine extracted from harvested Veratrum californicum which is taken through a series of alterations resulting in an analogue of the natural product cyclopamine, making IPI-926 the only compound in development/testing that is not fully synthetic.[2]

Saridegib is a member of a class of anti-cancer compounds known as hedgehog inhibitors (Hhi). Most of these compounds affect thehedgehog signaling pathway via inhibition of smoothened (Smo), a key component of the pathway. Depending on when a Hh inhibiting compound is approved by the U.S. Food and Drug Administration (FDA), there may be a perceived need for one to be differentiated over another for marketing purposes, which could lead to different nomenclature (e.g., a Hhi or an agonist of Smo).
This marketing technique is more of a differentiation strategy than a scientific property of these compounds, as the mechanism of action (MOA) in the end is inhibition of the Hh pathway, targeting cancer stem cells. However, as these new compounds are further studied, identification of differences in a compound’s MOA, could lead to hypotheses regarding the stage at which Smo is inhibited, where along the pathway the compound binds, or specific binding properties of a compound.
If these hypotheses are proven, claims could be made regarding a specific compound’s MOA and how it affects efficacy, safety, combinability with other cancer treatments, etc. Scientific data in support of such hypotheses have not been published to date.
SARIDEGIB

N-[(3R,3’R,3’aS,4aR,6’S,6aR,6bS,7’aR,9S,12aS,12bS)-3′,6′,11,12b-tetramethylspiro[1,2,3,4,4a,5,6,6a,6b,7,8,10,12,12a-tetradecahydronaphtho[2,1-a]azulene-9,2′-3a,4,5,6,7,7a-hexahydro-3H-furo[3,2-b]pyridine]-3-yl]methanesulfonamide
There are currently no drugs in the Hhi class FDA approved, however IPI-926 and GDC-0449 are the 2 leading compounds in the class. IPI-926, GDC-0449, and LDE-225 are the only compounds that have generic names passed by the United States Adopted Name (USAN) council (Infinity IPI-926/saridegib, Genentech GDC-0449/vismodegib, and Novartis LDE-225/erismodegib). Although Infinity is further along in chondrosarcoma, myelofibrosis, and AML, Roche/Genentech recently submitted an NDA for GDC-0449 for the treatment of adults with advanced basal cell carcinoma (BCC) when surgery is no longer an option, and the FDA has accepted and has filed the NDA, giving it priority review status. Thus it appears that Roche/Genentech will be the first Hhi to market with GDC-0449, if approved, for the treatment of advanced BCC, with Infinity second to market with IPI-926 for treatment in chondrosarcoma. It appears Infinity will not pursue an indication for BCC and focus on cancers with high unmet needs.[1][3][4][5][6]
Other Hhi-class compounds not as far along in development as IPI-926 and GDC-0449 include:[7]
- Novartis’ LDE-225 (USAN generic name erismodegib)
- Exelixis/Bristol-Myers Squibb’s BMS-833923 (XL139)
- Millennium Pharmaceuticals’s TAK-441
- Pfizer’s PF-04449913

Fig 1. Chemical structure comparison between IPI-926 and cyclopamine
IPI-926 is currently developed by Infinity Pharmaceuticals, Inc. Malignant activation of the Hedgehog pathway is implicated in multiple cancer settings and Infinity’s development strategy is designed to enable IPI-926 to target a broad range of critical oncology targets – from the tumor cell to the cancer microenvironment. This broadly applicable, targeted approach represents an innovative method for fighting cancer and has potential in treating a range of cancers, including pancreatic cancer, small cell lung cancer, ovarian cancer, bladder cancer, medulloblastoma, basal cell carcinoma, and certain hematological malignancies.
The hedgehog pathway inhibitor IPI-926 has been in clinical investigation for basal cell carcinoma, chondrosarcoma, and pancreatic cancer. In the final step of the synthesis of IPI-926 the drug substance (DS) is isolated as the hydrochloride salt of the 2-propanol (2-PrOH) solvate

A design of experiments (DoE) approach was taken to optimize purity and reaction yield of the final debenzylation and hydrochloride salt formation of IPI-926. The study involved a careful dissection of the different process steps to enable an independent investigation of these steps while ensuring that process streams were representative. The results enabled a streamlined process from the final chemical transformation to the salting and isolation and led to the elimination of variability in the process as well as a robust control of impurities. The optimized process was applied to production and demonstrated on the kilogram scale.
A Design of Experiments Approach to a Robust Final Deprotection and Reactive Crystallization of IPI-926, A Novel Hedgehog Pathway Inhibitor
The product was dried at a jacket temperature of 45 °C until an LOD <2.30% (w/w) was achieved. Yield: 11.5 kg (73% from compound 1, correcting for the seed). HPLC purity: 99.9% area (compound 2 content: 0.08% w/w). Assay: 83.7% w/w (as-is), 99.1% w/w (anhydrous, solvent-free). Moisture content: 1.6% w/w. Chlorine content: 5.72% w/w. Residual solvents: acetone (720 ppm); acetonitrile (<41 ppm); 2-MeTHF (none detected); 2-propanol (81 147 ppm); toluene (<90 ppm). Residual metals: palladium (0 ppm); platinum (0 ppm); ruthenium (0 ppm). Additional data for the IPI-926 free base:
1H NMR (400 MHz, CDCl3) 6.90 (br s, 1H), 3.31 (dt, J = 10.6, 3.8 Hz, 1H), 3.20 (br s, 1H), 3.10 (dd, J = 13.7, 4.5 Hz, 1H), 2.91 (s, 3H), 2.62 (dd,J = 9.9, 7.6 Hz, 1H), 2.33 (br d, J = 14.5 Hz, 1H), 2.27–2.15 (m, 1H), 2.10 (dd, J = 14.5, 6.9 Hz, 1H), 1.99–1.17 (m, 28H), 1.05 (q, J = 11.6 Hz, 1H), 0.93 (d, J = 7.4 Hz, 3H), 0.88 (d, J = 6.6 Hz, 3H), 0.86 (s, 3H) ppm.
13C NMR (100 MHz, CDCl3) 140.47, 124.53, 82.48, 76.97, 63.73, 54.08, 53.87, 50.12, 49.98, 47.19, 44.73, 42.27, 42.10, 40.24, 37.55, 37.44, 36.04, 34.44, 31.87, 31.33, 30.46, 29.79, 28.37, 27.94, 26.26, 24.19, 22.70, 18.92, 10.19 ppm;
MS: m/z = 505.29 [M + H]+.
PAPER
Tremblay, M. R.; Lescarbeau, A.; Grogan, M. J.; Tan, E.; Lin, G.; Austad, B. C.; Yu, L.-C.;Behnke, M. L.; Nair, S. J.; Hagel, M.; White, K.; Conley, J.; Manna, J. D.; Alvarez-Diez, T. M.; Hoyt, J.; Woodward, C. N.; Sydor, J. R.; Pink, M.; MacDougall, J.; Campbell, M. J.;Cushing, J.; Ferguson, J.; Curtis, M. S.; McGovern, K.; Read, M. A.; Palombella, V. J.;Adams, J.; Castro, A. C. J. Med. Chem. 2009, 52, 4400– 4418, DOI: 10.1021/jm900305z

Recent evidence suggests that blocking aberrant hedgehog pathway signaling may be a promising therapeutic strategy for the treatment of several types of cancer. Cyclopamine, a plant Veratrum alkaloid, is a natural product antagonist of the hedgehog pathway. In a previous report, a seven-membered D-ring semisynthetic analogue of cyclopamine, IPI-269609 (2), was shown to have greater acid stability and better aqueous solubility compared to cyclopamine. Further modifications of the A-ring system generated three series of analogues with improved potency and/or solubility. Lead compounds from each series were characterized in vitro and evaluated in vivo for biological activity and pharmacokinetic properties. These studies led to the discovery of IPI-926 (compound 28), a novel semisynthetic cyclopamine analogue with substantially improved pharmaceutical properties and potency and a favorable pharmacokinetic profile relative to cyclopamine and compound2. As a result, complete tumor regression was observed in a Hh-dependent medulloblastoma allograft model after daily oral administration of 40 mg/kg of compound 28.
28 (4.06 g, 8.05 mmol, 95% for two steps). NMR δH (400 MHz, CDCl3) 6.90 (br s, 1H), 3.31 (dt, J = 10.6, 3.8 Hz, 1H), 3.20 (br s, 1H), 3.10 (dd, J = 13.7, 4.5 Hz, 1H), 2.91 (s, 3H), 2.62 (dd, J = 9.9, 7.6 Hz, 1H), 2.33 (br d, J = 14.5 Hz, 1H), 2.27−2.15 (m, 1H), 2.10 (dd, J = 14.5, 6.9 Hz, 1H), 1.99−1.17 (m, 28H), 1.05 (q, J = 11.6 Hz, 1H), 0.93 (d, J = 7.4 Hz, 3H), 0.88 (d, J = 6.6 Hz, 3H), 0.86 (s, 3H); NMR δC (100 MHz, CDCl3) 140.47, 124.53, 82.48, 76.97, 63.73, 54.08, 53.87, 50.12, 49.98, 47.19, 44.73, 42.27, 42.10, 40.24, 37.55, 37.44, 36.04, 34.44, 31.87, 31.33, 30.46, 29.79, 28.37, 27.94, 26.26, 24.19, 22.70, 18.92, 10.19; m/z = 505.29 [M + H]+; HPLC 99.1 a/a % at 215 nm.

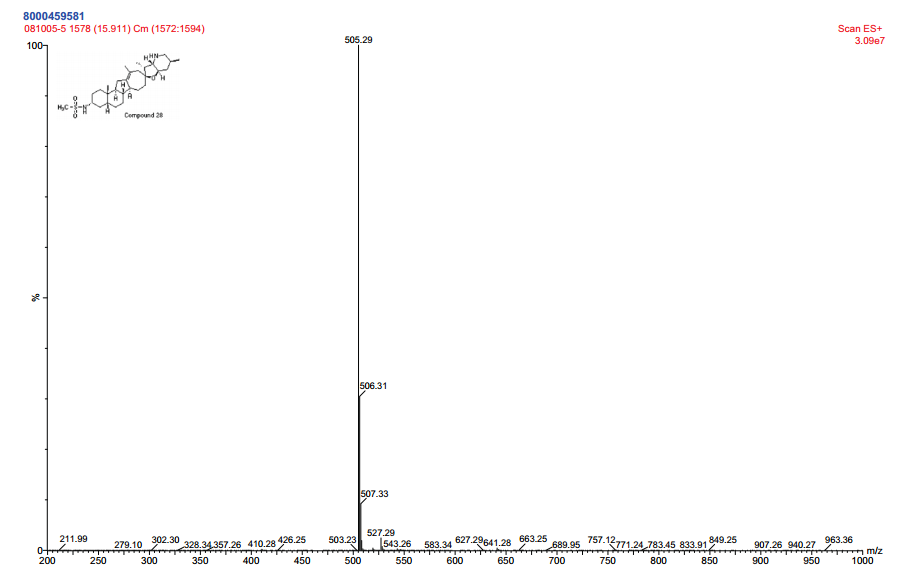

Click on images for clear view……………..
Paper
A design of experiments (DoE) approach was taken to optimize purity and reaction yield of the final debenzylation and hydrochloride salt formation of IPI-926. The study involved a careful dissection of the different process steps to enable an independent investigation of these steps while ensuring that process streams were representative. The results enabled a streamlined process from the final chemical transformation to the salting and isolation and led to the elimination of variability in the process as well as a robust control of impurities. The optimized process was applied to production and demonstrated on the kilogram scale.
Development of a Multi Kilogram-Scale, Tandem Cyclopropanation Ring-Expansion Reaction en Route to Hedgehog Antagonist IPI-926

The formation of the d-homocyclopamine ring system in IPI-926 is the key step in its semisynthesis and proceeds via a chemoselective cyclopropanation followed by a stereoselective acid-catalyzed carbocation rearrangement. In order to perform large-scale cyclopropanation reactions, we developed new iodomethylzinc bis(aryl)phosphate reagents that were found to be both effective and safe. These soluble reagents can be prepared under mild conditions and are stable during the course of the reaction. Importantly, they have favorable energetics relative to other cyclopropanating agents such as EtZnCH2I. Herein, we describe the process optimization studies that led to successful large-scale production of the d-homocyclopamine core necessary for IPI-926.
References
- “Pipeline: IPI-926”. Infinity Pharmaceuticals.
- Tremblay, MR; Lescarbeau, A; Grogan, MJ; Tan, E; Lin, G; Austad, BC; Yu, LC; Behnke, ML et al. (2009). “Discovery of a potent and orally active hedgehog pathway antagonist (IPI-926)”. Journal of Medical Chemistry 52 (14): 4400–18. doi:10.1021/jm900305z. PMID 19522463.
- “Pipeline”. Infinity Pharmaceuticals.
- “Genentech Pipeline”. Genentech.
- “USAN Stem List” (PDF). AMA.
- “Names under consideration”. AMA.
- “Search results for Hh clinical trials”. United National Institute of Health’s ClinicalTrials.gov.
- 1. Tremblay MR, Lescarbeau A, Grogan MJ, Tan E, Lin G, Austad BC, Yu LC, Behnke ML, Nair SJ, Hagel M et al.. (2009)
Discovery of a potent and orally active hedgehog pathway antagonist (IPI-926).
J. Med. Chem., 52 (14): 4400-18.
 |
|
 |
|
| Names | |
|---|---|
| IUPAC name
N-((2S,3R,3aS,3′R,4a′R,6S,6a′R,6b′S,7aR,12a&prmie;S,12b′S)-3,6,11′,12b′-tetramethyl-2′,3a,3′,4,4′,4a′,5,5&prmie;,6,6′,6a′,6b′,7,7a,7′,8′,10′,12′,12a′,12b′-icosahydro-1′H,3H-spiro[furo[3,2-b]pyridine-2,9′-naphtho[2,1-a]azulen]-3′-yl)methanesulfonamide
|
|
| Other names
saridegib
|
|
| Identifiers | |
| 1037210-93-7 |
|
| ChEMBL | ChEMBL538867 |
| ChemSpider | 26353073 |
| 8198 | |
| Jmol-3D images | Image |
| PubChem | 25027363 |
| UNII | JT96FPU35X |
| Properties | |
| C29H48N2O3S | |
| Molar mass | 504.77 g·mol−1 |
| Pharmacology | |
| Legal status |
|
/////Saridegib, IPI-926
EU: New GMP Implementing Act published
DRUG REGULATORY AFFAIRS INTERNATIONAL

The EU Commission has published a new public consultation on an Implementing Act on GMP principles and guidelines for medicinal products for human use.
The EU Commission has published a new public consultation on an Implementing Act on Principles and guidelines on good manufacturing practices for medicinal products for human use.
The reason is that once Regulation (EU) No 536/2014 on clinical trials becomes applicable, manufacture and import of Investigational Medicinal Products (IMPs) for the use in clinical trials carried out under that Regulation cannot follow GMP for IMPs set out in Directive 2003/94/EC. They then have to be manufactured or imported under regulations laid down by the Delegated Act or other specified regulation. It is therefore necessary that Directive 2003/94/EC is revised by a new Implementing Directive on principles and guidelines of good manufacturing practice for medicinal products for human use (without IMPs).
The EU Commission…
View original post 60 more words
WO 2015129603, NEW PATENT, Daiichi Sankyo Co Ltd, Edoxaban

HIGH-PURITY CRYSTALS OF ACTIVE BLOOD COAGULATION FACTOR X (FXA) INHIBITOR
DAIICHI SANKYO COMPANY,LIMITED [JP/JP]; 3-5-1,Nihonbashi Honcho,Chuo-ku, Tokyo 1038426 (JP)
Claims highly pure crystalline form of edoxaban p-toluenesulfonate monohydrate. Useful for treating thrombotic diseases. Daiichi Sankyo had developed and launched edoxaban for treating non-valvular atrial fibrillation, deep vein thrombosis and pulmonary embolism, the drug was recently launched in US (in February 2015) and approved in Europe (in June 2015).
The present invention addresses the problem of providing high-purity crystals of a compound which is represented by formula (1a) and is an active blood coagulation factor X (FXa) inhibitor. High-purity crystals of a compound represented by formula (1a) which: are characterised by being obtained by a step for dissolving crystals in a solvent and thereafter performing recrystallisation; have a 0.03% or less maximum content of one impurity as the impurity content by percentage; and have a 0.13% or less total impurity content.
In N represented 1 – (5-Chloro-2-yl) -N 2 – ((1S, 2R, 4S) -4 – [(dimethylamino) carbonyl] -2 – {[(5-methyl-4 , 5,6,7-tetrahydro thiazolone [5,4-c] pyridin-2-yl) carbonyl] amino} cyclohexyl) Etanjiamido p- toluenesulfonic acid monohydrate [hereinafter, may be referred to as compound (1a) is there
The obtained compound, in analysis using HPLC, as impurities, a peak of more impurities (both 0.03 wt%) is confirmed, the total of the impurities was 0.16 wt.% Since, its purity was 99.84% (Note that the content of% refers to% of the HPLC area value of the free form of formula (1a) compound).1 H-NMR (DMSO-d6) delta: 1.45-1.54 (1H, M), 1.66-1.78 (3H, M), 2.03-2.10 (2H, M), 2.28 (3H, s), 2.79 (3H, s), 2.91-3.02 (1H, m), 2.93 (3H, s), 2.99 (3H, s), 3 .13-3.24 (2H, m), 3.46-3.82 (2H, m), 3.98-4.04 (1H, m), 4.43-4.80 (3H, m) , 7.11 (2H, d, J = 7.8Hz), 7.46 (2H, d, J = 8.2Hz), 8.01 (2H, d, J = 1.8Hz), 8.46 ( 1H, t, J = 1.8Hz), 8.75 (1H, d, J = 6.9Hz), 9.10-9.28 (1H, br.s), 10.18 (1H, br.s ), 10.29 (1H, s).
Found: C; 50.25%, H; 5.36%, N; 13.32%

/////////////WO 2015129603, NEW PATENT, Daiichi Sankyo Co Ltd, Edoxaban
deleted
deleted
Chi-Med Says Fruquintinib Successful in Lung Cancer Trial

Fruquintinib
Phase 3…cancer
Hutchison Medipharma Enterprises Limited
Hutchison MediPharma for the treatment of locally advanced or metastatic colorectal cancer
C21H19N3O5
Exact Mass: 393.1325
cas 1194506-26-7, 6 ((6,7-dimethoxyquinazolin-4-yl) oxy) – N, 2-dimethylbenzofuran-3-carboxamide,
3-Benzofurancarboxamide, 6-[(6,7-dimethoxy-4-quinazolinyl)oxy]-N,2-dimethyl-
Synonym: Fruquintinib; HMPL-013; HMPL 013; HMPL013.
HPLC.http://www.medkoo.com/Product-Data/Fruquintinib/QC-Fruquintinib-CRB50706web.pdf
Fruquintinib, also known as HMPL-013, is an orally available, small molecule inhibitor of vascular endothelial growth factor receptors (VEGFRs), with potential anti-angiogenic and antineoplastic activities.
HMPL-013, a novel small molecule compound that selectively inhibits vascular endothelial growth factor receptor (VEGFR), is in phase III clinical studies at Hutchison MediPharma for the treatment of locally advanced or metastatic colorectal cancer. Phase II clinical trials are also ongoing for the treatment of non-squamous non-small cell lung cancer.
Early clinical development is under way at the company for the treatment of gastric cancer in combination with paclitaxel.
Fruquintinib’s mechanism of action is the inhibition of all three forms of VEGF receptors (VEGFR-1, 2, 3). Competitive advantages over currently marketed therapies are the compound’s unique kinase profile, a highly potent efficacy and excellent kinase selectivity, large safety margin, a broad spectrum antitumor activity and a low cost of goods.
Upon oral administration, fruquintinib inhibits VEGF-induced phosphorylation of VEGFRs 1, 2, and 3 which may result in the inhibition of migration, proliferation and survival of endothelial cells, microvessel formation, the inhibition of tumor cell proliferation, and tumor cell death. Expression of VEGFRs may be upregulated in a variety of tumor cell types.
In 2013, the company entered into a licensing, co-development, and commercialization agreement in China with Eli Lilly.
Angiogenesis is a physiological process of growing new blood vessels from pre-existing vessels. It takes place in a healthy subject to heal wounds, i.e., restoring blood flow to tissues after injury or insult.
Excessive angiogenesis may be triggered by certain pathological conditions such as cancer, age-related macular degeneration, and chronic inflammatory disease. As a result, new blood vessels feed diseased tissues and destroy normal tissues. In cancer, new blood vessels also allow tumor cells to escape into the circulation and lodge in other organs.
Vascular endothelial growth factor (VEGF), a homodimeric glycoprotein, and its receptors, e.g., kinase insert domain receptor (KDR), constitute an important angiogenic pathway. Studies have shown that inhibition of KDR resulted in endothelial cell apoptosis and, thus, suppression of angiogenesis. See Rubin M. Tuder, Chest, 2000; 117: 281. KDR inhibitors are therefore potential candidates for treating an angiogenesis-related disorder.
Chi-Med Says Fruquintinib Successful in Lung Cancer Trial
Written by Richard Daverman, PhD, Executive Editor, Greg B. Scott.
Hutchison MediPharma, a division of Chi-Med reported that fruquintinib met its primary endpoint in a second proof-of-concept China trial, this time as a treatment for advanced non-squamous non-small cell lung cancer. The company said fruquintinib “clearly” met its primary endpoint of progression-free survival, though specific data are being held for a scientific meeting. In 2013, Hutchison out-licensed China rights for the drug to Lilly. In May, the first proof-of-concept trial triggered two payments from Lilly to HMP totaling $18 million. More details…. http://www.chinabiotoday.com/articles/20150904
………….
Patent
US 20090281130
https://www.google.com.ar/patents/US20090281130
EXAMPLE 1 Synthesis of 6-(6,7-dimethoxyquinazolin-4-yloxy)-N,2-dimethylbenzofuran-3-carboxamide:

To a solution of 4-chloro-6,7-dimethoxyquinazoline (1 equiv.) in 2 ml CH3CN were added 6-hydroxy-N,2-dimethylbenzofuran-3-carboxamide (1 equiv.) and K2CO3 (1.5 equiv.). The mixture was refluxed under stirring for 10 hr. After the solvent was evaporated, the residue was washed with water, dried over MgSO4, filtered, concentrated, and purified by column chromatography to give the title compound in a yield of 85%.
1H NMR (DMSO-d6, 400 MHz) δ: 2.49 (s, 3H), 2.81 (d, J=8.4 Hz, 3H,10), 3.97 (s, 3H), 3.98 (s, 3H), 7.24 (dd, J=2.0, 8.4 Hz, 1H), 7.38 (s, 1H), 7.58 (s, 1H), 7.61 (d, J=2.0 Hz, 1H), 7.79 (d, J=8.4 Hz, 1H), 7.96 (m, 1H), 8.52 (s, 1H).
MS(m/e): 394.1 (M+1).
………………
WO 2009137797
https://www.google.com/patents/WO2009137797A2
……………….
CN 101575333
Example a: 6- (6,7-dimethoxy-quinazolin-4-oxo) -N, 2- dimethyl-benzofuran-3-carboxamide
[0048]

[0049] 4-Chloro-6,7-dimethoxy-quinazoline (1 mmol) was dissolved in 2 ml of acetonitrile, followed by addition of 6-hydroxy -N, 2- dimethyl-benzofuran-3- amide (1 mmol) and potassium carbonate (1.5 mmol). The reaction mixture was heated at reflux for 10 hours, concentrated to dryness, washed with water, and purified to give the desired product, yield 85%.
[0050] 1H NMR (DMS0-d6,400MHz) δ ppm:. 2 49 (s, 3H); 2.81 (d, J = 8. 4Hz; 3H, 10); 3.97 (s; 3H); 3.98 (s, 3H);. 7 24 (dd, J = 2. 0,8 4Hz;. 1H);. 7 38 (s, lH);. 7 58 (s, lH); 7.61 (d, J = 2. OHz; 1H);. 7 79 (d, J = 8. 4Hz; 1H);. 7 96 (m, 1H);. 8 52 (s, 1H).
[0051] MS (m / e)::. 394 1 (M + l).
………..
| EP1265874A2 * | Jan 23, 2001 | Dec 18, 2002 | Gödecke Gmbh | Method for the simplified production of (3-chloro-4-fluoro-phenyl)- 7-(3-morpholino-4-yl-propoxy)-6-nitro-quinazoline-4-yl]-amine or (3-chloro-4-fluoro-phenyl)- 7-(3-morpholino-4-yl-propoxy)-6-amino-quinazoline-4-yl]-amine |
| US20070208056 * | Jan 23, 2007 | Sep 6, 2007 | Bristol-Myers Squibb Company | Piperidinyl derivatives as modulators of chemokine receptor activity |
| US20080033000 * | May 15, 2007 | Feb 7, 2008 | Senex Biotechnology, Inc. | Identification of CDKI pathway inhibitors |
| 2 | See also references of EP2297115A2 | |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8212033 * | Sep 29, 2010 | Jul 3, 2012 | Hutchison Medipharma Enterprises Limited | Use of substituted quinazoline compounds in treating angiogenesis-related diseases |
| US8497372 | Jun 4, 2012 | Jul 30, 2013 | Hutchison Medipharma Enterprises Limited | Use of substituted quinazoline compounds in treating age-related macular degeneration |
| US8575184 | Sep 1, 2010 | Nov 5, 2013 | Bristol-Myers Squibb Company | Quinazolines as potassium ion channel inhibitors |
Hutchison Medipharma Enterprises Limited

Simon To, M.B.A.
Chairman

Mr To has been a Director since 2000 and an Executive Director and Chairman since 2006. He is also Chairman of the Remuneration Committee and a member of the Technical Committee of the Company. He is managing director of Hutchison Whampoa (China) Limited (“Hutchison China”) and has been with Hutchison China for over thirty years, building its business from a small trading company to a billion dollar investment group. He has negotiated major transactions with multinationals such as Procter & Gamble, Lockheed, Pirelli, Beiersdorf, United Airlines and British Airways.
Mr To’s career in China spans more than thirty years and he is well known to many of the top Government leaders in China. Mr To is the original founder of Hutchison Whampoa Limited’s healthcare business and has been instrumental in the acquisitions made to date. He received a First Class Honours Bachelor’s Degree in Mechanical Engineering from Imperial College, London and an MBA from Stanford University’s Graduate School of Business.
Christian Hogg, M.B.A.
Chief Executive Officer, Hutchison China MediTech Limited and Director, Hutchison MediPharma Holdings Limited

Mr Hogg has been an Executive Director and Chief Executive Officer since 2006. He is also a member of the Technical Committee of the Company. He joined Hutchison Whampoa (China) Limited in 2000 and has since led all aspects of the creation, implementation and management of the Company’s strategy, business and listing. This includes the creation of the Company’s start-up businesses and the acquisition and operational integration of assets that led to the formation of the Company’s China joint ventures.
Prior to joining Hutchison China, Mr Hogg spent ten years with Procter & Gamble starting in the US in Finance and then Brand Management in the Laundry and Cleaning Products Division. Mr Hogg then moved to China to manage P&G’s detergent business followed by a move to Brussels to run P&G’s global bleach business. Mr Hogg received a Bachelor’s degree in Civil Engineering from the University of Edinburgh and an MBA from the University of Tennessee.
Weiguo Su, Ph.D.
Executive Vice President and Chief Scientific Officer

Dr. Su has headed all drug discovery and research since he joined, including creating our R&D strategy, the formation and growth of research platform, and the research and discovery of each and every small molecule drug candidate in the Company’s portfolio.
Prior to joining in 2005, Dr. Su spent 15 years with Pfizer’s US R&D organization. Dr. Su delivered several high quality new drug candidates during his time with Pfizer, most recently as a director in the Medicinal Chemistry Department.
He received his Ph.D. and post-doctoral fellowship in Chemistry from Harvard University under the guidance of Nobel Laureate Professor E. J. Corey, and his Bachelor’s degree in Chemistry from Fudan University in Shanghai, China.
| R & D Center Address (A): Building 4, 720 Cailun Road Zhangjiang Hi-Tech Park Pudong, Shanghai, China Postal Code: 201203, China |
Head Office Address (B): Building 4, 917 Halei Road Zhangjiang Hi-Tech Park Pudong, Shanghai, China Postal Code: 201203, China |
Tel: +86 21 2067 3000 Email: BD@hmplglobal.com |
Addresses in Chinese:
R & D Center ( A): Chinese Cai Lun Road, Zhangjiang Hi-Tech Park in Pudong New Area, Shanghai, Lane 720 (intermediate哈雷路爱迪way out), Building 4
Head Office (B): Harley Road, Zhangjiang Hi-Tech Park, Pudong New Area, China, Shanghai, Lane 917, Building 4

![]()

///////
Indian pharma’s struggle to tighten standards paves way for M&A deals

MUMBAI – India’s smaller generic drugmakers, struggling to cope with a bruised reputation and tougher regulation in the United States, are under pressure to consider branching out to new, less-profitable markets or sell out to larger rivals.
Two years after its most high-profile regulatory setback to date in the United States – Ranbaxy’s $500 million U.S. fine for drug safety violations – India’s $15 billion a year generic drug industry is still rebuilding its image in its biggest market.
Many of its top firms are facing sanctions at some of their factories, as the U.S. Food and Drug Administration (FDA) tightens checks and its approvals process.
Combined with government-mandated price controls on drugs at home, that is piling pressure on smaller players.
“If they want to have a presence globally, they have to make investments. If they can’t, then they’ll have to focus on other markets or scale back their ambition outside of India, and that’s probably what will happen,” said Subhanu Saxena, CEO of Cipla , India’s fourth-largest drugmaker by revenue.
Ashok Anand, president of Hikal Ltd , a Mumbai-based drugmaker with a market value of $167 million, said some peers were putting themselves on the block.
“If they cannot deal with the stricter regulations, they might just prefer to sell out,” he said.
Pressure on U.S. sales has been felt across the Indian industry, with all drugmakers hit by delays in FDA approvals as the U.S. safety body overhauls its review process. Growth in U.S. revenue for drugmakers slowed to 14 percent in the year to March 2015, less than half what it was in the year to March 2012, according to brokerage Edelweiss.

But for larger players who want to plug gaps or, for the likes of Glenmark and Aurobindo who aim to grow in the United States, this pressure has lowered prices and could pave the way for attractive deals, bankers said.
“Now that some of the smaller companies are reeling under intensive regulatory scrutiny and want to cash out on their investments, valuations would be much more realistic,” said the head of India M&A at a large European bank in Mumbai.
SPENDING SPREE
Indian manufacturers say they have spent millions in high-end testing equipment, improved training and have hired larger teams in quality control since Ranbaxy was fined for manipulating clinical data.
Some consultants estimate spending on compliance has more than doubled to reach about 6 to 7 percent of sales for the larger companies.
But while the number of U.S. export bans issued to Indian companies fell to eight in 2014 from 21 in 2013, according to FDA data, the agency continues to find manufacturing violations at the plants of some of the biggest drugmakers in the country, an indication of the pervasiveness of the problem.
Sun Pharmaceutical Industries , Wockhardt , Dr Reddy’s Laboratories and Cadila Healthcarehave all faced FDA rebukes over the past year.
Smaller firms Ipca and Aarti Drugs faced FDA bans on their plants this year.
These failures – which executives blame on India’s “quick fix” culture and consultants blame on a failure to prioritize compliance – have clouded short-term growth prospects and added to pressure on smaller players, pushing some to look elsewhere.
“They can choose to be in lesser-regulated markets, such as Latin America, where there is a lot of demand. But they will have to live with much thinner margins,” said the finance director of a small Indian drugmaker, who did not want to be named. “It’s survival of the fittest.” REUTERS
http://m.todayonline.com/business/indian-pharmas-struggle-tighten-standards-paves-way-ma-deals
///////
China Generic Drugmakers Poaching Indian Execs

China Generic Drugmakers Poaching Indian Execs
Written by Richard Daverman, PhD, Executive Editor, Greg B. Scott.
In the competition between China and India pharmas, China’s generic drug industry leads in the supply of APIs to global drugmakers, but India supplies more finished generic drugs to the world’s marketplace. That may be changing. According to press reports,
China drugmakers have begun hiring experienced Indian pharma execs, offering them two to three times their present salaries.
The China companies are willing to pay at these levels because the Indian professionals have two skills the Chinese want: drug formulation experience and English.
China’s drugmakers want help as they target the western world’s lucrative generic drug market.
More details…. http://www.chinabiotoday.com/articles/20150903

///////////
