
Home » Articles posted by DR ANTHONY MELVIN CRASTO Ph.D (Page 188)
Author Archives: DR ANTHONY MELVIN CRASTO Ph.D
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
SILICO LINEZOLID, SILINEZOLID, NDS 10024

Therapeutic options for brain infections caused by pathogens with a reduced sensitivity to drugs are limited. Recent reports on the potential use of linezolid in treating brain infections prompted us to design novel compounds around this scaffold. Herein, we describe the design and synthesis of various oxazolidinone antibiotics with the incorporation of silicon.
Our findings in preclinical species suggest that silicon incorporation is highly useful in improving brain exposures. Interestingly, three compounds from this series demonstrated up to a 30-fold higher brain/plasma ratio when compared to linezolid thereby indicating their therapeutic potential in brain associated disorders
Design, Synthesis, and Identification of Silicon Incorporated Oxazolidinone Antibiotics with Improved Brain Exposure


Examples from patent

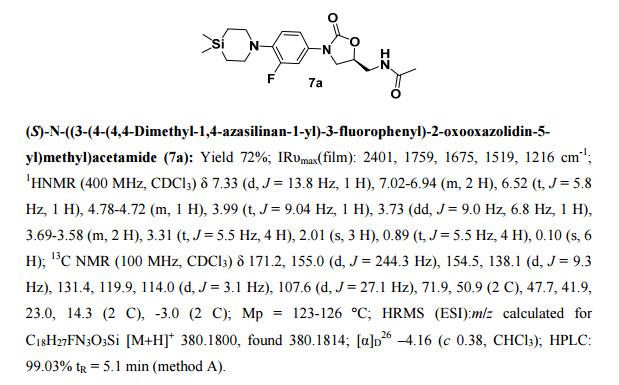
- (S)—N((3-(4-(4,4-dimethyl-1,4-azasilinan-1-yl)-3-fluorophenyl)-2 oxooxazolidin-5-yl)methyl)acetamide
- NDS 10024
- Preparation of (S)—N((3-(4-(4,4-dimethyl-1,4-azasilinan-1-yl)-3-fluorophenyl)-2 oxooxazolidin-5-yl)methyl)acetamide (12)
-
-
To a solution of 8 (50 mg, 0.135 mmol) in dimethylformamide (DMF), lithium-t-butoxide (LiOtBu) (32.3 mg, 0.4 mmol) is added. The mixture is stirred at 25° C. for 15 min, followed by the addition of MeOH (0.01 mL, 0.27 mmol). 6 (52 mg, 0.27 mmol) is then added and the reaction mixture is allowed to stir at 25° C. for 24 h. Glacial acetic acid is then added and the organic phase is extracted with EtOAc and washed with brine solution. The crude material is purified by column chromatography on silica gel using hexane-EtOAC mixtures to furnish the pure product 12. The analogous procedure for the corresponding morpholine analogue was adapted from Lu, C. V.; Chen, J. J.; Perrault, W. R.; Conway, B. G.; Maloney, M. T.; Wang, Y. Org. Pro. Res. and Development. 2006, 10, 272-277.
-
1H NMR (200 MHz, CDCl3): δ 7.33 (d, J=13.8 Hz, 1H), 7.02-6.94 (m, 2H), 6.52 (t, J=5.8 Hz, 1H), 4.77-4.73 (m, 1H), 3.99 (t, J=9.04 Hz, 1H), 3.72 (dd, J=9.0 Hz, 6.8 Hz, 1H), 3.69-3.58 (m, 2H), 3.31 (t, J=5.5 Hz, 4H), 2.01 (s, 3H), 0.89 (t, J=5.5 Hz, 4H), 0.10 (s, 6H). 13C NMR (100 MHz, CDCl3): δ171.2, 155.0 (d, J=244.3 Hz), 154.5, 138.2 (d, J=9.3 Hz), 131.5, 119.9, 114.0 (d, J=3.4 Hz), 107.6 (d, J=27.1 Hz), 71.9, 50.9, 47.7, 41.9, 23.0, 14.3, −2.9.
- Preparation of Bis(bromomethyl)dimethylsilane (2) (as per scheme 2)
-

-
HBr gas is bubbled to a solution of dimethyl divinylsilane 1 (10.0 g, 89.28 mmols), and dibenzoylperoxide (DBP, 100 mg), in heptane (100 mL) at 0° C. for 30 min. The Reaction mixture (RM) is allowed to stir at room temperature (25° C.) for 18 h, water (200 mL) is added to the reaction mixture and the organic layer is separated. The heptane layer is washed with 2N NaOH (2 100 mL), dried and concentrated to obtain the product 2 as a colourless liquid (24.5 g) in 100% yield.
-
1H NMR (200 MHz, CDCl3): δ 3.58-3.49 (m, 4H), 1.45-1.40 (m, 4H), 0.09 (s, 6H).
-

-
Benzylamine (20 mL, 182 mmol) and Et3N (15.2 mL, 109 mmol) are added to a solution of bis-(bromomethyl) dimethylsilane 2 (10 g, 36.5 mmol) in chloroform (100 mL). The mixture is then refluxed for 16 h. 5% sodiumhydroxide solution (150 mL) is then added and the aqueous layer is extracted with dichloromethane (DCM, 2×100 mL). It is then washed with brine (200 mL), dried and concentrated. The product is purified by column chromatography on silica gel using hexane-EtOAc mixtures to obtain the product 3 as a light yellow liquid (4.3 g) in 54% yield.
-
1H NMR (200 MHz, CDCl3): δ 7.23-7.35 (m, 5H), 3.66 (s, 2H), 2.68 (t, J=6.3 Hz, 4H), 0.75 (t, J=6.3 Hz, 4H), 0.04 (s, 6H).
- Preparation of 1-benzyl-4,4-dimethyl-1,4-azasilinane (3)
Preparation of 4,4-dimethyl-1,4-azasilinane hydrochloride (4)
-

-
To a solution of 4,4-dimethyl-1,4-azasilinane 3 (2.3 g, 10.5 mmol) in EtOH (20 mL), 6N hydrochloricacid (1.75 mL, 10.5 mmol) is added and the solvent is removed under reduced pressure. The reaction mixture is co-evaporated with EtOH (2×10 mL) and recrystallized from EtOH-diethyl ether. To a slurry of Pd/C (50 mg) in EtOH (15 mL) an ethanolic solution of above prepared HCl salt is added drop wise and stirred at 25° C. under hydrogen atmosphere for 20 h. The reaction mixture is filtered through celite and washed with 2×20 mL of MeOH. The filtrate is then concentrated under reduced pressure to give viscous oil which was triturated with diethyl ether to obtain the product 4 as a white solid (950 mg) in 70% yield.
Preparation of 1-(2-fluoro-4-nitrophenyl)-4,4-dimethyl-1,4-azasilinane (9)
-

-
To a solution of 4,4-dimethyl-1,4-azasilinane hydrochloride 4 (500 mg, 3.85 mmol) in EtOAc (15 mL), triethylamine (1.3 mL, 9.63 mmol) is added and stirred at 25° C. for 10 min. The reaction mixture is cooled to 0° C. and 3,4-difluoronitrobenzene (612 mg, 3.85 mmol) is added drop wise and allowed to stir at 25° C. for 6 h. Water is then added and the organic layer is separated. The aqueous layer is extracted with EtOAc (2×10 mL) and the solvent is removed under reduced pressure. The product is purified by column chromatography using hexane-EtOAc mixtures and a crystalline yellow solid 9 (721 mg) is obtained in 70% yield.
-
1H NMR (200 MHz, CDCl3): δ 7.93-7.84 (m, 2H), 6.86 (t, J=4 Hz, 1H), 3.70-3.67 (m, 4H), 0.91-0.85 (m, 4H), 0.12 (s, 6H). 13C NMR (50 MHz, CDCl3): δ 151.1 (d, J=246.71 Hz), 144.4 (d, J=7.13 Hz), 137.8 (d, J=8.59 Hz), 121.4, 115.9 (d, J=4.61 Hz), 113.2 (J=27.78 Hz), 49.4, 13.8, −2.8. IR (CHCl3): ν 2948, 2894, 1603, 1523, 1492, 1400, 1342, 1223, 983, 832, 742 cm−1′. M.P: 70-72° C.
Preparation of benzyl 4-(4,4-dimethyl-1,4-azasilinan-1-yl)-3-fluorophenylcarbamate (10)
-

-
To a solution of compound 9 (610 mg, 2.28 mmol) in THF (25 mL), Pd/C (30 mg) is added and hydrogenated under a pressure of 35 psi in a par hydrogenator for 8 h. The reaction mixture is filtered through celite. Celite pad is washed with THF (2×20 mL). To the filtrate, saturated NaHCO3 (420 mg, 5.01 mmol) and CBzCl (427 mg, 2.5 mmol) are added at 0° C. and stirred at 25° C. for 5 h. 10 mL water is added to reaction mixture and the aqueous layer is extracted with EtOAc (2×20 mL). The crude mixture is then subjected to column chromatography on silica gel using hexane-EtOAc mixtures to afford the product as a viscous liquid 10 (690 mg) in 82% yield.
-
1H NMR (200 MHz, CDCl3): δ 7.41-7.37 (m, 5H), 6.94-6.93 (m, 2H), 6.68 (s, 1H), 5.21 (s, 1H), 3.3 (t, J=6.38 Hz, 4H), 0.93 (t, J=6.08 Hz, 4H), −0.13 (s, 6H). 13C NMR (50 MHz, CDCl3): 155.4 (d, 244.4 Hz), 153.6, 136.1, 135.9, 128.6, 128.5, 128.3, 120.4, 117.2 (d, 18.7 Hz), 114.7, 108.3 (20.5 Hz), 67.1, 51.4, 14.4, −3.0. IR (CHCl3): ν 3317, 2953, 2803, 1706, 1594, 1521, 1271, 1221, 1058, 869, 759 cm−1. M.P: 80-82° C.
Preparation of (S)-5-(aminomethyl)-3-(4-(4,4-dimethyl-1,4-azasilinan-1-yl)-3-fluorophenyl)oxazolidin-2-one (11) (NDS-10057)
-

-
To a solution of 10 (1.20 g, 3.23 mmol) and (S)-tert-butyl 3-chloro-2-hydroxypropylcarbamate (1.35 g, 6.47 mmol) in DMF (10 mL), LiOtBu (1.03 g, 12.94 mmol) is added at 0° C. The mixture is stirred at 25° C. for 45 h. The starting material 10 is not consumed completely. Saturated NH4Cl is then added; the organic phase is extracted with EtOAc (2×20 mL), washed with brine solution, dried and concentrated. The crude residue is dissolved in 20 mL of DCM-TFA mixture (8:2) and stirred at 25° C. for 3 h. RM is concentrated and dissolved in water (10 mL), the aqueous layer is washed with diethyl ether (2×50 mL), basified with saturated NaHCO3 and extracted with DCM (2×50 mL). The DCM layer is dried and concentrated. The crude is purified by column chromatography on silica gel using hexane-EtOAc mixtures to obtain the product as an off-white solid (500 mg) in 45% (based on recovery of starting material) over 2 steps.
-
1H NMR (400 MHz, CDCl3): δ 7.36 (dd, J=14.2 Hz, 2.3 Hz, 1H), 7.09 (dd, J=8.8 Hz, 1.7 Hz, 1H), 6.96 (t, J=9.5 Hz, 1H), 4.72-4.59 (m, 1H), 4.00 (t, J=8.3 Hz, 1H), 3.79 (dd, J=8.7 Hz, 6.8 Hz, 1H), 3.30 (t, J=6.2 Hz, 4H), 3.03 (dq, J=13.6 Hz, 4.2 Hz, 2H), 0.90 (t, J=6.2 Hz, 4H), 0.10 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 155.1 (d, J=244.3 Hz), 154.7, 137.9 (d, J=9.0 Hz), 132.1 (d, J=10.3 Hz), 112.0 (d, J=4.3 Hz), 113.8 (d, J=3.2 Hz), 107.4 (d, J=26.9 Hz), 73.8, 51.0, 47.8, 45.01, 14.4, −2.9. IR (CHCl3): ν 3685, 3021, 2955, 2809, 2401, 1747, 1515, 1416, 1219, 1029, 991, 870, 771, 667 cm−1. M.P: 94-96° C. ESI-MS: 360.11 (M+Na).
Preparation of (S)—N-((3-(4-(4,4-dimethyl-1,4-azasilinan-1-yl)-3-fluorophenyl)-2-oxooxazolidin-5-yl)methy)acetamide (12) (NDS 10024)
-

-
To solution of amine 11 (300 mg, 0.9 mmol) and DIPEA (0.3 mL, 1.78 mmol) in dry THF (4.0 mL), acetylchloride (0.08 mL, 1.07 mmol) is added at 0° C., and stirred at 25° C. for 3 h. Further, saturated NaHCO3 (5.0 mL) is added to the reaction mixture and extracted with EtOAc (2×5 mL). The organic layer is washed with brine, dried and concentrated. The product is purified by column chromatography on silica gel using hexane-EtOAc mixtures to obtain the product as an off-white solid (170 mg) in 50% yield.
-
1HNMR (400 MHz, CDCl3): δ 7.33 (d, J=13.8 Hz, 1H), 7.02-6.94 (m, 2H), 6.52 (t, J=5.8 Hz, 1H), 4.77-4.73 (m, 1H), 3.99 (t, J=9.04 Hz, 1H), 3.72 (dd, J=9.0 Hz, 6.8 Hz, 1H), 3.69-3.58 (m, 2H), 3.31 (t, J=5.5 Hz, 4H), 2.01 (s, 3H), 0.89 (t, J=5.5 Hz, 4H), 0.10 (s, 6H). 13C NMR (100 MHz, CDCl3): δ171.2, 155.0 (d, J=244.3 Hz), 154.5, 138.2 (d, J=9.3 Hz), 131.5, 119.9, 114.0 (d, J=3.4 Hz), 107.6 (d, J=27.1 Hz), 71.9, 50.9, 47.7, 41.9, 23.0, 14.3, −2.9. IR (CHCl3): ν 2401, 1759, 1675, 1519, 1216, 759, 669 cm−1 M.P: 123-126° C. ESI-MS: 380.10 (M+H).


SCHEME2


SCHEME 3

SCHEME 4



Dr. D. Srinivasa Reddy of NCL winner Shanti Swarup Bhatnagar Award 2015
see
http://oneorganichemistoneday.blogspot.in/2015/02/dr-d-srinivasa-reddy.html
Dr. Srinivasa Reddy of CSIR-NCL bags the
prestigious Shanti Swarup Bhatnagar Prize

AN INTRODUCTION
Ph.D., University of Hyderabad, 2000 (Advisor: Professor Goverdhan Mehta).
Post-doctoral with Profs. Sergey A. Kozmin(University of Chicago, USA) and Prof.
Jeffrey Aubé (University of Kansas, USA)
Experienced in leading drug discovery programs (Dr. Reddy’s & TATA Advinus – 7
years of pharma experience)
Acquired skills in designing novel small molecules and lead optimization
Experienced in planning and execution of total synthesis of biologically active
molecules with moderate complexity
One of the molecules is currently in human clinical trials.
MYSELF WITH HIM

OTHER AUTHORS




////////
C[Si]1(C)CCN(CC1)c2ccc(cc2F)N3C[C@H](CNC(C)=O)OC3=O
CEP 18770, Delanzomib

CEP-18770, Delanzomib
cas 847499-27-8
Chemical Formula: C21H28BN3O5
Exact Mass: 413.21220, UNII-6IF28942WO;
CT-47098
NPH 007098
NPH007098
[(1R)-1-[[(2S,3R)-3-Hydroxy-2-[[(6-phenylpyridin-2-yl)carbonyl]amino]-1-oxobutyl]amino]-3-methylbutyl]boronic acid
[(lR)-l-[[(2S,3R)-3-hydroxy-2- [6-phenyl-pyridine-2-carbonyl)amino]-l-oxobutyl]amino]-3-methylbutylboronic acid,
Boronic acid, ((1R)-1-(((2S,3R)-3-hydroxy-1-oxo-2-(((6-phenyl-2-pyridinyl)carbonyl)amino)butyl)amino)-3-methylbutyl)-
In phase 2, multiple mylenoma, Ethical Oncology Science (EOS), licensee
CEP-18770 was discovered through collaboration between Cephalon and Novuspharma/CTI.
Cephalon, Inc., 145 Brandywine Parkway, West Chester, Pennsylvania 19380, and Cell Therapeutics Europe S.r.l., Via L. Ariosto, 23, I-20091 Bresso, Italy
Cephalon was acquired by Teva in October 2011. In 2013, EOS was acquired by Clovis Oncology.
Chemical Process Research and Development, Teva Branded Pharmaceutical Products R&D Inc., 383 Phoenixville Pike, Malvern, Pennsylvania 19355, United States

CEP-18770 is a reversible P2 threonine boronic acid inhibitor of the chymotrypsin-like activity of the proteasome. Displays anti-multimyeloma (MM) effect.

HPLC………http://www.apexbt.com/downloader/document/A4009/HPLC.pdf
NMR………http://www.apexbt.com/downloader/document/A4009/NMR.pdf


CLICK ON IMAGE FOR CLEAR VIEW
Delanzomib, also known as CEP-18770, is An orally bioavailable synthetic P2 threonine boronic acid inhibitor of the chymotrypsin-like activity of the proteasome, with potential antineoplastic activity. Proteasome inhibitor CEP 18770 represses the proteasomal degradation of a variety of proteins, including inhibitory kappaBalpha (IkappaBalpha), resulting in the cytoplasmic sequestration of the transcription factor NF-kappaB; inhibition of NF-kappaB nuclear translocation and transcriptional up-regulation of a variety of cell growth-promoting factors; and apoptotic cell death in susceptible tumor cell populations. In vitro studies indicate that this agent exhibits a favorable cytotoxicity profile toward normal human epithelial cells, bone marrow progenitors, and bone marrow-derived stromal cells relative to the proteasome inhibitor bortezomib. The intracellular protein IkappaBalpha functions as a primary inhibitor of the proinflammatory transcription factor NF-kappaB
New series of dipeptidyl boronate inhibitors of 20S proteasome were identified to be highly potent drug-like candidates with IC50 values of 1.2 and 1.6 nM, respectively, which showed better activities than the drug bortezomib on the market
ref
The potent, selective, and orally bioavailable threonine-derived 20S human proteasome inhibitor that has been advanced to preclinical development, [(1R)-1-[ [ (2S,3R)- 3-hydroxy-2-[ (6-phenylpyridine- 2-carbonyl) amino]-1 -oxobutyl] amino]- 3-methylbutyl] boronic acid (CEP-18770, has been reported
ref .
Dorsey BD, Iqbal M, Chatterjee S, Menta E, Bernardini R, Bernareggi A, et al. Discovery of a potent, selective, and orally active proteasome inhibitor for the treatment of cancer. J Med Chem. 2008;51:1068–1072. [PubMed]
Further, the anti-multiple myeloma protea-some inhibitor CEP-18770 enhanced the anti-myeloma activity of bortezomib and melphalan. The combination of anti-multiple myeloma proteasome inhibitor CEP-18770 intravenously and bortezomib exhibited complete regression of bortezomib-sensitive tumours. Moreover, this combination markedly delayed progression of bortezomib-resistant tumours compared to treatment with either agent alone
Paper
Development and scale-up of an optimized route to the peptide boronic acid, CEP-18770
Org Process Res Dev 2013, 17(3): 422
http://pubs.acs.org/doi/abs/10.1021/op400010u
 USED AS PRODRUG
USED AS PRODRUGCEP-18770 is an unstable peptide boronic acid and an amorphous solid, making it a challenging synthetic target. Process R&D led to a new process that avoided chromatography through crystalline intermediates, increased atom and volume efficiency, provided a chromophore, and gave higher yields and purity. A stable, crystalline diethanolamine adduct was discovered that has the potential to be used as a prodrug.

Compound 8 proved to be a direct substitute for delanzomib in the formulation process. In the first step of the IV formulation process, delanzomib is dissolved in water along with several excipients. Predictably, the delanzomib degrades during this process. It was found that upon dissolution in the lyophilization medium, 8 hydrolyzes to delanzomib,
N-[(1S,2R)-1-[[[(1R)-1–1[(3aS,4S,6S,7aR)-hexahydro-3a,5,5-trimethyl-4,6-methano-1,3,2-benzodioxaborol-2-yl]-3-methylbutyl]amino]carbonyl]-2-hydroxypropyl]-6-phenyl-2-pyridinecarboxamide (5)
PAPER
Discovery of a Potent, Selective, and Orally Active Proteasome Inhibitor for the Treatment of Cancer
http://pubs.acs.org/doi/abs/10.1021/jm7010589

The ubiquitin−proteasome pathway plays a central role in regulation of the production and destruction of cellular proteins. These pathways mediate proliferation and cell survival, particularly in malignant cells. The successful development of the 20S human proteasome inhibitor bortezomib for the treatment of relapsed and refractory multiple myeloma has established this targeted intervention as an effective therapeutic strategy. Herein, the potent, selective, and orally bioavailable threonine-derived 20S human proteasome inhibitor that has been advanced to preclinical development, [(1R)-1-[[(2S,3R)-3-hydroxy-2-[(6-phenylpyridine-2-carbonyl)amino]-1-oxobutyl]amino]-3-methylbutyl]boronic acid 20 (CEP-18770), is disclosed.
[(1R)-1-[[(2S,3R)-3-Hydroxy-2-[(6-phenylpyridine-2-carbonyl)amino]-1-oxobutyl]amino]-3-methylbutyl]boronic Acid (20)
Patent
http://www.google.com/patents/WO2010056733A1?cl=en
Preferred among these compounds is [(lR)-l-[[(2S,3R)-3-hydroxy-2- [6-phenyl-pyridine-2-carbonyl)amino]-l-oxobutyl]amino]-3-methylbutylboronic acid, also known as CEP- 18770, which has the following structure:

PATENT
http://www.google.co.in/patents/WO2005021558A2
NOT SAME BUT SIMILAR
Example E.4 Boronic acid, [(lR)-l-[[(2S,3R)-3-hydroxy-2-[[4-(3-pyridyl)benzoyl]amino]-l- oxobutyI]amino]-3-methyIbutyl].

[00275] A mixture of 4-(pyridin-3-yl)benzamide, N-[(1S,2R)-1-[[[(1R)-1-
[(3aS,4S,6S,7aR)-hexahydro-3a,5,5-trimethyl-4,6-methano-l,3,2-benzodioxaborol-2- yl]-3-methylbutyl]amino]carbonyl]-2-hydroxypropyl]- of Example D.8.3 (155 mg, 0.283 mmol), 2-methylpropylboronic acid (81 mg, 0.793 mmol) and 2N aqueous hydrochloric acid (0.3 ml) in a heterogeneous mixture of methanol (3 ml) and hexane (3 ml) was stirred at room temperature for 24 hours. The hexane layer was removed and the methanolic layer was washed with fresh hexane (about 5 ml). Ethyl acetate (10 ml) was added to the methanol layer which was then concentrated. The residue was taken up with ethyl acetate and the mixture was concentrated. This step was repeated (2-3 times) until an amorphous white solid was obtained. The solid was then triturated with diethyl ether (5 ml) and the surnatant was removed by decantation. This step was repeated. The residue (126 mg) was combined with the product of a similar preparation (140 mg) and dissolved in ethyl acetate (about 40 ml) and a small amount of methanol (2-3 ml). The solution was washed with a mixture of NaCl saturated solution (7 ml) and 10% NaHCO3 (2 ml). The layers were separated and the aqueous phase was further washed with ethyl acetate (2 x 20 ml). The combined organic phases were dried over sodium sulfate and concentrated. The residue was taken up with ethyl acetate (about 20 ml) and the minimum amount of methanol, and then concentrated to small volume (about 5 ml). The resulting white was collected by filtration and dried under vacuum at 50°C (160 mg, 65% overall yield).
1H NMR (MeOH-d4): 8.90 (IH, s); 8.49 (IH, d, J=4.0); 8.20 (IH, d, J=8.1); 8.06 (2H, d, J=8.1); 7.85 (2H, d, J=8.1); 7.58 (IH, t br., J=6.0); 4.80 (IH, d, J=3.9); 4.40-4.29 (IH, m); 2.78 (IH, t, J=7.5); 1.73-1.61 (IH, m); 1.38 (2H, t, J=6.9); 1.31 (3H, d, J=6.3); 0.94 (6H, d, J=6.31). [00276] Further compounds prepared according to the above procedure for
Example E.4 are reported in Table E-4. Table E-4

E.4.3 IS THE COMPD
D.8.12 Chemical Name: 6-Phenyl-2-pyridinecarboxamide,N-[(lS,2R)-l-[[[(lR)- l-[(3aS,4S,6S,7aR)-hexahydro-3a,5,5-trimethyl-4,6-
 THIS IS PRECURSOR OF FINAL PDT
THIS IS PRECURSOR OF FINAL PDTmethano-l,3,2-benzodioxaborol-2-yl]-3- methylbutyl]amino]carbonyl]-2-hydroxypropyl]. Analytical Data: Η -NMR (DMSO-d6): 9.20-8.95 (IH, m); 8.76 (IH, d, J=8.55 Hz); 8.26-8.16 (4H, m); 8.12 (IH, t, J= 7.77 Hz); 8.02 (IH, d, J= 7.56 Hz); 7.60-7.47 (4H, m); 5.27 (IH, d, J= 4.97 Hz); 4.50 (IH, dd, J= 4.22 Hz, J= 8.50 Hz); 4.16-4.07 (2H, m); 2.65-2.56 (IH, m); 2.25-2.15 (IH, m); 2.09-1.98 (IH, m); 1.84 (IH, t, J= 5.62 Hz); 1.79- 1.73 (IH, m); 1.73-1.66 (IH, m); 1.66-1.59 (IH, m); 1.40-1.26 (4H, m); 1.23 (7H, d, J= 10.89 Hz); 1.15-1.10 (4H, m); 0.85 (7H, d, J= 6.56 Hz); 0.79 (IH, bs).
|
References |
1. Fuchs, Ota. Proteasome inhibition as a therapeutic strategy in patients with multiple myeloma. Multiple Myeloma (2009), 101-125. CODEN: 69MVM2 AN 2010:737549
2. Genin, E.; Reboud-Ravaux, M.; Vidal, J. Proteasome inhibitors: recent advances and new perspectives in medicinal chemistry. Current Topics in Medicinal Chemistry (Sharjah, United Arab Emirates) (2010), 10(3), 232-256. CODEN: CTMCCL ISSN:1568-0266. CAN 152:516315 AN 2010:423458
3. Sanchez, Eric; Li, Mingjie; Steinberg, Jeffrey A.; Wang, Cathy; Shen, Jing; Bonavida, Benjamin; Li, Zhi-Wei; Chen, Haiming; Berenson, James R. The proteasome inhibitor CEP-18770 enhances the anti-myeloma activity of bortezomib and melphalan. British Journal of Haematology (2010), 148(4), 569-581. CODEN: BJHEAL ISSN:0007-1048. AN 2010:353952
4. Dick, Lawrence R.; Fleming, Paul E. \Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy. Drug Discovery Today (2010), 15(5/6), 243-249. CODEN: DDTOFS ISSN:1359-6446. AN 2010:318415
5. Ruggeri, Bruce; Miknyoczki, Sheila; Dorsey, Bruce; Hui, Ai-Min. The development and pharmacology of proteasome inhibitors for the management and treatment of cancer. Advances in Pharmacology (San Diego, CA, United States) (2009), 57(Contemporary Aspects of Biomedical Research: Drug Discovery), 91-135. CODEN: ADPHEL ISSN:1054-3589. AN 2010:62762
6. Chen-Kiang, Selina; Di Liberto, Maurizio; Huang, Xiangao. Targeting CDK4 and CDK6 kinases or genes thereof in cancer therapy for sensitizing drug-resistant tumors. PCT Int. Appl. (2009), 149pp. CODEN: PIXXD2 WO 2009061345 A2 20090514 CAN 150:531264 AN 2009:586623
7. Rickles, Richard; Lee, Margaret S. Use of adenosine A2A receptor agonists and phosphodiesterase (PDE) inhibitors for the treatment of B-cell proliferative disorders, and combinations with other agents. PCT Int. Appl. (2009), 70 pp. CODEN: PIXXD2 WO 2009011893 A2 20090122 CAN 150:160095 AN 2009:86451
8. Rickles, Richard; Pierce, Laura; Lee, Margaret S. Combinations for the treatment of B-cell proliferative disorders. PCT Int. Appl. (2009), 79pp. CODEN: PIXXD2 WO 2009011897 A1 20090122 CAN 150:160094 AN 2009:83374
9. Hoveyda, Hamid; Fraser, Graeme L.; Benakli, Kamel; Beauchemin, Sophie; Brassard, Martin; Drutz, David; Marsault, Eric; Ouellet, Luc; Peterson, Mark L.; Wang, Zhigang. Preparation and methods of using macrocyclic modulators of the ghrelin receptor. U.S. Pat. Appl. Publ. (2008), 178pp. CODEN: USXXCO US 2008194672 A1 20080814 CAN 149:288945 AN 2008:975261
10. Piva, Roberto; Ruggeri, Bruce; Williams, Michael; Costa, Giulia; Tamagno, Ilaria; Ferrero, Dario; Giai, Valentina; Coscia, Marta; Peola, Silvia; Massaia, Massimo; Pezzoni, Gabriella; Allievi, Cecilia; Pescalli, Nicoletta; Cassin, Mara; di Giovine, Stefano; Nicoli, Paola; de Feudis, Paola; Strepponi, Ivan; Roato, Ilaria; Ferracini, Riccardo; Bussolati, Benedetta; Camussi, Giovanni; Jones-Bolin, Susan; Hunter, Kathryn; Zhao, Hugh; Neri, Antonino; Palumbo, Antonio; Berkers, Celia; Ovaa, Huib; Bernareggi, Alberto; Inghirami, Giorgio. CEP-18770: a novel, orally active proteasome inhibitor with a tumor-selective pharmacologic profile competitive with bortezomib. Blood (2008), 111(5), 2765-2775. CODEN: BLOOAW ISSN:0006-4971. CAN 149:486154 AN 2008:292777
11. Dorsey, Bruce D.; Iqbal, Mohamed; Chatterjee, Sankar; Menta, Ernesto; Bernardini, Raffaella; Bernareggi, Alberto; Cassara, Paolo G.; D’Arasmo, Germano; Ferretti, Edmondo; De Munari, Sergio; Oliva, Ambrogio; Pezzoni, Gabriella; Allievi, Cecilia; Strepponi, Ivan; Ruggeri, Bruce; Ator, Mark A.; Williams, Michael; Mallamo, John P. Discovery of a Potent, Selective, and Orally Active Proteasome Inhibitor for the Treatment of Cancer. Journal of Medicinal Chemistry (2008), 51(4), 1068-1072. CODEN: JMCMAR ISSN:0022-2623. CAN 148:345774 AN 2008:146611
12. Dorsey, Bruce D.; Menta, Ernesto; Bernardini, Raffaella; Bernareggi, Alberto; Casara, Paolo G.; D’Arasmo, Germano; Ferretti, Edmondo; De Munari, Sergi; Oliva, Ambrogio; Iqbal, Mohamed; Chatterjee, Sankar; Ruggeri, Bruce; Ator, Mark A.; Williams, Michael; Mallamo, John P. CEP-18770: Discovery of a Potent, Selective and Orally Active Proteasome Inhibitor for the Treatment of Cancer. Frontiers in CNS and Oncology Medicinal Chemistry, ACS-EFMC, Siena, Italy, October 7-9 (2007), COMC-027. CODEN: 69KAR2 AN 2007:1171000
13. Marblestone Jeffrey G Ubiquitin Drug Discovery & Diagnostics 2009 – First Annual Conference. IDrugs : the investigational drugs journal (2009), 12(12), 750-3.
| Patent | Submitted | Granted |
|---|---|---|
| Proteasome inhibitors and methods of using the same [US7576206] | 2005-05-19 | 2009-08-18 |
| PROTEASOME INHIBITORS AND METHODS OF USING THE SAME [US7915236] | 2009-11-26 | 2011-03-29 |
| BORONATE ESTER COMPOUNDS AND PHARMACEUTICAL COMPOSITIONS THEREOF [US2009325903] | 2009-12-31 |
| US7442830 * | 6 Aug 2007 | 28 Oct 2008 | Millenium Pharmaceuticals, Inc. | Proteasome inhibitors |
| US7687662 * | 2 Jul 2008 | 30 Mar 2010 | Millennium Pharmaceuticals, Inc. | Proteasome inhibitors |
| US8003819 * | 12 Feb 2010 | 23 Aug 2011 | Millennium Pharmaceuticals, Inc. | Proteasome inhibitors |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8962572 | 4 Oct 2011 | 24 Feb 2015 | Fresenius Kabi Usa, Llc | Bortezomib formulations |
| WO2012177835A1 | 21 Jun 2012 | 27 Dec 2012 | Cephalon, Inc. | Proteasome inhibitors and processes for their preparation, purification and use |
/////CEP-18770, delanzomib
B(C(CC(C)C)NC(=O)C(C(C)O)NC(=O)C1=CC=CC(=N1)C2=CC=CC=C2)(O)O
This blog New Drug Approvals will touch 10 lakh views soon……..as on 7 NOV 2015

This blog New Drug Approvals will touch 10 lakh views soon……..as on 7 NOV 2015
////////
SCYX 7158
SCYX-7158
[4-fluoro-N-(1-hydroxy-3,3-dimethyl-1,3-dihydro-benzo[c]oxaborol-6-yl-2-trifluoromethyl benzamide]
- C17H14BF4NO3
- Average mass 367.103 Da
Human African trypanosomiasis (HAT) is an important public health problem in sub-Saharan Africa, affecting hundreds of thousands of individuals. An urgent need exists for the discovery and development of new, safe, and effective drugs to treat HAT, as existing therapies suffer from poor safety profiles, difficult treatment regimens, limited effectiveness, and a high cost of goods. We have discovered and optimized a novel class of small-molecule boron-containing compounds, benzoxaboroles, to identify SCYX-7158 as an effective, safe and orally active treatment for HAT.
The presence of a boron atom in the heterocyclic core structure has been found essential for trypanocidal activity of orally active series of benzoxaborole-6-carboxamides in murine models of human African trypanosomiasis. SCYX-7158 has been identified as an effective, safe and orally active treatment for human African trypanoso-miasis to enter preclinical studies, with expected progression to phase 1 clinical trials in 2011 ………http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3764666/
A drug discovery project employing integrated biological screening, medicinal chemistry and pharmacokinetic characterization identified SCYX-7158 as an optimized analog, as it is active in vitro against relevant strains of Trypanosoma brucei, including T. b. rhodesiense and T. b. gambiense, is efficacious in both stage 1 and stage 2 murine HAT models and has physicochemical and in vitro absorption, distribution, metabolism, elimination and toxicology (ADMET) properties consistent with the compound being orally available, metabolically stable and CNS permeable.
In a murine stage 2 study,SCYX-7158 is effective orally at doses as low as 12.5 mg/kg (QD×7 days). In vivo pharmacokinetic characterization of SCYX-7158 demonstrates that the compound is highly bioavailable in rodents and non-human primates, has low intravenous plasma clearance and has a 24-h elimination half-life and a volume of distribution that indicate good tissue distribution.
Most importantly, in rodents brain exposure of SCYX-7158 is high, with Cmax >10 µg/mL and AUC0–24 hr >100 µg*h/mL following a 25 mg/kg oral dose. Furthermore, SCYX-7158 readily distributes into cerebrospinal fluid to achieve therapeutically relevant concentrations in this compartment.

Medicinal Chemistry Synthesis of SCYX-7158 SCHEME1
While the original route was eff ective for producing multi-gram quantities of the API, it was not amenable to scale-up. The route started with 2, a relatively expensive aryl boronic acid. This was protected as borocan 3 and halogen-lithium exchange followed by reaction with acetone and subsequent deprotection provided the oxaborole 4. This protection/alkylation/deprotection sequence added two steps to the overall synthesis and the metalation was not reliable. However, the biggest concern in the sequence was nitration of 4 to give 5. This was accomplished by adding a concentrated solution of 4 to cold fuming nitric acid. Besides the signifi cant safety considerations, the reaction did not scale well. Reduction of the nitro group to give aniline 6 was followed by amide formation to provide 1. While this end game was effi cient, the material produced was dark in color. The colored impurities were not removed by crystallization of 1 and furthermore a mixture of two polymorphs was formed under the original conditions.

The process chemistry route to SCYX-7158 is shown in Scheme 2. When considering alternative routes to 1, the readily available and inexpensive methyl 2-bromobenzoate (8) was identifi ed as an attractive starting point. Gratifyingly, treatment of 8 with methylmagnesium bromide aff orded 2-bromocumyl alcohol (9) in high yield using simple operating conditions. Lithiumhalogen exchange followed by reaction with triisopropyl borate and acidic work-up provided benzoxaborole 4, along with cumyl alcohol (10). While this conversion was not completely atom-effi cient, it was easily scalable and several strategies are available to suppress the by-product in the future.
With benzoxaborole 4 in hand, attention turned to the introduction of a nitrogen-linked amide at the C(6) position. This was accomplished using the same nitration/reduction/acylation strategy used in Scheme 1. Yet signifi cant changes to the chemistry were required for safety and reliability reasons. The fi rst task was introduction of the nitrogen. Nitration was demonstrated using acetic anhydride/nitric acid. However, due to slow rates of nitration and potential for accumulation of a reactive intermediate, alternative conditions had to be identifi ed. These limitations were overcome by use of trifl uoroacetic anhydride/nitric acid, which provided a more reactive nitrating intermediate, thus improving the rate of nitration and aff ording a process in which nitric acid was slowly added until 4 was consumed. Full safety assessment of the nitration reaction, including extensive calorimetry studies, demonstrated the safety of this reaction. This process was used to prepare kilogram quantities of 5.
Following reduction of nitrobenzoxaborole 5 to aniline 6 under standard catalytic hydrogenation conditions, acylation with 7 provided the fi nal drug candidate in high chemical yield. Two challenges remained which needed to be addressed through further optimization of the process. The fi rst challenge was color and purity of the API, which derived from a highly colored impurity generated in the nitration reaction which carried through to fi nal product and was not removed by crystallization. The second challenge was to consistently obtain a single polymorph of the API. Both challenges were addressed by isolation of crystalline isopropyl boronate 11 which rejected colored impurities, followed by regeneration of 1 through addition of water and azeotropic removal of isopropanol. This crystallization provided the API as a single polymorph. The API was isolated in good yield, very high purity and was white in color.
PATENT
https://www.google.co.in/patents/WO2011019616A1?cl=en
N-(3,3-Dimethyl-l-phenyl-2,3-dihvdro-lH-benzotblborol-6-yl)-4-fluoro-2- trifluoromethylbenzatnide
HNO3

 This is not the compd, see precursor
This is not the compd, see precursorTo a suspension of 2-bromophenylboronic acid (75.Og, 373.4 mmol) in toluene (525 niL) was added JV-butyldiethanolamine (64.ImL, 392.1 mmol, 1.05 equiv.) via a syringe. The mixture was heated at 50 0C for two hours. After cooling to room temperature, the toluene was evaporated under reduced pressure and the remaining clear colorless oil was treated with heptanes (500 mL). The heptanes mixture was then sonicated for 5 min and the resulting suspension was allowed to stand at room temperature overnight. The solid that precipitated was collected by filtration, washed with heptanes, and dried in a vacuum oven overnight to yield 2-(2′- bromophenyl)-6-butyl[l,3,6,2]dioxazaborocan as a white solid. Data: 1H NMR (400 MHz, CHLOROFORM-^) δ ppm 0.86 (t, J=7.4 Hz, 3 H) 1.14 – 1.25 (m, 2 H) 1.51 – 1.62 (m, 2 H) 2.61 – 2.70 (m, 2 H) 3.01 – 3.11 (m, 2 H) 3.26 – 3.37 (m, 2 H) 4.09 – 4.26 (m, 4 H) 7.10 (td, J=7.6, 2.0 Hz, 1 H) 7.24 (td, J=7.3, 1.1 Hz, 1 H) 7.51 (d, J=7.9 Hz, 1 H) 7.81 (dd, J=IA, 1.9 Hz, 1 H). Amount obtained, 123.7 g (98.6% yield).
To a solution of 2-(2′-bromophenyl)-6-butyl[l,3,6,2]dioxazaborocan (30.0g, 89.2 mmol) in THF (740 mL) at -78 0C was added /?-BuLi (42.8 mL, 2.5M in hexane, 107.0 mmol, 1.2 equiv.) dropwise via a syringe over a period of 10 min while maintaining reaction temperature at -78 0C. After the addition the reaction solution was stirred for 20 min at -78 0C before acetone (7.5 mL, 124.8 mmol, 1.4 equiv.) was added dropwise via a syringe over a period of 10 min while maintaining the reaction temperature at -78 0C. The resulting mixture was allowed to stir for 20 min at -78 0C then warm to room temperature gradually. Once the reaction vessel reached room temperature, 6N HCl solution (150 mL) was added and the mixture was stirred for an additional 30 min. The mixture was extracted with EtOAc (3X). The EtOAc extracts were dried over Na2SO4, filtered and concentrated under reduced pressure. The light yellow oil was then subjected to flash chromatography (Isco Companion, 8Og SiO2 cartridge, solid loaded SiO2, neat heptanes to 20:80 EtOAc gradient at 60 ml/min for 90 min). 3,3-Dimethyl-3H-benzo[c][l,2]oxaborol-l-ol was recovered as clear colorless oil. 1H NMR (400 MHz, DMSO-J6) δ ppm 1.44 (s, 6 H) 7.31 (d, J=Ll Hz, 1 H) 7.38 – 7.47 (m, 2 H) 7.66 (d, J=7.2 Hz, 1 H) 8.99 (s, 1 H). Amount obtained: 9.4O g (65.2 % yield).
To 60 mL fuming HNO3 at -45 0C was slowly added a solution of 3,3- dimethyl-3H-benzo[c][l,2]oxaborol-l-ol (9.4 g, 58.0 mmol) in 11.9 mL nitrobenzene via a syringe while maintaining the reaction temperature between -40 to -45 0C. Once the addition was complete the resulting solution was allowed to stir at -45 ° C for an additional 45 min before poured into crushed ice. The ice mixture was allowed to melt and the aqueous solution was extracted with DCM (3X). The combined DCM extracts were dried over Na2SO4 then evaporated. The crude oil remaining was mixed with one liter 1 : 1 DCM/heptanes. The volume of the solution was reduced under reduced pressure by half and the resulting solution was allowed to stand overnight in a -20 0C freezer. The precipitate formed was filtered out, washed with heptanes and vacuum dried to give 3,3-dimethyl-6-nitro-3H-benzo[c][1.2]oxaborol-l-ol as a white solid. 1H NMR (400 MHz, DMSO-J6) δ ppm 1.46 (s, 6 H) 7.69 (d, J=8.4 Hz, 1 H) 8.28 (dd, J=8.4, 2.3 Hz, 1 H) 8.48 (d, J=2.2 Hz, 1 H) 9.41 (br. s., 1 H). Amount obtained: 7.31 g (60.4 % yield).
To a solution of 3,3-dimethyl-6-nitro-3H-benzo[c][l .2]oxaborol-l-ol (6.98 g, 33.3 mol) in THF ( 277 mL) was added 6N HC1( 16.6 mL, 100.2 mmol, 3.0 equiv.). The vessel was vacuum/N2 purged three times and 5% Pd/C (3.5 g) was added. The mixture was again vacuum/N2 purged three times then vacuum purged again. H2 was then introduced from a balloon and the reaction was allowed to stir at room
temperature over night. The reaction solution was filtered through a short pad of celite and the filtrate was evaporated to yield 6-amino-3, 3 -dimethyl -3H- benzo[c][l,2]oxaborol-l-ol HCl salt as a dark brown foamy solid. 1H NMR (400 MHz, DMSO-J6) δ ppm 1.36 (s, 6 H) 4.94 (s, 2 H) 6.66 (dd, J=8.1, 2.2 Hz, 1 H) 6.79 (d, J=2.0 Hz, 1 H) 7.01 (d, J=8.1 Hz, 1 H) 8.72 (s, 1 H). Amount obtained: 8.29 g (100% yield).
To a solution of 6-amino-3, 3 -dimethyl -3H-benzo[c][l,2]oxaborol-l-ol HCl salt (8.29 g, 33.3 mmol) in DCM (170 mL) was added Et3N (11.6 mL, 83.2 mmol, 2.5 equiv.). The mixture was cooled to 0 0C and 2-trifluoromethyl-4- fluorobenzoyl chloride (6.1 mL, 39.9 mmol, 1.2 equiv.) was added slowly via a syringe. The resulting solution was allowed to warm to room temperature gradually and stir for 2 hours. The reaction solution was diluted with DCM, washed with IN HCl, H2O, brine and then dried over Na2SO4, filtered and the filtrate was concentrated under reduced pressure to give an off- white solid. The solid was recrystallized from DCM/heptanes to give 4-fluoro-N-(l-hydroxy-3,3-dimethyl-l,3-dihydro- benzo[c][l,2]oxaborol-6-yl-2-trifluoromethyl benzamide as a white solid. LCMS (M/Z) : 368 (M+H); 1H NMR (DMSO-d6) δ: 10.58 (s, IH), 9.11 (s, IH), 8.02 (d, J = 1.7 Hz, IH), 7.75 – 7.83 (m, 2H), 7.60 – 7.71 (m, 2H), 7.38 (d, J = 8.2 Hz, IH), 1.44 (s, 6H). Amount obtained: 11.7 g (96% yield)………IS SCYX 7158
BELOW NOT SCYX 7158
The title compound was prepared using a similar procedure to that of N-(I- phenyl- 1 ,3 -dihydrobenzo [c] [ 1 ,2]oxaborol-6-yl)-2-trifluoromethylbenzamide with phenyl magnesium bromide replacing p-to IyI magnesium bromide and 4-fluoro-iV-(l- hydroxy-3,3-dimethyl-2,3-dihydro-lH-benzo[b]borol-6-yl)-2-trifluoromethyl benzamide replacing N-(I -hydroxy-1 ,3-dihydrobenzo[c] [ 1 ,2]oxaborol-6-yl)-2- trifiuoromethylbenzamide. Data: LCMS m/e: 428 (M+H); 1H NMR (400 MHz, DMSO-J6) δ ppm 1.59 (s, 6 H) 7.46 – 7.62 (m, 4 H) 7.71 (td, J=8.5, 2.7 Hz,l H) 7.77 – 7.90 (m, 3 H) 8.00 – 8.09 (m, 2 H) 8.39 (d, J=2.0 Hz, 1 H) 10.66 (s, 1 H). 10 N-fl-p-Tolyl-lJ-dihydro-benzofcIflJIoxaborol-ό-vD-benzatnide
PATENT
82 4-Fluow-N-(l-hydwxy-3,3-dimethyl-l,3-dihydw-benzofcIfl,2Ioxabowl-6- yl-2-trifluoromethyl benzamide

To a suspension of 2-bromophenylboronic acid (10. Og, 49.7 mmol) in toluene (70 niL) was added N-butyldiethanolamine (8.5 mL, 52.2 mmol, 1.05 equiv.) via a syringe. The mixture was heated at 50 0C for two hours. After cooling to room temperature, the toluene was evaporated under reduced pressure and the remaining clear colorless crude oil was treated with heptanes (~ 500 mL). The heptanes mixture was then sonicated ~ 5 min and the resulting suspension was allowed to stand at room temperature overnight. The solid that precipitated was collected by filtration, washed with heptanes, and dried in a vacuum oven overnight to yield a white solid as the titled compound. 1U NMR (400 MHz, CHLOROFORM-J) δ ppm 0.86 (t, J=7.4 Hz, 3 H) 1.14 – 1.25 (m, 2 H) 1.51 – 1.62 (m, 2 H) 2.61 – 2.70 (m, 2 H) 3.01 – 3.11 (m, 2 H) 3.26 – 3.37 (m, 2 H) 4.09 – 4.26 (m, 4 H) 7.10 (td, J=7.6, 2.0 Hz, 1 H) 7.24 (td, J=7.3, 1.1 Hz, 1 H) 7.51 (d, J=7.9 Hz, 1 H) 7.81 (dd, J=IA, 1.9 Hz, 1 H). Amount obtained, 16.0 g, (98 % yield).
To a solution of 2-(2′-bromophenyl)-6-butyl[l,3,6,2]dioxazaborocan (3.0g, 9.2 mmol) in THF (76 mL) at -78 0C was added /?-BuLi (4.4 mL, 2.5M in hexane, 11.0 mmol, 1.2 equiv.) dropwise via a syringe over a period of 10 min while maintaining reaction temperature at -78 0C. After the addition the reaction solution was stirred 20 min at -78 0C before acetone (946 μL, 12.8 mmol, 1.4 equiv.) was added dropwise via a syringe over a period of 10 min while maintaining the reaction temperature at -78 0C. The resulting mixture was allowed to stir for 20 min at -78 0C then warm to room temperature gradually. Once the reaction vessel reached room temperature, 6M HCl solution (30 mL) was added and the mixture was stirred for 30 min. The mixture was extracted with EtOAc (3X). The EtOAc extracts were dried over Na2SO4, filtered and concentrated under reduced pressure. The crude slightly yellow in color residual oil remaining was then subjected to flash chromatography (Isco Companion, 8Og SiO2 cartridge, solid loaded SiO2, neat heptane to 20:80 EtOAc gradient at 60 ml/min for 90 min). The product was recovered as clear colorless oil. 1H NMR (400 MHz, DMSO-J6) δ ppm 1.44 (s, 6 H) 7.31 (d, J=Ll Hz, 1 H) 7.38 – 7.47 (m, 2 H) 7.66 (d, J=7.2 Hz, 1 H) 8.99 (s, 1 H). Amount obtained: 1.76 g (61%).
To 14.2 ml fuming HNO3 at -45 0C was added a solution of 3,3-dimethyl- 3H-benzo[c][l,2]oxaborol-l-ol (2.28 g, 14.1 mmol) in 3.0 ml nitrobenzene slowly via a syringe while maintaining the reaction temperature between -40 to -45 0C. Once the addition was complete the resulting solution was allowed to stir at -45 ° C for an additional 45 min before poured into crushed ice (600 g). The ice mixture was allowed to melt and the aqueous solution was extracted with dichloromethane. The combined dichloromethane extracts were dried over Na2SO4 then evaporated. The crude oil remaining was mixed with one liter 1 : 1 DCM:heptane. The volume of the solution was reduced on a rotovap by half and the resulting solution was allowed to stand overnight in a -20 0C freezer overnight. The precipitate formed was filtered out, washed with heptanes and vacuum dried to give the titled compound as a white solid. 1H NMR (400 MHz, DMSO-J6) δ ppm 1.46 (s, 6 H) 7.69 (d, J=8.4 Hz, 1 H) 8.28 (dd, J=8.4, 2.3 Hz, 1 H) 8.48 (d, J=2.2 Hz, 1 H) 9.41 (br. s., 1 H). Amount obtained: 2.01 g (68%).
To a solution of 3,3-dimethyl-6-nitro-3H-benzo[c][1.2]oxaborol-l-ol (790 mg, 3.8 mmol) in THF ( 20 mL) was added HOAc (1.7 mL, 30 mmol). The vessel was vacuum/N2 purged three times and 5% Pd/C (200 mg) was added. The mixture was again vacuum/N2 purged three times then vacuum purged again. H2 was then introduced from a balloon and the reaction was allowed to stir for 2.5 hours. The reaction solution was filtered through a short pad of celite and the filtrate was evaporated to yield the title compound as a dark brown foamy solid. 1H NMR (400 MHz, DMSO-J6) δ ppm 1.36 (s, 6 H) 4.94 (s, 2 H) 6.66 (dd, J=8.1, 2.2 Hz, 1 H) 6.79 (d, J=2.0 Hz, 1 H) 7.01 (d, J=8.1 Hz, 1 H) 8.72 (s, 1 H). Amount obtained: 670 mg (89%). [0382] To a solution of 6-amino-3, 3 -dimethyl -3H-benzo[c][l,2]oxaborol-l-ol acetate salt (100 mg, 0.42 mmol) in DCM (2 niL) was added Et3N ( 117.3 μL, 0.84 mmol). The mixture was cooled to 0 0C and the 2-trifluoromethyl-4-fluorobenzoyl chloride (70.0 μL, 0.46 mmol) was added slowly via a syringe. The resulting solution was allowed to warm to room temperature gradually and stir for 2 hours. The reaction solution was diluted with DCM, washed with IN HCl, H2O and then dried over Na2SO4, filtered and the filtrate was concentrated under reduced pressure and the crude material was subjected to flash chromatography (Isco Companion, 4 g SiO2 cartridge, SiO2 solid load, neat heptanes to neat EtOAc gradient over 45 min, flow rate = 18 ml/min). The title compound was recovered as a white foam. LCMS (M/Z) : 368 (M+H); 1H NMR (DMSO-d6) δ: 10.58 (s, IH), 9.11 (s, IH), 8.02 (d, J = 1.7 Hz, IH), 7.75 – 7.83 (m, 2H), 7.60 – 7.71 (m, 2H), 7.38 (d, J = 8.2 Hz, IH), 1.44 (s, 6H). Amount obtained: 144.6 mg (93% yield).
Alternate Synthesis

82e
82b
A 500 mL round-bottomed-flask equipped with a magnetic stir bar and ice- H2O bath was charged with 82a (18.4g, 85.5 mmol) and anhydrous THF (200 mL). MeMgCl (68 mL, 3.0M in 2-methylTHF) was added dropwise through an additional funnel. The mixture was allowed to warm to rt. gradually and stirred overnight. After cooling back to 0 0C, the white milky suspension was carefully treated with HCl (3M) until the upper layer turned clear with white precipitate at the bottom of the flask (pH = 6). The upper clear solution was decanted into a separatory funnel. The precipitate was rinsed with methyl tert-butyl ether (MTBE) (100 mL) 3 times. Combined MTBE with the clear solution and the mixture was washed with H2O (100 mL) 3 times, brine (100 niL), dried over MgSO4, filtered and concentrated under reduced pressure to give 82b as a light yellow oil (20.2g, 100%).
82c
A 50 mL round-bottomed-flask equipped with a magnetic stir bar and ice- H2O bath was charged with 82b (860 mg, 4.0 mmol) and anhydrous THF (20 mL). MeMgBr (1.3 mL, 2.0 M in THF) was slowly added via a syringe. The mixture was stirred at 0 0C for 10 minute and the ice bath was replaced with a dry ice-acetone bath at -40 0C. BuLi (1.9 mL, 2.5 M in hexanes) was added dropwise via a syringe. The resulting mixture was stirred at -40 0C for another 2h before B(O-ipr)3 (1.4 mL, 4.8 mmol) was added dropwise. The mixture was allowed to warm up to rt gradually and stirred overnight. After carefully quenched the reaction with H2O (1 mL), HCl (3M, 10 mL) was added and the mixture was stirred at rt for Ih. The mixture was extracted with EtOAc (20 mL) 3 times. Combined extracts was washed with H2O (20 mL), brine (20 mL), dried over MgSO4, filtered and concentrated under reduced pressure to give a clear oil. The oil solidified overnight to give 82c as a pale yellow waxy solid (544mg, 82.4%).
82d
A 3 L round-bottomed-flask equipped with a mechanical stirrer, thermocouple and ice bath was charged with 82c (86.2 g of 58 wt%, 309 mmol) and trifluoroacetic acid (259 mL). Trifluoroacetic anhydride (129 mL, 926 mmol) was added in one portion. An exotherm of 18 0C was observed. The solution was again cooled to 0 0C and 90% nitric acid (18.0 mL, 386 mmol) was added via syringe pump over 2 h. After the addition was complete, the solution was aged for 1 h. Water (1.75 L) was added. Note: Initially the quench is quite exothermic. Add the water in 5 mL aliquots until the exotherm subsides. The resulting suspension was stirred for 16 h while warming to rt. The solids were collected on a frit, rinsed with water (2 x 500 mL), and air dried to constant weight to provide 50.3 g of crude 82d as a free-flowing orange solid. Note: the crude 82d can be carried forward without recrystallization. The solid was charged to a IL three-necked round-bottomed-flask equipped with a nitrogen inlet adapter, thermocouple, heating mantle and mechanical stirrer.
Isopropylacetate (IPAc, 75 mL) was added and the resulting slurry was warmed to 75 0C and heptanes (250 mL) was added over 15 min while maintaining an internal temp of > 65 0C. The slurry was allowed to cool to rt over night. The solids were collected on a frit and rinsed with 10% IP Ac/heptanes (100 mL) and then heptanes (100 rnL). The product was air dried to constant weight to provide a tan solid (31.7 g, 58%).
82e
A 500 mL round-bottomed-flask equipped with a magnetic stir bar, thermocouple and septum was charged with 82d (29.7 g, 192 mmol) and THF (150 mL, anhydrous stabilizer free). The vessel was inerted by cycling vacuum the nitrogen three times and 5% Pd/C (6.0 g, 50% wet, Degussa type NO/W) was added. The vessel was again inerted by cycling vacuum then nitrogen three times. A hydrogen filled balloon was attached via needle and the atmosphere was changed by cycling vacuum the hydrogen three times. The slurry was stirred vigorously for 16 h. The atmosphere was changed again to nitrogen by cycling vacuum then nitrogen three times. The mixture was filtered through a 1″ pad of celite and the cake was rinsed with THF (50 mL). Concentration in vacuo provided a light tan powder (26.82 g). In a 500 mL round bottomed-flask, the solids were slurried in IPAc (50 mL) and warmed in an 80 0C water bath. Heptanes (150 mL) were added over 10 min. The resulting slurry was allowed to cool to rt and stir for 16 h. The solids were collected on a frit, rinsed with heptanes (50 mL) and air dried to provide an off- white solid (24.39 g, 96%).
4-Fluoro-N-(l-hvdroxy-3,3-dimethyl-l,3-dihvdro-benzofcJfl,2Joxaborol-6-yl-2- triβuoromethyl benzatnide
A lL three-necked round-bottomed-flask equipped with a nitrogen inlet adapter, mechanical stirrer and thermocouple was charged with 82e (15.7g, 88.4 mmol), THF (160 mL, anhydrous, stabilizer free) and K2CO3 (14.7g, 106 mmol). The suspension was stirred at rt and 4-fluoro-2-(trifluoromethyl)benzoyl chloride (22.Og, 97.3 mmol) was added over 10 min. The resulting suspension was aged for 24 h at rt. Water (80 mL) and isopropyl acetate (160 mL) were added and the phases were partitioned. The organic phase was further extracted with water (80 mL) and then brine (50 mL). The organic phase was dried over MgSO4 (20 g) and concentrated in vacuo to provide a tan solid (34.26 g). The solid was dissolved with acetone (195 mL) and transferred to a mechanically stirred IL round-bottomed-flask. Distilled water (113 mL) was added in one portion and the mixture was stirred for 30 min to produce a seed bed and then additional distilled water (60 mL) was added over 30 min. The suspension was stirred at rt overnight and the solids were collected on a frit. The cake was rinsed with 1 : 1 acetone/water (100 rnL) and air dried to constant weight to provide an off-white solid (30.5 g, 94%).
Alternate Synthesis
HNO3 CF3CO2H (CF3CO)2O

to RT


1-1
A 72 L round-bottomed-flask was equipped with a cold bath, mechanical stirrer, nitrogen inlet adaptor, oxygen sensor, thermowell and 2 L dropping funnel. The flask was charged with methyl 2-bromobenzoate (2513 g, 11.7 mol) and the system was flushed with nitrogen to <0.1% O2. THF (18L, anhydrous, inhibitor free) was added and the cold bath was charged with ice and acetone. When the internal temp reached -4 0C, MeMgBr (11.6 L of a 3M solution in ether, 34.8 mol) was added via dropping funnel over 3 h. The internal temp was maintained below 15 0C throughout. At the end of addition, the cold bath was drained and the reaction was aged overnight at ambient temperature. The bath was again charged with ice and acetone and the suspension cooled to below 15 0C. HPLC indicated incomplete conversion (92:8 product, starting ester), so additional MeMgBr (2.3L of a 3M solution in ether) was added. After Ih, HPLC showed the conversion to be >99: 1. The reaction was quenched by slow addition of IN HCl (42 L) keeping the internal temp below 15 0C throughout. At the end of the quench, the pH was adjusted to 6 with IN HCl. The mixture was extracted with MTBE (10 L then 2x5L). The combined organic phases were dried over MgSO4, filtered and concentrated via rotary evaporation to provide 2482 g of 2-(2-bromophenyl)-propan-2-ol as a pale yellow oil. 1H NMR (CHLOPvOFORM-d) δ: 7.62 – 7.67 (m, IH), 7.53 – 7.58 (m, IH), 7.24 – 7.30 (m, IH), 7.03 – 7.10 (m, IH), 1.70 – 1.75 (m, 6H). 1-2
A 72L round-bottomed-flask was equipped with a mechanical stirrer, O2 sensor, thermowell, 2L dropping funnel, N2inlet adaptor, and cold bath. The vessel was inerted to 0.01% O2 and charged with THF (27L, anhydrous, inhibitor free). The resulting solution was cooled to -70 0C using dry ice and acetone and n-BuLi (8.2 L of a 2.5M solution in heptane, 20.5 mol) was added over Ih. 2-(2-Bromophenyl)- propan-2-ol (1994 g, 9.27 mol) was dissolved in THF (9L) and the solution was added to the BuLi via dropping funnel over 2h, keeping the internal temp below -70 0C. The resulting thin yellow suspension was aged for 30 min then B(OiPr)3 (244 Ig, 13.0 mol) was added rapidly via addition funnel. The cold bath was drained and the misture was allowed to warm to room temperature while aging over night. HPLC analysis shows an 81 :19 ratio of desired product: 2-phenyl-2-propanol. The mixture was cooled to -10 0C and 2N HCl (9.3 L) was added via dropping funnel over 30 min, keeping the reaction mixture below 10 0C. After 3 h, the pH was adjusted to 4 with additional HCl. The reaction mixture was extracted with MTBE (2 x 4L). The combined organic phases were concentrated to provide 2028 g of a heavy oil. The oil was dissolved in MTBE (14L) and extracted with IN NaOH (4.6, then 5, then 4L). The aqueous phases were combined and acidified with 2N HCl (6.8 L) to a pH of 4-5. The mixture was extracted with MTBE (5L). The organic phase was dried over MgSO4 (282 g) and concentrated to provide 1450 g (ca 60 wt%) of 3,3-dimethyl-3H- benzo[c][l,2]oxaborol-l-ol as a waxy white solid. LC/MS: m/z 163 (M+H)+; 1H NMR (DMSO-de) δ: 8.96 (br. s., IH), 7.62 (d, J = 7.2 Hz, IH), 7.33 – 7.45 (m, 2H), 7.25 – 7.30 (m, IH), 1.40 (s, 6H).
1-3
A 22 L round-bottomed-flask equipped with a mechanical stirrer, thermocouple, 2 L dropping funnel and cold bath was charged with 3,3-dimethyl-3H- benzo[c][l,2]oxaborol-l-ol (508 g, 300 g contained, 1.85 mol) and trifluoroacetic acid (1.54 L). The solution was cooled to 5 0C. Trifluoroacetic anhydride (722 mL, 5.56 mol, 3.00 eq) was added via dropping funnel over 15 min. After aging at 0 – 3 0C for 30 min, nitric acid (90% fuming, 108 mL, 2.31 mol, 1.5 eq) was added dropwise over 2h 50 min keeping the internal temp below 5 0C. After aging for 1 h, icewater (10.4L) was added over 50 min maintaining the reaction temp below 15 0C to provide a slurry. The slurry was aged at 0 0C overnight to provide an orange suspension. The solids were collected on a frit, rinsed with cold water (5L) and air dried under a stream of air to constant weight (ca 24h) to provide 364 g of 3,3- dimethyl-6-nitro-3H-benzo[c][l,2]oxaborol-l-ol as a 92.4 wt% pure solid (88%). LC/MS : m/z 208 (M+H)+; 1H NMR (DMSO-d6) δ: 8.52 (d, J = 2.2 Hz, IH), 8.32 (dd, J = 8.4, 2.2 Hz, IH), 7.74 (d, J = 8.4 Hz, IH), 1.50 (s, 6H)
1-4
A 2 gallon stirred pressure vessel was charged with 3,3-dimethyl-6-nitro- 3H-benzo[c][l,2]oxaborol-l-ol (966 g, 812 g corrected, 3.92 mol), 5% Pd/C (193 g, 50% wet, Degussa type 101 NO/W) and THF (4.83 L, inhibitor free). The vessel was sealed, the atmosphere was changed to H2 (5 psi) and the reaction was fun for 16 h. An exotherm to 30 0C was observed over about 30 min. The vessel was purged with N2, and completion of reaction was determined by HPLC. The reaction was vacuum filtered through a pad of celite (very slow filtration) and the filter cake was rinsed with THF (2L). The filtrate was concentrated via rotary evaporation to provide 982 g of a dark brown solid. This was transferred to a 22L round-bottomed-flask and warmed to 80 0C in iPAc (1.83 L) to provide a dark brown slurry. The slurry was cooled to 60 0C and heptanes (5.49L) were added over 2 h. The slurry was allowed to age with stirring over night while cooling to room temperature. The solids were collected on a frit, rinsed with heptanes (4L) and air dried to provide a dark brown solid (747 g).
The solids (747 g) were transferred to a 22L rbf and slurried in iPAc (3 L) at 70 0C. The batch was allowed to cool to 40 0C and heptanes (3L) were added over 5 h. The slurry was aged at room temperature over night and the solids were collected on a frit, rinsed with 1 : 1 iP Ac/heptanes (2L) then heptanes (IL) and air dried to provide 554 g of 6-amino-3,3-dimethyl-3H-benzo[c][l,2]oxaborol-l-ol as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 1.36 (s, 6 H) 4.94 (s, 2 H) 6.66 (dd, J=8.1, 2.2 Hz, 1 H) 6.79 (d, J=2.0 Hz, 1 H) 7.01 (d, J=8.1 Hz, 1 H) 8.72 (s, 1 H). 4-Fluow-N-(l-hvdwxy-3,3-dimethyl-l,3-dihvdw-benzofcJfl,2Joxabowl-6-yl)-2- triβuoromethyl benzatnide
A 22L four-necked round-bottomed-flask equipped with a nitrogen inlet adapter, mechanical stirrer and thermocouple was charged with 6-amino-3,3- dimethyl-3H-benzo[c][l,2]oxaborol-l-ol (554g, 3.13 mol), THF (5.5 L, anhydrous, stabilizer free) and K2CO3 (865 g, 6.26 mol). The suspension was stirred at room temperature for 30 min and 4-fluoro-2-(trifluoromethyl)benzoyl chloride (780 g, 3.44 mol) was added over 30 min. The resulting suspension was aged for 24 h at room temperature. HPLC showed unreacted 6-amino-3,3-dimethyl-3H-benzo[c][l,2] oxaborol-1-ol so an additional 42 niL of the acid chloride was added. After 30 min, water (2.8 L) and isopropyl acetate (5.5 L) were added and the phases were partitioned. The organic phase was further extracted with water (2.8 L) and then brine (2.8 L). The organic phase was dried over MgSO4 and concentrated in vacuo to provide a tan solid. The solid was dissolved with acetone (3.0 L) and transferred to a mechanically stirred 5OL round-bottomed-flask. Distilled water (2.0 L) was added in one portion and the mixture was stirred for 30 min to produce a seed bed and then additional water (1.0 L) was added over 30 min. The suspension was stirred at room temperature overnight and the solids were collected on a frit. The cake was rinsed with 1 : 1 acetone/water (1.0 L) and air dried to constant weight to provide 4-fluoro-N- ( 1 -hydroxy-3 ,3 -dimethyl- 1 ,3 -dihydro-benzo [c] [ 1 ,2]oxaborol-6-yl)-2-trifluoromethyl benzamide as a dark tan solid (1.3 kg).
Recrystallization of4-Fluoro-N-(l-hydroxy-3,3-dimethyl-l,3-dihvdro- benzotcl t 1,21 oxaborol-6-yl)-2-trifluoromethyl benzamide
A 22 L round-bottomed-flask was charged with the dark tan crude 4- fluoro-N-(l -hydroxy-3, 3 -dimethyl- 1 ,3-dihydro-benzo[c] [ 1 ,2]oxaborol-6-yl)-2- trifluoromethyl benzamide (1.3 kg), acetone (8L) and Darco G-60 (55 g, 400 mesh) and water (5.3L). The resulting suspension was stirred for 15 min, filtered through a pad of celite (ca 500 g) to provide a brown solution. The celite pad was washed with 60% acetone/water (8L). The combined filtrate and rinse were transferred to a 50 L round-bottomed-flask and water (2L) was added. The solution was seeded (5 g) to initiate crystallization and additional water (2.2 L) was added slowly via addition funnel. After aging at room temperature overnight, the solids were collected and the filter cake was rinsed with 30% acetone/water (4L). The solids were air dried for 24 h then dried in a room temperature vacuum oven for 5 days to constant weight to provide 969 g (72% recovery) of 4-fluoro-N-(l-hydroxy-3,3-dimethyl-l,3-dihydro- benzo[c][l,2]oxaborol-6-yl)-2-trifluoromethyl benzamide as a light tan solid.
LC/MS: m/z 368 (M+H)+;
1H NMR (400 MHz, DMSO-d6) δ ppm 1.44 (s, 5 H) 1.49 (s, 2 H) 7.39 (d, J=8.2 Hz, 1 H) 7.61 – 7.76 (m, 2 H) 7.77 – 7.84 (m, 2 H) 7.86 – 7.90 (m, 0 H) 8.03 (d, J=I.7 Hz, 1 H) 9.09 (s, 1 H) 10.58 (s, 1 H).
POTASSIUM SALT
Formation of potassium salt

To a 50OmL three-neck flask fitted with a mechanical stirrer was charged KOH (1.51 g, 26.9 mmol, 1.0 eq.). Under a nitrogen atmosphere, anhydrous acetone (140 mL) and H2O (2.5 mL, 5 eq.) were added via syringe. A solution of 4-fluoro-N- (l-hydroxy-3,3-dimethyl-l,3-dihydro-benzo[c][l,2]oxaborol-6-yl-2-trifluoromethyl benzamide (10.0 g, 27.2 mmol, 1.0 eq.) in anhydrous acetone (60 mL) was added to the flask with vigorous stirring. The resulting clear solution was stirred at room temperature. The potassium salt precipitated from the solution over ca. 4 hours to afford a thick suspension. The precipitate was collected by filtration, washed with acetone (200 mL) and dried in a vacuum oven overnight to afford a white solid (10.6g, 91.9% yield). 1H NMR (methanol-d4) δ: 7.70 – 7.76 (m, IH), 7.53 – 7.60 (m, 2H), 7.47 – 7.53 (m, IH), 7.33 – 7.36 (m, IH), 7.01 – 7.06 (m, IH), 1.46 (s, 6H); M.P. (range) 197 – 200 0C; Elemental analysis: Theory: C 48.25%, H 3.57%, N 3.31%, K 9.24%; Found: C 48.70%, H 3.41%, N 3.25%, K 9.19%.
REFERENCES
http://journals.plos.org/plosntds/article?id=10.1371/journal.pntd.0001151
- touguia J, Costa J (1999) Therapy of human African trypanosomiasis: current situation. Mem Inst Oswaldo Cruz 94: 221–224
- Barrett MP, Boykin DW, Brun R, Tidwell RR (2007) Human African trypanosomiasis: pharmacological re-engagement with a neglected disease. Br J Pharmacol 152: 1155–1171.
- 1986. Epidemiology and control of African trypanosomiasis. Report of a WHO expert committee. World Health Organization. Geneva, Switzerland. Technical Report Series, No. 739. 126 pp.
- Benzoxaboroles: a new class of potential drugs for human African trypanosomiasis. Robert T Jacobs, Jacob J Plattner, Bakela Nare, Stephen A Wring, Daitao Chen, Yvonne Freund, Eric G Gaukel, Matthew D Orr, Joe B Perales, Matthew Jenks, Robert A Noe, Jessica M Sligar, Yong-Kang Zhang, Cyrus J Bacchi, Nigel Yarlett, and Robert Don. Future Medicinal Chemistry. August 2011. Vol. 3, No. 10. Pages 1259-1278.
http://www.swisstph.ch/fileadmin/user_upload/Pdfs/Events/2010_09_Jacobs.pdf ……….POWERPOINT
Lead optimization investigation of oxaboroles for the treatment of human African trypanosomiasis
238th Am Chem Soc (ACS) Natl Meet (August 16-20, Washington) 2009, Abst MEDI 345
LINK
https://www.acsmedchem.org/ama/orig/abstracts/mediabstractf2009.pdf
Robert Jacobs, bob.jacobs@scynexis.com
Daitao Chen1 , Matt Orr1 , Jessica Sligar1 , Matt. Jenks1 , Andy Noe1 , Bakela Nare2 , Luke T. Mercer2 , Tana S. Bowling2 , Cindy Rewerts1 , Stephen Wring1 , Cyrus Bacchi3 , Nigel Yarllet3 , Charles Ding4 , Yvonne Freund5 , Kurt Jarnagin5 , Jacobs Plattner5 , and Robert Don6 . (1) Scynexis Inc, Duhram, NC 27713, (2) SCYNEXIS, Inc, Research Triangle Park, NC 27709-2878, (3) Pace University, New York, NY, (4) Anacor Pharmaceuticals, Inc, Palo Alto, CA, (5) Anacor Pharmaceuticals, Inc, (6) Drugs for Neglected Diseases initiative, Geneva, Switzerland

///////////SCYX-7158
FDA approves new treatment for HIV

November 5, 2015
Release
The U.S. Food and Drug Administration today approved Genvoya (a fixed-dose combination tablet containing elvitegravir, cobicistat, emtricitabine, and tenofovir alafenamide) as a complete regimen for the treatment of HIV-1 infection in adults and pediatric patients 12 years of age and older.
The CDC estimates that 1.2 million persons ages 13 years and older are living with HIV infection, and that more than another 150,000 persons in this age range have HIV but are unaware of their infection. Over the past decade, the number of people living with HIV has increased, while the annual number of new HIV infections has remained relatively stable.
“Today’s approval of a fixed dose combination containing a new form of tenofovir provides another effective, once daily complete regimen for patients with HIV-1 infection,” said Edward Cox, M.D., director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research.
Genvoya is approved for use in HIV-infected adults and children ages 12 years and older weighing at least 35 kilograms (77 pounds) who have never taken HIV therapy (treatment-naïve) and HIV-infected adults whose HIV-1 virus is currently suppressed. While Genvoya is not recommended for patients with severe renal impairment, those with moderate renal impairment can take Genvoya.
Genvoya’s safety and efficacy in adults were evaluated in 3,171 participants enrolled in four clinical trials. Depending on the trial, participants were randomly assigned to receive Genvoya or another FDA approved HIV treatment. Results showed Genvoya was effective in reducing viral loads and comparable to the other treatment regimens.
Genvoya contains a new form of tenofovir that has not been previously approved. This new form of tenofovir provides lower levels of drug in the bloodstream, but higher levels within the cells where HIV-1 replicates. It was developed to help reduce some drug side effects. Genvoya appears to be associated with less kidney toxicity and decreases in bone density than previously approved tenofovir containing regimens based on laboratory measures. Patients receiving Genvoya experienced greater increases in serum lipids (total cholesterol and low-density lipoprotein) than patients receiving other treatment regimens in the studies.
Genvoya carries a Boxed Warning alerting patients and health care providers that the drug can cause a buildup of lactic acid in the blood and severe liver problems, both of which can be fatal. The Boxed Warning also states that Genvoya is not approved to treat chronic hepatitis B virus infection. The most common side effect associated with Genvoya is nausea. Serious side effects include new or worsening kidney problems, decreased bone mineral density, fat redistribution and changes in the immune system (immune reconstitution syndrome). Health care providers are advised to monitor patients for kidney and bone side effects. Genvoya should not be given with other antiretroviral products and may have drug interactions with a number of other commonly used medications.
Genvoya is marketed by Gilead Sciences Inc. based in Foster City, California.
/////////
SERTINDOLE
 .
.
SERTINDOLE
Sertindole (brand names: Serdolect, and Serlect) is an antipsychotic medication. Sertindole was developed by the Danish pharmaceutical company H. Lundbeck and marketed under license by Abbott Labs. Like other atypical antipsychotics, it has activity at dopamine and serotonin receptors in the brain. It is used in the treatment of schizophrenia. It is classified chemically as a phenylindole derivative.
Sertindole is not approved for use in the United States.
Medical Uses
Sertindole appears effective as an antipsychotic in schizophrenia.[4]
Safety and status
USA
Abbott Labs first applied for U.S. Food and Drug Administration (FDA) approval for sertindole in 1996,[10] but withdrew this application in 1998 following concerns over the increased risk of sudden death from QTc prolongation.[11] In a trial of 2000 patients on taking sertindole, 27 patients died unexpectedly, including 13 sudden deaths.[12] Lundbeck cites the results of the Sertindole Cohort Prospective (SCoP) study of 10,000 patients to support its claim that although sertindole does increase the QTc interval, this is not associated with increased rates of cardiac arrhythmias, and that patients on sertindole had the same overall mortality rate as those on risperidone.[13] Nevertheless in April 2009 an FDA advisory panel voted 13-0 that sertindole was effective in the treatment of schizophrenia but 12-1 that it had not been shown to be acceptably safe.[14] As of October 2010, the drug has not been approved by the FDA for use in the USA.[15]
Europe
In Europe, sertindole was approved and marketed in 19 countries from 1996,[12] but its marketing authorization was suspended by the European Medicines Agency in 1998[16] and the drug was withdrawn from the market. In 2002, based on new data, the EMA’s CHMP suggested that Sertindole could be reintroduced for restricted use in clinical trials, with strong safeguards including extensive contraindications and warnings for patients at risk of cardiac dysrhythmias, a recommended reduction in maximum dose from 24 mg to 20 mg in all but exceptional cases, and extensive ECG monitoring requirement before and during treatment.[17][18]
Synthesis

PAPER
Identification and synthesis of impurities formed during sertindole preparation
 , and
, and 2Institute of Science and Technology, JNTU, Hyderabad-500072, India
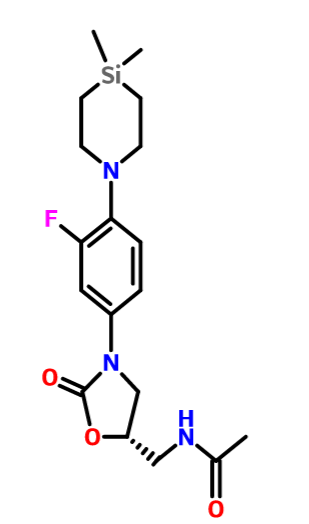
Corresponding author emailSertindole is designated chemically as 1-[2-[4-[5-chloro-1-(4-fluorophenyl)-1H-indol-3-yl]-1-piperidinyl]ethyl]-2-imidazolidinone. Its literature synthesis (Scheme 1) [1-5] involves the copper catalyzed N-arylation of 5-chloroindole (11) with 4-fluorobromobenzene (12). The product, 5-chloro-1-(4-fluorophenyl)indole (13), on treatment with 4-piperidinone hydrochloride monohydrate (14) under acidic conditions affords 5-chloro-1-(4-fluorophenyl)-3-(1,2,3,6-tetrahydropyridin-4-yl)-1H-indole hydrochloride (15). Reduction of 15 in the presence of platinum oxide yields 5-chloro-1-(4-fluorophenyl)-3-(4-piperdinyl)-1H-indole (9) which on condensation with 1-(2-chloroethyl)imidazolidinone (16) in the presence of a base gives sertindole (1).
![[1860-5397-7-5-i1]](http://beilstein-journals.org/bjoc/content/inline/1860-5397-7-5-i1.png?scale=2.0&max-width=1024&background=FFFFFF)
During the laboratory optimization of sertindole (1), many process related impurities were identified. The guidelines recommended by ICH state that the acceptable levels for a known and unknown compound (impurity) in the drug should be less than 0.15 and 0.10%, respectively [6]. In order to meet the stringent regulatory requirements, the impurities present in the drug substance must be identified and characterized. Literature reports [5,7-9] include impurities formed due to either over reduction (e.g., 2, 3 and 6) [5,7], incomplete reduction (e.g., 4 and 5) [5,8] or due to incomplete alkylation (e.g., 9 and 10) [5,7]. However, no synthetic details have been reported. In this context, the present study describes identification, synthesis and characterization of impurities formed during sertindole synthesis.
References
- Karamatskos, E; Lambert, M; Mulert, C; Naber, D (November 2012). “Drug safety and efficacy evaluation of sertindole for schizophrenia”. Expert Opinion on Drug Safety 11 (6): 1047–1062. doi:10.1517/14740338.2012.726984. PMID 22992213.
- “PRODUCT INFORMATION SERDOLECT® TABLETS” (PDF). TGA eBusiness Services. Lundbeck Australia Pty Ltd. 16 January 2013. Retrieved 27 October 2013.
- Juruena, MF; de Sena, EP; de Oliveira, IR (May 2011). “Sertindole in the Management of Schizophrenia” (PDF). Journal of Central Nervous System Disease 3: 75–85. doi:10.4137/JCNSD.S5729. PMC 3663609. PMID 23861640.
- Lewis, R; Bagnall, AM; Leitner, M (Jul 20, 2005). “Sertindole for schizophrenia.”. Cochrane database of systematic reviews (Online) (3): CD001715. doi:10.1002/14651858.CD001715.pub2. PMID 16034864.
- Taylor, D; Paton, C; Shitij, K (2012). The Maudsley prescribing guidelines in psychiatry. West Sussex: Wiley-Blackwell. ISBN 978-0-470-97948-8.
- Leucht, S; Cipriani, A; Spineli, L; Mavridis, D; Orey, D; Richter, F; Samara, M; Barbui, C; Engel, RR; Geddes, JR; Kissling, W; Stapf, MP; Lässig, B; Salanti, G; Davis, JM (September 2013). “Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis.”. Lancet 382 (9896): 951–962. doi:10.1016/S0140-6736(13)60733-3. PMID 23810019.
- Roth, BL; Driscol, J (12 January 2011). “PDSP Ki Database”. Psychoactive Drug Screening Program (PDSP). University of North Carolina at Chapel Hill and the United States National Institute of Mental Health. Retrieved 27 October 2013.
- Brunton, L; Chabner, B; Knollman, B (2010). Goodman and Gilman’s The Pharmacological Basis of Therapeutics (12th ed.). New York: McGraw-Hill Professional. ISBN 978-0-07-162442-8.
- http://www.trc-canada.com/detail.php?CatNum=D230095&CAS=173294-84-3&Chemical_Name=Dehydrosertindole&Mol_Formula=C24H24ClFN4O&Synonym=1-%5B2-%5B4-%5B5-Chloro-1-(4-fluorophenyl)-1H-indol-3-yl%5D-1-piperidinyl%5Dethyl%5D-1,3-dihydro-2H-Imidazol-2-one;%20Lu%2028-092
- Zeneca’s Seroquel Nears Market Approval – The Pharma Letter, 16 July 1997
- Abbott Labs Withdraws Sertindole NDA Sertindole – The Pharma Letter, 12 Jan 1998
- “WHO Pharmaceuticals Newsletter 1998, No. 03&04: Regulatory actions: Sertindole – approval application withdrawn”.
- FDA Advisory Committee provides opinion on Serdolect for the treatment of schizophrenia – Lundbeck press release, 8 Apr 2009
- Food and Drug Administration; Minutes of the Psychphamacological Drugs Advisory Committee, 7 Apr 2009
- [1]
- EU CHMP recommends lifting ban on atypical antipsychotic Serdolect (sertindole) – National electronic Library for Medicines, NHS
- COMMITTEE FOR PROPRIETARY MEDICINAL PRODUCTS OPINION FOLLOWING AN ARTICLE 36 REFERRAL: SERTINDOLE – European Medicines Agency, 13 Sep 2002
- Restricted re-introduction of the atypical antipsychotic sertindole (Serdolect) – MHRA, 2002
Perregaard, J.; Arnt, J.; Boegesoe, K. P.; Hyttel, J.; Sanchez, C. (1992). “Noncataleptogenic, centrally acting dopamine D-2 and serotonin 5-HT2 antagonists within a series of 3-substituted 1-(4-fluorophenyl)-1H-indoles”. Journal of Medicinal Chemistry 35 (6): 1092. doi:10.1021/jm00084a014.
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
1-[2-[4-[5-chloro-1-(4-fluorophenyl)-indol-3-yl]-1-piperidyl]ethyl]imidazolidin-2-one
|
|
| Clinical data | |
| AHFS/Drugs.com | International Drug Names |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 75%[1] |
| Protein binding | 99.5%[1] |
| Metabolism | Hepatic (mostly via CYP2D6 and CYP3A4)[2][3] |
| Biological half-life | 3 days[2] |
| Excretion | Faecal (the majority), Renal (4% metabolites; 1% unchanged)[2] |
| Identifiers | |
| CAS Registry Number | 106516-24-9 |
| ATC code | N05AE03 |
| PubChem | CID: 60149 |
| IUPHAR/BPS | 98 |
| DrugBank | DB06144 |
| ChemSpider | 54229 |
| UNII | GVV4Z879SP |
| KEGG | D00561 |
| ChEBI | CHEBI:9122 |
| ChEMBL | CHEMBL12713 |
| Chemical data | |
| Formula | C24H26ClFN4O |
| Molecular mass | 440.941 |
///////
HCV NS5A Inhibitor from Theravance, Inc. to treat hepatitis C virus infection

((S)-1-{(S)-2-[4-(4′-{[6-((2R,5S)-2,5-dimethyl-4-methylcarbamoyl-piperazin-1-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester
N-[(1S)-1-[[(2S)-2-[5-[4′-[[[6-[(2R,5S)-2,5-dimethyl-4-[(methylamino)carbonyl]-1-piperazinyl]-3-pyridinyl]carbonyl]amino]-2′-(trifluoromethoxy)[1,1′-biphenyl]-4-yl]-1H-imidazol-2-yl]-1-pyrrolidinyl]carbonyl]-2-methylpropyl]-, Carbamic acid, methyl ester
Carbamic acid, N-[(1S)-1-[[(2S)-2-[5-[4′-[[[6-[(2R,5S)-2,5-dimethyl-4-[(methylamino)carbonyl]-1-piperazinyl]-3-pyridinyl]carbonyl]amino]-2′-(trifluoromethoxy)[1,1′-biphenyl]-4-yl]-1H-imidazol-2-yl]-1-pyrrolidinyl]carbonyl]-2-methylpropyl]-, methyl ester
CAS 1374883-22-3, 819.87, C41 H48 F3 N9 O6
CAS of DIHCl 1480448-59-6
CAS of DIHCl, H2O 1480448-63-2
Theravance, Inc. INNOVATOR
To treat hepatitis C virus infection

-
((S)-1-{(S)-2-[4-(4′-{[6-((2R,5S)-2,5-dimethyl-4-methylcarbamoyl-piperazin-1-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester (compound 1):
-
 Recent estimates place the number of people infected with the hepatitis C virus (HCV) worldwide at more than 170 million, including 3 million people in the United States. The infection rate is thought to be roughly 4 to 5 times that of the human immunodeficiency virus (HIV). While in some individuals, the natural immune response is able to overcome the virus, in the majority of cases, a chronic infection is established, leading to increased risk of developing cirrhosis of the liver and hepatocellular carcinomas. Infection with hepatitis C, therefore, presents a serious public health problem.The virus responsible for HCV infection has been identified as a positive-strand RNA virus belonging to the family Flaviviridae. The HCV genome encodes a polyprotein that during the viral lifecycle is cleaved into ten individual proteins, including both structural and non-structural proteins. The six non-structural proteins, denoted as NS2, NS3, NS4A, NS4B, NS5A, and NS5B have been shown to be required for RNA replication. In particular, the NS5A protein appears to play a significant role in viral replication, as well as in modulation of the physiology of the host cell. Effects of NS5A on interferon signaling, regulation of cell growth and apoptosis have also been identified. (Macdonald et al., Journal of General Virology (2004), 85, 2485-2502.) Compounds which inhibit the function of the NS5A protein are expected to provide a useful approach to HCV therapy.Commonly-assigned U.S. Provisional Application Nos. 61/410,267, filed on Nov. 4, 2010, 61/444,046, filed on Feb. 17, 2011, and 61/492,267, filed on Jun. 1, 2011, and U.S. application Ser. No. 13/288,216, filed on Nov. 3, 2012 disclose pyridyl-piperazinyl compounds
Recent estimates place the number of people infected with the hepatitis C virus (HCV) worldwide at more than 170 million, including 3 million people in the United States. The infection rate is thought to be roughly 4 to 5 times that of the human immunodeficiency virus (HIV). While in some individuals, the natural immune response is able to overcome the virus, in the majority of cases, a chronic infection is established, leading to increased risk of developing cirrhosis of the liver and hepatocellular carcinomas. Infection with hepatitis C, therefore, presents a serious public health problem.The virus responsible for HCV infection has been identified as a positive-strand RNA virus belonging to the family Flaviviridae. The HCV genome encodes a polyprotein that during the viral lifecycle is cleaved into ten individual proteins, including both structural and non-structural proteins. The six non-structural proteins, denoted as NS2, NS3, NS4A, NS4B, NS5A, and NS5B have been shown to be required for RNA replication. In particular, the NS5A protein appears to play a significant role in viral replication, as well as in modulation of the physiology of the host cell. Effects of NS5A on interferon signaling, regulation of cell growth and apoptosis have also been identified. (Macdonald et al., Journal of General Virology (2004), 85, 2485-2502.) Compounds which inhibit the function of the NS5A protein are expected to provide a useful approach to HCV therapy.Commonly-assigned U.S. Provisional Application Nos. 61/410,267, filed on Nov. 4, 2010, 61/444,046, filed on Feb. 17, 2011, and 61/492,267, filed on Jun. 1, 2011, and U.S. application Ser. No. 13/288,216, filed on Nov. 3, 2012 disclose pyridyl-piperazinyl compounds

SYNTHESIS
CLICK ON IMAGES FOR CLEAR VIEW
………………………..
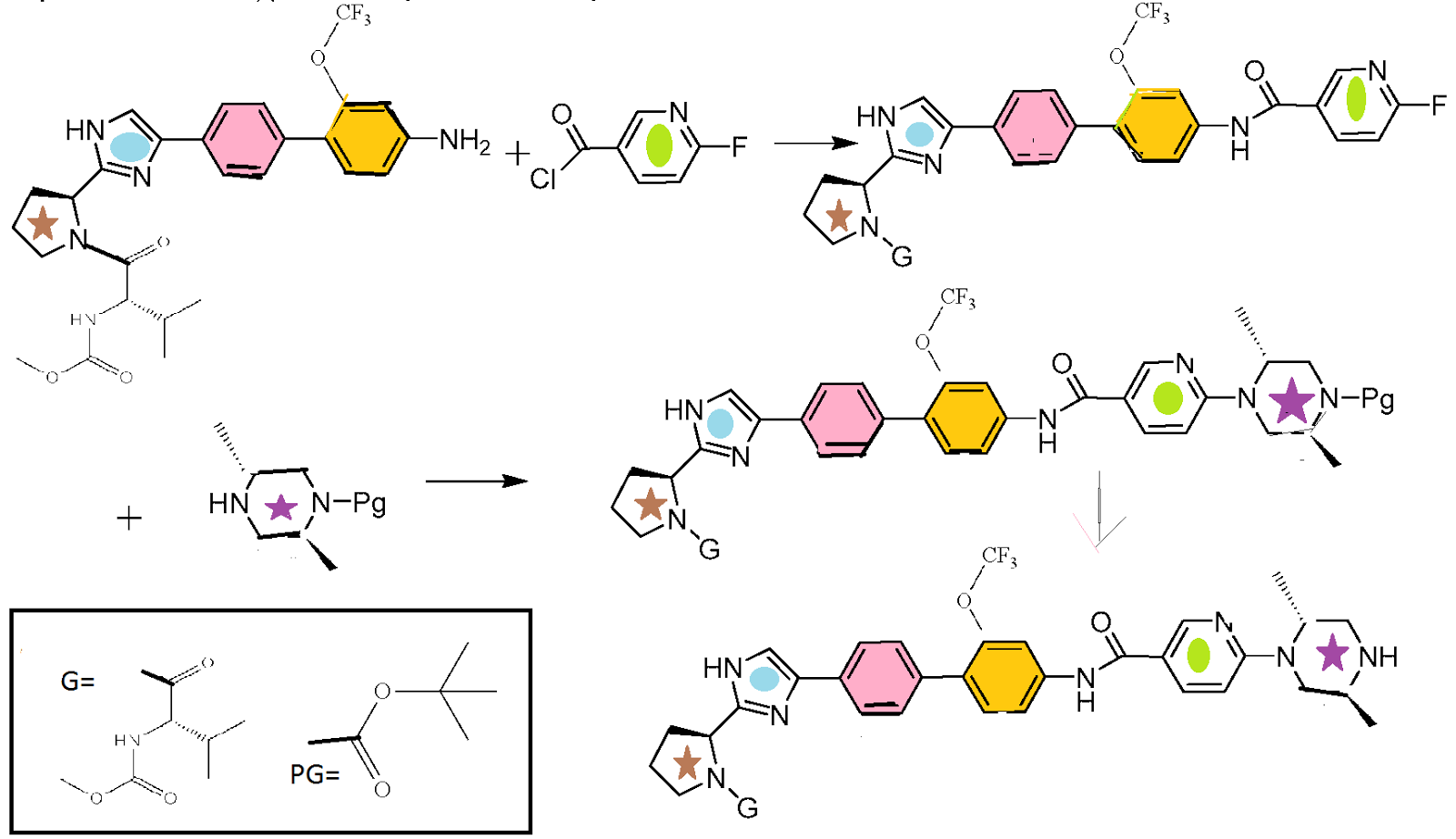
……………………………….
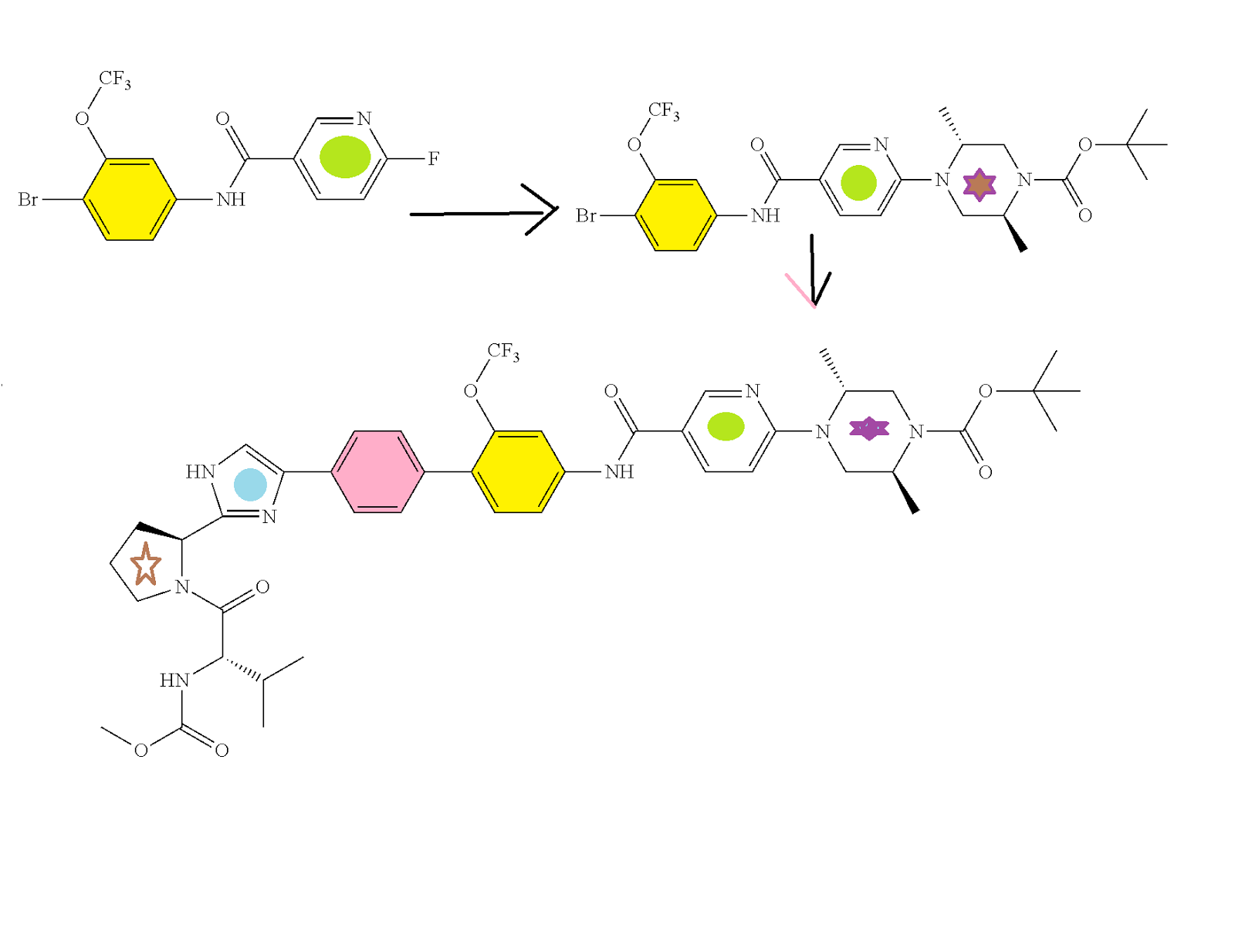
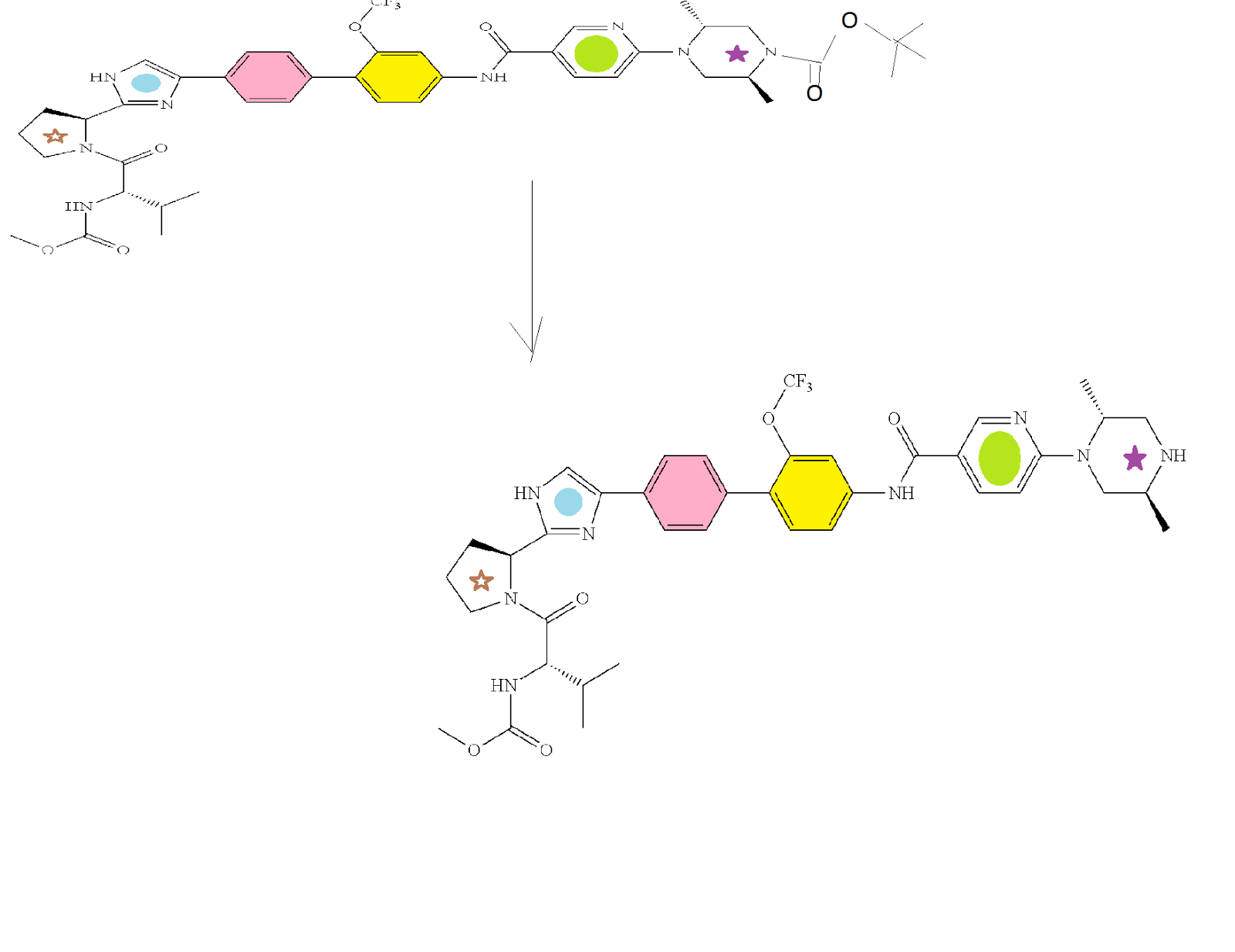
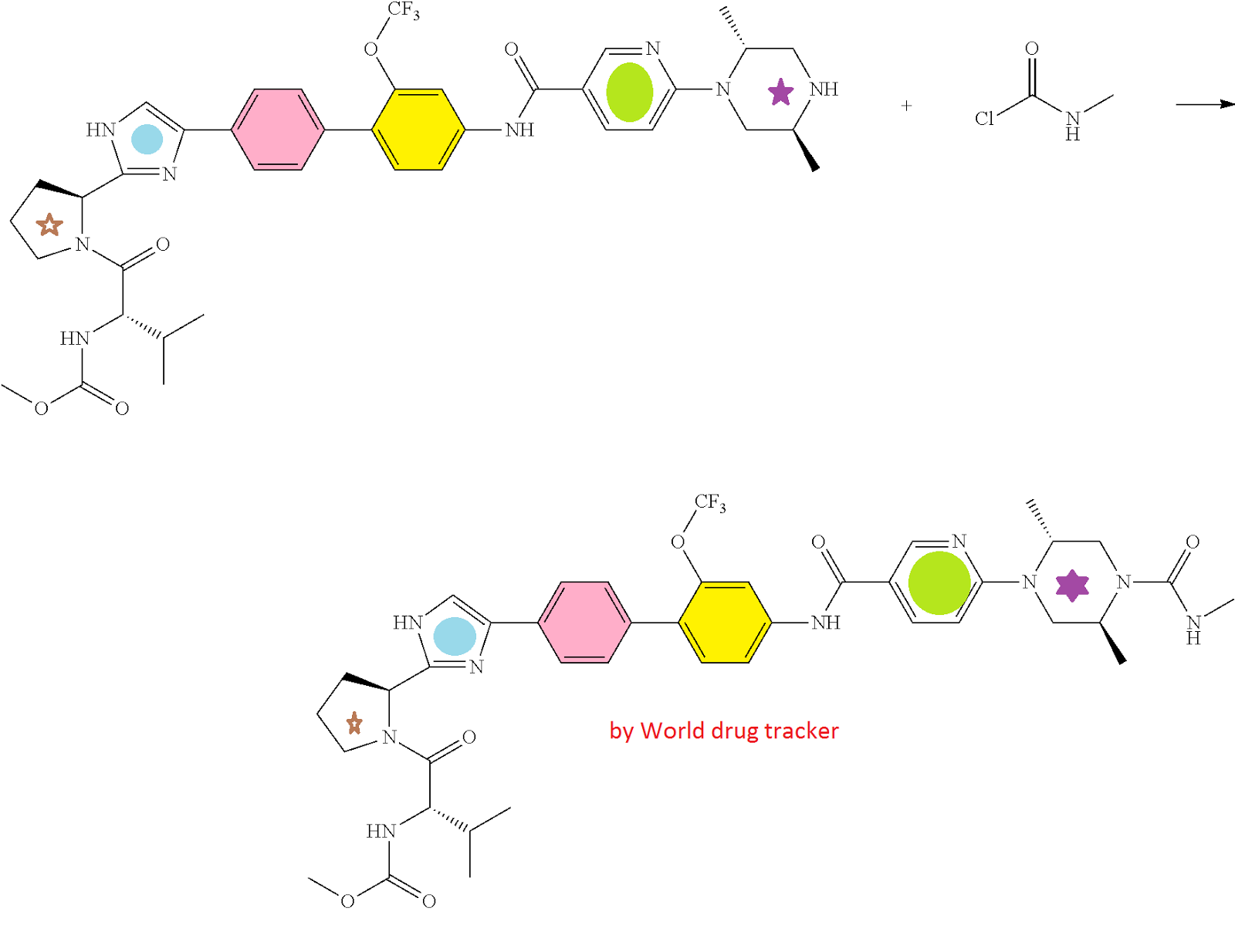
PATENT
WO-2013/165796
https://www.google.co.in/patents/WO2013165796A1?cl=en
PATENT
http://www.google.com/patents/US20130295048
crystalline compound 1 is advantageously prepared directly from the crude product of the final step of the synthesis of compound 1, illustrated in the following scheme, without purification of the amorphous form.

As described in Example 3 below, ((S)-1-{(S)-2-[4-(4′-{[6-(2R,5S)-2,5-dimethyl-piperazin-1-yl)-pyridine-3-carbonyl}-amino]-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester (2) is reacted with methylaminoformyl chloride to provide a crude product, which is recovered by conventional extraction and drying. The reaction is typically performed in the presence of an excess of base, in an inert diluent such as dichloromethane. Next, methanol is added to the crude product followed by the slow addition of water in a ratio of methanol:water of about 2.5:1 to about 2.7:1 to form a crystallization mixture. Seeds of crystalline compound 1 are added about halfway through the water addition. The crystallization mixture is stirred for a period of several days to form crystalline compound 1. To increase purity, the product can be recrystallized by a similar process: the crystalline compound is dissolved in methanol, water and seeds are added, such that the ratio of methanol to water in the mixture is about 2.5:1, and the mixture is stirred for a period of at least 12 hours to provide crystalline compound 1, which is recovered conventionally
-

-
To a solution of 4-bromo-3-trifluoromethoxy-phenylamine (3.15 g, 12.3 mmol) and triethylamine (3.43 mL, 24.6 mmol) in DCM (25 mL) was slowly added a solution of 2-fluoropyridine-5-carbonyl chloride (2.36 g, 14.8 mmol) in DCM (10 mL). After 2 h at RT, MTBE (90 mL) was added and the reaction mixture was washed with water, brine, and saturated sodium carbonate, dried, and evaporated to give a solid (5.4 g). Ethanol (43 mL) was added to the solid and then water (43 mL) was slowly added. The reaction mixture was stirred for 1.5 h, filtered, and washed with 1:4 ethanol:water (2×25 mL) to give the title intermediate as a white solid (3.87 g). Analytical HPLC: Retention time=21.3 min.
- Preparation 1: (2S,5R)-4-[5-(4-Bromo-3-trifluoromethoxy-phenylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester(a) N-(4-Bromo-3-trifluoromethoxy-phenyl)-6-fluoro-nicotinamide

(b) (2S,5R)-4-[5-(4-Bromo-3-trifluoromethoxy-phenylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester
-

-
The product of the previous step (3.86 g, 10.2 mmol) (2S,5R)-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester (2.62 g, 12.2 mmol) and N,N-diisopropylethylamine (5.32 mL, 30.5) was dissolved in DMSO (12 mL). The reaction mixture heated at 120° C. for 3 h, diluted with EtOAc (100 mL), washed with water, and saturated NH4Cl, water, and brine. The reaction mixture was evaporated to about 40% volume and 3 M HCl in cyclopentyl methyl ether (4.24 mL, 12.7 mmol) was added slowly. Seeds from a previous run at smaller scale were added and the reaction mixture was stirred for 2 days and filtered to provide the HCl salt of the title intermediate (5.15 g, 83% yield). Analytical HPLC: Retention time=21.1 min.

Preparation 2: (2S,5R)-4-[5-(4′-{2-[(S)-1-((S)-2-Methoxycarbonylamino-3-methyl-butyryl)-pyrrolidin-2-yl]-1H-imidazol-4-yl}-2-trifluoromethoxy-biphenyl-4-ylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester
-

-
To a solution of ((S)-1-{(S)-2-[4-(4-bromo-phenyl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester (3.05 g, 6.8 mmol), bis(pinacolato)diboron (1.81 g, 7.1 mmol) and potassium acetate (1.00 g, 10.2 mmol) was added nitrogen sparged toluene (15 mL). The resulting mixture was sparged with nitrogen and 1,1′-bis(diphenylphosphino)ferrocene-palladium(II)dichloride dichloromethane (Pd catalyst) (0.17 g, 0.204 mmol) was added. The reaction mixture was stirred at 90° C. overnight.
-
The reaction mixture was cooled to RT and to this mixture was added nitrogen sparged water (7.6 mL), potassium carbonate (5.16 g, 37.3 mmol), and (2S,5R)-4-[5-(4-bromo-3-trifluoromethoxy-phenylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester (4.35 g, 7.13 mmol). The reaction mixture was stirred at 95° C. overnight.
-
Another portion of the Pd catalyst used above (0.08 g, 0.10 mmol) was added to the reaction mixture. After 5 h, the reaction mixture was cooled to RT, diluted with EtOAc (150 mL), washed with water (150 mL) and brine (100 mL), dried over sodium sulfate, and evaporated to give a black residue (6.7 g), which was purified by silica gel chromatography (eluted with 50-100% EtOAc/hexane) to provide the title intermediate (5.3 g, 90% yield). Analytical HPLC: Retention time=14.7 min.

Preparation 3: ((S)-1-{(S)-2-[4-(4′-{[6-((2R,5S)-2,5-Dimethyl-piperazin-1-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester
-

-
Acetyl chloride (63.2 mL, 888 mmol) was added to ethanol (360 mL) and stirred at RT for 1 h. To the resulting HCl solution was added a solution of (2S,5R)-4-[5-(4′-{2-[(S)-1-((S)-2-methoxycarbonylamino-3-methyl-butyryl)-pyrrolidin-2-yl]-1H-imidazol-4-yl}-2-trifluoromethoxy-biphenyl-4-ylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester (73 g, 84 mmol) in ethanol (360 mL). The reaction mixture was stirred at RT overnight.
-
The reaction mixture was concentrated to dryness (124 g crude). Water 500 mL) was added and the mixture was extracted with EtOAc (2×500 mL). The aqueous layer was adjusted to pH 4 with 1:1 NaOH:water. Ethyl acetate (400 mL) and sat. aq. Na2CO3 (100 mL) were added and the layers were separated. The organic layer was dried over Na2SO4 and evaporated to give the title intermediate (62.8 g; 88% yield). Analytical HPLC: Retention time=10.0 min.

Example 1Amorphous ((S)-1-{(S)-2-[4-(4′-{[6-((2R,5S)-2,5-Dimethyl-4-methylcarbamoyl-piperazin-1-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester

(a) ((S)-1-{(S)-2-[4-(4′-{[6-((2R,5S)-2,5-Dimethyl-piperazin-1-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester tri-HCl
-
Acetyl chloride (0.71 mL, 10.0 mmol) was added to ethanol (7 mL) and stirred at RT for 1 h. The resulting HCl solution was added to a solution of (2S,5R)-4-[5-(4′-{2-[(5)-1-((S)-2-methoxycarbonylamino-3-methyl-butyryl)-pyrrolidin-2-yl]-1H-imidazol-4-yl}-2-trifluoromethoxy-biphenyl-4-ylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester (1.55 g, 1.8 mmol) in ethanol (7 mL). The reaction mixture was warmed to 35° C. and stirred overnight. The mixture was concentrated to dryness, and chased with DCM to provide the crude tri-HCl salt of the title intermediate (1.57 g) which was used directly in the next step. HPLC method C: Retention time=10.0 min.
(b) Amorphous ((S)-1-{(S)-2-[4-(4′-{[6-((2R,5S)-2,5-Dimethyl-4-methylcarbamoyl-piperazin-1-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester
-
To a solution of the product of the previous step (1.57 g crude, ca. 1.80 mmol) and N,N-diisopropylethylamine (3.14 mL, 18.0 mmol) in DCM (24 mL) was slowly added 1 M methylaminoformyl chloride in DMA (1.8 mL). The reaction mixture stirred at RT for 1 h, and then 1 M methylaminoformyl chloride in DMA (1.8 mL) was added. The reaction was quenched with sat. aq. NaHCO3 and the reaction mixture was stirred for 20 min. The layers were separated and the organic layer was dried and evaporated to give a residue. To the residue was added methanol (15 mL) followed by 2 N LiOH/water (3 mL). The reaction mixture was stirred at RT for 1 h, diluted with water, extracted with DCM (80 mL), dried, and evaporated to give a crude product which was purified by silica gel chromatography (40 g silica, 2-8% MeOH/DCM) to provide the title compound (0.93 g, 63% yield). Analytical HPLC: Retention time=11.0 min.
HPLC
- Analytical HPLC Method
-
-
- Column: Zorbax Bonus-RP 3.5 μm. 4.6×150 mm
- Column temperature: 35° C.
- Flow rate: 1.0 mL/min
- Mobile Phases: A=Water/ACN (98:2)+0.1% TFA
- B=Water/ACN (10:90)+0.1% TFA,
- Injection volume: 100-1500 μL
- Detector wavelength: 214 nm
- Sample preparation: Dissolve in 1:1 ACN:water
- Gradient: 29 min total (time (min)/% B): 0.5/10, 24/90, 25/90, 26/10, 29/10
-
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2010094977A1 * | 22 Feb 2010 | 26 Aug 2010 | Arrow Therapeutics Limited | Novel biphenyl compounds useful for the treatment of hepatitis c |
| WO2012061552A1 * | 3 Nov 2011 | 10 May 2012 | Theravance, Inc. | Novel inhibitors of hepatitis c virus |
| US201113288216 |
About Theravance Biopharma
The mission of Theravance Biopharma (NASDAQ: TBPH) is to create value from a unique and diverse set of assets: an approved product; a development pipeline of late-stage assets; and a productive research platform designed for long-term growth.
Our pipeline of internally discovered product candidates includes potential best-in-class opportunities in underserved markets in the acute care setting, representing multiple opportunities for value creation. VIBATIV® (telavancin), our first commercial product, is a once-daily dual-mechanism antibiotic approved in the U.S., Europe and certain other countries for certain difficult-to-treat infections. Revefenacin (TD-4208) is an investigational long-acting muscarinic antagonist (LAMA) being developed as a potential once-daily, nebulized treatment for COPD. Axelopran (TD-1211) is an investigational potential once-daily, oral treatment for opioid-induced constipation (OIC). Our earlier-stage clinical assets represent novel approaches for potentially treating diseases of the lung and gastrointestinal tract and infectious disease. In addition, we have an economic interest in future payments that may be made by GlaxoSmithKline plc pursuant to its agreements with Theravance, Inc. relating to certain drug development programs, including the combination of fluticasone furoate, umeclidinium and vilanterol (the “Closed Triple”).
With our successful drug discovery and development track record, commercial infrastructure, experienced management team and efficient corporate structure, we believe that we are well positioned to create value for our shareholders and make a difference in the lives of patients.
For more information, please visit www.theravance.com.
THERAVANCE®, the Cross/Star logo, MEDICINES THAT MAKE A DIFFERENCE® and VIBATIV® are registered trademarks of the Theravance Biopharma group of companies.
Journal of Medicinal Chemistry (2014), 57(5), 1643-1672……….

(e)Thalladi, V. R.; Nzerem, J.; Huang, X.; Zhang, W. Crystalline form of a pyridyl-piperazinyl hepatitis C virus inhibitor. World Patent Application WO-2013/165796, November 7, 2013.
PATENT
WO 2012061552
http://www.google.com.ar/patents/WO2012061552A1?cl=en
Preparation 28: ((S)-l-{(S)-2-[4-(4′-{[6-((2JR,5S)-2,5-Dimethyl-piperazin-l- yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-lH-imidazol-2- yl]-pyrrolidine-l-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester

A mixture of [(5)-2-methyl-l-((5)-2- {4-[4-(4,4,5,5-tetramethyl- [l,3,2]dioxaborolan-2-yl)-phenyl]-lH-imidazol-2-yl}-pyrrolidine-l-carbonyl)-propyl]- carbamic acid methyl ester (86 mg, 0.17 mmol) and (25′,5R)-4-[5-(4-bromo-3- trifluoromethoxy-phenylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-l-carboxylic acid tert-butyl ester (100 mg, 0.2 mmol, Preparation 27) was dissolved in 1,4-dioxane (1.8 mL, 23 mmol) and water (0.25 mL, 14 mmol). Cesium carbonate (170 mg, 0.52 mmol) was added. The reaction mixture was sparged with nitrogen and then
tetrakis(triphenylphosphine)palladium(0) (12.1 mg, 0.011 mmol) was added. The reaction mixture was sealed under nitrogen and heated at 95 °C overnight. The reaction mixture was extracted with ethyl acetate/water, the organic layer was dried over sodium sulfate and concentrated to produce an orange oil.
The oil from the previous step was treated with 4 M HCl in 1,4-dioxane (2 mL, 7 mmol) and stirred at room temperature for 1 h. The reaction mixture was concentrated and evaporated with ethyl acetate (2 x) to produce the HCl salt of the title compound as a yellow solid which was purified by preparative HPLC to provide the tri-TFA salt of the title compound (150 mg, 30 % overall yield), (m/z): [M+H] calcd for
763.35 found 763.7.
Example 29 ((S)-l-{(S)-2-[4-(4′-{[6-((2JR,5S)-2,5-Dimethyl-4- methylcarbamoyl-piperazin-l-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy- biphenyl-4-yl)-lH-imidazol-2-yl]-pyrrolidine-l-carbonyl}-2-methyl-propyl)- carbamic a

To a solution of ((5)- l – {(5)-2-[4-(4′- { [6-((2R,55)-2,5-dimethyl-piperazin- l-yl)- pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-lH-imidazol-2-yl]- pyrrolidine- l-carbonyl} -2-methyl-propyl)-carbamic acid methyl ester tri-TFA (1 1.4 mg, 0.01 1 mmol; Preparation 28) and N,N-diisopropylethylamine (18 uL, 0.1 1 mmol) dissolved in DMA (0.4 mL, 4 mmol) was added 1.0 M methyl isocyanate in toluene (10 uL, 0.01 mmol). The reaction mixture was stirred at RT overnight, concentrated, dissolved in 1 : 1 acetic acid:water (1.5 mL) and purified by preparative HPLC to provide the di-TFA salt of the title compound (7.1 mg). (m/z): [M+H]+ calcd for C41H48F3N906 820.37 found 820.5.
Alternative synthesis of ((5)-1-{(5)-2-[ -(4′-{[6-((2Λ,ί» 2,5- Dimethyl-4-methylcarbamoyl-piperazin-l-yl)-pyridine-3-carbonyl]-amino}-2′- trifluoromethoxy-biphenyl-4-yl)-lH-imidazol-2-yl]-pyrrolidine-l-carbonyl}-2- methyl-propyl)-carbamic acid methyl ester
(a) N-(‘4-Bromo-3-trifluoromethoxy-phenyl -6-fluoro-nicotinamide

To a solution of 4-bromo-3-trifluoromethoxy-phenylamine (3.15 g, 12.3 mmol) and triethylamine (3.43 mL, 24.6 mmol) in DCM (25 mL) was slowly added a solution of 2-fluoropyridine-5-carbonyl chloride (2.36 g, 14.8 mmol) in DCM (10 mL). After 2 h at RT, MTBE (90 mL) was added and the reaction mixture was washed with water, brine, and saturated sodium carbonate, dried, and evaporated to give a solid (5.4 g). Ethanol (43 mL) was added to the solid and then water (43 mL) was slowly added. The reaction mixture was stirred for 1.5 h, filtered, and washed with 1 :4 ethanohwater (2 x 25 mL) to give the title intermediate as a white solid (3.87 g). HPLC method C: Retention time = 21.3 min.
(b) (25,,5R)-4-r5-(4-Bromo-3-trifluoromethoxy-phenylcarbamoyl) -pyridin-2-yl1-2,5- dimethyl-piperazin – 1 -carboxylic acid fe/t-butyl ester

The product of the previous step (3.86 g, 10.2 mmol) (2S,5R)-2,5-dimethyl- piperazine-1 -carboxylic acid tert-butyl ester (2.62 g, 12.2 mmol) and N,N- diisopropylethylamine (5.32 mL, 30.5) was dissolved in DMSO (12 mL). The reaction mixture heated at 120 °C for 3 h, diluted with EtOAc (100 mL), washed with water, and saturated NH4C1, water, and brine. The reaction mixture was evaporated to about 40% volume and 3 M HCl in cyclopentyl methyl ether (4.24 mL, 12.7 mmol) was added slowly. Seeds from a previous run at smaller scale were added and the reaction mixture was stirred for 2 days and filtered to provide the HCl salt of the title intermediate (5.15 g, 83 % yield). HPLC method C: Retention time = 21.1 min (c) (2 .5R)-4 5-(4′-{2 ffl -((5f)-2-Methoxycarbonylamino-3-methyl-butyrvn- pyiTolidin-2-yl1-lH-imidazol-4-yl}-2-tri
pyridin-2- -2,5-dimethyl-piperazine-l-carboxylic acid fert-butyl ester

To a solution of ((5′)-l-{(5,)-2-[4-(4-bromo-phenyl)-lH-imidazol-2-yl]- pyrrolidine-l-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester (3.05 g, 6.8 mmol;), bis(pinacolato)diboron (1.81 g, 7.1 mmol) and potassium acetate (1,00 g, 10.2 mmol) was added nitrogen sparged toluene (15 mL). The resulting mixture was sparged with nitrogen and l,l’-bis(diphenylphosphino)ferrocene-palladium(II)dichloride dichloromethane (Pd catalyst) (0.17 g, 0.204 mmol) was added. The reaction mixture was stirred at 90 °C overnight.
The reaction mixture was cooled to RT and to this mixture was added nitrogen sparged water (7.6 mL), potassium carbonate (5.16 g, 37.3 mmol). The reaction mixture was stirred at 95°C overnight.
Another portion of the Pd catalyst used above (0.08 g, 0.10 mmol) was added to the reaction mixture. After 5 h, the reaction mixture was cooled to RT, diluted with EtOAc (150 mL), washed with water (150 mL) and brine (100 mL), dried over sodium sulfate, and evaporated to give a black residue (6.7 g), which was purified by silica gel chromatography (eluted with 50-100 % EtOAc/hexane) to provide the title intermediate (5.3 g, 90 % yield). HPLC method C: Retention time = 14.7 min.
(d) (ffl ffl-2 4-(4′ r6-((2R,5^-2,5-Dimethyl-piperazin-l-vn-pyridine-3-carbonyl1- amino}-2′ rifluoromethoxy-biphenyl-4-yl -lH-imidazol-2-yl1-pyrrolidine-l- carbonyl}-2-methyl-propyl)-carbamic acid methyl ester

Acetyl chloride (2.57 mL, 36.2 mmol) was added to ethanol (18 mL) and stirred at RT for 1 h. To the resulting HQ solution was added a solution of the product of the previous step (3.90 g, 4.5 mmol) in ethanol (18 mL). The reaction mixture was warmed to 35 °C and stirred overnight. Acetyl chloride (1.28 mL, 18.1 mmol) was added to ethanol (7.8 mL) and stirred for 30 min. The resulting HC1 solution was added to the reaction mixture at 35 °C. The temperature was raised to 40 °C. The mixture was concentrated to dryness chased by dichloromethane to provide the crude tri-HCl salt of the title intermediate (5.4 g) which was used directly in the next step. HPLC method C: Retention time = 10.1 min.
(e) ((^-l- {(^-2-r4-(4′- {r6-((2R.5^-2.5-Dimethyl-4-methylcarbamoyl-piperazin-l-yl)- pyridine-3-carbonyl1-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-lH-imidazol-2-yl1- pyrrolidine-l-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester

To a solution of the product of the previous step (5.4 g crude, ca. 3.96 mmol) and N,N-diisopropylethylamine (6.89 mL, 39.6 mmol) in DCM (52 mL) was slowly added 1 M methylaminoformyl chloride in DMA (4.3 mL). The reaction mixture stirred at room temperature for 1 h, and then water (50 mL) was added. The organic layer was washed with saturated NH4C1 and then brine, dried over Na2S04 and evaporated to give 5.2 g crude product, which was purified by silica gel chromatography (133 g silica, 2 to 8 % methanol/DCM for 15 min then 8 % methanol/DCM for 40 min) to provide the title compound (2.4 g, 74 % yield). HPLC method C: Retention time 1 1.2 min
Synthesis of intermediates
http://www.google.com.ar/patents/WO2012061552A1?cl=en
Preparation 1: 4-(4-bro -phenyl)-2-(S)-pyrrolidin-2-yl-lH-imidazole

(a) 2-Bromo-l-(4-bromo-phenyl)-ethanone
Bromine (80 g, 500 mmol) was added dropwise to a solution of l-(4-bromo- phenyl)-ethanone (100 g, 500 mmol) in dichloromethane (1500 mL) at ambient temperature. The reaction mixture was stirred for 3 h and then concentrated. The residue was washed with dichloromethane (100 mL) to give the crude title compound (120 g, 86
% yield) as a white solid. XH NMR (CDC13, 400 MHz) δ (ppm): 7.78 (d, J=8.4 Hz, 2H), 7.57 (d, J=8.4 Hz, 2H), 4.32 (s, 2H).
(b) (^-pyrrolidine- 1 ,2-dicarboxylic acid 2-r2-(4-bromo-phenyl)-2-oxo-ethyl1 ester \-tert- butyl ester
Diisopropylethylamine (67 g, 518 mmol) was added dropwise to a solution of the product of the previous step (120 g, 432 mmol) and (5)-pyrrolidine-l,2-dicarboxylic acid 1-tert-butyl ester ( -Boc proline) (102 g, 475 mmol) in acetonitrile (2 L) at room temperature. The reaction mixture was stirred overnight and concentrated to dryness. The residue was dissolved in ethyl acetate (2 L) and washed with water (2 L). The organic layer was dried over sodium sulfate and concentrated to give crude title compound (178 g, 100 % yield).
(c) (5f)-2-r4-(4-bromo-phenyl -lH-imidazol-2-yl1-pyrrolidine-l-carboxylic acid fe/t-butyl ester
A solution of the product of the previous step (178 g, 432 mmol) and ammonium acetate (500 g, 6.5 mol) in toluene (2 L) was heated at reflux overnight. The solvent was removed and the residue was dissolved in ethyl acetate (2 L) and washed with water (2 L). The organic layer was dried over sodium sulfate and concentrated. The residue was purified by silica gel column chromatography in 1:3 petroleum ether: ethyl acetate to give the title compound (120 g, 71 % yield) as a yellow solid. ¾ NMR (CDC13, 400 MHz) δ (ppm): 7.56 (s, 1H), 7.39 (d, J=8.0 Hz, 2H), 7.24 (m, 1H), 7.14 (s, 1H), 4.88 (m, 1H), 3.33 (m, 2H), 2.94 (s, 1H), 2.07 (m, 2H), 1.88 (m, 1H), 1.42 (s, 9H).
(d) 4-(4-bromo-phenyl)-2-(5f)-pyrrolidin-2-yl-lH-imidazole
To a solution of (5)-2-[4-(4-bromo-phenyl)-lH-imidazol-2-yl]-pyrrolidine-l- carboxylic acid tert-butyl ester (3 g, 7.6 mmol) in methanol (3 mL) was added 4N HQ in methanol (60 mL) at 0 °C. The reaction mixture was stirred for 2 h and then concentrated to give crude hydrochloride salt of the title compound (2.51 g 100 % yield) as a yellow solid.
Preparation 2: (5)-2-Methoxycarbonylamino-3-methyl-butyric acid

A mixture of (5)-2-amino-3 -methyl-butyric acid (10 g, 85 mmol), NaOH (10.3 g, 255 mmol) in water (100 mL) was treated with methylchloridocarbonate (8 g, 85 mmol) at 0 0 C. The reaction mixture was stirred for 24 h at room temperature and then 5 N aqueous HC1 was added to the reaction mixture to adjust pH to 4. The mixture was filtered through a pad of Celite to give the product (10 g, 67% yield) as a white solid. ¾ NMR (CH3OD, 400 MHz) δ (ppm) 4.05(d, 1H), 3.65(s, 3H), 2.14(m, 1H), 0.95(m, 6H). Preparation 3: ((S)-l-{(S)-2-[4-(4-bromo-phenyl)-lH-imidazol-2-yl]- pyrrolidine-l-carbonyl}-2-met methyl ester

Triethylamine (2.3 g, 11.4 mmol) was added to a solution of 4-(4-bromo-phenyl)- 2-(5)-pyrrolidin-2-yl-lH- imidazole hydrochloride (2 g, 11.4 mol), (5)-2- methoxycarbonylamino-3-methyl-butyric acid (2.5 g, 7.6 mmol), and HATU (4.3 g, 11.4 mmol) in dimethylformamide (50 mL) at 0 °C under nitrogen. The reaction mixture was stirred at room temperature overnight and treated with ethyl acetate (100 mL) and water (1000 mL). The organic layer was washed with water (2 x 100 mL) and brine (100 mL), dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel column chromatography in 1 : 1 petroleum ether: ethyl acetate to give the title compound (2.5 g 74 % yield) as a yellow solid. XH NMR (i¾-DMSO, 400 MHz) δ (ppm) 7.63 (d, J=8.8 Hz, 2H), 7.54 (m, IH), 7.47 (m, 2H), 7.26 (d, J=8.4 Hz, IH), 5.03 (m, IH), 4.02 (t, J =8.4 Hz, IH), 3.76 (m, 2H), 3.51 (s, 3H), 2.10 (m, 2H), 1.93 (m, 3H), 0.85 (d, J=6.8 Hz, 3H), 0.81 (d, J=6.8 Hz, 3H).
I AM NOT SURE OF BELOW DATA, It is a cut paste for TD 6450 , NOT ABLE TO CONNECT CAS 1374883-22-3 WITH TD 6450
IF YOU HAVE A STRUCTURE PIC FOR THE SAME MAIL ME amcrasto@gmail.com, call +919323115463
POSTER
50th Annu Meet Eur Assoc Study Liver (EASL) (April 22-26, Vienna) 2015, Abst P0898
http://ilc-congress.eu/abstract_25_04/ILC2015-abstract-book-25-04-Saturday.pdf
P0898
TD-6450,
A NEXT GENERATION ONCE-DAILY NS5A INHIBITOR, HAS POTENT ANTIVIRAL ACTIVITY FOLLOWING A 3-DAY MONOTHERAPY STUDY IN GENOTYPE 1 HCV INFECTION
E. Lawitz1, M. Rodriguez-Torres2, R. Kohler3, A. Amrite3, C. Barnes3, M.L.C. Pecoraro3, J. Budman3, M. McKinnell3, C.B. Washington3. 1Texas Liver Institute, University of Texas Health Science Center, San Antonio, TX, United States; 2Fundacion de Investigacion, San Juan, Puerto Rico; 3Theravance Biopharma, South San Francisco, CA, United States
E-mail: cwashington@theravance.com
Background and Aims: TD-6450 is a next generation HCV NS5A inhibitor with superior in vitro potency against resistanceassociated variants (RAVs) encountered with first-generation NS5A inhibitors. This study evaluated the safety, pharmacokinetics (PK) and antiviral activity of TD-6450 following multiple oral doses in HCV patients
Theravance Biopharma Announces Positive Results From Phase 1 Proof-of-Concept Study of TD-6450, an NS5A Inhibitor to Treat Hepatitis C
240 mg Achieved a Median Maximal Viral Load Decline of 4.9 Log10 IU/mL Following Three Daily Doses in Genotype 1a Patients
SOUTH SAN FRANCISCO, CA — (Marketwired) — 11/03/14 — Theravance Biopharma (NASDAQ: TBPH), through its U.S. operating subsidiary, Theravance Biopharma US, Inc., today announced positive results from the first three cohorts of Study 0110, a Phase 1 proof-of-concept study of TD-6450, a next-generation investigational NS5A inhibitor in development to treat patients with hepatitis C virus infection (HCV).
TD-6450 was evaluated in three cohorts of eight genotype 1a (GT-1a) patients each at doses of 60, 120 and 240 mg, administered once-daily for three days. TD-6450 demonstrated dose-dependent antiviral activity with median maximal declines of HCV RNA of 3.87, 4.63 and 4.89 log10 IU/mL for doses of 60, 120 and 240 mg, respectively.
In the 120 and 240 mg dose groups, three days of once-daily oral treatment resulted in levels of serum HCV RNA below the limit of detection (LOD) in 43% (3/7) and 57% (4/7) of patients treated with TD-6450, respectively. Three of the seven LOD patients went on to show no measurable virus at Day 14, and two of these patients still had no measurable virus at Day 28. At a two-month time point in a long-term follow-up study, the viral load in these two patients was measurable, but both remained more than three logs below their baseline.
None of the patients in the three dose groups had virologic breakthrough during their three-day treatment course, and 100% of the treated GT-1a patients in the study achieved at least a three log10 IU/mL reduction of HCV RNA. At the 120 and 240 mg doses, 71% (5/7) and 86% (6/7) of treated patients achieved at least a four log10 IU/mL reduction in HCV RNA, respectively.
All doses of TD-6450 were generally well tolerated after three doses and for the 28-day observation period. There were no serious adverse events and no patient discontinuations. There was no pattern of clinical adverse events or laboratory abnormalities related to treatment.
“We see diverse responses to direct antivirals in genotype 1 populations. Despite recent advances in HCV therapy, significant treatment challenges remain, including the required length of drug therapy. The robust activity of TD-6450 in genotype 1a patients suggests that this potentially best-in-class NS5A inhibitor could be a component of short and highly active combination therapy regimens,” said Eric Lawitz, MD, Vice President of Scientific and Research Development at the Texas Liver Institute and Clinical Professor of Medicine, The University of Texas Health Science Center San Antonio, and one of the principal investigators on the Phase 1 study.
“TD-6450, created using the principles of multivalent design, has a heterodimeric structure distinct from other NS5A inhibitors. We believe this unique structure allows it to bind asymmetrically across the NS5A protein interface, providing high in vitro potency against clinically encountered resistance-associated variants. We believe the potency of TD-6450 against both wild type virus and these resistance-associated variants enables the robust antiviral activity that we reported today,” said Mathai Mammen, MD, Senior Vice President, Research and Development, Theravance Biopharma. “We look forward to analyzing the full set of results from this Phase 1 study and evaluating the next steps in the development strategy for TD-6450.”
About the Phase 1 Proof-of-Concept Study (Study 0110)
This Phase 1 study is a double-blind, randomized, placebo-controlled, multiple-dose study to evaluate the safety, tolerability, pharmacokinetics and antiviral activity of orally administered TD-6450 in non-cirrhotic, treatment-naive patients with GT-1, 2, or 3 chronic HCV infection. The study includes seven cohorts. The first three cohorts enrolled eight GT-1a patients each (7 active; 1 placebo) and tested once-daily oral doses of 60, 120 or 240 mg, respectively. Patients were dosed for three days and followed for up to 28 days for viral load quantification. The limit of detection for the viral load quantification assay is 15 IU/mL.
Safety evaluations include monitoring for adverse events, routine laboratory assessments, vital signs and 12-lead ECG tracings.
In cohorts 4 through 6, patients with GT-1b, GT-2 and GT-3 are dosed once-daily at 240 mg. An additional cohort (cohort 7) of GT-1a patients is dosed twice daily with 240 mg. Data generation and analysis of results for cohorts 4 through 7 is ongoing. An interim analysis of those cohorts showed antiviral activity for GT-1b similar to that for GT-1a, but minimal antiviral activity for GT-2 and GT-3.
The Company anticipates presenting further data on all cohorts at a future scientific conference.
About TD-6450
TD-6450 is an internally discovered multivalent NS5A inhibitor designed to have improved antiviral activity against GT-1 resistance-associated variants (RAV) resistant to first generation NS5A inhibitors. TD-6450’s heterodimeric structure permits an asymmetric binding mode to NS5A relative to structurally symmetric inhibitors. TD-6450 has demonstrated additive activity with other classes of anti-HCV agents in replicon assays, and no cross-resistance with RAVs that confer resistance to other anti-HCV agents. The Company believes that the antiviral activity of TD-6450, in combination with other antivirals, may help improve cure rates and/or reduce treatment times for appropriate patients.
TD-6450 was previously evaluated in a single-ascending dose and a 14-day multiple-ascending dose study in healthy subjects (study 0094). This randomized, double-blind, placebo-controlled study evaluated the safety, tolerability and pharmacokinetics of TD-6450. Single doses (up to 500 mg) and multiple doses of TD-6450 (up to 240 mg daily for 14 days) were evaluated in healthy subjects. Following single and multiple doses, TD-6450 was generally well-tolerated and no subjects discontinued due to adverse events. Headache was the most commonly reported adverse event following multiple doses (n=4). TD-6450 pharmacokinetics were linear up to 240 mg following single and multiple doses and its long half-life supports once-daily dosing.
About Hepatitis C and the NS5A Inhibitor Class
Hepatitis C is an infectious disease of the liver. Worldwide, health experts estimate that 130 – 150 million people have chronic hepatitis C, with as many as four million of those cases in the United States. Hepatitis C, like all forms of hepatitis, can damage the liver. Of people infected, 55 to 85 percent will develop chronic infection, and 75 percent of those with chronic infection will develop chronic liver disease.
The hepatitis C non-structural 5A (NS5A) protein of HCV has emerged as an attractive drug target and inhibitors of NS5A have a central role in all-oral HCV therapy. The multi-functional NS5A protein is required for ribonucleic acid (RNA) replication and virion assembly, and a number of investigational and approved NS5A inhibitors have shown antiviral efficacy in HCV-infected patients.
Theravance Biopharma and Trek Therapeutics Announce Initiation of Phase 2a Trial of TD-6450, an NS5A Inhibitor to Treat Hepatitis C
Study Being Conducted by Trek Therapeutics Following Licensing of Worldwide Rights to Drug Candidate From Theravance Biopharma
DUBLIN, IRELAND and CAMBRIDGE, MA — (Marketwired) — 10/27/15 — Theravance Biopharma, Inc. (NASDAQ: TBPH) (“Theravance Biopharma”) and Trek Therapeutics (“TREKtx”) today announced that TREKtx has initiated a Phase 2a clinical trial of TD-6450, a next-generation investigational NS5A inhibitor in development to treat patients with hepatitis C virus (HCV). Theravance Biopharma recently granted TREKtx an exclusive worldwide license for the development, manufacturing, use, marketing and sale of TD-6450 as a component in combination HCV products. Other terms of the transaction have not been disclosed.
The Phase 2a clinical trial will evaluate faldaprevir (FDV), an HCV protease inhibitor, combined with TD-6450 and ribavirin (RBV) in patients infected with HCV genotype 4. The trial is being conducted in the United States.
Mathai Mammen, M.D., Ph.D., Senior Vice President of Research and Development at Theravance Biopharma commented, “We are pleased to see the initiation of this Phase 2a clinical trial with TD-6450. This NS5A inhibitor has shown robust antiviral activity in a Phase 1 trial in patients with HCV genotype 1, as well as preclinical potency against both wild type HCV and resistance-associated variants. We believe that its antiviral activity, in combination with other antivirals, may help improve cure rates and/or reduce treatment times for appropriate patients. We are especially pleased to collaborate with TREKtx and support their commitment to delivering novel and accessible combination HCV treatments to patients worldwide.”
“We are very excited about dosing our first genotype 4 patients in this combination study. If safety and efficacy are demonstrated, the goal is to initiate clinical trials in Egyptnext year, where the need is enormous,” said Dr. Robert Hindes, Chief Medical Officer of Trek Therapeutics.
About TD-6450
Theravance Biopharma discovered TD-6450, a multivalent NS5A inhibitor designed to have improved antiviral activity against genotype 1 resistance-associated variants (RAV) resistant to first generation NS5A inhibitors. TD-6450 has successfully completed Phase 1 studies in both healthy volunteers and HCV patients.
About Faldaprevir
Faldaprevir is a protease inhibitor that TREKtx acquired from Boehringer Ingelheim. FDV has completed Phase 3 studies in combination with pegylated interferon and RBV.
About HCV
Hepatitis C is an infectious disease of the liver. Of people infected, 55 to 85 percent will develop chronic infection, and 75 percent of those with chronic infection will develop chronic liver disease.
The U.S. Centers for Disease Control and Prevention estimates 2.7 million individuals in the United States have active hepatitis C virus (HCV) infection, most of whom are “baby boomers.” In the United States, chronic HCV infection is the leading cause of cirrhosis and liver cancer and the most common reason for liver transplantation. Worldwide, more than 135 million people have chronic HCV infection and most are undiagnosed.
About Trek Therapeutics
TREKtx is a private, clinical stage public benefit corporation developing treatments for serious infections. Its mission is to profitably develop affordable and accessible medicines to treat infectious diseases and to commercialize them for global populations. The company’s founders collectively participated in the development of seven approved antiviral drugs.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2008021927A2 * | 9 Aug 2007 | 21 Feb 2008 | Squibb Bristol Myers Co | Hepatitis c virus inhibitors |
| WO2010094977A1 * | 22 Feb 2010 | 26 Aug 2010 | Arrow Therapeutics Limited | Novel biphenyl compounds useful for the treatment of hepatitis c |
| US201161492267 |
| WO2013067267A1 * | 2 Nov 2012 | 10 May 2013 | Theravance, Inc. | Rod -like hepatitis c virus inhibitors containing the fragement {2- [4- (bi phenyl – 4 – yl) – 1h – imidazo – 2 – yl] pyrrolidine – 1 – carbonlymethyl} amine |
| WO2013163270A1 * | 24 Apr 2013 | 31 Oct 2013 | Theravance, Inc. | Hepatitis c virus inhibitors |
| WO2013165796A1 * | 25 Apr 2013 | 7 Nov 2013 | Theravance, Inc. | Crystalline form of a pyridyl-piperazinyl hepatitis c virus inhibitor |
//////////////
SOFOSBUVIR HPLC


SEE http://www.google.co.in/patents/US20100298257
Example 28
-
Chemical Purity Determination by HPLC
-
Various HPLC conditions can be used to determine the chemical purity of the compounds disclosed herein. One such example is disclosed above in relation to the thermodynamic aqueous solubility studies. Another example is disclosed below.
-
HPLC Conditions:
-
- LC: Waters Alliance 2695 Separations Module, Waters 2996 PDA detector and Waters Empower 2 Software (Version 6.00)
- Column: Phenomenex Luna C18(2); 4.6×50 mm; 3 μm
- Flow rate: 1.2 mL/min
- Injection Volume: 10 μL
- Mobile phase: Solvent A: 95% Water with 5% Methanol and 10 mM Ammonium Acetate; pH˜5.3
- Gradient Solvent B: MeOH with 10 mM Ammonium Acetate hold at 0% B 3 min
- 0-47% B 3-4 min
- hold at 47% B 4-10 min
- 47%-74% B 10-11 min
- hold at 74% B 11-13.5 min
- return to 0% B 13.5-13.6 min
- hold at 0% B 13.6-15.5 min
-
Under these conditions, the purity of 4, RP-4, and SP-4 was determined to be ˜99.6, ˜99%, and ˜99.5%, respectively. It is noted that higher purities can be realized by optimizing the methods disclosed above.
-
Inspection of the XRPD diffractograms shows that the two crystalline single diastereoisomers gave clearly different XRPD patterns. Additionally, there was a clear difference in the melting point of the two crystalline diastereoisomers, with RP-4 having a considerably higher onset than SP-4 (136° C. vs. 94° C.).
-
Example 29Additional Separation Methods
-
The following SFC separation (conditions listed below) yielded adequate separation of a mixture of the diastereomers, RP-4 and SP-4.
-
Preparative Method: Analytical Method: Chiralpak AS-H (2 × 25 cm) SN# 07-8656 Chiralpak AS-H (25 × 0.46 cm) 20% methanol/CO2 (100 bar) 20% methanol/CO2 (100 bar) 50 ml/min, 220 nm. 3 ml/min, 220 nm. Conc.: 260 mg/30 ml methanol, inj vol.: 1.5 ml -
The following SFC separation (conditions listed below) yielded adequate separation of a mixture of the diastereomers, RP-4 and SP-4.
-
Preparative Method: Analytical Method: Chiralpak IA(2 × 15 cm) 802091 Chiralpak IA(15 × 0.46 cm) 30% isopropanol(0.1% DEA)/CO2, 40% methanol(DEA)/CO2, 100 bar 100 bar 60 mL/min, 220 nm. 3 mL/min, 220 nm. inj vol.: 2 mL, 20 mg/mL methanol -
TABLE 16 Summary of results from the batch characterization of RP-4, 4, and SP-4. Analysis RP-4 4 SP-4 Proton NMR Single diastereoisomer 1:1 Mixture of Single diastereoisomer diastereoisomers XRPD Crystalline – different Amorphous Crystalline – different DSC from SP-4 Endotherm; 59° C. from RP-4 Endotherm; melt – 136° C. Endotherm; melt – 94° C. TGA No wt loss, No wt loss, decomposition No wt loss, decomposition >240° C. >240° C. decomposition >240° C. IR See above See above See above Aq Solubility 1.58 6.11 5.65 (mg · ml−1) HPLC Purity 96.9% 99.6% 99.5% 40° C./75% RH No form change Deliquescence inside 1.5 h Deliquescence inside 4.5 h 25° C./53% RH — Deliquescence No form change GVS Non-hygroscopic up to 90% — Non-hygroscopic up to 60% RH RH
- Example 27Thermodynamic Aqueous Solubility
-
Aqueous solubility was determined by suspending a sufficient amount of compound in water to give a maximum final concentration of ≧10 mg.ml−1 of the parent free-form of the compound. The suspension was equilibrated at 25° C. for 24 hours then the pH was measured. The suspension was then filtered through a glass fiber C filter into a 96 well plate. The filtrate was then diluted by a factor of 101. Quantitation was by HPLC with reference to a standard solution of approximately 0.1 mg.ml−1 in DMSO. Different volumes of the standard, diluted and undiluted sample solutions were injected. The solubility was calculated using the peak areas determined by integration of the peak found at the same retention time as the principal peak in the standard injection.
-
TABLE 14 HPLC Method Parameters for Solubility Measurements Type of method: Reverse phase with gradient elution Column: Phenomenex Luna, C18 (2) 5 μm 50 × 4.6 mm Column Temperature 25 (° C.): Standard Injections (μl): 1, 2, 3, 5, 7, 10 Test Injections (μl): 1, 2, 3, 10, 20, 50 Detection: 260, 80 Wavelength, Bandwidth (nm): Flow Rate (ml · min−1): 2 Phase A: 0.1% TFA in water Phase B: 0.085% TFA in acetonitrile Time (min) % Phase A % Phase B Timetable: 0.0 95 5 1.0 80 20 2.3 5 95 3.3 5 95 3.5 95 5 4.4 95 5 -
[0306]Analysis was performed under the above-noted conditions on an Agilent HP1100 series system equipped with a diode array detector and using ChemStation software vB.02.01-SR1.
-
TABLE 15 Aqueous solubility result for RP-4, 4, and SP-4. pH of Unfiltered Sample ID mixture Solubility/mg · ml−1 Comments RP-4 7.12 1.58 Suspension 4 7.03 6.11 Residual solid SP-4 6.88 5.65 Residual solid

FIG 1
Chemical structures of RBV, BOC, TVR, and VRT-127394. Shown are the chemical structures of the anti-HCV drugs RBV {1-[(2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1,2,4-triazole-3-carboxamide)} (A), BOC {(1R,2S,5S)-N-(4-amino-1-cyclobutyl-3,4-dioxobutan-2-yl)-3-[(2S)-2(tertbutylcarbamoylamino)-3,3-dimethylbutanoyl]-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2-carboxamide} (B), TVR {(3S,3aS,6aR)-2-[(2S)-2-[[(2S)-2-cyclohexyl-2-(pyrazine-2-carbonylamino)acetyl]amino]-3,3-dimethylbutanoyl]-N-[(3S)-1-(cyclopropylamino)-1, 2-dioxohexan-3-yl]-3,3a,4,5,6,6a-hexahydro-1H-cyclopenta[c]pyrrole-3-carboxamide} (C), and VRT-127394 (R diastereoisomer of TVR) (D).
Blank plasma samples used for matrix effect (ME) assessment and for the preparation of calibration and control samples were obtained from citrated blood (1,850 × g, 10 min, +4°C, Beckman J6B centrifuge) collected from Vaquez disease patients on the occasion of their regular phlebotomy.
The blank plasma used for the preparation of the calibration and quality control (QC) samples was acidified with 10% FA (50 μl of 10% FA added to 950 μl of plasma). The acidification of plasma aims at preventing the conversion of TVR to its epimer VRT-127394 that occurs in vivo and in vitro. (Tibotec-Janssen, personal communication).
Equipment.The LC system used consisted of Rheos Allegro quaternary pumps equipped with an online degasser and an HTS PAL autosampler (CTC Analytics AG, Zwingen, Switzerland) controlled by Janeiro-CNS 1.1 software (Flux Instruments AG, Thermo Fischer Scientific Inc., Waltham, MA). Separations were done on a Hypercarb 3-μm column (2.1 mm ID by 100 mm; Thermo Fischer Scientific) placed in a column oven thermostat regulated at +80°C (HotDog 5090; ProLab GmbH, Reinach, Switzerland). The chromatographic system was coupled to a triple-stage quadrupole quantum mass spectrometer (Thermo Fischer Scientific) equipped with an electrospray ionization (ESI) Ion Max interface and operated with the Xcalibur software package (version 2.0; Thermo Fischer Scientific).
READ
http://www.us.edu.pl/uniwersytet/jednostki/wydzialy/chemia/acta/ac14/zrodla/14_AC14.pdf
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2291777/
UPDATE………….DEC2015
SOFOSBUVIR
NEW PATENT WO2015188782,
(WO2015188782) METHOD FOR PREPARING SOFOSBUVIR
CHIA TAI TIANQING PHARMACEUTICAL GROUP CO., LTD [CN/CN]; No. 8 Julong North Rd., Xinpu District Lianyungang, Jiangsu 222006 (CN)
Sofosbuvir synthesis routes currently used include the following two methods:
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015188782&redirectedID=true

![]()
//////
////
AN-2718, a New Borole in the pipeline
AN-2718,
2,1-Benzoxaborole, 5-chloro-1,3-dihydro-1-hydroxy-
5-chloro-1,3-dihydro-l-hydroxy-2,1- benzoxaborole
5-Chloro-2,1-benzoxaborol-1(3H)-ol
CAS 174672-06-1
UNII: 810U6C2DGG
MW 168.3864, MF C7 H6 B Cl O2
MP…150-154 °C [WO 9533754]
MP 142-144 °C [ jmc 2006, 49(15) 447]
M.p. 147- 149 °C. WO2013050591
Anacor Pharmaceuticals Inc, INNOVATOR
Onychomycosis is a disease of the nail caused by yeast, dermatophytes, or other molds, and represents approximately 50% of all nail disorders. Toenail infection accounts for approximately 80% of onychomycosis incidence, while fingernails are affected in about 20% of the cases. Dermatophytes are the most frequent cause of nail plate invasion, particularly in toenail onychomycosis. Onychomycosis caused by a dermatophyte is termed Tinea unguium. Trichophyton rubrum is by far the most frequently isolated dermatophyte, followed by T. mentagrophytes. Distal subungual onychomycosis is the most common presentation of tinea unguium, with the main site of entry through the hyponychium (the thickened epidermis underneath the free distal end of a nail) progressing in time to involve the nail bed and the nail plate. Discoloration, onycholysis, and accumulation of subungual debris and nail plate dystrophy characterize the disease. The disease adversely affects the quality of life of its victims, with subject complaints ranging from unsightly nails and discomfort with footwear, to more serious complications including secondary bacterial infections.
Many methods are known for the treatment of fungal infections, including the oral and topical use of antibiotics (e.g., nystatin and amphotericin B), imidazole anti-fungal agents such as miconazole, clotrimazole, fluconazole, econazole and sulconazole, and non-imidazole fungal agents such as the allylamine derivatives terbinafme and naftifϊne, and the benzylamine butenafine. However, onychomycosis has proven to be resistant to most treatments. Nail fungal infections reside in an area difficult to access by conventional topical treatment and anti-fungal drugs cannot readily penetrate the nail plate to reach the infection sites under the nail. Therefore, onychomycosis has traditionally been treated by oral administration of anti-fungal drugs; however, clearly this is undesirable due to the potential for side effects of such drugs, in particular those caused by the more potent anti-fungal drugs such as itraconazole and ketoconazole. An alternative method of treatment of onychomycosis is by removal of the nail before treating with a topically active anti-fungal agent; such a method of treatment is equally undesirable. Systemic antimycotic agents require prolonged use and have the potential for significant side effects. Topical agents have usually been of little benefit, primarily because of poor penetration of the anti-fungal agents into and through the nail mass.
- 51. Hui X, Baker SJ, Wester RC, In Vitro penetration of a novel oxaborole antifungal (AN2690) into the human nail plate. J Pharm Sci 2007;96(10):2622-31 [CrossRef], [PubMed], [Web of Science ®]
- 55. Mao W, Seiradake E, Crepin T, AN2718 has Broad Spectrum Antifungal Activity Necessary for the Topical Treatment of Skin and Nail Fungal Infections. J Am Acad Dermatol 2007:56(2 Suppl):AB124
- 56. ClinicalTrials.gov. Cumulative Irritation Test. Available from: http://clinicaltrials.gov/ct2/show/NCT00781664 [Cited 11 December 2011]
SYNTHESIS

Reduction of 2-bromo-5-chlorobenzoic acid with BH3/THF in THF gives 2-bromo-5-chlorobenzyl alcohol , which is protected as the methoxymethyl derivative by treatment with MOM-Cl in the presence of DIEA in CH2Cl2. Metalation and boronylation of aryl bromide either with t-BuLi or BuLi (1,2,3,4) and (i-PrO)3B (4) or B(OMe)3 in THF affords the title compound .
Alternatively, reaction of 3-chlorobenzaldehyde with p-toluenesulfonylhydrazide in MeOH provides tosyl hydrazone which undergoes thermal decomposition in the presence of BBr3 and catalytic FeCl3 in refluxing CH2Cl2, followed by heating with aqueous NaOH to produce the title oxaborole compound .
You can construct from this…………….
 OH A
OH A
(2-bromo-5-chloro-phenyl)methanol (A)
 B
B
l-bromo-4-chloro-2-(methoxymethoxymethyl)benzene (B)
 1
1
5-chloro-l-hydroxy-3H-2,l-benzoxaborole (1)
PATENT
WO 9533754
https://www.google.com/patents/WO1995033754A1?cl=en
Example 1 Preparation of 5-chloro-1,3-dihydro-l-hydroxy-2,1- benzoxaborole (Method B).
a)
Preparation of 3-chlorobenzaldehyde tosyl hydrazide
A solution of 3-chlorobenzaldehyde (15.56 parts; 0.109M;
Aldrich) in methylated spirits (40 ml) was added slowly at below 10°C to a stirred suspension of p-toluene-sulphonylhydrazide (20.7 parts;
0.108M) in methylated spirits (150 ml). The reaction mass was then stirred at 20 to 25°C for 1 hour and then heated at 60-70°C for 1M hours when the reactants and products dissolved. The solvent was then removed by rotary evaporation and the product was obtained as a solid which was slurried with ether and washed with n-hexane. Yield = 27.2 parts (81.5% theory) mpt 122-3°C. Elemental analysis
Theory 54.5%C; 4.2%H; 9.1%N
Found 54.5%C; 4.3%H; 9.1%N
Proton NMR (CDCl3:ppm)
8.5, s, 1H(-NH-); 7.9, d, 2H(Tosyl aromatic); 7.7,s, 1H(CH=N); 7.5, s, 1H (aromatic); 7.2-7.4, m, 5H(Tosyl aromatic); 2.3, s, 3H(-CH3) b) Preparation of title compound
A suspension of anhydrous ferric chloride catalyst (0.75 parts, Fisons) in dry dichloromethane (20 ml) was added at 20 to 25°C simultaneously with boron tribromide (25 parts, 0.1M, Aldrich) in dry dichloromethane (100 mis) to a stirred suspension of the hydrazide from a) above (10.18 part, 0.033M) in dry dichloromethane (160 mis) under a nitrogen blanket. The reactants were then stirred under reflux and the evolved hydrogen bromide trapped under aqueous sodium hydroxide. After 3 hours stirring at reflux, the reactants were allowed to stand at 20- 25°C for 48 hours and then stirred under reflux for a further 4 hours. The reaction mass was then cooled and the solvent removed by rotary evaporation. The solid obtained was then stirred under reflux with 2N sodium hydroxide solution (160 ml) for 3 hours. The brown aqueous suspension was extracted with dichloromethane (50 ml), screened and then acidified to about pH 2 by addition of 2N hydrochloric acid. The solid was filtered, slurried with dichloromethane (400 ml) and then washed with a saturated solution of sodium bicarbonate followed by water.
Yield = 24 parts (43% theory). The solid was slurried in hot
dichloromethane and filtered to give 0.36 parts oxaborole mp 140-45°C. The dichloromethane solution was cooled and the solid filtered giving a further 0.35 parts oxaborole mp 146-8°C. The solids were combined and recrystallised from methylated spirits.
Yield = 0.51 parts (9.2% theory) mp 150-4°C.
Elemental Analysis
Theory 49.8%C, 3.5%H, 21.06%C1
Found 49.5%C, 3.5%H, 21.0%C1
Proton NMR (CDCl3) ppm
9.3, s, 1H(-OH); 7.5, d, s, d, 3H(aromatic);
5.0, s, 2H(-CH2-O).
PATENT
WO 2006089067
http://www.google.co.in/patents/WO2006089067A2?cl=en

Analytical data for exemplary compounds of structure I are provided below.
4.2. a 5-Chloro-1.3-dihydro-l -hvdroxy-2J-benzoxaborole (Cl) M.ρ. 142-15O0C. MS (ESI): m/z = 169 (M+l, positive) and 167 (M-I, negative). HPLC (220 nm): 99% purity. 1H NMR (300 MHz, DMSO-d6): δ 9.30 (s, IH), 7.71 (d, J = 7.8 Hz, IH), 7.49 (s, IH), 7.38 (d, J = 7.8 Hz, IH) and 4.96 (s, 2H) ppm.
PAPER
http://pubs.acs.org/doi/abs/10.1021/jm0603724?source=chemport
COMPD IS 19d
5-Chloro-1,3-dihydro-1-hydroxy-2,1-benzoxaborole (19d) This compound was made from 18d in the same manner as compound 19b (triturated with hexane, 75% yield):: mp 142-144°C. 1 H NMR (300MHz, DMSO-d6) δ (ppm) 4.96 (s, 2H), 7.38 (d, J = 7.8 Hz, 1H), 7.49 (s, 1H), 7.71 (d, J = 7.8 Hz, 1H), 9.30 (s, 1H); ESI-MS m/z 167 (M-H)- ; HPLC purity 99.0%; Anal (C7H6BClO2 ⋅ 0.1H2O) C, H
precursor 18d
2-Bromo-5-chloro-1-(methoxymethoxymethyl)benzene (18d) To a solution of 2-bromo-5-chlorobenzoic acid (5.49 g, 23.3 mmol) in anhydrous THF (70 mL) under nitrogen was added dropwise a BH3 THF solution (1.0 M, 55 mL) at 0o C and the reaction mixture was stirred overnight at room temperature. Then the mixture was cooled on an ice bath and MeOH (20 mL) was added dropwise to decompose excess BH3. The resulting mixture was stirred until no bubble was released and then 10% NaOH (10 mL) was added. The mixture was concentrated and the residue was S6 mixed with water (200 mL) and extracted with EtOAc. The residue from rotary evaporation was purified by silica gel column chromatography (5:1 hexane/EtOAc) to give 2-bromo-5-chlorobenzyl alcohol as a white solid (4.58 g, 88%): 1 H NMR (300 MHz, DMSO-d6): δ (ppm) 7.57 (d, J = 8.7 Hz, 1H), 7.50-7.49 (m, 1H), 7.28-7.24 (m, 1H), 5.59 (t, J = 6.0 Hz, 1H), 4.46 (d, J = 6.0 Hz, 2H). 2-Bromo-5-chlorobenzyl alcohol obtained above was dissolved in CH2Cl2 (150 mL) and cooled to 0o C on an ice bath. To this solution under nitrogen were added in sequence i-Pr2NEt (5.4 mL, 31 mmol) and chloromethyl methyl ether (2.0 mL, 26 mmol). The reaction mixture was stirred overnight at room temperature and washed with NaHCO3-saturated water and then brine. The residue after rotary evaporation was purified by silica gel column chromatography (5:1 hexane/EtOAc) to give 18d (4.67 g, 85%) as a colorless oil: 1 H NMR (300 MHz, DMSO-d6): δ (ppm) 3.30 (s, 3H), 4.53 (s, 2H), 4.71 (s, 2H), 7.32 (dd, J = 8.4, 2.4 Hz, 1H), 7.50 (dd, J = 2.4, 0.6 Hz, 1H), 7.63 (d, J = 8.7 Hz, 1H).
PATENT
http://www.google.com/patents/WO2013050591A2?cl=en
EXAMPLES Example 1 : Preparation of 5-chloro-l-hydroxy-3H-2,l-benzoxaborole (1)
Step 1 : Preparation of (2-bromo-5-chloro-phenyl)methanol (A)
A solution of borane-tetrahydrofuran complex in THF (0.15 L, 1.5 eq) was added dropwise to a solution of 2-bromo-5-chlorobenzoic acid (24 g) in anhydrous tetrahydrofuran (0.24 L) at 0°C and under argon atmosphere. The reaction mixture was stirred at room temperature for 16 h, before being slowly poured onto 0.10 L of a 2N aqueous solution of hydrogen chloride at 0°C. The mixture was stirred for 15 minutes and the volatiles were removed under reduced pressure. The aqueous layer was extracted with ethyl acetate and the combined organic layers were washed with a IN aqueous solution of sodium hydroxide and then water. After drying over sodium sulfate, filtration and concentration under reduced pressure, the crude product was purified by column chromatography; 23.2 g; M.p. 79-80 °C.
Step 2: Preparation of l-bromo-4-chloro-2-(methoxymethoxymethyl)benzene (B)
(2-bromo-5-chloro-phenyl)methanol (A, 12 g) was dissolved in dichloromethane (0.35 mL) and cooled to 0 °C. Under argon atmosphere, diisopropylethylamine (14 mL, 1.5 eq) and chloromethyl methyl ether (5.0 mL, 1.2 eq) were added. After 1 night of stirring at room temperature, the crude reaction mixture was washed with a saturated solution of sodium hydrogen carbonate, dried over sodium sulfate and evaporated under reduced pressure. Purification by column chromatography afforded 10.5 g of l-bromo-4-chloro-2- (methoxymethoxymethyl)benzene (B) as an oil.
Step 3: Preparation of 5-chloro-l-hydroxy-3H-2,l-benzoxaborole (1)
To a solution of (B) (6.0 g) in anhydrous tetrahydrofuran (120 mL) at -78°C was added dropwise a solution of butyllithium in hexane (15.6 mL, 1.1 eq). To the resulting yellow- brown solution trimethyl borate (2.5 mL, 1.0 eq) was injected in one portion and the cooling bath was removed. The mixture was warmed gradually for 30 minutes. After stirring at room temperature for 2 hours, 8.0 ml of a 6N aqueous solution of hydrogen chloride were added and the reaction mixture was left stirring overnight at room temperature. Evaporation of the volatiles gave a residue which was taken up in ethyl acetate, washed with water, brine, dried over sodium sulfate and then evaporated. The crude product was crystallized from ethyl acetate to give 1.4 g of 5-chloro-l-hydroxy-3H-2,l-benzoxaborole (1) as a white powder. Purification of the filtrate by column chromatography afforded 1.2 g more of 1. M.p. 147- 149 °C.
PATENT
http://www.google.fr/patents/WO2010110400A1?cl=en&hl=fr
Reference Example 187
5-chloro-2,1-benzoxazine ball roll -1 (3H) – All

5-chloro-2- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) benzaldehyde 8.50g synthesized in Reference Example 185 was dissolved in methanol 100ml, borohydride Sodium 2.40g was added, and after stirring for 30 minutes at room temperature and stirred for 2 hours at 60 ℃. The reaction solution was concentrated, and the organic layer after the layers were separated with ethyl acetate and saturated aqueous ammonium chloride and then concentrated under reduced pressure. The residue was added 100ml of tetrahydrofuran, and 6N hydrochloric acid 60ml, and stirred for 8 hours at room temperature.After the reaction mixture was dried and the organic layer was extracted with ethyl acetate, and concentrated. The residue was purified by silica gel column chromatography to give the title compound 9.6g. 1 H-NMR (400MHz, DMSO-d 6) δ: 4.95 (2H, s), 7.36 (1H, dd, J = 8.0,1.6Hz), 7.47 (1H, s) , 7.70 (1H, d, J = 8.0Hz), 9.28 (1H, s).
PATENT
http://www.google.com/patents/WO2008070257A2?cl=en
5-Fluoro-l,3-dihvdro-l-hvdroxy-2, 1-benzoxaborole (Cl)
1-Hydroxy-dihydrobenzoxaboroles, such as Cl, were synthesized as shown in Scheme 1. The protected o-bromobenzyl alcohol derivative (18), prepared from 16 or 17, was converted into the corresponding phenyl boronic acid. Deprotection of the methoxymethyl ether using hydrochloric acid followed by spontaneous cyclization gave the target compounds.
Scheme 7

Conditions (a) NaBH4, MeOH, rt (when X = H ), or BH3-THF, THF, rt (when X = OH), (b) MeOCH2CI, /-Pr2NEt, CH2CI2, rt, (c) MeMgBr, THF, -78 0C to rt , (d) NBS, AIBN, CCI4, reflux, (e) NaOAc, DM F, 70 0C, (f) NaOH, MeOH, reflux, (g) n-BuLι, (/-PrO)3B, THF, -780C to rt, (h) 6N HCI, THF, rt
References
- Hui, Xiaoying; Journal of Pharmaceutical Sciences 2007, 96(10), Pg2622-2631
- Baker, Stephen J.; Journal of Medicinal Chemistry 2006, 49(15), Pg4447-4450
- Austin, Peter William; WO 9533754 A1 1995 CAPLUS
| US5880188 * | 26 May 1995 | 9 Mar 1999 | Zeneca Limited | Oxaboroles and salts thereof, and their use as biocides |
| US6083903 * | 16 May 1995 | 4 Jul 2000 | Leukosite, Inc. | Boronic ester and acid compounds, synthesis and uses |
| WO2005013892A2 | 15 Jun 2004 | 17 Feb 2005 | Tsutomu Akama | Hydrolytically-resistant boron-containing therapeutics and methods of use |
| Reference | ||
|---|---|---|
| 1 | * | Austin et al., 1996, CAS: 124:234024. |
| 2 | * | fungicide: definition from Answre.com, 1998. |
| 3 | S. J. Baker, et al., “Progress on New Therapeutics for Fungal Nail Infections,“Annual Reports in Medicinal Chemistry, 40:323-335 (2005). | |
| 4 | Sudaxshina Murdan, “Drug Delivery to the Nail Following Topical Application,” International Journal of Pharmaceutics, 236:1-26 (2002). | |
see full series on boroles
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
do not miss out
//////////AN-2718, Borole, PHASE 2
B1(c2ccc(cc2CO1)Cl)O
FDA Says Chinese Pfizer Plant Hid Failures, Used Old Ingredients
DRUG REGULATORY AFFAIRS INTERNATIONAL


October 31, 2015 — 2:22 AM IST,
The Pfizer Ltd. research and development plant in Dalian, China.
Bernardo De Niz/Bloomberg
A Pfizer Inc. plant in China that was being inspected by Food and Drug Administration regulators in order to ship drugs to the U.S. kept a second set of quality and manufacturing records that didn’t match official ones, according to an FDA review of the facility.
read
![]()
/////
